
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 17 March 2025
Sec. Autoimmune and Autoinflammatory Disorders : Autoimmune Disorders
Volume 16 - 2025 | https://doi.org/10.3389/fimmu.2025.1537428
 Marvin Tigges1
Marvin Tigges1 Sören Dräger2
Sören Dräger2 Ilaria Piccini1
Ilaria Piccini1 Katja Bieber3
Katja Bieber3 Artem Vorobyev2
Artem Vorobyev2 Janin Edelkamp1Marta Bertolini1*
Janin Edelkamp1Marta Bertolini1* Ralf J. Ludwig1,2,3*
Ralf J. Ludwig1,2,3*Pemphigoid diseases constitute a group of organ-specific autoimmune diseases characterized and caused by autoantibodies targeting autoantigens expressed in the skin and mucous membranes. Current therapeutic options are still based on unspecific immunosuppression that is associated with severe adverse events. Biologics, targeting the IL4-pathway or IgE are expected to change the treatment landscape of pemphigoid diseases. However, clinical studies demonstrated that targeting these pathways alone is most likely not sufficient to meet patient and healthcare partitioners expectations. Hence, model systems are needed to identify and validate novel therapeutic targets in pemphigoid diseases. These include pre-clinical animal models, in vitro and ex vivo model systems, hypothesis-driven drug repurposing, as well as exploitation of real-world-data. In this review, we will highlight the medical need for pemphigoid diseases, and in-depth discuss the advantages and disadvantages of the available pemphigoid disease model systems. Ultimately, we discuss how rapid translation can be achieved for the benefit of the patients.
Pemphigoid diseases (PD) constitute a group of rare autoimmune skin disorders. Based on target antigen, autoantibody isotype and clinical presentation, seven PD subtypes are differentiated (1). Of note, despite recent advances (2) there are still unrecognized autoantigens in a minority of PD patients (3, 4). PD are characterized by autoantibodies targeting components of the dermal-epidermal junction, resulting in split formation and inflammation. This process typically necessitates the activation of myeloid cells, but events triggered by target engagement of autoantibodies have also been directly linked to subepidermal blister formation (Figure 1). The subepidermal split formation is characteristic for PD and differentiates them from other autoimmune bullous dermatoses (AIBD) (5). In this section, we provide a brief introduction into each PD and highlight the specific unmet medical need.
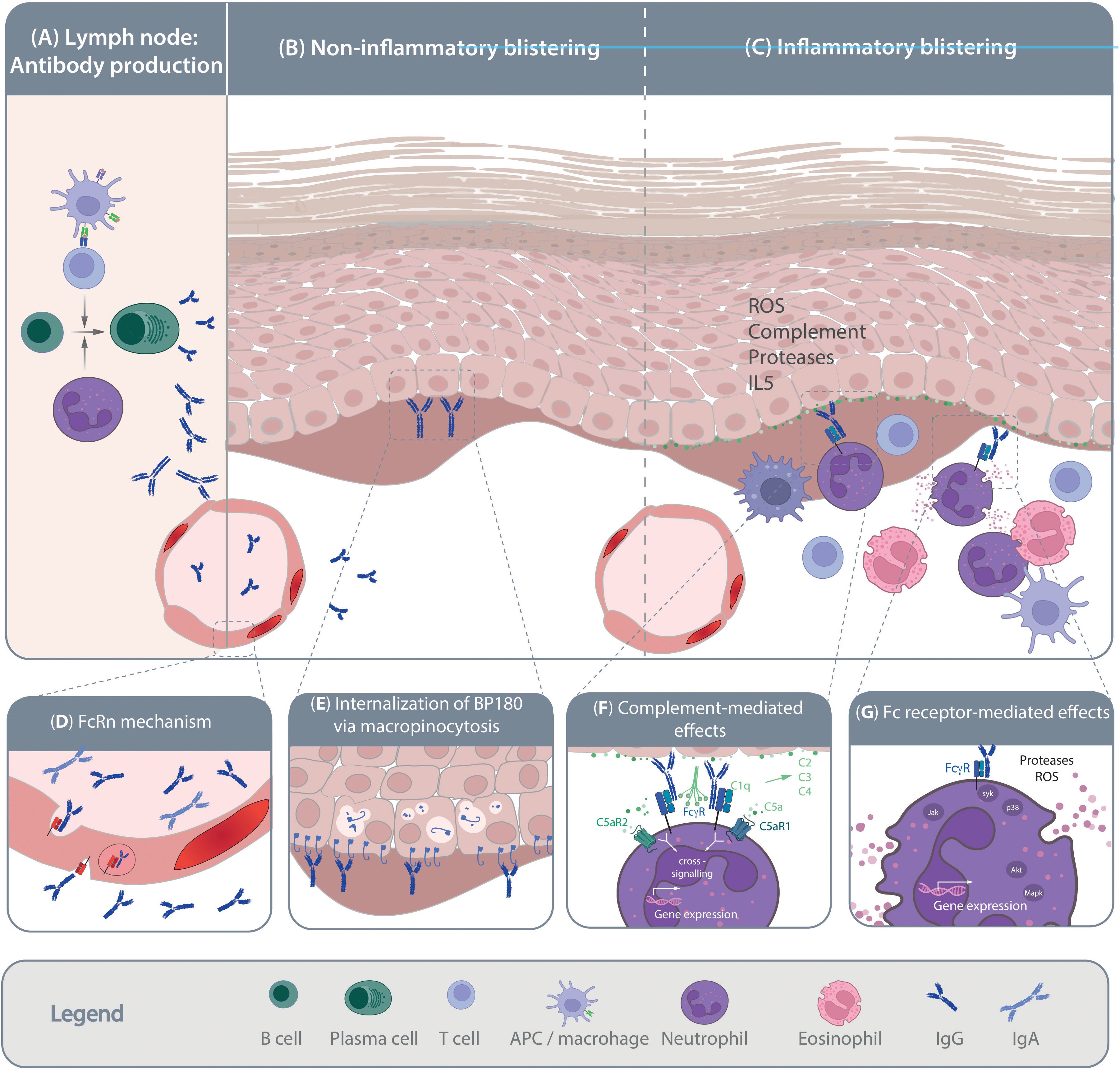
Figure 1. Pathogenesis of pemphigoid diseases. Schematic of the current understanding of pemphigoid disease pathogenesis. (A) Loss of tolerance leads to the generation of autoantigen-specific B cells. Activation, proliferation and maturation of B cells occurs in a T cell-depended manner and is further promoted by the presence of neutrophils. Mechanisms leading to the shift from the production of non-pathogenic towards pathogenic autoantibodies are so far poorly understood. Pathogenic autoantibodies are then released into the circulation and reach the skin through the vasculature. Autoantibody binding to the target antigens in the skin and/or mucous membranes can trigger blister formation through (B) non-inflammatory and/or (C) inflammatory mechanisms. Non-inflammatory blistering antibody binding to the target antigen, specifically BP180, induces the internalization of BP180, leading to destabilization of keratinocyte adherence to the basement-membrane. Inflammatory mechanism leading to blister formation in pemphigoid disease is relatively well-understood. These include release of pro-inflammatory mediators from the targeted cells and immune cells, which facilitates myeloid cell migration into the skin. Within the skin, myeloid cells bind to the tissue bound immune complexes and mediate blistering trough protease- and reactive oxygen species (ROS) release. The inserts (D-H) provide some more details on the highlighted pathogenic events. (D) Within the circulation the half-life of IgG autoantibodies is prolonged by the neonatal Fc receptor (FcRn). (E) After binding of anti-BP180 antibodies, BP180 is internalized through micropinocytosis. (F) Complement, especially C5 is release from both keratinocytes and myeloid cells. Cleavage of C5 into C5a recruits and activates myeloid cells to the site of autoantibody deposits in the skin that is mediated through both C5aR1 and C5aR2. (G) Ultimately, myeloid cells engage the tissue-bound immune complexes in a Fc gamma receptor-mediated fashion, leading to protease- and ROS-release. These processes are mediated by downstream intracellular signaling following Fc gamma receptor engagement.
Bullous pemphigoid (BP) is the most common pemphigoid disease, with an incidence of 20 cases per million in Germany (6). However, this incidence varies globally and is increasing over time (7). A significant factor contributing to this rise is the aging population and the use of medications that can trigger BP (8). BP predominantly manifests in individuals in their late 70s. In those aged over 80 years, the incidence surges to 150-330 cases per million per year (1, 9–14). The clinical presentation of BP is heterogenous, ranging from pruritus without evidence of skin lesions to dense blistering on inflamed skin (15) and is characterized by the autoimmune response targeting two structural components of hemidesmosomes, namely BP180 (type XVII collagen, COL17) and BP230. These proteins serve to link the cytoskeleton of basal keratinocytes to structures in the papillary dermis (16). Antibodies are usually of the immunoglobulin (Ig)G subclass, but IgA- and other subclasses have also been documented to cause BP (16). Autoantibodies in PD develop in a T-cell dependent B cell response (17). As outlined in Figure 1, binding of autoantibodies to their target epitopes initiates pathology through inflammation- dependent and -independent mechanisms (18, 19). Diagnosis of BP is based on clinical presentation and histopathological analysis. The diagnosis is confirmed by detection of Ig or complement C3 deposition at the dermal-epidermal junction using direct immunofluorescence (DIF), and the detection of circulating BP180 and/or BP230 autoantibodies (20). Treatment of BP centers on topical or systemic corticosteroid treatment and/or systemic immunosuppression. Corticosteroid treatment is highly effective with over 90% achieved remissions within 4-6 weeks (21–25). The major challenge in BP is maintenance of this initial very effective treatment response. Depending on diseases severity, 30%-50% of the patients experience a relapse within 6 months after the initial diagnosis (26). This, in turn, necessitates prolonged corticosteroid treatment, which contributes to the high morbidity and increased mortality in BP (27, 28).
Mucous membrane pemphigoid (MMP) is defined as a PD with predominant mucosal involvement. Studies estimate its incidence at 2.0 per 1 million people per year in the state of Franconia (Germany) with a prevalence of 24.6 per million people in Germany (1, 11, 29, 30). The disease typically manifests at a mean age of 60 to 65 years (31). MMP primarily affects mucous membranes in the mouth, nose, eyes, anogenital area, pharynx, larynx, and esophagus, but can also involve the skin. The most common affected site is the oral mucosa, followed by eyes and nose (32). Severity varies highly among patients and has a range from subtle lesions to devastating esophageal and conjunctival lesions that are extremely painful and may lead to esophageal strictures and blindness (31, 33, 34). MMP can be caused by autoantibodies targeting several different antigens, including BP180, BP230, laminin 332, COL7, and/or integrin α6β4. Among these, autoantibodies against BP180 are the most common (1, 32, 35, 36). If clinically suspected, MMP diagnosis is confirmed by the detection of immunoglobulin and/or C3 deposits using DIF (1). Of note, in cases where clinical suspicion of MMP is high, but DIF is negative, repeat biopsies should be conducted to increase the sensitivity of DIF microscopy (37). Although sometimes challenging, detecting circulating autoantibodies and determining their specificity should be attempted (30). MMP is among the more challenging pemphigoid diseases to treat, often requiring a combination of immunosuppressants to achieve remission. In MMP with autoantibodies against laminin 332, a 25-30% prevalence of mainly solid tumors has been described (38). The prognosis of MMP varies depending on the organs affected. Overall, mortality in MMP patients is increased 1.7-fold compared to matched controls (28).
Epidermolysis bullosa acquisita (EBA) is an orphan disease with an annual incidence of 0.2.-0.5 new cases per million people (11, 39, 40). EBA can occur at any age, with a mean age at onset of 46.7 years (29, 41–43). Like BP, the clinical presentations varies greatly in EBA patients. The two most common forms are the inflammatory (or non-mechano-bullous) and the mechano-bullous form. The mechano-bullous EBA presents with skin fragility, blisters and erosions on non-inflamed skin or scarred skin and millia formation. These symptoms predominantly occur on trauma-prone areas such as the hands, feet, elbows, and knees (18). Inflammatory EBA presents with profuse skin lesions on inflamed skin, similar to BP, and trauma induced lesions around non-inflamed skin (18, 44–46). Mucosal involvement is common in EBA (34, 47). EBA is caused by IgG or IgA autoantibodies against type VII collagen (COL7), commonly targeting the immunodominant NC1 region (48). In approximately 1/3rd of EBA patients IgA autoantibodies are detected, and in 10% of the patients this is the sole detected immunoglobulin subclass. Similar to BP, binding of these autoantibodies to their target antigen initiates an inflammatory cascade resulting blistering and inflammation. Mechanisms of non-inflammatory blistering in EBA are less well characterized (18). EBA diagnosis is confirmed by linear Ig and/or complement C3 deposits the dermal-epidermal junction of perilesional skin in DIF. Contrasting all other pemphigoid diseases, Ig deposition at the dermal-epidermal junction shows a distinct, so-called u-serrated binding pattern while all other PDs display an n-serrated pattern in DIF microscopy. Indirect IF microscopy on human salt-split skin or ELISA further supports the diagnosis by detecting COL7 autoantibodies or its immunodominant domains (1, 48, 49), but should only be used in addition of serration analysis because in a large proportion of EBA patients circulating autoantibodies cannot be detected (50, 51). If serration analysis is not possible, alternatives are fluorescent overlay antigen mapping or immunoelectron microscopy (18). The treatment of EBA is notably difficult. Typically, achieving remission necessitates extensive immunosuppression (43). On average, a continuous immunosuppressive treatment regimen of nine months is required to achieve remission (52). If remission is achieved, in many cases, continued immunosuppression is needed to maintain the therapeutic effects. Data on mortality in EBA are scant. However, a recent retrospective cohort study found that EBA patients have an approximate 2.5-fold increased risk of mortality compared to matched controls (28).
Anti-p200 pemphigoid is a rare autoimmune blistering disease characterized by subepidermal blisters and erosions primarily on the skin and sometimes on mucosal surfaces. The disease is defined by autoantibodies targeting the 200-kDa protein lamininβ4, a key component of the basement membrane zone (2). Clinically, anti-p200 pemphigoid presents with tense blisters, erythematous plaques, and urticarial lesions, predominantly affecting the hands and feet (53). Diagnosis involves detecting linear IgG deposits along the basement membrane zone via DIF and identifying circulating autoantibodies against laminin β4 (53). The presence of these autoantibodies is critical for differentiating anti-p200 pemphigoid from other similar blistering diseases. Treatment typically includes topical or systemic corticosteroids and immunosuppressive agents to manage inflammation and autoantibody production. The prognosis for patients with anti-p200 pemphigoid varies, with early and aggressive treatment potentially improving outcomes and reducing the risk of complications (53, 54).
Pemphigoid gestationis (PG) is a pregnancy-caused PD with an estimated incidence of 1 in 20,000 to 50,000 pregnancies per year (1, 55, 56). Onset of the disease is typically between the second trimester and the postpartum period (57). PG manifests with pruritus and polymorphic inflammatory skin lesions spreading from the umbilical region to the abdomen and extremities. In most cases, PG resolves after delivery. However, relapses during subsequent pregnancies are common (1, 57). PG is caused by autoantibodies targeting BP180. Similar to other PD, PG is diagnosed by the detection of tissue-bound immunoglobulins and/or complement C3 in DIF. Circulating BP180 antibodies can be detected using indirect IF with salt-split skin as substrate or with specific ELISAs. Topical corticosteroids can treat most cases of PG effectively. Interdisciplinary care involving gynecology is recommended due to the increased risk of adverse embryonic/fetal and maternal pregnancy outcomes associated with PG (58–60). Of note, approximately 10% of newborns exhibit PG-typical skin lesions due to the transplacental transfer of maternal IgG antibodies (61, 62), which may require supportive neonatal care.
