 Sarah Jesse1
Sarah Jesse1 Marie Riemann1Hauke Schneider2Marius Ringelstein3,4
Marie Riemann1Hauke Schneider2Marius Ringelstein3,4 Nico Melzer3Niklas Vogel3Lena Kristina Pfeffer5
Nico Melzer3Niklas Vogel3Lena Kristina Pfeffer5 Manuel A. Friese5
Manuel A. Friese5 Kurt-Wolfram Sühs6
Kurt-Wolfram Sühs6 Dominica Hudasch6
Dominica Hudasch6 Philipp Schwenkenbecher6
Philipp Schwenkenbecher6 Albrecht Günther7
Albrecht Günther7 Christian Geis7
Christian Geis7 Jonathan Wickel7Martin Lesser8Annika Kather8
Jonathan Wickel7Martin Lesser8Annika Kather8 Frank Leypoldt9,10Justina Dargvainiene9
Frank Leypoldt9,10Justina Dargvainiene9 Robert Markewitz9Klaus-Peter Wandinger9
Robert Markewitz9Klaus-Peter Wandinger9 Franziska S. Thaler11,12
Franziska S. Thaler11,12 Joseph Kuchling13Katharina Wurdack13
Joseph Kuchling13Katharina Wurdack13 Lidia Sabater14,15
Lidia Sabater14,15 Carsten Finke12
Carsten Finke12 Jan Lewerenz1* on behalf of the German Network for Research on Autoimmune Encephalitis (GENERATE)
Jan Lewerenz1* on behalf of the German Network for Research on Autoimmune Encephalitis (GENERATE)- 1Department of Neurology, University Hospital Ulm, Ulm, Germany
- 2Department of Neurology, Augsburg University, Augsburg, Germany
- 3Department of Neurology, Medical Faculty, Heinrich Heine University of Düsseldorf, Düsseldorf, Germany
- 4Department of Neurology, Centre for Neurology and Neuropsychiatry, LVR-Klinikum, Heinrich-Heine-University Düsseldorf, Düsseldorf, Germany
- 5Institute of Neuroimmunology and Multiple Sclerosis and Department of Neurology, University Medical Center Hamburg-Eppendorf, Hamburg, Germany
- 6Department of Neurology, Hannover Medical School, Hannover, Germany
- 7Section of Translational Neuroimmunology, Department of Neurology, Jena University Hospital, Jena, Germany
- 8Department of Neurology, Carl Gustav Carus University Dresden, Dresden, Germany
- 9Institute of Clinical Chemistry, University Hospital Schleswig-Holstein, Kiel/Lubeck, Germany
- 10Department of Neurology, University Hospital Schleswig-Holstein, Kiel, Germany
- 11Institute of Clinical Neuroimmunology, LMU University Hospital, LMU Munich, Munich, Germany
- 12Biomedical Center (BMC), Faculty of Medicine, LMU Munich, Munich, Germany
- 13Department of Neurology and Experimental Neurology, Charité – Universitätsmedizin Berlin, Berlin, Germany
- 14Fundació de Recerca Biomèdica Clínic Barcelona-Institut d’Investigacions August Pi i Sunyer-Caixa Research Institute, Universitat de Barcelona, Barcelona, Spain
- 15Spanish National Network for Research on Rare Diseases (CIBERER), Madrid, Spain
Introduction: Very rarely, adult NMDAR antibody-associated encephalitis (NMDAR-E) leads to persistent cerebellar atrophy and ataxia. Transient cerebellar ataxia is common in pediatric NMDAR-E. Immune-mediated cerebellar ataxia may be associated with myelin oligodendrocyte glycoprotein (MOG), aquaporin-4 (AQP-4), kelch-like family member 11 (KLHL11), and glutamate kainate receptor subunit 2 (GluK2) antibodies, all of which may co-occur in NMDAR-E. Here, we aimed to investigate the frequency, long-term outcome, and immunological concomitants of ataxia in NMDAR-E.
Methods: In this observational study, patients with definite NMDAR-E with a follow-up of >12 months were recruited from the GENERATE registry. Cases with documented ataxia were analyzed in detail.
Results: In 12 of 62 patients (19%), ataxia was documented. Bilateral cerebellar ataxia without additional focal CNS findings was found in four (one child and three adults); one of these was previously reported as a case with persistent cerebellar atrophy and ataxia. Two patients with bilateral cerebellar ataxia had additional focal neurological symptoms, optic neuritis and facial palsy. Two patients developed hemiataxia: one with diplopia suggesting brainstem dysfunction and the other probably resulting from cerebellar diaschisis due to contralateral status epilepticus. In all but the one developing cerebellar atrophy, cerebellar ataxia was transient and not associated with a worse long-term outcome. In all five patients with cerebellar ataxia tested, MOG, AQP-4, GluK2, and KLHL11 antibodies were negative. In two additional patients negative for both MOG and AQP-4 antibodies, ataxia was sensory and explained by cervical myelitis as part of multiple sclerosis (MS) manifesting temporal relation to NMDAR-E. One of the patients with bilateral ataxia with focal neurological deficits was also diagnosed with MS upon follow-up. Finally, in two patients, ataxia was explained by cerebral hypoxic damage following circulatory failure during an ICU stay with severe NMDAR-E.
Discussion: Ataxia of different types is quite common in NMDAR-E. Cerebellar ataxia in NMDAR-E is mostly transient. NMDAR-E followed by persistent ataxia and cerebellar atrophy is very rare. Cerebellar ataxia in NMDAR-E may not be explained by concomitant KLHL11, MOG, AQP-4, or GluK2 autoimmunity. Of note, ataxia in NMDAR-E may result from treatment complications and, most interestingly, from MS manifesting in temporal association with NMDAR-E.
1 Introduction
N-methyl D-aspartate receptor (NMDAR) antibody-associated encephalitis (NMDAR-E) is an autoimmune-encephalitis subtype characterized by the intrathecal synthesis of IgG antibodies against the NMDAR subunit NR1 (1). Anti-NMDAR IgG in NMDAR-E results in immunoglobulin-induced NMDAR internalization and thus inhibition of NMDAR signaling (2). Consequently, some symptoms as well as the changes in cerebral glucose metabolism upon positron-emission tomography resemble pharmacological NMDAR inhibition (3). Unless early immunosuppression is initiated, the clinical course of NMDAR-E follows a characteristic pattern: after a flu-like prodromal phase, psychotic symptoms develop, followed by speech disturbances and epileptic seizures, then—interpreted as generalized encephalitis—loss of consciousness, stereotypic movements, and autonomic dysfunction as well as central hypoventilation (4). The clinical picture of NMDAR-E varies with age: in infants and young children, movement disorders might predominate, and autonomic dysfunction and hypoventilation are less common (5). In addition, cerebellar ataxia and focal neurological deficits have been reported to be much more common in children than in adults with NMDAR-E (6). However, NMDAR-E in adulthood may present initially as cerebellar ataxia (7).
Of note, NMDAR subunit NR1 is expressed in the human (8) and rodent cerebellum (9). NMDAR antagonists are known to induce impaired motor coordination in mice (10) and non-human primates (11). In rats, prominent changes of cerebellar NMDAR subunit expression from birth to adulthood were demonstrated (9). Whether similar changes occur during childhood in humans is unknown. However, this could explain the high frequency of ataxia reported for childhood NMDAR-E.
We recently reported a case with severe NMDAR-E, which resulted in persistent severe cerebellar ataxia associated with progressive cerebellar atrophy (12). A few similar cases had been reported previously by Iizuka et al. (13). This rare phenotype of unknown pathophysiology, herewith coined a Iizuka phenotype, is characterized by cerebellar atrophy associated with post-acute reversal of supratentorial atrophy (12, 13). Severe persistent atrophy explains poor functional long-term outcome in these patients (12, 13).
NMDAR-E, including cases with focal neurological deficits suggestive of demyelinating events, has been associated with MOG and AQP-4 antibodies in some patients (14, 15). Neuromyelitis optica spectrum disorders (NMOSD) with AQP-4 antibodies as well as MOG-antibody associated diseases (MOGAD) may present with ataxia (16). Recently, two antibodies, directed against kelch-like family member 11 (KLHL11) and glutamate kainate receptor subunit 2 (GluK2), also associated with cerebellar ataxia, have been described (17, 18). Interestingly, KLHL11 antibodies share their association with ovarian teratomas with NMDAR antibodies (17, 18).
Here, we investigated the frequency of ataxia in mostly adult NMDAR-E patients enrolled in the multicentric registry of the German Network for Research on Autoimmune Encephalitis (GENERATE). In addition, we analyzed the detailed clinical characteristics, the immunological accompaniments, and the long-term functional outcome of ataxia associated with NMDAR-E.
2 Methods
Study design and participants
GENERATE is a multicentric, combined retrospective and prospective registry for autoimmune encephalitis patients in Germany (https://generate-net.de/) recruiting since 2013. The inclusion criteria for this analysis were as follows: (a) diagnosis of definite NMDAR-E according to current diagnostic criteria with detection of anti-NMDAR IgG (19) with the modification that patients with consistently negative anti-NMDAR IgG in cerebrospinal fluid were excluded; (b) available information on initial and predominating clinical symptoms; and (c) available long-term functional outcome (≥12 months) as a modified Rankin scale (mRS) score. A study plan was sent to the GENERATE sites in April 2017. Thirty-one sites agreed to participate. The recruitment period ended in February 2018. Clinical information was extracted from the database of the GENERATE registry. For patients with documented ataxia, more detailed information was collected in close collaboration with the respective GENERATE sites. This detailed information is summarized as case vignettes to be found in the online appendix. MRIs of all ataxia patients were centrally re-analyzed by experts in NMDAR-E and MS imaging (J.K., K.W., and C.F.) to detect ataxia-associated abnormalities as well as to confirm the diagnostic criteria for MS (20).
