
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 06 January 2025
Sec. Cancer Immunity and Immunotherapy
Volume 15 - 2024 | https://doi.org/10.3389/fimmu.2024.1494206
 Oana Hangiu1,2,3Rocío Navarro1Susana Frago1Laura Rubio-Pérez2,3,4Antonio Tapia-Galisteo2,3,4Laura Díez-Alonso2,3,4Marina Gómez-Rosel1,2,3Noelia Silva-Pilipich5,6
Oana Hangiu1,2,3Rocío Navarro1Susana Frago1Laura Rubio-Pérez2,3,4Antonio Tapia-Galisteo2,3,4Laura Díez-Alonso2,3,4Marina Gómez-Rosel1,2,3Noelia Silva-Pilipich5,6 Lucía Vanrell7
Lucía Vanrell7 Cristian Smerdou5,6
Cristian Smerdou5,6 Kenneth A. Howard8
Kenneth A. Howard8 Laura Sanz9
Laura Sanz9 Luis Álvarez-Vallina2,3,4*
Luis Álvarez-Vallina2,3,4* Marta Compte1*
Marta Compte1*Background: Immune checkpoint inhibitors have revolutionized cancer therapy, but many patients fail to respond or develop resistance, often due to reduced T cell activity. Costimulation via 4-1BB has emerged as a promising approach to enhance the effector function of antigen-primed T cells. Bispecific T cell-engaging (TCE) antibodies are an effective way to provide tumor-specific T cell receptor-mediated signaling to tumor-infiltrating lymphocytes. mRNA-based delivery of bispecific antibodies, offer a novel approach to enhance tumor-specific immune responses while minimizing adverse effects.
Methods: Two bispecific antibodies were generated: the EGFR x CD3 TCE antibody (LiTE) and the PD-L1 x 4-1BB costimulatory antibody (LiTCo), which was further fused to a high FcRn albumin variant (Albu-LiTCo). The mRNA encoding these bispecific antibodies contains an N1-methylpseudouridine modified nucleoside and regulatory sequences to ensure proper expression and stability. A series of in vitro assays and cell-based analyses were performed to characterize both antibodies. The in vivo efficacy of the mRNA-encoded bispecific antibodies was evaluated in xenograft tumor models expressing EGFR.
Results: We investigated the combined effect of two mRNA-encoded Fc-free bispecific antibodies with complementary mechanisms of action: an EGFR-targeting TCE and a half-life extended PD-L1 x 4-1BB costimulatory antibody. The mRNAs encoding both bispecific LiTERNA and Albu-LiTCoRNA, showed similar binding specificity and in vitro function to their protein analogues. Pharmacokinetic studies demonstrated sustained expression of both bispecific antibodies following intravenous administration of the mRNAs formulated using a polymer/lipid-based nanoparticle (LNP) but different pharmacokinetic profiles, shorter for the TCE and longer for the PD-L1 x 4-1BB. When administered as a mRNA-LNP combination (ComboRNA), the growth of EGFR-positive tumors in immunocompetent mice was significantly inhibited, resulting in tumor regression in 20% of cases with no associated toxicity. Histological analysis confirmed increased T cell infiltration in the tumors treated with LITERNA and ComboRNA. Repeated administration resulted in sustained production of bispecific antibodies with different exposure cycles and potent antitumor activity with a favorable safety profile.
Conclusions: These results highlight the potential of combining two mRNA-encoded bispecific antibodies with different mechanisms of action and programmable half-life for cancer immunotherapy.
