
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol., 21 March 2025
Sec. Cancer Immunity and Immunotherapy
Volume 16 - 2025 | https://doi.org/10.3389/fimmu.2025.1493329
This article is part of the Research TopicThe Next Stage of Immune Cell Design: Selective Targeting of Multi-Antigen ProfilesView all 7 articles
 Breanna DiAndreth
Breanna DiAndreth Pavlo A. NesterenkoAaron G. Winters
Pavlo A. NesterenkoAaron G. Winters Aaron D. FlynnClaudia A. JetteVasantika SuryawanshiSanam Shafaattalab
Aaron D. FlynnClaudia A. JetteVasantika SuryawanshiSanam Shafaattalab Sara MartireMark DarisElizabeth MooreRyan Elshimali
Sara MartireMark DarisElizabeth MooreRyan Elshimali Tanveer Gill
Tanveer Gill Timothy P. RileySara Miller
Timothy P. RileySara Miller Chawita NetirojjanakulAgnes E. Hamburger
Chawita NetirojjanakulAgnes E. Hamburger Alexander Kamb*
Alexander Kamb*Introduction: Despite advances in treatment of blood cancers, several—including acute myeloid leukemia (AML)—continue to be recalcitrant. Cell therapies based on chimeric antigen receptors (CARs) have emerged as promising approaches for blood cancers. However, current CAR-T treatments suffer from on-target, off-tumor toxicity, because most familiar blood cancer targets are also expressed in normal lineages. In addition, they face the common problem of relapse due to target-antigen loss. Cell therapeutics engineered to integrate more than one signal, often called logic-gated cells, can in principle achieve greater selectivity for tumors.
Methods: We applied such a technology, a NOT gated system called Tmod™ that is being developed to treat solid-tumor patients, to the problem of therapeutic selectivity for blood cancer cells.
Results: Here we show that Tmod cells can be designed to target 2-4 antigens to provide different practical and conceptual options for a blood cancer therapy: (i) mono- and bispecific activating receptors that target CD33, a well-known AML antigen expressed on the majority of AML tumors (as well as healthy myeloid cells) and CD43 (SPN), an antigen expressed on many hematopoietic cancers (and normal blood lineages); and (ii) mono- and bispecific inhibitory receptors that target CD16b (FCGR3B) and CLEC9A, antigens expressed on key normal blood cells but not on most blood cancers.
Discussion: These results further demonstrate the robust modularity of the Tmod system and generalize the Tmod approach beyond solid tumors.
Certain blood cancers such as AML are deadly diseases, with no broadly effective therapies other than chemotherapy preconditioning combined with stem cell transplant (1, 2). One of the fundamental challenges is the lack of genes expressed uniquely in AML and other blood cancers that can be exploited as molecular targets for selective elimination of malignant cells (3, 4). Most (if not all) AML molecular targets under consideration are also expressed in subsets of normal lineages. A good example is CD33, a lectin that is expressed on >90% of AML tumors but also on normal immune cells, especially myeloid cells such as monocytes, promyelocytes and neutrophils (5–8). Despite the relative appeal of CD33 as an AML tumor-associated antigen (TAA), therapeutics targeting CD33 have substantial clinical toxicity, presumably caused at least in part by expression on healthy myeloid cells (9–11). For instance, an FDA-approved antibody-drug conjugate (Mylotarg™) has serious dose-limiting toxicity that limits its use and efficacy, some of which is likely caused by on-target toxicity (12–15). CD33-directed T cell engagers have encountered similar problems in the clinic, with the additional challenge of cytokine release syndrome (CRS), a potentially serious immune overreaction caused by antigen expression on normal cells (16).
Although cell therapies have emerged as a powerful new weapon against certain types of blood cancer including Non Hodgkin’s lymphoma (NHL) and multiple myeloma (17, 18), so far they have not demonstrated the same success in AML (19). In contrast to NHL and certain other blood cancers, AML does not possess any genes that are expressed: (i) in the large majority of tumors indicated for treatment; and (ii) only in normal cells that are not required for survival in the short term (e.g., B cells) (7, 8). Indeed, there is little to suggest that TAA-directed AML therapeutics, including CD33 CAR-Ts, can avoid the toxicities caused by loss of key normal immune cells. This poses immediate safety issues and longer-term risk of infection (20). Though the precise cause of toxicities in individual patients can be complex and difficult to elucidate, CD33-directed investigational cell therapies can be severely toxic, and potentially fatal (21). This places drug developers in a quandary: How can toxicities be mitigated while maintaining the advantages of TAAs such as CD33?
One possibility involves cell therapeutics that respond to more than one antigen to achieve better selectivity toward AML cells. An interesting option is to engineer a NOT gate in immune cells to protect normal CD33-expressing cells without compromising AML cytotoxicity (22, 23). The best studied NOT gate is Tmod, a system that has demonstrated robust activity in multiple preclinical settings (24–26). Tmod is being developed to treat patients whose tumors have lost one or both alleles at the HLA locus via deletion (25, 27). This system employs T cells engineered to express an activating receptor (i.e. CAR or TCR) paired with a “blocker” derived from the LIR-1 (LILRB1) inhibitory receptor, the natural ligands of which are HLA-I molecules (28) (Figure 1A). Tmod therapeutic constructs have been shown to work in many tumor models of HLA deletion.
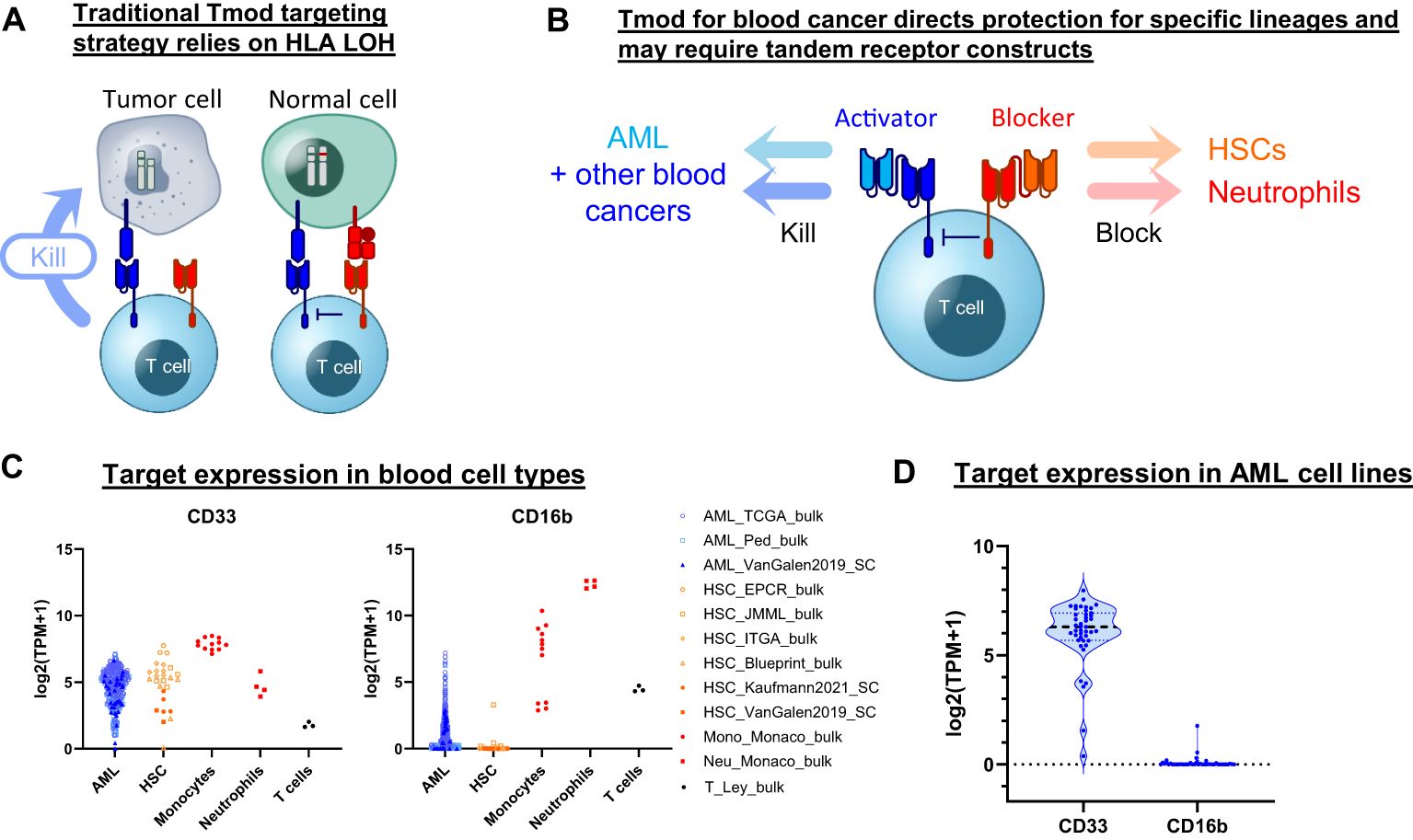
Figure 1. Tmod can be adapted for blood cancer. (A) Diagram for Tmod system showing the two receptors that comprise the NOT gate targeting HLA loss of heterozygosity (LOH) in solid tumors. (B) Diagram for Tmod utilizing tandem receptors for blood cancer. (C) CD33 and CD16b mRNA expression in primary AML and healthy blood cells including T cells, neutrophils, monocytes, and hematopoietic stem cells (HSC) (data from sources shown; see Supplementary Table 1). (D) mRNA expression of targets in AML cell lines (n=43; DepMap).
