 Allan Feng1,2
Allan Feng1,2 Michael V. Gonzalez3
Michael V. Gonzalez3 Muge Kalaycioglu1,2
Muge Kalaycioglu1,2 Xihui Yin1,2
Xihui Yin1,2 Melanie Mumau3Saishravan Shyamsundar3Mateo Sarmiento Bustamante3Sarah E. Chang1,2Shaurya Dhingra1,2Tea Dodig-Crnkovic4
Melanie Mumau3Saishravan Shyamsundar3Mateo Sarmiento Bustamante3Sarah E. Chang1,2Shaurya Dhingra1,2Tea Dodig-Crnkovic4 Jochen M. Schwenk4Tarun Garg5Kazuyuki Yoshizaki6
Jochen M. Schwenk4Tarun Garg5Kazuyuki Yoshizaki6 Frits van Rhee5
Frits van Rhee5 David C. Fajgenbaum3*
David C. Fajgenbaum3* Paul J. Utz1,2*
Paul J. Utz1,2*- 1Department of Medicine, Division of Immunology and Rheumatology, Stanford University School of Medicine, Stanford, CA, United States
- 2Institute for Immunity, Transplantation and Infection, Stanford University School of Medicine, Stanford, CA, United States
- 3Center for Cytokine Storm Treatment & Laboratory, University of Pennsylvania, Philadelphia, PA, United States
- 4Science for Life Laboratory, Department of Protein Science, KTH Royal Institute of Technology, Stockholm, Sweden
- 5Myeloma Center, University of Arkansas for Medical Sciences, Little Rock, AR, United States
- 6Department of Biomolecular Science and Regulation, The Institute of Scientific and Industrial Research, Osaka University, Osaka, Japan
Introduction: Idiopathic Multicentric Castleman Disease (iMCD) is a polyclonal lymphoproliferative disorder involving cytokine storms that can lead to organ failure and death. The cause of iMCD is unknown, but some clinical evidence suggests an autoimmune etiology. For example, connective tissue disorders (CTDs) and iMCD share many clinical features, and autoantibodies have been anecdotally reported in individual iMCD patients. This study investigates whether common autoantibodies are shared across iMCD patients.
Methods: We assembled custom bead-based protein arrays consisting of 52 autoantigens traditionally associated with CTDs and 38 full-length cytokines and screened serum samples from 101 iMCD patients for IgG autoantibodies. We also screened samples with a 1,103-plex array of recombinant human protein fragments to identify additional autoantibody targets. Finally, we performed receptor blocking assays on select samples with anti-cytokine autoantibodies (ACAs) identified by array.
Results: We found that an increased proportion of iMCD patients (47%) tested positive for at least one CTD-associated autoantibody compared to healthy controls (HC) (17%). Commonly detected CTD-associated autoantibodies were associated with myositis and overlap syndromes as well as systemic lupus erythematosus (SLE) and Sjögren’s Syndrome (SS). ACAs were also detected in a greater proportion of iMCD patients (38%) compared to HC (10%), while the protein fragment array identified a variety of other autoantibody targets. One iMCD sample tested positive for receptor blocking against interferon-ω (IFNω).
Discussion: IgG autoantibodies binding autoantigens associated with common CTDs and cytokines are elevated in iMCD patients compared to HC, suggesting that autoimmunity may be involved in iMCD pathogenesis.
Introduction
Human herpesvirus-8 (HHV-8)-negative/idiopathic multicentric Castleman disease (iMCD) is a rare, hematologic disorder involving multiple enlarged lymph nodes with characteristic histopathology, cytopenias, and systemic inflammation due to a cytokine storm often including interleukin-6 (IL-6) (1–3). Beyond these unifying features, iMCD is clinically heterogenous and is further categorized into three subtypes: (i) iMCD with thrombocytopenia, anasarca, fever, renal failure/reticulin fibrosis and organomegaly (iMCD-TAFRO), (ii) iMCD with idiopathic plasmacytic lymphadenopathy (iMCD-IPL) involving hypergammaglobulinemia and thrombocytosis, and (iii) iMCD-not otherwise specified (iMCD-NOS) which does not meet the criteria for other subtypes (4). Whereas multicentric Castleman disease (MCD) can be caused by uncontrolled infection with HHV-8 (HHV-8-associated MCD) or monoclonal plasma cells (POEMS-associated MCD), the etiology of all three subtypes of iMCD is unknown.
Several hypotheses for the etiology of iMCD have been proposed including infection, autoimmunity, autoinflammation, and a neoplastic process. Given the heterogeneity of clinical subtypes and variability of response to anti-IL-6 therapy, iMCD pathogenesis may involve multiple mechanisms. Recent studies suggest that an acute infectious etiology is unlikely to be the cause of iMCD and limited genomic studies have failed to identify shared molecular aberrations (5, 6). However, other studies suggest that investigation of autoimmunity may be warranted given anecdotal reports of autoantibodies in iMCD (7–11) and the clinical overlap between iMCD and connective tissue disorders (CTDs) like systemic lupus erythematosus (SLE). In this study, we investigate the role autoimmunity may play in iMCD pathogenesis by quantifying levels of serum immunoglobulin G (IgG) autoantibodies using large scale protein arrays.
Materials and methods
Cohort selection and study approval
Samples from iMCD patients were obtained from the University of Pennsylvania (UP, n = 44 samples from 38 patients; six patients with flare/remission samples), University of Arkansas for Medical Sciences (UAMS, n = 51 samples from 45 patients; five patients with flare/remission samples), and Osaka University (OU, n = 26 samples from 18 patients; eight patients with flare/remission samples). Flare and remission designations have been used previously. Disease flare was determined based on clinical features and laboratory test results, including hypoalbuminemia (<3.5 g/dL), elevated CRP (>10 mg/L), anemia (hemoglobin < 13.5 g/dL), renal dysfunction (creatinine > 1.3 mg/dL), constitutional symptoms, and fluid accumulation. Remission was defined as CRP < 10 mg/L, albumin > 3.5 g/dL, hemoglobin > 11.5 g/dL and currently not undergoing hospitalization (12). Hodgkin Lymphoma (HL) samples (n = 20) were obtained from UP. HC (n = 30) were obtained from Stanford Blood Bank and Stanford Hospital, UP, and OU. Five positive control plasma samples from patients with CTDs were purchased from Immunovision, while the positive control sample with anti-IFNγ autoantibodies was collected from a patient with atypical mycobacterial infection (AMI) at Stanford Hospital. Serum samples from all centers were obtained following informed consent and IRB approval (Penn cohort: IRB# 824758, Osaka University: IRB# 2021390, University of Arkansas for Medical Sciences: IRB# 249901).
