 Marco Puthenparampil1,2,3*
Marco Puthenparampil1,2,3* Marta Gaggiola1,2Francesca Rinaldi2
Marta Gaggiola1,2Francesca Rinaldi2 M. Nosadini3,4
M. Nosadini3,4 S. Sartori3,4Paola Perini2
S. Sartori3,4Paola Perini2 Paolo Gallo1,2
Paolo Gallo1,2- 1Department of Neurosciences, University of Padua, Padua, Italy
- 2Multiple Sclerosis Centre, Azienda Ospedaliera di Padova, Padua, Italy
- 3Immune-Mediated Nervous System Disease Study Group, Fondazione Istituto di Ricerca Pediatrica Città della Speranza, Padova, Italy
- 4Pediatric Neurology and Neurophysiology Unit, Department of Women’s and Children’s Health, University Hospital of Padova, Padua, Italy
Pediatric-Onset Multiple Sclerosis (POMS) is characterized by both white and grey matter inflammation, as well as by a higher risk of long-term physical and cognitive disability. The peculiar immunopathogenic mechanisms of POMS suggests that the use of induction therapies, including alemtuzumab (ALTZ), might be a promising approach, at least for postpuberal (> 11 yo) POMS. Although no data on the use of induction therapies in POMS are available from clinical trials currently, case series or case reports on the effect of alemtuzumab (ALTZ) have been recently published. In this review we have briefly revised the immunopathogenic features of POMS, as well as on how ALTZ might impact on them, reporting its efficacy observed in different POMS cohorts.
Pediatric-onset multiple sclerosis: clinical aspects
Acquired demyelinating syndrome (ADS) in children under 18 years of age may represent various neuroinflammatory conditions. Among them, Multiple sclerosis (MS) is a chronic autoimmune, demyelinating, and neurodegenerative disease of the central nervous system (CNS). Pediatric-onset MS (POMS), defined when the disease presents clinically under the age of 18, identifies 3–5% of patients with MS, while less than 2% of POMS have an onset under 10 years of age (1, 2). Therefore, pediatric MS is a rare disease, much less common than adult-onset MS (AOMS).
Clinical onset of POMS is often characterized by optic neuritis, transverse myelitis, brainstem syndromes, or an acute disseminated encephalomyelitis (ADEM)-like event. Although many of the focal or multifocal neurologic presentations of POMS resemble those seen in AOMS, brainstem and cerebellar syndromes are particularly common in young children and adolescent patients (3, 4). Moreover, POMS are also much more likely to present with encephalopathy with fever, seizures, and/or polyfocal symptoms, thus mimicking ADEM (5). Although 50% of POMS patients enter the secondary progressive phase of MS after a median period of 23 years, i.e., a time 10 years longer than the observed in AOMS (2), POMS are likely to experience progressive disability at a younger age. In addition, cognitive sequelae of POMS can develop earlier during the disease course, are not associated to physical disability, and are characterized by an impairment in working memory, executive function, and processing speed (6–8).
The above summarized clinical characteristics of POMS define an unfavorable prognosis, that, however, can be influenced by the quality of treatment. Indeed, a reduction of 50-70% of persistent disability has been described in a study that recruited 3198 POMS followed for 21.8 ± 11.7 years (9). The risk of disability (i.e., reaching EDSS score ≥4.0 or ≥6.0) was associated with the disease duration at first EDSS evaluation, the male sex and the availability of new high efficacy therapies at the time of assessment (before 1993 vs 1993-1999 vs 2000-2006 vs 2007-2013) (9). These findings indicate the need of appropriate therapeutic algorithms to prevent disability and disease progression in POMS.
Beside MS, ADS in children under 18 years of age may also include anti-aquaporin-4-associated neuromyelitis optica spectrum disorder (AQP4-NMOSD), myelin oligodendrocyte glycoprotein antibody-associated disorder (MOGAD), or acute disseminated encephalomyelitis (ADEM) with encephalopathy (10). Although studies over the past decade have established that these are distinct entities, clinical phenotypic overlaps can occur between MOGAD, AQP4-NMOSD, and MS. However, cumulative biological, clinical, and pathological evidence allows for differentiation between these conditions (11). Notably, accurate diagnosis at the onset of ADS is crucial, as several studies have shown that baseline disease-modifying therapies (DMTs) for MS, such as interferon beta, glatiramer acetate, and natalizumab, are ineffective in preventing relapses in MOGAD and NMOSD (12). Therefore, to rule out these pathologies is mandatory and constitute the first step for choosing the best therapeutic option in children with MS.
Different immunological mechanisms between pre- and post- puberal POMS
Although studies on the pathology of POMS are extremely rare, the available literature data highlight that prepuberal and post-puberal POMS have relevant pathological differences. Compared to AOMS, brain biopsy and autopsy samples from 19 children with POMS disclosed a more pronounced acute axonal damage in inflammatory demyelinating lesions, that was more pronounced in pre-pubertal (before 11 y.o.) than in post-puberal age (13). In both cases, axonal damage associated with macrophage rather lymphocyte (CD3-positive T cells or CD8-positive cytotoxic T cells) count, but the highest number of macrophages was measured in early active demyelinating lesions of pre-pubertal patients.
Consistent with these pathological findings, differences in clinical presentation, laboratory and imaging findings between prepubertal and pubertal MS onset were reported. Children with disease onset before puberty are more likely to present with a moderate to severe clinical pictures characterized by encephalopathy and/or multifocal symptoms (14–18). Furthermore, fever and impaired cognitive functioning are more common in younger children (14).