Here we describe a Tmod construct designed to improve the safety of CD33 targeted therapeutics. The engineered Tmod cells respond to CD33 only if a second antigen, CD16b, is absent. Because CD16b is expressed on normal myeloid lineages and not on AML, this approach offers the possibility of mitigating myeloid compartment depletion and CRS by specifically blocking healthy-cell-mediated activation of the CD33 | CD16b Tmod cells by normal myeloid cells. We build on this construct by adding OR-gating capability through additional ligand-binding domains to broaden the potential applicability of Tmod beyond monospecific AML designs. These designs include bispecific receptors that target 2 activator antigens (CD33 and SPN) and 2 blocker antigens (CD16b and CLEC9A) (Figure 1B). The results support further investigation of the Tmod approach to improve the therapeutic window of future blood cancer medicines and provide further proofs of concept for the use of non-HLA-I proteins as blocker antigens for Tmod constructs.
We sought to identify a blocker antigen compatible with Tmod constructs that incorporate a CD33 CAR activator. A bioinformatic search of RNA-Seq databases (29–40)(Supplementary Table 1) revealed that CD16b (FCGR3B) expression not only corresponds with CD33 expression in neutrophils but also is largely absent in primary AML specimens (Figure 1C). CD16b, first identified as Human Neutrophil Antigen-1, is a glycosylphosphatidylinositol-anchored protein that functions as a low-affinity receptor for the Fc region of IgG, with high surface expression limited to granulocytes (41). The absence of FCGR3B expression in AML is not the consequence of genetic deletion, but rather the epigenetic state of the malignant cell (Supplementary Figure 1). Low expression of FCGR3B is also observed in the large majority of AML cell lines, suggesting that its transcriptional off-state is robustly maintained in cell culture—much as the CD33 on-state is maintained (Figure 1D). Thus, based on expression profile alone, CD16b is an attractive blocker antigen for a Tmod construct directed at CD33.
To create CD33 | CD16b Tmod constructs, we began with a set of 4 CD33 CARs (CAR1-4) that were sensitive enough to respond to CD33 in AML-derived cell lines and two other cells lines (HeLa and K562) transfected with CD33 mRNA (Supplementary Figure 2; see Methods). We combined CAR1 with a blocker that uses an scFv derived from a commercially available monoclonal antibody (mAb; 3G8) (42) that binds both human CD16 paralogs (CD16a and CD16b) with high affinity. CD33 Tmod constructs using this CD16-directed scFv demonstrated selective response in both Jurkat reporter assays (Supplementary Figure 3A) and in primary T cell cytotoxicity assays (Supplementary Figure 3B). These results supported the feasibility of targeting CD16 via CD33 | CD16 Tmod constructs. However, because CD16a is expressed in a higher percentage of AML samples than CD16b (Supplementary Figure 4), a search for CD16b-specific binders was undertaken.
Despite the high similarity of the two CD16 paralogs (98% amino acid identity), a mouse immunization campaign, using soluble recombinant CD16b protein as the antigen, yielded a panel of CD16b-selective antibodies (Supplementary Figure 5A). Selectivity was confirmed in ELISA assays and via flow cytometry with engineered K562 cells that overexpress either CD16a or CD16b (Supplementary Figure 5B,C). Diverse VH and VL sequences were recovered from 32 CD16b-selective antibodies (Supplementary Figure 5D; see Methods). The VH and VL sequences were used to construct scFvs and fused to the LIR-1 (LILRB1) backbone to create blocker receptors. Tmod constructs encoding the CD33 CARs and CD16b blockers were tested in assays using engineered Jurkat cells cocultured with K562 target cells that overexpressed CD33. mRNA titration was used to estimate ligand-dependent inhibition (Figures 2A, B). The initial screen focused on a single CD33 CAR as the activator component of Tmod to measure percent-inhibition in K562 cells. The 10 top-ranked blockers in this assay were selected based on percent blocking and IC50 for further analysis (Supplementary Figure 6A). These 10 blockers were screened independently with 3 additional CD33 CARs (4 total) to identify the most robust and modular blockers (Supplementary Figure 6B). Finally, the top 6 blockers from this assay, plus the 3G8 clone benchmark, were combined for further testing with a set of 3 top CARs. These CD33 | CD16b Tmod combinations were profiled using 8-point mRNA dose-response titrations. Fitted sigmoid curves allowed selection of the CD33 | CD16b Tmod receptor pairs with optimal performance, based on a combination of high sensitivity (low IC50) and high maximum percent inhibition (Figure 2C, Supplementary Figure 6C). From this analysis, 9 pairs with varied functional profiles were advanced for further testing in T cells.
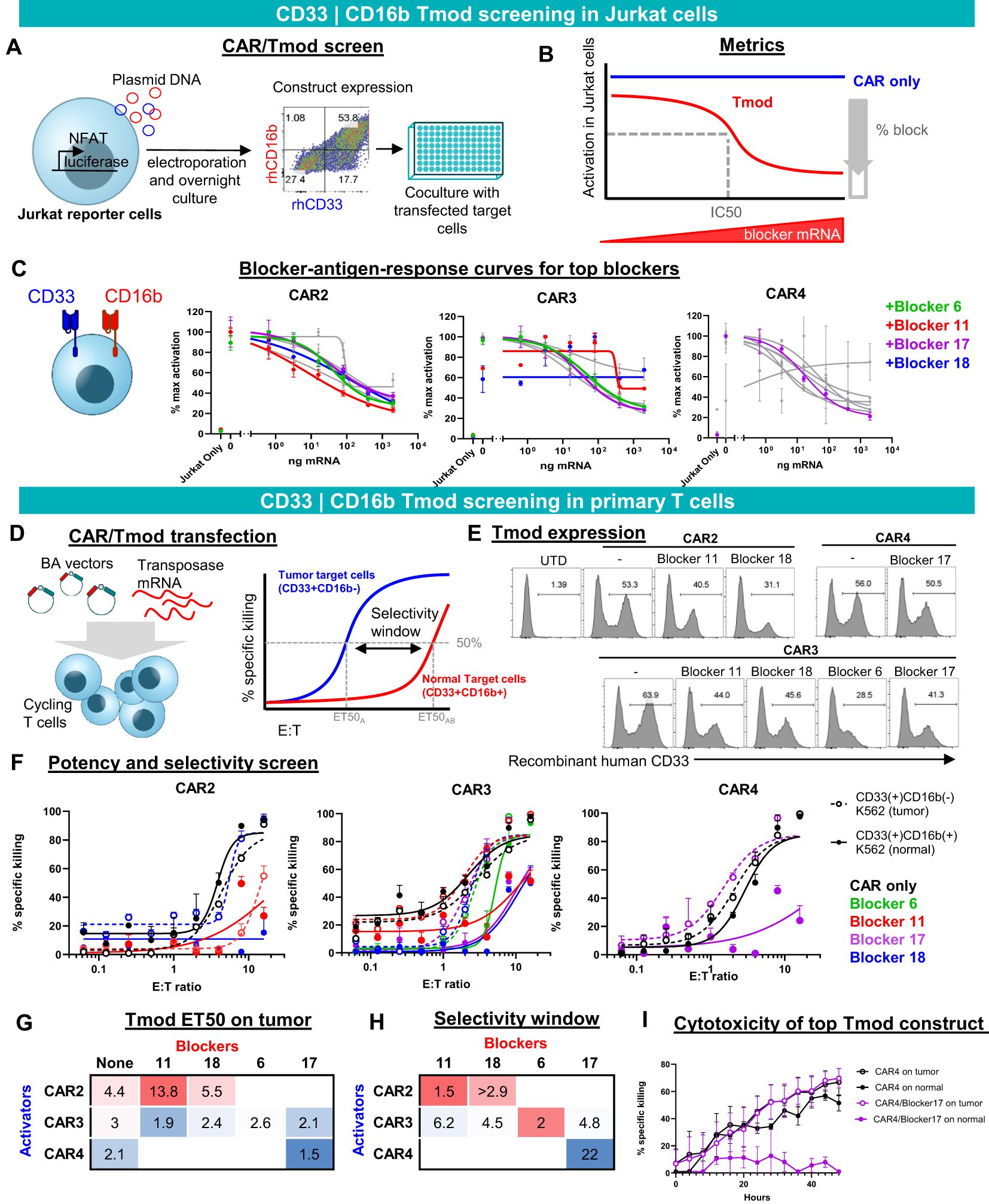
Figure 2. CD33 | CD16b Tmod functions robustly in Jurkat and primary T cells. (A) Diagram of functional screen in Jurkat reporter cell line cocultured with K562 target cells transfected with different amounts of CD16b mRNA. Tmod transgene expression in Jurkat cells was detected by staining with recombinant human (rh) CD16b and CD33. (B) Diagram of functional parameters estimated from the Jurkat cell assay data. (C) Functional readout from 8-point mRNA titration curves. Three CARs combined with 4 blockers, that were selected for further analysis, are shown in color. Data are shown as mean ± standard deviation of technical replicates (n=2), normalized to each sample’s maximum activation. (D) Left, diagram of non-viral construct-screening in primary T cells using PiggyBac transposase and single vectors (BA vectors). Right, metrics used to quantify the potency and selectivity of the Tmod pair. (E) Flow cytometry analysis of stable integrants via staining with labeled recombinant human CD33 (see Methods). (E, F) T cell cytotoxicity curves generated from GFP signal at 48 hour time point with each well normalized to the zero time point. Tumor (CD33(+)CD16(-)) target-cell curves are shown with dashed lines and “normal” (CD33(+)CD16(+)) target-cell curves with solid lines. Black lines are CAR constructs and colored lines are Tmod constructs. Tumor cells are K562 cells engineered with CD33 and normal cells are K562 cells engineered to overexpress CD33 and CD16b. Data are shown as mean ± standard deviation of technical replicates (n=3). (G) Potency calculated as ET50 of Tmod cells cocultured with tumor cells. (H) Selectivity ratios are calculated as ET50 on normal cells divided by ET50 on tumor cells. (I) Kinetic cytotoxicity analysis of the most selective and potent construct compared to the CAR-T. GFP(+) area was used as proxy for target cell viability. Data are shown as mean ± standard deviation of technical replicates (n=3).
