 Zhenlin Li1,2,3,4
Zhenlin Li1,2,3,4 Fangqi Lu1,2,3,4Fujin Zhou1,2,3
Fangqi Lu1,2,3,4Fujin Zhou1,2,3 Dekun Song1,2,3
Dekun Song1,2,3 Lunhui Chang1,2,3,4Weiying Liu5*
Lunhui Chang1,2,3,4Weiying Liu5* Guorong Yan2,3,4*Guolong Zhang1,2,3,4*
Guorong Yan2,3,4*Guolong Zhang1,2,3,4*- 1School of Medicine, Anhui University of Science and Technology, Huainan, China
- 2Department of Phototherapy, Shanghai Skin Disease Hospital, School of Medicine, Tongji University, Shanghai, China
- 3Skin Cancer Center, Shanghai Skin Disease Hospital, School of Medicine, Tongji University, Shanghai, China
- 4Institute of Photomedicine, School of Medicine, Tongji University, Shanghai, China
- 5Department of Dermatology, Hunan Aerospace Hospital, Changsha, China
Cutaneous squamous cell carcinoma (cSCC) is the second most common non-melanoma skin cancer, among which 82% arise from actinic keratosis (AK) characterized by lesions of epidermal keratinocyte dysplasia. It is of great significance to uncover the progression mechanisms from AK to cSCC, which will facilitate the early therapeutic intervention of AK before malignant transformation. Thus, more and more studies are trying to ascertain the potential transformation mechanisms through multi-omics, including genetics, transcriptomics, and epigenetics. In this review, we gave an overview of the specific biomarkers and signaling pathways that may be involved in the pathogenesis from AK to cSCC, pointing out future possible molecular therapies for the early intervention of AK and cSCC. We also discussed current interventions on AK and cSCC, together with future perspectives.
1 Introduction
Cutaneous squamous cell carcinoma (cSCC) is a malignant tumor originating from epidermal keratinocytes. It is one of the most commonly diagnosed non-melanoma skin cancers (NMSC), with its incidence gradually increasing worldwide in recent years, accounting for approximately 20% to 50% of all skin tumors (1, 2). In the United States, there are approximately 1.1 million new cases of cSCC annually, with an overall increase in incidence of 263% between 1976-1984 and 2000-2010 (3, 4). In western China, cSCC has the highest incidence rate among all skin cancers, at 29.4%, and this rate continues to rise annually by 2.6% (5). In European countries, it is estimated that by 2030, the incidence of cSCC will double (6). With a large number of cases, cSCC presents a significant burden on public health, with 3% to 7% of patients experiencing metastasis, leading to notable morbidity and mortality, thus imposing substantial societal public health burdens (3, 7, 8).
Actinic keratosis (AK) is a precancerous skin lesion caused by long-term chronic exposure to ultraviolet radiation (UVR), predominantly affecting sun-exposed areas such as the head and face, and commonly occurring in the elderly. AK carries a high risk of developing into cSCC and can present with multiple clinical and subclinical lesions simultaneously. The progression rate and probability of AK to cSCC vary among individuals, with an estimated annual progression rate of 0% to 0.075% per lesion (9). Among elderly patients with multiple AK lesions, the annual rate of progression to invasive cSCC is 0.6%, increasing to 2.57% four years after diagnosis (10).
The increasing incidence and mortality rates of cSCC have significant implications for public health. Understanding the malignant transformation from AK to cSCC is crucial for the prevention, early detection, and effective management strategies of cSCC. Additionally, a deeper understanding of the molecular mechanisms underlying the transition from AK to cSCC may facilitate the development of novel targets for the intervention of AK or cSCC.
2 Etiology, prevention, and early diagnosis of AK
The etiological factors of AK include environmental and individual factors. Environmental factors primarily refer to exposure to UVR, with the incidence of AK being positively correlated with cumulative UVR exposure (11, 12). Populations exposed to more sunlight (e.g., Australia and the United Kingdom) have a higher incidence of AK (13). UVR can induce cellular genetic mutations, chronic skin inflammation, and immune suppression, ultimately leading to abnormal proliferation of keratinocytes. The more sunlight the skin absorbs, the higher risk of AK (14). Five independent risk factors for AK are age (>60 years), gender (male), skin type, history of skin tumors, and outdoor work history (14). Individuals with fair skin (Fitzpatrick types I and II) are more prone to AK, while the incidence of AK is similar in Fitzpatrick types III and IV skin (12, 15). These conditions are influenced by various exposome factors such as air pollution, nutrition, and psychological stress, all of which can exacerbate skin carcinogenesis and aging (16). Additionally, immunosuppressed populations, such as 32.5% solid organ transplant recipients (SOTRs) (17) or patients taking long-term cytotoxic drugs, are at higher risk of AK (18–21).
Prevention of AK is strongly recommended, with evidence supporting using sunscreen and protective clothing to block initial and recurrent UVR radiation (22, 23). Randomized controlled trial (RCT) has shown that the use of sunscreen can reduce the incidence and progression of additional AK lesions (24). In addition to the significant role of sunscreen in AK prevention, oral medications also have a preventive effect on AK, such as oral nicotinamide (25–27), and acitretin (28, 29). However, a recent double-blind, randomized, placebo-controlled trial of oral nicotinamide for the prevention of AK in kidney transplant recipients showed that nicotinamide did not reduce the progression of AK in this population (30).
The diagnosis of AK emphasizes early detection and prediction of the risk of AK progression to cSCC. Non-invasive and highly efficient diagnostic techniques such as dermatoscopy, reflectance confocal microscopy (RCM), optical coherence tomography (OCT) (31), aminolevulinic acid (1%) (32), and high-frequency ultrasound of the skin (33) are employed for the diagnosis of AK. Dermatoscopy has good sensitivity and specificity for diagnosing AK, with a sensitivity of 95.6% and specificity of 95.0% for features such as the red pseudonetwork pattern combined with follicular pore enlargement (34). Dermatoscopic features of AK correlate with histological manifestations such as acanthosis, hyperkeratosis, and vascular proliferation. The “strawberry pattern” characteristic of non-pigmented AK can be detected by dermatoscopy and differentiated from early cSCC (35, 36). The most specific indications for diagnosing AK with RCM are the observation of disordered arrangements of keratinocyte layers and cellular atypia, with RCM having a unique diagnostic value for pigmented AK (12, 34). Histopathological examination remains the gold standard for diagnosing AK, especially when clinical diagnosis is uncertain or when progression to invasive cSCC is suspected. The main indications for pathological examination are uncertain clinical diagnosis, lesions > 1 cm in diameter, bleeding, ulceration, or induration, and rapid growth of skin lesions; secondary indications include lesions accompanied by severe itching, pain, and obvious hyperkeratosis (37). Skin lesions on certain special sites (such as the lips) are at high risk of invasion and require pathological examination. In addition, pathological subtyping helps guide the treatment and prognosis assessment of AK. Common pathological subtypes of AK include hypertrophic type, atrophic type, dyskeratotic type, mossy type, pigmented type, Bowenoid type, and actinic cheilitis type. Although AK histological subtypes have their characteristics, they mostly manifest as dysplastic keratinocytes arranged disorderly from mild basal layer to full-thickness epidermal structure disorder, appearing as a continuous spectrum structure of in situ squamous cell carcinoma, often accompanied by hyperkeratosis and dyskeratosis.
3 Etiology, prevention, and early diagnosis of cSCC
cSCC shares similar risk factors with AK, including exposure to UVR, age, fair skin, and immunosuppression (38). UVR exposure stands out as the primary risk factor for cSCC. Prolonged sun exposure, cumulative exposure in specific areas, and incidents of sunburn are closely correlated with cSCC incidence, particularly among individuals with fair skin (38). The TP53 mutations induced by UVR represent the most common genetic alterations in cSCC (39). This malignancy is more prevalent among Caucasians, with an average onset age of approximately 60 years, and the risk increases with age, with a male-to-female ratio of about 3:1 (40). Immunocompromised individuals, such as SOTRs, face a significantly higher risk of developing cSCC, with SOTRs being 65 to 250 times more susceptible than healthy adults (41–43). Additionally, human papillomavirus (HPV) has been implicated in cSCC pathogenesis, particularly in cases affecting the perianal or genital regions (44). Environmental factors contributing to cSCC include arsenic exposure, polycyclic aromatic hydrocarbons, nitrosamines, and exposure to ionizing radiation (45, 46). Early signs of arsenic exposure include the occurrence of palmoplantar keratosis (45). Genodermatoses such as xeroderma pigmentosum, epidermodysplasia verruciformis, and epidermolysis bullosa dystrophica are also risk factors for cSCC development (47).
The “Interdisciplinary Guidelines for Invasive cSCC Based on European Consensus: Part 1 – Diagnosis and Prevention - 2023 Update” emphasizes primary and secondary prevention strategies for cSCC (48). Primary prevention involves avoiding sunbathing, using protective clothing, applying sunscreen, and seeking shade to minimize UVR exposure (48). Chemical prevention aims to reduce the incidence of new cSCC cases and includes agents such as nicotinamide, retinoids, 5-FU, and nonsteroidal anti-inflammatory drugs (NSAIDs). Nicotinamide can repair DNA damage and immunosuppression caused by intense UVR (49). However, a 12-month RCT involving SOTRs showed that oral nicotinamide did not reduce the incidence of cSCC (50). Several studies have demonstrated that oral retinoids, particularly acitretin and isotretinoin, can lower the incidence of new cSCC cases, especially among high-risk individuals (51, 52). An RCT on 5-FU showed that topical treatment reduced the expected surgical risk of cSCC by 75% (53). Population-based case-control studies suggest an association between NSAID use and a reduced risk of cSCC, with frequent NSAID use being a protective factor against cSCC development (54).
Digital cSCC lacks distinctive clinical features, leading to a potential misdiagnosis rate of up to 82% if solely based on clinical manifestations (55). Histopathological examination remains the gold standard for cSCC diagnosis, with well-differentiated histopathological subtypes exhibiting low metastatic potential, including keratoacanthoma and verrucous carcinoma, while aggressive subtypes associated with poor prognosis include desmoplastic cSCC, adenosquamous cSCC, and cSCC arising in association with scars (56, 57). Dermoscopy provides early diagnostic clues for cSCC and aids in the approximate assessment of its pathological staging, playing a crucial role in diagnosis, subsequent treatment, and management (58). cSCC lesions exhibit two vascular patterns under dermoscopy: dotted vessels and glomerular vessels. In situ pigmented cSCC may also present with small brown globules and homogeneous brown pigmentation under dermoscopy. Invasive cSCC often displays circular or serpentine vessels. RCM enables non-invasive diagnostic assessment of cSCC or AK skin lesions, with cSCC lesions showing a disordered arrangement of epidermal cells or atypical honeycomb patterns, along with circular vessels in the papillary dermis (59, 60).
4 Established progressed-associated genes from AK to cSCC
Due to the fact that more than 70% of cSCC cases arise from AK (61), understanding the mechanisms underlying the progression from AK to cSCC is of paramount importance as it could aid in early therapeutic interventions before malignant transformation occurs. Identifying specific genomic changes that drive the transition from normal skin (NS) to AK and subsequently to invasive cSCC is challenging, given the substantial burden of UVR-induced mutations characteristic of all stages of this progression (Figure 1). Molecular genetic studies have examined AK and cSCC for known cancer gene expressions, chromosomal instability, and mutation levels (Figure 2). These studies generally indicate that AK and cSCC share similar genetic alterations (Table 1).
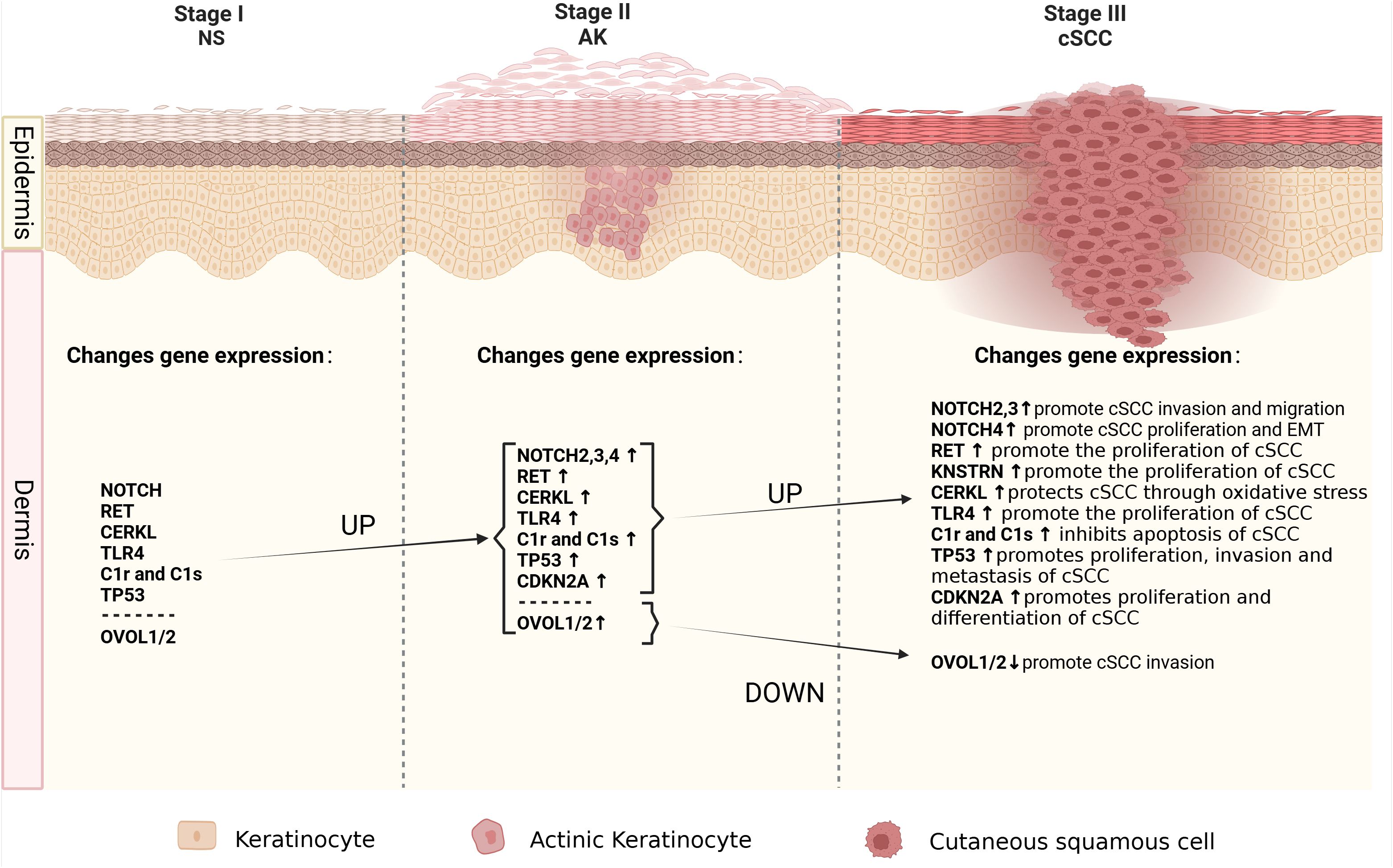
Figure 1. The molecular alterations associated with cancer progression from normal skin to AK to cSCC. Schematic depicting the spectrum of morphologies of NS-AK-cSCC lesions, with relevant changes in gene expression.
