
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 18 December 2024
Sec. Inflammation
Volume 15 - 2024 | https://doi.org/10.3389/fimmu.2024.1502242
 Agata Lesiak1
Agata Lesiak1 Paulina Paprocka1Urszula Wnorowska2Angelika Mańkowska1Grzegorz Król1Katarzyna Głuszek1
Paulina Paprocka1Urszula Wnorowska2Angelika Mańkowska1Grzegorz Król1Katarzyna Głuszek1 Ewelina Piktel3
Ewelina Piktel3 Jakub Spałek1,4Sławomir Okła1,4Krzysztof Fiedoruk2Bonita Durnaś1,5
Jakub Spałek1,4Sławomir Okła1,4Krzysztof Fiedoruk2Bonita Durnaś1,5 Robert Bucki2*
Robert Bucki2*Acne vulgaris (AV) is a chronic inflammatory condition of the pilosebaceous units characterized by multiple immunologic, metabolic, hormonal, genetic, psycho-emotional dysfunctions, and skin microbiota dysbiosis. The latter is manifested by a decreased population (phylotypes, i.e., genetically distinct bacterial subgroups that play different roles in skin health and disease) diversity of the predominant skin bacterial commensal - Cutinbacterium acnes. Like in other dysbiotic disorders, an elevated expression of endogenous antimicrobial peptides (AMPs) is a hallmark of AV. AMPs, such as human β-defensins, cathelicidin LL-37, dermcidin, or RNase-7, due to their antibacterial and immunomodulatory properties, function as the first line of defense and coordinate the host-microbiota interactions. Therefore, AMPs are potential candidates for pharmaceutical prophylaxis or treating this condition. This study outlines the current knowledge regarding the importance of AMPs in AV pathomechanism in light of recent transcriptomic studies. In particular, their role in improving the tight junctions (TJs) skin barrier by activating the fundamental cellular proteins, such as PI3K, GSK-3, aPKC, and Rac1, is discussed. We hypothesized that the increased expression of AMPs and their patterns in AV act as a compensatory mechanism to protect the skin with an impaired permeability barrier. Therefore, AMPs could be key determinants in regulating AV development and progression, linking acne-associated immune responses and metabolic factors, like insulin/IGF-1 and PI3K/Akt/mTOR/FoxO1 signaling pathways or glucotoxicity. Research and development of anti-acne AMPs are also addressed.
In 2010, acne vulgaris (AV) was in the top 10 most prevalent diseases worldwide, affecting 85% of adolescents in Westernized populations (1). Clinically, AV (ICD-11, ED80; according to the International Classification of Diseases 11th Revision; https://icd.who.int/) is considered as a chronic and multifactorial inflammatory disease of the pilosebaceous unit (PSU), i.e., a structure composed of the hair follicle, sebaceous gland, and arrector pili muscle, and associated with excessive sebum production (hyperseborrhea). However, from a physiological perspective, AV is perceived as a metabolic syndrome (MetS) of the sebaceous follicle, and like obesity, type 2 diabetes (T2D), or insulin resistance, belongs to the mechanistic target of rapamycin complex 1 (mTORC1)-driven diseases or so-called ‘Western civilization diseases’ (2, 3). mTORC1, as part of insulin/insulin-like growth factor-1 (IGF1) signaling pathways, is a key regulator of lipid and energy metabolism. Western dietary model, particularly a high hyperglycemic load and dairy product intake, contributes to AV development by overstimulating these pathways (2–4). Microbiologically, AV is a condition of imbalanced microbial skin colonization, i.e., dysbiosis, specifically by Cutibacterium acnes (5). Therefore, complex reciprocal interactions between dysregulated (i) lipid metabolism of the sebaceous gland (manifested as hyperseborrhea), (ii) hormonal homeostasis (linked with follicular hyperkeratinization, (iii) microbial skin colonization (specifically by C. acnes), and (iv) immune responses, are behind AV pathomechanism. Overall, these factors contribute to the formation of acne lesions, ranging from non-inflammatory comedones to inflammatory papules, pustules, and nodules (2).
The skin’s epidermis is a physical, chemical, and immune barrier against external infectious and non-infectious insults, also serving as an ecological niche for various microorganisms, collectively known as skin microbiota (Figure 1) (7, 8). In between 104 - 106 of bacteria inhabit each square centimeter of the skin (9), which represent 13 phyla with 622 prokaryotic species, where Actinomycetota (or Actinobacteria) (37.5%), Proteobacteria (25.4%), Firmicutes (25.1%), and Bacteroidota (8.8%) are the predominant ones (6). Their composition and diversity vary significantly between subjects and across sites, with sebaceous sites dominated by lipophilic Cutibacterium species (Actinomycetota). In contrast, bacteria that thrive in humid environments, such as Staphylococcus (Firmicutes) and Corynebacterium (Actinobacteria) species, are preferentially abundant in moist areas. The dry regions show the highest diversity, with variable colonization of the four main phyla.
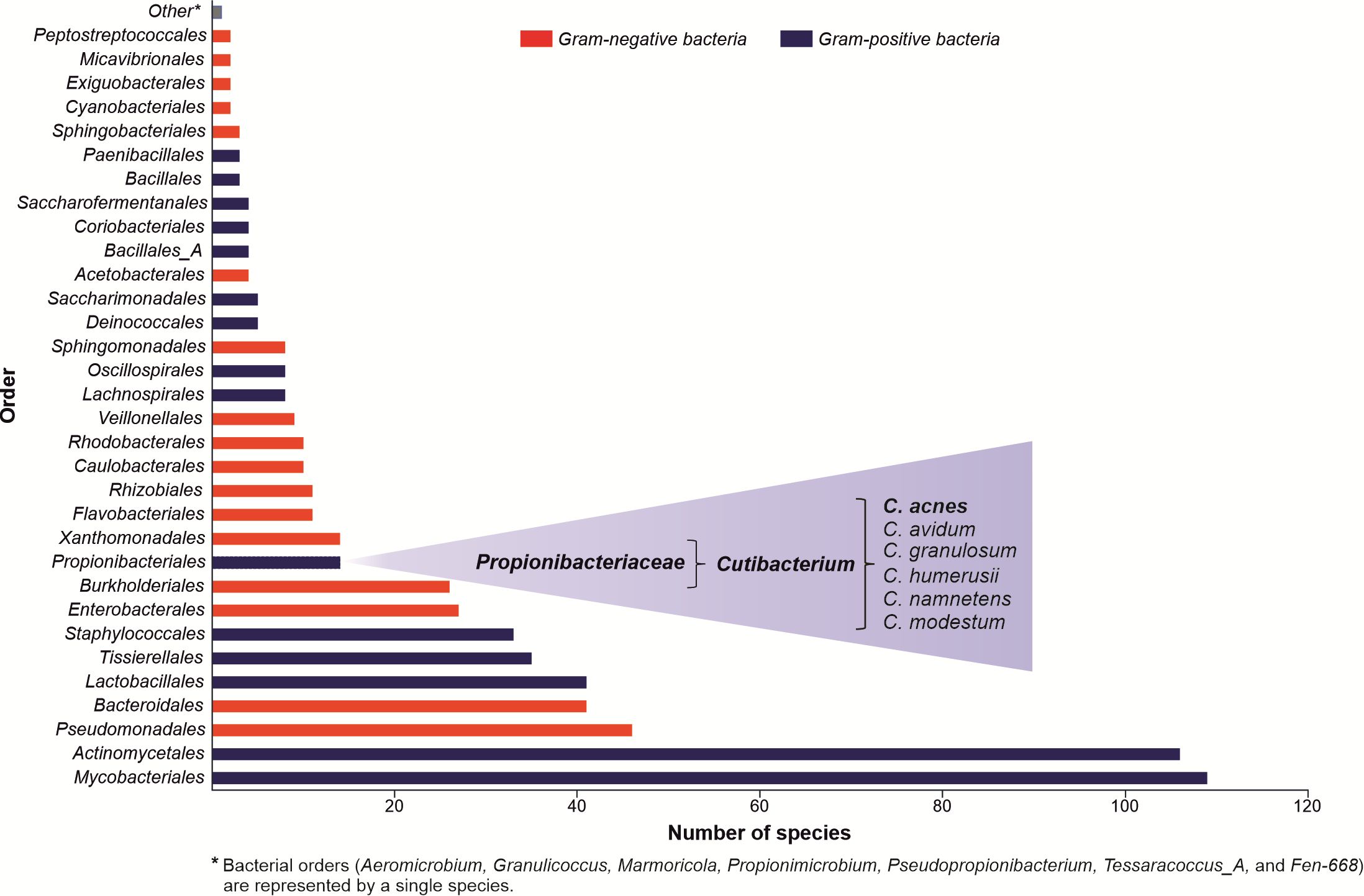
Figure 1. Composition of the skin microbiota. The graph was created based on data collected by the Skin Microbial Genome Collection (SMGC) project. The figure’s information summarizes the published experimental work results (6).
The skin microbiota coexists in a symbiotic or commensal relation with the human host, i.e., in a eubiosis state, and acting as a colonization resistance barrier supports a nonspecific skin defense system. Specifically, C. acnes and Staphylococcus epidermidis are critical for skin homeostasis (10). Dysbiosis, a condition of imbalanced microbiota manifested by qualitative and/or quantitative changes in its composition, is a hallmark of several skin disorders, including AV (5, 9, 11–13). Several mechanisms of C. acnes-induced acne aggravation have been proposed, such as augmentation of lipogenesis, alteration of lipid composition in sebum, or exaggeration of host immune responses (5, 14). Multiple innate and adaptive immune mediators mediate the latter, especially an abnormal production of endogenous antimicrobial peptides (AMPs), a hallmark of various dermatoses, such as rosacea, psoriasis, atopic dermatitis, and AV (15–20). According to the transcriptomic analysis of AV lesions, genes encoding AMPs belonging to human β-defensins (hBDs) and S100 family proteins are among the ten most overexpressed ones (Figure 2) (21, 22). AMPs are small, mainly cationic peptides with potent antimicrobial and immunomodulatory properties; hence, they are also known as host-defense peptides (HDPs). AMPs contribute to skin homeostasis, serving as (i) the first line of defense against microbial invasion, (ii) coordinators of host-microbiota interactions, and (iii) agents promoting skin integrity and regeneration (23). Specifically, due to the sequestration of pro-inflammatory bacterial factors, such as lipooligosaccharide (LPS) or lipoteichoic acids (LTAs), and suppression of pro-inflammatory cytokines secretion, AMPs act as sensors of microbial load and control microbiota-induced immune defense. However, since various bacterial species- or strain-specific factors, e.g., C. acnes virulence factors, can trigger the production of AMPs, an imbalanced proportion or relative abundance of specific bacterial species in skin microbiota or strains may cause an uncontrolled inflammatory response (15–20).
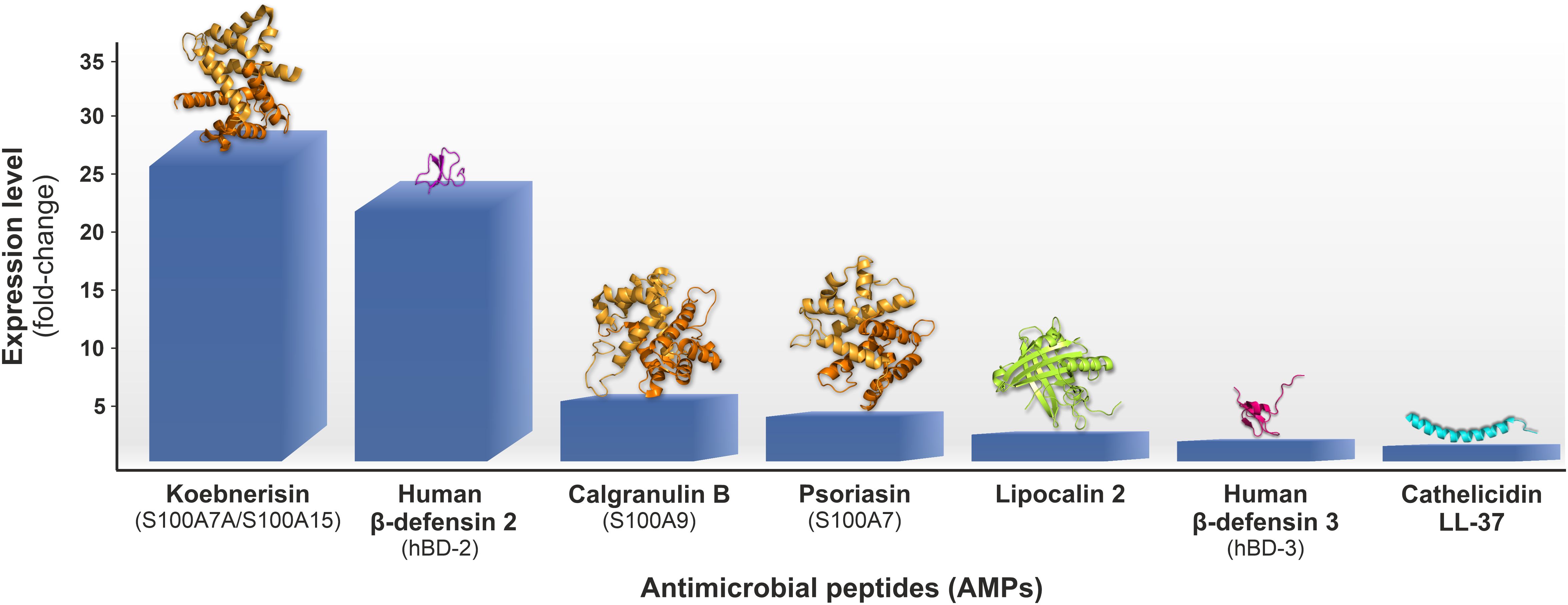
Figure 2. Antimicrobial peptides (AMPs) genes upregulation fold change (AV involved vs. non-involved skin) estimated by the microarray transcriptomic analysis of biopsies from early inflammatory AV lesions, i.e., comedones shading into small red papules, collected from twenty subjects with moderate to severe AV reported by Kelhälä et al. (21); 3D protein models were obtained from RCSB Protein Data Bank (https://www.rcsb.org).
The use of antibiotics, such as clindamycin or macrolides, in treating AV leads not only to the emergence of antibiotic-resistant strains of C. acnes but also to other skin commensals. For example, ~30% of S. epidermidis isolates from AV patients show resistance to erythromycin, roxithromycin, and clindamycin (24). Therefore, AMPs are promising candidates for novel anti-AV therapies. However, understanding the relationship between AMPs and eubiotic/dysbiotic skin conditions, as well as the host metabolic, hormonal, and genetic factors associated with AV pathophysiology, is essential for developing safe and efficient AMPs-based medications against AV and other dermatoses (25, 26). Here, we summarize the current knowledge regarding the significance of AMPs in AV pathomechanism and progress in research and development of anti-acne AMPs.
Antimicrobial peptides (AMPs) are cell membrane-targeting, low-molecular-weight molecules with fewer than 100 amino acid residues, characterized by a broad-spectrum activity against various bacterial, fungal, parasitic, and viral agents (27, 28). AMPs are a part of innate immunity and the first line of defense against microbial pathogens, while prokaryotes use them for interspecies competition, e.g., competitive exclusion (29). Additionally, AMPs regulate the host’s pro-inflammatory and anti-inflammatory responses, either with infectious and non-infectious etiology, as well as a variety of cellular processes, such as chemotaxis, autophagy, apoptosis, cell differentiation, or wound healing (29). Hence, AMPs commonly function as ‘alarmins’ or damage-associated molecular patterns (DAMPs), i.e., molecules produced by injured or dying cells that initiate signaling pathways responsible for various physiological and pathological processes. Thus, the term host defense peptides (HDPs) has emerged to encompass their pleiotropy (30).
The secondary structure divides AMPs into (i) α-helical- (e.g., cathelicidins), (ii) β-sheet- (e.g., human α- and β-defensins) or (iii) αβ-containing peptides (e.g., θ-defensins), and (iv) elongated or loop and rich in proline, tryptophan or glycine AMPs (e.g., indolicidin) (31). In aqueous solutions, the conformation of α-helical AMPs is disordered and adopts an amphipathic helical structure upon contact with biological membranes. This process compromises their integrity and increases permeability, which ultimately causes cell death (32). Several factors influence AMPs action, including peptide size, amino acid sequence, charge, conformation, hydrophobicity, and amphipathic characteristics. The biological activity of AMPs depends on various parameters, including peptide size, amino acid sequence, charge, conformation, hydrophobicity, and amphipathic nature.
Despite their distinct origin and various structural and physicochemical features AMPs share common characteristics (33). The vast majority of AMPs are positively charged (charge from +2 to +11) and composed of 5 to 100 amino acids oligopeptides, rich in leucine, glycine, and lysine (>8%), but not in methionine and tryptophan (<2%) (34). Thus, they are commonly known as cationic antimicrobial peptides (CAMPs). The electrostatic interactions between positively charged peptides and negatively charged bacterial surfaces, e.g., lipopolysaccharides (LPSs) or teichoic acids (TAs), are responsible for their antimicrobial activity. Moreover, AMPs exhibit an amphipathic structure due to hydrophobic and hydrophilic regions, which dictates their structural flexibility and, along with hydrophobicity, determines the membranolytic properties of these compounds and selective toxicity toward microbial cells (35).
Three distinct processes are involved in the production of AMPs: (i) classical ribosomal synthesis, (ii) non-ribosomal synthesis, and (iii) proteolytic processing. The former is utilized to synthesize human β-defensins (hBds) or histatins. In contrast, the non-ribosomal synthesis is typical for bacteria, and it is performed by non-ribosomal peptide synthetases, allowing for the incorporation of non-proteinogenic amino acids into AMPs. Several protein modifications, such as hydroxylation, glycosylation, and cyclization, are also common in AMPs synthesized by this pathway. Finally, certain AMPs, so-called ‘mystery peptides’, can be formed through the proteolytic cleavage of larger proteins with different functions (36). In addition, many AMPs are produced as inactive precursors, requiring proteolytic cleavage to gain functional activity.
The expression of AMPs can be constitutive or inducible. Constitutively expressed AMPs accumulate in high concentrations as inactive precursors in cell granules and are released locally at infection and/or inflammation sites. Inducible AMP expression is triggered by microbial-associated-molecular patterns (MAMPs) and host immune effector factors, such as cytokines (37).
The electrostatic interactions between positively charged peptides and negatively charged bacterial surfaces, e.g., LPSs or TAs, are responsible for their antimicrobial activity. In eukaryotic cell membranes, sphingomyelin and phosphatidylcholine due to their neutral charge at physiological pH, prevent interactions with AMPs. Hence, membrane lipid composition substantially affects the interaction between AMPs and microbial cell envelopes, determining their specificity and activity toward microbial cells. For instance, the cell wall of Gram-positive bacteria, such as C. acnes, comprises multiple layers of peptidoglycan (murein), 40-80 nm in diameter, stabilizing bacterial cell shape. Peptidoglycan is a polymer of the disaccharide N-acetylglucosamine and N-acetylmuramic acid cross-linked by peptide bridges with teichoic and lipoteichoic acids (LTAs), linked to N-acetylmuramic acid and membrane lipids, respectively (38). In addition, Gram-positive bacteria have more negatively charged phosphatidylglycerol with saturated, unsaturated, and branched fatty acids than Gram-negative species (39). Although plasma membrane phospholipid content in Gram-positive bacteria varies by species., a high concentration of phosphatidylglycerol and its derivatives, including lysyl phosphatidylglycerol, cardiolipins, and phosphatidylethanolamine, is characteristic of this group (40).
Subsequently, AMPs are classified as ‘membrane-acting’ and ‘non-membrane-acting’ peptides. The former AMPs disrupt microbial membranes, leading to cell death by osmotic shock via (i) the barrel stave mode., (ii) the carpet model, or (iii) the toroidal pore model (31). In contrast, ‘non-membrane-acting peptides’ can penetrate membranes without damaging them and target basic metabolic processes like protein or nucleic acid synthesis and metabolic activity (41). AMPs can also inhibit bacterial cell wall synthesis by binding with lipid II, a glycolipid precursor for this process, or by inhibiting its formation, e.g., by a lipoglycopeptide rhamoplanin (29, 42). Likewise, the biosynthesis of TAs and LTAs AMPs can be affected by AMPs (43).
Due to the cationic and amphiphilic nature of AMPs, they can directly interact with cell membranes with high anionic phospholipid content, accumulating continuously at the membrane surface and inducing structural or conformational changes (44). In sufficient concentrations, AMPs increase cell membrane permeability, breaking the membrane and releasing cellular content (45). In general, the cell membrane-directed activity of AMPs is determined by membrane structural/conformational changes and the peptide-lipid ratio (46). When the latter is high, AMPs gain access to the membrane’s hydrophobic interior, leading to cell death. Otherwise, AMPs remain stacked in a parallel orientation to the cell membrane’s surface (47).