To create constructs suitable for expression in primary human T cells, we generated PiggyBac vectors with inserts that produce a single transcript encoding bicistronic CD33 | CD16b Tmod. We term these BA vectors because the blocker is encoded upstream of the activator (Figure 2D), which helps to prevent activator-only expression (Supplementary Figure 8A). Of 9 pairs that were chosen to advance to the primary T cell stage, 7 expressed well as measured by staining with labeled CD33 soluble protein. Though some variability in the percent Tmod(+) cells was observed, the expression system consistently produced 30-50% Tmod(+) cells, depending on the CD33 | CD16b pair (Figure 2E).
We focused on two key parameters of Tmod function in cytotoxicity assays: potency and selectivity (Figure 2D, right). To measure these parameters, E:T titration experiments were performed using two variants of K562 as the target cells: (i) CD33(+)CD16b(-) cells that overexpress CD33, intended to represent tumor cells; and (ii) CD33(+)CD16b(+) cells that overexpress both CD33 and CD16b, intended to model normal myeloid cells in this experiment (Figure 2F). Potency was estimated by the E:T ratio at 50% maximum killing (ET50). As a rough gauge of potency, ET50s of Tmod cells cocultured with CD33(+)CD16(-) target cells were compared to the corresponding CAR T cells without the blocker. A lead pair (CAR4 | Blocker17) was selected for further study based on potency of the Tmod cells similar to the CAR control (Figure 2G).
We defined selectivity as a ratio of ET50 using surrogate normal CD33(+)CD16b(+) cells divided by ET50 using tumor CD33(+)CD16b(-) cells. The construct that had the best potency (CAR4/Blocker17) also had the best selectivity window (Figure 2H). This CD33 | CD16b Tmod construct also displayed robust on-target activity and good normal cell selectivity in kinetic assays of cytotoxicity (Figure 2I). Thus, the CAR4 | Blocker17 construct displayed several desirable features of a CD33 | CD16b Tmod candidate: high potency (low ET50) and high selectivity window (22x).
To test CD33 | CD16b Tmod cells in vivo, we utilized a xenograft model based on growth of the AML line MV-4-11 in NSG mice after intravenous infusion of tumor cells (Figure 3A). MV-4-11 cells express endogenous CD33 at levels similar to primary AML samples and within the sensitivity range of our lead CAR (Supplementary Figure 7). Because these cells do not express CD16b, we engineered a variant line to model normal cells by exogenous expression of CD16b (Supplementary Figure 8B). Both variants were further modified to express firefly luciferase and green fluorescent protein (GFP) for detection. The pair of MV-4-11 cell lines, when cocultured in vitro with a titration of effector CD33 | CD16b Tmod cells, elicited the expected pattern of cytotoxicity; i.e., a selectivity window of 7x (Figure 3B). CD33 | CD16b Tmod cells generated via PiggyBac and cultured for 31 days showed high expression of the Tmod components (Figure 3C).

Figure 3. CD33 | CD16b Tmod cells selectively kill tumor but not “normal” cells in vivo. (A) Schema for in vivo experiment. 2 million MV-4-11 AML cells or MV-4-11 cells that overexpress CD16b were injected into NSG-SGM3 mice and 6 days later 7.5 million T cells were injected. (B) Selectivity in vitro using MV-4-11 cells. Surrogate normal cells were generated by overexpression of CD16b in the AML cells. E:T cytotoxicity curves were generated from firefly luciferase bioluminescence at 48 hours. Data are shown as mean ± standard deviation of technical replicates (n=3). Inset: ET50 values of depicted curves. Data shown are interpolated values with 95% CI. (C) Flow cytometry analysis of construct expression by staining with labeled recombinant human CD16b and CD33. (D) Bioluminescence imaging (BLI) at 20 days post target-cell injection. (E) Flow cytometry analysis of MV-4-11 cells in the bone marrow 27 days post target-cell injection. (F) Quantification of data shown in panel. (E) Statistics were calculated using a non-parametric Kruskal-Wallis H test; *: 0.01 < adjusted p < 0.05; **: adjusted p value < 0.01; ns: not significant (adjusted p > 0.05).
Six days after the grafts were introduced, T cells were injected intravenously and MV-4-11 cells were monitored by BLI (Figure 3A). As observed in vitro, CD33 | CD16b Tmod cells killed the CD33(+)CD16b(-) tumor cells but not the CD33(+)CD16b(+) variant line in vivo (Figure 3D). As an independent readout of graft cell number, bone marrow was isolated from the mice at the end of the study (day 27 post MV-4-11 graft injection) and cells were analyzed by flow cytometry. In mice bearing CD33(+)CD16b(+) surrogate normal cells, numerous GFP(+) MV-4-11 cells were detected (Figures 3E, F). In contrast, very few graft cells were detected in mice from the CD33(+)CD16b(-) cohort, suggesting high selective killing in vivo of tumor vs. surrogate normal cells. Whereas CD33 CARs killed both MV-4-11 variant lines equally, CD33 | CD16b Tmod cells exhibited over 100x-fold increased killing of tumor cells compared to surrogate normal. Thus, in vivo AML tumor cells were killed by CD33 | CD16b Tmod cells while the surrogate normal cells, differing only by their expression of CD16b, were protected. These results provide a preclinical proof of concept for a potential CD33-targeted cell therapy gated by CD16b expression intended to eliminate AML cells in patients while sparing healthy myeloid cell types.
To extend the potential utility of Tmod beyond monospecific designs, we first tested Tmod constructs with tandem activators and blockers using previously validated binders (Figure 4). As an activator, we tested a CD19-CD20 bispecific CAR (43) in Jurkat cell assays and demonstrated that it was effectively inhibited by a previously studied monospecific HLA-A*02 blocker (24, 25, 27) in a ligand-dependent fashion (Figure 4A). The HLA-A*02 blocker inhibited the tandem CAR as effectively as monospecific CARs. Importantly blocking of the tandem CAR was maintained with either one or both activator antigens present. Next, to explore bi-specific blockers of more familiar design, we showed that a mesothelin (MSLN) CAR (25) was blocked by a bispecific blocker composed of one scFv directed at HLA-A*02 and a second scFv in tandem directed at HLA-A*03 (25) (Figure 4B). Finally, these two bispecific receptors were combined in a single cell to demonstrate that the CD19-CD20 bispecific CAR was blocked by the tandem HLA-A*02-A*03 blocker (Figure 4C). Thus, the double-tandem Tmod NOT gate successfully integrated signals from 4 antigens—2 activator and 2 blocker antigens—to regulate effector cell activation and inhibition.
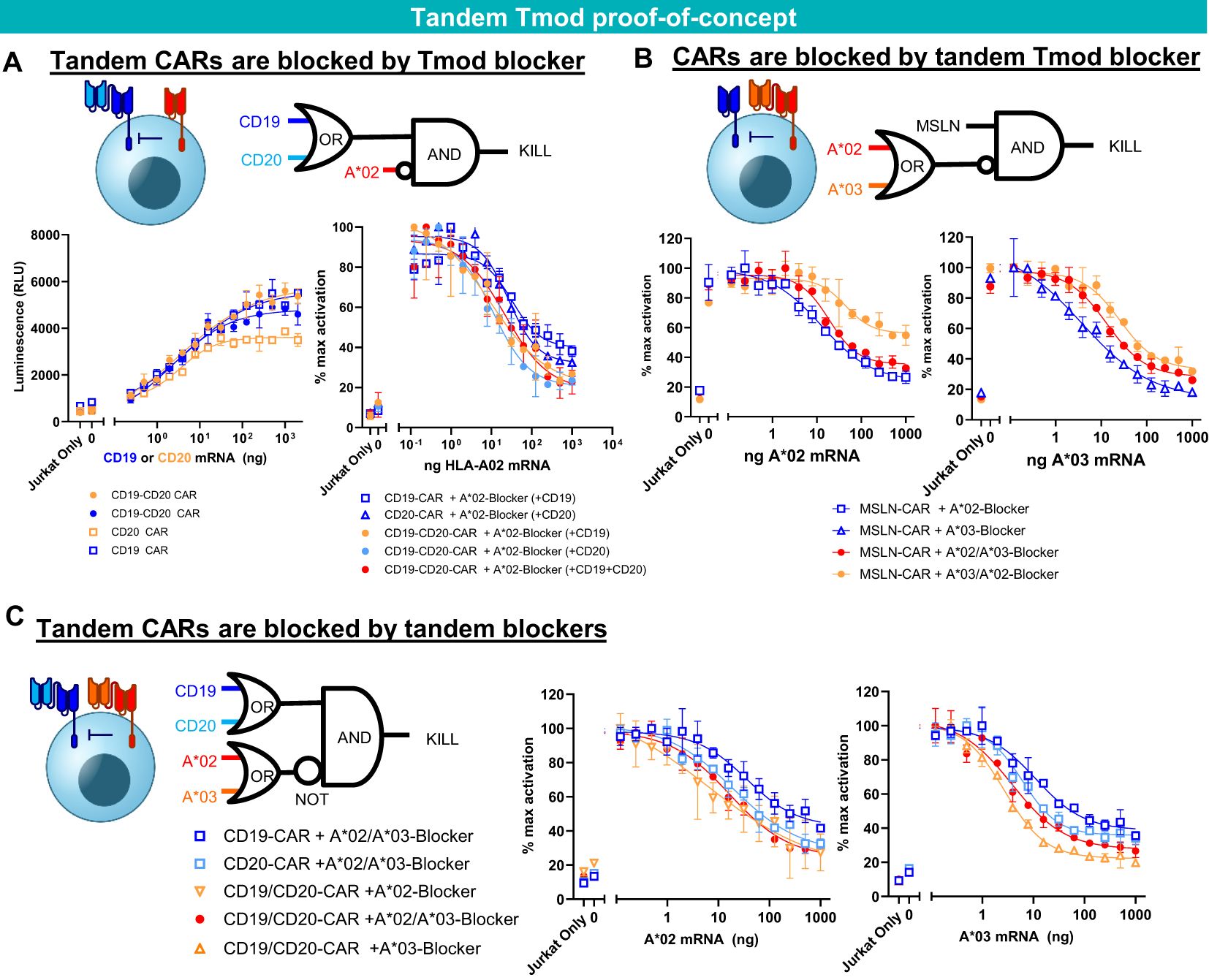
Figure 4. Tandem construct proof of concept. (A) A*02 blocker inhibits CD19-CD20 bispecific CAR. Jurkat functional readout from activator titration curves and blocker titration curves. (B) A*02-A*03 tandem blocker inhibits monospecific MSLN CAR. Jurkat (B2M KO) functional readout from blocker titration curves (either A*02 or A*03). (C) Tandem A*02-A*03 blocker inhibits tandem CD19-CD20 CAR. Jurkat (B2M KO) functional readout from blocker titration curves (either A*02 or A*03) with constant amounts of CD19 and CD20 mRNA. Data are shown as mean ± standard deviation of technical replicates (n=2).