Cerebrospinal fluid (CSF) profile is also modified by age at disease onset (15): patients under 11 years of age have a higher percentage of polynuclear cells and monocytes, as well as a lower percentage of lymphocyte in the CSF. Furthermore, post-puberal POMS are more likely to show intrathecal synthesis of IgG oligoclonal bands or elevated IgG index than post-puberal POMS (19, 20). Abnormalities in T cell phenotype and function have been reported in AOMS (21), whereas relatively limited cellular immunology data are available in POMS. An early study suggested that children with early MS exhibited abnormally heightened circulating T cell responses to CNS autoantigens. A subsequent study assessed responses of T cells of both AOMS and POMS to Myelin Basic Protein (MBP) and Myelin Oligodendrocyte Glycoprotein (MOG) and found that both groups mounted preferential but similar responses to particular antigenic epitopes, including MBP83–102, MBP139–153, and MBP146–162 and MOG1–26, MOG38–60, and MOG63–87 (22). Intriguingly, T-cell was determined in peripheral blood mononuclear cells without any cell-sorting, suggesting a high frequency of self-reactive T-cell clones. POMS also exhibited higher frequencies of proliferating memory CD4+ T cells and higher levels of interleukin-17 secretion in response to myelin peptides than healthy children, suggesting that this T cell population may be relevant to pathogenesis of POMS (23). Taken all together, literature data indicate that POMS patients have an increased frequency of self-reactive T-cells in blood, independently of the puberal status. In addition, pre-puberal POMS patients have a higher activation of innate immunity. Therefore, induction therapies that target different immunopathological mechanisms have a rational in POMS. Since a shift in the incidence of MS after puberty, from a 1:1 ratio to a distinct female predominance, a pivotal role of sex hormones in the etiopathogenesis of the disease has been indicated (24).
Therapeutic strategies for POMS
Although an increasing number of disease-modifying treatment options are available for patients with POMS, the above reported differences from AOMS in immunopathological, clinical, and radiological features question whether the efficacy and safety of DMT used to treat AOMS should be uncritically applied to POMS (25). Indeed, the efficacy of recently approved drugs (fingolimod, teriflunomide, dimethyl fumarate), that are considered treatment having a moderate efficacy in AOMS, was limited on the appearance of new/enlarging white matter lesions in POMS (i.e., annualized rate in Fingolimod-treated POMS 4.397; number per scan in Teriflunomide-treated POMS: 4.78; new/enlarging T2 lesion at week 96 in Dimethyl Fumarate-treated POMS: 12.49), thus calling for the use of more effective treatments.) (26–28). On the other hand, the high effect of natalizumab on clinical and radiological outcomes has been largely described, suggesting its use as first treatment in POMS in an escalation view of treatment (29, 30). Although the relevance of highly effective treatments in POMS have been already supported, clinical trials on ocrelizumab and ofatumumab are ongoing (a Phase 1, NCT02200718 and a Phase III Clinical trial, NCT05123703), while the experience on natalizumab is mainly derived from cohort studies (31–34).
Nevertheless, the peculiar immunological background of POMS might differentiate the effect of a drug between AOMS and POMS. This is particularly relevant for induction therapies (i.e., alemtuzumab (ALTZ), cladribine and autologous hematopoietic stem cell transplantation), whose mechanisms of action imply a marked and possibly long-lasting effect on adaptive immune system (i.e., B and T-cell receptor repertoire and network) (35).
The effect of alemtuzumab on adaptive immune system
ALTZ is a humanized monoclonal antibody that targets CD52, a surface molecule mainly expressed by T-and B-cells (36), inducing the death of these cell by an antibody-dependent cellular cytotoxicity mechanism (37).
Administered in two courses of respectively 5 and 3 days distanced by 12 months, ALTZ determines an extensive depletion of lymphocytes, followed by a so called ‘reconstitution phase’ (38–41). The effect of ALTZ on the immune system persists in absence of further drug-exposure, determining long-term control on disease reactivation risk (42–44).
A great emphasis was initially given to the effect of ALTZ on the quantitative difference between B- and T-cell repopulation (45). Indeed B-cell repopulation is fast and determines a rapid, progressive increase of B-cell count, that returns to baseline values after 6 months and increases up to 20-30% from baseline values at month 12 (45, 46). On the other hand, T-cell reconstitution takes longer time. Indeed, CD4+ and CD8+ T-cells are still reduced 12 months after alemtuzumab infusion (-70% and -50% compared to baseline values respectively). T-Helper-1 and -17 are particularly reduced by ALTZ (47), but then T-cell count progressively increases partially for thymic output, mainly for homeostatic proliferation (48). T-reg subset reconstitution is faster than T-naïve and T-memory cells (49), leading to a hypothesized window (between month 6 and 12 after ALTZ infusion) during which the activation of survived (and potentially self-reactive) T-cells is slowed down (50). Interestingly, it was demonstrated that T-regs reconstitution is not driven by thymic output, but again by homeostatic proliferation of survived T-regs and by conversion from residual T-cells during the early post-treatment phase (49). Indeed, the more rapid increase of Tregs compared with conventional T-Helper cells determine a higher percentage of Treg on CD4+ T-cells, that progressively increase from month 6 to month 12. The effect of alemtuzumab on T-reg subset was also evaluated in vitro, where ALTZ-exposed T cells displayed functional regulatory characteristics (anergy to stimulation with allogeneic dendritic cells and ability to suppress the allogeneic response of autologous T cells both with cell–cell contact and Interleukin-2 consumption) (49).