Immune checkpoint inhibitors (ICIs) unleash the immune system’s full antitumor potential for robust responses (1). However, a significant proportion of cancer patients do not respond or develop resistance over time (2). Reduced functional activity of tumor infiltrating lymphocytes (TILs) and/or PD-1/PD-L1 expression are factors contributing to response heterogeneity (3). Agonistic antibodies to co-stimulatory receptors, such as 4–1BB (CD137), have emerged as a promising strategy to enhance T cell function and have demonstrated synergistic effects with ICIs, improving antitumor responses in preclinical studies (4, 5). 4-1BB is a member of the tumor necrosis factor receptor (TNFR) superfamily that is expressed on antigen-activated T cells but not on resting T cells, which could restrict stimulation to tumor-reactive T cells (6–8). In preclinical models, anti-4–1BB agonistic mAbs restored CD8+ T cell function and promoted antitumor responses (9). However, off-tumor toxicities have significantly hindered the clinical development of anti-4-1BB agonistic IgGs (10–12). The anti-human 4-1BB human IgG4 urelumab (BMS-663513) demonstrated clinical activity but caused fatal liver toxicity (13, 14). Several studies suggest that toxicity is likely dependent on further crosslinking provided by Fc-Fc gamma receptor (FcγR) interaction (10, 11, 15, 16). New therapeutic strategies aim to limit 4-1BB costimulation to the tumor microenvironment. These approaches include Fc-free or Fc-silent bispecific antibodies targeting 4-1BB as well as tumor or stromal cells (10, 17–21). Previous studies from our research group have demonstrate lack of liver toxicity associated with other Fc-less constructs derived from the same anti-4-1BB moiety (10, 22), in contrast to the off-tumor toxicities observed in rodents treated with anti-4-1BB IgG. Several clinical trials combining such costimulatory antibodies with anti-PD-1/PD-L1 moieties are ongoing, and preliminary data suggest that these strategies are well tolerated, enhance TIL functions, and exhibit antitumor efficacy, supporting further investigation in advanced solid tumors (3, 23–25).
Beyond immune checkpoint blockade and costimulatory agonists, bispecific T cell-redirecting antibodies have gained momentum in recent years, with ten T cell engagers (TCE) in the market as of December 2024. TCE simultaneously bind to a tumor-associated antigen (TAA) and to CD3 in the TCR complex, redirecting T cells to kill tumor cells regardless of TCR specificity (26–29). However, potent T cell responses can result in off-target effects and systemic cytokine release-associated toxicities (30). Various Fc-free formats such as the diabody, the tandem single-chain variable fragment (ta-scFv), and the tandem of a single-domain antibody (VHH) and a scFv (LiTE, Light T cell Engager) have been developed to enhance their efficacy and reduce side effects (31, 32). TCE have shown great potential in hematological cancers, with seven FDA (Food and Drug Administration)-approved bispecific antibodies for the treatment of leukemia, non-Hodgkin lymphoma (NHL), and multiple myeloma (33, 34). However, their efficacy in solid tumors is still rather limited (35). In May 2024, the Food and Drug Administration (FDA) granted accelerated approval for tarlatamab, a DLL3xCD3 half-life-extended (HLE) TCE with a silenced Fc region for small cell lung cancer (SCLC) (36). Alternatively, TAA-targeted HLE Fc-free bispecifics have been generated by fusion to an engineered human serum albumin variant with high binding (albuminHB) to human FcRn29 such as the Albu-LiTE (37) and the 4-1BB agonist Albu-LiTCo (Light T cell Costimulatory) antibody (22).
Another strategy to avoid frequent dosing of Fc-free bispecific antibodies while overcoming problems associated with recombinant protein production is the use of mRNA. The advantages of mRNA-based gene delivery over viral vectors and plasmid DNA have led to its increased popularity in numerous therapeutic areas (38, 39). Synthetic mRNA allows for rapid and transient protein production without nuclear entry, avoiding genome integration. A preclinical study demonstrated that intravenous administration of mRNA encoding Fc-free bispecific TAAxCD3 sustained functional antibody production and resulted in tumor regression in mice (40). In addition, the first phase 1 clinical trial of mRNA-encoded TCE demonstrated safety and the potential to achieve therapeutic levels of circulating antibodies (41).
In the present study, we generated mRNA-encoded EGFRxCD3 LiTE and PD-L1x4-1BB Albu-LiTCo. Both mRNAs promoted EGFR-specific tumor cell lysis and PD-L1 -dependent costimulatory activity in vitro, respectively, and exhibited favorable pharmacokinetic properties in vivo. In addition, the combination of LiTERNA and Albu-LiTCoRNA enhanced T cell activation and tumor cell lysis and significantly delayed tumor growth in immunocompetent mice, offering a promising approach for cancer immunotherapy.
Seven-week-old female BALB/c were purchased from Envigo (Envigo Rms Spain S.L). Animals were housed in controlled conditions of temperature (21 ± 1°C), humidity (50 ± 5%), and 12 hours light and dark cycles. Animals were maintained under specific-pathogen-free conditions, and sterilized water and food were available ad libitum. All animal procedures were performed in accordance with European Union Directive 86/609/EEC and Recommendation 2007/526/EC, enforced in Spanish law under RD 1201/2005. Animal protocols were performed in strict adherence to the guidelines stated in the International Guiding Principles for Biomedical Research Involving Animals, established by the Council for International Organizations of Medical Sciences (CIOMS) and were approved by the Ethics Committee of Animal Experimentation of the Instituto Investigación Sanitaria Puerta de Hierro-Segovia de Arana (Hospital Universitario Puerta de Hierro Majadahonda, Madrid, Spain). Procedures were also approved by the Animal Welfare Division of the Environmental Affairs Council of the Government of Madrid (076/19).
