 Jianping Zhou
Jianping Zhou Yuting Xi2
Yuting Xi2 Xiaoyu Zeng
Xiaoyu Zeng Jun Yuan
Jun Yuan Ce Zhou
Ce Zhou- 1Hospital of Chengdu University of Traditional Chinese Medicine, Chengdu, China
- 2Zigong Hospital of Traditional Chinese Medicine, Zigong, China
Mitochondria are important organelles that regulate cellular energy and biosynthesis, as well as maintain the body’s response to environmental stress. Their dynamics and autophagy influence occurrence of cellular function, particularly under stressful conditions. They can generate reactive oxygen species (ROS) which is a major contributor to inflammatory diseases such as ulcerative colitis (UC). In this review, we discuss the key effects of mitochondrial dynamics and mitophagy on the pathogenesis of UC, with a particular focus on the cellular energy metabolism, oxidative stress, apoptosis, and immunoinflammatory activities. The therapeutic efficacy of existing drugs and phytochemicals targeting the mitochondrial pathway are discussed to reveal important insights for developing therapeutic strategies for treating UC. In addition, new molecular checkpoints with therapeutic potential are identified. We show that the integration of mitochondrial biology with the clinical aspects of UC may generate ideas for enhancing the clinical management of UC.
1 Introduction
Ulcerative Colitis (UC) is an inflammatory bowel disease (IBD) affecting the rectum and extending to the proximal colon parts (1). In 2023, the global prevalence of UC was estimated to be 5 million cases, with over 400 diagnoses per 100,000 people reported in North America (2–4). The development of UC involves multiple pathways including changes in genetic mutations, environmental influences, impaired gut microbiota, and imbalance in the mucosal immune system (5). The primary clinical symptoms of UC patients are blood in the stool, diarrhea, and abdominal pain, fever, dehydration, weight loss, and possibly depression, anxiety, sleep disorders, and sexual dysfunction (6–9). If untreated, UC can potentially increase the risk of colon cancer (10). Several treatments have been proposed for UC, which include 5-aminosalicylic acid (5-ASA), corticosteroids, immunosuppressants, biologics, and even surgical procedures. However, the maximum response to these treatments is estimated at 30% to 60% (11). Moreover, for patients with UC, the rate of colon resection are 12%-19% at 12 months despite treatment with sequential therapy (12). In recent year, UC has become an intractable clinical challenge due to the lack of safe and long-lasting treatment options.
The available treatments focus on alleviating symptoms, preventing complications and improving the patients’ quality of life (13). Evidence from previous studies has shown that the intestinal epithelial barrier is impaired in UC patients, accompanied with dysbiosis of gut microbiota, and a dysregulated immune response (4). Furthermore, mucosal destruction and oxidative-antioxidant imbalance have been recognized as the primary factors influencing the recurrence of UC (14). Oxidative stress (OS), mediated by ROS, plays a crucial role in the initiation of inflammatory response in the colon through positive feedback mechanisms (15). The mitochondria facilitate the production of ROS, which can be extremely harmful to cells at excessive levels. Uncontrolled ROS production from damaged mitochondria increases inflammatory reactions. Studies have demonstrated that mitochondria are cellular hubs for infection (16).
Roediger et al. reported that the UC can be classified as a metabolic disorder arising from mitochondrial dysfunction. For instance, impaired mitochondrial dynamics and mitophagy were detected in DSS- or TNBS-induced mouse models of enterocolitis and in UC patients (17–19). Moreover, the mitochondrial dynamics and mitophagy have been extensively investigated as important sources of disease biomarkers, including UC. Research has uncovered that, besides the environmental factors, cytokines changes in antimicrobial pathways, and autophagy form part of the pathomechanisms of UC, driven by numerous pathways (20–22). Currently, drugs such as mitochondrial fission antagonist P110 and Mdivi-1 are being investigated for their potential to treat UC, but their therapeutic efficacy is unknown and some side effects have been reported (18, 23). Phytochemicals are extracts from natural products with numerous advantages such as multi-targeting properties, few side effects and less costly (24). Several agents targeting mitochondrial dynamics and mitophagy have been explored for the management of degenerative neurological diseases, tumors, and osteoarthritis (25–27). Therefore, we aimed to discuss the alterations in mitophagy and mitochondrial dynamics in UC as reported the available studies. The treatment of UC using natural ingredients is discussed to provide new ideas for better management of UC in the future.
2 Mitochondrial dynamics
Mitochondrial dynamics comprises fusion, fission, and transport processes (28). Mitochondrial fission and fusion, along with their distribution along cytoskeletal trajectories, are highly coordinated mechanisms involved in the regulation of the mitochondrial network (Figure 1). Proper mitochondrial dynamics are driven by the biogenesis, turnover, distribution of mitochondrial DNA(mtDNA), and metabolic status (29). Studies have demonstrated that mitochondrial fission and fusion are essential components of cell survival, playing important roles in the maintenance of cellular health and disease development.
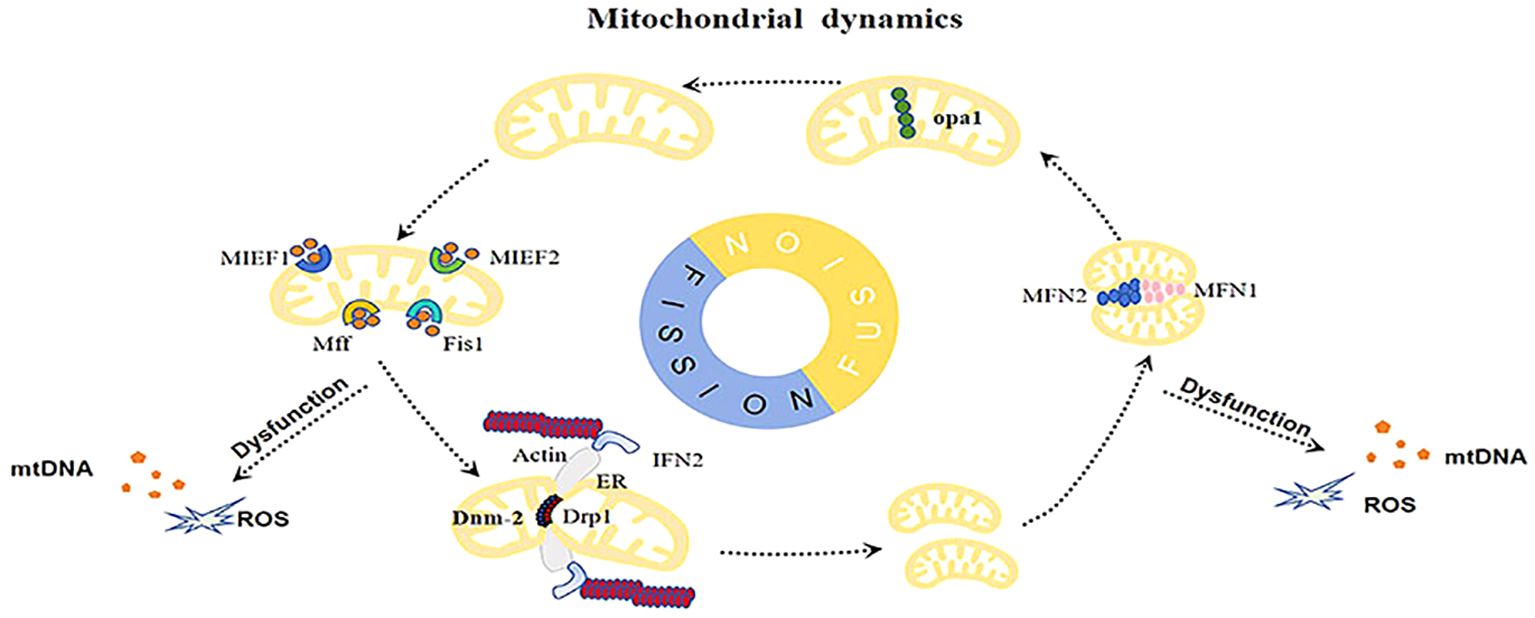
Figure 1. Abnormal mitochondrial dynamics increase the accumulation of damaged mitochondria, resulting in the buildup of mtDNA, ROS, and other damage-associated molecular patterns (DAMPs), inducing inflammation and cellular stress.
2.1 Mechanisms of mitochondrial fission
Mitochondrial division predominantly occurs during the S, G2, and M phases of the cell cycle (30). This process is initiated by stress stimuli and is aimed at eliminating damaged mitochondria from the cell through mitophagy. Therefore, mitochondrial division prevents the buildup of damaged mitochondria, alleviating the excessive production of ROS and the subsequent cellular stress (31).
On the other hand, mitochondrial fission is driven by the dynamin-related protein 1 (Drp1), which functions at the contractile site established by the interaction of actin and the endoplasmic reticulum (ER) (32). Phosphorylation-activated Drp1 translocates to the outer mitochondrial membrane (OMM) where it oligomerizes to form helices that drive OMM contraction and cleavage, thereby facilitating mitochondrial fission (33, 34). Given the Drp1 lacks the pleckstrin homology (PH) structural domain required for the direct interaction with the phospholipid membrane, its activity requires adaptor proteins. In mammals, four proteins regulate the recruitment of Drp1 to the OMM: fission protein 1 (Fis1), mitochondrial fission factor (Mff), mitochondrial elongation factor 1 (MIEF1), and mitochondrial elongation factor 2 (MIEF2), that serve as Drp1 receptors to improve the fission process (35). activation of fission following the enclosement of mitochondria by the ER leads to the Mff-dependent assembly of Drp1 (36). Inactivation of the Mff gene then inhibits mitochondrial recruitment of Drp1, mitochondrial and peroxisomal elongation, while Mff overexpression can fragment the network (37, 38). It has been shown that MIEF1 binds to the adenosine diphosphate (ADP) cofactor, whereas MIEF2 recruits Drp1 to the mitochondrial surface (39). Mff and MIEF1/MIEF2 function are important receptors of Drp1, which mediate it role on mitochondrial fission. Researchers have demonstrated that overexpression of Mff or MIEF is overexpressed, they significantly attract cytoplasmic Drp1 to the mitochondria, independent of hFis1, which enhances the process of mitochondrial fission (40). These receptors work together to help Drp1 divide mitochondria. In mammals, Fis1 usually plays a minor role in recruiting Drp1. However, during cell death or autophagy, Fis1 becomes more important, significantly boosting mitochondrial division. The role of Fis1 in humans is still unclear (41). Finally, the GTPase dynamin 2 (Dnm2) is transiently recruited to the ER contact site downstream of Drp1 where it facilitates the fission process (42, 43). Subsequently, Drp1 is transported back to the cytoplasm, creating a dynamic cycle between the cytoplasmic space and the mitochondrial membrane. The Drp1’s activation, movement, and oscillation between cellular compartments are tightly regulated by several post-translational modifications. These include phosphorylation, ubiquitination, sumoylation, and glycosylation. The mechanisms by which these modifications alter the functionality of Drp1 are described in details in later sections of this review (44–47).
2.2 Mechanisms of mitochondrial fusion
Mitochondrial fusion is commonly detected in the early S and G1 phases, and is essential to the generation of sufficient ATP needed to sustain respiration and protein synthesis (48). Moreover, the fusion modulates the exchange of materials such as mtDNA, proteins, and metabolites, enhancing the repair of damaged molecules, inhibiting phagocytosis of elongated mitochondria, which may result from site-blocking during nutrient deprivation and the induction of autophagy (48, 49).
The process of mitochondrial fusion involves the fusion of the inner mitochondrial membrane (IMM) and the OMM, occurring in multiple steps beginning with the activation of dynamin-associated GTPases, including mitofusins (MFN1 and MFN2) on the OMM, and optic atrophic protein 1 (Opa1) on the IMM. This is followed by the GTP hydrolysis-induced fusion of the OMM (50). Structurally, the MFNs are embedded in the OMM via two transmembrane regions, divided by a short loop. This structure allows the N-terminal region, containing the GTPase domain, the coiled-coil heptad repeat 1 (HR1), and the C-terminal region that harbors the HR2 domain, to orient towards the cytoplasmic side (51). This is followed by the IMM fusion, orchestrated by Opa1 in the IMM and endosomal lipid components (52). During fusion, two Opa1 proteins in the IMM interact to form oligomers that assemble into flexible helices, causing membrane swelling and bringing the two IMMs into close proximity. The nucleotide binding organizes and tightens the helix assembly, pulling the IMM closer together to initiate fusion. At the end of the fusion, the helical structure of the Opa1 oligomerization is uncoiled (53). This mechanism is crucial for Opa1 to form a helical structure by dimerizing the GTPase domain. Furthermore, the membrane-bending oligomers of Opa1 undergoes conformational changes, which retract the membrane insertion loop from the outer leaflet, causing remodeling (54). OMM fusion is usually coordinated with inner membrane fusion, but can sometimes occur independently. This phenomenon may be triggered by occurrence of mutations or reduced membrane potential which impairs the fusion of the IMM, but the OMM fusion is not affected (55).
2.3 Factors regulating mitochondrial dynamics
2.3.1 Drp1
The function of Drp1, a key regulator of mitochondrial fission, is influenced by multiple post-translational modifications, such as phosphorylation, ubiquitination, and sumoylation. Its activity is modulated through phosphorylation at three main sites: ser616, ser637, and ser693 (56). The ser616 site is phosphorylated by protein kinase Cδ (PKCδ), Rock kinase, or Pink1, which then activates fission and promotes the binding to other fission proteins (57, 58). On the other hand, the ser637 site is phosphorylated by protein kinase A, which leads to its inactivation (59). Phosphorylation of the ser693 site by GSK3β serves to inhibit mitochondrial division (60). Furthermore, the membrane-associated E3 ligase March5 can regulate Drp1 through ubiquitination (61). Likewise, the E3 ligase Mulan, which is also membrane-bound, was reported to influence Drp1 by promoting sumoylation (62). Functionally, Drp1 that functions without involvement in nitrosylation is thought to trigger mitochondrial fission (63, 64). Ubiquitination, particularly by March5, is crucial for regulating mitochondrial fission. However, while March5 ubiquitinates Drp1, it does not affect its stability. Instead, the process of ubiquitination may affect the dynamics of the Drp1’s interaction through the mitochondrial membrane. In this context, the attachment of ubiquitin alters the subcellular transport, promotes Drp1 assembly, and modulates the mitochondrial fission (61).
