 Shishir Upadhyay1†
Shishir Upadhyay1† Lewis Murugu
Lewis Murugu Lena Svensson
Lena Svensson- 1Department of Molecular Biology, Umeå University, Umeå, Sweden
- 2Umeå Centre for Microbial Research, Umeå University, Umeå, Sweden
During tumor immunosurveillance, leukocytes play a crucial role in the cellular defense system, working collaboratively with other immune components to recognize and eliminate aberrant cells. Integral to this process is the integrin Lymphocyte Function-Associated Antigen 1 (LFA-1). LFA-1 facilitates adhesion during leukocyte migration and helps establish stable cell-to-cell contacts between leukocytes and their targets. Additionally, as a receptor, LFA-1 signaling activates leukocytes, promoting their differentiation and effector functions against cancer. However, tumors can develop mechanisms to evade immune clearance by disrupting LFA-1 functions or hijacking its pathways. In this review, we first detail how leukocytes utilize LFA-1 during immunosurveillance and then explore how tumors counteract this process in the tumor microenvironment (TME) by either altering LFA-1 functions or exploiting it to drive tumorigenesis. Moreover, we discuss therapeutic strategies targeting LFA-1, including inhibitors tested in laboratory studies and animal models, highlighting their potential as anticancer interventions and the need for further research to evaluate their clinical utility.
1 Introduction
Leukocytes serve as the immune system’s primary cellular defense against cancer, performing distinct roles across the immune response, from tumor recognition to destruction. To execute these functions effectively, leukocytes depend heavily on integrins. Integrins are transmembrane receptors composed of alpha and beta subunits that assemble into heterodimers essential for cell adhesion and signal transduction (1–4). Among the integrins expressed in leukocytes, several, including αLβ2, αMβ2, αXβ2, αDβ2, α4β1, α4β7, and αEβ7, have been characterized and extensively reviewed elsewhere (1, 2, 5, 6). Of these, αLβ2, or Lymphocyte Function-Associated Antigen 1 (LFA-1), is a key leukocyte-specific integrin predominantly expressed in lymphocytes, with lower levels in other leukocytes (7–13). Due to its essential roles in leukocyte function, LFA-1 is the primary focus of our research and this review.
LFA-1 is pivotal to the immune system, mediating the migration and effector functions of leukocytes at sites of infection and inflammation. This function is primarily facilitated through its interaction with ICAM-1, its main ligand (8, 14). However, beyond ICAM-1, LFA-1 can bind to other ligands, including ICAM-2, ICAM-3, ICAM-4, ICAM-5, and JAM-A, which are expressed on various cell types such as endothelial cells and immune cells (15–19). These interactions are critical during the immune response and have been discussed in greater detail in the literature (15–22).
In T cells, LFA-1 supports adhesion to the vascular endothelium, enabling their exit from the bloodstream and subsequent entry into tissues (23). It also guides T cells into lymph nodes through high endothelial venules (HEVs) and plays an essential role in forming immunological synapses with antigen-presenting cells (APCs), a key step in initiating the adaptive immune response (24, 25). In cytotoxic T cells, LFA-1 stabilizes contact with infected or abnormal cells, including tumor cells, thereby enhancing their ability to eliminate these targets. For LFA-1 to function, it must transition from an inactive, bent-closed conformation to an active, extended-open state capable of ligand binding. This activation is mainly driven by inside-out signaling, initiated by signals from other transmembrane proteins such as chemokine receptors, T cell receptors, and selectin ligands (1, 3, 26, 27). Inside-out signaling involves the activation and recruitment of RAP-1, which facilitates the association of RAPL/MST1 with the LFA-1 αL subunit (28–31). RAP-1 and RIAM further recruit talin to the LFA-1 β chain, where talin, in conjunction with kindlin-3, stabilizes the integrin in its high-affinity state, promoting effective ligand binding and initiating outside-in signaling cascades that drive T cell adhesion and migration (32–34). Interestingly, recent findings from Springer’s group suggest that ligand binding itself may induce the conformational change from the bent-closed to the extended-open form, potentially playing a more significant role than talin in stabilizing this state (35). This challenges the previously understood sequence, implying that outside-in signaling may precede and facilitate inside-out activation (35, 36).
Although LFA-1 is a major integrin for T cells, it also plays a role in the function of other leukocyte subsets. For instance, LFA-1 facilitates natural killer (NK) cell activation and the formation of cytotoxic immune synapses with target cells (37–40). Additionally, it facilitates dendritic cell (DC) transmigration from tissues into afferent lymphatic vessels (41), and supports the migration and endothelial patrolling of non-classical monocytes (42), as observed in murine models. Given its central role in the optimal functioning of lymphocytes, particularly T cells and NK cells, LFA-1 is indispensable for effective tumor immunosurveillance. However, as cancer progresses, tumor cells can adopt mechanisms to evade immune detection (43–45). One such strategy involves disrupting LFA-1 function, thereby compromising the ability of leukocytes to mount an effective defense. Notably, impaired activation or conformation of LFA-1 has been linked to certain forms of cancer (46, 47).
This review paper will examine how modulation of LFA-1 in the tumor microenvironment (TME) contributes to immune evasion and tumor progression. We will first describe how LFA-1 generally promotes immune surveillance through its interactions with ligands, primarily its major ligand, ICAM-1, leading to leukocyte activation and tumor cell killing. Next, we will broadly outline three tumor immune-escape strategies involving LFA-1: altered LFA-1-mediated migration and infiltration of leukocytes into the tumor microenvironment, disrupted LFA-1 functionality in leukocytes within the tumor microenvironment, and LFA-1-mediated tumor invasiveness. These mechanisms not only highlight the multifaceted role of LFA-1 in tumor biology but also represent potential targets for immunotherapeutic interventions. In this context, we will discuss inhibitors and drugs, evaluated in in vitro and in vivo systems, that target various stages of LFA-1 modulation and show potential as anticancer therapies.
2 LFA-1 mediates antitumor immunosurveillance
Tumor cells often display abnormal or mutated proteins, known as tumor-associated antigens (TAAs), which are absent in healthy cells. These TAAs are recognized as foreign by the body’s defense mechanisms, initiating a process called immunosurveillance (43, 45). This process involves a coordinated series of events where innate immune cells detect TAAs, eventually triggering the activation, migration, and infiltration of tumor-specific cytotoxic lymphocytes into the tumor microenvironment. Once within the TME, these lymphocytes identify and destroy tumor cells by releasing cytotoxic mediators. LFA-1 plays a pivotal role throughout this antitumor immune response, facilitating critical steps such as immune cell activation, adhesion, migration, and target cell elimination. This section explores how immunocompetent cells rely on LFA-1 for effective tumor eradication. Figure 1 provides a general overview of the immune response cascade, while Figure 2 emphasizes the specific stages where LFA-1 is indispensable.
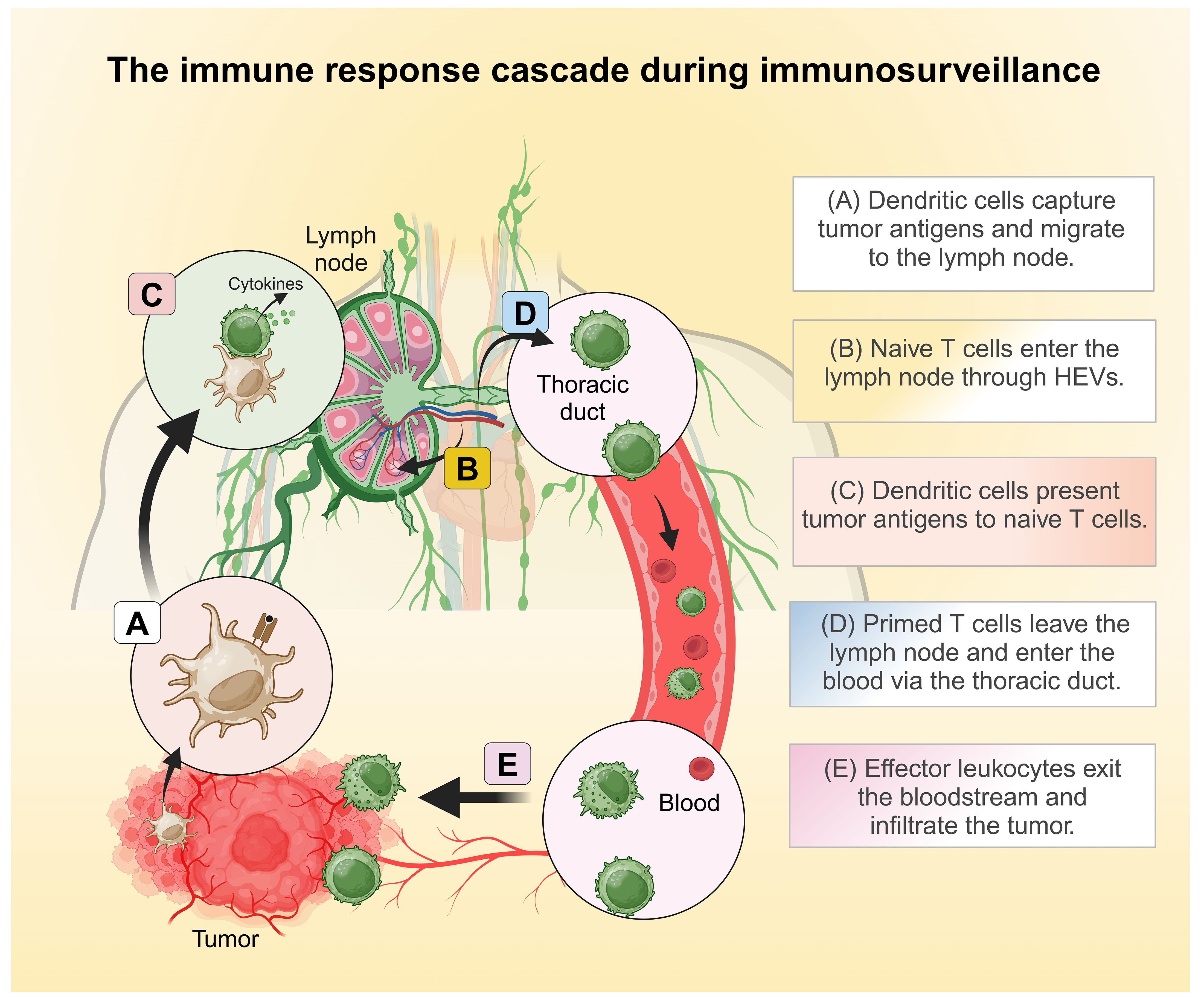
Figure 1. The general immune response cascades during immunosurveillance. (A) Dendritic cells capture tumor antigens and shuttle to the draining lymph nodes. (B) Naïve T cells enter the lymph nodes through high endothelial venules (HEVs). (C) In the lymph nodes, dendritic cells present tumor antigens to naïve T cells, thereby initiating T cell activation. (D) Primed T cells leave the lymph nodes and enter circulation via the thoracic duct. (E) Effector leukocytes exit the bloodstream and infiltrate the tumor. Created in BioRender. Murugu, L. (2024) https://BioRender.com/f91b170.
