 Yannick Bulliard1
Yannick Bulliard1 Robert Freeborn1
Robert Freeborn1 Molly Javier Uyeda1
Molly Javier Uyeda1 Daryl Humes1Ryan Bjordahl1David de Vries1
Daryl Humes1Ryan Bjordahl1David de Vries1 Maria Grazia Roncarolo1,2*
Maria Grazia Roncarolo1,2*- 1Department of Research and Development, Tr1X, Inc., San Diego, CA, United States
- 2Division of Hematology, Oncology, Stem Cell Transplantation, and Regenerative Medicine, Department of Pediatrics, Stanford University School of Medicine, Stanford, CA, United States
Autoimmune diseases, characterized by the immune system’s attack on the body’s own tissues, affect millions of people worldwide. Current treatments, which primarily rely on broad immunosuppression and symptom management, are often associated with significant adverse effects and necessitate lifelong therapy. This review explores the next generation of therapies for immune-mediated diseases, including chimeric antigen receptor (CAR) T cell and regulatory T cell (Treg)-based approaches, which offer the prospect of targeted, durable disease remission. Notably, we highlight the emergence of CD19-targeted CAR T cell therapies, and their ability to drive sustained remission in B cell-mediated autoimmune diseases, suggesting a possible paradigm shift. Further, we discuss the therapeutic potential of Type 1 and FOXP3+ Treg and CAR-Treg cells, which aim to achieve localized immune modulation by targeting their activity to specific tissues or cell types, thereby minimizing the risk of generalized immunosuppression. By examining the latest advances in this rapidly evolving field, we underscore the potential of these innovative cell therapies to address the unmet need for long-term remission and potential tolerance induction in individuals with autoimmune and immune-mediated diseases.
1 Introduction to autoimmune and other immune-mediated diseases
1.1 Overview of autoimmune diseases and current treatment approaches
Autoimmune and inflammatory diseases, which encompass a wide range of disorders with complex pathology and etiology, affect between 5-10% of the population in industrialized regions, and their burden continues to increase (1). Although heterogeneous, autoimmunity generally arises from a combination of genetic predisposition and environmental co-factors leading to initiation and potentiation of attacks by the body’s own immune cells, specifically autoreactive T and B cells (2). Conventional first-line approaches for the treatment of autoimmune diseases have historically involved either the suppression of general immune function to modulate uncontrolled inflammation via corticosteroids or immunosuppressants such as azathioprine and methotrexate, or the use of disease-modifying anti-rheumatic drugs (DMARDs). This broad, non-specific efficacy, combined with lifelong treatment requirements, leads to frequently intolerable side effects, including increased risk of infection due to continued immunosuppression (3). More recently, biologics focused specifically on certain components of the immune system, such as cytokines or cell surface proteins, have been applied successfully to these types of diseases. Similar to steroids and DMARDs, these newer biologics and small molecules, which include TNF-alpha inhibitors, other monoclonal antibodies, and JAK or TYK2 inhibitors, target downstream inflammatory mediators but generally do not address the underlying disease drivers. Unfortunately, many patients eventually experience primary treatment failure or lose response to these therapies over time due to the development of anti-drug antibodies (4).
Autoimmune diseases are largely characterized by an aberrant immune response directed against self-antigens, often mediated by dysregulated T and B lymphocytes. T cells can inflict direct tissue damage or orchestrate inflammatory cascades via cytokine release (5), while B cells contribute to pathology through both production of pro-inflammatory cytokines and through production of autoantibodies, which can bind to self-antigens on tissues or form immune complexes leading to complement activation and inflammation (6). Therapeutic strategies aimed at depleting these pathogenic lymphocytes seek to interrupt the self-enhancing damage they cause and have demonstrated clinical success across several indications.
1.2 Dual B and T cell targeting: mechanistic rationale to treat autoimmunity
While not curative, lymphocyte depletion or suppression has demonstrated efficacy in managing a range of autoimmune conditions, albeit with a concomitant risk of opportunistic infections due to transient generalized immunosuppression. Rituximab, a chimeric monoclonal antibody targeting the CD20 antigen on B cells, exemplifies the clinical utility of B cell depletion in autoimmune disease management. It has been shown to reduce disease activity in rheumatoid arthritis, systemic sclerosis and multiple sclerosis, among other diseases (7). Conversely, abatacept, an IgG1/CTLA4 fusion protein, works to inhibit T-cell activation by binding to CD80 and CD86 on antigen-presenting cells, preventing their interaction with CD28 on effector T cells and is approved in several autoimmune indications including rheumatoid arthritis and psoriatic arthritis. Combination therapies, aiming to target both cell subsets, including using rituximab and abatacept simultaneously have been attempted in autoimmune diseases such as rheumatoid arthritis. The results have demonstrated good efficacy but a mixed safety profile due to broad immunosuppression and associated increased infection rate (8). This dual targeting approach is also being explored in trials evaluating anti CD20 therapies in combination with different cell therapies, including NK cells, as further described below.
Newer agents have sought to improve on the specificity and safety profile of previous generation biologics by focusing on the mechanisms and signaling driving inflammation, tissue damage and autoantibody production, rather than broadly depleting B cells or suppressing T cell proliferation. Specifically, antibodies targeting more mature B cells than anti-CD20 (9) or both mature B and T cell subsets (e.g. anti-CD52 antibodies) (10) are being investigated in a number of autoimmune disease settings. Further studies are ongoing to identify bi-specific antibodies capable of an even more robust and precise B cell depletion than traditional monoclonal antibodies, by driving improved lymph node and tissue penetration (11). Beyond depletion, therapeutic antibodies and small molecules that can act on B cell and T cell signaling have also gained traction in treating a wide range of autoimmune disorders, such as those focusing on intracellular pathways (e.g complement inhibitors, S1PR modulators, TYK2 inhibitors, IL-12/23p40 inhibitors, and JAK inhibitors, among others) (12–14). In particular, JAK inhibitors have been successful in tackling a number of autoimmune and inflammatory diseases across rheumatological, dermatological, gastrointestinal and transplant-related disorders. Their efficacy in these multiple settings is somewhat hindered by their less-than-ideal toxicity profile, including the increased risk of infections, cardiovascular risk, hematological effects and malignancies, resulting in a boxed warning from the FDA.
Crucially, not all patients respond equally well, both in terms of depth and duration of response within the same indication to the same class of drug. This highlights the importance of understanding both the unique pathophysiology of disease in each individual, and the need for identification of biological markers predictive of response in heterogeneous patient populations. A recent example is the use of a companion diagnostic to identify likely responders to anti-tumor necrosis factor-like cytokine 1A (TL1A) antibodies for patients with inflammatory bowel disease (15)
Yet, despite these improvements in precision and reduction in overall toxicity profile, these novel therapies still only serve to manage, rather than cure autoimmune and immune-mediated diseases. Cell therapies, in particular CAR-T cells, which involve the manipulation and administration of engineered T cells to treat disease, have revolutionized the treatment landscape in oncology and are now being adapted for autoimmune and inflammatory disorders. In addition to conventional CD3+ T cells, other cell therapies, including hematopoietic stem cell transplant (HSCT), regulatory T (Treg) cells (16), natural killer (NK) cells and mesenchymal stem cells (MSCs) are being investigated for their ability to modulate immune responses and promote tolerance. These therapies aim to restore immune homeostasis by either suppressing autoreactive immune cells or promoting regulatory pathways that prevent tissue damage (17). The goal of these therapies is to induce long-term remission via the restoration of immune tolerance.
2 Cell therapies in autoimmunity and immune-mediated diseases: a new frontier
2.1 HSCT: the last line of defense
The use of cell therapy to treat autoimmune diseases traces back to the 1990s with the first confirmed use of hematopoietic stem cell transplantation (HSCT) for connective tissue disease and severe pulmonary hypertension (18). Since those early cases, HSCT has evolved to become a viable option for various severe, treatment-resistant autoimmune conditions, including multiple sclerosis, systemic sclerosis, and Crohn’s disease (19). HSCT involves the replacement of a patient’s hematopoietic and immune system with stem cells from a healthy donor (allogeneic HSCT), or more commonly in autoimmune diseases, from the patient themselves (autologous HSCT).
The mobilization and collection of peripheral stem cells for HSCT is a multi-step process that requires careful monitoring and management. Mobilization of stem cells from the bone marrow of patients into the peripheral blood is achieved primarily through the administration of granulocyte colony-stimulating factor (G-CSF), or in combination with cyclophosphamide as pre-treatment (20). Once mobilized, the stem cells are collected from the peripheral blood via leukapheresis. After collection, these cells can undergo further processing or manipulation based on the specific HSCT protocol being followed. Various protocols are currently used, and the optimal protocol for maximum therapeutic benefit has not been definitively established. For instance, in patients with systemic sclerosis treated with autologous HSCT, enrichment of CD34+ progenitor cells does not appear to provide additional clinical benefit (21).
The rationale behind HSCT in autoimmune diseases lies on the potential to ‘reset’ the aberrant immune system, fostering the regeneration of a self-tolerant immune repertoire (22). The precise mechanisms by which HSCT orchestrates immunological reset remain an area of active investigation. The process involves a multi-faceted interplay of immune cell depletion via lymphoablative conditioning, lymphopenia-induced proliferation, differentiation of new T cell progenitors in the thymus, and the modulation of Treg cells (23). Importantly, Treg cells, key players in immune tolerance, undergo significant changes following HSCT, with studies reporting an increase and more diverse Treg cell repertoire after autologous HSCT. This is associated with greater suppressive activity in immune mediated diseases such as Crohn’s, multiple sclerosis, systemic lupus erythematosus (SLE), and systemic sclerosis (24–28). In the context of allogeneic HSCT, a high frequency of donor Treg cells has been associated with a lower risk of Graft-versus-Host Disease (GvHD) mediated by donor effector T cells, which remains a problematic and frequently lethal side effect of HSCT (29).
Importantly, the procedure is not without risks. HSCT is an onerous process for patients that involves multiple complexities, including the need for taxing immunoablative pre-conditioning regimens, which increases treatment-related morbidity and mortality due to severe infections after immunosuppression and cytopenia (30). In the last decade, the safety of the procedure has shown remarkable improvement, thanks to better selection of the most appropriate patients to transplant, better donor matching, and decreased intensity of conditioning regimens, including use of novel agents. For patients with a relapse-remitting form of multiple sclerosis for instance, treatment related mortality due to autologous HSCT has decreased to <1% (31). For systemic sclerosis, where treatment-related mortality mostly stemmed from cardiac complications, careful patient selection has enabled a decrease mortality rate to <6% (19). While less frequently used in autoimmune disease compared to oncology, HSCT can be with is only really considered as a treatment option for life-threatening forms of these diseases or relapsing courses of disease occurring after autologous transplantation. Allogeneic HSCT carries a greater risk of death and complications compared to autologous HSCT (23). Thus, though HSCT has demonstrated benefit in treating conditions including multiple and systemic sclerosis, its use remains restricted to a limited number of patients with severe disease who have not responded to conventional or approved therapies.
2.2 Mesenchymal stem cell therapies
Research into the immuno-modulating and regenerative properties of MSCs has sparked interest in this primarily allogeneic stem cell-based modality for the treatment of autoimmune diseases (32). MSCs are believed to exert their therapeutic effects through paracrine signaling, releasing cytokines that can suppress pro-inflammatory responses, promote tissue repair, and modulate immune cell function (33). MSCs can also be engineered to deliver a therapeutic payload. For instance, IL-10-expressing MSCs have been shown to decrease inflammation and improve fine motor function after traumatic brain injury in rats (34). Clinical trials investigating the use of MSCs in autoimmune diseases such as Crohn’s disease and ulcerative colitis (35–37), GvHD (38), multiple sclerosis (39, 40) and severe idiopathic nephrotic syndrome (41) have so far reported encouraging, albeit mixed results. Likely contributors to these mixed results include the heterogeneity of MSC populations across tissues (42), the variability in isolation and expansion protocols (43), their limited persistence and trafficking (44), and uncertainty regarding the optimal route and dosing regimens to use across indications (45).
2.3 Moving beyond stem cells
The utilization of stem cells of hematopoietic or mesenchymal origin in autoimmune diseases, while groundbreaking, has encountered limitations such as high toxicity and inconsistent efficacy in a limited number of patients. Moreover, these therapies are not specifically tailored to target or eliminate disease-inducing immune cell subsets, such as autoreactive B or T lymphocytes. These limitations have spurred the exploration of alternative cell-based therapies that offer a more targeted and potentially safer approach to modulating the immune system in Treg cells and conventional T cells armored with a B-cell targeting moiety, such as a CD19-targeting chimeric antigen receptor (CAR).
While outside the scope of this review, additional differentiated cell subsets are also in development, such as NK cell therapy, an emerging modality in cancer immunotherapy and autoimmune disease. NK cells are innate immune effector cells that possess potent cytolytic function while exhibiting minimal risk of GvHD in the context of an allogeneic, donor-derived therapeutic due to both low allo-reactivity and limited persistence. Thus, NK cells do not have the need for strict HLA matching, a limitation of allogeneic effector T cell-based therapies. Engineering NK cells to express CARs targeting autoantigens or B-cell specific receptors enable precise targeting and elimination of pathogenic cell populations (46, 47). The efficacy of NK-based cell therapy, both with and without CAR modifications, in treating autoimmune diseases like SLE is currently being investigated in ongoing clinical trials both as a standalone therapy and in combination with more traditional B cell depleting agents. The results of these trials will provide valuable insights into the potential of this therapeutic approach for managing immune-mediated conditions.
3 CAR T cells to treat autoimmunity
3.1 CAR and TCR T cell therapy in oncology
The development of CAR T cells engineered to target the CD19 cell-surface antigen has revolutionized the treatment of B cell malignancies. Clinical trials have demonstrated their remarkable efficacy, with complete response rates ranging from 40-54% in multiple B cell lymphoma subtypes (48–50), 71-81% in B cell acute lymphoblastic leukemia (51, 52) and 18% in chronic lymphocytic leukemia (53). The durability of these responses suggests curative potential for some patients receiving these cells. The success of CD19-targeted CAR T cells has led to their approval by the FDA, either as a standalone therapy or as a bridge to HSCT (Axicabtagene ciloleucel, Tisagenlecleucel, Lisocabtagene maraleucel, Brexucabtagene autoleucel). In addition, CAR T cells targeting BCMA (Idecabtagene vicleucel, Ciltacabtagene autoleucel) have demonstrated efficacy in difficult to treat multiple myeloma, further expanding the therapeutic landscape for CAR T cell therapy among hematological cancers. Development of TCR-engineered T cells and tumor-infiltrating lymphocyte (TIL)-derived therapeutics have followed closely behind CAR T cell development. Recently, a MAGE-A4-targeted TCR-T cell therapy was granted accelerated approval by the FDA for the treatment of synovial sarcoma (uzatresgene autoleucel; NCT03132922) (54). Likewise, in 2024, approval was granted to a patient-derived TIL cell therapy for the treatment of melanoma (lifileucel; NCT02360579) (55).
3.2 CAR T cell therapy in autoimmunity
Application of CAR T cells in autoimmunity has largely followed the path laid out by hematological malignancies, namely using autologous CAR T cells targeting BCMA and CD19 antigens on the surface of B cells. The rationale for employing B-cell targeted CAR T cell therapy in autoimmune diseases is grounded in the critical role played by pathogenic B cells and autoreactive antibodies in the pathogenesis of many such conditions. The aim of B-cell targeted CAR T cell therapy is to disrupt these pathogenic processes and restore immune balance by depleting both circulating B cells, as well as those in the tissues and lymph nodes that are not reached by traditional anti-CD20 therapies. Early clinical trials have yielded promising results, demonstrating the safety and efficacy of B-cell targeted CAR T cell therapy in various autoimmune diseases, including SLE, myasthenia gravis, idiopathic inflammatory myopathy, systemic sclerosis, neuromyelitis optica spectrum disorder, and multiple sclerosis (56) (Tables 1, 2) (56). In a study involving five patients with refractory SLE who received anti-CD19 CAR T cell therapy, all patients achieved durable drug-free remission and cessation of nephritis (61). Mild cytokine release syndrome (CRS) occurred in all patients, with no concomitant immune effector cell-associated neurotoxicity syndrome (ICANS). Importantly, the B cell compartment reconstituted in these patients over a median of 8 months without disease flare or relapse. A follow-up study evaluating this product in fifteen severely ill patients with various autoimmune diseases, including eight with SLE, four with systemic sclerosis and three with inflammatory myopathies further confirmed these preliminary results. At a median follow-up of fifteen months, all the SLE patients were in drug-free remission without symptoms (62). The three treated patients with myositis also showed a major clinical response at six months, while the four patients with systemic sclerosis experienced decreases in disease severity scales and successfully discontinued all immunosuppressive medications during the follow-up period. Most autoantibodies were undetectable during up to twenty nine months of follow-up. Separately, in a Phase 1b/2a clinical trial of an anti-BCMA mRNA CAR T therapy in patients with generalized myasthenia gravis, eleven patients received six outpatient cell infusions (70). Patients receiving weekly infusions showed clinically meaningful benefits across three established clinical scoring systems and responses were maintained through the six-month follow-up. Serum titers of anti-AChR autoantibodies decreased by 22% at week 8 in patients with anti-AChR+ disease. Confirming these initial promising datasets, another anti-BCMA CAR T therapy was studied in twelve patients with neuromyelitis optica spectrum disorder (68). Eleven of twelve patients exhibited drug-free remission with no relapse during a median 5.5 months of follow-up. It is important to note that in these trials, all patients received lymphodepletion chemotherapy before CAR T infusion, and all experienced Grade 1 or 2 CRS.