After initial electrostatic and/or hydrophobic interactions, AMPs self-organization on the cell membrane adapts one of the aforementioned models (48, 49). In the barrel-stave pore model, AMPs initially oriented parallel to the cell membrane are inserted perpendicular to the lipid bilayer, forming trans-membrane channels through aggregation and conformational change. The amphipathic structure, α-helical or β-sheet, is crucial for pore formation since hydrophobic regions interface with membrane lipids, whereas hydrophilic residues generate channel lumens. A limited number of AMPs, such as protegrin and alamethicin, may exploit this mechanism due to their scant α-helical and β-sheet residues (50). In contrast, the toroidal pore model is not based on peptide-peptide interactions, as peptide helices attach to membrane lipids to form pores, inducing bend deformation in the lipid molecules. In the carpet model, AMPs, after reaching their maximum concentration, cover the entire membrane surface, causing degradation in a surfactant-like manner, which finally degrades the membrane by generating micelles. Several peptides utilize this model, including cathelicidin LL-37, indolicidin, and aureins (47, 48). The aggregate model is similar to the carpet mechanism in that AMPs are inserted into the cell membrane after they reach a threshold concentration. This interaction triggers a spatial change in the AMPs, allowing micelle-like complexes to form with the lipids and transverse the lipid bilayer in a peptide-lipid complex. These transmembrane aggregates of lipids, AMPs, and water can create channels for ions and intracellular content leakage, causing cell death. This mechanism also allows AMPs to be delivered into the cell, resulting in intracellular killing (51). Multiple intracellular processes can be affected by AMPs, including protein biosynthesis, nucleic acid synthesis, or cell division (52). AMPs binding to nucleic acids results in conformation changes followed by inhibition of DNA, RNA, and protein synthesis utilizing a mechanism similar to histone-DNA interactions. For instance, buforin II and indolicidin interfere with nucleic acids (52). The latter, for example, has a high affinity for the double-stranded binding of ds[AT], ds[CG], ds[AG] and a lower affinity for ds[GT] as well as prevents DNA relaxation via inactivation of DNA topoisomerase I (53, 54). In contrast, human neutrophil peptide-1 (HNP-1) inactivates the DNA damage response system, inducing programmed bacterial cell death, by blocking the interaction of RecA with single-stranded DNA (ssDNA) (55). Similarly, proline-rich AMPs interfere with bacterial protein folding by inhibiting the bacterial heat shock protein (DnaK), and human alpha-defensin 5 (HD5) inhibits bacterial cell division (56, 57).
Clinically, skin lesions, such as open comedones (blackheads) and closed comedones (whiteheads), papules, pustules, cysts, and nodules, on the face, back, chest, and other sebaceous gland-rich areas are typical manifestations of AV (58), observed in ~80% of young adults and adolescents (59, 60). Open and closed comedones are generally benign and non-inflammatory, whereas papular and pustular lesions are mild to moderately inflammatory, and nodules represent the most severe type of lesions. However, this classification may not accurately reflect the role of inflammation in AV pathogenesis, which appears to be significant at all stages of acne development, perhaps even before comedo formation (61).
On the transcriptome level, depending on the study, AV upregulates 211 or 904 genes and downregulates 18 or 395 genes (21, 22). The upregulated genes are primarily involved in inflammation, including mediators of innate/adaptive immunity and matrix remodeling processes (22). The former genes include interleukins 8 (IL-8) and 1 (IL-1), chemokine (C-C motif) ligands (CCLs), cytokines associated with the IL-17/Th17 pathway activation, such as IL-23, IL-6, and transforming growth factor β (TGF-β), as well as various AMPs (21). In fact, genes encoding specific AMPs, i.e., S100 family proteins and hBDs, were among the top ten overexpressed in both studies (21, 22). For instance, in the study by Kelhälä et al., the gene encoding S100A15 (S100A7A) protein, also known as koebnerisin, was the most upregulated one, with a fold change value of 29 (Figure 2) (21). Furthermore, the genes encoding matrix metalloproteinases (MMPs) and protease inhibitors (PIs), like elafin (peptidase inhibitor 3, PI3) or skin-derived antileukoprotease (SKALP), represented the leading overexpressed matrix remodeling factors (21, 22).
In general, the pathophysiology of AV involves the sequence of (i) androgen-induced sebum hypersecretion (hyperseborrhea), (ii) hyperkeratinization and hyperproliferation along with abnormal keratinocyte differentiation in hair follicles, (iii) dysbiosis of skin microbiota characterized by a decrease in population (phylotypes, i.e., genetically distinct bacterial subgroups that play different roles in skin health and disease) diversity of lipophilic skin commensal C. acnes, and (iv) aberrant host inflammatory response (Figure 3) (62). Recently, a mechanical skin barrier impairment has also been proposed as a novel explanatory variable of AV pathomechanism (63). Notably, AMPs appear to be key players in the enhancement of impaired, e.g., by C. acnes, skin barrier function through the activation of various fundamental cellular proteins, such as phosphoinositide 3-kinases (PI3K), glycogen synthase kinase-3 (GSK3), atypical protein kinase C (aPKC), and Ras-related C3 botulinum toxin substrate 1 (Rac1) (Figure 4) (63).
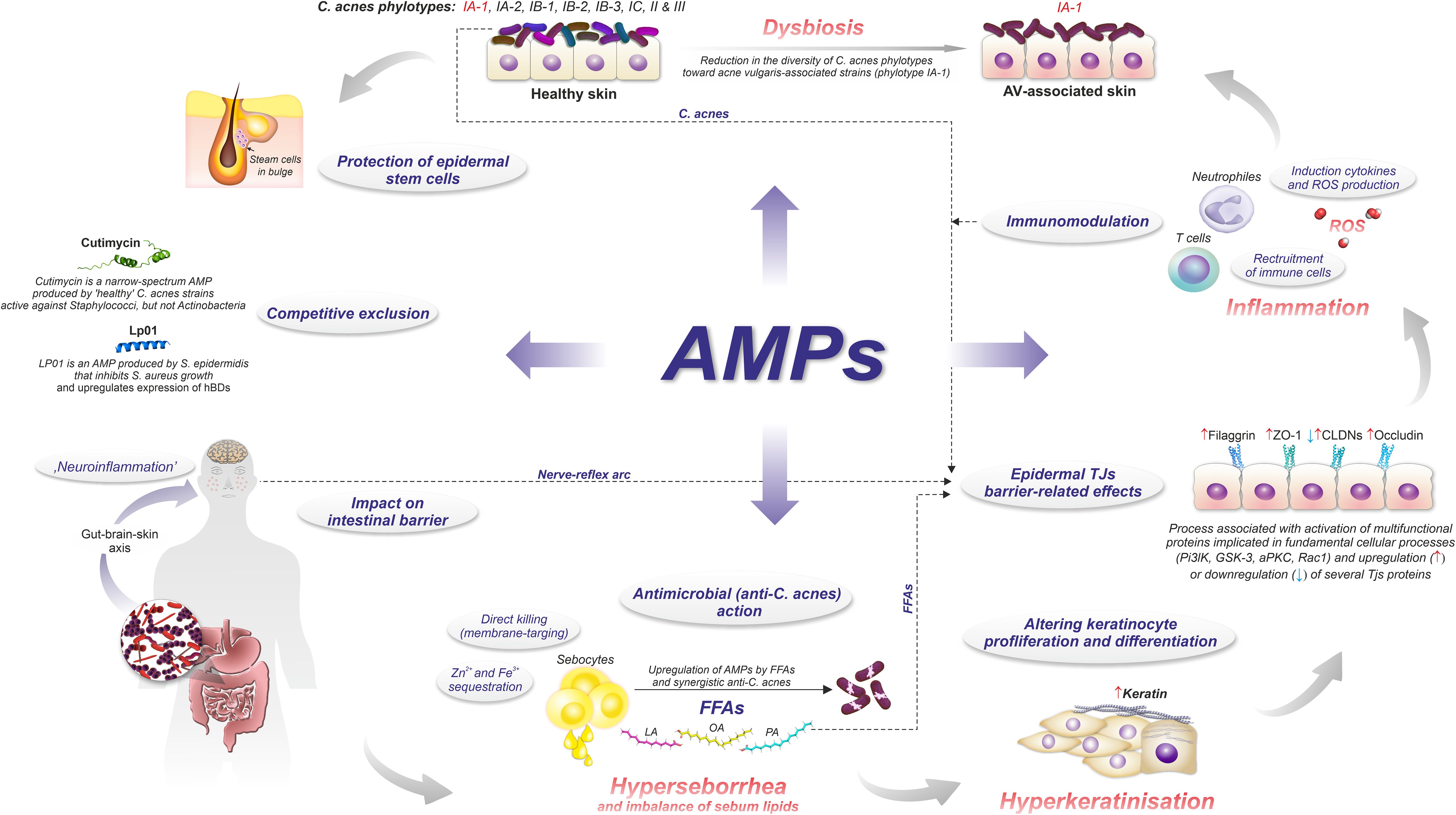
Figure 3. Role of AMPs in AV pathomechanism.
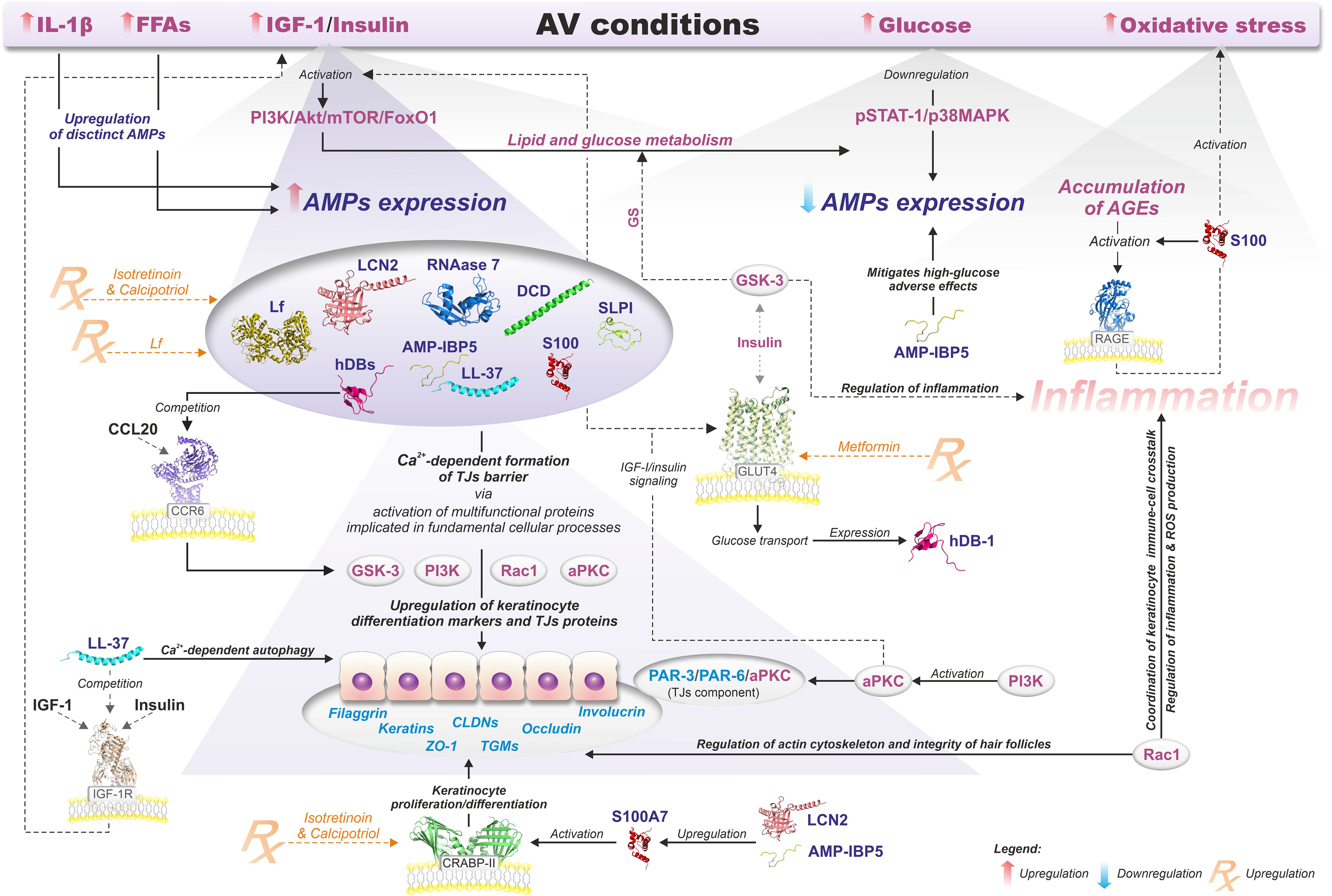
Figure 4. Relationship between AMPs and the modulation of epidermal TJs barrier function in AV metabolic background context. Details are discussed in the text with the use of citations. All information presented in the figure summarizes the results of the published experimental work (19, 63–112).
The androgen-induced hyperseborrhea is mediated via overstimulation of insulin, insulin-like growth factor (IGF-1), kinase Akt, and mTORC1 signaling pathways (insulin/IGF-1/Akt/mTORC1) (113–116). Accordingly, a decreased responsiveness of the sebaceous glands to insulin-mediated signals, i.e., (insulin resistance), is compensated by the upregulation of IGF-1, which stimulates the production of androgens (115).
Activation of mTORC1 causes the accumulation of triacylglycerols inducing adipo- and lipogenesis and inhibiting lipid catabolism, e.g., lipolysis and β-oxidation. Moreover, it promotes sebaceous lipogenesis via the PI3K/Akt/FoxO1/mTORC1 pathway and induction of the sterol regulatory element binding protein 1 (SREBP1) (3, 64, 65). In this process, Akt-mediated phosphorylation of nuclear forkhead transcription factors O1 and O3 (FoxO1 and FoxO3) initiates their extrusion into the cytoplasm, followed by the stimulation of lipogenic and proinflammatory transcription factors, such as androgen receptor (AR), sterol regulatory element-binding transcription factor 1 (SREBF1), peroxisome proliferator-activated receptor γ (PPARγ), and signal transducer and activator of transcription 3 (STAT3). At the same time, FoxO1-dependent expression of GATA binding protein 6 (GATA6) is decreased. GATA6 is a transcription factor vital for keratinocyte homeostasis, which prevents hyperkeratinization of the infundibulum. Accordingly, its downregulation in PSUs of AV patients has been reported (117). Additionally, the Akt-mediated phosphorylation of mouse-double minute 2 (MDM2) promotes the degradation of the transcription factor p53, resulting in decreased p53-mediated expression of FoxO1, FoxO3, and other p53 target genes (117).
Furthermore, hyperglycemic carbohydrates and insulinotropic dairy products also provoke the insulin/IGF-1/Akt/mTORC1 signaling, linking AV with a Western diet and the gut microbiota via gut-skin and gut-brain-skin axes (14, 118, 119). For instance, in animal models, long-term stress induces dysbiosis of the gut microbiota, e.g., characterized by a deficit of probiotic genera like Lactobacillus and Bifidobacterium (64, 65, 120). The intestinal microbiota-derived metabolites, such as short-chain fatty acids (SCFAs), could modulate innate and adaptive immunity through multiple signaling pathways, including mTOR (121, 122). Likewise, the gut microbiota, through the production and release into the blood neurotransmitters, such as acetylcholine, serotonin, norepinephrine, and increasing intestinal permeability, may contribute to stress-induced acne exacerbation by gut-brain-skin axis (14, 123). Specifically, emotional stresses appear to affect the function of PSUs, contributing to the development and/or aggravation of pre-existing acne via stimulation of (i) hormone production, (ii) neuropeptides, such as corticotropin-releasing hormone (CRH), (iii) melanocortins, such as alpha-melanocyte-stimulating hormone (α-MSH) and adrenocorticotropic hormone (ACTH), and substance P, as well as (iv) proinflammatory cytokines (124). Hormones, for example, via the CRH receptor 1, enhance sebaceous lipid synthesis and the release of IL-6 and IL-8 by sebaceous glands. In addition, CRH stimulates the secretion of adrenocorticotropic hormone (ACTH), hence, the production of dehydroepiandrosterone by the adrenal glands (125, 126). Furthermore, CRH activates the hypothalamic-pituitary-adrenal (HPA) axis resulting in cortisol release from the adrenal glands. Cortisol as a potent insulin-antagonistic hormone inhibits insulin secretion, promotes insulin resistance and hyperglycemia (127). Increased levels of cortisol can also suppress hypothalamic-pituitary-gonadal (HPG) axis and gonadotropin-releasing hormone (GnRH) secretion leading to lowers androgen production. Similarly, Substance P participates in the regulation of glucose metabolism via insulin signaling-associated pathways, and in rats its intravenous administration leads to hypoinsulinemia, hyperglucagonemia, and subsequently to hyperglycemia (128, 129). Melanocortins regulate glucose homeostasis via central nervous system pathways, primarily in the hypothalamus, via melanocortin receptors (MC1R–MC5R) (130). Activation of these receptors, especially MC4R, has been associated with improved insulin sensitivity and glucose homeostasis (130). Moreover, insulin-dependent regulation of intracellular glucose levels significantly impacts the expression of hBDs (Figure 4) (66).
Apart from hyperseborrhea per se, an imbalanced proportion of sebum-specific lipids plays a pivotal role in AV pathomechanism. The sebum comprises a diverse array of lipids, such as triglycerides and free fatty acids (FFAs) (40%–60%), wax esters (20%–30%), squalene (10%–20%), and cholesterol and its esters (2%–10%) (115). Accordingly, sebum of patients with AV is deficient in essential FFAs, like linoleic acid (LA), and relatively abundant in proinflammatory lipids, such as monounsaturated fatty acids (MUFAs) and lipoperoxides. The latter are products of squalene peroxidation and significantly contribute to the hyperkeratinization of the PSUs, inducing keratinocyte proliferation/differentiation and can initiate an inflammatory response, activating PPARs-dependent signaling pathways and triggering the secretion of pro-inflammatory cytokines (61, 131, 132). Likewise, palmitic acid (PA) is a powerful stimulator of the NLRP3 inflammasomes in macrophages (122). In contrast, LA has anti-inflammatory properties, and in human monocytes, it can suppress NF-κB signaling by a PPARγ-dependent mechanism (133). In addition, β-oxidated derivatives of linoleic acid (LA) serve as precursors for acetyl-CoA - a key metabolite for the biosynthesis of lipids. Therefore, a deficiency of LA in the hair follicle disturbs the composition of squalene, wax esters, and sphingolipids. Furthermore, FFAs, by promoting the expression of human β-defensin 2 (hBD-2) in sebocytes, may contribute to the skin’s natural barrier (67). Similarly, sphingolipids, such as ceramides, as essential epidermal barrier components controlling transepidermal water loss and directing processes of keratinocyte proliferation, differentiation, and apoptosis, are implicated in numerous dermatoses (134). In AV, sphingolipid deficiency stimulates follicular hyperkeratosis and abnormal desquamation of epithelium, resulting in pores clogging and comedone formation. Overall, sebum-rich and hypoxic conditions of comedone act as a dysbiotic factor that promotes their colonization by anaerobic and lipophilic species, like specific phylotypes of C. acnes (see below). This shift in the C. acnes population toward so-called ‘acne-associated’ phylotypes (or strains), characterized by enhanced inflammatory potential compared to the ‘healthy’ phylotypes, ultimately leads to intra-species dysbiosis (10, 135–138). As an illustration, Dagnelie et al. reported that the innate immune response of healthy skin explants exposed to a mix of three C. acnes phylotypes was weaker when compared to their individual applications. Therefore, the diversity of the C. acnes population appears to play a vital role in maintaining the cutaneous microbiota in eubiosis and may serve as a biomarker of healthy skin (139).
C. acnes (formerly Propionibacterium acnes) is a lipophilic, slow-growing, aerotolerant, anaerobic, rod-shaped Gram-positive bacterium important for human health as a predominant cutaneous and mucous membrane commensal, as well as an opportunistic pathogen, mainly involved in device- and biofilm-associated infections (140).
As a vital member of the skin microbiota, C. acnes contributes to skin homeostasis via (i) lipid metabolism (ii) skin acidification, (iii) colonization resistance and niche competition with other skin microbiota (competitive exclusion), (iv) antioxidant effects, and (v) immunomodulatory properties (Figures 3, 5) (10, 11, 139, 141–143). C. acnes substantially raises the amount of several sebum-specific lipids, such as triglycerides, FFAs, ceramides, and cholesterol (141). This activity is mediated mainly by its metabolites, like SCFAs, which interfere with the expression of multiple lipid synthesis genes, such as glycerol-3-phosphate-acyltransferase (GPAT), in the PPARα-dependent mechanism (Figure 3) (141). Overall, C. acnes promotes sebum secretion along with changes in its composition by the formation of FFAs, squalene oxidation, and increasing activity of diacylglycerol acyltransferase, resulting in higher levels of MUFAs and a decrease of LA abundance (136).

Figure 5. Role of C. acnes in skin health and AV pathomechanism.
C. acnes-derived SCFAs contribute to its colonization resistance and competitive strategies. Maintaining skin pH at 5 to 6, mainly by producing propionic acid, prevents its colonization by potentially harmful species, such as Staphylococcus aureus or Streptococcus pyogenes. SCFAs promote the growth of other commensal species and the activity of pH-dependent lipid-synthesis enzymes (141, 144, 145). Also, the skin barrier is improved by C. acnes, through the upregulation of several keratinocyte differentiation markers, such as filaggrin, involucrin, and transglutaminases (146). Conversely, C. acnes prevents biofilm formation by S. epidermidis and inhibits the growth of Staphylococci, producing an antimicrobial thiopeptide - cutimycin (Figures 3, 5) (142, 147).
On the other hand, C. acnes-derived SCFAs under hypoxic conditions inhibit histone deacetylases (HDACs) in human sebocytes and keratinocytes, exerting pro-inflammatory effects due to their enhanced responsiveness to Toll-like receptor-2 (TLR2)-dependent activation by cytokines expression and upregulation of free fatty acid receptors (FFARs), e.g., G protein-coupled receptors (GPRs) (148). Likewise, overstimulation of immune responses by C. acnes may lead to the reduction of claudin-1 (CLDN1) expression, which is a critical component of epidermal tight junctions (TJs) and substantially contributes to skin barrier function (146). Specifically, in keratinocyte monolayer cultures, C. acnes 889 decreased and increased levels of CLDN1 and CLDN4, respectively. Since CLDN4 is a tightening claudin, these opposite changes in CLDN1 and CLDN4 levels were explained as a compensatory mechanism that counteracts the impairment of the epidermal barrier due to CLDN1 downregulation. Similarly, C. acnes extracellular vesicles (EVs) may decrease epidermal keratin-10 and desmocollin through TLR2-dependent signaling (149). These observations suggest that AV pathogenesis may involve additional, beyond immune and inflammatory responses, mechanisms linked to mechanical skin barrier impairment (Figures 3, 4) (63).