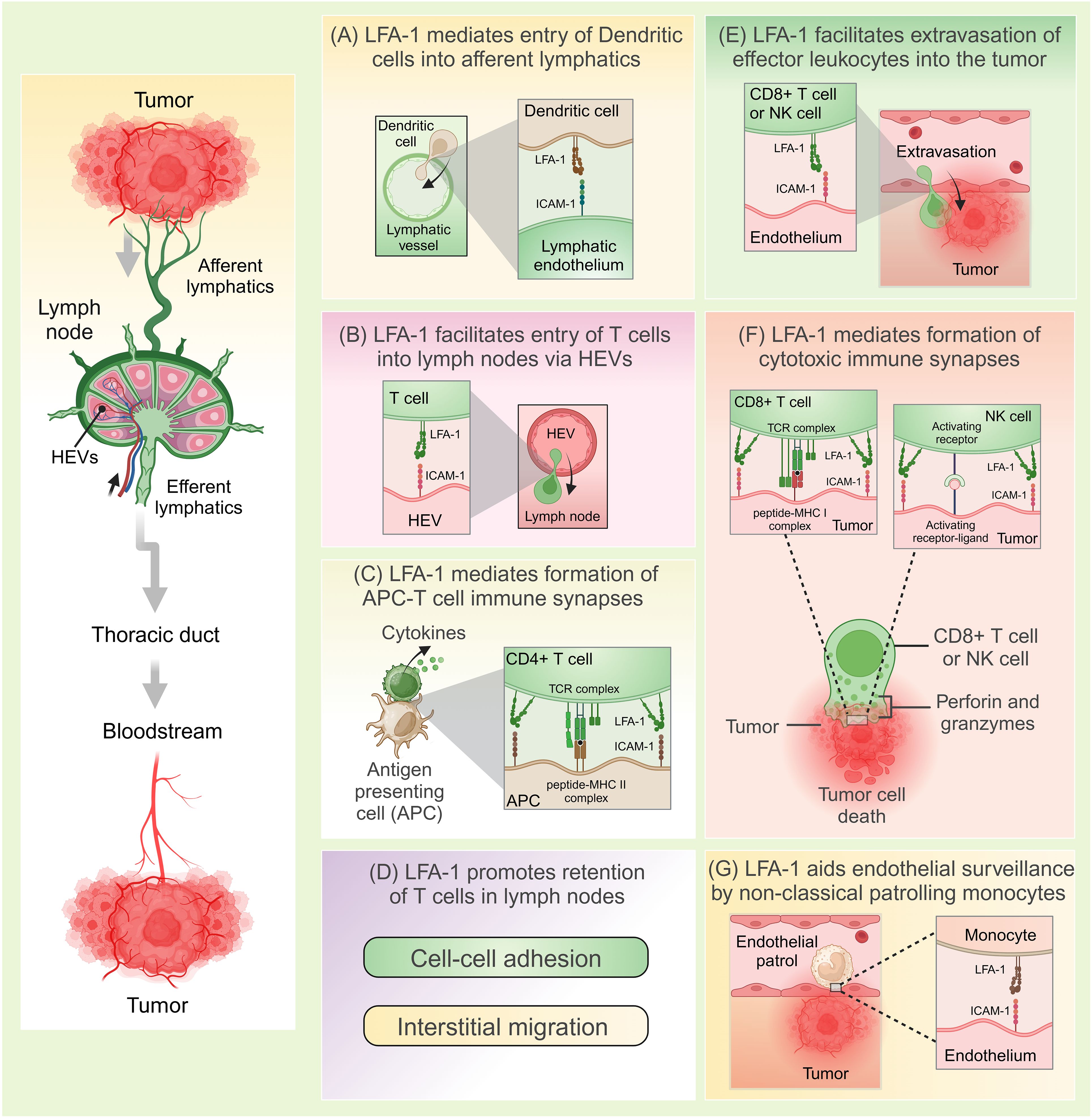
Figure 2. LFA-1-mediated anti-tumor immunosurveillance. (A) LFA-1 interacts with ICAM-1 to facilitate entry of dendritic cells from the tumor sites into the afferent lymphatics. (B) LFA-1 mediates migration of naïve T cells into the lymph nodes via high endothelial venules (HEVs) through its interactions with ICAM-1. (C) LFA-1 mediates formation of immunological synapses between antigen presenting cells (APCs) such as dendritic cells and T cells. Following antigen presentation, T cells are activated and release cytokines. (D) Through its interactions with ICAM-1, LFA-1 promotes retention of T cells in the lymph nodes optimizing their chances of encountering cognate antigens. This happens through efficient interstitial migration and formation of cell-to-cell attachments that function to delay T cell egression. (E) LFA-1 facilitates extravasation of effector leukocytes into the tumor. (F) LFA-1 mediates formation of cytolytic immune synapses and the eventual killing of tumors. (G) LFA-1 aids endovascular surveillance by patrolling non-classical monocytes. Created in BioRender. Murugu, L. (2024) https://BioRender.com/g87c843.
2.1 Recognition of tumor associated antigens in the TME
The initial step in immunosurveillance is the recognition of tumor associated antigens in the TME. Dendritic cells (DCs) play a key role in this process. After engulfing TAAs, DCs process and present these peptides on their surface MHC molecules before migrating to draining lymph nodes to function as antigen-presenting cells (APCs) (44). The migratory process is initiated by chemotaxis and is guided by the CCR7 receptor on DCs, which binds to CCL21—a ligand constitutively expressed by lymphatic endothelial cells. Ongoing inflammation in the TME releases TNF-α, which further elevates CCL21 levels. This increase in CCL21 in turn, promotes LFA-1 activation in DCs, mediating their transendothelial migration into the afferent lymphatic vessels, ultimately guiding them to the draining lymph nodes (41).
2.2 Priming of T cells in the draining lymph nodes
Once in the lymph nodes, DCs orchestrate an effective antitumor response by priming and activating naïve tumor-specific CD4+ and CD8+ T cells. These naïve T cells rely on LFA-1 to migrate and enter the lymph nodes through HEVs. Chemokines CCL19 and CCL21 from the lymph nodes as well as within HEVs, facilitate T cell migration by binding to CCR7 on T cells, triggering inside-out signaling that activates LFA-1. Once activated, LFA-1 on T cells binds to ICAM-1 on HEVs enabling their transendothelial entry into the lymph nodes (19). Within the lymph nodes, LFA-1 further aids effective antigen scanning by T cells, helping them locate their cognate antigen presented by APCs (48). Here, the adhesive state of LFA-1 in T cells is regulated by bidirectional signaling—inside-out signaling adjusts its affinity, and outside-in signaling provides feedback from LFA-1 engagement (49, 50). This dual signaling mechanism modulates T cell adhesion, ensuring that T cells migrate effectively within lymphoid tissue, optimizing their surveillance and interactions with tumor antigens. Notably, unlike chemokine-induced inside-out signaling, which alone can activate LFA-1 to a high-affinity state in T cells, full LFA-1 activation through TCR signaling requires LFA-1 to bind to ICAM-1 on APCs (51). In DCs, however, regulation of LFA-1’s activity plays a key role in DC/T cell conjugate formation, with mature DCs expressing elevated levels of cytohesin-1–interacting protein (CYTIP) to maintain LFA-1 in an inactive state (48). This inactivity is essential for the release of T cells and their subsequent priming. Additionally, the lower lateral mobility of ICAM-1 on mature DCs compared to immature DCs increases the likelihood of binding between LFA-1 on naïve T cells and ICAM-1 on mature DCs (52). The strength of LFA-1 binding to ICAM-1 on DCs, combined with CD2/CD58 interactions, improves T cell antigen discrimination (53). This process likely ensures that T cells bind specifically to non-self-antigens, including tumor antigens, through their TCR. Furthermore, LFA-1 binding to ICAM-1 within the lymphoid parenchyma increases T cell retention in the lymph nodes, preventing their immediate exit and allowing more time for T cells to encounter their cognate antigen (54). While our review paper focuses on the role of integrins, it is important to note that interstitial migration of leukocytes within lymph nodes may occur independently of integrins, as has been extensively demonstrated in other studies (55–58).
In vitro studies indicate that LFA-1/ICAM-1 interactions may promote CD4+ T cell differentiation towards the Th1 phenotype (59). Th1 cells are crucial for tumor elimination as they produce TNF-α and IFN-γ, which stimulate the recruitment and activation of cytotoxic CD8+ T cells (CTLs), NK cells, and monocytes to the tumor microenvironment, while also negatively regulating tumor-promoting Th17 cells (60). CD4+ T cells further support CD8+ T cell expansion by forming LFA-1-dependent transient homotypic synapses with adjacent activating CD8+ T cells (61, 62). Following differentiation, tumor-specific effector T cells exit the lymph nodes and migrate to peripheral tissues to perform their immune functions.
Unlike T cells though, NK cells can directly identify tumor cells without prior priming by APCs. NK cells become “educated,” meaning they gain the ability to recognize tumor cells during their development. Mature NK cells are characterized by high LFA-1 expression, as well as the expression of receptors such as NKp46, NKG2D, CD16, and elevated levels of perforin and granzyme B (63, 64). LFA-1, along with other cell-surface adhesion receptors including Mac-1, regulate the priming of NK cells and facilitates their homing and migration to both lymphoid and non-lymphoid organs (65, 66).
2.3 Entry and action of effector leukocytes in the TME
T cells exit the lymph nodes via the efferent lymphatic vessels and drain into the bloodstream through the thoracic duct (67). Their exit from the bloodstream into the TME is dependent on the interaction between LFA-1 and ICAM-1 on endothelial cells. A recent study in mice models highlighted the intriguing influence of the circadian clock on the transmigration of CTLs into tumors, with increased ICAM-1 expression on endothelial cells being observed in the morning, thereby enhancing T cell infiltration during this time (68). Similarly, NK cells are recruited to the tumor site through inflammation-induced signaling in response to interleukins (IL-12, IL-15, and IL-18) from target cells, with LFA-1 playing a crucial role in their migration (69, 70).
In addition, tumor-associated HEVs exhibit elevated ICAM-1 expression (71, 72), which may further facilitate lymphocyte infiltration into tumors and support the formation of tertiary lymphoid structures. Notably, a study of resected primary human breast carcinomas revealed that the density of ICAM-1-expressing HEVs predicted both the extent of T and B cell infiltration and clinical outcomes in breast cancer (72).
Within the TME, migrated CTLs recruit LFA-1 to engage with ICAM-1 on tumor cells, forming a cytolytic synapse ultrastructure that facilitates their effector functions. This includes the directed delivery of cytotoxic granules, which contain granzymes and perforin, through secretory clefts of the mature lytic synapse. The efficient delivery of these cytolytic granules depends on the arrangement of the microtubule-organizing center (MTOC), which is also influenced by Mg2+ and Ca2+ (73–75). Importantly, the conformational activation of LFA-1 is essential for licensing the exocytosis process, which enhances the local concentration of cytotoxic molecules to optimize tumor cell recognition and potentiate the antitumor immune response (74). What is more, earlier studies have underscored the importance of functional CTLs in antitumor responses. In particular, a study involving LFA-1-deficient mice demonstrated that these mice were unable to clear tumors injected into their footpads, which was attributed to a defective CTL response against the tumor cells (76). LFA-1 is also crucial for the mechanosensitive recognition of tumor cells by NK cells and facilitates the formation of lytic synapses between them (65). Upon successful synapse formation, NK cells release lytic granules, including perforin and granzymes (77, 78). Perforin creates pores in the target cell membrane, allowing granzymes to enter and trigger apoptosis, effectively killing tumor cells. The efficiency of NK cell degranulation and cytotoxicity is influenced by LFA-1 and a high Ca2+ influx (79). Moreover, activation with IL-2 further enhances NK cells’ broad antitumor cytotoxicity. NK cells can also destroy tumors through Antibody-Dependent Cellular Cytotoxicity (ADCC), where Fc receptor-bearing NK cells recognize and kill antibody-coated tumor cells. However, in the absence of LFA-1, NK cells are unable to effectively destroy their targets. A study of four patients with LFA-1 deficiency supports this, showing that their NK cells exhibited impaired cytotoxicity in cellular assays, despite retaining functional Fc receptors (80).
From a functional perspective, the role of LFA-1 in mediating immune surveillance is heavily dependent on its proper activation within T cells. Several proteins and factors regulate LFA-1 activation, conformational changes, and functions. For instance, layilin, a membrane glycoprotein expressed by certain tumor-specific CTLs, enhances LFA-1 activation by stabilizing its interaction with ICAM-1. Together with talin, layilin supports LFA-1-mediated adhesiveness and the cytotoxic activity of CTLs, playing a critical role in promoting immune synapse formation and tumor cell killing, as demonstrated in melanoma (81). However, further research is needed to fully elucidate the layilin-mediated pathways affecting LFA-1 expression and activation before these findings can be applied to therapeutic interventions. Also, certain co-receptors, such as CD226 (DNAM-1), regulate LFA-1 function by enhancing its adhesive properties through the regulation of its affinity conformation. Evidence shows that the loss of CD226 impairs LFA-1’s high-affinity conformation in CTLs, a change associated with malignancy, including multiple myeloma and lung adenocarcinoma (82). In CLL, T cells exhibit significantly reduced LFA-1 expression or impaired LFA-1 function, leading to their failure to adhere to and transmigrate across VCAM-1, ICAM-1, and CXCL12-expressing endothelium, which results in diminished trafficking to lymph nodes (83, 84). Taken together, it is apparent that proper functioning of LFA-1 is crucial for optimal antitumor CTL responses during immunosurveillance.
The role of LFA-1 in mediating immune surveillance against cancer by innate immune cells beyond NK cells remains underexplored. Nevertheless, some studies suggest that certain tumors are targeted by specific subsets of monocytes and neutrophils, despite differences in LFA-1 regulation and activation between these cell types (85). For example, through intravital microscopy studies in mice, non-classical monocytes have been shown to patrol the vasculature and maintain endothelial homeostasis by scavenging microparticles (42). This patrolling behaviour is facilitated by the expression of LFA-1 on these cells, enabling interactions with ICAM-1-rich endothelial cells (42). In lung cancer mouse models, these “intravascular housekeepers” have been observed removing metastatic tumor cells from vascularized tissues (86). Upon detecting primary tumor invasion, non-classical monocytes decrease their patrolling activity, migrate toward tumor particles, and engulf them (86). Similarly, tumor-entrained neutrophils (TENs) can suppress metastatic seeding in the premetastatic lung in mice underscoring their roles in limiting tumor spread (87). In addition, ICAM-1 expression on breast cancer cells in mice facilitates interactions with neutrophils that suppress lung metastasis, independent of cytotoxic T cell activity (88).