HEK293 (CRL-1573), CT26 (CRL-2638) and CHO-K1 (CCL-61) were obtained from American Type Culture Collection (ATCC) (Rockville, MD, USA). Mouse CT26 cells (CRL-2638) infected with p-BABEpuro-hEGFR expressing human EGFR (CT26EGFR) were provided by Dr M. Rescigno (42) (European Institute of Oncology, Milan). Production of lentiviral vectors for the generation of CT26 and CT26EGFR cells expressing the firefly luciferase (Luc) gene has been described previously (43). CHO-K1 cells stably expressing human PD-L1 (CHOPD-L1) were obtained from Genlantis (xCELLerateTMPD-L1 Stable Cell Line, XCL-PDL1) and CHO-K1 cells stably expressing human EGFR (CHOEGFR), or both (CHOPD-L1/EGFR) were generated using hEGFR encoding commercial lentiviral particles (G&P Biosciences, cat# LTV0169). Both cell lines were infected overnight at a final multiplicity of infection (MOI) of 10. Cells were supplemented with 500 µg/ml of Hygromycin B (Sigma-Aldrich) or 500 µg/ml of Hygromycin B+100 µg/ml of Blasticidin S HCl (Thermo Fisher Scientific), respectively. HEK293 cells expressing mouse 4-1BB (HEK2934-1BB) were obtained from Prof I. Melero (CIMA, Pamplona, Spain). The cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 2 mM L-glutamine (Lonza, USA, cat# BE17-605E), 10% (vol/vol) heat inactivated fetal bovine serum (FBS) (Gibco, USA, cat# 10500-064) and antibiotics (100 units/mL penicillin, 100 mg/mL streptomycin) (Gibco, USA, cat# 15140163), referred to as DMEM complete medium (DCM), at 37 °C in 5% CO2 humidity. T cell line, Jurkat, clone E6-1 (Human acute T cell leukemia; TIB-152) was obtained from American Type Culture Collection (ATCC) and T cell line 2B4 (Mouse T-cell hybridoma; CVCL_4Z38) was provided by Prof. J. R. Regueiro (UCM, Madrid, Spain). Cells were grown in Roswell Park Memorial Institute medium (RPMI) supplemented with 2 mM L-glutamine, 10% (vol/vol) heat inactivated FBS, and antibiotics (100 units/mL penicillin, 100 mg/mL streptomycin), referred to as RPMI complete medium (RPMI-C), at 37 °C in 5% CO2 humidity. The cell lines were routinely screened for mycoplasma contamination by PCR using the Mycoplasma Gel Detection Kit (Biotools, Spain, cat# 90.021-4542).
The FLAG-strepII-EGa1-2C11 DNA construct was created by obtaining the anti-EGFR VHH EGa1 from a previously published anti-EGFRxanti-CD3 LiTE (44), and the mouse anti-CD3ϵ VH and VL sequences of 2C11 scFv (45), were synthesized by GeneArt. Sequences were inserted into the pCR3.1-EGa1 expression vector using NotI/BglII restriction enzymes, resulting in the anti-EGFR x anti-mouse CD3ϵ bispecific antibody, hereafter referred to as LiTE (Light T cell Engager). The 6P VHH anti-PD-L1 DNA sequence (46) was inserted into the pCR3.1-EGa1 expression vector using HindIII/NotI restriction enzymes. The DNA sequence FLAG-strepII-6P-1D8, from GeneArt, was subcloned as AgeI/SnaBI into the vector containing the high affinity FcRn binding human albumin (AlbuminHB) (47) resulting in the FLAG-strepII-6P-1D8-AlbuminHB construct, anti-m/hPD-L1 x anti-m4-1BB bispecific antibody, referred to as Albu-LiTCo (Light T cell Costimulatory). All the sequences were verified using primers FwCMV and RvBGH (Supplementary Table 1). Constructed vectors were stably transfected into HEK293 cells using Lipofectamine 3000 (Invitrogen, USA, cat# L3000001) and selected with 500 μg/ml Geneticin (G418) to generate stable HEK293-LiTE and HEK293-Albu-LiTCo cell lines. Antibodies were purified from free-cell culture by affinity chromatography Strep-Tactin® purification system (IBA Lifesciences, Germany) using an ÄKTA Go system (Cytiva). Purified antibodies were dialyzed overnight at 4°C against PBS with 150 mM NaCl at pH 7.0. (10-20% Tris-Glycine gel) and analytical size exclusion chromatography (SEC) (Superdex 200 increase 10/300 GL; Cytiva Spain S.L.U.) were performed to determine the size and the purity of the bispecific antibodies.
