 Shuai Wang
Shuai Wang Chenghui Cao
Chenghui Cao Daoquan Peng
Daoquan Peng
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 27 February 2025
Sec. Inflammation
Volume 16 - 2025 | https://doi.org/10.3389/fimmu.2025.1462508
This article is part of the Research Topic Immunity, Atherosclerosis and Cardiovascular Disease: An Interdisciplinary Approach to Cardiometabolic Health View all 10 articles
Triggering receptor expressed on myeloid cells-2 (TREM2) is a transmembrane immune receptor that is expressed mainly on macrophages. As a pathology-induced immune signaling hub, TREM2 senses tissue damage and activates immune remodeling in response. Previous studies have predominantly focused on the TREM2 signaling pathway in Alzheimer’s disease, metabolic syndrome, and cancer. Recent research has indicated that TREM2 signaling is also activated in various cardiovascular diseases. In this review, we summarize the current understanding and the unanswered questions regarding the role of TREM2 signaling in mediating the metabolism and function of macrophages in atherosclerosis and various models of heart failure. In the context of atherosclerosis, TREM2 signaling promotes foam cell formation and is crucial for maintaining macrophage survival and plaque stability through efferocytosis and cholesterol efflux. Recent studies on myocardial infarction, sepsis-induced cardiomyopathy, and hypertensive heart failure also implicated the protective role of TREM2 signaling in cardiac macrophages through efferocytosis and paracrine functions. Additionally, we discuss the clinical significance of elevated soluble TREM2 (sTREM2) in cardiovascular disease and propose potential therapies targeting TREM2. The overall aim of this review is to highlight the various roles of TREM2 in cardiovascular diseases and to provide a framework for therapeutic strategies targeting TREM2.
Macrophages represent one of the most numerous and diverse leukocyte types in the body (1). They are important regulators of many cardiovascular diseases. Macrophages not only trigger damaging inflammatory responses but also mediate tissue repair and cardiac regeneration (2). Recent molecular techniques, such as single-cell mass cytometry by time-of-flight, have revealed significant complexity within macrophage populations (3). Distinct macrophage populations may mediate inflammatory or reparative macrophage behaviors (4). A deeper understanding of the diverse functions of cardiac macrophages will be essential for the development of targeted therapies to mitigate injury and orchestrate recovery.
Recently, via single-cell RNA sequencing, a subpopulation of nonproinflammatory foamy macrophages highly expressing triggering receptor expressed on myeloid cells-2 (TREM2) was identified in atherosclerotic plaques, but its importance remains to be elucidated (5, 6). Interestingly, single-cell RNA sequencing of cardiac immune cells also revealed upregulated Trem2 expression in subpopulations of macrophages in animal models of myocardial infarction (MI), hypertension-induced heart failure with preserved function (HFpEF), and septic cardiomyopathy (7–9). Trem2+ macrophages were found to promote recovery from cardiac injury and improve cardiac function in the above disease models (8–11).
TREM2 is a transmembrane receptor belonging to the immunoglobin superfamily. As a hub of myeloid cell immune activity, TREM2 signaling helps to maintain normal myeloid cell differentiation and survival under physiological conditions, whereas under conditions of acute or chronic injury, TREM2 regulates the phenotype and function of myeloid cells in response to injury (12, 13). In different scenarios, TREM2 signaling mediates various physiological processes, including phagocytosis, resistance to proinflammatory stimuli, the promotion of myeloid cell survival under conditions of stress, and phenotypic transformation (14).
Prior studies have mainly concentrated on the function and regulation of TREM2 in the development of neurodegenerative disorders, particularly Alzheimer’s disease (14). Nonetheless, the involvement of TREM2 in cardiovascular disease has not been thoroughly explored. A recent review has highlighted TREM2’s protective role in the heterogenous population of macrophages during inflammation following myocardial infarction (15). However, there is conflicting evidence regarding the importance of the TREM2 signaling pathway in macrophages related to atherosclerosis. Additionally, the implications of TREM2 signaling in macrophages during cardiac injury responses across various heart failure models still require further investigation. This review aims to compile existing knowledge and identify gaps regarding the Trem2 signaling pathway in both atherosclerosis and cardiomyopathy, while also considering the potential for therapeutic modulation of TREM2 signaling in cardiovascular disease.
Plaque stability is a key determinant of the prognosis of atherosclerotic cardiovascular diseases (16). Currently, therapeutic approaches for atherosclerotic diseases focus primarily on controlling risk factors (such as intensive lipid lowering). Although the combination of statins and proprotein convertase subtilisin/kexin (PCSK9) inhibitors can significantly reduce low-density lipoprotein cholesterol (LDL-C), plaque regression is limited (16). Thus, patients with atherosclerotic cardiovascular disease (ASCVD) still face a high residual risk (17). Macrophage-mediated plaque inflammation is recognized as an important factor influencing plaque progression and regression (18–21).
It was previously thought that macrophages within plaques possess both anti-inflammatory and proinflammatory phenotypes and play different roles in the progression and regression of plaques. M1 macrophages, which are activated in vitro by stimulation of Toll-like receptor ligands, lipopolysaccharide (LPS) and interferon gamma (INF-γ). M1 macrophages express the proinflammatory transcription factors NF-κB and STAT-1; secrete the proinflammatory cytokines IL-1β, IL-6, and TNF-α; and promote plaque progression. Alternatively, macrophages activated in vitro by IL-4 and IL-13 known as M2 macrophages. M2 macrophages highly express CD163, mannose receptor 1, resestin like-β, and arginase-1; secrete anti-inflammatory factors such as the IL-1 receptor antagonist IL-10 and collagen; and promote plaque regression (4). The traditional dichotomy of macrophage phenotypes is overly simplistic. With the application of mass cytometry and single-cell RNA sequencing technologies, more complex subgroups of macrophages have been discovered in atherosclerotic plaques (5, 6, 22).
Recently, a subset of macrophages TREM2hi, which are characterized by markers of Trem2, Cd9, Ctsd, and Spp1, was identified in human and mouse atherosclerotic plaques (5, 6, 23–25). TREM2, which acts as a lipid-sensing receptor, can bind to lipoprotein ligands such as apolipoprotein (ApoE) or phospholipids (26, 27). After TREM2 binds to lipid ligands, the p38/mitogen-activated protein kinase (MAPK)-proliferator-activated receptor γ (PPARγ) signaling pathway is activated, and CD36 expression is upregulated, which is a receptor that mediates cholesterol uptake (28). Previous studies have shown that TREM2hi macrophages are lipid-rich foam cells with a low inflammatory state (5, 6, 29). A recent study employing trajectory analysis, ligand-receptor interaction analysis, and functional experiments has confirmed that Trem2hi macrophage can transform into pro-inflammatory lipid-laden macrophages (PLIN2hi/TREM1hi), which are implicated in the progression of atherosclerotic lesions (30).The overexpression of TREM2 in cultured vascular smooth muscle cells (SMCs) and macrophages promote lipid uptake, exacerbates lipid influx and foam cell formation (28, 31). In an atherosclerotic mouse model, the density of Trem2-positive foam cells in aortic plaques increased in a time-dependent manner after ApoE knockout (ApoE-/-) mice were fed a high-fat diet (HFD) (28). Moreover, compared with ApoE-/- mice, Trem2-/-/ApoE-/- double-knockout mice presented significantly reduced atherosclerotic lesion sizes, foam cell numbers, and lipid burdens after HFD feeding (28). Therefore, TREM2 is speculated to exacerbate atherosclerosis development. However, the role of TREM2hi macrophages in atherosclerosis is controversial since other evidence suggests that TREM2hi macrophages may play a role in stabilizing plaques and promoting plaque regression (Figure 1).