C. acnes produces numerous enzymes, such as lipases, hyaluronidases, proteases, polyunsaturated fatty acid isomerases, glycosidases, and sialidases, acting as spreading and damage-associated molecular patterns (DAMPs)-inducing factors. As spreading factors, they accelerate extracellular matrix decomposition (ECMs) and, in turn, infiltration of hair follicles by inflammatory cells, such as neutrophils or monocytes, followed by their damage and releasing bacteria, keratin, and sebum into the dermis, initiating the scarring process (150). Accordingly, Trivedi et al. reported that genes implicated in matrix remodeling, such as MMPs and PIs, are among the most upregulated in AV patients (22). C. acnes can increase the production of several MMPs, such as MMP-1, MMP-9, and MMP-13, and a correlation between elevated levels of MMP-9 and the number of acne-induced skin lesions, e.g., pustules, has been reported (14, 151, 152). In addition, C. acnes can stimulate CD44 and TRL2 signaling pathways due to the degradation of hyaluronic acid (HA) by its hyaluronate lyase (HYL-IA) into fragments acting as ligands these cellular receptors (150). Likewise, pro-inflammatory responses could be triggered by other C. acnes virulence factors, such as porphyrins, CAMP (Christie-Atkins-Munch-Peterson) factors, dermatan-sulfate adhesins DsA1 and DsA2, HtaA iron acquisition protein (4). For example, CAMP1 upregulates multiple cytokines by a TLR2-dependent mechanism (153). In contrast, porphyrins contribute to the formation of acne skin lesions, initiating an inflammatory cascade via CD36 activation and stimulation of reactive oxygen species (ROS) production in keratinocytes (50). In addition, C. acnes induces secretion of IL-1β, a key inflammatory mediator, via NLRP3 and caspase-1 activation, also implicating inflammasome-mediated inflammation in AV pathogenesis (14). It should be emphasized that IL-1β is a crucial cytokine in AV pathogenesis, severity, and post-acne scar formation (68, 69).
Finally, C. acnes triggers the secretion of Th effector cytokines, such as IL-17 and interferon-γ (INF-γ), through the activation of CD4+ Th lymphocytes, including Th1 and Th17 cells (14, 154). The pathomechanism of AV is summarized in Figure 6.
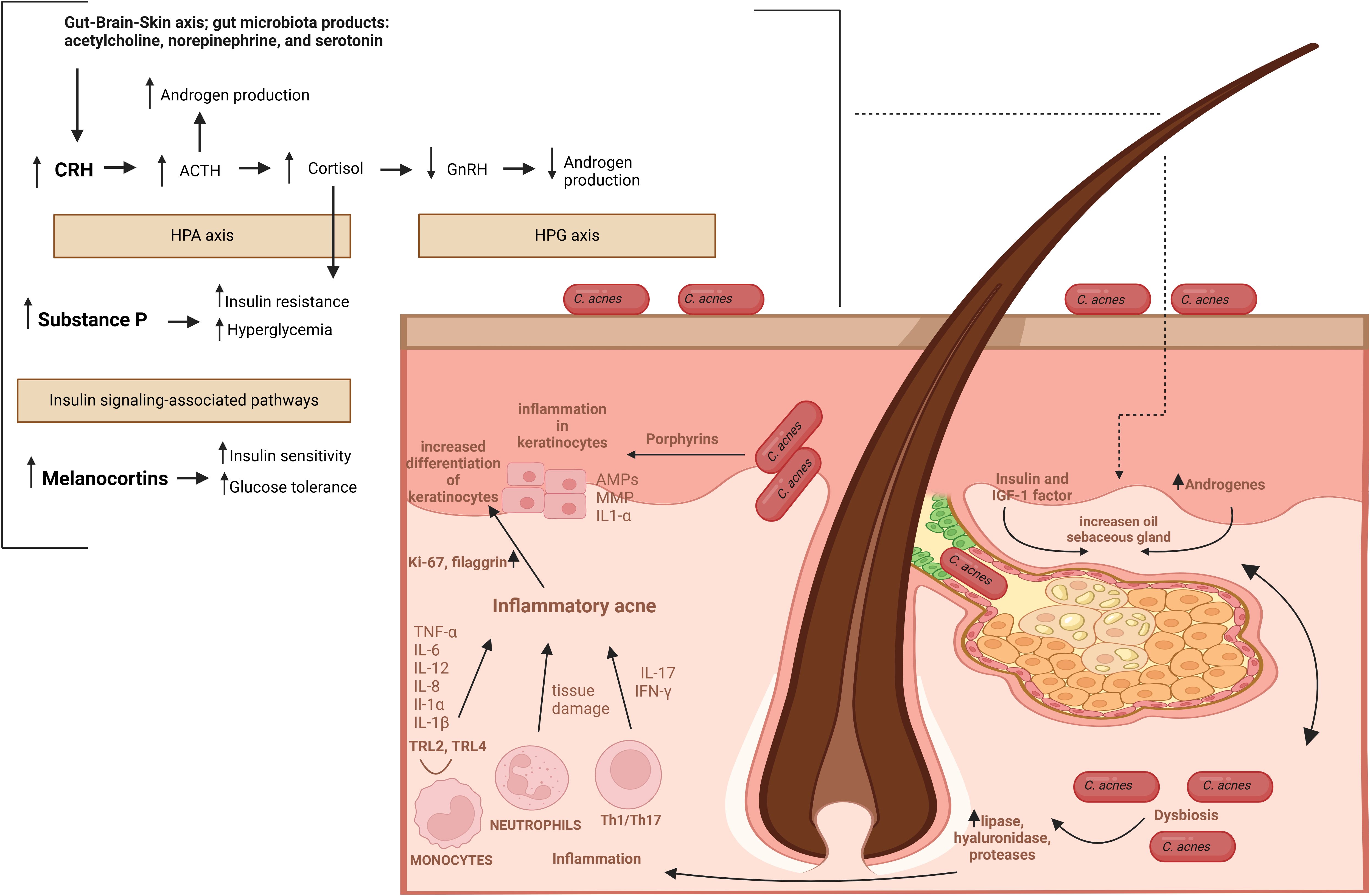
Figure 6. The pathomechanism of AV. Abbreviations: CRH, corticotropin-releasing hormone; ACTH, adrenocorticotropic hormone; HPA axis, hypothalamic-pituitary-adrenal axis; HPG axis, hypothalamic-pituitary-gonadal axis; GnRH, gonadotropin-releasing hormone; MC4R, melanocortin receptor 4.
Phylogenetically, C. acnes has been classified into three phylotypes (I-III) with subspecies status, i.e., C. acnes subsp. acnes (I), C. acnes subsp. defendens (II) and C. acnes subsp. elongatum (III) (14, 155, 156). Furthermore, based on gene sequence typing (ST), the phylotype I was divided into clades IA-1, IA-2, IB, IC (Belfast MLST scheme) or I-1a, I-1b, I-2 (Aarhus MLST scheme) (14, 157), and subsequently subdivided by the whole genome sequencing (WGS) into IA-1, IA-2, IB-1, IB-2, IB-3, IC (14, 158). The C. acnes phylotypes exhibit distinct biological properties, including a different composition of the cell wall polysaccharides, lipase activity, patterns of virulence factors, susceptibility to bacteriophages, and varied inflammatory potential; hence, they have different impacts on human health. Specifically, the phylotype IA-1 (or IA-2, depending on the region) C. acnes strains (so-called acne-associated C. acnes strains) show a significant positive correlation with AV when compared with other, so-called ‘healthy’ C. acnes phylotypes (14, 50, 155, 159–161). Therefore, transitioning from a mixed C. acnes cutaneous population to one dominated by acne-associated strains is responsible for skin microbiota dysbiosis (10, 135–138).
The acne-associated C. acnes strains carry specific virulence genes, and exhibit increased biofilm formation and survival capacity in acne inflammatory milieu (Figure 5) (14, 50, 155, 159, 160). For example, the presence of a virulence linear plasmid with a tight adhesion locus associated with biofilm formation is a phylotype I-specific trait (162). Moreover, porphyrins-dependent ROS-related inflammatory response in keratinocytes is promoted explicitly by acne-associated C. acnes isolates due to the lack of a porphyrin biosynthesis repressor gene (deoR) (14, 50, 155, 159, 160). Notably, FFAs, like linoleic acid, significantly suppress ROS activity, highlighting the role of oxidative stress (OS) in AV pathomechanism (70, 71). Acne-associated C. acnes strains may also directly modulate keratinocyte proliferation and differentiation by overexpression of IGF-1/IGF-1R, increasing the Ki67 proliferation index, and expression of filaggrin and multiple integrins (α-3, α-6, and vβ-6) in the epidermis (163, 164). They also upregulate IL-10, a pro-inflammatory cytokine associated with chronic inflammation and the development of nodular lesions (139). Furthermore, a high rate of biofilm development by C. acnes phylotype IA strains, along with hyperkeratinization and excessive sebum production, causes a blockage of the PSUs (143, 165).
Over 20 AMPs have been linked with skin defense and grouped based on their activity into (i) antimicrobials, (ii) protease inhibitors, (iii) chemokines, and (iv) neuropeptides (23). The human β-defensins, S100 proteins, RNases, cathelicidin LL-37 (LL-37), and dermicidin represent the best-studied skin-derived AMPs (166). In human skin, AMPs are constitutively or inducibly produced by numerous (i) resident cells, such as keratinocytes, sebocytes, sweat glands, and mast cells, as well as (ii) immune system cells, e.g., neutrophils and natural killer cells, recruited in response to injury, inflammation or skin infections (23, 72, 167, 168). Besides the direct antimicrobial activity, the skin-derived AMPs participate in maintaining skin physiological function through (i) coordination of the immune response, (ii) improvement of its permeability, (iii) angiogenesis, and (iv) re-epithelialization. Thus, dysregulated expression of AMPs, e.g., due to injury, infection, or abnormal inflammatory response, is a well-recognized trait in several chronic inflammatory skin diseases, like psoriasis, atopic dermatitis, and rosacea (15, 23, 167, 169). For instance, the upregulation of LL-37 correlates with skin inflammation in rosacea (170). Likewise, deficiency of hBD-2, hBD-3, LL-37, and dermicidin predisposes patients with atopic dermatitis to skin infections (19, 20, 171). On the contrary, cutaneous infections are rare in patients with psoriasis due to the upregulation of LL-37, hBD-2, and S100 proteins (172, 173).
Several studies have revealed overexpression of multiple AMPs in AV patients, such as α- and β-defensins (HNPs and hBDs), S100 proteins, LL-37, RNase 7, lipocalin 2 (LCN2), lactoferrin (Lf), or dermcidin (DCD). In fact, according to transcriptomic studies of AV patients, the genes encoding specific AMPs, i.e., representing S100 family proteins and hBDs, are the top overexpressed ones (21, 22). For instance, in the microarray transcriptomic analysis of biopsies from early inflammatory AV lesions collected from twenty subjects with moderate to severe AV reported by Kelhälä et al., the genes encoding S100A15 (S100A7A) protein and human β-defensin 2 (hBD-2) were the most upregulated ones, respectively with 29- and 25-fold change in expression (Figure 2) (21). Also, in the study by Trivedi et al., hBD-2 was the fourth most upregulated gene in inflammatory papule biopsies collected from six AV patients (22).
The precise function of AMPS in AV pathomechanisms remains to be clarified. However, AMPs (i) antimicrobial, (ii) anti- and (iii) pro-inflammatory, (iv) neuromodulatory (e.g., via gut-brain-skin axis), as well as (v) epidermal TJs barrier-related effects, are considered (Figure 3) (15, 17, 63, 166, 169, 174–176). In particular, TJs barrier-related effects of AMPs may shed new light on this process, linking AMPs with AV metabolic (as well as immune and microbial) background via activation of essential for cellular metabolism and signaling proteins, such as phosphoinositide 3-kinases (PI3K), atypical protein kinase C (aPKC), glycogen synthase kinase-3 (GSK3), and Ras-related C3 botulinum toxin substrate 1 (Rac1) (Figures 3, 4, 7).
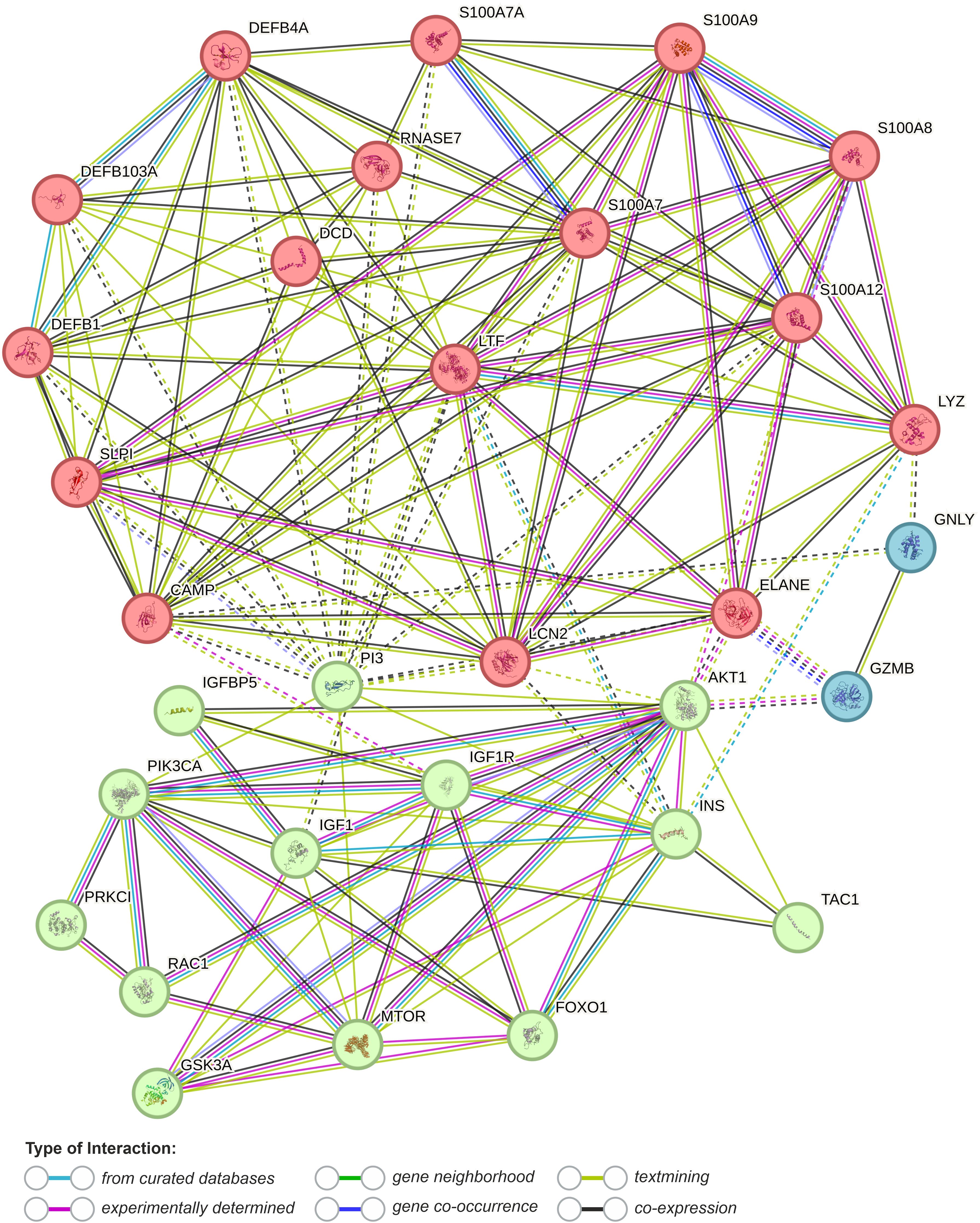
Figure 7. Functional association networks between AMPs and AV metabolic background obtained from String database with cluster analysis (k-means clustering, dotted lines represent edges between clusters) (last accessed 17.03.2024) (177). DEFB1, human β-defensin 1 (hBD-1); DEFB4A, human β-defensin 2 (hBD-2); DEFB103, human β-defensin 3 (hBD-3); S100A7, psoriasin; S100A8, calgranulin A; S100A9, calgranulin B; S100A12, calgranulin C; S100A7A, S100A15 (koebnerisin); SLPI, secretory leukocyte protease inhibitor; PI3, elafin; LTF, lactoferrin; DCD, dermcidin; LYZ, lysozyme; RNASE7, Ribonuclease 7; CAMP, cathelicidin LL-37; LCN2, lipocalin 2 (neutrophil gelatinase-associated lipocalin); GNLY, granulysin; GZMB, granzyme B; ELANE, neutrophil elastase; TAC1, substance B; PI3KCA, phosphoinositide 3-kinase; PRKC1, atypical protein kinase C (aPKC); RAC1, Ras-related C3 botulinum toxin substrate 1; INS, insulin; IGF1 - insulin-like growth factor 1; IFGF1R, insulin-like growth factor 1 receptor; AKT1, RAC-alpha serine/threonine-protein kinase; MTOR, Serine/threonine-protein kinase mTOR; FOXO1, forkhead box protein O1.
For instance, Boronova et al. published an interesting study on the relationship between AMPs and isotretinoin (13-cis retinoic acid, 13-cis RA) treatment in AV patients (178). The authors examined the expression levels of fifteen AMPs in acne skin biopsies across six months of isotretinoin therapy (178). Compared to healthy controls, an increased expression of LL-37, hBD-2, psoriasin (S100A7), koebnerisin (S100A15), RNase 7, lactoferrin, and lysozyme (Lyz) was observed in untreated acne lesions. While dermcidin, granulysin (GNLY), RANTES (CCL5), perforin, CXCL9, and two neuropeptides (substance P and chromogranin B) remained unaffected either in untreated acne patients as well as by isotretinoin treatment. Furthermore, AV patients showed reduced α-defensin-1 (HNP-1) expression levels before treatment. However, this observation is in contradiction to the study by Aidsen et al., who demonstrated a significant reduction in the perivascular and interstitial HNP 1-3 expression of pustular lesions after isotretinoin treatment; hence, the possibility of technical issues was considered (179).
In addition, isotretinoin treatment suppressed the upregulated AMPs to varied degrees, except for lysozyme and RNase 7. However, only LL-37 and koebnerisin (S100A15) returned to baseline levels, indicating their potential as biomarkers of acne treatment efficacy. The continued overexpression of lactoferrin, hBD-2, and psoriasin (S100A7) during isotretinoin treatment implies their involvement in both active and healed (subclinical) AV. Therefore, the authors concluded that their beneficial effects, e.g., anti-C. acnes action of hBD-2, may outweigh any pro-inflammatory action, addressing their acne-associated regulation mechanisms. On the other hand, hDBs may perpetuate inflammation by arresting Th17 cells on inflamed sites (Figure 3). Accordingly, regulation of Tregs/Th17 responses through TGF-β-dependent generation of Foxp3 is a likely mechanism of isotretinoin anti-acne properties (73, 180). These mechanisms might also be behind the anti-acne action of calcipotriol (a vitamin-D derivative) (17, 74, 75). However, compared to isotretinoin, calcipotriol strongly downregulates psoriasin (S100A7) and koebnerisin (S100A15) while it upregulates LL-37 expression, highlighting the complexity of regulatory mechanisms behind the expression of AMPs in AV (74, 75). Furthermore, isotretinoin therapy did not impact lysozyme and RNase 7, suggesting their, especially RNAse 7, mainly antibacterial rather than pro-inflammatory role in AV pathogenesis. RNase 7 shows high in vitro activity against C. acnes (LD90 = 4 μM) (16, 178, 181). RNase 7 is secreted by keratinocytes on the skin surface and in PSUs, where it may control microbial colonization (16, 17, 178). For instance, its low expression increases the risk of S. aureus cutaneous infection (18). In contrast, lysozyme cannot kill C. acnes, which may explain its minor upregulation in acne skin biopsies (178, 181).The expression of AMPs can also be affected by several AV-associated factors, such as FFAs, glucose, insulin, or IGF-1 levels (67, 72, 76, 77). In human sebocytes, FFAs, such as lauric acid, palmitic acid, or oleic acid, can significantly increase the production of hBD-2, but not hBD-1, hBD-3, or LL-37, via CD36 and the NF-κB signaling pathways (67). Furthermore, the anti-C. acnes activity of the supernatant from FFA-incubated sebocyte culture can be neutralized by anti-hBD-2, suggesting that this β-defensin is responsible for the antibacterial properties of sebum (174). Notably, in vitro hBD-2 at concentrations ≤2.5 μM does not exert anti-C. acnes action (78). Whereas in combination with a sublethal dose (25 μM) of lauric acid (but not with palmitic or oleic acid), it shows the dose-dependent killing of C. acnes, implicating their synergistic action (67). Therefore, without the context of the inflamed pilosebaceous milieu, the results produced by in vitro antimicrobial tests may lead to misleading conclusions regarding the antimicrobial efficiency of individual AMPs (78).