2.3.2 MFN1/2
MFN1/2 undergoes multiple post-translational modifications, including oxidation, ubiquitination, and phosphorylation. The redox state of specific residues in MFN can regulate its activity and promote membrane fusion by triggering oligomerization (65). During stress conditions, March5 activates the ubiquitination of acetylated MFN1, which marks it for degradation via the proteasome (66). In contrast, the histone deacetylase 6 augments MFN1-mediated mitochondrial fusion, particularly in response to oxidative stress (67). March5 specifically targets mitochondrial MFN2 for ubiquitination, promoting its oligomerization and strengthening mitochondria-ER tethering (68). In the context of cell apoptotic, the E3 ubiquitin ligase Parkin targets MFN1 for ubiquitination (69) and the activity of the deubiquitinating enzyme USP30 can reverse this process, which reactivates the mitochondrial fission process (70). Phosphorylation of MFNs can either promote or inhibit mitochondrial fusion, depending on the specific site and kinase involved. For example, ERK-mediated phosphorylation of the HR1 domain in MFN1 can suppress fusion and promote its interaction with Bak, triggering apoptosis (71). Cellular stress stimulates Jnk to phosphorylate MFN2 and the subsequent recruitment ofHuwe1 to MFN2, a critical step required for its proteasomal degradation and subsequent apoptosis activation (72).
2.3.3 Opa1
Studies have shown that the Opa1 is the only dynamin-like GTPase detected within the IMM. The MFN1 is by Opa1 to enhance mitochondrial fusion (52). Similarly, the Opa1’s activity is influenced by several post-translational modifications. However, the exact mechanisms need to be clarified through further studies.
The balance between mitochondrial fusion and fission benefits cells by regulating mitochondrial shape, enabling content exchange, ensuring fair mitochondrial inheritance, preserving healthy mtDNA, and eliminating damaged mitochondria (73). These structural changes in mitochondria may lead to the development of diseases by impairing the expression levels of proteins involved in mitochondrial dynamics. Moreover, abnormal activation of the signaling pathways can alter the mitochondrial dynamics (74). Previous studied investigations have shown that bacterial, viral, and parasitic pathogens can also modify host mitochondrial dynamics upon cell infection, facilitating their proliferation, significantly influencing disease outcomes (75). The subsequent mitochondrial dysfunction may activate intracellular inflammatory signaling pathways triggering the release of inflammatory factors (76). Mitochondrial dynamics contribute to the activation of immune cells (77). Specifically, the Drp1-mediated mitochondrial fission alters the T-cell activity, making it an important regulator of diverse autoimmune inflammatory diseases (78).
3 Mitophagy
Mitophagy is a selective autophagy mechanism known to maintain cellular homeostasis by eliminating damaged mitochondria. This process is initiated by mitochondrial depolarization and is especially critical for highly differentiated post-mitotic cells, which are largely dependent on aerobic metabolism (79). This process is a major regulator of mitochondrial quality control, which prevents the accumulation of potentially harmful mitochondria that initiates excessive inflammatory responses (80). Defective mitophagy can lead to inflammatory and autoimmune diseases by disrupting inflammatory cytokine secretion and immune cell function. Currently, there are two types of mitophagy pathways: the ubiquitin-dependent pathway and the non-ubiquitin-dependent pathway (Figure 2).
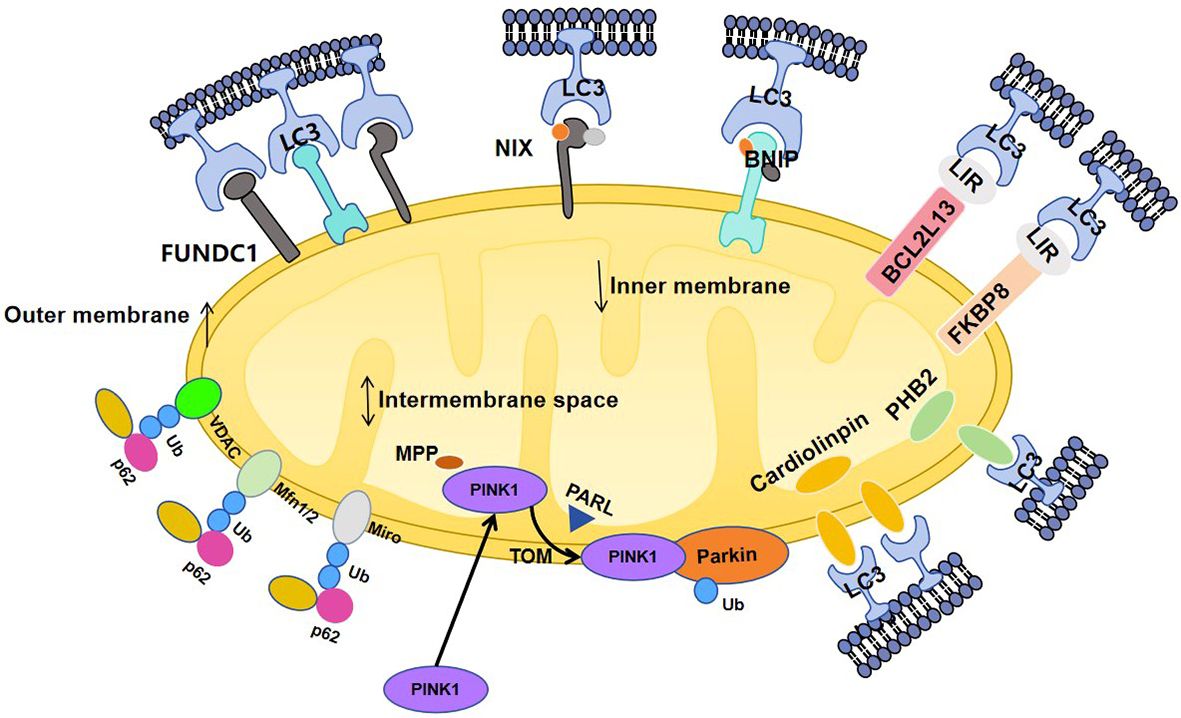
Figure 2. 1) Pink1 is sequentially targeted to the mitochondria via a targeting sequence and is degraded by the matrix processing peptidase (MPP) and subsequently cleaved by the IMM protease progerin-associated rhodopsin-like enzyme (PARL). Pink1 accumulates on the OMM via the regulation of the enzyme translocase of the outer membrane (TOM). Accumulated pink1 is autophosphorylated and activated through autophosphorylation which then phosphorylates ubiquitin on serine 65 (Ser65), triggering the recruitment of Parkin to the mitochondrial membrane. PINK1 and its substrate, ubiquitinates, phosphorylates and activates Parkin. Specifically, polyubiquitination of Parkin substrates, such as voltage-dependent anion channel-1 (VDAC1), mfn1/2, and Miro 1, induces their degradation by the proteasome. 2) Bcl2 like 13 (BCL2L13) is the mammalian homolog of atg32. In mammalian cells, BCL2L13 facilitates mitosis independently of Parkin. Like other LC3 receptors, BCL2L13 is located on the outer mitochondrial membrane and binds to LC3 via the LIR motif. Specifically, phosphorylation of the Ser272 site enhances the binding of BCL2L13 to the LC3.FK506-binding protein 8 (FKBP8). FKBP8 is located on the outer mitochondrial membrane and stimulates mitochondrial autophagy by interacting with LC3A; 3) Cardiolipin is an inner mitochondrial membrane lipid, and PHB2 are IMM proteins, which bind to the LC3 receptor to initiate mitophagy.
3.1 Pink1-Parkin
Autophagy receptors involved in the mitochondrial protein ubiquitination function through the ubiquitin-dependent mitophagy pathway, specifically the serine/threonine kinase Pink1/Parkin pathway (81). Once Pink1 is exposed on damaged mitochondria, it initiates Pink1/Parkin-dependent ubiquitination, which is often triggered by a loss of mitochondrial membrane potential (82). The expression of Pink1 is kept at low levels under physiological conditions, however, during mitochondrial damage, including mtDNA mutations, elevated mitochondria ROS (mtROS), and the buildup of misfolded proteins, the Pink1 accumulates at the OMM. This is followed by autophosphorylation and activation, as well as phosphorylation of ubiquitin on serine 65, promoting the recruiting Parkin from the cytoplasm to the mitochondrial membrane (83). Next, the Parkin is activated inducing the ubiquitination of mitochondrial proteins and triggering mitophagy (84). Even being absence in Pink1, Parkin could still be attracted to depolarized mitochondria, aiding in mitophagy. For example, HtrA2/Omi or LRRK2 can phosphorylate mitochondrial proteins or parkin itself to promote mitophagy under conditions of pink1 deficiency (85, 86). Alternatively, overexpressed or hyperactive FUNDC1 can recruit parkin to initiate autophagy (87). Among the known targets of Parkin ligase on the OMM are MFN1/2, voltage-dependent anion channel protein 1 (VDAC1), and mitochondrial GTPases (88–90). Proteomics analyses on the degradation of OMM components and the reorganization of the OMM proteome are highly advocated to increase our understanding of the process of mitophagy. Being the most abundant OMM protein, VDAC1 forms the mitochondrial pore and is a key modulator of metabolites, ions, and water transport under physiological conditions and influences mitochondrial homeostasis (91–93). The VDAC1 participates in the regulation of mitophagy, interacting with Parkin to facilitate polyubiquitination and recruitment of Parkin to induce mitophagy (94). By energizing the Pink1/Parkin mitophagy pathway, VDAC1 in conjunction with BNIP3 alters the mitophagic flux process (95). Hypoxia-induced GPCPD1 depalmitoylation has been reported to initiate mitophagy by regulating the PRKN-mediated VDAC1 ubiquitination (96). Application of the antidepressant drug sertraline was found to alter the VDAC1 protein, decrease ATP levels, activate AMPK, and inhibit the MTOR signaling pathway to induce autophagy (97).
3.2 NIX/BNIP3/FUNDC1 receptor-mediated
NIX and BNIP3 belong to the Bcl-2 protein family which is located on the OMM. The Bcl-2 family members on the OMM participate in the initiation of cell apoptosis, influencing the mitophagy process (98, 99). One of the mechanisms by which autophagy activates mitophagy is through the direct binding to the light chain 3 (LC3) via the BH3 structural motif. This motif functions as a molecular effector of mitochondrial membrane depolarization-induced hypoxia which promotes occurrence of mitophagy (100). NIX was reported to act as an adaptor protein that transports the components of the autophagy machinery to the mitochondria to trigger mitophagy. Another proposed model suggests that BNIP3 or NIX competes with Beclin-1 for binding to Bcl-xl. During erythropoiesis, the expression of NIX is upregulated, disrupting the Bcl-xl-Beclin-1 interaction and freeing Beclin-1 to trigger autophagy (101). Notably, the BNIP3 was demonstrated to lower the mTOR activity and regulate autophagy by increasing LC3 expression. Similar to BNIP3 and NIX, FUNDC1 is a mitochondrial junction protein induced by hypoxia that bind with LC3 via its LIR motif. Under hypoxic environments, the ULK1 kinase relocates to mitochondria, phosphorylating FUNDC1, enhancing its binding with LC3 to promote mitophagy (102).
Most researchers agree that mitophagy is closely linked to mitochondrial dynamics. Inhibiting fission with DRP1K38A or FIS1 RNAi reduces mitophagy, while overexpressing Opa1 also inhibits autophagy (103). Impaired fusion results in a reduction in IMM potential, triggering pink1 accumulation and parkin activation. However, the inverse dependence of fusion and autophagy on membrane potential makes them complementary rather than competing processes for daughter mitochondria after a fission event (104). Therefore, we suggest that mitochondrial fission is a prerequisite for mitophagy and that fusion will inhibit autophagy. However, inhibiting mitochondrial hyperfusion by silencing Drp1 or Mff does not affect mitophagy induced by Fis1 loss (105). In future, researchers should aim to explore the crosstalk between mitochondrial dynamics and mitochondrial autophagy. Moreover, several proteins involved in mitochondrial dynamics also participate in mitochondrial autophagy process. MFN1/2 is extracted from the OMM using ubiquitin-dependent chaperones and is subsequently degraded by the proteasome (106). The deletion of ubiquitination of MFN1/2 prevents the fusion of damaged mitochondria, while enhancing the fission, thereby promoting mitophagy (107). Additionally, Pink1 phosphorylates MFN2, which acts as a Parkin receptor to eliminate impaired mitochondria (108). However, it is unclear whether the OMM proteins coordinate both mitochondrial fission and mitophagy. The precise interplay between mitochondrial dynamics and autophagy requires further investigation.
4 Relationship between mitochondrial dynamics and UC
Previous studies have demonstrated changes in GTPase mRNA expression, such as Drp1, Opa1, and mitophagy in UC. Mancini et al. reported that in DSS-versus DNBS-treated mice with intestinal inflammation, the mRNA levels of Drp1 and Fis1 were elevated, triggering excessive mitochondrial fission (18). The expression levels of MFN1, MFN2, and Opa1 proteins were found to be downregulated in the intestinal epithelial cells of UC patients and DSS-induced mice, which weakens the mitochondrial fusion capacity and triggers mitochondrial dysfunction (109). Restoring the expression of these proteins may improve mitochondrial function in UC. In UC mice, mitochondria in the subnuclear region of inflammatory cells appear swollen and fragmented. A strong correlation exists between inflammation and the mitochondrial network disruption in colitis. The mitochondrial network is also disrupted in non-inflamed colonic regions, suggesting that the mitochondrial structure may be an early event in UC (17). Excessive mitochondrial fission and reduced fusion can stimulate the development of UC by altering energy metabolism, oxidative stress, and apoptosis (Figure 3).