Bead-based antigen arrays
Two custom antigen panels were used to generate bead arrays as previously described (13–20). The CTD array consisted of 52 antigens associated with categories of traditional CTDs including Scleroderma, Myositis/Overlap Syndromes, SLE/Sjögren’s Syndrome (SS), gastrointestinal(GI)/Endocrine, DNA-Associated, and Inflammation/Stress. The ACA array included 38 cytokines and cell surface proteins. Briefly, antigens were coupled to color-coded carboxylated magnetic beads (MagPlex, Luminex Corp.). 8 μg of antigen or control antibody was diluted in phosphate-buffered saline (PBS) and transferred to 96-well plates. One bead ID was processed without coupling to any antigen to quantify bead surface binding. Beads were distributed into 96-well plates (Greiner BioOne) and washed in PBS using a 96-well plate washer (Biotek). Each bead was activated by incubating with 0.5 mg 1-ethyl-3(3 dimethylaminopropyl)carbodiimide (Pierce) and 0.5 mg N-hydroxysuccinimide (Pierce) in 100 μl of phosphate buffer for 20 min. After incubation, beads were washed and resuspended in activation buffer (0.05 M 2-N-Morpholino Ethane Sulfonic acid, MES, pH 5.0). Diluted antigens and control antibodies were incubated with beads for 2 hours at room temperature. Beads were washed three times in 100 μl PBS-Tween, re-suspended in 60 μl storage buffer (Blocking reagent for Enzyme-Linked Immunosorbent Assay, ELISA, Roche), and stored at 4°C. Prototype human plasma samples were used for validation of bead arrays. Beads were combined in storage buffer (Blocking reagent for ELISA, Roche) to create each respective array. A comprehensive list of antigens, vendors, and catalogue numbers is in Supplementary Tables S1, S2.
To explore a broader set of antigens, a custom human protein fragment array (25-150 amino acids) was constructed in collaboration with the KTH Royal Institute of Technology as previously described (21) and similarly to the full-length CTD and ACA arrays with minor changes. In short, protein epitope signature tag (PrEST) antigens were coupled to carboxylated magnetic beads (MagPlex-C, Luminex Corp.), which were distributed in 96-well plates (Greiner BioOne, Longwood, FL), washed and re-suspended in phosphate buffer (0.1 M NaH2PO4, pH 6.2) using a plate magnet (Dexter) and a plate washer (EL406, Biotek, Winooski, VT). Beads were activated with 0.5 mg 1-ethyl-3(3 dimethylaminopropyl)carbodiimide (Pierce) and 0.5 mg N-hydroxysuccinimide (Pierce) in 100 μl phosphate buffer and incubated for 20 min incubation on a shaker (Grant Bio). After washing antigens were added at a concentration of 40 μg/ml in activation buffer. Activated beads and antigens were incubated together for coupling for 2 hours at room temperature. Conjugated beads were washed in PBS-T and then combined in storage buffer (Blocking reagent for ELISA, Roche).
Array probing
Serum samples were diluted 1:100 in 0.05% PBS-Tween supplemented with 1% (w/v) bovine serum albumin and transferred into 96-well plates. 5 µl of bead array were distributed into each well of a 384-well plate (Greiner BioOne). 45 µl of 1:100 diluted sera were transferred in duplicate into the 384-well plate containing the bead array. Samples and beads were incubated for 60 min on a shaker at room temperature and then washed with 3 × 60 µl PBS-Tween on a plate washer (EL406, Biotek). 50 µl of 1:1000 diluted R-phycoerythrin (R-PE) conjugated Fc-γ-specific goat anti-human IgG F(ab’)2 fragment (Jackson ImmunoResearch) were added to each well for detection of bound human IgG. After 30 min of incubation with the secondary antibody, the plate was washed with 3 × 60 µl PBS-Tween and re-suspended in 50 µl PBS-Tween. Samples were analyzed using a FlexMap3D™ instrument (Luminex Corp.). Binding events were measured as Median Fluorescence Intensity (MFI).
Cell-based receptor blocking assays
Serum samples positive by array for anti-IFNα2 and anti-IFNγ were assessed using blocking assays and U937 (ATCC CRL1593) cell lines as previously described (22, 23). Cells (400,000 cells/condition) were incubated with media, 10% HC serum, 10% patient serum, or blocking serum from a prototype patient with atypical mycobacterial infection (AMI) for 15 mins. Cells were then stimulated with 2.5 ng/mL of either IFNα2 (HumanKine, HZ-1066) or IFNγ (HumanKine, HZ-1301). Cells were analyzed on a BD LSR-II flow cytometer, and FACS data were analyzed using FlowJo.
To evaluate the blocking activity of anti–IFNλ2, anti–IFNβ, anti–IFNω, and anti-IL-31 autoantibodies, we used SEAP-reporter HEK293 cell lines stably expressing human receptors specific to these antigens, along with necessary signaling proteins. For TNFα blocking assays, a luciferase-reporter HVEM-293 cell line was employed. We determined the effective concentration (EC75) of the antigens and used this concentration at 10% in a mixture with either 10% healthy control serum or patient serum/plasma, making up a total volume of 50 µL with the cells. Blocking activity was measured by absorbance at 620 nm using a BioTek Synergy HTX Multimode Reader. The results were normalized to the negative controls (cells with only antigen, without any antibody) to demonstrate the antigen stimulation index as an inverse representation of blocking activity. For samples showing blocking activity, additional serum dilutions were prepared to identify the endpoint of the blocking effect.
Clinical-grade autoantibody assays
Data from clinical grade assays were extracted from medical records and performed by diagnostic laboratories (primarily Quest Diagnostics and Labcorp) as part of clinical evaluation and diagnosis. A list of clinical tests and associated catalogue numbers are available in Supplementary Table S3. The clinical autoantibody assays (e.g., Anti-nuclear antibody, ANA titers) evaluated are expressed as a ratio of a serial dilution indicating the amount of a given antibody in a sample.
High throughput protein quantification
Somalogic’s SOMAscan technology was used to quantify 6,383 proteins in serum from select iMCD patients in the UP cohort (n = 10). Proteins overlapping between the bead-based arrays and SOMAscan were used to investigate quantitative differences between samples with positive and negative autoantibody signals (24).
Extraction of CHA score as a measure of disease severity
A C-reactive protein (CRP), Hemoglobin (Hgb), and Albumin (Alb) score was calculated for each iMCD patient as a measure of clinical severity and disease activity (CHA score) (25). Peak values were selected for inclusion in the CHA score regardless of lab test timing.
Statistical analysis
R v4.2.2 and RStudio v2023.06.2 were used to perform analyses (26, 27). MFI values were normalized by subtracting MFI for “bare bead” IDs from MFI values for conjugated bead IDs. Replicate MFI values were averaged.