Accordingly, Chronnell et al. correlated the constitutive expression of hBD-1 and hBD-2 across various hair follicle compartments with microbial exposure (174). Specifically, the distal parts of the outer root sheath, hair follicle stem cell areas, and the pilosebaceous ducts showed higher expression of the hBDs when compared to the inner compartments, such as the proximal outer and inner root sheaths, along with the hair follicle bulb (174). The authors suggested that hBD-2 protects the population of epidermal stem cells in the hair follicle from microbial invasion (175). Indeed, hBD-2 has the most potent antibacterial activity among hBDs (182). Furthermore, the expression of hBD-1 was only moderately induced in most acne lesions (comedones, papules, pustules) compared to non-lesional skin of the same patient and healthy back skin and pilosebaceous follicles of controls. In contrast, hBD-2 was moderately to strongly upregulated in all acne lesions. Overall, the expression of both hBDs in acne lesions was summarized as follows: hBD-1 - healthy follicular skin ≤ pustule ≤ comedo < papule; hBD-2 - healthy follicular skin ≤ comedo < papule < pustule, and interpreted as a secondary response to the perilesional infiltration by immune cells and secretion of pro-inflammatory cytokines, such as IL-1β (175). The expression of hBDs and other AMPs is a keratinocyte differentiation-dependent process. Therefore, keratinocytes may serve as sensors of abnormal colonization of PSUs by C. acnes acne-associated strains due to TLR2- and TLR4-induced secretion of hBD-2 and IL-8 (78, 175). This may be a critical inflammatory event in the development of AV, since both compounds are potent leukocytes and neutrophils chemoattractants (78). Specifically, hBDs can recruit dendritic cells (iDCs) and T cells via interaction with C-C motif chemokine receptors 2 and 6 (CCR2 and CCR6) (17, 19, 78, 79). Moreover, hBDs (and LL-37) can increase the release of mast cell inflammatory mediators and vascular permeability via activation of Mas-related G-protein-coupled receptor X (MrgX2) and G-protein-coupled receptor GPCR and mitogen-activated protein kinase (MAPK) signaling pathways (80). In addition, hDBs, due to arresting Th17 cells (but not Th1 or Th2 cells) on inflamed sites in a CCR6-dependent mechanism, may potentially contribute to the perpetuation of inflammation (180). Likewise, hBD-2 and hBD-3 via CCR6 play a positive and negative regulatory role in the development and proliferation of human effector CD4+ T cells, and they may shift their surface marker expression to regulatory phenotype (CD4+ CD25+) (183). In detail, the authors observed that co-culture with hBD-2 and hBD-3 increases or decreases CD4+ T cell proliferation after 72 or 96 hours, respectively (183). HBDs also utilize CCR6 to enhance the epidermal TJ barrier (Figures 3, 4). Therefore, several either beneficial or potentially harmful pro-inflammatory effects can be exerted by hBDs in the CCR6-dependent mechanism, likely limiting its interaction with the canonical ligand, namely C-C motif chemokine ligand 20 (CCL20) (81). Upregulation of hBD-2 and CCR6 (along with LL-37) was also reported in psoriasis, highlighting the importance of the implicated CCR6-dependent signaling pathways in the pathomechanism of these dermatoses (82). Finally, hBDs (and LL-37) may also support anti-inflammatory responses, promoting keratinocyte migration and proliferation as well as wound healing via activation of epidermal growth factor receptor (EGFR) and signal transducer and activator of transcription (STAT) signaling pathways (80).
Individuals with insulin resistance are predisposed to AV, and it was suggested as an independent contributing factor in AV development that should be considered when diagnosing and treating acne (184, 185). Similarly, AV patients are at risk group for developing metabolic syndrome (MetS), i.e., a multisystem condition that raises diabetes, stroke, and cardiovascular disease risk (186). Increased blood glucose levels may contribute to AV development due to the stimulation of insulin release. Insulin, as a structural homolog of IGF-1, competes for its cellular receptor and, in turn, promotes IGF-1-mediated keratinocyte proliferation (83). Additionally, insulin enhances sebum production, upregulating androgen secretion. Insulin is a key regulator of hBD-1 expression through enhancing glucose uptake by the cells in a mechanism involving upregulation of the insulin-responsive glucose transporter (GLUT4) and the human sodium-glucose cotransporter (hSGLT1) expression (66). These observations may provide a link between metformin, a drug upregulating GLUT4 expression used to treat type 2 diabetes, and its application in AV therapy (84, 85). Hence, levels of hBDs, and possibly other AMPs, should be viewed in light of intracellular glucose concentration and the insulin transcriptional activity as vital explanatory variables of their expression in AV patients. For instance, hyperglycemia also decreases IL-6-mediated psoriasin (S100A7) expression in the urinary bladder, compromising uroepithelial barrier function and increasing susceptibility to Escherichia coli infection (86). Furthermore, Eicher et al. reported the induction of RNase 7 production in uroepithelial cells by insulin (via PI3K/Akt signaling pathway) as a protective mechanism against invasion by uropathogenic E. coli (77). Similarly, Yin et al. demonstrated that high glucose suppresses the wounding-induced upregulation of LL-37 in cultured human corneal epithelial cells (HCECs), and correlated wound healing activity of LL-37 with the activation of the heparin-binding EGF-like growth factor (HB-EGF)→EGFR→PI3K→Akt signaling pathway (87).
High-glucose and/or enhanced oxidative stress (OS) conditions promote advanced glycation endproducts (AGEs) accumulation, which contributes to several diabetic complications and AV progression (Figure 4) (71, 88, 89). In general, the toxicity of AGEs is mediated by their interaction with the transmembrane receptor for advanced glycation end products (RAGE), which increases OS and inflammatory processes by dysregulating multiple intracellular signaling pathways. Also, by enhancing OS, S100 proteins might be implicated in RAGE-induced inflammation (Figure 4) (90). The latter has been proposed as a biomarker index for AV activity and treatment monitoring (88). In fact, the therapeutic effectiveness of certain antimicrobial agents, such as tetracycline, macrolides, and metronidazole, is attributed mainly to their antioxidant effects, especially metronidazole as C. acnes is intrinsically resistant to this drug (70, 187). Similarly, FFAs, like linoleic acid, significantly suppress ROS (70). Accordingly, Xu et al. demonstrated ex vivo in cultured porcine and human corneas that high glucose delays corneal epithelial wound healing, likely in a ROS-dependent suppression of EGFR-PI3K/Akt signaling pathway (188). In detail, high glucose inhibited ROS-sensitive Akt phosphorylation. Thus, antioxidants, in combination with EGFR ligands, have been suggested as promising candidates for diabetic keratopathy treatments (188).
HBDs, especially hBD-3, and other AMPs, like LL-37 and S100A7, may further influence AV pathogenesis by upregulating TJs proteins, as demonstrated based on transepithelial electrical resistance (TERs) measurement (19). For example, Kiatsurayanon et al. noted that hBD-3 (but not hBD-1, hBD-2, and hBD-4) increases the expression and localization of several claudins (CLDNs) at cell-cell borders of human keratinocytes (63). Additionally, the upregulation of (i) occludin and zonulin (ZO-1) by hBD-3 and (ii) occludin and mucin-2 (MUC2) by hBD-2, in human epithelial Caco-2 cells was reported by Fusco et al. (189).
Notably, the improvement of TJs function by hDB-3 is mediated via activation of PI3K, atypical protein kinase C (aPKC), glycogen synthase kinase-3 (GSK3), and Ras-related C3 botulinum toxin substrate 1 (Rac1), in CCR6-dependent signaling (63). As aforementioned, IGF-1 and insulin promote lipogenesis in sebaceous glands by modulating PI3K/Akt/mTOR/FoxO1 signaling pathway. PI3K also activates aPKC, which, in a complex with PAR (partition defective) proteins, i.e., PAR-3 and PAR-6 (PAR-3/PAR-6/aPKC), is a vital component of TJs, responsible for establishing and maintaining cell polarity (64, 65, 91, 92). aPKC is also involved in the IGF-I/insulin signaling pathways to regulate various metabolic processes, such as GLUT4-dependent glucose transport and lipogenesis. Moreover, it can modulate the inflammatory response via activation of NF-κB signaling pathways (91). Similarly, GSK3 controls glucose metabolism, controlling glycogen synthase (GS) in response to insulin stimulation. However, its role in AV might be more versatile. GSK3, due to its interaction with 40 targets and more than 500 substrates, is implicated in virtually every central biological process in the cell and is a potent regulator of inflammation (93–95). Subsequently, GSK3 contributes to several inflammatory and metabolic disorders, e.g., diabetes mellitus (93). Likewise, Rac1, a small GTPase, as a regulator of the actin cytoskeleton and other fundamental cellular processes, including cellular plasticity, migration, invasion, adhesion, proliferation, apoptosis, ROS production, and inflammation, is implicated in multiple pathological conditions (96, 97). Specifically, Rac1 coordinates a keratinocyte immune-cell crosstalk, and other AMPs, e.g., LL-37, also enhance its activity. Thus, it is essential for skin homeostasis, regulating (i) epidermal TJs barrier function, (ii) wound re-epithelialization, and (iii) inflammation (98). Rac1’s contribution to AV pathogenesis might be particularly associated with (i) maintaining hair follicle integrity and (ii) regulation of actin reorganization in insulin-induced recruitment of GLUT4, i.e., a mechanism controlling the expression of hBD-1 (66).
In human skin, LL-37 is primarily produced by keratinocytes (and sebocytes) in both constitutive and inducible mechanisms. It is crucial for maintaining skin barrier homeostasis due to direct antimicrobial activity and also an acceleration of the skin regeneration processes and wound healing process through the promotion of cell migration and proliferation as well as angiogenesis (99, 190). LL-37 is proteolytically processed into smaller peptides, such as RK-31 and KS-30, with enhanced activity against specific microbes, e.g., staphylococcal and candidal species, and acting synergistically with other AMPs, such as hBD-2. LL-37 is a well-known DAMP agent that regulates the immune surveillance system via interaction with multiple cellular receptors, including TLR4, EGFR, MrgX2, glyceraldehyde-3-phosphate dehydrogenase (GAPDH), and formyl-peptide-receptor-like-1 (FPRL-1) (191, 192). For instance, it chemoattracts neutrophils, monocytes, and T lymphocytes by binding to the FPRL-1 (192). In addition, LL-37 induces calcium (Ca2+) mobilization in cells through purinergic receptor P2X7, the process that controls cell migration (193). Moreover, LL-37 is a biased agonist of IGF-1R, i.e., an agonist that stabilizes distinct receptor active states, activating only specific downstream signaling cascades, which preferentially promotes the phosphorylation of ERK1/2 over Akt (100). The pro-inflammatory actions of LL-37 include decreased expression of IL-10, increased expression of IL-1β, IL-18, mast cell degranulation, and release of inflammatory mediators. In contrast, inhibition of interferon-induced protein formation AIM2, tumor necrosis factor TNF-α, IL-12, and IL-4 are central mechanisms behind its anti-inflammatory properties, mediated by neutralization of bacterial PAMPs, such as LPS, or LTAs (194).
Furthermore, like hBDs, LL-37 contributes to cutaneous immunity by improving the TJs barrier function and keratinocyte differentiation (99). LL-37 can increase in epidermal keratinocyte the mRNA and protein levels of numerous TJ proteins, including claudin-1, 3, 4, 7 and occluding, by the activation of the aPKC, Rac1, GSK-3, and PI3K signaling pathways (99). Also, multiple keratinocyte differentiation markers, such as filaggrin, involucrin, keratin 1, keratin 10, and transglutaminase 1 (TGM1), but not loricrin or transglutaminase 3 (TGM3), are markedly induced by LL-37 (78, 79, 99). Interestingly, the LL-37-mediated improvement of the TJ barrier appears to be connected with its ability to activate autophagy, i.e., a process associated with skin diseases manifested by a defective epidermal barrier (101, 193). Additionally, in psoriasis, LL-37 suppresses keratinocyte apoptosis via upregulation of apoptosis-related genes, such as cyclooxygenase- 2 (COX-2), an inhibitor of apoptosis-2 (IAP-2) (195). LL-37 anti-apoptotic activity may also contribute to AV pathogenesis, as only acne-associated C. acnes isolates induce keratinocyte/sebocyte proliferation and differentiation (78, 79). Similarly, the anti-inflammatory role of LL-37 associated with blocking the activation of the DNA-sensing inflammasomes might be a common trait for psoriasis and AV (14, 194).
Although LL-37 can directly kill C. acnes, the bactericidal concentration (4 μM) is 100 times higher than its amount in extracts from sebocytes (0.038 μM) (196). However, its combination with psoriasin (S100A7), at a concentration of 10 μg/mL, other overexpressed in acne lesions AMP, reduces its C. acnes killing concentration to 0.5 μM (195).
Psoriasin, named for its discovery in psoriatic patients, belongs to the vertebrate-specific and Ca2+-binding S100 family proteins (197, 198). The S100 family involves 25 proteins implicated with various intracellular and extracellular functions. Intracellularly, S100 proteins are responsible for (i) calcium homeostasis, (ii) energy metabolism, (iii) regulation of cell cytoskeleton as well as (iv) proliferation, differentiation, and apoptosis via interacting with multiple nuclear proteins (198, 199). Extracellularly, S100 proteins as signal molecules modulate the inflammatory response and act as AMPs (200). The former action is mediated via interaction with multiple cellular receptors (201). For instance, calprotectin (the complex of S100A8 and S100A9) as an agonist for TLR4 and RAGE acts as a DAMP molecule in various inflammatory responses and serves as a biomarker in several immunomodulatory, antiproliferative, and infectious diseases (202, 203). Recently, a significant positive correlation was also reported between serum calprotectin levels and acne severity and duration (204, 205). In the skin, S100 proteins are expressed by keratinocytes, sebaceous glands, and hair follicles and regulate epidermal differentiation and proliferation (102, 206, 207). In fact, S100 protein genes are located in the epidermal maturation region (Epidermal Differentiation Complex; human chromosome 1q21), involving genes crucial for epidermis maturation, such as involucrin, filaggrin, trichoyalin, and repetin (208). The production of S100A proteins is stimulated by inflammatory cytokines (IL-1, IL-17, and TNFα) and in response to infection; hence, they serve as markers in several dermatoses manifested by inflammation and keratinocyte hyperproliferation (17, 102, 207, 209).
The AMP function has been identified for psoriasin (S100A7), calgranulin A (S100A8), calgranulin B (S100A9), calgranulin C (S100A12), and koebnerisin (S100A15, S100A7A) (198, 200, 207), and it associated with their Zn2+ sequestration (172, 210). Neutrophils extensively express and release S100A8/A9 (calprotectin) and calgranulin C (S100A12) during infection, which account for ~50% of their total cytoplasmic proteins (201). However, Zn2+ sequestration by S100 is markedly restricted by low pH. Therefore, Wang et al. suggested that Ca2+ binding is a mechanism enhancing the antimicrobial action of S100 proteins (S100A12 and S100A8/A9), due to conformational changes which increase their affinity to Zn2+ even under the physiologically relevant sub-neutral pH conditions (between pH 5.5 – 6.0) expected for activated neutrophils (210). Furthermore, S100A8/S100A9 tetramers are produced at high extracellular Ca2+ concentrations, which act as an autoinhibitory mechanism that modulates S100A8/9 biological activity exerted by TLR4 (201).
Also, TJs barrier formation is affected by S100 proteins in a Ca2+-dependent mechanism (211). For example, psoriasin (S100A7) increases the expression of keratinocyte differentiation markers (filaggrin, involucrin, keratin 1, keratin 2, loricrin, TGM1, and TGM3). Moreover, it upregulates the skin’s TJ proteins (claudin-1, claudin-3, claudin-4, claudin-7, claudin-9, claudin-14, and occluding) via GSK-3 and MAPK pathways. Subsequently, psoriasin (S100A7) promotes the accumulation of controlled by GSK-3 components of adherens junctions - β-catenin and E-cadherin at cell-cell contacts (212). Therefore, depletion of Ca2+ reversibly disturbs the assembly of TJs proteins, such as occludin, claudin-1, claudin-4, or E-cadherin, and, in consequence, weakens the permeability barrier (211).
Furthermore, psoriasin (S100A7) activates cellular retinoic acid-binding protein 2 (CRABP-II), which regulates human skin cell proliferation and retinoic acid (RA)-mediated differentiation (102, 103). Levels of retinoids (vitamin A derivatives) and vitamin D are vital for epidermal cell development; thus, the above observations may explain the effectiveness of isotretinoin and calcipotriol as AV topical treatment, which upregulate and downregulate psoriasin’s (and koebnerisin’s) expression (17, 74, 75, 213). Moreover, in AV, psoriasin (S100A7) is upregulated in sebaceous glands and acne lesions, acting as a potent and selective chemotactic inflammatory factor for CD4+ T lymphocytes and neutrophils (214).
Interestingly, according to the transcriptomic study by Kelhälä et al. koebnerisin (S100A15) was the most among 509 overexpressed genes in AV patients (21). Similar to psoriasin, koebnerisin was initially discovered in psoriatic skin, and subsequently, it has also been recognized as a new factor in the pathophysiology of rosacea (208, 215). Indeed, in human keratinocytes, its expression is stimulated by cytokines typical for the acne inflammatory milieu, such as TNF-α, IFN-γ, and IL-1β, indicating that inflamed skin may trigger its production in the epidermis. In fact, psoriasin (S100A7) and koebnerisin (S100A15) evolved by gene duplication and share >90% sequence identity; hence, they are difficult to differentiate (208). Nevertheless, both S100 proteins show distinct tissue distribution, regulation, and function. The expression of psoriasin (S100A7) is limited to the granular/cornified layers of the interfollicular epidermis and hair follicles, whereas koebnerisin (S100A15) is also expressed by basal epidermal and dendritic cells. In psoriasis, IL-17A is a primary inducer of both S100 proteins, especially koebnerisin, whereas other Th17-related cytokines, such as TNF-α and IL-22, differently regulate their expression in epidermal keratinocytes (216). The presence of IL-17A-positive T cells and Th17-related cytokines (IL-1β, IL-6, TGF-β, IL23p19) is also characteristic of acne lesions (21). Nonetheless, both proteins are co-upregulated under related pathological conditions by similar epidermotropic and microbial pro-inflammatory mediators, indicating their collaboration in the inflammatory response, e.g., they act synergistically as DAMPs in leukocyte recruitment to exacerbate inflammation in psoriasis (208, 216). In addition, S100A15 has two splicing isoforms, i.e., short (S100A15-S) and long (S100A15-L), characterized by distinct regulation. For instance, S100A15-L exhibits a more pronounced response to pro-inflammatory Th1 cytokines, such as TNF-α, IFN-γ, and IL-1β, than S100A15-S, which may explain its ~25 times greater expression than S100A150-S in acne lesions, as suggested for psoriasis (178, 208, 216).
In acne lesions, S100 proteins may also be upregulated by lipocalin 2 (LCN2), which, promoting their keratinocyte secretion, aggravates psoriasiform skin inflammation in a Th17-dependent manner (104). LCN2, known as neutrophil gelatinase-associated lipocalin (NGAL), is an antimicrobial protein and multifunctional adipokine linked with insulin resistance, obesity, and atherosclerotic disease, and a potential biomarker for infection inflammation, ischemia, or kidney injury (217). In health, its skin and serum expression is low; however, keratinocytes and neutrophils overexpress LCN2 by TLR2- and TLR4-dependent signaling in various skin disorders, including AV (21, 218). Indeed, Al Hashimi et al. reported elevated LCN2 serum levels in AV patients compared to healthy controls (218, 219). Likewise, Watanabe et al. suggested LCN2 as an objective biomarker of acne symptoms, correlating its decreasing stratum corneum levels on the cheeks of patients with AV with symptom alleviation (203).
In sebocytes, LCN2 expression rises in response to C. acnes and IL-1β, as well as isotretinoin (220). For instance, in a study by Nelson et al., LCN2 was among the top genes most upregulated by isotretinoin in the skin and cultured human sebaceous gland cells (SEB-1 sebocytes) (221). Moreover, the authors found that LCN2 is substantially raised in the skin of AV patients during the first week of isotretinoin treatment but not eight weeks later (222). Since isotretinoin reduces sebum production through partly controlled by LCN2 induction of sebocyte apoptosis, an isotretinoin-induced increase of LCN2 precedes a decrease in sebum and a reduction in C. acnes abundance (220).
The antimicrobial activity of LCN2 activity is mediated by the sequestration of bacterial siderophores, i.e., iron-binding proteins. Hence, it acts with lactoferrin (LF), a potent iron chelator and broad-spectrum AMP, to restrict this essential element and impede bacterial growth. Indeed, LF can suppress C. acnes-induced inflammation in both in vitro and animal model studies (223). Lactoferrin, also known as lactotransferrin (Ltf), is an 80-kDa iron-binding glycoprotein of the transferrin family with multiple biological functions, including antimicrobial, immunomodulatory, anti-inflammatory, antioxidant, and enzymatic activities. Along with lysozyme, LL-37, and α-defensin-1, LF is a component of neutrophil granules, and it is secreted into numerous body fluids, including sweat (105). Therefore, LF has excellent potential as a topical or oral treatment for skin infections and dermatoses (105). For instance, in the study by Chan et al., oral administration of LF with vitamin E and zinc significantly reduced acne lesions in patients with mild to moderate AV in a randomized, double-blind, placebo-controlled trial involving 168 subjects (106). Although the mechanism of action for orally administered LF is uncertain, its ingestion is safe and (i) promotes oral and intestinal homeostasis, (ii) regulates glucose and lipid metabolism, (iii) reduces systemic inflammation, and (iv) iron absorption and balance (107). Therefore, it is a promising nontoxic adjuvant for the long-term prevention of metabolic illnesses, including insulin resistance, T2D, and metabolic syndrome (107).
Furthermore, LCN2 significantly influences intestinal and metabolic inflammation (217). It is overexpressed in individuals with type 2 diabetes mellitus (T2DM), obesity, or nonalcoholic steatohepatitis (NASH). Accordingly, LCN2 regulates the gut microbiota composition and intestinal permeability, i.e., factors that may lead to systemic inflammation, insulin resistance, metabolic syndrome, and related characteristics (217). Therefore, LF and LCN2 may represent AMPs linking intestinal (gut dysbiosis) and metabolic inflammation with AV, e.g., via the gut-brain-skin axis, emotional stresses, and neuroinflammation. The latter is controlled by certain neuropeptides possessing also antimicrobial and immunomodulatory activities, such as substance P and chromogranin B (secretogranin-I). For instance, a positive correlation between stress scale and serum level of substance P was reported in AV patients, and acne skin is highly innervated due to the abundance of SP-containing nerves (224–227). It also stimulates sebaceous gland growth and differentiation, increases lipid synthesis in sebocytes, promotes mast cell proliferation and degranulation, and the release of pro-inflammatory cytokines (224, 225, 227). Thus, SP may bridge AV pathomechanism with its neurogenic and psychogenic aspects (225, 226). However, the overexpression of substance P and chromogranin B is not universally recognized in acne skin (178). Recently, Kwiecinska et al. revealed a novel, SLPI-mediated mechanism responsible for maintaining skin homeostasis through a nerve-reflex arc, preventing excessive skin dryness in psoriasis and possibly also in other dermatoses with compromised skin barrier function (228). SLPI is a ~12 kDa cationic AMP protein and an essential regulator of innate and adaptive immunity, anti-inflammatory properties in allergy and autoimmunity, as well as a component of tissue regeneration programs (229). Like LCN2, SLPI may be responsible for the interplay between microbiota and epithelial cells, regulating the threshold for epithelial activation and microbial signals (229).