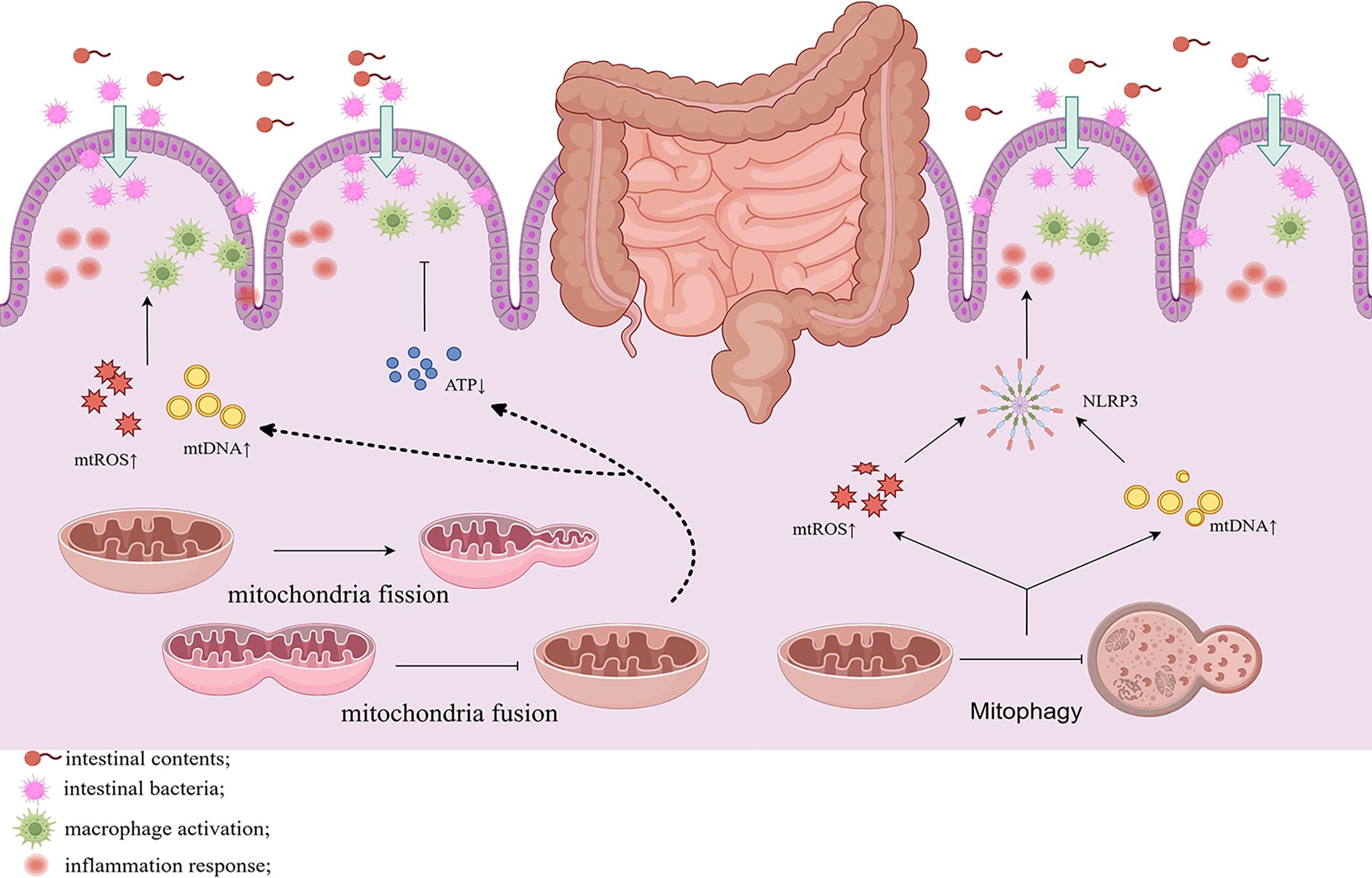
Figure 3. Altered mitochondrial dynamics of the intestinal cells in UC induces mitochondrial fission and inhibits fusion while attenuating mitochondrial autophagy. This results in the reduction of ATP production and promotes the release of mtDNA, mtROS, and activation of NLRP3 inflammasome, leading to inflammation and disruption of the intestinal barrier. Chronic inflammation and gut barrier disruption also activate mitochondrial fission and reduces autophagy, establishing a vicious cycle.
4.1 Changes in energy metabolism
As early as 1980, Roediger et al. postulated that IBD, particularly UC, may be an energy-deficient disease (19). Since then, several studies have demonstrated that patients with UC exhibited reduced mitochondrial function, including structural abnormalities, mtDNA mutations, reduced electron transport chain activity, decreased oxidative phosphorylation, and lower ATP concentrations (110–113). Mitochondrial dysfunction and imbalance in dynamics are often accompanied with impaired energy metabolism, which triggers inflammation and gastrointestinal symptoms (114). The colonic epithelial cells consume a high amount of energy to execute various processes, including the electrolyte exchange in the intestinal epithelium, glycoprotein synthesis in the mucus layer, lipid synthesis in cell membranes, structural protein synthesis, and detoxification. Therefore, energy deprivation may impair these processes, causing epithelial cell atrophy in the short term and colonic mucosal barrier damage in the long-run, leading to the development of UC (115, 116). Compared to healthy donors, oxidative phosphorylation metabolism in UC patients shifts towards glycolysis as a strategy to compensate for ATP production defects. This increases the cellular levels of lactate acid within colonic cells, whereas bacteria in the intestinal lumen consume epithelial oxygen, inducing ecological dysregulation which promotes inflammation (117). Cells with inflammatory colitis may use glycolysis to produce ATP as an adaptive response to butyrate transport and oxidative stress. A reduction in ATP levels induce detrimental effects on the actin cytoskeleton, whereas actin irregularities may impair the localization and function of cellular junctions, increased gaps between cells (118, 119). Lan A et al. demonstrated that in DSS-induced enterocolitis mouse model, the ATP activity was enhanced, and the expression of energy-dependent differentiation markers was upregulated. This was accompanied by enhanced epithelial repair, requiring large amounts of energy to enhance the structural and functional recovery of the colonic mucosa. This process has been linked to the increase in expression levels of regulators of mitochondrial mass and biogenesis, such as Sirt3, FoxO3, and PGCα. It is also characterized by enhanced fusion, as evidenced by the upregulated Mfn2 gene expression and good mitochondrial morphology (120). In previous studies, creatine supplementation or administration of energy sensors improved intestinal epithelial repair. Therefore, strategies for promoting cellular energy metabolism via modulating mitochondrial dynamics and intestinal barrier are potential treatments for UC (121, 122).
4.2 Increased oxidative stress
Impaired mitochondrial dynamics have been linked to the development of oxidative stress in UC patients. Mitochondrial aerobic metabolism promotes the proliferation and differentiation of intestinal stem cells within colonic crypts and supports the synthesis of intestinal epithelial cells (123). However, excessive production of ROS may occur in damaged mitochondria. Under normal conditions, mitochondria exhibit a well-coordinated self-repair mechanism. However, in cases of a dynamics imbalance, oxidative stress levels increase within the cells, triggering an inflammatory response (124). Excessive mitochondrial fission reduces energy production and leads to the generation of oxidative stress, increasing mitochondrial fission, and reducing cell viability (125). Mitochondrial fusion may trigger the mixing of mitochondrial contents, including DNA and respiratory chain complexes, which are involved in the repair of damaged mitochondria and reduce localized oxidative stress levels. However, a decrease in mitochondrial fusion may increase the development of oxidative stress (126). Studies have shown that gut microbiota and their metabolites participate in this process. Colic acid, a major biofilm component of Escherichia coli (E. coli), increases mitochondrial fission in enterocytes in a Drp1-dependent manner, enhancing the stress-activated transcription factor-mediated unfolded protein response (UPR) in response to mitochondrial stress (127). DSS can cause damage to the intestinal epithelial respiration and stimulate the mitochondrial complexes I, II, and IV, leading to the accumulation of damaged mitochondria and ROS, both of which cause cellular damage (128).
The mitochondrial chaperone protein Prohibitin 1 (PHB1), an endomembrane protein, regulates Opa1-mediated fusion within the IMM (129). Mitochondrial dysfunction in intestinal epithelial cells, induced by PHB1 deletion was found to trigger spontaneous ileitis in mice (130, 131). In the study, it was observed that epithelial cells showed mitochondrial dysfunction, crypt cell death, and Paneth cell abnormalities, which are also observed in the intestinal epithelium of patients with IBD. Treatment with the mitochondria-targeted antioxidant Mito-Tempo ameliorated the Paneth cell defects and reduced intestinal epithelial inflammation in mice lacking PHB1. The role of Paneth cell mitochondrial impairment in the development of ileitis has been documented in multiple studies on PHB1-deficient mice (131).
4.3 Cell apoptosis
The mitochondrial dynamics have been implicated in the regulation of apoptosis. Excessive mitochondrial fission can induce morpho-functional changes in mitochondria, which triggers the production of intrinsic apoptotic signals. Studies have demonstrated that inhibition of mitochondrial fusion stimulates that intracellular stress response and enhances apoptosis (132). The pathogenic E. coli secreted effector proteins, mitochondria-associated protein(Map), and E. coli secreted protein (EapF) are within the mitochondrial matrix. Map induces mitochondrial fission, while EspF promotes the permeabilization of the mitochondrial outer membrane (MOMP) in the OMM (133). Changes in mitochondrial membrane permeabilization may be accompanied by the release of pro-apoptotic factors, such as cytochrome c, which activate the apoptotic signaling pathways (134). Mitochondrial fission resulting from MFN deficiency in HeLa cells protects against Lactobacillus monocytogenes, suggesting that Listeria hemolysin O can disrupt mitochondrial dynamics and promote apoptosis via disrupting calcium influx. In multiple cell types, enhanced mitochondrial fusion inhibits the propagation of apoptotic signals, delaying cellular apoptosis (135). This phenomenon provides a potential alternative approach for treating UC.
In neurological diseases, dysregulation of Drp1 disrupts mitochondrial dynamics, stimulating inflammation and exacerbating disease severity (136). In pancreatic islet inflammatory cells, NF-κB activation was reported to increase Opa1 expression, while interleukin-6 (IL-6) was found to increase Fis1 and downregulate MFN2, enhancing disease progression (137). In colitis, abnormal mitochondrial dynamics stimulate multiple inflammatory pathways, such as the NF-κB pathway, which activated the inflammatory response. Mitochondrial fission can also trigger apoptosis, compromising intestinal barrier function and promoting cellular inflammation. In future, researchers should explore the role of mitochondrial dynamics in UC to identify robust therapeutic disease.
5 Relationship between mitophagy and UC
Mitophagy provides an important mechanism for eliminating damaged mitochondria to maintain normal mitochondrial function. Excessive mitochondrial damage and mitophagy levels have been detected in the intestinal tissues of UC patients, which are correlated with high disease severity (138). In epithelial tissues of DSS-induced enterocolitis mice, the number of damaged mitochondria, autophagy and mtROS were recorded, which further activated the NLRP3 inflammasomes to promote inflammation (139). This indicates that the role of mitophagy in apoptosis is bidirectional. Excessive mitophagy can trigger energy depletion, thereby inducing apoptosis, whereas, suppression of mitophagy may potentially prevent the clearance of damaged mitochondria, increasing the generation of ROS levels and inflammation.
5.1 Autophagy gene and development of UC
Genome-wide association studies (GWAS) have identified several autophagy genes, including Atg16L1, Lrrk2, and Irgm, which influence the genetic susceptibility of IBD (140–142). Atg16L1 is a critical component of the mitophagy machinery involved in the regulation of immune responses and inflammation. It is part of the complex that cleaves the ubiquitin-like protein LC3 via a lipolytic mechanism, promoting autophagosome formation and activity. The Atg16L1 risk variant (Atg16L1T300A), was found to influence the risk of IBD, creating a caspase cleavage site that weakens the stability of the protein, thereby diminishing autophagy, particularly in the presence of TNF-α (143). Irgm, a human immune-associated GTPase, translocates to mitochondria, where it regulates mitochondrial division and induces mitophagy (144). Irgm1, a homologue of Irgm, participated in the development of intestinal inflammation in mice. Irgm1 knockout mice exhibited increased severity of inflammation following exposure to DSS. Mice with DSS knockout exhibited significant disruptions in Paneth cell positioning and granule structure, which contributed to mitophagy and autophagy impairment within the Irgm1-deficient enterocytes, including Paneth cells. These findings demonstrate that Irgm1 regulates acute inflammatory response in mouse intestines, probably by regulating autophagy, which modulates the normal Paneth cell function (145). Lrrk2 is a multifunctional protein with kinase and GTPase activity; with mutations in the Lrrk2 gene found to decrease mitochondrial numbers, impaired mitochondrial dynamics, and inhibition of the mitophagy pathway (146). In individuals with UC, the expression level of Lrrk2 were upregulated in peripheral blood samples. In preclinical animal models of UC, Lrrk2 knockout ameliorated the progression of DSS-induced enterocolitis, which was accompanied with the transition of the intestinal macrophages (MФs) to alternatively activated macrophages, promoting probiotic colonization and attenuating the onset and progression of colitis (147). This indicates that Lrrk2 plays a crucial role in the development of UC.