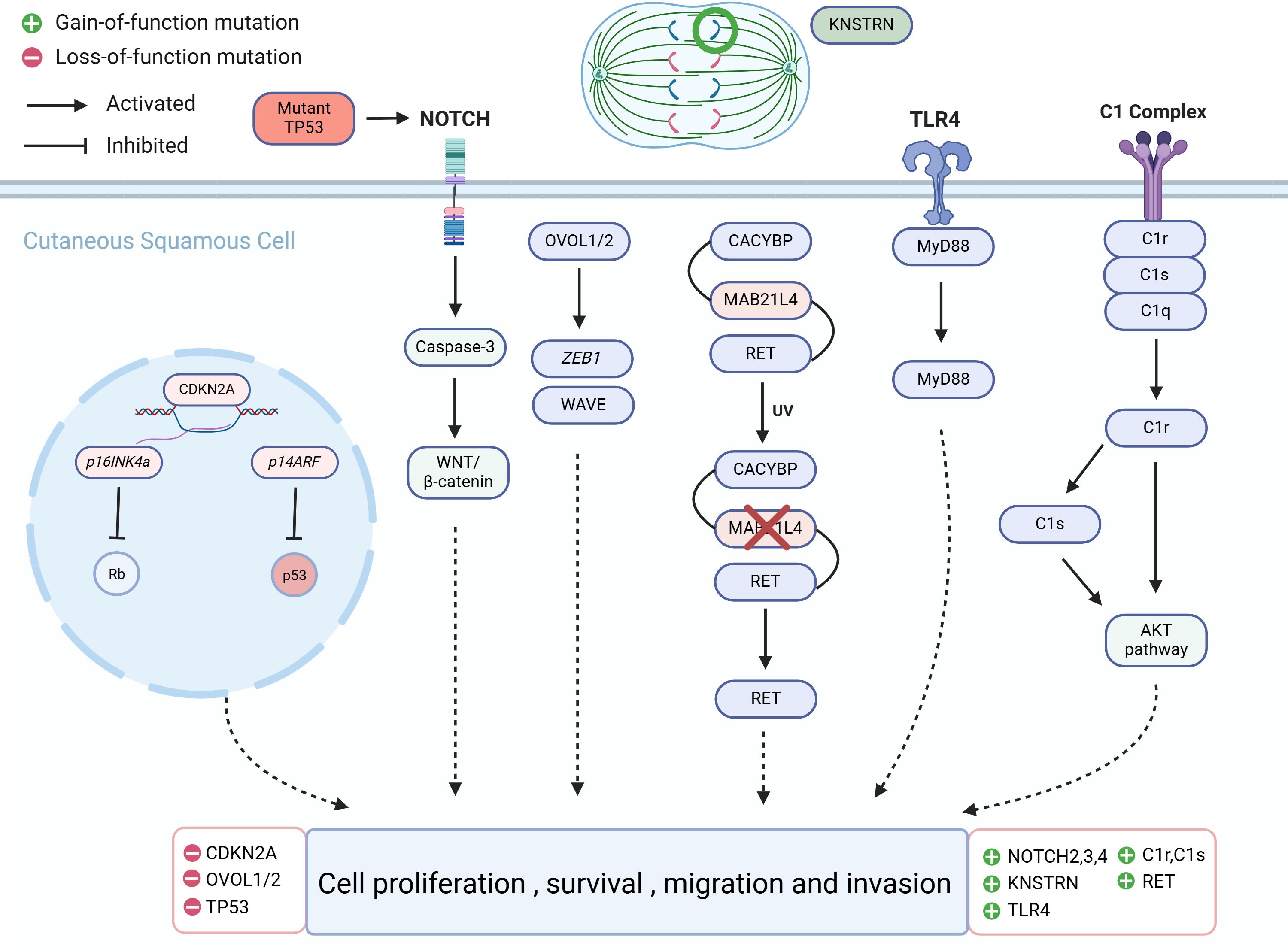
Figure 2. Molecular alterations that drive cutaneous squamous cell carcinoma (cSCC) proliferation.
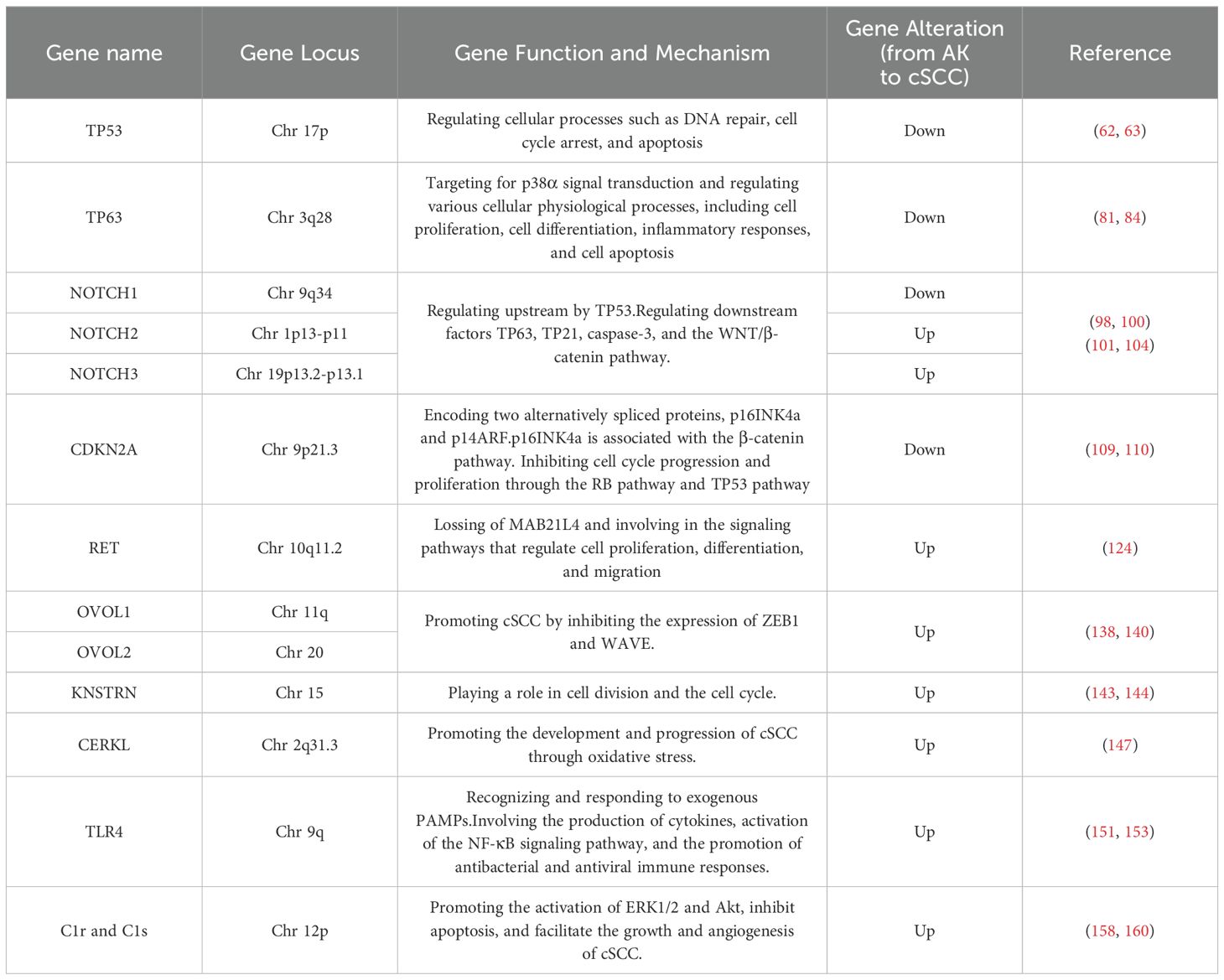
Table 1. The mutations and mechanisms in cSCC and AK genes across published studies.
4.1 TP53
The tumor protein p53 (TP53) is situated on the short arm of chromosome 17 and encodes a 53-kilodalton protein involved in regulating cellular processes such as DNA repair, cell cycle arrest, and apoptosis (62, 63). TP53 is widely recognized as the most common mutated gene in cancer, being a pivotal tumor suppressor gene, whose abnormalities are associated with most malignancies (62). UVR-induced TP53 mutations are early events in cSCC development, leading to genomic instability, followed by mutations in other tumor suppressor genes and oncogenes (64). The mutation frequency of TP53 in primary cSCC ranges from 50% to 60%; in metastatic cSCC, TP53 mutations are present in nearly 95% of patients (65, 66). TP53 mutations are also present in normal skin exposed to UVR, AK, in situ, and invasive cSCC, indicating that TP53 mutations are not only early manifestations of UVR damage but also associated with cSCC progression (65, 66). Targeted deep sequencing of 160 cancer-related genes in 17 AK and cSCC patients revealed driver mutations in TP53 in 81% of cases, suggesting widespread TP53 mutations in AK and cSCC lesions (67). TP53 gene mutations have been found in a significant proportion of human skin malignancies, not only participate in the malignant transformation from AK to cSCC but also serving as biomarkers for predicting skin cancer (68). Skin cells exposed to ionizing radiation, such as solar UVR, incur DNA damage, leading to destabilization and inactivation of TP53 (69). TP53 gene mutations or alterations in the TP53 signaling pathway during the progression from AK to cSCC result in functional impairment of TP53, shifting its role from tumor suppression to oncogenesis, manifested as clonal proliferation of keratinocytes. The most typical pattern in AK is CC>TT base changes in abnormally proliferating keratinocytes, leading to further genetic instability and eventual cSCC development (65, 70). This clonal proliferation is considered a critical step in cSCC development (71). A deep sequencing study of skin clonal mutations demonstrated that TP53 R248W and G245D hotspot mutations are highly recurrent, potentially representing early stages of cSCC development (65).
Immunohistochemical results showed that the expression level of TP53 in cSCC was slightly lower than that of AK, and the expression level was significantly lower than that of NS (72, 73). Missense mutations occur in TP53 in cSCC, with common mutation hotspots such as TP53 R248W. Mutant TP53 gains oncogenic activity, serving as a potent early driver of cSCC progression. Additionally, missense mutations of TP53, such as p53 R172H, can lead to metastatic cSCC development (74). Besides missense mutations in TP53, some mutations result in nonsense mutations, thereby prematurely introducing a termination codon (75). Truncated isoforms produced by TP53 nonsense mutations include tumor-specific expressions characterized by increased metastasis, which may be a feature of tumor metastasis (76, 77). The functional mechanisms of TP53 mutant variants, including activation of cell proliferation, promotion of cell invasion and metastasis, and the presence and potential functions of different TP53 subtypes in cSCC development, are not yet fully understood.
Transcription factor (TF) TP63 is a p53 family member and plays a key role in the development of keratinocyte transformation induced by basal cell and ectodermal appendage carcinoma genes in stratified cSCC cell survival (78–82). p63 is a major regulator of epidermal development and maintenance and is also a key target for p38α signal transduction (81). The level of p63 protein detected in a large number of human cancer cell lines is negatively correlated with phosphorylated p38 signaling (81). P38α is an important signal-transducing protein kinase that belongs to the mitogen-activated protein kinase (MAPK) family (83). It plays a role in regulating various cellular physiological processes, including cell proliferation, cell differentiation, inflammatory responses, and cell apoptosis (79–81, 84). In cSCC biopsies, a significant reduction in p38 phosphorylation in the tumor parenchyma links AK with SCC progression and p38 inactivity (85). p38α plays a role in the transition from AK to cSCC, and p38α kinase is involved in the suppression of cSCC by regulating p63 and stem cell frequency (85).
TP53, as a pivotal gene, holds significant clinical relevance and applications in AK and cSCC. TP53 mutations are found in up to 100% of AK cases and 90% of cSCC cases, indicating its central role in the pathogenesis of these skin lesions (86). TP53 mutations are an early event that leads to the inactivation of its tumor suppressor function, facilitating the accumulation of additional oncogenic mutations and progression from AK to invasive cSCC (65). These mutations often result in a loss of normal p53 function and can lead to more aggressive tumor phenotypes (87). TP53 serves as a diagnostic and prognostic biomarker, the presence of TP53 mutations can serve as a diagnostic marker for AK and cSCC, as well as a prognostic indicator of disease progression and potential resistance to therapy (88). Reduced p53 staining in AK is associated with a higher likelihood of progression to cSCC, particularly in common and hypertrophic types of AK (86). Additionally, strategies to target TP53 mutations include the development of small molecules that reactivate mutant p53 or prevent its degradation, thereby restoring its tumor suppressor function (89). These approaches are promising in enhancing the efficacy of existing treatments and overcoming resistance in TP53-mutated tumors (90). However, not all TP53 mutations have the same biological impact. The loss of E-cadherin and increased expression of p53 are associated with the progression of AK to cSCC (91). The loss of E-cadherin and increased p53 are associated with AK progression, but it is important to consider the broader context of these molecular changes (91). While TP53 mutations are a hallmark of AK and cSCC, their role extends beyond tumor suppression to influencing therapeutic outcomes. The development of targeted therapies that address the specific mutations and pathways involved in TP53 dysfunction holds promise for improving patient outcomes. However, the complexity and variability of TP53 mutations necessitate a nuanced approach to treatment and research.
4.2 NOTCH
The NOTCH signaling pathway serves as a convergence point for numerous essential signaling pathways and is one of the most critical pathways determining cell fate. Signal transduction between adjacent cells via NOTCH receptors regulates cell differentiation, proliferation, and apoptosis, playing a widespread role in the occurrence and development of malignant tumors. Loss-of-function mutations in NOTCH1, NOTCH2, and NOTCH3 are commonly found in cSCC and sun-exposed skin, including AK, which does not exhibit microscopically evident dysplasia (66). NOTCH1, NOTCH2, and NOTCH3 genes are located on chromosomes 9q34, 1p13-p11, 19p13.2-p13.1, respectively (92). NOTCH signaling maintains the homeostasis of keratinocytes, and in AK, NOTCH1 mutations can occur in extracellular or transmembrane domains, as well as in intracellular domains. These mutations can disrupt the cellular microenvironment signaling both inside and outside the cell, leading to a predisposition to cancer (93–97). A genomic profiling study of AK revealed that NOTCH1 and NOTCH2 are significantly mutated genes, with mutation levels positively correlated with UVR intensity (98, 99). This study also found that NOTCH1 mRNA is significantly upregulated in AK compared to healthy skin, suggesting that NOTCH1 may have a carcinogenic role in AK, contrary to its presumed tumor-suppressive role in cSCC, where NOTCH1 is typically inactivated (98, 100). Further research is needed to elucidate the distinct functional roles of NOTCH1 in AK and cSCC progression.
Approximately 40% of cSCC patients exhibit inactivating mutations in NOTCH1, making it the second most common mutated gene after TP53 (101, 102). Studies indicate upregulation of NOTCH1 expression in cSCC, suggesting that NOTCH1 may function as both a tumor suppressor and a facilitator, exerting both tumor-suppressive and tumorigenic effects in cSCC (101). NOTCH2, NOTCH3, and NOTCH4 may act as oncogenes in cSCC, with studies showing high expression of NOTCH2 and NOTCH3 in cSCC tumor tissues. Upregulation of NOTCH2 expression may promote tumor cell migration and invasion, while high expression of NOTCH3 may be positively correlated with tumor cell proliferation and volume (101). NOTCH4 is upregulated in cSCC through the NOTCH4/HEY1 pathway, inducing tumor cell proliferation, resistance to cisplatin, and promoting epithelial-to-mesenchymal transition (EMT) (103). The expression of NOTCH genes is associated with upstream gene TP53 and downstream regulatory factors TP63, TP21, caspase3, and the WNT/β-catenin pathway, although the specific mechanisms remain unclear (66, 101).
The clinical application of targeting the NOTCH gene in AK and cSCC involves understanding its dual role as both an oncogene and a tumor suppressor, depending on the cellular context (104). This dual role complicates the therapeutic targeting of NOTCH, as its inhibition could potentially have opposing effects depending on the specific cancer type and stage (105). Gamma-secretase inhibitors (GSIs) are a class of drugs that inhibit the activation of NOTCH receptors by preventing the cleavage of the NOTCH intracellular domain (106). Furthermore, given the complexity of NOTCH signaling, combination therapies that target multiple pathways are being investigated. For instance, combining NOTCH inhibitors with other signaling pathway inhibitors, such as those targeting the PI3K/Akt or VEGF pathways, may enhance therapeutic efficacy (105). These inhibitors are being explored in clinical trials for their potential to suppress tumor growth in cancers with aberrant NOTCH signaling (107). Nevertheless, inhibiting NOTCH signaling can have unintended consequences due to its role in normal cellular processes (104). Therefore, careful consideration of the potential side effects and the development of selective inhibitors are crucial for successful clinical application.