3 LFA-1 functions in engineered T cells
LFA-1 and its interaction with ICAM-1 have essential roles in the antitumor responses of Chimeric Antigen Receptor (CAR) T cells, much like their involvement in CTLs (89–91). CAR T cells are engineered from allogenic T cells, genetically modified to express a Chimeric Antigen Receptor (CAR) that recognizes the patient’s tumor-associated antigens (89, 90). These cells are expanded in vitro and infused back into the patient.
The LFA-1-ICAM-1 axis is particularly important for CAR T cell function, as evidenced by the differential effectiveness of CAR T cells in hematological versus solid tumors (74, 89–95). In solid tumors, CAR T cells often struggle to migrate to the tumor site and instead accumulate in non-target tissues. This phenomenon necessitates the infusion of large quantities of highly activated T cells, which raises both the risk of T cell toxicity and systemic cytokine release syndrome (CRS) while also significantly increasing the manufacturing costs of CAR T cell therapies (89). To better understand why in vitro-activated CD8+ T cells become sequestered in off-target tissue sites following intravenous transfer, Hong et al. used intravital microscopy in mice and a CRISPR-Cas9 screen, discovering that ST3GAL1-mediated glycosylation of CD18 disrupts LFA-1 recycling, leading to nonspecific sequestration of T cells in off-target tissues (93). Enhancing the expression of βII-spectrin in the CAR T cells counteracted this effect, improving tumor-specific homing and reduced tumor growth in mice. These findings suggest that targeting the ST3GAL1–βII-spectrin axis could enhance CAR T cell therapies by improving migration to tumor sites (93).
Moreover, ICAM-1 expression on tumor cells is critical for CAR T-cell functionality (94, 95). A CRISPR-based screen in a multiple myeloma cell line revealed that knockout of the ICAM-1 gene in tumor cells led to resistance to B-cell maturation antigen (BCMA) CAR T cells (95). Conversely, increased expression of ICAM-1 on target tissues has been identified as a limiting factor for CAR T cell efficacy. In one study, ICAM-1 expression was significantly associated with advanced stages of gastric cancer and poorer survival rates in human patients (94). Furthermore, ICAM-1-targeting CAR T cells exhibited substantial efficacy in vitro, with their effectiveness correlating to the level of ICAM-1 expression in target cells. In animal models, these CAR T cells successfully eliminated lung tumors but were less effective against peritoneal tumors. To enhance their efficacy, the study explored combinations with paclitaxel or CAR activation-dependent IL-12 release, which significantly increased anti-tumor activity and improved survival (94). These observations suggest that ICAM-1-targeting CAR T cells, alone or combined with chemotherapy, could be a promising strategy for treating patients with ICAM-1 positive advanced gastric cancer.
Another major challenge hindering CAR T cell efficacy is their limited ability to effectively leverage accessory receptors like LFA-1. A study demonstrated that CAR T cells exhibit over 100-fold lower antigen sensitivity than TCRs when antigens are presented on APCs, a disparity not observed with purified proteins (96). This is because CARs inefficiently utilize receptors such as CD2 and LFA-1, which significantly enhance TCR sensitivity but have minimal impact on CARs. Engineering approaches, such as fusing CARs to the TCR CD3ϵ subunit (TRuCs) or incorporating TCR αβ variable domains (STARs/HITs), restored sensitivity by improving accessory receptor engagement (96). These findings highlight the potential of enhancing CAR T cells’ interaction with LFA-1 to improve their therapeutic efficacy.
Together, these studies underscore the critical role of LFA-1 in CAR T cell efficacy and suggest that overcoming barriers related to migration, ICAM-1 expression, and receptor utilization could significantly enhance CAR T therapies, particularly in solid tumors.
4 Tumors escape immunosurveillance by altering LFA-1-mediated functions
Tumors employ several strategies to evade immune clearance by disrupting LFA-1-mediated processes. As broadly depicted in Figure 3, these disruptions generally affect anti-tumor immune functions by impairing leukocyte adhesion to tumor vasculature, infiltration into tumors, the formation of functional immunological synapses, and the destruction of malignant cells, while also exploiting LFA-1 to promote metastasis. Understanding these evasion mechanisms is crucial for developing therapeutic strategies that enhance leukocyte infiltration and function within the TME. This section will delve into how tumors manipulate LFA-1, focusing on its implications for cancer progression.
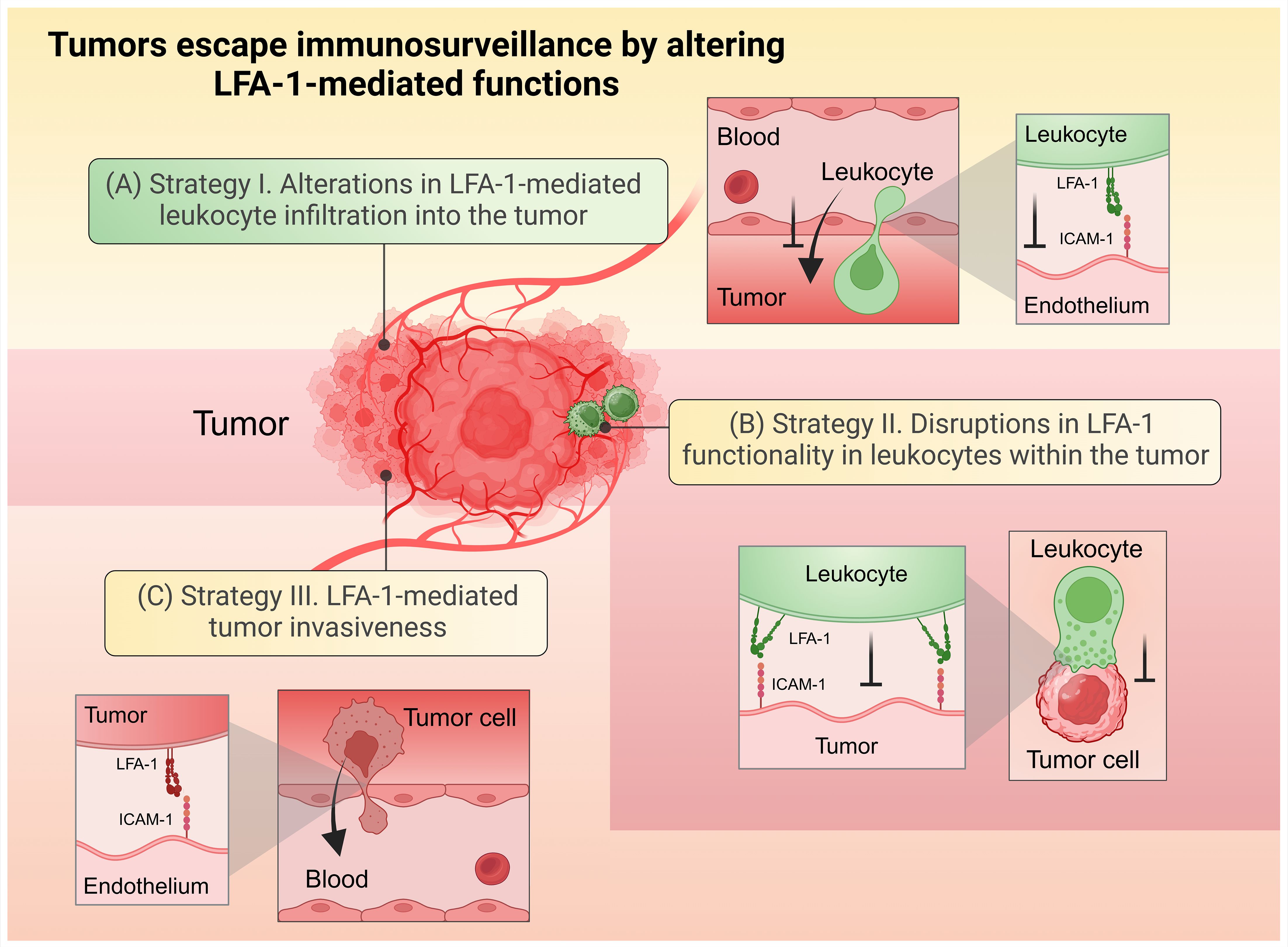
Figure 3. Tumors escape immunosurveillance by altering LFA-1-mediated functions. (A) Strategy (I) Alterations in LFA-1-mediated leukocyte infiltration into the tumor. (B) Strategy II. Disruptions in LFA-1 functionality in leukocytes within the tumor environment. (C) Strategy III. LFA-1-mediated tumor invasiveness. Created in BioRender. Murugu, L. (2024) https://BioRender.com/z47w979.
4.1 Strategy I: Alterations in LFA-1-mediated migration and infiltration of leukocytes into the tumor microenvironment
Leukocyte trafficking into the TME relies heavily on cell movement toward chemokine gradients and the use of integrins like LFA-1 for adhesion to vasculature and subsequent extravasation into the tumor (43, 97). However, there is evidence that tumors can disrupt these processes. For instance, non-inflammatory tumors with inactivated LFA-1 exhibit impaired T cell priming and reduced recruitment into the TME (98, 99). Although the exact mechanisms of LFA-1 inactivation are not fully understood, these tumors are known to release chemokines such as vascular endothelial growth factor (VEGF), which suppress the expression of endothelial ICAM-1, a crucial ligand for LFA-1 (100–102). This suppression hinders LFA-1-mediated leukocyte recruitment into the TME. Moreover, reduced secretion of chemokines like CXCL12 further impairs T cell infiltration by decreasing the inside-out signaling required for effective LFA-1 activation (98). Normally, these chemokines promote LFA-1 expression on the cell surface and enhance the ability of T cells to infiltrate the TME (98). A recent study on human melanoma patients revealed that tumors secrete the cytokine growth differentiation factor 15 (GDF-15), which hinders LFA-1-mediated adhesion of T cells to activated endothelial cells—a crucial step for T cell extravasation (103). GDF-15 does this by curtailing the interaction of LFA-1 with the actin cytoskeleton via the protein talin. The study also emphasized the significance of LFA-1 in the success of immunotherapy, showing that elevated serum GDF-15 levels in cancer patients were linked to poor outcomes following PD-1 therapy, and that inhibiting GDF-15 improved T cell recruitment and enhanced therapeutic efficacy (103).
The complement receptor C3aR has also been shown to interfere with LFA-1 activation in T cells and other leukocytes, disrupting their trafficking to tissues (104). In line with this, research by Nandagopal et al. revealed that high levels of the inflammatory factor C3a, released within the TME, bind to C3aR, causing the accumulation of high-affinity LFA-1 at the uropod (tail region) of NK cells (105). This suboptimally increases NK cell adhesion to the endothelium, thereby preventing effective infiltration into the TME (105). Also, impaired LFA-1 function in non-classical monocytes results in inefficient killing of metastatic tumor cells during vasculature patrolling (86). These monocytes fail to express Kindlin-3, a key intermediary molecule required for LFA-1 activation, leading to dysfunctional LFA-1 signaling. Consequently, the weakened LFA-1:ICAM-1 interactions cause the monocytes to ‘slip’ rather than firmly adhere to the endothelium, reducing their effectiveness in infiltrating and killing metastatic cells (86).
Regulating cell adhesion and motility is a complex process that involves the intracellular trafficking of integrins to and from adhesion sites, particularly in fast-moving cells like leukocytes (84, 106). In CLL, T cell movement is disrupted due to impaired LFA-1 resulting from continuous direct contact with the tumor cells. Although the precise receptors on T cells and ligands on CLL tumors involved in transmitting these immunosuppressive signals are not fully understood, this contact alters Rho GTPase signaling in T cells by downregulating RhoA and Rac1 and upregulating Cdc42, leading to defective T cell adhesion and motility (84). Further research is needed to fully understand how these impaired T cells migrate into secondary solid tumor niches and whether this process is equally impaired in CLL patients.
4.2 Strategy II: Disruptions in LFA-1 functionality in leukocytes within the tumor microenvironment
After infiltrating the tumor microenvironment, leukocytes are met with a barrage of dysregulating factors from the tumor that impair their ability to effectively use LFA-1 for immunological synapse formation and cytotoxic functions. These disruptions occur through multiple mechanisms, including altered LFA-1 activation and clustering, impaired shifting of LFA-1 to its high affinity conformation, and changes in the mechanical properties of the tumor cell surface (46, 47, 107, 108). Collectively, these factors significantly hinder the leukocyte’s capacity to engage and destroy tumor cells via LFA-1 within the TME.