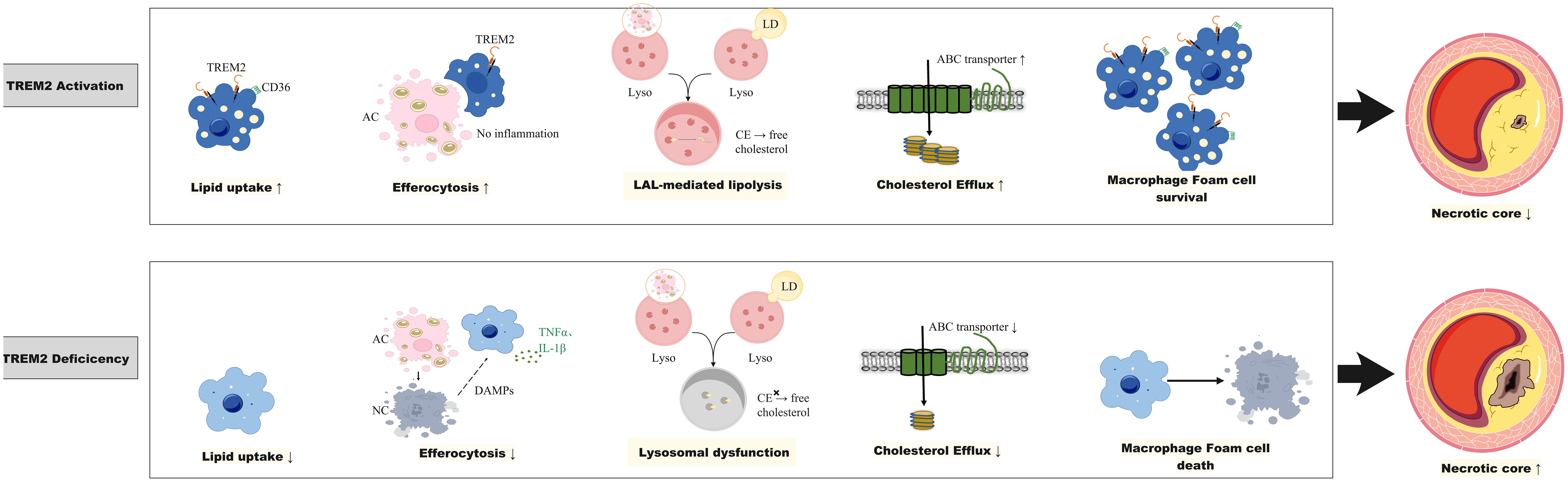
Figure 1. The role of Trem2 in atherosclerosis. TREM2 in plaque macrophages is involved in lipid uptake and foam cell formation, the efferocytosis of apoptotic or necrotic cells to limit plaque inflammation, maintaining lysosomal cholesterol homeostasis to prevent cholesterol ester (CE) accumulation and promoting cholesterol efflux through the upregulation of ATP-binding cassette A1 (ABCA1)/ABCG1. TREM2 activation promotes foam cell survival and reduces necrotic core, whereas TREM2 deficiency leads to foam cell death and an increase in the necrotic core. ABC transporter, ATP binding cassette transporter; CE, cholesterol ester; DAMPs, damage-associated molecular patterns; LAL, lysosomal acid lipase; LD, Lipid droplet; IL-1β, interleukin-1β; Lyso, lysosome; TNF-α, tumor necrosis factor alpha; TREM2, triggering receptor expressed on myeloid cells-2.
The process by which cholesterol in macrophages is transported to high-density lipoprotein (HDL) through the cholesterol transporters ATP binding Cassette A1 and G1 (ABCA1/ABCG1) is known as cholesterol efflux. Promoting cholesterol efflux from macrophages decreases cholesterol accumulation in foam cells, suppresses inflammation and promotes plaque stability and plaque regression (32, 33). Research on chronic demyelinating neurological diseases, ischemic stroke and neurotoxicity has shown that TREM2 promotes cholesterol efflux in microglia (27, 34–36). Single-cell transcriptomic and chromatin accessibility analysis techniques revealed that in human aortic plaques, TREM2hi macrophages highly express the cholesterol efflux transporters ABCA1 and ABCG1 (5). In addition, TREM2-dependent Abca1 and Abcg1 expression and cholesterol efflux have been reported to explain the sexually dimorphic effect of serum amyloid A3 (SAA3) on atherosclerosis (37). SAA3 is predominantly an extrahepatically expressed acute-phase protein in response to acute and chronic inflammatory stimuli (38, 39). A previous report suggested that SAA3 is proatherogenic in male ApoE-/- mice, as evidenced by worsened atherosclerosis when Saa3 was overexpressed and by improved atherosclerosis when Saa3 was silenced (40). However, these effects have been reported only in male mice but not in female mice (40). Chait et al. (37) showed that Saa3 deficiency differentially altered Trem2 expression in macrophages from male and female mice. Although Saa3 deficiency in male Ldlr-/- mice reduced aortic and aortic sinus atherosclerosis and increased Trem2 transcript levels in bone marrow-derived macrophages (BMDMs), Saa3-/- knockout in female Ldlr-/- mice increased aortic atherosclerosis and decreased Trem2 expression in BMDMs. In addition, BMDMs from male Saa3-/- mice presented increased expression of the cholesterol transporters Abca1 and Abcg1, as well as increased cholesterol efflux to HDL. In contrast, Saa3 deficiency in female BMDMs had no effect on cholesterol transporter gene expression or cholesterol efflux. The results of the study by Chait et al. support the notion that TREM2 in macrophages promotes cholesterol efflux and plays a role in combating atherosclerosis (37).
Efferocytosis is a process through which diseased and dying cells are engulfed by phagocytes for clearance (41). In healthy arteries, cells undergo apoptosis as a result of lipid ingestion after foam cell formation or due to homeostatic cell turnover (41). Macrophage efferocytosis is impaired in atherosclerotic cardiovascular disease (42). Efferocytosis failure accelerates atherogenesis by permitting the accumulation of debris and apoptotic cells within the arterial subintima and media (42, 43). The resulting plaque growth not only increases the degree of stenosis of the affected vessel but also increases plaque vulnerability to rupture given that the accumulation of this inflammatory tissue occurs within the necrotic core (41, 44, 45). Recent studies have shown that macrophage efferocytosis is dependent on TREM2 and that the absence or downregulation of TREM2 results in defects in tissue macrophage efferocytosis (46, 47). In addition, a recent study demonstrated that hematopoietic or global deficiency of TREM2 increased necrotic core formation in early atherosclerosis, while TREM2 agonism decreased it (48). In atherosclerosis-prone mice, treatment with Trem2 agonism AL002a promote macrophage survival and improves feature of plaque stability (49).These findings underscore the essential role of TREM2 in maintaining the balance between foam cell death and the clearance of dead cells in atherosclerotic lesions (48, 49). Further mechanistic studies on how TREM2 regulates efferocytosis revealed that TREM2 can bind to phosphatidylserine and phosphatidylcholine, both of which are “eat me” signaling molecules that are released by apoptotic cells, suggesting that TREM2 itself may be a scavenger receptor responsible for recognizing and clearing apoptotic cells (50). In addition, the TREM2-mediated signaling pathway can upregulate the expression of macrophage “recognition” receptors for apoptotic cells, such as CD36 (28, 51). Therefore, the observed TREM2hi macrophages in mouse models and human atherosclerotic lesions may reflect the active efferocytotic function of plaque macrophages.
Lysosomes are at the center of the cholesterol homeostasis regulatory network (52). When macrophages take up modified lipoproteins or ingest apoptotic cells, they face a substantial cholesterol load. Excessive cholesterol must be processed within lysosomes to avoid cytotoxic cholesterol accumulation, which may cause oxidative stress, mitochondrial dysfunction and inflammasome activation. Intracellular cholesterol needs to be re-esterified for storage or exported via the cholesterol efflux pathway. In lysosomes, cholesteryl ester is hydrolyzed into free cholesterol by lysosomal acid lipase (LAL) and is then distributed among various organelles via the Niemann−Pick C (NPC) 1 and NPC2 proteins (52). Lysosomal cholesterol hydrolysis, which is mediated by LAL, leads to the timely production of oxysterols, which are endogenous liver X receptor (LXR) ligands. LXR, a nuclear receptor, plays pivotal roles in the transcriptional regulation of lipid metabolism and efferocytosis (53–55). A deficiency in LAL impaired the production of LXR ligands, resulting in reduced expression of the cholesterol transporters ABCA1 and ABCG1 and the efferocytosis recognition receptor MerTK, thereby impeding subsequent macrophage cholesterol efflux and efferocytosis (56).