Also, a rapid increase of B-regs, expressed as both absolute number and percentage, was observed and paralleled T-regs expansion (51). In addition, B cells from treated patients secreted higher levels of Interleukin-10 and Brain-derived neurotrophic factor and were able to inhibit the proliferation of autologous conventional T-Helper cells (52).
Nevertheless, the dynamic of B- and T-cell repopulation was not able to explain the clinical and radiological disease reactivation, as well as the risk of autoimmune adverse events, questioning whether the clinical relevance of the repopulation dynamic was based on “quality” rather than on “quantity” (53). Indeed, B-cell reconstitution is mainly driven by the high bioavailability of B-cell activating factor (BAFF) that occurs immediately after ALTZ administration and complete B-cell depletion (54). BAFF bioavailability increases B-cell survival in bone-marrow and spleen and determines a rapid and progressive differentiated repopulation of all B-cell subsets: transitional B-cells repopulate first, followed by naïve B-cells and then memory B-cells, paralleling a progressive decrease of BAFF serum concentration (54). This repopulation mechanism explains why after ALTZ the variability of B-cell receptor repertoire is significantly increased. On the other hand, since T-cells repopulation is mainly driven by homeostatic proliferation of survived T-cells, T-cell Receptor repertoire is narrowed after ALTZ administration, and this effect is particularly relevant to predict the autoimmune adverse events (55–57), but might also explain the long-term effect on MS (48, 56).
Efficacy and safety of alemtuzumab in POMS
The major limitation of effect of ALTZ in pre-pubertal POMS consists in the absence of any activity on natural immunity cells that do not express CD52. Moreover, no data are available in the literature on pre-pubertal POMS, but a clinical trial (NCT03368664) is ongoing recruiting patients between 10 and 18 y.o.
In a case series with two post-puberal patients, the authors report a stable EDSS after a short follow-up (37 month, 20 months, i.e. 24 months and 8 months after ALTZ last infusion), in absence of any relevant systemic autoimmune adverse event, radiological worsening or clinical relapse (58). In both cases patients were initially treated with teriflunomide 14 mg and then switched to ALTZ within the first year of treatment. Notably, six months after starting ALTZ, one patient developed a syndrome of presumed viral origin, characterized by fatigue and mild headache, in the absence of fever or signs of meningeal involvement, with complete recovery after two weeks (58).
In a different cohort, 2 POMS started ALTZ following natalizumab. One patient (patient 2 of the case series, disease onset: 9 y.o.) had three relapses in the 18 months on natalizumab and then switched to ALTZ (59). Four months later he had a severe relapse (EDSS 5.0), with complete recovery in 2 weeks. After the second cycle he was clinically and radiologically stable (follow-up: 7 months). The second patient (patient 5 of the case series, disease onset: 16 y.o.) was treated with fingolimod and rapidly switched to natalizumab for radiological activity. He developed two relapses and 4 new white matter lesions during the first 7 months of natalizumab, and thus switched to ALTZ (no data on anti-natalizumab antibody is reported). After the first ALTZ cycle, he had no evidence of clinical or radiological disease activity for 32 months.
Since treatments before ALTZ might influence its efficacy we have recently evaluated the efficacy and safety of ALTZ in a cohort of POMS that discontinued NTZ (60). Survival analysis revealed that only 1 patient (10% of the whole cohort) developed a clinical relapse 12 months after last ALTZ infusion, while 4 patients (40%) developed asymptomatic radiological disease activity during the follow up. No serious adverse event was observed. Considering the high disease activity rate before NTZ, the administration of ALTZ determined 36 months of No Evidence of Disease Activity 3 (NEDA-3) condition in 37.5% of patients, describing a good profile of efficacy and safety of ALTZ in post-puberal POMS. These data suggest that in POMS a maintenance therapy should be administrated earlier than in adults. On the other hand, the efficacy of ALTZ after NTZ was higher in AOMS. Indeed, only 2 patients developed clinical relapse during the follow up (one had the diagnosis of MS when she was 19 y.o.). Moreover, a higher percentage of AOMS (85.7%) achieved NEDA-3 at month 36 compared to POMS (p=0.05) (Table 1). In addition, the qualitative effect on the immunopathogenic mechanisms could be also hypothesized since the disease reactivation were mainly radiological, in line with the high impact on inflammatory parameters already observed in AOMS (61–63).