Autoantibody testing in a clinical setting typically involves setting a standard cutoff for what is considered “positive” for each antigen. In many cases, this is set at a level of at least 3 standard deviations (SD) above the mean for healthy controls who are known to not have this reactivity. We elected to use a much more stringent 5SD above the HC mean cutoff to reduce false positivity rates for individual autoantibodies. Highly-stringent cutoffs also facilitate identification of samples with higher levels of autoantibodies that may be prioritized for mechanistic studies such as the ACA neutralization assays in this study. Thus, samples were considered “positive” for autoantibodies if normalized MFI was greater than 5SD above the average MFI for HC and greater than 3,000 MFI (14). Samples run on the protein fragment array were considered autoantibody-positive if the normalized MFI value was >5SD above the HC mean and greater than 500 MFI. Patients with longitudinal samples were deemed autoantibody-positive if the mean MFI of all timepoints was >5SD above the HC mean and MFI was >3,000.
Statistical differences in MFI were determined using two-sided Wilcoxon rank-sum tests with Bonferroni correction for multiple comparisons. Statistical differences in MFI between paired samples was determined using one-tailed signed rank tests. Fisher’s exact test was used to determine associations between autoantibody positivity and disease status. Spearman’s correlation was used to determine the relationship between CHA score and autoantibody prevalence. A Fisher’s exact test was used to compare overall prevalence of autoantibodies between experimental groups. GraphPad Prism was used for dot and line plots. R package Complexheatmap was used to visualize heatmaps (28).
Results
Autoantibodies targeting autoantigens traditionally associated with CTDs are elevated in iMCD
We investigated autoantibody prevalence in six categories of CTD antigens: Scleroderma, Myositis/Overlap Syndromes, SLE/SS, GI/Endocrine, DNA-Associated, and Inflammation/Stress across three cohorts of iMCD patients. There were no sex-based differences (p = 0.38) between cohorts, but there were statistically significant differences between cohorts for ethnicity, age, iMCD subtype, and therapy prior to blood draw (Table 1).

Table 1. Clinical characteristics of iMCD patients from the University of Pennsylvania (UP), University of Arkansas for Medical Sciences (UAMS), and Osaka University (OU) cohorts.
Overall, significantly more iMCD patients were positive for autoantibodies targeting at least one autoantigen on the CTD array (n = 47 of 101, 47%) than HCs (n = 5 of 30, 17%; P = 0.003; Figures 1A, B, Supplementary Figures S1, S2A). The most frequently detected CTD-associated autoantibodies in iMCD patients were associated with Myositis/Overlap Syndromes (n = 22, 22%; P = 0.026) and SLE/SS (n = 18, 18%; P = 0.073), although they did not reach statistical significance compared to HCs after Bonferroni correction. The most common Myositis/Overlap Syndrome autoantibodies in iMCD patients were anti-signal recognition particle 54 (SRP54; n = 7, 7%), anti-Mi-2 (n = 6, 6%), and anti-EJ (n = 4, 4%) (Figure 1C). Most SLE/SS-associated autoantibodies detected in iMCD patients targeted La (n = 10, 10%), Ro60 (n = 5, 5%), and Sm/RNP (n = 4, 4%) (w 1D). None of these autoantibodies were detected in HCs. Autoantibodies binding DNA-associated proteins, such as histones and nucleolin, were also found in six (6%) iMCD patients but not in HCs (Figure 1A, fifth horizontal panel). When comparing iMCD and HL, which has been previously found to have increased autoantibodies compared to HCs and solid malignancies (33), there were similar proportions of CTD-associated autoantibody positive patients (Figure 1B). However, several autoantibodies found in high frequencies in iMCD samples (e.g., anti-SRP54, anti-EJ, anti-Ro60, anti-Sm/RNP) were not detected in any HL samples (Figures 1C, D). We also screened five plasma samples from patients with CTDs using the CTD array with more focused antigen content and detected specific autoantibodies associated with each patient’s CTD (Supplementary Figure S3A). Taken together, a variety of autoantibodies are more commonly identified in iMCD than HC, and most of them are associated with SLE, SS, myositis, and overlap syndromes.

Figure 1. Autoantibodies associated with CTDs are prevalent in iMCD patients. (A) Heatmap displaying serum IgG AAbs identified by a 52-plex array of autoantigens associated with traditional CTDs in iMCD patients (n = 101), healthy controls (HC, n = 30) and Hodgkin’s Lymphoma (HL, n = 20) samples. (B) Proportion of samples that were positive for at least one autoantibody target in the CTD array (* = P < 0.05). A Fisher’s exact test was used to determine significance between groups. (C) Dot plots comparing MFI values for three of the most targeted autoantigens associated with myositis and overlap syndromes in iMCD patients. (D) Dot plots displaying three of the most commonly targeted autoantigens associated with SLE and SS in iMCD patients.
Clinical-grade autoantibody lab tests correlate with the research-grade CTD array
34 samples from 29 iMCD patients from the UP cohort were also screened with a clinical-grade ANA (Anti-nuclear antibody) test or other clinical-grade autoantibody screened for by the CTD array (Ro, La, Sm, and RNP) as part of routine clinical care. Results from clinical-grade assays largely correlated with those from the CTD array (Supplementary Table S4). Supplementary Table S4 shows direct comparisons between clinical autoantibody tests and array based results. Concordance was observed in 15/16 patients (94%) for anti-SSA, 12/14 patients (86%) for anti-SSB, and 9/9 patients (100%) for anti-Smith and anti-RNP assays.
Prevalence of clinical autoantibodies in iMCD
To further interrogate autoantibody burden in iMCD, we queried the ACCELERATE registry (29) to identify 89 iMCD patients who had clinical autoantibodies measured. Thirty-five patients (38%) had a positive ANA result (Table 2), and ANA titers, when available, were generally mild-to-moderately elevated across iMCD. No significant differences were observed between clinical subtypes (iMCD-TAFRO, iMCD-NOS, iMCD-IPL, Supplementary Figure S4). Among other autoantibodies, for which there were >30 patients assessed, a notable proportion were positive (e.g., anti-SSA, 22%; anti-SSB, 15%; anti-dsDNA, 6%; anti-Sm, 6%; Direct Coombs test, 44%, Table 2). A similar prevalence of these autoantibodies was found among the UP cohort of patients who were tested for autoantibodies by clinical-grade assays and research-grade CTD and ACA arrays (Table 2).
