 Qin Ying Lim1,2†
Qin Ying Lim1,2† Daniel Leung2†
Daniel Leung2† Crystal K. Lam3Xingtian Yang2Kai N. Cheong2,4
Crystal K. Lam3Xingtian Yang2Kai N. Cheong2,4 Andrew K. H. Yik2Jing Yang2
Andrew K. H. Yik2Jing Yang2 Koon-Wing Chan2Pamela P. W. Lee1,2,4,5
Koon-Wing Chan2Pamela P. W. Lee1,2,4,5 Miyuki Tsumura6†Elaine Y. L. Au3
Miyuki Tsumura6†Elaine Y. L. Au3 Jaime S. Rosa Duque1,2,4*†‡
Jaime S. Rosa Duque1,2,4*†‡ Satoshi Okada6*†‡
Satoshi Okada6*†‡ Yu Lung Lau1,2,4†
Yu Lung Lau1,2,4†- 1Department of Paediatrics and Adolescent Medicine, Queen Mary Hospital, Hong Kong, Hong Kong SAR, China
- 2Department of Paediatrics and Adolescent Medicine, The University of Hong Kong, Hong Kong, Hong Kong SAR, China
- 3Division of Clinical Immunology, Department of Pathology, Queen Mary Hospital, Hong Kong, Hong Kong SAR, China
- 4Department of Paediatrics and Adolescent Medicine, Hong Kong Children’s Hospital, Hong Kong, Hong Kong SAR, China
- 5Department of Paediatrics, The University of Hong Kong – Shenzhen Hospital, Shenzhen, China
- 6Department of Paediatrics, Hiroshima University Graduate School of Biomedical and Health Sciences, Hiroshima, Japan
Signal transducer and activator of transcription 1 (STAT1) gene mutations have broad clinical phenotypes, classified by the inheritance pattern and functional state. Individuals with autosomal dominant STAT1 deficiency are more susceptible to intracellular bacteria, the hallmark of which is Mendelian susceptibility to mycobacterial diseases (MSMDs) that are associated with increased risks of invasive disease by weakly virulent mycobacteria. We report a novel de novo heterozygous missense mutation in exon 23 of the STAT1 gene (NM_007315.4):c.2129C>T(p.Ser710Phe) (S710F), located in the transactivation domain (TAD) for two Chinese siblings, whereby the index patient presented with multifocal osteomyelitis after Bacillus Calmette–Guerin (BCG) vaccine, while the younger sibling was spared the infection, as BCG vaccination was withheld at birth. STAT1 loss-of-function was confirmed by the gamma-activated sequence reporter assay, representing the first loss-of-function mutation in the TAD of the STAT1 gene. Both parents did not have the same mutation, and this finding is suggestive of gonadal mosaicism.
Introduction
Signal transducer and activator of transcription 1 (STAT1) is an important molecule involved in interferon (IFN)-mediated immunity as part of the well-known Janus activation kinases (JAK)-STAT pathway. IFN-γ stimulation leads to phosphorylation of the STAT1 tyrosine 701 residue (Y701) by Janus kinase 1 and 2 in the cytoplasm, forming the IFN-γ activated factor (GAF) homodimer. The homodimer is translocated into the nucleus, where it binds to the gamma-activated sequence (GAS) to induce transcription of target genes involved in defense against intracellular pathogens. In contrast, stimulation by IFN-α and IFN-β induces phosphorylation of STAT1 and STAT2 that bind to interferon regulating factor 9 (IRF9) to form the IFN-stimulated gene factor 3 (ISGF3) heterotrimer. The heterotrimer translocates into the nucleus and binds to the IFN-stimulated response element (ISRE) to induce target gene transcription involved in defense against viruses. This extensive signaling and activation pathway that mediate regulation of IFN-responsive target gene transcription play a crucial role in human immune defense (1).
Over 150 disease-causing STAT1 gene mutations with broad clinical phenotypes have been described, classified by the inheritance pattern and functional state (2). STAT1 loss-of-function diseases include autosomal dominant (AD) STAT1 deficiency and autosomal recessive (AR) complete and partial STAT1 deficiency. The hallmark of AD STAT1 deficiency is Mendelian susceptibility to mycobacterial diseases (MSMD), and individuals with this disease are susceptible to invasive infections caused by weakly virulent mycobacteria (3). Those with AR STAT1 deficiency have increased susceptibility to intracellular organisms and viruses in addition to increased susceptibility to mycobacteria, of which AR complete STAT1 deficiency often has more severe clinical phenotypes compared to AR partial STAT1 deficiency. On the other hand, those with AD STAT1 gain-of-function (GOF) mutations present with chronic mucocutaneous candidiasis (CMC) (4).
We present a pair of Chinese siblings with novel de novo heterozygous missense mutation in exon 23 of the STAT1 gene, which was identified after the index patient presented with multifocal osteomyelitis following Bacillus Calmette–Guerin (BCG) vaccination. Subsequently, his younger sister was found to have the same mutation on family screening while the parents were unaffected, suggesting gonadal mosaicism. This mode of inheritance is seldom recognized or reported in patients with inborn errors of immunity (IEI), which can lead to missed diagnoses. The mutation is the first reported loss-of-function STAT1 located in the transactivation domain (TAD).
Case description
Patient 1
Patient 1 was a 2-year-old Chinese boy with non-consanguineous parents and no personal or family history of recurrent infections or early neonatal death, who presented with left wrist and forearm swelling in the past 2 months. Additionally, there was history of pustule formation over his neonatal intradermally administered live-attenuated Mycobacterium bovis Bacille Calmette–Guerin (BCG) vaccine scar site on his left upper arm and, thereafter, intermittent discharge from the same site for approximately 1 year. The patient otherwise remained well and had satisfactory growth parameters, with no fever, chronic respiratory symptoms, unusual environmental exposures, or challenges in accessing healthcare since birth (Figure 1A). His parents had no history of mycobacterial infections.