TREM2 was recently reported to play an important role in maintaining normal lysosomal function and cholesterol metabolism homeostasis (57, 58). TREM2 loss-of-function mutations lead to significant lysosomal dysfunction and downregulated expression of genes involved in lipid metabolism in induced pluripotent stem cell (iPSC)-derived microglia from patients with Nasu-Hakola disease (NHD) (57). In animal studies, knocking out Trem2 in mice led to reduced expression of microglial lysosomal function-related genes and the accumulation of cholesterol esters (58). These findings suggest TREM2-dependent expression of genes involved in lysosomal cholesteryl ester hydrolysis and cholesterol export. Although this has not been confirmed in the context of atherosclerosis, the high expression of TREM2 on foam cells is hypothesized to be an adaptive response to cholesterol load, through which normal lysosomal cholesterol processing is maintained to prevent toxic cholesterol accumulation.
In summary, the significance of TREM2hi macrophages in atherosclerosis is still under debate since opposite evidence exists. Given that foam cell formation reflects the scavenging role of macrophages to clear subintimal lipid accumulation and that TREM2 is important for maintaining macrophage cholesterol efflux, efferocytosis, and lysosomal cholesterol metabolism, it is speculated that high expression of TREM2 is protective in atherosclerosis. However, further studies in different animal models of atherosclerosis are needed.
Macrophages play a central role in the response to injury after MI. Post MI, macrophages follow a biphasic response to injury, transiting from a proinflammatory phenotype toward a reparative phenotype, which has been classically defined as M1-to-M2 polarization (59). However, macrophages in the post-MI heart exhibit heterogeneous subtypes of surface markers and functions that lie outside the M1/M2 spectrum (59, 60).
Recent studies utilizing spatial transcriptomics and single-cell RNA sequencing in a mouse model of MI identified a group of macrophages with high Trem2 expression (Trem2hi macrophages). These cells highly express anti-inflammatory genes, cytokines, and chemokines such as Il10, Tgfb1, Cx3cr1, Alox15, and Arg1 (7). Trem2hi macrophages that appear in the myocardium 3–7 days post-MI are thought to be recruited from the circulation because the expression of Trem2 in circulating monocytes increases synchronously (10, 11). In addition, the use of a CCR2 chemokine antagonist inhibits the migration of TREM2hi-BMDMs into the heart (11).
A key function of Trem2hi macrophages in the infarcted myocardium is efferocytosis. It is well known that efferocytosis after MI is an essential aspect of healing following cardiac injury and that it stabilizes the myocardial wall to prevent wall rupture (2, 61, 62). Specifically, knocking out Trem2 in mice significantly suppressed the efferocytosis of dying cells, aggravated cardiac dysfunction and increased mortality, whereas the overexpression of Trem2 or local myocardial injection of an adenoviral vector expressing Trem2 restored the clearance of tissue debris and cardiac function (10, 11).
TREM2 also plays a critical role in mediating efferocytosis-dependent intracellular signaling. As a transmembrane receptor, TREM2 mediates intracellular signaling by associating with the adaptor proteins DAP12 and DAP10 to recruit the intracellular signaling molecules spleen tyrosine kinase (Syk) and phosphoinositide 3-kinase (PI3K) for signal transduction (63). Gong et al. (11) recently showed in an MI mouse model accompanied by efferocytosis that the TREM2-SYK-SMAD signaling pathway was activated, resulting in remodeling of cardiac macrophage metabolism and the secretion of protective cytokines. Activation of TREM2-SYK-SMAD signaling suppressed the transcription of the mitochondrial inner membrane NAD+ transporter solute carrier 25 member 53 (SLC25A53), thus impeding the entry of NAD+ into mitochondria, which caused reduced generation of α-ketoglutarate from isocitrate via the tricarboxylic acid (TCA) cycle. The consequence of this metabolic remodeling is the accumulation of the bypass derivative itaconate in the cytosol, which is secreted into the microenvironment and acts on adjacent cardiomyocytes and fibroblasts (11). Itaconate has been demonstrated to exert a protective effect on myocardial infarction by inhibiting the apoptosis of cardiomyocytes in an ischemic environment, promoting the proliferation of fibroblasts, and suppressing inflammation (11, 64–67).
Overall, these studies suggest that TREM2hi macrophages play a protective role in the response to myocardial infarction by promoting healing and tissue remodeling through efferocytosis of apoptotic and necrotic cardiomyocytes and secretion of anti-inflammatory factors that promote fibrocyte proliferation.
Sepsis can lead to cardiac pump dysfunction, commonly referred to as sepsis-induced cardiomyopathy (SICM). Although there is no consensus on the definition of SICM, a recent systematic review and meta-analysis comprising 16 studies defined SICM as new-onset left ventricular (LV) systolic dysfunction occurring with the onset of sepsis, with a pooled prevalence of 20% (68). The mechanism underlying sepsis-induced myocardial injury involves cardiac mitochondrial dysfunction (69). A recent study has demonstrated that cardiomyocytes can expel damaged mitochondria in the form of vesicles, known as exophers, into the extracellular space of the heart (70). Given that free mitochondria and mitochondrial DNA can induce cardiac damage, the elimination of exophers containing cardiomyocyte-derived mitochondria, a process termed heterophage, is crucial for maintaining cardiac homeostasis and restoring optimal cardiac function (70, 71).
In a cecal ligation and puncture (CLP) sepsis mouse model, single-cell RNA sequencing with fate mapping techniques identified a subgroup of cardiac-resident macrophages highly expressing Trem2 (Trem2hi-CD163+RETNLA+ macrophages). This group of Trem2hi macrophages highly expresses a transcriptome related to phagocytosis according to Gene Ontology (GO) enrichment analysis. Confocal and three-dimensional (3D) images revealed that TREM2hi macrophages engulfed exophers containing cardiomyocyte-derived mitochondria in septic hearts. In addition, TREM2hi macrophages robustly eliminate cardiomyocyte-derived dysfunctional mitochondria through lysosomes (9).
To determine the physiological role of TREM2hi macrophages in SICM, CLP was performed in Trem2 knockout (Trem2-/-) mice and wild-type littermates as controls. Cardiac macrophages from Trem2-/-mice had reduced phagocytotic gene signatures, as evidenced by scRNA-seq data analysis. In addition, Trem2-/- septic mice exhibited impaired ability to clear damaged mitochondria. In addition, compared with WT controls, Trem2-/- septic mice had notably increased mortality and worse cardiac function (9). These results suggest that TREM2 is essential for macrophages to protect cardiac function after sepsis. Interestingly, the intraperitoneal injection of Trem2hi macrophages into Trem2-/- septic mice significantly enhanced cardiac function and improved cardiac injury and inflammation (9).
Taken together, these results suggest that TREM2hi cardiac macrophages contribute to the recovery of cardiac function during SICM through the scavenging of defective mitochondria. The administration of TREM2hi macrophages or the induction of TREM2 expression in macrophages may be a potential therapeutic approach for preventing and rescuing cardiac dysfunction in sepsis patients.
Increasing evidence suggests that HFpEF is associated with a proinflammatory state (72, 73). Previous studies have suggested that cardiac macrophages contribute to diastolic dysfunction through the secretion of profibrotic cytokines in an HFpEF mouse model and that blockade of macrophage recruitment prevents diastolic dysfunction in a pressure overload mode (74–77). However, embryonic-derived cardiac-resident macrophages are important for initiating adaptive responses and preserving cardiac function during hypertensive stress (78). Therefore, a deeper understanding of the diverse functions of cardiac macrophages will be essential for the development of targeted therapies for HFpEF.