Antibody testing
Sera of atactic patients with serum samples still available in the GENERATE biomaterial banks were tested using in-house live cell–based assays for MOG and GluK2, in-house paraformaldehyde-fixed in-house cell-based assay for KLHL11 antibodies, all three with transiently transfected HEK293 cells, and a commercial HEK293-based assay from Euroimmun (Luebeck, Germany) for AQP4 antibodies as described previously (17, 18, 21, 22). NMDAR antibodies and all antineuronal antibodies obtained during the initial diagnostic workup of the patients with ataxia and NMDARE were obtained from the clinical files of the patients. Serum of Patient 10 had been tested by commercial cell-based assays from Euroimmun in serum only, but never in CSF. Thus, we confirmed anti-NMDAR IgG using paraformaldehyde-fixed rat brain sections as described previously (23).
Statistics
Statistical analysis was performed using GraphPad Prism (Graph Pad Inc., La Jolla, USA). Categorical variables were analyzed using Fisher’s exact test. For ordinal and continuous variables, the median and the interquartile range (IQR) were calculated, and the Mann-Whitney U test was used to compare two groups. A two-sided p-value of <0.05 was regarded as statistically significant. Due to the explorative nature of this study, all results from statistical tests must be interpreted as hypothesis generating.
Data availability
The datasets generated and/or analyzed during the current study are not publicly available but can be obtained by qualified researchers from the corresponding author upon reasonable request.
Ethics
All patients or their legal representatives gave their informed consent. The study was approved by the institutional review board of the University of Schleswig-Holstein (#13-162).
3 Results
Study cohort
At database cut, 689 patients had been enrolled in the GENERATE registry (Figure 1). Of those, 190 (28%) had been documented positive for NMDAR antibodies. Twenty-four patients were excluded as they did not meet the diagnostic criteria of definite NMDAR-E as described in the inclusion criteria. Anti-NMDAR IgG in CSF was repeatedly negative, although serum anti-NMDAR IgG was positive in one patient (4%). Other reasons for exclusion were anti-NMDAR IgA or IgM only (n = 23, 12%). Overall, definite NMDAR-E was confirmed in 166 cases (85%). In 62 of these patients (37%), follow-up data for functional impairment as mRS score were available for at least 12 months after initial hospital admission. In our patient collective, six patients (10%) in the total cohort were younger than 18 years; only one (2%) was younger than 12 years. The basic demographic variables of this cohort can be found in Table 1.
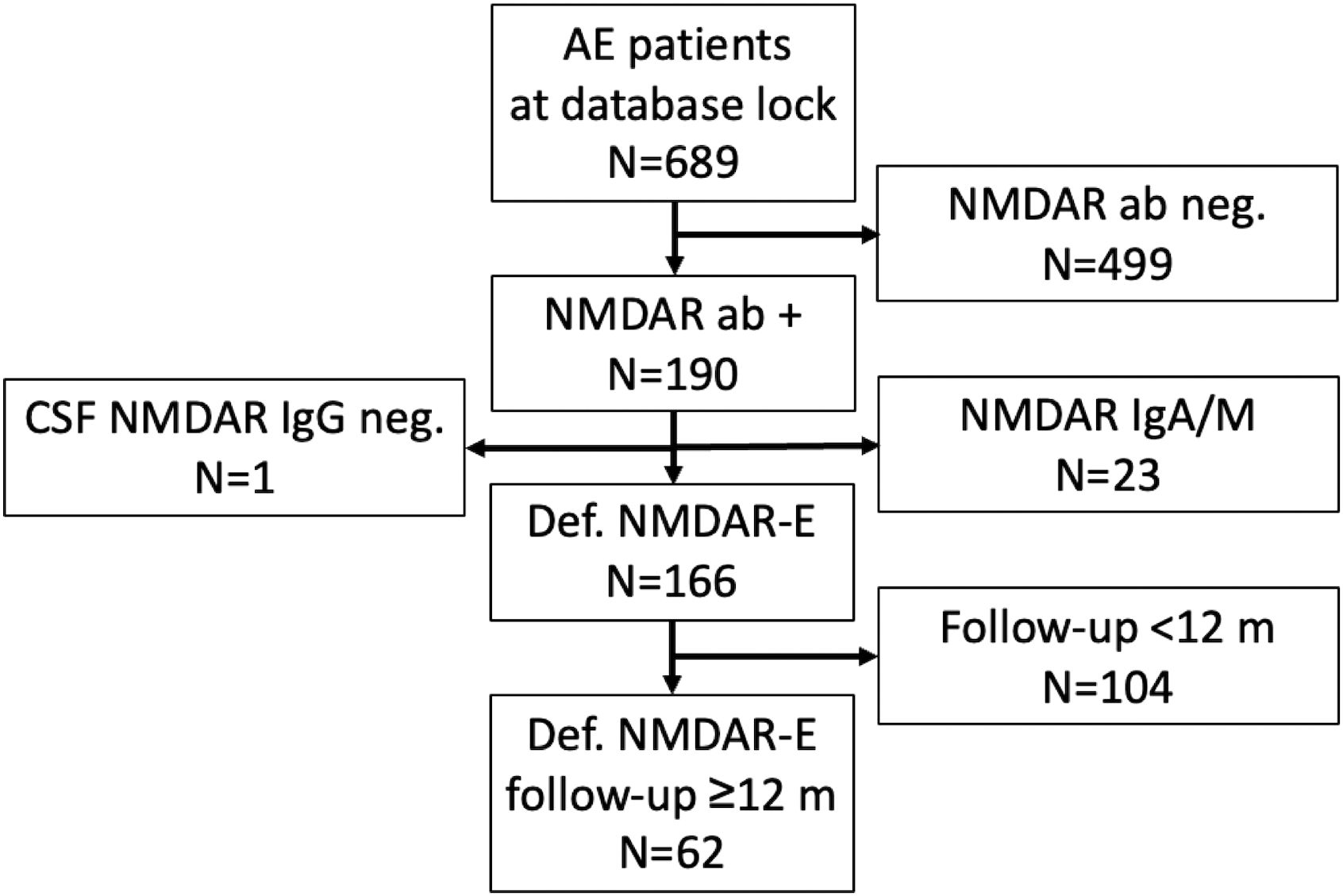
Figure 1. Study recruitment. AE, autoimmune encephalitis; ab, antibody; CSF, cerebrospinal fluid; NMDAR-E, N-methyl D-aspartate receptor antibody-associated encephalitis.
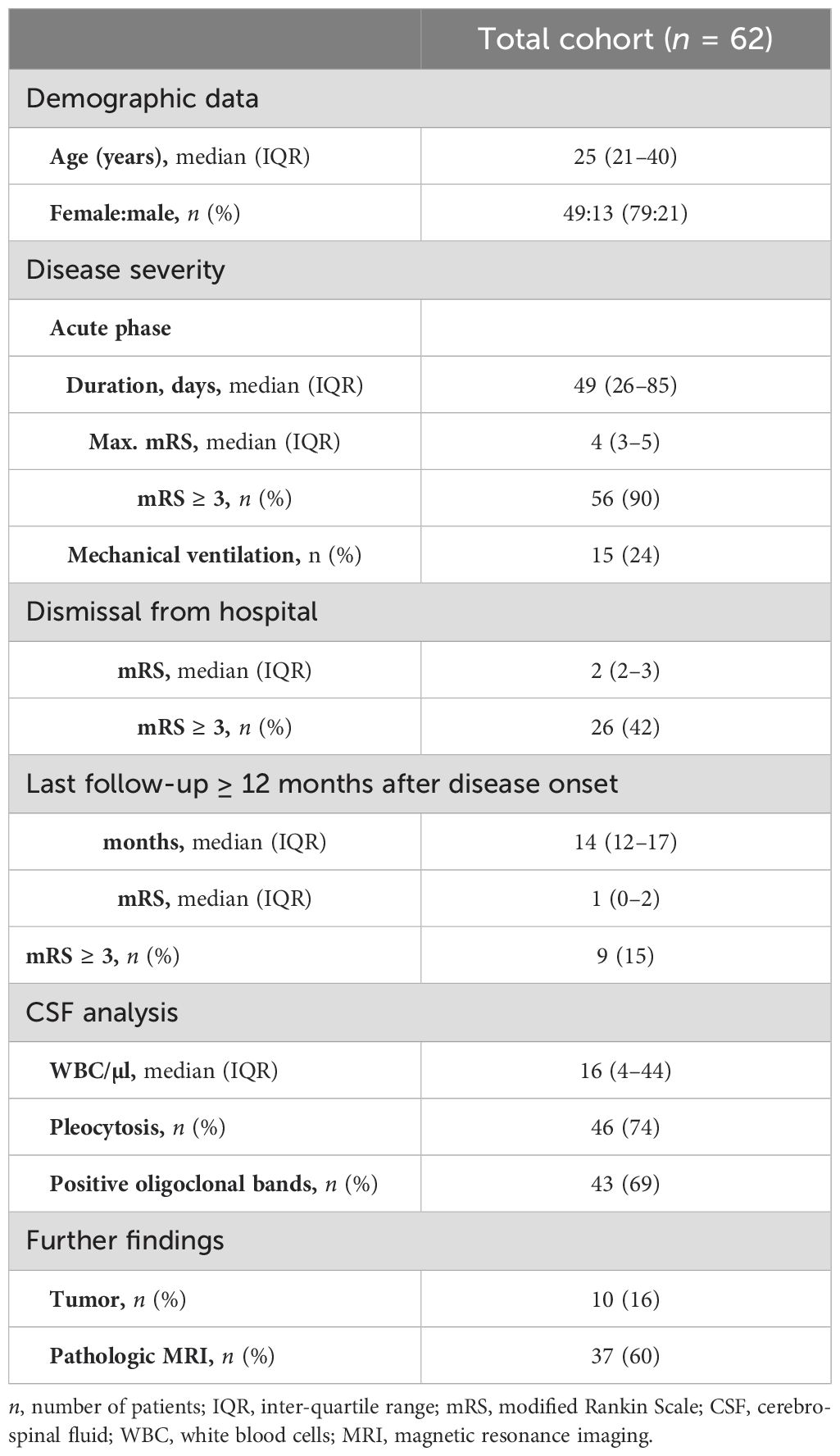
Table 1. Demographic data of total patient cohort.