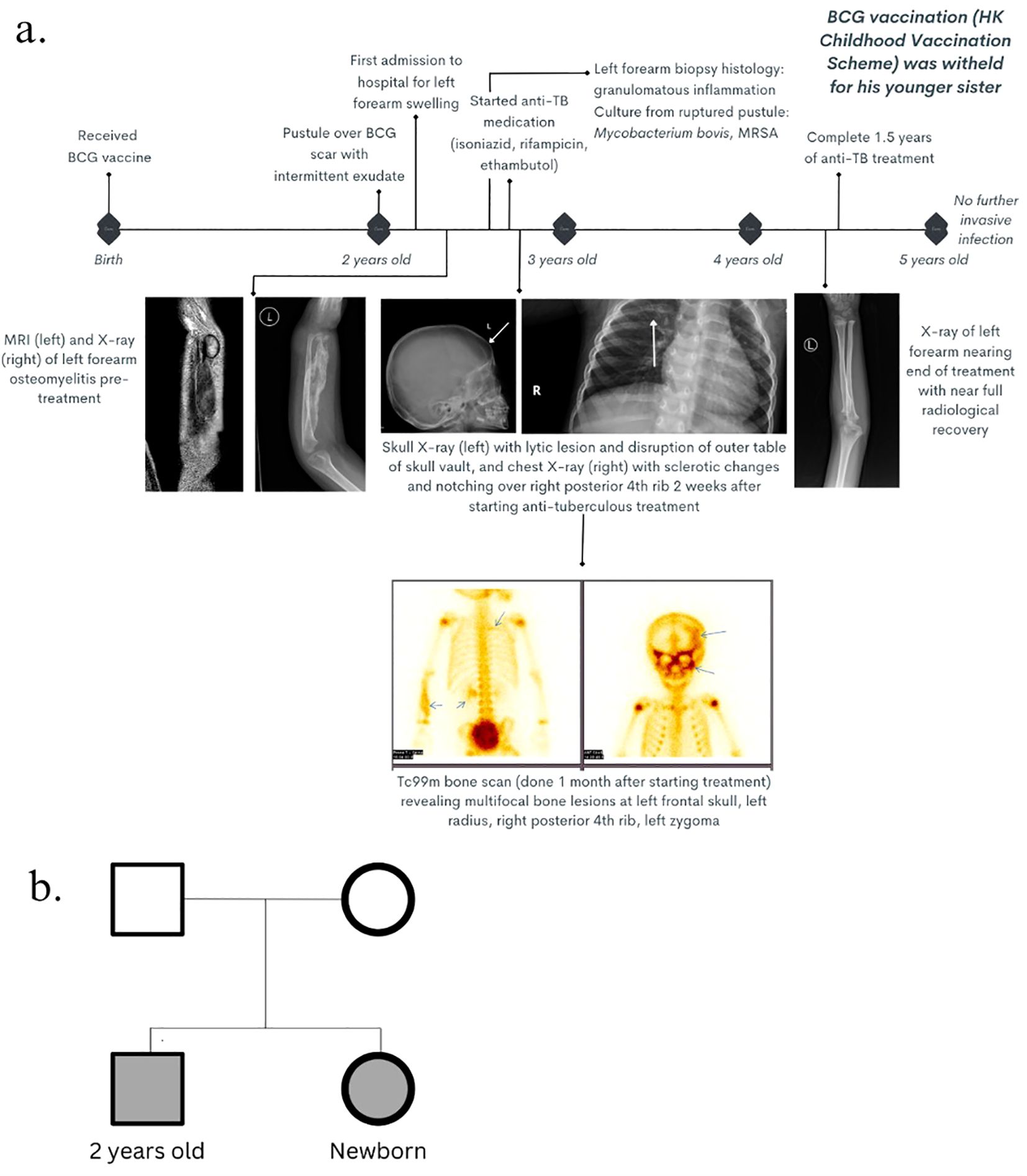
Figure 1. Clinical history. (A) Timeline of clinical course, radiological features, and management from birth until 5 years old. (B) Genogram of the two patients with unaffected parents.  : male (affected);
: male (affected);  : male (unaffected);
: male (unaffected);  : female (affected);
: female (affected);  : female (unaffected).
: female (unaffected).
Patient 2
Patient 2 was patient 1’s newborn younger sister (Figure 1B).
Diagnostic assessment
Patient 1
Ultrasonography showed a non-vascular left distal radial lesion, with osseous and soft tissue component changes, and axillary lymphadenopathy, consistent with osteomyelitis of low-grade infection. X-ray of the left forearm showed features of acute osteomyelitis. Magnetic resonance imaging (MRI) of the left forearm was performed 4 months later, which was delayed due to his inadequate fasting period required for sedation and the waiting queue for public radiological services in this region, and the findings confirmed acute osteomyelitis and sinus formation. Further assessment with MRI of the brain and bone scan revealed disseminated osteomyelitis involving the skull, left zygoma and right posterior fourth rib (Figure 1B). Biopsy of the left radius was taken, and the histological examination revealed focal granuloma formation, dense lymphoplasmacytic infiltrates, scattered eosinophils, and multinucleated giant histiocytes engulfing the calcified and transparent foreign material, consistent with a granulomatous lesion, while the microbiological culture yielded Mycobacterium bovis. Culture of the purulent discharge from the ulcerated BCG injection site was positive for attenuated Mycobacterium bovis and methicillin-resistant Staphylococcus aureus (MRSA).