Recently, in a mouse model of HFpEF driven by hypertension, Trem2-expressing macrophages were found to be protective in cardiac remodeling (8). The mouse model of hypertension induced by deoxycorticosterone acetate (DOCA) salt displayed clinical features of HFpEF, including diastolic dysfunction, reduced exercise tolerance, and pulmonary congestion (8). Cellular indexing of transcriptomes and epitopes by sequencing (CITE-Seq) revealed that DOCA-salt-treated mice exhibited significant upregulation of Trem2 gene expression in cardiac macrophages (8). To determine the role of Trem2 in HFpEF, Trem2+/+ and Trem2-/- mice were subjected to DOCA-salt treatment. Compared with wild-type control mice, mice with genetic deletion of the Trem2 gene exhibited more severe cardiac hypertrophy and diastolic dysfunction, and no evidence of wall thinning or dilation was observed. Trem2-deficient mice also exhibit exacerbated cardiac hypertrophy and a decrease in cardiac capillary density (8). In addition, Trem2-deficient macrophages exhibit reduced expression of proangiogenic genes and increased expression of proinflammatory cytokines (8). These results suggest a protective role of Trem2 in hypertension-induced HFpEF (Figure 2).
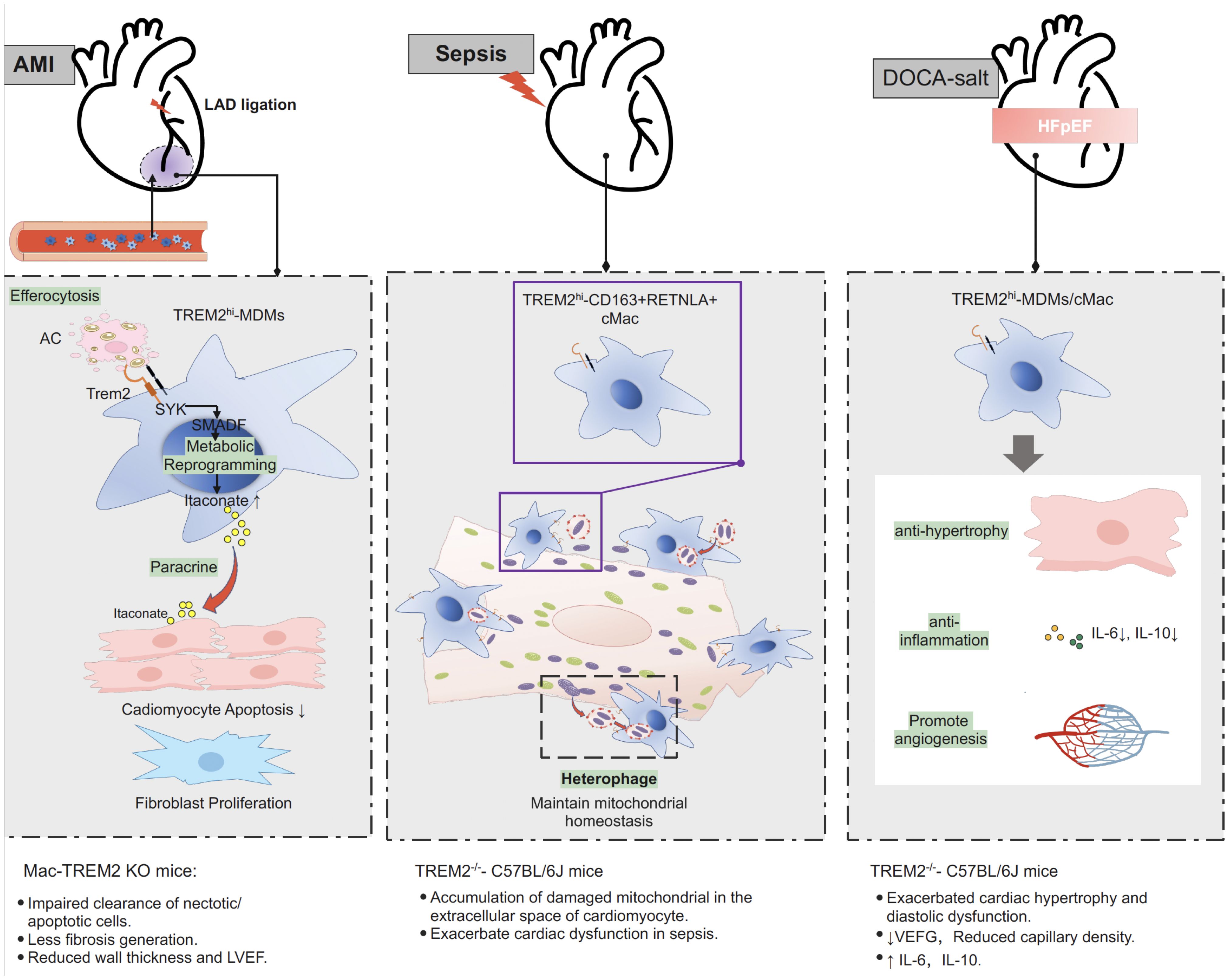
Figure 2. Protective role of Trem2 in different models of myocardial injury. After myocardial infarction, Trem2hi macrophage is responsible for efferocytosis of apoptotic and necrotic cardiomyocytes. Efferocytosis-dependent activation of TREM2-SYK-SMAD signaling pathway remold the metabolism of macrophage and promote section of itaconate, inhibiting the apoptosis of cardiomyocytes in an ischemic environment, promoting the proliferation of fibroblasts, and suppressing inflammation. Specific knocking out Trem2 in macrophage (LysMCreTREMflox/flox, also termed Mac-TREM2 KO) resulted in impaired clearance of necrotic/apoptotic cell after MI, less fibrosis generation, reduced wall thickness and LVEF. In sepsis-induced cardiomyopathy, TREM2hi macrophage contribute to the maintaining of mitochondrial homeostasis and recovery of cardiac function through the scavenging of defective mitochondria. Mice with complete knockout of the TREM2 gene (TREM2-/–C57BL/6J) resulted in accumulation of damaged mitochondria in the extracellular space of cardiomyocyte and exacerbated cardiac function in sepsis. In animal model of DOCA-salt induced hypertensive HFpEF, TREM2 expression was significantly upregulated in macrophage. Deletion of Trem2 (TREM2-/–C57BL/6J) lead to exacerbated cardiac hypertrophy and diastolic dysfunction, reduced capillary density, reduced expression of proangiogenic genes and increased expression of proinflammatory cytokines in macrophage. AMI, acute myocardial infarction; DOCA, deoxycorticosterone acetate; HFpEF, heart failure with preserved ejection fraction; LAD, left anterior descending coronary artery; MDMs, monocyte derived macrophages; cMac, cardiac macrophage; LVEF, left ventricular ejection fraction; PI3K, phosphoinositide 3-kinase; SYK, signaling molecules spleen tyrosine kinase; Trem2, triggering receptor expressed on myeloid cells-2.
The extracellular domain of TREM2 is cleaved at H157-S158 by the proteases a-secretases disintegrin and metalloproteinase domain-containing protein 10 (ADAM10) and ADAM17, which results in the production of soluble TREM2 (sTREM2) (14). The remaining C-terminal fragment on the cell membrane is removed via γ-secretase processing. Moreover, sTREM2 can also be derived from the translation of another transcript lacking a transmembrane domain (79).
Currently, the binding of TREM2 to its ligand is believed to trigger the cleavage of sTREM2 and the removal of TREM2 from the cell membrane. Cells need to continuously mature and transport newly synthesized TREM2 proteins to the cell surface to maintain sustained receptor activity. The levels of sTREM2 in cerebrospinal fluid and plasma may reflect the contact, activation, and cleavage of the receptor TREM2 with its ligand, as well as the flow of new TREM2 proteins through the transport pathway (80).
Therefore, an increase in sTREM2 levels may reflect the extent of the macrophage response to injury and may serve as a biomarker for disease diagnosis and prognosis. For example, in nonalcoholic fatty liver disease and neuroinflammatory diseases, an increase in sTREM2 levels in blood or cerebrospinal fluid is correlated with disease severity. Therefore, sTREM2 may be a potential biochemical marker for disease diagnosis and severity assessment (81–84). In cardiovascular diseases, serum sTREM2 levels are significantly greater in patients with coronary heart disease than in healthy controls (85), and an increase in serum sTREM2 is associated with an increased risk of cardiovascular death in patients with coronary heart disease; thus, sTREM2 could serve as a marker for predicting plaque rupture (86). In patients with HFpEF, serum sTREM2 levels are significantly greater than those in patients without heart failure, and sTREM2 levels are independently associated with heart failure status (8).