Linear IgA disease (LAD) manifests across all age groups, with the highest incidence observed during adolescence, early adulthood, and the sixth decade of life. In pediatric populations, it is the most prevalent autoimmune blistering disorder, with an average onset age of 4.5 years. The annual incidence is estimated to range from 0.2 to 2.3 cases per million individuals (63). Clinically, LAD presents with tense blisters and vesicles, often forming in a characteristic “crown of jewels” or “string of pearls” pattern, particularly in children. Lesions can appear on both the skin and mucous membranes, causing significant discomfort due to associated pruritus (1, 64). LAD is caused by IgA autoantibodies targeting the extracellular 97 kDa portion of BP180 and the 120 kDa ectodomain of BP180, known as LAD-1 (63, 65, 66). Diagnosis of LAD is primarily based on DIF microscopy, which shows linear IgA deposits along the basement membrane zone. Identification of circulating antibodies targeting LAD-1 supports the diagnosis (67). Dapsone is the treatment of choice for LAD, which, in most cases rapidly alleviates itch, inflammation and blister formation. In children, the disease often resolves spontaneously within a few years. In adults, however, LAD may follow a more chronic course, requiring prolonged treatment (68, 69).
Lichen planus pemphigoides (LPP) is an orphan AIBD that combines features of lichen planus (70) and bullous pemphigoid. It is characterized by the presence of lichenoid papules and plaques, which subsequently develop tense blisters. LPP is at least partially caused by autoantibodies targeting BP180. Clinically, LPP presents with intensely pruritic, violaceous, polygonal papules and plaques that are often located on the extremities. The blisters usually develop on skin areas that were previously unaffected by lichen planus lesions. Diagnosis is confirmed through DIF, which shows linear deposits of IgG and/or C3 at the basement membrane zone, and the presence of circulating autoantibodies targeting BP180. The pathogenesis of LPP is incompletely understood. It potentially involves an initial lichenoid inflammation that may promote an autoimmune response against components of the epidermal basement membrane, particularly BP180. This could lead to the formation of BP180-specific autoantibodies that ultimately lead to subepidermal blistering. Treatment options for LPP include systemic corticosteroids, which are often the first line of treatment. LPP usually responds well to treatment, but more definite data on LPP prognosis is scant due to the rarity of the disease (71).
PD impose a significant morbidity and dramatically impair the quality-of-life of the affected patients. Furthermore, PD patients bear an increased mortality risk, e.g., a 1-year mortality ranging from 20% to 40% in BP (13, 14, 28, 72). While PG, LAD, and p200 pemphigoid generally respond favorably to therapeutic interventions, BP, MMP and EBA are notably more challenging to manage (73). These latter PD often relapse and typically require prolonged courses of immunosuppressive therapy to achieve remission (1, 73). Initial treatment of PDs mostly consists of systemically or (in BP) topically applied corticosteroids (27). The administration of systemic corticosteroids, such as prednisone, in elderly patients links to significant adverse effects and an increased risk of mortality (21, 22, 74). Potent topical corticosteroids, such as clobetasol propionate, exhibit fewer side effects compared to systemic corticosteroids. However, their use is limited by corticosteroid-induced skin atrophy and practical challenges, particularly in elderly patients (21, 75). Long term treatment is usually centered on corticosteroid sparing immunosuppressive drugs (73).
Second- and third-line treatments for PD include immunosuppressants, high doses of intravenous immunoglobulins, rituximab, or more recently use of biologics such as dupilumab or omalizumab. For most PD treatment guidelines are available. For detailed insights we refer to those guidelines (23, 76–79). The use of biologics in PD is detailed below when describing drug repurposing to develop novel treatment options for PD.
The study from Lamberts and colleagues (80) revealed that patients, clinicians and researchers agreed that the most urgent need was improvement of therapeutic options for pemphigoid diseases. Furthermore, half of the patients were unsatisfied with patient care during the diagnostic process due to misdiagnosis and long diagnostic delay. PD patients also express the desire for increased disease awareness among healthcare professionals to facilitate more accurate and timely diagnosis. Researchers, on the other hand, seek an increase in clinical trials, a deeper understanding of disease pathophysiology to aid in drug development, and an exploration of trigger factors. Besides unmet patient needs, the increasing incidence of BP and limitations of currently available treatment options, e.g. incomplete response rates, severe side effects and costs underline the need for improvement in the aforementioned areas and the need to keep developing novel, more efficient and specific treatments. Use of pre-clinical models for target identification and -validation is one possibility to improve long-term therapeutic outcomes in PD.
Pre-clinical studies of PDs are mostly reliant on in vivo animal models. These models allow to investigate autoantibody-induced interactions of specific molecules underlying the pathomechanisms and in identifying suitable therapeutic target candidates (81–84). The following section summarizes the available pre-clinical animal models for PDs, their applications, their respective advantages and disadvantages. In principle, three different types of animal models are utilized in PD research (Table 1): (Auto)-antibody transfer models, lymphocyte-transfer models and immunization-induced models. Spontaneous PD, found primarily in dogs and occasionally in other domestic animals, is a rare occurrence and is only rarely used for drug development (83, 85). In addition, the deletion of the NC14A domain in mice leads to the spontaneous loss of tolerance and the mice subsequently develop BP-like symptoms (86).
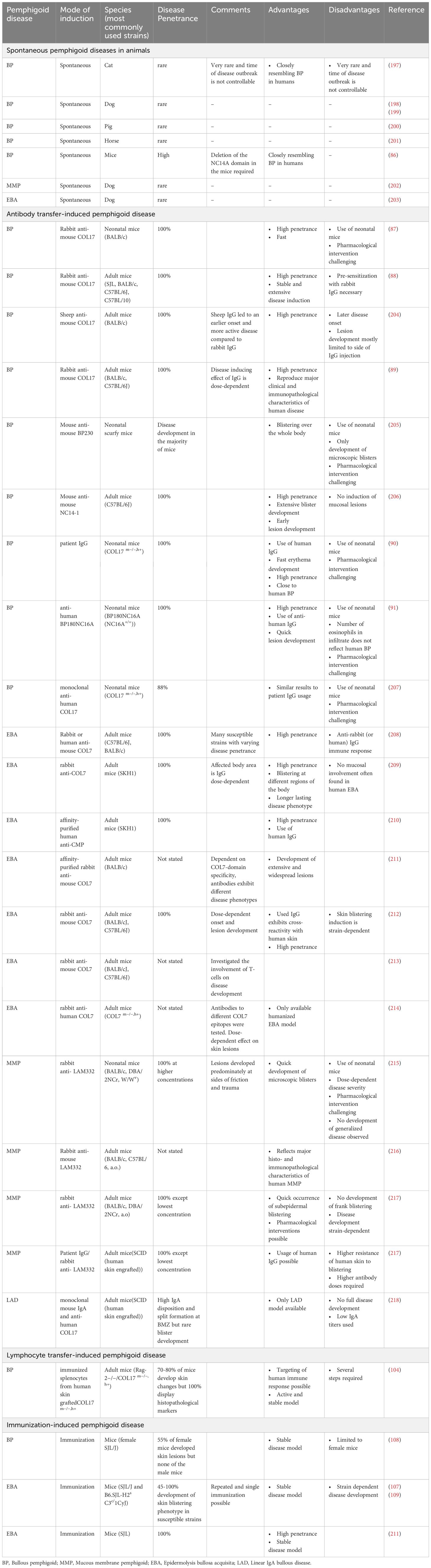
Table 1. Pre-clinical models of pemphigoid disease.
Antibody transfer-induced mouse models have been established for BP, EBA, MMP and LAD (83). The injection of pathogenic (auto)-antibodies into mice induces symptoms duplicating features of the corresponding human disease. In the case of BP, injection of rabbit IgG targeting the murine-COL17 (mCOL17) domain corresponding to the NC16A human-COL17 (hCOL17) domain, have been found to induce dermal-epidermal separation in neonatal wild-type mice as early as 1993 (87). Further development of this model led to the usage of adult mice that can reflect that BP develops in elderly patients (88, 89). Moreover, transgenic mice either carrying hCOL17 or mCOL17 expressing the human NC16A domain have been introduced and develop BP upon injection of human IgG autoantibodies (90, 91). Based on this principle, antibody transfer models have been developed for EBA and MMP (92, 93). These models show a high disease penetrance in most inbred mouse strains (94, 95).
Data from antibody transfer-induced models of EBA have been the basis to obtain orphan designation for dimethyl fumarate and coversin/nomacopan in BP (96–99). The dual complement factor 5 (C5) and leukotriene B4 (LTB4) inhibitor nomacopan was successfully evaluated in a phase 2a nonrandomized controlled trial (100, 101). More recently, again based on data in an antibody transfer model of MMP (101), a patent on the use of CXCL8 inhibitors for the treatment of MMP was filed (102). Collectively, this illustrates the applicability of antibody transfer PD mouse models for clinical translation.
These models are based on the transfer of lymphocytes from an antigen-deficient mouse strain (with or without immunization with the respective antigen) into immunodeficient mice with the expression of the respective antigen (103). A lymphocyte transfer model has been established for BP (Table 1). Here, wild-type mice are immunized with human COL17 by grafting skin from hCOL17 transgenic mice. Transfer of lymphocytes from these mice into immunodeficient mice expressing the hCOL17 transgene in the skin induces a strong anti-hCOL17-specific humoral immune response. These mice develop a clinical phenotype resembling the clinical, histological, and immunopathological features of the human disease (104). This model has mainly been used to unravel the pathogenesis of autoantibody production in BP, for example, the requirement of CD4 T cells (105). Notably, IVIG treatment, a second- or third-line treatment option for BP, is also effective in this model (106). This demonstrates that the lymphocyte transfer model of BP is in principle well suited to identify and validate novel treatment strategies in BP.
Immunization-induced PD model systems have been established for BP and EBA (107–109) (Table 1). Immunization-induced BP shows a low disease penetrance and almost exclusively manifests in female mice. Due to these limitations, this model is not widely used. By contrast, the immunization-induced EBA mouse model based on a single immunization with recombinant mCOL7 (109) induces clinical disease manifestation in 60%-80% of the mice within 4-10 weeks. Mouse strains that are susceptible to immunization-induced EBA are SJL/J and B6.SJL-H2s C3c/1CyJ mice. The latter are C57Bl6/J mice that carry the H2s haplotype from the SJL/J strain. After single immunization, peak disease severity is observed after around 10 weeks, reaching a plateau that is maintained for at least 10 months (109). Based on these characteristics, immunization-induced EBA has been used to evaluate the effects of established and emerging treatments of PD (92, 96, 108, 110, 111).
All murine models duplicate key aspects of human PD. Furthermore, parallel use of antibody transfer-induced models and lymphocyte transfer- or immunization-induced models also allows to disentangle the afferent (loss of tolerance, autoantibody production) from the effector (autoantibody-induced tissue damage) phase. In the antibody transfer-induced models, especially when the antibodies are injected locally into the skin of the ear, clinical symptoms manifest within days following the transfer of pathogenic IgG, allowing for timely analysis. A significant limitation of these models, however, is its restriction to studying only the effector phase of the disease, as symptoms abate within days post-autoantibody transfer. Furthermore, these models are not conducive to examining the mechanisms of tolerance loss or the production of autoantibodies (83). As these models are still almost exclusively based on the transfer of rabbit or human IgG into mice, an antibody response towards rabbit or human IgG limits long-term observations (112). Conversely, lymphocyte transfer- and especially immunization-induced PD mouse models are characterized by a prolonged manifestation of a clinical PD phenotype, enabling the evaluation of long-term interventions and the investigation of factors influencing tolerance loss and autoantibody production. Yet, these models are significantly more time-consuming and resource-intensive compared to the autoantibody transfer models. There are also significant disadvantages associated with both models. Firstly, both models rely on murine signaling pathways and cell interactions, which may not accurately translate to human physiology, thus limiting their predictive value for clinical trials (83). Secondly, animal trials inherently involve animal suffering and death. Third, a significant proportion of drug development centers on specific antibodies. Often, these do not cross-react between species, which needs to be addressed and may be challenging. Hence, the potential advantages have to be carefully weighed against the burden and death of experimental animals.
These models have significantly contributed to our understanding of PD pathogenesis and have led to the identification and validation of several new therapeutic targets. Equally, these models also excluded several molecules as targets, which is at least equally important. Table 2 summarizes the cellular and molecular targets investigated in preclinical PD mouse models. The following paragraphs highlight those targets that have emerged as most promising.
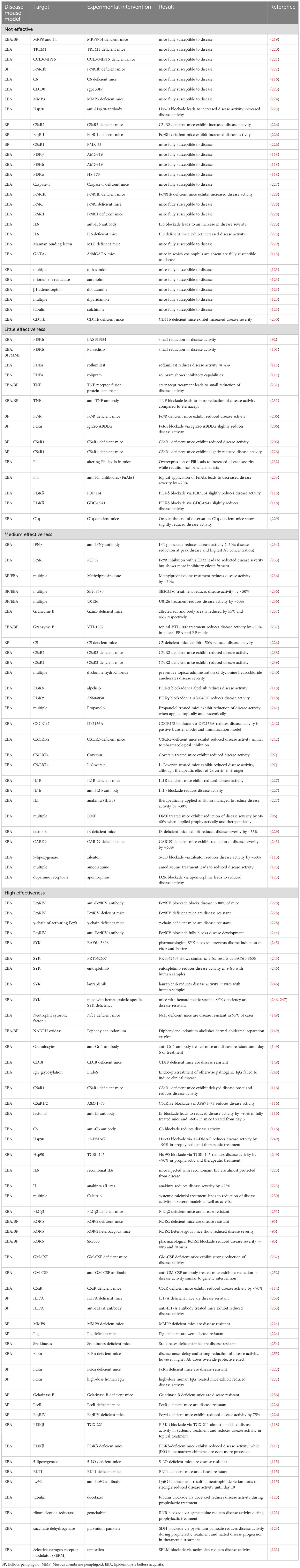
Table 2. Experimental interventions in pre-clinical animal models of PD.
Dimethyl fumarate (DMF) is approved for psoriasis (Skilarence®) and multiple sclerosis (Tecfidera®). In the antibody transfer-induced EBA mouse model, DMF reduced EBA severity by more than 50%, and, even more strikingly, led to resolving of lesions when used in therapeutic settings in the immunization-induced EBA mouse model. Investigating of the mode of action (MOA) demonstrated that DMF impairs neutrophil activation in vitro as well as tissue disruption ex vivo in an ERK1/2, p38 MAPK and Akt-dependent manner (96). Based on these results, orphan designation (98) was granted by the European Medicines Agency (EMA). Following this publication, a BP patient was successfully treated with DMF (113). The planned phase 2 clinical trial aiming to evaluate the safety and efficacy of DMF in BP could, however, not be initiated. Nomacopan, also known as Coversin, received orphan designation for the treatment of BP, again based on data from antibody transfer-induced EBA (97, 99). As a dual inhibitor of leukotriene B4 (LTB4) and complement component C5, its efficacy was validated in the antibody transfer-induced EBA model, following the identification of LTB4 and the C5a/C5aR1-axis as key factors for autoantibody-induced tissue damage in the model (97, 114–116). The subsequent phase 2a clinical trial including 9 BP patients showed that nomacopan is safe and may have therapeutic benefits for suppressing acute disease flares (100). The ß-isoform of phosphoinositide 3-kinase (PI3Kß) was identified as a target in a study that showed that genetic and pharmacological inhibition of PI3Kß leads to substantial disease protection in antibody transfer-induced EBA. Regarding the MOA, PI3Kß mediates several immune complex (IC)-elicited neutrophil responses in vitro, including release of reactive oxygen species (ROS) (117). A follow-up study, investigating the impact of PI3K-inhibitors with different selectivity for PI3Kα, β, γ or δ, supported these findings. Here, only the Pi3Kβ-selective TGX-221 impaired the clinical disease manifestation of antibody transfer-induced EBA – of note, also when topically applied. In parallel, the impact of different PI3K-inhibtors on neutrophil functions was evaluated. In these experiments, TGX-221 impaired IL-8-induced neutrophil migration, spreading of neutrophils on immobilized IC and IC-induced ROS release from neutrophils (118). Given that PI3Kβ-selective inhibitors are in clinical trials for other indications (119, 120), targeting this pathway (preferably by topical application) in PD seems valid.