Immunological workup was not suggestive of chronic granulomatous disease (CGD); the neutrophil function by nitroblue tetrazolium test (NBT) was normal, and although the initial dihydrorhodamine reduction test (DHR) performed during the initial infection showed suppressed oxidative burst activity with low mean fluorescent intensity (MFI), oxidative burst activity normalized on repeat testing. The lymphocyte subset was not suggestive of severe combined immunodeficiency (SCID). The clinical or biochemical condition was not consistent with hemophagocytic lymphohistiocytosis.
Subsequent to these initial diagnostic testing methods, genetic workup by whole exome sequencing (WES) to survey for disease-causing coding genes, followed by confirmation using Sanger sequencing (SS), found a novel de novo heterozygous missense mutation in exon 23 of the STAT1 gene (NM_007315.4): c.2129C>T (p.Ser710Phe) (Supplementary Material), which has not been previously reported. The mutation was located in the transactivation domain of the STAT1 molecule.
Patient 2
Patient 2 was identified upon further screening of immediate family members by targeted gene SS, which revealed the same STAT1 mutation (Figure 1B).
Genetic investigation and functional testing
Although the clinical phenotype of patient 1 was compatible with AD STAT1 deficiency, the same STAT1 mutation by targeted gene SS was not found in either parent, suggestive of gonadal mosaicism. This variant found in the two siblings has a high pathogenicity score, with a combined annotation-dependent depletion (CADD) score of 6.988 and a deleterious annotation of genetic variants using neural networks (DANN) score of 0.999. The mutation is located at a highly conserved position, with a genomic evolutionary rate profiling (GERP) score of 5.95. According to the ACMG guidelines, the variant was classified as likely pathogenic.
In the cytokine response assay (Supplementary Material), there was normal response in the production of IFN-γ to IL-12 with or without phytohemagglutinin (PHA) or lipopolysaccharide (LPS) for both siblings (Figure 2A). However, the two siblings had reduced production of tumor necrosis factor alpha (TNF-α) with IFN-γ stimulation, which normalized with the addition of LPS (Figure 2B). These findings were supportive of IFN-γ response defect, such as IFN-γ receptor deficiency and STAT1 deficiency due to markedly reduced interleukin-12 (IL-12) p40 and p70 production in response to IFN-γ stimulation of peripheral blood mononuclear cells (PBMCs), which was more obvious in response to combined lipopolysaccharide (LPS) and IFN-γ (Figure 2C).
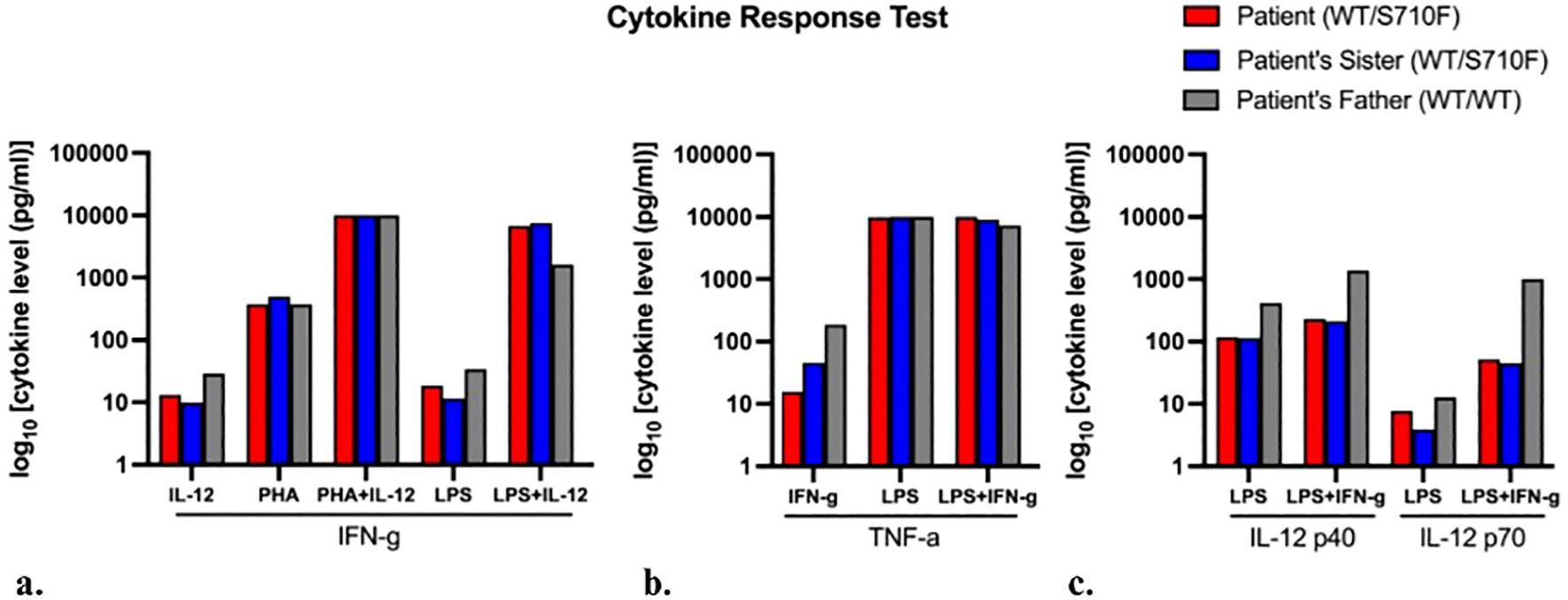
Figure 2. Cytokine response test. Cytokine response levels produced by blood mononuclear cells when stimulated by agents listed on the x-axis, in the production of (A) interferon-γ (IFN-γ), (B) tumor necrosis factor-α (TNF-α), and (C) interleukin 12 (IL-12), respectively. Levels in patient’s father (control sample) shown in gray. Each bar represents an individual’s single assay result; hence, statistical comparison was non-applicable.
