 Alberto L. Horenstein1
Alberto L. Horenstein1 Angelo C. Faini1,2
Angelo C. Faini1,2 Fabio Morandi3
Fabio Morandi3 Erika Ortolan1
Erika Ortolan1 Paola Storti4
Paola Storti4 Nicola Giuliani4,5
Nicola Giuliani4,5 Paul G. Richardson6
Paul G. Richardson6 Fabio Malavasi1,7*
Fabio Malavasi1,7*- 1Lab of Immunogenetics, Department of Medical Sciences, University of Torino, Torino, Italy
- 2Immunogenetics and Transplant Biology, University Hospital “Città della Salute e della Scienza di Torino”, Torino, Italy
- 3UOSD Laboratorio di Terapie Cellulari, IRCCS Istituto Giannina Gaslini, Genova, Italy
- 4Department of Medicine and Surgery, University of Parma, Parma, Italy
- 5Department of Medicine and Surgery, University of Parma & Multiple Myeloma Program, AOU, Parma, Italy
- 6Jerome Lipper Multiple Myeloma Center, Dana-Farber Cancer Institute, Boston, MA, United States
- 7Fondazione Ricerca Molinette Ets, Torino, Italy
Monoclonal antibody therapy using CD38 as a target remains central to managing human multiple myeloma (MM). CD38 was selected early on as a target for mAb-mediated therapy for MM, driven by findings from an early Cluster of Differentiation (CD) Workshop. The first CD38-targeting antibody to be approved yielded strong trial results, significantly improving survival rates and earning widespread patient acceptance. However, resistance to the therapy later emerged, complicating treatment management. Despite CD38’s still central role in MM therapy, too little attention has been paid to its broader roles–not only as a myeloma marker but also as an enzyme and adhesion molecule in physiology. This review, a collaborative effort between basic scientists and clinical experts, explores some of the lesser-known mechanisms of antibody action and interactions with CD38 at key stages of treatment. The review also highlights the relevance of the MM environment, focusing on the importance of the bone marrow (BM) niche. The goal is to identify new agents whose unique properties may enhance tumor eradication. By gaining a deeper understanding of interactions between therapeutic antibodies, myeloma cells, and the tumor microenvironment (TME), it is hoped that previously unrecognized vulnerabilities within the disease may be revealed, paving the way to more effective treatment strategies.
1 Introduction
In vivo therapy based on monoclonal antibodies (mAbs) represents the natural extension of their widespread use in vitro. The unique specificity of mAbs is complemented by their adaptability to industrial production, which forms the basis for an optimal biopharmaceutical.
The selection of CD38 as a target molecule was a key step in the design of a mAb-mediated therapy for human multiple myeloma (MM). This choice was primarily influenced by the main characteristics of CD38, which were initially identified at an early Cluster of Differentiation (CD) Workshop as a T-cell activation antigen. Subsequent research confirmed its high expression in MM cells. Additionally, the molecule is particularly well conserved within the Caucasoid population, exhibiting limited genetic polymorphisms.
CD38 is expressed by the majority of MM patients and is generally maintained during the in vivo development of the disease (1). Mainly in virtue of these characteristics, CD38 has been adopted by pharmaceutical companies as an ideal target for antibody-mediated in vivo therapy of MM, a diagnosis that is becoming increasingly common worldwide. Since the inception of the project, attention has predominantly focused on CD38 as a marker, while its roles as an enzyme and an adhesion molecule have received more limited consideration (vide infra). The first therapeutic mAb, Daratumumab–a fully human immunoglobulin (IgG)–delivered strong trial results and was rapidly approved by the American FDA for in vivo use, with Janssen as the drug provider. The results obtained confirmed the effectiveness of CD38 as a target, showing a marked improvement in survival rates (2). The enthusiasm derived from the success of clinical applications was matched by its warm acceptance by patients, as side effects were relatively mild. However, early cases of resistance to the antibody therapy began to emerge, posing problems for treatment management.
This review aims to describe some of the lesser-known mechanisms of action of the antibodies and their interactions with CD38 at key stages of treatment. Furthermore, we will re-examine the enzymatic functions of CD38 in the context of the immune response. Additionally, we will address aspects initially not considered in detail, such as the role of different families of IgG Fc receptors (FcRs), which may influence interactions among mAbs, the environment, and MM cells.
Further structural analysis of the therapeutic antibodies will also be conducted to identify potential areas for improving their efficacy. Currently, the number of approved therapeutic reagents is significantly growing in the last years with Daratumumab and Isatuximab (from Sanofi) leading the way. Felzartamab (from I-Mab), already approved for treating membranous nephropathy, is undergoing trials for additional diseases. For more details, see reference (3).
The final section will focus on the MM environment, with particular attention to the bone marrow (BM) niche, aiming to identify new players and leverage their unique properties to enhance tumor eradication.
The medical community widely expects that deeper insights into the interplay between therapeutic antibodies, myeloma cells, and the tumor microenvironment (TME) will uncover previously unrecognized vulnerabilities of the disease, opening new avenues for treatment.
2 Available evidence
2.1 The CD38 molecule
CD38 is almost ubiquitously expressed, found at varying levels across most tissues, and present both on the cell surface and within cytoplasmic compartments. The characterization of CD38 has been an extensive process, revealing its high expression on the plasma membranes of both normal cells and malignant myeloma cells. Although the tissue distribution of CD38 has been widely studied and described, its functions remain only partially understood. Initially discovered in connection with the T cell receptor (TCR), early research was shaped by this association (4). CD38 was first classified as a signaling molecule, with its cell activation capability suggesting the existence of a natural ligand, later identified as CD31 (and also hyaluronic acid) (1).
Further analysis of the CD38 sequence revealed a remarkable homology in amino acid structure with a cytoplasmic adenosine diphosphate (ADP)-ribosyl cyclase enzyme previously purified from Aplysia (4), suggesting a potential similar function in mammals. This hypothesis was confirmed when human CD38 was found to act as an enzyme with NAD+-glycohydrolase activity, consuming nicotinamide adenine dinucleotide (NAD+) and producing ADP ribose (ADPR), cyclic ADPR (cADPR) and nicotinamide (NAM) under neutral conditions, and nicotinic acid adenine dinucleotide phosphate (NAADP+) at low pH when consuming phosphorylated NAD+ (NADP+) (4, 5) (vide infra).
2.1.1 Cell surface expression of human CD38
Extracellular CD38 is a type 2 transmembrane molecule, approximately 46 kDa in size, featuring a short cytoplasmic tail that grants high mobility within the liquid bilayer, facilitating the formation of soluble forms of CD38 (6). This flexible anchorage is complemented by selective localization in specific domains of the cell membrane, which varies according to lineage (Figure 1). The properties of membrane-bound CD38 and its interactions with different surface receptors are summarized in reference (1).
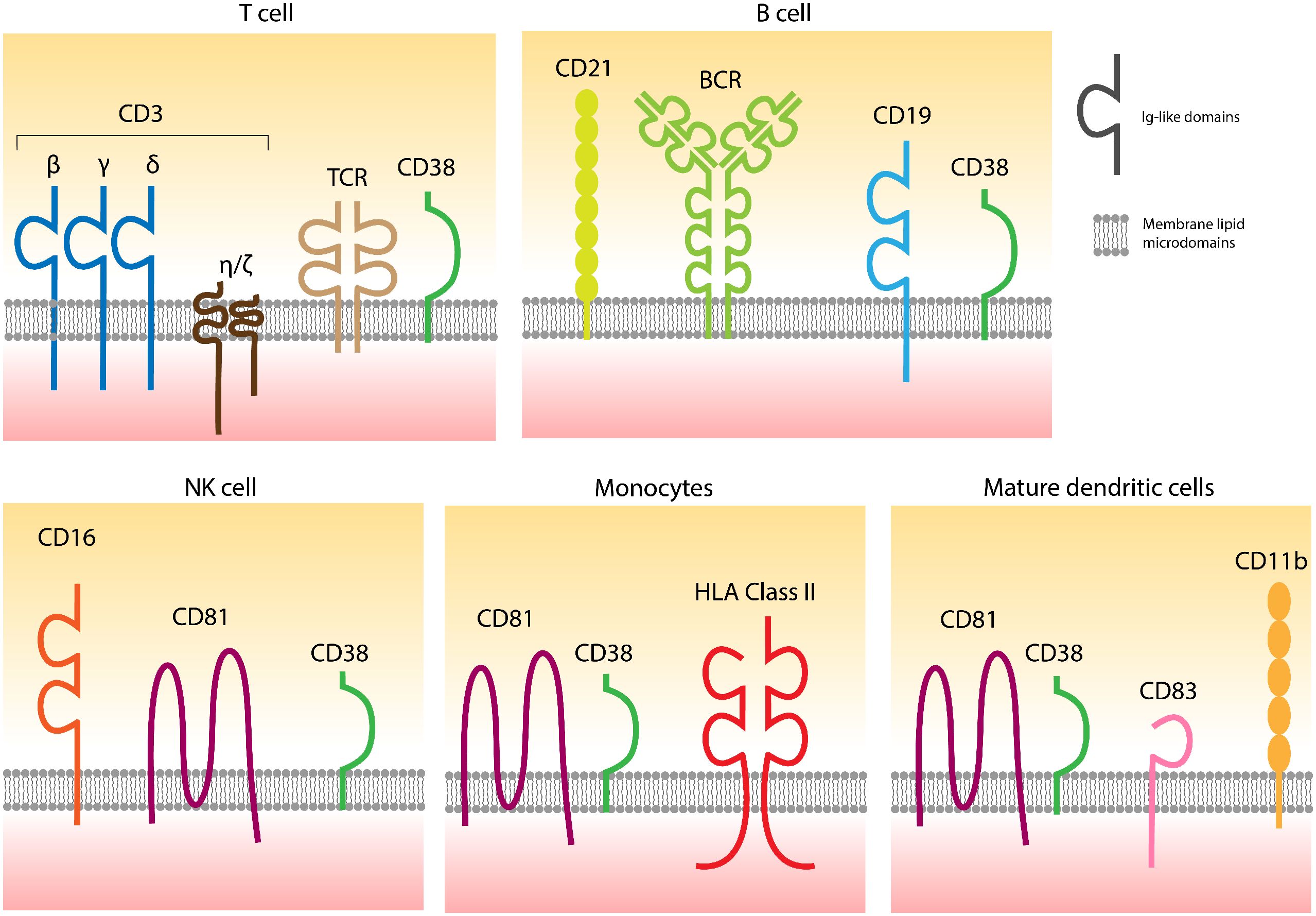
Figure 1. CD38 is localized in specific domains of the cell surfaces containing different receptors, also varying according to the lineage. The links between CD38 and distinct receptors are confirmed, while functional connections are partially hypothesized. Adapted from (1).
Cell surface expression of CD38 can fluctuate in response to physiological events, such as cellular activation or pharmacological interventions. The interaction between therapeutic antibodies and CD38 on neoplastic cells is inherently dependent on the availability of molecular targets. Consequently, early strategies to enhance the efficacy of MM therapies focused on increasing the number of CD38 targets available for antibody binding.
Various approaches and modalities have been employed to regulate CD38 surface expression on target cells.