Another AMP that connects AV with its metabolic background could be AMP-IBP5, a 22 antimicrobial peptide generated from insulin-like growth factor-binding protein 5 (IGFBP-5). This 22-amino acid peptide shows antibacterial activity even better than LL-37 or hBD-2. In addition, it stimulates various keratinocyte and fibroblast functions via the receptor low-density lipoprotein receptor-related protein-1 (LRP1) and MrgX1-X4 receptors (108, 230–232). In addition, AMP-IBP5 suppresses the expression of Th2 cytokines such as IL-4, IL-13, IL-31, IL-33, and thymic stromal lymphopoietin (TSLP), stimulates the production of IL-8 and VEGF (108, 232). Furthermore, AMP-IBP5 improves the skin’s barrier function by upregulating and distributing TJs proteins, such as claudin-1, -4 and -7, occluding and ZO-1, through aPKC and Rac1 pathways (108). AMP-IBP5, unlike other AMPs, such as hBDs, LL-37, and S100A7, is downregulated in psoriatic skin tissues (109). Furthermore, AMP-IBP5 mitigates the harmful effects of the high glucose (HG) environment on keratinocyte proliferation and migration, as well as accelerates delayed angiogenesis and wound healing in diabetic mice via the EGFR, STAT, and MAPK pathways. Therefore, AMP-IBP5 may also be implicated in AV pathomechanism due to its protective effect against glucotoxicity (110).
The MAPK metabolic pathway is also activated by dermcidin (DCD), stimulating keratinocytes to generate cytokines and chemokines (19, 111, 112). In contrast to other AMPs, DCD is only constitutively expressed, i.e., regardless of the inflammatory conditions, in the eccrine sweat glands and secreted with sweat on the epidermal surface, followed by its proteolytic cleaving into active DCD-1 and DCD-1L (233, 234). Bactericidal concentrations of DCD against 68% and 83% of C. acnes isolates were estimated at 50 μg/mL and 270 μg/mL, respectively (235). Furthermore, Nakano et al. found reduced DCD levels in sweat from AV patients (median 9.8 μg/ml, range 6.9–95.3) compared to healthy volunteers (median 136.7 μg/ml, range 45.4–201.6 μg/ml) (235). Hence, DCD deficiency in the sweat of AV patients may allow C. acnes to colonize and multiply in the pilosebaceous unit, contributing to AV development (235).
Finally, skin microbiota-produced AMPs might also be implicated in AV pathogenesis as competitive exclusion or immunomodulatory factors, as in other dysbiotic disorders (236, 237). Figure 4 summarizes the relationship between AMPs and the modulation of epidermal TJs barrier function in the context of AV metabolic background.
Anti-acne therapeutic potential of human, non-human, semi-, and synthetic AMPs, associated with their antibacterial and anti-inflammatory activities, have been reported in several studies (238). For example, NAI003 peptide has completed a phase 1 clinical trial as an AV topical treatment candidate (EudraCT No. 2005-005531-99; https://www.clinicaltrialsregister.eu/) (239). NAI003 is a protein synthesis inhibitor targeting elongation factor Tu (EF-Tu) derived from Planobispora rosea-produced thiopeptide GE2270A, with a potent, selective action against C. acnes (MIC range 0.007 - 0.25 μg/mL) but not to other skin commensals, such as staphylococci (239). Also, the anti-acne activity of a synthetic lipohexapeptide HB-1345 and omiganan (CLS001/MBI 594AN, MBI 226 or MX 226), a 12 amino acid indolicidin-derivative, was assessed in clinical trials (ClinicalTrials.gov ID: NCT02571998; NCT00211523; NCT00211497) (240–243). Briefly, in a six-week, randomized, double-blind phase IIa study involving 75 subjects with facial AV omiganan reduced inflammatory lesions (papules and pustules; 39% vs. 21% reduction) in mild to moderate AV patients and non-inflammatory lesions (comedones; (10% vs. 25% reduction) compared to the placebo group, and improved physician’s Global Severity Assessment scores. However, in a longer, 12-week phase IIb randomized, double-blind, involving 241 participants, no statistically significant differences were noted beyond six weeks between the groups (242).
A reduction of acne severity after 12 weeks of topical treatment was also reported for a combination of 20 granulysin-derived peptides (GDP 20) in a study involving 30 AV patients (244). Similarly, combined therapy of GDP-20 with isotretinoin was superior over low-dose systemic isotretinoin alone in treating patients with mild-to-moderate AV (245). Furthermore, McInturff et al. designed five GDPs, and one of them, the D-type amino acid of peptide D-31–50v44w, effectively killed C. acnes in vitro, either in the growth media and in sebaceous microcomedome extracts as well as decreased C. acnes-stimulated production of cytokines and chemokines (246). Granulysin is a unique AMP since it is released by T cells instead of epithelial cells in the skin; hence, it serves as the adaptive immune agent rather than the innate immune system. T cells in early and late acne lesions imply granulysin’s importance for acne pathogenesis (246). Indeed, the presence of IL-17A-positive T cells and Th17-related cytokines in acne lesions suggests that the Th17 pathway is activated and may be crucial to the disease process (21).
In addition, anti-C. acnes and anti-inflammatory properties of non-human AMPs were investigated in several studies. For instance, Popovic et al. reported inhibition of C. acnes growth (MIC = 3-12.5 μM) and stimulation of anti-inflammatory cytokines (IL-10, TGF-β, and IL-4) production in peripheral blood mononuclear (PBM) cells by five frog skin-derived antimicrobial peptides ([D4k]ascaphin-8, [G4K]XT-7, [T5k]temporin-DRa, brevinin-2GU, and B2RP-ERa) (247). Moreover, modulation of adaptive immune defense associated with decreased T cell responses in favor of the protective function of Th2 cells, e.g., via suppression of pro-inflammatory IL-12 and IFN-γ cytokines by IL-10, appears to be an attractive trait of these AMPs (247). Accordingly, Ryu et al. showed that P5, a synthetic hybrid of cecropin A/magainin 2, efficiently kills C. acnes (MBC = 0.2 μM) and reduces the expression of pro-inflammatory cytokines IL-8 and TNF-α in C. acnes-treated human keratinocytes, likely via neutralization of its lipoteichoic acid, and has no cytotoxicity to skin cells (248). Likewise, Han et al. showed anti-C. acnes and anti-inflammatory activity of CEN1HC-Br, a 28 amino acid peptide isolated from the green sea urchin (249). In general, CEN1HC-Br was more active than clindamycin against 15 clinical C. acnes isolates with MIC range from 0.125 µg/mL to 32 µg/mL. In rats, CEN1HC-Br and clindamycin reduced C. acnes-induced ear swelling and the level of several pro-inflammatory factors (IL-8, TNF-α, MMP-2, and TLR2). However, only CEN1HC-Br significantly reduced in a TLR-dependent mechanism the expression of several pro-inflammatory cytokines, such as IL-12p40, IL-6, IL-1β, and TNF-α, in monocytes (249). Similarly, Bombinin-like peptide 7 (BLP-7) from Bombina orientalis has been shown to inhibit C. acnes growth (MIC = 5 μM) and to suppress the production of IL-8 and granulocyte-macrophage colony-stimulating factor (GM-CSF) by normal human epidermal keratinocytes (NHEKs) co-cultured with C. acnes (250). Additionally, in the rat ear edema model, BLP-7 efficiently reduced C. acnes-induced skin inflammation compared to the controls (250). Moronecidin, a 22 amino acid antimicrobial peptide derived from hybrid striped bass, was found to reduce C. acnes-induced inflammation in a rat model by these authors, and its MIC value against C. acnes was estimated at 10 μM (250). Furthermore, the study by Lee et al. revealed that melittin, an AMP isolated from honey bee venom, may suppress inflammatory cytokines, particularly TNF-a and IL-1β, by modulating NF-kB and AP-1 transcription factors, significantly reducing heat-killed C. acnes-induced inflammatory responses in keratinocytes. In addition, when administered intradermally to mice’s ears, melittin substantially reduced swelling and granulomatous responses as opposed to ears injected exclusively with living C. acnes (251). Likewise, a study by Wang et al. found that cathelicidin-BF, produced from snake Bungarus fasciatus, venom, has significant antibacterial action against C. acnes in an experimental mice skin colonization model. Cathelicidin-BF MIC values against two C. acnes strains (ATCC 6919 and ACTC 11827) were lower than those obtained for LL-37 and clindamycin (1.3 μM vs 2.2 μM vs 5.2 μM). Furthermore, in human monocytic cells, cathelicidin-BF significantly reduced secretion of the pro-inflammatory factors (TNF-a, IL-8, IL-1β, and MCP-1) as well as O2.− production by human HaCaT keratinocyte cells triggered by C. acnes. The anti-inflammatory action was also confirmed in vivo with C. acnes-induced mice ear swelling and granulomatous inflammation (252). Anti-oxidant, anti-inflammatory, and anti-C. acnes (MIC = 400 µg/mL; MBC = 600 µg/mL) properties characterize also extracellular peptides (YTCY-Eps) isolated from Weizmannia coagulans strain YTCY, a probiotic gram-positive rod from in the family Bacillacea (253). These 9 - 18 amino acids peptides can in vitro reduce C. acnes-induced ROS level ~3 times and downregulated expression of inflammatory cytokines, chemokines, and MMPs genes by diminishing activation of TLR2 by C. acnes and subsequent NF-kB and MAPK/AP-1 signaling pathways. YTCY-Eps also reduced the expression of inflammatory cytokines and MMPs, as well as improved keratinization, on the rabbit ear acne model, suggesting their application as a potential anti-acne raw material in cosmetics (253). A relatively high MIC value - 200 μg/mL against C. acnes strain BCRC #10723 was also reported for a synthetic, 21 amino acids in length, antimicrobial peptide derived from the marine organism Epinephelus coioides epinecidin-1 by Pan et al. (254).
Synthetic or designed antimicrobial peptides (dAMPs), i.e., engineering analogs of naturally occurring AMPs characterized by a reduced risk of developing bacterial resistance, represent another approach to developing anti-acne AMPs (255–257). For example, Zhang et al. designed a 15 amino acid residues peptide named LZ1 characterized by potent, four times lower compared to clindamycin, antimicrobial activity against C. acnes (MIC = 0.6 µg/mL) and staphylococci (MIC values from 2.3 to 4.7 µg/mL), with little cytotoxic and hemolytic activity (255). Additionally, in the mice skin colonization model, ear swelling, inflammatory cell infiltration, and C. acnes colonization were significantly reduced by LZ1 by inhibiting the secretion of pro-inflammatory cytokines, IL-1β, and TNF-α (255). Similarly, Woodburn and colleagues developed five dAMPs (RP444, RP551, RP554, RP556, and RP557) with potent in vitro anti-C. acnes. One of them, RP556, as a topical agent (5 mg/mL), was successfully used to treat intradermal murine infection caused by multidrug-resistant C. acnes (256). Finally, Dong et al., using a series of deep learning (DL) models trained toward the prediction of antimicrobial and hemolytic activity, designed a set of 42 novel linear peptides. Five of them (14-15 amino acid residues) exhibited high potency (MIC = 2–4 µg/mL) and selectivity against C. acnes without simultaneous hemolytic and cytotoxic action (257).
To summarize, the developed anti-acne AMPs represent a diverse group of either cationic and anionic peptides (charge at pH 7.0 ranging from -3.1 to +10.9), with length ranging from 5 to 35 residues, and various physicochemical properties, such as aliphatic index (range from 26.1 to 214.0), Grand average of hydropathicity index) (range from -1.9 to 1.824), and instability index (range from -26.0 to 131.9) (Figure 8; Supplementary Table S1). Nonetheless, several AMPs share specific amino acid motifs with human endogenous AMPs, including hBDs, LL-37, dermcidin, RNAse 7, or granulysin, which may explain their anti-acne properties (Table 1). For instance, LL-37 antimicrobial and immunomodulatory activities are linked to amino acid residues 13-32 and 17-29, respectively (258). Accordingly, AMP-29 shares the KI(I/G)K motif with LL-37, a part of its ‘antimicrobial’ region.
Figure 8. The physicochemical parameters of the developed anti-acne AMPs (n=42) discussed in the text; the graphs were created based on the AMPs parameters calculated using the ProtParam tool, using the website (https://web.expasy.org/protparam/), additional information included in Supplementary Table S1. The information in the figure summarizes the results of published experimental work (21, 238–257).
Table 1. Amino acid sequence motifs shared by AMPs developed to target C. acnes (highlighted in bold), i.e., AMP-29, AMP-31, and AMP-33 (257); P5 (248); [G4K]XT-7 and Brevinin-2GU (247); Cathelicidin-BF (252); RPM556 and RMP557 (256); YTCY_B (253), and human endogenous AMPs.
These observations undoubtedly identify AMPs as crucial factors in the pathomechanism of AV. However, the complicated network of interactions between AMPs and other host factors as well as skin microbiota, remains to be deciphered. In addition, significant variation in the intensity of AMPs expression among patients, even when matched for sex and age, and between different body regions and hair follicles indicate that individual factors, possibly genetic or environmental, must be considered in such investigations (175, 259). For instance, polymorphism in cutaneous androgen metabolism-regulated genes HSD3B1 and HSD17B3, cytochrome P450 family genes (CYP17 and CYP19A1), and in genes involved in immune responses is correlated with a risk of developing AV (152, 259–262). It highlights the intricate nature of AV as a multifactorial disease, involving a network of reciprocal interactions between hormonal, metabolic, immunological, microbiological, genetic, and psycho-emotional factors, with AMPs as their central regulatory and ‘effector nodes.’ In particular, hBD-2, and psoriasin (S100A7), appear to be its ‘critical nodes’, involved either in active and healed (subclinical) stages of AV. Furthermore, lipocalin 2 (LCN2) and lactoferrin (LF) may constitute AMPs linking intestinal (gut dysbiosis) and metabolic inflammation with AV, e.g., via the gut-brain-skin axis, emotional stress, and neuroinflammation. Therefore, deciphering complex mechanisms behind their expression and activated cellular pathways is crucial to elucidate their role in AV development and progression. For instance, the importance of AMPs competing with the canonical ligands (and likely between each other) for several cellular receptors associated with immune (e.g., CCR6) and metabolic (e.g., IGF-1R) responses is noteworthy, as AMPs may function as their biased agonists.
Also, the role of AMPs in maintaining epidermal TJs homeostasis deserves further investigation. The skin’s permeability and antibacterial barriers are inextricably linked, as disturbance of the mechanical barrier induces the expression of AMPs while regeneration reduces their level (212, 263). Since the formation of TJs is a Ca2+-dependent process, the role of calcium-binding and highly elevated in acne lesions S100A proteins, such as psoriasin (S100A7), S100A8/A9 (calprotectin), S100A12 (calgranulin C), and koebnerisin (S100A15, S100A7A), appear to be fundamental. In this light, the increased expression of AMPs in AV may reflect a compensatory mechanism to protect the skin with an impaired permeability barrier via activation of core cellular proteins, such as PI3K, GSK-3, aPKC, and Rac1. Therefore, AMPs may be key determinants in developing and progressing acne-associated immune responses, skin barrier integrity, and metabolic factors, like insulin/IGF-1 and PI3K/Akt/mTOR/FoxO1 signaling pathways or high glucose levels. From this perspective, AMP-IBP5, an antimicrobial peptide derived from IGF-binding protein 5, with superior antibacterial activity over hBD-2 or LL-37, which reduces the expression of Th2-specific cytokines, mitigates adverse effects of glucotoxicity and accelerates angiogenesis and wound healing in diabetic mice, is a promising candidate for further researches in this area. However, the antibacterial activity of AMPs must be carefully balanced in future anti-AV therapies to target specifically acne-associated C. acnes strains or to promote the ‘healthy’ ones rather than indiscriminately eradicate all C. acnes populations.