The template for mRNA IVT (in vitro transcription) was carried out by GenScript Biotech (Netherlands). The mRNA-encoding bispecific antibodies incorporates a 5’ type I capping structure (5’ Cap: Cap1) and N1-Methylpseudouridine modified nucleosides (N1-me-Ψ), to increase stability and reduce its immunogenicity (48). The IVT template (linearized plasmids), contains a 5’UTR, Kozak sequence, 3’UTR and a Poly(A)tail containing more than 110 successive adenines and the coding sequence. For in vitro and in vivo studies, mRNA-encoded LiTE (LiTERNA) and mRNA-encoded Albu-LiTCo (Albu-LiTCoRNA) were formulated with the polymer/lipid-based formulation TransIT™-mRNA Transfection Kit (Mirus Bio, USA, cat# MIR2304), in accordance with the manufacturer’s protocol. Briefly, HEK293 cells cultured in DCM were seeded at 3 × 105 cells/well on a 6-well plate. The day after, LiTERNA and Albu-LiTCoRNA (1 µg each) were diluted in 50 µl of Opti-MEM, followed by the addition of 2 µl of TransIT Boost reagent and 2 µl of TransIT-mRNA reagent. After two minutes of incubation, the reagent mixture complexes were added to the cell culture. The transfection was performed in duplicate, and after 48 hours incubation at 37°C, 5% CO2, the cell culture supernatants were harvested by centrifugation (2000 rpm, 5 min), pooled, and stored at -20°C until analysis. An EGFP mRNA (EGFPRNA) (GenScript; # SC2325-1mg) which incorporates a 5’ Cap: Cap1 structure and N1-me-Ψ, as the mRNA encoding bispecific antibodies, was used as a control. Regarding in vivo experiments, a modification was made to the protocol originally proposed by Stadler et al. (40) as follows. 10 micrograms of mRNA were diluted in 171.60 µl of DMEM with 4.5 g/l glucose (Thermo Fisher) and mixed with 11.20 µl of TransIT-mRNA reagent and 7.20 µl of TransIT Boost reagent. mRNA reactions were immediately vortexed, incubated for two minutes at room temperature (RT), and injected into mice within five minutes. 200 µl was the total volume of 10 µg of mRNA complex.
Protein samples were separated under reducing conditions on 10–20% Tris-glycine gels and transferred to nitrocellulose membranes and probed either with anti-FLAG (clone M2; Merck KGaA, Germany, cat# F3165) monoclonal antibody (mAb) (1 μg/ml), followed by incubation with goat anti-mouse DyLight 680 (1:5000 dilution) (Rockland, cat# 800-656-002), or Horseradish peroxidase (HRP)-conjugated MonoRab™ Rabbit Anti-Camelid VHH Cocktail antibody (anti-VHH Cocktail mAb) (GenScript, cat#A02016) at 1:2500 dilution working dilution. Visualization of protein bands was performed with the Amersham ImageQuant™ 800 (Cytiva). Images were analyzed with Image Studio Lite software.
Maxisorp immunoplates (NUNC Brand Products, Denmark, cat# 439454) were coated overnight at 4°C with human (h) EGFR-hu IgG1 Fc chimera (hEGFR) (Bio-Techne R&D Systems; cat# 344-ER), mouse (m) 4-1BB-hIgG1 Fc chimera (m4-1BB) (SinoBiological Europe GmbH, Germany, cat# 50811-M02H), recombinant mPD-L1 (mPD-L1) (clone B7-H1) or recombinant hPD-L1 (hPD-L1) (clone B7-H1) (Bio-Techne R&D Systems; cat# 1019-B7 and cat# 156-B7) at 3 µg/ml in PBS. Plates were washed and blocked with 5% bovine serum albumin (BSA) or 5% nonfat dry milk (NFDM) in PBS, and 100 μl of supernatant from transfected HEK293 cells or purified antibodies at 3-fold serial dilutions, were added in triplicates and incubated for 1 hour at room temperature. The wells were washed three times with PBS + 0.05% Tween20, followed by three washes with PBS. For detection, anti-FLAG mAb (1μg/ml) was added for 1 hour incubation at room temperature. The plates were washed as above described, and 100 μl of HRP-conjugated goat anti-mouse (GAM) (Jackson ImmunoResearch Europe Ltd., UK, cat# 115-085-166) or HRP-conjugated sheep anti-human serum albumin (HSA-HRP) (Abcam, UK, cat# ab8941), were diluted at a working concentration of 1:2000 in PBS + 1% BSA or, PBS + 1% NFDM, respectively.