As detailed above, investigations using pre-clinical model systems of PD additionally employed in vitro methods to identify the MOA of the tested compounds. Although not systematically addressed, a large proportion of the compounds shown to inhibit key pathogenic pathways in PD (Figure 1) were also able to impair the onset of antibody transfer-induced PD or have therapeutic effects in the immunization-induced models. There is a clear political commitment in the European Union to accelerate phasing out of animal testing (121), and the FDA modernization act “authorizes the use of certain alternatives to animal testing, including cell-based assays and computer models, to obtain an exemption from the Food and Drug Administration to investigate the safety and effectiveness of a drug” (https://www.congress.gov/bill/117th-congress/senate-bill/5002). To address this and to strictly implement the 3R principle (refine, reduce, replace) (122), we trust that routine and innovative new human-based in vitro or ex vivo models allowing testing of potential compounds will - to a certain extend - lead to prediction of efficacious therapeutics. These will not only replicate the outcome of in vivo experiments and thus significantly reduce the need for animal experimentation (Figure 2), but also provide assays in which human-targeting drug can be evaluated. This principle has recently partially been implemented in a large-scale screening endeavor to repurpose drugs for modulation of innate and acquired immune responses (123). In this paragraph, we thus will discuss alternative strategies to replace rodent-utilizing, preclinical model systems to align with the 3R principles (124). The below proposed in vitro, ex vivo and ex vivo systems align with the current understanding of PD pathogenesis outlined in Figure 1.
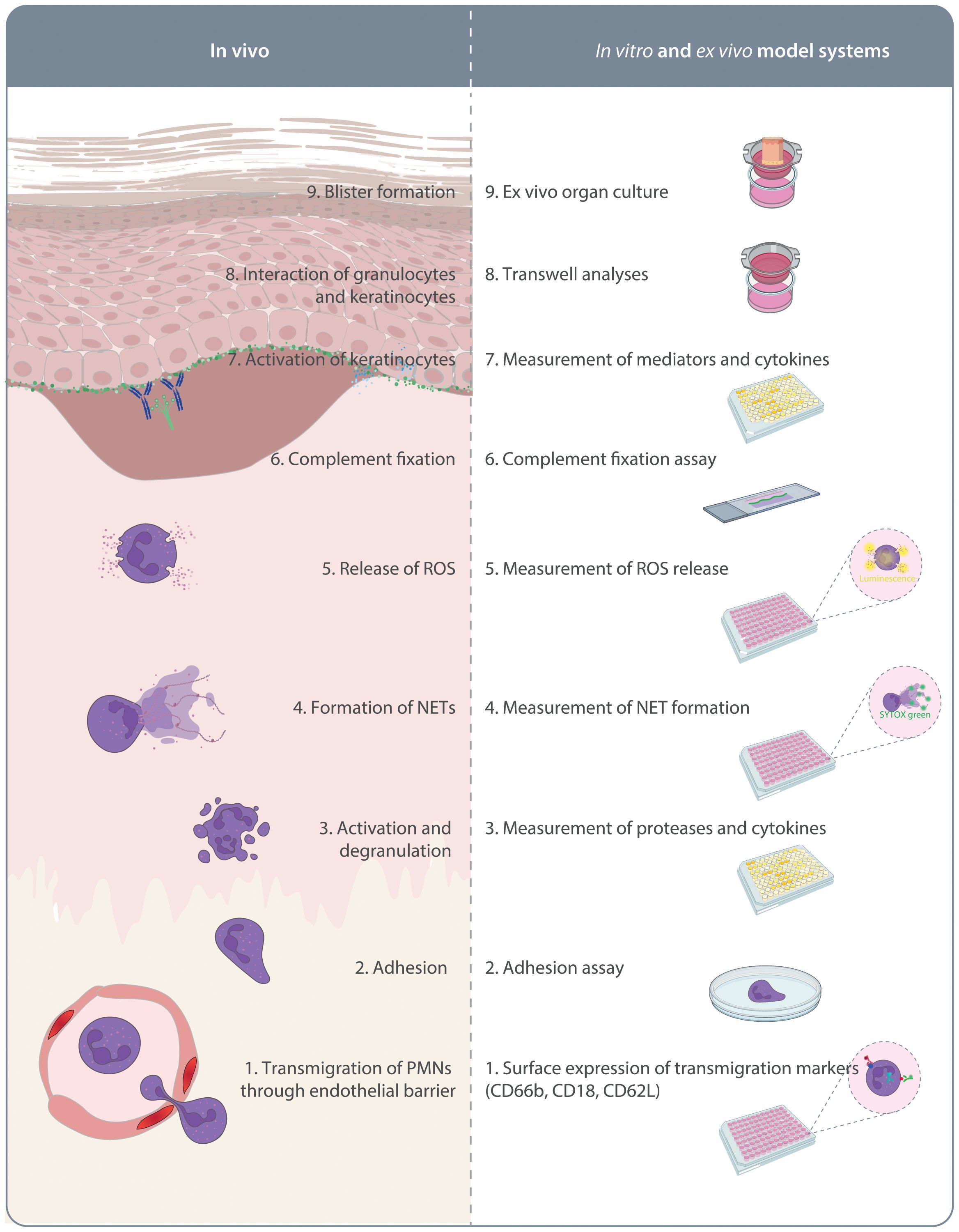
Figure 2. In vitro and ex vivo model systems of pemphigoid diseases. On the left side the pathogenesis of autoantibody-mediated tissue pathology in pemphigoid diseases is indicated. This is initiated by migration of polymorphonuclear leukocytes (PMN) into the skin. On the right side, assays mimicking these aspects of pemphigoid disease pathogenesis are indicated. These assays are described in detail in the text.
Most investigations used non-antigen-specific T cell stimulations using anti-CD3 and anti-CD28 antibodies (123). Results from these investigations can potentially predict the impact on autoreactive T cells. However, as pan-T cell targeting treatments are expected to be associated with considerable adverse events (125), this, relative basic method may be used to screen larger libraries to select compounds for further in vitro testing.
Flow cytometry can be used to detect antigen-specific T cells in PD (126, 127). One can envision combining this with specific assays targeting these antigen-specific T cells (128). Yet, these assays need to be established for BP.
The ARTE technology allows investigating antigen-specific T cell responses and has recently been used to in-depth characterize autoantigen-specific CD4 T cells in several autoimmune diseases, including BP (126). Whilst ARTE primarily aims to decipher the phenotype of antigen specific T cells from human samples, it could also be used to characterize these cells following an experimental intervention in vitro. The latter, to be developed, assays would be a potential asset to predict the efficacy of these intervention in pre-clinical model systems.
B cells can be stimulated in vitro by IL-21 and anti-CD40. This unspecific B cell stimulation has been used to identify B cell inhibitory compounds in a drug screening attempt. Here 1,200 compounds were screened for their potential B cell inhibitory activity. Screening and in vitro validation identified five drugs that were subsequently tested in pre-clinical PD mouse models. Three of the five compounds were indeed able to impair the induction of immunization-induced EBA (123). This highlights that this assay is indeed able to identify B cell modulatory compounds that are effective in vivo. These compounds are likely to non-specifically suppress B cell activation, which could lead to potential adverse events, thereby limiting their clinical applicability.
Similar considerations, as outlined above for T cells, apply. Immunophenotyping of PD patients’ B cell populations has been described (129), and these methods may be modified to detect changes upon treatment. In addition, ELISpot may be used to determine B cell functions following an in vitro manipulation (130).
One of the main functions of the neonatal Fc receptor (FcRn) is to maintain high levels of circulating IgG concentrations in the blood. This is achieved by protecting IgG from lysosomal degradation after uptake by endothelial cells (131, 132). Inhibition of the FcRn lowers circulating IgG concentrations, including those of IgG autoantibodies, and this has been demonstrated to be effective in autoantibody-mediated diseases (133). Assays to determine the impact of compounds targeting the FcRn have been developed (134), but so far, have not been used in PD research. These cellular recycling assays would be, however, valuable to determine if compounds to be tested in pre-clinical PD model systems have an impact on IgG turnover.
The current understanding of pemphigoid disease pathogenesis considers keratinocytes to be key effector cells in mediating autoantibody-induced tissue pathology (135). This understanding is based on the discovery of IL-6 and IL-8 release from keratinocytes incubated with anti-BP180 antibodies (136). The principle of this assay is still in use today. However, systematic investigations on the impact of drugs on autoantibody-induced mediator release and their subsequent effects in keratinocyte model systems is so far lacking.
Another direct impact of PD autoantibodies on keratinocytes is the internalization of autoantigens (137). This weakening of the adhesion of hemidesmosomes to the lamina densa, followed by the inflammatory events triggered by autoantibody binding could be the reason for the ultrastructural site of the split formation. Like with many of the in vitro model systems in PD, a systematic approach aiming to correlate in vitro observations to efficacy in pre-clinical models is lacking.
Initially developed as a diagnostic assay for PG (138), modifications of the complement (C)-fixation assay can be used to model complement activation in PD. For this, cryosections of human skin are incubated with PD antibodies. After washing, a complement source is added. Endpoints include determination of C3 deposits at the dermal-epidermal junction and evaluation of complement cleavage products in the supernatant (110, 139). So far, no insight on the prediction of outcomes regarding in vivo model systems has been obtained. However, compounds positively evaluated in the complement fixation assay showed efficacy in phase 1 clinical trials including BP patients (140), or other pre-clinical disease models (141).
Spreading is one of the first events after binding of neutrophils to IC. The assay duplicating this key event has been adopted for PD in 2010 (142). This assay has been used in several publications relating to PD. Yet, the prognostic value of this test for predicting efficacy in pre-clinical model systems has not been systematically evaluated.
Changes in expression of adhesion molecules is another hallmark of IC-induced neutrophil activation, indicating altered migratory capabilities (e.g., CD18, CD62L) or degranulation (e.g., CD66) (143). Regarding PD, flow cytometry has been used to address IC-induced changes in neutrophils (111). Due to the relatively limited number of publications utilizing this method, the predictive value of altered surface molecule expression on IC-activated neutrophils regarding effects in pre-clinical PD model systems remains to be elucidated.
Mediator release is another hallmark of neutrophil activation (143). In PD, lipid mediators, cytokines and complement are key soluble mediators (18). So far, the cellular source of these mediators remains to be determined. Given the central role of soluble mediators in neutrophil activity regulation and resolving inflammation as well as the relative straight-forward and highly up-scalable methods for determining these soluble mediators (143–145), analysis of these for prediction of treatment outcomes holds a high potential.
The “ROS-release assay” is widely used to investigate IC-induced neutrophil activation in PD (146). In this assay, immune complexes are generated in 96-well plates which are incubated with freshly isolated human or (more rarely) mouse neutrophils. These, in an Fc-gamma, or Fc-alpha-receptor mediated fashion, become activated (147, 148) and release ROS, which are detected by chemiluminescence. The “ROS-release assay” has been the basis to identify drug candidates to inhibit IC-induced neutrophil activation in a repurposing study. This in vitro screening identified six from a total of 1,200 compounds. All six were then evaluated for their safety and efficacy in the antibody transfer-induced EBA mouse model, where 3 of 6 compounds impaired induction of experimental EBA (123). Overall, this indicates that the “ROS-release assay” can be used to drastically reduce the requirement for animal experimentation. However, half of the compounds identified by the “ROS-release assay” as potential new treatment options of PD, failed validation in pre-clinical animal models.
The cryosection assay duplicates the subepidermal blistering observed in PD. Here, cryosections of normal human (and rarely mouse) skin are incubated with PD autoantibodies. After washing, neutrophils from healthy donors are added. Again, these are activated by binding of Fc-gamma, or Fc-alpha-receptor to the tissue-bound IC. Ultimately, this leads to neutrophil spreading (see above), ROS- and protease-release, which mediate the dermal-epidermal separation (149, 150). The cryosection assay is not applicable to high throughput and is thus mainly used to validate findings from the ROS-release assay. With regard to implementation of the 3R principles, the cryosection assay may be used following the ROS-release assay to further limit the number of compounds to be used in vivo.
A recent paper described a 3D human skin equivalent that was incubated with BP180-affinity-purified IgG from BP patients. This polyclonal anti-BP180 induced BP180 internalization and led to subepidermal split formation (151). Of note, addition of an FcRn inhibitor reduced IgG deposition along the basement membrane of the keratinocytes, indicating that this assay may also be used to test for FcRn function. We believe that this model holds the potential to investigate autoantibody-induced tissue damage in pemphigoid diseases in great depth. Modifications, for example, addition of leukocytes would allow to investigate the interplay between keratinocytes, autoantibodies and leukocytes in vitro. Use of skin biopsies from healthy donors or pemphigoid disease patients would allow to additionally investigate the impact of resident immune cells within the skin. Especially these ex vivo models could significantly reduce the need for animal testing when investigating autoantibody-induced tissue damage.
IL-8 (and its’ mouse homologues) and C5a are central in PD pathogenesis (36, 114, 116). In PD, IL-8- or C5a-induced neutrophil migration has been used in several studies (95, 96). In some, but not all of the studies, the migratory ability of neutrophils in vitro correlated well with those observed in pre-clinical model systems. Thus, the use of these assays to reduce or replace animal experimentation needs to be elucidated.
In summary, a systematic evaluation of the above-described in vitro models is warranted to determine whether one, or more likely a combination of several in vitro systems, can effectively predict in vivo effects of molecules or drugs. This approach is expected to significantly reduce the need for animal experimentation. Thus, a systematic investigation to address the predictive value of PD in vitro assays for compound efficacy in pre-clinical models is urgently needed.
Advances in the availability and analysis of real-world data (RWD) have expanded the possibilities and applicability to generate evidence and thereby allowing regulatory agencies, such as the FDA, to base decisions on RWD. Thus, the FDA is putting more emphasis on RWD to enhance therapeutic drug development and strengthen regulatory oversight throughout the medical product lifecycle (152). The data quantity and quality of some RWD databases allows to predict disease onset and allows to stimulate clinical trials (153). In the context of PD, a recent study compared the risk of death and relapse in BP patients treated either with topical or systemic corticosteroids. Here, risk of death was increased in patients with BP exposed to any dose of systemic corticosteroids versus BP patients treated with topical clobetasol propionate (22). This study can in principle be adopted to any other drug. As a limitation, however, insights into efficacy are limited because disease severity scores are not available and drug dosages are not recorded.
Drugs approved for other indications may be repurposed for PD. An excellent example for this within the field of pemphigus and pemphigoid is the use and licensing of rituximab in pemphigus. The CD-20 antibody rituximab was initially licensed for the treatment of patients with relapsed or refractory B cell non-Hodgkin’s lymphoma in 1997 (154). Due to the drug’s MOA, specifically B cell depletion, it was given to a 30-year-old woman with refractory pemphigus vulgaris, resulting in partial remission (155). Following two randomized controlled clinical trials, rituximab in combination with prednisolone is now the standard of care in pemphigus (156, 157). For PD, controlled clinical trials of the safety and efficacy of rituximab are so far lacking. However, data from retrospective analyses indicate a moderate effect of rituximab in PD (158–160).
In PD, three drugs, in addition to DMF which is discussed above, licensed for other indications seem to be effective: The anti-IL4Rα antibody dupilumab, the anti-IgE antibody omalizumab and Janus kinase (JAK) inhibitors (JAKi). Of these, dupilumab holds the largest promise. In 2017, dupilumab was licensed for atopic dermatitis (161, 162). Based on the Th2-phenotype of T cells in BP (163), and the intense pruritus in BP, dupilumab was successfully used off-label in several BP patients by different medical practitioners (164, 165). Recently, a multicenter, ambispective cohort study investigated the safety and efficacy of dupilumab in 103 BP patients. Overall, dupilumab was safe and effective: Adverse events were observed in 13 of 103 patients and were mostly mild. Complete remission was achieved in 53.4% of BP patients within 4 weeks and 95.7% by week 52 (166). A recent press release from Sanofi on their randomized, phase 2/3, double-blind, placebo-controlled study evaluating dupilumab in BP reported that the study met all primary endpoints. Specifically at week 36 of the study, 20% of BP patients in the treatment arm experienced sustained disease remission, compared to 4% in the control arm (167). Similar findings were made in a large retrospective cohort study from China (168). Based on these considerations, approval for dupilumab in BP is expected within the next 1-2 years. Of note, a rapid and sustained response to the IL-13 targeting antibody tralokinumab has been reported in one patient with BP (169), that replicated in a larger case-series (170). At this point, this data does not allow to draw any final conclusions. However, the precise contribution of IL-4 and IL-13 to PD pathogenesis remains to be fully elucidated.