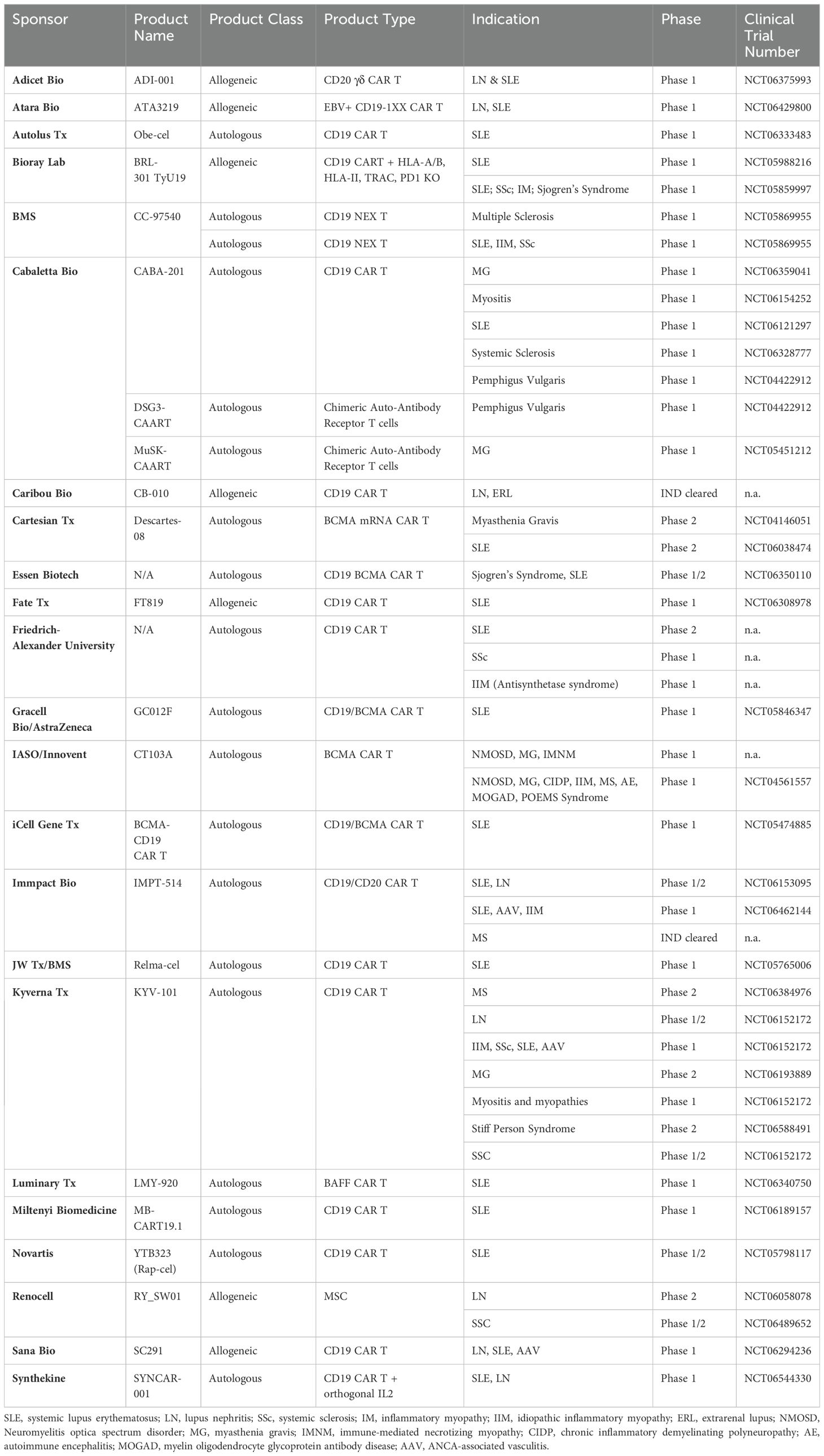
Table 1. Ongoing clinical trials using CAR T Cell therapies in autoimmune diseases.
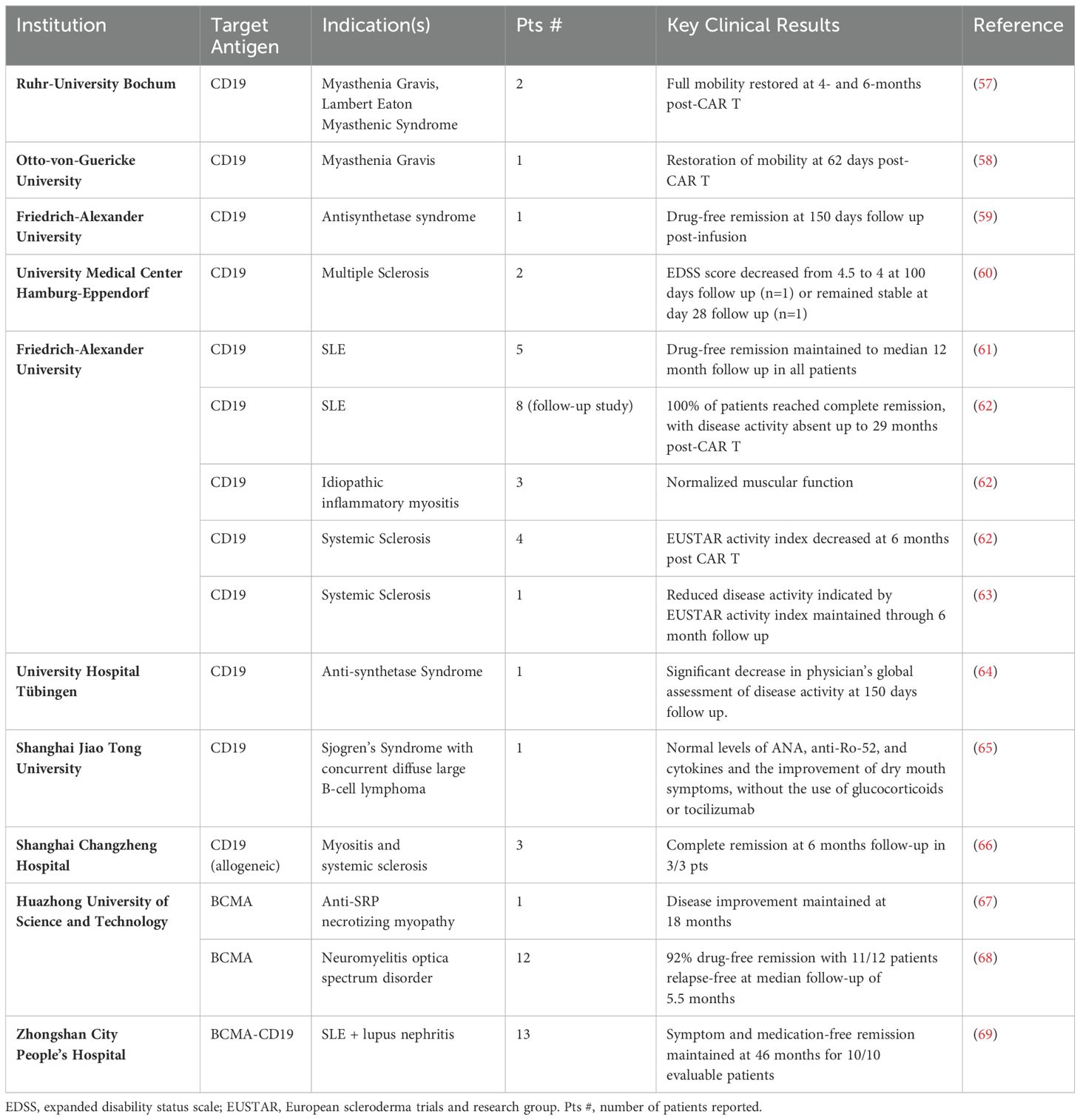
Table 2. Published clinical results for CAR T cell therapies in autoimmunity.
Of note, CD19-targeted CAR T cells appear to be more effective compared to other systemically administered B cell depleting agents like rituximab and obinutuzumab, in SLE and lupus nephritis (9, 71, 72). This difference in efficacy could be attributed to several factors: CD20, the target of rituximab and obinutuzumab, has a more restricted expression pattern along the B cell development axis compared to CD19 (73). Thus, the broader expression of CD19 allows CD19-targeted CAR T cells to deplete a wider range of B-cell populations, including CD19+CD20- plasma cells. These cells may play an important role in the pathogenesis of certain diseases, as they have been implicated in increased disease severity in preclinical models of multiple sclerosis (74). Additionally, the ability of CAR T cells to expand upon antigen-encounter, infiltrate tissues and induce deep in situ B-cell depletion, including tissue-resident B cells that are less sensitive to anti-CD20 antibody-mediated depletion, may contribute to their enhanced efficacy (75–77). The use of a lymphodepleting conditioning regimen prior to CAR T infusion may also contribute to deeper B cell depletion than antibodies alone.
Although CD19-targeted CAR T cell therapy effectively eliminates most autoantibodies against DNA and nucleosomal structures in SLE, certain autoantibodies, like those against Ro60 and Scl-70, persist (62). A possible hypothesis in need of confirmation is that B cell subsets responsible for their production do not express CD19. Despite a broader range of CD19 expression across B cell lineages compared to CD20, CD19-targeted CAR T cell therapies preserve the protective effect of prior vaccination in patients (62), possibly due to the preservation of long-lived, CD19-negative plasma cells responsible for vaccine-induced humoral immunity (78).
BCMA, expressed on mature B cells and plasma cells, presents another promising target for conditions where autoantibody production is driven by CD19-negative, BCMA+ plasma cells. Depleting BCMA-expressing cells with CAR T cell therapy has shown encouraging efficacy in neuromyelitis optica spectrum disorder, anti-signal recognition particle (SRP) necrotizing myopathy, and SLE (67, 68, 79). BCMA-targeted therapies may lead to a more extensive B-cell depletion and a greater alteration of the autoantibody repertoire, possibly increasing clinical benefit compared to CD19 CAR T (80). However, this broader targeting also results in a more significant depletion of protective antibodies, increasing the rate of infections and impairing pre-existing vaccine responses (81–83). BCMA CAR T cell therapy recipients have lower seroprotection to vaccine-preventable infections compared to CD19 CAR T-cell recipients, likely due to depletion of antibody-producing plasma cells (82). In a study investigating the impact of teclistamab, a BCMA-targeted T-cell engager antibody, on humoral immunity, the therapy was found to reduce the levels of polyclonal immunoglobulins and impair humoral immune response after vaccination (81). The high rate of serious infections in this patient group was attributed, at least in part, to the development of hypogammaglobulinemia and failure to generate new humoral immune responses. The use of IVIG supplementation was associated with a significantly lower risk of serious infections, supporting the use of immunoglobulin supplementation as primary prophylaxis in patients receiving a BCMA-targeting therapies. The development of bi-specific CAR T cells capable of targeting both CD19 and BCMA aims to further enhance B-cell depletion and prevent antigen escape (69, 84), but likewise might further elevate the risk of severe infections due to profound B cell depletion and suppressed vaccine-induced immunity.
To improve specificity, chimeric autoantigen receptor (CAAR) T cells have also been developed, intending to directly target autoantibody-producing pathogenic B cells. This is achieved by replacing the CAR’s targeting moiety with the autoantigen itself, while keeping the architecture of the CAR intracellular domain intact, in theory leading to the specific elimination of those B cells (85). CAAR T cell therapy is particularly relevant for diseases driven by well-defined autoantigens, such as muscle-specific tyrosine kinase (MuSK) in myasthenia gravis or desmoglein 3 (DSG3) in pemphigus vulgaris (86, 87). This approach is currently being tested in two clinical trials (NCT04422912, NCT05451212). It is important to note, however, that CAAR T cells primarily target B cells expressing a membrane-bound B-cell receptor (BCR) and may not be effective against diseases driven by long-lived plasma cells devoid of a BCR (86).
(85) In contrast to autoimmune diseases with a dominant pathogenic B cell component, chronic inflammatory diseases such as psoriasis and other inflammatory skin diseases, inflammatory bowel disease, and spondyloarthritis are unlikely to respond to B cell-targeted CAR T cell therapies. Indeed, these diseases are instead characterized by dysregulated T cell activation and IL-23/IL-17-mediated inflammation (88–90). Therefore, T cell-directed CAR T cells, such as those targeting the T cell-specific antigens CD5 and CD7, represent a promising alternative for patients with these predominantly T-cell mediated diseases. Several such products are currently undergoing clinical evaluation for the treatment of T cell malignancies with encouraging results (91–94), while their efficacy in autoimmune and chronic inflammatory diseases remains to be demonstrated.
A key limitation of current CAR and CAAR T cell therapies is their inability to simultaneously target multiple pathogenic cell types. This constraint poses a challenge in treating autoimmune diseases where the underlying pathophysiology involves a complex interplay of dysregulated B and T lymphocytes, along with inflammation driven by myeloid-derived innate immune cells. For example, while ANCA-associated vasculitis and myasthenia gravis both have a clear B cell component, neutrophils and thymus-derived T cells also play critical roles in their pathogenesis (95, 96). Similarly, in multiple sclerosis, while autoreactive B cells contribute to disease pathology, as evidenced by the efficacy of B cell-depleting antibodies (97, 98), definitive disease-causing autoantibodies have not been identified. Instead, a complex interplay between pathogenic B cells, autoreactive T cells, and CNS-localized inflammation is believed to drive demyelination, neurotoxicity, and disease progression (99–101). As discussed in the next section, FOXP3+ and Tr1-based Treg cell therapies are well-suited to dampen T cell-driven autoimmunity and inflammation, but lack the ability to specifically deplete B cells. Combining these therapies with B cell-modulating agents, such as anti-CD20 antibodies or Bruton tyrosine kinase (BTK) inhibitors, may represent the next step in the evolution of these treatments, conferring multimodal activity for complex autoimmune diseases. Crucially, any combination therapy must maintain an acceptable safety and tolerability profile without inducing generalized immunosuppression that could increase the risk of opportunistic infections.
4 Regulatory T cells to treat immune-mediated diseases
Regulatory T cells represent a diverse subset of lymphocytes responsible for maintaining immunological tolerance and homeostasis to both self and foreign peptides in healthy individuals. Treg cells are frequently underrepresented or dysregulated in patients with autoimmune conditions. Here we highlight the immunomodulatory mechanisms of two major CD4+ regulatory T cell types: FOXP3+ Treg cells and Tr1 Treg cells, and how their functionality can be harnessed to treat autoimmunity (Figure 1).
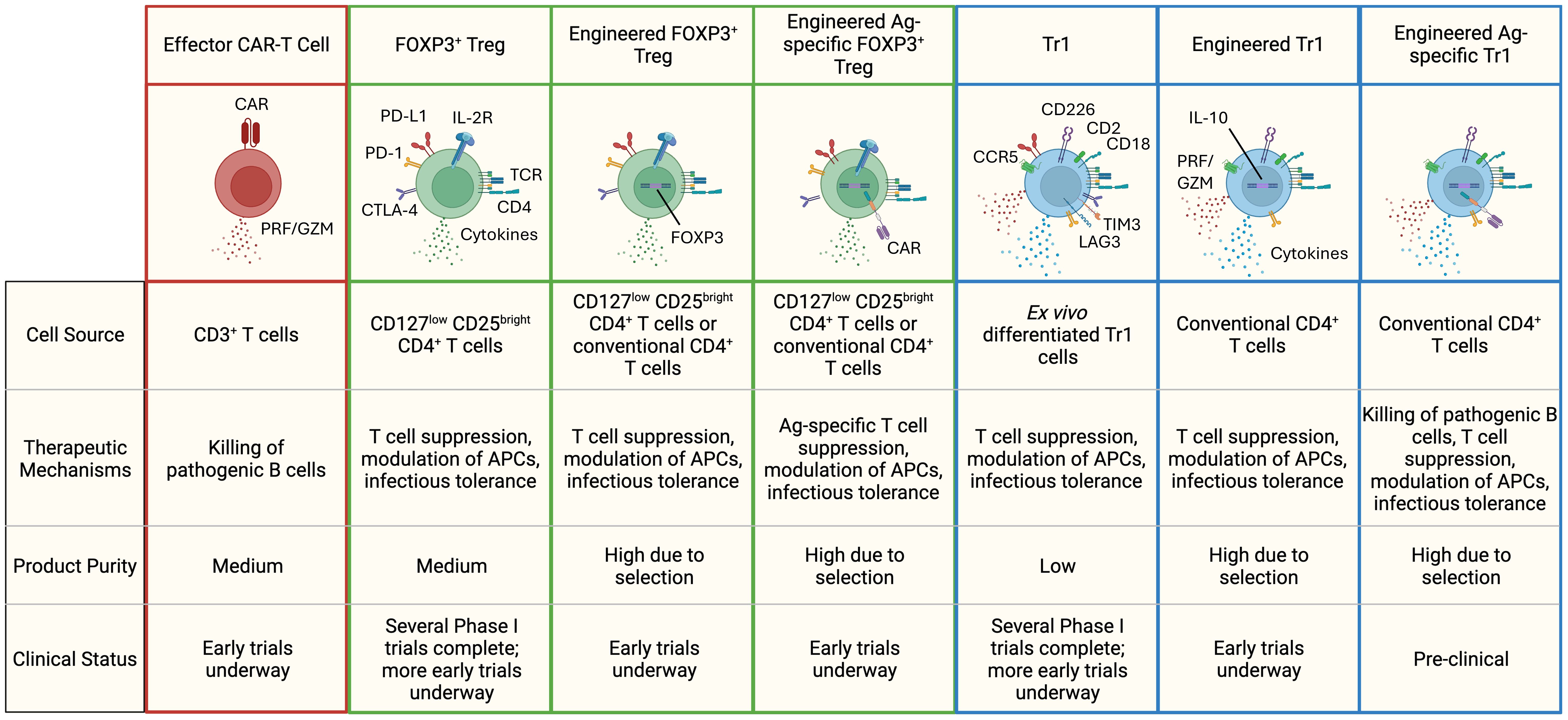
Figure 1. Different types of T-cell based cell therapies for the treatment of autoimmune diseases. Highlighted are some of the hallmark features of each cell therapy subset. FOXP3+ Tregs secrete immuno-regulatory cytokines like TGF-β and IL-35, while Tr1 cells produce high levels of IL-10 and TGF-β, along with moderate amounts of IFN-γ, IL-22, and IL-5. FOXP3+ Tregs are identified by elevated expression of the high-affinity IL-2 receptor and co-inhibitory receptors (PD-L1, PD-1, CTLA-4). Tr1 cells exhibit higher levels of CCR5, CD2, CD18, CD226, and co-inhibitory receptors (LAG-3, PD-1, TIM-3, CTLA-4, PD-L1). In therapies lacking an engineered surface marker (e.g., CAR, truncated-non-signaling forms of NGFR or EGFR), a combination of markers is used to define product purity. PRF, perforin; GZM, granzymes.