2.1.1.1 All-trans retinoic acid (ATRA)
CD38 expression can be modulated both in vitro and in vivo through the nuclear retinoic acid receptor α (7). ATRA, a vitamin A derivative approved for in vivo use, has proven effective in enhancing CD38 levels in several ex vivo myeloma cell preparations, leading to improved efficacy of therapeutic antibodies (8). However, its in vivo efficacy in combination with Daratumumab was called into question in a clinical trial involving relapsed/refractory (RR) MM patients. In this trial, CD38 re-expression was too transient to significantly impact clinical outcomes, at least in the patient cohort studied (9).
2.1.1.2 Histone deacetylases (HDAC)
Another compound investigated for its capacity to regulate CD38 expression is the HDAC inhibitor Panobinostat. Unlike ATRA, Panobinostat upregulates CD38 expression on myeloma and on lymphoma cells, while sparing normal cells. When combined with Daratumumab, Panobinostat boosts antibody-dependent cellular cytotoxicity (ADCC) in both cell lines and primary MM cells. In therapy, Panobinostat induces CD38 upregulation, thereby improving the anti-myeloma effects of Daratumumab (10). Similarly, Ricolinostat, another HDAC inhibitor, increases CD38 expression on MM cells without affecting normal cells. However, Ricolinostat does not affect myeloid-derived suppressor cells (MDSCs), which are depleted by this inhibitor (11).
2.1.1.3 JAK-STAT signaling pathway
The JAK-STAT pathway can influence CD38 regulation through its signaling activities. The CD38 gene features an atypical promoter responsive to retinoids, vitamin D, and various cytokines in circulating cells. In MM, IL-6 activates the JAK-STAT pathway. JAK-STAT3 signaling down-regulates CD38 expression, whereas JAK-STAT1 signaling promotes its upregulation. Although these effects are less pronounced than those seen with ATRA, STAT3 inhibitors or STAT1 activators are being explored as promising candidates for combination therapies with Daratumumab (12).
2.1.1.4 Degradation of CD38 mRNA
Research involving microRNA (miR) has demonstrated that noncoding RNA can influence CD38 expression, with miR-26a and miR-140-3p being key candidates for investigation. In MM cells, miR-26a is downregulated. Using mimics or antisense nucleotides to inhibit miR-26a can elevate CD38 expression in MM cells with low surface levels of CD38. Conversely, manipulating these miRs can enhance CD38 expression via cell effectors (13).
2.1.1.5 Immunomodulatory imide Drugs (IMiDs)
IMiDs are key components of standard myeloma therapy, especially in combination with mAbs. Their effectiveness is largely attributed to the significant upregulation of surface CD38, achieved by degrading Ikaros and Aiolos. This leads to increased CD38 mRNA transcription, which in turn favors surface CD38 expression and, consequently, improved binding and lytic potential of the antibody (14).
2.1.1.6 Other potential modulators of CD38 surface expression
Surface levels of CD38 rapidly decline following in vivo treatment with mAbs, displaying prolonged recovery times that contradict the expected rapid re-synthesis of the molecule or mobilization from internal stores (1). This behavior may also vary according to the normal CD38+ effectors.
These effects may be due to the unique localization of CD38 within specific membrane domains of effector cells. Indeed, CD38 has been implicated in the formation of immune synapses in T lymphocytes. Early experiments, later confirmed through various technical approaches, identified two distinct pools of CD38: one on the cell membrane and another within recycling endosomes (15). This distribution pattern is also observed in monocytes (16), mature dendritic cells (15, 17), and NK cells, where CD38 co-localizes with CD16, a key molecule for their activation (18). Comparable effects are also noted with CD28H (19).
Overall, these observations indicate that CD38 influences multiple functional events in NK cells, T lymphocytes, monocytes, and dendritic cells by interacting with various immune receptor systems.
2.1.1.7 Antibody-induced CD38 modulation
CD38 expression during antibody therapy can be studied by examining how the antibody binds the target. The very short cytoplasmic tail of CD38 allows for flexible movement within the cell membrane. At 37°C, therapeutic antibody binding increases the molecule’s ability to float within the membrane and - as known from traditional immunology - the antigen/antibody complex tends to accumulate at one pole of the cell. This polar aggregation is also known as “capping” (20). The consequence is that antibody ligation is followed by the release of microvesicles (MVs) containing membrane rafts with CD38 and the bound antibody, thereby reducing surface levels of CD38 (21). This mechanism may contribute to antibody refractoriness. Polar aggregation is most pronounced when therapeutic antibodies are presented to the target as being bound by a FcR present on normal effector cells. In vitro models using polyclonal anti-human IgG mimic the so-called “armed mAb” scenario observed in vivo. First, polar aggregation induced by an armed mAb results in a significant re-distribution of CD38 on the membrane and a marked reduction in its expression. Second, molecules accumulating at the pole tend to be extruded through protrusions in the myeloma membrane and eventually released into biological fluids as MVs. These MVs carry not only CD38, but also CD39, CD73 and CD203a ectoenzymes, inhibitory complement receptors CD55 and CD59, the checkpoint inhibitor PD-L1, and the myeloma differentiation marker CD138 (21). The fate of these MVs is determined by specific constraints such as environmental conditions, molecular composition, or cellular interactions. Deriving from cell membranes, they are able to cross vessels and tissues, interacting with monocytes and MDSC (both rich in high affinity FcRs), T lymphocytes, and NK cells, the latter expressing CD16 (low affinity FcR), where they are internalized and release their cargo. These interactions may partially explain the development of antibody refractoriness and resistance.
In addition to reducing surface CD38, antibody-induced modulation may involve two other mechanisms: i) signal transduction that activates intracellular signaling pathways, and ii) cargo transfer. MVs can carry a variety of molecular components, among which coding genes (DNA), non-coding RNAs (ncRNAs), RNAs [including messenger RNA (mRNAs) and microRNAs (miRNAs)], proteins (including enzymes, structural proteins and signaling molecules) nucleotides and other metabolites.
NK cells, in particular, show a marked and sustained decrease in surface CD38 following antibody treatment. Studies with Isatuximab have demonstrated that the cell population appears to be initially activated but is then modulated by the action of a gene set involved in the control of the cell cycle. This was confirmed through molecular analysis of the content of NK cells (22). As a result, NK cells tend to have a modified phenotype and lose surface CD38 while retaining partial cytotoxic activity (23).
2.2 The IgG Fc receptors
Another important aspect to explore in detail is the structure of the therapeutic antibody itself. As an IgG, the antibody has two main functional domains. In addition to the domain responsible for binding the specific epitope [i.e., monovalent Fab or bivalent F(ab’)2], the second domain (i.e., Fc) interacts with cell surface receptors (i.e., IgG FcRs) with varying affinities and specificities. The interactions between the Fc region of the therapeutic IgG and FcRs on surrounding effector cells are particularly relevant in the MM environment. Most effector cells express different FcRs, while only the MM cells are FcR-negative.
The signals triggered by the antibody in normal effectors, which are predominantly CD38+ and FcR+, are one of the most important findings obtained from in vivo MM therapy. FcR-mediated functions control a broad array of antibody-driven therapeutic mechanisms, including complement-dependent cytotoxicity (CDC), antibody-dependent cell cytotoxicity (ADCC), antibody-dependent cell phagocytosis (ADCP), and the induction of cell death by apoptosis. These cytotoxic activities have been extensively analyzed in references (2, 24, 25)
Paiva et al. analyzed the effects of Isatuximab, the second anti-CD38 reagent approved by the FDA, on NK cells, which express CD16, a low affinity FcR (22). By using Fc blockers, they demonstrated that NK cell activation is significantly influenced by transmembrane signaling mediated through antibody ligation. The ability of CD38 to transduce signals aligns with early observations of the CD38 molecule (1) and supports the hypothesis that a number of the therapeutic effects, at least in the case of Daratumumab, is reliant on extracellular signaling channeled by CD38. A recent version of Isatuximab with an improved Fc domain, for increased affinity for CD16 and CD32, has recently been tested (26).
Overall, these results suggest that some of the therapeutic actions of the antibody are mediated by direct signaling through the CD38 molecule, without the effects driven by FcRs, at least in the NK cells tested in vitro. The potential influence of the FCGR3AF158V polymorphism, which affects FcRs binding to the Fc fragment of antibodies and is more prevalent among patients with MM and MGUS, remains to be investigated (27).
2.2.1 Neonatal IgG Fc receptor
The Neonatal IgG Fc Receptor (FcRn) was characterized years ago from maternal milk. The roles of FcRn are distinct from those of receptors binding the Fc domains of different classes of immunoglobulins. Its primary functions identified so far are related to the metabolism of circulating IgG, including therapeutic monoclonal IgG, and albumin. These molecules are internalized into cytoplasmic compartments and then into endosomes. Similar internal processing occurs for antibodies, which are eventually extruded and released into biological fluids at neutral pH. This step is crucial for regulating the stability of therapeutic antibodies and directly determining their in vivo half-life (28).
FcRn also differs from other FcRs in terms of structure, regulation, and tissue distribution. FcRn are heterodimers composed of heavy and light molecular weight chains (29). The light chain consists of β2-microglobulin (β2m), a monomorphic component also shared with HLA Class I molecules. The heavy chain is encoded by the Fcϒ receptor and transporter (FCGRT) gene, located on chromosome 19q13.3. Although reminiscent of HLA-controlled products, suggesting potential yet unidentified functions, FcRn is not capable of presenting peptides. Its known ligands include IgG, albumin, and pan-echovirus. Upregulation of FcRn is primarily associated with stimulation via tumor necrosis factor (TNF), while other pathways have been identified in non-human models. Down-regulation is primarily achieved through JAK/STAT1 signaling mediated by interferon γ.
Among immune cells, FcRn is expressed by monocytes, macrophages, dendritic cells, and neutrophils, and is present at low levels in B lymphocytes but not detectable on T lymphocytes and NK cells. FcRn maintains the tissue distribution of IgG by virtue of its ability to mediate transcytosis, transporting IgG across polarized endothelial and some epithelial cells (30).
FcRn serves multiple active and passive roles in immunity. Its passive functions include recycling and transcytosis, which protect monomeric IgG from degradation and facilitate its transport across external cell membranes. Its active functions are more varied, linked to the involvement of IgG in triggering various immune responses. These physiological characteristics can be leveraged to enhance the persistence of IgG and albumin in the circulation.
To date, a comprehensive analysis of FcRn’s influence has not been conducted in the context of the anti-CD38 antibodies approved for in vivo use, whose Fc regions are not modified. Only Nguyen et al. have analyzed the in vivo dynamics of Isatuximab (31, 32).
2.3 CD38-controlled cellular activities and metabolic adaptation during MM
2.3.1 CD38 as an enzyme
CD38 has been studied in MM therapy primarily due to its function as an ectoenzyme. Initially, the mechanisms of action of this enzyme were considered paradoxical because CD38 is predominantly localized on the cell surface, yet its final products are utilized within cytoplasmic compartments. Only recently has CD38 been described within the cytoplasm and internal organelles (5).
The amino acid sequence of the CD38 enzyme is highly conserved throughout evolution, indicating that its enzymatic function is likely its original role. Functions related to immunity and cell biology were probably acquired over its long evolutionary history (at least 750 million years), eventually leading to its duplication and the emergence of the CD157 gene. CD157 encodes a molecule that differs significantly in structure and tissue distribution, retaining only some of the enzymatic functions attributed to CD38 (33).
