 Qian Zhu
Qian Zhu Jin Yang1,2†
Jin Yang1,2† Jieying Zhang
Jieying Zhang Junlong Li
Junlong Li- 1First Teaching Hospital of Tianjin University of Traditional Chinese Medicine, Tianjin, China
- 2National Clinical Research Center for Chinese Medicine Acupuncture and Moxibustion, Tianjin, China
Migraine is a complex neurovascular disorder whose pathogenesis involves activation of the trigeminal vascular system, central and peripheral sensitization, and neuroinflammation. Calcitonin gene-related peptide (CGRP) plays a dominant role and activation of MAPK and NF-κB signaling pathways regulates neuropeptide release, glial cell activation, and amplification of nociceptive signals. Aberrant activation of these pathways drives migraine onset and chronicity. The ubiquitin-proteasome system (UPS) is involved in neurological and inflammatory disorders. ubiquitination in the UPS is achieved through a cascade of enzymes, including Ub-activating enzyme (E1), Ub-coupling enzyme (E2), and Ub-ligase (E3). The aim of this review is to systematically explore the role of ubiquitination in the regulation of MAPK and NF-κB signaling pathways, with a focus on the mechanisms of ubiquitinating enzymes in neuroinflammation and pain signal amplification, and to explore their potential as diagnostics, biomarkers, predictors of response to therapy, and monitoring of chronicity in migraine disease.
1 Introduction
Migraine is now the sixth most prevalent disease worldwide and a leading cause of disability (1). According to the Global Burden of Disease Study (GBD) 2019, approximately 8-15% of migraine sufferers have at least one attack per year. The pathophysiology of migraine is thought to involve abnormal activation of the trigeminal vascular system, peripheral and central sensitization, and neuroinflammation (2). Among them, calcitonin gene-related peptide (CGRP) plays a dominant role. Related regulatory pathways, such as the mitogen-activated protein kinase (MAPK) and nuclear factor-kappa B (NF-κB) pathways, regulate neuropeptide release, glial cell activation, and pain signaling, mediating inflammatory and sensitizing responses. These may drive is the development of migraine pathogenesis and chronicity.
As a type of protein post-translational modification (PTM), the ubiquitin-proteasome system (UPS) is a pathway for protein ubiquitination and degradation. Ubiquitination involves a cascade reaction of three enzymes: Ub-activating enzymes (E1s), Ub-coupling enzymes (E2s) and Ub-ligases (E3s); deubiquitination is mediated by deubiquitinating enzymes (DUBs) (3, 4). Disturbances in the UPS have been shown to be associated with the induction and severity of a variety of neurologic and inflammatory disorders, suggesting its possible involvement in the pathogenesis of migraine headaches.
This paper systematically explores the role of ubiquitination in the regulation of MAPK and NF-κB signaling pathways, focusing on the regulatory mechanisms of ubiquitinating enzymes in neuroinflammation and nociceptive signal amplification, as well as exploring its feasibility as a biomarker for disease diagnosis, prediction of therapeutic response, and chronicity monitoring.
2 Pathogenesis of migraine and association of MAPK and NF-κB signaling pathways
Migraine is a complex neurovascular disorder whose pathogenesis involves multilevel interactions between the nervous and vascular systems. Recent studies have shown that the core mechanisms of migraine include activation of the trigeminal vascular system, central sensitization, peripheral sensitization and neuroinflammation. Together, these mechanisms form the pathophysiologic basis of migraine (Figure 1A).
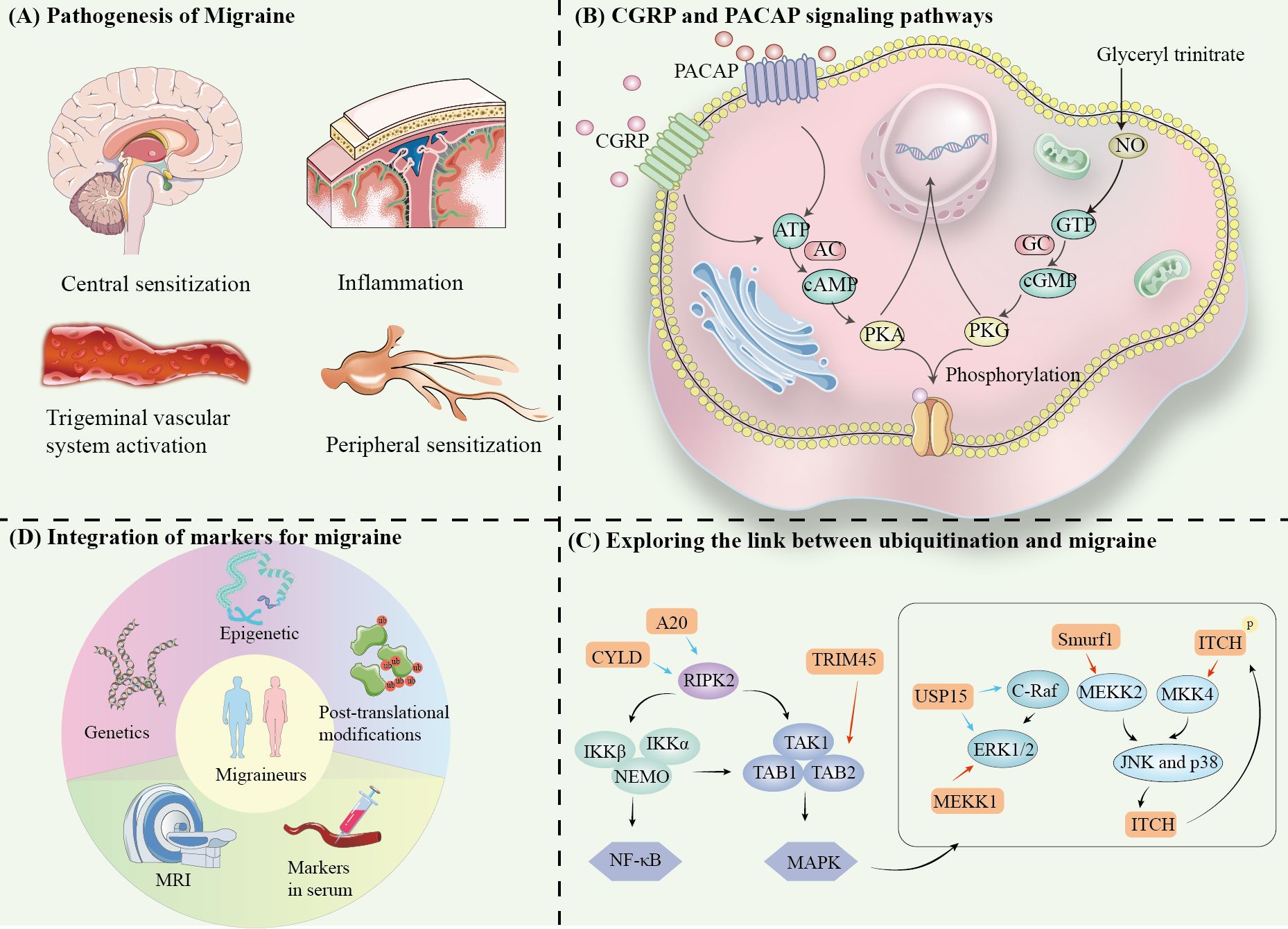
Figure 1. (A) Pathogenesis of Migraine. (B) CGRP and PACAP signaling pathways. (C) Exploring the link between ubiquitination and migraine. (D) Integration of markers for migraine.