AL: Conceptualization, Investigation, Software, Visualization, Writing – original draft, Writing – review & editing, Resources. PP: Investigation, Writing – original draft. UW: Investigation, Project administration, Writing – original draft. AM: Investigation, Writing – original draft. GK: Investigation, Writing – original draft. KG: Investigation, Writing – original draft. EP: Investigation, Project administration, Writing – original draft, Funding acquisition. JS: Investigation, Writing – original draft. SO: Investigation, Writing – original draft. KF: Conceptualization, Investigation, Project administration, Software, Visualization, Writing – original draft, Writing – review & editing. BD: Investigation, Writing – original draft. RB: Conceptualization, Funding acquisition, Investigation, Project administration, Resources, Supervision, Writing – original draft, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was financially supported by Medical University of Białystok B.SUB.23.328 (EP).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2024.1502242/full#supplementary-material
1. Hay RJ, Johns NE, Williams HC, Bolliger IW, Dellavalle RP, Margolis DJ, et al. The global burden of skin disease in 2010: an analysis of the prevalence and impact of skin conditions. J Invest Dermatol. (2014) 134:1527–34. doi: 10.1038/jid.2013.446
2. Clatici VG, Voicu C, Voaides C, Roseanu A, Icriverzi M, Jurcoane S. Diseases of civilization - cancer, diabetes, obesity and acne - the implication of milk, IGF-1 and mTORC1. Maedica. (2018) 13:273–81. doi: 10.26574/maedica.2018.13.4.273
3. Melnik BC. Acne vulgaris: The metabolic syndrome of the pilosebaceous follicle. Clinics Dermatol. (2018) 36:29–40. doi: 10.1016/j.clindermatol.2017.09.006
4. Tuchayi SM, Makrantonaki E, Ganceviciene R, Dessinioti C, Feldman SR, Zouboulis CC. Acne vulgaris. Nat Rev Dis Primers. (2015) 1:15029. doi: 10.1038/nrdp.2015.29
5. Ramasamy S, Barnard E, Dawson TL Jr., Li H. The role of the skin microbiota in acne pathophysiology. Br J Dermatol. (2019) 181:691–9. doi: 10.1111/bjd.v181.4
6. Saheb Kashaf S, Proctor DM, Deming C, Saary P, Hölzer M, Taylor ME, et al. Integrating cultivation and metagenomics for a multi-kingdom view of skin microbiome diversity and functions. Nat Microbiol. (2022) 7:169–79. doi: 10.1038/s41564-021-01011-w
7. Byrd AL, Belkaid Y, Segre JA. The human skin microbiome. Nat Rev Microbiol. (2018) 16:143–55. doi: 10.1038/nrmicro.2017.157
8. Swaney MH, Kalan LR. Living in your skin: microbes, molecules, and mechanisms. Infect Immun. (2021) 89. doi: 10.1128/IAI.00695-20
9. Carmona-Cruz S, Orozco-Covarrubias L, Sáez-de-Ocariz M. The human skin microbiome in selected cutaneous diseases. Front Cell Infect Microbiol. (2022) 12:834135. doi: 10.3389/fcimb.2022.834135
10. Dreno B, Dekio I, Baldwin H, Demessant AL, Dagnelie MA, Khammari A, et al. Acne microbiome: From phyla to phylotypes. J Eur Acad Dermatol Venereol. (2023). doi: 10.1111/jdv.19540
11. Dréno B, Dagnelie MA, Khammari A, Corvec S. The skin microbiome: A new actor in inflammatory acne. Am J Clin Dermatol. (2020) 21:18–24. doi: 10.1007/s40257-020-00531-1
12. Cavallo I, Sivori F, Truglio M, De Maio F, Lucantoni F, Cardinali G, et al. Skin dysbiosis and Cutibacterium acnes biofilm in inflammatory acne lesions of adolescents. Sci Rep. (2022) 12:21104. doi: 10.1038/s41598-022-25436-3
13. Condrò G, Guerini M, Castello M, Perugini P. Acne vulgaris, atopic dermatitis and rosacea: the role of the skin microbiota-A review. Biomedicines. (2022) 10. doi: 10.3390/biomedicines10102523
14. Lee YB, Byun EJ, Kim HS. Potential role of the microbiome in acne: A comprehensive review. J Clin Med. (2019) 8:987. doi: 10.3390/jcm8070987
15. Marcinkiewicz M, Majewski S. The role of antimicrobial peptides in chronic inflammatory skin diseases. Postepy Dermatol Alergol. (2016) 33:6–12. doi: 10.5114/pdia.2015.48066
16. Harder J, Schroder JM. RNase 7, a novel innate immune defense antimicrobial protein of healthy human skin. J Biol Chem. (2002) 277:46779–84. doi: 10.1074/jbc.M207587200
17. Harder J, Tsuruta D, Murakami M, Kurokawa I. What is the role of antimicrobial peptides (AMP) in acne vulgaris? Exp Dermatol. (2013) 22:386–91. doi: 10.1111/exd.12159
18. Simanski M, Köten B, Schröder JM, Gläser R, Harder J. Antimicrobial RNases in cutaneous defense. J Innate Immun. (2012) 4:241–7. doi: 10.1159/000335029
19. Niyonsaba F, Kiatsurayanon C, Chieosilapatham P, Ogawa H. Friends or Foes? Host defense (antimicrobial) peptides and proteins in human skin diseases. Exp Dermatol. (2017) 26:989–98. doi: 10.1111/exd.2017.26.issue-11
20. Takahashi T, Gallo RL. The critical and multifunctional roles of antimicrobial peptides in dermatology. Dermatol Clin. (2017) 35:39–50. doi: 10.1016/j.det.2016.07.006
21. Kelhälä HL, Palatsi R, Fyhrquist N, Lehtimäki S, Väyrynen JP, Kallioinen M, et al. IL-17/Th17 pathway is activated in acne lesions. PloS One. (2014) 9:e105238. doi: 10.1371/journal.pone.0105238
22. Trivedi NR, Gilliland KL, Zhao W, Liu W, Thiboutot DM. Gene array expression profiling in acne lesions reveals marked upregulation of genes involved in inflammation and matrix remodeling. J Invest Dermatol. (2006) 126:1071–9. doi: 10.1038/sj.jid.5700213
23. Schauber J, Gallo RL. Antimicrobial peptides and the skin immune defense system. J Allergy Clin Immunol. (2008) 122:261–6. doi: 10.1016/j.jaci.2008.03.027
24. McDowell A, Barnard E, Nagy I, Gao A, Tomida S, Li H, et al. An expanded multilocus sequence typing scheme for propionibacterium acnes: investigation of ‘pathogenic’, ‘commensal’ and antibiotic resistant strains. PloS One. (2012) 7:e41480. doi: 10.1371/journal.pone.0041480
25. Santiago-Rodriguez TM, Le François B, Macklaim JM, Doukhanine E, Hollister EB. The skin microbiome: current techniques, challenges, and future directions. Microorganisms. (2023) 11. doi: 10.3390/microorganisms11051222
26. Grice EA. The skin microbiome: potential for novel diagnostic and therapeutic approaches to cutaneous disease. Semin Cutan Med Surg. (2014) 33:98–103. doi: 10.12788/j.sder.0087
27. Pasupuleti M, Schmidtchen A, Malmsten M. Antimicrobial peptides: key components of the innate immune system. Crit Rev Biotechnol. (2012) 32:143–71. doi: 10.3109/07388551.2011.594423
28. Zhang K, Li X, Yu C, Wang Y. Promising therapeutic strategies against microbial biofilm challenges. Front Cell Infect Microbiol. (2020) 10:359. doi: 10.3389/fcimb.2020.00359
29. Li X, Zuo S, Wang B, Zhang K, Wang Y. Antimicrobial mechanisms and clinical application prospects of antimicrobial peptides. Molecules. (2022) 27. doi: 10.3390/molecules27092675
30. Prasad SV, Fiedoruk K, Daniluk T, Piktel E, Bucki R. Expression and function of host defense peptides at inflammation sites. Int J Mol Sci. (2019) 21. doi: 10.3390/ijms21010104
31. Bin Hafeez A, Jiang X, Bergen PJ, Zhu Y. Antimicrobial peptides: an update on classifications and databases. Int J Mol Sci. (2021) 22. doi: 10.3390/ijms222111691
32. Bhunia A, Saravanan R, Mohanram H, Mangoni ML, Bhattacharjya S. NMR structures and interactions of temporin-1Tl and temporin-1Tb with lipopolysaccharide micelles: mechanistic insights into outer membrane permeabilization and synergistic activity. J Biol Chem. (2011) 286:24394–406. doi: 10.1074/jbc.M110.189662
33. Moretta A, Scieuzo C, Petrone AM, Salvia R, Manniello MD, Franco A, et al. Antimicrobial peptides: A new hope in biomedical and pharmaceutical fields. Front Cell Infect Microbiol. (2021) 11:668632. doi: 10.3389/fcimb.2021.668632
34. Wang G, Vaisman II, van Hoek ML. Machine learning prediction of antimicrobial peptides. Methods Mol Biol. (2022) 2405:1–37. doi: 10.1007/978-1-0716-1855-4_1
35. Aoki W, Ueda M. Characterization of antimicrobial peptides toward the development of novel antibiotics. Pharm (Basel). (2013) 6:1055–81. doi: 10.3390/ph6081055
36. Buda De Cesare G, Cristy SA, Garsin DA, Lorenz MC. Antimicrobial peptides: a new frontier in antifungal therapy. mBio. (2020) 11. doi: 10.1128/mBio.02123-20
37. Mahlapuu M, Håkansson J, Ringstad L, Björn C. Antimicrobial peptides: an emerging category of therapeutic agents. Front Cell Infect Microbiol. (2016) 6:194. doi: 10.3389/fcimb.2016.00194
38. Brown S, Santa Maria JP Jr., Walker S. Wall teichoic acids of gram-positive bacteria. Annu Rev Microbiol. (2013) 67:313–36. doi: 10.1146/annurev-micro-092412-155620
39. Malanovic N, Lohner K. Antimicrobial peptides targeting gram-positive bacteria. Pharm (Basel). (2016) 9. doi: 10.3390/ph9030059
40. Mitchell NJ, Seaton P, Pokorny A. Branched phospholipids render lipid vesicles more susceptible to membrane-active peptides. Biochim Biophys Acta. (2016) 1858:988–94. doi: 10.1016/j.bbamem.2015.10.014
41. Boparai JK, Sharma PK. Mini review on antimicrobial peptides, sources, mechanism and recent applications. Protein Pept Lett. (2020) 27:4–16. doi: 10.2174/18755305MTAwENDE80
42. Somner EA, Reynolds PE. Inhibition of peptidoglycan biosynthesis by ramoplanin. Antimicrob Agents Chemother. (1990) 34:413–9. doi: 10.1128/AAC.34.3.413
43. Hussein M, Karas JA, Schneider-Futschik EK, Chen F, Swarbrick J, Paulin OKA, et al. The Killing Mechanism of Teixobactin against Methicillin-Resistant Staphylococcus aureus: an Untargeted Metabolomics Study. mSystems. (2020) 5. doi: 10.1128/msystems.00077-20
44. Matsuzaki K, Sugishita K, Ishibe N, Ueha M, Nakata S, Miyajima K, et al. Relationship of membrane curvature to the formation of pores by magainin 2. Biochemistry. (1998) 37:11856–63. doi: 10.1021/bi980539y
45. Kumar P, Kizhakkedathu JN, Straus SK. Antimicrobial peptides: diversity, mechanism of action and strategies to improve the activity and biocompatibility in vivo. Biomolecules. (2018) 8. doi: 10.3390/biom8010004
46. Abrunhosa F, Faria S, Gomes P, Tomaz I, Pessoa JC, Andreu D, et al. Interaction and lipid-induced conformation of two cecropin-melittin hybrid peptides depend on peptide and membrane composition. J Phys Chem B. (2005) 109:17311–9. doi: 10.1021/jp051572e
47. Marquette A, Bechinger B. Biophysical investigations elucidating the mechanisms of action of antimicrobial peptides and their synergism. Biomolecules. (2018) 8. doi: 10.3390/biom8020018
48. Zhang QY, Yan ZB, Meng YM, Hong XY, Shao G, Ma JJ, et al. Antimicrobial peptides: mechanism of action, activity and clinical potential. Mil Med Res. (2021) 8:48. doi: 10.1186/s40779-021-00343-2
49. Epand RM, Walker C, Epand RF, Magarvey NA. Molecular mechanisms of membrane targeting antibiotics. Biochim Biophys Acta. (2016) 1858:980–7. doi: 10.1016/j.bbamem.2015.10.018
50. Johnson T, Kang D, Barnard E, Li H. Strain-level differences in porphyrin production and regulation in propionibacterium acnes elucidate disease associations. mSphere. (2016) 1. doi: 10.1128/mSphere.00023-15
51. Hale JD, Hancock RE. Alternative mechanisms of action of cationic antimicrobial peptides on bacteria. Expert Rev Anti Infect Ther. (2007) 5:951–9. doi: 10.1586/14787210.5.6.951
52. Le CF, Fang CM, Sekaran SD. Intracellular targeting mechanisms by antimicrobial peptides. Antimicrob Agents Chemother. (2017) 61. doi: 10.1128/AAC.02340-16
53. Hsu CH, Chen C, Jou ML, Lee AY, Lin YC, Yu YP, et al. Structural and DNA-binding studies on the bovine antimicrobial peptide, indolicidin: evidence for multiple conformations involved in binding to membranes and DNA. Nucleic Acids Res. (2005) 33:4053–64. doi: 10.1093/nar/gki725
54. Marchand C, Krajewski K, Lee HF, Antony S, Johnson AA, Amin R, et al. Covalent binding of the natural antimicrobial peptide indolicidin to DNA abasic sites. Nucleic Acids Res. (2006) 34:5157–65. doi: 10.1093/nar/gkl667
55. Xie Q, Wang Y, Zhang M, Wu S, Wei W, Xiao W, et al. Recombinant HNP-1 Produced by Escherichia coli Triggers Bacterial Apoptosis and Exhibits Antibacterial Activity against Drug-Resistant Bacteria. Microbiol Spectr. (2022) 10:e0086021. doi: 10.1128/spectrum.00860-21
56. Chileveru HR, Lim SA, Chairatana P, Wommack AJ, Chiang IL, Nolan EM. Visualizing attack of Escherichia coli by the antimicrobial peptide human defensin 5. Biochemistry. (2015) 54:1767–77. doi: 10.1021/bi501483q
57. Otvos L, Insug O, Rogers ME, Consolvo PJ, Condie BA, Lovas S, et al. Interaction between heat shock proteins and antimicrobial peptides. Biochemistry. (2000) 39:14150–9. doi: 10.1021/bi0012843
58. Shaheen B, Gonzalez M. Acne sans P. acnes. J Eur Acad Dermatol Venereol. (2013) 27:1–10. doi: 10.1111/j.1468-3083.2012.04516.x
59. Shamban AT, Narurkar VA. Multimodal treatment of acne, acne scars and pigmentation. Dermatol Clin. (2009) 27:459–71, vi. doi: 10.1016/j.det.2009.08.010
60. Fox L, Csongradi C, Aucamp M, du Plessis J, Gerber M. Treatment modalities for acne. Molecules. (2016) 21. doi: 10.3390/molecules21081063
61. Tanghetti EA. The role of inflammation in the pathology of acne. J Clin Aesthetic Dermatol. (2013) 6:27–35.
62. Williams HC, Dellavalle RP, Garner S. Acne vulgaris. Lancet. 2012 379:361–72. doi: 10.1016/S0140-6736(11)60321-8
63. Kiatsurayanon C, Niyonsaba F, Smithrithee R, Akiyama T, Ushio H, Hara M, et al. Host defense (Antimicrobial) peptide, human β-defensin-3, improves the function of the epithelial tight-junction barrier in human keratinocytes. J Invest Dermatol. (2014) 134:2163–73. doi: 10.1038/jid.2014.143
64. Luu M, Visekruna A. Short-chain fatty acids: Bacterial messengers modulating the immunometabolism of T cells. Eur J Immunol. (2019) 49:842–8. doi: 10.1002/eji.201848009
65. Karagianni F, Pavlidis A, Malakou LS, Piperi C, Papadavid E. Predominant role of mTOR signaling in skin diseases with therapeutic potential. Int J Mol Sci. (2022) 23. doi: 10.3390/ijms23031693
66. Barnea M, Madar Z, Froy O. Glucose and insulin are needed for optimal defensin expression in human cell lines. Biochem Biophys Res Commun. (2008) 367:452–6. doi: 10.1016/j.bbrc.2007.12.158
67. Nakatsuji T, Kao MC, Zhang L, Zouboulis CC, Gallo RL, Huang CM. Sebum free fatty acids enhance the innate immune defense of human sebocytes by upregulating beta-defensin-2 expression. J Invest Dermatol. (2010) 130:985–94. doi: 10.1038/jid.2009.384
68. ElAttar Y, Mourad B, Alngomy HA, Shams El Deen A, Ismail M. Study of interleukin-1 beta expression in acne vulgaris and acne scars. J Cosmetic Dermatol. (2022) 21:4864–70. doi: 10.1111/jocd.v21.10
69. Kistowska M, Gehrke S, Jankovic D, Kerl K, Fettelschoss A, Feldmeyer L, et al. IL-1β drives inflammatory responses to propionibacterium acnes in vitro and in vivo. J Invest Dermatol. (2014) 134:677–85. doi: 10.1038/jid.2013.438
70. Akamatsu H, Horio T. The possible role of reactive oxygen species generated by neutrophils in mediating acne inflammation. Dermatol (Basel Switzerland). (1998) 196:82–5. doi: 10.1159/000017876
71. Bungau AF, Radu AF, Bungau SG, Vesa CM, Tit DM, Endres LM. Oxidative stress and metabolic syndrome in acne vulgaris: Pathogenetic connections and potential role of dietary supplements and phytochemicals. Biomedicine Pharmacotherapy. (2023) 164:115003. doi: 10.1016/j.biopha.2023.115003
72. Sørensen OE, Cowland JB, Theilgaard-Mönch K, Liu L, Ganz T, Borregaard N. Wound healing and expression of antimicrobial peptides/polypeptides in human keratinocytes, a consequence of common growth factors. J Immunol. (2003) 170:5583–9. doi: 10.4049/jimmunol.170.11.5583
73. Sardana K, Verma G. Propionibacterium acnes and the th1/th17 axis, implications in acne pathogenesis and treatment. Indian J Dermatol. (2017) 62:392–4. doi: 10.4103/ijd.IJD_483_16
74. Abdel-Wahab HM, Ali AK, Ragaie MH. Calcipotriol: A novel tool in treatment of acne vulgaris. Dermatologic Ther. (2022) 35:e15690. doi: 10.1111/dth.15690
75. Mahran A, Ghazally A, Ali AS, Bakr RM. Efficacy and safety of calcipotriol as a potential topical treatment of acne vulgaris: a randomized, controlled, triple blinded, split-face clinical trial. Clin Exp Dermatol. (2023) 49:348–55. doi: 10.1093/ced/llad371
76. Dommisch H, Winter J, Götz W, Miesen J, Klein A, Hierse L, et al. Effect of growth factors on antimicrobial peptides and pro-inflammatory mediators during wound healing. Clin Oral Investigations. (2015) 19:209–20. doi: 10.1007/s00784-014-1239-9
77. Eichler TE, Becknell B, Easterling RS, Ingraham SE, Cohen DM, Schwaderer AL, et al. Insulin and the phosphatidylinositol 3-kinase signaling pathway regulate Ribonuclease 7 expression in the human urinary tract. Kidney Int. (2016) 90:568–79. doi: 10.1016/j.kint.2016.04.025
78. Nagy I, Pivarcsi A, Koreck A, Széll M, Urbán E, Kemény L. Distinct strains of Propionibacterium acnes induce selective human beta-defensin-2 and interleukin-8 expression in human keratinocytes through toll-like receptors. J Invest Dermatol. (2005) 124:931–8. doi: 10.1111/j.0022-202X.2005.23705.x
79. Nagy I, Pivarcsi A, Kis K, Koreck A, Bodai L, McDowell A, et al. Propionibacterium acnes and lipopolysaccharide induce the expression of antimicrobial peptides and proinflammatory cytokines/chemokines in human sebocytes. Microbes Infection. (2006) 8:2195–205. doi: 10.1016/j.micinf.2006.04.001
80. Niyonsaba F, Ushio H, Hara M, Yokoi H, Tominaga M, Takamori K, et al. Antimicrobial peptides human beta-defensins and cathelicidin LL-37 induce the secretion of a pruritogenic cytokine IL-31 by human mast cells. J Immunol. (2010) 184:3526–34. doi: 10.4049/jimmunol.0900712
81. Hirota K, Yoshitomi H, Hashimoto M, Maeda S, Teradaira S, Sugimoto N, et al. Preferential recruitment of CCR6-expressing Th17 cells to inflamed joints via CCL20 in rheumatoid arthritis and its animal model. J Exp Med. (2007) 204:2803–12. doi: 10.1084/jem.20071397
82. Li D, Li J, Duan Y, Zhou X. Expression of LL-37, human beta defensin-2, and CCR6 mRNA in patients with psoriasis vulgaris. J Huazhong Univ Sci Technol Med Sci = Hua zhong ke ji da xue xue bao Yi xue Ying wen ban = Huazhong keji daxue xuebao Yixue Yingdewen ban. (2004) 24:404–6. doi: 10.1007/BF02861879
83. Kim MS, Lee DY. Insulin-like growth factor (IGF)-I and IGF binding proteins axis in diabetes mellitus. Ann Pediatr Endocrinol Metab. (2015) 20:69–73. doi: 10.6065/apem.2015.20.2.69
84. Sadowska-Przytocka A, Gruszczyńska M, Ostałowska A, Antosik P, Czarnecka-Operacz M, Adamski Z, et al. Insulin resistance in the course of acne - literature review. Postepy Dermatologii I Alergologii. (2022) 39:231–8. doi: 10.5114/ada.2021.107101
85. Sadati MS, Yazdanpanah N, Shahriarirad R, Javaheri R, Parvizi MM. Efficacy of metformin vs. doxycycline in treating acne vulgaris: An assessor-blinded, add-on, randomized, controlled clinical trial. J Cosmetic Dermatol. (2023) 22:2816–23. doi: 10.1111/jocd.v22.10
86. Mohanty S, Kamolvit W, Scheffschick A, Björklund A, Tovi J, Espinosa A, et al. Diabetes downregulates the antimicrobial peptide psoriasin and increases E. coli burden in the urinary bladder. Nat Commun. (2022) 13:4983. doi: 10.1038/s41467-022-32636-y
87. Yin J, Yu FS. LL-37 via EGFR transactivation to promote high glucose-attenuated epithelial wound healing in organ-cultured corneas. Invest Ophthalmol Visual Sci. (2010) 51:1891–7. doi: 10.1167/iovs.09-3904
88. Al-Shobaili HA. Oxidants and anti-oxidants status in acne vulgaris patients with varying severity. Ann Clin Lab Sci. (2014) 44:202–7.