Different types of ataxia due to diverse pathophysiologies occur with different temporal association to NMDAR-E
In addition to our index case with the progressive cerebellar atrophy associated with severe and persistent cerebellar ataxia, following severe NMDAR-E (Case 4) (12), a phenotype that due its first describer (13) we suggest to name Iizuka-type NMDAR-E, another 11 patients were documented as exhibiting ataxia during the acute NMDAR-E episode in the GENERATE database. None of these tested positive for other antibodies than anti-NMDAR during routine work-up (Supplementary Table S1).
A detailed analysis of these cases, including the index case, allowed categorization into five different groups (Table 2). The first group consisted of four patients who presented with bilateral cerebellar ataxia in close temporal relationship to the onset of symptoms typical for the NMDAR-E episode without additional focal CNS symptoms beyond ataxia. In the youngest patient of our cohort, a 7-year-old child, bilateral symmetric cerebellar ataxia was the first symptom of NMDAR-E (Case 1). Two other adult patients developed bilateral cerebellar ataxia after the onset of typical NMDAR-E symptoms (Cases 2–3). All of these patients had a good prognosis, including complete remission of the ataxia. In contrast, the patient with Iizuka-type NMDAR-E upon long-term follow-up never reached an mRS<4 due to severe persistent cerebellar ataxia associated within progressive cerebellar atrophy (Figure 2A) after she had recovered from all other symptoms directly related to the NMDAR-E episode. Initially, prolonged analgesia was required with numerous complications during intensive care (12).
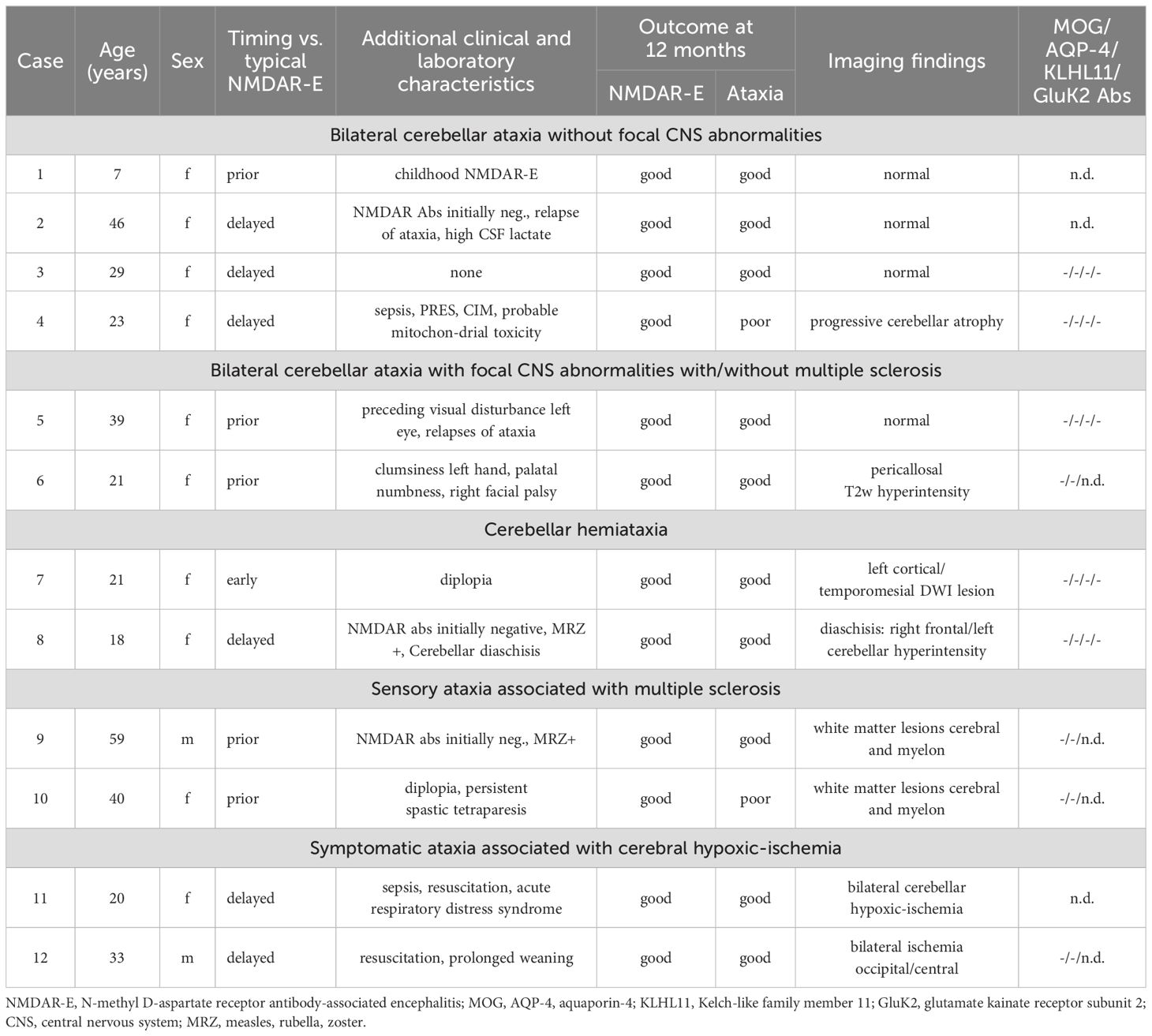
Table 2. Groups of NMDAR-E patients with documented ataxia.
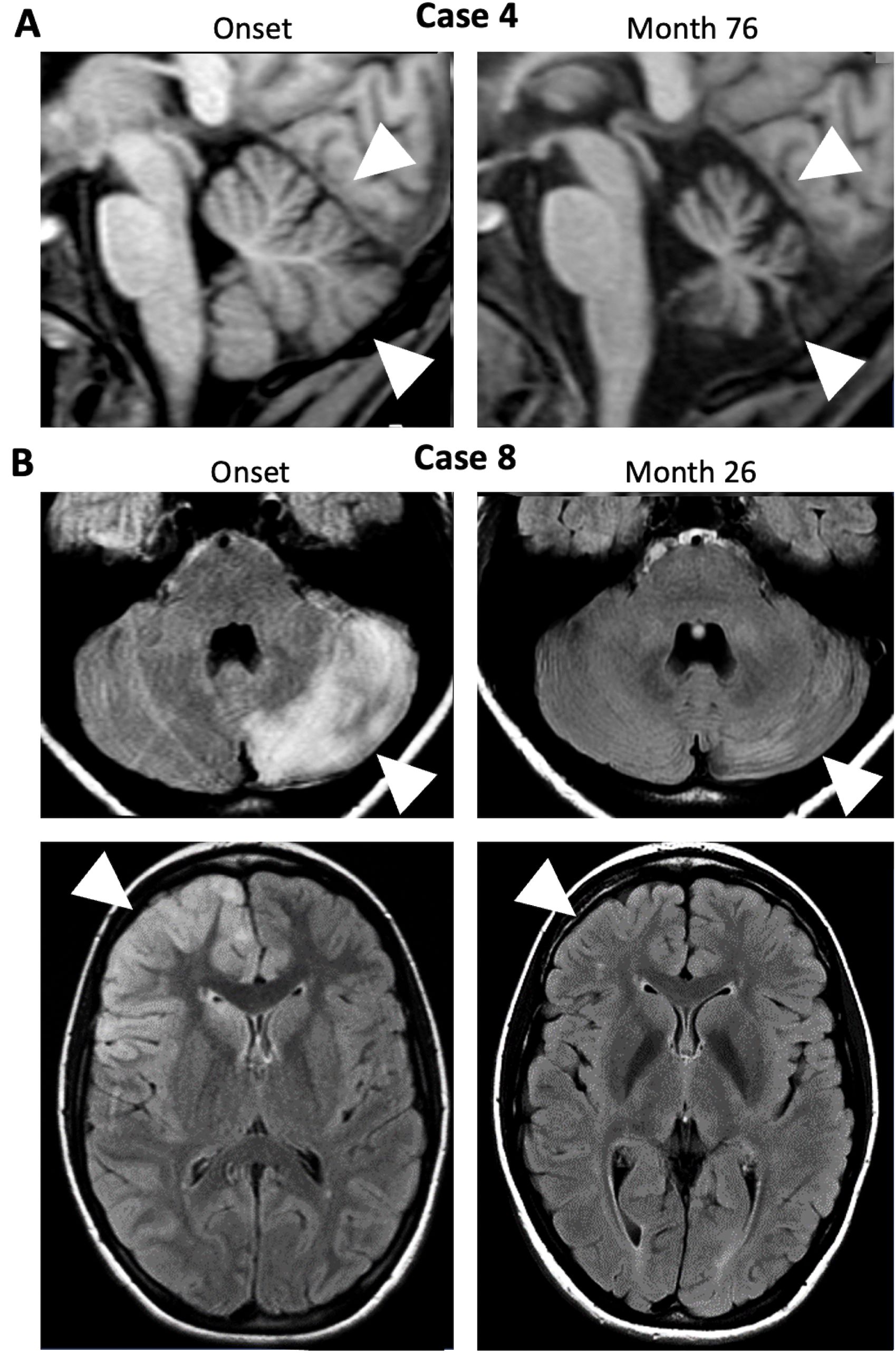
Figure 2. MRI changes in NMDAR-E patients with cerebellar ataxia are rare. (A) Sagittal 1-weighted MRI of Case 4 at onset (left panel) and after recovery from severe NMDAR- E but with persisting bilateral cerebellar ataxia (month 76; arrows indicate cerebellar atrophy). (B) FLAIR images of Case 8 with reversible left-sided hemiataxia in the acute phase (onset, left panels) show diffuse, mostly cortical left cerebellar T2-hyperintensity (upper left panel, arrowhead) possibly resulting from cerebellar diaschisis following right frontal status epilepticus associated with right frontal cortical hyperintensities (lower panel, arrowhead). T2-hyperintensities were reversible (Month 26, right panels.