4.1 Immunosuppressive functionalities of FOXP3+ and Tr1 Treg cells
FOXP3+ Treg cells are primarily identified by their robust expression of the transcription factor Forkhead Box Protein 3 (FOXP3) alongside high levels of the high-affinity IL-2 receptor α (CD25) and low expression of the IL-7 receptor (CD127) (102). Additional markers associated with FOXP3+ Treg cells include elevated expression of the transcription factor Helios (103), high levels of the co-inhibitory surface molecule CTLA-4 (104), and specific demethylation patterns within the Treg-specific demethylation region (TSDR) (105). These phenotypic attributes underpin the potent immunomodulatory functions of FOXP3+ Treg cells, which are mediated through a combination of indirect and direct mechanisms (106) and include:
1. IL-2 deprivation: FOXP3+ Treg cells express high levels of CD25, enabling them to efficiently consume IL-2, thereby limiting the availability of this cytokine for effector T cells and curbing their proliferation (107).
2. Co-inhibitory signaling: FOXP3+ Treg cells express co-inhibitory receptors such as CTLA-4, which can modulate APCs and alter their antigen-presentation functionality (108).
3. Adenosine production: Through the sequential action of the ectoenzymes CD39 and CD73, FOXP3+ Treg cells convert extracellular ATP to adenosine, an immunosuppressive molecule that promotes a tolerogenic environment (109).
4. Inhibitory cytokine secretion: FOXP3+ Treg cells secrete a range of inhibitory cytokines, especially TGF-β and IL-35, which suppress immune cell activation and foster anti-inflammatory responses (110, 111).
The critical role of FOXP3+ Treg cells in immune regulation is underscored in disease states where their function or presence is compromised. For instance, patients harboring mutations in the FOXP3 gene are diagnosed with immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome (112). These individuals often exhibit a constellation of autoimmune manifestations, including enteropathy, type 1 diabetes, and dermatitis (113). In other autoimmune diseases with more complex etiologies, such as SLE (114) and rheumatoid arthritis, the relationship between FOXP3+ Treg cells and disease pathogenesis is less clear. Studies have reported inconsistent findings regarding FOXP3+ Treg cell phenotype and frequency in these conditions, with observations ranging from increased to decreased numbers, to no discernible changes compared to healthy donors or in relation to disease severity (115), though there is clear evidence of FOXP3+ Treg dysregulation (116). Whether FOXP3+ Treg defects are a causative factor in these systemic autoimmune diseases, a consequence of the ongoing inflammatory process, or a combination of both, remains an area of active investigation (115, 117).
Tr1 Treg cells are a distinct subset of regulatory T cells that do not constitutively express high levels of the transcription factor FOXP3. These cells are induced in the periphery and play a crucial role in maintaining tolerance to non-pathogenic antigens. Tr1 Treg cells are characterized by the co-expression of the surface markers CD49b and LAG3, and the production of high levels of IL-10 and IL-22, with low levels of IL-2 and IL-4 (118). IL-10, a key immunoregulatory cytokine produced by Tr1 Treg cells, can suppress the activation and proliferation of effector T cells directly, or indirectly by downregulating the function of antigen-presenting cells (119–121). Furthermore, high IL-10 secretion by Tr1 Treg cells has been shown to downregulate the NLRP3 inflammasome in monocytes, reducing the production of pro-inflammatory cytokines such as IL-1β (122). IL-22 secretion by Tr1 cells contributes to the protection of epithelial barrier integrity from inflammatory damage (123). In addition to cytokine-mediated suppression, Tr1 Treg cells also exert cell-to-cell contact-mediated suppression through the engagement of co-inhibitory receptors such as PD-1, CTLA-4, and TIM-3 (124).
Tr1 cells were first identified in patients with severe combined immunodeficiency (SCID) receiving mismatched HSCT (125). These patients showed long term engraftment of donor-derived T cells with no GvHD, despite complete HLA-mismatch, providing evidence for peripherally-induced tolerance. Subsequent studies on patient-derived material identified antigen-specific IL-10-producing T cells, later termed Tr1 Treg cells, as crucial in achieving transplant tolerance (126, 127). Dysregulated Tr1 Treg cell function has been strongly associated with autoimmune and inflammatory diseases such as multiple sclerosis, Crohn’s Disease, and type 1 diabetes (128). Studies characterizing the immune systems of patients with these diseases consistently show a reduced frequency or impaired suppressive function of Tr1 Treg, or IL-10-producing CD4+ T cells, compared to healthy individuals (128).
4.2 Ex-vivo expanded FOXP3+ Treg cells as therapy to treat immune-mediated diseases
The immunomodulatory properties of regulatory T cells have generated considerable interest in their use as a therapeutic. However, harnessing their potential faces challenges, including the low frequency of FOXP3+ Tregs in the blood, which complicates their isolation and expansion for therapeutic use. Manufacturing polyclonal FOXP3+ Treg-based cell therapies typically involves a multi-step process: first, Treg cells are isolated from peripheral blood based on surrogate cell surface markers such as CD4, CD127, and CD25. This step may also include the depletion of CD8+ T or CD19+ B lymphocytes. Next, the isolated FOXP3+ Treg cells are activated and expanded in vitro, typically using anti-CD3 and anti-CD28 antibodies in the presence of high-dose IL-2 (129). Rapamycin, an mTOR inhibitor, which has been shown to promote the expansion of FOXP3+ Tregs, is often added to selectively promote Treg expansion while limiting the growth of contaminating non-Treg cells (130, 131).
Early clinical trials have employed ex-vivo expanded polyclonal FOXP3+ Treg cells for the prevention or treatment of GvHD in the context of allogeneic HSCT, or graft rejection in the context of solid organ transplants. In 2009, a first of its kind clinical study used ex-vivo expanded polyclonal FOXP3+ Treg cells for the treatment of GvHD (132). Another study of polyclonal FOXP3+ Treg cells in kidney transplant proved safe, with increased levels of circulating Treg cells following the cell therapy’s administration (NCT02145325) (133). In the context of solid organ transplantation, donor-antigen-specific FOXP3+ Treg cells, generated by activation with donor-derived antigen-presenting cells, have shown superior efficacy in promoting allograft survival in preclinical models compared to polyclonal FOXP3+ Treg cells (134–137). A clinical study involving liver transplant patients receiving donor-specific Treg cells demonstrated the safety of this approach, with some patients able to discontinue immunosuppressive drugs for over 24 months (138). An alternative method of activation, distinct from CD3 or APC stimulation, utilizes the HIV envelope glycoprotein gp120’s ability to stimulate these cells (139). The interaction of gp120 with CD4 on Treg cells triggers their activation and boosts their suppressive function, demonstrating protection against GvHD in humanized mice. This approach (Actileucel) has progressed to clinical trials, with the first patient treated earlier this year in a GvHD prevention trial. Beyond GvHD, multiple trials have assessed or are actively investigating the safety and efficacy of polyclonal FOXP3+ Treg cell therapy in various autoimmune disease settings, including type 1 diabetes (NCT02691247) (140–143), lupus (144), multiple sclerosis (145), Crohn’s (146, 147), and Pemphigus vulgaris (NCT03239470), among others. These trials have collectively demonstrated the safety of FOXP3+ Treg cell therapy, while additional research is ongoing to further establish their efficacy (Table 3).

Table 3. Ongoing clinical trials using non-engineered Treg and Tr1 cells in autoimmunity.
Umbilical cord blood has been used as an alternative source of FOXP3+ Treg cells for cell therapy. A small-scale trial for GvHD prevention investigated the use of partially matched cord-blood FOXP3+ Treg products in five patients undergoing stem cell transplantation (NCT02423915). Although GvHD occurred in all four surviving patients, symptoms resolved, and patients were able to discontinue immunosuppressive medications after a median follow-up of 25 months (148). The safety and potential efficacy of allogeneic cord-blood-derived FOXP3+ Treg cells are also being evaluated in an ongoing clinical trial involving six patients with amyotrophic lateral sclerosis (ALS) without HLA matching (NCT05695521). A subsequent Phase 1b trial is currently underway, aiming to assess 60 additional patients in a randomized, controlled study.
A key challenge of natural FOXP3+ Treg cell manufacturing has been the potential for these cells to lose FOXP3 expression, particularly when exposed to inflammatory conditions (149), and convert to an undesired effector T cell phenotype (150–153). Conventional CD4+ T cells engineered to constitutively express FOXP3 present a compelling alternative to ex-vivo expanded natural FOXP3+ Treg cells. Enforced expression of FOXP3 effectively establishes a stable FOXP3+ Treg phenotype and associated suppressive functions, even under inflammatory conditions (154–156). The efficacy of these cells, generated using lentiviral-mediated transduction of FOXP3, has been demonstrated in preclinical models of GvHD and IPEX, and is currently being evaluated in a clinical study of patients with IPEX syndrome (NCT05241444) (154, 157). Gene editing technologies, such as CRISPR or TALEN, have also been employed to drive FOXP3 expression and generate engineered FOXP3+ Treg cells with a stable phenotype. In the context of IPEX patients, these techniques have been employed to restore functional FOXP3 expression by substituting the mutated and deficient endogenous sequence with a functional version of the gene (155). Alternative gene editing techniques include insertion of a robust enhancer/promoter near the FOXP3 locus (156), or utilization of a dual knock-in/knock-out approach to insert FOXP3 into a specific locus, while simultaneously disrupting expression of the targeted gene (158).
4.3 Tr1 Treg cell therapies to treat immune-mediated diseases
In a 2009 study, antigen-specific, clinical-grade Tr1 Treg cell clones were successfully isolated and expanded in vitro, and used to treat patients with refractory moderate-to-severe Crohn’s disease (159). This Phase 1/2a trial evaluated the safety and efficacy of autologous, ovalbumin-specific Tr1 Treg cell clones in patients fed with an ovalbumin-rich diet, to ensure cell activation specifically in the gut (160). The therapy was well-tolerated, and early efficacy signals were noted despite documented product impurities and cell exhaustion following an extensive and time-consuming expansion and activation protocol. Other allo-antigen-specific Tr1-enriched cell therapies have been developed and tested in the prevention of GvHD. CD4+ T cells co-cultured with CD3-depleted allogeneic PBMCs were manufactured in presence of IL-10, resulting in a product containing 3-5% of Tr1 Treg cells. This cell product was infused in patients undergoing haploidentical HSCT to facilitate immune reconstitution without GvHD (161). The treatment resulted in long term tolerance and cure (follow up > 7 years) in a subset of patients, without inducing general immune suppression, as tolerized patients mounted normal immune responses against microbes and vaccines. More recently, another host alloantigen-specific donor-autologous Tr1-enriched cell product containing 9-13% of Tr1 cells, was used to treat children and adults with hematologic malignancies receiving unmanipulated (NCT03198234) or TCRαβ+ T cell and CD19+ B cell depleted (NCT04640987) HSCT from mismatched unrelated or haploidentical donors (162). In these two clinical trials, no dose limiting toxicity has been observed in any of the patients treated so far. Patients displayed good T-cell immune reconstitution without increased risk of GvHD. Preliminary analyses suggested a reduced incidence of severe infections compared to historical controls. Furthermore, Tr1 Treg cells were detectable in the peripheral blood shortly after product infusion, reaching the highest levels at day +7 post-infusion and persisting for up to one year after administration (163).
While clinical trials have demonstrated the safety of patient- or donor-derived Tr1 Treg cell therapies, just like with natural FOXP3+ Treg cells, generating large numbers of highly pure Tr1 Treg cells from a low-frequency population remains a challenge. To address this limitation, recent efforts have focused on engineering allogeneic CD4+ T cells to express IL-10 and inducing cellular reprogramming through specific activation and expansion protocols. The resulting engineered cells effectively recapitulate many aspects of natural Tr1 cell biology, including high IL-10 secretion, low IL-4 production, in vitro suppression of T cells, and in vivo protection from GvHD in humanized mouse models (164–166). This efficient manufacturing of Tr1-like cells has enabled the development of novel polyclonal, allogeneic, IL-10-engineered Tr1 Treg cell therapies. These therapies are currently in clinical development for the prevention of GvHD following mismatched HSCT (NCT06462365), as well as for the treatment of refractory Crohn’s disease (167).
4.4 Opportunities for antigen-targeted, engineered FOXP3+ Treg and Tr1 Treg cell and Tr1 cell therapies
To achieve localized immune modulation, improve efficacy, and avoid the risk of systemic, generalized immunosuppression, engineered regulatory T cells can be equipped with a targeting moiety to focus their activity on specific tissues or cell types. The advantage of this approach is supported by evidence that antigen-specific Treg cells exhibit greater potency compared to their polyclonal counterparts (134–137). However, an inherent challenge lies in the typically low frequency of antigen-specific Treg cells, which can complicate the manufacturing process and limit its scalability.
As an alternative, antigen-specific regulatory T cells can be generated by incorporating a CAR or antigen-specific TCR, which facilitates the generation of large numbers of cells. For instance, conventional CD4+ T cells have been reprogrammed to co-express FOXP3, an engineered IL-2 receptor responsive to rapamycin, and a pancreatic islet antigen-specific TCR (168). This TCR-FOXP3+ Treg cell therapy product is being developed for the treatment of type 1 diabetes, aiming to prevent T cell-mediated destruction of pancreatic islet β cells. Another example comes from HLA-A2-directed CAR FOXP3+ Treg cells, which have demonstrated superior potency compared to polyclonal FOXP3+ Treg cells, particularly in solid organ transplantation (169–173). Clinical trials are currently underway to evaluate autologous HLA-A2-specific CAR Treg cell therapy in kidney and liver transplantation, aiming to induce immunological tolerance to the transplanted organ and eliminate the need for lifelong immunosuppression (NCT04817774; NCT05234190) (174). An alternative target, CD6, is a cell surface antigen expressed on T lymphocytes and NK cells. An allogeneic, CD6-CAR FOXP3+ Treg cell therapy is in development for the treatment of chronic GvHD after HSCT (NCT05993611). Beyond transplantation and GvHD, several other autologous CAR- and TCR-redirected FOXP3+ Treg cell therapies are being developed for various autoimmune diseases, including multiple sclerosis (NCT06566261), SLE and lupus nephritis (175, 176), rheumatoid arthritis (NCT06201416), and hidradenitis suppurativa (NCT06361836). Several donor-derived, allogeneic CAR-Treg cell therapies are also in earlier stages of development and are anticipated to enter clinical trials soon. Tr1 Treg cells possess both regulatory and cytotoxic functions (66, 165). Another tantalizing, yet theoretical strategy would be to equip Tr1 Treg cells with a B cell-targeting CAR, enabling them to eliminate autoreactive B cells, while simultaneously suppressing pathogenic T cells and inflammation.
A comprehensive overview of clinical trials involving antigen-targeted, engineered regulatory T cells in autoimmune and immune-mediated diseases is presented in Table 4. The relatively small, but growing number of these efforts highlight an increasing interest in harnessing the therapeutic potential of autologous or allogeneic engineered regulatory T cells to treat a wider range of immune disorders.