Can cleaved sTREM2 bind to other cell surface transmembrane ligands to perform its biological functions? Recent research on Alzheimer’s disease suggested that sTREM2 can bind to microglia and promote their proliferation, migration, and aggregation around plaques, thus playing a neuroprotective role (87, 88). In vitro studies have shown that sTREM2 can affect the expression of inflammatory factors in THP-1-derived macrophages but has different effects on macrophages of different phenotypes: sTREM2 promotes the expression of proinflammatory factors in steady-state M0 and anti-inflammatory phenotype M2 macrophages while inhibiting the expression of inflammatory factors in proinflammatory phenotype (M1) macrophages (89). These findings suggest that sTREM2 may activate the macrophage response to damage, thereby limiting excessive inflammatory damage. In an MI mouse model, sTrem2 levels began to increase on the third day after myocardial infarction, reaching a peak level on the seventh day (7). In vitro experiments revealed that sTrem2 affects the phenotype of thioglycolate-elicited peritoneal macrophages. sTrem2 treatment reduces the expression of the proinflammatory genes Il1b and Nos2 and increases Arg1 expression (7). An in vivo study revealed that the injection of sTrem2 into the peri-infarct area of MI model mice significantly improved heart function and ventricular remodeling (7).
In the context of CVD and HF, macrophages play active roles in the healing process following a wide range of injuries, ranging from inflammation to fibrosis and tissue remodeling. As the hub of pathological immune signaling, TREM2 is an attractive therapeutic target for cardiovascular disease (90). Recent preclinical studies have shown that immediate intramyocardial injection of recombinant adenovirus encoding full-length mouse TREM2 or sTREM2 significantly improves cardiac function in mice with myocardial infarction following ligation of the left anterior descending (LAD) coronary artery (7, 10, 11). Additionally, the injection of TREM2hiMac1 cells into the pericardial cavity following CLP significantly alleviated cardiac dysfunction in mice with sepsis (9).
Recent studies have demonstrated that activated microglia regulate the progression of diseases such as hypertension, myocardial infarction, and ischemia/reperfusion injury by modulating autonomic nervous system activity following neuroinflammation (91). The overexpression of microglial TREM2 mitigates the inflammatory response of microglia and prevents the continuous activation of the sympathetic nervous system, a primary contributor to pathological cardiac remodeling. This suggests that targeting microglial TREM2 may represent a promising therapeutic approach for cardiovascular diseases (92).
Future strategies targeting TREM2 include the following: 1. Directly targeting the receptor’s active domain via specific monoclonal antibodies or small molecules to activate downstream signaling. A project led by Alector, LLC, using an agonistic monoclonal antibody against TREM2 (AL002) has completed phase I clinical trials (93). 2. The targeting conditions and/or tissue-specific TREM2 ligands can be used to direct therapeutics to the precise location at which treatment is needed. However, further understanding of the specific ligands of TREM2 in different tissues is still needed. 3. As TREM2 is continuously cleaved by ADAMs, inhibiting this cleavage is another potential target of treatment. A research team from Germany targeted the cleavage site of TREM2, thereby interfering with ADAM receptor cleavage, increasing the concentration of the receptor on the membrane, and enhancing its signaling activity. The team developed an antibody, 4D9, that binds to the stalk region of TREM2, reduces its proteolytic cleavage, and activates TREM2 signaling (94).
In summary, this review summarizes recent studies on the significance of TREM2 signals in cardiovascular disease, including 1) the impact of the TREM2 cellular pathway on macrophage metabolism and function, as well as the conflicting evidence concerning its role in atherosclerosis; 2) the latest research progress on the importance of macrophages with high TREM2 expression in the recovery of myocardial injury in different models of heart failure; 3) soluble TREM2 (sTREM2) as a potential prognostic biomarker in cardiovascular diseases; and 4) the prospects and challenges of potential treatments targeting TREM2.
The challenge for future interventions targeting TREM2 lies in how to specifically upregulate TREM2 expression in cardiac or plaque macrophages while avoiding potential side effects in other tissues, such as the harmful anti-inflammatory and immunosuppressive activities of TREM2, which promote tumor growth and immune escape (95–97).
SW: Conceptualization, Funding acquisition, Investigation, Project administration, Resources, Writing – original draft, Writing – review & editing. CC: Project administration, Writing – original draft, Writing – review & editing. DP: Conceptualization, Formal analysis, Resources, Supervision, Writing – review & editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by the Natural Science Foundation of Hunan Province (grant number: 2023JJ40855).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Pinto AR, Ilinykh A, Ivey MJ, Kuwabara JT, D’Antoni ML, Debuque R, et al. Revisiting cardiac cellular composition. Circ Res. (2016) 118:400–9. doi: 10.1161/CIRCRESAHA.115.307778
2. Lavine KJ, Pinto AR, Epelman S, Kopecky BJ, Clemente-Casares X, Godwin J, et al. The macrophage in cardiac homeostasis and disease: JACC macrophage in CVD series (Part 4). J Am Coll Cardiol. (2018) 72:2213–30. doi: 10.1016/j.jacc.2018.08.2149
3. Yona S, Kim KW, Wolf Y, Mildner A, Varol D, Breker M, et al. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity. (2013) 38:79–91. doi: 10.1016/j.immuni.2012.12.001
4. Kopecky BJ, Lavine KJ. Cardiac macrophage metabolism in health and disease. Trends Endocrinol Metab. (2024) 35:249–62. doi: 10.1016/j.tem.2023.10.011
5. Depuydt MAC, Prange KHM, Slenders L, Ord T, Elbersen D, Boltjes A, et al. Microanatomy of the human atherosclerotic plaque by single-cell transcriptomics. Circ Res. (2020) 127:1437–55. doi: 10.1161/CIRCRESAHA.120.316770
6. Cochain C, Vafadarnejad E, Arampatzi P, Pelisek J, Winkels H, Ley K, et al. Single-cell RNA-seq reveals the transcriptional landscape and heterogeneity of aortic macrophages in murine atherosclerosis. Circ Res. (2018) 122:1661–74. doi: 10.1161/CIRCRESAHA.117.312509
7. Jung SH, Hwang BH, Shin S, Park EH, Park SH, Kim CW, et al. Spatiotemporal dynamics of macrophage heterogeneity and a potential function of Trem2(hi) macrophages in infarcted hearts. Nat Commun. (2022) 13:4580. doi: 10.1038/s41467-022-32284-2