4.2.1 Impairment of LFA-1 activation and clustering
LFA-1 clustering, and activation are crucial for stabilizing interactions between T cells and their target cells (46, 84, 109, 110). A recent ex vivo study by Lacouture et al. demonstrated that human CD8+ T cell activation and degranulation are regulated by the availability of discrete LFA-1 nanoclusters within the cytolytic synapse with P815 mastocytoma target cells (109). However, certain solid tumors can impair LFA-1 activation in cytotoxic T lymphocytes by releasing specific secretory factors (47, 103). One such factor is galectin, which has been shown to coat tumor-infiltrating CD8+ T cells, disrupting the lateral diffusion, recruitment, and activation of LFA-1 at the immune synapse (47). These galectin-covered T cells exhibit defective actin reorganization and reduced adhesion to LFA-1’s ligand, ICAM-1, compromising their ability to secrete cytokines at the synaptic interface. Markedly, the removal of galectin from the surface of these T cells alleviates such defects and restores proper synaptic function (47).
A study by Ramsay et al. found that healthy CD8+ and CD4+ T cells cocultured with CLL B cells exhibited significantly reduced LFA-1 clustering at the immunological synapse compared to those cocultured with healthy B cells (46). This reduction in clustering was associated with a marked decrease in the active form of LFA-1 and impaired recruitment of the tyrosine kinase Lck to the T cell synapse, leading to altered F-actin dynamics (46). Similar LFA-1 impairments have also been observed in T cells isolated from CLL patients (46, 111, 112), highlighting a key mechanism by which CLL cells evade immunosurveillance.
In advanced stages of solid tumors, interleukin-2 (IL-2) cytokine levels are substantially reduced, which diminishes the effectiveness of anti–programmed death 1 (anti-PD-1) antibody therapy (113). IL-2 is crucial for activating STAT5, which in turn enhances LFA-1 activation and expression (62, 113). This further aids in the formation of immunological synapses necessary for effective CD8+ T cell antitumor responses. To address this issue, studies have shown that adoptive transfer therapy—where CD8+ T cells with elevated LFA-1 expression are reinfused into mice—can counteract the reduced IL-2 levels. Enhancing LFA-1 expression or IL-2/STAT5 signaling pathways boosts antitumor efficacy in CD8+ T cells and improves the effectiveness of PD-1 blockade therapy, benefiting both early and late-stage tumors (113).
4.2.2 Impairment of LFA-1’s high affinity conformation
The extended high-affinity conformation of LFA-1 is essential for forming mature immunological synapses between T cells or NK cells and their targets. Blockade of high-affinity LFA-1 in human CD8+ T cells has been shown ex vivo to significantly impair their adhesion to and cytotoxic activity against P815 mastocytoma target cells (109). In a study by Weulersse et al., defective activation of LFA-1 into its high-affinity conformation was identified as a key factor impairing CD8+ T cell responses across several human malignancies, including multiple myeloma, lung adenocarcinoma, breast carcinoma, and ovarian carcinoma (82). By combining experimental approaches using human and mouse samples, this impairment was linked to the loss of CD226—a critical coreceptor that enhances LFA-1 activation—in tumor-infiltrating CD8+ T cells. This loss was driven by the transcription factor Eomes; however, the exact signaling pathways involved in the Eomes-CD226-LFA-1 axis remain an area of active investigation. The resulting defect in LFA-1 activation was associated with CD8+ T cell hyporesponsiveness, evidenced by diminished production of key anti-tumor mediators such as TNF-α and IFN-γ (82).
Extracellular divalent ions such as magnesium (Mg²+) are crucial in regulating LFA-1’s conformational dynamics. In CD8+ T cells, Mg²+ induces a high-affinity conformational change in LFA-1, enhancing calcium flux and promoting signal transduction, which supports immune synapse formation and cytotoxicity (75). Moreover, in a study by Van Kooyk et al., using the human CD4+ T cell clone JS136 and peripheral blood T lymphocytes, Mg²+ regulated LFA-1’s affinity for ICAM-1, while calcium (Ca²+) modulated its clustering and surface distribution, highlighting the distinct roles of these divalent ions in T cell adhesion (110). Hypomagnesemia, a condition characterized by low serum magnesium levels, is commonly observed in inflammatory conditions, including cancer (107). Additionally, chronic hypomagnesemia can predispose individuals to cancer (107, 114). Alterations in ion homeostasis within the TME are common in many malignancies, and tumors may outcompete immune cells for essential micronutrients, including ions like Mg²+, due to their altered cellular metabolism and rapid division (115, 116). In this context, depletion or reduction of divalent ions in the TME could potentially impair LFA-1’s high-affinity conformation in leukocytes, contributing to tumor immune evasion. However, whether primary tumor development induces hypomagnesemia leading to immune escape, or whether it results from other underlying chronic inflammatory factors, remains a complex research question with significant implications for translational cancer research. Nonetheless, Nasulewicz et al. previously observed that metastatic dissemination of lung carcinoma cells was heightened in mice fed a low-magnesium diet (117). More recently, Lötscher et al. demonstrated that extracellular Mg²+ is sensed through LFA-1 and is required for LFA-1-mediated activation and antitumor effector functions of CD8+ T cells (75). They also found that hypomagnesemia in B cell lymphoma patients was associated with increased disease progression and worse outcomes following CAR T cell therapy or immune checkpoint inhibitor treatment (75).
4.2.3 Alterations in tumor cell surface mechanics
Recent cellular biophysical assays by Wang et al. highlighted the importance of mechanical forces in LFA-1 function by demonstrating that regions of active force exertion through LFA-1, within the immune synapse are where lytic granule exocytosis occurs in CD8+ T cells (118). Disrupting these forces, such as by depleting the adaptor molecule talin, significantly reduces CTL cytotoxicity (118). Cancer cells, particularly those in metastatic breast, glioma, or ovarian cancer, often develop softer mechanical properties compared to their non-malignant counterparts (119–122). This reduction in rigidity may play a key role in immune evasion. Supporting this, an in vitro study using both mouse and human tumor cell lines from melanoma, colon cancer, breast cancer, and hepatocellular carcinoma revealed that tumor-repopulating cells evade cytolytic CD8+ T-cell killing by exploiting their mechanical softness (123). This softness hinders the formation of perforin pores at the immune synapse, thereby impairing effective T-cell-mediated cytotoxicity. For NK and T cells to effectively form LFA-1-mediated immunological synapses and spread symmetrically around cancer cells, they require a gradual, continuous flow of actin and proper polarization of the microtubule-organizing center (108, 124). These processes rely on the cancer cell surface maintaining optimal stiffness to allow efficient mechanical forces through LFA-1 (108).
In a similar manner, NK cells may fail to respond effectively to softer targets, leading to reduced production of proinflammatory cytokines such as IFN-γ and TNF-α. Using cell-sized alginate beads of varying stiffness as target models, research by Friedmann et al. showed that NK cell activation, including degranulation and cytokine secretion, is enhanced on stiffer targets through LFA-1 (125). This increased stiffness promoted better cell spreading, microtubule-organizing center polarization, and efficient lytic granule delivery at the immune synapse (125). Conversely, interactions with softer targets led to unstable synapses and impaired NK cell cytotoxicity. These findings emphasize the critical role of mechanical tension at the interface between NK cells and their targets in facilitating immune synapse formation and function. The results highlight how changes in cell or tissue stiffness—such as tumor cells becoming softer—could potentially serve as a novel immune evasion strategy in disease contexts. However, further research involving actual tumor-NK cell interactions is needed to confirm and expand on these observations.
4.3 Strategy III: LFA-1-mediated tumor invasiveness
In solid tumors, metastatic extravasation involves increased invasiveness through adhesion and transendothelial migration of disseminated cancer cells (126). Tumor-associated leukocytes, such as pro-tumorigenic macrophages, can exacerbate this process. For example, in nasopharyngeal carcinoma, tumor-associated macrophages (TAMs) interact with interferon-stimulated gene 15 (ISG15) via LFA-1 (127). This interaction triggers SRC family kinase (SFK) signaling in the TAMs, leading to the secretion of CCL18 into the TME, which plays a crucial role in promoting tumor cell dissemination (127). Equally, tumor-associated neutrophils (TANs) are known to promote tumor growth and angiogenesis in breast cancer. A study using murine models showed that estradiol increased pro-tumorigenic neutrophil recruitment to the tumor site, in a process facilitated by the overexpression of TGFβ1 and LFA-1 (128).
Microglia, the resident immune cells in the brain that express LFA-1, also play a crucial role in the TME, contributing to tumorigenesis and neoplastic cell growth in low-grade gliomas (LGGs) (129). Although the exact mechanism by which LFA-1 interaction mediates metastasis in LGGs is unclear, genetic ablation of CD11a (a subunit of LFA-1) has been shown to reduce microglia infiltration, proliferation, and CCL5 secretion, thereby decreasing the likelihood of LGG progression (129).
In CLL, leukemic B cells exhibit enhanced adhesion due to LFA-1 activation, which is triggered by CXCL12, leading to the dissemination of CLL cells to secondary lymphoid organs (130, 131). During the pathogenesis of CLL, CXCL12 activates protein tyrosine kinase (PTK), Bruton’s tyrosine kinase (BTK), and Janus Kinase (JAK), which in turn activate the small GTPase RhoA (132). This signaling cascade shifts LFA-1 from an inactive to a high-affinity conformation, thereby promoting adhesion to ICAM-1 and spreading of CLL B cells into secondary niches (131). Elsewhere, human peripheral blood mononuclear cells (PBMCs) from healthy donors or CLL patients were injected into non-irradiated mice, and three hours later, cells were collected from lymph nodes, bone marrow, and spleen. Using flow cytometry, the study found that CLL B cells had reduced LFA-1 expression, impairing homing to lymph nodes but allowing re-entry into the bone marrow via VLA-4 (83). Both normal and CLL B cells could home to the spleen independently of these integrins, suggesting that integrin targeting may block CLL cells from survival niches in lymph nodes and the bone marrow (83).
T regulatory cells (Tregs) serve as an obstacle to effective antitumor immunity (133), and there is evidence suggesting that LFA-1 may influence tumorigenesis through them. A study using LFA-1 knockout mice demonstrated that the absence of LFA-1 significantly inhibited tumor growth in both subcutaneous melanoma and intestinal adenocarcinoma models (134). This tumor suppression was linked to a substantial decrease in Treg cell numbers in the spleen, blood, and mesenteric lymph nodes. Similarly, treatment of tumor-bearing wild-type mice with the LFA-1 inhibitor BIRT377 reduced both tumor growth and Treg cell accumulation. Moreover, analysis of tumor databases revealed a positive correlation between LFA-1 expression, Treg cell infiltration, and tumor progression. These findings suggest that LFA-1 contributes to a pro-tumorigenic microenvironment by promoting Treg cell-mediated immune suppression, drawing attention to its potential as a therapeutic target for cancer immunotherapy.
Interestingly, certain tumors progress and metastasize by leveraging LFA-1’s interaction with its other ligands, particularly ICAM-3 (135, 136). Shen et al. demonstrated that in human lung carcinoma and breast adenocarcinoma, signaling through ICAM-3 promoted cancer cell stemness (135). Their investigation revealed that ICAM-3 upregulation in these tumors correlated with higher tumor grades and played a critical role in driving metastasis via LFA-1 (136). They further showed that ICAM-3 binds LFA-1 through its extracellular domain and associates with the structural protein ERM, while its intracellular domain links to lamellipodia. This interaction generates mechanical tension, facilitating cell separation and metastasis (136).
ICAM-2 has also been implicated in angiogenesis, as shown in a study using ICAM-2–deficient mice and endothelial cells. The absence of ICAM-2 impaired angiogenesis both in vitro and in vivo by disrupting homophilic interactions that likely contributed to endothelial tube formation (137). ICAM-2–deficient cells exhibited defective migration and increased apoptosis in response to stressors such as serum deprivation, anti-Fas antibody, or staurosporine, underlining its critical role in supporting endothelial cell function during angiogenesis (137). In contrast to its role in promoting angiogenesis, ICAM-2 expression in neuroblastoma has been linked to a reduced metastatic potential. A study using neuroblastoma cells from mouse models demonstrated that ICAM-2 interacted with α-actinin, which strengthened the membrane-cytoskeleton link (138). This interaction reduced cellular invasiveness and motility, thereby supporting a more favorable clinical outcome, suggesting that ICAM-2 might act as a tumor suppressor in neuroblastoma by limiting metastasis. While both studies show ICAM-2’s involvement in regulation of cellular adhesion and actin dynamics, they highlight its context-dependent functions—supporting tumor suppression in neuroblastoma while aiding endothelial migration and survival in angiogenesis.
Altogether, these studies underscore the dual role of LFA-1 in immunosurveillance. They reveal the intricate dependence of leukocytes on this integrin, while also highlighting how tumors and pro-tumorigenic leukocytes, including Tregs, can exploit LFA-1 and its ligands to drive tumorigenesis and metastasis. This duality poses several challenges in understanding LFA-1 dynamics in tumors and leukocytes and complicates the development of LFA-1-targeting therapeutics in oncology.