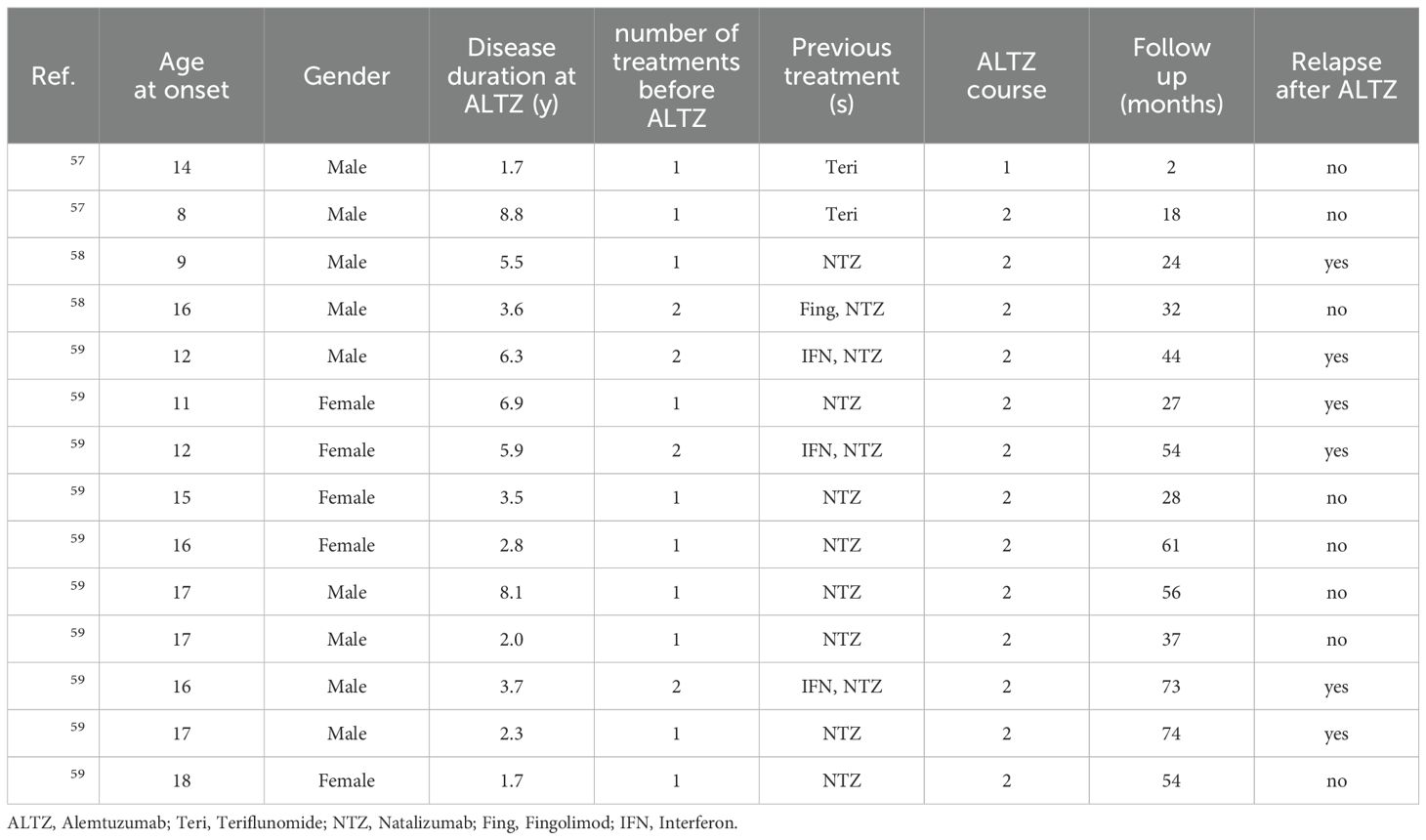
Table 1. Demographic variables of POMS treated with Alemtuzumab.
The different efficacy of ALTZ after NTZ in POMS might be explained by the high percentage of circulating self-reactive lymphocytes in POMS, which might limit the impact of induction therapies in these patients. Moreover, in post-NTZ POMS auto-proliferation may cause a significant expansion of the self-reactive repertoire, increasing the probability of survival of self-reactive T-cell after ALTZ. Interestingly, a more rapid reconstitution of the T cell repertoire is also observed in children compared to adults after autologous haemopoietic stem cell (64). Taken all together, these considerations suggest a more rapid homeostatic proliferation of survived T-cells (including self-reactive T-cell whose percentage is increased also by auto-proliferation) in POMS after ALTZ.
Piecing together the 14 POMS treated with alemtuzumab, we can observe that the risk of disease reactivation in higher in POMS treated with more than one drug before ALTZ (80%) compared with those who had ALTZ as second treatments (11.1%, Odds Ratio 32.00 95%IC 1.294 - 421.3, p=0.023).
Future prospective
Post-puberal POMS might be eligible for ALTZ treatment, but clinical trials will define whether ALTZ might be equally effective in both pre- and post-puberal POMS. Specific condition (e.g, previous treatment with NTZ) might expand self-reactive repertoire, reducing the probability of eliminating all autoreactive lymphocytes. Biomarkers, such as self TCR/BCR expansion, are warranted to optimize treatment response, especially in POMS, tailoring personalized therapy in POMS.
Conclusions
The high inflammatory activity that characterizes POMS requires the administration of highly efficacy treatments as soon as possible. While the use in ALTZ following NTZ should be planned with caution, its use in naïve post-pubertal POMS may have a relevant clinical impact. More data from RCT are needed in order to set more effective and safe therapeutic protocols.
Author contributions
MP: Conceptualization, Data curation, Supervision, Writing – original draft, Writing – review & editing. MG: Data curation, Writing – original draft. FR: Conceptualization, Writing – review & editing. MN: Conceptualization, Writing – review & editing. SS: Conceptualization, Writing – review & editing. PP: Conceptualization, Writing – review & editing. PG: Conceptualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. Open Access funding provided by Università degli Studi di Padova | University of Padua, Open Science Committee. The pubblication was supported by our Institution (università degli Studi di Padova).