To incorporate these ideas and findings in the context of AML and other blood cancers, we tested a variety of bispecific constructs targeting CD33, CD16b and other blood-related antigens (Figures 5, 6A). These were intended to target antigens expressed potentially on a broader population of blood cancers, while protecting other key normal lineages, primarily by sparing hematopoietic stem cells (HSCs). For AML, this approach also provided an opportunity to address AML relapse caused by CD33 antigen loss. To this end, we identified one additional activator antigen (SPN) and blocker antigen (CLEC9A) with expression profiles consistent with our aims (Figure 6B, C, Supplementary Table 1). SPN (CD43 or sialophorin) is a surface glycoprotein of poorly understood function highly expressed on most hematopoietic cells other than red blood cells (44). CLEC9A (CD370) is a C-type lectin known to be expressed on myeloid lineages (45) including HSCs (46, 47).
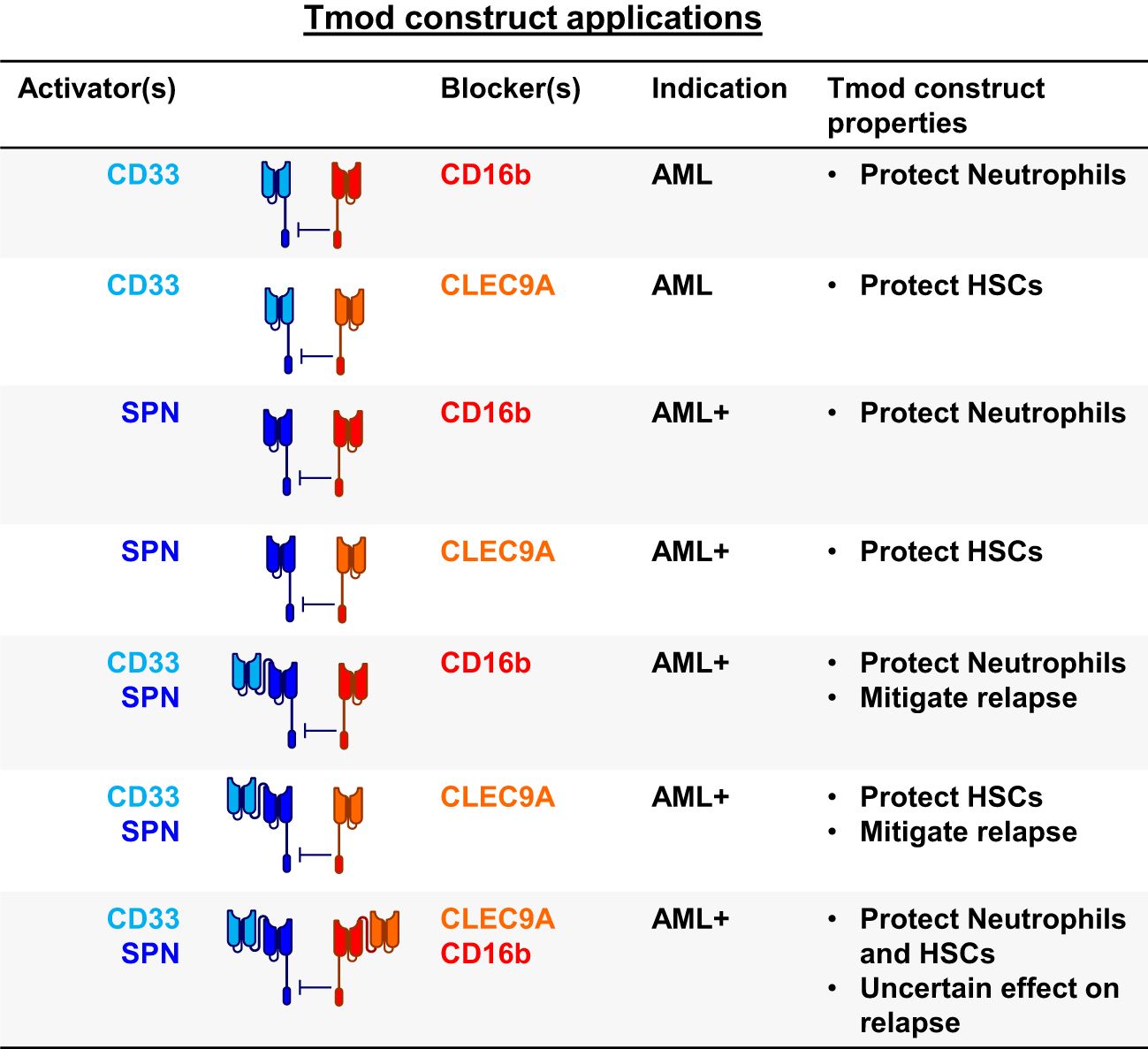
Figure 5. Blood cancer applications for Tmod constructs. SPN (CD43 protein); FCGR3B (CD16b protein); HSCs, hematopoietic stem cells; AML+ refers to blood cancers beyond AML.

Figure 6. Tandem Tmod constructs for blood cancer. (A) Diagram of a Tmod cell with bispecific activator to target AML (CD33) and other blood cancers (SPN) and bispecific blocker to protect HSCs (CLEC9A) and neutrophils (CD16b). (B) Target expression in primary blood cancers and healthy blood cells (data from sources shown; see Supplementary Table 1). (C) Target expression in blood cancer cell lines (DepMap). (D) Jurkat cell (SPN KO) functional readout of SPN | CD16b Tmod with blocker titration curves. (E) Jurkat cell (SPN KO) functional readout of SPN-CD33 tandem CAR activation and blocking by CD16b blocker in the presence of SPN and CD33 antigens. (F) Jurkat functional readout of binders cloned as CARs with titration of primary HSCs. (G) Jurkat functional readout of CD33 CAR4 blocked by tandem CLEC9A-CD16b blocker. (H) Jurkat functional readout of CD33 and/or SPN monospecific or bispecific activators paired with CD16b and/or CLEC9A monospecific or bispecific blockers. Data are shown as mean ± standard deviation of technical replicates (n=2).
For SPN, two CARs with different scFvs directed at SPN were tested against 4 CD16b blockers (Figure 6D). All the constructs performed well (>50% block). One of the SPN CARs was converted to bispecific SPN-CD33 formats and tested with various binders in different orientations (Supplementary Figure 9A). The top-ranked 3 tandem CARs were paired with CD16b Blocker17 (Supplementary Figure 9B) and shown to regulate Jurkat-cell function effectively (see data from best-performing pair in Figure 6E).
For CLEC9A, novel CLEC9A binders were identified and screened as CARs (Supplementary Figure 10; see Methods). Two top-ranked binders were then tested for responsiveness to primary HSCs (Figure 6F). While controls and CD16b CAR-T cells did not show HSC-dependent activation, the CLEC9A CAR-Ts showed responses similar to a CD33 CAR-T, suggesting sufficient target expression and binder sensitivity for response. These top CLEC9A binders were then cloned as blockers and, in tandem with CD16b Blocker17 scFv, shown to function well (see best-performing construct in Figure 6G). Constructs that contained the tandem activators and blockers (CD33-SPN | CD16b-CLEC9A Tmod) expressed in Jurkat cells and functioned as expected from the behavior of the individual components; i.e., each scFv responded to antigen titration without interference from the others (Figure 6H). Together, these data showed that Tmod functions with tandem activators and blockers directed at novel non-HLA-I antigens relevant to blood cancer therapy.