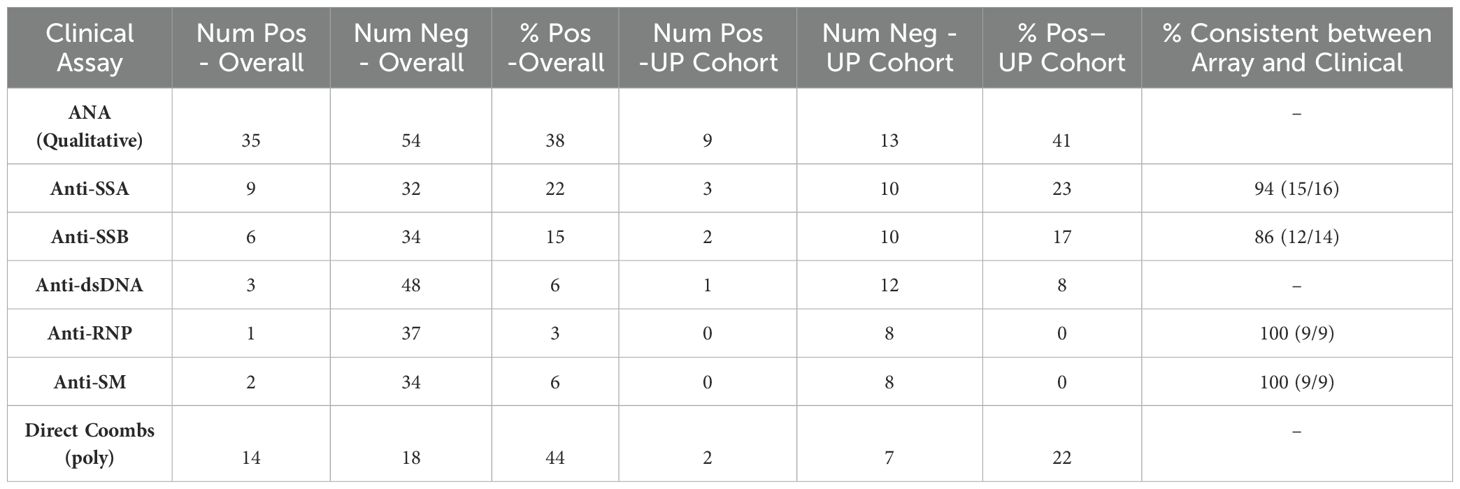
Table 2. Autoantibody clinical assay results in ACCELERATE iMCD patient cohort.
Prevalence of autoantibodies targeting secreted and cell-surface proteins in iMCD
Next, we measured ACAs in iMCD patients using a 38-plex array. We first investigated anti-IL-6 antibodies given that a large portion of iMCD patients with known anti-IL-6 therapy status (n = 76) were on anti-IL-6 therapy with siltuximab or had received siltuximab within a year of serum collection (n = 30, 39%). As expected, a high proportion of iMCD patients on siltuximab (n = 27, 90%) tested positive for anti-IL-6 by the ACA array. Anti-IL-6 was then removed from further analyses.
Overall, ACAs were detected in significantly more iMCD patients (n = 38, 38%) than HCs (n = 3, 10%; P = 0.004) (Figures 2A, B, Supplementary Figures S2B, S5). The most common ACAs identified in iMCD bound oncostatin M (OSM) (n = 8, 8%), tumor necrosis factor (TNFα; n = 5, 5%), integral membrane protein 2B (ITM2B; n = 5, 5%), IFNϵ (n = 4, 4%), IFNω (n = 4, 4%), and IFNλ2 (n = 3, 3%; Figure 2C). There was no significant difference in the proportions of ACA positive iMCD and HL patients with 10 HL patients (50%) positive for at least one ACA (Figure 2B). However, certain ACAs such as anti-ITM2B, anti-TNFα, and anti-IFNλ2 were commonly detected in iMCD patients but in no HL or HC samples (Figure 2C). We also screened a positive control sample from a patient with atypical mycobacterial infection (AMI) with known anti-IFNγ autoantibodies, which tested positive for anti-IFNγ by the ACA array as expected (Supplementary Figure S3B).
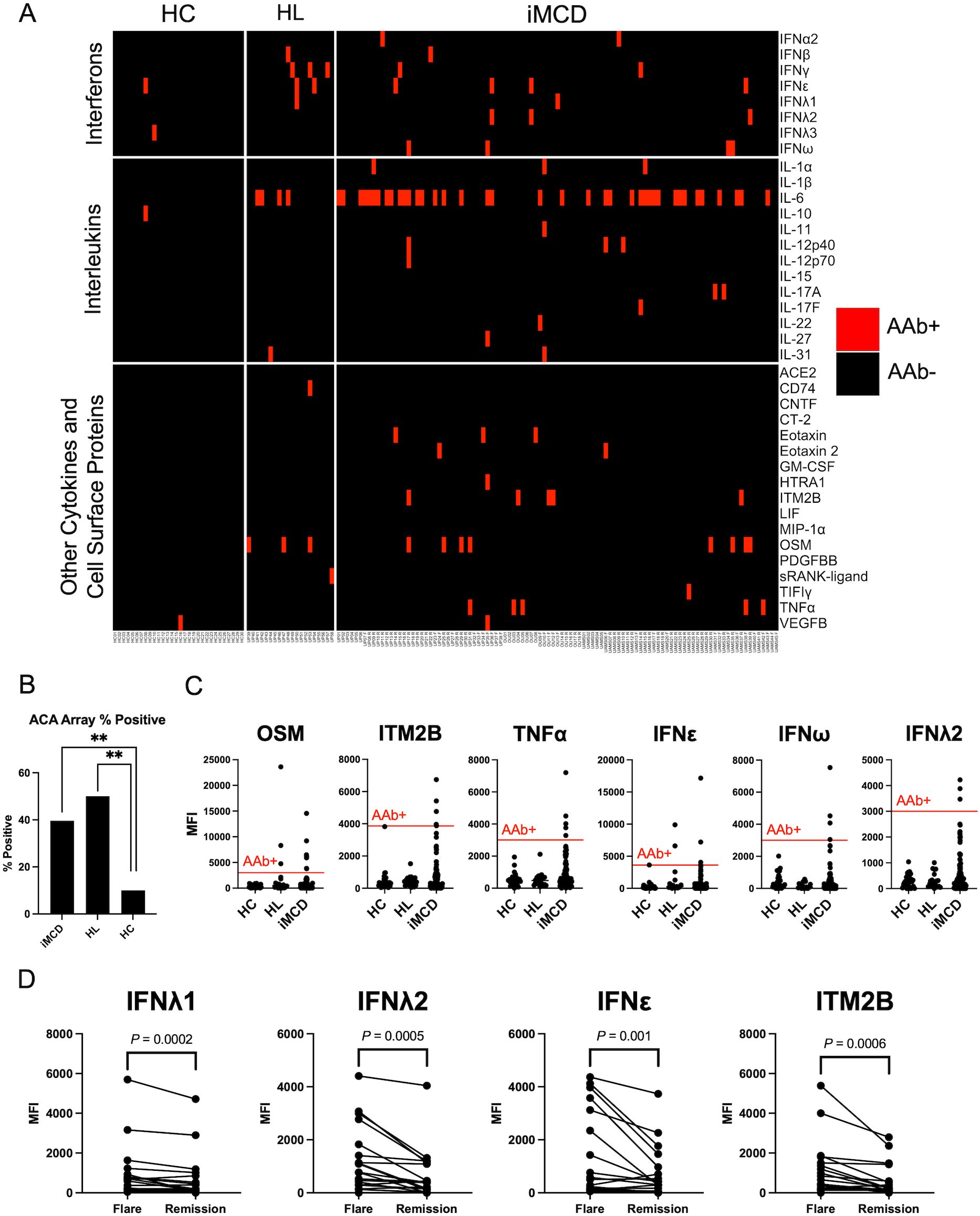
Figure 2. IgG ACAs are common in iMCD patients. (A) Heatmap displaying serum IgG ACAs identified by a 38-plex array of secreted and cell-surface proteins in iMCD patients (n = 101), healthy controls (HC, n = 30) and Hodgkin’s Lymphoma (HL, n = 20) samples. (B) Proportion of samples that were positive for at least one ACA. iMCD and Lymphoma cohorts were significantly enriched for ACAs (** = P < 0.01) compared to HC. A Fisher’s exact test was used to determine significance between groups. (C) Dotplots comparing MFI values for six cytokines and cell surface proteins that were most commonly targeted by ACA in iMCD patients. (D) Longitudinal analysis comparing flare and remission disease state MFI values. Only comparisons that surpassed a Bonferroni-corrected P-value cutoff after Wilcoxon signed rank tests are shown.