The anti-IgE antibody omalizumab was first licensed in 2003 for the treatment of asthma (171). Subsequently, it was also licensed for other type 2 inflammatory diseases including chronic spontaneous urticaria, chronic rhinosinusitis with nasal polyps and most recently also for food allergies (172–174). There is a body of evidence pointing towards a potential contribution of IgE in PD, especially BP: Elevated levels of total IgE are observed in BP. In some patients, antigen-specific IgE can be detected. Last but not least, in an antibody-transfer model, antigen-specific IgE antibodies elicited experimental BP when transferred into mice (175, 176). In 2009, a BP patient was successfully treated with off-label use of omalizumab (177). These results were confirmed in several case reports. More recently, a multicenter retrospective study conducted by the French Study Group on Autoimmune Bullous Diseases investigated the effectiveness and safety of omalizumab in BP patients. The study included 100 BP patients. Omalizumab led to complete remission in close to 80% of the patients and displayed a favorable safety profile. Of note, complete remission was more frequently observed in patients with an increased serum baseline level of antigen-specific IgE autoantibodies targeting BP180 (178). Taken together, omalizumab is another promising candidate for the treatment of BP. This may also be applicable to other pemphigoid diseases, e.g., MMP (179), but data on IgE and/or omalizumab is rather scant for PD other than BP.
More recently, JAKi have been licensed for several non-communicable inflammatory diseases, including their topical administration in atopic dermatitis and vitiligo (180, 181). Given the boxed warning of JAKi concerning their cardiovascular risk profile (182), topical application in PD, especially BP, would be the preferred application route. The recent case reports on the off-label use of JAKi in PD is mostly based on their increased expression at the site of inflammation (183–185). So far, six patients with BP (186–190), four patients with MMP (191, 192), and one patient with LPP (193) have been reported to have responded to off-label JAKi treatment. More data on the efficacy, and most notably on the safety of JAKi in PD are warranted. However, based on the evidence available so far, topical JAKi may be useful in BP, whilst their systemic application may best be applicable for treatment refractory MMP or EBA.
Please note that most of these studies originated from Europe or the US, and that ethnicity is seldom indicated. As we have recently demonstrated, consideration of racial disparities in autoimmune skin blistering diseases is, however, quite important (194).
As seen in other dermatological disorders (161, 162), biologics, specifically dupilumab and omalizumab, are most likely to change the treatment landscape of PD significantly. Although current available treatments, i.e. dupilumab and omalizumab, seem to be less efficient compared the gold standard corticosteroid treatment, their main advantage is the far more favorable adverse event profile. Thus, corticosteroids will be potentially used to induce rapid remission, whilst remission is maintained by long-term treatment with either dupilumab or omalizumab.
Methotrexate (MTX) has long been used as an adjuvant treatment for BP (195). A randomized trial from the French Study Group on Autoimmune Bullous Diseases compared the efficacy and safety of topical corticosteroid therapy with or without low dose MTX, where the topical corticosteroid treatment was stopped after 4-6 weeks in the MTX arm, and was continued in the control arm for nine months. A total of 300 patients were screened, but only a small fraction of BP patients was recruited due to MTX-related exclusion criteria. The remission rates were 75% in the MTX arm and 57% in the topical corticosteroid arm, which reached statistical significance. The number of severe adverse events and mortality rate was similar between the groups (196). Taken together, this indicates that MTX is a good alternative to continued (topical) corticosteroid treatment, that is, however, only applicable to 10-15% of BP patients.
In conclusion, the number of potential targets and molecules identified (Table 2) significantly exceeds the compounds that have been evaluated to date and hold promise for future PD treatment. In addition, those emerging PD treatments are so far exclusively limited to BP management. For all other PD, the pipeline for new treatments is practically non-existent. Arguably, some of the compounds effective in BP can potentially be repurposed for other PD. In the authors’ opinion, overcoming this challenge requires a joint effort from industry and academia, supported by robust model systems, including also in vitro and ex vivo human models, meticulous clinical observations, and a shared commitment to improving patient well-being through translational research.
MT: Writing – original draft, Writing – review & editing. SD: Visualization, Writing – review & editing. IP: Writing – review & editing. KB: Visualization, Writing – review & editing. AV: Writing – review & editing. JE: Writing – review & editing. MB: Conceptualization, Funding acquisition, Visualization, Writing – review & editing. RL: Conceptualization, Funding acquisition, Writing – original draft.
The author(s) declare that financial support was received for the research and/or publication of this article. QIMA Monasterium GmbH, and from the Deutsche Forschungsgemeinschaft: Cluster of Excellence “Precision Medicine in Chronic Inflammation” (EXC 2167), and Collaborative Research Center “PANTAU” (SFB 1526), and the Schleswig-Holstein Excellence-Chair Program from the State of Schleswig Holstein.
MT, IP, JE, MB, RL are full-or part-time employees of QIMA Monasterium GmbH.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
The author(s) declare that Generative AI was used in the creation of this manuscript. To edit text.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Schmidt E, Zillikens D. Pemphigoid diseases. Lancet. (2013) 381:320–32. doi: 10.1016/S0140-6736(12)61140-4
2. Goletz S, Pigors M, Lari TR, Hammers CM, Wang Y, Emtenani S, et al. Laminin β4 is a constituent of the cutaneous basement membrane zone and additional autoantigen of anti-p200 pemphigoid. J Am Acad Dermatol. (2024) 90:790–7. doi: 10.1016/j.jaad.2023.11.014
3. Chan LS, Fine JD, Briggaman RA, Woodley DT, Hammerberg C, Drugge RJ, et al. Identification and partial characterization of a novel 105-kDalton lower lamina lucida autoantigen associated with a novel immune-mediated subepidermal blistering disease. J Invest Dermatol. (1993) 101:262–7. doi: 10.1111/1523-1747.ep12365189
4. Venning VA. Linear IgA disease: clinical presentation, diagnosis, and pathogenesis. Dermatologic Clinics. (2011) 29:453–458, ix. doi: 10.1016/j.det.2011.03.013
6. van Beek N, Weidinger A, Schneider SW, Kleinheinz A, Gläser R, Holtsche MM, et al. Incidence of pemphigoid diseases in Northern Germany in 2016 - first data from the Schleswig-Holstein Registry of Autoimmune Bullous Diseases. J Eur Acad Dermatol Venereology: JEADV. (2021) 35:1197–202. doi: 10.1111/jdv.17107
7. Persson MSM, Begum N, Grainge MJ, Harman KE, Grindlay D, Gran S. The global incidence of bullous pemphigoid: a systematic review and meta-analysis. Br J Dermatol. (2022) 186:414–25. doi: 10.1111/bjd.20743
8. Kridin K, Ludwig RJ. The growing incidence of bullous pemphigoid: overview and potential explanations. Front Med. (2018) 5:220. doi: 10.3389/fmed.2018.00220
9. Jung M, Kippes W, Messer G, Zillikens D, Rzany B. Increased risk of bullous pemphigoid in male and very old patients: A population-based study on incidence. J Am Acad Dermatol. (1999) 41:266–8. doi: 10.1016/s0190-9622(99)70061-7
10. Marazza G, Pham HC, Schärer L, Pedrazzetti PP, Hunziker T, Trüeb RM, et al. Incidence of bullous pemphigoid and pemphigus in Switzerland: a 2-year prospective study. Br J Dermatol. (2009) 161:861–8. doi: 10.1111/j.1365-2133.2009.09300.x
11. Bertram F, Bröcker EB, Zillikens D, Schmidt E. Prospective analysis of the incidence of autoimmune bullous disorders in Lower Franconia, Germany. J der Deutschen Dermatologischen Gesellschaft = J German Soc Dermatology: JDDG. (2009) 7:434–40. doi: 10.1111/j.1610-0387.2008.06976.x
12. Gudi VS, White MI, Cruickshank N, Herriot R, Edwards SL, Nimmo F, et al. Annual incidence and mortality of bullous pemphigoid in the Grampian Region of North-east Scotland. Br J Dermatol. (2005) 153:424–7. doi: 10.1111/j.1365-2133.2005.06662.x
13. Joly P, Baricault S, Sparsa A, Bernard P, Bédane C, Duvert-Lehembre S, et al. Incidence and mortality of bullous pemphigoid in France. J Invest Dermatol. (2012) 132:1998–2004. doi: 10.1038/jid.2012.35
14. Langan SM, Smeeth L, Hubbard R, Fleming KM, Smith CJP, West J. Bullous pemphigoid and pemphigus vulgaris–incidence and mortality in the UK: population based cohort study. BMJ (Clinical Res ed.). (2008) 337:a180. doi: 10.1136/bmj.a180
15. Moro F, Fania L, Sinagra JLM, Salemme A, Zenzo GD. Bullous pemphigoid: trigger and predisposing factors. Biomolecules. (2020) 10(10):1432. doi: 10.3390/biom10101432
16. Kasperkiewicz M, Zillikens D. The pathophysiology of bullous pemphigoid. Clin Rev Allergy Immunol. (2007) 33:67–77. doi: 10.1007/s12016-007-0030-y
17. Hertl M. Humoral and cellular autoimmunity in autoimmune bullous skin disorders. Int Arch Allergy Immunol. (2000) 122:91–100. doi: 10.1159/000024364
18. Koga H, Prost-Squarcioni C, Iwata H, Jonkman MF, Ludwig RJ, Bieber K. Epidermolysis bullosa acquisita: the 2019 update. Front Med. (2019) 5:362. doi: 10.3389/fmed.2018.00362
19. Hiroyasu S, Ozawa T, Kobayashi H, Ishii M, Aoyama Y, Kitajima Y, et al. Bullous pemphigoid IgG induces BP180 internalization via a macropinocytic pathway. Am J Pathology. (2013) 182:828–40. doi: 10.1016/j.ajpath.2012.11.029
20. van Beek N, Holtsche MM, Atefi I, Olbrich H, Schmitz MJ, Pruessmann J, et al. State-of-the-art diagnosis of autoimmune blistering diseases. Front Immunol. (2024) 15:1363032. doi: 10.3389/fimmu.2024.1363032
21. Joly P, Roujeau JC, Benichou J, Picard C, Dreno B, Delaporte E, et al. A comparison of oral and topical corticosteroids in patients with bullous pemphigoid. New Engl J Med. (2002) 346:321–7. doi: 10.1056/NEJMoa011592
22. Kridin K, Bieber K, Vorobyev A, Moderegger EL, Hernandez G, Schmidt E, et al. Risk of death, major adverse cardiac events and relapse in patients with bullous pemphigoid treated with systemic or topical corticosteroids. Br J Dermatol. (2024) 191:539–47. doi: 10.1093/bjd/ljae219
23. Ujiie H, Iwata H, Yamagami J, Nakama T, Aoyama Y, Ikeda S, et al. Japanese guidelines for the management of pemphigoid (including epidermolysis bullosa acquisita). J Dermatol. (2019) 46:1102–35. doi: 10.1111/1346-8138.15111
24. Patel PM, Jones VA, Murray TN, Amber KT. A review comparing international guidelines for the management of bullous pemphigoid, pemphigoid gestationis, mucous membrane pemphigoid, and epidermolysis bullosa acquisita. Am J Clin Dermatol. (2020) 21:557–65. doi: 10.1007/s40257-020-00513-3
25. Guignant M, Tedbirt B, Murrell DF, Amagai M, Aoki V, Bauer J, et al. How do experts treat patients with bullous pemphigoid around the world? An international survey. JID innovations: skin Sci molecules to population Health. (2022) 2:100129. doi: 10.1016/j.xjidi.2022.100129
26. Joly P, Roujeau JC, Benichou J, Delaporte E, D’Incan M, Dreno B, et al. A comparison of two regimens of topical corticosteroids in the treatment of patients with bullous pemphigoid: a multicenter randomized study. J Invest Dermatol. (2009) 129:1681–7. doi: 10.1038/jid.2008.412
27. Ujiie H, Rosmarin D, Schön MP, Ständer S, Boch K, Metz M, et al. Unmet medical needs in chronic, non-communicable inflammatory skin diseases. Front Med. (2022) 9:875492. doi: 10.3389/fmed.2022.875492
28. Boch K, Zirpel H, Thaci D, Mruwat N, Zillikens D, Ludwig RJ, et al. Mortality in eight autoimmune bullous diseases: A global large-scale retrospective cohort study. J Eur Acad Dermatol Venereology: JEADV. (2023) 37:e535–7. doi: 10.1111/jdv.18700
29. Hübner F, Recke A, Zillikens D, Linder R, Schmidt E. Prevalence and age distribution of pemphigus and pemphigoid diseases in Germany. J Invest Dermatol. (2016) 136:2495–8. doi: 10.1016/j.jid.2016.07.013
30. Holtsche MM, Zillikens D, Schmidt E. Schleimhautpemphigoid. Der Hautarzt. (2018) 69:67–83. doi: 10.1007/s00105-017-4089-y
31. Thorne JE, Anhalt GJ, Jabs DA. Mucous membrane pemphigoid and pseudopemphigoid. Ophthalmology. (2004) 111:45–52. doi: 10.1016/j.ophtha.2003.03.001
32. van Beek N, Kridin K, Bühler E, Kochan AS, Ständer S, Ludwig RJ, et al. Evaluation of site- and autoantigen-specific characteristics of mucous membrane pemphigoid. JAMA Dermatol. (2022) 158:84–9. doi: 10.1001/jamadermatol.2021.4773
33. Ahmed AR, Kurgis BS, Rogers RS. Cicatricial pemphigoid. J Am Acad Dermatol. (1991) 24:987–1001. doi: 10.1016/0190-9622(91)70159-y
34. Chan LS, Ahmed AR, Anhalt GJ, Bernauer W, Cooper KD, Elder MJ, et al. The first international consensus on mucous membrane pemphigoid: definition, diagnostic criteria, pathogenic factors, medical treatment, and prognostic indicators. Arch Dermatol. (2002) 138:370–9. doi: 10.1001/archderm.138.3.370
35. Domloge-Hultsch N, Gammon WR, Briggaman RA, Gil SG, Carter WG, Yancey KB. Epiligrin, the major human keratinocyte integrin ligand, is a target in both an acquired autoimmune and an inherited subepidermal blistering skin disease. J Clin Invest. (1992) 90:1628–33. doi: 10.1172/JCI116033
36. Schmidt E, Reimer S, Kruse N, Bröcker EB, Zillikens D. The IL-8 release from cultured human keratinocytes, mediated by antibodies to bullous pemphigoid autoantigen 180, is inhibited by dapsone. Clin Exp Immunol. (2001) 124:157–62. doi: 10.1046/j.1365-2249.2001.01503.x
37. Shimanovich I, Nitz JM, Zillikens D. Multiple and repeated sampling increases the sensitivity of direct immunofluorescence testing for the diagnosis of mucous membrane pemphigoid. J Am Acad Dermatol. (2017) 77:700–705.e3. doi: 10.1016/j.jaad.2017.05.016
38. Hofmann SC, Günther C, Böckle BC, Didona D, Ehrchen J, Gaskins M, et al. S2k Guideline for the diagnosis and treatment of mucous membrane pemphigoid. J der Deutschen Dermatologischen Gesellschaft = J German Soc Dermatology: JDDG. (2022) 20:1530–50. doi: 10.1111/ddg.14905
39. Bernard P, Vaillant L, Labeille B, Bedane C, Arbeille B, Denoeux JP, et al. Incidence and distribution of subepidermal autoimmune bullous skin diseases in three French regions. Bullous Dis French Study Group Arch Dermatol. (1995) 131:48–52. doi: 10.1001/archderm.1995.01690130050009
40. Wong SN, Chua SH. Spectrum of subepidermal immunobullous disorders seen at the National Skin Centre, Singapore: a 2-year review. Br J Dermatol. (2002) 147:476–80. doi: 10.1046/j.1365-2133.2002.04919.x
41. Trigo-Guzmán FX, Conti A, Aoki V, Maruta CW, Santi CG, Silva CMR, et al. Epidermolysis bullosa acquisita in childhood. J Dermatol. (2003) 30:226–9. doi: 10.1111/j.1346-8138.2003.tb00376.x
42. Marzano AV, Cozzani E, Fanoni D, De Pità O, Vassallo C, Berti E, et al. Diagnosis and disease severity assessment of epidermolysis bullosa acquisita by ELISA for anti-type VII collagen autoantibodies: an Italian multicentre study. Br J Dermatol. (2013) 168:80–4. doi: 10.1111/bjd.12011
43. Iwata H, Vorobyev A, Koga H, Recke A, Zillikens D, Prost-Squarcioni C, et al. Meta-analysis of the clinical and immunopathological characteristics and treatment outcomes in epidermolysis bullosa acquisita patients. Orphanet J Rare Diseases. (2018) 13:153. doi: 10.1186/s13023-018-0896-1
44. Briggaman RA, Gammon WR, Woodley DT. Epidermolysis bullosa acquisita of the immunopathological type (dermolytic pemphigoid). J Invest Dermatol. (1985) 85:79s–84s. doi: 10.1111/1523-1747.ep12275505
46. Woodley DT, Briggaman RA, Gammon WT. Review and update of epidermolysis bullosa acquisita. Semin Dermatol. (1988) 7:111–22.