Tumor vs. normal selectivity is arguably the key problem in cancer therapy and many approaches have been developed to address it. These efforts include: (i) targeting critical functional dependencies in tumors such as HER2 in breast cancer and BCR-ABL in CML (48–51); (ii) targeting TAAs such as CD19 in NHL and BCMA in multiple myeloma (52, 53); (iii) targeting tumor-specific antigens such as HPV E7 and other neoantigens in solid tumors (54–58); (iv) protease-cleavable masked cytotoxic proteins designed to activate selectively in solid tumors (59); and (v) logic-gated cell therapies that respond to antigen profiles rather than single antigens (60–63).
The Tmod system was developed to address the problem of tumor vs. normal tissue selectivity via a NOT gate (26). The NOT gate conceptually allows a different set of antigens to be engaged to confer selectivity—those that are absent from the tumor but present in key normal tissues (26). In its original form, Tmod is intended to exploit a situation that arises in numerous cancer patients where solid tumors lose one HLA-I allele via loss of heterozygosity (LOH) in the founding clone of the tumor (24, 64). HLA LOH in these patients confers a genetically identifiable, homogeneous difference on the tumors that can in principle direct Tmod cells gated by, for example, HLA-A*02 to selectively kill tumor cells that express a TAA but have lost expression of HLA-A*02 (25, 27).
Here we extend this approach to tumor types such as AML that do not undergo HLA LOH. To do this, we have developed a blocker that targets a non-HLA-I molecule, in this case CD16b, and paired it with a CD33 CAR to create a CD33 | CD16b Tmod construct that selectively kills CD33(+)CD16b(-) AML cells, but spares isogenic cells that express CD16b, in vitro and in vivo. The expression profile of CD16b (very low in AML and high in myeloid cells) suggests that CD33 | CD16b constructs can create a therapeutic window in AML patients where CD33 is expressed on key normal tissues. In addition, though the cause of CRS is debated, it is possible that engagement of CD16b to block T cell activation by myeloid cells may mitigate the component of CRS that arises from reaction to CD33-expressing normal cells (65, 66).
Creation of a functional CD33 | CD16b Tmod construct required solution of two technical problems: (i) selection of a blocker antigen that is compatible with blocker function and the inverse of an AML TAA (i.e., with expression that is low in AML and high in normal blood cells); and (ii) identification of an scFv that can distinguish CD16b from its very close paralog, CD16a, which is expressed in a subset of AML malignancies and thus could limit efficacy. These challenges were overcome by a combination of antigen selection with screens for binding and function.
To improve the CD33 | CD16b construct and address matters that include relapse from single-antigen-loss in AML, blood cancers beyond AML, and protection of HSCs, we created proof-of-concept Tmod designs that incorporate 2 additional antigen-binding domains directed at SPN and CLEC9A placed in tandem with the CD33 scFv for the SPN activator and the CD16b scFv for the CLEC9A blocker. Others have shown that a NOT gate targeting endomucin (EMCN) to protect HSCs and a tandem CD33-FLT3 CAR to target AML cells functions in preclinical experiments (67). Here we extend these concepts to include tandem blockers with broader protection of normal cells and activators capable in principle of targeting blood cancers beyond AML, including NHL and multiple myeloma that express SPN.
It is not clear a priori which of the many possible multi-targeted designs are optimal for AML and other blood cancers (see Figure 5). Targeting 2 activator antigens should mitigate relapse, a common problem with CD19 CAR-Ts in NHL and all AML therapies. Indeed CD19-C20 bispecific CAR-Ts are under development in NHL for this purpose (68). The advantage of using a broadly expressed antigen such as SPN, whose expression is also maintained in most blood-cancer-derived cell lines, is to not only address relapse in AML but also potentially extend the product to patients with other blood cancers. These additional antigens compound the risk of on-target, off-tumor toxicity, challenges that may be addressed by multi-antigen-targeted blockers. However, depending on the propensities of tumors to up-regulate, rather than lose, expression of genes, such blockers may introduce an additional mechanism for tumor relapse. Note also that inclusion of activators that target antigens expressed in T cells (e.g., SPN) may necessitate other measures to reduce fratricide, e.g., inclusion of shRNA modules or gene-disruption to reduce/eliminate expression of the target antigen in the engineered T cells.
The flexibility and modularity of Tmod demonstrated here and in other publications support exploration of the Tmod technology in tumors that do not exhibit LOH. Although LIR-1 (LILRB1) evolved to specialize in HLA-I antigens (69), it is now clear that, providing certain design rules are followed, non-HLA-I antigens can also function as redirecting ligands for LIR-1 (70, 71). These results are important because many tumor types including blood tumors and certain solid tumors such as prostate carcinoma (see GISTIC database) do not exhibit high rates of HLA LOH. In addition, even among solid tumors that exhibit significant rates of LOH, a large segment (>75%) remains that do not have HLA LOH. Patients with these tumors require a different therapeutic approach. The demonstration that non-HLA-I antigens can robustly control CAR activation in Tmod cells suggests that it may be possible to exploit instances where antigen expression is absent in tumors for reasons other than HLA LOH.
All cell lines were purchased from ATCC, except for the NFAT-firefly luciferase reporter Jurkat cell line that was purchased from BPS bioscience (#60621). K562 cells were maintained in RPMI supplemented with 10% heat inactivated FBS and 1% penicillin-streptomycin. HeLa cells were maintained in EMEM with 10% FBS. Jurkat cells were cultured in RPMI supplemented with 10% heat-inactivated FBS, 1% penicillin-streptomycin and 0.4% geneticin to maintain reporter expression. MV-4-11 cells were cultured in RPMI with 10% heat-inactivated FBS and 1% penicillin-streptomycin.
PBMCs were obtained from STEMCELL. Enriched T cells were derived from Leukopaks apheresed from healthy donors (Charles River), followed by enrichment using CD56 depletion and CD4/CD8 enrichment using a CliniMACS Prodigy prior to freezing. T cells were cultured in X-VIVO15 (Lonza) supplemented with 5% human AB serum, 1% penicillin-streptomycin and 300 IU/ml of IL-2 (STEMCELL). For functional assays IL-2 was not included. For HSC experiments, human CD34(+) HSPCs derived from bone marrow were purchased from Charles River Laboratory. Cryopreserved cells were thawed and cultured in StemSpan SFEMII medium (STEMCELL) supplemented with StemSpan™ CC110 (STEMCELL).
CAR and blocker constructs were synthesized (IDT) and cloned via Golden Gate assembly into lentivirus or PiggyBac transposon vectors containing flanking inverted terminal repeats for transposase recognition and insulators to prevent epigenetic silencing of transgene expression. CAR and blocker transgenes were designed as described previously (25, 72). Briefly, third-generation CARs were created by fusing nucleotide sequences encoding VL-(G4S)3GG-VH scFvs to sequences encoding a CD8a hinge, CD28 transmembrane domain, and CD28, 4-1BB, and CD3z intracellular domains. Blockers were generated by fusing VL-(G4S)3GG-VH scFvs to the hinge, transmembrane, and intracellular domains of LIR-1 (LILRB1). For tandem designs, additional scFvs were fused to these constructs using a (G4S)4 linker in Fv orientations as described in Supplementary Figure 9. The 2 receptors were cloned into single “BA” vectors with the blocker (B) in the first position, T2A cleavage site, followed by the CAR (A). When indicated, a puromycin resistance gene was included to enable puromycin-driven selection of stably integrated transgenes. Plasmid DNA maxipreps were performed by Aldevron. Cloned products were verified using Sanger sequencing (Azenta) or Nanopore sequencing (Primordium). Antigen mRNA was synthesized by in vitro transcription as previously described (25, 72).
CD1 mice (n=5; Charles River) were immunized with either recombinant CD16b (NA2) (Acro Biosystems) or Clec9a-Fc (R&D) emulsified in Complete Freund’s Adjuvant (Thermo) and/or Sigma Adjuvant System (Sigma) according to a modified Repetitive Immunization at Multiple Sites (RIMMS) schedule targeting the inguinal, axial, brachial, and popliteal lymph nodes. Lymph nodes were harvested, non B cells depleted using a B cell isolation kit (STEMCELL) and fused to P3X.653 myeloma cells to generate hybridomas. 384-well based ELISA screens utilizing soluble CD16b, Clec9a-Fc, CD16a (AcroBiosystems) or KLRG1-Fc (R&D) biotinylated with NHS-PEG12-biotin (Thermo) were completed 12 days after hybridoma fusion. Primary ELISA hits were subsequently screened in mixed-culture flow cytometry binding assays. For CD16b specific binders, K562 cells were transfected with CD16b and loaded with 1uM CMFDA (Thermo #C2925) following manufacturer protocols. These cells were then mixed in the same well with WT K562 cells transfected with CD16a. For Clec9a binders, K562 cells were transfected with Clec9a and mixed together with CMFDA dye loaded WT K562 cells. Hybridoma supernatants were added to the appropriate antigen expressing cell wells, incubated, washed, and detected with a polyclonal Goat anti-mouse IgG Fc-specific AF 647 conjugate (Jackson) by flow cytometry (BD Fortessa). Results were analyzed using FlowJo software to generate MFI values and plotted using GraphPad Prism
Hybridomas were sequenced using Mouse BCR profiling kit (Takara) and cDNA was sequenced using next generation sequencing (Azenta).