Conflict of interest
MP, report grants from Almirall, Teva, Sanofi Genzyme, Merck Serono, Biogen Italy, Novartis; consultancy for Novartis, Biogen Italy, Sanofi Genzyme; board membership Sanofi Genzyme, Novartis, Biogen Italy, and Sandoz. FR report grants from Almirall, Teva, Sanofi Genzyme, Merck Serono, Biogen Italy, Novartis; consultancy for Novartis, Biogen Italy, Sanofi Genzyme. PP reports grants from Almirall, Teva, Sanofi Genzyme, Merck Serono, Biogen Italy, Novartis, Roche; consultancy for Novartis, Biogen Italy, Sanofi Genzyme, Roche. PG reports grant from Almirall, Teva, Sanofi Genzyme, Merck Serono, Biogen Italy, Novartis, Roche, Bristol Myers Squibb; consultancy for Novartis, Biogen Italy, Sanofi Genzyme, Roche, Bristol Myers Squibb; board membership Sanofi Genzyme, Novartis, Biogen Italy, Roche, Merck Serono, Bristol Myers Squibb.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Bigi S, Banwell B. Pediatric multiple sclerosis. J Child Neurol. (2012) 27:1378–83. doi: 10.1177/0883073812452784
2. Boiko A, Vorobeychik G, Paty D, Devonshire V, Sadovnick D, Hashimoto S, et al. Early onset multiple sclerosis: A longitudinal study. Neurology. (2002) 59:1006–10. doi: 10.1212/WNL.59.7.1006
3. Ghezzi A, Deplano V, Faroni J, Grasso MG, Liguori M, Marrosu G, et al. Multiple sclerosis in childhood: clinical features of 149 cases. Mult Scler. (1997) 3:43–6. doi: 10.1177/135245859700300105
4. Bellantonio P, Iuliano G, Di Blasio F, Ruggieri S. Prevalence and incidence of multiple sclerosis in campobasso (Molise region chieftown, southern Italy). Clin Neurol Neurosurg. (2013) 115:1806–8. doi: 10.1016/j.clineuro.2013.05.001
5. Ghezzi A, Pozzilli C, Liguori M, Marrosu MG, Milani N, Milanese C, et al. Prospective study of multiple sclerosis with early onset. Mult Scler. (2002) 8:115–8. doi: 10.1191/1352458502MS786OA
6. Till C, Noguera A, Verhey LH, O’Mahony J, Yeh EA, Mah JK, et al. Cognitive and behavioral functioning in childhood acquired demyelinating syndromes. J Int Neuropsychol Soc. (2016) 22:1050–60. doi: 10.1017/S1355617716000308
7. Amato MP, Goretti B, Ghezzi A, Hakiki B, Niccolai C, Lori S, et al. Neuropsychological features in childhood and juvenile multiple sclerosis: five-year follow-up. Neurology. (2014) 83:1432–8. doi: 10.1212/WNL.0000000000000885
8. Banwell BL, Anderson PE. The cognitive burden of multiple sclerosis in children. Neurology. (2005) 64:891–4. doi: 10.1212/01.WNL.0000152896.35341.51
9. Baroncini D, Simone M, Iaffaldano P, Morra VB, Lanzillo R, Filippi M, et al. Risk of persistent disability in patients with pediatric-onset multiple sclerosis. JAMA Neurol. (2021) 78:726–35. doi: 10.1001/JAMANEUROL.2021.1008
10. Abdel-Mannan O, Hacohen Y. Pediatric inflammatory leukoencephalopathies. Handb Clin Neurol. (2024) 204:369–98. doi: 10.1016/B978-0-323-99209-1.00001-6
11. Armangue T, Capobianco M, de Chalus A, Laetitia G, Deiva K, Bruijstens AL, et al. E.U. Paediatric MOG consortium consensus: part 3 - biomarkers of paediatric myelin oligodendrocyte glycoprotein antibody-associated disorders. Eur J Paediatr Neurol. (2020) 29:22–31. doi: 10.1016/J.EJPN.2020.11.001
12. Jakimovski D, Awan S, Eckert SP, Farooq O, Weinstock-Guttman B. Multiple sclerosis in children: differential diagnosis, prognosis, and disease-modifying treatment. CNS Drugs. (2022) 36:45–59. doi: 10.1007/S40263-021-00887-W
13. Bar-Or A, Hintzen RQ, Dale RC, Rostasy K, Brück W, Chitnis T. Immunopathophysiology of pediatric CNS inflammatory demyelinating diseases. Neurology. (2016) 87:S12–9. doi: 10.1212/WNL.0000000000002821
14. Banwell B, Ghezzi A, Bar-Or A, Mikaeloff Y, Tardieu M. Multiple sclerosis in children: clinical diagnosis, therapeutic strategies, and future directions. Lancet Neurol. (2007) 6:887–902. doi: 10.1016/S1474-4422(07)70242-9
15. Chabas D, Strober J, Waubant E. Pediatric multiple sclerosis. Curr Neurol Neurosci Rep. (2008) 8:434–41. doi: 10.1007/S11910-008-0067-1
16. Dale RC, Brilot F, Banwell B. Pediatric central nervous system inflammatory demyelination: acute disseminated encephalomyelitis, clinically isolated syndromes, neuromyelitis optica, and multiple sclerosis. Curr Opin Neurol. (2009) 22:233–40. doi: 10.1097/WCO.0B013E32832B4C47