The functional expression of LiTERNA and Albu-LiTCoRNA in supernatants were assayed by coating MaxiSorp plates with hEGFR, m4-1BB, and mPD-L1 (3 µg/ml) in PBS overnight at 4°C. After washing and blocking with 5% BSA or 5% NFDM, duplicates of supernatants were added at room temperature for 1 hour. Plates were incubated for 1 hour at room temperature with HRP-conjugated anti-VHH Cocktail (1:2000) diluted in PBS + 0.01% Tween20, or HSA-HRP (1:2000) diluted in PBS + 1% NFDM for detection. The estimated concentration of LiTERNA (3.3 µg/ml) and Albu-LiTCoRNA (3.9 µg/ml) was determined using the same procedure, with 3-fold serial dilutions of purified antibodies (as standard controls) and detection by HRP-conjugated anti-VHH cocktail (1:2000). Plates were washed and developed using Tetramethylbenzidine (TMB) (Merck KGaA cat# T0440), stopped with 4N H2SO4, and measured at 450-570 nm using a Bio-Rad microplate-reader. Data was analyzed and plotted in GraphPad Prism 8.4.0.
HEK293, HEK2934-1BB, CHO, CHOPD-L1, CT26, CT26EGFR, Jurkat or 2B4 cells (2.5 x 105 cells/well) were incubated on ice for 30 min with conditioned media from transfected HEK293 cells or purified antibodies (5 μg/ml), washed and incubated with anti-FLAG mAb (5 μg/ml) for 30 min and detected with a R-phycoerythrin (PE) F(ab’)2 Fragment Goat Anti-Mouse (GAM-PE) IgG, Fcγ fragment specific (Jackson ImmunoResearch, cat# 115-116-071) at 1:500 working dilution. Anti-4-1BB IgG (clone 3H3; BioXcell; cat# BE0239), anti-PD-L1 IgG (atezolizumab), anti-EGFR IgG (cetuximab), and anti-CD3 IgG (clone 145-2C11; Biolegend, USA, cat# 100303) were used as controls, at 5 μg/ml. Binding was detected after incubation with R-Phycoerythrin (PE) F(ab’)2 Fragment Goat Anti-Human (GAH) IgG, Fcγ fragment specific (Jackson ImmunoResearch, cat# 109-116-170) or GAM-PE at 1:500 working dilutions, as needed.
The binding analysis of LiTERNA and Albu-LiTCoRNA was also performed using the anti-FLAG mAb followed by GAM-PE at a working dilution of 1:500. The cell surface expression of mPD-L1 was analyzed on CT26 and CT26EGFR cells, before and after 24 hours incubation with soluble recombinant human IFNγ (20 ng/ml) (#300-02, PeproTech) at 37°C. Cells were stained with PE-conjugated anti-mPD-L1 mAb (clone 10F.9G2, cat# 124308 BioLegend) (2 μg/ml). A minimum of 20,000 events were acquired for each sample with a FACSCanto II cytometer (BD Biosciences). Collected data was evaluated using the FlowJo X (BD Biosciences) software.
CT26 and CT26EGFR cells (0.3 x 105 cells/well) were plated in triplicate in flat-bottom 96-well plates overnight at 37°C. Total splenocytes from 8-week-old female BALB/c mice were resuspended in RPMI supplemented with 10% FBS and 50 μM β-mercaptoethanol (Life Technologies) and added to the plate (1.5 x 105 cells/well) at a effector/target ratio of 5:1. LiTE and purified anti-mouse CD3ε IgG (clone 145-2C11) were added at 3.34 nM (equivalent to 0.175 µg/mL and 0.5 µg/mL respectively) equimolar concentration. After 24 hours supernatants were collected and assayed for IFNγ and IL-2 (Diaclone, France, cat# 861.050.005, and cat# 861.000.005) secretion by ELISA following manufacturer’s protocol. Cells were collected and incubated on ice for 30 min with fluoresceine (FITC) conjugated-Hamster anti-mouse CD69 IgG1 (clone H1.2F3, cat# 561929, BD Biosciences, USA) or FITC-conjugated Hamster IgG1, Isotype Control (clone G235-2356, cat# 553953 BD Biosciences) (3 μg/ml), both diluted in PBS + 1%FCS and washed. Samples were analyzed with FACSCanto II cytometer and FlowJo X software was used for analyzing flow cytometry data.