Quantification of CD33 levels on the surface was done using QIFIKIT quantitative analysis kit (Agilent) with the CD33 antibody clone WM53 (BD Biosciences). The protocol was previously described (25, 27).
The Jurkat functional assay was performed as previously described (72). Briefly, NFAT-luciferase reporter Jurkat cells were harvested and washed in PBS and transfected with DNA plasmids using the Neon transfection system for 100 µL reactions (# MPK10096). Electroporation conditions were set to 3 pulses at 1500V for 10 ms. Cells were rested overnight in 1 mL of media in 24 well plates. The next day Jurkat cells were combined with target cells and cocultured for 6 hrs before the ONE™ step luciferase firefly assay system (BPS Bioscience) was used to determine luminescence intensity on a Tecan Infinite M1000. For mRNA titration experiments, target cells were either HeLa or K562 cells that were also transfected one day before with mRNA titration using the 4D nucleofector (Lonza). For HSC cell titration experiments, target human CD34(+) HSPCs were thawed, rested overnight, collected, counted, and seeded ranging from 675 to 15,000 cells/well in a 384-well plate in a 2-fold dilution series.
Human PBMCs or enriched T cells were thawed into warm T cell media, washed and resuspended in T cell media supplemented with recombinant human IL-2 and stimulated with TransAct (Miltenyi Biotec) at 1:100 titer according to manufacturer recommendations. T cells were plated into a 24 well plate at 1 million/mL in 2 mL/well for 48 hrs. After 48 hrs, cells were harvested, washed with PBS and resuspended in Lonza P3 buffer (# V4XP-3032) at 20 μL/reaction. 1 μg of PiggyBac transposase mRNA (Hera Biolabs) and 1.6 μg of transposon plasmid DNA were added to 1 million T cells which were then electroporated using the 4D nucleofector (Lonza) program EO-115 in 16-well cuvette strip. Cells were immediately transferred to 200 μL of warm media in a 96 well round-bottom plate and cultured overnight, re-stimulated with TransAct at 1:100 titer, then transferred to 500 μL in 48 well plates. After 1-3 days, cells were transferred to GREX24 (Wilson Wolf) for expansion until use in assays. For the in vivo study, transgene positive cells were enriched with puromycin selection (0.5ug/mL) beginning 4 days after transfection.
K562 cells were transfected with transposon plasmids encoding CD33 and CD16b in the presence of transposase mRNA to establish stable cell lines. Two million K562 cells, 1.6 μg of DNA plasmid and (0.125-1) μg of transposase mRNA were combined in 20 μL of Lonza SF buffer (# V4XC-2032) and then electroporated using the 4D nucleofector (Lonza) program FF-120 in 16-well cuvette strip. Cells were transferred to warm media in a 12-well plate for recovery. CD16b+ MV-4-11 cells were created by lentiviral transduction followed by cell sorting using a FACSMelody (BD). CD33 and CD16b expression was confirmed by flow cytometry using CD33 antibody clone WM53 (BD Biosciences) and CD16 antibody clone 3G8 (BD Biosciences). GFP-firefly luciferase or GFP-renilla luciferase was introduced to engineered MV-4-11 or engineered K562, respectively, via lentiviral transduction followed by cell sorting using a FACSMelody. To generate B2M-, SPN-, or CD33- variants of cell lines, genetic modification using CRISPR-Cas9 was performed. Streptococcus pyogenes HiFi Cas9 protein (IDT) were mixed at 1:3 molar ratio to form ribonucleoprotein complex then transfected into desired cell lines using 4D-Nucleofector (Lonza). Knockout cells were enriched by cell sorting using a FACSMelody using appropriate antibodies (B2M: W6/32, SPN: 1G10, CD33: WM53).
Cytotoxicity was assessed via imaging on the IXM system (Molecular Devices) and endpoint luciferase activity was recorded as a secondary measurement (26). Target cells, K562 or MV-4-11, were plated in a 384-well plate with transparent bottom coated with poly-D-Lysine (Greiner #781946) in T cell media and allowed to adhere for 1 hr. Engineered T cells were profiled via flow cytometry for construct expression using recombinant human CD33 (Acro Biosystems) and recombinant human CD16b (NA2) (Acro Biosystems). T cells were normalized to a single transgene-positive percentage by addition of untransposed cells. T cells were washed with PBS, resuspended in T cell media without IL-2, and combined with the target cells at a titration of effector to target (E:T) cell ratios.
NSG-SGM3 mice were purchased from Jackson Laboratories and housed at the Charles River Accelerator and Development Lab. Post acclimation, mice were injected with 2 million target cells in 100uL via tail vein. MV-4-11 target cells were harvested during log-phase growth, washed with HBSS and resuspended in HBSS at 2 million cells/100 μL. Six days later, engineered T cells were injected at 7.5 million cells per mouse in 200 μL of HBSS. In vivo Bioluminescence imaging (BLI) was performed twice a week. Briefly, 150 μL of D-luciferin (150 μg/ml, Perkin Elmer) was injected intraperitoneally into each animal. After 15 minutes, and additional time points, mice were imaged belly up. At the end of the study femur and tibia bones were collected and bone marrow was extracted from one bone per mouse for flow cytometry analysis. The following antibodies were used to profile bone marrow by flow cytometry: anti-mouse CD45-BV421 (Biolegend), anti-human CD8-PerCP-Cy5.5 (Biolegend), anti-human CD4-PE-Cy7 (Biolegend) and anti-human CD3-APC-e780 (Thermo Fisher). Recombinant human CD33 and CD16b were also included to stain transgene-positive T cells. Xenograft MV-4-11 cells were measured via GFP fluorescence.
Statistical analyses were performed using GraphPad Prism 10.1. Data from Jurkatcell-based experiments are shown as mean ± standard deviation of technical replicates. Curve fitting was performed using four-parameter non-linear regression analysis, with EC50 and IC50 values calculated directly from best-fit curves. Maximum % inhibition values are calculated from interpolated values of these best-fit curves at the top blocker mRNA titration point. All Jurkat assay summary metrics are reported in Supplementary Table 2. In vivo data were analyzed using a Kruskal-Wallis H test followed by a post hoc Dunn’s multiple comparisons test to correct for multiple comparisons; adjusted p-values were reported and the null hypothesis was rejected for adjusted p <0.05.
The data presented in the study are deposited in the GenBank reposity, accession numbers PV220986-92.
The animal study was approved by Institutional Animal Care and Use Committee (IACUC)-. The study was conducted in accordance with the local legislation and institutional requirements.
BD: Conceptualization, Formal analysis, Investigation, Methodology, Project administration, Supervision, Writing – original draft, Writing – review & editing. PN: Formal analysis, Investigation, Methodology, Writing – original draft, Writing – review & editing. AW: Conceptualization, Formal analysis, Investigation, Methodology, Resources, Supervision, Writing – review & editing. AF: Formal analysis, Investigation, Methodology, Writing – review & editing. CJ: Investigation, Methodology, Writing – review & editing. VS: Formal analysis, Methodology, Software, Writing – review & editing. SS: Investigation, Methodology, Writing – review & editing. SMa: Formal analysis, Investigation, Methodology, Supervision, Writing – review & editing. MD: Investigation, Methodology, Resources, Writing – review & editing. EM: Investigation, Resources, Writing – review & editing. RE: Investigation, Resources, Writing – review & editing. TG: Investigation, Resources, Writing – review & editing. TR: Software, Resources, Writing – review & editing. SMi: Investigation, Writing – review & editing. CN: Supervision, Writing – review & editing. AH: Conceptualization, Project administration, Supervision, Writing – review & editing. AK: Conceptualization, Project administration, Supervision, Writing – original draft, Writing – review & editing.
The author(s) declare that no financial support was received for the research and/or publication of this article.
We thank Dr. Mark Sandberg for setting up the AML tumor model. We also thank Alexandre Zampieri for helping with BLI assay oversight. Drs. Armen Mardiros and John Welch provided advice about AML disease progression and mouse models. We are grateful to Casey Gahrs for cloning assistance; Kristian Bolanos for key data in Figure 4; Drs. Yuta Ando and Diane Manry for generating first proofs-of-concept for CLEC9A targeting.