2.1 Trigeminal vascular system activation
The trigeminal vascular system is the anatomical and physiological basis of migraine attacks, and a key mechanism of migraine nociceptive perception (5–7) (Figure 1B). Stimulation of the trigeminal ganglion (TG) leads to vascular release of neurotransmitters and inflammatory mediators, such as CGRP, PACAP, and substance P upon activation (8). These molecules bind to receptors and induce intracranial vasodilation, neuroinflammatory activation and enhanced pain signaling. Then, it leads to sensitization of secondary neurons in the trigeminal cervical complex (TCC) of the brainstem, and ultimately activate tertiary neurons in the thalamus (9).
CGRP plays a key role in neural sensitization and amplification of pain signals in migraine by binding to its receptor, which consists of CLR and RAMP1. After binding, CGRP activates Gs proteins, initiating the production of cAMP by adenylyl cyclase (AC). It further activates protein kinase A (PKA), which regulates sodium and potassium ion channel (10–12). Currently, CGRP receptor antagonists (gepants) have become effective acute treatments (13, 14).
PACAP acts mainly by binding to PAC1, VPAC1 and VPAC2 receptors (15–18). Similar to CGRP, PACAP also activates the cAMP-PKA pathway leading to migraine headaches (19). In addition, PACAP appears to have unique downstream effects, such as acting through direct activation of the EPAC pathway (20, 21). Furthermore, through the Ras-Raf-MEK-ERK signaling pathway, PKA contributes to extracellular signal-regulated kinase (ERK) phosphorylation and activation, which in turn combine peripheral sensitization and central sensitization mechanisms (15, 22).
2.2 Peripheral sensitization
Peripheral sensitization refers to a decreased threshold of damage receptors and an increased sensitivity of injury receptors. In response to repetitive stimuli or inflammatory states, excitability of sensory neurons in the periphery increases, which enhances the transmission of pain signals (23–25).
Transient receptor potential (TRP) channels, such as TRPV1, TRPA1 and TRPM8, have been shown to be closely associated with neuropathic pain. Studies have shown that blocking TRPV1 and TRPA1 significantly attenuates neurogenic hypersensitivity reaction (26–31).
In the inflammatory state, TRPV1 receptors are activated, triggering a massive inward flow of sodium and calcium ions, which triggers depolarization of injury receptors, thereby amplifying pain signals (32). Activation of TRPV1 also stimulates the release of CGRP and substance P from the nerve endings, leading to vasodilation (33).Vasodilation leads to an increase in the mechanical pressure on the local tissues, thereby further stimulating the sensory neurons of the trigeminal nerve fiber system, causing a decrease in the nociceptive threshold of injury receptors.
Notably, TRPV1 not only causes acute pain, but also persistent pain, especially pain associated with inflammation (34–36). It was found that hormone level changes may indirectly regulate the release and physiological roles of CGRP by affecting the expression and activity of TRPV1 (37). In particular, TRPV1 activation not only promotes CGRP release, but also enhances synaptic transmission efficiency and exacerbates central sensitization of neurons by promoting the release of glutamatergic vesicles (38).
Through the cAMP-PKA pathway, CGRP modulates ion channels on neuronal cell membranes, enhancing peripheral nerve sensitivity. Upon tissue injury, CGRP acts synergistically with substance P, leading to an increase in vascular permeability of local tissues and the release of inflammatory factors (39–42). Inflammatory response further activates primary afferent neurons, forming a positive feedback loop that amplifies pain signals through inflammation.
2.3 Central sensitization
Central sensitization is an important mechanism of chronic pain and is characterized by CNS hyperresponsiveness to injurious stimuli (43–45). It involves increased presynaptic neurotransmitter release and persistent neuroinflammation (45–47). Enhanced synaptic transmission in the caudate of the TNC is the neural basis for central sensitization in a CM rat model (45, 48, 49). The release of glutamate (Glu) is a key step in central sensitization, the regulation of which is dependent on the activation of ERK and p38 signaling pathways (50, 51). At presynaptic sites, the central terminals of injurious primary afferent nerves, activation of cytokine receptors and chemokine receptors leads to phosphorylation of ERK and p38 (P-ERK, P-p38) and glutamate (Glu) release. This synaptic vesicle release is associated with activation of the ion channels TRPV1, Na channel (52, 53).
At postsynaptic sites, phosphorylation of AMPA and NMDA glutamate receptors significantly enhances neuronal responsiveness to excitatory signals. Phosphorylation of AMPA receptors by PKA)and Ca2+/calmodulin-dependent kinase II (CaMKII) results in increased insertion into synaptic membranes, a significant elevation in open probability, and enhanced response to glutamate response (54, 55). phosphorylation of NMDA receptors in response to Src kinase enhances calcium ion permeability and amplifies postsynaptic Ca2+ signaling (56, 57).
In addition, P-ERK reduces the repolarization capacity of postsynaptic neurons by inhibiting their potassium channel activity, leaving them in a state of hyperexcitability (58, 59). Notably, P-ERK translocates to the nucleus and promotes the phosphorylation of the cAMP response element-binding protein (CREB), which activates proinflammatory factors, such as c-Fos and NK-1, as well as the nociceptive regulation-related gene expression, further consolidating the molecular basis of central sensitization (60, 61).
2.4 Neuroinflammation
By activating the NF-κB signaling pathway, microglia and astrocytes are activated in response to injurious stimuli and release a series of proinflammatory factors and chemokines, including tumor necrosis factor (TNF-α), interleukin-1β (IL-1β), and chemokines (62–64). Release of these factors enhances the inflammatory response and further exacerbates neuronal excitability. It was shown that upregulation of miR-155-5p activated the NF-κB signaling pathway by inhibiting SIRT1, which in turn exacerbated the release of microglial pro-inflammatory factors and neuroinflammation by inhibiting miR-155-5p, it was able to activate SIRT1 in the TNC region of CM mice, which effectively reduced neuroinflammation (60). In addition, activation of NLRP3 inflammatory vesicles was also involved in the release of pro-inflammatory factors, which further contributed to the onset of central sensitization and the chronicity of migraine (65, 66).