CT26 and CT26EGFR Luc (0.3 x 105 cells/well) gene-modified for the expression of luciferase, were seeded in triplicate in flat-bottom 96-well plates overnight at 37°C. Next day, target cells were incubated with increasing amounts of LiTE (0 to 600 nM) or LiTERNA-HEK293 undiluted supernatant (3.3 µg/ml). Total splenocytes were isolated from 8-week-old female BALB/c mice, resuspended in RPMI supplemented with 10% FBS and were added to the plate (1.5 x 105 cells/well) at 1:5 target:effector ratio. After 48 hours of incubation, D-luciferin substrate (20 μg/well, Promega E1602) was added, and relative light units (RLU) were measured with the luminescence plate reader Infinite 1200 (Tecan Trading). Percent tumor cell viability was calculated by dividing the mean bioluminescence of each sample by the mean bioluminescence of the input number of control target cells, multiplied by 100. Specific lysis is the difference in tumor cell viability relative to control (0%). Wells with target and effector cells in the absence of antibodies were set as 100% of viability.
Potency to neutralize PD-L1 binding to PD-1 was assessed in competition ELISA where 4 µg/ml recombinant hPD1-hIgG1 Fc chimera (hPD-1) (SinoBiological Europe GmbH, cat# 10377-H02H) was coated. Three-fold serial dilutions of anti-PD-L1 IgG (atezolizumab) or purified Albu-LiTCo were simultaneously incubated with 1 μg/ml of recombinant biotinylated hPD-L1/B7-H1 Protein His-tagged (SinoBiological Europe GmbH, cat# 10084-H08H-B) for 1 hour at room temperature, washed and detected by streptavidin-HRP (Sigma-Aldrich, cat# RABHRP3) (1:1000) followed by TMB. The reaction was stopped with 4N H2SO44 and the absorbance was measured at 450-570 nm using a Bio-Rad microplate-reader. Data was analyzed and plotted using IC50 values calculated by fitting the concentration–response curves with a four-parameter logistic regression model using GraphPad Prism 8.4.0 software.
Jurkat T cells stably expressing human PD-1 and NFAT-induced luciferase (JurkatNFAT-RE−luc/PD-1, referred as JurkatPD-1) and CHO-K1 cells stably expressing human PD-L1 (PD-L1 aAPC/CHO-K1) were obtained from Promega (cat# J1250) and were used following the manufacturer’s instructions. Briefly, 0.3 × 105 PD-L1 aAPC/CHO-K1 cells/well were seeded in 96-well white flat-bottom plates (Merck KGaA, cat# CLS3922) in DCM and incubated overnight at 37 °C. Next day, medium was removed and anti-PD-L1 IgG or Albu-LiTCo were added equimolarly at two-fold serial dilutions in 30 µl of RPMI+ 1% FBS/well. Then, 1.5 × 105 JurkatPD-1 cells/well were added in 30 µl RPMI + 1% FBS and incubated for 6 hours at 37°C. Bio-Glo™ Luciferase Assay Reagent (Promega; cat# G7941) was added and luciferase activity was assessed using a Tecan Infinite F200 plate-reading luminometer (Tecan Trading). The experiments were performed in triplicates and data are reported as x-fold of induction relative to the values obtained from unstimulated cells. Data was analyzed and plotted using GraphPad Prism 8.4.0 software.