The gamma-activated sequence (GAS) reporter assay was performed (Supplementary Material). The S710F mutant in our patients, along with Y701C, a known loss-of-function mutation identified in patients with MSMD (5), exhibited decreased activation of GAS compared to the wild type (WT) upon stimulation by IFN-γ (Figure 3A). In contrast, the R274Q mutant, a known gain-of-function mutation found in patients with chronic mucocutaneous candidiasis, showed excess GAS activation in response to IFN-γ.
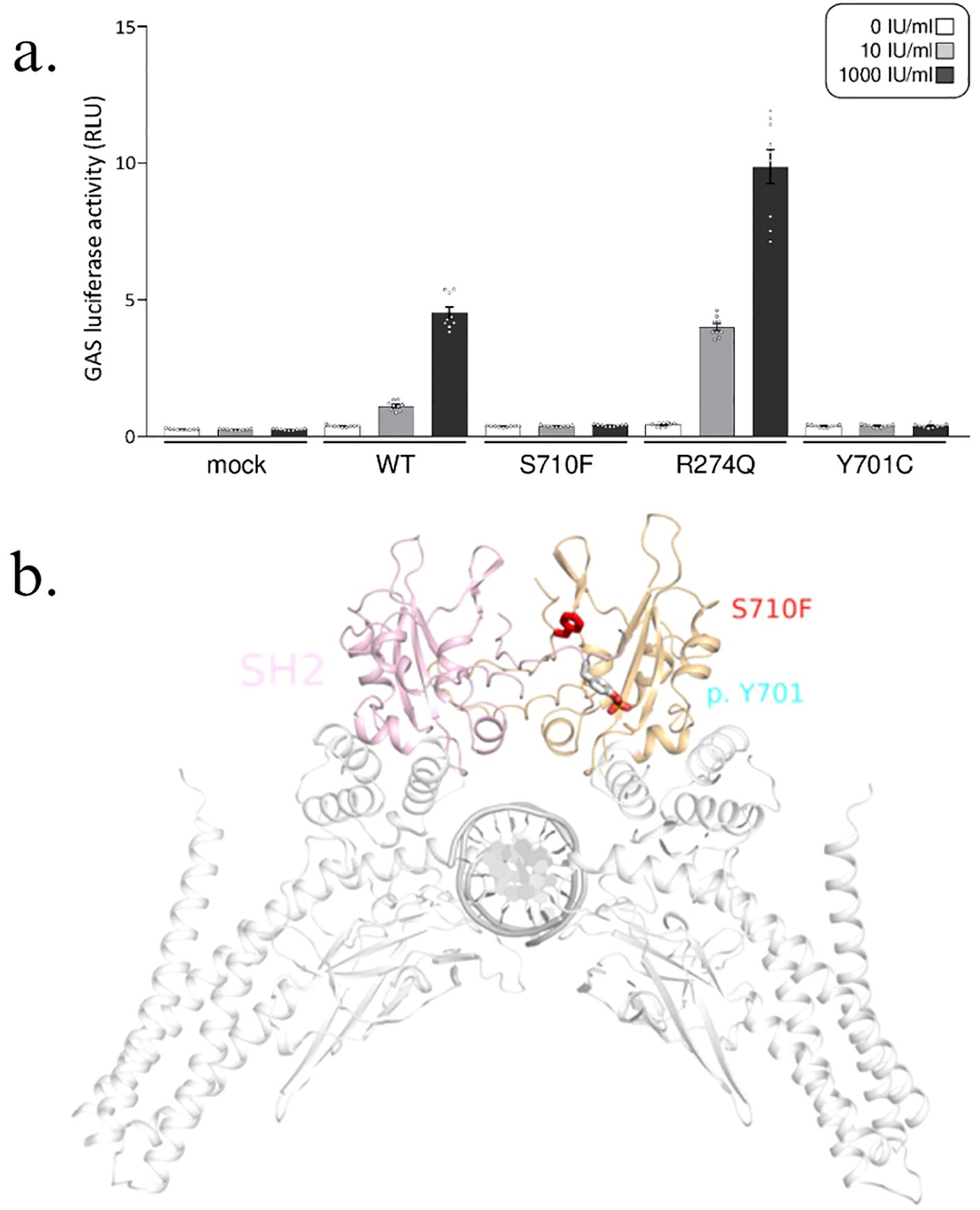
Figure 3. STAT1 S710F mutation. (A) Gamma-activated sequence (GAS) reporter assay with human cells completely lacking STAT1 (U3C cells) that transiently express wild-type (WT) or mutant STAT1 were subjected to a GAS reporter assay. Both the S710F (mutation in our patients) and Y701C (known loss-of-function mutation) STAT1 mutants exhibited reduced GAS activation, whereas the R274Q (known gain-of-function mutation) mutant resulted in increased GAS activation upon IFN-γ stimulation. This GAS reporter assay confirmed that the two siblings had a STAT1 loss-of-function mutation. The bar graphs display the means with standard errors. (B) STAT1 protein with the mutation site S710F (red); activation of STAT1 by phosphorylation at Y701 (blue).
Therapeutic intervention
Patient 1 was treated for disseminated BCG osteomyelitis with a prolonged course of anti-mycobacterial regimen using isoniazid, rifampicin, and ethambutol for 1 year and 8 months.
BCG vaccination according to the Hong Kong Childhood Vaccination Scheme was withheld for patient 2 after her birth. She was advised to avoid high-risk mycobacterial environmental and sick-contact exposures, while pharmacological mycobacterial prophylaxis was deemed unnecessary.