Further studies revealed that autophagy plays an important role in the regulation of neuroinflammatory and oxidative stress processes in astrocytes. By inhibiting autophagy, the binding of TRAF6 to K63 ubiquitinated proteins could be promoted, which increased the activities of p-MAPK8/JNK and NF-κB, thereby exacerbating the release of pro-inflammatory factors (e.g., TNF-α, IL-1β). Conversely, activation of autophagy can significantly reduce neuroinflammatory levels (67).
3 Ubiquitination and migraine
Multiple family and twin studies have shown that common migraine is heritable, with heritability estimates ranging from 30% to 60%, suggesting the presence of genetic factors that predispose individuals to migraine (68, 69). Migraine has a heritability of 42%, and relatives of people with migraine are 2- to 3-fold more likely to have the disorder (70). Although the risk of migraine is predominantly polygenic, pathogenic variants in a single gene can lead to monogenic migraine disorders (e.g., familial hemiplegic migraine FHM) and the gene is dominant, this suggests that the susceptibility and complexity of migraines may be based on genetics and may be subject to different gene-gene and gene-environment interactions (71).
The susceptibility and pathophysiological aspects of migraine have been explained from a locus perspective, but recently there has been a developing interest in investigating the role of gene regulatory mechanisms in the predisposition and chronicity of migraine, particularly epigenetic regulation. The number of studies on the role of epigenetic mechanisms in migraine is now found to be increasing yearly. Epigenetic mechanisms regulate cell cycle development by controlling the expression of individual genes, including acetylation, phosphorylation, etc (72, 73). TRP channels can convert injurious stimuli into pain signals, and the expression of TRPA1, TRPA1 encoding gene, has been demonstrated to be affected by pain-related syndromes. Acetylation modifications, this process may enhance neural excitability and facilitate pain transmission by altering electrical activity or localization (74, 75).
Similar to acetylation, ubiquitination also acts at the protein level. Ubiquitination occurs by adding single or multiple ubiquitin molecules to a target protein, modulating its stability, activity, or degradation. Studies demonstrating the relevance of the ubiquitination system to migraine are extremely limited. Ubiquitin C terminal hydrolase 1 (UCHL1), an enzyme with both ligase and hydrolase activities, is present in almost all neurons (76, 77). Serum levels of UCHL1 were significantly elevated during acute attacks in migraine patients; also, before treatment, UCHL1 levels were significantly and positively correlated with visual analog scores (VAS) (78). This suggests that UCHL1 can be used to assess seizure severity and response to treatment. Although there is conclusive evidence providing the relevance of ubiquitination system-associated proteins in acute attacks of migraine, there is little direct evidence that the meso-ubiquitination system plays a role in migraine.
Ubiquitinating enzymes (e.g., MEKK1, Smurf1, ITCH, and TRIM45) and deubiquitinating enzymes (e.g., USP15, A20, and CYLD) may be involved in the occurrence and chronicity of migraine by regulating the ubiquitination of MAPK, JNK, and NF-κB signaling pathways.
Migraine mechanisms involve dynamic processes of central sensitization and peripheral sensitization, abnormal activation of the trigeminal-vascular system, and neurogenic inflammation. Each of the four mechanisms is associated with various enzymes of the ubiquitination system, given the important role of ubiquitination in other neuroinflammatory disorders, it is reasonable to hypothesize that ubiquitination is involved in migraine attacks and chronicity (79–81).
Various trigger molecules can induce migraine, including CGRP, PACAP, adenosine triphosphate-sensitive potassium (KATP) channel opener, and large conductance calcium-activated potassium (BKCa) channel opener. The epigenetic link of CGRP and its potential in migraine has been discussed (82). In isolated trigeminal ganglion neurons, CGRP stimulates pain-related intracellular signaling molecules such as cAMP, CREB, MAPK, p38, and ERK (83). In the following, we explore the ubiquitylation of these proteins to play a function in migraine Possibilities (Figure 1C).
3.1 Regulation of ERK1/2 by ubiquitination
The ERK1/2 signaling cascade was first identified in four MAPK signaling pathways (84, 85). ubiquitination of ERK1/2 is regulated by MEKK1, which has a RING finger structure and exhibits an E3 ligase function, and USP15, a deubiquitinating enzyme. ERK2 is deubiquitinated by USP15, but the stability of the protein is not affected. Not only associated with ubiquitination, USP15 also induces ERK1/2 phosphorylation (86). Interestingly, USP15 is also known to regulate C-Raf DUB, binding to C-Raf and protecting the protein from proteolytic degradation by the 26S proteasome 34688658. overexpression of C-Raf and activation of the ERK1/2 signaling pathway cause overexpression of USP15 expression, leading to cell proliferation and migration (87).
3.2 Regulation of p38, JNK by ubiquitination
Although MEKK1 has ubiquitinating enzyme properties, it is still a member of the MAP3K family. jNK1/2/3 and p38 signaling cascades share upstream regulators such as MEKK1-4 and MKK4. In the inflammatory response, the E3 ubiquitin ligase Smad ubiquitination regulatory factor 1 (Smurf1) ubiquitinates the K48-conjugated polyubiquitin chain of MEKK2, the same type of ubiquitinating enzyme, ITCH, participates in a negative feedback loop of JNK (88). ITCH regulates MKK4 (89). ITCH is a downstream substrate of JNK, and activation of JNK promotes ITCH phosphorylation, and phosphorylated ITCH induces ubiquitination of K140 and K143 of MKK4 (90).
3.3 Regulation of NF-κB by ubiquitination
RIPK1 is the first kinase found in the RIPK family (91). RIPK2 does not have any death structural domains and does not trigger cell death signaling, but has a cysteine asparaginase activating and recruiting structural domain (CARD) that contributes to function in the NOD-like receptor (NLR)-associated inflammatory signaling pathway (92, 93). K63 ubiquitination of RIPK2 interacts with LUBAC and the kinase complex TAK1 and promotes linear ubiquitination of RIPK2, initiating the MAPK signaling pathway (94). Interestingly, the kinase complex also triggers a separate pathway for activation of the IKK complex, which consists of NEMO, IKKα, and IKKβ, and activation of the protein complex leads to the activation of NF-kinase. The IKK complex consists of NEMO, IKKα, and IKKβ, and activation of this protein complex leads to NF-κB activation. In addition, deubiquitinating enzymes A20 and CYLD have linear bonding specificity that can counteract RIPK2 ubiquitination (95, 96). Linear ubiquitination is much more attractive for NEMO binding than normal polyubiquitination, so ubiquitinated RIP1 also attracts NEMO/IKK and TAB/TAKI bindings, and thus activates downstream NF-KB signaling pathways. NF-KB signaling pathway and JNK, P38/MAPK signaling pathway (97).
The E3 ligase tripartite motif-containing 45 (TRIM45) constitutively interacts with TAB2 and promotes polyubiquitination of the TAB2-Lys-63 linkage, leading to the formation of the TAB1-TAK1-TAB2 complex and activation of TAK1, and ultimately the activation of the nuclear factor-carbamyl B (NF-κB) signaling pathway (98).