The primary substrate of CD38 is NAD+, a key molecule in energy production and signaling. NAD+ plays a crucial role in a broad network of metabolic enzymes, such as nicotinamide phosphoribosyltransferase (NAMPT), nicotinate phosphorybosiltransferase (NAPRT), Sirtuins, and Sterile α-Toll/interleukin receptor (TIR), as well as the recently identified TIR motif- containing 1 (SARM1) (3).
CD38 is overexpressed by malignant plasma cells, significantly affecting i) the enzymatic reactions of CD38 within the BM niche, ii) the types of CD38-associated ectonucleotidases (e.g., CD203a, CD73, TRAP) involved in adenosine (ADO) production, and iii) the environmental metabolic conditions (e.g., pH, oxygen levels) within the BM niche (33). While the role of CD38’s enzymatic activity in MM therapy resistance is hypothesized, it has not yet been confirmed (34).
2.3.2 CD38 and extracellular NAD+ metabolism
Nucleotides have a second life as intercellular communicators and signal transducers in the BM niche (35).
NAD+ metabolism is governed by cell surface ectoenzymes, which work together to disassemble extracellular and intracellular nucleotides extruded from cells via active channeling mechanisms or by microvesicles (MVs). CD38 is the principal extracellular NAD+-consuming enzyme. In fact, CD38 knockout mice exhibit significant increases in both extra- and intracellular NAD+ levels (36, 37).
Because of its ability to use NAD+ as a substrate, CD38 has been hypothesized to act as a metabolic sensor, limiting the duration of NAD+ half-life and signaling in the extracellular compartment. Besides serving as a substrate for CD38, NAD+ may also function as a cytokine, eliciting rapid functional responses mediated by specific purinergic type 2 (P2) receptors (34, 38).
The products of NAD+ disassembly mediated by extracellular CD38, namely ADPR and NAM, may undergo further metabolic transformations [(e.g., to adenosine monophosphate (AMP) or adenosine (ADO)], particularly when co-expressed with other ectonucleotidases in certain pathological environments, such as the bone marrow (BM) niche (13). This niche is a seemingly closed system where MM cells are in direct contact with other cellular components, all expressing a range of surface enzymes. The plasma assures contact among the different cells, allowing the storage and exchange of substrates and final reaction products. In addition to CD38, ectoenzymes present in the niche (whether surface or soluble molecules) include CD39 (ectonucleoside triphosphate diphosphohydrolase-1), CD73 (ecto-5′-nucleotidase), and CD203a (ectonucleotide pyrophosphatase/phosphodiesterase-1). CD39 and CD73 run the canonical pathway of ADO production starting from ATP. CD203a, also known as Plasma Cell-1 (PC-1), was the first surface marker identified on plasma cells and is an enzyme that also uses ATP as a substrate (39). Recent reports suggest that CD38 can use phosphorylated NAD+ (NADP+) as a substrate and, in the presence of CD203a and CD73 under low pH conditions, produce ADO as a final product (40). Lowering environmental pH is one of the strategies employed by MM cells to alter their metabolism and promote immune evasion. In line with this, the canonical pathway of ADO production (CD39/CD73) operates at a very low rate in these conditions (40).
A key question is whether the products of the CD38-dependent chain of ectoenzymes play a role in physiology and especially in pathology. The MM niche provides an ideal model to explore this. ADO levels in BM plasma samples from MM patients were compared with those from patients with precursor forms of monoclonal gammopathy of undetermined significance (MGUS) and smoldering MM (SMM). The catabolism of NAD+, ADPR, and AMP to ADO was higher in samples from patients with severe MM than in those with MGUS and SMM (41).
These experiments lead to several key conclusions. First, ADO is produced in the BM niches where MM cells grow; second, ADO predominantly derives from the non-canonical pathway mediated by CD38/CD203a/CD73; third, disease severity is directly correlated with ADO levels, which are higher in patients with more severe prognoses. These observations highlight the presence of metabolic modifications within the MM niche that may facilitate immune evasion strategies (41).
The high level of ADO detected in the BM niche may contribute to the overall anergic state observed in MM patients. Relevant to this review is the link between NAD+ levels and T cell-mediated responses against tumors, an area that has gained increasing attention with the recent development of pharmacological inhibitors targeting CD38’s enzymatic activities (42). Several small-molecule inhibitors, such as 78C, suppress the enzymatic activity of CD38, thereby reducing NAD+ consumption (42). However, these compounds have not yet been used clinically due to potential off-target effects. In contrast, inhibition of CD38 exoenzymatic activity using the Ab68 mAb reduced the production of ADPR (43). These findings regarding ADO production warrant further research.
Recent research by Cea et al. showed that upregulation of surface CD38 leads to intracellular NAD+ depletion, which induces mitochondrial alterations and increases oxidative stress (44). Conversely, the availability of different NAD+ precursors appears to improve NAD+ levels, acting as adjuvants to boost T lymphocyte responses (44).
CD38 primarily functions as a NAD+-glycohydrolase (42). Its main product, ADPR, can either be metabolized by the CD203a/CD73 exoenzymatic tandem into ADO (40), which binds to purinergic type 1 (P1) receptors (particularly A2A and A2B), or facilitate Ca2+ influx through TRPM2 channels (45). These processes modulate lymphocyte proliferation and T cell activation (42, 46).
2.3.3 CD38 as an immune checkpoint in therapy
Based on this understanding, it is speculated that CD38 serves as an immune checkpoint regulating NAD+ and ADO homeostasis. Consequently, metabolic reprogramming of NAD+ regulation via CD38 inhibition has garnered attention as a potential strategy for immunotherapy. This hypothesis was validated in a preclinical melanoma model, where blocking CD38 expression in T lymphocytes boosted NAD+ levels, improving the effectiveness of adoptively transferred T cells (47). Targeting the ADO pathway may also increase the antitumor effects of drugs through various mechanisms, such as by boosting effector T and NK cell functions and inhibiting the immunosuppressive effects of myeloid-derived suppressor cells (MDSC) (48). Additionally, due to its participation in the CD38/CD203a/CD73 adenosinergic pathway, CD38 has been identified as an effective predictor of anti-PD-1 antibody-based checkpoint immunotherapy responses (49).
Beside reported pharmacological inhibitors (42), several new strategies aim to inhibit CD38 and increase NAD+ pool concentrations. These approaches include antibody-drug conjugates (ADCs), antibody recruiting molecules (ARMs), engineered toxin bodies (ETBs), bispecific T-cell engagers (BiTEs), and XmAb Fc domain technology. Preclinical studies and clinical trials evaluating CD38 inhibition are ongoing in patients with either solid tumors or hematological malignancies, including MM (3).
2.4 CD38 enzyme activity and therapeutic antibodies: potential influence on refractoriness
A crucial question is whether the binding of therapeutic CD38 antibodies interferes with the enzyme’s functional activity. The differing actions of Daratumumab and Isatuximab in vivo are summarized in recent reviews (50, 51), and their effects further analyzed in Horenstein A.L. et al. (2024, submitted).
The primary distinction between these antibodies lies in the epitopes of CD38 they target. Isatuximab interacts with a discontinuous epitope partially overlapping the catalytic site, whereas Daratumumab binds to two continuous amino acid sequences situated outside the enzymatic active site of CD38 (52, 53). These differences may reflect the methods by which the two antibodies were selected. Isatuximab was originally developed for its ability to operate via CDC, while Daratumumab was selected to enhance ADCC activity. Additionally, Isatuximab was chosen for its capacity to inhibit the ADP-ribosyl cyclase activity of CD38 (i.e., in vitro formation of cADPR from NAD+) through an allosteric antagonism (49–51). Further comparative analysis has shown that Daratumumab reduces in vitro cADPR production catalyzed by CD38’s ADP-ribosyl cyclase activity, while Isatuximab exhibits a stronger inhibitory effect, reducing cyclase activity by approximately 70% (54, 55).
These findings indicate a partial allosteric inhibition of CD38’s enzymatic activity, disrupting the catalytic site of extracellular CD38 responsible for in vitro formation of cADPR. However, the validity of these experimental conditions is limited by two factors, namely: i) CD38 produces multiple enzymatic products (56), and ii) antibody binding does not fully represent the enzymatic machinery. Notably, the inhibition of CD38’s extracellular cyclase activity accounts for only 2% of the molecule’s total enzymatic activity when targeted by therapeutic antibodies (4).
An alternative view suggests that the in vitro conditions used in these studies might not adequately represent in vivo dynamics. Nevertheless, the overarching conclusion is that while Daratumumab moderately affects CD38’s in vitro cyclase activity, Isatuximab exerts a stronger effect. Interestingly, both antibodies mildly activate CD38’s hydrolase activity (i.e., the formation of ADPR from cADPR as a substrate) (57, 58). Importantly, it remains unclear whether the in vitro reduction of cADPR levels produced by extracellular CD38 in the presence of therapeutic mAbs leads to reduced intracellular Ca2+ mobilization and decreased signaling potential. On the other hand, potentially increased ADPR levels could contribute to immune suppression through ADO. Further investigations are essential to understand these complex interactions, especially regarding in vivo NAD+ homeostasis and tumor survival in closed systems. Other potential strategies to address therapeutic resistance are summarized in (59).
2.4.1 Other potential methods for overcoming anti CD38 mAbs resistance: the role of CD47
CD47, an integrin-associated receptor, is widely expressed on the surface of many cell types, including red blood cells and cancer cells (60). Within the immunoglobulin family, it functions as a “don’t eat me” signal. Its ligands include thrombospondin-1 (TSP-1), signal-regulatory protein α (SIRPα), and integrins (61). When CD47 binds to SIRPα on dendritic cells and macrophages, it activates an immunoreceptor tyrosine-based inhibitory motif that suppresses phagocytosis (61). Blocking this signaling axis enhances the elimination of tumor cells by macrophages and neutrophils (62). CD47 is highly expressed in solid tumors and hematologic malignancies, and this overexpression correlates with poor prognosis in various cancers (60, 63). In the context of MM cancer cells upregulate CD47 to evade immune surveillance, exploiting this pathway to enhance their survival.
CD47 expression increases as the disease progresses from MGUS to MM (64). Numerous studies have shown promising preclinical results for anti-CD47 therapies in treating hematologic malignancies. These therapies are effective either as standalone treatments or in combination with clinically approved drugs, including those for MM (63, 65–67). Anti-CD47 therapy alone has shown efficacy against MM (64, 68), and its therapeutic impact is improved when combined with CD38-targeting agents. Storti et al. reported that treatment with daratumumab increases MM cell death, especially in the presence of a subset of CD14+/CD16+ monocytes, and that combining daratumumab with anti-CD47 enhances the elimination of MM cells that are resistant to daratumumab alone (66).
Bispecific antibodies (BsAbs) are also emerging as promising tools in cancer therapy. These antibodies exhibit higher affinity and binding activity for CD38 than for CD47, reducing potential on-target and off-tumor effects. A study by Li et al. explored various CD38/CD47 BsAbs, each with unique in vitro properties: IMM5605–26B4 displayed the most potent inhibitory effect on CD38 enzymatic activity, while IMM5605–12C10 was effective in directly killing MM cells and completely eradicating established tumors in mouse models (69). ISB 1442, a human bispecific antibody was recently reported. The antibody has one arm equipped with biparatopic sequences reacting with distinct epitopes of the CD38 molecule. The second arm is designed to react at high affinity with CD47. The basic strategy is to obtain a reagent focusing its activity prevalently around the cells expressing CD38, which enables avidity-induced blocking of CD47 on the same cells, in this way avoiding unwanted off-tumor effects. The reagent was also equipped with a modified domain with enhanced FcR functions. ISB 1442 showed enhanced tumor killing in vitro compared to daratumumab on myeloma cells with varying CD38 expression levels. IBS 1442 is currently in a Phase I clinical trial for relapsed refractory multiple myeloma (NCT05427812) (70).