4.3 CDKN2A
The CDKN2A gene is situated on chromosome 9p21.3, a region known for its involvement in tumorigenesis across different cancer types (108). It encodes two alternatively spliced proteins, p16INK4a and p14ARF, which inhibit cell cycle progression and proliferation through the RB pathway and TP53 pathway, respectively (98, 109, 110). When CDKN2A is highly expressed, it inhibits the proliferation of tumor cells. However, mutations or inactivation of CDKN2A can diminish its ability to suppress tumor cells, leading to rapid proliferation and differentiation (111). Exome sequencing studies of AK have identified CDKN2A as a non-candidate driver gene (98). Previous research also suggests that CDKN2A does not mutate in normal skin and may serve as a gatekeeper in the AK-cSCC transition (112). Genomic studies of somatic mutations and copy number alterations in AK and cSCC have found that CDKN2A driver mutations are more common in cSCC (30.8%) than in AK (11.1%), indicating fewer genetic mutations in CDKN2A in AK. However, CDKN2A is located on chromosome band 9p21, one of the most common genomic alteration regions in human cancers (113). Loss of heterozygosity at 9p21, leading to CDKN2A mutations or homozygous deletion, results in loss of function, which may contribute to the AK-to-cSCC transition (109, 114). Mutation analysis of CDKN2A loci in 26 AK specimens revealed new mutations, additional mutations, and missense mutations, corresponding to four different nucleotide changes, three in exon 1a of p16INK4a and one in exon 2 (115).
CDKN2A is strongly associated with the occurrence of skin malignancies. As a tumor suppressor gene, CDKN2A negatively regulates the cell cycle and promotes cellular senescence. Knockout mouse experiments suggest that p14ARF may play a role in the initiation of cSCC development, while p16INK4a may be involved in determining the differentiation status of cSCC (109, 116). Recurrent CDKN2A mutations have been found in skin malignancies, and inactivation of CDKN2A at the locus may be due to allelic loss, point mutations, and promoter hypermethylation. Inactivation of the CDKN2A locus leads to loss of control over the cell cycle and cell growth (117). Heterozygous loss or point mutations have been detected in 21% to 62% of cSCC cases, and promoter hypermethylation has been found in 35% to 78% of cases. Recently, immunohistochemical analysis of cSCC precursor lesions revealed a linear correlation between the immunostaining of p16INK4a and the membranous immunostaining of β-catenin in AK, indicating the importance of p16INK4a in the progression of cSCC precursor lesions (118).
CDKN2A mutation and expression levels serve as valuable prognostic markers and potential therapeutic targets. CDKN2A mutations are significantly associated with disease-specific death in metastatic cSCC, indicating its potential as a prognostic biomarker. Patients with CDKN2A mutations exhibit shorter disease-specific survival compared to those without such mutations (119). In cSCC, CDKN2A mutations are early genetic events, often occurring alongside TP53 mutations (119). These mutations are present in both primary tumors and metastases, suggesting their role in the early stages of tumorigenesis. The presence of CDKN2A mutations correlates with adverse clinical outcomes, such as increased tumor size and invasion depth, which are significant predictors of poor survival in cSCC patients (119). In the context of CDKN2A-associated therapeutic approaches, CDKN2A expression is linked to immune cell infiltration and immune regulation pathways, which could influence the tumor microenvironment and response to immunotherapy (120). The gene’s role in immune regulation and its association with immune checkpoints like PD-1 suggest that CDKN2A could guide immunotherapy strategies, potentially improving responses in cancers where it is highly expressed (121). The gene’s ability to influence tumor progression and response to therapies highlights its importance as a therapeutic target and biomarker. However, further research is needed to explore its specific applications in AK and cSCC, considering the unique pathophysiology of these skin conditions.
4.4 RET
RET (Rearranged during transfection) is located on chromosome 10q11.2 and is involved in the signaling pathways that regulate cell proliferation, differentiation, and migration (122). Through different splicing mechanisms, RET can undergo 3′-end cleavage to form various receptor tyrosine kinases, including RET9, RET43, and RET51, which constitute the single-pass transmembrane tyrosine kinase receptor protein RET family. These isoforms exhibit tissue-specific expression (123). The exact role of RET in AK and cSCC remains unclear. It is activated by binding with soluble glial cell-derived neurotrophic factor family ligand proteins, leading to the production of RET proteins with abnormal activity. These aberrant signals may influence processes such as cell growth, survival, invasion, and metastasis. In AK, RET is significantly expressed and can drive cSCC, and the activation mechanism of RET is associated with the loss of MAB21L4 (124). MAB21L4, also known as Male Abnormal 21-like 4 or C2orf54, plays a role in differentiating human progenitor epidermal keratinocytes (125). MAB21L4 is considered a bridging protein between CACYBP and RET. In the presence of MAB21L4, the binding affinity between CACYBP and RET increases by 44 times. CACYBP can bind to the E3 ubiquitin ligase SIAH1 and participate in various cellular processes, including cell differentiation, proliferation, cytoskeletal dynamics, and tumorigenesis (126). High expression of RET in AK is caused by the loss of MAB21L4, which is an early event in AK. This loss disrupts the proximity of RET to the CACYBP-SIAH1 complex, preventing RET ubiquitination (124).
While the primary focus of RET inhibitors has been on lung and thyroid cancers, their application in skin cancers like AK and cSCC is an area of interest. The application of RET inhibitors in treating these cancers has evolved significantly, with a focus on developing selective inhibitors to improve efficacy and reduce side effects. This approach is particularly relevant in the context of AK and cSCC, where targeted therapies are increasingly being explored (127). The development of selective RET inhibitors like selpercatinib and pralsetinib has marked a significant advancement in targeting RET-driven cancers (128). These inhibitors have shown high efficacy and favorable toxicity profiles compared to earlier multikinase inhibitors (MKIs), which had off-target effects and limited clinical activity (129). Selpercatinib and pralsetinib have been approved by the FDA for treating RET-altered cancers, demonstrating high response rates in clinical trials (130). These inhibitors are effective against RET gatekeeper mutations, which are common resistance mechanisms in cancer therapy (131). The exploration of RET inhibitors in skin cancers is still in its early stages. However, the success of these inhibitors in other cancer types suggests potential applicability, especially in cases where RET alterations are identified (132).
4.5 OVOL1/2
OVOL1 and OVOL2 (OVOL1/2) belong to the OVO gene family and are located on chromosomes 11 and 20, encoding a class of DNA-binding proteins that typically function as transcription factors regulating gene expression, acting as both transcriptional activators and repressors (84, 133–136). Three members of the OVO gene family have been identified: OVOL1, OVOL2, and OVOL3 (137). Among them, (OVOL1/2) have been studied most extensively. OVOL1/2 participate in suppressing EMT in tumor cells (133). They can inhibit the expression of Zinc finger E-box binding homeobox 1 (ZEB1) in tumor cells (138). OVOL1 levels are significantly elevated in non-metastatic oral squamous cell carcinoma tissue and negatively correlated with ZEB1 levels. OVOL2 is significantly downregulated in metastatic nasopharyngeal carcinoma cells, which exhibit strong invasive capabilities. Knockdown of ZEB1 can reduce the invasion capability of these cells (139). In 30 AK and 30 cSCC cells, knocking down OVOL1/2 increased the mRNA expression of ZEB1 and WAVE proteins. Specifically, knocking down OVOL1 significantly increased the expression of ZEB1 and WAVE proteins, while knocking down OVOL2 significantly increased the expression of ZEB1. Loss of OVOL2 significantly enhanced the invasion capability of cSCC cells (140). Immunohistochemical analysis of AK and cSCC cells showed that the expression of OVOL1 and OVOL2 was upregulated in AK but significantly downregulated in cSCC. Conversely, ZEB1 and WAVE proteins were upregulated in cSCC, indicating that the downregulation of OVOL1/2 and the upregulation of ZEB1 and WAVE proteins may be associated with the progression of AK to cSCC (140).
The application of OVOL1 and OVOL2 genes in the treatment of AK and cSCC is a promising area of research due to their roles in modulating EMT. This makes them attractive targets for therapeutic strategies aimed at halting or reversing the progression of these skin lesions. Targeting the OVOL2/ZEB1 axis could be a viable therapeutic approach to prevent the development of cSCC from AK. By inhibiting ZEB1, OVOL2 can potentially reduce the invasiveness of cSCC cells, making it a promising target for drug development (140). While the potential of OVOL1 and OVOL2 in treating AK and cSCC is promising, it is important to consider the complexity of cancer progression and the multitude of genetic and environmental factors involved. Therefore, while targeting OVOL1 and OVOL2 could be beneficial, a comprehensive approach that considers these other factors may be necessary for effective treatment.
4.6 KNSTRN
KNSTRN is a protein that binds to Astrin on the kinetochore in the late stage of cell mitosis and is closely related to cell division (141). The KNSTRN gene is located on chromosome 15 at the 15q15.1 locus. It encodes a protein that is essential for proper chromosome alignment and segregation during cell division, particularly mitosis (142). KNSTRN is believed to play a role in cell division and the cell cycle process. Recurrent mutations were detected in 19% of AK and cSCC, mainly concentrated in KNSTRN mutations (143). Exome sequencing of 112 cSCC genes showed that the mutation sites were located at the N-terminus. 21 out of 112 (18.7%) patients had somatic mutations in KNSTRN, with over half of the mutations occurring at codon 24, resulting in a change from serine to phenylalanine due to a C-T transition (143). Detection of the frequency of KNSTRN gene mutations in healthy skin, photo-damaged skin, AK, and invasive cSCC with different histological grades showed that recurrent somatic mutations in KNSTRN p.Ala40Glu were associated with basal proliferative AK in invasive squamous cell carcinoma (144). These studies also suggest that KNSTRN may play a driving role in the occurrence and development of cSCC and could be a useful predictor for cSCC development.
The KNSTRN gene has emerged as a significant player in the development and progression of AK and cSCC. Mutations in this gene, particularly the recurrent somatic mutations, have been linked to the acceleration of cSCC development and are associated with different histological classifications of AK. These findings suggest that targeting the KNSTRN gene could be a promising strategy for treating these skin conditions. Given the role of KNSTRN mutations in cSCC progression, therapies targeting these mutations could potentially halt or reverse the disease process. This could involve the development of small molecules or biologics that specifically inhibit the mutated forms of the KNSTRN protein (140). Techniques such as CRISPR-Cas9 could be employed to correct KNSTRN mutations in affected cells, potentially preventing the progression of AK to cSCC (140). While KNSTRN mutations are prevalent in cSCC, they are not exclusive to this cancer type, as they have also been found in other cutaneous tumors like malignant melanomas (140). This necessitates a careful approach to ensure that therapies targeting KNSTRN mutations do not adversely affect normal tissues.
4.7 CERKL
The equations should be inserted in editable format from the equation editor. Ceramide kinase-like (CERKL) is situated on chromosome 2q31.3, and it is an oxidative stress-related protein whose expression is typically limited to the retina, nervous tissue, kidneys, trachea, testes, and lungs (145, 146). Levels of CERKL in normal skin are relatively low, but CERKL mRNA and protein are highly expressed in AK and cSCC (147). Knocking down CERKL in cSCC cells reduces the content of cell sheath lipids and enhances the sensitivity of cSCC cells to oxidative stress, indicating that CERKL may protect cSCC cells through an oxidative stress protection mechanism (147). The mechanism of oxidative stress protection mediated by CERKL is currently unclear but it may involve CERKL-mediated regulation of cellular autophagy (148). The mechanisms of CERKL in AK and cSCC require further investigation.
Studies have identified various genes involved in cSCC progression, including CERKL, which could serve as biomarkers for early diagnosis and treatment strategies (149). CERKL is highly expressed in cSCC and AK compared to normal epidermis. It plays a role in maintaining cellular sphingolipids and protecting cells from oxidative stress, which are critical in the survival and proliferation of cancer cells (149). By enhancing resistance to oxidative stress, CERKL may contribute to the survival of cancer cells under hostile conditions, making it a potential target for therapies aimed at increasing cancer cell susceptibility to oxidative damage (149). While CERKL presents a promising target for cSCC and AK treatment, it is essential to consider the complex genetic and molecular landscape of these conditions.
4.8 TLR4
Toll-like receptor 4 (TLR4) is located on chromosome 9q in humans, and it also is a receptor protein in the human immune system (150). It belongs to the Toll-like receptors (TLRs) family and plays a major role in regulating immune responses and inflammatory responses. TLR4 protein is mainly expressed on the surface of immune cells such as macrophages, dendritic cells, and certain epithelial cells. Its main function is to recognize and respond to pathogen-associated molecular patterns (PAMPs) from exogenous pathogens. TLR4 binds to these PAMPs and activates immune cells, triggering a series of immune responses, including the production of cytokines, activation of the NF-κB signaling pathway, and promotion of antibacterial and antiviral immune responses. TLR4 is expressed in human skin, AK, and cSCC (151). TLR4 signal transduction promotes cSCC development in a MyD88-dependent manner and is essential for inflammatory cell recruitment during carcinogenesis (152). TLR4 may play a tumor-promoting role in cSCC, and immunohistochemical detection shows that the expression of TLR4 in cSCC is significantly higher than that in AK tissue, and its expression in low-differentiated cSCC is significantly lower than that in moderately differentiated and highly differentiated cSCC (153).
The clinical application of the TLR4 gene in AK and cSCC involves understanding its role in tumor progression and immune response modulation. TLR4 antagonists, such as eritoran and resatorvid, have been identified as potential therapeutics. These agents can suppress UV-induced inflammatory signaling, which is crucial in preventing skin photodamage and photocarcinogenesis (154). On top of that, TLR agonists have been used in immunotherapy for skin cancers, including melanoma and basal cell carcinoma, by enhancing dendritic cell recruitment and T-cell responses (155). This approach could be adapted for cSCC, targeting TLR4 to modulate immune responses (156).
4.9 C1r, C1s, and factor D
C1r and C1s are two important components of the complement system, which belong to the C1 complex. Both C1r and C1s genes are located on chromosome 12, specifically in the region 12p13 (157). The C1 complex is the initial enzyme complex of the complement system and participates in the regulation of immune responses and inflammatory reactions. C1r and C1s are both enzyme proteins that play a key role in the C1 complex. C1r plays the role of an initiator enzyme in the activation of the C1 complex. When C1r is activated, it catalyzes the activation of C1s (158). C1s, on the other hand, is responsible for further activating other components of the complement system, such as C4 and C2 (159). Compared with normal epidermal keratinocytes, the mRNA levels and production of C1r and C1s are significantly increased in cSCC cells (160). Reducing the expression of C1r and C1s in cSCC cells suppressed the activation of extracellular signal-regulated kinase 1/2 and Akt, promoting cSCC cell apoptosis and significantly inhibiting the growth and angiogenesis of human cSCC xenografts (158). Furthermore, C1r and C1s were highly expressed in aggressive disseminated cSCC and cSCC associated with pemphigus vulgaris, while they were expressed less in in situ cSCC, AK, and normal skin (160).
Given their role in promoting cSCC growth, C1r and C1s represent potential targets for clinical application therapeutic intervention. As markers of aggressive tumor behavior, C1r and C1s could help predict the likelihood of progression from AK to invasive cSCC, aiding in risk stratification and management decisions (160). Inhibitors or monoclonal antibodies against these proteins could be developed to suppress tumor growth and improve patient outcomes (160). The integration of C1r and C1s targeting into clinical practice will require further validation in larger cohorts and the development of specific inhibitors.
Complement factor D(FD) cleaves C3 to produce the active fragments C3a and C3b. C3a can induce inflammatory reactions, causing vasodilation and promoting the infiltration of inflammatory cells (161–163). In culture and in vivo, cSCC cells express FD significantly. The in vivo FD expression level in non-metastatic cSCC, metastatic cSCC, cSCC metastasis, and pemphigus vulgaris-related cSCC is significantly higher than that in normal skin, precancerous lesions, AK, and in situ cSCC. The strongest FD expression is limited to the invasive edge of the tumor, while the weak FD staining pattern in normal skin, AK, and cSCC in situ shows a diffuse and uniform distribution (164).
FD’s role in the alternative complement pathway makes it a promising target for therapeutic intervention. The small-molecule FD inhibitor Danicopan has been proposed as a specific drug candidate for advanced cSCC therapy (164). Inhibition of FD activity has been shown to attenuate cSCC cell proliferation, suggesting that targeting FD could be an effective strategy for managing cSCC progression (164). While the role of FD is promising in the treatment of cSCC, further research is needed to fully elucidate the complex interactions between these factors and to validate FD’s clinical utility in larger, diverse patient populations.
5 Nomenclature established progressed-associated non-coding RNAs from AK to cSCC
RNA, as an integral part of gene regulation, has garnered widespread attention, with non-coding RNA (ncRNA) emerging as a focal point of research in recent years. Variants such as long non-coding RNA (lncRNA) and microRNA (miRNA) have been particularly emphasized. The precise mechanisms and specific targets of ncRNA are still actively under investigation, as they serve as potential regulatory molecules with significant roles in skin cancer. Further research endeavors will contribute to a better understanding of the exact functions of RNA molecules in the progression from AK to cSCC and their potential clinical applications.
5.1 Long non-coding RNA
Long non-coding RNA (lncRNA) refers to a group of endogenous RNAs with lengths exceeding 200 nucleotides, lacking complete open reading frames, and thus not encoding proteins (165). However, lncRNA can interact with DNA, RNA, and proteins through various mechanisms, participating in related biological processes such as serving as protein scaffolds, directly interacting with proteins and RNA (166), participating in chromatin modification, regulating mRNA, transcription factors, and epigenetics, and acting as competing endogenous RNA for microRNA (167). A study employing RNA sequencing of two cases of AK and three cases of normal skin tissue identified lncRNA uc011fnr.2 negatively regulating SCIMP and TLR4, and being involved in activating the JAK-STAT3 signaling pathway in AK (168). Additionally, the dysregulation of lncRNAs in premalignant lesions like actinic keratosis (AK) suggests their involvement in the progression to invasive cSCC (168).
5.2 miRNA
miRNAs are short non-coding RNAs that regulate protein-coding gene expression at the post-transcriptional level. miRNAs play important roles in the initiation and progression of tumorigenesis by regulating the expression of tumor suppressor genes and oncogenes (169–171). The most significant changes in miRNA expression occur during the transition from “early” stages of normal epidermis, AK, and early photodamaged epidermis to the “late” stages of epidermal carcinogenesis, late-stage AK, and cSCC (172). By analyzing the differential expression of genes in 10 AK and 30 cSCC tissue samples, using microarray dataset GSE45216 obtained from the gene expression database, they identified 16 miRNAs and found that three co-expressed network modules EIF4EBP1, SNX17, PRPF4, NXT1, and UBA5 may be pathogenic genes contributing to the development of AK into cSCC (173). Recent studies have also demonstrated that miRNA regulation plays a critical role in the progression of epidermal keratinocytic neoplasms (174, 175).
miR-204 primarily functions as a tumor suppressor gene. It has been shown that DNA methylation in the upstream promoter region of TRPM3, a gene upstream of miR-204, is one of the mechanisms contributing to the silencing of miR-204 in cSCC (176). Comparison of miR-204 expression between cSCC, AK, and healthy skin tissues, revealed significantly downregulated miR-204 in cSCC. Overexpression of miR-204 inhibits the activation and translocation of transcription activator 3, thereby suppressing the development of cSCC. Protein tyrosine phosphatase non-receptor type 11 (PTPN11) has been identified as a direct target of miR-204 (176). miR-23b is downregulated in both AK and cSCC, and its expression is regulated by the MAPK signaling pathway. miR-23b reduces the expression of FGF2 at both mRNA and protein levels, impairing the ability of cSCC cells to induce angiogenesis. Overexpression of miR-23b inhibits colony and sphere formation in cSCC cells, while CRISPR/Cas-9-mediated deletion of miR-23b leads to increased colony and tumor sphere formation in vitro (177). Experimental studies have confirmed the overexpression of Ras-related protein RRAS2 in cSCC, and the interference of RRAS2 expression impairs angiogenesis, colony formation, and tumor sphere formation. Importantly, RRAS2 is a direct target of miR-23b in cSCC (177). miR-181a acts as a transcriptional driver factor in the progression from AK to cSCC. miR-181a promotes the formation of various proteins by targeting the TGFβ receptor 3 (TGFβ R3) in the TGFβ signaling pathway. In cSCC, the expression level of miR-181a is inversely correlated with the expression level of TGFβ R3. Overexpression of miR-181a or knockdown of TGFβ R3 inhibits apoptosis and enhances cell survival. Moreover, miR-181a enhances cell migration and invasion and upregulates epithelial-mesenchymal transition markers. miR-181a regulates these processes by directly interacting with TGFβ R3. Experimental results have demonstrated that miR-181a inhibits tumor growth, and reversing these effects can be achieved by increasing or decreasing TGFβ R3 expression (178). Additionally, a study has shown the involvement of miRNA-31 and MMP-1 in differentiation pathways associated with increased EMT and angiogenesis, which are important mechanisms in cSCC invasion. EMT is enhanced in AK (179). These findings hold significant implications for future research on targeted therapies.
6 Application of clinical drugs in AK and cSCC
The treatment of AK and cSCC involves a variety of clinical drugs and therapeutic approaches. These conditions, which are closely related, often require a combination of topical, systemic, and procedural treatments to manage effectively. The choice of treatment depends on factors such as the severity of the lesions, patient characteristics, and the potential for malignant transformation. Below is an overview of the current clinical drugs used in the management of AK and cSCC (Figure 3).
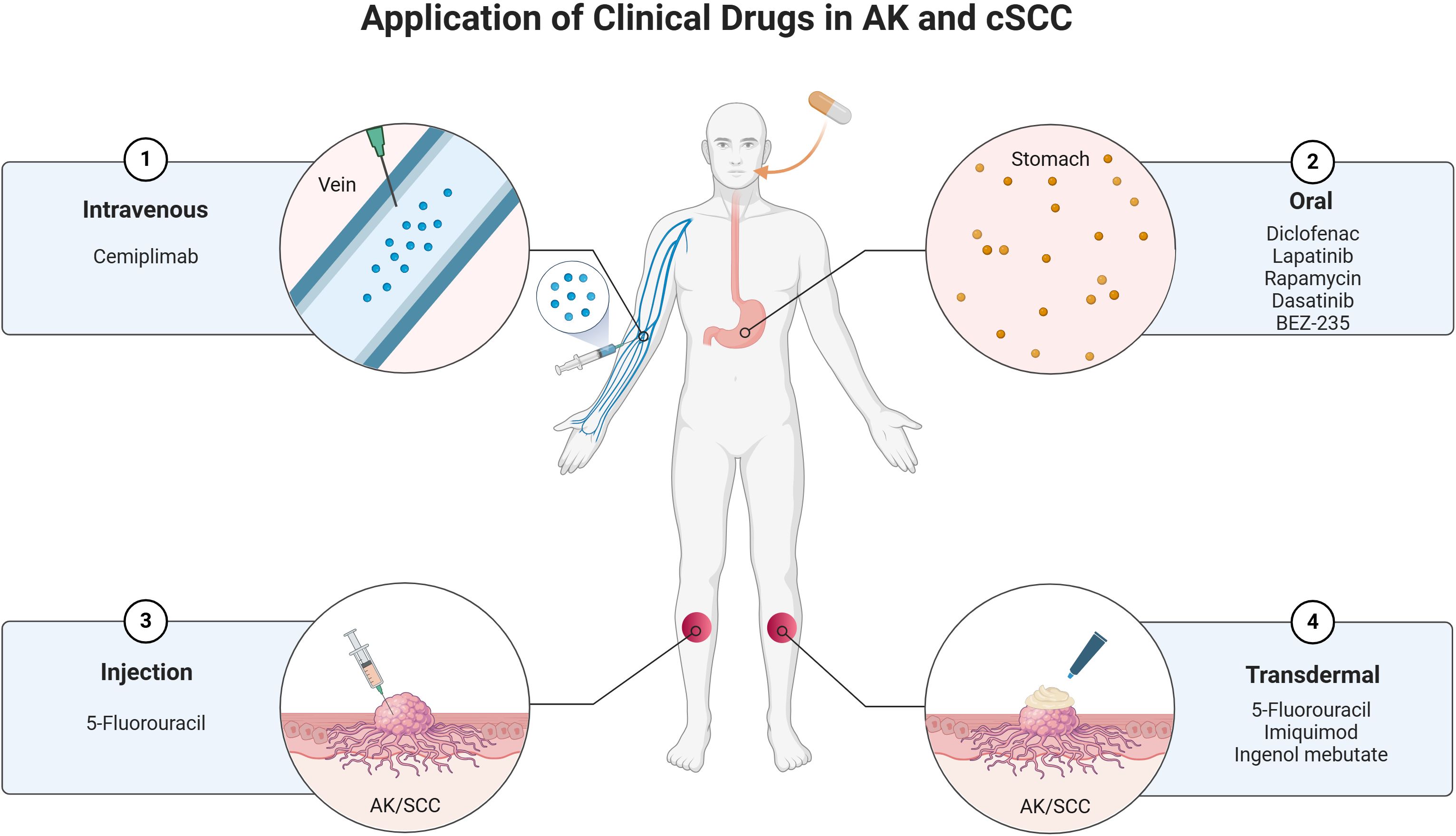
Figure 3. Current clinical drugs used in the management of AK and cSCC.
6.1 Topical treatments
6.1.1 5-fluorouracil
This is a widely used topical chemotherapeutic agent for AK and cSCC (180). A novel 4% formulation of 5-FU cream has shown high efficacy in treating multiple AKs, with significant reductions in the Actinic Keratosis Area and Severity Index (AKASI) scores observed at 6 and 12 weeks post-treatment (181). It works by inhibiting DNA synthesis in rapidly dividing cells, leading to the eradication of AK and cSCC lesions (182). Combining 5-FU with photodynamic therapy (PDT) has been shown to enhance lesion clearance rates (183). Pretreatment with 5-FU increases protoporphyrin IX accumulation, improving the efficacy of PDT in AK treatment (183). Unlike its use in the treatment of AK, 5-FU has alternative approaches for managing cSCC. Intralesional 5-FU at lower concentrations (10.0 mg/mL and 16.7 mg/mL) has been effective in treating cSCC and keratoacanthomas, achieving a 96% lesion clearance rate with minimal adverse effects (184). This approach strategically reduces the cumulative dose of 5-FU, thereby mitigating potential toxicities without compromising therapeutic efficacy. Furthermore, a combinative treatment strategy involving debulking surgery followed by intralesional 5-FU has evinced robust tumor clearance rates, particularly in instances of well-differentiated SCCs (185). This integrated method has proven especially efficacious for managing tumors located on the trunk and extremities, offering a tailored and effective treatment modality for such cases. While 5-FU has proven effective in treating AK and cSCC, its application requires careful consideration. The potential for combination therapies, such as with PDT, offers promising avenues for enhanced treatment outcomes. However, the variability in response rates and the need for individualized treatment plans underscore the complexity of managing these conditions. Further research is needed to optimize 5-FU use across diverse patient groups and to explore its full potential in preventing progression to invasive cSCC.
6.1.2 Imiquimod
Imiquimod is widely used for AK treatment due to its ability to modulate the immune system, leading to the clearance of lesions. This immune response modifier is used in 5% cream form for AK treatment (186). It works by inducing cytokine production, which promotes apoptosis in abnormal keratinocytes (187). Studies have shown clearance rates ranging from 66% to 87% when used as monotherapy (188). However, combining imiquimod with other agents like 5-fluorouracil has been explored to enhance efficacy (188). This combination has shown higher clearance rates and shorter treatment durations, suggesting a synergistic effect (188). In the treatment of cSCC, imiquimod has been used off-label for treating cSCC, particularly in cases where surgery or radiation is not feasible (189). A notable case involved the successful treatment of a locally advanced cSCC of the eyelid with imiquimod 3.75%, resulting in complete tumor regression without significant side effects (189). While imiquimod has demonstrated efficacy in treating AK and cSCC, its application is not without challenges. The variability in patient response, potential for local skin reactions, and the need for further studies to confirm long-term outcomes and optimal treatment regimens are areas that require attention. Overall, imiquimod remains a valuable tool in dermatological oncology, with ongoing research likely to expand its utility and effectiveness.
6.1.3 Diclofenac
Diclofenac, a non-steroidal anti-inflammatory drug (NSAID), has been explored for its application in treating AK and cSCC. Diclofenac’s mechanism involves the inhibition of cyclooxygenase-2 (COX-2), reducing prostaglandin E2 levels, which are implicated in tumorigenesis and inflammation associated with AK (190). And the drug also induces apoptosis and alters cell cycle profiles in cancerous cells, potentially through p53 induction and metabolic alterations, as observed in esophageal squamous cell carcinoma models (191). Diclofenac 3% gel, often combined with hyaluronic acid, is a topical treatment for AK. It has been shown to be effective, with a resolution of at least 50% of lesions in 78% of patients, and a high satisfaction rate of 85% (192). But in a comparative study, diclofenac was less effective than imiquimod in preventing the progression of AK to more severe forms or invasive SCC over a three-year period (192) While diclofenac offers a viable option for treating AK and potentially cSCC, its role is often complementary to other therapies. Its ability to modulate immune responses and cellular metabolism presents a unique advantage, but its efficacy in preventing progression to invasive SCC is less than some alternatives. Future research may focus on optimizing its use in combination therapies and exploring its full potential in cancer prevention and treatment.
6.1.4 Ingenol mebutate
Ingenol mebutate is FDA-approved for treating AK, a precursor to cSCC (193). It induces cell death through the PKC/MEK/ERK signaling pathway, leading to reduced cell viability and proliferation in keratinocyte-derived cells (194). Despite its efficacy, there are concerns about an increased risk of SCC in patients treated with ingenol mebutate for AK, as indicated by pharmacovigilance studies (195). Ingenol mebutate exhibits diverse therapeutic applications; however, its association with an increased risk of SCC and adverse reactions in certain treatments underscores the necessity for cautious application and further research to ensure safe and effective use.
6.2 Systemic treatments
6.2.1 Cemiplimab
Cemiplimab, a monoclonal antibody targeting the programmed cell death protein 1 (PD-1), has demonstrated high efficacy in treating advanced cSCC, with objective response rates (ORR) ranging from 32% to 77% in various studies (196). In a phase II trial, cemiplimab showed a 62% ORR with a complete response in 22% of patients (197). Real-world data indicate a clinical benefit rate of 54.3% and a median progression-free survival of 12.2 months (198). While cemiplimab has shown significant efficacy in treating advanced cSCC, its application in AK is less documented in the provided contexts. The focus remains on cSCC, where cemiplimab has established itself as a critical therapeutic option.
6.2.2 Lapatinib
Lapatinib, a tyrosine kinase inhibitor, has shown potential in the treatment of cutaneous squamous cell carcinoma cSCC and AK by targeting the EGFR and HER2 pathways (199). It disrupts the PI3K/AKT/mTOR signaling pathway, leading to cell cycle arrest in the G2/M phase and suppression of EMT in cSCC cells (200). A phase 2 clinical trial demonstrated that short-term lapatinib treatment resulted in tumor size reduction in 2 out of 8 patients with cSCC (199). Additionally, a mean regression of 30% in AK lesions was observed after 56 days of treatment (199). While lapatinib shows promise in treating cSCC and AK, its efficacy as a monotherapy is limited, and side effects such as hepatotoxicity pose challenges.
6.2.3 mTOR inhibitors
mTOR inhibitors like rapamycin work by blocking the mTOR pathway, which is crucial for cell growth and proliferation (201). This blockade leads to decreased phosphorylation of mTOR targets, resulting in apoptotic death of cancer cells and reduced growth and metabolic activity of surviving cells (201). Inhibition of mTOR also reduces the expression of mutant p53, a common mutation in SCC, further contributing to the reduction of tumor growth (201). Oral administration of mTOR inhibitors in UVB-induced mouse models has been effective in reducing the number and size of AK and cSCC lesions, indicating their potential in both treatment and prevention of these conditions (202). While mTOR inhibitors show significant promise in the treatment and prevention of AK and cSCC, there are challenges and considerations to address. Additionally, while preclinical models provide valuable insights, clinical trials are essential to confirm efficacy and safety in humans. The potential for topical formulations of mTOR inhibitors could mitigate systemic side effects and offer a more targeted approach, but further research is needed to optimize these treatments (203).
6.3 Emerging therapies
Agents like dasatinib and BEZ-235, these small molecule kinase inhibitors have shown efficacy in inducing regression of cSCC with minimal inflammation and side effects (180). They target kinases that promote keratinocyte growth, offering a promising therapeutic approach for AK and cSCC (180). The development of topical kinase inhibitors represents a significant advancement in dermatological therapeutics, particularly for conditions like AK and cSCC. These inhibitors provide a targeted approach that minimizes systemic exposure and associated risks, making them a valuable addition to the treatment landscape. However, it is important to consider the potential for adverse effects and the need for further research to optimize their use in clinical practice.
7 Current limitations and future perspectives
The progression from AK to cSCC is a multifactorial process influenced by genetic, environmental, and molecular mechanisms, with approximately in the progression from AK to cSCC, a comprehensive understanding of the precise molecular mechanisms remains limited, largely due to the complex interplay between genetic, epigenetic, and environmental factors. The identification of potential biomarkers for early detection and prognosis of cSCC remains a challenge, with many findings yet to be validated in clinical settings. Current therapeutic interventions for AK and cSCC are limited, with few targeted therapies available, particularly due to the heterogeneity of genetic mutations in key genes such as TP53 and NOTCH. Additionally, there is a scarcity of clinical trials focusing on early intervention in AK to prevent its progression to cSCC, with most trials concentrated on advanced stages of cSCC. The significant variability in progression rates among individuals, influenced by genetic predisposition, immune status, and environmental factors, further complicates the establishment of standardized treatment protocols. Integrating multi-omics data (e.g., genomics, transcriptomics, and proteomics) into a coherent framework to understand the progression from AK to cSCC remains a formidable challenge.
In the future, multi-omics approaches, integrating genomics, transcriptomics, and proteomics, can reveal complex molecular mechanisms and identify potential biomarkers for AK. This can aid in understanding the progression from AK to SCC and in developing targeted interventions (204). By analyzing the unique molecular profile of each patient, multi-omics can facilitate personalized treatment plans, potentially improving efficacy and reducing adverse effects. Implementing personalized treatment strategies based on patient genetic profiling may enhance therapeutic efficacy. There is an urgent need for more clinical trials focusing on the early stages of AK and evaluating the effectiveness of preventive strategies, including combination therapies targeting multiple pathways simultaneously. Public health initiatives should promote awareness and preventive measures against UV exposure to reduce the incidence of AK and cSCC, emphasizing education about sun protection and early skin assessments for early detection. Furthermore, further studies on the roles of non-coding RNAs, such as lncRNAs and miRNAs, in the progression from AK to cSCC may reveal new regulatory mechanisms and potential therapeutic targets.
8 Discussion
The progression from AK to cSCC is a multifactorial process influenced by genetic, environmental, and molecular mechanisms, with approximately 70% of cSCC cases originating from AK (205). Key drivers include chronic UVR exposure, which induces TP53 mutations, a frequent early event in the AK-to-cSCC pathway (206). TP53 mutations disrupt cell cycle regulation, DNA repair, and apoptosis, contributing to genomic instability and abnormal keratinocyte proliferation (207). Identifying TP53 mutations in normal skin exposed to UVR indicates their potential as biomarkers for early risk stratification and targeted prevention strategies, emphasizing the importance of genetic screening and early intervention to reduce cSCC burden (208).
In addition to TP53, genes such as NOTCH, CDKN2A, and RET play critical roles in the AK-to-cSCC transition. The NOTCH pathway, involved in cell differentiation and proliferation, exhibits dual functions as both a tumor suppressor and promoter in AK and cSCC, with mutations disrupting keratinocyte homeostasis, offering therapeutic potential (209). CDKN2A mutations contribute to the loss of cell cycle control, underscoring the importance of genomic integrity (210). Furthermore, dysregulation of lncRNAs and miRNAs influences gene expression and cellular behavior, with miR-204 acting as a tumor suppressor and miR-181a promoting survival and invasion, highlighting their roles as therapeutic targets in cSCC progression (176, 178).
Moreover, current treatments for AK and cSCC, such as topical agents like 5-fluorouracil and imiquimod, exhibit varied efficacy. Incorporating molecular markers and genetic profiling into clinical practice could optimize therapeutic selection and enhance patient outcomes, with specific TP53 mutations potentially benefiting from targeted therapies addressing underlying genetic alterations. Additionally, emerging systemic treatments like cemiplimab and mTOR inhibitors show promise in advanced cSCC, while topical kinase inhibitors provide a novel, minimally systemic approach to treating both AK and cSCC, potentially reducing adverse effects of traditional therapies.
Despite the progress made in understanding the molecular mechanisms underlying the transition from AK to cSCC, several challenges remain. The heterogeneity of tumors, variations in individual responses to UVR, and the influence of host factors complicate the development of universal treatment strategies. Moreover, the potential for resistance to therapies targeting specific mutations highlights the need for personalized approaches in managing patients with AK and cSCC.
9 Conclusion
cSCC remains a major public health challenge, and the progression from AK to cSCC in normal skin is still not well understood. However, the progression of AK to cSCC represents an example of the transformation of a precursor lesion into an aggressive malignancy. The comprehensive review of the development mechanisms from AK to cSCC has provided valuable insights into the complex processes underlying this progression. By deepening our knowledge and understanding of the underlying mechanisms, we can advance the development of targeted prevention strategies, early detection methods, and personalized therapeutic approaches. Ultimately, these efforts will contribute to reducing the incidence, improving prognoses, and enhancing the overall management of cSCC.
Author contributions
ZL: Data curation, Investigation, Writing – original draft, Writing – review & editing. FL: Software, Writing – review & editing. FZ: Data curation, Writing – review & editing. DS: Investigation, Writing – review & editing. LC: Investigation, Writing – review & editing. WL: Funding acquisition, Resources, Writing – review & editing. GY: Funding acquisition, Resources, Software, Supervision, Writing – original draft, Writing – review & editing. GZ: Formal analysis, Funding acquisition, Methodology, Project administration, Supervision, Writing – review & editing, Writing – original draft.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This research was funded by the National Natural Science Foundation of China, grant number 82303576, the Research Project of the Health Commission of Hunan Province, grant number B202304128140, and Sponsored by Science Foundation of Hunan Aerospace Hospital, grant number 2022yj01.
Acknowledgments
This is a short text to acknowledge the contributions of specific colleagues, institutions, or agencies that aided the efforts of the authors.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Fania L, Didona D, Di Pietro FR, Verkhovskaia S, Morese R, Paolino G, et al. Cutaneous squamous cell carcinoma: From pathophysiology to novel therapeutic approaches. Biomedicines. (2021) 9:171. doi: 10.3390/biomedicines9020171
2. Jiang R, Fritz M and Que SKT. Cutaneous squamous cell carcinoma: An updated review. Cancers (Basel). (2024) 16:1800. doi: 10.3390/cancers16101800
3. Rogers HW, Weinstock MA, Feldman SR and Coldiron BM. Incidence estimate of nonmelanoma skin cancer (keratinocyte carcinomas) in the u.S. Population, 2012. JAMA Dermatol. (2015) 151:1081–6. doi: 10.1001/jamadermatol.2015.1187
4. Karia PS, Han J and Schmults CD. Cutaneous squamous cell carcinoma: Estimated incidence of disease, nodal metastasis, and deaths from disease in the United States, 2012. J Am Acad Dermatol. (2013) 68:957–66. doi: 10.1016/j.jaad.2012.11.037
5. Gao TW, Sun DJ, Li CY, He H, Li Q, Liu YS, et al. retrospective analysis of 1905 patients with skin cancer from two general hospitals in western China from 1981 to 2000. Beijing Da Xue Xue Bao Yi Xue Ban. (2004) 36:469–72.
6. Stratigos AJ, Garbe C, Dessinioti C, Lebbe C, Bataille V, Bastholt L, et al. European interdisciplinary guideline on invasive squamous cell carcinoma of the skin: Part 1. Epidemiology, diagnostics and prevention. Eur J Cancer. (2020) 128:60–82. doi: 10.1016/j.ejca.2020.01.007
7. Schmults CD, Karia PS, Carter JB, Han J and Qureshi AA. Factors predictive of recurrence and death from cutaneous squamous cell carcinoma: A 10-year, single-institution cohort study. JAMA Dermatol. (2013) 149:541–7. doi: 10.1001/jamadermatol.2013.2139
8. Rees JR, Zens MS, Celaya MO, Riddle BL, Karagas MR and Peacock JL. Survival after squamous cell and basal cell carcinoma of the skin: A retrospective cohort analysis. Int J Cancer. (2015) 137:878–84. doi: 10.1002/ijc.29436
9. Werner RN, Sammain A, Erdmann R, Hartmann V, Stockfleth E and Nast A. The natural history of actinic keratosis: A systematic review. Br J Dermatol. (2013) 169:502–18. doi: 10.1111/bjd.12420
10. Criscione VD, Weinstock MA, Naylor MF, Luque C, Eide MJ, Bingham SF, et al. Actinic keratoses: Natural history and risk of Malignant transformation in the veterans affairs topical tretinoin chemoprevention trial. Cancer. (2009) 115:2523–30. doi: 10.1002/cncr.24284
11. Dreno B, Amici JM, Basset-Seguin N, Cribier B, Claudel JP, Richard MA, et al. Management of actinic keratosis: A practical report and treatment algorithm from akteam expert clinicians. J Eur Acad Dermatol Venereol. (2014) 28:1141–9. doi: 10.1111/jdv.12434
12. Chetty P, Choi F and Mitchell T. Primary care review of actinic keratosis and its therapeutic options: A global perspective. Dermatol Ther (Heidelb). (2015) 5:19–35. doi: 10.1007/s13555-015-0070-9
13. de Oliveira ECV, da Motta VRV, Pantoja PC, Ilha CSO, Magalhaes RF, Galadari H, et al. Actinic keratosis - review for clinical practice. Int J Dermatol. (2019) 58:400–7. doi: 10.1111/ijd.14147
14. Flohil SC, van der Leest RJ, Dowlatshahi EA, Hofman A, de Vries E, Nijsten T. Prevalence of actinic keratosis and its risk factors in the general population: The rotterdam study. J Invest Dermatol. (2013) 133:1971–8. doi: 10.1038/jid.2013.134
15. Fargnoli MC, Altomare G, Benati E, Borgia F, Broganelli P, Carbone A, et al. Prevalence and risk factors of actinic keratosis in patients attending italian dermatology clinics. Eur J Dermatol. (2017) 27:599–608. doi: 10.1684/ejd.2017.3126
16. Molina-Garcia M, Malvehy J, Granger C, Garre A, Trullas C and Puig S. Exposome and skin. Part 2. The influential role of the exposome, beyond uvr, in actinic keratosis, bowen’s disease and squamous cell carcinoma: A proposal. Dermatol Ther (Heidelb). (2022) 12:361–80. doi: 10.1007/s13555-021-00644-3
17. Shope C, Andrews L, Leath MN, Linkous C and Lee LW. Identifying risk factors for cutaneous disease among solid organ transplant recipients: A retrospective review. JAAD Int. (2023) 11:157–64. doi: 10.1016/j.jdin.2023.02.003
18. Ulrich C, Jurgensen JS, Degen A, Hackethal M, Ulrich M, Patel MJ, et al. Prevention of non-melanoma skin cancer in organ transplant patients by regular use of a sunscreen: A 24 months, prospective, case-control study. Br J Dermatol. (2009) 161 Suppl 3:78–84. doi: 10.1111/j.1365-2133.2009.09453.x
19. O’Reilly Zwald F, Brown M. Skin cancer in solid organ transplant recipients: Advances in therapy and management: Part i. Epidemiology of skin cancer in solid organ transplant recipients. J Am Acad Dermatol. (2011) 65:253–61. doi: 10.1016/j.jaad.2010.11.062
20. Nashan D, Meiss F and Muller M. Therapeutic strategies for actinic keratoses–a systematic review. Eur J Dermatol. (2013) 23:14–32. doi: 10.1684/ejd.2013.1923
21. Iannacone MR, Sinnya S, Pandeya N, Isbel N, Campbell S, Fawcett J, et al. Prevalence of skin cancer and related skin tumors in high-risk kidney and liver transplant recipients in queensland, Australia. J Invest Dermatol. (2016) 136:1382–6. doi: 10.1016/j.jid.2016.02.804
22. Eisen DB, Asgari MM, Bennett DD, Connolly SM, Dellavalle RP, Freeman EE, et al. Guidelines of care for the management of actinic keratosis. J Am Acad Dermatol. (2021) 85:e209–33. doi: 10.1016/j.jaad.2021.02.082
23. Thompson SC, Jolley D and Marks R. Reduction of solar keratoses by regular sunscreen use. N Engl J Med. (1993) 329:1147–51. doi: 10.1056/NEJM199310143291602
24. Darlington S, Williams G, Neale R, Frost C and Green A. A randomized controlled trial to assess sunscreen application and beta carotene supplementation in the prevention of solar keratoses. Arch Dermatol. (2003) 139:451–5. doi: 10.1001/archderm.139.4.451
25. Moloney F, Vestergaard M, Radojkovic B and Damian D. Randomized, double-blinded, placebo controlled study to assess the effect of topical 1% nicotinamide on actinic keratoses. Br J Dermatol. (2010) 162:1138–9. doi: 10.1111/j.1365-2133.2010.09659.x
26. Surjana D, Halliday GM, Martin AJ, Moloney FJ and Damian DL. Oral nicotinamide reduces actinic keratoses in phase ii double-blinded randomized controlled trials. J Invest Dermatol. (2012) 132:1497–500. doi: 10.1038/jid.2011.459
27. Chen AC, Martin AJ, Choy B, Fernandez-Penas P, Dalziell RA, McKenzie CA, et al. A phase 3 randomized trial of nicotinamide for skin-cancer chemoprevention. N Engl J Med. (2015) 373:1618–26. doi: 10.1056/NEJMoa1506197
28. Bavinck JN, Tieben LM, van der Woude FJ, Tegzess AM, Hermans J, ter Schegget J, et al. Prevention of skin cancer and reduction of keratotic skin lesions during acitretin therapy in renal transplant recipients: A double-blind, placebo-controlled study. J Clin Oncol. (1995) 13:1933–8. doi: 10.1200/JCO.1995.13.8.1933
29. Badri O, Schmults CD, Karia PS and Ruiz ES. Efficacy and cost analysis for acitretin for basal and squamous cell carcinoma prophylaxis in renal transplant recipients. Dermatol Surg. (2021) 47:125–6. doi: 10.1097/DSS.0000000000002423
30. Zhang H, George-Washburn EA, Hashemi KB, Cho E, Walker J, Weinstock MA, et al. Oral nicotinamide for actinic keratosis prevention in kidney transplant recipients: A pilot double-blind, randomized, placebo-controlled trial. Transplant Proc. (2023) 55:2079–84. doi: 10.1016/j.transproceed.2023.06.016
31. Pathak GN, Sanabria B, Caetano VD, Rafiq B and Rao BK. Evaluation and treatment monitoring of actinic keratosis using line-field confocal optical coherence tomography. Skin Res Technol. (2024) 30:e13726. doi: 10.1111/srt.13726
32. Casari A, Chester J and Pellacani G. Actinic keratosis and non-invasive diagnostic techniques: An update. Biomedicines. (2018) 6:8. doi: 10.3390/biomedicines6010008
33. Zhu AQ, Wang LF, Li XL, Wang Q, Li MX, Ma YY, et al. High-frequency ultrasound in the diagnosis of the spectrum of cutaneous squamous cell carcinoma: Noninvasively distinguishing actinic keratosis, bowen’s disease, and invasive squamous cell carcinoma. Skin Res Technol. (2021) 27:831–40. doi: 10.1111/srt.13028
34. Huerta-Brogeras M, Olmos O, Borbujo J, Hernandez-Nunez A, Castano E, Romero-Mate A, et al. Validation of dermoscopy as a real-time noninvasive diagnostic imaging technique for actinic keratosis. Arch Dermatol. (2012) 148:1159–64. doi: 10.1001/archdermatol.2012.1060
35. Alvarez-Salafranca M, Zaballos P. Dermoscopy of squamous cell carcinoma: From actinic keratosis to invasive forms. Actas Dermosifiliogr. (2024) 115:T883–T95. doi: 10.1016/j.ad.2024.03.023
36. Prow TW, Tan JM and Pellacani G. Reflectance confocal microscopy: Hallmarks of keratinocyte cancer and its precursors. Curr Probl Dermatol. (2015) 46:85–94. doi: 10.1159/000366541
37. Cockerell CJ. Histopathology of incipient intraepidermal squamous cell carcinoma (“actinic keratosis”). J Am Acad Dermatol. (2000) 42:11–7. doi: 10.1067/mjd.2000.103344
38. Schmults CD, Blitzblau R, Aasi SZ, Alam M, Amini A, Bibee K, et al. Basal cell skin cancer, version 2.2024, nccn clinical practice guidelines in oncology. J Natl Compr Canc Netw. (2023) 21:1181–203. doi: 10.6004/jnccn.2023.0056
39. Wikonkal NM, Brash DE. Ultraviolet radiation induced signature mutations in photocarcinogenesis. J Investig Dermatol Symp Proc. (1999) 4:6–10. doi: 10.1038/sj.jidsp.5640173
40. Xiang F, Lucas R, Hales S and Neale R. Incidence of nonmelanoma skin cancer in relation to ambient uv radiation in white populations, 1978-2012: Empirical relationships. JAMA Dermatol. (2014) 150:1063–71. doi: 10.1001/jamadermatol.2014.762
41. Jensen P, Hansen S, Moller B, Leivestad T, Pfeffer P, Geiran O, et al. Skin cancer in kidney and heart transplant recipients and different long-term immunosuppressive therapy regimens. J Am Acad Dermatol. (1999) 40:177–86. doi: 10.1016/s0190-9622(99)70185-4
42. Hartevelt MM, Bavinck JN, Kootte AM, Vermeer BJ, Vandenbroucke JP. Incidence of skin cancer after renal transplantation in the Netherlands. Transplantation. (1990) 49:506–9. doi: 10.1097/00007890-199003000-00006
43. Lindelof B, Sigurgeirsson B, Gabel H and Stern RS. Incidence of skin cancer in 5356 patients following organ transplantation. Br J Dermatol. (2000) 143:513–9. doi: 10.1111/j.1365-2133.2000.03703.x
44. Zur Hausen H. Papillomaviruses in the causation of human cancers - a brief historical account. Virology. (2009) 384:260–5. doi: 10.1016/j.virol.2008.11.046
45. Torchia D, Massi D, Caproni M and Fabbri P. Multiple cutaneous precanceroses and carcinomas from combined iatrogenic/professional exposure to arsenic. Int J Dermatol. (2008) 47:592–3. doi: 10.1111/j.1365-4632.2008.03547.x
46. Edwards MJ, Hirsch RM, Broadwater JR, Netscher DT and Ames FC. Squamous cell carcinoma arising in previously burned or irradiated skin. Arch Surg. (1989) 124:115–7. doi: 10.1001/archsurg.1989.01410010125024
47. Skin Tumor Research Center DaVB, Chinese Medical Association, Skin Tumor Group, Dermatology Branch, Chinese Medical Doctor Association. Expert consensus on the diagnosis and treatment of cutaneous squamous cell carcinoma (2021). Chin J Dermatol. (2021) 21,54:653–64. doi: 10.35541/cjd.20201140
48. Stratigos AJ, Garbe C, Dessinioti C, Lebbe C, van Akkooi A, Bataille V, et al. European consensus-based interdisciplinary guideline for invasive cutaneous squamous cell carcinoma. Part 1: Diagnostics and prevention-update 2023. Eur J Cancer. (2023) 193:113251. doi: 10.1016/j.ejca.2023.113251
49. Damian DL, Patterson CR, Stapelberg M, Park J, Barnetson RS, Halliday GM. Uv radiation-induced immunosuppression is greater in men and prevented by topical nicotinamide. J Invest Dermatol. (2008) 128:447–54. doi: 10.1038/sj.jid.5701058
50. Allen NC, Martin AJ, Snaidr VA, Eggins R, Chong AH, Fernandez-Penas P, et al. Nicotinamide for skin-cancer chemoprevention in transplant recipients. N Engl J Med. (2023) 388:804–12. doi: 10.1056/NEJMoa2203086
51. Nijsten TE, Stern RS. Oral retinoid use reduces cutaneous squamous cell carcinoma risk in patients with psoriasis treated with psoralen-uva: A nested cohort study. J Am Acad Dermatol. (2003) 49:644–50. doi: 10.1067/s0190-9622(03)01587-1
52. Anforth R, Blumetti TC, Clements A, Kefford R, Long GV, Fernandez-Penas P. Systemic retinoids for the chemoprevention of cutaneous squamous cell carcinoma and verrucal keratosis in a cohort of patients on braf inhibitors. Br J Dermatol. (2013) 169:1310–3. doi: 10.1111/bjd.12519
53. Nemer KM, Council ML. Topical and systemic modalities for chemoprevention of nonmelanoma skin cancer. Dermatol Clin. (2019) 37:287–95. doi: 10.1016/j.det.2019.02.004
54. Reinau D, Surber C, Jick SS and Meier CR. Nonsteroidal anti-inflammatory drugs and the risk of nonmelanoma skin cancer. Int J Cancer. (2015) 137:144–53. doi: 10.1002/ijc.29357
55. Gormley RH, Groft CM, Miller CJ and Kovarik CL. Digital squamous cell carcinoma and association with diverse high-risk human papillomavirus types. J Am Acad Dermatol. (2011) 64:981–5. doi: 10.1016/j.jaad.2009.10.044
56. Que SKT, Zwald FO and Schmults CD. Cutaneous squamous cell carcinoma: Incidence, risk factors, diagnosis, and staging. J Am Acad Dermatol. (2018) 78:237–47. doi: 10.1016/j.jaad.2017.08.059
57. Paolino G, Donati M, Didona D, Mercuri SR and Cantisani C. Histology of non-melanoma skin cancers: An update. Biomedicines. (2017) 5:71. doi: 10.3390/biomedicines5040071
58. Combalia A, Carrera C. Squamous cell carcinoma: An update on diagnosis and treatment. Dermatol Pract Concept. (2020) 10::e2020066. doi: 10.5826/dpc.1003a66
59. Palaniappan V, Karthikeyan K. Bowen’s disease. Indian Dermatol Online J. (2022) 13:177–89. doi: 10.4103/idoj.idoj_257_21
60. Rishpon A, Kim N, Scope A, Porges L, Oliviero MC, Braun RP, et al. Reflectance confocal microscopy criteria for squamous cell carcinomas and actinic keratoses. Arch Dermatol. (2009) 145:766–72. doi: 10.1001/archdermatol.2009.134
61. Jason Thomson CH, Strid J, Healy E, Wang J. O02 neoantigens from actinic keratosis are predicted to be more immunogenic than those from cutaneous squamous cell carcinoma – a strategy for immune escape. Br J Dermatol. (2023) 189:e4–5. doi: 10.1093/bjd/ljad174.002
62. Xiong Y, Zhang Y, Xiong S, Williams-Villalobo AE. A glance of p53 functions in brain development, neural stem cells, and brain cancer. Biol (Basel). (2020) 9:258. doi: 10.3390/biology9090285
63. Monti P, Menichini P, Speciale A, Cutrona G, Fais F, Taiana E, et al. Heterogeneity of tp53 mutations and p53 protein residual function in cancer: Does it matter? Front Oncol. (2020) 10:593383. doi: 10.3389/fonc.2020.593383
64. Lazarian G, Tausch E, Eclache V, Sebaa A, Bianchi V, Letestu R, et al. Tp53 mutations are early events in chronic lymphocytic leukemia disease progression and precede evolution to complex karyotypes. Int J Cancer. (2016) 139:1759–63. doi: 10.1002/ijc.30222
65. Piipponen M, Riihila P, Nissinen L and Kahari VM. The role of p53 in progression of cutaneous squamous cell carcinoma. Cancers (Basel). (2021) 13:4507. doi: 10.3390/cancers13184507
66. Hedberg ML, Berry CT, Moshiri AS, Xiang Y, Yeh CJ, Attilasoy C, et al. Molecular mechanisms of cutaneous squamous cell carcinoma. Int J Mol Sci. (2022) 23:3478. doi: 10.3390/ijms23073478
67. Kim YS, Jung SH, Park YM, Park GS, Kim HS, Maeng LS, et al. Targeted deep sequencing reveals genomic alterations of actinic keratosis/cutaneous squamous cell carcinoma in situ and cutaneous squamous cell carcinoma. Exp Dermatol. (2023) 32:447–56. doi: 10.1111/exd.14730
68. Nelson MA, Einspahr JG, Alberts DS, Balfour CA, Wymer JA, Welch KL, et al. Analysis of the p53 gene in human precancerous actinic keratosis lesions and squamous cell cancers. Cancer Lett. (1994) 85:23–9. doi: 10.1016/0304-3835(94)90234-8
69. Levine AJ. P53, the cellular gatekeeper for growth and division. Cell. (1997) 88:323–31. doi: 10.1016/s0092-8674(00)81871-1
70. Batinac T, Zamolo G, Jonjic N, Gruber F and Petrovecki M. P53 protein expression and cell proliferation in non-neoplastic and neoplastic proliferative skin diseases. Tumori. (2004) 90:120–7. doi: 10.1177/030089160409000124
71. Takata M, Saida T. Early cancers of the skin: Clinical, histopathological, and molecular characteristics. Int J Clin Oncol. (2005) 10:391–7. doi: 10.1007/s10147-005-0532-7
72. Ziegler A, Jonason AS, Leffell DJ, Simon JA, Sharma HW, Kimmelman J, et al. Sunburn and p53 in the onset of skin cancer. Nature. (1994) 372:773–6. doi: 10.1038/372773a0
73. Taguchi M, Watanabe S, Yashima K, Murakami Y, Sekiya T and Ikeda S. Aberrations of the tumor suppressor p53 gene and p53 protein in solar keratosis in human skin. J Invest Dermatol. (1994) 103:500–3. doi: 10.1111/1523-1747.ep12395643
74. Li Z, Gonzalez CL, Wang B, Zhang Y, Mejia O, Katsonis P, et al. Cdkn2a suppresses metastasis in squamous cell carcinomas induced by the gain-of-function mutant p53(r172h). J Pathol. (2016) 240:224–34. doi: 10.1002/path.4770
75. Miller M, Shirole N, Tian R, Pal D and Sordella R. The evolution of tp53 mutations: From loss-of-function to separation-of-function mutants. J Cancer Biol Res. (2016) 4:1091.
76. Senturk S, Yao Z, Camiolo M, Stiles B, Rathod T, Walsh AM, et al. P53psi is a transcriptionally inactive p53 isoform able to reprogram cells toward a metastatic-like state. Proc Natl Acad Sci U.S.A. (2014) 111:E3287–96. doi: 10.1073/pnas.1321640111
77. Shirole NH, Pal D, Kastenhuber ER, Senturk S, Boroda J, Pisterzi P, et al. Tp53 exon-6 truncating mutations produce separation of function isoforms with pro-tumorigenic functions. Elife. (2016) 5:e25532. doi: 10.7554/eLife.17929
78. Dolado I, Swat A, Ajenjo N, De Vita G, Cuadrado A and Nebreda AR. P38alpha map kinase as a sensor of reactive oxygen species in tumorigenesis. Cancer Cell. (2007) 11:191–205. doi: 10.1016/j.ccr.2006.12.013
79. Freshney NW, Rawlinson L, Guesdon F, Jones E, Cowley S, Hsuan J, et al. Interleukin-1 activates a novel protein kinase cascade that results in the phosphorylation of hsp27. Cell. (1994) 78:1039–49. doi: 10.1016/0092-8674(94)90278-x
80. Han J, Lee JD, Bibbs L and Ulevitch RJ. A map kinase targeted by endotoxin and hyperosmolarity in mammalian cells. Science. (1994) 265:808–11. doi: 10.1126/science.7914033
81. Freund A, Patil CK and Campisi J. P38mapk is a novel DNA damage response-independent regulator of the senescence-associated secretory phenotype. EMBO J. (2011) 30:1536–48. doi: 10.1038/emboj.2011.69
82. Yi M, Tan Y, Wang L, Cai J, Li X, Zeng Z, et al. Tp63 links chromatin remodeling and enhancer reprogramming to epidermal differentiation and squamous cell carcinoma development. Cell Mol Life Sci. (2020) 77:4325–46. doi: 10.1007/s00018-020-03539-2
83. Lee JC, Laydon JT, McDonnell PC, Gallagher TF, Kumar S, Green D, et al. A protein kinase involved in the regulation of inflammatory cytokine biosynthesis. Nature. (1994) 372:739–46. doi: 10.1038/372739a0
84. Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. (2009) 461:1071–8. doi: 10.1038/nature08467
85. Choo MK, Kraft S, Missero C and Park JM. The protein kinase p38alpha destabilizes p63 to limit epidermal stem cell frequency and tumorigenic potential. Sci Signal. (2018) 11:eaau0727. doi: 10.1126/scisignal.aau0727
86. Neto PD, Alchorne M, Michalany N, Abreu M and Borra R. Reduced p53 staining in actinic keratosis is associated with squamous cell carcinoma: A preliminary study. Indian J Dermatol. (2013) 58:325. doi: 10.4103/0019-5154.113935
87. Tominaga O, Hamelin R, Remvikos Y, Salmon RJ and Thomas G. P53 from basic research to clinical applications. Crit Rev Oncog. (1992) 3:257–82.
88. Velculescu VE, El-Deiry WS. Biological and clinical importance of the p53 tumor suppressor gene. Clin Chem. (1996) 42:858–68. doi: 10.1093/clinchem/42.6.858
89. Wiman KG. Strategies for therapeutic targeting of the p53 pathway in cancer. Cell Death Differ. (2006) 13:921–6. doi: 10.1038/sj.cdd.4401921
90. McCubrey JA, Lertpiriyapong K, Fitzgerald TL, Martelli AM, Cocco L, Rakus D, et al. Roles of tp53 in determining therapeutic sensitivity, growth, cellular senescence, invasion and metastasis. Adv Biol Regul. (2017) 63:32–48. doi: 10.1016/j.jbior.2016.10.001
91. Bakshi A, Shafi R, Nelson J, Cantrell WC, Subhadarshani S, Andea A, et al. The clinical course of actinic keratosis correlates with underlying molecular mechanisms. Br J Dermatol. (2020) 182:995–1002. doi: 10.1111/bjd.18338
92. Larsson C, Lardelli M, White I and Lendahl U. The human notch1, 2, and 3 genes are located at chromosome positions 9q34, 1p13-p11, and 19p13.2-p13.1 in regions of neoplasia-associated translocation. Genomics. (1994) 24:253–8. doi: 10.1006/geno.1994.1613
93. Demehri S, Turkoz A and Kopan R. Epidermal notch1 loss promotes skin tumorigenesis by impacting the stromal microenvironment. Cancer Cell. (2009) 16:55–66. doi: 10.1016/j.ccr.2009.05.016
94. Wang NJ, Sanborn Z, Arnett KL, Bayston LJ, Liao W, Proby CM, et al. Loss-of-function mutations in notch receptors in cutaneous and lung squamous cell carcinoma. Proc Natl Acad Sci U.S.A. (2011) 108:17761–6. doi: 10.1073/pnas.1114669108
95. Yokoyama A, Kakiuchi N, Yoshizato T, Nannya Y, Suzuki H, Takeuchi Y, et al. Age-related remodelling of oesophageal epithelia by mutated cancer drivers. Nature. (2019) 565:312–7. doi: 10.1038/s41586-018-0811-x
96. Zheng Q, Capell BC, Parekh V, O’Day C, Atillasoy C, Bashir HM, et al. Whole-exome and transcriptome analysis of uv-exposed epidermis and carcinoma in situ reveals early drivers of carcinogenesis. J Invest Dermatol. (2021) 141:295–307 e13. doi: 10.1016/j.jid.2020.05.116
97. Kim YS, Shin S, Jung SH, Park YM, Park GS, Lee SH, et al. Genomic progression of precancerous actinic keratosis to squamous cell carcinoma. J Invest Dermatol. (2022) 142:528–538 e8. doi: 10.1016/j.jid.2021.07.172
98. Thomson J, Bewicke-Copley F, Anene CA, Gulati A, Nagano A, Purdie K, et al. The genomic landscape of actinic keratosis. J Invest Dermatol. (2021) 141:1664–1674 e7. doi: 10.1016/j.jid.2020.12.024
99. Martincorena I, Roshan A, Gerstung M, Ellis P, Van Loo P, McLaren S, et al. Tumor evolution. High burden and pervasive positive selection of somatic mutations in normal human skin. Science. (2015) 348:880–6. doi: 10.1126/science.aaa6806
100. South AP, Purdie KJ, Watt SA, Haldenby S, den Breems N, Dimon M, et al. Notch1 mutations occur early during cutaneous squamous cell carcinogenesis. J Invest Dermatol. (2014) 134:2630–8. doi: 10.1038/jid.2014.154
101. Fukusumi T, Califano JA. The notch pathway in head and neck squamous cell carcinoma. J Dent Res. (2018) 97:645–53. doi: 10.1177/0022034518760297
102. Zhou B, Lin W, Long Y, Yang Y, Zhang H, Wu K, et al. Notch signaling pathway: Architecture, disease, and therapeutics. Signal Transduct Target Ther. (2022) 7:95. doi: 10.1038/s41392-022-00934-y
103. Fukusumi T, Guo TW, Sakai A, Ando M, Ren S, Haft S, et al. The notch4-hey1 pathway induces epithelial-mesenchymal transition in head and neck squamous cell carcinoma. Clin Cancer Res. (2018) 24:619–33. doi: 10.1158/1078-0432.CCR-17-1366
104. Aggarwal V, Tuli HS, Varol M, Tuorkey M, Sak K, Parashar NC, et al. Notch signaling: Journey of an evolutionarily conserved pathway in driving tumor progression and its modulation as a therapeutic target. Crit Rev Oncol Hematol. (2021) 164:103403. doi: 10.1016/j.critrevonc.2021.103403
105. Tamagnone L, Zacchigna S and Rehman M. Taming the notch transcriptional regulator for cancer therapy. Molecules. (2018) 23:431. doi: 10.3390/molecules23020431
106. Garber K. Notch emerges as new cancer drug target. J Natl Cancer Inst. (2007) 99:1284–5. doi: 10.1093/jnci/djm148
107. Miele L, Miao H and Nickoloff BJ. Notch signaling as a novel cancer therapeutic target. Curr Cancer Drug Targets. (2006) 6:313–23. doi: 10.2174/156800906777441771
108. Xing J, Griffith CC. Cdkn2a/p16 evaluation in cytology specimens. Cancer Cytopathol. (2023) 131:672–6. doi: 10.1002/cncy.22701
109. Brown VL, Harwood CA, Crook T, Cronin JG, Kelsell DP and Proby CM. P16ink4a and p14arf tumor suppressor genes are commonly inactivated in cutaneous squamous cell carcinoma. J Invest Dermatol. (2004) 122:1284–92. doi: 10.1111/j.0022-202X.2004.22501.x
110. Sherr CJ. The ink4a/arf network in tumour suppression. Nat Rev Mol Cell Biol. (2001) 2:731–7. doi: 10.1038/35096061
111. Martinez-Hervas S, Sanchez-Garcia V, Herrero-Cervera A, Vinue A, Real JT, Ascaso JF, et al. Type 1 diabetic mellitus patients with increased atherosclerosis risk display decreased cdkn2a/2b/2bas gene expression in leukocytes. J Transl Med. (2019) 17:222. doi: 10.1186/s12967-019-1977-1
112. Inman GJ, Wang J, Nagano A, Alexandrov LB, Purdie KJ, Taylor RG, et al. The genomic landscape of cutaneous scc reveals drivers and a novel azathioprine associated mutational signature. Nat Commun. (2018) 9:3667. doi: 10.1038/s41467-018-06027-1
113. Mortier L, Marchetti P, Delaporte E, Martin de Lassalle E, Thomas P, Piette F, et al. Progression of actinic keratosis to squamous cell carcinoma of the skin correlates with deletion of the 9p21 region encoding the p16(ink4a) tumor suppressor. Cancer Lett. (2002) 176:205–14. doi: 10.1016/s0304-3835(01)00757-1
114. Agrawal P, Barton I, Bianco RD, Hovig D, Sarley D and Yadav P. Moving medicine, moving minds: Helping developing countries overcome barriers to outsourcing health commodity distribution to boost supply chain performance and strengthen health systems. Glob Health Sci Pract. (2016) 4:359–65. doi: 10.9745/GHSP-D-16-00130
115. Kanellou P, Zaravinos A, Zioga M, Stratigos A, Baritaki S, Soufla G, et al. Genomic instability, mutations and expression analysis of the tumour suppressor genes p14(arf), p15(ink4b), p16(ink4a) and p53 in actinic keratosis. Cancer Lett. (2008) 264:145–61. doi: 10.1016/j.canlet.2008.01.042
116. Child FJ, Scarisbrick JJ, Calonje E, Orchard G, Russell-Jones R, Whittaker SJ. Inactivation of tumor suppressor genes p15(ink4b) and p16(ink4a) in primary cutaneous b cell lymphoma. J Invest Dermatol. (2002) 118:941–8. doi: 10.1046/j.1523-1747.2002.01785.x
117. Smeds J, Berggren P, Ma X, Xu Z, Hemminki K and Kumar R. Genetic status of cell cycle regulators in squamous cell carcinoma of the oesophagus: The cdkn2a (p16(ink4a) and p14(arf)) and p53 genes are major targets for inactivation. Carcinogenesis. (2002) 23:645–55. doi: 10.1093/carcin/23.4.645
118. Vilcea AM, Stoica LE, Andreiana BC, Mirea CS, Tenea Cojan TS, Vilcea IC, et al. Immunoexpression of ki67, p16 and beta-catenin in precursor lesions of cutaneous squamous cell carcinoma. Rom J Morphol Embryol. (2024) 65:19–25. doi: 10.47162/RJME.65.1.03
119. Kusters-Vandevelde HV, Van Leeuwen A, Verdijk MA, de Koning MN, Quint WG, Melchers WJ, et al. Cdkn2a but not tp53 mutations nor hpv presence predict poor outcome in metastatic squamous cell carcinoma of the skin. Int J Cancer. (2010) 126:2123–32. doi: 10.1002/ijc.24871
120. Zhang D, Wang T, Zhou Y and Zhang X. Comprehensive analyses of cuproptosis-related gene cdkn2a on prognosis and immunologic therapy in human tumors. Med (Baltimore). (2023) 102:e33468. doi: 10.1097/MD.0000000000033468
121. Dong Y, Zheng M, Wang X, Yu C, Qin T and Shen X. High expression of cdkn2a is associated with poor prognosis in colorectal cancer and may guide pd-1-mediated immunotherapy. BMC Cancer. (2023) 23:1097. doi: 10.1186/s12885-023-11603-w
122. Hadoux J, Al Ghuzlan A, Lamartina L, Bani MA, Moog S, Attard M, et al. Patterns of treatment failure after selective rearranged during transfection (ret) inhibitors in patients with metastatic medullary thyroid carcinoma. JCO Precis Oncol. (2023) 7:e2300053. doi: 10.1200/PO.23.00053
123. Takahashi M, Ritz J and Cooper GM. Activation of a novel human transforming gene, ret, by DNA rearrangement. Cell. (1985) 42:581–8. doi: 10.1016/0092-8674(85)90115-1
124. Srivastava A, Tommasi C, Sessions D, Mah A, Bencomo T, Garcia JM, et al. Mab21l4 deficiency drives squamous cell carcinoma via activation of ret. Cancer Res. (2022) 82:3143–57. doi: 10.1158/0008-5472.CAN-22-0047
125. Ogami T, Tamura Y, Toss K, Yuki K, Morikawa M, Tsutsumi S, et al. Mab21l4 regulates the tgf-beta-induced expression of target genes in epidermal keratinocytes. J Biochem. (2022) 171:399–410. doi: 10.1093/jb/mvab141
126. Li Q, Liu Z, Ma L, Yin W and Zhang K. Cacybp knockdown inhibits progression of prostate cancer via p53. J Cancer Res Clin Oncol. (2023) 149:5761–72. doi: 10.1007/s00432-022-04497-x
127. Acharya B, Frett B. An updated patent review of rearranged during transfection (ret) kinase inhibitors (2016-present). Expert Opin Ther Pat. (2022) 32:1067–77. doi: 10.1080/13543776.2022.2132851
128. Gou Q, Gan X, Li L, Gou Q and Zhang T. Precious gene: The application of ret-altered inhibitors. Molecules. (2022) 27:8839. doi: 10.3390/molecules27248839
129. Thein KZ, Velcheti V, Mooers BHM, Wu J and Subbiah V. Precision therapy for ret-altered cancers with ret inhibitors. Trends Cancer. (2021) 7:1074–88. doi: 10.1016/j.trecan.2021.07.003
130. Saha D, Ryan KR, Lakkaniga NR, Acharya B, Garcia NG, Smith EL, et al. Targeting rearranged during transfection in cancer: A perspective on small-molecule inhibitors and their clinical development. J Med Chem. (2021) 64:11747–73. doi: 10.1021/acs.jmedchem.0c02167
131. Liu X, Hu X, Shen T, Li Q, Mooers BHM and Wu J. Ret kinase alterations in targeted cancer therapy. Cancer Drug Resist. (2020) 3:472–81. doi: 10.20517/cdr.2020.15
132. Santarpia L, Bottai G. Inhibition of ret activated pathways: Novel strategies for therapeutic intervention in human cancers. Curr Pharm Des. (2013) 19:864–82. doi: 10.2174/138161213804547240
133. Zhou BB, Elledge SJ. The DNA damage response: Putting checkpoints in perspective. Nature. (2000) 408:433–9. doi: 10.1038/35044005
134. Marechal A, Zou L. Rpa-coated single-stranded DNA as a platform for post-translational modifications in the DNA damage response. Cell Res. (2015) 25:9–23. doi: 10.1038/cr.2014.147
135. Chen Z, Dragan M, Sun P, Haensel D, Vu R, Cui L, et al. An ahr-ovol1-id1 regulatory axis in keratinocytes promotes skin homeostasis against atopic dermatitis. bioRxiv. (2024). doi: 10.1101/2024.01.29.577821
136. Jiang Y, Zhang Z. Ovol2: An epithelial lineage determiner with emerging roles in energy homeostasis. Trends Cell Biol. (2023) 33:824–33. doi: 10.1016/j.tcb.2023.05.008
137. Li B, Dai Q, Li L, Nair M, Mackay DR and Dai X. Ovol2, a mammalian homolog of drosophila ovo: Gene structure, chromosomal mapping, and aberrant expression in blind-sterile mice. Genomics. (2002) 80:319–25. doi: 10.1006/geno.2002.6831
138. Roca H, Hernandez J, Weidner S, McEachin RC, Fuller D, Sud S, et al. Transcription factors ovol1 and ovol2 induce the mesenchymal to epithelial transition in human cancer. PloS One. (2013) 8:e76773. doi: 10.1371/journal.pone.0076773
139. Qi XK, Han HQ, Zhang HJ, Xu M, Li L, Chen L, et al. Ovol2 links stemness and metastasis via fine-tuning epithelial-mesenchymal transition in nasopharyngeal carcinoma. Theranostics. (2018) 8:2202–16. doi: 10.7150/thno.24003
140. Murata M, Ito T, Tanaka Y, Yamamura K, Furue K and Furue M. Ovol2-mediated zeb1 downregulation may prevent promotion of actinic keratosis to cutaneous squamous cell carcinoma. J Clin Med. (2020) 9:618. doi: 10.3390/jcm9030618
141. Fang L, Seki A and Fang G. Skap associates with kinetochores and promotes the metaphase-to-anaphase transition. Cell Cycle. (2009) 8:2819–27. doi: 10.4161/cc.8.17.9514
142. Zhang W, Liao Y, Liu C, Liu L and Zhou X. Knstrn, a poor prognostic biomarker, affects the tumor immune microenvironment and immunotherapy outcomes in pan-cancer. Dis Markers. (2023) 2023:6729717. doi: 10.1155/2023/6729717
143. Lee CS, Bhaduri A, Mah A, Johnson WL, Ungewickell A, Aros CJ, et al. Recurrent point mutations in the kinetochore gene knstrn in cutaneous squamous cell carcinoma. Nat Genet. (2014) 46:1060–2. doi: 10.1038/ng.3091
144. Schmitz L, Grinblat B, Novak B, Hoeh AK, Handschke K, von Dobbeler C, et al. Somatic mutations in kinetochore gene knstrn are associated with basal proliferating actinic keratoses and cutaneous squamous cell carcinoma. J Eur Acad Dermatol Venereol. (2019) 33:1535–40. doi: 10.1111/jdv.15615
145. Bornancin F, Mechtcheriakova D, Stora S, Graf C, Wlachos A, Devay P, et al. Characterization of a ceramide kinase-like protein. Biochim Biophys Acta. (2005) 1687:31–43. doi: 10.1016/j.bbalip.2004.11.012
146. Avela K, Sankila EM, Seitsonen S, Kuuluvainen L, Barton S, Gillies S, et al. A founder mutation in cerkl is a major cause of retinal dystrophy in Finland. Acta Ophthalmol. (2018) 96:183–91. doi: 10.1111/aos.13551
147. Solomon B, Young RJ and Rischin D. Head and neck squamous cell carcinoma: Genomics and emerging biomarkers for immunomodulatory cancer treatments. Semin Cancer Biol. (2018) 52:228–40. doi: 10.1016/j.semcancer.2018.01.008
148. Hu X, Lu Z, Yu S, Reilly J, Liu F, Jia D, et al. Cerkl regulates autophagy via the nad-dependent deacetylase sirt1. Autophagy. (2019) 15:453–65. doi: 10.1080/15548627.2018.1520548
149. Zheng Y, Chi S and Li C. Identification of potential gene drivers of cutaneous squamous cell carcinoma: Analysis of microarray data. Med (Baltimore). (2020) 99:e22257. doi: 10.1097/MD.0000000000022257
150. Kilding R, Akil M, Till S, Amos R, Winfield J, Iles MM, et al. A biologically important single nucleotide polymorphism within the toll-like receptor-4 gene is not associated with rheumatoid arthritis. Clin Exp Rheumatol. (2003) 21:340–2.
151. Iotzova-Weiss G, Freiberger SN, Johansen P, Kamarachev J, Guenova E, Dziunycz PJ, et al. Tlr4 as a negative regulator of keratinocyte proliferation. PloS One. (2017) 12:e0185668. doi: 10.1371/journal.pone.0185668
152. Patot S, Imbert PR, Baude J, Martins Simoes P, Campergue JB, Louche A, et al. The tir homologue lies near resistance genes in staphylococcus aureus, coupling modulation of virulence and antimicrobial susceptibility. PloS Pathog. (2017) 13:e1006092. doi: 10.1371/journal.ppat.1006092
153. Mikami E, Kudo M, Ohashi R, Kawahara K, Kawamoto Y, Teduka K, et al. Toll−like receptor 4 plays a tumor−suppressive role in cutaneous squamous cell carcinoma. Int J Oncol. (2019) 54:2179–88. doi: 10.3892/ijo.2019.4790
154. Dickinson SE, Wondrak GT. Tlr4-directed molecular strategies targeting skin photodamage and carcinogenesis. Curr Med Chem. (2018) 25:5487–502. doi: 10.2174/0929867324666170828125328
155. Burns EM, Yusuf N. Toll-like receptors and skin cancer. Front Immunol. (2014) 5:135. doi: 10.3389/fimmu.2014.00135
156. Wenzel J, Tormo D and Tuting T. Toll-like receptor-agonists in the treatment of skin cancer: History, current developments and future prospects. Handb Exp Pharmacol. (2008) 183:201–20. doi: 10.1007/978-3-540-72167-3_10
157. Zahedi K, Prada AE, Davis AE 3rd. Structure and regulation of the c1 inhibitor gene. Behring Inst Mitt. (1993) 93:115–9.