The second group consisted of two patients, both of whom developed bilateral cerebellar ataxia in addition to other focal neurological deficits beyond typical NMDAR-E symptoms. One patient reported a monocular decrease in visual acuity many months before the onset of ataxia and then NMDAR-E-typical symptoms (Case 5). The other patient, a 21-year-old woman, developed mild facial paralysis, palatal hypoesthesia, and clumsiness of the left hand in close temporal association with cerebellar ataxia and NMDAR-E-typical symptoms (Case 6).
In the third group, patients also showed other focal neurological abnormalities, but cerebellar hemiataxia instead of bilaterally symmetric ataxia. In the first patient (Case 7), a 21-year-old woman, cerebellar hemiataxia developed in parallel with diplopia and NMDAR-E-typical symptoms. Although the combination of diplopia and hemiataxia suggests a brainstem lesion, cerebral MRI was unremarkable. In the other patient (Case 8), an 18-year-old woman, hemiataxia developed later in the disease course. Interestingly, in this patient, cerebral MRI showed a prominent T2-weighted lesion of the cerebellum involving almost the entire hemisphere ipsilateral to the ataxia (Figure 2B). This lesion was located contralateral to the right frontal T2-weighted hyperintensities, predominantly cortical, but also extending into the underlying white matter as an MRI correlate of focal status epilepticus. All lesions were transient. Thus, the hemiataxia can be interpreted as symptomatic cerebellar diaschisis secondary to status epilepticus (24) directly caused by NMDAR-E.
In the fourth group (Cases 9–10), atactic gait was explained by posterior column dysfunction resulting from cervical myelitis. Case 9 had multiple juxtacortical, periventricular, as well as spinal (Figure 3A); Case 10 had extensive periventricular and infratentorial white matter lesions (WML) (Figure 3B) suggestive for demyelination. Together with corresponding clinical signs and positive oligoclonal IgG in CSF, the diagnosis of MS was made based on current diagnostic criteria (20). Case 10 also exhibited a positive MRZ (measles, rubella, varicella zoster) reaction with elevated CSF/serum antibody indices for measles and varicella zoster typical for MS (25). Of note, Case 6, which had only shown unspecific pericallosal and subcortical WML, also developed progressive periventricular, juxtacortical, and subcortical T2-weighted hyperintense MRI lesions during a follow-up period of five years and was subsequently diagnosed with MS (Figure 3C).
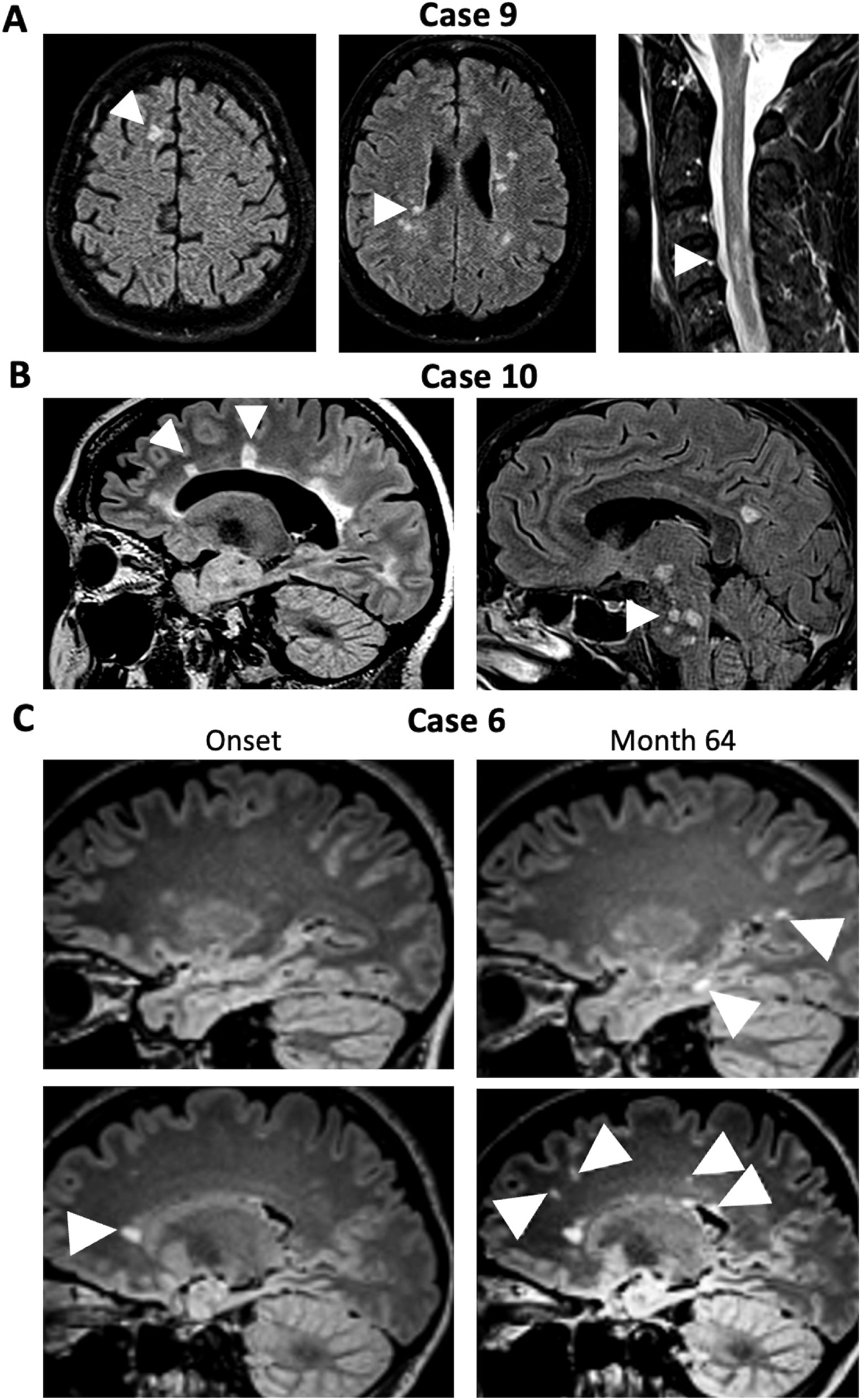
Figure 3. MRI in patients NMDAR-E with ataxia and concomitant or later diagnosis of multiple sclerosis. (A) Initial FLAIR cerebral MRI of Case 9 show juxtacortical (left panel, arrowhead) as well as periventricular lesions (middle panel, arrowhead) suggestive of demyelination. T2-weighted MRI of the cervical spinal cord also shows multiple demyelinating lesion (arrowhead). (B) Cerebral FLAIR MRI of Case 10 shows extensive periventricular T2-hyperintense lesions with Dawson finger-like orientation, highly suggestive of MS-like demyelination (left panel, arrowhead) as well as infratentorial lesions (right, arrowhead). (C) Initial cerebral FLAIR MRI of Case 6 (left panels) shows small unspecific subcortical white matter lesions but one periventricular lesion (arrowhead). Upon follow-up (Month 64, right panels) a juxtacortical T2-hyperintense lesion (upper right panel, lower arrowhead) as well as a new periventricular lesion (lower right panel, right arrowhead) in addition to multiple subcortical lesions (other arrowhead), some with radial orientation (lower right panel, left two arrowheads).
In the fifth group (Cases 11/12), the ataxia, one cerebellar, the other non-cerebellar, resulted from cerebral hypoxic ischemia acquired due to circulatory failure and resuscitation during ICU treatment of severe NMDAR-E (Figure 4).
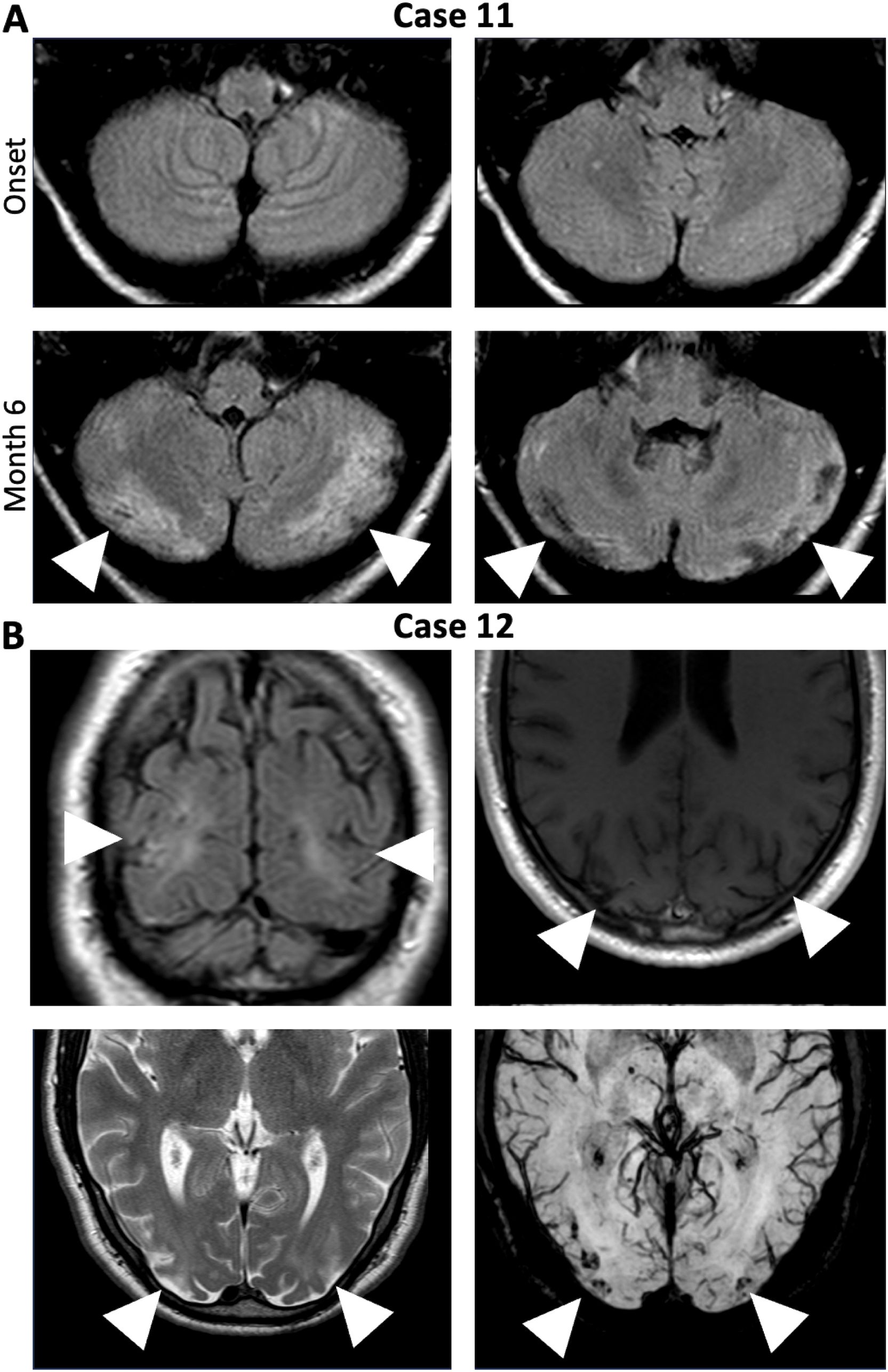
Figure 4. Symptomatic non-cerebellar and cerebellar ataxia due to ischemic hypoxic lesion acquired during circulatory failures in severe NMDAR-E. (A) Cerebral FLAIR MRI of Case 11 at onset did not show any cerebellar abnormalities (upper panels). After resuscitated 6 months after disease onset (lower panel, Month 6), repeat cerebral MRI showed bilateral subacute cerebellar infarctions (lower panels, arrowheads). (B) Bilateral hemodynamic ischemic lesions in the parietooccipital region (arrowheads) with hemorrhagic transformation (left upper panel: coronal FLAIR, right upper panel: transverse T1-weighted image, left lower panel: transversal T2-weighted image, right lower panel: Transverse susceptibility-weighted images (white arrows) in Case 12 after cardiopulmonary arrest and successful cardiopulmonary resuscitation.