5 Intervention strategies targeting LFA-1-mediated processes in cancer
Following the previous chapter’s examination of how tumors undermine LFA-1-mediated immune responses, this chapter shifts focus to therapeutic strategies that may potentially counteract these cancer immune evasion tactics. As summarized in Table 1, we will explore LFA-1-centered interventions that may potentiate: (i) leukocyte infiltration and activation in the TME, (ii) stable LFA-1-mediated synapse formation between leukocytes and tumors to boost cytokine production and cytotoxicity, and (iii) inhibition of LFA-1-driven tumor progression and metastasis.
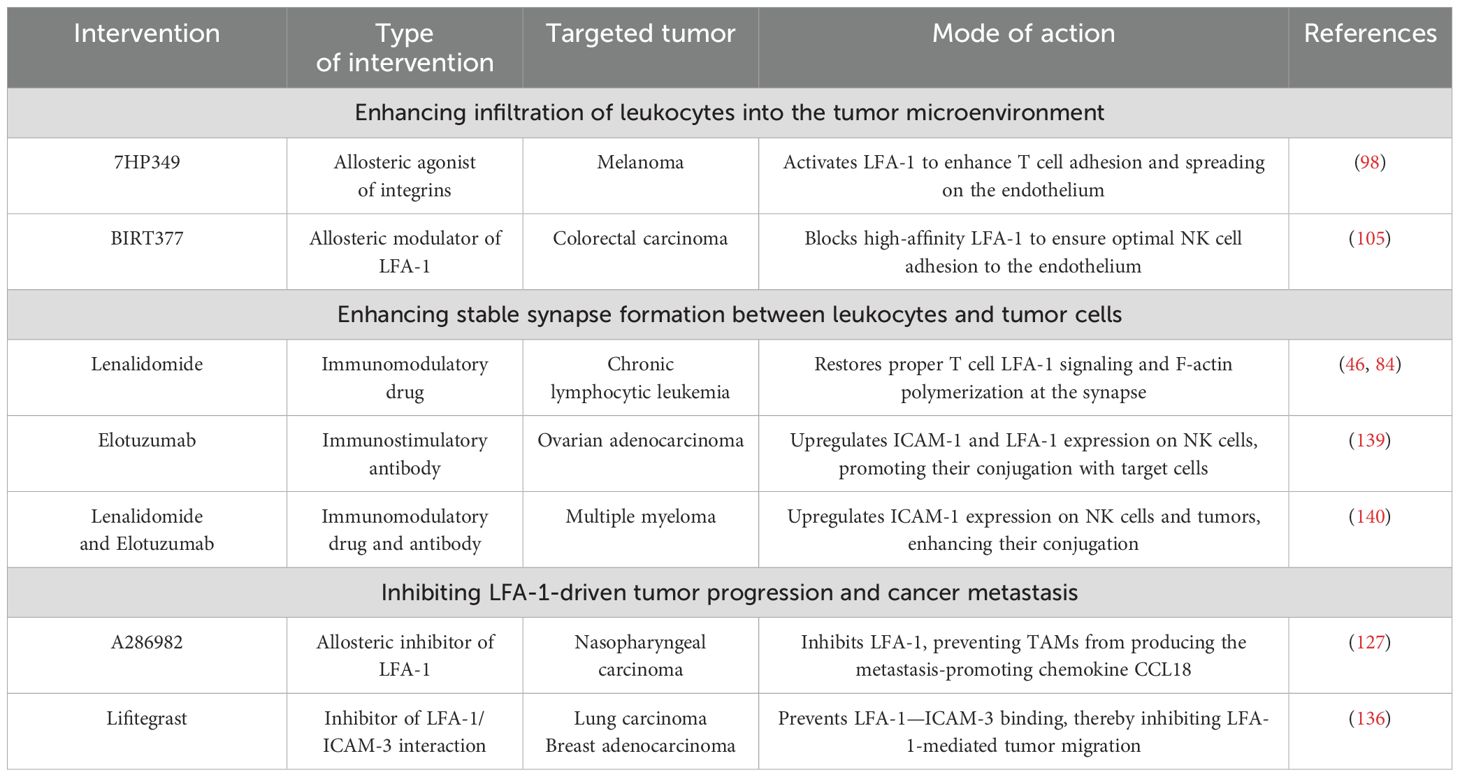
Table 1. A summary of interventions targeting LFA-1-mediated processes.
5.1 Enhancing LFA-1-mediated leukocyte infiltration into the tumor microenvironment
Increased leukocyte infiltration into tumors has long been linked to improved tumor clearance and better cancer prognosis, a correlation that holds true even following immunotherapy (97, 99, 141, 142). Proof-of-concept studies using mouse models have demonstrated that LFA-1-activating interventions can convert a T cell–exclusionary tumor microenvironment into one enriched with T cells (98). Hickman et al. showed that in melanoma, treatment with 7HP349, a small molecule that activates LFA-1, significantly increased the infiltration of tumor-specific CD8+ T cells into tumors by promoting chemoattraction via CXCL12. Furthermore, when 7HP349 was combined with anti–CTLA-4 therapy, infiltration of CD8+ effector T cells in anti–programmed death 1–resistant (anti–PD-1)-resistant tumors was enhanced, and facilitated tumor regression (98). By activating LFA-1, 7HP349 enhances T cell adhesion to ICAM-1, leading to improved cell spreading and migration from the vasculature into the tumor microenvironment.
In contrast, the drug BIRT377, which inhibits the high-affinity conformation of LFA-1 by blocking domain I in the β2 subunit, enhances NK cell infiltration into colorectal carcinoma and drives tumor regression in mice models (105). This mechanism is particularly important because, as discussed in the previous chapter, Nandagopal et al. demonstrated that C3a/C3aR signaling induces high-affinity LFA-1 in the uropods of NK cells, leading to reduced tumor infiltration, likely due to NK cells becoming trapped on the endothelium (105). By preventing this high-affinity LFA-1 state, BIRT377 effectively reduces excessive adhesion of NK cells to the endothelium and supports them to migrate into the tumor microenvironment.
5.2 Enhancing LFA-1-dependent stable synapse formation between leukocytes and tumors to promote cytokine production and cytotoxicity
The immunomodulatory drug lenalidomide has been shown to restore impaired immune synapse formation between T cells and CLL B cells, as demonstrated in in vitro studies using human samples (46). These defective synapses are primarily due to disruptions in the localization of signaling molecules, including the clustering of LFA-1 at the synapse, which impairs F-actin remodeling—a critical process for the formation of mature synapses (46). Lenalidomide normalizes Rho GTPase signaling, thereby restoring proper downstream signaling of LFA-1, which facilitates effective actin dynamics and the formation of functional immunological synapses (46, 84).
Elsewhere, treatment with the monoclonal antibody elotuzumab, which targets signaling lymphocytic activation molecule family member 7 (SLAMF7) on tumor cells, was shown to enhance conjugation between NK cells and ovarian adenocarcinoma target cells in cell culture (139). This was correlated with upregulated expression of active LFA-1 and ICAM-1 on NK cells, potentially accounting for the increased conjugation. Moreover, there was a notable elevation in the levels of the activating receptor NKG2D on NK cells. These changes collectively facilitated improved NK cell adhesion and cytotoxicity against tumor targets (139). On top of that, combining elotuzumab with lenalidomide significantly increased ICAM-1 expression in both NK cells and multiple myeloma cells, surpassing the effects of either treatment alone (140). This elevated ICAM-1 expression was associated with the formation of stable contacts with tumor cells, leading to enhanced NK cell-mediated cytotoxicity. Overall, these reports emphasize the fundamental role of ICAM-1/LFA-1 interactions in leukocyte-driven tumoricidal processes and demonstrate how LFA-1 functions can be enhanced through combinatorial therapy.
5.3 Inhibiting LFA-1-driven tumor progression and cancer metastasis
As discussed in the previous chapter, LFA-1 can adversely influence tumor progression through its interaction with interferon-stimulated gene 15 (ISG15), which activates tumor-associated macrophages (TAMs) (127). These macrophages subsequently release CCL18, a promiscuous chemokine known to drive tumor progression and metastasis (127). In this regard, Chen et al. demonstrated using in vitro assays and a murine model that inhibiting LFA-1 with the small allosteric inhibitor A286982 effectively blocked CCL18 secretion from macrophages. Additionally, the increased migration of nasopharyngeal carcinoma cells, induced by CCL18 from the supernatants of ISG15-treated macrophages, was significantly reduced following treatment with this LFA-1 inhibitor (127).
In human lung carcinoma and breast adenocarcinoma, tumor progression and metastasis have been linked to the interaction between ICAM-3 and LFA-1 (135, 136). Lifitegrast, a drug that disrupts the binding between LFA-1 and ICAM-3, was shown through in vitro assays and mouse models to inhibit cell migration in both tumor cell lines (136). Furthermore, combining Lifitegrast with an LFA-1 antibody resulted in a significant reduction in tumor cell migration (136).
Taken together, these studies, primarily based on in vitro experiments and in vivo work in murine models, highlight the critical role of targeting ICAM-1/LFA-1 interactions between leukocytes and tumors for therapeutic purposes. This approach can either enhance leukocyte-mediated tumoricidal activity or, conversely, inhibit LFA-1-driven tumorigenesis and metastasis. Moreover, the findings suggest that in certain cancers, LFA-1 functionality can be optimized through combinatorial therapies, offering a more effective treatment strategy. Nonetheless, to fully realize the clinical potential of these approaches, further studies are essential to validate their utility in clinical settings.
6 Conclusions and remarks
In summary, we have explored the role of LFA-1 in promoting immunosurveillance and how tumors subvert these processes by altering LFA-1 functions. We identified three strategies through which tumors can escape immune surveillance via LFA-1-mediated processes: (I) alterations in LFA-1-mediated leukocyte infiltration into the tumor, (II) disruptions in LFA-1 functionality within the tumor, and (III) LFA-1-driven tumor invasiveness. Finally, we reviewed various interventions aimed at overcoming these challenges and emphasized the need for further studies beyond in vitro and animal models.
Developing LFA-1-targeting cancer therapies remains complex due to the duality of LFA-1’s roles—beneficial for immune defense but also exploitable by cancer cells during tumorigenesis. This duality is highly context-dependent, and further research is needed to fully understand where LFA-1 can be most effectively leveraged. Therapeutic targeting of LFA-1 requires balancing its pro-tumor activity with its essential role in immune function. Focusing on LFA-1 signaling pathways rather than the protein itself may be promising, though these pathways are often redundant and intertwined with other cellular receptors, adding complexity.
Future research should focus on better understanding how tumors impair immune cells within the TME through LFA-1 and the associated signaling pathways. A deeper understanding of these mechanisms will enable the rational design of LFA-1-targeted interventions. For example, exploring negative regulatory pathways through LFA-1 in tumor-contacted leukocytes, along with the specific molecules involved, may uncover new molecular targets for future therapies.
Author contributions
SU: Conceptualization, Writing – original draft. LM: Conceptualization, Writing – original draft, Visualization, Writing – review & editing. LS: Conceptualization, Writing – original draft, Writing – review & editing, Funding acquisition, Supervision.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was supported by The Swedish Research Council (grant 2018-05229), The Swedish Cancer Society (grants CAN2018/696 and 21 1613), the Kempe Foundation (SMK-2060), Bäckströms Foundation, Sandströms Foundation, Insamlingsstiftelserna and Umeå University.