2.5 Next generation anti-CD38 antibodies
Advances in antibody therapy may also derive from the development of new anti-CD38 reagents that retain specificity but feature significant modifications in their structure and functions. Some modifications concern the antibody’s ability to react with the family of FcRs, to avoid mechanical trapping in tissues or the implementation of FcR-mediated signals interfering with the therapeutic process.
Of the numerous anti-CD38 reagents currently undergoing evaluation, summarized below are representative examples of antibodies now being designed for therapy.
1. Pre-clinical evaluation of the monoclonal antibody CM313. This conventional antibody was developed by selecting a precise CD38 sequence and identifying unique complementary-determining regions, which provided much higher sensitivity and improved performance compared to traditional reagents. The original murine antibody was humanized, and CM313 has been shown to inhibit some of the enzymatic activities of CD38. Toxicology studies in Cynomolgus monkeys and murine models indicate that CM313 is well tolerated (71).
2. The Danish company GenMab introduced specific mutations in the Fc domain of a human anti-CD38 antibody, leading to (or improving) hexamer formation upon binding to cell surface CD38. The strategy adopted is reported in a recent paper (72). This approach increased CDC function in MM cells (even in cells expressing low levels of surface CD38) and was also effective against other hematological malignancies. The hexabody did not compete in vitro with Daratumumab or Isatuximab, but strongly inhibited the cyclase activity of the target. Furthermore, the hexabody induced FcR-mediated signals in NK cells and macrophages, while other effectors involved in the immune response were apparently unaffected. Clinical trials evaluating the safety and efficacy of the reagent in vivo are ongoing (NCT04824794) (73).
3. Bispecific antibodies feature two distinct Fabs which can lead to the generation of effects similar to those evoked by CAR-T cells (which are beyond the scope of this review). Of the two arms, one is designed to provide tumor specificity, while the other has the role of triggering the signaling potential. This last step elicits an anti-tumor response by T lymphocytes or by NK cells. The use of bispecific reagents has found wide clinical applications because of its simplicity and–equally important–lower costs (74).
4. A further strategy was to increase the function of the T lymphocytes, by activating two distinct activation pathways. Sanofi (Europe and USA) designed SAR442257, a trispecific antibody where CD38 confers target specificity and CD3 and CD28 provide dual activation signals to optimize T lymphocyte effector functions and to ensure sustained proliferation. An original characteristic of this reagent is the choice of CD28-mediated signals to help prevent cell death, which is often observed after T-cell activation. The trispecific reagent was evaluated for its ability to induce activation and proliferation of T lymphocytes. Cytokine release syndrome (CRS) was avoided by using an IgG4 isotype, which does not activate complement fixation (67).
CD28 is not only expressed by T lymphocyte but also by myeloma cells (75). Preclinical tests have shown that 86% of RRMM patients express CD28, compared to newly diagnosed MM patients. The conclusion is that the trispecific SAR442257 operates via dual targeting of CD38 and CD28 on myeloma cells and of CD3 and CD28 on T lymphocytes, showing superior MM killing compared to bispecific antibodies. The study also highlighted the role of TNF-β in favoring resistance to the antibody. The TGF-β inhibitor Vactoserib enhances sensitivity of the target cells to the antibody (76).
The same antibody was analyzed ex vivo using Myeloma Drug Sensitivity Testing to identify suitable MM patients for this therapy. To this end, tests were conducted by challenging the patients’ endogenous T lymphocytes with primary MM cells. This novel approach enabled the identification of patients who responded to SAR442257. The association among T-cell phenotype, performance, disease state and sensitivity to this antibody is currently under investigation (77). The trispecific antibody was also recently tested on acute myeloid leukemia cell lines and patients (78).
5. An alternative strategy was to flank CD3 engager with co-targeting CD38 and BCMA. The ISB 2001 antibody is a recombinant trispecific complex: one arm contains a sequence reacting with CD38, while the second arm contains specificities for CD3 and BCMA. The results obtained by an international group are apparently superior to other trispecific antibodies, as shown in vivo in mouse models. The group has also devised a quantitative systems pharmacology model, which exploits clinical patient data to obtain a personalized posology (79).
6. Another strategy to improve overall binding efficiency involves biparatopic antibodies. These recombinant reagents use two heavy chains that react with distinct epitopes of the same molecule. The California-based company Teneobio targeted CD38 with the aim of developing an antibody capable of fully inhibiting the enzymatic activities of the molecule–not yet achieved with the anti-CD38 reagents currently used in in vivo therapy. The platform used for constructing these antibodies included two Fabs that react with two distinct sequences of the CD38 domain hosting the catalytic site. Additionally, the antibody was improved with a silenced IgG4 Fc, which does not deplete or interfere with the normal effectors (80). The TNB-T38 antibody is now in trials. The reagent has been instrumental in demonstrating a relationship between the products generated by the CD38 enzyme and multi-organ fibrosis (43).
7. Another original approach was adopted by Candelaria et al., who switched the class of Ig, using IgE instead of the conventional IgG. The potentially beneficial features of IgE include its affinity for the FcϵR, expressed by the immune effectors responsible for ADCC and ADCP, and the low levels of normal IgE in the blood, which avoids competition with the therapeutic IgE. In vitro results are promising, showing induction of degranulation in FcϵR-expressing cells. Increased survival was observed in mice when disseminated tumor cells were exposed to the IgE antibody and PBMC as a source of monocyte effectors (81).
2.6 Areas to be investigated to expand research in the field of CD38
2.6.1 Fate of unbound therapeutic antibody
One proposal is the selection of therapeutic antibodies that show negligible reactivity with red blood cells (RBC) and platelets, which are reported to express CD38 at a cell surface density too low to trigger lysis. Despite their reduced ability to capture the therapeutic antibodies, the high number of these cells suggests that they could act as carriers of anti-CD38 antibodies in biological fluids. Thus, RBC and likely platelets in biological fluids may sequester a portion of the antibody pool away from their therapeutic targets (82).
2.6.2 Tumor microenvironment
S. Paget’s “seed and soil theory” from 1889 predicted the role of TME in tumor growth. This concept has been supported by numerous studies across various types of cancer (83). The core concept is that normal components of the tissue (or of the niche where the tumor grows) may provide, either occasionally or as part of a precise neoplastic strategy, a set of soluble factors that contribute to, or even determine, the growth of the tumor itself or facilitate immune evasion.
The TME consists of multiple elements: in addition to epithelial, stromal, and immune cells, recent interest has been focused on cancer-associated fibroblasts (CAF) and adipocytes.
2.6.2.1 Cancer-associated fibroblasts (CAFs)
Traditionally considered as tumor-supporting, recent reports suggest they also play a role in inhibiting tumor development. This dual role may be attributable to the heterogeneity and plasticity of cancer-associated fibroblastsCAFs), largely dependent on their origin. Newly proposed classifications may help translate their functional characteristics into therapeutic strategies (84, 85). This therapeutic potential has been confirmed in lung (85) and breast cancers (86). The connection between MM and CAFs is increasingly recognized, as outlined in the following recent publications (87–91):
2.6.2.2 Adipocytes
Recently acknowledged as potential contributors to myeloma dynamics and immune cell functions, adipocytes are abundant in the BM and increase with age–a relevant factor given MM’s prevalence in older individuals. Unlike traditional classifications of brown, beige, and white fat, BM adipocytes exhibit distinct characteristics and functions (92, 93). Single-nucleus RNA sequencing (snRNA-seq) has identified a subpopulation of BM adipocytes involved in thermogenesis (94). Notably, forward-feedback loops between adipocytes and malignant plasma cells in the BM niche can modulate MM cell proliferation and support MM growth. Some of these interactions involve the upregulation of CD38, mediated by adipocyte-secreted angiotensin II (Ang II), which has been identified as the direct cause of increased expression of acetyl-CoA synthetase (ACSS2) in MM plasma cells, promoting growth by stabilizing the oncoprotein IRF4 in vitro and in vivo (95).
CD38 can also influence sirtuin activities, affecting histone acetylation and gene expression. This suggests a potential link between ACSS2 and CD38 in regulating gene expression through epigenetic mechanisms.
Moreover, extracellular CD38 is involved in the conversion of NAD+ to adenosine (ADO), leading to NAD+ depletion (40). ADO plays a role in regulating intercellular crosstalk as well as thermogenic and metabolic functions, such as adipocyte differentiation and lipid synthesis, in adipose tissues (96).
Recent evidence highlights the active and complex roles of BM adipocytes, suggesting that they are not merely passive energy stores but actively regulate multiple functions that can influence hematopoiesis, for instance, either positively or negatively (97).
Additionally, multiple reports confirm that adipocytes secrete a range of paracrine factors relevant to MM dynamics, notably stem cell factor, adiponectin, leptin, and IL-2β. MM plasma cells have also been shown to modulate inflammatory cytokine production, which can influence adipocyte function and lipolysis. For a comprehensive review, see reference (98).
Further, adipocytes are also studied in relation to myeloma progression. An analysis of an MGUS sample revealed a significant upregulation of adipogenic commitment. Moreover, adipocytes are reported to modulate responses to treatment. Specifically, Ochiai M et al. (99) demonstrated that adipocytes mediate drug resistance in MM plasma cells, contributing to recurrence and/or disease progression (99).
The literature indicates that MM may stimulate lipolysis in adipocytes through the activation of specific pathways. The resulting free fatty acids (FFA) are then taken up by MM cells via fatty acid transporter proteins, which support their proliferation within the BM niche (100–102). Figure 2 provides a schematic illustration of the homeostatic feedback circuits between adipocytes and MM cells.