We next investigated whether autoantibody levels were temporally associated with disease flare vs. remission states. Longitudinal flare and remission samples were available for 19 iMCD patients (n = 6, UP cohort; n = 5, UAMS cohort; n = 8, OU cohort). No differences were observed in the CTD array, but several ACAs targeting cytokines including IFNλ1 (P = 0.0002), IFNλ2 (P = 0.0005), IFNϵ (P = 0.001), and ITM2B (P = 0.0006) were significantly elevated in flare samples compared to matched remission samples (Figure 2D).
As an orthogonal approach to identify antibodies to secreted proteins, we utilized a 1,103-plex array of recombinant human protein fragments representing 556 secreted proteins. Sufficient bead pools allowed for analyses of the two largest US cohorts, UP (n = 38 patients) and UAMS (n = 45 patients), and HCs. Autoantibodies recognizing 36 protein fragments in the array were identified in at least one patient from both cohorts (Supplementary Figures S6A, B). The most common autoantibodies detected in iMCD samples but in no HCs included anti-JAM3 (7%), anti-LRG1 (7%), anti-HTRA1 (7%), and anti-AGGF1 (7%) (Supplementary Figures S6C, D). Overall, the protein fragment array identified additional candidate antigens, but there was little overlap with the ACA array built on full-length proteins.
Receptor blocking activity in iMCD sera
Next, we performed receptor blocking activity assays to determine whether the ACAs detected in this study are receptor blocking. Upon stimulation with IFNα2 and IFNy, cells treated with patient or control serum had similar levels of pSTAT1 activation, suggesting that anti-IFNα2 and anti-IFNy present in patient sera from two patients in this study are non-blocking (Supplementary Figure S7A). A similar approach involved culturing reporter cell-lines with antigen-serum complexes for 18 hours for TNFα and 24 hours for the other cytokines to assess the blocking activity of antibodies. ACA+ iMCD sera were found to be non-blocking for IFNλ2, IFNβ, IL-31, and TNFα (Supplementary Figure S7B). However, one sample exhibited robust blocking activity against IFNω, which continued to display blocking activity with serial dilutions until five-fold dilution (Supplementary Figure S7C).
Autoantibody levels do not correlate with clinical severity or serum protein levels
To determine if detected autoantibodies were correlated with iMCD subtypes, we re-analyzed the autoantibody results after separating patients into iMCD-TAFRO, iMCD-NOS, and iMCD-IPL. Each subtype had significantly higher proportions iMCD patients with CTD-associated autoantibodies and ACAs compared to HC, except for ACAs in iMCD-TAFRO (Supplementary Tables S5A, B). To examine whether these autoantibodies correlated with clinical severity, we measured CHA scores in the UP cohort where sufficient data existed (25) and found no correlation between CHA score and number of autoantibody positive iMCD patients (Supplementary Figures S8A, B). We also evaluated whether ACA+ patients had increased serum levels of target cytokines. We quantified serum levels of IFNϵ, and Eotaxin-2 from 10 patients and found no trend between presence or lack of ACA and serum cytokine levels (Supplementary Figure S9).
Discussion
A fundamental question in CD research is whether iMCD is an autoinflammatory disease, autoimmune disease, neoplastic disease, infectious disease, or some combination. Our aim was to determine whether autoimmune mechanisms may be involved in iMCD pathogenesis. Autoantibodies commonly found in certain CTDs were identified in a serum-based cohort of iMCD patients. Nearly half (47%) of iMCD patients tested positive for at least one CTD-associated autoantibody compared to only 17% of HCs. Most autoantibodies targeted proteins that complex with RNA and DNA and are linked to SLE and myositis. These nucleic acid-associated proteins contained on the CTD array include spliceosomal RNPs (e.g., Sm/RNP), Ro and La, nucleosomal proteins (e.g., Mi-2), aminoacyl-tRNA synthetases (e.g., EJ and Jo1), and SRP54. Interestingly, many of these autoantibodies were recently detected in patients with acute severe COVID-19 (14), and in pre-pandemic samples from patients with infections due to other pathogens in intensive care units (22). In iMCD patients, the increased prevalence of CTD-associated autoantibodies suggests that autoimmunity may partially explain a range of immune-mediated signs, symptoms, and clinical laboratory test abnormalities observed in iMCD that are also associated with autoimmune disorders such as SLE and myositis. Although our arrays did not contain platelet surface proteins or red blood cell (RBC) antigens, a recent article suggests immune-mediated destruction of platelets and RBCs occurs in some iMCD patients (30).
We also identified several ACAs in iMCD patients. ACAs have been increasingly recognized to be able to modulate autoimmune disease course. Neutralizing ACA against Type I IFNs such as IFNα, IFNβ, and IFNω are found in various autoimmune diseases such as SLE, SS, myasthenia gravis, autoimmune polyglandular syndrome type 1 (APS-1), and immunodysregulation polyendocrinopathy enteropathy X-linked syndrome (IPEX) (31). Interestingly, some ACAs do not neutralize but instead stabilize their cytokine targets as is the case with anti-IL-6 in APS-1 (32) and anti-IL-8 in ARDS (33). The ACAs we evaluated were found to be mostly non-receptor blocking, except for one patient with strong blocking activity against IFNω. The presence of anti-OSM in multiple iMCD samples is noteworthy, as OSM is an inflammatory cytokine from the IL-6 cytokine family. OSM is produced by activated T-cells, neutrophils, macrophages, and dendritic cells (34–37), and plays important roles in hematopoiesis, inflammation, fibrosis, mesenchymal stem cell differentiation, and cancer (38–42). TNF and IL-1β are other pro-inflammatory cytokines elevated in some iMCD patients (43, 44) and both cytokines stimulate IL-6 production (45, 46). Interestingly, we detected anti-TNFα in a high proportion of sera from iMCD patients (6%) but none of the samples displayed TNFα blocking activity.
Levels of several ACAs were elevated in flare versus remission states. We hypothesized that autoantibodies correlated with iMCD disease activity would be expected to be higher during disease flare and lower during remission. In total, we identified 4 ACAs, including anti-IFNλ (1 and 2), anti-IFNϵ, and anti-ITM2B, that were significantly upregulated during disease flare. Type-III interferons are critical for immune defense and promote inflammation in certain contexts. IFNλ is increased in several autoimmune rheumatic diseases, including SLE, and correlates with disease activity (47). Although Type-I IFN signatures have been identified in iMCD patients (12) and are hypothesized to work through a JAK dependent mechanism, Type-III IFNs have not been explored. Importantly, current biologic therapies targeting Type-I IFNs or their receptors do not block effects of IFNλ. Notably, TNF inhibition has been recently proposed as a therapy for iMCD (48).
In addition to helping with understanding of iMCD pathogenesis, this study may shed light on diagnosis. Currently, the diagnostic criteria for iMCD requires exclusion of known infectious, malignant, and autoimmune disorders that clinically mimic iMCD. These include autoimmune/autoinflammatory disorders such as SLE, rheumatoid arthritis (RA), systemic juvenile idiopathic arthritis (sJIA), and autoimmune lymphoproliferative syndrome (ALPS) (1). Importantly, all patients included in this study did not fulfill criteria for any of these CTDs. While we did not detect any autoantibody that was elevated across all iMCD patients to suggest it may be a diagnostic biomarker, future studies may identify autoantibodies that contribute to diagnosis. Moreover, increased levels of autoantibodies identified in this study suggest that autoimmune-mediated mechanisms may play a pathogenic role in a subset of iMCD patients. If autoimmune disease mechanisms drive disease, then treatments targeting CD19-positive cells such as bispecific antibodies and chimeric antigen receptor therapies should be considered.
Our study has several limitations. First, while autoantibodies suggest autoimmune involvement, the presence of autoantibodies does not directly implicate autoimmunity as a pathological mechanism. For example, Hodgkin’s disease and post-infectious etiologies can present with elevated autoantibodies. Autoantibody production could also be a response to hypercytokinemia during disease flares. Second, sample sizes were limited in both cases and healthy controls, especially in the longitudinal samples. Third, we chose one primary method for quantifying autoantibodies among several options. We chose to employ focused, bead-based arrays that would be more clinically valuable as a large majority of the autoantibodies we screened could be assessed by clinical lab assays, making results more rapidly translatable.
In summary, this study confirms the presence of autoantibodies in iMCD, which has been anecdotally reported in case studies (7–11). In fact, findings revealed autoantibodies targeting SLE and myositis-specific autoantigens and cytokines in iMCD. We also demonstrate that levels of specific ACA are increased in flare compared to remission. More research is needed to understand the role of these autoantibodies in iMCD pathogenesis. Future studies require expanded longitudinal sampling to ascertain changes in autoantibody levels between disease states. Further mechanistic work is also needed to understand potential functions of autoantibodies in iMCD.
Data availability statement
Deidentified array data are publicly available on the Gene Expression Omnibus (GEO) database with accession numbers GSE290521, GSE290527, GSE290532, GSE290549, and GSE290594. Code used for data analysis and figure generation may requested from the corresponding authors.
Ethics statement
The studies involving humans were approved by the IRBs of the University of Pennsylvania (IRB# 824758), Osaka University (IRB# 2021390), and the University of Arkansas for Medical Sciences (IRB# 249901). The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
Author contributions
AF: Data curation, Formal Analysis, Investigation, Methodology, Software, Visualization, Writing – original draft, Writing – review & editing. MG: Data curation, Formal Analysis, Writing – original draft, Writing – review & editing, Resources. MK: Data curation, Formal Analysis, Methodology, Visualization, Writing – original draft, Writing – review & editing. XY: Formal Analysis, Investigation, Writing – review & editing, Methodology. MM: Data curation, Writing – original draft, Writing – review & editing, Resources. SS: Data curation, Writing – review & editing, Resources. MB: Writing – review & editing. SC: Data curation, Investigation, Software, Writing – review & editing. SD: Data curation, Investigation, Writing – review & editing. TD: Data curation, Investigation, Methodology, Writing – review & editing. JS: Conceptualization, Methodology, Resources, Writing – review & editing. TG: Resources, Writing – review & editing. KY: Resources, Writing – review & editing. FR: Resources, Writing – review & editing. DF: Conceptualization, Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Writing – original draft, Writing – review & editing. PU: Conceptualization, Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. AF was funded by the Stanford University Vice Provost for Undergraduate Education’s (VPUE) Major Grant, a grant from the Stanford Medical Scholars Research Program, and the Berg Scholars Program. PU was supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health, R01 AI125197-04, NIH U01 AI150741-01S1, Penn Orphan Disease Center, Castleman Disease Collaborative Network, and the Henry Gustav Floren Trust. DF was supported by the National Heart, Lung, and Blood Institute (R01HL141408), US Food & Drug Administration (R01FD007632), and the Colton Center for Autoimmunity at the University of Pennsylvania.
Acknowledgments
The authors would like to acknowledge the Elana Amsterdam Health and Wellness Fund, Maria Elena Cobena and Peter Taylor, and Marjorie Raines for making this work possible. In addition, the authors would like to acknowledge Sharon Dickow for expert administrative support, Dr. Carl Ware at Sanford Burnham Prebys for gifting the cell line to us, and the patients and their families who participated in the research and provided samples for our studies.