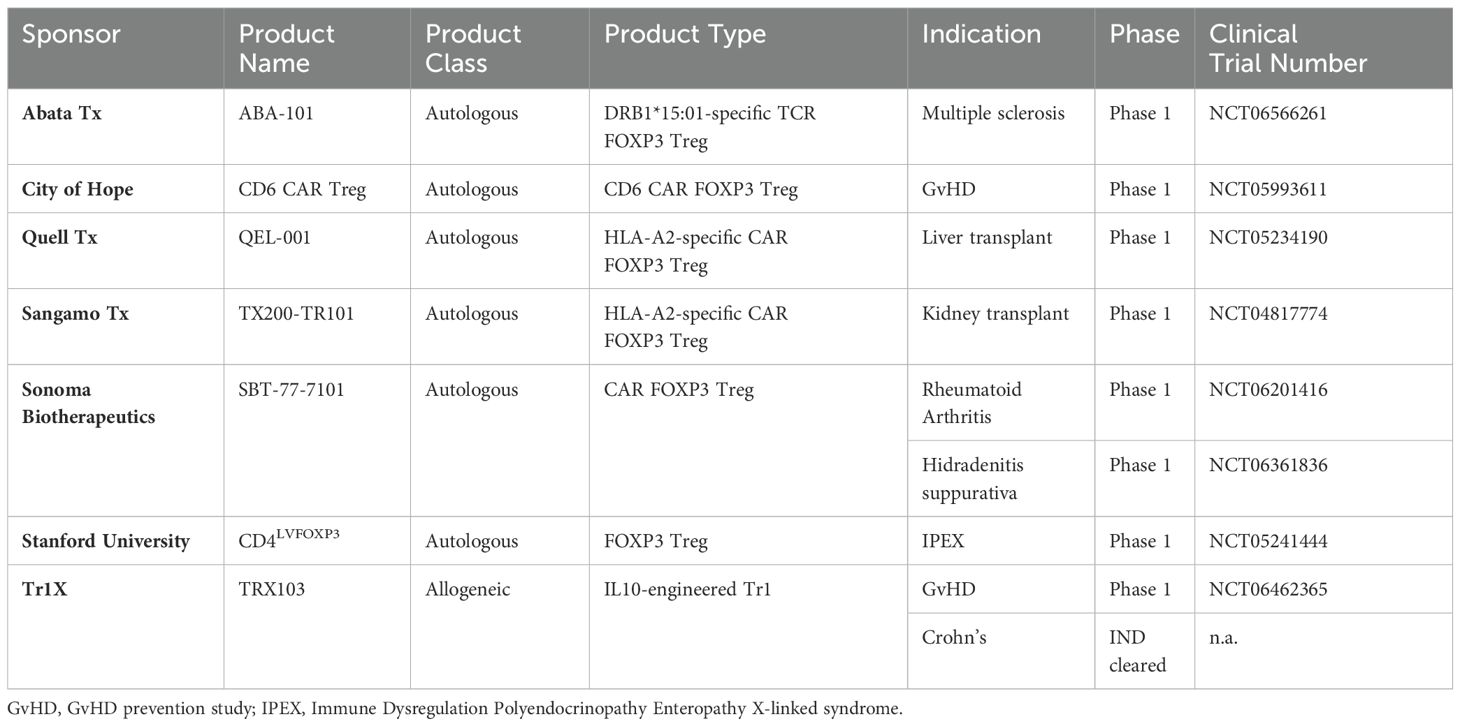
Table 4. Ongoing clinical trials using engineered Treg and Tr1 Cell therapeutics in autoimmunity.
5 Limitations and opportunities
5.1 Moving beyond lymphodepleting conditioning regimens
Lymphodepletion, commonly achieved through the combined administration of fludarabine and cyclophosphamide, is a standard conditioning regimen employed in CAR T cell therapies for both cancer and autoimmune diseases. This procedure is crucial for enhancing the engraftment, expansion, and persistence of the infused CAR T cells. However, it is associated with significant toxicity, including prolonged cytopenia and increased susceptibility to infections (177–183). While lymphodepletion is generally well-tolerated in the context of oncology, its application in autoimmune diseases raises concerns due to the potential for exacerbating the underlying condition through severe side effects. Interestingly, a case study involving a patient with SLE suggests that CAR T cell therapy may be effective in autoimmune diseases even with reduced doses of lymphodepleting agents (184). This observation may indicate that the threshold for achieving therapeutic efficacy in autoimmune diseases might be lower than in oncology, opening the door for lower, less aggressive lymphodepletion regimens. Reducing the intensity of conditioning regimens may necessitate strategies to enhance T cell expansion and compensate for lower peak CAR T cell numbers, a factor strongly correlated with efficacy in oncology (181, 183). Various engineering strategies aimed at augmenting T cell expansion and function will be explored below.
For Treg-based cell therapies, the IL-2 signaling pathway plays a critical role in the homeostasis and function of FOXP3+ Treg cells, therefore, augmenting IL-2 signaling represents a promising strategy for enhancing this type of cell therapy. Early clinical investigations employing IL-2 as an adjuvant for CAR T cell therapy in non-Hodgkin lymphoma demonstrated an increase in the frequency of FOXP3+ Treg cells (185, 186). This observation is consistent with the well-established role of IL-2 in promoting the survival, stability, and suppressive function of FOXP3+ Tregs (187, 188). Furthermore, clinical trials exploring low-dose IL-2 administration in patients with autoimmune diseases have consistently reported an expansion of FOXP3+ Treg populations, although the overall clinical responses have been variable (189). However, a significant limitation of systemic IL-2 administration, even at low doses, is its potential to activate other immune cell subsets and induce severe side effects, thus restricting its utility as an adjuvant for cell therapy (190). To circumvent this challenge, innovative orthogonal IL-2 systems have been developed. These systems utilize a modified IL-2 molecule that selectively binds to an engineered IL-2 receptor, enabling the targeted expansion of cells expressing this artificial receptor (191, 192). Preclinical studies have demonstrated the efficacy of this approach. FOXP3+ Tregs engineered to express the orthogonal IL-2 receptor can be selectively expanded in vivo using orthogonal, soluble IL-2, leading to improved donor hematopoietic stem cell engraftment and organ transplant tolerance (193). While this orthogonal IL-2 technology is currently undergoing clinical development in the field of oncology (NCT05665062), its therapeutic potential for autoimmune diseases warrants further investigation.
Beyond IL-2, other cytokine pathways offer promising targets for enhancing the in vivo proliferation and persistence of therapeutic effector and regulatory T cells. To mitigate the risk of exacerbating autoimmune pathology through systemic cytokine exposure, a favored strategy is to engineer T cells to co-opt the pathway in a cell-intrinsic fashion (194). Homeostatic cytokines such as IL-7 and IL-15 are well-known for their ability to augment the persistence and function of effector T cells. Notably, the efficacy of lymphodepletion is partially attributed to the increased availability of these cytokines (195). Preclinical studies have demonstrated that engineering CAR T cells to express a constitutively active form of the IL-7 receptor enhances their proliferation and persistence in vivo (196, 197). Similarly, co-expressing CAR with membrane-bound IL-15 has been shown to improve CAR T cell expansion and persistence in murine models (198), with encouraging clinical results reported in an oncology setting (199). Given the critical role of IL-15 in the maintenance and expansion of Tr1 Treg cells (200), IL-15 overexpression could also prove beneficial for Tr1-based therapies. While IL-12 and IL-18 have demonstrated potential in promoting CAR T cell proliferation and persistence without preconditioning in murine models (201, 202), their pro-inflammatory nature poses a significant challenge for their application in autoimmune diseases.
The pleiotropic nature of IL-10 presents a unique advantage for CAR T cell therapy in autoimmune diseases. IL-10 can enhance the persistence of engineered T cells through autocrine signaling while simultaneously promoting an anti-inflammatory response via paracrine signaling. Studies have shown that IL-10 improves CAR T cell proliferation and mitochondrial health, a critical determinant of T cell fitness (203). Moreover, IL-10 contributes to memory CD4+ T cell homeostasis and has been linked to the prolonged persistence of CD4+ T cells in the central nervous system during viral infections (204, 205). Importantly, Tr1 Treg cells, which naturally produce IL-10, can transfer a Tr1-like phenotype to bystander CD4+ T cells, a positive feedback loop known as infectious tolerance (206, 207). Through these combined mechanisms, it is plausible that Tr1-based therapies may require less expansion in vivo compared to effector CAR T cells to achieve comparable clinical benefit.
5.2 Long-term toxicity/safety of B-cell targeting effector CAR T and CAR Treg cells
Beyond the immediate risks associated with lymphodepletion, careful consideration must be given to the long-term safety of B-cell targeting CAR T cell therapies, particularly the potential for prolonged B cell aplasia and its impact on humoral immunity. In the context of cancer treatment, extended B cell depletion can increase the risk of infections and compromise B cell memory responses, with studies reporting a significant incidence of pneumonia requiring hospitalization months after CAR T cell infusion (208, 209). However, current treatment protocols for autoimmune diseases appear to result in the rapid clearance of CAR T cells, potentially mitigating this risk (61, 62). This observation is supported by clinical data demonstrating a low incidence of serious infections following CAR T cell therapy, despite profound and prolonged B cell depletion, lasting more than 100 days in some patients (62). It is crucial to recognize that these risks may be more pronounced with BCMA-targeting CARs compared to CD19-targeting CARs, due to the distinct B cell subpopulations expressing these antigens (81–83). Additionally, the risk of excessive immunosuppression warrants particular attention for non-targeted regulatory T cell therapies, as FOXP3+ Treg cells are known to suppress immune responses to various pathogens and malignancies (210, 211). Therefore, as the field progresses, it will be essential to develop strategies for controlling the proliferation of therapeutic FOXP3+ T cells, particularly those of autologous origin. Incorporating “kill switch” or suicide gene technology may enable the controlled elimination of these cells once the therapeutic objective is achieved.
One widely employed approach is the inducible caspase 9 (iCas9) system, which has been utilized in numerous clinical trials (212). Activation of iCas9 triggers apoptosis, leading to the elimination of the engineered T cells. Another strategy involves the herpes simplex virus thymidine kinase (HSV-tk) suicide gene, which converts the prodrug ganciclovir into a toxic metabolite, disrupting DNA synthesis and inducing cell death (213). However, the potential immunogenicity of HSV-tk can lead to premature elimination of the therapeutic cells by the host immune cells (214). Alternatively, co-expressing a truncated, inactive form of EGFR (EGFRt) or CD20 on the surface of engineered cells allows for their targeted elimination through antibody-dependent cellular cytotoxicity or complement activation upon administration of clinically approved monoclonal antibodies such as cetuximab or rituximab (215, 216).
While these safety mechanisms have primarily been explored in CAR T cell therapies for cancer, there is growing interest in incorporating them into engineered Treg cell therapies as well (217). This is exemplified by an ongoing Treg-based clinical trial employing an undisclosed safety switch technology (NCT05234190).
While suicide switch technologies offer effective elimination of engineered T cells, complete eradication may be disadvantageous for CAR Treg cell therapies, where maintaining a reservoir of potentially beneficial cells could be desirable. Positive regulators of CAR activity provide an alternative approach, enabling future reactivation of the therapeutic cells if needed. These systems utilize small molecules or drugs to control the expression or activity of the CAR transgene. This control can be implemented at the genetic level, using inducible promoters to regulate CAR expression, or at the protein level, through various mechanisms (comprehensively reviewed in (218, 219)). An example of such an inducible system is currently being evaluated in a clinical trial for acute myeloid leukemia (NCT05105152), where rapamycin acts as a dimerization agent to activate CAR functionality (220, 221). These inducible systems provide an “on-off” switch for CAR T cell activity, allowing for treatment to be tailored to the patient’s disease activity and offering greater control and flexibility. It is important to note that donor-derived allogeneic T cell therapies are naturally subject to rejection by the host immune system over time. Therefore, this class of therapeutics is expected to pose a lower risk of long-term adverse effects.
5.3 Scaling the production of cell therapies for autoimmunity
The scalability and cost of manufacturing autologous T cell therapies, including Treg, Tr1, and CAR T cell therapies, has been a major obstacle to their widespread application beyond rare diseases like acute lymphoblastic leukemia or multiple myeloma. The prospect of treating more prevalent autoimmune diseases, such as lupus and multiple sclerosis, which affect millions of patients worldwide, poses significant manufacturing, logistical, and financial hurdles. Furthermore, patients with autoimmune diseases often require long-term treatment with glucocorticoids or other immunosuppressive agents, particularly when their disease is poorly controlled. These medications can negatively impact T cell number and function, potentially compromising the collection, manufacture, and quality of the final autologous T cell product. Allogeneic, or “off-the-shelf”, cell therapies, where cells from universal donors are engineered for multiple patients, offer a potential solution to the inherent limitations of autologous cell therapy. However, allogeneic T cell therapy faces significant challenges, including the risk of GvHD due to the alloreactive TCR repertoire of the infused cells, and the potential for rapid rejection mediated by host CD8+ T cells and NK cells (222).
The advent of gene editing technologies, particularly CRISPR-Cas9 and its derivatives, has ushered in a new era for allogeneic T cell therapies. These tools enable precise modifications of T cells, mitigating the risk of GvHD by directly eliminating TCR expression and enhancing their persistence in non-HLA identical recipients by ablating HLA expression, thereby reducing the likelihood of rejection by the host immune system. The clinical potential of multiplexed gene editing with CRISPR-Cas9 has been demonstrated in several effector CAR T cell clinical trials in oncology (NCT04502446) (223–226). Consequently, numerous allogeneic CD19-targeting CAR T cell therapies are being actively developed for the treatment of autoimmune diseases (Table 1). One such product utilizes CRISPR-Cas9 to simultaneously knock-out the TCR α chain to prevent GvHD, as well as HLA-A/B, CIITA, and PD-1 to evade host-mediated immune rejection and enhance persistence (66). This therapy is currently in clinical development for myositis and systemic sclerosis, where it has shown promising results, achieving sustained remission in three out of three patients (NCT05859997). Other clinical studies with gene-edited CAR T cells are also underway in lupus and ANCA-associated vasculitis (NCT06308978, NCT06294236). It is worth noting that multiplexed gene editing using CRISPR-Cas9 may come with risks due to unpredictable translocation events between double-stranded breaks (227). Future strategies using Cas9-derived “base editors” hold promise to greatly improve the safety profile of edited allogenic products. These variants may enable the targeted editing of specific genetic loci without inducing genotoxic breaks in the DNA (228). The improved safety profile and efficacy of this approach has been demonstrated preclinically and in a Phase 1 study with quadruply-edited allogeneic CAR-T cells for relapsed leukemia showing undetectable translocation events (229, 230). As an alternative to using genetic editing to knock-out TCR expression, generating allogeneic CAR T cells with a virus-specific, non-alloreactive TCR may reduce the risk of GvHD and simplify the manufacturing process, an approach being tested clinically for patients with SLE (NCT06429800).
Unlike allogeneic effector CAR T cells, which require extensive genetic engineering to prevent GvHD and rejection, Tr1 and FOXP3+ Treg cells possess inherent suppressive properties that naturally mitigate their immunogenicity and potential to induce toxicity via GvHD. To date, there is no evidence to suggest that the transfer of allogeneic Tr1 or FOXP3+ Treg cells induces GvHD in humans. For instance, a recent Phase 1 study of HLA-mismatched, cord-blood derived Treg cells used to treat nine patients with bone marrow failure syndrome showed no signs of GvHD or other serious side effects (231). Moreover, IL-10 production by Tr1 cells can directly and indirectly suppress alloantigen-specific T cell responses (232), potentially enhancing their persistence in vivo (NCT06462365). Development of an allogeneic cell therapy that does not necessitate extensive gene editing would greatly simplify manufacturing complexities and dramatically lower its costs. This opens the door for broader development of allogeneic Treg-based cell therapies as a way to obviate manufacturing and scalability challenges.
6 Discussion
Over the last thirty years, the treatment of autoimmune and inflammatory diseases has undergone a remarkable transformation, progressing from broadly immunosuppressive drugs to targeted biologics and now to the promising field of cell therapies. HSCT, while effective in severe cases, carries significant risks and has limited applicability. MSCs offer a less invasive option but face challenges in standardization, persistence and in achieving consistent clinical outcomes.
The success of CAR T cell therapy in oncology has paved the way for its exploration in autoimmunity, with promising early clinical trials showcasing the safety and efficacy of B-cell-targeted CAR T cells in various conditions. The prospect of inducing long-term remission through an immune system reset offers hope for a long-lasting, if not curative, outcome. However, further validation is needed through larger-scale clinical trials with extended follow-up periods. These studies will be crucial for confirming the long-term safety and efficacy of this approach in a broader patient population and for assessing the durability of treatment responses. Treg and CAR Treg cell therapies, which suppress pathogenic T cells and inflammation, offer a potentially safer and more effective approach for treating diseases characterized by dysregulated T cell activation and inflammation, such as psoriasis or rheumatoid arthritis. Furthermore, combining the immunosuppressive properties of Treg cell therapies with a B-cell suppressing modality may offer a promising new avenue for treating diseases caused by both pathogenic B and T lymphocytes, such as multiple sclerosis.
The path forward for these promising therapies involves addressing challenges such as minimizing the need for lymphodepletion, ensuring long-term safety, and enhancing scalability, lower cost and increase accessibility. Onerous autologous approaches continue to pose a significant hurdle for their broader application in autoimmune diseases with higher prevalence than subtypes of cancer. Allogeneic therapies, coupled with gene engineering or gene editing technologies like CRISPR-Cas9, offer a potential solution by overcoming the limitations of personalized treatment and the risk of GvHD or rejection, although achieving sufficient cell persistence remains a concern.
The future of cell therapies in autoimmune and immune-mediated diseases holds immense promise in the quest to alleviate patients’ lifelong reliance on immunosuppressants. Ongoing research and clinical trials are laying the groundwork for the development of next-generation therapies that are safer, more effective, and scalable. The potential to achieve long-term remission or even cures, coupled with a deeper understanding of the complex mechanisms governing immunological tolerance offers hope for a transformative shift in the treatment of these diseases.