As MOG, AQP-4, KLHL11, and GluK2 antibodies have been identified in NMDAR-E patients and have been associated with neurological symptoms including ataxia (14, 17, 18), we tested available sera for all four antibodies. Serum was tested negative for MOG, AQP-4, KLHL11, and GluK2 antibodies in five of eight patients with cerebellar ataxia (Cases 3–5, 7, and 8). Among these were two of four patients with bilateral cerebellar ataxia without additional CNS findings (Cases 3/4), among those the patient with persistent ataxia, one of the two patients with bilateral cerebellar ataxia with focal CNS findings (Case 5), and both patients with hemiataxia (Cases 7/8, Table 1). The three patients meeting the diagnostic criteria for both NMDARE and MS (Cases 6, 9, and 10) were tested negative for both MOG and AQP-4 antibodies (Table 1).
In summary, in 4/12 (50%) NMDAR-E patients, ataxia was definitely of a different pathophysiology than the NMDAR-E. In two each, ataxia either was sensory resulting from myelitis in the context of MS or ataxia was explained by hypoxic brain damage as a treatment complication. In two additional patients, our data suggest an etiology distinct from NMDARE. Ataxia was either probably either explained by contralateral status epilepticus and crossed cerebellar diaschisis or, in retrospect, might have been a first manifestation of MS.
In 6/62 patients with cerebellar ataxia (10%), no definite or probable etiology than NMDAR-E itself was identified. These six patients with cerebellar ataxia not explained by other factors than NMDAR-E did not differ from the other 56 patients regarding age [cerebellar ataxia: median 26 years (IQR 18–41 years), other: median years 25 (21–39 years); p = 0.7580], sex distribution [female/male: cerebellar ataxia: 6/0 (100%/0%), other: 43/13 (77%/23%); p = 0.3280], or outcome [mRS ≤2: cerebellar ataxia: 5/6 (88%) vs. 49/56 (91%), p = 0.5799]. When dichotomized into adult and juvenile/childhood NMDAR-E, cerebellar ataxia was not more frequent in children than in adults [age ≥18 years: 5/56 (9%), age <18 years: 1/6 (17%), p = 0.4710].
4 Discussion
We intended to identify cases with clear NMDAR-E and ataxia in a cohort of predominantly adult NMDAR-E patients and investigated the immunological comorbidities and outcome of patients with ataxia associated with NMDAR-E over a 12-month period. Ataxia during acute NMDAR-E was documented in 12 cases. Cerebellar ataxia without any alternative pathology was identified in 6/12 cases. In the other six patients, definite or probably other pathophysiologies could be demonstrated. In two of these cases, we found sensory ataxia associated with posterior column dysfunction. Although apparently asymptomatic prior to their NMDAR-E diagnosis, both patients met diagnostic criteria for MS in close temporal relation to the NMDAR-E episode. In one additional case, cerebellar ataxia and concurrent focal neurological deficits fulfilled the diagnostic criteria for MS upon long-term follow-up. Thus, in retrospect, the features not typical for NMDAR including ataxia might have been the first manifestation of MS. In two other patients, the ataxia developed due to cerebral hypoxic ischemia, which was only indirectly related to NMDAR-E.
In contrast, among adult NMDARE patients in our total cohort, 10% had symptoms of cerebellar ataxia without another pathology but NMDAR-E explaining the cerebellar dysfunction. A high frequency of ataxia in childhood (15%–20%) has been previously reported, while it is considered rare in adult NMDAR-E patients (< 3%) (6). Our results suggest that cerebellar ataxias may be more common in adult NMDAR-E patients than expected. In more than half of our patients with NMDARE-associated ataxia, we were able to exclude the presence of MOG, AQP-4, KLHL11, and GluK2 antibodies, all of which have previously been associated with both NMDAR-E and CNS neurological deficits including ataxia (14–18). As NMDAR are expressed in the human cerebellum (8) and their inhibition causes cerebellar ataxia (10, 11), transient bilateral cerebellar ataxia in the context of NMDAR-E may result from anti-NMDAR IgG-mediated NMDAR downregulation.
Surprisingly, three of 12 NMDAR-E patients (25%) with documented ataxia developed MS. Demyelinating diseases associated with NMDAR-E have been reported previously (15), but were found to represent concurrent NMOSD or MOGAD. In all our three cases, both AQP-4-positive NMOSD and MOGAD could be excluded. One case with NMDAR-E/MS overlap (Case 9) had documented a positive MRZ reaction, but also a case without MS (Case 8). The MRZ reaction is thought to be the most specific laboratory marker for MS (25). MRZ positivity in NMDAR-E in a single case was first reported by us in 2015 (26). Subsequently, we could demonstrate that the MRZ reaction is positive in one-fifth to one-third of patients with NMDAR-E (27). In addition, isolated cases with both MS and NMDAR-E have been reported (28–32). In synopsis, these results strongly suggest that NMDAR-E is not only the second most common neurological disease in which a positive MRZ reaction occurs but also that co-occurrence of both NMDAR-E and MS may be far more common than expected. This may indicate immunological commonalities between both diseases that need to be investigated.
In some of our other patients with NMDAR-E and ataxia, additional neurological symptoms suggestive of brainstem and optic nerve dysfunction were present. In the absence of demyelinating lesions in these patients and in the absence of MOG, AQP-4, KLHL11, as well as GluK2 antibodies in those tested, the pathophysiological basis of these symptoms remains elusive. However, neurological deficits like hemiparesis have been reported rather frequently in childhood NMDAR-E (6) and thus might represent a less common and underappreciated NMDAR-E symptom in adults alike.
In one of our patients, cerebellar MRI abnormalities were highly suggestive for cerebellar diaschisis due to contralateral status epilepticus (24). Usually, crossed cerebellar diaschisis remains asymptomatic. However, single cases with hemiataxia, as in our case, have been reported (33). In addition to this case, we identified two other patients with ataxia caused by cerebral hypoxic infarction and two more cases with sensory ataxia due to myelitis. Consequently, although ataxia might be a common symptom of adult NMDAR-E, this illustrates that alternative additional causes must be excluded.
Finally, our study confirms that NMDAR-E of the Iizuka-type with persistent cerebellar ataxia and initially progressive cerebellar atrophy following severe NMDAR-E (13) is rare. With exception of our index case, which initiated this project (12), no other case with this peculiar phenotype was discovered. Cerebellar ataxia was transient and not associated with a poor functional outcome in all patients but this case. Thus, in general, cerebellar ataxia does not indicate a poor prognosis in NMDAR-E.
The limitations of our study are its retrospective nature and the limited size of our cohort. This was partially due to our decision to only include patients with at least one year of follow-up, as one of the primary goals was to identify additional patients with persisting cerebellar ataxia leading to a persistently poor functional status. However, our study may be a good starting point to explore the frequency of different phenotypes and origins of ataxia in larger cohorts of NMDAR-E patients. In addition, our unexpected finding that MS might co-occur more often with NMDAR-E than expected requires further investigation.
In summary, our study indicates that cerebellar ataxia occurs more often in adult NMDAR-E than expected previously. Cerebellar ataxia with and without additional neurological symptoms is commonly neither explained by concomitant AQP-4 antibody-positive NMOSD or MOGAD nor KLHL11 or GluK2 antibodies. Thus, it might be directly induced by anti-NMDAR antibodies. We demonstrate that it is mandatory to exclude alternative diagnoses as the origin of ataxia in NMDAR-E. These include complications of intensive care treatment, including resuscitation in severe NMDAR-E. In addition, ataxia and associated focal neurological impairments, including them, might represent the first manifestation of comorbid MS.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Ethics statement
The studies involving humans were approved by Ethikkommission der Universität zu Lübeck. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author contributions
SJ: Data curation, Supervision, Writing – original draft. MR: Data curation, Formal Analysis, Investigation, Writing – review & editing. HS: Data curation, Investigation, Writing – review & editing. MR: Data curation, Investigation, Writing – review & editing. NM: Data curation, Investigation, Writing – review & editing. NV: Data curation, Investigation, Writing – review & editing. LP: Data curation, Investigation, Writing – review & editing. MF: Data curation, Investigation, Supervision, Writing – review & editing. KS: Data curation, Investigation, Writing – review & editing. DH: Data curation, Investigation, Writing – review & editing. PS: Data curation, Investigation, Writing – review & editing. AG: Data curation, Investigation, Writing – review & editing. CG: Data curation, Investigation, Writing – review & editing. JW: Data curation, Investigation, Writing – review & editing. ML: Data curation, Investigation, Writing – review & editing. AK: Data curation, Investigation, Writing – review & editing. JD: Data curation, Investigation, Writing – review & editing. FL: Data curation, Investigation, Writing – review & editing. RM: Data curation, Investigation, Writing – review & editing. KW: Data curation, Investigation, Writing – review & editing. FT: Data curation, Investigation, Writing – review & editing. JK: Data curation, Investigation, Writing – review & editing. KW: Data curation, Investigation, Writing – review & editing. LS: Data curation, Investigation, Writing – review & editing. CF: Data curation, Investigation, Supervision, Writing – review & editing. JL: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Supervision, Validation, Writing – original draft, Writing – review & editing.