For studies with target cells, CHO and CHOPD-L1/EGFR cells (0.3 x 105 cells/well) were seeded overnight at 37°C and 5% CO2. Mouse CD8a+ T cells were purified from the spleens of 8-week-old BALB/c female mice using the mouse CD8a+ T Cell Isolation Kit (Miltenyi Biotec, Germany, cat# 130-095-236), resuspended in 100 µl of culture media (RPMI supplemented with 10% FBS and 50 μM β-mercaptoethanol (Life Technologies) and pre-activated with soluble LiTE or anti-CD3ε IgG (clone 145-2C11; Biolegend, cat# 100303) at 3.34 nM equimolar final concentration (0.175 µg/mL and 0.5 µg/mL, respectively) for at least 5 hours at 37°C. Mouse CD8a+ T cells were added to the plate (1.5 x 105 cells/well) at an effector/target ratio of 5:1, along with purified Albu-LiTCo or anti-4-1BB IgG (clone 3H3) diluted in 100 µl of culture media at a final concentration of 6.67 nM equimolar dose. As a negative control for costimulation, LiTE-pre-activated mouse CD8a+ T cells were added to the co-culture in the absence of both Albu-LiTCo and anti-4-1BB IgG. Additionally, non-pre-activated mouse CD8a+ T cells were included in the co-culture as a negative control for the assay. For studies with purified antigens, 96-well flat-bottom plates were coated with 3 µg/ml of recombinant mPD-L1/B7-H1 hIgG1 Fc chimera (R&D, cat# 1019-B7-100#) or BSA as a control, overnight at 4 °C. After washing, plates were blocked with RPMI + 5% BSA for 1 h at 37°C. Mouse CD8a+ T cells were isolated as described above, resuspended in 100 µl of culture media and pre-activated with anti-CD3ε IgG (3.34 nM) for at least 5 hours at 37°C. Mouse CD8a+ T cells (1.5 x 105 cells/well) were then added to the plates with Albu-LiTCo, anti-4-1BB IgG or purified mouse IgG1K isotype (clone MG1-45; Biolegend, cat# 401402) diluted in 100 µl of culture media at a final concentration of 6.67 nM equimolar dose. As a negative control for costimulation, CD3-pre-activated mouse CD8a+ T cells were added to the plates without Albu-LiTCo, anti-4-1BB IgG or IgG1K isotype. Additionally, non-pre-activated mouse CD8a+ T cells were included as a negative control for the assay.
For the functional analysis of LiTERNA-HEK293 supernatants (LiTERNA), CHO and CHOPD-L1/EGFR cells (0.3 x 105 cells/well) were seeded in 96-flat well plates overnight at 37°C. Mouse CD8a+ T cells were isolated as previously described, and directly resuspended at 1.5 x 105/well. in 100 µl of undiluted LiTERNA (3.3 µg/ml). Cells were pre-activated for at least 5 hours at 37°C and added to the co-culture at an E/T ratio of 5:1. Albu-LiTCo, anti-4-1BB IgG, anti-PD-L1 IgG (clone MPDL3280A; MCE, cat# HY-P9904) or IgG1K isotype were then added in 100 µl of culture medium at a final concentration of 6.67 nM equimolar dose. As a negative control for costimulation, LiTERNA pre-activated mouse CD8a+ T cells were added to the plates in the absence of the above antibodies. Additionally, non-pre-activated mouse CD8a+ T cells were included in the co-culture as a negative control for the assay.
For the functional analysis of Albu-LiTCoRNA-HEK293 supernatant (Albu-LiTCoRNA), CHO and CHOPD-L1/EGFR cells (0.3 x 105 cells/well) were seeded in 96-flat well plates overnight at 37°C. Mouse CD8a+ T cells were purified, resuspended in 100 µl of culture media (1.5 x 105 cells/well) and pre-activated with soluble LiTE at 3.34 nM equimolar final concentration for at least 5 hours at 37°C, and added to the coculture at an E/T ratio of 5:1. Afterwards, 100 µl of undiluted Albu-LiTCoRNA (3.9 µg/ml) was added to the co-culture. As a negative control for costimulation, LiTE-pre-activated mouse CD8a+ T cells were added to the plates in the presence of 100 µl of EGFPRNA-HEK293 supernatant (EGFPRNA). Additionally, non-pre-activated mouse CD8a+ T cells combined with 100 µl of EGFPRNA were included in the co-culture as a negative control for the assay.
In all cases, co-cultures were incubated for 72 hours, supernatants were collected and assayed for IFNγ secretion by ELISA (Diaclone, cat# 861.050.005) following manufacturer’s protocol. Results are expressed as a mean ± SD from one of at least three separate experiments. All the experiments were performed in triplicates in 200 µl final volume.