Follow-up and outcomes
Genetic counseling was provided to the family and long-term follow-up assessments to the two siblings.
There was clinical and radiological recovery of osteomyelitis for patient 1 (Figure 4). He remains well with no further invasive infection. Although this young child was unable to attend nursery and kindergarten during his anti-tuberculous therapy, our directly observed therapy (DOT) outpatient service aimed at improving daily life, psychosocial wellbeing, and drug adherence facilitated early hospital discharge and adequate treatment assurance, for which the parents expressed gratitude in easing the patient’s diagnostic and treatment journey.
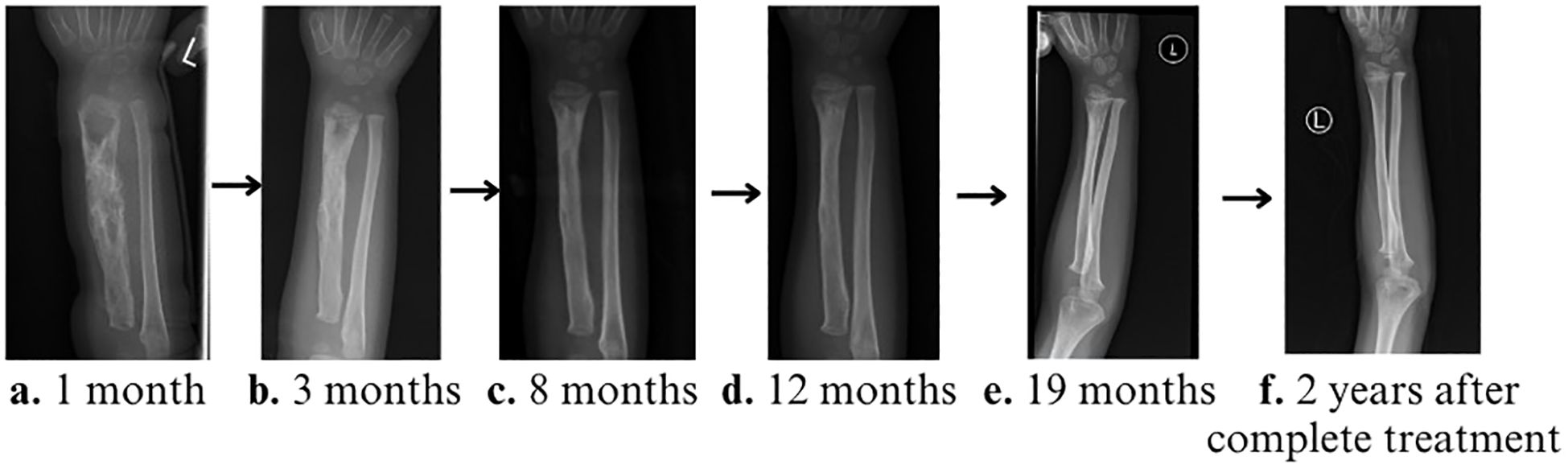
Figure 4. Serial X-rays of left forearm during treatment. (A) 1 month, (B) 3 months, (C) 8 months, (D) 12 months, and (E) 19 months (immediately prior to discontinuation of the anti-tuberculosis medications) showing progressive radiological disease improvement, with near full recovery and remodeling towards the end of the treatment. (F) X-ray of the left forearm 2 years after treatment completion, with full bone remodeling.
Although her older sibling had disseminated osteomyelitis, patient 2 has remained asymptomatic from birth to age 6 years at the time of her last clinical follow-up.
Discussion
We identified unique features in these two siblings that had a novel de novo heterozygous loss-of-function STAT1 mutation associated with disseminated BCG osteomyelitis. This genetic mutation likely resulted from gonadal mosaicism, and to the best of our knowledge, the siblings also represent the first reported loss-of-function mutation located in the transactivation domain (TAD, residues 710–750) of the STAT1 gene. The two above features in these siblings have not been reported in STAT1 LOF mutations previously.
The STAT1 protein is comprised of the N-terminal domain, coiled-coil domain (CCD), DNA binding domain, linker domain, SH2 domain, and tail segment domain, in addition to the TAD. The CCD, involved in protein–protein interactions and the SH2 domain, which is required for STAT1 recruitment to interferon receptors (6, 7), had been more frequently implicated in STAT1 LOF mutations (Asano et al., 2023, Ye et al., 2022). These mutations can lead to impairment of STAT1 dimerization and dephosphorylation of pSTAT1 (8–11). To date, four other mutations located in the TAD have been reported, but all had been gain-of-function mutations (12). For our mutation, changes in local conformation of the STAT1 protein due to the proximity of the mutation site to the phosphorylation site (Figure 3B) may explain the effect on STAT1 phosphorylation. This can result in impaired downstream signaling of the GAF homodimer and its critical role in host defense against Mycobacteria. In addition, serine 727 phosphorylation of STAT1 at the TAD has been implicated in its role in autoimmune and antipathogen responses (13), although further research and functional analyses are required to determine its mechanism in antimycobacterial defense.
The clinical phenotype in our mutation was most compatible with AD STAT1 deficiency, and functional studies demonstrated STAT1 loss-of-function. Identification of the same mutation in both siblings may indicate germline mosaicism, as both siblings are affected, in the parental gametes, although further investigation would be required to confirm this. Gonadal mosaicism is of higher possibility than gonosomal mosaicism, as both parents are asymptomatic, and they do not have the same mutation identified in their peripheral blood cells. Gene mosaicism has been recognized but reported rarely in patients with inborn errors of immunity (IEI), including X-linked severe combined immunodeficiency, X-linked agammaglobulinemia, hyper-IgE syndrome, X-linked chronic granulomatous disease, autoimmune lymphoproliferative disease, Blau syndrome, and cryopyrin-associated periodic syndrome, most of which have been somatic mosaicisms (14–19). In particular, germline mosaicism has been described in a case report previously, which was associated with a gain-of-function mutation of STAT1 (20). However, a recent study investigating gene mosaicism in families with IEIs reported an incidence of up to 25% overall, including somatic, gonadal, and gonosomal mosaicism, of which 3.3% was gonadal and revertant mosaicism (21), suggesting that gene mosaicism plays an underrecognized but hugely relevant role in patients with IEIs, and such findings warrant further research into the genetic mechanisms underlying the pathogenesis of IEIs and the clinical importance of genetic counseling for families affected.