5.2 Mitophagy and immune inflammation response in UC
Mitophagy influences the innate and adaptive immune responses, thereby maintaining intestinal homeostasis (148). In previous studies, most studies focused on the intestinal immunity of macrophages and T cells. In UC patients and mice with DSS-induced colitis, the mitophagy of intestinal macrophages was significantly decreased. It was found that the expression of proteins associated with the Pink1/Parkin and NIX-mediated autophagy pathways was decreased. This was accompanied by the accumulation of mtROS and mtDNA, which were then released into the cytoplasm, activating the NLRP3 inflammasome and enhancing the maturation of IL-18 and IL-1β (149, 150). In a mouse model of T-cell metastatic colitis, the expression the Th1 transcription factor TBX21 and Th1 cells in the lamina propria of Pink1 knockout (Pink1KO) mice was higher compared to the levels of naïve T cells. Treatment with the Urolithin A (UA), a mitophagy agonist, inhibited the Th1 differentiation, reducing the formation of IFN-γ and IL-17, which suppresses the levels of the associated inflammatory response (151). This finding suggests that inhibiting inflammation via enhancing mitophagy may be an attractive strategy for alleviating enteritis symptoms. Several cytokines have been reported to modulate mitophagy. IL-10, a key anti-inflammatory factor, inhibits the metabolic shifts associated with inflammatory stimuli in macrophages, while maintaining mitochondrial integrity and function by inhibiting mTOR (152).
Mitochondria are important regulators of the innate immune signaling, with several immune effectors clustered on the OMM (153). Among then, MAVS which targets the OMM through its C-terminal transmembrane domain was demonstrated to activate the downstream signaling pathways via NF-κB to regulate IFN production, and its activation is modulated by mitochondrial dynamics (154, 155). Furthermore, studies have shown that depletion of autophagy proteins, such as LC3B and Beclin 1, results in the accumulation of damaged mitochondria and the translocation of mtDNA into the cytoplasm, activating inflammatory gene transcription. The activities of TOMM20 and HSP60, proteins forming part of the OMM and mitochondrial matrix, respectively, negatively correlate with autophagy. In LPS-treated UC mouse models, MODE-K cells and colonic tissues displayed increased expression of p62, TOMM20, and HSP60, causing autophagy inhibition and enhancing inflammation (156). The absence of Nix/BNIP3L-mediated mitophagy during PHB1 protein deficiency causing excessive production of ROS and activation of inflammation, which suggests that mitochondrial dysfunction may impair mitophagy (157). Studies investigating the effect of polystyrene nanoplastics (PS-NPs) on enterocolitis development revealed that the buildup of these plastics in mitochondria induced mitochondrial stress, initiating Pink1/Parkin-mediated mitophagy. Additionally, LC3 dots in Caco-2 cells co-localized with mitotracker-labeled mitochondria, indicating the initiation of mitophagy (158).
6 Current studies of potential therapeutic approaches to modulate both mitochondrial dynamics and mitophagy in UC
The recent advancements in research investigating potential strategies for modulating mitochondrial dynamics and mitophagy in UC have achieved significant progress. Despite the advances in UC treatment, many patients show poor response to biological therapies (159). Proper mitochondrial fission, fusion, and mitophagy are essential for mitochondrial health. Inhibition of excessive fission has been shown to enhance mitophagy with beneficial effects in animal models of myocardial infarction, pulmonary hypertension, ischemia-reperfusion injury, multiple sclerosis, and Huntington’s disease (160–164). Modulating this mechanism may help to control mitochondrial dysfunction in colitis. Pharmacological or natural products have been proposed to restore mitophagy homeostasis, promoting the clearance of irreversibly dysfunctional mitochondria, making them promising therapeutic approaches for UC.
6.1 Mitochondria-targeted therapy for UC
Suppressing excessive mitochondrial fission has been shown to accelerate mucosal healing in mice. Patients with UC exhibit increased mitochondrial fission, along with higher butyrate exposure in the environment surrounding colonic stem cells. Excessive mitochondrial fission inhibits stem cell proliferation by disrupting butyrate metabolism in colonic organoids. Mechanistically, results from various enzyme activity assays in colonoids have shown that excessive fission delays mucosal repair by increasing ROS leading to the inhibition of mitochondrial acetoacetyl CoA thiolase activity, which impairs butyrate metabolism. Therefore, the failure of antibiotics to promote mucosal healing in mice was reversed following the treatment of the mitochondrial fission antagonist P110 and exogenous butyrate (165). Further findings show that P110 suppresses excessive fission by blocking Drp1 binding to Fis1, significantly alleviating intestinal inflammation and reducing UC symptoms (18). Unfortunately, few drugs effectively inhibit mitochondrial fission in vivo. Mdivi-1 is one such drug, targeting Drp1, but it may induce unintended side effects on oxidative phosphorylation and ROS levels (23). Leflunomide, an antirheumatic drug that inhibits pyrimidine synthesis has been found to mitigate mitochondrial fission by enhancing fusion to alleviate the associated symptoms (166). In addition, Chen W and colleagues described several chemotherapeutic agents that target different mechanisms of mitochondrial fission and fusion. However, evidence-based data to support the therapeutic benefits of these agents in the treatment of UC (50).
6.2 Natural products on UC-related tissues through mitophagy
Natural products, such as curcumin, can treat diseases by targeting mitochondrial function. For instance, curcumin protects cartilage in osteoarthritis by stimulating AMPK/PINK1/Parkin-mediated mitophagy (167). Berberine protects glomerular podocytes by inhibiting Drp1-mediated mitochondrial fission and dysfunction (168). Natural products can regulate mitophagy making them potential treatments for UC-associated mitochondrial dysfunction (Table 1). To assess the therapeutic efficacy of these natural products and their potential side effects on tissues or cells, UC-associated models and in vitro experiments need to be performed to accelerate the clinical translation of these findings. Dipak Kumar Sahoodeng et al. described the effects of various natural antioxidant components on IBD (169). In the following section, we provide a summary of natural products that regulate UC by modulating mitochondrial dynamics and mitophagy.
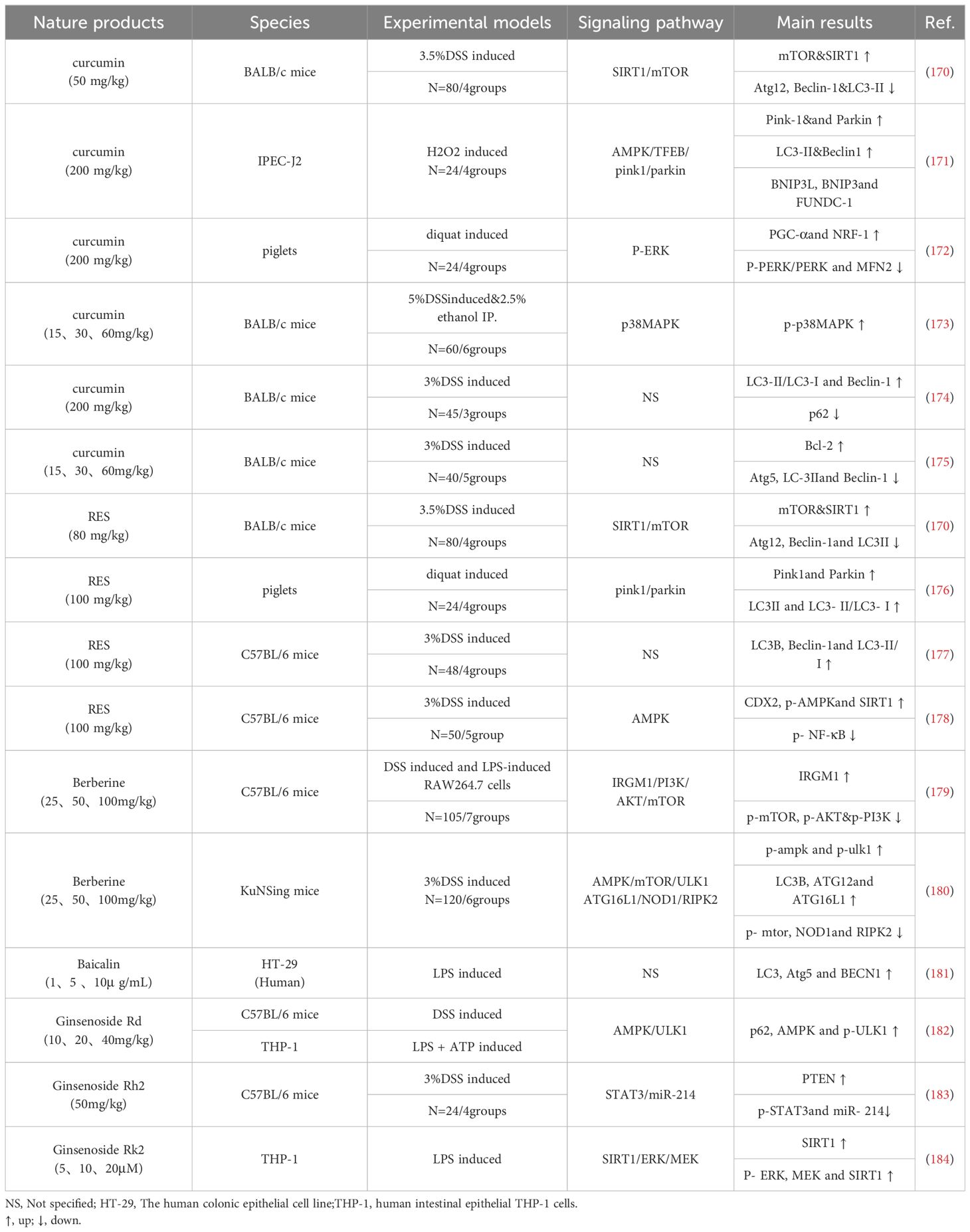
Table 1. Effects of natural products on UC-related tissues through mitophagy.
6.2.1 Curcumin with mitophagy on UC
Curcumin is the main active ingredient in dried, powdered rhizome of turmeric and has been found to exert beneficial effects against the development and progression of UC in animal and human trials (185, 186). A meta-analysis on the use of curcumin to treat UC comprising nine randomized controlled trials found that compared with the control group, the curcumin treatment increased the clinical remission rates (RR=2.28, 95%CI[1.43, 3.62], P=0.0005) and endoscopic remission rates (RR=1.66, 95%CI[1.07, 2.60], P=0.03), without inducing significant adverse events (187). Another study showed that it enhanced G2/M cell cycle arrest and autophagy and low doses of curcumin may activate adaptive stress responses, while high doses trigger acute responses (188). Shuting Cao et al. demonstrated that H2O2-induced epithelial barrier disruption and mitochondrial dysfunction models of UC in IPEC-J2 cells, curcumin treatment upregulated Pink1 and Parkin genes and proteins, but did not affect the expression of Nix, BNIP3, and FUNDC-1, necessitating the hypothesis that curcumin may activate mitophagy primarily via the Pink1-Parkin pathway (171). Other studies have demonstrated that curcumin abolished the diquat-induced oxidative stress and jejunal injury in piglets, enhanced the activity of complexes I-IV and suppressed the expression level of phosphorylated-PERK/PERK and phosphorylated-eIF2α/eIF2α, to improve the expression of mitochondrial function (172).
6.2.2 Resveratrol with mitophagy on UC
The RES is a natural polyphenol found in various plants and fruits (189). Dietary supplementation of RES improved intestinal barrier integrity, oxidative stress, and intestinal inflammation in a colitis model (190). The study by Shuting Cao et al. concluded that RES activated mitophagy which was indicated by the upregulated expression of Pink1, Parkin, and LC3-II/LC3-I relative to the piglets injected with diquat (176).
6.2.3 Berberine with mitophagy on UC
Berberine is a quaternary ammonium alkaloid primarily extracted from Coptis chinensis and Phellodendron amurense (191). It has the potential to treat various diseases such as cardiac aging and acute kidney injury via enhancing mitophagy (192, 193). Approximately, 211 potential targets of Berberine and 210 UC genes were predicted on the PharmMapper database whereas UC genes were determined on the GeneCards database and the OMIM database (194). Berberine targets IRGM1 to inhibit the PI3K/AKT/mTOR pathway, suppressing inflammatory response in UC (179). Similarly, Berberine was found to stimulate autophagy via the AMPK/MTOR/ULK1 pathway, as well as inhibit lysozyme and its secretion, to accelerate lysosomal maturation and expression, implying that it has the potential to treat inflammation (180).
6.2.4 Ginsenoside with mitophagy on UC
Ginsenoside is the most abundant active ingredient in the traditional Chinese medicine ginseng, with diverse structures. Evidence for its anti-fatigue, immunomodulation, and anti-tumor properties have been documented (195). In a meta-analysis comprising 15 studies with 300 animals, it was observed that ginsenosides significantly reduced the levels of pro-inflammatory factors (IL-β, IL-6, TNF-α) and upregulated the expression of the anti-inflammatory factors IL-10 and tight junction proteins (Zonula Occludens-1, occludin) (196). Elsewhere, oral administration of ginsenoside Rd alleviated the DSS-induced enteritis symptoms in a dose dependent-manner. In vitro, ginsenoside Rd significantly inhibited the NLRP3 inflammasome, enhancing the p62-dependent mitochondrial translocation and mitophagy predominantly through the AMPK/ULK1 signaling pathway (182).
These natural compounds have shown promising potential in treating UC. The cellular and animal models discussed in the above sections have uncovered the pivotal roles of natural products in enhancing mitophagy via pink1/parkin, AMPK/mTOR/ULK1, and SIRT1/ERK/MEK pathways, to prevent inflammation and enhance the repair of intestinal barrier. However, overstimulation or inhibition of mitochondrial function may result in detrimental effects, and thus, it is imperative to balance between mitochondrial dynamics and mitophagy to achieve UC treatment. Several animal model experiments have explored the mechanisms of chronic UC, identifying new targets for drug development. Compared with pharmaceutical drugs, natural ingredients have the advantages of being multi-target and eliciting fewer side effects. However, the efficacy of such drugs is based on findings from preclinical investigations, necessitating further well-designed clinical trials to verify their clinical efficacy and address the current limitations.