17. Huppke B, Ellenberger D, Rosewich H, Friede T, Gärtner J, Huppke P. Clinical presentation of pediatric multiple sclerosis before puberty. Eur J Neurol. (2014) 21:441–6. doi: 10.1111/ENE.12327
18. Waubant E, Chabas D. Pediatric multiple sclerosis. Curr Treat Options Neurol. (2009) 11:203–10. doi: 10.1007/S11940-009-0024-6/METRICS
19. Yeh EA, Chitnis T, Krupp L, Ness J, Chabas D, Kuntz N, et al. Pediatric multiple sclerosis. Nat Rev Neurol. (2009) 5:621–31. doi: 10.1038/NRNEUROL.2009.158
20. Coyle PK. Pediatric multiple sclerosis: just like their elders? Arch Neurol. (2011) 68:419–21. doi: 10.1001/ARCHNEUROL.2011.49
21. Chitnis T. The role of CD4 T cells in the pathogenesis of multiple sclerosis. Int Rev Neurobiol. (2007) 79:43–72. doi: 10.1016/S0074-7742(07)79003-7
22. Correale J, Tenembaum SN. Myelin basic protein and myelin oligodendrocyte glycoprotein T-cell repertoire in childhood and juvenile multiple sclerosis. Mult Scler. (2006) 12:412–20. doi: 10.1191/135248506MS1282OA
23. Vargas-Lowy D, Kivisäkk P, Gandhi R, Raddassi K, Soltany P, Gorman MP, et al. Increased th17 response to myelin peptides in pediatric MS. Clin Immunol. (2013) 146:176–84. doi: 10.1016/J.CLIM.2012.12.008
24. Waubant E. Effect of puberty on multiple sclerosis risk and course. Mult Scler. (2018) 24:32–5. doi: 10.1177/1352458517737393
25. GianFrancesco MA, Stridh P, Shao X, Rhead B, Graves JS, Chitnis T, et al. Genetic risk factors for pediatric-onset multiple sclerosis. Multiple Sclerosis J. (2018) 24:1825–34. doi: 10.1177/1352458517733551
26. Chitnis T, Arnold DL, Banwell B, Brück W, Ghezzi A, Giovannoni G, et al. Trial of fingolimod versus interferon beta-1a in pediatric multiple sclerosis. New Engl J Med. (2018) 379:1017–27. doi: 10.1056/NEJMOA1800149/SUPPL_FILE/NEJMOA1800149_DISCLOSURES.PDF
27. Chitnis T, Banwell B, Kappos L, Arnold DL, Gücüyener K, Deiva K, et al. Safety and efficacy of teriflunomide in paediatric multiple sclerosis (TERIKIDS): A multicentre, double-blind, phase 3, randomised, placebo-controlled trial. Lancet Neurol. (2021) 20:1001–11. doi: 10.1016/S1474-4422(21)00364-1
28. Vermersch P, Scaramozza M, Levin S, Alroughani R, Deiva K, Pozzilli C, et al. Effect of dimethyl fumarate vs interferon β-1a in patients with pediatric-onset multiple sclerosis: the CONNECT randomized clinical trial. JAMA Netw Open. (2022) 5:e2230439–e2230439. doi: 10.1001/JAMANETWORKOPEN.2022.30439
29. Benallegue N, Rollot F, Wiertlewski S, Casey R, Debouverie M, Kerbrat A, et al. Highly effective therapies as first-line treatment for pediatric-onset multiple sclerosis. JAMA Neurol. (2024) 81:273–82. doi: 10.1001/JAMANEUROL.2023.5566
30. Huppke P, Huppke B, Ellenberger D, Rostasy K, Hummel H, Stark W, et al. Therapy of highly active pediatric multiple sclerosis. Multiple Sclerosis J. (2019) 25:72–80. doi: 10.1177/1352458517732843/ASSET/IMAGES/LARGE/10.1177_1352458517732843-FIG3.JPEG
31. Kornek B, Aboul-Enein F, Rostasy K, Milos RI, Steiner I, Penzien J, et al. Natalizumab therapy for highly active pediatric multiple sclerosis. JAMA Neurol. (2013) 70:469–75. doi: 10.1001/JAMANEUROL.2013.923
32. Baroncini D, Ghezzi A, Guaschino C, Moiola L, Filippi M, Ianniello A, et al. Long-term follow-up (up to 11 years) of an italian pediatric MS cohort treated with natalizumab: A multicenter, observational study. Neurol Sci. (2022) 43:6415–23. doi: 10.1007/S10072-022-06211-8
33. Gontika M, Skarlis C, Markoglou N, Tzanetakos D, Vakrakou A, Toulas P, et al. Natalizumab therapy in patients with pediatric-onset multiple sclerosis in Greece: clinical and immunological insights of time-long administration and future directions-a single-center retrospective observational study. Naunyn Schmiedebergs Arch Pharmacol. (2022) 395:933–43. doi: 10.1007/S00210-022-02238-Y
34. Puthenparampil M, Gaggiola M, Ponzano M, Zanotelli G, Miscioscia A, Nosadini M, et al. High NEDA and no PIRA in natalizumab-treated patients with pediatric-onset multiple sclerosis. Neurology(R) neuroimmunology Neuroinflamm. (2024) 11:e200303. doi: 10.1212/NXI.0000000000200303