All authors are current or former employees and shareholders of A2 Biotherapeutics, Inc.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2025.1493329/full#supplementary-material
1. Kuykendall A, Duployez N, Boissel N, Lancet JE, Welch JS. Acute myeloid leukemia: the good, the bad, and the ugly. Am Soc Clin Oncol Educ Book. (2018) 38:555–73. doi: 10.1200/EDBK_199519
2. Dohner H, Estey E, Grimwade D, Amadori S, Appelbaum FR, Buchner T, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood. (2017) 129:424–47. doi: 10.1182/blood-2016-08-733196
3. Mardiros A, Dos Santos C, McDonald T, Brown CE, Wang X, Budde LE, et al. T cells expressing CD123-specific chimeric antigen receptors exhibit. specific cytolytic effector functions antitumor effects against Hum acute myeloid leukemia. Blood. (2013) 122:3138–48. doi: 10.1182/blood-2012-12-474056
4. Gill S, Tasian SK, Ruella M, Shestova O, Li Y, Porter DL, et al. Preclinical targeting of human acute myeloid leukemia and myeloablation using chimeric antigen receptor-modified T cells. Blood. (2014) 123:2343–54. doi: 10.1182/blood-2013-09-529537
5. Brinkman-Van-der-Linden EC, Angata T, Reynolds SA, Powell LD, Hedrick SM, Varki A. CD33/Siglec-3 binding specificity, expression pattern, and consequences of gene deletion in mice. Mol Cell Biol. (2003) 23:4199–206. doi: 10.1128/MCB.23.12.4199-4206.2003
6. Taussig DC, Pearce DJ, Simpson C, Rohatiner AZ, Lister TA, Kelly G, et al. Hematopoietic stem cells express multiple myeloid markers: implications for the origin and targeted therapy of acute myeloid leukemia. Blood. (2005) 106:4086–92. doi: 10.1182/blood-2005-03-1072
7. Levine JH, Simonds EF, Bendall SC, Davis KL, Amir el AD, Tadmor MD, et al. Data-driven phenotypic dissection of AML reveals progenitor-like cells that correlate with prognosis. Cell. (2015) 162:184–97. doi: 10.1016/j.cell.2015.05.047
8. Kenderian SS, Ruella M, Shestova O, Klichinsky M, Aikawa V, Morrissette JJ, et al. CD33-specific chimeric antigen receptor T cells exhibit potent preclinical activity against human acute myeloid leukemia. Leukemia. (2015) 29:1637–47. doi: 10.1038/leu.2015.52
9. Kim MY, Yu KR, Kenderian SS, Ruella M, Chen S, Shin TH, et al. Genetic inactivation of CD33 in hematopoietic stem cells to enable CAR T cell immunotherapy for acute myeloid leukemia. Cell. (2018) 173:1439–1453.e19. doi: 10.1016/j.cell.2018.05.013
10. Bohme M, Kayser S. Immune-based therapeutic strategies for acute myeloid leukemia. Cancers (Basel). (2021) 14:105.
11. Pizzitola I, Anjos-Afonso F, Rouault-Pierre K, Lassailly F, Tettamanti S, Spinelli O, et al. Chimeric antigen receptors against CD33/CD123 antigens efficiently target primary acute myeloid leukemia cells in vivo. Leukemia. (2014) 28:1596–605. doi: 10.1038/leu.2014.62
12. Gottardi M, Simonetti G, Sperotto A, Nappi D, Ghelli Luserna di Rora A, Padella A, et al. Therapeutic targeting of acute myeloid leukemia by gemtuzumab ozogamicin. Cancers (Basel). (2021) 13. doi: 10.3390/cancers13184566
13. Griffin JD, Linch D, Sabbath K, Larcom P, Schlossman SF. A monoclonal antibody reactive with normal and leukemic human myeloid progenitor cells. Leuk Res. (1984) 8:521–34. doi: 10.1016/0145-2126(84)90001-8
14. Castaigne S, Pautas C, Terre C, Raffoux E, Bordessoule D, Bastie JN, et al. Effect of gemtuzumab ozogamicin on survival of adult patients with de-novo acute myeloid leukaemia (ALFA-0701): a randomised, open-label, phase 3 study. Lancet. (2012) 379:1508–16. doi: 10.1016/S0140-6736(12)60485-1
15. Leopold LH, Berger MS, Feingold J. Acute and long-term toxicities associated with gemtuzumab ozogamicin (Mylotarg) therapy of acute myeloid leukemia. Clin Lymphoma. (2002) 2 Suppl 1:S29–34. doi: 10.3816/CLM.2002.s.006
16. Farhad Ravandi M, Anthony S. Stein MD, Hagop M. Kantarjian MD, Roland B. Walter MD, PhD MS, Peter Paschka MD, et al. A phase 1 first-in-human study of AMG 330, an anti-CD33 bispecific T-cell engager (BiTE®) antibody construct, in relapsed/refractory acute myeloid leukemia (R/R AML). in. Am Soc Hematology.(2018) 132(Supplement 1):25.
17. Zhang X, Zhu L, Zhang H, Chen S, Xiao Y. CAR-T cell therapy in hematological Malignancies: current opportunities and challenges. Front Immunol. (2022) 13:927153. doi: 10.3389/fimmu.2022.927153
18. Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N Engl J Med. (2017) 377:2531–44. doi: 10.1056/NEJMoa1707447
19. Wang QS, Wang Y, Lv HY, Han QW, Fan H, Guo B, et al. Treatment of CD33-directed chimeric antigen receptor-modified T cells in one patient with relapsed and refractory acute myeloid leukemia. Mol Ther. (2015) 23:184–91. doi: 10.1038/mt.2014.164
20. Hansen BA, Wendelbo O, Bruserud O, Hemsing AL, Mosevoll KA, Reikvam H. Febrile neutropenia in acute leukemia. Epidemiology, etiology, pathophysiology and treatment. Mediterr J Hematol Infect Dis. (2020) 12:e2020009.
21. 2seventy bio Announces Clinical Study Pause of PLAT-08 Trial of SC-DARIC33 in Acute Myeloid Leukemia (2023). Available online at: https://ir.2seventybio.com/news-releases/news-release-details/2seventy-bio-announces-clinical-study-pause-plat-08-trial-sc (Accessed March 2, 2025).
22. Fedorov VD, Themeli M, Sadelain M. PD-1- and CTLA-4-based inhibitory chimeric antigen receptors (iCARs) divert off-target immunotherapy responses. Sci Transl Med. (2013) 5:215ra172. doi: 10.1126/scitranslmed.3006597
23. Richards RM, Zhao F, Freitas KA, Parker KR, Xu P, Fan A, et al. NOT-gated CD93 CAR T cells effectively target AML with minimized endothelial cross-reactivity. Blood Cancer Discovery. (2021) 2:648–65. doi: 10.1158/2643-3230.BCD-20-0208
24. Hamburger AE, DiAndreth B, Cui J, Daris ME, Munguia ML, Deshmukh K, et al. Engineered T cells directed at tumors with defined allelic loss. Mol Immunol. (2020) 128:298–310. doi: 10.1016/j.molimm.2020.09.012
25. Tokatlian T, Asuelime GE, Mock JY, DiAndreth B, Sharma S, Toledo Warshaviak D, et al. Mesothelin-specific CAR-T cell therapy that incorporates an HLA-gated safety mechanism selectively kills tumor cells. J Immunother Cancer. (2022) 10. doi: 10.1136/jitc-2021-003826
26. DiAndreth B, Hamburger AE, Xu H, Kamb A. The Tmod cellular logic gate as a solution for tumor-selective immunotherapy. Clin Immunol. (2022) 241:109030. doi: 10.1016/j.clim.2022.109030
27. Sandberg ML, Wang X, Martin AD, Nampe DP, Gabrelow GB, Li CZ, et al. A carcinoembryonic antigen-specific cell therapy selectively targets tumor cells with HLA loss of heterozygosity in vitro and in vivo. Sci Transl Med. (2022) 14:eabm0306. doi: 10.1126/scitranslmed.abm0306
28. Borges L, Hsu ML, Fanger N, Kubin M, Cosman D. A family of human lymphoid and myeloid Ig-like receptors, some of which bind to MHC class I molecules. J Immunol. (1997) 159:5192–6. doi: 10.4049/jimmunol.159.11.5192
29. Kramer MH, Zhang Q, Sprung R, Day RB, Erdmann-Gilmore P, Li Y, et al. Proteomic and phosphoproteomic landscapes of acute myeloid leukemia. Blood. (2022) 140:1533–48. doi: 10.1182/blood.2022016033
30. Monaco G, Lee B, Xu W, Mustafah S, Hwang YY, Carré C, et al. RNA-seq signatures normalized by mRNA abundance allow absolute deconvolution of human immune cell types. Cell Rep. (2019) 26:1627–1640.e7. doi: 10.1016/j.celrep.2019.01.041
31. Grossman RL, Heath AP, Ferretti V, Varmus HE, Lowy DR, Kibbe WA, et al. Toward a shared vision for cancer genomic data. New Engl J Med. (2016) 375:1109–12. doi: 10.1056/NEJMp1607591
32. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. New Engl J Med. (2013) 368:2059–74. doi: 10.1056/NEJMoa1301689
33. D’Angelo SP, Mahoney MR, Van Tine BA, Atkins J, Milhem MM, Jahagirdar BN, et al. Nivolumab with or without ipilimumab treatment for metastatic sarcoma (Alliance A091401): two open-label, non-comparative, randomised, phase 2 trials. Lancet Oncol. (2018) 19:416–26. doi: 10.1016/S1470-2045(18)30006-8
34. van Galen P, Hovestadt V, Wadsworth Ii MH, Hughes TK, Griffin GK, Battaglia S, et al. Single-cell RNA-seq reveals AML hierarchies relevant to disease progression and immunity. Cell. (2019) 176:1265–1281 e24. doi: 10.1016/j.cell.2019.01.031
35. Zhang J, Bajari R, Andric D, Gerthoffert F, Lepsa A, Nahal-Bose H, et al. The international cancer genome consortium data portal. Nat Biotechnol. (2019) 37:367–9. doi: 10.1038/s41587-019-0055-9
36. Kaufmann KB, Zeng AGX, Coyaud E, Garcia-Prat L, Papalexi E, Murison A, et al. A latent subset of human hematopoietic stem cells resists regenerative stress to preserve stemness. Nat Immunol. (2021) 22:723–34. doi: 10.1038/s41590-021-00925-1
37. Fares I, Chagraoui J, Lehnertz B, MacRae T, Mayotte N, Tomellini E, et al. EPCR expression marks UM171-expanded CD34(+) cord blood stem cells. Blood. (2017) 129:3344–51. doi: 10.1182/blood-2016-11-750729
38. Tomellini E, Fares I, Lehnertz B, Chagraoui J, Mayotte N, MacRae T, et al. Integrin-alpha3 is a functional marker of ex vivo expanded human long-term hematopoietic stem cells. Cell Rep. (2019) 28:1063–1073.e5. doi: 10.1016/j.celrep.2019.06.084
39. Solman M, Blokzijl-Franke S, Piques F, Yan C, Yang Q, Strullu M, et al. Inflammatory response in hematopoietic stem and progenitor cells triggered by activating SHP2 mutations evokes blood defects. Elife. (2022) 11. doi: 10.7554/eLife.73040.sa2
40. Chen L, Kostadima M, Martens JHA, Canu G, Garcia SP, Turro E, et al. Transcriptional diversity during lineage commitment of human blood progenitors. Science. (2014) 345:1251033. doi: 10.1126/science.1251033
41. Flesch BK, Reil A. Molecular genetics of the human neutrophil antigens. Transfus Med Hemother. (2018) 45:300–9. doi: 10.1159/000491031
42. Fleit HB, Wright SD, Unkeless JC. Human neutrophil Fc gamma receptor distribution and structure. Proc Natl Acad Sci U.S.A. (1982) 79:3275–9.