7 Concluding remarks and future perspectives
In conclusion, a strong link exists between intestinal barrier damage, chronic inflammation, and mitochondrial dysfunction in ulcerative colitis. Natural products targeting these processes show promise as potential treatments. However, several challenges remain to be solved. 1) Although the roles of mitochondrial dynamics and autophagy in UC have been recognized, the precise regulation of the balance between mitochondrial fission and autophagy is poorly understood. In addition, several differences exist mitochondrial function and autophagic response across different species, making it difficult to formulate appropriate targeting strategies. Most of the current studies are based on animal models, with few studies conducted in actual UC patients. 2) Although several natural products have been found to modulate mitochondrial autophagy, most of such studies involved in vitro experiments and small-scale animal models. Therefore, the clinical efficacy of natural products need to be validated because data from preclinical trials may not accurately reflect the clinical settings, in terms of the dosage, safety, and side effects. 3) Current treatments for UC often target mitochondrial fission or autophagy, but these approaches may not be specific to individual patient needs. How to select appropriate therapeutic targets and avoid over-regulation of mitochondrial function remains to be clarified.
In the future, clinical trials for various mitochondria-targeted therapies should be conducted to confirm the therapeutic efficacy of natural products discussed in this review. The trials should integrate a wide spectrum of data, including clinical profiles, gene mutations, and gut molecular signatures, to identify patient subgroups that are most likely to benefit. Moreover, an integrated approach is essential to address the inherent heterogeneity of UC and enhance the application of personalized treatment. This primary focus of this review was to enhance the current understanding of the complexity of UC and discuss the available treatments for specific patient subgroups.
Author contributions
JZ: Writing – original draft, Writing – review & editing. YX: Writing – review & editing. TW: Writing – review & editing. XZ: Writing – review & editing. JY: Writing – review & editing. LP: Writing – review & editing. HF: Writing – original draft, Writing – review & editing. CZ: Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the National Natural Science Foundation of China (82305250), the Natural Science Foundation of Sichuan Province of China (2022NSFSC1418).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Kobayashi T, Siegmund B, Le Berre C, Wei SC, Ferrante M, Shen B, et al. Ulcerative colitis. Nat Rev Dis Primers. (2020) 6:73. doi: 10.1038/s41572-020-00215-4
2. Agrawal M, Jess T. Implications of the changing epidemiology of inflammatory bowel disease in a changing world. United Eur Gastroenterol J. (2022) 10:1113–20. doi: 10.1002/ueg2.12317
3. Gros B, Kaplan GG. Ulcerative colitis in adults: A review. JAMA. (2023) 330:951–65. doi: 10.1001/jama.2023.15389
4. Le Berre C, Honap S, Peyrin-Biroulet L. Ulcerative colitis. Lancet. (2023) 402:571–84. doi: 10.1016/s0140-6736(23)00966-2
5. Nakase H, Sato N, Mizuno N, Ikawa Y. The influence of cytokines on the complex pathology of ulcerative colitis. Autoimmun Rev. (2022) 21:103017. doi: 10.1016/j.autrev.2021.103017
6. Amiot A, Chaibi S, Bouhnik Y, Serrero M, Filippi J, Roblin X, et al. Prevalence and determinants of fatigue in patients with IBD: A cross-sectional survey from the GETAID. J Crohns Colitis. (2023) 17:1418–25. doi: 10.1093/ecco-jcc/jjad060
7. Trujillo de la Fuente K, López Goméz J, Cortes Espinosa T, Perez-Cabeza de Vaca R, Paredes Amenabar C, Romero Lozania J, et al. P061 Impact of Inflammatory Bowel Disease on sleep quality in a Mexican population attended in a referral center. Am J Gastroenterol. (2021) 116:S16. doi: 10.14309/01.ajg.0000798844.44337.f4
8. Zhang J, Liu C, An P, Chen M, Wei Y, Li J, et al. Psychological symptoms and quality of life in patients with inflammatory bowel disease in China: A multicenter study. United Eur Gastroenterol J. (2024) 12:374–89. doi: 10.1002/ueg2.12532
9. Zhang J, Nie J, Zou M, Zeng Q, Feng Y, Luo Z, et al. Prevalence and associated factors of sexual dysfunction in patients with inflammatory bowel disease. Front Endocrinol (Lausanne). (2022) 13:881485. doi: 10.3389/fendo.2022.881485
11. Wangchuk P, Yeshi K, Loukas A. Ulcerative colitis: clinical biomarkers, therapeutic targets, and emerging treatments. Trends Pharmacol Sci. (2024) 45:892–903. doi: 10.1016/j.tips.2024.08.003
12. Barnes EL, Zhang X, Long MD, Herfarth HH, Kappelman MD. Rate of colectomy increases within 12 months of sequential therapy for ulcerative colitis. Am J Gastroenterol. (2023) 118:2080–3. doi: 10.14309/ajg.0000000000002331
13. Paik J. Ozanimod: A review in ulcerative colitis. Drugs. (2022) 82:1303–13. doi: 10.1007/s40265-022-01762-8
14. Sun T, Ren K, Xu G, Ma R, Wang X, Min T, et al. Plasma-activated solutions mitigates DSS-induced colitis via restoring redox homeostasis and reversing microbiota dysbiosis. Adv Sci (Weinh). (2023) 10:e2304044. doi: 10.1002/advs.202304044
15. Larabi A, Barnich N, Nguyen HTT. New insights into the interplay between autophagy, gut microbiota and inflammatory responses in IBD. Autophagy. (2020) 16:38–51. doi: 10.1080/15548627.2019.1635384
16. Andrieux P, Chevillard C, Cunha-Neto E, Nunes JPS. Mitochondria as a cellular hub in infection and inflammation. Int J Mol Sci. (2021) 22:11338. doi: 10.3390/ijms222111338
17. Chojnacki AK, Navaneetha Krishnan S, Jijon H, Shutt TE, Colarusso P, McKay DM. Tissue imaging reveals disruption of epithelial mitochondrial networks and loss of mitochondria-associated cytochrome-C in inflamed human and murine colon. Mitochondrion. (2023) 68:44–59. doi: 10.1016/j.mito.2022.10.004
18. Mancini NL, Goudie L, Xu W, Sabouny R, Rajeev S, Wang A, et al. Perturbed mitochondrial dynamics is a novel feature of colitis that can be targeted to lessen disease. Cell Mol Gastroenterol Hepatol. (2020) 10:287–307. doi: 10.1016/j.jcmgh.2020.04.004
19. Roediger WE. The colonic epithelium in ulcerative colitis: an energy-deficiency disease? Lancet. (1980) 2:712–5. doi: 10.1016/s0140-6736(80)91934-0
20. Graham DB, Xavier RJ. Pathway paradigms revealed from the genetics of inflammatory bowel disease. Nature. (2020) 578:527–39. doi: 10.1038/s41586-020-2025-2
21. Lahiri A, Hedl M, Abraham C. MTMR3 risk allele enhances innate receptor-induced signaling and cytokines by decreasing autophagy and increasing caspase-1 activation. Proc Natl Acad Sci U.S.A. (2015) 112:10461–6. doi: 10.1073/pnas.1501752112
22. Lahiri A, Hedl M, Yan J, Abraham C. Human LACC1 increases innate receptor-induced responses and a LACC1 disease-risk variant modulates these outcomes. Nat Commun. (2017) 8:15614. doi: 10.1038/ncomms15614
23. Bordt EA, Clerc P, Roelofs BA, Saladino AJ, Tretter L, Adam-Vizi V, et al. The putative drp1 inhibitor mdivi-1 is a reversible mitochondrial complex I inhibitor that modulates reactive oxygen species. Dev Cell. (2017) 40:583–94.e6. doi: 10.1016/j.devcel.2017.02.020
24. Rodrigues T, Reker D, Schneider P, Schneider G. Counting on natural products for drug design. Nat Chem. (2016) 8:531–41. doi: 10.1038/nchem.2479
25. Malpartida AB, Williamson M, Narendra DP, Wade-Martins R, Ryan BJ. Mitochondrial dysfunction and mitophagy in parkinson’s disease: from mechanism to therapy. Trends Biochem Sci. (2021) 46:329–43. doi: 10.1016/j.tibs.2020.11.007
26. Panigrahi DP, Praharaj PP, Bhol CS, Mahapatra KK, Patra S, Behera BP, et al. The emerging, multifaceted role of mitophagy in cancer and cancer therapeutics. Semin Cancer Biol. (2020) 66:45–58. doi: 10.1016/j.semcancer.2019.07.015
27. Wang FS, Kuo CW, Ko JY, Chen YS, Wang SY, Ke HJ, et al. Irisin mitigates oxidative stress, chondrocyte dysfunction and osteoarthritis development through regulating mitochondrial integrity and autophagy. Antioxidants (Basel). (2020) 9:810. doi: 10.3390/antiox9090810
28. Dorn GW 2nd. Evolving concepts of mitochondrial dynamics. Annu Rev Physiol. (2019) 81:1–17. doi: 10.1146/annurev-physiol-020518-114358
29. Yapa NMB, Lisnyak V, Reljic B, Ryan MT. Mitochondrial dynamics in health and disease. FEBS Lett. (2021) 595:1184–204. doi: 10.1002/1873-3468.14077
30. Margineantu DH, Gregory Cox W, Sundell L, Sherwood SW, Beechem JM, Capaldi RA. Cell cycle dependent morphology changes and associated mitochondrial DNA redistribution in mitochondria of human cell lines. Mitochondrion. (2002) 1:425–35. doi: 10.1016/s1567-7249(02)00006-5
31. Amiri M, Hollenbeck PJ. Mitochondrial biogenesis in the axons of vertebrate peripheral neurons. Dev Neurobiol. (2008) 68:1348–61. doi: 10.1002/dneu.20668
32. Tilokani L, Nagashima S, Paupe V, Prudent J. Mitochondrial dynamics: overview of molecular mechanisms. Essays Biochem. (2018) 62:341–60. doi: 10.1042/ebc20170104
33. Breitzig MT, Alleyn MD, Lockey RF, Kolliputi N. A mitochondrial delicacy: dynamin-related protein 1 and mitochondrial dynamics. Am J Physiol Cell Physiol. (2018) 315:C80–c90. doi: 10.1152/ajpcell.00042.2018
34. Zhan M, Brooks C, Liu F, Sun L, Dong Z. Mitochondrial dynamics: regulatory mechanisms and emerging role in renal pathophysiology. Kidney Int. (2013) 83:568–81. doi: 10.1038/ki.2012.441
35. Yu R, Lendahl U, Nistér M, Zhao J. Regulation of mammalian mitochondrial dynamics: opportunities and challenges. Front Endocrinol (Lausanne). (2020) 11:374. doi: 10.3389/fendo.2020.00374
36. Rowland AA, Voeltz GK. Endoplasmic reticulum-mitochondria contacts: function of the junction. Nat Rev Mol Cell Biol. (2012) 13:607–25. doi: 10.1038/nrm3440
37. Gandre-Babbe S, van der Bliek AM. The novel tail-anchored membrane protein Mff controls mitochondrial and peroxisomal fission in mammalian cells. Mol Biol Cell. (2008) 19:2402–12. doi: 10.1091/mbc.e07-12-1287
38. Otera H, Wang C, Cleland MM, Setoguchi K, Yokota S, Youle RJ, et al. Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J Cell Biol. (2010) 191:1141–58. doi: 10.1083/jcb.201007152
39. Losón OC, Meng S, Ngo H, Liu R, Kaiser JT, Chan DC. Crystal structure and functional analysis of MiD49, a receptor for the mitochondrial fission protein Drp1. Protein Sci. (2015) 24:386–94. doi: 10.1002/pro.2629
40. Osellame LD, Singh AP, Stroud DA, Palmer CS, Stojanovski D, Ramachandran R, et al. Cooperative and independent roles of the Drp1 adaptors Mff, MiD49 and MiD51 in mitochondrial fission. J Cell Sci. (2016) 129:2170–81. doi: 10.1242/jcs.185165
41. Ciarlo L, Manganelli V, Garofalo T, Matarrese P, Tinari A, Misasi R, et al. Association of fission proteins with mitochondrial raft-like domains. Cell Death Differ. (2010) 17:1047–58. doi: 10.1038/cdd.2009.208
42. Lee JE, Westrate LM, Wu H, Page C, Voeltz GK. Multiple dynamin family members collaborate to drive mitochondrial division. Nature. (2016) 540:139–43. doi: 10.1038/nature20555
43. Ramachandran R. Mitochondrial dynamics: The dynamin superfamily and execution by collusion. Semin Cell Dev Biol. (2018) 76:201–12. doi: 10.1016/j.semcdb.2017.07.039