4 The potential of ubiquitination in migraine treatment
In recent years, the development of drugs targeting ubiquitinating and deubiquitinating enzymes has emerged as a research hotspot in the field of precision therapeutics (99). Although most studies have focused on cancer, the role of these drugs in ubiquitination regulation provides a potential reference for migraine treatment.
Inhibitors of ubiquitinating enzymes (TAK-243, an E1 ligase inhibitor, and MLN4924, an E3 ligase inhibitor) as potential drugs for precise regulation of inflammatory responses and cell signaling pathways (100–102).
The development of activators of USP family deubiquitinating enzymes could be a potential strategy in the treatment of migraine. USP25 inhibits the overactivation of the NF-κB and MAPK pathways by removing the K63 polyubiquitin chain on TAB2, thereby attenuating microglia-mediated neuroinflammation (103). Notably, it has been found that USP5 inhibits the expression of proinflammatory factors by maintaining NF-κB signaling pathway activation to promote the expression of pro-inflammatory factors, whereas its inhibitor Vialinin A was able to significantly reduce TNF-α and IL-1β-induced pro-inflammatory gene expression, suggesting that USP5 inhibitors may serve as drug candidates for the treatment of inflammatory diseases, such as Kawasaki disease (104). In addition, the metalloproteinase inhibitor THL significantly inhibited NLRP3 activation by blocking BRCC3 complex-mediated deubiquitination, alleviating inflammation and tissue damage in a variety of inflammatory disease models such as sepsis, autoimmune encephalomyelitis, and nonalcoholic fatty liver disease (105).
5 Conclusion
In recent years, studies on migraine mechanisms have revealed the important roles of inflammatory factors, neurogenic inflammation, and CGRP signaling pathways in the disease. As one of the important mechanisms of epigenetic regulation, ubiquitination plays a key role in the pathogenesis and chronicity of migraine by regulating protein degradation, cell signaling, and inflammatory responses. In terms of genetic markers, mutations in CACNA1A, ATP1A2, and SCN1A are closely associated with FHM, while variants in NOTCH3 and TREX1 genes have been linked to migraine and its associated cerebrovascular diseases (e.g., cerebral arteriolar dominant disorders) (106–109). In addition, DNA methylation analysis identified methylated regions specific to migraine patients, suggesting a role for epigenetic modifications in migraine susceptibility (110). In the blood, plasma CGRP levels were significantly elevated in patients during migraine attacks (111). On the imaging side, functional magnetic resonance imaging (fMRI) and diffusion tensor imaging (DTI) studies have shown that migraine patients have increased visual Increased thickness of the visual cortex, significantly altered functional connectivity in nociceptive pathways, and white matter hyperintensity are characteristic imaging hallmarks of migraine with aura (112, 113). These studies suggest the importance of plastic changes in neural structure and function in migraine.
Future studies need to further elucidate the specific link between ubiquitination and the above markers. For example, do ubiquitination modifications affect the degradation of key molecules in the CGRP signaling pathway? Does it mediate the chronicity of migraine by modulating inflammatory factors such as TNF and IL-6? By integrating multi-modal data from epigenetics, genetic markers, blood biomarkers, and neuroimaging, we aim to enhance the understanding of the molecular mechanisms underlying migraine, thus advancing marker-based precision medicine. This integrated approach may pave the way for more effective, individualized treatments for migraine patients (114) (Figure 1D).
Author contributions
QZ: Conceptualization, Supervision, Writing – original draft, Writing – review & editing. JY: Visualization, Writing – original draft, Writing – review & editing. LS: Data curation, Validation, Writing – original draft. JZ: Data curation, Validation, Writing – original draft. PZ: Supervision, Writing – original draft. JL: Visualization, Writing – original draft. XS: Conceptualization, Validation, Writing – original draft.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
3. Popovic D, Vucic D, Dikic I. Ubiquitination in disease pathogenesis and treatment. Nat Med. (2014) 20:1242–53. doi: 10.1038/nm.3739
4. Swatek KN, Usher JL, Kueck AF, Gladkova C, Mevissen TET, Pruneda JN, et al. Insights into ubiquitin chain architecture using Ub-clipping. Nature. (2019) 572:533–7. doi: 10.1038/s41586-019-1482-y
5. Charles A. The pathophysiology of migraine: implications for clinical management. Lancet Neurol. (2018) 17:174–82. doi: 10.1016/S1474-4422(17)30435-0
6. Ornello R, Andreou AP, De Matteis E, Jürgens TP, Minen MT, Sacco S. Resistant and refractory migraine: clinical presentation, pathophysiology, and management. EBioMedicine. (2024) 99:104943. doi: 10.1016/j.ebiom.2023.104943
7. Khan J, Asoom LIA, Sunni AA, Rafique N, Latif R, Saif SA, et al. Genetics, pathophysiology, diagnosis, treatment, management, and prevention of migraine. BioMed Pharmacother. (2021) 139:111557. doi: 10.1016/j.biopha.2021.111557
8. Edvinsson L, Haanes KA. Identifying new antimigraine targets: lessons from molecular biology. Trends Pharmacol Sci. (2021) 42:217–25. doi: 10.1016/j.tips.2021.01.002
9. Ashina M, Hansen JM, Do TP, Melo-Carrillo A, Burstein R, Moskowitz MA. Migraine and the trigeminovascular system-40 years and counting. Lancet Neurol. (2019) 18:795–804. doi: 10.1016/S1474-4422(19)30185-1
10. Egea SC, Dickerson IM. Direct interactions between calcitonin-like receptor (CLR) and CGRP-receptor component protein (RCP) regulate CGRP receptor signaling. Endocrinology. (2012) 153:1850–60. doi: 10.1210/en.2011-1459
11. Simonetta I, Riolo R, Todaro F, Tuttolomondo A. New insights on metabolic and genetic basis of migraine: novel impact on management and therapeutical approach. Int J Mol Sci. (2022) 23(6):3018. doi: 10.3390/ijms23063018
12. Edvinsson JCA, Warfvinge K, Krause DN, Blixt FW, Sheykhzade M, Edvinsson L, et al. C-fibers may modulate adjacent Aδ-fibers through axon-axon CGRP signaling at nodes of Ranvier in the trigeminal system. J Headache Pain. (2019) 20:105. doi: 10.1186/s10194-019-1055-3