43. Zah E, Lin MY, Silva-Benedict A, Jensen MC, Chen YY. T cells expressing CD19/CD20 bispecific chimeric antigen receptors prevent antigen escape by Malignant B cells. Cancer Immunol Res. (2016) 4:498–508. doi: 10.1158/2326-6066.CIR-15-0231
44. Remold-O’Donnell E, Zimmerman C, Kenney D, Rosen FS. Expression on blood cells of sialophorin, the surface glycoprotein that is defective in Wiskott-Aldrich syndrome. Blood. (1987) 70:104–9. doi: 10.1182/blood.V70.1.104.104
45. Huysamen C, Willment JA, Dennehy KM, Brown GD. CLEC9A is a novel activation C-type lectin-like receptor expressed on BDCA3+ dendritic cells and a subset of monocytes. J Biol Chem. (2008) 283:16693–701. doi: 10.1074/jbc.M709923200
46. Belluschi S, Calderbank EF, Ciaurro V, Pijuan-Sala B, Santoro A, Mende N, et al. Myelo-lymphoid lineage restriction occurs in the human haematopoietic stem cell compartment before lymphoid-primed multipotent progenitors. Nat Commun. (2018) 9:4100. doi: 10.1038/s41467-018-06442-4
47. Anjos-Afonso F, Bonnet D. Human CD34+ hematopoietic stem cell hierarchy: how far are we with its delineation at the most primitive level? Blood. (2023) 142:509–18. doi: 10.1182/blood.2022018071
48. Exman P, Tolaney SM. HER2-positive metastatic breast cancer: a comprehensive review. Clin Adv Hematol Oncol. (2021) 19:40–50.
50. Druker BJ, Talpaz M, Resta DJ, Peng B, Buchdunger E, Ford JM, et al. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med. (2001) 344:1031–7. doi: 10.1056/NEJM200104053441401
51. Lugo TG, Pendergast AM, Muller AJ, Witte ON. Tyrosine kinase activity and transformation potency of bcr-abl oncogene products. Science. (1990) 247:1079–82. doi: 10.1126/science.2408149
52. Gambella M, Carlomagno S, Raiola AM, Giannoni L, Ghiggi C, Setti C, et al. CD19-targeted immunotherapies for diffuse large B-cell lymphoma. Front Immunol. (2022) 13:837457. doi: 10.3389/fimmu.2022.837457
53. Kleber M, Ntanasis-Stathopoulos I, Terpos E. BCMA in multiple myeloma-A promising key to therapy. J Clin Med. (2021) 10:4088. doi: 10.3390/jcm10184088
54. Want MY, Bashir Z, Najar RA. T cell based immunotherapy for cancer: approaches and strategies. Vaccines (Basel). (2023) 11:835. doi: 10.3390/vaccines11040835
55. Norberg SM, Hinrichs CS. Engineered T cell therapy for viral and non-viral epithelial cancers. Cancer Cell. (2023) 41:58–69. doi: 10.1016/j.ccell.2022.10.016
56. Nagarsheth NB, Norberg SM, Sinkoe AL, Adhikary S, Meyer TJ, Lack JB, et al. TCR-engineered T cells targeting E7 for patients with metastatic HPV-associated epithelial cancers. Nat Med. (2021) 27:419–25. doi: 10.1038/s41591-020-01225-1
57. Linnemann C, van Buuren MM, Bies L, Verdegaal EM, Schotte R, Calis JJ, et al. High-throughput epitope discovery reveals frequent recognition of neo-antigens by CD4+ T cells in human melanoma. Nat Med. (2015) 21:81–5. doi: 10.1038/nm.3773
58. Pan Y, Phillips JW, Zhang BD, Noguchi M, Kutschera E, McLaughlin J, et al. IRIS: Discovery of cancer immunotherapy targets arising from pre-mRNA alternative splicing. Proc Natl Acad Sci U.S.A. (2023) 120:e2221116120. doi: 10.1073/pnas.2221116120
59. Lucchi R, Bentanachs J, Oller-Salvia B. The masking game: design of activatabl antibodies and mimetics for selective therapeutics and cell control. ACS Cent Sci. (2021) 7:724–38. doi: 10.1021/acscentsci.0c01448
60. Labanieh L, Mackall CL. CAR immune cells: design principles, resistance and the next generation. Nature. (2023) 614:635–48. doi: 10.1038/s41586-023-05707-3
61. Morsut L, Roybal KT, Xiong X, Gordley RM, Coyle SM, Thomson M, et al. Engineering customized cell sensing and response behaviors using synthetic notch receptors. Cell. (2016) 164:780–91. doi: 10.1016/j.cell.2016.01.012
62. Williams JZ, Allen GM, Shah D, Sterin IS, Kim KH, Garcia VP, et al. Precise T cell recognition programs designed by transcriptionally linking multiple receptors. Science. (2020) 370:1099–104. doi: 10.1126/science.abc6270
63. Tousley AM, Rotiroti MC, Labanieh L, Rysavy LW, Kim WJ, Lareau C, et al. Co-opting signalling molecules enables logic-gated control of CAR T cells. Nature. (2023) 615:507–16. doi: 10.1038/s41586-023-05778-2
64. Hwang MS, Mog BJ, Douglass J, Pearlman AH, Hsiue EH, Paul S, et al. Targeting loss of heterozygosity for cancer-specific immunotherapy. Proc Natl Acad Sci U.S.A. (2021) 118. doi: 10.1073/pnas.2022410118
65. Jain MD, Smith M, Shah NN. How I treat refractory CRS and ICANS after CAR T-cell therapy. Blood. (2023) 141:2430–42.
66. Norelli M, Camisa B, Barbiera G, Falcone L, Purevdorj A, Genua M, et al. Monocyte-derived IL-1 and IL-6 are differentially required for cytokine-release syndrome and neurotoxicity due to CAR T cells. Nat Med. (2018) 24:739–48. doi: 10.1038/s41591-018-0036-4
67. Frankel NW, Deng H, Yucel G, Gainer M, Leemans N, Lam A, et al. Precision off-the-shelf natural killer cell therapies for oncology with logic-gated gene circuits. Cell Rep. (2024) 43:114145. doi: 10.1016/j.celrep.2024.114145
68. Larson SM, Walthers CM, Ji B, Ghafouri SN, Naparstek J, Trent J, et al. CD19/CD20 bispecific chimeric antigen receptor (CAR) in naive/memory T cells for the treatment of relapsed or refractory non-hodgkin lymphoma. Cancer Discovery. (2023) 13:580–97. doi: 10.1158/2159-8290.CD-22-0964
69. Barclay AN, Hatherley D. The counterbalance theory for evolution and function of paired receptors. Immunity. (2008) 29:675–8. doi: 10.1016/j.immuni.2008.10.004
70. Partin AC, Bruno R, Shafaattalab S, Vander Mause E, Winters A, Daris M, et al. Geometric parameters that affect the behavior of logic-gated CAR T cells. Front Immunol. (2024) 15:1304765. doi: 10.3389/fimmu.2024.1304765
71. Martire S, Wang X, McElvain M, Suryawanshi V, Gill T, DiAndreth B, et al. High-throughput screen to identify and optimize NOT gate receptors for cell therapy. Cytometry Part A. (2024) 105(10):741–51. doi: 10.1002/cyto.a.24893
Keywords: CAR-T cells, CD33, CD16, SPN, AML, LIR-1 (LILRB1), logic gate, blood cancer
Citation: DiAndreth B, Nesterenko PA, Winters AG, Flynn AD, Jette CA, Suryawanshi V, Shafaattalab S, Martire S, Daris M, Moore E, Elshimali R, Gill T, Riley TP, Miller S, Netirojjanakul C, Hamburger AE and Kamb A (2025) Multi-targeted, NOT gated CAR-T cells as a strategy to protect normal lineages for blood cancer therapy. Front. Immunol. 16:1493329. doi: 10.3389/fimmu.2025.1493329
Received: 09 September 2024; Accepted: 21 February 2025;
Published: 21 March 2025.
Edited by:
Jamie Berta Spangler, Johns Hopkins University, United StatesReviewed by:
Ali Roghanian, University of Southampton, United KingdomCopyright © 2025 DiAndreth, Nesterenko, Winters, Flynn, Jette, Suryawanshi, Shafaattalab, Martire, Daris, Moore, Elshimali, Gill, Riley, Miller, Netirojjanakul, Hamburger and Kamb. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alexander Kamb, YWthbWJAYTJiaW90aGVyYXBldXRpY3MuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.