44. Figueroa-Romero C, Iñiguez-Lluhí JA, Stadler J, Chang CR, Arnoult D, Keller PJ, et al. SUMOylation of the mitochondrial fission protein Drp1 occurs at multiple nonconsensus sites within the B domain and is linked to its activity cycle. FASEB J. (2009) 23:3917–27. doi: 10.1096/fj.09-136630
45. Gawlowski T, Suarez J, Scott B, Torres-Gonzalez M, Wang H, Schwappacher R, et al. Modulation of dynamin-related protein 1 (DRP1) function by increased O-linked-β-N-acetylglucosamine modification (O-GlcNAc) in cardiac myocytes. J Biol Chem. (2012) 287:30024–34. doi: 10.1074/jbc.M112.390682
46. Ko AR, Hyun HW, Min SJ, Kim JE. The differential DRP1 phosphorylation and mitochondrial dynamics in the regional specific astroglial death induced by status epilepticus. Front Cell Neurosci. (2016) 10:124. doi: 10.3389/fncel.2016.00124
47. Nakamura N, Kimura Y, Tokuda M, Honda S, Hirose S. MARCH-V is a novel mitofusin 2- and Drp1-binding protein able to change mitochondrial morphology. EMBO Rep. (2006) 7:1019–22. doi: 10.1038/sj.embor.7400790
48. Mitra K, Wunder C, Roysam B, Lin G, Lippincott-Schwartz J. A hyperfused mitochondrial state achieved at G1-S regulates cyclin E buildup and entry into S phase. Proc Natl Acad Sci U.S.A. (2009) 106:11960–5. doi: 10.1073/pnas.0904875106
49. Xie JH, Li YY, Jin J. The essential functions of mitochondrial dynamics in immune cells. Cell Mol Immunol. (2020) 17:712–21. doi: 10.1038/s41423-020-0480-1
50. Chen W, Zhao H, Li Y. Mitochondrial dynamics in health and disease: mechanisms and potential targets. Signal Transduct Target Ther. (2023) 8:333. doi: 10.1038/s41392-023-01547-9
51. Brandt T, Cavellini L, Kühlbrandt W, Cohen MM. A mitofusin-dependent docking ring complex triggers mitochondrial fusion in vitro. Elife. (2016) 5:e14618. doi: 10.7554/eLife.14618
52. Cipolat S, Martins de Brito O, Dal Zilio B, Scorrano L. OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proc Natl Acad Sci U S A. (2004) 101:15927–32. doi: 10.1073/pnas.0407043101
53. Nyenhuis SB, Wu X, Strub MP, Yim YI, Stanton AE, Baena V, et al. OPA1 helical structures give perspective to mitochondrial dysfunction. Nature. (2023) 620:1109–16. doi: 10.1038/s41586-023-06462-1
54. von der Malsburg A, Sapp GM, Zuccaro KE, von Appen A, Moss FR 3rd, Kalia R, et al. Structural mechanism of mitochondrial membrane remodelling by human OPA1. Nature. (2023) 620:1101–8. doi: 10.1038/s41586-023-06441-6
55. Olichon A, Baricault L, Gas N, Guillou E, Valette A, Belenguer P, et al. Loss of OPA1 perturbates the mitochondrial inner membrane structure and integrity, leading to cytochrome c release and apoptosis. J Biol Chem. (2003) 278:7743–6. doi: 10.1074/jbc.C200677200
56. van der Bliek AM, Shen Q, Kawajiri S. Mechanisms of mitochondrial fission and fusion. Cold Spring Harb Perspect Biol. (2013) 5:a011072. doi: 10.1101/cshperspect.a011072
57. Han H, Tan J, Wang R, Wan H, He Y, Yan X, et al. PINK1 phosphorylates Drp1(S616) to regulate mitophagy-independent mitochondrial dynamics. EMBO Rep. (2020) 21:e48686. doi: 10.15252/embr.201948686
58. Taguchi N, Ishihara N, Jofuku A, Oka T, Mihara K. Mitotic phosphorylation of dynamin-related GTPase Drp1 participates in mitochondrial fission. J Biol Chem. (2007) 282:11521–9. doi: 10.1074/jbc.M607279200
59. Chang CR, Blackstone C. Cyclic AMP-dependent protein kinase phosphorylation of Drp1 regulates its GTPase activity and mitochondrial morphology. J Biol Chem. (2007) 282:21583–7. doi: 10.1074/jbc.C700083200
60. Chou CH, Lin CC, Yang MC, Wei CC, Liao HD, Lin RC, et al. GSK3beta-mediated Drp1 phosphorylation induced elongated mitochondrial morphology against oxidative stress. PloS One. (2012) 7:e49112. doi: 10.1371/journal.pone.0049112
61. Karbowski M, Neutzner A, Youle RJ. The mitochondrial E3 ubiquitin ligase MARCH5 is required for Drp1 dependent mitochondrial division. J Cell Biol. (2007) 178:71–84. doi: 10.1083/jcb.200611064
62. Calle X, Garrido-Moreno V, Lopez-Gallardo E, Norambuena-Soto I, Martínez D, Peñaloza-Otárola A, et al. Mitochondrial E3 ubiquitin ligase 1 (MUL1) as a novel therapeutic target for diseases associated with mitochondrial dysfunction. IUBMB Life. (2022) 74:850–65. doi: 10.1002/iub.2657
63. Prudent J, Zunino R, Sugiura A, Mattie S, Shore GC, McBride HM. MAPL SUMOylation of drp1 stabilizes an ER/mitochondrial platform required for cell death. Mol Cell. (2015) 59:941–55. doi: 10.1016/j.molcel.2015.08.001
64. Zemirli N, Pourcelot M, Ambroise G, Hatchi E, Vazquez A, Arnoult D. Mitochondrial hyperfusion promotes NF-κB activation via the mitochondrial E3 ligase MULAN. FEBS J. (2014) 281:3095–112. doi: 10.1111/febs.12846
65. Mattie S, Riemer J, Wideman JG, McBride HM. A new mitofusin topology places the redox-regulated C terminus in the mitochondrial intermembrane space. J Cell Biol. (2018) 217:507–15. doi: 10.1083/jcb.201611194
66. Park YY, Nguyen OT, Kang H, Cho H. MARCH5-mediated quality control on acetylated Mfn1 facilitates mitochondrial homeostasis and cell survival. Cell Death Dis. (2014) 5:e1172. doi: 10.1038/cddis.2014.142
67. Lee JY, Kapur M, Li M, Choi MC, Choi S, Kim HJ, et al. MFN1 deacetylation activates adaptive mitochondrial fusion and protects metabolically challenged mitochondria. J Cell Sci. (2014) 127:4954–63. doi: 10.1242/jcs.157321
68. Sugiura A, Nagashima S, Tokuyama T, Amo T, Matsuki Y, Ishido S, et al. MITOL regulates endoplasmic reticulum-mitochondria contacts via Mitofusin2. Mol Cell. (2013) 51:20–34. doi: 10.1016/j.molcel.2013.04.023
69. Escobar-Henriques M, Langer T. Dynamic survey of mitochondria by ubiquitin. EMBO Rep. (2014) 15:231–43. doi: 10.1002/embr.201338225
70. Cunningham CN, Baughman JM, Phu L, Tea JS, Yu C, Coons M, et al. USP30 and parkin homeostatically regulate atypical ubiquitin chains on mitochondria. Nat Cell Biol. (2015) 17:160–9. doi: 10.1038/ncb3097
71. Pyakurel A, Savoia C, Hess D, Scorrano L. Extracellular regulated kinase phosphorylates mitofusin 1 to control mitochondrial morphology and apoptosis. Mol Cell. (2015) 58:244–54. doi: 10.1016/j.molcel.2015.02.021
72. Leboucher GP, Tsai YC, Yang M, Shaw KC, Zhou M, Veenstra TD, et al. Stress-induced phosphorylation and proteasomal degradation of mitofusin 2 facilitates mitochondrial fragmentation and apoptosis. Mol Cell. (2012) 47:547–57. doi: 10.1016/j.molcel.2012.05.041
73. Chan DC. Mitochondrial dynamics and its involvement in disease. Annu Rev Pathol. (2020) 15:235–59. doi: 10.1146/annurev-pathmechdis-012419-032711
74. Whitley BN, Engelhart EA, Hoppins S. Mitochondrial dynamics and their potential as a therapeutic target. Mitochondrion. (2019) 49:269–83. doi: 10.1016/j.mito.2019.06.002
75. Khan S, Raj D, Jaiswal K, Lahiri A. Modulation of host mitochondrial dynamics during bacterial infection. Mitochondrion. (2020) 53:140–9. doi: 10.1016/j.mito.2020.05.005
76. de Oliveira LG, Angelo YS, Iglesias AH, Peron JPS. Unraveling the link between mitochondrial dynamics and neuroinflammation. Front Immunol. (2021) 12:624919. doi: 10.3389/fimmu.2021.624919
77. Mills EL, Kelly B, O’Neill LAJ. Mitochondria are the powerhouses of immunity. Nat Immunol. (2017) 18:488–98. doi: 10.1038/ni.3704
78. Song J, Yi X, Gao R, Sun L, Wu Z, Zhang S, et al. Impact of drp1-mediated mitochondrial dynamics on T cell immune modulation. Front Immunol. (2022) 13:873834. doi: 10.3389/fimmu.2022.873834
79. Doblado L, Lueck C, Rey C, Samhan-Arias AK, Prieto I, Stacchiotti A, et al. Mitophagy in human diseases. Int J Mol Sci. (2021) 22:3903. doi: 10.3390/ijms22083903
80. Lemasters JJ. Variants of mitochondrial autophagy: Types 1 and 2 mitophagy and micromitophagy (Type 3). Redox Biol. (2014) 2:749–54. doi: 10.1016/j.redox.2014.06.004
81. Johansen T, Lamark T. Selective autophagy: ATG8 family proteins, LIR motifs and cargo receptors. J Mol Biol. (2020) 432:80–103. doi: 10.1016/j.jmb.2019.07.016
82. Youle RJ. Mitochondria-Striking a balance between host and endosymbiont. Science. (2019) 365:eaaw9855. doi: 10.1126/science.aaw9855
83. Heo JM, Ordureau A, Paulo JA, Rinehart J, Harper JW. The PINK1-PARKIN mitochondrial ubiquitylation pathway drives a program of OPTN/NDP52 recruitment and TBK1 activation to promote mitophagy. Mol Cell. (2015) 60:7–20. doi: 10.1016/j.molcel.2015.08.016
84. Nguyen TN, Padman BS, Lazarou M. Deciphering the molecular signals of PINK1/parkin mitophagy. Trends Cell Biol. (2016) 26:733–44. doi: 10.1016/j.tcb.2016.05.008
85. Singh F, Ganley IG. Parkinson’s disease and mitophagy: an emerging role for LRRK2. Biochem Soc Trans. (2021) 49:551–62. doi: 10.1042/bst20190236
86. Torii S, Kasai S, Yoshida T, Yasumoto KI, Shimizu S. Mitochondrial E3 ubiquitin ligase parkin: relationships with other causal proteins in familial parkinson’s disease and its substrate-involved mouse experimental models. Int J Mol Sci. (2020) 21:1202. doi: 10.3390/ijms21041202
87. Sugo M, Kimura H, Arasaki K, Amemiya T, Hirota N, Dohmae N, et al. Syntaxin 17 regulates the localization and function of PGAM5 in mitochondrial division and mitophagy. EMBO J. (2018) 37:e98899. doi: 10.15252/embj.201798899
88. Eberhardt EL, Ludlam AV, Tan Z, Cianfrocco MA. Miro: A molecular switch at the center of mitochondrial regulation. Protein Sci. (2020) 29:1269–84. doi: 10.1002/pro.3839
89. McLelland GL, Goiran T, Yi W, Dorval G, Chen CX, Lauinger ND, et al. Mfn2 ubiquitination by PINK1/parkin gates the p97-dependent release of ER from mitochondria to drive mitophagy. Elife. (2018) 7:e32866. doi: 10.7554/eLife.32866
90. Shoshan-Barmatz V, Nahon-Crystal E, Shteinfer-Kuzmine A, Gupta R. VDAC1, mitochondrial dysfunction, and Alzheimer’s disease. Pharmacol Res. (2018) 131:87–101. doi: 10.1016/j.phrs.2018.03.010
91. Camara AKS, Zhou Y, Wen PC, Tajkhorshid E, Kwok WM. Mitochondrial VDAC1: A key gatekeeper as potential therapeutic target. Front Physiol. (2017) 8:460. doi: 10.3389/fphys.2017.00460
92. Lawen A, Ly JD, Lane DJ, Zarschler K, Messina A, De Pinto V. Voltage-dependent anion-selective channel 1 (VDAC1)–a mitochondrial protein, rediscovered as a novel enzyme in the plasma membrane. Int J Biochem Cell Biol. (2005) 37:277–82. doi: 10.1016/j.biocel.2004.05.013
93. Martel C, Wang Z, Brenner C. VDAC phosphorylation, a lipid sensor influencing the cell fate. Mitochondrion. (2014) 19 Pt A:69–77. doi: 10.1016/j.mito.2014.07.009
94. Ham SJ, Lee D, Yoo H, Jun K, Shin H, Chung J. Decision between mitophagy and apoptosis by Parkin via VDAC1 ubiquitination. Proc Natl Acad Sci U.S.A. (2020) 117:4281–91. doi: 10.1073/pnas.1909814117
95. Zhang Q, Chen C, Ma Y, Yan X, Lai N, Wang H, et al. PGAM5 interacts with and maintains BNIP3 to license cancer-associated muscle wasting. Autophagy. (2024) 20:1–16. doi: 10.1080/15548627.2024.2360340
96. Liu Y, Zhang H, Liu Y, Zhang S, Su P, Wang L, et al. Hypoxia-induced GPCPD1 depalmitoylation triggers mitophagy via regulating PRKN-mediated ubiquitination of VDAC1. Autophagy. (2023) 19:2443–63. doi: 10.1080/15548627.2023.2182482
97. Hwang HY, Shim JS, Kim D, Kwon HJ. Antidepressant drug sertraline modulates AMPK-MTOR signaling-mediated autophagy via targeting mitochondrial VDAC1 protein. Autophagy. (2021) 17:2783–99. doi: 10.1080/15548627.2020.1841953
98. Tummers B, Green DR. The evolution of regulated cell death pathways in animals and their evasion by pathogens. Physiol Rev. (2022) 102:411–54. doi: 10.1152/physrev.00002.2021