9-week-old female BALB/c mice were intravenously (i.v.) injected (tail vein) with a single dose of 10 μg mRNA-polymer/lipid (mRNA-LNP) formulated LiTERNA (n = 3) or, Albu-LiTCoRNA (n = 3) or PBS (n = 3). A similar assay was performed with Albu-LiTCo (n=4) and LiTE (n=4) purified antibodies injected i.v. once at 1 mg/kg into 8-week-old female BALB/C mice. In both experiments, blood samples were collected at 0, 4, 24, 48, 72, 98 and 168 hours in separation gel BD microtubes (BD Biosciences, USA, cat# 365968), centrifuged at 5000 rpm for 1.5 min to obtain serum and stored at -20 °C until use. Detection and quantification of LiTERNA and Albu-LiTCoRNA were determined by ELISA. Briefly, MaxiSorp plates were coated with hEGFR, m4-1BB, and mPD-L1 (3 µg/ml) in PBS overnight at 4°C. After washing and blocking with 5% BSA, duplicates of non-diluted sera were added at room temperature for 1 hour. Standards were 3-fold serial dilutions of purified recombinant LiTE or Albu-LiTCo antibodies. Plates were incubated for 1 hour at room temperature with HRP-conjugated anti-VHH Cocktail (1:2000) diluted in PBS + 0.01% Tween20 for detection. Plates were washed and developed using (TMB), stopped with 4N H2SO4, and measured at 450-570 nm using a Bio-Rad microplate-reader. Data was analyzed and plotted in GraphPad Prism 8.4.0.
Undiluted LiTE- and Albu-LiTCo-containing mouse serum collected at 4, 96, and 168 hours post-mRNA-LNP administration were added in triplicates to cocultures of mouse splenocytes (1.5 x 105 cells/well) with either EGFR-positive (CT26EGFRLuc) or EGFR-negative (CT26Luc) cells (0.3 x 105 cells/well) at an E/T ratio of 5:1. Translated protein levels peaked at approximately 100 ng/ml within 4 hours. LiTE stabilized at 40 ng/ml for up to 72 hours, while for Albu-LiTCo, remained at 30 ng/ml for up to 168 hours. Total splenocytes were isolated from 8-week-old female BALB/c mice and resuspended in RPMI supplemented with 10% FBS. After 48 hours of incubation, D-luciferin substrate (20 μg/well, Promega E1602) was added, and relative light units (RLU) were measured with the luminescence plate reader Infinite 1200 (Tecan Trading). Tumor cell viability was calculated by dividing the mean bioluminescence of each sample by the mean bioluminescence of the input number of control target cells, multiplied by 100. Specific lysis is the difference in tumor cell viability relative to control (0%). Wells with target and effector cells in the absence of antibodies were set as 100% viability.
CT26EGFR cells (1.5 x 106/mouse) were implanted subcutaneously into the dorsal space of 6-week-old female BALB/c mice. At day 4 animals were randomized into treatment groups (n=5). Tumor growth was monitored by caliper measurements three times a week, measurements were conducted randomly by a blinded investigator. Mice received an intravenous (i.v.) injection of PBS, LiTERNA formulated with 10 µg polymer/lipid, Albu-LiTCoRNA, or ComboRNA (LiTERNA and Albu-LiTCoRNA) through the tail vein every week for three weeks. On days 0 and 14 after treatment, mice were anesthetized and bled for serum collection, which was stored at -20°C until use. IFNγ levels in serum were analyzed by ELISA. Mice weights were measured twice a week to monitor toxicity. Mice were euthanized when tumor size reached a volume of 1.5 cm3, tumors ulcerated, or at signs of distress.
Tumors from different treatment groups were collected and fixed in 4% formalin solution (Sigma-Aldrich) for 48 hours. After extensive washing in PBS, tissues were embedded in paraffin (FFPE). 4μm-thick FFPE sections were processed on the Dako PT Link system for optimized staining consistency. Antigen retrieval was performed with EDTA pH9, and sections were incubated with CD8 FLEX (clone C8/144B, DAKO) or Perforin (clone 5B10, Abcam) antibodies on the Dako Autostainer Link 48 platform. Nuclei were counterstained with Harris’ hematoxylin. Positive control sections were included for each staining run. All slides were dehydrated, cleared, and mounted with a permanent mounting medium for microscopic evaluation. Whole digital slides were acquired with a Zeiss AxioScan Z1 slide scanner, and positive versus total cells were automatically quantified using QuPath v0.4.3 software (49).
Statistical analysis was performed using GraphPad Prism Software version 8.4.0. All in vitro experiments were done in triplicates, and values are presented as mean ± SD from one of at least three separate experiments. