 Yan Long1
Yan Long1 Chenghan Wang1Jie Xiao2Yunhua Huang1Xiaoting Ling1
Chenghan Wang1Jie Xiao2Yunhua Huang1Xiaoting Ling1 Chaoyu Huang1Ying Chen1Jiaqi Luo1Rongheng Tang1
Chaoyu Huang1Ying Chen1Jiaqi Luo1Rongheng Tang1 Faquan Lin1*
Faquan Lin1* Yifang Huang1*
Yifang Huang1*- 1Department of Clinical Laboratory, The First Affiliated Hospital of Guangxi Medical University, Key Laboratory of Clinical Laboratory Medicine of Guangxi Department of Education, Nanning, Guangxi, China
- 2Department of Pediatrics, Rong’an County People’s Hospital, Liuzhou, Guangxi, China
Immunodeficiency, centromeric instability, and facial anomalies syndrome (ICF) is a rare genetic disease characterized by hypogammaglobulinemia, T cell immune deficiency with age, pericentromeric hypomethylation, facial abnormalities, and intellectual disability. This study aimed to investigate the phenotype and immune function of a girl with ICF2, identify her genetic defect, and explore the potential pathogenic mechanisms of the disease. We identified a homologous deletion mutation in this girl, which involves exons 1-5 and part of introns 1 and 6 of the ZBTB24 gene (NG_029388.1: g.2831_18,995del). This ZBTB24 variant produces a severely truncated ZBTB24 protein that lacks the BTB, A-T hook and eight zinc fingers. The above changes may lead to abnormal transcriptional function of the ZBTB24 protein. Karyotype analysis showed fragile sites and entire arm deletions were detected on chromosomes 1 and 16 and triradials on chromosome 16. The novel multi-exon deletion of ZBTB24 causes immunodeficiency, severe pneumonia and centromeric instability in the patient. During the follow-up, the patient’s pneumonia continued to progress despite receiving intravenous immunoglobulin (IVIG) replacement and anti-infective therapy. These results indicated that this novel multi-exon deletion variant of ZBTB24 may be the genetic etiology of ICF2. The discovery of this novel mutation expands the mutation spectrum of the ZBTB24 gene and improves our understanding of the molecular mechanisms underlying ICF.
1 Introduction
Immunodeficiency, centromeric instability, and facial anomalies syndrome (ICF) is a rare autosomal recessive genetic disease and one of the earliest congenital diseases discovered to be caused by DNA methylation defects (1, 2). To date, only approximately 120 cases of ICF have been reported worldwide (3). ICF is characterized by a significant reduction in immunoglobulin levels that can lead to recurrent infections in respiratory and gastrointestinal systems, facial abnormalities (such as ocular hypertelorism, epicanthic folds, low-set ears, and flat nose) and intellectual disability (4). According to the molecular genetic evidence, ICF can be divided into five subtypes: about 60% of patients carry DNMT3B variants, known as ICF syndrome 1 (ICF1, OMIM 242860); nearly 30% of ICF individuals belong to ICF syndrome 2 (ICF2, OMIM 614069), caused by variants in ZBTB24; ICF syndrome 3 (ICF3, OMIM 616910) and ICF syndrome 4 (ICF4, OMIM 616911) are respectively associated with variants in CDCA7 and HELLS; Only very few cases of ICF with unknown causative genes are classified as ICF syndrome X (ICFX) (1, 5).
The human ZBTB24 gene is located on chromosome 6q21 and contains 7 exons. The protein encoded by this gene is a member of the ZBTB family of transcriptional regulators. ZBTB24 is composed of a BTB (broad-complex, tram-track, and bric-a-brac) domain, an A-T hook domain, and eight C2H2-type zinc fingers (6). Previous study found that low expression of ZBTB24 was significantly associated with decreased yields and impaired differentiation of mature B lymphocytes, resulting in a higher incidence of hypogammaglobulinemia in ICF2 patients (7). Although ICF patients with different subtypes show similar clinical phenotypes, the clinical outcomes can vary depending on the severity and frequency of infection (5, 8, 9). Weemaes et al. found that a majority of the 44 patients with ICF suffered from severe pneumonia, resulting in the mortality rate as high as 34.09%. The age range at death for ICF1 patients was 0.75-19 years, and for ICF2 patients it was 4-13 years (10). The study above highlights the importance of early detection and intervention in cases of ICF due to its rapid progression and unfavorable prognosis. And ICF is often underdiagnosed because of the rarity and complexity of the disease. Therefore, more and more studies are devoted to revealing the immunological and genetic characteristics of ICF to help standardize the diagnosis and treatment of ICF (9, 11, 12). In this study, we continuously monitored the immune status and evolution of lung inflammation in an ICF2 patient and discovered a novel ZBTB24 variant that includes a homologous deletion of exons 1-5. The findings expand the mutation spectrum of ZBTB24 and reveal novel molecular defects associated with ICF2.
2 Materials and methods
2.1 Subjects
A 9-year-old girl diagnosed with ICF2 was enrolled in our study and her examination results and peripheral blood samples were collected. Written informed consent from the legal guardian of the child and informed assent from the child were obtained. This work was conducted in accordance with the guidelines of the Helsinki Declaration and approved by the First Affiliated Hospital of Guangxi Medical University Ethical Review Committee.
2.2 Whole-exome sequencing
Genomic DNA was extracted from the patient’s blood sample and fragmented according to the manufacturer’s instructions. The enriched exome library was sequenced on the Illumina HiSeq 2000 platform. After quality filtering the raw data using cutadapt (version 1.16), clean reads were mapped to human reference genome GRCh37/hg19 by the Burrows-Wheeler Aligner (BWA) software. Finally, the variants identified in the patient’s DNA were annotated using ANNOVAR software and the pathogenicity of each variant was assessed using REVEL.
2.3 Validation of ZBTB24 variant
The exon deletion was verified by quantification of the DNA copy number of exons 2-7 using real-time quantitative PCR (qPCR), and the data were normalized to GAPDH. Exon 1 with high GC content was amplified using the following PCR system: a total reaction volume of 25 μL containing LA Taq polymerase 1.25 U, 2×GC Buffer I 12.5 μL, dNTP Mixture 400 μM (Takara Bio, Dalian, China), 0.4 μM of each primer and 200 ng of genomic DNA template. Amplification parameters were as follows: pre-denaturation at 94°C for 1 min, denaturation at 94°C for 30 s, and annealing extension at 68°C for 5 min in the first 10 cycles. In the following 20 cycles, the annealing time was increased by 20 s per cycle. A final extension at 68°C for 10 min was performed. The products were separated by agarose gel electrophoresis and subjected to Sanger sequencing.
In order to locate the specific site of the deleted sequence, we designed primers targeting two short fragments (N1 and N2) of intron 1 and four short fragments (C1-C4) of intron 6 and performed qPCR. We selected the forward primer of fragment N1 and the reverse primer of fragment C4 for gap-PCR when only fragments N1 and C4 showed typical amplification curves. The reagents and experimental procedures were the same as those for exon 1 described above. All experiments used genomic DNA from normal individuals as controls. Primer sequences are shown in Supplementary Table S1. Primers for GAPDH were purchased from Biochuangyi Bio Inc. (Shanghai, China).
2.4 Protein mutation analysis
The translated amino acid sequence of this ZBTB24 variant was analyzed using SnapGene (v.6.0.2). The three-dimensional structural models of wild-type and mutant ZBTB24 proteins were predicted using SWISS-MODEL software (https://swissmodel.expasy.org/) for visualization and comparison.
2.5 G-banded karyotype analysis
Firstly, cell culture and phytohemagglutinin (PHA) stimulation to induce cell division were performed. Then, colchicine was added to stop the cells from dividing. After hypotonicity treatment and fixation, Giemsa staining was used. Cytogenetic analysis was performed on 100 metaphase chromosome images using Ikaros karyotyping software with a resolution of 400-550 bands. The karyotypes were described according to ISCN 2020 (13).
3 Results
3.1 Case information
The patient was a 9-year-old girl who had suffered from recurrent respiratory infections since the age of 3 months, with more than 7 episodes per year. The previous infections had improved with antibacterial treatment. In November 2023, the girl was transferred to our hospital because the recurrent infection progressed to severe pneumonia. Physical examination revealed that the child was 122 cm tall (<-3SD) and weighed 20 kg (<-2SD). She had slightly wider eyes, a flat nose, and low-set ears. It was also learned that the patient had difficulty understanding complex courses and was considered to have a mild intellectual disability according to a total score of 72 on the Chinese-Wechsler Intelligence Scale for Children (C-WISC). These results suggest that the girl has growth and cognitive delays, with motor and speech development similar to that of her peers.
Further family investigation (Figure 1A) revealed that the parents of this proband (II-5) had no immunodeficiency and were not consanguineous. However, the proband’s sister (II-3) had growth retardation and died of unknown causes when she was less than 10 years old. During the fourth spontaneous conception, her mother discovered that the fetus (II-4) had facial abnormalities by antenatal examination and underwent an induced abortion. After the birth of the proband, her mother gave birth to a healthy boy (II-6), who is now 7 years old.
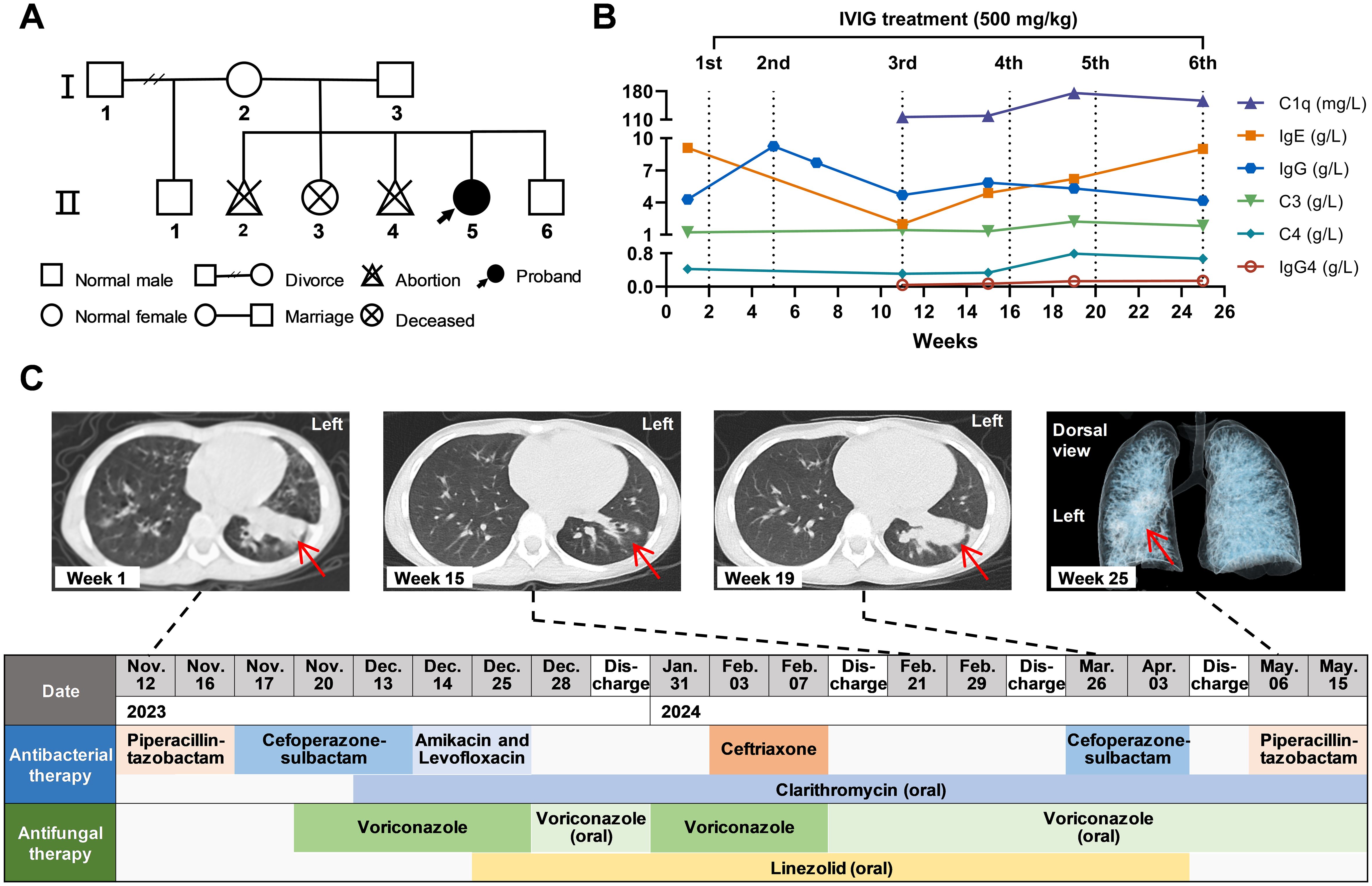
Figure 1. Clinical characteristics of the ICF2 patient. (A) Family pedigree of this proband. (B) The levels of various immunoglobulins and complement during follow-up. The number and dose of IVIG treatment are indicated above the line graphs. (C) Timeline of antibacterial and antifungal treatment. Antibiotics were given intravenously except for those marked “oral”. Above the timeline are chest CT at weeks 1, 15, and 19 and three-dimensional reconstruction of the bronchi at week 25. The red arrows point to the lung lesions.
3.2 Immunologic monitoring and replacement therapy
The immunological assessment of the patient before intravenous immunoglobulin (IVIG) treatment indicated immunoglobulin (Ig) G at 4.29 g/L (Figure 1B), IgA at 0.47 g/L, and low levels of IgM at 0.01 g/L. The anti-tetanus toxoid IgG content was 0.09 IU/mL, indicating a poor antibody response to the tetanus toxoid vaccine. Lymphocyte subpopulation analysis in the first week showed that the proportion of total T cells was normal (72.03%), the CD4+/CD8+ ratio was 1.35, the proportion of B cells was increased (25.37%), and the proportion of NK cells was significantly low (1.07%). The proportion of T cells increased to 83.90% (4692 cells/μL), and the proportion of B cells decreased (12.57%; 725 cells/μL) at week 11. The results of complement C3, complement C4, autoantibody testing, and allergen testing were essentially normal. The patient then received IVIG replacement treatment at a dose of 500 mg/kg each time at weeks 2, 5, 11, 16, 20, and 25, respectively. After the first IVIG treatment, the IgG level could rise to 9.25 g/L, but the IgG levels were lower than the normal ranges and fluctuated between 4.17-5.82 g/L since the third treatment. Specific values of laboratory tests are shown in Supplementary Table S2.
3.3 Manifestations and progression of infection
Blood tests on admission revealed an increased white blood cell (WBC) count of 15.79×109/L, aspartate aminotransferase (AST) level of 84 U/L, and alanine aminotransferase (ALT) level of 99 U/L (Supplementary Table S2). Computed tomography (CT) of the chest showed patchy and linear increased density shadows in the lower lingual segment of the upper lobe and the anterior inner basal segment of the lower lobe in the left lung (Figure 1C; Supplementary Figure S1). Unexpectedly, multiple Gram and acid-fast stains, bacterial and fungal cultures of sputum and bronchoalveolar lavage fluid (BALF) were negative. Tuberculin skin test and interferon-gamma release assay (IGRA) yielded negative results. The fifth G (1,3-β-D-glucan) test and GM (galactomannan) test of BALF were positive, but the previous four tests were negative. Moreover, metagenomic next-generation sequencing (mNGS) of BALF did not detect highly pathogenic microorganisms.
The patient’s infection symptoms improved, and the CT scan at week 15 showed lung inflammation subsided significantly after receiving IVIG, antibacterial, and antifungal treatment (Figure 1C). Antituberculosis therapy was not given because there was no definitive evidence of mycobacterium tuberculosis infection. However, this patient developed a persistent high fever (39°C ± 0.5°C) at week 19, and the CT scan at that time revealed severe lung inflammation compared with that at week 15. The reconstructed three-dimensional bronchial model at week 25 suggested retention of secretions in the left lower lobe bronchus.
3.4 Identification of novel genetic variant
WES was performed and identified a novel variant of homologous deletions of exons 2, 3, 4, and 5 on ZBTB24 gene (Figure 2A). Subsequently, qPCR was used to amplify the DNA fragment of exons 2-7 to verify the deletion of genomic DNA. The results of qPCR confirmed the homozygous deletion of exons 2-5, while exons 6 and 7 were not deleted (Figure 2B; Supplementary Figure S2). LA Taq polymerase with GC Buffer was employed to amplify exon 1, a GC-rich DNA region, to prevent missed detection from insufficient sequencing depth of WES. Agarose gel electrophoresis and Sanger sequencing results (Figure 2C) showed that the control group of the normal individual had the target band whose DNA sequence was consistent with the exon 1 sequence, while no band can be detected in the patient’s lane. These data complemented the WES results and suggested the deletion of exons 1-5 on ZBTB24.
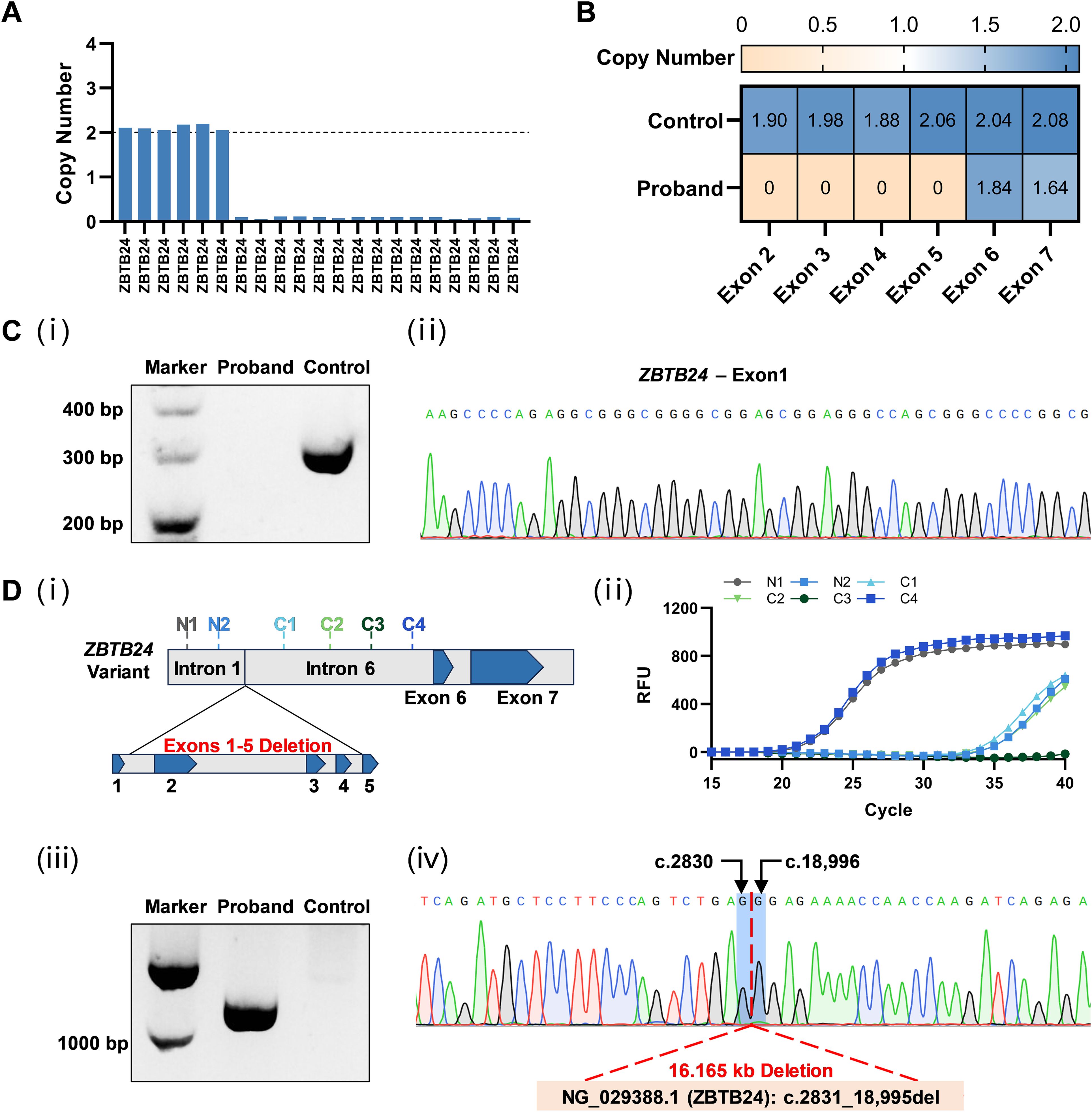
Figure 2. Identification of the novel ZBTB24 variant. (A) Copy number loss of multi-exons of ZBTB24 was detected by WES. (B) The deletion of exons 2-5 in ZBTB24 was confirmed by qPCR. (C) i: Agarose gel electrophoresis showed the expected band of about 300 bp in the control group while no band in the patient’s lane. ii: The PCR product of the control group was clearly found to contain exon 1 of ZBTB24 by Sanger sequencing. (D) i: Two pairs of primers were designed in intron 1 (N1 and N2), and four pairs of primers were designed in intron 6 (C1-C4). ii: The results of qPCR using these six primer pairs indicated that only fragments N1 and C4 were amplified normally. iii: Agarose gel electrophoresis showed that the patient had a DNA fragment (about 1000 bp) containing the deleted sequence information in gap-PCR, while the control group was negative. iv: Sanger sequencing results for the breakpoint position analysis using gap-PCR product.
Since the 1st and 6th introns of ZBTB24 are approximately 12 kb in length and the breakpoint positions are unknown, it is difficult to directly determine the deletion variant by gap-PCR. Therefore, as shown in Figure 2D, we first screened the expression of six regions using six pairs of qPCR primers and revealed that only the fragment N1 of intron 1 and the fragment C4 of intron 6 were expressed at normal levels. Subsequently, a gap-PCR product containing the deletion variant information was successfully amplified using the forward primer of fragment N1 and the reverse primer of fragment C4. After Sanger sequencing verification, we found that this ZBTB24 variant had a deletion length of 16.165 kb and was described as NG_029388.1: g.2831_18,995del. These results further confirmed the deletion of the whole of exons 1-5 on ZBTB24 of the patient. There is no record of this variant in current public databases or literature. The variant is classified as likely pathogenic according to the ACMG guidelines (14).
3.5 Characterization of mutant ZBTB24 protein
The wild-type ZBTB24 protein (NP_055612.2) has a total of 697 amino acids, which include the BTB, A-T hook, and eight zinc fingers. In our study, we found that this multi-exon homozygous deletion variant in ZBTB24 results in the deletion of the original start codon (ATG) located in exon 2 and the shutoff of subsequent series of amino acids synthesis. The new translation start point is likely to be located at the first ATG codon (g.21,989_21,991) in exon 7, encoding the 566th amino acid in the original sequence (Figure 3A). The mutant ZBTB24 protein showed a severe truncation with only 132 amino acids corresponding to the disordered region and was recorded as p.(Ala2_Met566del) (Figure 3B). Hence, this variant we report causes a transcriptional defect in the ZBTB24 protein because it lacks the necessary structure made up of a BTB, an A-T hook, and eight zinc fingers.
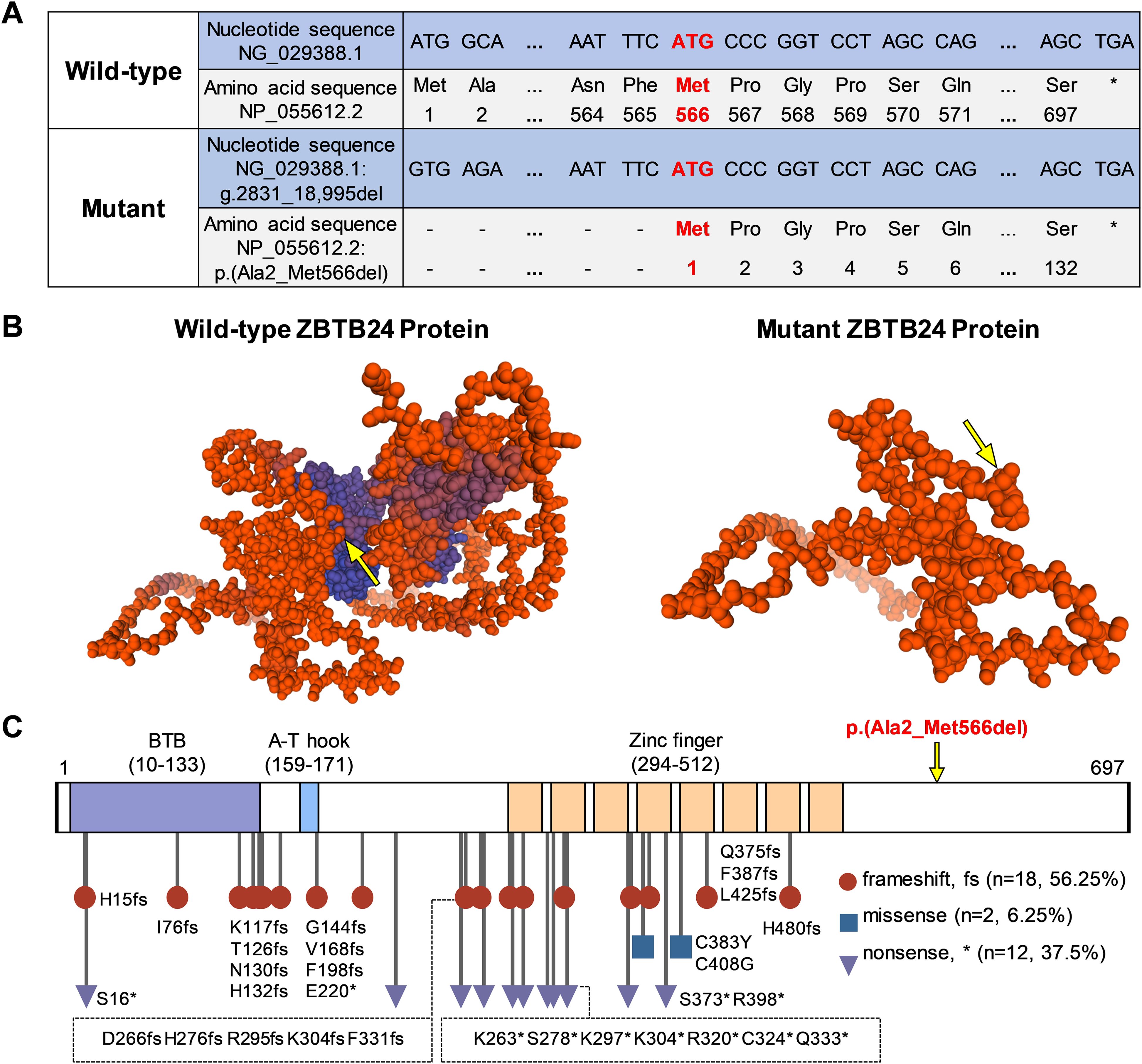
Figure 3. Effects of ZBTB24 variants on protein structure. (A) Through sequence comparison, we found that the start codon (ATG) of this ZBTB24 variant is located in exon 7, corresponding to the 566th amino acid (Met) of the wild-type, and the stop codon remains unchanged. (B) The three-dimensional predicted models of wild-type and mutant ZBTB24 proteins. The yellow arrow points to the original amino acid 566 (Met). (C) The amino acid sequence and known variant sites of ZBTB24. The symbol "*" marks the stop codon.
ZBTB24 variants in the ClinVar database and previous studies (4, 5, 9, 10, 15, 16) were summarized (Figure 3C; Supplementary Table S3). Among the 32 ZBTB24 variants, including the present study, frameshift variants accounted for 56.25%, nonsense variants accounted for 37.5%, and only 2 cases were missense variants. It could be observed that most of the known variants were located in the region of three major functional domains, and there were no obvious mutational hotspots. A review of known ZBTB24 variant cases revealed that different ZBTB24 variants may present similar phenotypes, such as hypogammaglobulinemia, centromeric instability, facial anomalies, motor development delay, and intellectual disability (Supplementary Table S4).
3.6 Abnormal chromosome configuration
It was found that fragile site (fra), triradial (tr), and deletion (del) detected on chromosome 16q11.2 in multiple metaphases. The deletion and fragile site were also presented on chromosome 1q12 (Figure 4). The patient’s karyotype was described as 46,XX,fra(16)(q11.2)[14]/46,XX,tr(16)(q11.2)[6]/46,XX,del(16)(q11.2)[5]/46,XX,del(1)(q12)[4]/46,XX,fra(1)(q12)[3]/46,XX[68], according to ISCN 2020 (13).
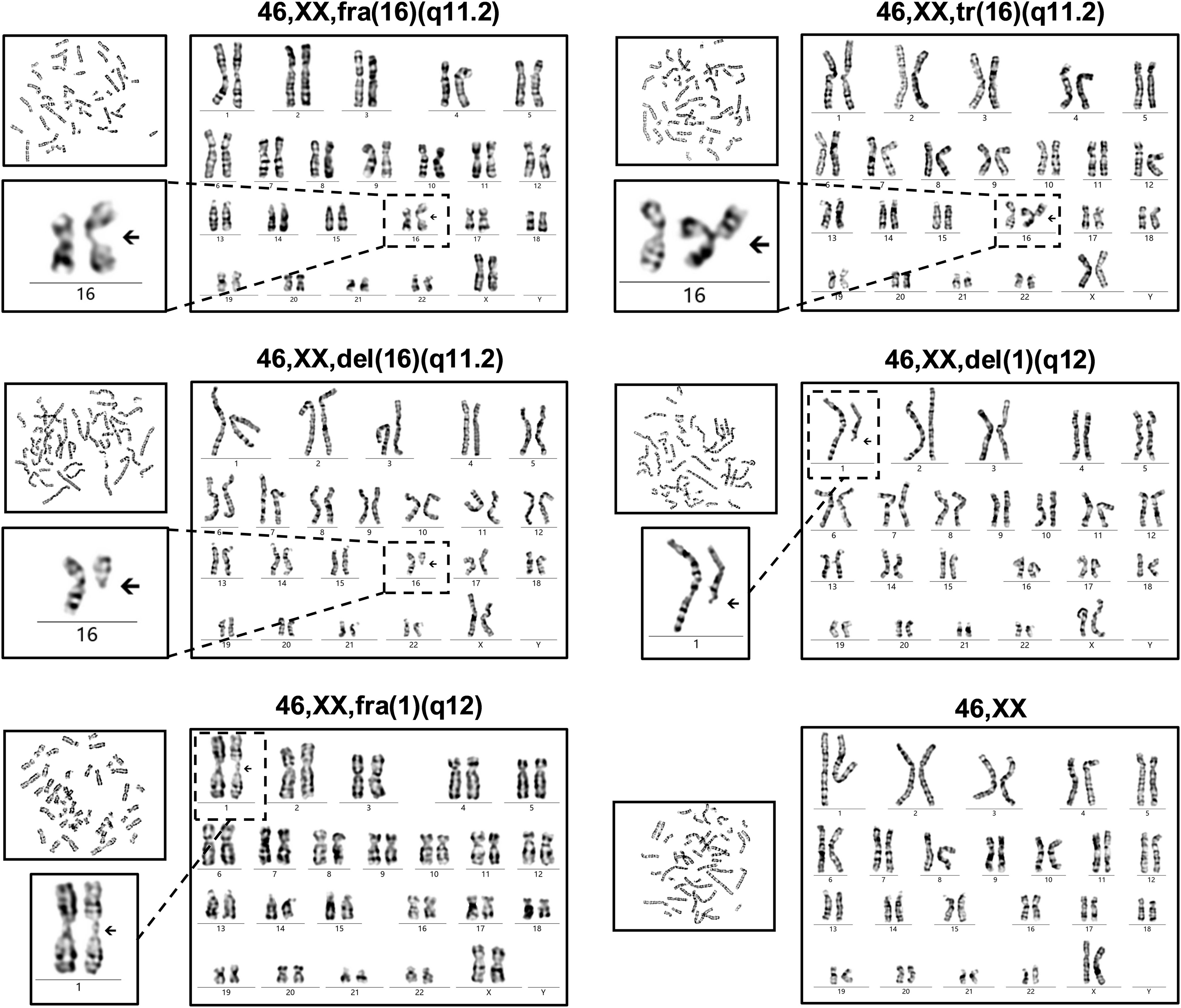
Figure 4. Chromosome karyotype of the ICF2 patient. The chromosome karyotype is described above each set of images. The patient had the characteristic karyotypes associated with DNA hypomethylation in cases with ICF, including fragile sites (fra), whole-arm deletions (del), and triradials (tr).
4 Discussion
Immunodeficiency, centromeric instability, and facial anomalies syndrome (ICF) is a rare genetic defect that is inherited in an autosomal recessive manner, and the pathogenic genes include DNMT3B, ZBTB24, CDCA7, and HELLS (1). Recurrent infections caused by hypogammaglobulinemia are the most prominent clinical symptoms of ICF that prompt these patients to seek medical attention (10). The known cases of ICF were diagnosed based on their symptoms, karyotyping, immune assessment, and genome sequencing results (4). In this study, we initially identified a variant of exons 2-5 deletion of ZBTB24 associated with ICF2 in a 9-year-old girl with severe pneumonia by WES. During validation, we found that exon 1 was also deleted using PCR targeting GC-rich DNA fragments. Further gap-PCR and Sanger sequencing revealed the exact ZBTB24 mutation site (NG_029388.1: g.2831_18,995del). Multiple exon deletions of ZBTB24 are the copy number variations (CNVs) at the exon level with a deletion length of about 16.165 kb. As far as we know, this ZBTB24 variant is a novel variant that has yet to be documented in public databases.
It is noteworthy that ICF is also one of the first identified diseases with DNA methylation defect (2). DNA methylation is ubiquitous in mammals and maintains genome stability by inhibiting the expression and translocation of genomic transposable elements (17). Unfortunately, ICF patients have widespread hypomethylation, primarily affecting the pericentromeric regions of chromosomes 1, 9, and 16, which are typical targets for hypomethylation of highly repetitive satellite DNA sequences that do not encode proteins (12, 18). Chromosome structural abnormalities can be observed in metaphase images of lymphocytes after stimulation with phytohemagglutinin (PHA) in vitro (19). Different chromosomal structural abnormalities were found in our studied case including fragile sites, whole-arm deletions, and triradials. These unusual karyotypes are typical manifestations of DNA hypomethylation and are considered reliable evidence for the diagnosis of ICF (12, 20).
Recent studies have shown that the ZBTB24 protein is a ubiquitously expressed nuclear protein and belongs to the transcription factor family, which is consist of a BTB region, an A-T hook domain, and eight tandem C2H2 zinc fingers (6, 21). The BTB mediates homodimerization, heterodimerization and interactions with other transcriptional coregulators (6). The A-T hook is a functional domain that preferentially binds to short AT-rich DNA fragments (22). The majority of nonsense mutations and frameshift mutations of ZBTB24 caused abnormalities recorded in the ClinVar database are located in the zinc finger region, indicating that zinc fingers may be the crucial structure for maintaining ZBTB24 protein function. It has been demonstrated that zinc fingers are the structural basis for the ZBTB24 protein to specifically recognize and bind to target DNA and exert its transcriptional regulatory functions (6). The multi-exon homozygous deletion variant of ZBTB24 that we report here results in the absence of the original start codon, and the shutoff synthesis of subsequent series of amino acids. The severely truncated ZBTB24 protein has only 132 disordered amino acids, which is likely to lead to the loss of its physiological function as a transcription factor.
The functional deficit of the ZBTB24 protein closely correlates with the occurrence and development of immunodeficiency. In general, ZBTB24 is highly expressed in primary B cells, and downregulation of ZBTB24 will hinder B cell cycle progression and reduce its proliferation (23). Low expression of ZBTB24 impedes B lymphocyte differentiation, leading to a significant decrease in immunoglobulin secretion and thus result in primary immunodeficiency (7). The T cell defects also observed in certain ICF2 patients may be due to the mechanism that the deficiency of ZBTB24 protein inhibits T cell proliferation through the CDCA7/TRAIL receptor axis (24). Moreover, the ZBTB24 protein affects multiple cellular pathways as a transcriptional factor, including amino acid metabolism, oxidative stress response, telomere function, apoptosis, and differentiation (21). Consequently, ICF2 patients develop immunodeficiency and are often accompanied by other systemic diseases such as intellectual disability, slow growth and development, facial abnormalities, inflammatory bowel disease (5), autoimmune (25), atrial septal defect (4), and brain cysts (26). Nevertheless, the type and extent of the structural defect in the ZBTB24 protein do not completely correspond to the severity of the immunodeficiency and the complexity of the symptoms (15, 25, 27). At present, it is still difficult to conclude mutation hotspots in ZBTB24 and genotype-phenotype associations in ICF2, probably due to the limited number of cases in ICF2.
The ICF2 patient we report can be classified as common variable immunodeficiency (CVID) because of recurrent infections, decreased IgG, IgA, and IgM, no secondary causes, a poor response to vaccine, and no profound T cell deficiency (28). The combination approach of intravenous immunoglobulin (IVIG) replacement and anti‐infective is considered to be the first-line treatment option for this immunodeficiency (4, 10, 12). The patient’s parents and physician indicated that the patient’s condition had improved and the frequency of infections had decreased during the early stages of treatment. Regrettably, the patient’s long-term prognosis is unfavorable as primary immunodeficiency cannot be cured with medication. Hematopoietic stem cell transplantation (HSCT) is a radical curative regimen for correcting immunodeficiency in patients with ICF, especially those who have sustained severe infections in infancy or persistent infections that are insufficient with drug treatment (29). There are about 10 cases of ICF children whose life expectancy has been extended through successful HSCT treatment worldwide till now (30, 31).
In summary, we identified a novel pathogenic variant (exons 1-5 del) of ZBTB24 that is highly correlated with ICF2. A review of previous literature related to ZBTB24 variants provides a deeper understanding of the underlying mechanisms of ICF and helps to clarify the association between ICF genotype and phenotype.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
The studies involving humans were approved by First Affiliated Hospital of Guangxi Medical University Ethical Review Committee. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article. Written informed consent was obtained from the participant/patient(s) for the publication of this case report.
Author contributions
YL: Conceptualization, Methodology, Writing – original draft, Writing – review & editing. CW: Data curation, Validation, Writing – original draft. JX: Data curation, Formal analysis, Investigation, Writing – review & editing. YHH: Investigation, Visualization, Writing – review & editing. XL: Investigation, Visualization, Writing – review & editing. CH: Formal analysis, Investigation, Writing – review & editing. YC: Formal analysis, Investigation, Writing – review & editing. JL: Formal analysis, Investigation, Writing – review & editing. RT: Formal analysis, Investigation, Writing – review & editing. FL: Conceptualization, Methodology, Resources, Supervision, Writing – review & editing. YFH: Conceptualization, Funding acquisition, Methodology, Resources, Supervision, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. The work was supported by the “Medical Excellence Award” Funded by the Creative Research Development Grant from the First Affiliated Hospital of Guangxi Medical University.
Acknowledgments
We thank the patient and her parents for participating in this study.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2025.1517417/full#supplementary-material
References
1. Vukic M, Daxinger L. DNA methylation in disease: Immunodeficiency, Centromeric instability, Facial anomalies syndrome. Essays Biochem. (2019) 63:773–83. doi: 10.1042/EBC20190035
2. Maraschio P, Zuffardi O, Dalla Fior T, Tiepolo L. Immunodeficiency, centromeric heterochromatin instability of chromosomes 1, 9, and 16, and facial anomalies: the ICF syndrome. J Med Genet. (1988) 25:173–80. doi: 10.1136/jmg.25.3.173
3. Kiaee F, Zaki-Dizaji M, Hafezi N, Almasi-Hashiani A, Hamedifar H, Sabzevari A, et al. Clinical, immunologic and molecular spectrum of patients with immunodeficiency, centromeric instability, and facial anomalies (ICF) syndrome: A systematic review. Endocrine Metab Immune Disord Drug Targets. (2021) 21:664–72. doi: 10.2174/1871530320666200613204426
4. de Greef JC, Wang J, Balog J, den Dunnen JT, Frants RR, Straasheijm KR, et al. Mutations in ZBTB24 are associated with immunodeficiency, centromeric instability, and facial anomalies syndrome type 2. Am J Hum Genet. (2011) 88:796–804. doi: 10.1016/j.ajhg.2011.04.018
5. Conrad MA, Dawany N, Sullivan KE, Devoto M, Kelsen JR. Novel ZBTB24 mutation associated with immunodeficiency, centromere instability, and facial anomalies type-2 syndrome identified in a patient with very early onset inflammatory bowel disease. Inflammatory Bowel Dis. (2017) 23:2252–5. doi: 10.1097/MIB.0000000000001280
6. Ren R, Hardikar S, Horton JR, Lu Y, Zeng Y, Singh AK, et al. Structural basis of specific DNA binding by the transcription factor ZBTB24. Nucleic Acids Res. (2019) 47:8388–98. doi: 10.1093/nar/gkz557
7. Blanco-Betancourt CE, Moncla A, Milili ML, Jiang YL, Viegas-PéQuignot EM, Roquelaure B, et al. Defective B-cell-negative selection and terminal differentiation in the ICF syndrome. Blood. (2004) 103:2683–90. doi: 10.1182/blood-2003-08-2632
8. Hagleitner MM, Lankester A, Maraschio P, Hulten M, Fryns JP, Schuetz C, et al. Clinical spectrum of immunodeficiency, centromeric instability and facial dysmorphism (ICF syndrome). J Med Genet. (2007) 45:93–9. doi: 10.1136/jmg.2007.053397
9. Kamae C, Imai K, Kato T, Okano T, Honma K, Nakagawa N, et al. Clinical and immunological characterization of ICF syndrome in Japan. J Clin Immunol. (2018) 38:927–37. doi: 10.1007/s10875-018-0559-y
10. Weemaes CM, van Tol MJ, Wang J, van Ostaijen-ten Dam MM, van Eggermond MC, Thijssen PE, et al. Heterogeneous clinical presentation in ICF syndrome: correlation with underlying gene defects. Eur J Hum Genetics: EJHG. (2013) 21:1219–25. doi: 10.1038/ejhg.2013.40
11. Alghamdi HA, Tashkandi SA, Alidrissi EM, Aledielah RD, Alsaidi KA, Alharbi ES, et al. Three types of immunodeficiency, centromeric instability, and facial anomalies (ICF) syndrome identified by whole-exome sequencing in saudi hypogammaglobulinemia patients: clinical, molecular, and cytogenetic features. J Clin Immunol. (2018) 38:847–53. doi: 10.1007/s10875-018-0569-9
12. Sterlin D, Velasco G, Moshous D, Touzot F, Mahlaoui N, Fischer A, et al. Genetic, cellular and clinical features of ICF syndrome: a french national survey. J Clin Immunol. (2016) 36:149–59. doi: 10.1007/s10875-016-0240-2
13. McGowan-Jordan J, Hastings RJ, Moore S eds. ISCN 2020: an international system for human cytogenomic nomenclature. Basel: Karger (2020).
14. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med: Off J Am Coll Med Genet. (2015) 17:405–24. doi: 10.1038/gim.2015.30
15. Nitta H, Unoki M, Ichiyanagi K, Kosho T, Shigemura T, Takahashi H, et al. Three novel ZBTB24 mutations identified in Japanese and Cape Verdean type 2 ICF syndrome patients. J Hum Genet. (2013) 58:455–60. doi: 10.1038/jhg.2013.56
16. Chouery E, Abou-Ghoch J, Corbani S, El Ali N, Korban R, Salem N, et al. A novel deletion in ZBTB24 in a Lebanese family with immunodeficiency, centromeric instability, and facial anomalies syndrome type 2. Clin Genet. (2012) 82:489–93. doi: 10.1111/j.1399-0004.2011.01783.x
17. Chen Z, Zhang Y. Role of mammalian DNA methyltransferases in development. Annu Rev Biochem. (2020) 89:135–58. doi: 10.1146/annurev-biochem-103019-102815
18. Hardikar S, Ren R, Ying Z, Zhou J, Horton JR, Bramble MD, et al. The ICF syndrome protein CDCA7 harbors a unique DNA binding domain that recognizes a CpG dyad in the context of a non-B DNA. Sci Adv. (2024) 10:eadr0036. doi: 10.1126/sciadv.adr0036
19. Ehrlich M. The ICF syndrome, a DNA methyltransferase 3B deficiency and immunodeficiency disease. Clin Immunol (Orlando Fla). (2003) 109:17–28. doi: 10.1016/S1521-6616(03)00201-8
20. Bilgic Eltan S, Nain E, Catak MC, Ezen E, Sefer AP, Karimi N, et al. Evaluation of clinical and immunological alterations associated with ICF syndrome. J Clin Immunol. (2023) 44:26. doi: 10.1007/s10875-023-01620-6
21. Thompson JJ, Kaur R, Sosa CP, Lee JH, Kashiwagi K, Zhou D, et al. ZBTB24 is a transcriptional regulator that coordinates with DNMT3B to control DNA methylation. Nucleic Acids Res. (2018) 46:10034–51. doi: 10.1093/nar/gky682
22. Garabedian A, Jeanne Dit Fouque K, Chapagain PP, Leng F, Fernandez-Lima F. AT-hook peptides bind the major and minor groove of AT-rich DNA duplexes. Nucleic Acids Res. (2022) 50:2431–9. doi: 10.1093/nar/gkac115
23. Liang J, Yan R, Chen G, Feng J, Wu WW, Ren W, et al. Downregulation of ZBTB24 hampers the G0/1- to S-phase cell-cycle transition via upregulating the expression of IRF-4 in human B cells. Genes Immun. (2016) 17:276–82. doi: 10.1038/gene.2016.18
24. Qin XY, Feng J, Chen G, Dou XW, Dai XQ, Dong HL, et al. ZBTB24 regulates the apoptosis of human T cells via CDCA7/TRAIL-receptor axis. Biochem Biophys Res Commun. (2019) 514:259–65. doi: 10.1016/j.bbrc.2019.04.147
25. von Bernuth H, Ravindran E, Du H, Fröhler S, Strehl K, Krämer N, et al. Combined immunodeficiency develops with age in Immunodeficiency-centromeric instability-facial anomalies syndrome 2 (ICF2). Orphanet J Rare Dis. (2014) 9:116. doi: 10.1186/s13023-014-0116-6
26. Cerbone M, Wang J, van der Maarel SM, D’Amico A, D’Agostino A, Romano A, et al. Immunodeficiency, centromeric instability, facial anomalies (ICF) syndrome, due to ZBTB24 mutations, presenting with large cerebral cyst. Am J Med Genet. (2012) 158a:2043–6. doi: 10.1002/ajmg.a.v158a.8
27. van den Boogaard ML, Thijssen PE, Aytekin C, Licciardi F, Kıykım AA, Spossito L, et al. Expanding the mutation spectrum in ICF syndrome: Evidence for a gender bias in ICF2. Clin Genet. (2017) 92:380–7. doi: 10.1111/cge.2017.92.issue-4
28. Lee TK, Gereige JD, Maglione PJ. State-of-the-art diagnostic evaluation of common variable immunodeficiency, Annals of allergy, asthma & immunology: official publication of the American College of Allergy. Asthma Immunol. (2021) 127:19–27. doi: 10.1016/j.anai.2021.03.005
29. Harnisch E, Buddingh EP, Thijssen PE, Brooks AS, Driessen GJ, Kersseboom R, et al. Hematopoietic stem cell transplantation in a patient with ICF2 syndrome presenting with EBV-induced hemophagocytic lymphohystiocytosis. Transplantation. (2016) 100:e35–6. doi: 10.1097/TP.0000000000001210
30. Gennery AR, Slatter MA, Bredius RG, Hagleitner MM, Weemaes C, Cant AJ, et al. Hematopoietic stem cell transplantation corrects the immunologic abnormalities associated with immunodeficiency-centromeric instability-facial dysmorphism syndrome. Pediatrics. (2007) 120:e1341–4. doi: 10.1542/peds.2007-0640
Keywords: case report, ZBTB24, ICF syndrome 2, nonsense variant, intravenous immunoglobulin
Citation: Long Y, Wang C, Xiao J, Huang Y, Ling X, Huang C, Chen Y, Luo J, Tang R, Lin F and Huang Y (2025) Case report: Novel multi-exon homozygous deletion of ZBTB24 causes immunodeficiency, centromeric instability, and facial anomalies syndrome 2. Front. Immunol. 16:1517417. doi: 10.3389/fimmu.2025.1517417
Received: 26 October 2024; Accepted: 13 January 2025;
Published: 31 January 2025.
Edited by:
Silvia Clara Giliani, University of Brescia, ItalyReviewed by:
Faranaz Atschekzei, Hannover Medical School, GermanyUwe Kölsch, IMD Laboratory Berlin, Germany
Copyright © 2025 Long, Wang, Xiao, Huang, Ling, Huang, Chen, Luo, Tang, Lin and Huang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yifang Huang, MDUwMWh5ZkAxNjMuY29t; Faquan Lin, ZnFsaW4xOTk4QDE2My5jb20=