47. Prost-Squarcioni C, Caux F, Schmidt E, Jonkman MF, Vassileva S, Kim SC, et al. International Bullous Diseases Group: consensus on diagnostic criteria for epidermolysis bullosa acquisita. Br J Dermatol. (2018) 179:30–41. doi: 10.1111/bjd.16138
48. Chen M, Chan LS, Cai X, O’Toole EA, Sample JC, Woodley DT. Development of an ELISA for rapid detection of anti-type VII collagen autoantibodies in epidermolysis bullosa acquisita. J Invest Dermatol. (1997) 108:68–72. doi: 10.1111/1523-1747.ep12285634
49. Chen M, Kim GH, Prakash L, Woodley DT. Epidermolysis bullosa acquisita: autoimmunity to anchoring fibril collagen. Autoimmunity. (2012) 45:91–101. doi: 10.3109/08916934.2011.606450
50. Buijsrogge JJA, Diercks GFH, Pas HH, Jonkman MF. The many faces of epidermolysis bullosa acquisita after serration pattern analysis by direct immunofluorescence microscopy. Br J Dermatol. (2011) 165:92–8. doi: 10.1111/j.1365-2133.2011.10346.x
51. Vodegel RM, Jonkman MF, Pas HH, de Jong MCJM. U-serrated immunodeposition pattern differentiates type VII collagen targeting bullous diseases from other subepidermal bullous autoimmune diseases. Br J Dermatol. (2004) 151:112–8. doi: 10.1111/j.1365-2133.2004.06006.x
52. Kim JH, Kim YH, Kim SC. Epidermolysis bullosa acquisita: a retrospective clinical analysis of 30 cases. Acta Dermato-Venereologica. (2011) 91:307–12. doi: 10.2340/00015555-1065
53. Kridin K, Ahmed AR. Anti-p200 pemphigoid: A systematic review. Front Immunol. (2019) 10:2466. doi: 10.3389/fimmu.2019.02466
54. Goletz S, Hashimoto T, Zillikens D, Schmidt E. Anti-p200 pemphigoid. J Am Acad Dermatol. (2014) 71:185–91. doi: 10.1016/j.jaad.2014.02.036
55. Jenkins RE, Hern S, Black MM. Clinical features and management of 87 patients with pemphigoid gestationis. Clin Exp Dermatol. (1999) 24:255–9. doi: 10.1046/j.1365-2230.1999.00472.x
56. Ambros-Rudolph CM, Müllegger RR, Vaughan-Jones SA, Kerl H, Black MM. The specific dermatoses of pregnancy revisited and reclassified: results of a retrospective two-center study on 505 pregnant patients. J Am Acad Dermatol. (2006) 54:395–404. doi: 10.1016/j.jaad.2005.12.012
57. Genovese G, Derlino F, Cerri A, Moltrasio C, Muratori S, Berti E, et al. A systematic review of treatment options and clinical outcomes in pemphigoid gestationis. Front Med. (2020) 7:604945. doi: 10.3389/fmed.2020.604945
58. Preuß SL, Vorobyev A, Moderegger EL, Terheyden P, Bieber K, Kridin K, et al. Pemphigoid gestationis is associated with an increased risk for adverse pregnancy outcomes: A large-scale propensity-matched retrospective cohort study. J Am Acad Dermatol. (2024) 91(4):748–50. doi: 10.1016/j.jaad.2024.05.087
59. Cordel N, Flament J, Jouen F, Seta V, Tancrède-Bohin E, Dahan CP, et al. Anti-BP180 IgG antibody ELISA values correlate with adverse pregnancy outcomes in pemphigoid gestationis. J Eur Acad Dermatol Venereology: JEADV. (2023) 37:1207–14. doi: 10.1111/jdv.18973
60. Sävervall C, Sand FL, Thomsen SF. Pemphigoid gestationis: current perspectives. Clinical Cosmetic Investigational Dermatol. (2017) 10:441–9. doi: 10.2147/CCID.S128144
61. Intong LRA, Murrell DF. Pemphigoid gestationis: pathogenesis and clinical features. Dermatologic Clinics. (2011) 29:447–452, ix. doi: 10.1016/j.det.2011.03.002
62. Al-Mutairi N, Sharma AK, Zaki A, El-Adawy E, Al-Sheltawy M, Nour-Eldin O. Maternal and neonatal pemphigoid gestationis. Clin Exp Dermatol. (2004) 29:202–4. doi: 10.1111/j.1365-2230.2004.01481.x
63. Bernett CN, Fong M, Yadlapati S, Rosario-Collazo JA. Linear IGA dermatosis, in: StatPearls (2024). Treasure Island (FL: StatPearls Publishing. Available online at: http://www.ncbi.nlm.nih.gov/books/NBK526113/ (Accessed 6th November 2024).
64. Wojnarowska F, Marsden RA, Bhogal B, Black MM. Chronic bullous disease of childhood, childhood cicatricial pemphigoid, and linear IgA disease of adults. A comparative study demonstrating clinical and immunopathologic overlap. J Am Acad Dermatol. (1988) 19:792–805. doi: 10.1016/s0190-9622(88)70236-4
65. Zone JJ, Taylor TB, Kadunce DP, Meyer LJ. Identification of the cutaneous basement membrane zone antigen and isolation of antibody in linear immunoglobulin A bullous dermatosis. J Clin Invest. (1990) 85:812–20. doi: 10.1172/JCI114508
66. Marinkovich MP, Taylor TB, Keene DR, Burgeson RE, Zone JJ. LAD-1, the linear IgA bullous dermatosis autoantigen, is a novel 120-kDa anchoring filament protein synthesized by epidermal cells. J Invest Dermatol. (1996) 106:734–8. doi: 10.1111/1523-1747.ep12345782
67. Juratli HA, Sárdy M. Lineare igA-dermatose. Der Hautarzt. (2019) 70:254–9. doi: 10.1007/s00105-019-4377-9
68. Shin L, Gardner JT, Dao H. Updates in the diagnosis and management of linear igA disease: A systematic review. Medicina (Kaunas Lithuania). (2021) 57:818. doi: 10.3390/medicina57080818
69. Mori F, Saretta F, Liotti L, Giovannini M, Castagnoli R, Arasi S, et al. Linear immunoglobulin a bullous dermatosis in children. Front Pediatrics. (2022) 10:937528. doi: 10.3389/fped.2022.937528
70. Boch K, Langan EA, Kridin K, Zillikens D, Ludwig RJ, Bieber K. Lichen planus. Front Med. (2021) 8:737813. doi: 10.3389/fmed.2021.737813
71. Hübner F, Langan EA, Recke A. Lichen planus pemphigoides: from lichenoid inflammation to autoantibody-mediated blistering. Front Immunol. (2019) 10:1389. doi: 10.3389/fimmu.2019.01389
72. Cortés B, Marazza G, Naldi L, Combescure C, Borradori L, Autoimmune Bullous Disease Swiss Study Group. Mortality of bullous pemphigoid in Switzerland: a prospective study. Br J Dermatol. (2011) 165:368–74. doi: 10.1111/j.1365-2133.2011.10413.x
73. Bieber K, Ludwig RJ. Drug development in pemphigoid diseases. Acta Dermato-Venereologica. (2020) 100:5663. doi: 10.2340/00015555-3400
74. Rzany B, Partscht K, Jung M, Kippes W, Mecking D, Baima B, et al. Risk factors for lethal outcome in patients with bullous pemphigoid: low serum albumin level, high dosage of glucocorticosteroids, and old age. Arch Dermatol. (2002) 138:903–8. doi: 10.1001/archderm.138.7.903
75. Bernard P, Antonicelli F. Bullous pemphigoid: A review of its diagnosis, associations and treatment. Am J Clin Dermatol. (2017) 18:513–28. doi: 10.1007/s40257-017-0264-2
76. Borradori L, Van Beek N, Feliciani C, Tedbirt B, Antiga E, Bergman R, et al. Updated S2 K guidelines for the management of bullous pemphigoid initiated by the European Academy of Dermatology and Venereology (EADV). J Eur Acad Dermatol Venereology: JEADV. (2022) 36:1689–704. doi: 10.1111/jdv.18220
77. Rashid H, Lamberts A, Borradori L, Alberti-Violetti S, Barry RJ, Caproni M, et al. European guidelines (S3) on diagnosis and management of mucous membrane pemphigoid, initiated by the European Academy of Dermatology and Venereology - Part I. J Eur Acad Dermatol Venereology: JEADV. (2021) 35:1750–64. doi: 10.1111/jdv.17397
78. Schmidt E, Rashid H, Marzano AV, Lamberts A, Di Zenzo G, Diercks GFH, et al. European Guidelines (S3) on diagnosis and management of mucous membrane pemphigoid, initiated by the European Academy of Dermatology and Venereology - Part II. J Eur Acad Dermatol Venereology: JEADV. (2021) 35:1926–48. doi: 10.1111/jdv.17395
79. Caux F, Patsatsi A, Karakioulaki M, Antiga E, Baselga E, Borradori L, et al. S2k guidelines on diagnosis and treatment of linear IgA dermatosis initiated by the European Academy of Dermatology and Venereology. J Eur Acad Dermatol Venereology. (2024) 38:1006–23. doi: 10.1111/jdv.19880
80. Lamberts A, Yale M, Grando S, Horváth B, Zillikens D, Jonkman M. Unmet needs in pemphigoid diseases: an international survey amongst patients, clinicians and researchers. Acta Dermato Venereologica. (2019) 99:224–5. doi: 10.2340/00015555-3052
81. Nishie W. Update on the pathogenesis of bullous pemphigoid: an autoantibody-mediated blistering disease targeting collagen XVII. J Dermatol Science. (2014) 73:179–86. doi: 10.1016/j.jdermsci.2013.12.001
82. Kasperkiewicz M, Sadik CD, Bieber K, Ibrahim SM, Manz RA, Schmidt E, et al. Epidermolysis bullosa acquisita: from pathophysiology to novel therapeutic options. J Invest Dermatol. (2016) 136:24–33. doi: 10.1038/JID.2015.356
83. Bieber K, Koga H, Nishie W. In vitro and in vivo models to investigate the pathomechanisms and novel treatments for pemphigoid diseases. Exp Dermatol. (2017) 26:1163–70. doi: 10.1111/exd.13415
84. Ludwig RJ. Signalling and targeted therapy of inflammatory cells in epidermolysis bullosa acquisita. Exp Dermatol. (2017) 26:1179–86. doi: 10.1111/exd.13335
85. Olivry T, Chan LS. Autoimmune blistering dermatoses in domestic animals. Clinics Dermatol. (2001) 19:750–60. doi: 10.1016/s0738-081x(00)00197-8
86. Hurskainen T, Kokkonen N, Sormunen R, Jackow J, Löffek S, Soininen R, et al. Deletion of the major bullous pemphigoid epitope region of collagen XVII induces blistering, autoimmunization, and itching in mice. J Invest Dermatol. (2015) 135:1303–10. doi: 10.1038/jid.2014.443
87. Liu Z, Diaz LA, Troy JL, Taylor AF, Emery DJ, Fairley JA, et al. A passive transfer model of the organ-specific autoimmune disease, bullous pemphigoid, using antibodies generated against the hemidesmosomal antigen, BP180. J Clin Invest. (1993) 92:2480–8. doi: 10.1172/JCI116856
88. Oswald E, Sesarman A, Franzke CW, Wölfle U, Bruckner-Tuderman L, Jakob T, et al. The flavonoid luteolin inhibits Fcγ-dependent respiratory burst in granulocytes, but not skin blistering in a new model of pemphigoid in adult mice. PloS One. (2012) 7:e31066. doi: 10.1371/journal.pone.0031066
89. Schulze FS, Beckmann T, Nimmerjahn F, Ishiko A, Collin M, Köhl J, et al. Fcγ receptors III and IV mediate tissue destruction in a novel adult mouse model of bullous pemphigoid. Am J Pathology. (2014) 184:2185–96. doi: 10.1016/j.ajpath.2014.05.007
90. Nishie W, Sawamura D, Goto M, Ito K, Shibaki A, McMillan JR, et al. Humanization of autoantigen. Nat Med. (2007) 13:378–83. doi: 10.1038/nm1496
91. Liu Z, Sui W, Zhao M, Li Z, Li N, Thresher R, et al. Subepidermal blistering induced by human autoantibodies to BP180 requires innate immune players in a humanized bullous pemphigoid mouse model. J Autoimmunity. (2008) 31:331–8. doi: 10.1016/j.jaut.2008.08.009
92. Koga H, Kasprick A, López R, Aulí M, Pont M, Godessart N, et al. Therapeutic effect of a novel phosphatidylinositol-3-kinase δ Inhibitor in experimental epidermolysis bullosa acquisita. Front Immunol. (2018) 9:1558. doi: 10.3389/fimmu.2018.01558
93. Shi L, Li X, Qian H. Anti-laminin 332-type mucous membrane pemphigoid. Biomolecules. (2022) 12:1461. doi: 10.3390/biom12101461
94. Iwata H, Bieber K, Hirose M, Ludwig RJ. Animal models to investigate pathomechanisms and evaluate novel treatments for autoimmune bullous dermatoses. Curr Pharm Design. (2015) 21:2422–39. doi: 10.2174/1381612821666150316122502
95. Sadeghi H, Gupta Y, Möller S, Samavedam UK, Behnen M, Kasprick A, et al. The retinoid-related orphan receptor alpha is essential for the end-stage effector phase of experimental epidermolysis bullosa acquisita. J Pathology. (2015) 237:111–22. doi: 10.1002/path.4556
96. Müller S, Behnen M, Bieber K, Möller S, Hellberg L, Witte M, et al. Dimethylfumarate impairs neutrophil functions. J Invest Dermatol. (2016) 136:117–26. doi: 10.1038/JID.2015.361
97. Sezin T, Murthy S, Attah C, Seutter M, Holtsche MM, Hammers CM, et al. Dual inhibition of complement factor 5 and leukotriene B4 synergistically suppresses murine pemphigoid disease. JCI Insight. (2019) 4:e128239. doi: 10.1172/jci.insight.128239
98. EU/3/16/1698 - orphan designation for treatment of bullous pemphigoid . European Medicines Agency (EMA. Available online at: https://www.ema.europa.eu/en/medicines/human/orphan-designations/eu-3-16-1698 (Accessed 26th November 2024).
99. EU/3/20/2289 - orphan designation for treatment of bullous pemphigoid . European Medicines Agency (EMA. Available online at: https://www.ema.europa.eu/en/medicines/human/orphan-designations/eu-3-20-2289 (Accessed 26th November 2024).