Conflict of interest
DCF received consultancy fees and research funding from EUSA Pharma. PJU serves on the scientific advisory boards of SeraNova, 4DMT, Yolo Therapeutics, and the Arthritis National Research Foundation. He is co-founder and member of the Board of Directors of the Physician Scientist Support Foundation.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2025.1528465/full#supplementary-material
References
1. Fajgenbaum DC, Uldrick TS, Bagg A, Frank D, Wu D, Srkalovic G, et al. International, evidence-based consensus diagnostic criteria for HHV-8-negative/idiopathic multicentric Castleman disease. Blood. (2017) 129:1646–57. doi: 10.1182/blood-2016-10-746933
2. Fajgenbaum DC, van Rhee F, Nabel CS. HHV-8-negative, idiopathic multicentric Castleman disease: novel insights into biology, pathogenesis, and therapy. Blood. (2014) 123:2924–33. doi: 10.1182/blood-2013-12-545087
3. Dispenzieri A, Fajgenbaum DC. Overview of castleman disease. Blood. (2020) 135:1353–64. doi: 10.1182/blood.2019000931
4. Nishikori A, Nishimura MF, Nishimura Y, Otsuka F, Maehama K, Ohsawa K, et al. Idiopathic plasmacytic lymphadenopathy forms an independent subtype of idiopathic multicentric castleman disease. Int J Mol Sci. (2022) 23:10301. doi: 10.3390/ijms231810301
5. Nabel CS, Sameroff S, Shilling D, Alapat D, Ruth JR, Kawano M, et al. Virome capture sequencing does not identify active viral infection in unicentric and idiopathic multicentric Castleman disease. PloS One. (2019) 14:e0218660. doi: 10.1371/journal.pone.0218660
6. Carbone A, Borok M, Damania B, Gloghini A, Polizzotto MN, Jayanthan RK, et al. Castleman disease. Nat Rev Dis Primer. (2021) 7:1–18. doi: 10.1038/s41572-021-00317-7
7. Sun D-P, Chen W-M, Wang L, Wang Z, Liang JH, Zhu HY, et al. Clinical characteristics and immunological abnormalities of Castleman disease complicated with autoimmune diseases. J Cancer Res Clin Oncol. (2021) 147:2107–15. doi: 10.1007/s00432-020-03494-2
8. Pan Y, Cui Z, Wang S, Zheng D, Deng Z, Tian X, et al. Idiopathic multicentric Castleman disease with Sjögren’s syndrome and secondary membranous nephropathy: a case report and review of the literature. BMC Nephrol. (2020) 21:528. doi: 10.1186/s12882-020-02191-z
9. Saleh M, Hallbeck M, Sjöwall C. A rare case of idiopathic multicentric Castleman disease in a patient with long-standing systemic autoimmunity. Scand J Rheumatol. (2022) 51:161–3. doi: 10.1080/03009742.2021.1947591
10. Tabata S, Higuchi T, Tatsukawa S, Narimatsu K, Takeo H, Matsukuma S, et al. Idiopathic multicentric castleman disease with autoimmune hemolytic anemia and production of anti-drug antibody against tocilizumab. Intern Med. (2019) 58:3313–8. doi: 10.2169/internalmedicine.2989-19
11. Liu AY, Nabel CS, Finkelman BS, Ruth JR, Kurzrock R, van Rhee F, et al. Idiopathic multicentric Castleman’s disease: a systematic literature review. Lancet Haematol. (2016) 3:e163–175. doi: 10.1016/S2352-3026(16)00006-5
12. Pai R-AL, Japp AS, Gonzalez M, Rasheed RF, Okumura M, Arenas D, et al. Type I IFN response associated with mTOR activation in the TAFRO subtype of idiopathic multicentric Castleman disease. JCI Insight. (2020) 5:e135031. doi: 10.1172/jci.insight.135031
13. Price JV, Haddon DJ, Kemmer D, Delepine G, Mandelbaum G, Jarrell JA, et al. Protein microarray analysis reveals BAFF-binding autoantibodies in systemic lupus erythematosus. J Clin Invest. (2013) 123:5135–45. doi: 10.1172/JCI70231
14. Chang SE, Feng A, Meng W, Apostolidis SA, Mack E, Artandi M, et al. New-onset IgG autoantibodies in hospitalized patients with COVID-19. Nat Commun. (2021) 12:5417. doi: 10.1038/s41467-021-25509-3
15. Ayoglu B, Mitsios N, Kockum I, Khademi M, Zandian A, Sjöberg R, et al. Anoctamin 2 identified as an autoimmune target in multiple sclerosis. Proc Natl Acad Sci U.S.A. (2016) 113:2188–93. doi: 10.1073/pnas.1518553113
16. Sng J, Ayoglu B, Chen JW, Schickel JN, Ferre EMN, Glauzy S, et al. AIRE expression controls the peripheral selection of autoreactive B cells. Sci Immunol. (2019) 4:eaav6778. doi: 10.1126/sciimmunol.aav6778
17. Walter JE, Rosen LB, Csomos K, Rosenberg JM, Mathew D, Keszei M, et al. Broad-spectrum antibodies against self-antigens and cytokines in RAG deficiency. J Clin Invest. (2015) 125:4135–48. doi: 10.1172/JCI80477
18. Price JV, Jarrell JA, Furman D, Kattah NH, Newell E, Dekker CL, et al. Characterization of influenza vaccine immunogenicity using influenza antigen microarrays. PloS One. (2013) 8:e64555. doi: 10.1371/journal.pone.0064555
19. Furman D, Jojic V, Kidd B, Shen-Orr S, Price J, Jarrell J, et al. Apoptosis and other immune biomarkers predict influenza vaccine responsiveness. Mol Syst Biol. (2013) 9:659. doi: 10.1038/msb.2013.15
20. Rosenberg JM, Utz PJ. Protein microarrays: A new tool for the study of autoantibodies in immunodeficiency. Front Immunol. (2015) 6:138. doi: 10.3389/fimmu.2015.00138
21. Ayoglu B, Häggmark A, Khademi M, Olsson T, Uhlén M, Schwenk JM, et al. Autoantibody profiling in multiple sclerosis using arrays of human protein fragments. Mol Cell Proteomics MCP. (2013) 12:2657–72. doi: 10.1074/mcp.M112.026757
22. Feng A, Yang EY, Moore AR, Dhingra S, Chang SE, Yin X, et al. Autoantibodies are highly prevalent in non–SARS-CoV-2 respiratory infections and critical illness. JCI Insight. (2023) 8:e163150. doi: 10.1172/jci.insight.163150
23. Bastard P, Rosen LB, Zhang Q, Michailidis E, Hoffmann HH, Zhang Y, et al. Autoantibodies against type I IFNs in patients with life-threatening COVID-19. Science. (2020) 370:eabd4585. doi: 10.1126/science.abd4585
24. Candia J, Daya GN, Tanaka T, Ferrucci L, Walker KA. Assessment of variability in the plasma 7k SomaScan proteomics assay. Sci Rep. (2022) 12:17147. doi: 10.1038/s41598-022-22116-0
25. Fujimoto S, Koga T, Kawakami A, Kawabata H, Okamoto S, Mizuki M, et al. Tentative diagnostic criteria and disease severity classification for Castleman disease: A report of the research group on Castleman disease in Japan. Mod Rheumatol. (2018) 28:161–7. doi: 10.1080/14397595.2017.1366093
26. R Core Team. R: A Language and Environment for Statistical Computing. Available online at: https://www.R-project.org (Accessed December 9, 2023).
27. Posit team. RStudio: Integrated Development Environment for R (2023). Available online at: http://www.posit.co/ (Accessed December 9, 2023).