8. Smart CD, Fehrenbach DJ, Wassenaar JW, Agrawal V, Fortune NL, Dixon DD, et al. Immune profiling of murine cardiac leukocytes identifies triggering receptor expressed on myeloid cells 2 as a novel mediator of hypertensive heart failure. Cardiovasc Res. (2023) 119:2312–28. doi: 10.1093/cvr/cvad093
9. Zhang K, Wang Y, Chen S, Mao J, Jin Y, Ye H, et al. TREM2(hi) resident macrophages protect the septic heart by maintaining cardiomyocyte homeostasis. Nat Metab. (2023) 5:129–46. doi: 10.1038/s42255-022-00715-5
10. Fu C, Xu Q, Liu J, Tang S, Liu C, Cao Y. Triggering receptor expressed on myeloid cells-2 promotes survival of cardiomyocytes after myocardial ischemic injury through PI3K/AKT pathway. Cardiovasc Diagn Ther. (2022) 12:24–36. doi: 10.21037/cdt-21-490
11. Gong S, Zhai M, Shi J, Yu G, Lei Z, Shi Y, et al. TREM2 macrophage promotes cardiac repair in myocardial infarction by reprogramming metabolism via SLC25A53. Cell Death Differ. (2024) 31:239–53. doi: 10.1038/s41418-023-01252-8
12. Kirschenbaum D, Xie K, Ingelfinger F, Katzenelenbogen Y, Abadie K, Look T, et al. Time-resolved single-cell transcriptomics defines immune trajectories in glioblastoma. Cell. (2024) 187:149–65 e23. doi: 10.1016/j.cell.2023.11.032
13. Park MD, Reyes-Torres I, LeBerichel J, Hamon P, LaMarche NM, Hegde S, et al. TREM2 macrophages drive NK cell paucity and dysfunction in lung cancer. Nat Immunol. (2023) 24:792–801. doi: 10.1038/s41590-023-01475-4
14. Deczkowska A, Weiner A, Amit I. The physiology, pathology, and potential therapeutic applications of the TREM2 signaling pathway. Cell. (2020) 181:1207–17. doi: 10.1016/j.cell.2020.05.003
15. Kim SH, Lee KY, Chang K. The protective role of TREM2 in the heterogenous population of macrophages during post-myocardial infarction inflammation. Int J Mol Sci. (2023) 24. doi: 10.3390/ijms24065556
16. Sarraju A, Nissen SE. Atherosclerotic plaque stabilization and regression: a review of clinical evidence. Nat Rev Cardiol. (2024) 21(7):487–97. doi: 10.1038/s41569-023-00979-8
17. Sabatine MS, Giugliano RP, Keech AC, Honarpour N, Wiviott SD, Murphy SA, et al. Evolocumab and clinical outcomes in patients with cardiovascular disease. N Engl J Med. (2017) 376:1713–22. doi: 10.1056/NEJMoa1615664
18. Nidorf SM, Fiolet ATL, Mosterd A, Eikelboom JW, Schut A, Opstal TSJ, et al. Colchicine in patients with chronic coronary disease. N Engl J Med. (2020) 383:1838–47. doi: 10.1056/NEJMoa2021372
19. Vaidya K, Arnott C, Martinez GJ, Ng B, McCormack S, Sullivan DR, et al. Colchicine therapy and plaque stabilization in patients with acute coronary syndrome: A CT coronary angiography study. JACC Cardiovasc Imaging. (2018) 11:305–16. doi: 10.1016/j.jcmg.2017.08.013
20. Tardif JC, Kouz S, Waters DD, Bertrand OF, Diaz R, Maggioni AP, et al. Efficacy and safety of low-dose colchicine after myocardial infarction. N Engl J Med. (2019) 381:2497–505. doi: 10.1056/NEJMoa1912388
21. Barrett TJ. Macrophages in atherosclerosis regression. Arterioscler Thromb Vasc Biol. (2020) 40:20–33. doi: 10.1161/ATVBAHA.119.312802
22. Zernecke A, Erhard F, Weinberger T, Schulz C, Ley K, Saliba AE, et al. Integrated single-cell analysis-based classification of vascular mononuclear phagocytes in mouse and human atherosclerosis. Cardiovasc Res. (2023) 119:1676–89. doi: 10.1093/cvr/cvac161
23. Deng H, Sun Y, Zeng W, Li H, Guo M, Yang L, et al. New classification of macrophages in plaques: a revolution. Curr Atheroscler Rep. (2020) 22:31. doi: 10.1007/s11883-020-00850-y
24. Willemsen L, de Winther MP. Macrophage subsets in atherosclerosis as defined by single-cell technologies. J Pathol. (2020) 250:705–14. doi: 10.1002/path.5392
25. Mocci G, Sukhavasi K, Ord T, Bankier S, Singha P, Arasu UT, et al. Single-cell gene-regulatory networks of advanced symptomatic atherosclerosis. Circ Res. (2024) 134:1405–23. doi: 10.1161/CIRCRESAHA.123.323184
26. Jaitin DA, Adlung L, Thaiss CA, Weiner A, Li B, Descamps H, et al. Lipid-associated macrophages control metabolic homeostasis in a trem2-dependent manner. Cell. (2019) 178:686–98 e14. doi: 10.1016/j.cell.2019.05.054
27. Nugent AA, Lin K, van Lengerich B, Lianoglou S, Przybyla L, Davis SS, et al. TREM2 regulates microglial cholesterol metabolism upon chronic phagocytic challenge. Neuron. (2020) 105:837–54 e9. doi: 10.1016/j.neuron.2019.12.007
28. Guo X, Li B, Wen C, Zhang F, Xiang X, Nie L, et al. TREM2 promotes cholesterol uptake and foam cell formation in atherosclerosis. Cell Mol Life Sci. (2023) 80:137. doi: 10.1007/s00018-023-04786-9
29. Zernecke A, Winkels H, Cochain C, Williams JW, Wolf D, Soehnlein O, et al. Meta-analysis of leukocyte diversity in atherosclerotic mouse aortas. Circ Res. (2020) 127:402–26. doi: 10.1161/CIRCRESAHA.120.316903
30. Dib L, Koneva LA, Edsfeldt A, Zurke YX, Sun J, Nitulescu M, et al. Lipid-associated macrophages transition to an inflammatory state in human atherosclerosis increasing the risk of cerebrovascular complications. Nat Cardiovasc Res. (2023) 2:656–72. doi: 10.1038/s44161-023-00295-x
31. Patterson MT, Firulyova MM, Xu Y, Hillman H, Bishop C, Zhu A, et al. Trem2 promotes foamy macrophage lipid uptake and survival in atherosclerosis. Nat Cardiovasc Res. (2023) 2:1015–31. doi: 10.1038/s44161-023-00354-3
32. Westerterp M, Fotakis P, Ouimet M, Bochem AE, Zhang H, Molusky MM, et al. Cholesterol efflux pathways suppress inflammasome activation, NETosis, and atherogenesis. Circulation. (2018) 138:898–912. doi: 10.1161/CIRCULATIONAHA.117.032636
33. Groenen AG, Halmos B, Tall AR, Westerterp M. Cholesterol efflux pathways, inflammation, and atherosclerosis. Crit Rev Biochem Mol Biol. (2021) 56:426–39. doi: 10.1080/10409238.2021.1925217
34. Wei W, Zhang L, Xin W, Pan Y, Tatenhorst L, Hao Z, et al. TREM2 regulates microglial lipid droplet formation and represses post-ischemic brain injury. BioMed Pharmacother. (2024) 170:115962. doi: 10.1016/j.biopha.2023.115962
35. Li W, Meng X, Peng K, Han Y, Liu H, Zhao W, et al. Boosting microglial lipid metabolism via TREM2 signaling by biomimetic nanoparticles to attenuate the sevoflurane-induced developmental neurotoxicity. Adv Sci (Weinh). (2024) 11:e2305989. doi: 10.1002/advs.202305989
36. Bosch-Queralt M, Cantuti-Castelvetri L, Damkou A, Schifferer M, Schlepckow K, Alexopoulos I, et al. Diet-dependent regulation of TGFbeta impairs reparative innate immune responses after demyelination. Nat Metab. (2021) 3:211–27. doi: 10.1038/s42255-021-00341-7
37. Chait A, Wang S, Goodspeed L, Gomes D, Turk KE, Wietecha T, et al. Sexually dimorphic relationships among saa3 (Serum amyloid A3), inflammation, and cholesterol metabolism modulate atherosclerosis in mice. Arterioscler Thromb Vasc Biol. (2021) 41:e299–313. doi: 10.1161/ATVBAHA.121.316066
38. Reardon CA. Saa3 deficiency identifies a sexually dimorphic effect on atherosclerosis that may be mediated in part by alterations in trem2 expression in macrophages. Arterioscler Thromb Vasc Biol. (2021) 41:1890–2. doi: 10.1161/ATVBAHA.121.316292