Author contributions
YB: Writing – original draft, Writing – review & editing. RF: Writing – original draft. MU: Writing – original draft. DH: Writing – original draft. RB: Writing – original draft. DdV: Writing – original draft, Writing – review & editing. MR: Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by a grant from the California Institute for Regenerative Medicine (Grant Number CLIN1-14840). The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of CIRM or and other agency of the State of California.
Conflict of interest
All authors are employed by Tr1X, Inc.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Miller FW. The increasing prevalence of autoimmunity and autoimmune diseases: an urgent call to action for improved understanding, diagnosis, treatment, and prevention. Curr Opin Immunol. (2023) 80:102266. doi: 10.1016/j.coi.2022.102266
2. Pisetsky DS. Pathogenesis of autoimmune disease. Nat Rev Nephrol. (2023) 19:509–24. doi: 10.1038/s41581-023-00720-1
3. Glück T, Kiefmann B, Grohmann M, Falk W, Straub RH, Schölmerich J. Immune status and risk for infection in patients receiving chronic immunosuppressive therapy. J Rheumatol. (2005) 32:1473–80.
4. Singh S, George J, Boland BS, Casteele NV, Sandborn WJ. Primary non-response to tumor necrosis factor antagonists is associated with inferior response to second-line biologics in patients with inflammatory bowel diseases: A systematic review and meta-analysis. J Crohn’s Colitis. (2018) 12:635–43. doi: 10.1093/ecco-jcc/jjy004
5. Skapenko A, Leipe J, Lipsky PE, Schulze-Koops H. The role of the T cell in autoimmune inflammation. Arthritis Res Ther. (2005) 7 Suppl 2:S4–14. doi: 10.1186/ar1703
6. Hampe CS. B cells in autoimmune diseases. Scientifica. (2012) 2012:215308. doi: 10.6064/2012/215308
7. Kaegi C, Wuest B, Schreiner J, Steiner UC, Vultaggio A, Matucci A, et al. Systematic review of safety and efficacy of rituximab in treating immune-mediated disorders. Front Immunol. (2019) 10:1990. doi: 10.3389/fimmu.2019.01990
8. Rigby WFC, Mease PJ, Olech E, Ashby M, Tole S. Safety of rituximab in combination with other biologic disease-modifying antirheumatic drugs in rheumatoid arthritis: an open-label study. J Rheumatol. (2013) 40:599–604. doi: 10.3899/jrheum.120924
9. Furie R, Petri M, Zamani O, Cervera R, Wallace DJ, Tegzová D, et al. randomized, placebo-controlled study of belimumab, a monoclonal antibody that inhibits B lymphocyte stimulator, in patients with systemic lupus erythematosus. Arthritis Rheum. (2011) 63:3918–30. doi: 10.1002/art.30613
10. Coles AJ, Twyman CL, Arnold DL, Cohen JA, Confavreux C, Fox EJ, et al. Alemtuzumab for patients with relapsing multiple sclerosis after disease-modifying therapy: a randomised controlled phase 3 trial. Lancet. (2012) 380:1829–39. doi: 10.1016/s0140-6736(12)61768-1
11. Zhao Q. Bispecific antibodies for autoimmune and inflammatory diseases: clinical progress to date. BioDrugs. (2020) 34:111–9. doi: 10.1007/s40259-019-00400-2
12. Guo J, Zhang H, Lin W, Lu L, Su J, Chen X. Signaling pathways and targeted therapies for psoriasis. Signal Transduct Target Ther. (2023) 8:437. doi: 10.1038/s41392-023-01655-6
13. Misselwitz B, Juillerat P, Sulz MC, Siegmund B, Brand S. Gastroenterology SIbd an official working group of the Swiss Society of. Emerging Treatment Options in Inflammatory Bowel Disease: Janus Kinases, Stem Cells, and More. Digestion. (2020) 101:69–82. doi: 10.1159/000507782
14. Burg N, Salmon JE, Hla T. Sphingosine 1-phosphate receptor-targeted therapeutics in rheumatic diseases. Nat Rev Rheumatol. (2022) 18:335–51. doi: 10.1038/s41584-022-00784-6
15. Feagan BG, Sands B, Siegel CA, Dubinsky M, Longman R, Sabinho J, et al. DOP87 the anti-TL1A antibody PRA023 demonstrated proof-of-concept in crohn’s disease: phase 2a APOLLO-CD study results. J Crohn’s Colitis. (2023) 17:i162–4. doi: 10.1093/ecco-jcc/jjac190.0127
16. Eggenhuizen PJ, Ng BH, Ooi JD. Treg enhancing therapies to treat autoimmune diseases. Int J Mol Sci. (2020) 21:7015. doi: 10.3390/ijms21197015
17. Műzes G, Sipos F. CAR-based therapy for autoimmune diseases: A novel powerful option. Cells. (2023) 12:1534. doi: 10.3390/cells12111534
18. Tamm M, Gratwohl A, Tichelli A, Perruchoud AP, Tyndall A. Autologous haemopoietic stem cell transplantation in a patient with severe pulmonary hypertension complicating connective tissue disease. Ann Rheum Dis. (1996) 55:779. doi: 10.1136/ard.55.10.779
19. Alexander T, Greco R. Hematopoietic stem cell transplantation and cellular therapies for autoimmune diseases: overview and future considerations from the Autoimmune Diseases Working Party (ADWP) of the European Society for Blood and Marrow Transplantation (EBMT). Bone Marrow Transplant. (2022) 57:1055–62. doi: 10.1038/s41409-022-01702-w
20. Wright DE, Cheshier SH, Wagers AJ, Randall TD, Christensen JL, Weissman IL. Cyclophosphamide/granulocyte colony-stimulating factor causes selective mobilization of bone marrow hematopoietic stem cells into the blood after M phase of the cell cycle. Blood. (2001) 97:2278–85. doi: 10.1182/blood.v97.8.2278
21. Oliveira MC, Labopin M, Henes J, Moore J, Papa ND, Cras A, et al. Does ex vivo CD34+ positive selection influence outcome after autologous hematopoietic stem cell transplantation in systemic sclerosis patients? Bone Marrow Transplant. (2016) 51:501–5. doi: 10.1038/bmt.2015.299
22. Couzin-Frankel J. Replacing an immune system gone haywire. Science. (2010) 327:772–4. doi: 10.1126/science.327.5967.772
23. Swart JF, Delemarre EM, Wijk FV, Boelens J-J, Kuball J, Laar JM, et al. Haematopoietic stem cell transplantation for autoimmune diseases. Nat Rev Rheumatol. (2017) 13:244–56. doi: 10.1038/nrrheum.2017.7
24. Delemarre EM, Van Den Broek T, Mijnheer G, Meerding J, Wehrens EJ, Olek S, et al. Autologous stem cell transplantation aids autoimmune patients by functional renewal and TCR diversification of regulatory T cells. Blood. (2016) 127:91–101. doi: 10.1182/blood-2015-06-649145
25. Baraut J, Grigore EI, Jean-Louis F, Khelifa SH, Durand C, Verrecchia F, et al. Peripheral blood regulatory T cells in patients with diffuse systemic sclerosis (SSc) before and after autologous hematopoietic SCT: a pilot study. Bone Marrow Transplant. (2014) 49:349–54. doi: 10.1038/bmt.2013.202
26. Alexander T, Thiel A, Rosen O, Massenkeil G, Sattler A, Kohler S, et al. Depletion of autoreactive immunologic memory followed by autologous hematopoietic stem cell transplantation in patients with refractory SLE induces long-term remission through de novo generation of a juvenile and tolerant immune system. Blood. (2009) 113:214–23. doi: 10.1182/blood-2008-07-168286
27. Burman J, Fransson M, Tötterman TH, Fagius J, Mangsbo SM, Loskog ASI. T-cell responses after haematopoietic stem cell transplantation for aggressive relapsing–remitting multiple sclerosis. Immunology. (2013) 140:211–9. doi: 10.1111/imm.12129
28. Abrahamsson SV, Angelini DF, Dubinsky AN, Morel E, Oh U, Jones JL, et al. Non-myeloablative autologous haematopoietic stem cell transplantation expands regulatory cells and depletes IL-17 producing mucosal-associated invariant T cells in multiple sclerosis. Brain. (2013) 136:2888–903. doi: 10.1093/brain/awt182
29. Rezvani K, Mielke S, Ahmadzadeh M, Kilical Y, Savani BN, Zeilah J, et al. High donor FOXP3-positive regulatory T-cell (Treg) content is associated with a low risk of GVHD following HLA-matched allogeneic SCT. Blood. (2006) 108:1291–7. doi: 10.1182/blood-2006-02-003996
30. Daikeler T, Tichelli A, Passweg J. Complications of autologous hematopoietic stem cell transplantation for patients with autoimmune diseases. Pediatr Res. (2012) 71:439–44. doi: 10.1038/pr.2011.57
31. Cencioni MT, Genchi A, Brittain G, De Silva TI, Sharrack B, Snowden JA, et al. Immune reconstitution following autologous hematopoietic stem cell transplantation for multiple sclerosis: A review on behalf of the EBMT autoimmune diseases working party. Front Immunol. (2022) 12:813957. doi: 10.3389/fimmu.2021.813957
32. Blanc KL, Rasmusson I, Sundberg B, Götherström C, Hassan M, Uzunel M, et al. Treatment of severe acute graft-versus-host disease with third party haploidentical mesenchymal stem cells. Lancet. (2004) 363:1439–41. doi: 10.1016/s0140-6736(04)16104-7
33. Alvites R, Branquinho M, Sousa AC, Lopes B, Sousa P, Maurício AC. Mesenchymal stem/stromal cells and their paracrine activity—Immunomodulation mechanisms and how to influence the therapeutic potential. Pharmaceutics. (2022) 14:381. doi: 10.3390/pharmaceutics14020381
34. Peruzzaro ST, Andrews MMM, Al-Gharaibeh A, Pupiec O, Resk M, Story D, et al. Transplantation of mesenchymal stem cells genetically engineered to overexpress interleukin-10 promotes alternative inflammatory response in rat model of traumatic brain injury. J Neuroinflamm. (2019) 16:2. doi: 10.1186/s12974-018-1383-2
35. Eiro N, Fraile M, González-Jubete A, González LO, Vizoso FJ. Mesenchymal (Stem) stromal cells based as new therapeutic alternative in inflammatory bowel disease: basic mechanisms, experimental and clinical evidence, and challenges. Int J Mol Sci. (2022) 23:8905. doi: 10.3390/ijms23168905
36. Shi X, Chen Q, Wang F. Mesenchymal stem cells for the treatment of ulcerative colitis: a systematic review and meta-analysis of experimental and clinical studies. Stem Cell Res Ther. (2019) 10:266. doi: 10.1186/s13287-019-1336-4
37. Garcia-Olmo D, Gilaberte I, Binek M, D´Hoore AJL, Lindner D, Selvaggi F, et al. Follow-up study to evaluate the long-term safety and efficacy of darvadstrocel (Mesenchymal stem cell treatment) in patients with perianal fistulizing crohn’s disease: ADMIRE-CD phase 3 randomized controlled trial. Dis Colon Rectum. (2022) 65:713–20. doi: 10.1097/dcr.0000000000002325
38. Introna M, Lucchini G, Dander E, Galimberti S, Rovelli A, Balduzzi A, et al. Treatment of graft versus host disease with mesenchymal stromal cells: A phase I study on 40 adult and pediatric patients. Biol Blood Marrow Transplant. (2014) 20:375–81. doi: 10.1016/j.bbmt.2013.11.033
39. Connick P, Kolappan M, Patani R, Scott MA, Crawley C, He X-L, et al. The mesenchymal stem cells in multiple sclerosis (MSCIMS) trial protocol and baseline cohort characteristics: an open-label pre-test: post-test study with blinded outcome assessments. Trials. (2011) 12:62. doi: 10.1186/1745-6215-12-62
40. Islam MA, Alam SS, Kundu S, Ahmed S, Sultana S, Patar A, et al. Mesenchymal stem cell therapy in multiple sclerosis: A systematic review and meta-analysis. J Clin Med. (2023) 12:6311. doi: 10.3390/jcm12196311
41. Vivarelli M, Colucci M, Algeri M, Zotta F, Emma F, L’Erario I, et al. A phase 1 study of autologous mesenchymal stromal cells for severe steroid-dependent nephrotic syndrome. JCI Insight. (2023) 8:e169424. doi: 10.1172/jci.insight.169424
42. Li J, Wu Z, Zhao L, Liu Y, Su Y, Gong X, et al. The heterogeneity of mesenchymal stem cells: an important issue to be addressed in cell therapy. Stem Cell Res Ther. (2023) 14:381. doi: 10.1186/s13287-023-03587-y
43. Calcat-i-Cervera S, Rendra E, Scaccia E, Amadeo F, Hanson V, Wilm B, et al. Harmonised culture procedures minimise but do not eliminate mesenchymal stromal cell donor and tissue variability in a decentralised multicentre manufacturing approach. Stem Cell Res Ther. (2023) 14:120. doi: 10.1186/s13287-023-03352-1
44. Zhang J, Huang X, Wang H, Liu X, Zhang T, Wang Y, et al. The challenges and promises of allogeneic mesenchymal stem cells for use as a cell-based therapy. Stem Cell Res Ther. (2015) 6:234. doi: 10.1186/s13287-015-0240-9
45. Markovic BS, Kanjevac T, Harrell CR, Gazdic M, Fellabaum C, Arsenijevic N, et al. Molecular and cellular mechanisms involved in mesenchymal stem cell-based therapy of inflammatory bowel diseases. Stem Cell Rev Rep. (2018) 14:153–65. doi: 10.1007/s12015-017-9789-2
46. Meng H, Sun X, Song Y, Zou J, An G, Jin Z, et al. La/SSB chimeric autoantibody receptor modified NK92MI cells for targeted therapy of autoimmune disease. Clin Immunol. (2018) 192:40–9. doi: 10.1016/j.clim.2018.04.006
47. Reighard SD, Cranert SA, Rangel KM, Ali A, Gyurova IE, de la Cruz-Lynch AT, et al. Therapeutic targeting of follicular T cells with chimeric antigen receptor-expressing natural killer cells. Cell Rep Med. (2020) 1:100003. doi: 10.1016/j.xcrm.2020.100003
48. Abramson JS, Palomba ML, Gordon LI, Lunning MA, Wang M, Arnason J, et al. Lisocabtagene maraleucel for patients with relapsed or refractory large B-cell lymphomas (TRANSCEND NHL 001): a multicentre seamless design study. Lancet. (2020) 396:839–52. doi: 10.1016/s0140-6736(20)31366-0
49. Schuster SJ, Bishop MR, Tam CS, Waller EK, Borchmann P, McGuirk JP, et al. Tisagenlecleucel in adult relapsed or refractory diffuse large B-cell lymphoma. N Engl J Med. (2018) 380:45–56. doi: 10.1056/nejmoa1804980
50. Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N Engl J Med. (2017) 377:2531–44. doi: 10.1056/nejmoa1707447
51. Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N Engl J Med. (2018) 378:439–48. doi: 10.1056/nejmoa1709866
52. Shah BD, Ghobadi A, Oluwole OO, Logan AC, Boissel N, Cassaday RD, et al. KTE-X19 for relapsed or refractory adult B-cell acute lymphoblastic leukaemia: phase 2 results of the single-arm, open-label, multicentre ZUMA-3 study. Lancet. (2021) 398:491–502. doi: 10.1016/s0140-6736(21)01222-8
53. Siddiqi T, Maloney DG, Kenderian SS, Brander DM, Dorritie K, Soumerai J, et al. Lisocabtagene maraleucel in chronic lymphocytic leukaemia and small lymphocytic lymphoma (TRANSCEND CLL 004): a multicentre, open-label, single-arm, phase 1–2 study. Lancet. (2023) 402:641–54. doi: 10.1016/s0140-6736(23)01052-8
54. D’Angelo SP, Araujo DM, Razak ARA, Agulnik M, Attia S, Blay J-Y, et al. Afamitresgene autoleucel for advanced synovial sarcoma and myxoid round cell liposarcoma (SPEARHEAD-1): an international, open-label, phase 2 trial. Lancet. (2024) 403:1460–71. doi: 10.1016/s0140-6736(24)00319-2
55. Schoenfeld AJ, Lee SM, de Speville BD, Gettinger SN, Hafliger S, Sukari A, et al. Lifileucel, an autologous tumor-infiltrating lymphocyte monotherapy, in patients with advanced non-small cell lung cancer resistant to immune checkpoint inhibitors. Cancer Discovery. (2024) 14:1389–402. doi: 10.1158/2159-8290.cd-23-1334
56. Schett G, Müller F, Taubmann J, Mackensen A, Wang W, Furie RA, et al. Advancements and challenges in CAR T cell therapy in autoimmune diseases. Nat Rev Rheumatol. (2024) 20:531–44. doi: 10.1038/s41584-024-01139-z
57. Motte J, Sgodzai M, Schneider-Gold C, Steckel N, Mika T, Hegelmaier T, et al. Treatment of concomitant myasthenia gravis and Lambert-Eaton myasthenic syndrome with autologous CD19-targeted CAR T cells. Neuron. (2024) 112:1757–1763.e2. doi: 10.1016/j.neuron.2024.04.014
58. Haghikia A, Hegelmaier T, Wolleschak D, Böttcher M, Desel C, Borie D, et al. Anti-CD19 CAR T cells for refractory myasthenia gravis. Lancet Neurol. (2023) 22:1104–5. doi: 10.1016/s1474-4422(23)00375-7
59. Taubmann J, Knitza J, Müller F, Völkl S, Aigner M, Kleyer A, et al. Rescue therapy of antisynthetase syndrome with CD19-targeted CAR-T cells after failure of several B-cell depleting antibodies. Rheumatology. (2023) 59:e12–4. doi: 10.1093/rheumatology/kead330
60. Fischbach F, Richter J, Pfeffer LK, Fehse B, Berger SC, Reinhardt S, et al. CD19-targeted chimeric antigen receptor T cell therapy in two patients with multiple sclerosis. Med. (2024) 5:550–558.e2. doi: 10.1016/j.medj.2024.03.002
61. Mackensen A, Müller F, Mougiakakos D, Böltz S, Wilhelm A, Aigner M, et al. Anti-CD19 CAR T cell therapy for refractory systemic lupus erythematosus. Nat Med. (2022) 28:2124–32. doi: 10.1038/s41591-022-02017-5
62. Müller F, Taubmann J, Bucci L, Wilhelm A, Bergmann C, Völkl S, et al. CD19 CAR T-cell therapy in autoimmune disease — A case series with follow-up. N Engl J Med. (2024) 390:687–700. doi: 10.1056/nejmoa2308917
63. Bergmann C, Müller F, Distler JHW, Györfi A-H, Völkl S, Aigner M, et al. Treatment of a patient with severe systemic sclerosis (SSc) using CD19-targeted CAR T cells. Ann Rheumatic Dis. (2023) 82:1117–20. doi: 10.1136/ard-2023-223952. ard-2023-223952.