99. Youle RJ, Narendra DP. Mechanisms of mitophagy. Nat Rev Mol Cell Biol. (2011) 12:9–14. doi: 10.1038/nrm3028
100. Li Y, Zheng W, Lu Y, Zheng Y, Pan L, Wu X, et al. BNIP3L/NIX-mediated mitophagy: molecular mechanisms and implications for human disease. Cell Death Dis. (2021) 13:14. doi: 10.1038/s41419-021-04469-y
101. Zhang J, Ney PA. Role of BNIP3 and NIX in cell death, autophagy, and mitophagy. Cell Death Differ. (2009) 16:939–46. doi: 10.1038/cdd.2009.16
102. Wu W, Tian W, Hu Z, Chen G, Huang L, Li W, et al. ULK1 translocates to mitochondria and phosphorylates FUNDC1 to regulate mitophagy. EMBO Rep. (2014) 15:566–75. doi: 10.1002/embr.201438501
103. Twig G, Elorza A, Molina AJ, Mohamed H, Wikstrom JD, Walzer G, et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. (2008) 27:433–46. doi: 10.1038/sj.emboj.7601963
104. Twig G, Shirihai OS. The interplay between mitochondrial dynamics and mitophagy. Antioxid Redox Signal. (2011) 14:1939–51. doi: 10.1089/ars.2010.3779
105. Xian H, Yang Q, Xiao L, Shen HM, Liou YC. STX17 dynamically regulated by Fis1 induces mitophagy via hierarchical macroautophagic mechanism. Nat Commun. (2019) 10:2059. doi: 10.1038/s41467-019-10096-1
106. Escobar-Henriques M, Joaquim M. Mitofusins: disease gatekeepers and hubs in mitochondrial quality control by E3 ligases. Front Physiol. (2019) 10:517. doi: 10.3389/fphys.2019.00517
107. Tanaka A, Cleland MM, Xu S, Narendra DP, Suen DF, Karbowski M, et al. Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J Cell Biol. (2010) 191:1367–80. doi: 10.1083/jcb.201007013
108. Chen Y, Dorn GW 2nd. PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science. (2013) 340:471–5. doi: 10.1126/science.1231031
109. Zhang YF, Fan MY, Bai QR, Zhao R, Song S, Wu L, et al. Precision therapy for ulcerative colitis: insights from mitochondrial dysfunction interacting with the immune microenvironment. Front Immunol. (2024) 15:1396221. doi: 10.3389/fimmu.2024.1396221
110. Boyapati RK, Dorward DA, Tamborska A, Kalla R, Ventham NT, Doherty MK, et al. Mitochondrial DNA is a pro-inflammatory damage-associated molecular pattern released during active IBD. Inflammation Bowel Dis. (2018) 24:2113–22. doi: 10.1093/ibd/izy095
111. Ho GT, Aird RE, Liu B, Boyapati RK, Kennedy NA, Dorward DA, et al. MDR1 deficiency impairs mitochondrial homeostasis and promotes intestinal inflammation. Mucosal Immunol. (2018) 11:120–30. doi: 10.1038/mi.2017.31
112. Kameyama J, Narui H, Inui M, Sato T. Energy level in large intestinal mucosa in patients with ulcerative colitis. Tohoku J Exp Med. (1984) 143:253–4. doi: 10.1620/tjem.143.253
113. Özsoy M, Stummer N, Zimmermann FA, Feichtinger RG, Sperl W, Weghuber D, et al. Role of energy metabolism and mitochondrial function in inflammatory bowel disease. Inflammation Bowel Dis. (2022) 28:1443–50. doi: 10.1093/ibd/izac024
114. Alula KM, Jackson DN, Smith AD, Kim DS, Turner K, Odstrcil E, et al. Targeting mitochondrial damage as a therapeutic for ileal crohn’s disease. Cells. (2021) 10:1349. doi: 10.3390/cells10061349
115. Ahmad MS, Krishnan S, Ramakrishna BS, Mathan M, Pulimood AB, Murthy SN. Butyrate and glucose metabolism by colonocytes in experimental colitis in mice. Gut. (2000) 46:493–9. doi: 10.1136/gut.46.4.493
116. Guerbette T, Boudry G, Lan A. Mitochondrial function in intestinal epithelium homeostasis and modulation in diet-induced obesity. Mol Metab. (2022) 63:101546. doi: 10.1016/j.molmet.2022.101546
117. Lee JY, Cevallos SA, Byndloss MX, Tiffany CR, Olsan EE, Butler BP, et al. High-fat diet and antibiotics cooperatively impair mitochondrial bioenergetics to trigger dysbiosis that exacerbates pre-inflammatory bowel disease. Cell Host Microbe. (2020) 28:273–84.e6. doi: 10.1016/j.chom.2020.06.001
118. Mandel LJ, Doctor RB, Bacallao R. ATP depletion: a novel method to study junctional properties in epithelial tissues. II. Internalization of Na+,K(+)-ATPase and E-cadherin. J Cell Sci. (1994) 107:3315–24. doi: 10.1242/jcs.107.12.3315
119. Söderholm JD, Olaison G, Peterson KH, Franzén LE, Lindmark T, Wirén M, et al. Augmented increase in tight junction permeability by luminal stimuli in the non-inflamed ileum of Crohn’s disease. Gut. (2002) 50:307–13. doi: 10.1136/gut.50.3.307
120. Lan A, Guerbette T, Andriamihaja M, Magnin B, Bordet M, Ferron PJ, et al. Mitochondrial remodeling and energy metabolism adaptations in colonic crypts during spontaneous epithelial repair after colitis induction in mice. Free Radic Biol Med. (2023) 205:224–33. doi: 10.1016/j.freeradbiomed.2023.06.007
121. Olivier S, Diounou H, Pochard C, Frechin L, Durieu E, Foretz M, et al. Intestinal epithelial AMPK deficiency causes delayed colonic epithelial repair in DSS-induced colitis. Cells. (2022) 11:590. doi: 10.3390/cells11040590
122. Wallimann T, Hall CHT, Colgan SP, Glover LE. Creatine supplementation for patients with inflammatory bowel diseases: A scientific rationale for a clinical trial. Nutrients. (2021) 13:1429. doi: 10.3390/nu13051429
123. Rehman J. Empowering self-renewal and differentiation: the role of mitochondria in stem cells. J Mol Med (Berl). (2010) 88:981–6. doi: 10.1007/s00109-010-0678-2
124. Pokharel MD, Garcia-Flores A, Marciano D, Franco MC, Fineman JR, Aggarwal S, et al. Mitochondrial network dynamics in pulmonary disease: Bridging the gap between inflammation, oxidative stress, and bioenergetics. Redox Biol. (2024) 70:103049. doi: 10.1016/j.redox.2024.103049
125. Wang DK, Zheng HL, Zhou WS, Duan ZW, Jiang SD, Li B, et al. Mitochondrial dysfunction in oxidative stress-mediated intervertebral disc degeneration. Orthop Surg. (2022) 14:1569–82. doi: 10.1111/os.13302
126. Ikeda Y, Sciarretta S, Nagarajan N, Rubattu S, Volpe M, Frati G, et al. New insights into the role of mitochondrial dynamics and autophagy during oxidative stress and aging in the heart. Oxid Med Cell Longev. (2014) 2014:210934. doi: 10.1155/2014/210934
127. Han B, Sivaramakrishnan P, Lin CJ, Neve IAA, He J, Tay LWR, et al. Microbial genetic composition tunes host longevity. Cell. (2017) 169:1249–62.e13. doi: 10.1016/j.cell.2017.05.036
128. Santhanam S, Rajamanickam S, Motamarry A, Ramakrishna BS, Amirtharaj JG, Ramachandran A, et al. Mitochondrial electron transport chain complex dysfunction in the colonic mucosa in ulcerative colitis. Inflammation Bowel Dis. (2012) 18:2158–68. doi: 10.1002/ibd.22926
129. Hsieh SY, Shih TC, Yeh CY, Lin CJ, Chou YY, Lee YS. Comparative proteomic studies on the pathogenesis of human ulcerative colitis. Proteomics. (2006) 6:5322–31. doi: 10.1002/pmic.200500541
130. Ho GT, Theiss AL. Mitochondria and inflammatory bowel diseases: toward a stratified therapeutic intervention. Annu Rev Physiol. (2022) 84:435–59. doi: 10.1146/annurev-physiol-060821-083306
131. Jackson DN, Panopoulos M, Neumann WL, Turner K, Cantarel BL, Thompson-Snipes L, et al. Mitochondrial dysfunction during loss of prohibitin 1 triggers Paneth cell defects and ileitis. Gut. (2020) 69:1928–38. doi: 10.1136/gutjnl-2019-319523
132. Cheng M, Lin N, Dong D, Ma J, Su J, Sun L. PGAM5: A crucial role in mitochondrial dynamics and programmed cell death. Eur J Cell Biol. (2021) 100:151144. doi: 10.1016/j.ejcb.2020.151144
133. Kenny B. Mechanism of action of EPEC type III effector molecules. Int J Med Microbiol. (2002) 291:469–77. doi: 10.1078/1438-4221-00155
134. Bock FJ, Tait SWG. Mitochondria as multifaceted regulators of cell death. Nat Rev Mol Cell Biol. (2020) 21:85–100. doi: 10.1038/s41580-019-0173-8
135. Stavru F, Bouillaud F, Sartori A, Ricquier D, Cossart P. Listeria monocytogenes transiently alters mitochondrial dynamics during infection. Proc Natl Acad Sci U.S.A. (2011) 108:3612–7. doi: 10.1073/pnas.1100126108
136. Cai P, Li W, Xu Y, Wang H. Drp1 and neuroinflammation: Deciphering the interplay between mitochondrial dynamics imbalance and inflammation in neurodegenerative diseases. Neurobiol Dis. (2024) 198:106561. doi: 10.1016/j.nbd.2024.106561
137. Baltrusch S. Mitochondrial network regulation and its potential interference with inflammatory signals in pancreatic beta cells. Diabetologia. (2016) 59:683–7. doi: 10.1007/s00125-016-3891-x
138. Liu Z, Ren Z, Zhang J, Chuang CC, Kandaswamy E, Zhou T, et al. Role of ROS and nutritional antioxidants in human diseases. Front Physiol. (2018) 9:477. doi: 10.3389/fphys.2018.00477
139. Liang H, Zhang F, Wang W, Zhao W, Zhou J, Feng Y, et al. Heat shock transcription factor 2 promotes mitophagy of intestinal epithelial cells through PARL/PINK1/parkin pathway in ulcerative colitis. Front Pharmacol. (2022) 13:893426. doi: 10.3389/fphar.2022.893426
140. Barrett JC, Hansoul S, Nicolae DL, Cho JH, Duerr RH, Rioux JD, et al. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn’s disease. Nat Genet. (2008) 40:955–62. doi: 10.1038/ng.175
141. Lees CW, Barrett JC, Parkes M, Satsangi J. New IBD genetics: common pathways with other diseases. Gut. (2011) 60:1739–53. doi: 10.1136/gut.2009.199679
142. Tsianos VE, Kostoulas C, Gazouli M, Frillingos S, Georgiou I, Christodoulou DK, et al. ATG16L1 T300A polymorphism is associated with Crohn’s disease in a Northwest Greek cohort, but ECM1 T130M and G290S polymorphisms are not associated with ulcerative colitis. Ann Gastroenterol. (2020) 33:38–44. doi: 10.20524/aog.2019.0434
143. Lassen KG, Kuballa P, Conway KL, Patel KK, Becker CE, Peloquin JM, et al. Atg16L1 T300A variant decreases selective autophagy resulting in altered cytokine signaling and decreased antibacterial defense. Proc Natl Acad Sci U.S.A. (2014) 111:7741–6. doi: 10.1073/pnas.1407001111
144. Singh SB, Ornatowski W, Vergne I, Naylor J, Delgado M, Roberts E, et al. Human IRGM regulates autophagy and cell-autonomous immunity functions through mitochondria. Nat Cell Biol. (2010) 12:1154–65. doi: 10.1038/ncb2119
145. Liu B, Gulati AS, Cantillana V, Henry SC, Schmidt EA, Daniell X, et al. Irgm1-deficient mice exhibit Paneth cell abnormalities and increased susceptibility to acute intestinal inflammation. Am J Physiol Gastrointest Liver Physiol. (2013) 305:G573–84. doi: 10.1152/ajpgi.00071.2013
146. Kania E, Long JS, McEwan DG, Welkenhuyzen K, La Rovere R, Luyten T, et al. LRRK2 phosphorylation status and kinase activity regulate (macro)autophagy in a Rab8a/Rab10-dependent manner. Cell Death Dis. (2023) 14:436. doi: 10.1038/s41419-023-05964-0
147. Yan J, Yu W, Wang G, Lu C, Liu C, Jiang L, et al. LRRK2 deficiency mitigates colitis progression by favoring resolution of inflammation and restoring homeostasis of gut microbiota. Genomics. (2022) 114:110527. doi: 10.1016/j.ygeno.2022.110527
148. Vincent G, Novak EA, Siow VS, Cunningham KE, Griffith BD, Comerford TE, et al. Nix-mediated mitophagy modulates mitochondrial damage during intestinal inflammation. Antioxid Redox Signal. (2020) 33:1–19. doi: 10.1089/ars.2018.7702
149. Mai CT, Wu MM, Wang CL, Su ZR, Cheng YY, Zhang XJ. Palmatine attenuated dextran sulfate sodium (DSS)-induced colitis via promoting mitophagy-mediated NLRP3 inflammasome inactivation. Mol Immunol. (2019) 105:76–85. doi: 10.1016/j.molimm.2018.10.015