100. Sadik CD, Rashid H, Hammers CM, Diercks GFH, Weidinger A, Beissert S, et al. Evaluation of nomacopan for treatment of bullous pemphigoid: A phase 2a nonrandomized controlled trial. JAMA Dermatol. (2022) 158:641–9. doi: 10.1001/jamadermatol.2022.1156
101. Ghorbanalipoor S, Emtenani S, Parker M, Kamaguchi M, Osterloh C, Pigors M, et al. Cutaneous kinase activity correlates with treatment outcomes following PI3K delta inhibition in mice with experimental pemphigoid diseases. Front Immunol. (2022) 13:865241. doi: 10.3389/fimmu.2022.865241
102. Schmidt E, Ludwig R, Patzelt S, Aramini A, Brandolini L, Cocchiaro P, et al. Cxcl8 inhibitors for use in the treatment of ocular mucous membrane pemphigoid and/or oral mucous membrane pemphigoid (2024). Available online at: https://patents.google.com/patent/WO2024146918A1/en (Accessed 25th November 2024).
103. Amagai M, Tsunoda K, Suzuki H, Nishifuji K, Koyasu S, Nishikawa T. Use of autoantigen-knockout mice in developing an active autoimmune disease model for pemphigus. J Clin Invest. (2000) 105:625–31. doi: 10.1172/JCI8748
104. Ujiie H, Shibaki A, Nishie W, Sawamura D, Wang G, Tateishi Y, et al. A novel active mouse model for bullous pemphigoid targeting humanized pathogenic antigen. J Immunol. (2010) 184:2166–74. doi: 10.4049/jimmunol.0903101
105. Ujiie H, Shibaki A, Nishie W, Shinkuma S, Moriuchi R, Qiao H, et al. Noncollagenous 16A domain of type XVII collagen-reactive CD4+ T cells play a pivotal role in the development of active disease in experimental bullous pemphigoid model. Clin Immunol (Orlando Fla.). (2012) 142:167–75. doi: 10.1016/j.clim.2011.10.002
106. Sasaoka T, Ujiie H, Nishie W, Iwata H, Ishikawa M, Higashino H, et al. Intravenous igG reduces pathogenic autoantibodies, serum IL-6 levels, and disease severity in experimental bullous pemphigoid models. J Invest Dermatol. (2018) 138:1260–7. doi: 10.1016/j.jid.2018.01.005
107. Sitaru C, Chiriac M, Mihai S, Büning J, Ishiko A, Zillikens D. Induction of complement-fixing autoantibodies against type VII collagen results in subepidermal blistering in mice1. J Immunol (Baltimore Md. : 1950). (2006) 177:3461–8. doi: 10.4049/jimmunol.177.5.3461
108. Hirose M, Recke A, Beckmann T, Shimizu A, Ishiko A, Bieber K, et al. Repetitive immunization breaks tolerance to type XVII collagen and leads to bullous pemphigoid in mice. J Immunol (Baltimore Md.: 1950). (2011) 187:1176–83. doi: 10.4049/jimmunol.1100596
109. Iwata H, Bieber K, Tiburzy B, Chrobok N, Kalies K, Shimizu A, et al. B cells, dendritic cells, and macrophages are required to induce an autoreactive CD4 helper T cell response in experimental epidermolysis bullosa acquisita. J Immunol (Baltimore Md.: 1950). (2013) 191:2978–88. doi: 10.4049/jimmunol.1300310
110. Kasprick A, Holtsche MM, Rose EL, Hussain S, Schmidt E, Petersen F, et al. The anti-C1s antibody TNT003 prevents complement activation in the skin induced by bullous pemphigoid autoantibodies. J Invest Dermatol. (2018) 138:458–61. doi: 10.1016/j.jid.2017.08.030
111. Koga H, Recke A, Vidarsson G, Pas HH, Jonkman MF, Hashimoto T, et al. PDE4 inhibition as potential treatment of epidermolysis bullosa acquisita. J Invest Dermatol. (2016) 136:2211–20. doi: 10.1016/j.jid.2016.06.619
112. Niebuhr M, Kasperkiewicz M, Maass S, Hauenschild E, Bieber K, Ludwig RJ, et al. Evidence for a contributory role of a xenogeneic immune response in experimental epidermolysis bullosa acquisita. Exp Dermatol. (2017) 26:1207–13. doi: 10.1111/exd.13439
113. Bilgic-Temel A, Das S, Murrell DF. Successful management of bullous pemphigoid with dimethyl fumarate therapy: A case report. Int J Women’s Dermatol. (2019) 5:179–80. doi: 10.1016/j.ijwd.2019.02.001
114. Karsten CM, Pandey MK, Figge J, Kilchenstein R, Taylor PR, Rosas M, et al. Galactosylated IgG1 links FcγRIIB and Dectin-1 to block complement-mediated inflammation. Nat Med. (2012) 18:1401–6. doi: 10.1038/nm.2862
115. Sezin T, Krajewski M, Wutkowski A, Mousavi S, Chakievska L, Bieber K, et al. The leukotriene B4 and its receptor BLT1 act as critical drivers of neutrophil recruitment in murine bullous pemphigoid-like epidermolysis bullosa acquisita. J Invest Dermatol. (2017) 137:1104–13. doi: 10.1016/j.jid.2016.12.021
116. Mihai S, Hirose M, Wang Y, Thurman JM, Holers VM, Morgan BP, et al. Specific inhibition of complement activation significantly ameliorates autoimmune blistering disease in mice. Front Immunol. (2018) 9:535. doi: 10.3389/fimmu.2018.00535
117. Kulkarni S, Sitaru C, Jakus Z, Anderson KE, Damoulakis G, Davidson K, et al. PI3Kβ plays a critical role in neutrophil activation by immune complexes. Sci Signaling. (2011) 4:ra23. doi: 10.1126/scisignal.2001617
118. Zillikens H, Kasprick A, Osterloh C, Gross N, Radziewitz M, Hass C, et al. Topical application of the PI3Kβ-selective small molecule inhibitor TGX-221 is an effective treatment option for experimental epidermolysis bullosa acquisita. Front Med. (2021) 8:713312. doi: 10.3389/fmed.2021.713312
119. Nastoupil LJ, Neelapu SS, Davis RE, Samaniego F, Fowler NH, Westin J, et al. Preclinical and phase I studies of KA2237, a selective and potent inhibitor of PI3K β/δ in relapsed refractory B cell lymphoma. Leukemia Lymphoma. (2021) 62:3452–62. doi: 10.1080/10428194.2021.1957874
120. Nylander S, Wågberg F, Andersson M, Skärby T, Gustafsson D. Exploration of efficacy and bleeding with combined phosphoinositide 3-kinase β inhibition and aspirin in man. J Thromb haemostasis: JTH. (2015) 13:1494–502. doi: 10.1111/jth.13027
121. Commission acts to accelerate phasing out of animal testing . Available online at: https://ec.europa.eu/commission/presscorner/detail/en/ip_23_3993 (Accessed 25th November 2024).
122. Tannenbaum J, Bennett BT. Russell and Burch’s 3Rs then and now: the need for clarity in definition and purpose. J Am Assoc Lab Anim Science: JAALAS. (2015) 54:120–32.
123. Ghorbanalipoor S, Matsumoto K, Gross N, Heimberg L, Krause M, Veldkamp W, et al. High throughput screening identifies repurposable drugs for modulation of innate and acquired immune responses. J Autoimmunity. (2024) 148:103302. doi: 10.1016/j.jaut.2024.103302
124. Törnqvist E, Annas A, Granath B, Jalkesten E, Cotgreave I, Öberg M. Strategic focus on 3R principles reveals major reductions in the use of animals in pharmaceutical toxicity testing. PloS One. (2014) 9:e101638. doi: 10.1371/journal.pone.0101638
125. Bardazzi F, Filippi F, Chessa MA, Iommi M, Loi C, Campanati A, et al. Mortality and prognostic factors in patients with bullous pemphigoid: a retrospective multicenter Italian study. J Eur Acad Dermatol Venereology: JEADV. (2022) 36:2473–81. doi: 10.1111/jdv.18441
126. Saggau C, Bacher P, Esser D, Rasa M, Meise S, Mohr N, et al. Autoantigen-specific CD4+ T cells acquire an exhausted phenotype and persist in human antigen-specific autoimmune diseases. Immunity. (2024) 57:2416–2432.e8. doi: 10.1016/j.immuni.2024.08.005
127. Thoma-Uszynski S, Uter W, Schwietzke S, Schuler G, Borradori L, Hertl M. Autoreactive T and B cells from bullous pemphigoid (BP) patients recognize epitopes clustered in distinct regions of BP180 and BP230. J Immunol (Baltimore Md.: 1950). (2006) 176:2015–23. doi: 10.4049/jimmunol.176.3.2015
128. Kiesgen S, Messinger JC, Chintala NK, Tano Z, Adusumilli PS. Comparative analysis of assays to measure CAR T-cell-mediated cytotoxicity. Nat Protoc. (2021) 16:1331–42. doi: 10.1038/s41596-020-00467-0
129. Berkani N, Joly P, Golinski ML, Colliou N, Lim A, Larbi A, et al. Author Correction: B-cell depletion induces a shift in self antigen specific B-cell repertoire and cytokine pattern in patients with bullous pemphigoid. Sci Rep. (2019) 9:18991. doi: 10.1038/s41598-019-54421-6
130. Rockstroh C, Hintz K, Kannenberg J, Jassoy C. Measurement of the memory B cell response via antibodies from activated cells. Antibodies (Basel Switzerland). (2024) 13:81. doi: 10.3390/antib13040081
131. Roopenian DC, Akilesh S. FcRn: the neonatal Fc receptor comes of age. Nat Rev Immunol. (2007) 7:715–25. doi: 10.1038/nri2155
132. Patel DD, Bussel JB. Neonatal Fc receptor in human immunity: Function and role in therapeutic intervention. J Allergy Clin Immunol. (2020) 146:467–78. doi: 10.1016/j.jaci.2020.07.015
133. Howard JF, Bril V, Burns TM, Mantegazza R, Bilinska M, Szczudlik A, et al. Randomized phase 2 study of FcRn antagonist efgartigimod in generalized myasthenia gravis. Neurology. (2019) 92:e2661–73. doi: 10.1212/WNL.0000000000007600
134. Grevys A, Nilsen J, Sand KMK, Daba MB, Øynebråten I, Bern M, et al. A human endothelial cell-based recycling assay for screening of FcRn targeted molecules. Nat Commun. (2018) 9:621. doi: 10.1038/s41467-018-03061-x
135. Yan T, Zhang Z. Adaptive and innate immune pathogenesis of bullous pemphigoid: A review. Front Immunol. (2023) 14:1144429. doi: 10.3389/fimmu.2023.1144429
136. Schmidt E, Reimer S, Kruse N, Jainta S, Bröcker EB, Marinkovich MP, et al. Autoantibodies to BP180 associated with bullous pemphigoid release interleukin-6 and interleukin-8 from cultured human keratinocytes. J Invest Dermatol. (2000) 115:842–8. doi: 10.1046/j.1523-1747.2000.00141.x
137. Iwata H, Kamio N, Aoyama Y, Yamamoto Y, Hirako Y, Owaribe K, et al. IgG from patients with bullous pemphigoid depletes cultured keratinocytes of the 180-kDa bullous pemphigoid antigen (type XVII collagen) and weakens cell attachment. J Invest Dermatol. (2009) 129:919–26. doi: 10.1038/jid.2008.305
138. Huilaja L, Mäkikallio K, Tasanen K. Gestational pemphigoid. Orphanet J Rare Diseases. (2014) 9:136. doi: 10.1186/s13023-014-0136-2
139. Gutjahr A, Heck F, Emtenani S, Hammers AK, Hundt JE, Muck P, et al. Bullous pemphigoid autoantibody-mediated complement fixation is abolished by the low-molecular-weight heparin tinzaparin sodium. Br J Dermatol. (2019) 181:593–4. doi: 10.1111/bjd.18156
140. Freire PC, Muñoz CH, Derhaschnig U, Schoergenhofer C, Firbas C, Parry GC, et al. Specific inhibition of the classical complement pathway prevents C3 deposition along the dermal-epidermal junction in bullous pemphigoid. J Invest Dermatol. (2019) 139:2417–2424.e2. doi: 10.1016/j.jid.2019.04.025
141. Liu F, Ryan ST, Fahnoe KC, Morgan JG, Cheung AE, Storek MJ, et al. C3d-Targeted factor H inhibits tissue complement in disease models and reduces glomerular injury without affecting circulating complement. Mol Therapy: J Am Soc Gene Ther. (2024) 32:1061–79. doi: 10.1016/j.ymthe.2024.02.001
142. Yu X, Holdorf K, Kasper B, Zillikens D, Ludwig RJ, Petersen F. FcγRIIA and FcγRIIIB are required for autoantibody-induced tissue damage in experimental human models of bullous pemphigoid. J Invest Dermatol. (2010) 130:2841–4. doi: 10.1038/jid.2010.230
143. Burn GL, Foti A, Marsman G, Patel DF, Zychlinsky A. The neutrophil. Immunity. (2021) 54:1377–91. doi: 10.1016/j.immuni.2021.06.006
144. Wang H, Zhao T, Zeng J, Zhang R, Pu L, Qian S, et al. Methods and clinical biomarker discovery for targeted proteomics using Olink technology. Proteomics. Clin Applications. (2024) 18(5):e2300233. doi: 10.1002/prca.202300233
145. Cui M, Cheng C, Zhang L. High-throughput proteomics: a methodological mini-review. Lab Investigation; J Tech Methods Pathology. (2022) 102:1170–81. doi: 10.1038/s41374-022-00830-7
146. Kasprick A, Bieber K, Ludwig RJ. Drug discovery for pemphigoid diseases. Curr Protoc Pharmacol. (2019) 84:e55. doi: 10.1002/cpph.55
147. Recke A, Trog LM, Pas HH, Vorobyev A, Abadpour A, Jonkman MF, et al. Recombinant human IgA1 and IgA2 autoantibodies to type VII collagen induce subepidermal blistering ex vivo. J Immunol (Baltimore Md.: 1950). (2014) 193:1600–8. doi: 10.4049/jimmunol.1400160
148. Thornton AJ, Morley CJ, Cole TJ, Green SJ, Walker KA, Rennie JM. Field trials of the Baby Check score card in hospital. Arch Dis Childhood. (1991) 66:115–20. doi: 10.1136/adc.66.1.115
149. Chiriac MT, Roesler J, Sindrilaru A, Scharffetter-Kochanek K, Zillikens D, Sitaru C. NADPH oxidase is required for neutrophil-dependent autoantibody-induced tissue damage. J Pathology. (2007) 212:56–65. doi: 10.1002/path.2157
150. Shimanovich I, Mihai S, Oostingh GJ, Ilenchuk TT, Bröcker EB, Opdenakker G, et al. Granulocyte-derived elastase and gelatinase B are required for dermal-epidermal separation induced by autoantibodies from patients with epidermolysis bullosa acquisita and bullous pemphigoid. J Pathology. (2004) 204:519–27. doi: 10.1002/path.1674
151. Bao L, Perez White BE, Chang RC, Li J, Vanderheyden K, Verheesen P, et al. Neonatal fc receptor inhibition reduces bullous pemphigoid anti-basement membrane zone igG deposition and blistering in 3-dimensional human skin equivalents. J Invest Dermatol. (2024) 144(12):2809–12.e2. doi: 10.1016/j.jid.2024.05.013. S0022-202X(24)01504-5.
152. Real-world evidence . FDA. Available online at: https://www.fda.gov/science-research/science-and-research-special-topics/real-world-evidence (Accessed 25th November 2024).