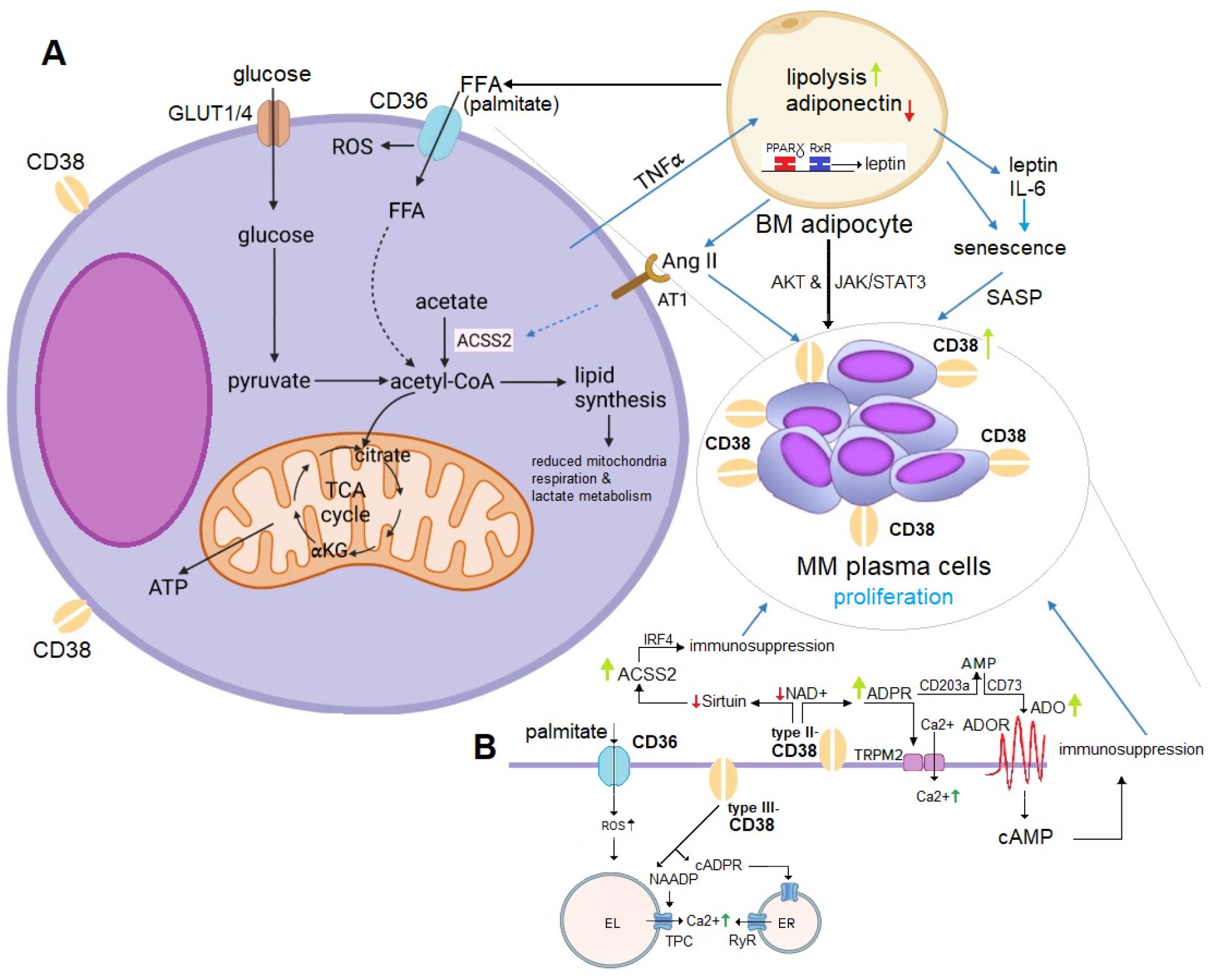
Figure 2. (A) Mechanistic Role of Adipocyte-Derived ACSS2 in MM Proliferation. Adipocyte-derived ACSS2 generates acetyl-CoA, fueling the TCA cycle and driving fatty acid (FFA) synthesis (e.g., palmitate). This promotes MM proliferation through multiple mechanisms: (1) stabilizing the oncogenic protein IRF4 via sirtuin-mediated histone acetylation, and (2) supporting lipogenesis, which induces metabolic changes favoring MM cell survival. Adiponectin inhibits MM growth but is downregulated by malignant plasma cells via TNFα signaling. Conversely, leptin and IL-6 stimulate proliferation through the AKT and JAK/STAT3 pathways. IL-6 also induces a SASP phenotype in BM adipocytes, leading to upregulation of CD38 expression in MM plasma cells. Additionally, lipolysis in BM adipocytes releases FFA, which MM plasma cells uptake to further enhance their proliferation. Elevated Ang II levels from BM adipocytes activate CD38 overexpression, promoting ADPR-to-ADO conversion (detailed in panel B), and contributing to immunosuppression and IRF4-mediated MM growth. (B) CD36-CD38 Crosstalk in Lipid and NAD+ Metabolism. ACSS2-derived palmitate induces ER stress and lipotoxicity by activating NF-κB, producing ROS, and activating the NLRP3 inflammasome. The uptake of palmitate via CD36 influences substrate availability for NAD+ synthesis, affecting CD38 activity and disrupting metabolic homeostasis. CD36 and CD38 expression are regulated by PPARγ, which forms a heterodimer with RXRα to control genes involved in FFA uptake, adipocyte differentiation, and inflammation. PPARγ activation upregulates CD38, increasing NAADP production (via type III-CD38) and intracellular Ca2+ levels to support MM cell survival. CD38-overexpressing MM cells deplete NAD+, impairing sirtuin activity and disrupting histone acetylation and gene expression. This interaction emphasizes the interplay between lipid metabolism and epigenetic regulation in MM proliferation. ACSS2, acetyl-CoA synthetase 2; ADO, adenosine; Ang II, angiotensin II; AT1, angiotensin II receptor type 1; EL, endolysosomes; ER, endoplasmic reticulum; FFA, free fatty acids; GLUT1/4, glucose transporter 1/4; PPARγ, proliferator-activated receptor-γ; RXRα, retinoic X receptor-α; ROS, reactive oxygen species; SASP; senescence-associated secretory phenotype; TCA cycle, tricarboxylic acid cycle; TNFα, tumor necrosis factor alpha.
The link between obesity and MM remains largely correlative. However, research in this area is rapidly expanding, including studies that investigate the role of senescence. Beyond scientific interest, these findings have translational relevance, as modulating adipocyte function or addressing eating behaviors could serve as potential targets for adjunctive therapies in MM (95, 101). A recent review by C. Marques-Mourlet et al. provides a comprehensive overview of the mechanistic contributions to MM, summarized in a clear model [see Figure 2, reference (103)]. Although these observations are primarily based on murine studies (104), they suggest a promising path toward rapid confirmation and potential translation into clinical settings.
3 Conclusions
CD38 antibody therapy has marked a significant advancement in managing myeloma, providing a valuable proof-of-concept for exploring in vivo questions derived from basic science. The positive clinical outcomes were accompanied by unforeseen effects that had not been predicted by conventional in vitro immunology. These complexities are further underscored by a recent report by Soriani et al., which suggests that CD38 can act as a decoy for extracellular cGAMP, a mediator of innate immunity. The relevance of these observations is currently being validated in in vivo models (105).
One critical challenge in clinical settings has been the emergence of antibody refractoriness in MM patients. This resistance was initially thought to be secondary to a significant decrease in surface CD38 following antibody exposure. Here, basic science has provided insights into the molecular regulation of CD38, as well as a link between CD38 expression, retinoids, and other emerging drug combinations.
A deeper understanding of how the antibody delivers its lethal hits or channels signals is also key. Antibody action on cell targets is mediated by FcRs, which then activate CDC, ADCC, ADCP, and direct signaling. This system, however, is more complex than initially thought. For instance, complement activation is regulated by inhibitory receptors, adding another layer of complexity to the process.
The benefits observed in antibody therapy derive from the multiple effects induced by IMiDs, including a synergistic enhancement of surface CD38 expression and the simultaneous inhibition of complement inhibitory receptors. Additional inhibitory actions are exerted by IMiDs on suppressor regulatory cells, and their influence may also vary according to the antibody used (50, 51).
However, observations from in vivo studies complicate the picture even further. For example, patients refractory to IMiDs in earlier lines of therapy regained sensitivity to lenalidomide or pomalidomide following in vivo treatment with Daratumumab (106). This evidence suggests a multifaceted scenario, where the antibody not only targets and eliminates tumor cells through specific mechanisms but also simultaneously activates various pathways in killer cells while inhibiting and depleting suppressor cells.
Further evidence supporting CD38’s role as a signaling molecule comes from studies targeting CD38 with soluble agonists, derived from traditional Chinese medicine, as was the case for the family of retinoids. These promising results are still awaiting confirmation in human in vivo models (107).
A unifying hypothesis may come from the notion that the effects driven by an antibody result from a combination of target binding via the Fab domains and simultaneous interaction of the Fc domain with specific receptors. The mechanisms underlying this heterotypic cross-talk are not yet fully understood, nor are the various effects observed in normal and pathologic conditions (e.g., inflammation, autoimmunity and–relevant to this review–responsiveness to antibody therapy) (108).
When applying these observations to the MM model, which lacks FcR, the lytic effects must be mediated by cells expressing FcR. The action of the therapeutic antibody on effectors, which often display both the CD38 target and FcRs, is harder to dissect. At high in vivo concentrations, the therapeutic antibody may shift the balance toward these effector cells. As a result, the effectors are influenced by signals from the ligation of the target molecule by the Fab-binding site of the antibody and the simultaneous engagement of the Fc domain of the IgG by the different FcRs.
The balance between activating and inhibitory receptors ultimately determines the therapeutic outcome. While the reaction of the therapeutic antibody seems relatively straightforward on the myeloma side (as MM cells are FcR-negative), more complex interactions occur with effector cells (FcR+), where the antibody can engage both the Fab-binding site and one of the FcRs on the same cell (Figure 3) (110).
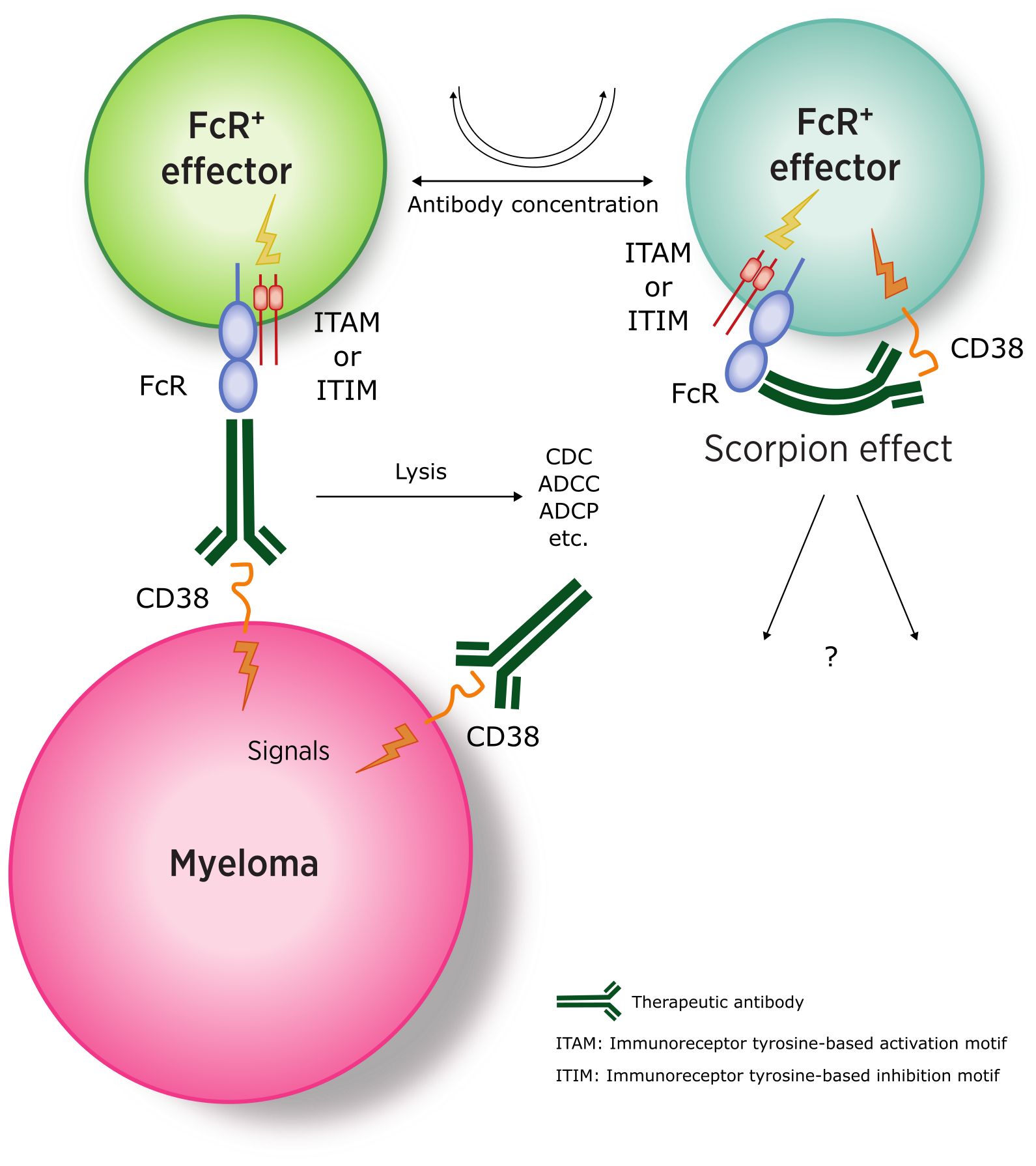
Figure 3. Interaction between anti-CD38 antibodies and myeloma (FcR-negative) and effector cells (CD38-and FcR-positive). (ITAM, immunoreceptor tyrosine-based activation motif; ITIM, immunoreceptor tyrosine-based inhibitory motif). Adapted from (109).