Acknowledgments
We extend our special thanks to the Department of Molecular Biology at Umeå University for their invaluable support. The figures used in this paper were created in BioRender.com by Lewis Murugu.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Barczyk M, Carracedo S, Gullberg D. Integrins. Cell Tissue Res. (2010) 339:269–80. doi: 10.1007/s00441-009-0834-6
2. Springer TA. Adhesion receptors of the immune system. Nature. (1990) 346:425–34. doi: 10.1038/346425a0
3. Springer TA. Traffic signals for lymphocyte recirculation and leukocyte emigration: the multistep paradigm. Cell. (1994) 76:301–14. doi: 10.1016/0092-8674(94)90337-9
4. Tamkun JW, DeSimone DW, Fonda D, Patel RS, Buck C, Horwitz AF, et al. Structure of integrin, a glycoprotein involved in the transmembrane linkage between fibronectin and actin. Cell. (1986) 46:271–82. doi: 10.1016/0092-8674(86)90744-0
5. Hyun YM, Lefort CT, Kim M. Leukocyte integrins and their ligand interactions. Immunol Res. (2009) 45:195–208. doi: 10.1007/s12026-009-8101-1
6. Sanchez-Madrid F, Nagy JA, Robbins E, Simon P, Springer TA. A human leukocyte differentiation antigen family with distinct alpha-subunits and a common beta-subunit: the lymphocyte function-associated antigen (Lfa-1), the C3bi complement receptor (Okm1/mac-1), and the P150,95 molecule. J Exp Med. (1983) 158:1785–803. doi: 10.1084/jem.158.6.1785
7. Berlin-Rufenach C, Otto F, Mathies M, Westermann J, Owen MJ, Hamann A, et al. Lymphocyte migration in lymphocyte function-associated antigen (Lfa)-1-deficient mice. J Exp Med. (1999) 189:1467–78. doi: 10.1084/jem.189.9.1467
8. Evans BJ, McDowall A, Taylor PC, Hogg N, Haskard DO, Landis RC. Shedding of lymphocyte function-associated antigen-1 (Lfa-1) in a human inflammatory response. Blood. (2006) 107:3593–9. doi: 10.1182/blood-2005-09-3695
9. Gay D, Coeshott C, Golde W, Kappler J, Marrack P. The major histocompatibility complex-restricted antigen receptor on T cells. Ix. Role of accessory molecules in recognition of antigen plus isolated ia. J Immunol. (1986) 136:2026–32. doi: 10.4049/jimmunol.136.6.2026
10. Henderson RB, Lim LH, Tessier PA, Gavins FN, Mathies M, Perretti M, et al. The use of lymphocyte function-associated antigen (Lfa)-1-deficient mice to determine the role of lfa-1, mac-1, and alpha4 integrin in the inflammatory response of neutrophils. J Exp Med. (2001) 194:219–26. doi: 10.1084/jem.194.2.219
11. Lawrence MB, Springer TA. Leukocytes Roll on a Selectin at Physiologic Flow Rates: Distinction from and Prerequisite for Adhesion through Integrins. Cell. (1991) 65:859–73. doi: 10.1016/0092-8674(91)90393-d
12. Salas A, Shimaoka M, Kogan AN, Harwood C, von Andrian UH, Springer TA. Rolling adhesion through an extended conformation of integrin alphalbeta2 and relation to alpha I and beta I-like domain interaction. Immunity. (2004) 20:393–406. doi: 10.1016/s1074-7613(04)00082-2
13. Shamri R, Grabovsky V, Gauguet JM, Feigelson S, Manevich E, Kolanus W, et al. Lymphocyte arrest requires instantaneous induction of an extended lfa-1 conformation mediated by endothelium-bound chemokines. Nat Immunol. (2005) 6:497–506. doi: 10.1038/ni1194
14. Marlin SD, Springer TA. Purified intercellular adhesion molecule-1 (Icam-1) is a ligand for lymphocyte function-associated antigen 1 (Lfa-1). Cell. (1987) 51:813–9. doi: 10.1016/0092-8674(87)90104-8
15. Staunton DE, Dustin ML, Springer TA. Functional cloning of icam-2, a cell adhesion ligand for lfa-1 homologous to icam-1. Nature. (1989) 339:61–4. doi: 10.1038/339061a0
16. Fawcett J, Holness CL, Needham LA, Turley H, Gatter KC, Mason DY, et al. Molecular cloning of icam-3, a third ligand for lfa-1, constitutively expressed on resting leukocytes. Nature. (1992) 360:481–4. doi: 10.1038/360481a0
17. Bailly P, Tontti E, Hermand P, Cartron JP, Gahmberg CG. The red cell lw blood group protein is an intercellular adhesion molecule which binds to cd11/cd18 leukocyte integrins. Eur J Immunol. (1995) 25:3316–20. doi: 10.1002/eji.1830251217
18. Tian L, Kilgannon P, Yoshihara Y, Mori K, Gallatin WM, Carpén O, et al. Binding of T lymphocytes to hippocampal neurons through icam-5 (Telencephalin) and characterization of its interaction with the leukocyte integrin cd11a/cd18. Eur J Immunol. (2000) 30:810–8. doi: 10.1002/1521-4141(200003)30:3<810::AID-IMMU810>3.0.CO;2-X
19. Ostermann G, Weber KS, Zernecke A, Schröder A, Weber C. Jam-1 is a ligand of the beta(2) integrin lfa-1 involved in transendothelial migration of leukocytes. Nat Immunol. (2002) 3:151–8. doi: 10.1038/ni755
20. Barber DF, Faure M, Long EO. Lfa-1 contributes an early signal for nk cell cytotoxicity. J Immunol. (2004) 173:3653–9. doi: 10.4049/jimmunol.173.6.3653
21. Hayflick JS, Kilgannon P, Gallatin WM. The intercellular adhesion molecule (Icam) family of proteins. New members and novel functions. Immunol Res. (1998) 17:313–27. doi: 10.1007/bf02786454
22. Smith CW. 3. Adhesion molecules and receptors. J Allergy Clin Immunol. (2008) 121:S375–9; quiz S414. doi: 10.1016/j.jaci.2007.07.030
23. Shi H, Shao B. Lfa-1 activation in T-cell migration and immunological synapse formation. Cells. (2023) 12:1136. doi: 10.3390/cells12081136
24. Gérard A, Cope AP, Kemper C, Alon R, Köchl R. Lfa-1 in T cell priming, differentiation, and effector functions. Trends Immunol. (2021) 42:706–22. doi: 10.1016/j.it.2021.06.004
25. Springer TA, Dustin ML. Integrin inside-out signaling and the immunological synapse. Curr Opin Cell Biol. (2012) 24:107–15. doi: 10.1016/j.ceb.2011.10.004
26. Kim M, Carman CV, Springer TA. Bidirectional transmembrane signaling by cytoplasmic domain separation in integrins. Science. (2003) 301:1720–5. doi: 10.1126/science.1084174
27. Varga G, Nippe N, Balkow S, Peters T, Wild MK, Seeliger S, et al. Lfa-1 contributes to signal I of T-cell activation and to the production of T(H)1 cytokines. J Invest Dermatol. (2010) 130:1005–12. doi: 10.1038/jid.2009.398
28. Katagiri K, Maeda A, Shimonaka M, Kinashi T. Rapl, a rap1-binding molecule that mediates rap1-induced adhesion through spatial regulation of lfa-1. Nat Immunol. (2003) 4:741–8. doi: 10.1038/ni950
29. Raab M, Wang H, Lu Y, Smith X, Wu Z, Strebhardt K, et al. T cell receptor “inside-out” Pathway via signaling module skap1-rapl regulates T cell motility and interactions in lymph nodes. Immunity. (2010) 32:541–56. doi: 10.1016/j.immuni.2010.03.007
30. Sebzda E, Bracke M, Tugal T, Hogg N, Cantrell DA. Rap1a positively regulates T cells via integrin activation rather than inhibiting lymphocyte signaling. Nat Immunol. (2002) 3:251–8. doi: 10.1038/ni765
31. Tohyama Y, Katagiri K, Pardi R, Lu C, Springer TA, Kinashi T. The critical cytoplasmic regions of the alphal/beta2 integrin in rap1-induced adhesion and migration. Mol Biol Cell. (2003) 14:2570–82. doi: 10.1091/mbc.e02-09-0615
32. Han J, Lim CJ, Watanabe N, Soriani A, Ratnikov B, Calderwood DA, et al. Reconstructing and deconstructing agonist-induced activation of integrin alphaiibbeta3. Curr Biol. (2006) 16:1796–806. doi: 10.1016/j.cub.2006.08.035
33. Lefort CT, Rossaint J, Moser M, Petrich BG, Zarbock A, Monkley SJ, et al. Distinct roles for talin-1 and kindlin-3 in lfa-1 extension and affinity regulation. Blood. (2012) 119:4275–82. doi: 10.1182/blood-2011-08-373118
34. Ménasché G, Kliche S, Chen EJ, Stradal TE, Schraven B, Koretzky G. Riam links the adap/skap-55 signaling module to rap1, facilitating T-cell-receptor-mediated integrin activation. Mol Cell Biol. (2007) 27:4070–81. doi: 10.1128/mcb.02011-06
35. Li J, Jo MH, Yan J, Hall T, Lee J, López-Sánchez U, et al. Ligand binding initiates single-molecule integrin conformational activation. Cell. (2024) 187:2990–3005.e17. doi: 10.1016/j.cell.2024.04.049
36. Vicente-Manzanares M, Choi CK, Horwitz AR. Integrins in cell migration–the actin connection. J Cell Sci. (2009) 122:199–206. doi: 10.1242/jcs.018564
37. Gross CC, Brzostowski JA, Liu D, Long EO. Tethering of intercellular adhesion molecule on target cells is required for lfa-1-dependent nk cell adhesion and granule polarization. J Immunol. (2010) 185:2918–26. doi: 10.4049/jimmunol.1000761
38. Mace EM, Monkley SJ, Critchley DR, Takei F. A dual role for talin in nk cell cytotoxicity: activation of lfa-1-mediated cell adhesion and polarization of nk cells. J Immunol. (2009) 182:948–56. doi: 10.4049/jimmunol.182.2.948
39. Mace EM, Zhang J, Siminovitch KA, Takei F. Elucidation of the integrin lfa-1-mediated signaling pathway of actin polarization in natural killer cells. Blood. (2010) 116:1272–9. doi: 10.1182/blood-2009-12-261487
40. Urlaub D, Höfer K, Müller ML, Watzl C. Lfa-1 activation in nk cells and their subsets: influence of receptors, maturation, and cytokine stimulation. J Immunol. (2017) 198:1944–51. doi: 10.4049/jimmunol.1601004
41. Rouzaut A, Garasa S, Teijeira Á, González I, Martinez-Forero I, Suarez N, et al. Dendritic cells adhere to and transmigrate across lymphatic endothelium in response to ifn-A. Eur J Immunol. (2010) 40:3054–63. doi: 10.1002/eji.201040523
42. Carlin LM, Stamatiades EG, Auffray C, Hanna RN, Glover L, Vizcay-Barrena G, et al. Nr4a1-dependent ly6clow monocytes monitor endothelial cells and orchestrate their disposal. Cell. (2013) 153:362–75. doi: 10.1016/j.cell.2013.03.010
43. Ribatti D. The concept of immune surveillance against tumors. First Theories Oncotarget. (2017) 8:7175–80. doi: 10.18632/oncotarget.12739
44. Smyth MJ, Godfrey DI, Trapani JA. A fresh look at tumor immunosurveillance and immunotherapy. Nat Immunol. (2001) 2:293–9. doi: 10.1038/86297
45. Swann JB, Smyth MJ. Immune surveillance of tumors. J Clin Invest. (2007) 117:1137–46. doi: 10.1172/jci31405
46. Ramsay AG, Johnson AJ, Lee AM, Gorgün G, Le Dieu R, Blum W, et al. Chronic lymphocytic leukemia T cells show impaired immunological synapse formation that can be reversed with an immunomodulating drug. J Clin Iinvest. (2008) 118:2427–37. doi: 10.1172/JCI35017
47. Petit A-E, Demotte N, Scheid B, Wildmann C, Bigirimana R, Gordon-Alonso M, et al. A major secretory defect of tumour-infiltrating T lymphocytes due to galectin impairing lfa-1-mediated synapse completion. Nat Commun. (2016) 7:12242. doi: 10.1038/ncomms12242
48. Balkow S, Heinz S, Schmidbauer P, Kolanus W, Holzmann B, Grabbe S, et al. Lfa-1 activity state on dendritic cells regulates contact duration with T cells and promotes T-cell priming. Blood. (2010) 116:1885–94. doi: 10.1182/blood-2009-05-224428
49. Katakai T, Habiro K, Kinashi T. Dendritic cells regulate high-speed interstitial T cell migration in the lymph node via lfa-1/icam-1. J Immunol. (2013) 191:1188–99. doi: 10.4049/jimmunol.1300739
50. Verma NK, Kelleher D. Not just an adhesion molecule: lfa-1 contact tunes the T lymphocyte program. J Immunol. (2017) 199:1213–21. doi: 10.4049/jimmunol.1700495