64. Pecher A-C, Hensen L, Klein R, Schairer R, Lutz K, Atar D, et al. CD19-targeting CAR T cells for myositis and interstitial lung disease associated with antisynthetase sxyndrome. JAMA. (2023) 329:2154–62. doi: 10.1001/jama.2023.8753
65. Sheng L, Zhang Y, Song Q, Jiang X, Cao W, Li L, et al. Concurrent remission of lymphoma and Sjögren’s disease following anti-CD19 chimeric antigen receptor-T cell therapy for diffuse large B-cell lymphoma: a case report. Front Immunol. (2023) 14:1298815. doi: 10.3389/fimmu.2023.1298815
66. Wang X, Wu X, Tan B, Zhu L, Zhang Y, Lin L, et al. Allogeneic CD19-targeted CAR-T therapy in patients with severe myositis and systemic sclerosis. Cell. (2024) 18:P4890–904. doi: 10.1016/j.cell.2024.06.027
67. Qin C, Dong M-H, Zhou L-Q, Wang W, Cai S-B, You Y-F, et al. Single-cell analysis of refractory anti-SRP necrotizing myopathy treated with anti-BCMA CAR-T cell therapy. Proc Natl Acad Sci. (2024) 121:e2315990121. doi: 10.1073/pnas.2315990121
68. Qin C, Tian D-S, Zhou L-Q, Shang K, Huang L, Dong M-H, et al. Anti-BCMA CAR T-cell therapy CT103A in relapsed or refractory AQP4-IgG seropositive neuromyelitis optica spectrum disorders: phase 1 trial interim results. Signal Transduct Target Ther. (2023) 8:5. doi: 10.1038/s41392-022-01278-3
69. Wang W, He S, Zhang W, Zhang H, DeStefano VM, Wada M, et al. BCMA-CD19 compound CAR T cells for systemic lupus erythematosus: a phase 1 open-label clinical trial. Ann Rheum Dis. (2024) 83:1304–14. doi: 10.1136/ard-2024-225785
70. Granit V, Benatar M, Kurtoglu M, Miljković MD, Chahin N, Sahagian G, et al. Safety and clinical activity of autologous RNA chimeric antigen receptor T-cell therapy in myasthenia gravis (MG-001): a prospective, multicentre, open-label, non-randomised phase 1b/2a study. Lancet Neurol. (2023) 22:578–90. doi: 10.1016/s1474-4422(23)00194-1
71. Merrill JT, Neuwelt CM, Wallace DJ, Shanahan JC, Latinis KM, Oates JC, et al. Efficacy and safety of rituximab in moderately-to-severely active systemic lupus erythematosus: The randomized, double-blind, phase ii/iii systemic lupus erythematosus evaluation of rituximab trial. Arthritis Rheum. (2010) 62:222–33. doi: 10.1002/art.27233
72. Furie RA, Aroca G, Cascino MD, Garg JP, Rovin BH, Alvarez A, et al. B-cell depletion with obinutuzumab for the treatment of proliferative lupus nephritis: a randomised, double-blind, placebo-controlled trial. Ann Rheum Dis. (2022) 81:100–7. doi: 10.1136/annrheumdis-2021-220920
73. Forsthuber TG, Cimbora DM, Ratchford JN, Katz E, Stüve O. B cell-based therapies in CNS autoimmunity: differentiating CD19 and CD20 as therapeutic targets. Ther Adv Neurol Disord. (2018) 11:1756286418761697. doi: 10.1177/1756286418761697
74. Chen D, Ireland SJ, Davis LS, Kong X, Stowe AM, Wang Y, et al. Autoreactive CD19+CD20– plasma cells contribute to disease severity of experimental autoimmune encephalomyelitis. J Immunol. (2016) 196:1541–9. doi: 10.4049/jimmunol.1501376
75. Thurlings RM, Vos K, Wijbrandts CA, Zwinderman AH, Gerlag DM, Tak PP. Synovial tissue response to rituximab: mechanism of action and identification of biomarkers of response. Ann Rheum Dis. (2008) 67:917. doi: 10.1136/ard.2007.080960
76. Kamburova EG, Koenen HJPM, Borgman KJE, Ten Berge IJ, Joosten I, Hilbrands LB. A single dose of rituximab does not deplete B cells in secondary lymphoid organs but alters phenotype and function. Am J Transplant. (2013) 13:1503–11. doi: 10.1111/ajt.12220
77. Tur C, Eckstein M, Velden J, Rauber S, Bergmann C, Auth J, et al. CD19-CAR T-cell therapy induces deep tissue depletion of B cells. Ann Rheum Dis. (2024) ard-2024-226142. doi: 10.1136/ard-2024-226142
78. Bhoj VG, Arhontoulis D, Wertheim G, Capobianchi J, Callahan CA, Ellebrecht CT, et al. Persistence of long-lived plasma cells and humoral immunity in individuals responding to CD19-directed CAR T-cell therapy. Blood. (2016) 128:360–70. doi: 10.1182/blood-2016-01-694356
79. Alexander T, Krönke J, Cheng Q, Keller U, Krönke G. Teclistamab-induced remission in refractory systemic lupus erythematosus. N Engl J Med. (2024) 391:864–6. doi: 10.1056/nejmc2407150
80. Bodansky A, Yu DJL, Rallistan AN, Kalaycioglu M, Boonyaratanakornkit J, Green DJ, et al. Unveiling the proteome-wide autoreactome enables enhanced evaluation of emerging CAR-T therapies in autoimmunity. J Clin Investig. (2024) 134:e180012. doi: 10.1172/jci180012
81. Frerichs KA, Verkleij CPM, Mateos MV, Martin TG, Rodriguez C, Nooka A, et al. Teclistamab impairs humoral immunity in patients with heavily pretreated myeloma: importance of immunoglobulin supplementation. Blood Adv. (2024) 8:194–206. doi: 10.1182/bloodadvances.2023011658
82. Walti CS, Krantz EM, Maalouf J, Boonyaratanakornkit J, Keane-Candib J, Joncas-Schronce L, et al. Antibodies to vaccine-preventable infections after CAR-T-cell therapy for B-cell Malignancies. JCI Insight. (2021) 6:e146743. doi: 10.1172/jci.insight.146743
83. Wang Y, Li C, Xia J, Li P, Cao J, Pan B, et al. Humoral immune reconstitution after anti-BCMA CAR T-cell therapy in relapsed/refractory multiple myeloma. Blood Adv. (2021) 5:5290–9. doi: 10.1182/bloodadvances.2021004603
84. Feng J HUY, Chang AH, Huang H. CD19/BCMA CAR-T cell therapy for refractory systemic lupus erythematosus - safety and preliminary efficacy data from a phase I clinical study. Blood. (2023) 142:4835. doi: 10.1182/blood-2023-186669
85. Ellebrecht CT, Bhoj VG, Nace A, Choi EJ, Mao X, Cho MJ, et al. Reengineering chimeric antigen receptor T cells for targeted therapy of autoimmune disease. Science. (2016) 353:179–84. doi: 10.1126/science.aaf6756
86. Oh S, Mao X, Manfredo-Vieira S, Lee J, Patel D, Choi EJ, et al. Precision targeting of autoantigen-specific B cells in muscle-specific tyrosine kinase myasthenia gravis with chimeric autoantibody receptor T cells. Nat Biotechnol. (2023) 353:179–84. doi: 10.1038/s41587-022-01637-z
87. Lee J, Lundgren DK, Mao X, Manfredo-Vieira S, Nunez-Cruz S, Williams EF, et al. Antigen-specific B cell depletion for precision therapy of mucosal pemphigus vulgaris. J Clin Invest. (2020) 130:6317–24. doi: 10.1172/jci138416
88. Liu T, Li S, Ying S, Tang S, Ding Y, Li Y, et al. The IL-23/IL-17 pathway in inflammatory skin diseases: from bench to bedside. Front Immunol. (2020) 11:594735. doi: 10.3389/fimmu.2020.594735
89. Schmitt H, Neurath MF, Atreya R. Role of the IL23/IL17 pathway in crohn’s disease. Front Immunol. (2021) 12:622934. doi: 10.3389/fimmu.2021.622934
90. Sieper J, Poddubnyy D, Miossec P. The IL-23–IL-17 pathway as a therapeutic target in axial spondyloarthritis. Nat Rev Rheumatol. (2019) 15:747–57. doi: 10.1038/s41584-019-0294-7
91. Pan J, Tan Y, Wang G, Deng B, Ling Z, Song W, et al. Donor-derived CD7 chimeric antigen receptor T cells for T-cell acute lymphoblastic leukemia: first-in-human, phase I trial. J Clin Oncol. (2021) 39:3340–51. doi: 10.1200/jco.21.00389
92. Lin H, Cheng J, Zhu L, Zeng Y, Dai Z, Zhang Y, et al. Anti-CD5 CAR-T cells with a tEGFR safety switch exhibit potent toxicity control. Blood Cancer J. (2024) 14:98. doi: 10.1038/s41408-024-01082-y
93. Hill L, Rouce RH, Smith TS, Yang L, Srinivasan M, Zhang H, et al. CD5 CAR T-cells for treatment of patients with relapsed/refractory CD5 expressing T-cell lymphoma demonstrates safety and anti-tumor activity. Biol Blood Marrow Transplant. (2020) 26:S237. doi: 10.1016/j.bbmt.2019.12.482
94. Hu Y, Zhang M, Yang T, Mo Z, Wei G, Jing R, et al. Sequential CD7 CAR T-cell therapy and allogeneic HSCT without GVHD prophylaxis. N Engl J Med. (2024) 390:1467–80. doi: 10.1056/nejmoa2313812
95. Dresser L, Wlodarski R, Rezania K, Soliven B. Myasthenia gravis: epidemiology, pathophysiology and clinical manifestations. J Clin Med. (2021) 10:2235. doi: 10.3390/jcm10112235
96. Nakazawa D, Masuda S, Tomaru U, Ishizu A. Pathogenesis and therapeutic interventions for ANCA-associated vasculitis. Nat Rev Rheumatol. (2019) 15:91–101. doi: 10.1038/s41584-018-0145-y
97. Hauser SL, Bar-Or A, Comi G, Giovannoni G, Hartung H-P, Hemmer B, et al. Ocrelizumab versus interferon beta-1a in relapsing multiple sclerosis. N Engl J Med. (2017) 376:221–34. doi: 10.1056/nejmoa1601277
98. Montalban X, Hauser SL, Kappos L, Arnold DL, Bar-Or A, Comi G, et al. Ocrelizumab versus placebo in primary progressive multiple sclerosis. N Engl J Med. (2017) 376:209–20. doi: 10.1056/nejmoa1606468
99. Oh J, Bar-Or A. Emerging therapies to target CNS pathophysiology in multiple sclerosis. Nat Rev Neurol. (2022) 18:466–75. doi: 10.1038/s41582-022-00675-0
100. Yong HYF, Yong VW. Mechanism-based criteria to improve therapeutic outcomes in progressive multiple sclerosis. Nat Rev Neurol. (2022) 18:40–55. doi: 10.1038/s41582-021-00581-x
101. Van Langelaar J, Rijvers L, Smolders J, van Luijn MM. B and T cells driving multiple sclerosis: identity, mechanisms and potential triggers. Front Immunol. (2020) 11:760. doi: 10.3389/fimmu.2020.00760
102. Ziegler SF. FOXP3: Of mice and men. Ann Rev Immunol. (2006) 24:209–26. doi: 10.1146/annurev.immunol.24.021605.090547
103. Thornton AM, Korty PE, Tran DQ, Wohlfert EA, Murray PE, Belkaid Y, et al. Expression of helios, an ikaros transcription factor family member, differentiates thymic-derived from peripherally induced foxp3+ T regulatory cells. J Immunol. (2010) 184:3433–41. doi: 10.4049/jimmunol.0904028
104. Takahashi T, Tagami T, Yamazaki S, Uede T, Shimizu J, Sakaguchi N, et al. Immunologic self-tolerance maintained by cd25+Cd4+Regulatory T cells constitutively expressing cytotoxic T lymphocyte–associated antigen 4. J Exp Med. (2000) 192:303–10. doi: 10.1084/jem.192.2.303
105. Baron U, Floess S, Wieczorek G, Baumann K, Grützkau A, Dong J, et al. DNA demethylation in the human FOXP3 locus discriminates regulatory T cells from activated FOXP3+ conventional T cells. Eur J Immunol. (2007) 37:2378–89. doi: 10.1002/eji.200737594
106. Schmidt A, Oberle N, Krammer PH. Molecular mechanisms of treg-mediated T cell suppression. Front Immunol. (2012) 3:51. doi: 10.3389/fimmu.2012.00051
107. Chinen T, Kannan AK, Levine AG, Fan X, Klein U, Zheng Y, et al. An essential role for the IL-2 receptor in Treg cell function. Nat Immunol. (2016) 17:1322–33. doi: 10.1038/ni.3540
108. Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z, et al. CTLA-4 control over foxp3+ Regulatory T cell function. Science. (2008) 322:271–5. doi: 10.1126/science.1160062
109. Deaglio S, Dwyer KM, Gao W, Friedman D, Usheva A, Erat A, et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med. (2007) 204:1257–65. doi: 10.1084/jem.20062512
110. Collison LW, Chaturvedi V, Henderson AL, Giacomin PR, Guy C, Bankoti J, et al. IL-35-mediated induction of a potent regulatory T cell population. Nat Immunol. (2010) 11:1093–101. doi: 10.1038/ni.1952
111. Levings MK, Sangregorio R, Sartirana C, Moschin AL, Battaglia M, Orban PC, et al. Human CD25+CD4+ T Suppressor Cell Clones Produce Transforming Growth Factor β, but not Interleukin 10, and Are Distinct from Type 1 T Regulatory Cells. J Exp Med. (2002) 196:1335–46. doi: 10.1084/jem.20021139
112. Bennett CL, Christie J, Ramsdell F, Brunkow ME, Ferguson PJ, Whitesell L, et al. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet. (2001) 27:20–1. doi: 10.1038/83713
113. Bacchetta R, Roncarolo MG. IPEX syndrome from diagnosis to cure, learning along the way. J Allergy Clin Immunol. (2024) 153:595–605. doi: 10.1016/j.jaci.2023.11.021
114. Tsai Y-G, Liao P-F, Hsiao K-H, Wu H-M, Lin C-Y, Yang KD. Pathogenesis and novel therapeutics of regulatory T cell subsets and interleukin-2 therapy in systemic lupus erythematosus. Front Immunol. (2023) 14:1230264. doi: 10.3389/fimmu.2023.1230264
115. Rajendiran A, Tenbrock K. Regulatory T cell function in autoimmune disease. J Transl Autoimmun. (2021) 4:100130. doi: 10.1016/j.jtauto.2021.100130
116. Buckner JH. Mechanisms of impaired regulation by CD4+CD25+FOXP3+ regulatory T cells in human autoimmune diseases. Nat Rev Immunol. (2010) 10:849–59. doi: 10.1038/nri2889
117. Schlöder J, Shahneh F, Schneider F-J, Wieschendorf B. Boosting regulatory T cell function for the treatment of autoimmune diseases – That’s only half the battle! Front Immunol. (2022) 13:973813. doi: 10.3389/fimmu.2022.973813
118. Gagliani N, Magnani CF, Huber S, Gianolini ME, Pala M, Licona-Limon P, et al. Coexpression of CD49b and LAG-3 identifies human and mouse T regulatory type 1 cells. Nat Med. (2013) 19:739–46. doi: 10.1038/nm.3179