To date, 22 patients from 14 kindreds with 14 heterozygous mutations have been reported to be associated with AD STAT1 deficiency, the majority of which were shown to be LOF mutations with dominant negative effects. Incomplete penetrance is a typical feature and has been quoted to be 88%, and good outcomes with mortality of 0% (12). However, asymptomatic carriers may also display functional defects (22). Although this report is limited by the small number of patients restricted to a single family and the lack of in vivo functional and animal model experiments, the novel finding of loss-of-function mutation in the TAD of STAT1 confirmed by the GAS reporter assay for both siblings further contributes to the current knowledge on STAT1 deficiency. In a recent systematic review, the clinical spectrum of AD STAT1 deficiency is diverse and most commonly manifests as MSMD, of which Mycobacterium bovis from BCG was the most common pathogen (23), and multifocal osteomyelitis is also a relatively common reported feature (24–26). However, increased susceptibility to viral infections has also been reported in rare instances (24). This is in contrast to AR complete and partial STAT1 deficiency, in which recurrent viral infections is a common feature in addition to susceptibility to intracellular bacteria (12, 23). This reflects impaired IFN-γ signaling with preserved IFN-α and IFN-β signaling in AD STAT1 deficiency. The most common anatomical site in AD STAT1 deficiency involved was the bone, followed by cutaneous and disseminated disease. When considering disease involvement of both AD STAT1 deficiency and AR STAT1 deficiency together, however, a recent study found that among 64 patients with AR or AD STAT1 deficiency, the most common sites were disseminated disease for 44.4%, followed by cutaneous disease for 41.3% and then bone disease for 39.7% (24). This is not surprising, as AR partial or complete STAT1 deficiency often has more severe clinical presentation compared to AD STAT1 deficiency. Treatment recommendations differ significantly within the spectrum of STAT1 deficiency due to the difference in clinical severity, in which hematopoietic stem cell transplant (HSCT) is recommended for those with AR complete STAT1 deficiency, while antimicrobial treatment is generally adequate for AR partial and AD STAT1 deficiency (4).
Aside from MSMD, BCGiosis can be caused by a myriad of other immunodeficiencies, and therefore, its clinical manifestation should raise concern for underlying immunodeficiency, followed by relevant investigations that should be arranged. In a recent single center review in Singapore, patients with this presentation was ultimately diagnosed with a multitude of underlying IEIs, including SCID, combined immunodeficiency (CID), MSMD, and anhidrotic ectodermal dysplasia with primary immunodeficiency (EDA-ID) (27). In terms of workup for immunodeficiency, the initial abnormal DHR result in our patient corroborated a previous study’s finding that acute infection can affect the reliability of DHR (28).
For this family, the timely diagnosis and implementation of family screening, with the help of WES, was crucial in the identification of the same mutation in his younger sister, for whom exposure to BCG was successfully prevented by omission of the mandatory BCG vaccine that is given as part of the Childhood Vaccination Program in Hong Kong. Under this vaccination program, infants with undiagnosed inborn errors of immunity can be exposed to BCG that is routinely given at birth. The year 2021 marked the commencement of IEI screening, with the implementation of newborn screening for severe combined immunodeficiency (NBS-SCID) by TREC and KREC analysis, in Hong Kong. With the growing trend of newborn screening programs for inborn errors of immunity globally, the importance and potential of screening programs for diagnoses of a broader range of IEIs is expected to be recognized and considered alongside other ethical, socioeconomical, and logistical implications.
In conclusion, the clinical presentation of BCGiosis should raise concern for an underlying immunodeficiency. STAT1 LOF disease should be considered in young children presenting with MSMD. Early genetic diagnosis can provide families with appropriate genetic counseling and medical interventions. Due to the possibility of parental germline mosaicism, genetic testing for siblings should be considered even when parents tested negative. Phenotypic classification and functional analyses of STAT1 LOF mutations play a pivotal role in guiding treatment decisions.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Ethics statement
The studies involving humans were approved by University of Hong Kong Institutional Review Board (UW 08-301). The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the minor(s)’ legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article. Written informed consent was obtained from the participant/patient(s) for the publication of this case report.
Author contributions
QL: Writing – original draft, Writing – review & editing, Data curation, Formal analysis, Investigation, Project administration, Visualization. DL: Writing – review & editing, Formal analysis, Investigation, Visualization. CL: Writing – review & editing, Formal analysis, Investigation, Methodology, Software. XY: Writing – review & editing, Formal analysis, Investigation, Methodology, Software. KNC: Writing – review & editing, Project administration. AY: Visualization, Writing – review & editing. JY: Formal analysis, Investigation, Methodology, Software, Writing – review & editing. K-WC: Formal analysis, Investigation, Writing – review & editing. PL: Project administration, Writing – review & editing. MT: Formal analysis, Investigation, Methodology, Software, Visualization, Writing – review & editing. EA: Formal analysis, Investigation, Writing – review & editing, Funding acquisition. JR: Data curation, Formal analysis, Investigation, Project administration, Supervision, Writing – review & editing. SO: Formal analysis, Investigation, Methodology, Software, Visualization, Writing – review & editing. YL: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Project administration, Resources, Supervision, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. Funding for genetic diagnoses of the inborn error of immunity was supported by the Society for the Relief of Disabled Children, Hong Kong through YL. Professor SO was supported by the Japan Society for the Promotion of Science (JSPS) KAKENHI (grant numbers 22H03041 and 22KK0113) and the Japan Agency for Medical Research and Development (AMED) (grant numbers JP23ek0109623 and JP24ek019754).