13. Russo AF, Hay DL. CGRP physiology, pharmacology, and therapeutic targets: migraine and beyond. Physiol Rev. (2023) 103:1565–644. doi: 10.1152/physrev.00059.2021
14. Edvinsson L. Calcitonin gene-related peptide (CGRP) is a key molecule released in acute migraine attacks-Successful translation of basic science to clinical practice. J Intern Med. (2022) 292:575–86. doi: 10.1111/joim.v292.4
15. Fyfe I. PACAP - an alternative target to CGRP? Nat Rev Neurol. (2022) 18:187. doi: 10.1038/s41582-022-00639-4
16. Akerman S, Goadsby PJ. Neuronal PAC1 receptors mediate delayed activation and sensitization of trigeminocervical neurons: Relevance to migraine. Sci Transl Med. (2015) 7:308ra157. doi: 10.1126/scitranslmed.aaa7557
17. Tasma Z, Rees TA, Guo S, Tan S, O’Carroll SJ, Faull RLM, et al. Pharmacology of PACAP and VIP receptors in the spinal cord highlights the importance of the PAC(1) receptor. Br J Pharmacol. (2024) 181:2655–75. doi: 10.1111/bph.v181.15
18. Guo S, Rasmussen RH, Hay-Schmidt A, Ashina M, Asuni AA, Jensen JM, et al. VPAC1 and VPAC2 receptors mediate tactile hindpaw hypersensitivity and carotid artery dilatation induced by PACAP38 in a migraine relevant mouse model. J Headache Pain. (2024) 25:126. doi: 10.1186/s10194-024-01830-2
19. Rees TA, Labastida-Ramírez A, Rubio-Beltrán E. Calcitonin/PAC(1) receptor splice variants: a blind spot in migraine research. Trends Pharmacol Sci. (2023) 44:651–63. doi: 10.1016/j.tips.2023.07.003
20. Robichaux WG 3rd., Cheng X. Intracellular cAMP sensor EPAC: physiology, pathophysiology, and therapeutics development. Physiol Rev. (2018) 98:919–1053. doi: 10.1152/physrev.00025.2017
21. Hucho TB, Dina OA, Levine JD. Epac mediates a cAMP-to-PKC signaling in inflammatory pain: an isolectin B4(+) neuron-specific mechanism. J Neurosci. (2005) 25:6119–26. doi: 10.1523/JNEUROSCI.0285-05.2005
22. Kuburas A, Russo AF. Shared and independent roles of CGRP and PACAP in migraine pathophysiology. J Headache Pain. (2023) 24:34. doi: 10.1186/s10194-023-01569-2
23. Eliav T, Benoliel R, Korczeniewska OA. Post-traumatic trigeminal neuropathy: neurobiology and pathophysiology. Biol (Basel). (2024) 13(3):167. doi: 10.3390/biology13030167
24. Costigan M, Scholz J, Woolf CJ. Neuropathic pain: a maladaptive response of the nervous system to damage. Annu Rev Neurosci. (2009) 32:1–32. doi: 10.1146/annurev.neuro.051508.135531
25. Finnerup NB, Kuner R, Jensen TS. Neuropathic pain: from mechanisms to treatment. Physiol Rev. (2021) 101:259–301. doi: 10.1152/physrev.00045.2019
26. Bamps D, Vriens J, de Hoon J, Voets T. TRP channel cooperation for nociception: therapeutic opportunities. Annu Rev Pharmacol Toxicol. (2021) 61:655–77. doi: 10.1146/annurev-pharmtox-010919-023238
27. Spekker E, Körtési T, Vécsei L. TRP channels: recent development in translational research and potential therapeutic targets in migraine. Int J Mol Sci. (2022) 24(1):700. doi: 10.3390/ijms24010700
28. Huang Y, Zhao M, Chen X, Zhang R, Le A, Hong M, et al. Tryptophan metabolism in central nervous system diseases: pathophysiology and potential therapeutic strategies. Aging Dis. (2023) 14:858–78. doi: 10.14336/AD.2022.0916
29. Zhang M, Ma Y, Ye X, Zhang N, Pan L, Wang B. TRP (transient receptor potential) ion channel family: structures, biological functions and therapeutic interventions for diseases. Signal Transduct Target Ther. (2023) 8:261. doi: 10.1038/s41392-023-01464-x
30. Koivisto AP, Belvisi MG, Gaudet R, Szallasi A. Advances in TRP channel drug discovery: from target validation to clinical studies. Nat Rev Drug Discovery. (2022) 21:41–59. doi: 10.1038/s41573-021-00268-4
31. Yue WWS, Yuan L, Braz JM, Basbaum AI, Julius D. TRPV1 drugs alter core body temperature via central projections of primary afferent sensory neurons. Elife. (2022) 11:e80139. doi: 10.7554/eLife.80139.sa2
32. Premkumar LS, Abooj M. TRP channels and analgesia. Life Sci. (2013) 92:415–24. doi: 10.1016/j.lfs.2012.08.010
33. Helyes Z, Németh J, Thán M, Bölcskei K, Pintér E, Szolcsányi J. Inhibitory effect of anandamide on resiniferatoxin-induced sensory neuropeptide release in vivo and neuropathic hyperalgesia in the rat. Life Sci. (2003) 73:2345–53. doi: 10.1016/S0024-3205(03)00651-9
34. Selvaggi F, Melchiorre E, Casari I, Cinalli S, Cinalli M, Aceto GM, et al. Perineural invasion in pancreatic ductal adenocarcinoma: from molecules towards drugs of clinical relevance. Cancers (Basel). (2022) 14(23):5793. doi: 10.3390/cancers14235793
35. Hofmann NA, Barth S, Waldeck-Weiermair M, Klec C, Strunk D, Malli R, et al. TRPV1 mediates cellular uptake of anandamide and thus promotes endothelial cell proliferation and network-formation. Biol Open. (2014) 3:1164–72. doi: 10.1242/bio.20149571
36. Sidwell AB, McClintock C, Beča KI, Campbell SE, Girard BM, Vizzard MA. Repeated variate stress increased voiding frequency and altered TrpV1 and TrpV4 transcript expression in lower urinary tract (LUT) pathways in female mice. Front Urol. (2023) 2. doi: 10.3389/fruro.2022.1086179
37. Jamaluddin A, Chuang CL, Williams ET, Siow A, Yang SH, Harris PWR, et al. Lipidated calcitonin gene-related peptide (CGRP) peptide antagonists retain CGRP receptor activity and attenuate CGRP action in vivo. Front Pharmacol. (2022) 13:832589. doi: 10.3389/fphar.2022.832589
38. Zhang M, Ruwe D, Saffari R, Kravchenko M, Zhang W. Effects of TRPV1 activation by capsaicin and endogenous N-arachidonoyl taurine on synaptic transmission in the prefrontal cortex. Front Neurosci. (2020) 14:91. doi: 10.3389/fnins.2020.00091
39. Iyengar S, Ossipov MH, Johnson KW. The role of calcitonin gene-related peptide in peripheral and central pain mechanisms including migraine. Pain. (2017) 158:543–59. doi: 10.1097/j.pain.0000000000000831
40. Gold MS, Gebhart GF. Nociceptor sensitization in pain pathogenesis. Nat Med. (2010) 16:1248–57. doi: 10.1038/nm.2235
41. Hucho T, Levine JD. Signaling pathways in sensitization: toward a nociceptor cell biology. Neuron. (2007) 55:365–76. doi: 10.1016/j.neuron.2007.07.008
42. Ji RR, Xu ZZ, Gao YJ. Emerging targets in neuroinflammation-driven chronic pain. Nat Rev Drug Discovery. (2014) 13:533–48. doi: 10.1038/nrd4334
43. Zeng X, Mai J, Xie H, Yang L, Liu X. Activation of CB1R alleviates central sensitization by regulating HCN2-pNR2B signaling in a chronic migraine rat model. J Headache Pain. (2023) 24:44. doi: 10.1186/s10194-023-01580-7