In conclusion, understanding these complex interactions is key to advancing myeloma therapy and for achieving significant improvements in patient outcomes. Although a definitive cure has yet to be found, the treatment landscape has dramatically changed over the past decade, with monoclonal antibodies playing a central role in this progress.
Author contributions
AH: Writing – review & editing. AF: Writing – review & editing. FMo: Data curation, Conceptualization, Writing – review & editing. EO: Conceptualization, Writing – review & editing. PS: Writing – review & editing, Investigation. NG: Investigation, Writing – original draft, Conceptualization. PR: Writing – review & editing. FMa: Writing – review & editing, Conceptualization, Funding acquisition.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Malavasi F, Deaglio S, Funaro A, Ferrero E, Horenstein AL, Ortolan E, et al. Evolution and function of the ADP ribosyl cyclase/CD38 gene family in physiology and pathology. Physiol Rev. (2008) 88:841–86. doi: 10.1152/physrev.00035.2007
2. van de Donk NWCJ, Richardson PG, Malavasi F. CD38 antibodies in multiple myeloma: back to the future. Blood. (2018) 131:13–29. doi: 10.1182/blood-2017-06-740944
3. Yong J, Cai S, Zeng Z. Targeting NAD+ metabolism: dual roles in cancer treatment. Front Immunol. (2023) 14:1269896. doi: 10.3389/fimmu.2023.1269896
4. Lee HC. Structure and enzymatic functions of human CD38. Mol Med. (2006) 12:317–23. doi: 10.2119/2006–00086.Lee
5. Lee HC, Deng QW, Zhao YJ. The calcium signaling enzyme CD38 - a paradigm for membrane topology defining distinct protein functions. Cell Calcium. (2021) 101:102514. doi: 10.1016/j.ceca.2021.102514
6. Funaro A, Horenstein AL, Calosso L, Morra M, Tarocco RP, Franco L, et al. Identification and characterization of an active soluble form of human CD38 in normal and pathological fluids. Int Immunol. (1996) 8:1643–50. doi: 10.1093/intimm/8.11.1643
7. Drach J, McQueen T, Engel H, Andreeff M, Robertson KA, Collins SJ, et al. Retinoic acid-induced expression of CD38 antigen in myeloid cells is mediated through retinoic acid receptor-alpha. Cancer Res. (1994) 54:1746–52.
8. Nijhof IS, Groen RWJ, Lokhorst HM, van Kessel B, Bloem AC, van Velzen J, et al. Upregulation of CD38 expression on multiple myeloma cells by all-trans retinoic acid improves the efficacy of daratumumab. Leukemia. (2015) 29:2039–49. doi: 10.1038/leu.2015.123
9. Frerichs KA, Minnema MC, Levin M-D, Broijl A, Bos GMJ, Kersten MJ, et al. Efficacy and safety of daratumumab combined with all-trans retinoic acid in relapsed/refractory multiple myeloma. Blood Adv. (2021) 5:5128–39. doi: 10.1182/bloodadvances.2021005220
10. García-Guerrero E, Gogishvili T, Danhof S, Schreder M, Pallaud C, Pérez-Simón JA, et al. Panobinostat induces CD38 upregulation and augments the antimyeloma efficacy of daratumumab. Blood. (2017) 129:3386–8. doi: 10.1182/blood-2017-03-770776
11. Wang H-F, Ning F, Liu Z-C, Wu L, Li Z-Q, Qi Y-F, et al. Histone deacetylase inhibitors deplete myeloid-derived suppressor cells induced by 4T1 mammary tumors. Vivo vitro Cancer Immunol Immunother. (2017) 66:355–66. doi: 10.1007/s00262-016-1935-1
12. Ogiya D, Liu J, Ohguchi H, Kurata K, Samur MK, Tai Y-T, et al. The JAK-STAT pathway regulates CD38 on myeloma cells in the bone marrow microenvironment: therapeutic implications. Blood. (2020) 136:2334–45. doi: 10.1182/blood.2019004332
13. Hu Y, Liu H, Fang C, Li C, Xhyliu F, Dysert H, et al. Targeting of CD38 by the tumor suppressor miR-26a serves as a novel potential therapeutic agent in multiple myeloma. Cancer Res. (2020) 80(10):2031–44. doi: 10.1158/0008-5472.CAN-19-1077
14. Fedele PL, Willis SN, Liao Y, Low MS, Rautela J, Segal DH, et al. IMiDs prime myeloma cells for daratumumab-mediated cytotoxicity through loss of Ikaros and Aiolos. Blood. (2018) 132:2166–78. doi: 10.1182/blood-2018-05-850727
15. Muñoz P, Mittelbrunn M, de la Fuente H, Pérez-Martínez M, García-Pérez A, Ariza-Veguillas A, et al. Antigen-induced clustering of surface CD38 and recruitment of intracellular CD38 to the immunologic synapse. Blood. (2008) 111:3653–64. doi: 10.1182/blood-2007-07-101600
16. Zilber M-T, Setterblad N, Vasselon T, Doliger C, Charron D, Mooney N, et al. MHC class II/CD38/CD9: a lipid-raft-dependent signaling complex in human monocytes. Blood. (2005) 106:3074–81. doi: 10.1182/blood-2004-10-4094
17. Frasca L, Fedele G, Deaglio S, Capuano C, Palazzo R, Vaisitti T, et al. CD38 orchestrates migration, survival, and Th1 immune response of human mature dendritic cells. Blood. (2006) 107:2392–9. doi: 10.1182/blood-2005-07-2913
18. Deaglio S, Zubiaur M, Gregorini A, Bottarel F, Ausiello CM, Dianzani U, et al. Human CD38 and CD16 are functionally dependent and physically associated in natural killer cells. Blood. (2002) 99:2490–8. doi: 10.1182/blood.v99.7.2490
19. Leau R, Duplouye P, Huchet V, Nerrière-Daguin V, Martinet B, Néel M, et al. Correct stimulation of CD28H arms NK cells against tumor cells. Eur J Immunol. (2024) 54(11):e2350901. doi: 10.1002/eji.202350901
20. Ceppellini R, Garotta G, Malavasi F, Trucco M. Modulation of expression of HLA components at the cell surface induced by anti-beta 2m reagents. Tissue Antigens. (1981) 17:28–36. doi: 10.1111/j.1399-0039.1981.tb00663.x
21. Malavasi F, Faini AC, Morandi F, Castella B, Incarnato D, Oliviero S, et al. Molecular dynamics of targeting CD38 in multiple myeloma. Br J Haematol. (2021) 193(3):581–51. doi: 10.1111/bjh.17329
22. Moreno L, Perez C, Zabaleta A, Manrique I, Alignani D, Ajona D, et al. The mechanism of action of the anti-CD38 monoclonal antibody isatuximab in multiple myeloma. Clin Cancer Res. (2019) 25:3176–87. doi: 10.1158/1078-0432.CCR-18-1597
23. Viola D, Dona A, Caserta E, Troadec E, Besi F, McDonald T, et al. Daratumumab induces mechanisms of immune activation through CD38+ NK cell targeting. Leukemia. (2021) 35:189–200. doi: 10.1038/s41375-020-0810-4
24. Zhu C, Song Z, Wang A, Srinivasan S, Yang G, Greco R, et al. Isatuximab acts through fc-dependent, independent, and direct pathways to kill multiple myeloma cells. Front Immunol. (2020) 11:1771. doi: 10.3389/fimmu.2020.01771
25. Chung S, Lin YL, Reed C, Ng C, Cheng ZJ, Malavasi F, et al. Characterization of in vitro antibody-dependent cell-mediated cytotoxicity activity of therapeutic antibodies — Impact of effector cells. J Immunol Methods. (2014) 407:63–75. doi: 10.1016/j.jim.2014.03.021
26. Kapoor P, Nathwani N, Jelinek T, Pour L, Perrot A, Dimopoulos M-A, et al. An open-label, first-in-human, single agent, dose escalation study for the evaluation of safety and efficacy of SAR442085 in patients with relapsed or refractory multiple myeloma. Eur J Haematol. (2024) 113:593–605. doi: 10.1111/ejh.14270
27. Constantinides M, Robert N, Multrier C, Coënon L, Campos-Mora M, Jacquard C, et al. FCGR3A F158V alleles frequency differs in multiple myeloma patients from healthy population. OncoImmunology. (2024) 13:2388306. doi: 10.1080/2162402X.2024.2388306
28. Chung S, Lin YL, Nguyen V, Liu C. Methods for Functional Characterization of FcRn Interactions with Therapeutic Antibodies and Fc-Fusion Proteins. In: Houen G, editor. Therapeutic Antibodies. Methods in Molecular Biology. Springer US, New York, NY (2022). p. 295–303. doi: 10.1007/978-1-0716-1450-1_18
29. Pyzik M, Kozicky LK, Gandhi AK, Blumberg RS. The therapeutic age of the neonatal Fc receptor. Nat Rev Immunol. (2023) 23:415–32. doi: 10.1038/s41577-022-00821-1
30. Roopenian DC, Akilesh S. FcRn: the neonatal Fc receptor comes of age. Nat Rev Immunol. (2007) 7:715–25. doi: 10.1038/nri2155
31. Fau J-B, El-Cheikh R, Brillac C, Koiwai K, Mace N, Campana F, et al. Drug-disease interaction and time-dependent population pharmacokinetics of isatuximab in relapsed/refractory multiple myeloma patients. CPT Pharmacom Syst Pharmacol. (2020) 9:649–58. doi: 10.1002/psp4.12561
32. Zhou Y, Jiang S. Roles of fcRn in antigen-presenting cells during autoimmunity and a clinical evaluation of efgartigimod as an fcRn blocker. Pathogens. (2023) 12:817. doi: 10.3390/pathogens12060817
33. Ferrero E, Faini AC, Malavasi F. A phylogenetic view of the leukocyte ectonucleotidases. Immunol Lett. (2019) 205:51–8. doi: 10.1016/j.imlet.2018.06.008
34. Horenstein AL, Chillemi A, Quarona V, Zito A, Roato I, Morandi F, et al. NAD+-metabolizing ectoenzymes in remodeling tumor-host interactions: the human myeloma model. Cells. (2015) 4:520–37. doi: 10.3390/cells4030520
35. Horenstein AL, Ferri V, Bolzoni M, Quarona V, Chillemi A, Zaccarello G, et al. Ectoenzymes and their products as communicators in the human myeloma niche. Blood. (2014) 124:2064–4. doi: 10.1182/blood.V124.21.2064.2064
36. Aksoy P, White TA, Thompson M, Chini EN. Regulation of intracellular levels of NAD: a novel role for CD38. Biochem Biophys Res Commun. (2006) 345:1386–92. doi: 10.1016/j.bbrc.2006.05.042
37. Young GS, Choleris E, Lund FE, Kirkland JB. Decreased cADPR and increased NAD+ in the Cd38–/– mouse. Biochem Biophys Res Commun. (2006) 346:188–92. doi: 10.1016/j.bbrc.2006.05.100
38. Grahnert A, Grahnert A, Klein C, Schilling E, Wehrhahn J, Hauschildt S. Review: NAD + : A modulator of immune functions. Innate Immun. (2011) 17:212–33. doi: 10.1177/1753425910361989
39. Goding JW, Terkeltaub R, Maurice M, Deterre P, Sali A, Belli SI. Ecto-phosphodiesterase/pyrophosphatase of lymphocytes and non-lymphoid cells: structure and function of the PC-1 family. Immunol Rev. (1998) 161:11–26. doi: 10.1111/j.1600-065x.1998.tb01568.x
40. Horenstein AL, Chillemi A, Zaccarello G, Bruzzone S, Quarona V, Zito A, et al. A CD38/CD203a/CD73 ectoenzymatic pathway independent of CD39 drives a novel adenosinergic loop in human T lymphocytes. OncoImmunology. (2013) 2:e26246. doi: 10.4161/onci.26246