119. Saraiva M, Vieira P, O’Garra A. Biology and therapeutic potential of interleukin-10. J Exp Med. (2020) 217:e20190418. doi: 10.1084/jem.20190418
120. Neumann C, Scheffold A, Rutz S. Functions and regulation of T cell-derived interleukin-10. Semin Immunol. (2019) 44:101344. doi: 10.1016/j.smim.2019.101344
121. Malefyt R de W, Haanen J, Spits H, Roncarolo MG, te Velde A, Figdor C, et al. Interleukin 10 (IL-10) and viral IL-10 strongly reduce antigen-specific human T cell proliferation by diminishing the antigen-presenting capacity of monocytes via downregulation of class II major histocompatibility complex expression. J Exp Med. (1991) 174:915–24. doi: 10.1084/jem.174.4.915
122. Yao Y, Vent-Schmidt J, McGeough MD, Wong M, Hoffman HM, Steiner TS, et al. Tr1 Cells, but Not Foxp3 + Regulatory T Cells, Suppress NLRP3 Inflammasome Activation via an IL-10–Dependent Mechanism. J Immunol. (2015) 195:488–97. doi: 10.4049/jimmunol.1403225
123. Cook L, Stahl M, Han X, Nazli A, MacDonald KN, Wong MQ, et al. Suppressive and gut-reparative functions of human type 1 T regulatory cells. Gastroenterology. (2019) 157:1584–98. doi: 10.1053/j.gastro.2019.09.002
124. Brockmann L, Soukou S, Steglich B, Czarnewski P, Zhao L, Wende S, et al. Molecular and functional heterogeneity of IL-10-producing CD4 + T cells. Nat Commun. (2018) 9:5457–7. doi: 10.1038/s41467-018-07581-4
125. Roncarolo MG, Yssel H, Touraine JL, Betuel H, Vries JED, Spits H. Autoreactive T cell clones specific for class I and class II HLA antigens isolated from a human chimera. J Exp Med. (1988) 167:1523–34. doi: 10.1084/jem.167.5.1523
126. Groux H, O’Garra A, Bigler M, Rouleau M, Antonenko S, Vries JED, et al. A CD4+ T-cell subset inhibits antigen-specific T-cell responses and prevents colitis. Nature. (1997) 389:737–42. doi: 10.1038/39614
127. Bacchetta R, Bigler M, Touraine JL, Parkman R, Tovo PA, Abrams J, et al. High levels of interleukin 10 production in vivo are associated with tolerance in SCID patients transplanted with HLA mismatched hematopoietic stem cells. J Exp Med. (1994) 179:493–502. doi: 10.1084/jem.179.2.493
128. Freeborn RA, Strubbe S, Roncarolo MG. Type 1 regulatory T cell-mediated tolerance in health and disease. Front Immunol. (2022) 13:1032575. doi: 10.3389/fimmu.2022.1032575
129. Baron KJ, Turnquist HR. Clinical manufacturing of regulatory T cell products for adoptive cell therapy and strategies to improve therapeutic efficacy. Organogenesis. (2023) 19:2164159. doi: 10.1080/15476278.2022.2164159
130. Theil A, Tuve S, Oelschlägel U, Maiwald A, Döhler D, Oßmann D, et al. Adoptive transfer of allogeneic regulatory T cells into patients with chronic graft-versus-host disease. Cytotherapy. (2015) 17:473–86. doi: 10.1016/j.jcyt.2014.11.005
131. Battaglia M, Stabilini A, Migliavacca B, Horejs-Hoeck J, Kaupper T, Roncarolo MG. Rapamycin promotes expansion of functional CD4+CD25+FOXP3+ regulatory T cells of both healthy subjects and type 1 diabetic patients. J Immunol. 177:8338–47. doi: 10.4049/jimmunol.177.12.8338
132. Trzonkowski P, Bieniaszewska M, Juścińska J, Dobyszuk A, Krzystyniak A, Marek N, et al. First-in-man clinical results of the treatment of patients with graft versus host disease with human ex vivo expanded CD4+CD25+CD127– T regulatory cells. Clin Immunol. (2009) 133:22–6. doi: 10.1016/j.clim.2009.06.001
133. Mathew JM, H.-Voss J, LeFever A, Konieczna I, Stratton C, He J, et al. A phase I clinical trial with ex vivo expanded recipient regulatory T cells in living donor kidney transplants. Sci Rep. (2018) 8:7428. doi: 10.1038/s41598-018-25574-7
134. Sagoo P, Ali N, Garg G, Nestle FO, Lechler RI, Lombardi G. Human regulatory T cells with alloantigen specificity are more potent inhibitors of alloimmune skin graft damage than polyclonal regulatory T cells. Sci Transl Med. (2011) 3:83ra42. doi: 10.1126/scitranslmed.3002076
135. Raimondi G, Sumpter TL, Matta BM, Pillai M, Corbitt N, Vodovotz Y, et al. Mammalian target of rapamycin inhibition and alloantigen-specific regulatory T cells synergize to promote long-term graft survival in immunocompetent recipients. J Immunol. (2010) 184:624–36. doi: 10.4049/jimmunol.0900936
136. Sánchez-Fueyo A, Sandner S, Habicht A, Mariat C, Kenny J, Degauque N, et al. Specificity of CD4+CD25+ Regulatory T cell function in alloimmunity. J Immunol. (2006) 176:329–34. doi: 10.4049/jimmunol.176.1.329
137. Tsang JY-S, Tanriver Y, Jiang S, Xue S-A, Ratnasothy K, Chen D, et al. Conferring indirect allospecificity on CD4+CD25+ Tregs by TCR gene transfer favors transplantation tolerance in mice. J Clin Investig. (2008) 118:3619–28. doi: 10.1172/jci33185
138. Todo S, Yamashita K, Goto R, Zaitsu M, Nagatsu A, Oura T, et al. A pilot study of operational tolerance with a regulatory T-cell-based cell therapy in living donor liver transplantation. Hepatology. (2016) 64:632–43. doi: 10.1002/hep.28459
139. Becker C, Taube C, Bopp T, Becker C, Michel K, Kubach J, et al. Protection from graft-versus-host disease by HIV-1 envelope protein gp120-mediated activation of human CD4+CD25+ regulatory T cells. Blood. (2009) 114:1263–9. doi: 10.1182/blood-2009-02-206730
140. Marek-Trzonkowska N, Myśliwiec M, Dobyszuk A, Grabowska M, Techmańska I, Juścińska J, et al. Administration of CD4+CD25highCD127– regulatory T cells preserves β-cell function in type 1 diabetes in children. Diabetes Care. (2012) 35:1817–20. doi: 10.2337/dc12-0038
141. Bluestone JA, Buckner JH, Fitch M, Gitelman SE, Gupta S, Hellerstein MK, et al. Type 1 diabetes immunotherapy using polyclonal regulatory T cells. Sci Trans Med. (2015) 7. doi: 10.1126/scitranslmed.aad4134
142. Marek-Trzonkowska N, Myśliwiec M, Dobyszuk A, Grabowska M, Derkowska I, Juścińska J, et al. Therapy of type 1 diabetes with CD4+CD25highCD127-regulatory T cells prolongs survival of pancreatic islets — Results of one year follow-up. Clin Immunol. (2014) 153:23–30. doi: 10.1016/j.clim.2014.03.016
143. Dong S, Hiam-Galvez KJ, Mowery CT, Herold KC, Gitelman SE, Esensten JH, et al. The effects of low-dose IL-2 on Treg adoptive cell therapy in patients with Type 1 diabetes. JCI Insight. (2021) 6:e147474. doi: 10.1172/jci.insight.147474
144. Dall’Era M, Pauli ML, Remedios K, Taravati K, Sandova PM, Putnam AL, et al. Adoptive treg cell therapy in a patient with systemic lupus erythematosus. Arthritis Rheumatol. (2019) 71:431–40. doi: 10.1002/art.40737
145. Chwojnicki K, Iwaszkiewicz-Grześ D, Jankowska A, Zieliński M, Łowiec P, Gliwiński M, et al. Administration of CD4+CD25highCD127–foxP3+ Regulatory T cells for relapsing-remitting multiple sclerosis: A phase 1 study. BioDrugs. (2021) 35:47–60. doi: 10.1007/s40259-020-00462-7
146. Canavan JB, Scottà C, Vossenkämper A, Goldberg R, Elder MJ, Shoval I, et al. Developing in vitro expanded CD45RA+ regulatory T cells as an adoptive cell therapy for Crohn’s disease. Gut. (2016) 65:584. doi: 10.1136/gutjnl-2014-306919
147. Voskens C, Stoica D, Rosenberg M, Vitali F, Zundler S, Ganslmayer M, et al. Autologous regulatory T-cell transfer in refractory ulcerative colitis with concomitant primary sclerosing cholangitis. Gut. (2023) 72:49–53. doi: 10.1136/gutjnl-2022-327075
148. Kellner JN, Delemarre EM, Yvon E, Nierkens S, Boelens JJ, McNiece I, et al. Third party, umbilical cord blood derived regulatory T-cells for prevention of graft versus host disease in allogeneic hematopoietic stem cell transplantation: feasibility, safety and immune reconstitution. Oncotarget. (2018) 9:35611–22. doi: 10.18632/oncotarget.26242
149. Samanta A, Li B, Song X, Bembas K, Zhang G, Katsumata M, et al. TGF-β and IL-6 signals modulate chromatin binding and promoter occupancy by acetylated FOXP3. Proc Natl Acad Sci. (2008) 105:14023–7. doi: 10.1073/pnas.0806726105
150. Zhou X, Bailey-Bucktrout SL, Jeker LT, Penaranda C, Martínez-Llordella M, Ashby M, et al. Instability of the transcription factor Foxp3 leads to the generation of pathogenic memory T cells. vivo. Nat Immunol. (2009) 10:1000–7. doi: 10.1038/ni.1774
151. Bailey-Bucktrout SL, Martinez-Llordella M, Zhou X, Anthony B, Rosenthal W, Luche H, et al. Self-antigen-driven activation induces instability of regulatory T cells during an inflammatory autoimmune response. Immunity. (2013) 39:949–62. doi: 10.1016/j.immuni.2013.10.016
152. Komatsu N, Okamoto K, Sawa S, Nakashima T, Oh-hora M, Kodama T, et al. Pathogenic conversion of Foxp3+ T cells into TH17 cells in autoimmune arthritis. Nat Med. (2014) 20:62–8. doi: 10.1038/nm.3432
153. Noval Rivas M, Burton OT, Wise P, Charbonnier L-M, Georgiev P, Oettgen HC, et al. Regulatory T cell reprogramming toward a th2-cell-like lineage impairs oral tolerance and promotes food allergy. Immunity. (2015) 42:512–23. doi: 10.1016/j.immuni.2015.02.004
154. Passerini L, Mel ER, Sartirana C, Fousteri G, Bondanza A, Naldini L, et al. CD4+ T cells from IPEX patients convert into functional and stable regulatory T cells by FOXP3 gene transfer. Sci Trans Med. (2013) 5:1–11. doi: 10.1126/scitranslmed.3007320
155. Goodwin M, Lee E, Lakshmanan U, Shipp S, Froessl L, Barzaghi F, et al. CRISPR-based gene editing enables FOXP3 gene repair in IPEX patient cells. Sci Adv. (2020) 6. doi: 10.1126/sciadv.aaz0571
156. Honaker Y, Hubbard N, Xiang Y, Fisher L, Hagin D, Sommer K, et al. Gene editing to induce FOXP3 expression in human CD4+ T cells leads to a stable regulatory phenotype and function. Sci Trans Med. (2020) 12. doi: 10.1126/scitranslmed.aay6422
157. Sato Y, Passerini L, Piening BD, Uyeda MJ, Goodwin M, Gregori S, et al. Human-engineered Treg-like cells suppress FOXP3-deficient T cells but preserve adaptive immune responses. vivo. Clin Transl Immunol. (2020) 9:e1214–4. doi: 10.1002/cti2.1214
158. Hunt MS, Yang SJ, Mortensen E, Boukhris A, Buckner J, Cook PJ, et al. Dual-locus, dual-HDR editing permits efficient generation of antigen-specific regulatory T cells with robust suppressive activity. Mol Ther. (2023) 10:P2872–86. doi: 10.1016/j.ymthe.2023.07.016
159. Brun V, Bastian H, Neveu V, Foussat A. Clinical grade production of IL-10 producing regulatory Tr1 lymphocytes for cell therapy of chronic inflammatory diseases. Int Immunopharmacol. (2009) 9:609–13. doi: 10.1016/j.intimp.2009.01.032
160. Desreumaux P, Foussat A, Allez M, Beaugerie L, Hébuterne X, Bouhnik Y, et al. Safety and efficacy of antigen-specific regulatory T-cell therapy for patients with refractory Crohn’s disease. Gastroenterology. (2012) 143:1207–17. doi: 10.1053/j.gastro.2012.07.116
161. Bacchetta R, Lucarelli B, Sartirana C, Gregori S, Stanghellini MTL, Miqueu P, et al. Immunological outcome in haploidentical-HSC transplanted patients treated with IL-10-anergized donor T Cells. Front Immunol. (2014) 5:16. doi: 10.3389/fimmu.2014.00016
162. Chen PP, Cepika A, Agarwal-hashmi R, Saini G, Uyeda MJ, Louis DM, et al. Alloantigen-specific type 1 regulatory T cells suppress through CTLA-4 and PD-1 pathways and persist long-term in patients. Sci Trans Med. (2021) 13:eabf5264. doi: 10.1126/scitranslmed.abf5264
163. Bertaina A, Bacchetta R, Shy BR, Saini G, Lee J, Kristovich K, et al. T-allo10 Infusion after αβ depleted-HSCT in Children and Young Adults with Hematologic Malignancies: Improved Immune Reconstitution in the Absence of Severe GvHD. Transpl Cell Ther. (2023) 29:S209–10. doi: 10.1016/S2666-6367(23)00340-8
164. Andolfi G, Fousteri G, Rossetti M, Magnani CF, Jofra T, Locafaro G, et al. Enforced IL-10 expression confers type 1 regulatory T cell (Tr1) phenotype and function to human CD4+ T cells. Mol Ther. (2012) 20:1778–90. doi: 10.1038/mt.2012.71
165. Locafaro G, Andolfi G, Russo F, Cesana L, Spinelli A, Camisa B, et al. IL-10-engineered human CD4+ Tr1 cells eliminate myeloid leukemia in an HLA class I-dependent mechanism. Mol Ther. (2017) 25:2254–69. doi: 10.1016/j.ymthe.2017.06.029
166. Liu JM-H, Chen P, Uyeda MJ, Cieniewicz B, Sayitoglu EC, Thomas BC, et al. Pre-clinical development and molecular characterization of an engineered type 1 regulatory T-cell product suitable for immunotherapy. Cytotherapy. (2021) 23:10174–1028. doi: 10.1016/j.jcyt.2021.05.010
167. Neurath MF, Sands BE, Rieder F. Cellular immunotherapies and immune cell depleting therapies in inflammatory bowel diseases: the next magic bullet? Gut. (2024). doi: 10.1136/gutjnl-2024-332919. gutjnl-2024-332919.