Members of the GENERATE consortium
Achim Berthele, München TU Neurologie. Aiden Haghikia, Magdeburg UK Neurologie. Aleksandra Juranek, Osnabrück Neurologie. Alexander Emmer, Halle, UK Neurologie. Alexander Finke, Lüneburg Neurologie. Alexandra Philipsen, Bonn, UK Psychotherapie. Amelie Bohn, Aachen UK RTHW Neurologie. Andeas Linsa, Hoyerswerda, Neurologie. Andeas Steinbrecher, Erfurt, Neurologie. Andre Dik, Münster UK Neurologie/Schlafmedizin. Andrea Kraft, Halle Martha-Maria Neurologie. Andreas Binder, Saarbrücken, Neurologie. Andreas van Baalen, Kiel UKSH, Pädiatrie. Anja Tietz, Kiel, UKSH Neuroimmunolo./klein.Chem. Anna Gorsler, Beelitz-Heilstätten, Neuroreha. Anne-Katrin Pröbstel, Basel UK, Neurologie. Annette Baumgartner, Basel UK, Neurologie. Annika Kather, Dresden UK Neurologie. Antonia Harms, Bonn Epileptoloige. Armin Grau, Ludwighafen, Neurologie. Astrid Blaschek, München Haunersches Pädiatrie. Bettina Balint, Zürich, UK Neurologie. Birgit Berger, Rostock, Neurologie. Brigitte Wildemann, Heidelberg, Neurologie. Britta Greshake, Ulm UK Neurologie. Carlos Martinez Quesada, Essen, Neurologie. Carolin Baade-Büttner, Jena UK, Neurologie. Carsten Finke, Berlin Charité, Neurologie/Neurocure. Catharina Groß, Münster UK Neurologie/Schlafmedizin. Christian Bien, Bielefeld Mara, Epilepsiezentrum. Christian Geis, Jena UK, Neurologie. Christian Hofmann, Ulm UK Neurologie. Christian Urbanek, Ludwigshafen, Neurologie. Christine Strippel, Münster UK Neurologie/Schlafmedizin. Christoph Kellinghaus, Osnabrück Neurologie. Christoph Lehrich, Münster UK Neurologie/Schlafmedizin. Christos Krogias, Bochum, St. Josef-Hospital, Neurologie. Chung Ha-Yeun, Jena UK, Neurologie. Claudia Sommer, Würzburg Neurologie. Constanze Kerin (geb. Mönig), Münster UK Neurologie/Schlafmedizin. Corinna Bien, Bad Salzuflen, MVZ Labor Krone. Corinna Trebst, Hannover, MHH Neurologie. Daniel Bittner, Nordhausen, Neurologie. Daniel Rapp, Ulm UK Neurologie. Daniela Esser, Kiel UKSH, Neuroimm./klein. Chemie. Dietrich Sturm. Dirk Fitzner, Göttingen, Neurologie. Dominica Ratuszny, Hannover, MHH Neurologie. Dominique Endres, Freiburg Psychatrie. Duygu Pul, Düsseldorf UK Neurologie. Ellen Knierim, ohne Zentrum (ehem. Berlin, Charite; Pädiatrie). Fabian Möller, Hamburg Asklepios Barmbek, Neurologie. Fatme Seval Ismail, Bochum, KnappschaftsKH, Neurologie. Felix Rosenow, Frankfurt, Epilepsiezentrum & Neurologie. Felix Fischbach, Hamburg Eppendorf, Neurologie. Felix von Poderwils, Greifswald, Neurologie. Florian Then Bergh, Leipzig Neurologie. Frank Hoffmann, Halle Martha-Maria Neurologie. Frank Leypoldt, Kiel, UKSH Neuroimmunolo./klein.Chem. Frank Seifert, Erlangen, Neurologie. Franz Blaes, Gummersbach KKH Neurologie. Franziska Thaler, München LMU Neuroimmunologie/Neurologie. Friedrich Ebinger, Paderborn Neurologie. George Trendelenburg, Mühlhausen, Neurologie. Gerd Meyer zu Hörste, Münster UK Neurologie/Schlafmedizin. Gernot Reimann, Dortmund, Klinikum, Neurologie. Gesa Schreyer, Kiel UKSH, Neuroimm./klein. Chemie. Gregor Kuhlenbäumer, Kiel UKSH, Neuroimm./klein. Chemie. Günter Seidel, Hamburg Nord, Neurologie. Hagen Huttner, Giessen, Neurologie. Harald Prüß, Berlin Charité, Neurologie/Neurocure. Hauke Schneider, Augsburg UK, Neurologie. Hayrettin Tumani, Ulm UK Neurologie. Henning Stolze, Flensburg, Neurologie. Henrik Rohner, Bonn, UK Psychotherapie. Ilya Ayzenberg, Bochum, St. Josef-Hospital, Neurologie. Ina Reichen, Zürich, UK Neurologie. Ina Schröder, Kiel UKSH, Neuroimm./klein. Chemie. Ina-Isabelle Schmütz, Kiel UKSH, Neuroimm./klein. Chemie. Jan Lewerenz, Ulm UK Neurologie. Jan Lünemann, Münster UK Neurologie/Schlafmedizin. Jan Wagner, Ulm UK Neurologie. Jaqueline Klausewitz, Bochum, St. Joseph, Neurologie. Jens Harmel, Kempen Hospital Heiligen Geist, Neurologie. Jens Schaumberg, Lübeck Sana Kliniken, Neurologie. Jens Schmidt, Göttingen, Neurologie. Joachim Havla, München LMU Neuroimmunologie/Neurologie. Johanna Maria Helena Rau, Münster UK Neurologie/Schlafmedizin. Johannes Piepgras, Mainz, UK Neurologie. Jonathan Wickel, Jena UK, Neurologie. Josef Priller, Berlin Charité, Pschologie/Psychatrie. Jost Obrocki, Elmshorn, Psychatrie. Judith Wagner, Gelsenkirchen, Neurologie. Julia Maren Decker, Rüdersdorf, Neurologie. Julian Dominik, LMU München/Großraden. Juliane Spiegler, Würzburg Pädiatrie. Jürgen Hartmut Faiss, Teupitz Neurologie. Justina Dargvainiene, Kiel UKSH, Neuroimm./klein. Chemie. Kai Siebenbrodt, Frankfurt, Epilepsiezentrum & Neurologie. Karin Storm van`s Gravesande, Freiburg, Pädiatrie. Karsten Witt, Oldenburg, Neurologie. Kartharina Wurdack, Berlin Charité, Neurologie/Neurocure. Katharina Eisenhut, München LMU Neuroimmunologie/Neurologie. Kathrin Doppler, Würzburg Neurologie. Katrin Thies, Klinikum Kassel, Neurologie. Kerstin Hellwig, Bochum, St. Josef Hospital, Neurologie. Kevin Rostasy, Datteln, Neuropädiatrie. Kim Kristin Falk, Kiel UKSH, Neuroimm./klein. Chemie. Klarissa Hanja Stürner, Kiel, UKSH Neuroimmunolo./klein. Chem. Klaus-Peter Wandinger, Lübeck UKSH, Neuroimmunolog./klein. Chem.. Kristin Stefanie Melzer, Münster UK Neurologie/Schlafmedizin. Kurt-Wolfram Sühs, Hannover, MHH Neurologie. Lara Zieger, Marburg UK Neurologie. Laura Ehrhardt, Mainz, UK Pädiatrie. Lena Edelhoff, Hamburg Marienkrankhaus, Neurologie. Lena Kristina Pfeffer, Hamburg Eppendorf, Neurologie. Loana Penner, Ulm UK Neurologie. Luise Appeltshauser, Würzburg Neurologie. Makbule Senel, Ulm UK Neurologie. Manuel Friese, Hamburg Eppendorf Neurologie. Marc Nikolaus, Berlin; Charité Pädiatrie. Marcel Gebhard, Halle, Martha-Maria, Neurologie. Marco Gallus, Münster UK Neurologie/Schlafmedizin. Mareike Jansen, Kiel, UKSH Neuroimmunolo./klein.Chem. Mareike Schimmel, Augsburg UK, Neuropädiatrie. Marie Madlener, Köln UK Neurologie. Marie-Luise Mono, Zürich Stadtspital Neurologie. Marina Entscheva, Lübben, Neurologie. Marina Flotats-Bastardas, Homburg/Saar Pädiatrie. Marius Ringelstein, Düsseldorf UK Neurologie. Markus Krämer, Essen Alfried-Krupp KH Neurologie. Markus Rauchenzauner, Vogtareuth, Schön Klinik. Martin Berghoff, Giessen, Neurologie. Martin Häusler, Aachen UK RTHW , Neuropädiatrie. Martin Lesser, Dresden UK Neurologie. Martina Jansen, Kiel UKSH, Neuroimm./klein. Chemie. Mathias Fousse, Homburg/Saar, Neurologie. Matthias von Mering, Bremen Klinikum Nord, Neurologie. Max Kaufmann, Hamburg Eppendorf, Neurologie. Max Vogtmann, Halle, UK Neurologie. Methap Türedi, Ulm UK Neurologie. Michael Adelmann, Weilmünster Neurologie. Michael Karenfort, Düsseldorf UK, Pädiatrie. Michael Malter, Erkelenz, Neurologie. Mona Dreesmann, Potsdam, Pädiatrie. Monika Meister, Ulm, Bundeswehrkrankenhaus. Mosche Pompsch, Essen, Alfried-Krupp Krankenhaus, Neurologie. Muriel Stoppe, Leipzig Neurologie. Nele Retzlaff, Rostock, Neurologie. Nico Melzer, Düsseldorf UK Neurologie. Niels Hansen, Göttingen, Psychatrie. Niels Margraf, UKSH Kiel, Neurologie. Niklas Vogel, Düsseldorf UK Neurologie. Njiku Melchior Wellmer, Ulm UK Neurologie. Olga Simova, Hamburg Alsterdorf, Epilepsie-Zentrum. Oliver Bähr, Aschaffenburg, Neurologie. Oliver Grauer, Münster UK Neurologie/Schlafmedizin. Oliver Stammel, Hamburg Barnbek Neurologie. Patrick Schramm, Giessen, Neurologie. Paul Friedemann, Berlin Charité, Neurologie/Neurocure. Pawel Tacik, Bonn, Neurodegeneration/Gerondopsych. Peter Huppke, Jena UK Pädiatrie. Peter Körtvélyessy, Berlin Charité, Neurologie/Neurocure. Philip Hillebrand, Bonn UK, Pädiatrie. Raphael Reinecke, Frankfurt, Epilepsiezentrum & Neurologie. Regina Trollmann, Erlangen, Pädiatrie. Robert Berger, Hamburg Altona, Neurologie. Robert Handreka, Cottbus, Carl-Thime Klinikum, Neurologie. Robert