41. Horenstein AL, Quarona V, Toscani D, Costa F, Chillemi A, Pistoia V, et al. Adenosine generated in the bone marrow niche through a CD38-mediated pathway correlates with progression of human myeloma. Mol Med. (2016) 22:694–704. doi: 10.2119/molmed.2016.00198
42. Hogan KA, Chini CCS, Chini EN. The multi-faceted ecto-enzyme CD38: roles in immunomodulation, cancer, aging, and metabolic diseases. Front Immunol. (2019) 10:1187. doi: 10.3389/fimmu.2019.01187
43. Shi B, Amin A, Dalvi P, Wang W, Lukacs N, Kai L, et al. Heavy-chain antibody targeting of CD38 NAD+ hydrolase ectoenzyme to prevent fibrosis in multiple organs. Sci Rep. (2023) 13:22085. doi: 10.1038/s41598-023-49450-1
44. Becherini P, Soncini D, Ravera S, Gelli E, Martinuzzi C, Giorgetti G, et al. CD38-induced metabolic dysfunction primes multiple myeloma cells for NAD+-lowering agents. Antioxid (Basel). (2023) 12:494. doi: 10.3390/antiox12020494
45. Froghi S, Grant CR, Tandon R, Quaglia A, Davidson B, Fuller B. New insights on the role of TRP channels in calcium signalling and immunomodulation: review of pathways and implications for clinical practice. Clin Rev Allergy Immunol. (2021) 60:271–92. doi: 10.1007/s12016-020-08824-3
46. Morandi F, Marimpietri D, Horenstein AL, Bolzoni M, Toscani D, Costa F, et al. Microvesicles released from multiple myeloma cells are equipped with ectoenzymes belonging to canonical and non-canonical adenosinergic pathways and produce adenosine from ATP and NAD. Oncoimmunology. (2018) 7:e1458809. doi: 10.1080/2162402X.2018.1458809
47. Chatterjee S, Daenthanasanmak A, Chakraborty P, Wyatt MW, Dhar P, Selvam SP, et al. CD38-NAD+Axis regulates immunotherapeutic anti-tumor T cell response. Cell Metab. (2018) 27:85–100.e8. doi: 10.1016/j.cmet.2017.10.006
48. Allard B, Allard D, Buisseret L, Stagg J. The adenosine pathway in immuno-oncology. Nat Rev Clin Oncol. (2020) 17:611–29. doi: 10.1038/s41571-020-0382-2
49. Ng HHM, Lee RY, Goh S, Tay ISY, Lim X, Lee B, et al. Immunohistochemical scoring of CD38 in the tumor microenvironment predicts responsiveness to anti-PD-1/PD-L1 immunotherapy in hepatocellular carcinoma. J Immunother Cancer. (2020) 8:e000987. doi: 10.1136/jitc-2020-000987
50. Leleu X, Martin T, Weisel K, Schjesvold F, Iida S, Malavasi F, et al. Anti-CD38 antibody therapy for patients with relapsed/refractory multiple myeloma: differential mechanisms of action and recent clinical trial outcomes. Ann Hematol. (2022) 101:2123–37. doi: 10.1007/s00277-022-04917-5
51. Bisht K, Fukao T, Chiron M, Richardson P, Atanackovic D, Chini E, et al. Immunomodulatory properties of CD38 antibodies and their effect on anticancer efficacy in multiple myeloma. Cancer Med. (2023) 12:20332–52. doi: 10.1002/cam4.6619
52. Lee HT, Kim Y, Park UB, Jeong TJ, Lee SH, Heo Y-S. Crystal structure of CD38 in complex with daratumumab, a first-in-class anti-CD38 antibody drug for treating multiple myeloma. Biochem Biophys Res Commun. (2021) 536:26–31. doi: 10.1016/j.bbrc.2020.12.048
53. Lammerts van Bueren J, Jakobs D, Kaldenhoven N, Roza M, Hiddingh S, Meesters J, et al. Direct in vitro comparison of daratumumab with surrogate analogs of CD38 antibodies MOR03087, SAR650984 and ab79. Blood. (2014) 124:3474–4. doi: 10.1182/blood.V124.21.3474.3474
54. Martin TG, Corzo K, Chiron M, van de Velde H, Abbadessa G, Campana F, et al. Therapeutic opportunities with pharmacological inhibition of CD38 with isatuximab. Cells. (2019) 8:1522. doi: 10.3390/cells8121522
55. van de Donk NWCJ. Immunomodulatory effects of CD38-targeting antibodies. Immunol Lett. (2018) 199:16–22. doi: 10.1016/j.imlet.2018.04.005
56. Horenstein AL, Faini AC, Malavasi F. CD38 in the age of COVID-19: a medical perspective. Physiol Rev. (2021) 101:1457–86. doi: 10.1152/physrev.00046.2020
57. Baum N, Fliegert R, Bauche A, Hambach J, Menzel S, Haag F, et al. Daratumumab and Nanobody-Based Heavy Chain Antibodies Inhibit the ADPR Cyclase but not the NAD+ Hydrolase Activity of CD38-Expressing Multiple Myeloma Cells. Cancers. (2020) 13:76. doi: 10.3390/cancers13010076
58. Horenstein AL, Faini AC, Morandi F, Bracci C, Lanza F, Giuliani N, et al. The circular life of human CD38: from basic science to clinics and back. Molecules. (2020) 25:4844. doi: 10.3390/molecules25204844
59. Vijayan D, Young A, Teng MWL, Smyth MJ. Targeting immunosuppressive adenosine in cancer. Nat Rev Cancer. (2017) 17:709–24. doi: 10.1038/nrc.2017.86
60. Willingham SB, Volkmer J-P, Gentles AJ, Sahoo D, Dalerba P, Mitra SS, et al. The CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. Proc Natl Acad Sci USA. (2012) 109:6662–7. doi: 10.1073/pnas.1121623109
61. Barclay AN, Van Den Berg TK. The interaction between signal regulatory protein alpha (SIRPα) and CD47: structure, function, and therapeutic target. Annu Rev Immunol. (2014) 32:25–50. doi: 10.1146/annurev-immunol-032713-120142
62. Tseng D, Volkmer J-P, Willingham SB, Contreras-Trujillo H, Fathman JW, Fernhoff NB, et al. Anti-CD47 antibody-mediated phagocytosis of cancer by macrophages primes an effective antitumor T-cell response. Proc Natl Acad Sci U.S.A. (2013) 110:11103–8. doi: 10.1073/pnas.1305569110
63. Eladl E, Tremblay-LeMay R, Rastgoo N, Musani R, Chen W, Liu A, et al. Role of CD47 in hematological Malignancies. J Hematol Oncol. (2020) 13:96. doi: 10.1186/s13045-020-00930-1
64. Sun J, Muz B, Alhallak K, Markovic M, Gurley S, Wang Z, et al. Targeting CD47 as a novel immunotherapy for multiple myeloma. Cancers. (2020) 12:305. doi: 10.3390/cancers12020305
65. Maute R, Xu J, Weissman IL. CD47–SIRPα-targeted therapeutics: status and prospects. Immuno-Oncol Technol. (2022) 13:100070. doi: 10.1016/j.iotech.2022.100070
66. Storti P, Vescovini R, Costa F, Marchica V, Toscani D, Dalla Palma B, et al. CD14 + CD16 + monocytes are involved in daratumumab-mediated myeloma cells killing and in anti-CD47 therapeutic strategy. Br J Haematol. (2020) 190:430–6. doi: 10.1111/bjh.16548
67. Russ A, Hua AB, Montfort WR, Rahman B, Riaz IB, Khalid MU, et al. Blocking “don’t eat me” signal of CD47-SIRPα in hematological Malignancies, an in-depth review. Blood Rev. (2018) 32:480–9. doi: 10.1016/j.blre.2018.04.005
68. Kim D, Wang J, Willingham SB, Martin R, Wernig G, Weissman IL. Anti-CD47 antibodies promote phagocytosis and inhibit the growth of human myeloma cells. Leukemia. (2012) 26:2538–45. doi: 10.1038/leu.2012.141
69. Li S, Chen D, Yang Y, Guo H, Liu D, Sun N, et al. Combining CD38 antibody with CD47 blockade is a promising strategy for treating hematologic Malignancies expressing CD38. Front Immunol. (2024) 15:1398508. doi: 10.3389/fimmu.2024.1398508
70. Grandclément C, Estoppey C, Dheilly E, Panagopoulou M, Monney T, Dreyfus C, et al. Development of ISB 1442, a CD38 and CD47 bispecific biparatopic antibody innate cell modulator for the treatment of multiple myeloma. Nat Commun. (2024) 15:2054. doi: 10.1038/s41467-024-46310-y
71. Liu W, Yu J, Sun K, Song Q, Li Y, He Y, et al. Preclinical characterization of a novel investigational monoclonal antibody CM313 with potent CD38-positive cell killing activity. Front Immunol. (2024) 15:1410457. doi: 10.3389/fimmu.2024.1410457
72. Van Der Horst HJ, Mutis T. Enhancing Fc -mediated effector functions of monoclonal antibodies: The example of HexaBodies. Immunol Rev. (2024) 328(1):456–65. doi: 10.1111/imr.13394
73. Hiemstra IH, Santegoets KCM, Janmaat ML, De Goeij BECG, Ten Hagen W, van Dooremalen S, et al. Preclinical anti-tumour activity of HexaBody-CD38, a next-generation CD38 antibody with superior complement-dependent cytotoxic activity. EBioMedicine. (2023) 93:104663. doi: 10.1016/j.ebiom.2023.104663
74. Pouleau B, Estoppey C, Suere P, Nallet E, Laurendon A, Monney T, et al. Pre-clinical characterization of ISB 1342, a CD38xCD3 T-cell engager for relapsed/refractory multiple myeloma. Blood J. (2023) 142(3):260–73. doi: 10.1182/blood.2022019451
75. Bahlis NJ, King AM, Kolonias D, Carlson LM, Liu HY, Hussein MA, et al. CD28-mediated regulation of multiple myeloma cell proliferation and survival. Blood. (2007) 109:5002–10. doi: 10.1182/blood-2006-03-012542
76. Grab AL, Kim PS, John L, Bisht K, Wang H, Baumann A, et al. Pre-clinical assessment of SAR442257, a CD38/CD3xCD28 trispecific T cell engager in treatment of relapsed/refractory multiple myeloma. Cells. (2024) 13:879. doi: 10.3390/cells13100879
77. Keller AL, Reiman LT, Perez De Acha O, Parzych SE, Forsberg PA, Kim PS, et al. Ex vivo efficacy of SAR442257 anti-CD38 trispecific T-cell engager in multiple myeloma relapsed after daratumumab and BCMA-targeted therapies. Cancer Res Commun. (2024) 4:757–64. doi: 10.1158/2767-9764.CRC-23-0434
78. Martín-Sánchez E, Blanco L, Kim PS, Bisht K, Wang H, Van De Velde H, et al. Targeting CD38 with isatuximab and a novel CD38/CD3×CD28 trispecific T-cell engager in older patients with acute myeloid leukemia. Blood Adv. (2024) 8:3875–9. doi: 10.1182/bloodadvances.2024013212
79. Carretero-Iglesia L, Hall OJ, Berret J, Pais D, Estoppey C, Chimen M, et al. ISB 2001 trispecific T cell engager shows strong tumor cytotoxicity and overcomes immune escape mechanisms of multiple myeloma cells. Nat Cancer. (2024) 5:1494–514. doi: 10.1038/s43018-024-00821-1
80. Ugamraj HS, Dang K, Ouisse L-H, Buelow B, Chini EN, Castello G, et al. TNB-738, a biparatopic antibody, boosts intracellular NAD+ by inhibiting CD38 ecto-enzyme activity. MAbs. (2022) 14:2095949. doi: 10.1080/19420862.2022.2095949
81. Candelaria PV, Nava M, Daniels-Wells TR, Penichet ML. A fully human igE specific for CD38 as a potential therapy for multiple myeloma. Cancers. (2023) 15:4533. doi: 10.3390/cancers15184533
82. Zhang G, Guo C, Wang Y, Zhang X, Liu S, Qu W, et al. FTL004, an anti-CD38 mAb with negligible RBC binding and enhanced pro-apoptotic activity, is a novel candidate for treatments of multiple myeloma and non-Hodgkin lymphoma. J Hematol Oncol. (2022) 15:177. doi: 10.1186/s13045-022-01395-0