51. Feigelson SW, Pasvolsky R, Cemerski S, Shulman Z, Grabovsky V, Ilani T, et al. Occupancy of lymphocyte lfa-1 by surface-immobilized icam-1 is critical for tcr- but not for chemokine-triggered lfa-1 conversion to an open headpiece high-affinity state. J Immunol. (2010) 185:7394–404. doi: 10.4049/jimmunol.1002246
52. Comrie WA, Burkhardt JK. Action and traction: cytoskeletal control of receptor triggering at the immunological synapse. Front Immunol. (2016) 7:68. doi: 10.3389/fimmu.2016.00068
53. Pettmann J, Awada L, Różycki B, Huhn A, Faour S, Kutuzov M, et al. Mechanical forces impair antigen discrimination by reducing differences in T-cell receptor/peptide–mhc off-rates. EMBO J. (2022) 42(7):e111841. doi: 10.15252/embj.2022111841
54. Reichardt P, Patzak I, Jones K, Etemire E, Gunzer M, Hogg N. A role for lfa-1 in delaying T-lymphocyte egress from lymph nodes. EMBO J. (2013) 32:829–43. doi: 10.1038/emboj.2013.33
55. Feigelson SW, Solomon A, Biram A, Hatzav M, Lichtenstein M, Regev O, et al. Icams are not obligatory for functional immune synapses between naive cd4 t cells and lymph node dcs. Cell Rep. (2018) 22:849–59. doi: 10.1016/j.celrep.2017.12.103
56. Lämmermann T, Bader BL, Monkley SJ, Worbs T, Wedlich-Söldner R, Hirsch K, et al. Rapid leukocyte migration by integrin-independent flowing and squeezing. Nature. (2008) 453:51–5. doi: 10.1038/nature06887
57. Woolf E, Grigorova I, Sagiv A, Grabovsky V, Feigelson SW, Shulman Z, et al. Lymph node chemokines promote sustained T lymphocyte motility without triggering stable integrin adhesiveness in the absence of shear forces. Nat Immunol. (2007) 8:1076–85. doi: 10.1038/ni1499
58. Yanguas A, Garasa S, Teijeira Á, Aubá C, Melero I, Rouzaut A. Icam-1-lfa-1 dependent cd8+ T-lymphocyte aggregation in tumor tissue prevents recirculation to draining lymph nodes. Front Immunol. (2018) 9:2084. doi: 10.3389/fimmu.2018.02084
59. Salomon Bt, Bluestone JA. Cutting edge: lfa-1 interaction with icam-1 and icam-2 regulates th2 cytokine production1. J Immunol. (1998) 161:5138–42. doi: 10.4049/jimmunol.161.10.5138
60. Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, et al. Interleukin 17–producing cd4+ Effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol. (2005) 6:1123–32. doi: 10.1038/ni1254
61. Tay RE, Richardson EK, Toh HC. Revisiting the role of cd4+ T cells in cancer immunotherapy—New insights into old paradigms. Cancer Gene Ther. (2021) 28:5–17. doi: 10.1038/s41417-020-0183-x
62. Sabatos CA, Doh J, Chakravarti S, Friedman RS, Pandurangi PG, Tooley AJ, et al. A synaptic basis for paracrine interleukin-2 signaling during homotypic T cell interaction. Immunity. (2008) 29:238–48. doi: 10.1016/j.immuni.2008.05.017
63. Barao I, Hudig D, Ascensao JL. Il-15-mediated induction of lfa-1 is a late step required for cytotoxic differentiation of human nk cells from cd34+Lin– bone marrow cells1. J Immunol. (2003) 171:683–90. doi: 10.4049/jimmunol.171.2.683
64. Huntington ND, Vosshenrich CAJ, Di Santo JP. Developmental pathways that generate natural-killer-cell diversity in mice and humans. Nat Rev Immunol. (2007) 7:703–14. doi: 10.1038/nri2154
65. Santoni G, Amantini C, Santoni M, Maggi F, Morelli MB, Santoni A. Mechanosensation and mechanotransduction in natural killer cells. Front Immunol. (2021) 12:688918. doi: 10.3389/fimmu.2021.688918
66. Orange JS, Harris KE, Andzelm MM, Valter MM, Geha RS, Strominger JL. The mature activating natural killer cell immunologic synapse is formed in distinct stages. Proc Natl Acad Sci U.S.A. (2003) 100:14151–6. doi: 10.1073/pnas.1835830100
67. Hunter MC, Teijeira A, Halin C. T cell trafficking through lymphatic vessels. Front Immunol. (2016) 7:613. doi: 10.3389/fimmu.2016.00613
68. Wang C, Zeng Q, Gül ZM, Wang S, Pick R, Cheng P, et al. Circadian tumor infiltration and function of cd8(+) T cells dictate immunotherapy efficacy. Cell. (2024) 187:2690–702.e17. doi: 10.1016/j.cell.2024.04.015
69. Paul S, Lal G. The molecular mechanism of natural killer cells function and its importance in cancer immunotherapy. Front Immunol. (2017) 8:1124. doi: 10.3389/fimmu.2017.01124
70. Boieri M, Ulvmoen A, Sudworth A, Lendrem C, Collin M, Dickinson AM, et al. Il-12, il-15, and il-18 pre-activated nk cells target resistant T cell acute lymphoblastic leukemia and delay leukemia development in vivo. Oncoimmunology. (2017) 6:e1274478. doi: 10.1080/2162402x.2016.1274478
71. Asrir A, Tardiveau C, Coudert J, Laffont R, Blanchard L, Bellard E, et al. Tumor-associated high endothelial venules mediate lymphocyte entry into tumors and predict response to pd-1 plus ctla-4 combination immunotherapy. Cancer Cell. (2022) 40:318–34.e9. doi: 10.1016/j.ccell.2022.01.002
72. Martinet L, Garrido I, Filleron T, Le Guellec S, Bellard E, Fournie JJ, et al. Human solid tumors contain high endothelial venules: association with T- and B-lymphocyte infiltration and favorable prognosis in breast cancer. Cancer Res. (2011) 71:5678–87. doi: 10.1158/0008-5472.Can-11-0431
73. Capece T, Walling BL, Lim K, Kim KD, Bae S, Chung HL, et al. A novel intracellular pool of lfa-1 is critical for asymmetric cd8(+) T cell activation and differentiation. J Cell Biol. (2017) 216:3817–29. doi: 10.1083/jcb.201609072
74. Franciszkiewicz K, Le Floc’h A, Boutet M, Vergnon I, Schmitt A, Mami-Chouaib F. Cd103 or lfa-1 engagement at the immune synapse between cytotoxic T cells and tumor cells promotes maturation and regulates T-cell effector functions. Cancer Res. (2013) 73:617–28. doi: 10.1158/0008-5472.Can-12-2569
75. Lötscher J, i Líndez A-AM, Kirchhammer N, Cribioli E, Attianese GMPG, Trefny MP, et al. Magnesium sensing via lfa-1 regulates cd8+ T cell effector function. Cell. (2022) 185:585–602.e29. doi: 10.1016/j.cell.2021.12.039
76. Schmits R, Kündig TM, Baker DM, Shumaker G, Simard JJ, Duncan G, et al. Lfa-1-deficient mice show normal ctl responses to virus but fail to reject immunogenic tumor. J Exp Med. (1996) 183:1415–26. doi: 10.1084/jem.183.4.1415
77. Mace EM, Wu WW, Ho T, Mann SS, Hsu H-T, Orange JS. Nk cell lytic granules are highly motile at the immunological synapse and require F-actin for post-degranulation persistence. J Immunol. (2012) 189:4870–80. doi: 10.4049/jimmunol.1201296
78. Orange JS. Formation and function of the lytic nk-cell immunological synapse. Nat Rev Immunol. (2008) 8:713–25. doi: 10.1038/nri2381
79. Poggi A, Carosio R, Spaggiari GM, Fortis C, Tambussi G, Dell’Antonio G, et al. Nk cell activation by dendritic cells is dependent on lfa-1-mediated induction of calcium-calmodulin kinase ii: inhibition by hiv-1 tat C-terminal domain. J Immunol. (2002) 168:95–101. doi: 10.4049/jimmunol.168.1.95
80. Kohl S, Springer TA, Schmalstieg FC, Loo LS, Anderson DC. Defective natural killer cytotoxicity and polymorphonuclear leukocyte antibody-dependent cellular cytotoxicity in patients with lfa-1/okm-1 deficiency. J Immunol. (1984) 133:2972–8. doi: 10.4049/jimmunol.133.6.2972
81. Mahuron KM, Moreau JM, Glasgow JE, Boda DP, Pauli ML, Gouirand V, et al. Layilin augments integrin activation to promote antitumor immunity. J Exp Med. (2020) 217(9):e20192080. doi: 10.1084/jem.20192080
82. Weulersse M, Asrir A, Pichler AC, Lemaitre L, Braun M, Carrié N, et al. Eomes-dependent loss of the co-activating receptor cd226 restrains cd8(+) T cell anti-tumor functions and limits the efficacy of cancer immunotherapy. Immunity. (2020) 53:824–39.e10. doi: 10.1016/j.immuni.2020.09.006
83. Hartmann TN, Grabovsky V, Wang W, Desch P, Rubenzer G, Wollner S, et al. Circulating B-cell chronic lymphocytic leukemia cells display impaired migration to lymph nodes and bone marrow. Cancer Res. (2009) 69:3121–30. doi: 10.1158/0008-5472.Can-08-4136
84. Ramsay AG, Evans R, Kiaii S, Svensson L, Hogg N, Gribben JG. Chronic lymphocytic leukemia cells induce defective lfa-1–directed T-cell motility by altering rho gtpase signaling that is reversible with lenalidomide. Blood. (2013) 121:2704–14. doi: 10.1182/blood-2012-08-448332
85. Thomas G, Tacke R, Hedrick CC, Hanna RN. Nonclassical patrolling monocyte function in the vasculature. Arterioscler Thromb Vasc Biol. (2015) 35:1306–16. doi: 10.1161/atvbaha.114.304650
86. Marcovecchio PM, Zhu YP, Hanna RN, Dinh HQ, Tacke R, Wu R, et al. Frontline science: kindlin-3 is essential for patrolling and phagocytosis functions of nonclassical monocytes during metastatic cancer surveillance. J Leukoc Biol. (2020) 107:883–92. doi: 10.1002/JLB.4HI0420-098R
87. Granot Z, Henke E, Comen EA, King TA, Norton L, Benezra R. Tumor entrained neutrophils inhibit seeding in the premetastatic lung. Cancer Cell. (2011) 20:300–14. doi: 10.1016/j.ccr.2011.08.012
88. Regev O, Kizner M, Roncato F, Dadiani M, Saini M, Castro-Giner F, et al. Icam-1 on breast cancer cells suppresses lung metastasis but is dispensable for tumor growth and killing by cytotoxic T cells. Front Immunol. (2022) 13:849701. doi: 10.3389/fimmu.2022.849701
89. Espie D, Donnadieu E. New insights into car T cell-mediated killing of tumor cells. Front Immunol. (2022) 13:1016208. doi: 10.3389/fimmu.2022.1016208
90. Kantari-Mimoun C, Barrin S, Vimeux L, Haghiri S, Gervais C, Joaquina S, et al. Car T-Cell Entry into Tumor Islets Is a Two-Step Process Dependent on Ifnγ and Icam-1car T-Cell Entry into Tumor Islets. Cancer Immunol Res. (2021) 9:1425–38. doi: 10.1158/2326-6066.CIR-20-0837
91. Larson RC, Kann MC, Bailey SR, Haradhvala NJ, Llopis PM, Bouffard AA, et al. Car T cell killing requires the ifnγr pathway in solid but not liquid tumours. Nature. (2022) 604:563–70. doi: 10.1038/s41586-022-04585-5
92. Dong E, Yue XZ, Shui L, Liu BR, Li QQ, Yang Y, et al. Ifn-Γ Surmounts pd-L1/pd1 inhibition to car-T cell therapy by upregulating icam-1 on tumor cells. Signal Transduct Target Ther. (2021) 6:20. doi: 10.1038/s41392-020-00357-7
93. Hong Y, Walling BL, Kim HR, Serratelli WS, Lozada JR, Sailer CJ, et al. St3gal1 and Bii-spectrin pathways control car T cell migration to target tumors. Nat Immunol. (2023) 24:1007–19. doi: 10.1038/s41590-023-01498-x
94. Jung M, Yang Y, McCloskey JE, Zaman M, Vedvyas Y, Zhang X, et al. Chimeric antigen receptor T cell therapy targeting icam-1 in gastric cancer. Mol Ther Oncol. (2020) 18:587–601. doi: 10.1016/j.omto.2020.08.009
95. Ramkumar P, Abarientos AB, Tian R, Seyler M, Leong JT, Chen M, et al. Crispr-based screens uncover determinants of immunotherapy response in multiple myeloma. Blood Adv. (2020) 4:2899–911. doi: 10.1182/bloodadvances.2019001346
96. Burton J, Siller-Farfán JA, Pettmann J, Salzer B, Kutuzov M, van der Merwe PA, et al. Inefficient exploitation of accessory receptors reduces the sensitivity of chimeric antigen receptors. PNAS USA. (2023) 120:e2216352120. doi: 10.1073/pnas.2216352120