153. Jia K, Kundrot S, Palchuk MB, Warnick J, Haapala K, Kaplan ID, et al. A pancreatic cancer risk prediction model (Prism) developed and validated on large-scale US clinical data. EBioMedicine. (2023) 98:104888. doi: 10.1016/j.ebiom.2023.104888
154. Grillo-López AJ, White CA, Dallaire BK, Varns CL, Shen CD, Wei A, et al. Rituximab: the first monoclonal antibody approved for the treatment of lymphoma. Curr Pharm Biotechnol. (2000) 1:1–9. doi: 10.2174/1389201003379059
155. Salopek TG, Logsetty S, Tredget EE. Anti-CD20 chimeric monoclonal antibody (rituximab) for the treatment of recalcitrant, life-threatening pemphigus vulgaris with implications in the pathogenesis of the disorder. J Am Acad Dermatol. (2002) 47:785–8. doi: 10.1067/mjd.2002.126273
156. Joly P, Maho-Vaillant M, Prost-Squarcioni C, Hebert V, Houivet E, Calbo S, et al. First-line rituximab combined with short-term prednisone versus prednisone alone for the treatment of pemphigus (Ritux 3): a prospective, multicentre, parallel-group, open-label randomised trial. Lancet (London England). (2017) 389:2031–40. doi: 10.1016/S0140-6736(17)30070-3
157. Werth VP, Joly P, Mimouni D, Maverakis E, Caux F, Lehane P, et al. Rituximab versus mycophenolate mofetil in patients with pemphigus vulgaris. New Engl J Med. (2021) 384:2295–305. doi: 10.1056/NEJMoa2028564
158. Bohelay G, Alexandre M, Le-Roux-Villet C, Sitbon I, Doan S, Soued I, et al. Rituximab therapy for mucous membrane pemphigoid: A retrospective monocentric study with long-term follow-up in 109 patients. Front Immunol. (2022) 13:915205. doi: 10.3389/fimmu.2022.915205
159. Polansky M, Eisenstadt R, DeGrazia T, Zhao X, Liu Y, Feldman R. Rituximab therapy in patients with bullous pemphigoid: A retrospective study of 20 patients. J Am Acad Dermatol. (2019) 81:179–86. doi: 10.1016/j.jaad.2019.03.049
160. Lourari S, Herve C, Doffoel-Hantz V, Meyer N, Bulai-Livideanu C, Viraben R, et al. Bullous and mucous membrane pemphigoid show a mixed response to rituximab: experience in seven patients. J Eur Acad Dermatol Venereology: JEADV. (2011) 25:1238–40. doi: 10.1111/j.1468-3083.2010.03889.x
161. Beck LA, Thaçi D, Hamilton JD, Graham NM, Bieber T, Rocklin R, et al. Dupilumab treatment in adults with moderate-to-severe atopic dermatitis. New Engl J Med. (2014) 371:130–9. doi: 10.1056/NEJMoa1314768
162. Thaçi D, Simpson EL, Beck LA, Bieber T, Blauvelt A, Papp K, et al. Efficacy and safety of dupilumab in adults with moderate-to-severe atopic dermatitis inadequately controlled by topical treatments: a randomised, placebo-controlled, dose-ranging phase 2b trial. Lancet (London England). (2016) 387:40–52. doi: 10.1016/S0140-6736(15)00388-8
163. Schinner J, Cunha T, Mayer JU, Hörster S, Kind P, Didona D, et al. Skin-infiltrating T cells display distinct inflammatory signatures in lichen planus, bullous pemphigoid and pemphigus vulgaris. Front Immunol. (2023) 14:1203776. doi: 10.3389/fimmu.2023.1203776
164. Kaye A, Gordon SC, Deverapalli SC, Her MJ, Rosmarin D. Dupilumab for the treatment of recalcitrant bullous pemphigoid. JAMA Dermatol. (2018) 154:1225–6. doi: 10.1001/jamadermatol.2018.2526
165. Seidman JS, Eichenfield DZ, Orme CM. Dupilumab for bullous pemphigoid with intractable pruritus. Dermatol Online J. (2019) 25:13030/qt25q9w6r9. doi: 10.5070/D32511046147
166. Planella-Fontanillas N, Bosch-Amate X, Jiménez Antón A, Moreno-Vílchez C, Guerrero MG, Del Mar Blanes Martínez M, et al. Real-world evaluation of the effectiveness and safety of dupilumab in bullous pemphigoid: an ambispective multicenter case series. Br J Dermatol. (2024) 192(3):501–9. doi: 10.1093/bjd/ljae403. ljae403.
167. Press Release: Dupixent is the first and only biologic to achieve significant improvements in disease remission and symptoms in bullous pemphigoid positive pivotal study . Available online at: https://www.sanofi.com/en/media-room/press-releases/2024/2024-09-11-05-00-00-2944237 (Accessed 27th November 2024).
168. Zhao L, Wang Q, Liang G, Zhou Y, Yiu N, Yang B, et al. Evaluation of dupilumab in patients with bullous pemphigoid. JAMA Dermatol. (2023) 159:953–60. doi: 10.1001/jamadermatol.2023.2428
169. Maglie R, Baffa ME, Senatore S, Pipitò C, Caproni M, Solimani F, et al. Rapid and sustained response to tralokinumab in a patient with severe bullous pemphigoid and end-stage kidney disease. Clin Exp Dermatol. (2024) 49:161–3. doi: 10.1093/ced/llad331
170. Maglie R, Didona D, Solimani F, Pipitò C, Baffa ME, Hertl M, et al. Off-label use of tralokinumab in the treatment of bullous pemphigoid- a case series. Br J Dermatol. (2025) 29:ljaf035. doi: 10.1093/bjd/ljaf035
171. Busse W, Corren J, Lanier BQ, McAlary M, Fowler-Taylor A, Cioppa GD, et al. Omalizumab, anti-IgE recombinant humanized monoclonal antibody, for the treatment of severe allergic asthma. J Allergy Clin Immunol. (2001) 108:184–90. doi: 10.1067/mai.2001.117880
172. Maurer M, Rosén K, Hsieh HJ, Saini S, Grattan C, Gimenéz-Arnau A, et al. Omalizumab for the treatment of chronic idiopathic or spontaneous urticaria. New Engl J Med. (2013) 368:924–35. doi: 10.1056/NEJMoa1215372
173. Gevaert P, Omachi TA, Corren J, Mullol J, Han J, Lee SE, et al. Efficacy and safety of omalizumab in nasal polyposis: 2 randomized phase 3 trials. J Allergy Clin Immunol. (2020) 146:595–605. doi: 10.1016/j.jaci.2020.05.032
174. Wood RA, Togias A, Sicherer SH, Shreffler WG, Kim EH, Jones SM, et al. Omalizumab for the treatment of multiple food allergies. New Engl J Med. (2024) 390:889–99. doi: 10.1056/NEJMoa2312382
175. Lin L, Hwang BJ, Culton DA, Li N, Burette S, Koller BH, et al. Eosinophils mediate tissue injury in the autoimmune skin disease bullous pemphigoid. J Invest Dermatol. (2018) 138:1032–43. doi: 10.1016/j.jid.2017.11.031
176. Messingham KAN, Holahan HM, Fairley JA. Unraveling the significance of IgE autoantibodies in organ-specific autoimmunity: lessons learned from bullous pemphigoid. Immunologic Res. (2014) 59:273–8. doi: 10.1007/s12026-014-8547-7
177. Fairley JA, Baum CL, Brandt DS, Messingham KAN. Pathogenicity of IgE in autoimmunity: successful treatment of bullous pemphigoid with omalizumab. J Allergy Clin Immunol. (2009) 123:704–5. doi: 10.1016/j.jaci.2008.11.035
178. Chebani R, Lombart F, Chaby G, Dadban A, Debarbieux S, Viguier MA, et al. Omalizumab in the treatment of bullous pemphigoid resistant to first-line therapy: a French national multicentre retrospective study of 100 patients. Br J Dermatol. (2024) 190:258–65. doi: 10.1093/bjd/ljad369
179. Alexandre M, Bohelay G, Gille T, Le-Roux-Villet C, Soued I, Morin F, et al. Rapid disease control in first-line therapy-resistant mucous membrane pemphigoid and bullous pemphigoid with omalizumab as add-on therapy: A case series of 13 patients. Front Immunol. (2022) 13:874108. doi: 10.3389/fimmu.2022.874108
180. Kim BS, Howell MD, Sun K, Papp K, Nasir A, Kuligowski ME, et al. Treatment of atopic dermatitis with ruxolitinib cream (JAK1/JAK2 inhibitor) or triamcinolone cream. J Allergy Clin Immunol. (2020) 145:572–82. doi: 10.1016/j.jaci.2019.08.042
181. Rosmarin D, Pandya AG, Lebwohl M, Grimes P, Hamzavi I, Gottlieb AB, et al. Ruxolitinib cream for treatment of vitiligo: a randomised, controlled, phase 2 trial. Lancet (London England). (2020) 396:110–20. doi: 10.1016/S0140-6736(20)30609-7
182. Ytterberg SR, Bhatt DL, Mikuls TR, Koch GG, Fleischmann R, Rivas JL, et al. Cardiovascular and cancer risk with tofacitinib in rheumatoid arthritis. New Engl J Med. (2022) 386:316–26. doi: 10.1056/NEJMoa2109927
183. Juczynska K, Wozniacka A, Waszczykowska E, Danilewicz M, Wagrowska-Danilewicz M, Wieczfinska J, et al. Expression of the JAK/STAT signaling pathway in bullous pemphigoid and dermatitis herpetiformis. Mediators Inflammation. (2017) 2017:6716419. doi: 10.1155/2017/6716419
184. Klein AL, Chan KL, Walley V. A new paraspinal window in the echocardiographic diagnosis of descending aortic dissection. Am Heart J. (1987) 114:902–4. doi: 10.1016/0002-8703(87)90805-2
185. James H, Paley GL, Brasington R, Custer PL, Margolis TP, Paley MA. Tofacitinib for refractory ocular mucous membrane pemphigoid. Am J Ophthalmol Case Rep. (2021) 22:101104. doi: 10.1016/j.ajoc.2021.101104
186. Zhou W, Tan Y, Chen X, Zhang W, Sun Z, Shen Y, et al. Successful treatment of infantile refractory bullous pemphigoid with baricitinib. Australas J Dermatol. (2024) 65(8):642–6. doi: 10.1111/ajd.14345
187. Su F, Wang T, Qin Q, Xie Z. Upadacitinib for the management of bullous pemphigoid coexisting with psoriasis vulgaris: a case report and literature review. J Dermatol Treat. (2024) 35:2302394. doi: 10.1080/09546634.2024.2302394
188. Li H, Wang H, Qiao G, Liu Y, Zhang F, Pan F. Concurrent bullous pemphigoid and psoriasis vulgaris successfully treated with Janus kinase inhibitor tofacitinib: A case report and review of the literature. Int Immunopharmacology. (2023) 122:110591. doi: 10.1016/j.intimp.2023.110591
189. Nash D, Kirchhof MG. Bullous pemphigoid treated with Janus kinase inhibitor upadacitinib. JAAD Case Rep. (2023) 32:81–3. doi: 10.1016/j.jdcr.2022.12.006
190. Youssef S, Gallitano S, Bordone LA. Two cases of bullous pemphigoid effectively treated with oral tofacitinib. JAAD Case Rep. (2023) 32:77–80. doi: 10.1016/j.jdcr.2022.10.028
191. Teng Y, Ren M, Yang X, Lu W, Tao X. Real-time experience of abrocitinib for the treatment of mucous membrane pemphigoid: A case report. Patient Preference Adherence. (2024) 18:503–6. doi: 10.2147/PPA.S451007
192. Burningham KM, Cao J, Dominguez AR. Successful treatment of recalcitrant mucous membrane pemphigoid with multisystem involvement with baricitinib and methotrexate. JAAD Case Rep. (2022) 27:67–9. doi: 10.1016/j.jdcr.2022.07.013
193. Balighi K, Sharifi A, Hesari KK, Ansari MS. A successful experience in treating lichen planus pemphigoides with tofacitinib: a case report. Int J Dermatol. (2024). doi: 10.1111/ijd.17472
194. Kasperkiewicz M, Elbuluk N, Ngo BT, Kridin K, Ludwig RJ. Differing features of pemphigus in skin of colour versus white skin: A broad-scale cohort study. J Eur Acad Dermatol Venereology: JEADV. (2025) 39:e54–6. doi: 10.1111/jdv.20094
195. Paul MA, Jorizzo JL, Fleischer AB, White WL. Low-dose methotrexate treatment in elderly patients with bullous pemphigoid. J Am Acad Dermatol. (1994) 31:620–5. doi: 10.1016/s0190-9622(94)70227-6
196. Dereure O, Bernard P, Oro S, Chosidow O, Machet L, Duvert-Lehembre S, et al. Corticothérapie locale brève + méthotrexate vs corticothérapie locale seule prolongée dans la pemphigoïde bulleuse: essai national multicentrique. Annales Dermatologie Vénéréologie. (2017) 144:S86. doi: 10.1016/j.annder.2017.09.087
197. Olivry T, Chan LS, Xu L, Chace P, Dunston SM, Fahey M, et al. Novel feline autoimmune blistering disease resembling bullous pemphigoid in humans: IgG autoantibodies target the NC16A ectodomain of type XVII collagen (BP180/BPAG2). Veterinary Pathology. (1999) 36:328–35. doi: 10.1354/vp.36-4-328
198. Iwasaki T, Olivry T, Lapiere JC, Chan LS, Peavey C, Liu YY, et al. Canine bullous pemphigoid (BP): identification of the 180-kd canine BP antigen by circulating autoantibodies. Veterinary Pathology. (1995) 32:387–93. doi: 10.1177/030098589503200407
199. Xu L, O’Toole EA, Olivry T, Hernandez C, Peng J, Chen M, et al. Molecular cloning of canine bullous pemphigoid antigen 2 cDNA and immunomapping of NC16A domain by canine bullous pemphigoid autoantibodies. Biochim Et Biophys Acta. (2000) 1500:97–107. doi: 10.1016/s0925-4439(99)00092-7
200. Olivry T, Mirsky ML, Singleton W, Dunston SM, Borrillo AK, Xu L, et al. A spontaneously arising porcine model of bullous pemphigoid. Arch Dermatol Res. (2000) 292:37–45. doi: 10.1007/pl00007459
201. Olivry T, Borrillo AK, Xu L, Dunston SM, Slovis NM, Affolter VK, et al. Equine bullous pemphigoid IgG autoantibodies target linear epitopes in the NC16A ectodomain of collagen XVII (BP180, BPAG2). Veterinary Immunol Immunopathology. (2000) 73:45–52. doi: 10.1016/s0165-2427(99)00151-8
202. Olivry T, Dunston SM, Schachter M, Xu L, Nguyen N, Marinkovich MP, et al. A spontaneous canine model of mucous membrane (cicatricial) pemphigoid, an autoimmune blistering disease affecting mucosae and mucocutaneous junctions. J Autoimmunity. (2001) 16:411–21. doi: 10.1006/jaut.2001.0510
203. Bizikova P, Linder KE, Wofford JA, Mamo LB, Dunston SM, Olivry T. Canine epidermolysis bullosa acquisita: a retrospective study of 20 cases. Veterinary Dermatol. (2015) 26:441–450, e102-103. doi: 10.1111/vde.12255
204. Chiriac MT, Licarete E, Sas AG, Rados AM, Lupan I, Chiriac AM, et al. Passive transfer of collagen XVII-specific antibodies induces sustained blistering disease in adult mice. Orphanet J Rare Diseases. (2013) 8:17. doi: 10.1186/1750-1172-8-17
205. Haeberle S, Wei X, Bieber K, Goletz S, Ludwig RJ, Schmidt E, et al. Regulatory T-cell deficiency leads to pathogenic bullous pemphigoid antigen 230 autoantibody and autoimmune bullous disease. J Allergy Clin Immunol. (2018) 142:1831–1842.e7. doi: 10.1016/j.jaci.2018.04.006
206. Pigors M, Patzelt S, Reichhelm N, Dworschak J, Khil’chenko S, Emtenani S, et al. Bullous pemphigoid induced by IgG targeting type XVII collagen non-NC16A/NC15A extracellular domains is driven by Fc gamma receptor- and complement-mediated effector mechanisms and is ameliorated by neonatal Fc receptor blockade. J Pathology. (2024) 262:161–74. doi: 10.1002/path.6220