150. Yu T, Lu X, Liang Y, Yang L, Yin Y, Chen H. Ononin alleviates DSS-induced colitis through inhibiting NLRP3 inflammasome via triggering mitophagy. Immun Inflammation Dis. (2023) 11:e776. doi: 10.1002/iid3.776
151. Mei X, Lei Y, Ouyang L, Zhao M, Lu Q. Deficiency of Pink1 promotes the differentiation of Th1 cells. Mol Immunol. (2023) 160:23–31. doi: 10.1016/j.molimm.2023.06.005
152. Ip WKE, Hoshi N, Shouval DS, Snapper S, Medzhitov R. Anti-inflammatory effect of IL-10 mediated by metabolic reprogramming of macrophages. Science. (2017) 356:513–9. doi: 10.1126/science.aal3535
153. Weinberg SE, Sena LA, Chandel NS. Mitochondria in the regulation of innate and adaptive immunity. Immunity. (2015) 42:406–17. doi: 10.1016/j.immuni.2015.02.002
154. Castanier C, Garcin D, Vazquez A, Arnoult D. Mitochondrial dynamics regulate the RIG-I-like receptor antiviral pathway. EMBO Rep. (2010) 11:133–8. doi: 10.1038/embor.2009.258
155. Seth RB, Sun L, Ea CK, Chen ZJ. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell. (2005) 122:669–82. doi: 10.1016/j.cell.2005.08.012
156. Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol. (2011) 12:222–30. doi: 10.1038/ni.1980
157. Alula KM, Delgado-Deida Y, Callahan R, Till A, Underwood L, Thompson WE, et al. Inner mitochondrial membrane protein Prohibitin 1 mediates Nix-induced, Parkin-independent mitophagy. Sci Rep. (2023) 13:18. doi: 10.1038/s41598-022-26775-x
158. Xu D, Ma Y, Peng C, Gan Y, Wang Y, Chen Z, et al. Differently surface-labeled polystyrene nanoplastics at an environmentally relevant concentration induced Crohn’s ileitis-like features via triggering intestinal epithelial cell necroptosis. Environ Int. (2023) 176:107968. doi: 10.1016/j.envint.2023.107968
159. Reinglas J, Gonczi L, Kurt Z, Bessissow T, Lakatos PL. Positioning of old and new biologicals and small molecules in the treatment of inflammatory bowel diseases. World J Gastroenterol. (2018) 24:3567–82. doi: 10.3748/wjg.v24.i32.3567
160. Bugger H, Pfeil K. Mitochondrial ROS in myocardial ischemia reperfusion and remodeling. Biochim Biophys Acta Mol Basis Dis. (2020) 1866:165768. doi: 10.1016/j.bbadis.2020.165768
161. Li A, Gao M, Liu B, Qin Y, Chen L, Liu H, et al. Mitochondrial autophagy: molecular mechanisms and implications for cardiovascular disease. Cell Death Dis. (2022) 13:444. doi: 10.1038/s41419-022-04906-6
162. Mantle D, Hargreaves IP. Mitochondrial dysfunction and neurodegenerative disorders: role of nutritional supplementation. Int J Mol Sci. (2022) 23:12603. doi: 10.3390/ijms232012603
163. Pokharel MD, Marciano DP, Fu P, Franco MC, Unwalla H, Tieu K, et al. Metabolic reprogramming, oxidative stress, and pulmonary hypertension. Redox Biol. (2023) 64:102797. doi: 10.1016/j.redox.2023.102797
164. Tabrizi SJ, Flower MD, Ross CA, Wild EJ. Huntington disease: new insights into molecular pathogenesis and therapeutic opportunities. Nat Rev Neurol. (2020) 16:529–46. doi: 10.1038/s41582-020-0389-4
165. Fu SC, Qu JY, Li LX, Yang XX, Li YQ, Zuo XL. Excessive mitochondrial fission suppresses mucosal repair by impairing butyrate metabolism in colonocytes. Inflammation Bowel Dis. (2024) 30:114–24. doi: 10.1093/ibd/izad132
166. Miret-Casals L, Sebastián D, Brea J, Rico-Leo EM, Palacín M, Fernández-Salguero PM, et al. Identification of new activators of mitochondrial fusion reveals a link between mitochondrial morphology and pyrimidine metabolism. Cell Chem Biol. (2018) 25:268–78.e4. doi: 10.1016/j.chembiol.2017.12.001
167. Jin Z, Chang B, Wei Y, Yang Y, Zhang H, Liu J, et al. Curcumin exerts chondroprotective effects against osteoarthritis by promoting AMPK/PINK1/Parkin-mediated mitophagy. BioMed Pharmacother. (2022) 151:113092. doi: 10.1016/j.biopha.2022.113092
168. Qin X, Zhao Y, Gong J, Huang W, Su H, Yuan F, et al. Berberine protects glomerular podocytes via inhibiting drp1-mediated mitochondrial fission and dysfunction. Theranostics. (2019) 9:1698–713. doi: 10.7150/thno.30640
169. Sahoo DK, Heilmann RM, Paital B, Patel A, Yadav VK, Wong D, et al. Oxidative stress, hormones, and effects of natural antioxidants on intestinal inflammation in inflammatory bowel disease. Front Endocrinol (Lausanne). (2023) 14:1217165. doi: 10.3389/fendo.2023.1217165
170. Zhang L, Xue H, Zhao G, Qiao C, Sun X, Pang C, et al. Curcumin and resveratrol suppress dextran sulfate sodium−induced colitis in mice. Mol Med Rep. (2019) 19:3053–60. doi: 10.3892/mmr.2019.9974
171. Cao S, Wang C, Yan J, Li X, Wen J, Hu C. Curcumin ameliorates oxidative stress-induced intestinal barrier injury and mitochondrial damage by promoting Parkin dependent mitophagy through AMPK-TFEB signal pathway. Free Radic Biol Med. (2020) 147:8–22. doi: 10.1016/j.freeradbiomed.2019.12.004
172. Li X, Zhu J, Lin Q, Yu M, Lu J, Feng J, et al. Effects of curcumin on mitochondrial function, endoplasmic reticulum stress, and mitochondria-associated endoplasmic reticulum membranes in the jejunum of oxidative stress piglets. J Agric Food Chem. (2022) 70:8974–85. doi: 10.1021/acs.jafc.2c02824
173. Li CP, Li JH, He SY, Chen O, Shi L. Effect of curcumin on p38MAPK expression in DSS-induced murine ulcerative colitis. Genet Mol Res. (2015) 14:3450–8. doi: 10.4238/2015.April.15.8
174. Hong J. Protective effects of curcumin-regulated intestinal epithelial autophagy on inflammatory bowel disease in mice. Gastroenterol Res Pract. (2022) 2022:2163931. doi: 10.1155/2022/2163931
175. Yue W, Liu Y, Li X, Lv L, Huang J, Liu J. Curcumin ameliorates dextran sulfate sodium-induced colitis in mice via regulation of autophagy and intestinal immunity. Turk J Gastroenterol. (2019) 30:290–8. doi: 10.5152/tjg.2019.18342
176. Cao S, Shen Z, Wang C, Zhang Q, Hong Q, He Y, et al. Resveratrol improves intestinal barrier function, alleviates mitochondrial dysfunction and induces mitophagy in diquat challenged piglets(1). Food Funct. (2019) 10:344–54. doi: 10.1039/c8fo02091d
177. Pan HH, Zhou XX, Ma YY, Pan WS, Zhao F, Yu MS, et al. Resveratrol alleviates intestinal mucosal barrier dysfunction in dextran sulfate sodium-induced colitis mice by enhancing autophagy. World J Gastroenterol. (2020) 26:4945–59. doi: 10.3748/wjg.v26.i33.4945
178. Zhang B, Zhang Y, Liu X, Yin J, Li X, Zhang X, et al. Differential protective effect of resveratrol and its microbial metabolites on intestinal barrier dysfunction is mediated by the AMPK pathway. J Agric Food Chem. (2022) 70:11301–13. doi: 10.1021/acs.jafc.2c04101
179. Meng G, Li P, Du X, Feng X, Qiu F. Berberine alleviates ulcerative colitis by inhibiting inflammation through targeting IRGM1. Phytomedicine. (2024) 133:155909. doi: 10.1016/j.phymed.2024.155909
180. Xu X, Li W, Yu Z, Zhang L, Duo T, Zhao Y, et al. Berberine ameliorates dextran sulfate sodium-induced ulcerative colitis and inhibits the secretion of gut lysozyme via promoting autophagy. Metabolites. (2022) 12:676. doi: 10.3390/metabo12080676
181. Rizzo V, Ferlazzo N, Currò M, Isola G, Matarese M, Bertuccio MP, et al. Baicalin-induced autophagy preserved LPS-stimulated intestinal cells from inflammation and alterations of paracellular permeability. Int J Mol Sci. (2021) 22:2315. doi: 10.3390/ijms22052315
182. Liu C, Wang J, Yang Y, Liu X, Zhu Y, Zou J, et al. Ginsenoside Rd ameliorates colitis by inducing p62-driven mitophagy-mediated NLRP3 inflammasome inactivation in mice. Biochem Pharmacol. (2018) 155:366–79. doi: 10.1016/j.bcp.2018.07.010
183. Chen X, Xu T, Lv X, Zhang J, Liu S. Ginsenoside Rh2 alleviates ulcerative colitis by regulating the STAT3/miR-214 signaling pathway. J Ethnopharmacol. (2021) 274:113997. doi: 10.1016/j.jep.2021.113997
184. Huang X, Xiao J, Wen M, Liang J. Ginsenoside Rk2 Protects against Ulcerative Colitis via Inactivating ERK/MEK Pathway by SIRT1. J Environ Pathol Toxicol Oncol. (2022) 41:89–98. doi: 10.1615/JEnvironPatholToxicolOncol.2021039648
185. Grammatikopoulou MG, Gkiouras K, Theodoridis X, Asteriou E, Forbes A, Bogdanos DP. Oral adjuvant curcumin therapy for attaining clinical remission in ulcerative colitis: A systematic review and meta-analysis of randomized controlled trials. Nutrients. (2018) 10:1737. doi: 10.3390/nu10111737
186. Kumar S, Ahuja V, Sankar MJ, Kumar A, Moss AC. Curcumin for maintenance of remission in ulcerative colitis. Cochrane Database Syst Rev. (2012) CD008424. doi: 10.1002/14651858.CD008424.pub2
187. Zeng L, Yang T, Yang K, Yu G, Li J, Xiang W, et al. Curcumin and curcuma longa extract in the treatment of 10 types of autoimmune diseases: A systematic review and meta-analysis of 31 randomized controlled trials. Front Immunol. (2022) 13:896476. doi: 10.3389/fimmu.2022.896476
188. Rainey NE, Moustapha A, Petit PX. Curcumin, a multifaceted hormetic agent, mediates an intricate crosstalk between mitochondrial turnover, autophagy, and apoptosis. Oxid Med Cell Longev. (2020) 2020:3656419. doi: 10.1155/2020/3656419
189. Chen X, Zhang J, Yin N, Wele P, Li F, Dave S, et al. Resveratrol in disease prevention and health promotion: A role of the gut microbiome. Crit Rev Food Sci Nutr. (2024) 64:5878–95. doi: 10.1080/10408398.2022.2159921
190. Mayangsari Y, Suzuki T. Resveratrol ameliorates intestinal barrier defects and inflammation in colitic mice and intestinal cells. J Agric Food Chem. (2018) 66:12666–74. doi: 10.1021/acs.jafc.8b04138
191. Xu X, Yi H, Wu J, Kuang T, Zhang J, Li Q, et al. Therapeutic effect of berberine on metabolic diseases: Both pharmacological data and clinical evidence. BioMed Pharmacother. (2021) 133:110984. doi: 10.1016/j.biopha.2020.110984
192. Wang L, Tang XQ, Shi Y, Li HM, Meng ZY, Chen H, et al. Tetrahydroberberrubine retards heart aging in mice by promoting PHB2-mediated mitophagy. Acta Pharmacol Sin. (2023) 44:332–44. doi: 10.1038/s41401-022-00956-w
193. Yang Y, Jiang S, Mu Y, Liu C, Han Y, Jiang J, et al. Berberine alleviated contrast-induced acute kidney injury by mitophagy-mediated NLRP3 inflammasome inactivation in a mice model. Toxicol Appl Pharmacol. (2024) 486:116952. doi: 10.1016/j.taap.2024.116952
194. Jiang Y, Zhao L, Chen Q, Zhou L. Exploring the mechanism of berberine intervention in ulcerative colitis from the perspective of inflammation and immunity based on systemic pharmacology. Evid Based Complement Alternat Med. (2021) 2021:9970240. doi: 10.1155/2021/9970240
195. Song X, Wang L, Fan D. Insights into recent studies on biotransformation and pharmacological activities of ginsenoside rd. Biomolecules. (2022) 12:512. doi: 10.3390/biom12040512
Keywords: UC, mitochondrial dynamics, mitophagy, targeted therapy, nature products
Citation: Zhou J, Xi Y, Wu T, Zeng X, Yuan J, Peng L, Fu H and Zhou C (2025) A potential therapeutic approach for ulcerative colitis: targeted regulation of mitochondrial dynamics and mitophagy through phytochemicals. Front. Immunol. 15:1506292. doi: 10.3389/fimmu.2024.1506292
Received: 04 October 2024; Accepted: 02 December 2024;
Published: 07 January 2025.
Edited by:
Chunping Wan, First Affiliated Hospital of Yunnan University of Traditional Chinese Medicine, ChinaReviewed by:
Dongdong Qin, Yunnan University of Chinese Medicine, ChinaChaojun Yan, Wuhan University, China
Copyright © 2025 Zhou, Xi, Wu, Zeng, Yuan, Peng, Fu and Zhou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ce Zhou, NTYxNkBjZHV0Y20uZWR1LmNu; Hao Fu, eGpoZ2N5anNAMTI2LmNvbQ==