Rehmann, Dortmund, Klinikum, Neurologie. Robert Weissert, Regensburg, UK Neurologie. Rolf Kern, Worms, Neurologie. Ruth Kerkhoff, Mönchenglatbach, Maria-Hilf. Ruth Schilling, Leipzig, Neurologie. Sarah Bernsen, Bonn, Neurodegeneration/Gerondopsych. Sascha Berning, Osnabrück Neurologie. Saskia Jania Räuber, Münster UK Neurologie/Schlafmedizin. Sebastian Baatz, Altenburg Neurologie. Sebastian Bauer, Frankfurt, Epilepsiezentrum & Neurologie. Sergio Castro-Gomez, Bonn, Neurodegeneration/Gerondopsych. Sigrid Mues, Bochum, KnappschaftsKH, Neurologie. Simon Schuster, Kiel UKSH, Neuroimm./klein. Chemie. Simone Tauber, Aachen UK RTHW Neurologie. Sonka Benesch, Aschaffenburg, Neurologie. Stefan Bittner, Mainz, UK Neurologie. Stefanie Becker, Ulm Neurologie. Steffen Pfeuffer, Giessen, Neurologie. Steffen Syrbe, Heidelberg, Pädiatrie. Stephan Rüegg, Basel UK, Neurologie. Stephan Schreiber, Henningsdorf, Oberhavel Kliniken. Stjepana Kovac, Münster UK Neurologie/Schlafmedizin. Susanne Knake, Marburg UK Neurologie. Sven Ehrlich, Ohne Zentrum (ehem. Wermsdorf). Sven Meuth, Düsseldorf UK Neurologie. Tanja Kümpfel, München LMU Neuroimmunologie/Neurologie. Thanos Tsaktanis, Erlangen, Neurologie. Theodor Rüber, Bonn Epileptoloige. Thomas Grüter, Bochum, St. Joseph, Neurologie. Thomas Pfefferkorn, Ingolstadt, Neurologie. Thomas Seifert-Held, Graz Neurologie. Thorleif Etgen, Traunstein, Neurologie. Til Menge, Düsseldorf, LVR, Neurologie. Timo Deba, Münster, Pädiatrie. Tobias Freilinger, Passau Neurologie. Ulrich Hofstadt-van Oy, Dortmund, Westphalen, Neurologie. Valentin Held, Mannheim UK Neurologie. Verena Kraus, München TU Pädiatrie. Walid Fazeli, Bonn UK, Pädiatrie. Wolfgang Heide, Celle Allg. KH, Neurologie. Yannic Saathoff. Yetzenia Dubraska Haro Alizo, Ingolstadt, Neurologie.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. For the research, authorship, and publication of this article the authors were supported by the German Ministry of Education and Research (CONNECT-GENERATE/CONNECT-GENERATE 2.0, 01GM1908A und 01GM2208A to FL, 01GM1908C und 01GM2208B to JL, 01GM1908D and 01GM2208C to CF).
Conflict of interest
MF received honoraria as speaker and for consultation from Alexion, Biogen, Kyverna, Lundbeck, Merck KGaA, Novartis, Roche and Sudo Biosciences, as well as from the Gemeinnützige Hertie-Stiftung and Wings for Life. His research is funded by the German Federal Ministry of Education and Research (BMBF), Deutsche Forschungsgemeinschaft (DFG), Landesforschungsförderung Hamburg, Gemeinnützige Hertie-Stiftung, Else Kröner-Fresenius-Stiftung, Deutsche Multiple Sklerose Gesellschaft, Fritz-Thyssen-Stiftung, Werner-Otto-Stiftung, Walter und Ilse Rose-Stiftung, Stiftung zur Bekämpfung neuroviraler Erkrankungen and Research Fund of the University Medical Center Hamburg-Eppendorf; AG received speaker’s honoraria from Boehringer Ingelheim, Daichii Sankyo, Pfizer, Merz, Shionogi, Occlutech and Ipsen Pharma as well as research grants from MERZ and Ipsen Pharma; FL discloses speaker honoraria from Grifols, Teva, Biogen, Bayer, Roche, Novartis, Fresenius, travel funding from Merck, Grifols and Bayer and serving on advisory boards for Roche, Biogen and Alexion. His research FL is supported by German Ministry of Education and Research (BMBF), 01GM1908A und 01GM2208A, CONNECT-GENERATE, by E-Rare Joint Transnational research support ERA-Net, LE3064/2-1, Stiftung Pathobiochemie of the German Society for Laboratory Medicine and HORIZON MSCA 2022 Doctoral Network 101119457 — IgG4-TREAT; JL reports travel honoraria and speaker’s fees from the Cure Huntington’s Disease Initiative (CHDI), the Movement Disorders Society as the German Society for Cerebrospinal Fluid Diagnostic and Clinical Neurochemistry (DGLN), Alexion, Biogen, Sanofi, Teva, Merck, Novartis, Janssen, Fujirebio, Roche, and Neuraxpharm. His institution received financial compensation for clinical trials with JL as principal investigator from CHDI and SOM Biotech. He is member of the executive board of the DGLN. He received research funding from the German Federal Ministry of Education and Research (BMBF), CONNECT-GENERATE, 01GM1908B und 01GM2208B; and the Boehringer Ingelheim University Biocenter; NM has received honoraria for lecturing and travel expenses for attending meetings from Biogen Idec, GlaxoSmith Kline, Teva, Novartis Pharma, Bayer Healthcare, Genzyme, Alexion Pharmaceuticals, Fresenius Medical Care, Diamed, UCB Pharma, AngeliniPharma, BIAL and Sanofi-Aventis, has received royalties for consulting from UCB Pharma, Alexion Pharmaceuticals; and Sanofi-Aventis and has received financial research support from Euroimmun, Fresenius Medical Care, Diamed, Alexion Pharmaceuticals, and Novartis Pharma; MR received speaker honoraria from Novartis, Bayer Vital GmbH, Roche, Alexion, Horizon/Amgen; and Ipsen and travel reimbursement from Bayer Schering, Biogen Idec, Merz, Genzyme, Teva, Roche, Horizon, Alexion; and Merck, none related to this study; K-WS reports honoraria for lectures or travel reimbursements for attending meetings from Biogen, Merck, Mylan, Roche, Bavarian Nordic, Viatris and Bristol-Myers Squibb as well as research support from Bristol-Myers Squibb.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2024.1500904/full#supplementary-material
References
1. Leypoldt F, Armangue T, Dalmau J. Autoimmune Encephalopathies. Ann N Y Acad Sci. (2015) 1338:94–114. doi: 10.1111/nyas.12553
2. Moscato EH, Peng X, Jain A, Parsons TD, Dalmau J, Balice-Gordon RJ. Acute Mechanisms Underlying Antibody Effects in Anti-N-Methyl-D-Aspartate Receptor Encephalitis. Ann Neurol. (2014) 76(1):108–19. doi: 10.1002/ana.24195
3. Leypoldt F, Buchert R, Kleiter I, Marienhagen J, Gelderblom M, Magnus T, et al. Fluorodeoxyglucose Positron Emission Tomography in Anti-N-Methyl-D-Aspartate Receptor Encephalitis: Distinct Pattern of Disease. J Neurol Neurosurg Psychiatry. (2012) 83(7):681–6. doi: 10.1136/jnnp-2011-301969
4. Dalmau J, Armangue T, Planaguma J, Radosevic M, Mannara F, Leypoldt F, et al. An Update on Anti-NMDA Receptor Encephalitis for Neurologists and Psychiatrists: Mechanisms and Models. Lancet Neurol. (2019) 18(11):1045–57. doi: 10.1016/S1474-4422(19)30244-3
5. Florance NR, Davis RL, Lam C, Szperka C, Zhou L, Ahmad S, et al. Anti-N-Methyl-D-Aspartate Receptor (NMDAR) Encephalitis in Children and Adolescents. Ann Neurol. (2009) 66(1):11–8. doi: 10.1002/ana.21756
6. Titulaer MJ, McCracken L, Gabilondo I, Armangue T, Glaser C, Iizuka T, et al. Treatment and Prognostic Factors for Long-Term Outcome in Patients with Anti-NMDA Receptor Encephalitis: An Observational Cohort Study. Lancet Neurol. (2013) 12(2):157–65. doi: 10.1016/S1474-4422(12)70310-1
7. Poorthuis MHF, van Rooij JLM, Koch AH, Verdonkschot AEM, Leembruggen MM, Titulaer MJ. Cerebellar Ataxia as a Presenting Symptom in a Patient with Anti-NMDA Receptor Encephalitis. Neurol Neuroimmunol Neuroinflamm. (2019) 6(4):e579. doi: 10.1212/NXI.0000000000000579
8. Schmitt A, Koschel J, Zink M, Bauer M, Sommer C, Frank J, et al. Gene Expression of NMDA Receptor Subunits in the Cerebellum of Elderly Patients with Schizophrenia. Eur Arch Psychiatry Clin Neurosci. (2010) 260(2):101–11. doi: 10.1007/s00406-009-0017-1
9. Akazawa C, Shigemoto R, Bessho Y, Nakanishi S, Mizuno N. Differential Expression of Five N-Methyl-D-Aspartate Receptor Subunit mRNAs in the Cerebellum of Developing and Adult Rats. J Comp Neurol. (1994) 347(1):150–60. doi: 10.1002/cne.903470112
10. Carter AJ. Many Agents That Antagonize the NMDA Receptor-Channel Complex in Vivo Also Cause Disturbances of Motor Coordination. J Pharmacol Exp Ther. (1994) 269(2):573–80.
11. Rupniak NM, Boyce S, Tye S, Cook G, Iversen SD. Anxiolytic-Like and Antinociceptive Effects of MK-801 Accompanied by Sedation and Ataxia in Primates. Pharmacol Biochem Behav. (1993) 44(1):153–6. doi: 10.1016/0091-3057(93)90293-3
12. Jesse S, Wagner J, Gastl R, Steinacker P, Otto M, Kassubek J, et al. On Razor's Edge: Managing Analgosedation During Severe Anti-NMDA Receptor Encephalitis. Neurol Neuroimmunol Neuroinflamm. (2019) 6(1):e522. doi: 10.1212/NXI.0000000000000522
13. Iizuka T, Kaneko J, Tominaga N, Someko H, Nakamura M, Ishima D, et al. Association of Progressive Cerebellar Atrophy with Long-Term Outcome in Patients with Anti-N-Methyl-D-Aspartate Receptor Encephalitis. JAMA Neurol. (2016) 73(6):706–13. doi: 10.1001/jamaneurol.2016.0232
14. Titulaer MJ, Hoftberger R, Iizuka T, Leypoldt F, McCracken L, Cellucci T, et al. Overlapping Demyelinating Syndromes and Anti-N-Methyl-D-Aspartate Receptor Encephalitis. Ann Neurol. (2014) 75(3):411–28. doi: 10.1002/ana.24117
15. Martinez-Hernandez E, Guasp M, Garcia-Serra A, Maudes E, Arino H, Sepulveda M, et al. Clinical Significance of Anti-NMDAR Concurrent with Glial or Neuronal Surface Antibodies. Neurology. (2020). doi: 10.1212/WNL.0000000000009239
16. Banks SA, Morris PP, Chen JJ, Pittock SJ, Sechi E, Kunchok A, et al. Brainstem and Cerebellar Involvement in MOG-IgG-Associated Disorder Versus Aquaporin-4-IgG and MS. J Neurol Neurosurg Psychiatry. (2020). doi: 10.1136/jnnp-2020-325121
17. Maudes E, Landa J, Munoz-Lopetegi A, Armangue T, Alba M, Saiz A, et al. Clinical Significance of Kelch-Like Protein 11 Antibodies. Neurol Neuroimmunol Neuroinflamm. (2020) 7(3). doi: 10.1212/NXI.0000000000000666
18. Landa J, Guasp M, Miguez-Cabello F, Guimaraes J, Mishima T, Oda F, et al. Encephalitis with Autoantibodies against the Glutamate Kainate Receptors Gluk2. Ann Neurol. (2021) 90(1):101–17. doi: 10.1002/ana.26098
19. Graus F, Titulaer MJ, Balu R, Benseler S, Bien CG, Cellucci T, et al. A Clinical Approach to Diagnosis of Autoimmune Encephalitis. Lancet Neurol. (2016) 15(4):391–404. doi: 10.1016/S1474-4422(15)00401-9
20. Thompson AJ, Banwell BL, Barkhof F, Carroll WM, Coetzee T, Comi G, et al. Diagnosis of Multiple Sclerosis: 2017 Revisions of the Mcdonald Criteria. Lancet Neurol. (2018) 17(2):162–73. doi: 10.1016/S1474-4422(17)30470-2
21. Di Pauli F, Mader S, Rostasy K, Schanda K, Bajer-Kornek B, Ehling R, et al. Temporal Dynamics of Anti-MOG Antibodies in CNS Demyelinating Diseases. Clin Immunol. (2011) 138(3):247–54. doi: 10.1016/j.clim.2010.11.013
22. Jarius S, Probst C, Borowski K, Franciotta D, Wildemann B, Stoecker W, et al. Standardized Method for the Detection of Antibodies to Aquaporin-4 Based on a Highly Sensitive Immunofluorescence Assay Employing Recombinant Target Antigen. J Neurol Sci. (2010) 291(1-2):52–6. doi: 10.1016/j.jns.2010.01.002
23. Ances BM, Vitaliani R, Taylor RA, Liebeskind DS, Voloschin A, Houghton DJ, et al. Treatment-Responsive Limbic Encephalitis Identified by Neuropil Antibodies: MRI and PET Correlates. Brain. (2005) 128(Pt 8):1764–77. doi: 10.1093/brain/awh526
24. Miyazaki D, Fukushima K, Nakahara A, Kodaira M, Mochizuki K, Kaneko K, et al. Crossed Cerebellar Diaschisis in Status Epilepticus. Intern Med. (2016) 55(12):1649–51. doi: 10.2169/internalmedicine.55.6689
25. Jarius S, Eichhorn P, Franciotta D, Petereit HF, Akman-Demir G, Wick M, et al. The Mrz Reaction as a Highly Specific Marker of Multiple Sclerosis: Re-Evaluation and Structured Review of the Literature. J Neurol. (2017) 264(3):453–66. doi: 10.1007/s00415-016-8360-4
26. Gahr M, Lauda F, Wigand ME, Connemann BJ, Rosenbohm A, Tumani H, et al. Periventricular White Matter Lesion and Incomplete MRZ Reaction in a Male Patient with Anti-N-Methyl-D-Aspartate Receptor Encephalitis Presenting with Dysphoric Mania. BMJ Case Rep. (2015) 2015. doi: 10.1136/bcr-2014-209075
27. Durr M, Nissen G, Suhs KW, Schwenkenbecher P, Geis C, Ringelstein M, et al. CSF Findings in Acute Nmdar and LGI1 Antibody-Associated Autoimmune Encephalitis. Neurol Neuroimmunol Neuroinflamm. (2021) 8(6). doi: 10.1212/NXI.0000000000001086
28. Uzawa A, Mori M, Takahashi Y, Ogawa Y, Uchiyama T, Kuwabara S. Anti-N-Methyl D-Aspartate-Type Glutamate Receptor Antibody-Positive Limbic Encephalitis in a Patient with Multiple Sclerosis. Clin Neurol Neurosurg. (2012) 114(4):402–4. doi: 10.1016/j.clineuro.2011.10.047
29. Fleischmann R, Pruss H, Rosche B, Bahnemann M, Gelderblom H, Deuschle K, et al. Severe Cognitive Impairment Associated with Intrathecal Antibodies to the NR1 Subunit of the N-Methyl-D-Aspartate Receptor in a Patient with Multiple Sclerosis. JAMA Neurol. (2015) 72(1):96–9. doi: 10.1001/jamaneurol.2014.1817
30. Ramberger M, Bsteh G, Schanda K, Hoftberger R, Rostasy K, Baumann M, et al. NMDA Receptor Antibodies: A Rare Association in Inflammatory Demyelinating Diseases. Neurol Neuroimmunol Neuroinflamm. (2015) 2(5):e141. doi: 10.1212/NXI.0000000000000141
31. Baheerathan A, Brownlee WJ, Chard DT, Shields K, Gregory R, Trip SA. Antecedent Anti-NMDA Receptor Encephalitis in Two Patients with Multiple Sclerosis. Mult Scler Relat Disord. (2017) 12:20–2. doi: 10.1016/j.msard.2016.12.009
32. Gulec B, Kurucu H, Bozbay S, Dikmen Y, Sayman H, Tuzun E, et al. Co-Existence of Multiple Sclerosis and Anti-NMDA Receptor Encephalitis: A Case Report and Review of Literature. Mult Scler Relat Disord. (2020) 42:102075. doi: 10.1016/j.msard.2020.102075
Keywords: NMDAR-encephalitis, ataxia, outcome, cerebellum, multiple sclerosis, MOG antibody, aquaporin-4 antibody
Citation: Jesse S, Riemann M, Schneider H, Ringelstein M, Melzer N, Vogel N, Pfeffer LK, Friese MA, Sühs K-W, Hudasch D, Schwenkenbecher P, Günther A, Geis C, Wickel J, Lesser M, Kather A, Leypoldt F, Dargvainiene J, Markewitz R, Wandinger K-P, Thaler FS, Kuchling J, Wurdack K, Sabater L, Finke C and Lewerenz J (2024) Frequency, characteristics, and immunological accompaniments of ataxia in anti-NMDAR antibody-associated encephalitis. Front. Immunol. 15:1500904. doi: 10.3389/fimmu.2024.1500904
Received: 24 September 2024; Accepted: 18 November 2024;
Published: 13 December 2024.
Edited by:
Moussa Antoine Chalah, GHU Paris Psychiatrie et Neurosciences, FranceReviewed by:
Mariano Marrodan, Fundación Para la Lucha Contra las Enfermedades Neurológicas de la Infancia (FLENI), ArgentinaGregorio Spagni, University of Florence, Italy
Copyright © 2024 Jesse, Riemann, Schneider, Ringelstein, Melzer, Vogel, Pfeffer, Friese, Sühs, Hudasch, Schwenkenbecher, Günther, Geis, Wickel, Lesser, Kather, Leypoldt, Dargvainiene, Markewitz, Wandinger, Thaler, Kuchling, Wurdack, Sabater, Finke and Lewerenz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jan Lewerenz, amFuLmxld2VyZW56QHVuaS11bG0uZGU=