39. Meek RL, Eriksen N, Benditt EP. Murine serum amyloid A3 is a high density apolipoprotein and is secreted by macrophages. Proc Natl Acad Sci U.S.A. (1992) 89:7949–52. doi: 10.1073/pnas.89.17.7949
40. Thompson JC, Wilson PG, Shridas P, Ji A, de Beer M, de Beer FC, et al. Serum amyloid A3 is pro-atherogenic. Atherosclerosis. (2018) 268:32–5. doi: 10.1016/j.atherosclerosis.2017.11.011
41. Adkar SS, Leeper NJ. Efferocytosis in atherosclerosis. Nat Rev Cardiol. (2024) 21:762–79. doi: 10.1038/s41569-024-01037-7
42. Schrijvers DM, De Meyer GR, Kockx MM, Herman AG, Martinet W. Phagocytosis of apoptotic cells by macrophages is impaired in atherosclerosis. Arterioscler Thromb Vasc Biol. (2005) 25:1256–61. doi: 10.1161/01.ATV.0000166517.18801.a7
43. Wang Z, Su J, Gong F, Xue L, Su Z. The impaired mechanism and facilitated therapies of efferocytosis in atherosclerosis. J Cardiovasc Pharmacol. (2022) 80:407–16. doi: 10.1097/FJC.0000000000001311
44. Cominacini L, Garbin U, Mozzini C, Stranieri C, Pasini A, Solani E, et al. The atherosclerotic plaque vulnerability: focus on the oxidative and endoplasmic reticulum stress in orchestrating the macrophage apoptosis in the formation of the necrotic core. Curr Med Chem. (2015) 22:1565–72. doi: 10.2174/0929867322666150311150829
45. Wang Y, Liu XY, Wang Y, Zhao WX, Li FD, Guo PR, et al. NOX2 inhibition stabilizes vulnerable plaques by enhancing macrophage efferocytosis via MertK/PI3K/AKT pathway. Redox Biol. (2023) 64:102763. doi: 10.1016/j.redox.2023.102763
46. Wang X, He Q, Zhou C, Xu Y, Liu D, Fujiwara N, et al. Prolonged hypernutrition impairs TREM2-dependent efferocytosis to license chronic liver inflammation and NASH development. Immunity. (2023) 56:58–77 e11. doi: 10.1016/j.immuni.2022.11.013
47. Hu M, Lin Y, Men X, Wang S, Sun X, Zhu Q, et al. High-salt diet downregulates TREM2 expression and blunts efferocytosis of macrophages after acute ischemic stroke. J Neuroinflamm. (2021) 18:90. doi: 10.1186/s12974-021-02144-9
48. Piollet M, Porsch F, Rizzo G, Kapser F, Schulz DJJ, Kiss MG, et al. TREM2 protects from atherosclerosis by limiting necrotic core formation. Nat Cardiovasc Res. (2024) 3:269–82. doi: 10.1038/s44161-024-00429-9
49. Patterson MT, Xu Y, Hillman H, Osinski V, Schrank PR, Kennedy AE, et al. Trem2 agonist reprograms foamy macrophages to promote atherosclerotic plaque stability-brief report. Arterioscler Thromb Vasc Biol. (2024) 44:1646–57. doi: 10.1161/ATVBAHA.124.320797
50. Shirotani K, Hori Y, Yoshizaki R, Higuchi E, Colonna M, Saito T, et al. Aminophospholipids are signal-transducing TREM2 ligands on apoptotic cells. Sci Rep. (2019) 9:7508. doi: 10.1038/s41598-019-43535-6
51. Kim SM, Mun BR, Lee SJ, Joh Y, Lee HY, Ji KY, et al. TREM2 promotes Abeta phagocytosis by upregulating C/EBPalpha-dependent CD36 expression in microglia. Sci Rep. (2017) 7:11118. doi: 10.1038/s41598-017-11634-x
52. Meng Y, Heybrock S, Neculai D, Saftig P. Cholesterol handling in lysosomes and beyond. Trends Cell Biol. (2020) 30:452–66. doi: 10.1016/j.tcb.2020.02.007
53. Parikh M, Patel K, Soni S, Gandhi T. Liver X receptor: a cardinal target for atherosclerosis and beyond. J Atheroscler Thromb. (2014) 21:519–31. doi: 10.5551/jat.19778
54. Guo S, Li L, Yin H. Cholesterol homeostasis and liver X receptor (LXR) in atherosclerosis. Cardiovasc Hematol Disord Drug Targets. (2018) 18:27–33. doi: 10.2174/1871529X18666180302113713
55. Ag N, Bensinger SJ, Hong C, Beceiro S, Bradley MN, Zelcer N, et al. Apoptotic cells promote their own clearance and immune tolerance through activation of the nuclear receptor LXR. Immunity. (2009) 31:245–58. doi: 10.1016/j.immuni.2009.06.018
56. Viaud M, Ivanov S, Vujic N, Duta-Mare M, Aira LE, Barouillet T, et al. Lysosomal cholesterol hydrolysis couples efferocytosis to anti-inflammatory oxysterol production. Circ Res. (2018) 122:1369–84. doi: 10.1161/CIRCRESAHA.117.312333
57. Filipello F, You SF, Mirfakhar FS, Mahali S, Bollman B, Acquarone M, et al. Defects in lysosomal function and lipid metabolism in human microglia harboring a TREM2 loss of function mutation. Acta Neuropathol. (2023) 145:749–72. doi: 10.1007/s00401-023-02568-y
58. Damisah EC, Rai A, Grutzendler J. TREM2: modulator of lipid metabolism in microglia. Neuron. (2020) 105:759–61. doi: 10.1016/j.neuron.2020.02.008
59. Peet C, Ivetic A, Bromage DI, Shah AM. Cardiac monocytes and macrophages after myocardial infarction. Cardiovasc Res. (2020) 116:1101–12. doi: 10.1093/cvr/cvz336
60. Lafuse WP, Wozniak DJ, Rajaram MVS. Role of cardiac macrophages on cardiac inflammation, fibrosis and tissue repair. Cells. (2020) 10. doi: 10.3390/cells10010051
61. DeBerge M, Yeap XY, Dehn S, Zhang S, Grigoryeva L, Misener S, et al. MerTK cleavage on resident cardiac macrophages compromises repair after myocardial ischemia reperfusion injury. Circ Res. (2017) 121:930–40. doi: 10.1161/CIRCRESAHA.117.311327
62. Tsujita K, Kaikita K, Hayasaki T, Honda T, Kobayashi H, Sakashita N, et al. Targeted deletion of class A macrophage scavenger receptor increases the risk of cardiac rupture after experimental myocardial infarction. Circulation. (2007) 115:1904–11. doi: 10.1161/CIRCULATIONAHA.106.671198
63. Peng Q, Malhotra S, Torchia JA, Kerr WG, Coggeshall KM, Humphrey MB. TREM2- and DAP12-dependent activation of PI3K requires DAP10 and is inhibited by SHIP1. Sci Signal. (2010) 3:ra38. doi: 10.1126/scisignal.2000500
64. Chen LL, Morcelle C, Cheng ZL, Chen X, Xu Y, Gao Y, et al. Itaconate inhibits TET DNA dioxygenases to dampen inflammatory responses. Nat Cell Biol. (2022) 24:353–63. doi: 10.1038/s41556-022-00853-8
65. Hooftman A, Angiari S, Hester S, Corcoran SE, Runtsch MC, Ling C, et al. The immunomodulatory metabolite itaconate modifies NLRP3 and inhibits inflammasome activation. Cell Metab. (2020) 32:468–78 e7. doi: 10.1016/j.cmet.2020.07.016
66. Lampropoulou V, Sergushichev A, Bambouskova M, Nair S, Vincent EE, Loginicheva E, et al. Itaconate links inhibition of succinate dehydrogenase with macrophage metabolic remodeling and regulation of inflammation. Cell Metab. (2016) 24:158–66. doi: 10.1016/j.cmet.2016.06.004
67. Runtsch MC, Angiari S, Hooftman A, Wadhwa R, Zhang Y, Zheng Y, et al. Itaconate and itaconate derivatives target JAK1 to suppress alternative activation of macrophages. Cell Metab. (2022) 34:487–501 e8. doi: 10.1016/j.cmet.2022.02.002
68. Hasegawa D, Ishisaka Y, Maeda T, Prasitlumkum N, Nishida K, Dugar S, et al. Prevalence and prognosis of sepsis-induced cardiomyopathy: A systematic review and meta-analysis. J Intensive Care Med. (2023) 38:797–808. doi: 10.1177/08850666231180526
69. Hollenberg SM, Singer M. Pathophysiology of sepsis-induced cardiomyopathy. Nat Rev Cardiol. (2021) 18:424–34. doi: 10.1038/s41569-020-00492-2