89. Popa GL, Mitran CI, Mitran MI, Tampa M, Matei C, Popa MI, et al. Markers of oxidative stress in patients with acne: A literature review. Life (Basel Switzerland). (2023) 13. doi: 10.3390/life13071433
90. Sorci G, Riuzzi F, Giambanco I, Donato R. RAGE in tissue homeostasis, repair and regeneration. Biochim Biophys Acta. (2013) 1833:101–9. doi: 10.1016/j.bbamcr.2012.10.021
91. Farese RV, Sajan MP. Metabolic functions of atypical protein kinase C: “good” and “bad” as defined by nutritional status. Am J Physiol Endocrinol Metab. (2010) 298:E385–94. doi: 10.1152/ajpendo.00608.2009
92. Heinemann U, Schuetz A. Structural features of tight-junction proteins. Int J Mol Sci. (2019) 20. doi: 10.3390/ijms20236020
93. Hoffmeister L, Diekmann M, Brand K, Huber R. GSK3: A kinase balancing promotion and resolution of inflammation. Cells. (2020) 9:820. doi: 10.3390/cells9040820
94. Jope RS, Yuskaitis CJ, Beurel E. Glycogen synthase kinase-3 (GSK3): inflammation, diseases, and therapeutics. Neurochemical Res. (2007) 32:577–95. doi: 10.1007/s11064-006-9128-5
95. Cortés-Vieyra R, Silva-García O, Gómez-García A, Gutiérrez-Castellanos S, Álvarez-Aguilar C, Baizabal-Aguirre VM. Glycogen synthase kinase 3β Modulates the inflammatory response activated by bacteria, viruses, and parasites. Front Immunol. (2021) 12. doi: 10.3389/fimmu.2021.675751
96. Marei H, Malliri A. Rac1 in human diseases: The therapeutic potential of targeting Rac1 signaling regulatory mechanisms. Small GTPases. (2017) 8:139–63. doi: 10.1080/21541248.2016.1211398
97. Ma N, Xu E, Luo Q, Song G. Rac1: A regulator of cell migration and a potential target for cancer therapy. Molecules. (2023) 28:2976. doi: 10.3390/molecules28072976
98. Winge MCG, Marinkovich MP. Epidermal activation of the small GTPase Rac1 in psoriasis pathogenesis. Small GTPases. (2019) 10:163–8. doi: 10.1080/21541248.2016.1273861
99. Akiyama T, Niyonsaba F, Kiatsurayanon C, Nguyen TT, Ushio H, Fujimura T, et al. The human cathelicidin LL-37 host defense peptide upregulates tight junction-related proteins and increases human epidermal keratinocyte barrier function. J Innate Immun. (2014) 6:739–53. doi: 10.1159/000362789
100. Bareja A, Patel S, Hodgkinson CP, Payne A, Dzau VJ. Understanding the mechanism of bias signaling of the insulin-like growth factor 1 receptor: Effects of LL37 and HASF. Cell Signalling. (2018) 46:113–9. doi: 10.1016/j.cellsig.2018.02.013
101. Ikutama R, Peng G, Tsukamoto S, Umehara Y, Trujillo-Paez JV, Yue H, et al. Cathelicidin LL-37 activates human keratinocyte autophagy through the P2X7, mechanistic target of rapamycin, and MAPK pathways. J Invest Dermatol. (2023) 143:751–61.e7. doi: 10.1016/j.jid.2022.10.020
102. Ganceviciene R, Fimmel S, Glass E, Zouboulis CC. Psoriasin and follicular hyperkeratinization in acne comedones. Dermatology. (2006) 213:270–2. doi: 10.1159/000095058
103. Boylan JF, Gudas LJ. Overexpression of the cellular retinoic acid binding protein-I (CRABP-I) results in a reduction in differentiation-specific gene expression in F9 teratocarcinoma cells. J Cell Biol. (1991) 112:965–79. doi: 10.1083/jcb.112.5.965
104. Hau CS, Kanda N, Tada Y, Shibata S, Uozaki H, Fukusato T, et al. Lipocalin-2 exacerbates psoriasiform skin inflammation by augmenting T-helper 17 response. J Dermatol. (2016) 43:785–94. doi: 10.1111/jde.2016.43.issue-7
105. Niaz B, Saeed F, Ahmed A, Imran M, Maan AA, Khan MKI, et al. Lactoferrin (LF): a natural antimicrobial protein. Int J Food Properties. (2019) 22:1626–41. doi: 10.1080/10942912.2019.1666137
106. Chan H, Chan G, Santos J, Dee K, Co JK. A randomized, double-blind, placebo-controlled trial to determine the efficacy and safety of lactoferrin with vitamin E and zinc as an oral therapy for mild to moderate acne vulgaris. Int J Dermatol. (2017) 56:686–90. doi: 10.1111/ijd.2017.56.issue-6
107. Ianiro G, Niro A, Rosa L, Valenti P, Musci G, Cutone A. To boost or to reset: the role of lactoferrin in energy metabolism. Int J Mol Sci. (2023) 24:15925. doi: 10.3390/ijms242115925
108. Nguyen HLT, Peng G, Trujillo-Paez JV, Yue H, Ikutama R, Takahashi M, et al. The antimicrobial peptide AMP-IBP5 suppresses dermatitis-like lesions in a mouse model of atopic dermatitis through the low-density lipoprotein receptor-related protein-1 receptor. Int J Mol Sci. (2023) 24:5200. doi: 10.3390/ijms24065200
109. Yoshiba S, Peng GE, Niyonsaba F. A skin-derived antimicrobial peptide derived from insulin-like growth factor-binding protein 5 (AMP-IBP5) as therapeutic candidate for psoriasis. Juntendo Iji Zasshi. (2023) 69:103–4. doi: 10.14789/jmj.JMJ22-0050-OT
110. Yue H, Song P, Sutthammikorn N, Umehara Y, Trujillo-Paez JV, Nguyen HLT, et al. Antimicrobial peptide derived from insulin-like growth factor-binding protein 5 improves diabetic wound healing. Wound Repair Regeneration. (2022) 30:232–44. doi: 10.1111/wrr.12997
111. Chopra D, Arens RA, Amornpairoj W, Lowes MA, Tomic-Canic M, Strbo N, et al. Innate immunity and microbial dysbiosis in hidradenitis suppurativa - vicious cycle of chronic inflammation. Front Immunol. (2022) 13:960488. doi: 10.3389/fimmu.2022.960488
112. Kopfnagel V, Wagenknecht S, Harder J, Hofmann K, Kleine M, Buch A, et al. RNase 7 strongly promotes TLR9-mediated DNA sensing by human plasmacytoid dendritic cells. J Invest Dermatol. (2018) 138:872–81. doi: 10.1016/j.jid.2017.09.052
113. Rosenfield RL, Deplewski D. Role of androgens in the developmental biology of the pilosebaceous unit. Am J Med. (1995) 98:S80–S8. doi: 10.1016/S0002-9343(99)80063-1
114. Faruga-Lewicka W, Kardas MJACM. The influence of insulin-like growth factor IGF-1 on the course of acne vulgaris. Aesthetic Cosmetology and Medicine - Aesthetic Cosmetology. (2022) 11:105–8. doi: 10.52336/acm.2022.016
115. Okoro OE, Camera E, Flori E, Ottaviani M. Insulin and the sebaceous gland function. Front Physiol. (2023) 14:1252972. doi: 10.3389/fphys.2023.1252972
116. Vora S, Ovhal A, Jerajani H, Nair N, Chakrabortty A. Correlation of facial sebum to serum insulin-like growth factor-1 in patients with acne. Br J Dermatol. (2008) 159:990–1. doi: 10.1111/j.1365-2133.2008.08764.x
117. Melnik BC. Acne transcriptomics: fundamentals of acne pathogenesis and isotretinoin treatment. Cells. (2023) 12:2600. doi: 10.3390/cells12222600
118. Melnik BC. Linking diet to acne metabolomics, inflammation, and comedogenesis: an update. Clinical Cosmetic Investigational Dermatol. (2015) 8:371–88. doi: 10.2147/CCID.S69135
119. Melnik BC, John SM, Schmitz G. Over-stimulation of insulin/IGF-1 signaling by Western diet may promote diseases of civilization: lessons learnt from Laron syndrome. Nutr Metab. (2011) 8:41. doi: 10.1186/1743-7075-8-41
120. Liu XF, Shao JH, Liao YT, Wang LN, Jia Y, Dong PJ, et al. Regulation of short-chain fatty acids in the immune system. Front Immunol. (2023) 14:1186892. doi: 10.3389/fimmu.2023.1186892
121. Bowe WP, Logan AC. Acne vulgaris, probiotics and the gut-brain-skin axis - back to the future? Gut Pathog. (2011) 3:1. doi: 10.1186/1757-4749-3-1
122. Sánchez-Pellicer P, Navarro-Moratalla L, Núñez-Delegido E, Ruzafa-Costas B, Agüera-Santos J, Navarro-López V. Acne, microbiome, and probiotics: the gut-skin axis. Microorganisms. (2022) 10. doi: 10.3390/microorganisms10071303
123. Gao Y, Tian T. mTOR signaling pathway and gut microbiota in various disorders: mechanisms and potential drugs in pharmacotherapy. Int J Mol Sci. (2023) 24. doi: 10.3390/ijms241411811
124. Jović A, Marinović B, Kostović K, Čeović R, Basta-Juzbašić A, Bukvić Mokos Z. The impact of pyschological stress on acne. Acta Dermatovenerologica Croatica: ADC. (2017) 25:1133–141.
125. Ganceviciene R, Böhm M, Fimmel S, Zouboulis CC. The role of neuropeptides in the multifactorial pathogenesis of acne vulgaris. Dermato-endocrinology. (2009) 1:170–6. doi: 10.4161/derm.1.3.8496
126. Zouboulis CC. Propionibacterium acnes and sebaceous lipogenesis: a love-hate relationship? J Invest Dermatol. (2009) 129:2093–6. doi: 10.1038/jid.2009.190
127. Schernthaner-Reiter MH, Wolf P, Vila G, Luger A. The interaction of insulin and pituitary hormone syndromes. Front Endocrinol. (2021) 12:. doi: 10.3389/fendo.2021.626427
128. Karagiannides I, Bakirtzi K, Kokkotou E, Stavrakis D, Margolis KG, Thomou T, et al. Role of substance P in the regulation of glucose metabolism via insulin signaling-associated pathways. Endocrinology. (2011) 152:4571–80. doi: 10.1210/en.2011-1170
129. Brown M, Vale W. Effects of neurotensin and substance p on plasma insulin, glucagon and glucose levels. Endocrinology. (1976) 98:819–22. doi: 10.1210/endo-98-3-819
130. Robinson GN, Pickering RJ. Melanocortins and their potential for the treatment, prevention and amelioration of complications of diabetes. Diabetology. (2024) 5:69–84. doi: 10.3390/diabetology5010006
131. Makrantonaki E, Ganceviciene R, Zouboulis C. An update on the role of the sebaceous gland in the pathogenesis of acne. Dermato-endocrinology. (2011) 3:41–9. doi: 10.4161/derm.3.1.13900
132. Ottaviani M, Camera E, Picardo M. Lipid mediators in acne. Mediators Inflammation. (2010) 2010:858176. doi: 10.1155/2010/858176
133. Zhao G, Etherton TD, Martin KR, Vanden Heuvel JP, Gillies PJ, West SG, et al. Anti-inflammatory effects of polyunsaturated fatty acids in THP-1 cells. Biochem Biophys Res Commun. (2005) 336:909–17. doi: 10.1016/j.bbrc.2005.08.204
134. Borodzicz S, Rudnicka L, Mirowska-Guzel D, Cudnoch-Jedrzejewska A. The role of epidermal sphingolipids in dermatologic diseases. Lipids Health Dis. (2016) 15:13. doi: 10.1186/s12944-016-0178-7
135. Yamamoto A, Takenouchi K, Ito M. Impaired water barrier function in acne vulgaris. Arch Dermatol Res. (1995) 287:214–8. doi: 10.1007/BF01262335
136. Okoro OE, Adenle A, Ludovici M, Truglio M, Marini F, Camera E. Lipidomics of facial sebum in the comparison between acne and non-acne adolescents with dark skin. Sci Rep. (2021) 11:16591. doi: 10.1038/s41598-021-96043-x
137. Tax G, Urbán E, Palotás Z, Puskás R, Kónya Z, Bíró T, et al. Propionic acid produced by propionibacterium acnes strains contri-butes to their pathogenicity. Acta Derm Venereol. (2016) 96:43–9. doi: 10.2340/00015555-2154
138. Shu M, Wang Y, Yu J, Kuo S, Coda A, Jiang Y, et al. Fermentation of Propionibacterium acnes, a commensal bacterium in the human skin microbiome, as skin probiotics against methicillin-resistant Staphylococcus aureus. PloS One. (2013) 8:e55380. doi: 10.1371/journal.pone.0055380
139. Dagnelie MA, Corvec S, Saint-Jean M, Nguyen JM, Khammari A, Dréno B. Cutibacterium acnes phylotypes diversity loss: a trigger for skin inflammatory process. J Eur Acad Dermatol Venereol. (2019) 33:2340–8. doi: 10.1111/jdv.v33.12
140. Mongaret C, Velard F, Reffuveille F. Cutibacterium acnes: the urgent need to identify diagnosis markers. Infect Immun. (2021) 89. doi: 10.1128/IAI.00753-20
141. Almoughrabie S, Cau L, Cavagnero K, O’Neill AM, Li F, Roso-Mares A, et al. Commensal Cutibacterium acnes induce epidermal lipid synthesis important for skin barrier function. Sci Adv. (2023) 9:eadg6262. doi: 10.1126/sciadv.adg6262
142. Claesen J, Spagnolo JB, Ramos SF, Kurita KL, Byrd AL, Aksenov AA, et al. A Cutibacterium acnes antibiotic modulates human skin microbiota composition in hair follicles. Sci Transl Med. (2020) 12. doi: 10.1126/scitranslmed.aay5445
143. Dréno B, Pécastaings S, Corvec S, Veraldi S, Khammari A, Roques C. Cutibacterium acnes (Propionibacterium acnes) and acne vulgaris: a brief look at the latest updates. J Eur Acad Dermatol Venereol. (2018) 32 Suppl 2:5–14. doi: 10.1111/jdv.15043
144. Platsidaki E, Dessinioti C. Recent advances in understanding Propionibacterium acnes (Cutibacterium acnes) in acne. F1000Res. (2018) 7. doi: 10.12688/f1000research
145. Goldenberger D, Søgaard KK, Cuénod A, Seth-Smith H, de Menezes D, Vandamme P, et al. Cutibacterium modestum and “Propionibacterium humerusii” represent the same species that is commonly misidentified as Cutibacterium acnes. Antonie van Leeuwenhoek. (2021) 114:1315–20. doi: 10.1007/s10482-021-01589-5
146. Szabó K, Bolla BS, Erdei L, Balogh F, Kemény L. Are the cutaneous microbiota a guardian of the skin’s physical barrier? The intricate relationship between skin microbes and barrier integrity. Int J Mol Sci. (2023) 24. doi: 10.3390/ijms242115962
147. Nakamura K, O’Neill AM, Williams MR, Cau L, Nakatsuji T, Horswill AR, et al. Short chain fatty acids produced by Cutibacterium acnes inhibit biofilm formation by Staphylococcus epidermidis. Sci Rep. (2020) 10:21237. doi: 10.1038/s41598-020-77790-9
148. Sanford JA, O’Neill AM, Zouboulis CC, Gallo RL. Short-chain fatty acids from cutibacterium acnes activate both a canonical and epigenetic inflammatory response in human sebocytes. J Immunol. (2019) 202:1767–76. doi: 10.4049/jimmunol.1800893
149. Choi EJ, Lee HG, Bae IH, Kim W, Park J, Lee TR, et al. Propionibacterium acnes-derived extracellular vesicles promote acne-like phenotypes in human epidermis. J Invest Dermatol. (2018) 138:1371–9. doi: 10.1016/j.jid.2018.01.007
150. Nazipi S, Stødkilde-Jørgensen K, Scavenius C, Brüggemann H. The skin bacterium propionibacterium acnes employs two variants of hyaluronate lyase with distinct properties. Microorganisms. (2017) 5. doi: 10.3390/microorganisms5030057
151. Jugeau S, Tenaud I, Knol AC, Jarrousse V, Quereux G, Khammari A, et al. Induction of toll-like receptors by Propionibacterium acnes. Br J Dermatol. (2005) 153:1105–13. doi: 10.1111/j.1365-2133.2005.06933.x
152. Firlej E, Kowalska W, Szymaszek K, Roliński J, Bartosińska J. The role of skin immune system in acne. J Clin Med. (2022) 11. doi: 10.3390/jcm11061579
153. Mayslich C, Grange PA, Castela M, Marcelin AG, Calvez V, Dupin N. Characterization of a cutibacterium acnes camp factor 1-related peptide as a new TLR-2 modulator in in vitro and ex vivo models of inflammation. Int J Mol Sci. (2022) 23. doi: 10.3390/ijms23095065
154. Mouser PE, Baker BS, Seaton ED, Chu AC. Propionibacterium acnes-reactive T helper-1 cells in the skin of patients with acne vulgaris. J Invest Dermatol. (2003) 121:1226–8. doi: 10.1046/j.1523-1747.2003.12550_6.x
155. Yu Y, Champer J, Agak GW, Kao S, Modlin RL, Kim J. Different propionibacterium acnes phylotypes induce distinct immune responses and express unique surface and secreted proteomes. J Invest Dermatol. (2016) 136:2221–8. doi: 10.1016/j.jid.2016.06.615
156. Dekio I, Culak R, Misra R, Gaulton T, Fang M, Sakamoto M, et al. Dissecting the taxonomic heterogeneity within Propionibacterium acnes: proposal for Propionibacterium acnes subsp. acnes subsp. nov. and Propionibacterium acnes subsp. elongatum subsp. nov. Int J Syst Evol Microbiol. (2015) 65:4776–87. doi: 10.1099/ijsem.0.000648
157. Dagnelie MA, Corvec S, Saint-Jean M, Bourdès V, Nguyen JM, Khammari A, et al. Decrease in diversity of propionibacterium acnes phylotypes in patients with severe acne on the back. Acta Derm Venereol. (2018) 98:262–7. doi: 10.2340/00015555-2847
158. Tomida S, Nguyen L, Chiu BH, Liu J, Sodergren E, Weinstock GM, et al. Pan-genome and comparative genome analyses of propionibacterium acnes reveal its genomic diversity in the healthy and diseased human skin microbiome. mBio. (2013) 4:e00003–13. doi: 10.1128/mBio.00003-13
159. Liu J, Cheng A, Bangayan NJ, Barnard E, Curd E, Craft N, et al. Draft genome sequences of propionibacterium acnes type strain ATCC6919 and antibiotic-resistant strain HL411PA1. Genome Announc. (2014) 2. doi: 10.1128/genomeA.00740-14
160. Fournière M, Latire T, Souak D, Feuilloley MGJ, Bedoux G. Staphylococcus epidermidis and Cutibacterium acnes: Two Major Sentinels of Skin Microbiota and the Influence of Cosmetics. Microorganisms. (2020) 8. doi: 10.3390/microorganisms8111752
161. Nakase K, Hayashi N, Akiyama Y, Aoki S, Noguchi N. Antimicrobial susceptibility and phylogenetic analysis of Propionibacterium acnes isolated from acne patients in Japan between 2013 and 2015. J Dermatol. (2017) 44:1248–54. doi: 10.1111/jde.2017.44.issue-11
162. Jahns AC, Lundskog B, Ganceviciene R, Palmer RH, Golovleva I, Zouboulis CC, et al. An increased incidence of Propionibacterium acnes biofilms in acne vulgaris: a case-control study. Br J Dermatol. (2012) 167:50–8. doi: 10.1111/j.1365-2133.2012.10897.x
163. Jarrousse V, Castex-Rizzi N, Khammari A, Charveron M, Dréno B. Modulation of integrins and filaggrin expression by Propionibacterium acnes extracts on keratinocytes. Arch Dermatol Res. (2007) 299:441–7. doi: 10.1007/s00403-007-0774-5
164. Isard O, Knol AC, Ariès MF, Nguyen JM, Khammari A, Castex-Rizzi N, et al. Propionibacterium acnes activates the IGF-1/IGF-1R system in the epidermis and induces keratinocyte proliferation. J Invest Dermatol. (2011) 131:59–66. doi: 10.1038/jid.2010.281
165. Cunliffe WJ, Holland DB, Clark SM, Stables GI. Comedogenesis: some new aetiological, clinical and therapeutic strategies. Br J Dermatol. (2000) 142:1084–91. doi: 10.1046/j.1365-2133.2000.03531.x
166. Gallo RL, Murakami M, Ohtake T, Zaiou M. Biology and clinical relevance of naturally occurring antimicrobial peptides. J Allergy Clin Immunol. (2002) 110:823–31. doi: 10.1067/mai.2002.129801
167. Harder J, Gläser R, Schröder JM. The role and potential therapeutical applications of antimicrobial proteins in infectious and inflammatory diseases. Endocr Metab Immune Disord Drug Targets. (2007) 7:75–82. doi: 10.2174/187153007780832091
168. Schittek B, Paulmann M, Senyürek I, Steffen H. The role of antimicrobial peptides in human skin and in skin infectious diseases. Infect Disord Drug Targets. (2008) 8:135–43. doi: 10.2174/1871526510808030135
169. Harder J, Dressel S, Wittersheim M, Cordes J, Meyer-Hoffert U, Mrowietz U, et al. Enhanced expression and secretion of antimicrobial peptides in atopic dermatitis and after superficial skin injury. J Invest Dermatol. (2010) 130:1355–64. doi: 10.1038/jid.2009.432
170. Yamasaki K, Di Nardo A, Bardan A, Murakami M, Ohtake T, Coda A, et al. Increased serine protease activity and cathelicidin promotes skin inflammation in rosacea. Nat Med. (2007) 13:975–80. doi: 10.1038/nm1616
171. Hata TR, Gallo RL. Antimicrobial peptides, skin infections, and atopic dermatitis. Semin Cutan Med Surg. (2008) 27:144–50. doi: 10.1016/j.sder.2008.04.002
172. Gläser R, Harder J, Lange H, Bartels J, Christophers E, Schröder JM. Antimicrobial psoriasin (S100A7) protects human skin from Escherichia coli infection. Nat Immunol. (2005) 6:57–64. doi: 10.1038/ni1142
173. Takahashi T, Yamasaki K. Psoriasis and antimicrobial peptides. Int J Mol Sci. (2020) 21. doi: 10.3390/ijms21186791
174. Chronnell CM, Ghali LR, Ali RS, Quinn AG, Holland DB, Bull JJ, et al. Human beta defensin-1 and -2 expression in human pilosebaceous units: upregulation in acne vulgaris lesions. J Invest Dermatol. (2001) 117:1120–5. doi: 10.1046/j.0022-202x.2001.01569.x
175. Philpott MP. Defensins and acne. Mol Immunol. (2003) 40:457–62. doi: 10.1016/S0161-5890(03)00154-8
176. Harder J, Schröder J-M. Antimicrobial Peptides: Role in Human Health and Disease. Switzerland: Springer International Publishing (2016).