207. Li Q, Ujiie H, Shibaki A, Wang G, Moriuchi R, Qiao HJ, et al. Human IgG1 monoclonal antibody against human collagen 17 noncollagenous 16A domain induces blisters via complement activation in experimental bullous pemphigoid model. J Immunol (Baltimore Md.: 1950). (2010) 185:7746–55. doi: 10.4049/jimmunol.1000667
208. Sitaru C, Mihai S, Otto C, Chiriac MT, Hausser I, Dotterweich B, et al. Induction of dermal-epidermal separation in mice by passive transfer of antibodies specific to type VII collagen. J Clin Invest. (2005) 115:870–8. doi: 10.1172/JCI21386
209. Woodley DT, Chang C, Saadat P, Ram R, Liu Z, Chen M. Evidence that anti-type VII collagen antibodies are pathogenic and responsible for the clinical, histological, and immunological features of epidermolysis bullosa acquisita. J Invest Dermatol. (2005) 124:958–64. doi: 10.1111/j.0022-202X.2005.23702.x
210. Chen M, Doostan A, Bandyopadhyay P, Remington J, Wang X, Hou Y, et al. The cartilage matrix protein subdomain of type VII collagen is pathogenic for epidermolysis bullosa acquisita. Am J Pathology. (2007) 170:2009–18. doi: 10.2353/ajpath.2007.061212
211. Csorba K, Chiriac MT, Florea F, Ghinia MG, Licarete E, Rados A, et al. Blister-inducing antibodies target multiple epitopes on collagen VII in mice. J Cell Mol Med. (2014) 18:1727–39. doi: 10.1111/jcmm.12338
212. Iwata H, Witte M, Samavedam UKSRL, Gupta Y, Shimizu A, Ishiko A, et al. Radiosensitive hematopoietic cells determine the extent of skin inflammation in experimental epidermolysis bullosa acquisita. J Immunol (Baltimore Md.: 1950). (2015) 195:1945–54. doi: 10.4049/jimmunol.1501003
213. Bieber K, Witte M, Sun S, Hundt JE, Kalies K, Dräger S, et al. T cells mediate autoantibody-induced cutaneous inflammation and blistering in epidermolysis bullosa acquisita. Sci Rep. (2016) 6:38357. doi: 10.1038/srep38357
214. Vorobyev A, Ujiie H, Recke A, Buijsrogge JJA, Jonkman MF, Pas HH, et al. Autoantibodies to multiple epitopes on the non-collagenous-1 domain of type VII collagen induce blisters. J Invest Dermatol. (2015) 135:1565–73. doi: 10.1038/jid.2015.51
215. Lazarova Z, Yee C, Darling T, Briggaman RA, Yancey KB. Passive transfer of anti-laminin 5 antibodies induces subepidermal blisters in neonatal mice. J Clin Invest. (1996) 98:1509–18. doi: 10.1172/JCI118942
216. Heppe EN, Tofern S, Schulze FS, Ishiko A, Shimizu A, Sina C, et al. Experimental laminin 332 mucous membrane pemphigoid critically involves C5aR1 and reflects clinical and immunopathological characteristics of the human disease. J Invest Dermatol. (2017) 137:1709–18. doi: 10.1016/j.jid.2017.03.037
217. Lazarova Z, Hsu R, Yee C, Yancey KB. Human anti-laminin 5 autoantibodies induce subepidermal blisters in an experimental human skin graft model. J Invest Dermatol. (2000) 114:178–84. doi: 10.1046/j.1523-1747.2000.00829.x
218. Zone JJ, Egan CA, Taylor TB, Meyer LJ. IgA autoimmune disorders: development of a passive transfer mouse model. J Invest Dermatol Symposium Proc. (2004) 9:47–51. doi: 10.1111/j.1087-0024.2004.00840.x
219. Akbarzadeh R, Yu X, Vogl T, Ludwig RJ, Schmidt E, Zillikens D, et al. Myeloid-related proteins-8 and -14 are expressed but dispensable in the pathogenesis of experimental epidermolysis bullosa acquisita and bullous pemphigoid. J Dermatol Science. (2016) 81:165–72. doi: 10.1016/j.jdermsci.2015.12.001
220. Dräger S, Kalies K, Sidronio TB, Witte M, Ludwig RJ, Bieber K. Increased TREM-1 expression in inflamed skin has no functional impact on the pathogenesis of cutaneous disorders. J Dermatol Science. (2017) 88:152–5. doi: 10.1016/j.jdermsci.2017.05.016
221. Kasperkiewicz M, Kalies K, Pagel R, Bieber K, Zillikens D, Ludwig RJ. CCL3/MIP1α represents a biomarker but not a mandatory cytokine for disease development in experimental epidermolysis bullosa acquisita. J Dermatol Science. (2017) 88:248–50. doi: 10.1016/j.jdermsci.2017.06.019
222. Li N, Zhao M, Hilario-Vargas J, Prisayanh P, Warren S, Diaz LA, et al. Complete FcRn dependence for intravenous Ig therapy in autoimmune skin blistering diseases. J Clin Invest. (2005) 115:3440. doi: 10.1172/JCI24394
223. Samavedam UKSRL, Kalies K, Scheller J, Sadeghi H, Gupta Y, Jonkman MF, et al. Recombinant IL-6 treatment protects mice from organ specific autoimmune disease by IL-6 classical signalling-dependent IL-1ra induction. J Autoimmunity. (2013) 40:74–85. doi: 10.1016/j.jaut.2012.08.002
224. Liu Z, Li N, Diaz LA, Shipley M, Senior RM, Werb Z. Synergy between a plasminogen cascade and MMP-9 in autoimmune disease. J Clin Invest. (2005) 115:879–87. doi: 10.1172/JCI23977
225. Tukaj S, Mantej J, Sitko K, Zillikens D, Ludwig RJ, Bieber K, et al. Pathological relevance of anti-hsp70 igG autoantibodies in epidermolysis bullosa acquisita. Front Immunol. (2022) 13:877958. doi: 10.3389/fimmu.2022.877958
226. Karsten CM, Beckmann T, Holtsche MM, Tillmann J, Tofern S, Schulze FS, et al. Tissue destruction in bullous pemphigoid can be complement independent and may be mitigated by C5aR2. Front Immunol. (2018) 9:488. doi: 10.3389/fimmu.2018.00488
227. Sadeghi H, Lockmann A, Hund AC, Samavedam UK, Pipi E, Vafia K, et al. Caspase-1-independent IL-1 release mediates blister formation in autoantibody-induced tissue injury through modulation of endothelial adhesion molecules. J Immunol. (2015) 194(8):3656–63. doi: 10.4049/jimmunol.1402688
228. Kasperkiewicz M, Nimmerjahn F, Wende S, Hirose M, Iwata H, Jonkman MF, et al. Genetic identification and functional validation of FcγRIV as key molecule in autoantibody-induced tissue injury. J Pathology. (2012) 228:8–19. doi: 10.1002/path.4023
229. Mihai S, Chiriac MT, Takahashi K, Thurman JM, Holers VM, Zillikens D, et al. The alternative pathway of complement activation is critical for blister induction in experimental epidermolysis bullosa acquisita. J Immunol. (2007) 178:6514–21. doi: 10.4049/jimmunol.178.10.6514
230. Deng F, Chen Y, Zheng J, Huang Q, Cao X, Zillikens D, et al. CD11b-deficient mice exhibit an increased severity in the late phase of antibody transfer-induced experimental epidermolysis bullosa acquisita. Exp Dermatol. (2017) 26:1175–8. doi: 10.1111/exd.13434
231. Hirose M, Kasprick A, Beltsiou F, Dieckhoff Schulze K, Schulze FS, Samavedam UK, et al. Reduced skin blistering in experimental epidermolysis bullosa acquisita after anti-TNF treatment. Mol Med (Cambridge Mass.). (2017) 22:918–26. doi: 10.2119/molmed.2015.00206
232. Kopecki Z, Arkell RM, Strudwick XL, Hirose M, Ludwig RJ, Kern JS, et al. Overexpression of the Flii gene increases dermal-epidermal blistering in an autoimmune ColVII mouse model of epidermolysis bullosa acquisita. J Pathology. (2011) 225:401–13. doi: 10.1002/path.2973
233. Kopecki Z, Ruzehaji N, Turner C, Iwata H, Ludwig RJ, Zillikens D, et al. Topically applied flightless I neutralizing antibodies improve healing of blistered skin in a murine model of epidermolysis bullosa acquisita. J Invest Dermatol. (2013) 133:1008–16. doi: 10.1038/jid.2012.457
234. Gross N, Marketon J, Mousavi S, Kalies K, Ludwig RJ, Bieber K. Inhibition of interferon gamma impairs induction of experimental epidermolysis bullosa acquisita. Front Immunol. (2024) 15:1343299. doi: 10.3389/fimmu.2024.1343299
235. Iwata H, Pipi E, Möckel N, Sondermann P, Vorobyev A, van Beek N, et al. Recombinant soluble CD32 suppresses disease progression in experimental epidermolysis bullosa acquisita. J Invest Dermatol. (2015) 135:916–9. doi: 10.1038/jid.2014.451
236. Hellberg L, Samavedam UKSRL, Holdorf K, Hänsel M, Recke A, Beckmann T, et al. Methylprednisolone blocks autoantibody-induced tissue damage in experimental models of bullous pemphigoid and epidermolysis bullosa acquisita through inhibition of neutrophil activation. J Invest Dermatol. (2013) 133:2390–9. doi: 10.1038/jid.2013.91
237. Hiroyasu S, Zeglinski MR, Zhao H, Pawluk MA, Turner CT, Kasprick A, et al. Granzyme B inhibition reduces disease severity in autoimmune blistering diseases. Nat Commun. (2021) 12:302. doi: 10.1038/s41467-020-20604-3
238. Seiler DL, Kleingarn M, Kähler KH, Gruner C, Schanzenbacher J, Ehlers-Jeske E, et al. C5aR2 deficiency ameliorates inflammation in murine epidermolysis bullosa acquisita by regulating fcγ Receptor expression on neutrophils. J Invest Dermatol. (2022) 142:2715–2723.e2. doi: 10.1016/j.jid.2021.12.029
239. Seiler DL, Kähler KH, Kleingarn M, Sadik CD, Bieber K, Köhl J, et al. The complement receptor C5aR2 regulates neutrophil activation and function contributing to neutrophil-driven epidermolysis bullosa acquisita. Front Immunol. (2023) 14:1197709. doi: 10.3389/fimmu.2023.1197709
240. Wen L, Dong X, Li Q, Schramm G, Zhang B, Zillikens D, et al. Preventive but not therapeutic topical application of local anesthetics can inhibit experimental epidermolysis bullosa acquisita in mice. Front Immunol. (2021) 12:750160. doi: 10.3389/fimmu.2021.750160
241. Stüssel P, Schulze Dieckhoff K, Künzel S, Hartmann V, Gupta Y, Kaiser G, et al. Propranolol is an effective topical and systemic treatment option for experimental epidermolysis bullosa acquisita. J Invest Dermatol. (2020) 140:2408–20. doi: 10.1016/j.jid.2020.04.025
242. Hirose M, Brandolini L, Zimmer D, Götz J, Westermann J, Allegretti M, et al. The allosteric CXCR1/2 inhibitor DF2156A improves experimental epidermolysis bullosa acquisita. Genet Syndromes Gene Ther. (2013) S3. doi: 10.4172/2157-7412.S3-005
243. Németh T, Futosi K, Sitaru C, Ruland J, Mócsai A. Neutrophil-specific deletion of the CARD9 gene expression regulator suppresses autoantibody-induced inflammation in vivo. Nat Commun. (2016) 7:11004. doi: 10.1038/ncomms11004
244. Haeger SC, Kridin K, Pieper M, Griewahn L, Nimmerjahn F, Zillikens D, et al. Therapeutic effects of Fc gamma RIV inhibition are mediated by selectively blocking immune complex-induced neutrophil activation in epidermolysis bullosa acquisita. Front Immunol. (2022) 13:938306. doi: 10.3389/fimmu.2022.938306
245. Samavedam UK, Mitschker N, Kasprick A, Bieber K, Schmidt E, Laskay T, et al. Whole-genome expression profiling in skin reveals SYK as a key regulator of inflammation in experimental epidermolysis bullosa acquisita. Front Immunol. (2018) 9:249. doi: 10.3389/fimmu.2018.00249
246. Németh T, Balogh L, Káposztás E, Szilveszter KP, Mócsai A. Neutrophil-specific syk expression is crucial for skin disease in experimental epidermolysis bullosa acquisita. J Invest Dermatol. (2023) 143:1147–56. doi: 10.1016/j.jid.2022.12.016
247. Németh T, Virtic O, Sitaru C, Mócsai A. The syk tyrosine kinase is required for skin inflammation in an in vivo mouse model of epidermolysis bullosa acquisita. J Invest Dermatol. (2017) 137:2131–9. doi: 10.1016/j.jid.2017.05.017
248. Hirose M, Vafia K, Kalies K, Groth S, Westermann J, Zillikens D, et al. Enzymatic autoantibody glycan hydrolysis alleviates autoimmunity against type VII collagen. J Autoimmunity. (2012) 39:304–14. doi: 10.1016/j.jaut.2012.04.002
249. Kasperkiewicz M, Müller R, Manz R, Magens M, Hammers CM, Somlai C, et al. Heat-shock protein 90 inhibition in autoimmunity to type VII collagen: evidence that nonmalignant plasma cells are not therapeutic targets. Blood. (2011) 117:6135–42. doi: 10.1182/blood-2010-10-314609
250. Tukaj S, Bieber K, Witte M, Ghorbanalipoor S, Schmidt E, Zillikens D, et al. Calcitriol treatment ameliorates inflammation and blistering in mouse models of epidermolysis bullosa acquisita. J Invest Dermatol. (2018) 138:301–9. doi: 10.1016/j.jid.2017.09.009
251. Szilveszter KP, Vikár S, Horváth ÁI, Helyes Z, Sárdy M, Mócsai A. Phospholipase Cγ2 is essential for experimental models of epidermolysis bullosa acquisita. J Invest Dermatol. (2022) 142:1114–25. doi: 10.1016/j.jid.2021.09.019
252. Samavedam UKSRL, Iwata H, Müller S, Schulze FS, Recke A, Schmidt E, et al. GM-CSF modulates autoantibody production and skin blistering in experimental epidermolysis bullosa acquisita. J Immunol (Baltimore Md.: 1950). (2014) 192:559–71. doi: 10.4049/jimmunol.1301556
253. Chakievska L, Holtsche MM, Künstner A, Goletz S, Petersen BS, Thaci D, et al. IL-17A is functionally relevant and a potential therapeutic target in bullous pemphigoid. J Autoimmunity. (2019) 96:104–12. doi: 10.1016/j.jaut.2018.09.003
254. Kovács M, Németh T, Jakus Z, Sitaru C, Simon E, Futosi K, et al. The Src family kinases Hck, Fgr, and Lyn are critical for the generation of the in vivo inflammatory environment without a direct role in leukocyte recruitment. J Exp Med. (2014) 211:1993–2011. doi: 10.1084/jem.20132496
255. Sesarman A, Sitaru AG, Olaru F, Zillikens D, Sitaru C. Neonatal Fc receptor deficiency protects from tissue injury in experimental epidermolysis bullosa acquisita. J Mol Med (Berlin Germany). (2008) 86:951–9. doi: 10.1007/s00109-008-0366-7
Keywords: pemphigoid diseases, model systems, pathogenesis, treatment, bullous pemphigoid, mucous membrane pemphigoid
Citation: Tigges M, Dräger S, Piccini I, Bieber K, Vorobyev A, Edelkamp J, Bertolini M and Ludwig RJ (2025) Pemphigoid disease model systems for clinical translation. Front. Immunol. 16:1537428. doi: 10.3389/fimmu.2025.1537428
Received: 16 December 2024; Accepted: 26 February 2025;
Published: 17 March 2025.
Edited by:
Seng-Lai Tan, TFC Therapeutics, United StatesReviewed by:
Jong Hoon Kim, Yonsei University, Republic of KoreaCopyright © 2025 Tigges, Dräger, Piccini, Bieber, Vorobyev, Edelkamp, Bertolini and Ludwig. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ralf J. Ludwig, cmFsZi5sdWR3aWdAdWtzaC5kZQ==; Marta Bertolini, bWFydGEuYmVydG9saW5pQHFpbWEuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.