44. Andreou AP, Edvinsson L. Mechanisms of migraine as a chronic evolutive condition. J Headache Pain. (2019) 20:117. doi: 10.1186/s10194-019-1066-0
45. Latremoliere A, Woolf CJ. Central sensitization: a generator of pain hypersensitivity by central neural plasticity. J Pain. (2009) 10:895–926. doi: 10.1016/j.jpain.2009.06.012
46. Hoegh M. Pain science in practice (Part 5): central sensitization II. J Orthop Sports Phys Ther. (2023) 53:55–8. doi: 10.2519/jospt.2023.11571
47. Woolf CJ, Salter MW. Neuronal plasticity: increasing the gain in pain. Science. (2000) 288:1765–9. doi: 10.1126/science.288.5472.1765
48. Tian R, Zhang Y, Pan Q, Wang Y, Wen Q, Fan X, et al. Calcitonin gene-related peptide receptor antagonist BIBN4096BS regulates synaptic transmission in the vestibular nucleus and improves vestibular function via PKC/ERK/CREB pathway in an experimental chronic migraine rat model. J Headache Pain. (2022) 23:35. doi: 10.1186/s10194-022-01403-1
49. Fried NT, Maxwell CR, Elliott MB, Oshinsky ML. Region-specific disruption of the blood-brain barrier following repeated inflammatory dural stimulation in a rat model of chronic trigeminal allodynia. Cephalalgia. (2018) 38:674–89. doi: 10.1177/0333102417703764
50. Yang Y, Zhou W, Xu X, Ge X, Wang F, Zhang GQ, et al. and p38/MAPK to attenuate inflammation and suppresses inflammatory pain. Front Pharmacol. (2021) 12:811584. doi: 10.3389/fphar.2021.811584
51. Qin F, Zhang H, Liu A, Wang Q, Sun Q, Lu S, et al. Analgesic effect of zanthoxylum nitidum extract in inflammatory pain models through targeting of ERK and NF-κB signaling. Front Pharmacol. (2019) 10:359. doi: 10.3389/fphar.2019.00359
52. Jin X, Gereau RW. Acute p38-mediated modulation of tetrodotoxin-resistant sodium channels in mouse sensory neurons by tumor necrosis factor-alpha. J Neurosci. (2006) 26:246–55. doi: 10.1523/JNEUROSCI.3858-05.2006
53. Ji RR, Samad TA, Jin SX, Schmoll R, Woolf CJ. p38 MAPK activation by NGF in primary sensory neurons after inflammation increases TRPV1 levels and maintains heat hyperalgesia. Neuron. (2002) 36:57–68. doi: 10.1016/S0896-6273(02)00908-X
54. Nowacka A, Getz AM, Bessa-Neto D, Choquet D. Activity-dependent diffusion trapping of AMPA receptors as a key step for expression of early LTP. Philos Trans R Soc Lond B Biol Sci. (2024) 379:20230220. doi: 10.1098/rstb.2023.0220
55. Kristensen AS, Jenkins MA, Banke TG, Schousboe A, Makino Y, Johnson RC, et al. Mechanism of Ca2+/calmodulin-dependent kinase II regulation of AMPA receptor gating. Nat Neurosci. (2011) 14:727–35. doi: 10.1038/nn.2804
56. Casili G, Lanza M, Filippone A, Cucinotta L, Paterniti I, Repici A, et al. Dimethyl fumarate (DMF) alleviated post-operative (PO) pain through the N-methyl-d-aspartate (NMDA) receptors. Antioxidants (Basel). (2022) 11(9):1774. doi: 10.3390/antiox11091774
57. Viviani B, Bartesaghi S, Gardoni F, Vezzani A, Behrens MM, Bartfai T, et al. Interleukin-1beta enhances NMDA receptor-mediated intracellular calcium increase through activation of the Src family of kinases. J Neurosci. (2003) 23:8692–700. doi: 10.1523/JNEUROSCI.23-25-08692.2003
58. Hu HJ, Carrasquillo Y, Karim F, Jung WE, Nerbonne JM, Schwarz TL, et al. The kv4.2 potassium channel subunit is required for pain plasticity. Neuron. (2006) 50:89–100. doi: 10.1016/j.neuron.2006.03.010
59. Park HR, Cai M, Yang EJ. Novel psychopharmacological herbs relieve behavioral abnormalities and hippocampal dysfunctions in an animal model of post-traumatic stress disorder. Nutrients. (2023) 15(17):3815. doi: 10.3390/nu15173815
60. Wen Q, Wang Y, Pan Q, Tian R, Zhang D, Qin G, et al. MicroRNA-155-5p promotes neuroinflammation and central sensitization via inhibiting SIRT1 in a nitroglycerin-induced chronic migraine mouse model. J Neuroinflamm. (2021) 18:287. doi: 10.1186/s12974-021-02342-5
61. Ji RR, Befort K, Brenner GJ, Woolf CJ. ERK MAP kinase activation in superficial spinal cord neurons induces prodynorphin and NK-1 upregulation and contributes to persistent inflammatory pain hypersensitivity. J Neurosci. (2002) 22:478–85. doi: 10.1523/JNEUROSCI.22-02-00478.2002
62. Zhang ZJ, Cao DL, Zhang X, Ji RR, Gao YJ. Chemokine contribution to neuropathic pain: respective induction of CXCL1 and CXCR2 in spinal cord astrocytes and neurons. Pain. (2013) 154:2185–97. doi: 10.1016/j.pain.2013.07.002
63. Braga AV, Costa S, Rodrigues FF, Melo ISF, Morais MI, Coelho MM, et al. Thiamine, riboflavin, and nicotinamide inhibit paclitaxel-induced allodynia by reducing TNF-α and CXCL-1 in dorsal root ganglia and thalamus and activating ATP-sensitive potassium channels. Inflammopharmacology. (2020) 28:201–13. doi: 10.1007/s10787-019-00625-1
64. Jiang BC, Cao DL, Zhang X, Zhang ZJ, He LN, Li CH, et al. CXCL13 drives spinal astrocyte activation and neuropathic pain via CXCR5. J Clin Invest. (2016) 126:745–61. doi: 10.1172/JCI81950
65. Sun S, Fan Z, Liu X, Wang L, Ge Z. Microglia TREM1-mediated neuroinflammation contributes to central sensitization via the NF-κB pathway in a chronic migraine model. J Headache Pain. (2024) 25:3. doi: 10.1186/s10194-023-01707-w
66. Wang H, Wang H, Zheng W, Wang D, Sun C, Dong J, et al. OTULIN’s influence on neuroinflammation and pain modulation in trigeminal neuralgia. CNS Neurosci Ther. (2024) 30:e70006. doi: 10.1111/cns.70006
67. Li J, Tian M, Hua T, Wang H, Yang M, Li W, et al. Combination of autophagy and NFE2L2/NRF2 activation as a treatment approach for neuropathic pain. Autophagy. (2021) 17:4062–82. doi: 10.1080/15548627.2021.1900498
68. Aguilar-Shea AL, Membrilla Md JA, Diaz-de-Teran J. Migraine review for general practice. Aten Primaria. (2022) 54:102208. doi: 10.1016/j.aprim.2021.102208
69. Mulder EJ, Van Baal C, Gaist D, Kallela M, Kaprio J, Svensson DA, et al. Genetic and environmental influences on migraine: a twin study across six countries. Twin Res. (2003) 6:422–31. doi: 10.1375/136905203770326420
70. Honkasalo ML, Kaprio J, Winter T, Heikkilä K, Sillanpää M, Koskenvuo M. Migraine and concomitant symptoms among 8167 adult twin pairs. Headache. (1995) 35:70–8. doi: 10.1111/j.1526-4610.1995.hed3502070.x
71. Jen JC. Familial hemiplegic migraine. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews(®). University of Washington, Seattle (1993). Copyright © 1993-2024.