Acknowledgments
The authors thank Mr. Tsun Ming Lau in the visualization of data. We also thank the Society for the Relief of Disabled Children, Hong Kong in supporting YL for the funding of genetic diagnoses of inborn errors of immunity, and the Japan Society for the Promotion of Science (JSPS) KAKENHI (grant numbers 22H03041 and 22KK0113) and the Japan Agency for Medical Research and Development (AMED) (grant Number: JP23ek0109623 and JP24ek019754) for their support for Professor Satoshi Okada.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2024.1504816/full#supplementary-material
References
1. Olbrich P, Freeman AF. STAT1 and STAT3 mutations: important lessons for clinical immunologists. Expert Rev Clin Immunol. (2018) 14:1029–41. doi: 10.1080/1744666X.2018.1531704
2. Boisson-Dupuis S, Kong XF, Okada S, Cypowyj S, Puel A, Abel L, et al. Inborn errors of human STAT1: allelic heterogeneity governs the diversity of immunological and infectious phenotypes. Curr Opin Immunol. (2012) 24:364–78. doi: 10.1016/j.coi.2012.04.011
3. Lee WI, Huang JL, Yeh KW, Jaing TH, Lin TY, Huang YC, et al. Immune defects in active mycobacterial diseases in patients with primary immunodeficiency diseases (PIDs). J Formos Med Assoc. (2011) 110:750–8. doi: 10.1016/j.jfma.2011.11.004
4. Mizoguchi Y, Okada S. Inborn errors of STAT1 immunity. Curr Opin Immunol. (2021) 72:59–64. doi: 10.1016/j.coi.2021.02.009
5. Hirata O, Okada S, Tsumura M, Kagawa R, Miki M, Kawaguchi H, et al. Heterozygosity for the Y701C STAT1 mutation in a multiplex kindred with multifocal osteomyelitis. Haematologica. (2013) 98:1641–9. doi: 10.3324/haematol.2013.083741
6. Ueki M, Yamada M, Ito K, Tozawa Y, Morino S, Horikoshi Y, et al. A heterozygous dominant-negative mutation in the coiled-coil domain of STAT1 is the cause of autosomal-dominant Mendelian susceptibility to mycobacterial diseases. Clin Immunol. (2017) 174:24–31. doi: 10.1016/j.clim.2016.11.004
7. Sampaio EP, Bax HI, Hsu AP, Kristosturyan E, Pechacek J, Chandrasekaran P, et al. A novel STAT1 mutation associated with disseminated mycobacterial disease. J Clin Immunol. (2012) 32:681–9. doi: 10.1007/s10875-012-9659-2
8. Chen X, Chen J, Chen R, Mou H, Sun G, Yang L, et al. Genetic and functional identifying of novel STAT1 loss-of-function mutations in patients with diverse clinical phenotypes. J Clin Immunol. (2022) 42:1778–94. doi: 10.1007/s10875-022-01339-w
9. Mertens C, Zhong M, Krishnaraj R, Zou W, Chen X, Darnell JE Jr. Dephosphorylation of phosphotyrosine on STAT1 dimers requires extensive spatial reorientation of the monomers facilitated by the N-terminal domain. Genes Dev. (2006) 20:3372–81. doi: 10.1101/gad.1485406
10. Braunstein J, Brutsaert S, Olson R, Schindler C. STATs dimerize in the absence of phosphorylation. J Biol Chem. (2003) 278:34133–40. doi: 10.1074/jbc.M304531200
11. Mao X, Ren Z, Parker GN, Sondermann H, Pastorello MA, Wang W, et al. Structural bases of unphosphorylated STAT1 association and receptor binding. Mol Cell. (2005) 17:761–71. doi: 10.1016/j.molcel.2005.02.021
12. Asano T, Utsumi T, Kagawa R, Karakawa S, Okada S. Inborn errors of immunity with loss- and gain-of-function germline mutations in STAT1. Clin Exp Immunol. (2022). doi: 10.1093/cei/uxac106
13. Chodisetti SB, Fike AJ, Domeier PP, Schell SL, Mockus TE, Choi NM, et al. Serine phosphorylation of the STAT1 transactivation domain promotes autoreactive B cell and systemic autoimmunity development. J Immunol. (2020) 204:2641–50. doi: 10.4049/jimmunol.2000170
14. O'Marcaigh AS, Puck JM, Pepper AE, De Santes K, Cowan MJ. Maternal mosaicism for a novel interleukin-2 receptor gamma-chain mutation causing X-linked severe combined immunodeficiency in a Navajo kindred. J Clin Immunol. (1997) 17:29–33. doi: 10.1023/A:1027332327827
15. Sakamoto M, Kanegane H, Fujii H, Tsukada S, Miyawaki T, Shinomiya N. Maternal germinal mosaicism of X-linked agammaglobulinemia. Am J Med Genet. (2001) 99:234–7. doi: 10.1002/1096-8628(2001)9999:9999<::AID-AJMG1159>3.0.CO;2-M
16. Holzelova E, Vonarbourg C, Stolzenberg MC, Arkwright PD, Selz F, Prieur AM, et al. Autoimmune lymphoproliferative syndrome with somatic Fas mutations. N Engl J Med. (2004) 351:1409–18. doi: 10.1056/NEJMoa040036