177. Szklarczyk D, Gable AL, Nastou KC, Lyon D, Kirsch R, Pyysalo S, et al. The STRING database in 2021: customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. (2021) 49:D605–d12. doi: 10.1093/nar/gkaa1074
178. Borovaya A, Dombrowski Y, Zwicker S, Olisova O, Ruzicka T, Wolf R, et al. Isotretinoin therapy changes the expression of antimicrobial peptides in acne vulgaris. Arch Dermatol Res. (2014) 306:689–700. doi: 10.1007/s00403-014-1477-3
179. Adişen E, Yüksek J, Erdem O, Aksakal FN, Aksakal AB. Expression of human neutrophil proteins in acne vulgaris. J Eur Acad Dermatol Venereol. (2010) 24:32–7. doi: 10.1111/j.1468-3083.2009.03347.x
180. Ghannam S, Dejou C, Pedretti N, Giot J-P, Dorgham K, Boukhaddaoui H, et al. CCL20 and β-defensin-2 induce arrest of human th17 cells on inflamed endothelium in vitro under flow conditions. J Immunol. (2011) 186:1411–20. doi: 10.4049/jimmunol.1000597
181. Webster GF, Leyden JJ, Musson RA, Douglas SD. Susceptibility of Propionibacterium acnes to killing and degradation by human neutrophils and monocytes in vitro. Infect Immun. (1985) 49:116–21. doi: 10.1128/iai.49.1.116-121.1985
182. van Wetering S, Sterk PJ, Rabe KF, Hiemstra PS. Defensins: key players or bystanders in infection, injury, and repair in the lung? J Allergy Clin Immunol. (1999) 104:1131–8. doi: 10.1016/s0091-6749(99)70004-7
183. Taefehshokr N, Isazadeh A, Oveisi A, Key YA, Taefehshokr S. Reciprocal role of hBD2 and hBD3 on the adaptive immune response by measuring T lymphocyte proliferation in terms of CD4 and CCR6 expression. Horm Mol Biol Clin Investig. (2018) 35:. doi: 10.1515/hmbci-2018-0023
184. Gruszczyńska M, Sadowska-Przytocka A, Szybiak W, Więckowska B, Lacka K. Insulin resistance in patients with acne vulgaris. Biomedicines. (2023) 11:2294. doi: 10.3390/biomedicines11082294
185. Emiroğlu N, Cengiz FP, Kemeriz F. Insulin resistance in severe acne vulgaris. Postepy Dermatologii I Alergologii. (2015) 32:281–5. doi: 10.5114/pdia.2015.53047
186. Chandak S, Singh A, Madke B, Jawade S, Khandelwal R. Acne vulgaris and metabolic syndrome: A possible association. Cureus. (2022) 14:e24750. doi: 10.7759/cureus.24750
187. Webster GF. Inflammation in acne vulgaris. J Am Acad Dermatol. (1995) 33:247–53. doi: 10.1016/0190-9622(95)90243-0
188. Xu KP, Li Y, Ljubimov AV, Yu FS. High glucose suppresses epidermal growth factor receptor/phosphatidylinositol 3-kinase/Akt signaling pathway and attenuates corneal epithelial wound healing. Diabetes. (2009) 58:1077–85. doi: 10.2337/db08-0997
189. Fusco A, Savio V, Donniacuo M, Perfetto B, Donnarumma G. Antimicrobial peptides human beta-defensin-2 and -3 protect the gut during candida albicans infections enhancing the intestinal barrier integrity: in vitro study. Front Cell Infect Microbiol. (2021) 11:666900. doi: 10.3389/fcimb.2021.666900
190. Niyonsaba F, Nagaoka I, Ogawa H, Okumura K. Multifunctional antimicrobial proteins and peptides: natural activators of immune systems. Curr Pharm Des. (2009) 15:2393–413. doi: 10.2174/138161209788682271
191. Tokajuk J, Deptuła P, Piktel E, Daniluk T, Chmielewska S, Wollny T, et al. Cathelicidin LL-37 in health and diseases of the oral cavity. Biomedicines. (2022) 10:1086. doi: 10.3390/biomedicines10051086
192. Yang D, Chertov O, Oppenheim JJ. Participation of mammalian defensins and cathelicidins in anti-microbial immunity: receptors and activities of human defensins and cathelicidin (LL-37). J Leukoc Biol. (2001) 69:691–7. doi: 10.1189/jlb.69.5.691
193. Dhiman A, Talukdar S, Chaubey GK, Dilawari R, Modanwal R, Chaudhary S, et al. Regulation of macrophage cell surface GAPDH alters LL-37 internalization and downstream effects in the cell. J Innate Immun. (2023) 15:581–98. doi: 10.1159/000530083
194. Dombrowski Y, Peric M, Koglin S, Kammerbauer C, Göss C, Anz D, et al. Cytosolic DNA triggers inflammasome activation in keratinocytes in psoriatic lesions. Sci Transl Med. (2011) 3:82ra38. doi: 10.1126/scitranslmed.3002001
195. Chamorro CI, Weber G, Grönberg A, Pivarcsi A, Ståhle M. The human antimicrobial peptide LL-37 suppresses apoptosis in keratinocytes. J Invest Dermatol. (2009) 129:937–44. doi: 10.1038/jid.2008.321
196. Lee DY, Yamasaki K, Rudsil J, Zouboulis CC, Park GT, Yang JM, et al. Sebocytes express functional cathelicidin antimicrobial peptides and can act to kill propionibacterium acnes. J Invest Dermatol. (2008) 128:1863–6. doi: 10.1038/sj.jid.5701235
197. Donato R, Cannon BR, Sorci G, Riuzzi F, Hsu K, Weber DJ, et al. Functions of S100 proteins. Curr Mol Med. (2013) 13:24–57. doi: 10.2174/156652413804486214
198. Sreejit G, Flynn MC, Patil M, Krishnamurthy P, Murphy AJ, Nagareddy PR. S100 family proteins in inflammation and beyond. Adv Clin Chem. (2020) 98:173–231. doi: 10.1016/bs.acc.2020.02.006
199. Xia P, Ji X, Yan L, Lian S, Chen Z, Luo Y. Roles of S100A8, S100A9 and S100A12 in infection, inflammation and immunity. Immunology. (2024) 171:365–76. doi: 10.1111/imm.v171.3
200. D’Amico F, Skarmoutsou E, Granata M, Trovato C, Rossi GA, Mazzarino MC. S100A7: A rAMPing up AMP molecule in psoriasis. Cytokine Growth Factor Rev. (2016) 32:97–104. doi: 10.1016/j.cytogfr.2016.01.002
201. Gonzalez LL, Garrie K, Turner MD. Role of S100 proteins in health and disease. Biochim Et Biophys Acta Mol Cell Res. (2020) 1867:118677. doi: 10.1016/j.bbamcr.2020.118677
202. Jukic A, Bakiri L, Wagner EF, Tilg H, Adolph TE. Calprotectin: from biomarker to biological function. Gut. (2021) 70:1978–88. doi: 10.1136/gutjnl-2021-324855
203. Schiopu A, Cotoi OS. S100A8 and S100A9: DAMPs at the crossroads between innate immunity, traditional risk factors, and cardiovascular disease. Mediators Inflammation. (2013) 2013:828354. doi: 10.1155/2013/828354
204. Fouda I, Obaid ZM, Hegazy SF, Samir Abd Al-Samie H, Nofal A. Calprotectin in acne vulgaris: A possible contributory role. J Cosmetic Dermatol. (2021) 20:621–5. doi: 10.1111/jocd.13574
205. Korkmaz S, Fıçıcıoğlu SK. Calprotectin can play an inflammatory role in acne vulgaris. Postepy Dermatologii I Alergologii. (2018) 35:397–9. doi: 10.5114/ada.2017.71286
206. Gläser R, Köten B, Wittersheim M, Harder J. Psoriasin: key molecule of the cutaneous barrier? J Dtsch Dermatol Ges. (2011) 9:897–902. doi: 10.1111/j.1610-0387.2011.07683.x
207. Kurpet K, Chwatko G. S100 proteins as novel therapeutic targets in psoriasis and other autoimmune diseases. Molecules. (2022) 27. doi: 10.3390/molecules27196640
208. Wolf R, Ruzicka T, Yuspa SH. Novel S100A7 (psoriasin)/S100A15 (koebnerisin) subfamily: highly homologous but distinct in regulation and function. Amino Acids. (2011) 41:789–96. doi: 10.1007/s00726-010-0666-4
209. Algermissen B, Sitzmann J, LeMotte P, Czarnetzki B. Differential expression of CRABP II, psoriasin and cytokeratin 1 mRNA in human skin diseases. Arch Dermatol Res. (1996) 288:426–30. doi: 10.1007/BF02505229
210. Wang Q, Aleshintsev A, Jose AN, Aramini JM, Gupta R. Calcium regulates S100A12 zinc sequestration by limiting structural variations. Chembiochem: Eur J Chem Biol. (2020) 21:1372–82. doi: 10.1002/cbic.201900623
211. Yuki T, Haratake A, Koishikawa H, Morita K, Miyachi Y, Inoue S. Tight junction proteins in keratinocytes: localization and contribution to barrier function. Exp Dermatol. (2007) 16:324–30. doi: 10.1111/j.1600-0625.2006.00539.x
212. Hattori F, Kiatsurayanon C, Okumura K, Ogawa H, Ikeda S, Okamoto K, et al. The antimicrobial protein S100A7/psoriasin enhances the expression of keratinocyte differentiation markers and strengthens the skin’s tight junction barrier. Br J Dermatol. (2014) 171:742–53. doi: 10.1111/bjd.2014.171.issue-4
213. Rosenthal DS, Griffiths CE, Yuspa SH, Roop DR, Voorhees JJ. Acute or chronic topical retinoic acid treatment of human skin in vivo alters the expression of epidermal transglutaminase, loricrin, involucrin, filaggrin, and keratins 6 and 13 but not keratins 1, 10, and 14. J Invest Dermatol. (1992) 98:343–50. doi: 10.1111/1523-1747.ep12499802
214. Jinquan T, Vorum H, Larsen CG, Madsen P, Rasmussen HH, Gesser B, et al. Psoriasin: a novel chemotactic protein. J Invest Dermatol. (1996) 107:5–10. doi: 10.1111/1523-1747.ep12294284
215. Batycka-Baran A, Hattinger E, Marchenkov A, Koziol M, Bieniek A, Szepietowski J, et al. Koebnerisin (S100A15): A novel player in the pathogenesis of rosacea. J Am Acad Dermatol. (2019) 80:1753–5. doi: 10.1016/j.jaad.2018.06.012
216. Hegyi Z, Zwicker S, Bureik D, Peric M, Koglin S, Batycka-Baran A, et al. Vitamin D analog calcipotriol suppresses the Th17 cytokine-induced proinflammatory S100 “alarmins” psoriasin (S100A7) and koebnerisin (S100A15) in psoriasis. J Invest Dermatol. (2012) 132:1416–24. doi: 10.1038/jid.2011.486
217. Moschen AR, Adolph TE, Gerner RR, Wieser V, Tilg H. Lipocalin-2: A master mediator of intestinal and metabolic inflammation. Trends Endocrinol Metabolism: TEM. (2017) 28:388–97. doi: 10.1016/j.tem.2017.01.003
218. Al Hashimi SHS, Zaki MS, Soltan MY. Evaluation of serum lipocalin-2 level and its relation to insulin resistance in patients with inflammatory acne vulgaris. QJM: Int J Med. (2023) 116. doi: 10.1093/qjmed/hcad069.242
219. Sorour NE, Hamed AM, Behery EG, Elsayed EM. Assessment of serum level of lipocalin2 in patients with acne vulgaris. J Benha J Appl Sci. (2019) 4:23–6. doi: 10.21608/bjas.2019.187225
220. Lumsden KR, Nelson AM, Dispenza MC, Gilliland KL, Cong Z, Zaenglein AL, et al. Isotretinoin increases skin-surface levels of neutrophil gelatinase-associated lipocalin in patients treated for severe acne. Br J Dermatol. (2011) 165:302–10. doi: 10.1111/j.1365-2133.2011.10362.x
221. Nelson AM, Zhao W, Gilliland KL, Zaenglein AL, Liu W, Thiboutot DM. Neutrophil gelatinase-associated lipocalin mediates 13-cis retinoic acid-induced apoptosis of human sebaceous gland cells. J Clin Invest. (2008) 118:1468–78. doi: 10.1172/JCI33869
222. Nelson AM, Zhao W, Gilliland KL, Zaenglein AL, Liu W, Thiboutot DM. Isotretinoin temporally regulates distinct sets of genes in patient skin. J Invest Dermatol. (2009) 129:1038–42. doi: 10.1038/jid.2008.338
223. Su Y, Cui W, Wei H. Influence of lactoferrin on Propionibacterium acnes-induced inflammation in vitro and in vivo. Dermatologic Ther. (2020) 33:e14483. doi: 10.1111/dth.14483
224. Jusuf NK, Putra IB, Sutrisno AR. Correlation between stress scale and serum substance P level in acne vulgaris. Int J Gen Med. (2021) 14:681–6. doi: 10.2147/IJGM.S294509
225. Lee WJ, Jung HD, Lee HJ, Kim BS, Lee SJ, Kim DW. Influence of substance-P on cultured sebocytes. Arch Dermatol Res. (2008) 300:311–6. doi: 10.1007/s00403-008-0854-1
226. Toyoda M, Nakamura M, Makino T, Kagoura M, Morohashi M. Sebaceous glands in acne patients express high levels of neutral endopeptidase. Exp Dermatol. (2002) 11:241–7. doi: 10.1034/j.1600-0625.2002.110307.x
227. Zouboulis CC, Coenye T, He L, Kabashima K, Kobayashi T, Niemann C, et al. Sebaceous immunobiology - skin homeostasis, pathophysiology, coordination of innate immunity and inflammatory response and disease associations. Front Immunol. (2022) 13:. doi: 10.3389/fimmu.2022.1029818
228. Kwiecinska P, Grygier B, Morytko A, Sanecka-Duin A, Majchrzak-Gorecka M, Kwitniewski M, et al. Secretory leukocyte protease inhibitor regulates nerve reflex-mediated skin barrier function in psoriasis. J Eur Acad Dermatol Venereol. (2022) 36:1266–74. doi: 10.1111/jdv.18065
229. Majchrzak-Gorecka M, Majewski P, Grygier B, Murzyn K, Cichy J. Secretory leukocyte protease inhibitor (SLPI), a multifunctional protein in the host defense response. Cytokine Growth Factor Rev. (2016) 28:79–93. doi: 10.1016/j.cytogfr.2015.12.001
230. MaChado M, Silva S, Costa EM. Are antimicrobial peptides a 21st-century solution for atopic dermatitis? Int J Mol Sci. (2023) 24:13460. doi: 10.3390/ijms241713460
231. Osaki T, Sasaki K, Minamino N. Peptidomics-based discovery of an antimicrobial peptide derived from insulin-like growth factor-binding protein 5. J Proteome Res. (2011) 10:1870–80. doi: 10.1021/pr101114a
232. Chieosilapatham P, Niyonsaba F, Kiatsurayanon C, Okumura K, Ikeda S, Ogawa H. The antimicrobial peptide derived from insulin-like growth factor-binding protein 5, AMP-IBP5, regulates keratinocyte functions through Mas-related gene X receptors. J Dermatol Sci. (2017) 88:117–25. doi: 10.1016/j.jdermsci.2017.05.008
233. Rieg S, Garbe C, Sauer B, Kalbacher H, Schittek B. Dermcidin is constitutively produced by eccrine sweat glands and is not induced in epidermal cells under inflammatory skin conditions. Br J Dermatol. (2004) 151:534–9. doi: 10.1111/j.1365-2133.2004.06081.x
234. Burian M, Schittek B. The secrets of dermcidin action. Int J Med Microbiol. (2015) 305:283–6. doi: 10.1016/j.ijmm.2014.12.012
235. Nakano T, Yoshino T, Fujimura T, Arai S, Mukuno A, Sato N, et al. Reduced expression of dermcidin, a peptide active against propionibacterium acnes, in sweat of patients with acne vulgaris. Acta Dermato-Venereologica. (2015) 95:783–6. doi: 10.2340/00015555-2068
236. Hibbing ME, Fuqua C, Parsek MR, Peterson SB. Bacterial competition: surviving and thriving in the microbial jungle. Nat Rev Microbiol. (2010) 8:15–25. doi: 10.1038/nrmicro2259
237. Kaliniak S, Fiedoruk K, Spałek J, Piktel E, Durnaś B, Góźdź S, et al. Remodeling of paranasal sinuses mucosa functions in response to biofilm-induced inflammation. J Inflammation Res. (2024) 17:1295–323. doi: 10.2147/JIR.S443420
238. Marta Guarna M, Coulson R, Rubinchik E. Anti-inflammatory activity of cationic peptides: application to the treatment of acne vulgaris. FEMS Microbiol Lett. (2006) 257:1–6. doi: 10.1111/j.1574-6968.2006.00156.x
239. Fabbretti A, He CG, Gaspari E, Maffioli S, Brandi L, Spurio R, et al. A Derivative of the Thiopeptide GE2270A Highly Selective against Propionibacterium acnes. Antimicrobial Agents Chemotherapy. (2015) 59:4560–8. doi: 10.1128/AAC.05155-14
240. Easton DM, Nijnik A, Mayer ML, Hancock RE. Potential of immunomodulatory host defense peptides as novel anti-infectives. Trends Biotechnol. (2009) 27:582–90. doi: 10.1016/j.tibtech.2009.07.004
241. Melo MN, Dugourd D, Castanho MA. Omiganan pentahydrochloride in the front line of clinical applications of antimicrobial peptides. Recent Patents Anti-Infective Drug Discovery. (2006) 1:201–7. doi: 10.2174/157489106777452638
242. Wiesner J, Vilcinskas A. Antimicrobial peptides: the ancient arm of the human immune system. Virulence. (2010) 1:440–64. doi: 10.4161/viru.1.5.12983
243. Zhang L, Scheicher S, Harris S eds. Lipohexapeptide HB1345: A novel anti-infective for acne. Chicago (IL: American Academy of Dermatology Meeting (2008).
244. Lim HS, Chun SM, Soung MG, Kim J, Kim SJ. Antimicrobial efficacy of granulysin-derived synthetic peptides in acne vulgaris. Int J Dermatol. (2015) 54:853–62. doi: 10.1111/ijd.2015.54.issue-7
245. Ma Z, Kochergin N, Olisova O, Snarskaya E. Topical antimicrobial peptides in combined treatment of acne patients. J Cosmetic Dermatol. (2022) 21:1533–8. doi: 10.1111/jocd.14300
246. McInturff JE, Wang SJ, Machleidt T, Lin TR, Oren A, Hertz CJ, et al. Granulysin-derived peptides demonstrate antimicrobial and anti-inflammatory effects against Propionibacterium acnes. J Invest Dermatol. (2005) 125:256–63. doi: 10.1111/j.0022-202X.2005.23805.x
247. Popovic S, Urbán E, Lukic M, Conlon JM. Peptides with antimicrobial and anti-inflammatory activities that have therapeutic potential for treatment of acne vulgaris. Peptides. (2012) 34:275–82. doi: 10.1016/j.peptides.2012.02.010
248. Ryu S, Han HM, Song PI, Armstrong CA, Park Y. Suppression of propionibacterium acnes infection and the associated inflammatory response by the antimicrobial peptide P5 in mice. PloS One. (2015) 10:e0132619. doi: 10.1371/journal.pone.0132619
249. Han R, Blencke H-M, Cheng H, Li C. The antimicrobial effect of CEN1HC-Br against Propionibacterium acnes and its therapeutic and anti-inflammatory effects on acne vulgaris. Peptides. (2018) 99:36–43. doi: 10.1016/j.peptides.2017.11.001
250. Wu Y, Zhang G, Zhou M. Inhibitory and anti-inflammatory effects of two antimicrobial peptides moronecidin and temporin-1Dra against Propionibacterium acnes in vitro and in vivo. J Pept Science: an Off Publ Eur Pept Soc. (2020) 26:e3255. doi: 10.1002/psc.v26.7
251. Lee W-R, Kim K-H, An H-J, J-y K, Chang Y-C, Chung H, et al. The protective effects of melittin on propionibacterium acnes–induced inflammatory responses in vitro and in vivo. J Invest Dermatol. (2014) 134:1922–30. doi: 10.1038/jid.2014.75
252. Wang Y, Zhang Z, Chen L, Guang H, Li Z, Yang H, et al. Cathelicidin-BF, a snake cathelicidin-derived antimicrobial peptide, could be an excellent therapeutic agent for acne vulgaris. PloS One. (2011) 6:e22120. doi: 10.1371/journal.pone.0022120
253. Zhang Y, Jiang Y, Zhao J, Mo Q, Wang C, Wang D, et al. Weizmannia coagulans extracellular proteins reduce skin acne by inhibiting pathogenic bacteria and regulating TLR2/TRAF6-mediated NF-κB and MAPKs signaling pathways. Probiotics Antimicrobial Proteins. (2023). doi: 10.1007/s12602-023-10175-2
254. Pan CY, Chen JY, Lin TL, Lin CH. In vitro activities of three synthetic peptides derived from epinecidin-1 and an anti-lipopolysaccharide factor against Propionibacterium acnes, Candida albicans, and Trichomonas vaginalis. Peptides. (2009) 30:1058–68. doi: 10.1016/j.peptides.2009.02.006
255. Zhang Z, Mu L, Tang J, Duan Z, Wang F, Wei L, et al. A small peptide with therapeutic potential for inflammatory acne vulgaris. PloS One. (2013) 8:e72923. doi: 10.1371/journal.pone.0072923
256. Woodburn KW, Jaynes J, Clemens LE. Designed antimicrobial peptides for topical treatment of antibiotic resistant acne vulgaris. Antibiotics (Basel). (2020) 9. doi: 10.3390/antibiotics9010023
257. Dong Q, Wang S, Miao Y, Luo H, Weng Z, Yu L. Novel antimicrobial peptides against Cutibacterium acnes designed by deep learning. Sci Rep. (2024) 14:4529. doi: 10.1038/s41598-024-55205-3
258. Tokajuk J, Deptuła P, Chmielewska SJ, Skłodowski K, Mierzejewska ŻA, Grądzka-Dahlke M, et al. Ceragenin CSA-44 as a means to control the formation of the biofilm on the surface of tooth and composite fillings. Pathogens. (2022) 11. doi: 10.3390/pathogens11050491
259. Li FJ, Surolia R, Li H, Wang Z, Liu G, Kulkarni T, et al. Citrullinated vimentin mediates development and progression of lung fibrosis. Sci Trans Med. (2021) 13:eaba2927. doi: 10.1126/scitranslmed.aba2927
260. Yang XY, Wu WJ, Yang C, Yang T, He JD, Yang Z, et al. Association of HSD17B3 and HSD3B1 polymorphisms with acne vulgaris in Southwestern Han Chinese. Dermatology. (2013) 227:202–8. doi: 10.1159/000353581
261. Chamaie-Nejad F, Saeidi S, Najafi F, Ebrahimi A, Rahimi Z, Shakiba E, et al. Association of the CYP17 MSP AI (T-34C) and CYP19 codon 39 (Trp/Arg) polymorphisms with susceptibility to acne vulgaris. Clin Exp Dermatol. (2018) 43:183–6. doi: 10.1111/ced.2018.43.issue-2
262. Szabó K, Tax G, Teodorescu-Brinzeu D, Koreck A, Kemény L. TNFα gene polymorphisms in the pathogenesis of acne vulgaris. Arch Dermatol Res. (2011) 303:19–27. doi: 10.1007/s00403-010-1050-7
Keywords: Cutibacterium acnes, acne vulgaris, antimicrobial peptides, skin dysbiosis, inflammation
Citation: Lesiak A, Paprocka P, Wnorowska U, Mańkowska A, Król G, Głuszek K, Piktel E, Spałek J, Okła S, Fiedoruk K, Durnaś B and Bucki R (2024) Significance of host antimicrobial peptides in the pathogenesis and treatment of acne vulgaris. Front. Immunol. 15:1502242. doi: 10.3389/fimmu.2024.1502242
Received: 26 September 2024; Accepted: 02 December 2024;
Published: 18 December 2024.
Edited by:
Philippe Saas, Etablissement Français du Sang AuRA, FranceReviewed by:
Juri Koizumi, Gifu Pharmaceutical University, JapanCopyright © 2024 Lesiak, Paprocka, Wnorowska, Mańkowska, Król, Głuszek, Piktel, Spałek, Okła, Fiedoruk, Durnaś and Bucki. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Robert Bucki, YnVja2lyb2JlcnRAZ21haWwuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.