70. Nicolas-Avila JA, Lechuga-Vieco AV, Esteban-Martinez L, Sanchez-Diaz M, Diaz-Garcia E, Santiago DJ, et al. A network of macrophages supports mitochondrial homeostasis in the heart. Cell. (2020) 183:94–109 e23. doi: 10.1016/j.cell.2020.08.031
71. Stanzani G, Duchen MR, Singer M. The role of mitochondria in sepsis-induced cardiomyopathy. Biochim Biophys Acta Mol Basis Dis. (2019) 1865:759–73. doi: 10.1016/j.bbadis.2018.10.011
72. Abernethy A, Raza S, Sun JL, Anstrom KJ, Tracy R, Steiner J, et al. Pro-inflammatory biomarkers in stable versus acutely decompensated heart failure with preserved ejection fraction. J Am Heart Assoc. (2018) 7. doi: 10.1161/JAHA.117.007385
73. Schiattarella GG, Rodolico D, Hill JA. Metabolic inflammation in heart failure with preserved ejection fraction. Cardiovasc Res. (2021) 117:423–34. doi: 10.1093/cvr/cvaa217
74. Kai H, Kuwahara F, Tokuda K, Imaizumi T. Diastolic dysfunction in hypertensive hearts: roles of perivascular inflammation and reactive myocardial fibrosis. Hypertens Res. (2005) 28:483–90. doi: 10.1291/hypres.28.483
75. Kuwahara F, Kai H, Tokuda K, Takeya M, Takeshita A, Egashira K, et al. Hypertensive myocardial fibrosis and diastolic dysfunction: another model of inflammation? Hypertension. (2004) 43:739–45. doi: 10.1161/01.HYP.0000118584.33350.7d
76. Hulsmans M, Sager HB, Roh JD, Valero-Munoz M, Houstis NE, Iwamoto Y, et al. Cardiac macrophages promote diastolic dysfunction. J Exp Med. (2018) 215:423–40. doi: 10.1084/jem.20171274
77. Martini E, Kunderfranco P, Peano C, Carullo P, Cremonesi M, Schorn T, et al. Single-cell sequencing of mouse heart immune infiltrate in pressure overload-driven heart failure reveals extent of immune activation. Circulation. (2019) 140:2089–107. doi: 10.1161/CIRCULATIONAHA.119.041694
78. Zaman R, Hamidzada H, Kantores C, Wong A, Dick SA, Wang Y, et al. Selective loss of resident macrophage-derived insulin-like growth factor-1 abolishes adaptive cardiac growth to stress. Immunity. (2021) 54:2057–71 e6. doi: 10.1016/j.immuni.2021.07.006
79. Del-Aguila JL, Benitez BA, Li Z, Dube U, Mihindukulasuriya KA, Budde JP, et al. TREM2 brain transcript-specific studies in AD and TREM2 mutation carriers. Mol Neurodegener. (2019) 14:18. doi: 10.1186/s13024-019-0319-3
80. Filipello F, Goldsbury C, You SF, Locca A, Karch CM, Piccio L. Soluble TREM2: Innocent bystander or active player in neurological diseases? Neurobiol Dis. (2022) 165:105630. doi: 10.1016/j.nbd.2022.105630
81. Hendrikx T, Porsch F, Kiss MG, Rajcic D, Papac-Milicevic N, Hoebinger C, et al. Soluble TREM2 levels reflect the recruitment and expansion of TREM2(+) macrophages that localize to fibrotic areas and limit NASH. J Hepatol. (2022) 77:1373–85. doi: 10.1016/j.jhep.2022.06.004
82. Kothari V, Savard C, Tang J, Lee SP, Subramanian S, Wang S, et al. sTREM2 is a plasma biomarker for human NASH and promotes hepatocyte lipid accumulation. Hepatol Commun. (2023) 7. doi: 10.1097/HC9.0000000000000265
83. Zhou W, Zhou Y, Li J. Association between cerebrospinal fluid soluble TREM2, alzheimer’s disease and other neurodegenerative diseases. J Clin Med. (2023) 12. doi: 10.3390/jcm12103589
84. Spanic Popovacki E, Babic Leko M, Langer Horvat L, Brgic K, Vogrinc Z, Boban M, et al. Soluble TREM2 concentrations in the cerebrospinal fluid correlate with the severity of neurofibrillary degeneration, cognitive impairment, and inflammasome activation in alzheimer’s disease. Neurol Int. (2023) 15:842–56. doi: 10.3390/neurolint15030053
85. Liu W, Weng S, Liu H, Cao C, Wang S, Wu S, et al. Serum soluble TREM2 is an independent biomarker associated with coronary heart disease. Clin Chim Acta. (2023) 548:117499. doi: 10.1016/j.cca.2023.117499
86. Cuciuc V, Tshori S, Grib L, Sella G, Tuvali O, Volodarsky I, et al. Circulating soluble TREM2 and cardiovascular outcome in cohort study of coronary atherosclerosis patients. Int J Mol Sci. (2022) 23. doi: 10.3390/ijms232113121
87. Zhong L, Chen XF, Wang T, Wang Z, Liao C, Wang Z, et al. Soluble TREM2 induces inflammatory responses and enhances microglial survival. J Exp Med. (2017) 214:597–607. doi: 10.1084/jem.20160844
88. Zhong L, Xu Y, Zhuo R, Wang T, Wang K, Huang R, et al. Soluble TREM2 ameliorates pathological phenotypes by modulating microglial functions in an Alzheimer’s disease model. Nat Commun. (2019) 10:1365. doi: 10.1038/s41467-019-09118-9
89. Arsenault R, Marshall S, Salois P, Li Q, Zhang W. sTREM2 differentially affects cytokine expression in myeloid-derived cell models via MAPK-JNK signaling pathway. Biol (Basel). (2024) 13. doi: 10.3390/biology13020087
90. Sansonetti M, Al Soodi B, Thum T, Jung M. Macrophage-based therapeutic approaches for cardiovascular diseases. Basic Res Cardiol. (2024) 119:1–33. doi: 10.1007/s00395-023-01027-9
91. Wang M, Pan W, Xu Y, Zhang J, Wan J, Jiang H. Microglia-mediated neuroinflammation: A potential target for the treatment of cardiovascular diseases. J Inflammation Res. (2022) 15:3083–94. doi: 10.2147/JIR.S350109
92. Xu X, Du L, Jiang J, Yang M, Wang Z, Wang Y, et al. Microglial TREM2 mitigates inflammatory responses and neuronal apoptosis in angiotensin II-induced hypertension in middle-aged mice. Front Aging Neurosci. (2021) 13:716917. doi: 10.3389/fnagi.2021.716917
93. Wang S, Mustafa M, Yuede CM, Salazar SV, Kong P, Long H, et al. Anti-human TREM2 induces microglia proliferation and reduces pathology in an Alzheimer’s disease model. J Exp Med. (2020) 217. doi: 10.1084/jem.20200785
94. Schlepckow K, Kleinberger G, Fukumori A, Feederle R, Lichtenthaler SF, Steiner H, et al. An Alzheimer-associated TREM2 variant occurs at the ADAM cleavage site and affects shedding and phagocytic function. EMBO Mol Med. (2017) 9:1356–65. doi: 10.15252/emmm.201707672
95. Zhang X, Wang W, Li P, Wang X, Ni K. High TREM2 expression correlates with poor prognosis in gastric cancer. Hum Pathol. (2018) 72:91–9. doi: 10.1016/j.humpath.2017.10.026
96. Wang XQ, Tao BB, Li B, Wang XH, Zhang WC, Wan L, et al. Overexpression of TREM2 enhances glioma cell proliferation and invasion: a therapeutic target in human glioma. Oncotarget. (2016) 7:2354–66. doi: 10.18632/oncotarget.6221
Keywords: macrophage, TREM2, atherosclerosis, myocardial infarction, sepsis-induced cardiomyopathy, HFpEF
Citation: Wang S, Cao C and Peng D (2025) The various roles of TREM2 in cardiovascular disease. Front. Immunol. 16:1462508. doi: 10.3389/fimmu.2025.1462508
Received: 10 July 2024; Accepted: 10 February 2025;
Published: 27 February 2025.
Edited by:
Jürgen Bernhagen, Ludwig Maximilian University of Munich, GermanyReviewed by:
Nicolau Beckmann, Novartis Institutes for BioMedical Research, SwitzerlandCopyright © 2025 Wang, Cao and Peng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Daoquan Peng, cGVuZ2RxQGNzdS5lZHUuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.