168. Uenishi GI, Repic M, Yam JY, Landuyt A, Saikumar-Lakshmi P, Guo T, et al. GNTI-122: an autologous antigen-specific engineered Treg cell therapy for type 1 diabetes. JCI Insight. (2024) 9:e171844. doi: 10.1172/jci.insight.171844
169. MacDonald KG, Hoeppli RE, Huang Q, Gillies J, Luciani DS, Orban PC, et al. Alloantigen-specific regulatory T cells generated with a chimeric antigen receptor. J Clin Investig. (2016) 126:1413–24. doi: 10.1172/jci82771
170. Boardman DA, Philippeos C, Fruhwirth GO, Ibrahim MAA, Hannen RF, Cooper D, et al. Expression of a chimeric antigen receptor specific for donor HLA class I enhances the potency of human regulatory T cells in preventing human skin transplant rejection. Am J Transplant. (2017) 17:931–43. doi: 10.1111/ajt.14185
171. Noyan F, Zimmermann K, Hardtke-Wolenski M, Knoefel A, Schulde E, Geffers R, et al. Prevention of allograft rejection by use of regulatory T cells with an MHC-specific chimeric antigen receptor. Am J Transplant. (2017) 17:917–30. doi: 10.1111/ajt.14175
172. Muller YD, Ferreira LMR, Ronin E, Ho P, Nguyen V, Faleo G, et al. Precision engineering of an anti-HLA-A2 chimeric antigen receptor in regulatory T cells for transplant immune tolerance. Front Immunol. (2021) 12:686439. doi: 10.3389/fimmu.2021.686439
173. Proics E, David M, Mojibian M, Speck M, Lounnas-Mourey N, Govehovitch A, et al. Preclinical assessment of antigen-specific chimeric antigen receptor regulatory T cells for use in solid organ transplantation. Gene Ther. (2023) 30:309–22. doi: 10.1038/s41434-022-00358-x
174. Schreeb K, Culme-Seymour E, Ridha E, Dumont C, Atkinson G, Hsu B, et al. Study design: human leukocyte antigen class I molecule A∗02-chimeric antigen receptor regulatory T cells in renal transplantation. Kidney Int Rep. (2022) 7:1258–67. doi: 10.1016/j.ekir.2022.03.030
175. Doglio M, Ugolini A, Bercher-Brayer C, Camisa B, Toma C, Norata R, et al. Regulatory T cells expressing CD19-targeted chimeric antigen receptor restore homeostasis in Systemic Lupus Erythematosus. Nat Commun. (2024) 15:2542. doi: 10.1038/s41467-024-46448-9
176. Eggenhuizen PJ, Cheong RMY, Lo C, Chang J, Ng BH, Ting YT, et al. Smith-specific regulatory T cells halt the progression of lupus nephritis. Nat Commun. (2024) 15:899. doi: 10.1038/s41467-024-45056-x
177. Shah NN, Lee DW, Yates B, Yuan CM, Shalabi H, Martin S, et al. Long-term follow-up of CD19-CAR T-cell therapy in children and young adults with B-ALL. J Clin Oncol. (2021) 39:1650–9. doi: 10.1200/jco.20.02262
178. Kochenderfer JN, Somerville RPT, Lu T, Yang JC, Sherry RM, Feldman SA, et al. Long-duration complete remissions of diffuse large B cell lymphoma after anti-CD19 chimeric antigen receptor T cell therapy. Mol Ther. (2017) 25:2245–53. doi: 10.1016/j.ymthe.2017.07.004
179. Turtle CJ, Hanafi L-A, Berger C, Hudecek M, Pender B, Robinson E, et al. Immunotherapy of non-Hodgkin’s lymphoma with a defined ratio of CD8+ and CD4+ CD19-specific chimeric antigen receptor–modified T cells. Sci Transl Med. (2016) 8:355ra116. doi: 10.1126/scitranslmed.aaf8621
180. Gauthier J, Bezerra ED, Hirayama AV, Fiorenza S, Sheih A, Chou CK, et al. Factors associated with outcomes after a second CD19-targeted CAR T-cell infusion for refractory B-cell Malignancies. Blood. (2021) 137:323–35. doi: 10.1182/blood.2020006770
181. Hirayama AV, Gauthier J, Hay KA, Voutsinas JM, Wu Q, Gooley T, et al. The response to lymphodepletion impacts PFS in patients with aggressive non-Hodgkin lymphoma treated with CD19 CAR T cells. Blood. (2019) 133:1876–87. doi: 10.1182/blood-2018-11-887067
182. Gardner RA, Finney O, Annesley C, Brakke H, Summers C, Leger K, et al. Intent-to-treat leukemia remission by CD19 CAR T cells of defined formulation and dose in children and young adults. Blood. (2017) 129:3322–31. doi: 10.1182/blood-2017-02-769208
183. Turtle CJ, Hanafi L-A, Berger C, Gooley TA, Cherian S, Hudecek M, et al. CD19 CAR–T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J Clin Investig. (2016) 126:2123–38. doi: 10.1172/jci85309
184. Taubmann J, Müller F, Mutlu MY, Völkl S, Aigner M, Bozec A, et al. CD19 chimeric antigen receptor T cell treatment: unraveling the role of B cells in systemic lupus erythematosus. Arthritis Rheumatol. (2024) 76:497–504. doi: 10.1002/art.42784
185. Till BG, Jensen MC, Wang J, Chen EY, Wood BL, Greisman HA, et al. Adoptive immunotherapy for indolent non-Hodgkin lymphoma and mantle cell lymphoma using genetically modified autologous CD20-specific T cells. Blood. (2008) 112:2261–71. doi: 10.1182/blood-2007-12-128843
186. Till BG, Jensen MC, Wang J, Qian X, Gopal AK, Maloney DG, et al. CD20-specific adoptive immunotherapy for lymphoma using a chimeric antigen receptor with both CD28 and 4-1BB domains: pilot clinical trial results. Blood. (2012) 119:3940–50. doi: 10.1182/blood-2011-10-387969
187. Abbas AK, Trotta E, Simeonov DR, Marson A, Bluestone JA. Revisiting IL-2: Biology and therapeutic prospects. Sci Immunol. (2018) 3. doi: 10.1126/sciimmunol.aat1482
188. Barron L, Dooms H, Hoyer KK, Kuswanto W, Hofmann J, O’Gorman WE, et al. Cutting edge: mechanisms of IL-2–dependent maintenance of functional regulatory T cells. J Immunol. (2010) 185:6426–30. doi: 10.4049/jimmunol.0903940
189. Harris F, Berdugo YA, Tree T. IL-2-based approaches to Treg enhancement. Clin Exp Immunol. (2022) 211:149–63. doi: 10.1093/cei/uxac105
190. Hartemann A, Bensimon G, Payan CA, Jacqueminet S, Bourron O, Nicolas N, et al. Low-dose interleukin 2 in patients with type 1 diabetes: a phase 1/2 randomised, double-blind, placebo-controlled trial. Lancet Diabetes Endocrinol. (2013) 1:295–305. doi: 10.1016/s2213-8587(13)70113-x
191. Sockolosky JT, Trotta E, Parisi G, Picton L, Su LL, Le AC, et al. Selective targeting of engineered T cells using orthogonal IL-2 cytokine-receptor complexes. Science. (2018) 359:1037–42. doi: 10.1126/science.aar3246
192. Aspuria P-J, Vivona S, Bauer M, Semana M, Ratti N, McCauley S, et al. An orthogonal IL-2 and IL-2Rβ system drives persistence and activation of CAR T cells and clearance of bulky lymphoma. Sci Transl Med. (2021) 13:eabg7565. doi: 10.1126/scitranslmed.abg7565
193. Hirai T, Ramos TL, Lin P-Y, Simonetta F, Su LL, Picton LK, et al. Selective expansion of regulatory T cells using an orthogonal IL-2/IL-2 receptor system facilitates transplantation tolerance. J Clin Investig. (2021) 131:e139991. doi: 10.1172/jci139991
194. Thomas S, Abken H. CAR T cell therapy becomes CHIC: “cytokine help intensified CAR” T cells. Front Immunol. (2023) 13:1090959. doi: 10.3389/fimmu.2022.1090959
195. Gattinoni L, Finkelstein SE, Klebanoff CA, Antony PA, Palmer DC, Spiess PJ, et al. Removal of homeostatic cytokine sinks by lymphodepletion enhances the efficacy of adoptively transferred tumor-specific CD8+ T cells. J Exp Med. (2005) 202:907–12. doi: 10.1084/jem.20050732
196. Shum T, Omer B, Tashiro H, Kruse RL, Wagner DL, Parikh K, et al. Constitutive signaling from an engineered IL7 receptor promotes durable tumor elimination by tumor-redirected T cells. Cancer Discovery. (2017) 7:1238–47. doi: 10.1158/2159-8290.cd-17-0538
197. Sharma S, Sauer T, Omer BA, Shum T, Rollins LA, Rooney CM. Constitutive interleukin-7 cytokine signaling enhances the persistence of epstein–barr virus-specific T-cells. Int J Mol Sci. (2023) 24:15806. doi: 10.3390/ijms242115806
198. Hurton LV, Singh H, Najjar AM, Switzer KC, Mi T, Maiti S, et al. Tethered IL-15 augments antitumor activity and promotes a stem-cell memory subset in tumor-specific T cells. Proc Natl Acad Sci. (2016) 113:E7788–97. doi: 10.1073/pnas.1610544113
199. Sun Y, Su Y, Wang Y, Liu N, Li Y, Chen J, et al. CD19 CAR-T cells with membrane-bound IL-15 for B-cell acute lymphoblastic leukemia after failure of CD19 and CD22 CAR-T cells: case report. Front Immunol. (2021) 12:728962. doi: 10.3389/fimmu.2021.728962
200. Bacchetta R, Sartirana C, Levings MK, Bordignon C, Narula S, Roncarolo MG. Growth and expansion of human T regulatory type 1 cells are independent from TCR activation but require exogenous cytokines. Eur J Immunol. (2002) 32:2237–45. doi: 10.1002/1521-4141(200208)32:8<2237::aid-immu2237>3.0.co;2-2
201. Pegram HJ, Lee JC, Hayman EG, Imperato GH, Tedder TF, Sadelain M, et al. Tumor-targeted T cells modified to secrete IL-12 eradicate systemic tumors without need for prior conditioning. Blood. (2012) 119:4133–41. doi: 10.1182/blood-2011-12-400044
202. Avanzi MP, Yeku O, Li X, Wijewarnasuriya DP, van Leeuwen DG, Cheung K, et al. Engineered tumor-targeted T cells mediate enhanced anti-tumor efficacy both directly and through activation of the endogenous immune system. Cell Rep. (2018) 23:2130–41. doi: 10.1016/j.celrep.2018.04.051
203. Zhao Y, Chen J, Andreatta M, Feng B, Xie Y-Q, Wenes M, et al. IL-10-expressing CAR T cells resist dysfunction and mediate durable clearance of solid tumors and metastases. Nat Biotechnol. (2024) 42:1–12. doi: 10.1038/s41587-023-02060-8
204. Harper J, Ribeiro SP, Chan CN, Aid M, Deleage C, Micci L, et al. Interleukin-10 contributes to reservoir establishment and persistence in SIV-infected macaques treated with antiretroviral therapy. J Clin Investig. (2022) 132:e155251. doi: 10.1172/jci155251
205. Puntambekar SS, Bergmann CC, Savarin C, Karp CL, Phares TW, Parra GI, et al. Shifting hierarchies of interleukin-10-producing T cell populations in the central nervous system during acute and persistent viral encephalomyelitis. J Virol. (2011) 85:6702–13. doi: 10.1128/jvi.00200-11
206. Jofra T, Fonte RD, Galvani G, Kuka M, Iannacone M, Battaglia M, et al. Tr1 cell immunotherapy promotes transplant tolerance via de novo Tr1 cell induction in mice and is safe and effective during acute viral infection. Eur J Immunol. (2018) 48:1389–99. doi: 10.1002/eji.201747316
207. Qin S, Cobbold SP, Pope H, Elliott J, Kioussis D, Davies J, et al. Infectious” Transplantation tolerance. Science. (1993) 259:974–7. doi: 10.1126/science.8094901
208. Cordeiro A, Bezerra ED, Hirayama AV, Hill JA, Wu QV, Voutsinas J, et al. Late events after treatment with CD19-targeted chimeric antigen receptor modified T cells. Biol Blood Marrow Transplant. (2020) 26:26–33. doi: 10.1016/j.bbmt.2019.08.003
209. Cappell KM, Kochenderfer JN. Long-term outcomes following CAR T cell therapy: what we know so far. Nat Rev Clin Oncol. (2023) 20:359–71. doi: 10.1038/s41571-023-00754-1
210. Goldmann O, Nwofor OV, Chen Q, Medina E. Mechanisms underlying immunosuppression by regulatory cells. Front Immunol. (2024) 15:1328193. doi: 10.3389/fimmu.2024.1328193
211. Tay C, Tanaka A, Sakaguchi S. Tumor-infiltrating regulatory T cells as targets of cancer immunotherapy. Cancer Cell. (2023) 41:450–65. doi: 10.1016/j.ccell.2023.02.014
212. Lu L, Xie M, Yang B, Zhao W, Cao J. Enhancing the safety of CAR-T cell therapy: Synthetic genetic switch for spatiotemporal control. Sci Adv. (2024) 10:eadj6251. doi: 10.1126/sciadv.adj6251
213. Bonini C, Ferrari G, Verzeletti S, Servida P, Zappone E, Ruggieri L, et al. HSV-TK gene transfer into donor lymphocytes for control of allogeneic graft-versus-leukemia. Science. (1997) 276:1719–24. doi: 10.1126/science.276.5319.1719
214. Berger C, Flowers ME, Warren EH, Riddell SR. Analysis of transgene-specific immune responses that limit the in vivo persistence of adoptively transferred HSV-TK–modified donor T cells after allogeneic hematopoietic cell transplantation. Blood. (2006) 107:2294–302. doi: 10.1182/blood-2005-08-3503
215. Wang Q, He F, He W, Huang Y, Zeng J, Zi F, et al. A transgene-encoded truncated human epidermal growth factor receptor for depletion of anti- B-cell maturation antigen CAR-T cells. Cell Immunol. (2021) 363:104342–2. doi: 10.1016/j.cellimm.2021.104342
216. Griffioen M, van Egmond EHM, Kester MGD, Willemze R, Falkenburg JHF, Heemskerk MHM. Retroviral transfer of human CD20 as a suicide gene for adoptive T-cell therapy. Haematologica. (2009) 94:1316–20. doi: 10.3324/haematol.2008.001677
217. Bluestone JA, McKenzie BS, Beilke J, Ramsdell F. Opportunities for Treg cell therapy for the treatment of human disease. Front Immunol. (2023) 14:1166135. doi: 10.3389/fimmu.2023.1166135
218. Celichowski P, Turi M, Charvátová S, Radhakrishnan D, Feizi N, Chyra Z, et al. Tuning CARs: recent advances in modulating chimeric antigen receptor (CAR) T cell activity for improved safety, efficacy, and flexibility. J Transl Med. (2023) 21:197. doi: 10.1186/s12967-023-04041-6
219. Neeser A, Ramasubramanian R, Wang C, Ma L. Engineering enhanced chimeric antigen receptor-T cell therapy for solid tumors. Immuno-Oncol Technol. (2023) 19:100385. doi: 10.1016/j.iotech.2023.100385
220. Leung W-H, Gay J, Martin U, Garrett TE, Horton HM, Certo MT, et al. Sensitive and adaptable pharmacological control of CAR T cells through extracellular receptor dimerization. JCI Insight. (2019) 4. doi: 10.1172/jci.insight.124430
221. Appelbaum J, Price AE, Oda K, Zhang J, Leung W-H, Tampella G, et al. Drug-regulated CD33-targeted CAR T cells control AML using clinically optimized rapamycin dosing. J Clin Investig. (2024) 134:e162593. doi: 10.1172/jci162593
222. Depil S, Duchateau P, Grupp SA, Mufti G, Poirot L. Off-the-shelf” allogeneic CAR T cells: development and challenges. Nat Rev Drug Discovery. (2020) 19:185–99. doi: 10.1038/s41573-019-0051-2
223. Li S, Wang X, Yuan Z, Liu L, Luo L, Li Y, et al. Eradication of T-ALL cells by CD7-targeted universal CAR-T cells and initial test of ruxolitinib-based CRS management. Clin Cancer Res. (2021) 27:1242–6. doi: 10.1158/1078-0432.ccr-20-1271
224. Hu Y, Zhou Y, Zhang M, Ge W, Li Y, Yang L, et al. CRISPR/cas9-engineered universal CD19/CD22 dual-targeted CAR-T cell therapy for relapsed/refractory B-cell acute lymphoblastic leukemia. Clin Cancer Res. (2021) 27:2764–72. doi: 10.1158/1078-0432.ccr-20-3863
225. Ottaviano G, Georgiadis C, Gkazi SA, Syed F, Zhan H, Etuk A, et al. Phase 1 clinical trial of CRISPR-engineered CAR19 universal T cells for treatment of children with refractory B cell leukemia. Sci Transl Med. (2022) 14:eabq3010. doi: 10.1126/scitranslmed.abq3010
226. Guo Y, Tong C, Su L, Zhang W, Jia H, Liu Y, et al. CRISPR/Cas9 genome-edited universal CAR T cells in patients with relapsed and refractory lymphoma. Blood Adv. (2022) 6:2695–9. doi: 10.1182/bloodadvances.2021006232
227. Nahmad AD, Reuveni E, Goldschmidt E, Tenne T, Liberman M, Horovitz-Fried M, et al. Frequent aneuploidy in primary human T cells after CRISPR–Cas9 cleavage. Nat Biotechnol. (2022) 40:1807–13. doi: 10.1038/s41587-022-01377-0
228. Komor AC, Kim YB, Packer MS, Zuris JA, Liu DR. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature. (2016) 533:420–4. doi: 10.1038/nature17946
229. Diorio C, Murray R, Naniong M, Barrera L, Camblin A, Chukinas J, et al. Cytosine base editing enables quadruple-edited allogeneic CART cells for T-ALL. Blood. (2022) 140:619–29. doi: 10.1182/blood.2022015825
230. Chiesa R, Georgiadis C, Syed F, Zhan H, Etuk A, Gkazi SA, et al. Base-edited CAR7 T cells for relapsed T-cell acute lymphoblastic leukemia. N Engl J Med. (2023) 389:899–910. doi: 10.1056/nejmoa2300709
231. Kadia TM, Huang M, Pemmaraju N, Abbas HA, Ly C, Masarova L, et al. Phase 1 study of CK0801 in treatment of bone marrow failure syndromes. NEJM Évid. (2024) 3:EVIDoa2300362. doi: 10.1056/evidoa2300362
Keywords: Tr1, Treg, autoimmunity, CAR T, cell therapy, Tr1 cells
Citation: Bulliard Y, Freeborn R, Uyeda MJ, Humes D, Bjordahl R, de Vries D and Roncarolo MG (2024) From promise to practice: CAR T and Treg cell therapies in autoimmunity and other immune-mediated diseases. Front. Immunol. 15:1509956. doi: 10.3389/fimmu.2024.1509956
Received: 11 October 2024; Accepted: 12 November 2024;
Published: 04 December 2024.
Edited by:
Asif Amin Dar, Children’s Hospital of Philadelphia, United StatesReviewed by:
Gerson D. Keppeke, Universidad Católica del Norte, ChileSaeed Mohammadi, Golestan University of Medical Sciences, Iran
Copyright © 2024 Bulliard, Freeborn, Uyeda, Humes, Bjordahl, de Vries and Roncarolo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maria Grazia Roncarolo, bWdyb25jYXJvbG9AdHIxeC5iaW8=