72. Hogg SJ, Beavis PA, Dawson MA, Johnstone RW. Targeting the epigenetic regulation of antitumour immunity. Nat Rev Drug Discovery. (2020) 19:776–800. doi: 10.1038/s41573-020-0077-5
73. Gibson F, Hanly A, Grbic N, Grunberg N, Wu M, Collard M, et al. Epigenetic dysregulation in autoimmune and inflammatory skin diseases. Clin Rev Allergy Immunol. (2022) 63:447–71. doi: 10.1007/s12016-022-08956-8
74. Fila M, Pawlowska E, Szczepanska J, Blasiak J. Epigenetic connections of the TRPA1 ion channel in pain transmission and neurogenic inflammation - a therapeutic perspective in migraine? Mol Neurobiol. (2023) 60:5578–91. doi: 10.1007/s12035-023-03428-2
75. Iannone LF, Nassini R, Patacchini R, Geppetti P, De Logu F. Neuronal and non-neuronal TRPA1 as therapeutic targets for pain and headache relief. Temperature (Austin). (2023) 10:50–66. doi: 10.1080/23328940.2022.2075218
76. Gao H, Antony R, Srinivasan R, Wu P, Wang X, Li Y. UCHL1 regulates oxidative activity in skeletal muscle. PloS One. (2020) 15:e0241716. doi: 10.1371/journal.pone.0241716
77. Bilguvar K, Tyagi NK, Ozkara C, Tuysuz B, Bakircioglu M, Choi M, et al. Recessive loss of function of the neuronal ubiquitin hydrolase UCHL1 leads to early-onset progressive neurodegeneration. Proc Natl Acad Sci U.S.A. (2013) 110:3489–94. doi: 10.1073/pnas.1222732110
78. Atescelik M, Yilmaz M. Ubiquitin C-terminal hydrolase 1 is increased in migraine attack. Rev Neurol (Paris). (2023) 179:201–7. doi: 10.1016/j.neurol.2022.06.010
79. Xu Y, Lin F, Liao G, Sun J, Chen W, Zhang L. Ripks and neuroinflammation. Mol Neurobiol. (2024) 61:6771–87. doi: 10.1007/s12035-024-03981-4
80. Mishra R, Upadhyay A, Prajapati VK, Dhiman R, Poluri KM, Jana NR, et al. LRSAM1 E3 ubiquitin ligase: molecular neurobiological perspectives linked with brain diseases. Cell Mol Life Sci. (2019) 76:2093–110. doi: 10.1007/s00018-019-03055-y
81. Stemkowski P, García-Caballero A, Gadotti VM, M’Dahoma S, Huang S, Black SAG, et al. TRPV1 nociceptor activity initiates USP5/T-type channel-mediated plasticity. Cell Rep. (2016) 17:2901–12. doi: 10.1016/j.celrep.2016.11.047
82. Fila M, Sobczuk A, Pawlowska E, Blasiak J. Epigenetic connection of the calcitonin gene-related peptide and its potential in migraine. Int J Mol Sci. (2022) 23(11):6151. doi: 10.3390/ijms23116151
83. Walker CS, Raddant AC, Woolley MJ, Russo AF, Hay DL. CGRP receptor antagonist activity of olcegepant depends on the signalling pathway measured. Cephalalgia. (2018) 38:437–51. doi: 10.1177/0333102417691762
84. Kurtzeborn K, Kwon HN, Kuure S. MAPK/ERK signaling in regulation of renal differentiation. Int J Mol Sci. (2019) 20(7):1779. doi: 10.3390/ijms20071779
85. Servant MJ, Giasson E, Meloche S. Inhibition of growth factor-induced protein synthesis by a selective MEK inhibitor in aortic smooth muscle cells. J Biol Chem. (1996) 271:16047–52. doi: 10.1074/jbc.271.27.16047
86. Wang W, Zhu Y, Sun Z, Jin C, Wang X. Positive feedback regulation between USP15 and ERK2 inhibits osteoarthritis progression through TGF-β/SMAD2 signaling. Arthritis Res Ther. (2021) 23:84. doi: 10.1186/s13075-021-02456-4
87. Chen Q, Hang Y, Zhang T, Tan L, Li S, Jin Y. USP10 promotes proliferation and migration and inhibits apoptosis of endometrial stromal cells in endometriosis through activating the Raf-1/MEK/ERK pathway. Am J Physiol Cell Physiol. (2018) 315:C863–c872. doi: 10.1152/ajpcell.00272.2018
88. Ma X, Wang D, Li N, Gao P, Zhang M, Zhang Y. Hippo kinase NDR2 inhibits IL-17 signaling by promoting Smurf1-mediated MEKK2 ubiquitination and degradation. Mol Immunol. (2019) 105:131–6. doi: 10.1016/j.molimm.2018.10.005
89. Sala-Gaston J, Costa-Sastre L, Pedrazza L, Martinez-Martinez A, Ventura F, Rosa JL. Regulation of MAPK signaling pathways by the large HERC ubiquitin ligases. Int J Mol Sci. (2023) 24(5):4906. doi: 10.3390/ijms24054906
90. Ahn YH, Kurie JM. MKK4/SEK1 is negatively regulated through a feedback loop involving the E3 ubiquitin ligase itch. J Biol Chem. (2009) 284:29399–404. doi: 10.1074/jbc.M109.044958
91. Cuny GD, Degterev A. RIPK protein kinase family: Atypical lives of typical kinases. Semin Cell Dev Biol. (2021) 109:96–105. doi: 10.1016/j.semcdb.2020.06.014
92. Ellwanger K, Briese S, Arnold C, Kienes I, Heim V, Nachbur U, et al. XIAP controls RIPK2 signaling by preventing its deposition in speck-like structures. Life Sci Alliance. (2019) 2(4):e201900346. doi: 10.1101/545400
93. Trindade BC, Chen GY. NOD1 and NOD2 in inflammatory and infectious diseases. Immunol Rev. (2020) 297:139–61. doi: 10.1111/imr.v297.1
94. Kuss-Duerkop SK, Keestra-Gounder AM. NOD1 and NOD2 activation by diverse stimuli: a possible role for sensing pathogen-induced endoplasmic reticulum stress. Infect Immun. (2020) 88(7):e00898–19. doi: 10.1128/IAI.00898-19