97. Melssen MM, Sheybani ND, Leick KM, Slingluff CL Jr. Barriers to immune cell infiltration in tumors. J Immunother Cancer. (2023) 11(4):e006401. doi: 10.1136/jitc-2022-006401
98. Hickman A, Koetsier J, Kurtanich T, Nielsen MC, Winn G, Wang Y, et al. Lfa-1 activation enriches tumor-specific T cells in a cold tumor model and synergizes with ctla-4 blockade. J Clin Investig. (2022) 132(13):e154152. doi: 10.1172/JCI154152
99. Liu Y-T, Sun Z-J. Turning cold tumors into hot tumors by improving T-cell infiltration. Theranostics. (2021) 11:5365. doi: 10.7150/thno.58390
100. Bonaventura P, Shekarian T, Alcazer V, Valladeau-Guilemond J, Valsesia-Wittmann S, Amigorena S, et al. Cold tumors: A therapeutic challenge for immunotherapy. Front Immunol. (2019) 10:168. doi: 10.3389/fimmu.2019.00168
101. Griffioen AW. Anti-angiogenesis: making the tumor vulnerable to the immune system. Cancer Immunol Immunother. (2008) 57:1553–8. doi: 10.1007/s00262-008-0524-3
102. Griffioen AW, Damen CA, Blijham GH, Groenewegen G. Tumor angiogenesis is accompanied by a decreased inflammatory response of tumor-associated endothelium. Blood. (1996) 88:667–73. doi: 10.1182/blood.V88.2.667.bloodjournal882667
103. Haake M, Haack B, Schäfer T, Harter PN, Mattavelli G, Eiring P, et al. Tumor-derived gdf-15 blocks lfa-1 dependent T cell recruitment and suppresses responses to anti-pd-1 treatment. Nat Commun. (2023) 14:4253. doi: 10.1038/s41467-023-39817-3
104. Kolev M, West EE, Kunz N, Chauss D, Moseman EA, Rahman J, et al. Diapedesis-induced integrin signaling via lfa-1 facilitates tissue immunity by inducing intrinsic complement C3 expression in immune cells. Immunity. (2020) 52:513–27.e8. doi: 10.1016/j.immuni.2020.02.006
105. Nandagopal S, Li CG, Xu Y, Sodji QH, Graves EE, Giaccia AJ. C3ar signaling inhibits nk-cell infiltration into the tumor microenvironment in mouse models. Cancer Immunol Res. (2022) 10:245–58. doi: 10.1158/2326-6066.CIR-21-0435
106. Samuelsson M, Potrzebowska K, Lehtonen J, Beech JP, Skorova E, Uronen-Hansson H, et al. Rhob controls the rab11-mediated recycling and surface reappearance of lfa-1 in migrating T lymphocytes. Sci Signal. (2017) 10:eaai8629. doi: 10.1126/scisignal.aai8629
107. Workeneh BT, Uppal NN, Jhaveri KD, Rondon-Berrios H. Hypomagnesemia in the cancer patient. Kidney360. (2021) 2:154. doi: 10.34067/KID.0005622020
108. Wurzer H, Hoffmann C, Al Absi A, Thomas C. Actin cytoskeleton straddling the immunological synapse between cytotoxic lymphocytes and cancer cells. Cells. (2019) 8:463. doi: 10.3390/cells8050463
109. Lacouture C, Chaves B, Guipouy D, Houmadi R, Duplan-Eche V, Allart S, et al. Lfa-1 nanoclusters integrate tcr stimulation strength to tune T-cell cytotoxic activity. Nat Commun. (2024) 15:407. doi: 10.1038/s41467-024-44688-3
110. van Kooyk Y, Weder P, Heije K, Figdor CG. Extracellular ca2+ Modulates leukocyte function-associated antigen-1 cell surface distribution on T lymphocytes and consequently affects cell adhesion. J Cell Biol. (1994) 124:1061–70. doi: 10.1083/jcb.124.6.1061
111. Arruga F, Gyau BB, Iannello A, Vitale N, Vaisitti T, Deaglio S. Immune response dysfunction in chronic lymphocytic leukemia: dissecting molecular mechanisms and microenvironmental conditions. Int J Mol Sci. (2020) 21:1825. doi: 10.3390/ijms21051825
112. Roessner PM, Seiffert M. T-cells in chronic lymphocytic leukemia: guardians or drivers of disease? Leukemia. (2020) 34:2012–24. doi: 10.1038/s41375-020-0873-2
113. Shan J, Jing W, Ping Y, Shen C, Han D, Liu F, et al. Lfa-1 regulated by il-2/stat5 pathway boosts antitumor function of intratumoral cd8(+) T cells for improving anti-pd-1 antibody therapy. Oncoimmunology. (2024) 13:2293511. doi: 10.1080/2162402x.2023.2293511
114. Castiglioni S, Maier JA. Magnesium and cancer: A dangerous liason. Magnes Res. (2011) 24:S92–100. doi: 10.1684/mrh.2011.0285
115. Ginefra P, Carrasco Hope H, Spagna M, Zecchillo A, Vannini N. Ionic regulation of T-cell function and anti-tumour immunity. Int J Mol Sci. (2021) 22(24):13668. doi: 10.3390/ijms222413668
116. Li Y, Wu Y, Hu Y. Metabolites in the tumor microenvironment reprogram functions of immune effector cells through epigenetic modifications. Front Immunol. (2021) 12:641883. doi: 10.3389/fimmu.2021.641883
117. Nasulewicz A, Wietrzyk J, Wolf FI, Dzimira S, Madej J, Maier JA, et al. Magnesium deficiency inhibits primary tumor growth but favors metastasis in mice. Biochim Biophys Acta. (2004) 1739:26–32. doi: 10.1016/j.bbadis.2004.08.003
118. Wang MS, Hu Y, Sanchez EE, Xie X, Roy NH, de Jesus M, et al. Mechanically active integrins target lytic secretion at the immune synapse to facilitate cellular cytotoxicity. Nat Commun. (2022) 13:3222. doi: 10.1038/s41467-022-30809-3
119. Swaminathan V, Mythreye K, O’Brien ET, Berchuck A, Blobe GC, Superfine R. Mechanical stiffness grades metastatic potential in patient tumor cells and in cancer cell linesmechanical stiffness of cells dictates cancer cell invasion. Cancer Res. (2011) 71:5075–80. doi: 10.1158/0008-5472.CAN-11-0247
120. Zhang W, Kai K, Ueno NT. Qin L. A brief review of the biophysical hallmarks of metastatic cancer cells. Cancer Hallm. (2013) 1:59–66. doi: 10.1166/ch.2013.1010
121. Liu Z, Lee SJ, Park S, Konstantopoulos K, Glunde K, Chen Y, et al. Cancer cells display increased migration and deformability in pace with metastatic progression. FASEB J. (2020) 34:9307. doi: 10.1096/fj.202000101RR
122. Pepin KM, McGee KP, Arani A, Lake DS, Glaser KJ, Manduca A, et al. Mr elastography analysis of glioma stiffness and idh1-mutation status. Am J Neuroradiol. (2018) 39:31–6. doi: 10.3174/ajnr.A5415
123. Liu Y, Zhang T, Zhang H, Li J, Zhou N, Fiskesund R, et al. Cell softness prevents cytolytic T-cell killing of tumor-repopulating cells. Cancer Res. (2021) 81:476–88. doi: 10.1158/0008-5472.Can-20-2569
124. Kuhn JR, Poenie M. Dynamic polarization of the microtubule cytoskeleton during ctl-mediated killing. Immunity. (2002) 16:111–21. doi: 10.1016/s1074-7613(02)00262-5
125. Friedman D, Simmonds P, Hale A, Bere L, Hodson NW, White MR, et al. Natural killer cell immune synapse formation and cytotoxicity are controlled by tension of the target interface. J Cell Sci. (2021) 134:jcs258570. doi: 10.1242/jcs.258570
126. Cheng X, Cheng K. Visualizing cancer extravasation: from mechanistic studies to drug development. Cancer Metast Rev. (2021) 40:71–88. doi: 10.1007/s10555-020-09942-2
127. Chen R-H, Xiao Z-W, Yan X-Q, Han P, Liang F-Y, Wang J-Y, et al. Tumor cell-secreted isg15 promotes tumor cell migration and immune suppression by inducing the macrophage M2-like phenotype. Front Immunol. (2020) 11:594775. doi: 10.3389/fimmu.2020.594775
128. Vazquez Rodriguez G, Abrahamsson A, Jensen LDE, Dabrosin C. Estradiol promotes breast cancer cell migration via recruitment and activation of neutrophils. Cancer Immunol Res. (2017) 5:234–47. doi: 10.1158/2326-6066.CIR-16-0150
129. De Andrade Costa A, Chatterjee J, Cobb O, Sanapala S, Scheaffer S, Guo X, et al. Rna sequence analysis reveals itgal/cd11a as a stromal regulator of murine low-grade glioma growth. Neuro-oncology. (2022) 24:14–26. doi: 10.1093/neuonc/noab130
130. Montresor A, Toffali L, Fumagalli L, Constantin G, Rigo A, Ferrarini I, et al. Activation of protein tyrosine phosphatase receptor type Γ Suppresses mechanisms of adhesion and survival in chronic lymphocytic leukemia cells. J Immunol. (2021) 207:671–84. doi: 10.4049/jimmunol.2001462
131. Montresor A, Toffali L, Rigo A, Ferrarini I, Vinante F, Laudanna C. Cxcr4-and bcr-triggered integrin activation in B-cell chronic lymphocytic leukemia cells depends on jak2-activated bruton’s tyrosine kinase. Oncotarget. (2018) 9:35123. doi: 10.18632/oncotarget.26212
132. Montresor A, Toffali L, Mirenda M, Rigo A, Vinante F, Laudanna C. Jak2 tyrosine kinase mediates integrin activation induced by cxcl12 in B-cell chronic lymphocytic leukemia. Oncotarget. (2015) 6:34245. doi: 10.18632/oncotarget.v6i33
133. Ohue Y, Nishikawa H. Regulatory T (Treg) cells in cancer: can treg cells be a new therapeutic target? Cancer Sci. (2019) 110:2080–9. doi: 10.1111/cas.14069
134. Niu T, Li Z, Huang Y, Ye Y, Liu Y, Ye Z, et al. Lfa-1 knockout inhibited the tumor growth and is correlated with treg cells. Cell Commun Signal. (2023) 21:233. doi: 10.1186/s12964-023-01238-6
135. Shen W, Xie J, Zhao S, Du R, Luo X, He H, et al. Icam3 mediates inflammatory signaling to promote cancer cell stemness. Cancer Lett. (2018) 422:29–43. doi: 10.1016/j.canlet.2018.02.034
136. Shen W, Zhang X, Du R, Fan Y, Luo D, Bao Y, et al. Icam3 mediates tumor metastasis via a lfa-1-icam3-erm dependent manner. Biochim Biophys Acta. (2018) 1864:2566–78. doi: 10.1016/j.bbadis.2018.05.002
137. Huang MT, Mason JC, Birdsey GM, Amsellem V, Gerwin N, Haskard DO, et al. Endothelial intercellular adhesion molecule (Icam)-2 regulates angiogenesis. Blood. (2005) 106:1636–43. doi: 10.1182/blood-2004-12-4716
138. Yoon KJ, Phelps DA, Bush RA, Remack JS, Billups CA, Khoury JD. Icam-2 expression mediates a membrane-actin link, confers a nonmetastatic phenotype and reflects favorable tumor stage or histology in neuroblastoma. PloS One. (2008) 3:e3629. doi: 10.1371/journal.pone.0003629
139. Pazina T, James AM, Colby KB, Yang Y, Gale A, Jhatakia A, et al. Enhanced slamf7 homotypic interactions by elotuzumab improves nk cell killing of multiple myeloma. Cancer Immunol Res. (2019) 7:1633–46. doi: 10.1158/2326-6066.CIR-18-0579
140. Richardson K, Keam SP, Zhu JJ, Meyran D, D’Souza C, Macdonald S, et al. The efficacy of combination treatment with elotuzumab and lenalidomide is dependent on crosstalk between natural killer cells, monocytes and myeloma cells. Haematologica. (2023) 108:83–97. doi: 10.3324/haematol.2021.279930
141. Lohr J, Ratliff T, Huppertz A, Ge Y, Dictus C, Ahmadi R, et al. Effector T-cell infiltration positively impacts survival of glioblastoma patients and is impaired by tumor-derived tgf-B. Clin Cancer Res. (2011) 17:4296–308. doi: 10.1158/1078-0432.Ccr-10-2557
Keywords: LFA-1, leukocytes, cancer, TME, immunosurveillance, immune escape
Citation: Upadhyay S, Murugu L and Svensson L (2025) Tumor cells escape immunosurveillance by hampering LFA-1. Front. Immunol. 16:1519841. doi: 10.3389/fimmu.2025.1519841
Received: 30 October 2024; Accepted: 02 January 2025;
Published: 22 January 2025.
Edited by:
Luca Simeoni, University Hospital Magdeburg, GermanyReviewed by:
Alessandro Poggi, San Martino Hospital (IRCCS), ItalyRoxana Khazen, INSERM U1037 Centre de Recherche en Cancérologie de Toulouse, France
Copyright © 2025 Upadhyay, Murugu and Svensson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lena Svensson, bGVuYS5zdmVuc3NvbkB1bXUuc2U=
†These authors have contributed equally to this work and share first authorship