35. Sorensen PS, Sellebjerg F. Pulsed immune reconstitution therapy in multiple sclerosis. Ther Adv Neurol Disord. (2019) 12. doi: 10.1177/1756286419836913
36. Rao SP, Sancho J, Campos-Rivera J, Boutin PM, Severy PB, Weeden T, et al. Human peripheral blood mononuclear cells exhibit heterogeneous CD52 expression levels and show differential sensitivity to alemtuzumab mediated cytolysis. PloS One. (2012) 7. doi: 10.1371/JOURNAL.PONE.0039416
37. Wing MG, Moreau T, Greenwood J, Smith RM, Hale G, Isaacs J, et al. Mechanism of first-dose cytokine-release syndrome by CAMPATH 1-H: involvement of CD16 (FcgammaRIII) and CD11a/CD18 (LFA-1) on NK cells. J Clin Invest. (1996) 98:2819. doi: 10.1172/JCI119110
38. Coles AJ, Twyman CL, Arnold DL, Cohen JA, Confavreux C, Fox EJ, et al. Alemtuzumab for patients with relapsing multiple sclerosis after disease-modifying therapy: A randomised controlled phase 3 trial. Lancet. (2012) 380:1829–39. doi: 10.1016/S0140-6736(12)61768-1
39. Cohen JA, Coles AJ, Arnold DL, Confavreux C, Fox EJ, Hartung HP, et al. Alemtuzumab versus interferon beta 1a as first-line treatment for patients with relapsing-remitting multiple sclerosis: A randomised controlled phase 3 trial. Lancet. (2012) 380:1819–28. doi: 10.1016/S0140-6736(12)61769-3
40. Coles AJ, Compston DA, Selmaj KW, Lake SL, Moran S, Margolin DH, et al. Alemtuzumab vs. Interferon beta-1a in early multiple sclerosis. N Engl J Med. (2008) 359:1786–801. doi: 10.1056/NEJMOA0802670
41. Hill-Cawthorne GA, Button T, Tuohy O, Jones JL, May K, Somerfield J, et al. Long term lymphocyte reconstitution after alemtuzumab treatment of multiple sclerosis. J Neurol Neurosurg Psychiatry. (2012) 83:298–304. doi: 10.1136/jnnp-2011-300826
42. Steingo B, Al Malik Y, Bass AD, Berkovich R, Carraro M, Fernández Ó., et al. Long-term efficacy and safety of alemtuzumab in patients with RRMS: 12-year follow-up of CAMMS223. J Neurol. (2020) 267:3343–53. doi: 10.1007/S00415-020-09983-1
43. Coles AJ, Achiron A. Safety and efficacy with alemtuzumab over 13 years in relapsing-remitting multiple sclerosis: final results from the open-label TOPAZ study. Ther Adv Neurol Disord. (2023) 16. doi: 10.1177/17562864231194823/ASSET/IMAGES/LARGE/10.1177_17562864231194823-FIG5.JPEG
44. Coles A, Habek M, Bass A, Brinar V, Vladic A, Margolin D, et al. Durable efficacy of alemtuzumab over 10 years: long-term follow-up of patients with RRMS from the CAMMS223 study (P3.053). Neurology. (2016) 86. doi: 10.1212/WNL.86.16_SUPPLEMENT.P3.053
45. Baker D, Herrod SS, Alvarez-Gonzalez C, Giovannoni G, Schmierer K. Interpreting lymphocyte reconstitution data from the pivotal phase 3 trials of alemtuzumab. JAMA Neurol. (2017) 74:961–9. doi: 10.1001/JAMANEUROL.2017.0676
46. Baker D, Herrod SS, Alvarez-Gonzalez C, Zalewski L, Albor C, Schmierer K. Both cladribine and alemtuzumab may effect MS via B-cell depletion. Neurol - Neuroimmunology Neuroinflamm. (2017) 4:e360. doi: 10.1212/NXI.0000000000000360
47. Zhang X, Tao Y, Chopra M, Ahn M, Marcus KL, Choudhary N, et al. Differential reconstitution of T cell subsets following immunodepleting treatment with alemtuzumab (Anti-CD52 monoclonal antibody) in patients with relapsing-remitting multiple sclerosis. J Immunol. (2013) 191:5867–74. doi: 10.4049/JIMMUNOL.1301926
48. Jones JL, Thompson SAJ, Loh P, Davies JL, Tuohy OC, Curry AJ, et al. Human autoimmunity after lymphocyte depletion is caused by homeostatic T-cell proliferation. Proc Natl Acad Sci U.S.A. (2013) 110:20200–5. doi: 10.1073/PNAS.1313654110/-/DCSUPPLEMENTAL
49. Havari E, Turner MJ, Campos-Rivera J, Shankara S, Nguyen TH, Roberts B, et al. Impact of alemtuzumab treatment on the survival and function of human regulatory T cells in vitro. Immunology. (2014) 141:123. doi: 10.1111/IMM.12178
50. Haas J, Würthwein C, Korporal-Kuhnke M, Viehoever A, Jarius S, Ruck T, et al. Alemtuzumab in multiple sclerosis: short- and long-term effects of immunodepletion on the peripheral treg compartment. Front Immunol. (2019) 10:1204/BIBTEX. doi: 10.3389/FIMMU.2019.01204/BIBTEX
51. Kim Y, Kim G, Shin HJ, Hyun JW, Kim SH, Lee E, et al. Restoration of regulatory B cell deficiency following alemtuzumab therapy in patients with relapsing multiple sclerosis. J Neuroinflamm. (2018) 15:300. doi: 10.1186/S12974-018-1334-Y