17. Yamada M, Okura Y, Suzuki Y, Fukumura S, Miyazaki T, Ikeda H, et al. Somatic mosaicism in two unrelated patients with X-linked chronic granulomatous disease characterized by the presence of a small population of normal cells. Gene. (2012) 497:110–5. doi: 10.1016/j.gene.2012.01.019
18. Hsu AP, Sowerwine KJ, Lawrence MG, Davis J, Henderson CJ, Zarember KA, et al. Intermediate phenotypes in patients with autosomal dominant hyper-IgE syndrome caused by somatic mosaicism. J Allergy Clin Immunol. (2013) 131:1586–93. doi: 10.1016/j.jaci.2013.02.038
19. Nakagawa K, Gonzalez-Roca E, Souto A, Kawai T, Umebayashi H, Campistol JM, et al. Somatic NLRP3 mosaicism in Muckle-Wells syndrome. A genetic mechanism shared by different phenotypes of cryopyrin-associated periodic syndromes. Ann Rheum Dis. (2015) 74:603–10. doi: 10.1136/annrheumdis-2013-204361
20. Lee JS, An Y, Yoon CJ, Kim JY, Kim KH, Freeman AF, et al. Germline gain-of-function mutation of STAT1 rescued by somatic mosaicism in immune dysregulation-polyendocrinopathy-enteropathy-X-linked-like disorder. J Allergy Clin Immunol. (2020) 145:1017–21. doi: 10.1016/j.jaci.2019.11.028
21. Mensa-Vilaro A, Bravo Garcia-Morato M, de la Calle-Martin O, Franco-Jarava C, Martinez-Saavedra MT, Gonzalez-Granado LI, et al. Unexpected relevant role of gene mosaicism in patients with primary immunodeficiency diseases. J Allergy Clin Immunol. (2019) 143:359–68. doi: 10.1016/j.jaci.2018.09.009
22. Tsumura M, Okada S, Sakai H, Yasunaga S, Ohtsubo M, Murata T, et al. Dominant-negative STAT1 SH2 domain mutations in unrelated patients with Mendelian susceptibility to mycobacterial disease. Hum Mutat. (2012) 33:1377–87. doi: 10.1002/humu.22113
23. Zhang W, Chen X, Gao G, Xing S, Zhou L, Tang X, et al. Clinical relevance of gain- and loss-of-function germline mutations in STAT1: A systematic review. Front Immunol. (2021) 12:654406. doi: 10.3389/fimmu.2021.654406
24. Ye F, Zhang W, Dong J, Peng M, Fan C, Deng W, et al. A novel STAT1 loss-of-function mutation associated with Mendelian susceptibility to mycobacterial disease. Front Cell Infect Microbiol. (2022) 12:1002140. doi: 10.3389/fcimb.2022.1002140
25. Kagawa R, Fujiki R, Tsumura M, Sakata S, Nishimura S, Itan Y, et al. Alanine-scanning mutagenesis of human signal transducer and activator of transcription 1 to estimate loss- or gain-of-function variants. J Allergy Clin Immunol. (2017) 140:232–41. doi: 10.1016/j.jaci.2016.09.035
26. Boudjemaa S, Dainese L, Heritier S, Masserot C, Hachemane S, Casanova JL, et al. Disseminated bacillus calmette-guerin osteomyelitis in twin sisters related to STAT1 gene deficiency. Pediatr Dev Pathol. (2017) 20:255–61. doi: 10.1177/1093526616686255
27. Ong RYL, Chan SB, Chew SJ, Liew WK, Thoon KC, Chong CY, et al. Disseminated bacillus-calmette-guerin infections and primary immunodeficiency disorders in Singapore: A single center 15-year retrospective review. Int J Infect Dis. (2020) 97:117–25. doi: 10.1016/j.ijid.2020.05.117
Keywords: STAT1 loss-of-function, BCG osteomyelitis, Mendelian susceptibility to mycobacterial diseases, germline mosaicism, case report
Citation: Lim QY, Leung D, Lam CK, Yang X, Cheong KN, Yik AKH, Yang J, Chan K-W, Lee PPW, Tsumura M, Au EYL, Rosa Duque JS, Okada S and Lau YL (2025) Case report: A novel de novo germline loss-of-function mutation in the STAT1 transactivation domain in two Chinese siblings, with the elder sibling presenting with multifocal Bacillus Calmette–Guerin osteomyelitis. Front. Immunol. 15:1504816. doi: 10.3389/fimmu.2024.1504816
Received: 01 October 2024; Accepted: 09 December 2024;
Published: 07 January 2025.
Edited by:
Sudhir Gupta, University of California, Irvine, United StatesReviewed by:
Rajeev Kumar Pandey, Thermo Fisher Scientific, IndiaEleuterio A. Sánchez Romero, European University of Madrid, Spain
Angel Robles-Marhuenda, La Paz Hospital, Spain
Copyright © 2025 Lim, Leung, Lam, Yang, Cheong, Yik, Yang, Chan, Lee, Tsumura, Au, Rosa Duque, Okada and Lau. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Satoshi Okada, c29rYWRhQGhpcm9zaGltYS11LmFjLmpw; Jaime S. Rosa Duque, anNyZHVxdWVAaGt1Lmhr
†These authors have contributed equally to this work
‡ORCID: Qin Ying Lim, orcid.org/0009-0009-4479-5945
Daniel Leung, orcid.org/0000-0002-9360-6233
Miyuki Tsumura, orcid.org/0000-0002-8272-9588
Jaime S. Rosa Duque, orcid.org/0000-0002-5292-5510
Satoshi Okada, orcid.org/0000-0002-4622-5657
Yu Lung Lau, orcid.org/0000-0002-4780-0289