158. Almitairi JOM, Venkatraman Girija U, Furze CM, Simpson-Gray X, Badakshi F, Marshall JE, et al. Structure of the c1r-c1s interaction of the c1 complex of complement activation. Proc Natl Acad Sci U.S.A. (2018) 115:768–73. doi: 10.1073/pnas.1718709115
159. Venkatraman Girija U, Gingras AR, Marshall JE, Panchal R, Sheikh MA, Harper JA, et al. Structural basis of the c1q/c1s interaction and its central role in assembly of the c1 complex of complement activation. Proc Natl Acad Sci U.S.A. (2013) 110:13916–20. doi: 10.1073/pnas.1311113110
160. Riihila P, Viiklepp K, Nissinen L, Farshchian M, Kallajoki M, Kivisaari A, et al. Tumour-cell-derived complement components c1r and c1s promote growth of cutaneous squamous cell carcinoma. Br J Dermatol. (2020) 182:658–70. doi: 10.1111/bjd.18095
161. Ricklin D, Hajishengallis G, Yang K and Lambris JD. Complement: A key system for immune surveillance and homeostasis. Nat Immunol. (2010) 11:785–97. doi: 10.1038/ni.1923
162. Serna M, Giles JL, Morgan BP and Bubeck D. Structural basis of complement membrane attack complex formation. Nat Commun. (2016) 7:10587. doi: 10.1038/ncomms10587
163. Bohlson SS, Garred P, Kemper C and Tenner AJ. Complement nomenclature-deconvoluted. Front Immunol. (2019) 10:1308. doi: 10.3389/fimmu.2019.01308
164. Rahmati Nezhad P, Riihila P, Knuutila JS, Viiklepp K, Peltonen S, Kallajoki M, et al. Complement factor d is a novel biomarker and putative therapeutic target in cutaneous squamous cell carcinoma. Cancers (Basel). (2022) 14:305. doi: 10.3390/cancers14020305
165. Ran R, Gong CY, Wang ZQ, Zhou WM, Zhang SB, Shi YQ, et al. Long non−coding rna part1: Dual role in cancer. Hum Cell. (2022) 35:1364–74. doi: 10.1007/s13577-022-00752-y
166. Gong Z, Shen G, Huang C, Zhang J and Ji J. Downregulation of lncrna neat1 inhibits the proliferation of human cutaneous squamous cell carcinoma. Vivo vitro. Ann Transl Med. (2022) 10:79. doi: 10.21037/atm-21-6916
167. Zhao J, Jin W, Yi K, Wang Q, Zhou J, Tan Y, et al. Combination lsd1 and hotair-ezh2 inhibition disrupts cell cycle processes and induces apoptosis in glioblastoma cells. Pharmacol Res. (2021) 171:105764. doi: 10.1016/j.phrs.2021.105764
168. Luan C, Jin S, Hu Y, Zhou X, Liu L, Li R, et al. Whole-genome identification and construction of the lncrna-mrna co-expression network in patients with actinic keratosis. Transl Cancer Res. (2022) 11:4070–8. doi: 10.21037/tcr-22-842
169. Lee YS, Dutta A. Micrornas in cancer. Annu Rev Pathol. (2009) 4:199–227. doi: 10.1146/annurev.pathol.4.110807.092222
170. Ma L, Weinberg RA. Micrornas in Malignant progression. Cell Cycle. (2008) 7:570–2. doi: 10.4161/cc.7.5.5547
171. Schickel R, Boyerinas B, Park SM and Peter ME. Micrornas: Key players in the immune system, differentiation, tumorigenesis and cell death. Oncogene. (2008) 27:5959–74. doi: 10.1038/onc.2008.274
172. Mizrahi A, Barzilai A, Gur-Wahnon D, Ben-Dov IZ, Glassberg S, Meningher T, et al. Alterations of micrornas throughout the Malignant evolution of cutaneous squamous cell carcinoma: The role of mir-497 in epithelial to mesenchymal transition of keratinocytes. Oncogene. (2018) 37:218–30. doi: 10.1038/onc.2017.315
173. Zhang L, Qin H, Wu Z, Chen W and Zhang G. Pathogenic genes related to the progression of actinic keratoses to cutaneous squamous cell carcinoma. Int J Dermatol. (2018) 57:1208–17. doi: 10.1111/ijd.14131
174. Chiantore MV, Iuliano M, Mongiovi RM, Luzi F, Mangino G, Grimaldi L, et al. Micrornas differentially expressed in actinic keratosis and healthy skin scrapings. Biomedicines. (2023) 11:1719. doi: 10.3390/biomedicines11061719
175. Danczak-Pazdrowska A, Pazdrowski J, Polanska A, Basta B, Schneider A, Kowalczyk MJ, et al. Profiling of micrornas in actinic keratosis and cutaneous squamous cell carcinoma patients. Arch Dermatol Res. (2022) 314:257–66. doi: 10.1007/s00403-021-02221-2
176. Toll A, Salgado R, Espinet B, Diaz-Lagares A, Hernandez-Ruiz E, Andrades E, et al. Mir-204 silencing in intraepithelial to invasive cutaneous squamous cell carcinoma progression. Mol Cancer. (2016) 15:53. doi: 10.1186/s12943-016-0537-z
177. Sun C, Mahapatra KD, Elton J, Li C, Fernando W, Lohcharoenkal W, et al. Microrna-23b plays a tumor-suppressive role in cutaneous squamous cell carcinoma and targets ras-related protein rras2. J Invest Dermatol. (2023) 143:2386–96. doi: 10.1016/j.jid.2023.05.026
178. Chitsazzadeh V, Nguyen TN, de Mingo Pulido A, Bittencourt BB, Du L, Adelmann CH, et al. Mir-181a promotes multiple protumorigenic functions by targeting tgfbetar3. J Invest Dermatol. (2022) 142:1956–1965 e2. doi: 10.1016/j.jid.2021.09.040
179. Fernandez-Figueras MT, Carrato C, Saenz-Sarda X, Musulen E, Fuente MJ and Puig L. Microrna31 and mmp-1 contribute to the differentiated pathway of invasion -with enhanced epithelial-to-mesenchymal transition- in squamous cell carcinoma of the skin. Arch Dermatol Res. (2022) 314:767–75. doi: 10.1007/s00403-021-02288-x
180. Yang X, Daifallah AEM, Shankar S, Beer J, Marshall C, Dentchev T, et al. Topical kinase inhibitors induce regression of cutaneous squamous cell carcinoma. Exp Dermatol. (2019) 28:609–13. doi: 10.1111/exd.13902
181. Briatico G, Brancaccio G, Scharf C, Di Brizzi EV, Pellerone S, Caccavale S, et al. Real-world experience with topical 5-fluorouracil 4% (40 mg/g) cream for the treatment of actinic keratosis. Dermatol Pract Concept. (2023) 13:e2023151. doi: 10.5826/dpc.1302a151
182. Toffoli L, Dianzani C, Bonin S, Guarneri C, Guarneri F, Giuffrida R, et al. Actinic keratoses: A prospective pilot study on a novel formulation of 4% 5-fluorouracil cream and a review of other current topical treatment options. Cancers (Basel). (2023) 15:2956. doi: 10.3390/cancers15112956
183. Maytin EV, Anand S, Riha M, Lohser S, Tellez A, Ishak R, et al. 5-fluorouracil enhances protoporphyrin ix accumulation and lesion clearance during photodynamic therapy of actinic keratoses: A mechanism-based clinical trial. Clin Cancer Res. (2018) 24:3026–35. doi: 10.1158/1078-0432.CCR-17-2020
184. Marka A, Rodgers D, Zelaya Castillo L, Hoyt B, Chapman M and Carter J. Dilute intralesional 5-fluorouracil for the treatment of squamous cell carcinomas and keratoacanthomas: A case series. J Drugs Dermatol. (2023) 22:507–8. doi: 10.36849/JDD.5058
185. Hamad J, Jolly PS. Debulking followed by intralesional 5-fluorouracil for the treatment of cutaneous squamous cell carcinoma and keratoacanthoma: A retrospective analysis. Dermatol Ther. (2021) 34:e15139. doi: 10.1111/dth.15139
186. Tanghetti E, Werschler P. Comparison of 5% 5-fluorouracil cream and 5% imiquimod cream in the management of actinic keratoses on the face and scalp. J Drugs Dermatol. (2007) 6:144–7.
187. Paolino G, Cantoresi F, Mercuri SR and Cantisani C. Clinical evaluation and efficacy of imiquimod 3.75% cream for the treatment of actinic keratosis, pigmented basal cell carcinomas, and actinic cheilitis. Dermatol Ther. (2020) 33:e13356. doi: 10.1111/dth.13356
188. Maxwell Regester R, Cannella AC, Hirz J, Sayles B, Schlange S, Rau A, et al. Combination therapy with imiquimod and 5-fluorouracil cream for the treatment of actinic keratoses, a prospective open-label nonrandomized uncontrolled pilot trial. J Am Acad Dermatol. (2024) 90:1247–9. doi: 10.1016/j.jaad.2024.01.065
189. Toso F, Tronconi MC, Cortese A, Fiorillo G, Bressan A, Costanzo A, et al. Complete response of locally advanced cutaneous squamous cell carcinoma of the eyelid to topical imiquimod 3.75. Dermatol Ther. (2022) 35:e15800. doi: 10.1111/dth.15800
190. Thomas GJ, Herranz P, Cruz SB and Parodi A. Treatment of actinic keratosis through inhibition of cyclooxygenase-2: Potential mechanism of action of diclofenac sodium 3% in hyaluronic acid 2.5. Dermatol Ther. (2019) 32:e12800. doi: 10.1111/dth.12800
191. Kabir MF, Jackson JL, Fuller AD, Gathuka L, Karami AL, Conde DG, et al. Diclofenac exhibits cytotoxic activity associated with metabolic alterations and p53 induction in escc cell lines and decreases escc tumor burden. vivo. Carcinogenesis. (2023) 44:182–95. doi: 10.1093/carcin/bgad019
192. Gollnick H, Dirschka T, Ostendorf R, Kerl H and Kunstfeld R. Long-term clinical outcomes of imiquimod 5% cream vs. Diclofenac 3% gel for actinic keratosis on the face or scalp: A pooled analysis of two randomized controlled trials. J Eur Acad Dermatol Venereol. (2020) 34:82–9. doi: 10.1111/jdv.15868
193. Erlendsson AM. Exploring the use of ingenol mebutate to prevent non-melanoma skin cancer. Dan Med J. (2017) 64:B5368.
194. Freiberger SN, Cheng PF, Iotzova-Weiss G, Neu J, Liu Q, Dziunycz P, et al. Ingenol mebutate signals via pkc/mek/erk in keratinocytes and induces interleukin decoy receptors il1r2 and il13ra2. Mol Cancer Ther. (2015) 14:2132–42. doi: 10.1158/1535-7163.MCT-15-0023-T
195. Jedlowski PM. Ingenol mebutate is associated with increased reporting odds for squamous cell carcinoma in actinic keratosis patients, a pharmacovigilance study of the fda adverse event reporting system (faers). J Cutan Med Surg. (2023) 27:39–43. doi: 10.1177/12034754221143091
196. Mager L, Gardeen S, Carr DR and Shahwan KT. Cemiplimab for the treatment of advanced cutaneous squamous cell carcinoma: Appropriate patient selection and perspectives. Clin Cosmet Investig Dermatol. (2023) 16:2135–42. doi: 10.2147/CCID.S381471
197. Rischin D, Hughes BGM, Basset-Seguin N, SChadendorf D, Bowyer S, Trabelsi Messai S, et al. High response rate with extended dosing of cemiplimab in advanced cutaneous squamous cell carcinoma. J Immunother Cancer. (2024) 12:e008325. doi: 10.1136/jitc-2023-008325
198. Verkerk K, Geurts BS, Zeverijn LJ, van der Noort V, Verheul HMW, Haanen J, et al. Cemiplimab in locally advanced or metastatic cutaneous squamous cell carcinoma: Prospective real-world data from the drug access protocol. Lancet Reg Health Eur. (2024) 39:100875. doi: 10.1016/j.lanepe.2024.100875
199. Jenni D, Karpova MB, Muhleisen B, Mangana J, Dreier J, Hafner J, et al. A prospective clinical trial to assess lapatinib effects on cutaneous squamous cell carcinoma and actinic keratosis. ESMO Open. (2016) 1:e000003. doi: 10.1136/esmoopen-2015-000003
200. Yao M, Shang YY, Zhou ZW, Yang YX, Wu YS, Guan LF, et al. The research on lapatinib in autophagy, cell cycle arrest and epithelial to mesenchymal transition via wnt/erk/pi3k-akt signaling pathway in human cutaneous squamous cell carcinoma. J Cancer. (2017) 8:220–6. doi: 10.7150/jca.16850
201. Amornphimoltham P, Leelahavanichkul K, Molinolo A, Patel V and Gutkind JS. Inhibition of mammalian target of rapamycin by rapamycin causes the regression of carcinogen-induced skin tumor lesions. Clin Cancer Res. (2008) 14:8094–101. doi: 10.1158/1078-0432.CCR-08-0703
202. Booty MG, Komalo B, Hosny A, Headland SE, Fernandez-Figueras MT, Nguyen AM, et al. Oral mtor inhibition limits and reduces actinic keratosis and cutaneous squamous cell carcinoma in a uvb-induced mouse model. bioRxiv. (2022). doi: 10.1101/2022.11.03.513568
203. Fogel AL, Hill S and Teng JM. Advances in the therapeutic use of mammalian target of rapamycin (mtor) inhibitors in dermatology. J Am Acad Dermatol. (2015) 72:879–89. doi: 10.1016/j.jaad.2015.01.014
204. Hedberg M, Seykora JT. Clarifying progress on the genomic landscape of actinic keratosis. J Invest Dermatol. (2021) 141:1622–4. doi: 10.1016/j.jid.2021.02.761
205. Czarnecki D, Meehan CJ, Bruce F and Culjak G. The majority of cutaneous squamous cell carcinomas arise in actinic keratoses. J Cutan Med Surg. (2002) 6:207–9. doi: 10.1007/s10227-001-0041-x
206. Roshan A, Jones PH. Chronic low dose uv exposure and p53 mutation: Tilting the odds in early epidermal preneoplasia? Int J Radiat Biol. (2012) 88:682–7. doi: 10.3109/09553002.2012.699697
207. Klein AM, Brash DE, Jones PH and Simons BD. Stochastic fate of p53-mutant epidermal progenitor cells is tilted toward proliferation by uv b during preneoplasia. Proc Natl Acad Sci U.S.A. (2010) 107:270–5. doi: 10.1073/pnas.0909738107
208. Yang Y, Wu R, Sargsyan D, Yin R, Kuo HC, Yang I, et al. Uvb drives different stages of epigenome alterations during progression of skin cancer. Cancer Lett. (2019) 449:20–30. doi: 10.1016/j.canlet.2019.02.010
209. Zhang M, Biswas S, Qin X, Gong W, Deng W and Yu H. Does notch play a tumor suppressor role across diverse squamous cell carcinomas? Cancer Med. (2016) 5:2048–60. doi: 10.1002/cam4.731
Keywords: actinic keratosis, cutaneous squamous cell carcinoma, progression, malignant transformation, skin cancers, non-melanoma skin cancers
Citation: Li Z, Lu F, Zhou F, Song D, Chang L, Liu W, Yan G and Zhang G (2025) From actinic keratosis to cutaneous squamous cell carcinoma: the key pathogenesis and treatments. Front. Immunol. 16:1518633. doi: 10.3389/fimmu.2025.1518633
Received: 28 October 2024; Accepted: 09 January 2025;
Published: 24 January 2025.
Edited by:
Selma Ugurel, Essen University Hospital, GermanyReviewed by:
Daniela Pinto, Human Advanced Microbiome Project-HMAP, ItalyJenan Almatouq, Mohammed Al Mana College for Health Sciences (MACHS), Saudi Arabia
Mangala Hegde, Indian Institute of Technology Guwahati, India
Xinyi Zhang, Yale University, United States
Copyright © 2025 Li, Lu, Zhou, Song, Chang, Liu, Yan and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Weiying Liu, d3cwMDEwMEAxNjMuY29t; Guorong Yan, Z3Vvcm9uZ3lhbkB0b25namkuZWR1LmNu; Guolong Zhang, Z2x6aGFuZ3RqQHRvbmdqaS5lZHUuY24=