52. Kashani N, Kelland EE, Vajdi B, Anderson LM, Gilmore W, Lund BT. Immune regulatory cell bias following alemtuzumab treatment in relapsing-remitting multiple sclerosis. Front Immunol. (2021) 12:706278/FULL. doi: 10.3389/FIMMU.2021.706278/FULL
53. Wiendl H, Carraro M, Comi G, Izquierdo G, Kim HJ, Sharrack B, et al. Lymphocyte pharmacodynamics are not associated with autoimmunity or efficacy after alemtuzumab. Neurol Neuroimmunol Neuroinflamm. (2020) 7:E635. doi: 10.1212/NXI.0000000000000635
54. Thompson SAJ, Jones JL, Cox AL, Compston DAS. Coles, A.J. B-cell reconstitution and BAFF after alemtuzumab (Campath-1H) treatment of multiple sclerosis. J Clin Immunol. (2010) 30:99–105. doi: 10.1007/S10875-009-9327-3
55. von Essen MR, Chow HH, Holm Hansen R, Buhelt S, Sellebjerg F. Immune reconstitution following alemtuzumab therapy is characterized by exhausted T cells, increased regulatory control of proinflammatory T cells and reduced B cell control. Front Immunol. (2023) 14:1249201/FULL. doi: 10.3389/FIMMU.2023.1249201/FULL
56. Ruck T, Barman S, Schulte-Mecklenbeck A, Pfeuffer S, Steffen F, Nelke C, et al. Alemtuzumab-induced immune phenotype and repertoire changes: implications for secondary autoimmunity. Brain. (2022) 145:1711–25. doi: 10.1093/BRAIN/AWAC064
57. Rinaldi F, Federle L, Puthenparampil M, Perini P, Grassivaro F, Gallo P. Evidence of B-cell dysregulation in severe CNS inflammation after alemtuzumab therapy. Neurol - Neuroimmunology Neuroinflamm. (2018) 5:e420. doi: 10.1212/NXI.0000000000000420
58. Jure Hunt D, Traboulsee A. Short-term outcomes of pediatric multiple sclerosis patients treated with alemtuzumab at a canadian university multiple sclerosis clinic. Mult Scler J Exp Transl Clin. (2020) 6. doi: 10.1177/2055217320926613
59. Immovilli P, De Mitri P, Bazzurri V, Vollaro S, Morelli N, Biasucci G, et al. The impact of highly effective treatment in pediatric-onset multiple sclerosis: A case series. Children. (2022) 9:1698. doi: 10.3390/CHILDREN9111698
60. Puthenparampil M, Gaggiola M, Miscioscia A, Mauceri VA, De Napoli F, Zanotelli G, et al. Alemtuzumab following natalizumab is more effective in adult-onset than paediatric-onset multiple sclerosis. Ther Adv Neurol Disord. (2023) 16. doi: 10.1177/17562864231177196
61. Federle L, Puthenparampil M, Stenta G, Paolo G, Francesco P. Alemtuzumab as rescue therapy in case of multiple sclerosis rebound following natalizumab break: clinical case and literature review. Mult Scler Relat Disord. (2019) 30:262–4. doi: 10.1016/j.msard.2019.03.002
62. Akgün K, Metz I, Kitzler HH, Brück W, Ziemssen T. Rescue therapy with alemtuzumab in B cell/antibody-mediated multiple sclerosis. Ther Adv Neurol Disord. (2018) 11. doi: 10.1177/1756286418759895
63. Huhn K, Bayas A, Doerck S, Frank B, Gerbershagen K, Hellwig K, et al. Alemtuzumab as rescue therapy in a cohort of 50 relapsing-remitting MS patients with breakthrough disease on fingolimod: A multi-center observational study. J Neurol. (2018) 265:1521–7. doi: 10.1007/S00415-018-8871-2
64. Small TN, Papadopoulos EB, Boulad F, Black P, Castro-Malaspina H, Childs BH, et al. Comparison of immune reconstitution after unrelated and related T-cell–depleted bone marrow transplantation: effect of patient age and donor leukocyte infusions. Blood. (1999) 93:467–80. doi: 10.1182/BLOOD.V93.2.467
Keywords: pediatric-onset MS, POMS, alemtuzumab, induction therapy, multiple sclerosis
Citation: Puthenparampil M, Gaggiola M, Rinaldi F, Nosadini M, Sartori S, Perini P and Gallo P (2025) The immunological bases of alemtuzumab as induction-therapy in pediatric-onset multiple sclerosis. Front. Immunol. 15:1509987. doi: 10.3389/fimmu.2024.1509987
Received: 11 October 2024; Accepted: 18 December 2024;
Published: 08 January 2025.
Edited by:
Douglas Kazutoshi Sato, Pontifical Catholic University of Rio Grande do Sul, BrazilReviewed by:
Eslam Shosha, McMaster University, CanadaCopyright © 2025 Puthenparampil, Gaggiola, Rinaldi, Nosadini, Sartori, Perini and Gallo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marco Puthenparampil, bWFyY28ucHV0aGVucGFyYW1waWxAdW5pcGQuaXQ=