95. Takiuchi T, Nakagawa T, Tamiya H, Fujita H, Sasaki Y, Saeki Y, et al. Suppression of LUBAC-mediated linear ubiquitination by a specific interaction between LUBAC and the deubiquitinases CYLD and OTULIN. Genes Cells. (2014) 19:254–72. doi: 10.1111/gtc.2014.19.issue-3
96. Hitotsumatsu O, Ahmad RC, Tavares R, Wang M, Philpott D, Turer EE, et al. The ubiquitin-editing enzyme A20 restricts nucleotide-binding oligomerization domain containing 2-triggered signals. Immunity. (2008) 28:381–90. doi: 10.1016/j.immuni.2008.02.002
97. O’Donnell MA, Ting AT. RIP1 comes back to life as a cell death regulator in TNFR1 signaling. FEBS J. (2011) 278:877–87. doi: 10.1111/j.1742-4658.2011.08016.x
98. Xia Q, Zhan G, Mao M, Zhao Y, Li X. TRIM45 causes neuronal damage by aggravating microglia-mediated neuroinflammation upon cerebral ischemia and reperfusion injury. Exp Mol Med. (2022) 54:180–93. doi: 10.1038/s12276-022-00734-y
99. Deng L, Meng T, Chen L, Wei W, Wang P. The role of ubiquitination in tumorigenesis and targeted drug discovery. Signal Transduct Target Ther. (2020) 5:11. doi: 10.1038/s41392-020-0107-0
100. Sendo S, MaChado CRL, Boyle DL, Benschop RJ, Perumal NB, Choi E, et al. Dysregulated NUB1 and neddylation enhances rheumatoid arthritis fibroblast-like synoviocyte inflammatory responses. Arthritis Rheumatol. (2024) 76:1252–62. doi: 10.1002/art.42856
101. Hyer ML, Milhollen MA, Ciavarri J, Fleming P, Traore T, Sappal D, et al. A small-molecule inhibitor of the ubiquitin activating enzyme for cancer treatment. Nat Med. (2018) 24:186–93. doi: 10.1038/nm.4474
102. Bhattacharyya S, Oblinger JL, Beauchamp RL, Yin Z, Erdin S, Koundinya P, et al. Proteasomal pathway inhibition as a potential therapy for NF2-associated meningioma and schwannoma. Neuro Oncol. (2023) 25:1617–30. doi: 10.1093/neuonc/noad037
103. Li Z, Liu B, Lambertsen KL, Clausen BH, Zhu Z, Du X, et al. USP25 inhibits neuroinflammatory responses after cerebral ischemic stroke by deubiquitinating TAB2. Adv Sci (Weinh). (2023) 10:e2301641. doi: 10.1002/advs.202301641
104. Huang C, Wang W, Huang H, Jiang J, Ding Y, Li X, et al. Kawasaki disease: ubiquitin-specific protease 5 promotes endothelial inflammation via TNFα-mediated signaling. Pediatr Res. (2023) 93:1883–90. doi: 10.1038/s41390-022-02341-z
105. Ren GM, Li J, Zhang XC, Wang Y, Xiao Y, Zhang XY, et al. Pharmacological targeting of NLRP3 deubiquitination for treatment of NLRP3-associated inflammatory diseases. Sci Immunol. (2021) 6(58):eabe2933. doi: 10.1126/sciimmunol.abe2933
106. Polderman TJ, Benyamin B, de Leeuw CA, Sullivan PF, van Bochoven A, Visscher PM, et al. Meta-analysis of the heritability of human traits based on fifty years of twin studies. Nat Genet. (2015) 47:702–9. doi: 10.1038/ng.3285
107. Hansen JM, Lipton RB, Dodick DW, Silberstein SD, Saper JR, Aurora SK, et al. Migraine headache is present in the aura phase: a prospective study. Neurology. (2012) 79:2044–9. doi: 10.1212/WNL.0b013e3182749eed
108. de Boer I, van den Maagdenberg A, Terwindt GM. Advance in genetics of migraine. Curr Opin Neurol. (2019) 32:413–21. doi: 10.1097/WCO.0000000000000687
109. Russell MB, Olesen J. Increased familial risk and evidence of genetic factor in migraine. BMJ. (1995) 311:541–4. doi: 10.1136/bmj.311.7004.541
110. Gerring ZF, McRae AF, Montgomery GW, Nyholt DR. Genome-wide DNA methylation profiling in whole blood reveals epigenetic signatures associated with migraine. BMC Genomics. (2018) 19:69. doi: 10.1186/s12864-018-4450-2
111. Goadsby PJ, Edvinsson L, Ekman R. Release of vasoactive peptides in the extracerebral circulation of humans and the cat during activation of the trigeminovascular system. Ann Neurol. (1988) 23:193–6. doi: 10.1002/ana.410230214
112. Bashir A, Lipton RB, Ashina S, Ashina M. Migraine and structural changes in the brain: a systematic review and meta-analysis. Neurology. (2013) 81:1260–8. doi: 10.1212/WNL.0b013e3182a6cb32
113. Gaist D, Hougaard A, Garde E, Reislev NL, Wiwie R, Iversen P, et al. Migraine with visual aura associated with thicker visual cortex. Brain. (2018) 141:776–85. doi: 10.1093/brain/awx382
Keywords: ubiquitination, migraine, inflammation, biomarker, central sensitization, peripheral sensitization
Citation: Zhu Q, Yang J, Shi L, Zhang J, Zhang P, Li J and Song X (2025) Exploring the role of ubiquitination modifications in migraine headaches. Front. Immunol. 16:1534389. doi: 10.3389/fimmu.2025.1534389
Received: 25 November 2024; Accepted: 09 January 2025;
Published: 31 January 2025.
Edited by:
Wenyi Jin, City University of Hong Kong, Hong Kong SAR, ChinaReviewed by:
Zhe Pei, Virginia Tech, United StatesCopyright © 2025 Zhu, Yang, Shi, Zhang, Zhang, Li and Song. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaoli Song, ZG9jdG9yc29uZzExMDdAMTYzLmNvbQ==
†These authors share first authorship