28. Gu Z, Eils R, Schlesner M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinforma Oxf Engl. (2016) 32:2847–9. doi: 10.1093/bioinformatics/btw313
29. Pierson SK, Khor JS, Ziglar J, Liu A, Floess K, NaPier E, et al. ACCELERATE: A patient-powered natural history study design enabling clinical and therapeutic discoveries in a rare disorder. Cell Rep Med. (2020) 1:100158. doi: 10.1016/j.xcrm.2020.100158
30. Rubenstein AI, Pierson SK, Shyamsundar S, Bustamante MS, Gonzalez MV, Milller ID, et al. Immune-mediated thrombocytopenia and IL-6-mediated thrombocytosis observed in idiopathic multicentric Castleman disease. Br J Haematol. (2024) 204:921–30. doi: 10.1111/bjh.v204.3
31. Cheng A, Holland SM. Anti-cytokine autoantibodies: mechanistic insights and disease associations. Nat Rev Immunol. (2023) 24:161–77. doi: 10.1038/s41577-023-00933-2
32. Kärner J, Pihlap M, Ranki A, Krohn K, Trebusak Podkrajsek K, Bratanic N, et al. IL-6-specific autoantibodies among APECED and thymoma patients. Immun Inflammation Dis. (2016) 4:235–43. doi: 10.1002/iid3.109
33. Fudala R, Krupa A, Stankowska D, Allen TC, Kurdowska AK. Anti-interleukin-8 autoantibody:interleukin-8 immune complexes in acute lung injury/acute respiratory distress syndrome. Clin Sci Lond Engl 1979. (2008) 114:403–12. doi: 10.1042/CS20070272
34. Suda T, Chida K, Todate A, Ide K, Asada K, Nakamura Y, et al. Oncostatin M production by human dendritic cells in response to bacterial products. Cytokine. (2002) 17:335–40. doi: 10.1006/cyto.2002.1023
35. Brown TJ, Lioubin MN, Marquardt H. Purification and characterization of cytostatic lymphokines produced by activated human T lymphocytes. Synergistic antiproliferative activity of transforming growth factor beta 1, interferon-gamma, and oncostatin M for human melanoma cells. J Immunol Baltim Md 1950. (1987) 139:2977–83. doi: 10.4049/jimmunol.139.9.2977
36. Zarling JM, Shoyab M, Marquardt H, Hanson MB, Lioubin MN, Todaro GJ. Oncostatin M: a growth regulator produced by differentiated histiocytic lymphoma cells. Proc Natl Acad Sci U.S.A. (1986) 83:9739–43. doi: 10.1073/pnas.83.24.9739
37. Grenier A, Combaux D, Chastre J, Gougerot-Pocidalo MA, Gibert C, Dehoux M, et al. Oncostatin M production by blood and alveolar neutrophils during acute lung injury. Lab Invest. (2001) 81:133–41. doi: 10.1038/labinvest.3780220
38. Silver JS, Hunter CA. gp130 at the nexus of inflammation, autoimmunity, and cancer. J Leukoc Biol. (2010) 88:1145–56. doi: 10.1189/jlb.0410217
39. Matsuda M, Tsurusaki S, Miyata N, Saijou E, Okochi H, Miyajima A, et al. Oncostatin M causes liver fibrosis by regulating cooperation between hepatic stellate cells and macrophages in mice. Hepatol Baltim Md. (2018) 67:296–312. doi: 10.1002/hep.29421
40. West NR, Hegazy AN, Owens BMJ, Bullers SJ, Linggi B, Buonocore S, et al. Oncostatin M drives intestinal inflammation and predicts response to tumor necrosis factor–neutralizing therapy in patients with inflammatory bowel disease. Nat Med. (2017) 23:579–89. doi: 10.1038/nm.4307
41. Yu Z, Li Z, Wang C, Pan T, Chang X, Wang X, et al. Oncostatin M receptor, positively regulated by SP1, promotes gastric cancer growth and metastasis upon treatment with Oncostatin M. Gastric Cancer. (2019) 22:955–66. doi: 10.1007/s10120-019-00934-y
42. Guihard P, Danger Y, Brounais B, David E, Brion R, Delecrin J, et al. Induction of osteogenesis in mesenchymal stem cells by activated monocytes/macrophages depends on oncostatin M signaling. Stem Cells Dayt Ohio. (2012) 30:762–72. doi: 10.1002/stem.1040
43. Gherardi RK, Bélec L, Fromont G, Divine M, Malapert D, Gaulard P, et al. Elevated levels of interleukin-1β (IL-1β) and IL-6 in serum and increased production of IL-1β mRNA in lymph nodes of patients with polyneuropathy, organomegaly, endocrinopathy, M protein, and skin changes (POEMS) syndrome. Blood. (1994) 83:2587–93. doi: 10.1182/blood.V83.9.2587.2587
44. Pierson SK, Shenoy S, Oromendia AB, Gorzewski AM, Langan Pai RA, Nabel CS, et al. Discovery and validation of a novel subgroup and therapeutic target in idiopathic multicentric Castleman disease. Blood Adv. (2021) 5:3445–56. doi: 10.1182/bloodadvances.2020004016
45. Dinarello CA. Immunological and inflammatory functions of the interleukin-1 family. Annu Rev Immunol. (2009) 27:519–50. doi: 10.1146/annurev.immunol.021908.132612
46. Bradley JR. TNF-mediated inflammatory disease. J Pathol. (2008) 214:149–60. doi: 10.1002/path.v214:2
47. Goel RR, Kotenko SV, Kaplan MJ. Interferon lambda in inflammation and autoimmune rheumatic diseases. Nat Rev Rheumatol. (2021) 17:349–62. doi: 10.1038/s41584-021-00606-1
Keywords: iMCD, TAFRO, luminex, protein array, autoantibody, connective tissue disorders, autoimmunity
Citation: Feng A, Gonzalez MV, Kalaycioglu M, Yin X, Mumau M, Shyamsundar S, Bustamante MS, Chang SE, Dhingra S, Dodig-Crnkovic T, Schwenk JM, Garg T, Yoshizaki K, van Rhee F, Fajgenbaum DC and Utz PJ (2025) Common connective tissue disorder and anti-cytokine autoantibodies are enriched in idiopathic multicentric castleman disease patients. Front. Immunol. 16:1528465. doi: 10.3389/fimmu.2025.1528465
Received: 14 November 2024; Accepted: 24 February 2025;
Published: 13 March 2025.
Edited by:
Felipe Andrade, Johns Hopkins University, United StatesCopyright © 2025 Feng, Gonzalez, Kalaycioglu, Yin, Mumau, Shyamsundar, Bustamante, Chang, Dhingra, Dodig-Crnkovic, Schwenk, Garg, Yoshizaki, van Rhee, Fajgenbaum and Utz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: David C. Fajgenbaum, ZGF2aWRmYUBwZW5ubWVkaWNpbmUudXBlbm4uZWR1; Paul J. Utz, cGp1dHpAc3RhbmZvcmQuZWR1