83. Paget S. The distribution of secondary growths in cancer of the breast. Cancer Metast Rev. (1989) 8:98–101.
84. Cords L, De Souza N, Bodenmiller B. Classifying cancer-associated fibroblasts—The good, the bad, and the target. Cancer Cell. (2024) 42:1480–5. doi: 10.1016/j.ccell.2024.08.011
85. Yang D, Liu J, Qian H, Zhuang Q. Cancer-associated fibroblasts: from basic science to anticancer therapy. Exp Mol Med. (2023) 55:1322–32. doi: 10.1038/s12276-023-01013-0
86. Croizer H, Mhaidly R, Kieffer Y, Gentric G, Djerroudi L, Leclere R, et al. Deciphering the spatial landscape and plasticity of immunosuppressive fibroblasts in breast cancer. Nat Commun. (2024) 15:2806. doi: 10.1038/s41467-024-47068-z
87. Ding Z, Shi R, Hu W, Tian L, Sun R, Wu Y, et al. Cancer-associated fibroblasts in hematologic Malignancies: elucidating roles and spotlighting therapeutic targets. Front Oncol. (2023) 13:1193978. doi: 10.3389/fonc.2023.1193978
88. Miaomiao S, Xiaoqian W, Yuwei S, Chao C, Chenbo Y, Yinghao L, et al. Cancer-associated fibroblast-derived exosome microRNA-21 promotes angiogenesis in multiple myeloma. Sci Rep. (2023) 13:9671. doi: 10.1038/s41598-023-36092-6
89. Sakemura R, Hefazi M, Siegler EL, Cox MJ, Larson DP, Hansen MJ, et al. Targeting cancer-associated fibroblasts in the bone marrow prevents resistance to CART-cell therapy in multiple myeloma. Blood. (2022) 139:3708–21. doi: 10.1182/blood.2021012811
90. Saltarella I, Morabito F, Giuliani N, Terragna C, Omedè P, Palumbo A, et al. Prognostic or predictive value of circulating cytokines and angiogenic factors for initial treatment of multiple myeloma in the GIMEMA MM0305 randomized controlled trial. J Hematol Oncol. (2019) 12:4. doi: 10.1186/s13045-018-0691-4
91. Cheng Q, Li X, Liu J, Ye Q, Chen Y, Tan S, et al. Multiple Myeloma-Derived Exosomes Regulate the Functions of Mesenchymal Stem Cells Partially via Modulating miR-21 and miR-146a. Stem Cells Int. (2017) 2017:9012152. doi: 10.1155/2017/9012152
92. Austin MJ, Kalampalika F, Cawthorn WP, Patel B. Turning the spotlight on bone marrow adipocytes in haematological Malignancy and non-malignant conditions. Br J Haematol. (2023) 201:605–19. doi: 10.1111/bjh.18748
93. Caruso R, Chen GY. High fat stems tumor immune surveillance. Cell Rep Med. (2021) 2:100483. doi: 10.1016/j.xcrm.2021.100483
94. Sun W, Dong H, Balaz M, Slyper M, Drokhlyansky E, Colleluori G, et al. snRNA-seq reveals a subpopulation of adipocytes that regulates thermogenesis. Nature. (2020) 587:98–102. doi: 10.1038/s41586-020-2856-x
95. Li Z, Liu H, He J, Wang Z, Yin Z, You G, et al. Acetyl-coA synthetase 2: A critical linkage in obesity-induced tumorigenesis in myeloma. Cell Metab. (2021) 33:78–93.e7. doi: 10.1016/j.cmet.2020.12.011
96. Kwon SY, Park YJ. Function of NAD metabolism in white adipose tissue: lessons from mouse models. Adipocyte. (2024) 13:2313297. doi: 10.1080/21623945.2024.2313297
97. Fairfield H, Karam M, Schimelman A, Qiang Y-W, Reagan MR. Adipocytes and metabolism: Contributions to multiple myeloma. J Bone Oncol. (2024) 46:100609. doi: 10.1016/j.jbo.2024.100609
98. Randall TD, Meza-Perez S. Immunity in adipose tissues: Cutting through the fat. Immunol Rev. (2024) 324:4–10. doi: 10.1111/imr.13344
99. Ochiai M, Fierstein S, XsSali F, DeVito N, Purkey LR, May R, et al. Unlocking drug resistance in multiple myeloma: adipocytes as modulators of treatment response. Cancers. (2023) 15:4347. doi: 10.3390/cancers15174347
100. Panaroni C, Fulzele K, Mori T, Siu KT, Onyewadume C, Maebius A, et al. Multiple myeloma cells induce lipolysis in adipocytes and uptake fatty acids through fatty acid transporter proteins. Blood. (2022) 139:876–88. doi: 10.1182/blood.2021013832
101. Choa R, Panaroni C, Bhatia R, Raje N. It is worth the weight: obesity and the transition from monoclonal gammopathy of undetermined significance to multiple myeloma. Blood Adv. (2023) 7:5510–23. doi: 10.1182/bloodadvances.2023010822
102. El-Masri BM, Leka B, Mustapha F, Gundesen MT, Hinge M, Lund T, et al. Bone marrow adipocytes provide early sign for progression from MGUS to multiple myeloma. Oncotarget. (2024) 15:20–6. doi: 10.18632/oncotarget.28548
103. Marques-Mourlet C, Di Iorio R, Fairfield H, Reagan MR. Obesity and myeloma: Clinical and mechanistic contributions to disease progression. Front Endocrinol. (2023) 14:1118691. doi: 10.3389/fendo.2023.1118691
104. Lwin ST, Olechnowicz SWZ, Fowler JA, Edwards CM. Diet-induced obesity promotes a myeloma-like condition in vivo. Leukemia. (2015) 29:507–10. doi: 10.1038/leu.2014.295
105. Cuollo L, Di Cristofano S, Sandomenico A, Iaccarino E, Oliver A, Zingoni A, et al. CD38 restrains the activity of extracellular cGAMP in a model of multiple myeloma. iScience. (2024) 27:109814. doi: 10.1016/j.isci.2024.109814
106. Gavriatopoulou M, Kastritis E, Ntanasis-Stathopoulos I, Fotiou D, Roussou M, Migkou M, et al. The addition of IMiDs for patients with daratumumab-refractory multiple myeloma can overcome refractoriness to both agents. Blood. (2018) 131:464–7. doi: 10.1182/blood-2017-10-809293
107. Liu Y, Zhang L, Wang L, Tang X, Wan S, Huang Q, et al. Targeting CD38/ADP-ribosyl cyclase as a novel therapeutic strategy for identification of three potent agonists for leukopenia treatment. Pharmacol Res. (2024) 200:107068. doi: 10.1016/j.phrs.2024.107068
108. Bournazos S, Wang TT, Dahan R, Maamary J, Ravetch JV. Signaling by antibodies: recent progress. Annu Rev Immunol. (2017) 35:285–311. doi: 10.1146/annurev-immunol-051116-052433
109. Malavasi F, Faini AC. Mechanism of action of a new anti-CD38 antibody: enhancing myeloma immunotherapy. Clin Cancer Res. (2019) 25:2946–8. doi: 10.1158/1078-0432.CCR-19-0260
Keywords: antibody therapies, CD38 monoclonal antibodies, IgG Fc receptors, ectoenzymatic activity, multiple myeloma
Citation: Horenstein AL, Faini AC, Morandi F, Ortolan E, Storti P, Giuliani N, Richardson PG and Malavasi F (2025) Monoclonal anti-CD38 therapy in human myeloma: retrospects and prospects. Front. Immunol. 16:1519300. doi: 10.3389/fimmu.2025.1519300
Received: 29 October 2024; Accepted: 23 January 2025;
Published: 12 February 2025.
Edited by:
John Williams, City of Hope, United StatesReviewed by:
Federico Simonetta, Hôpitaux Universitaires de Genève (HUG), SwitzerlandNora Heisterkamp, City of Hope, United States
Copyright © 2025 Horenstein, Faini, Morandi, Ortolan, Storti, Giuliani, Richardson and Malavasi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fabio Malavasi, ZmFiaW8ubWFsYXZhc2lAdW5pdG8uaXQ=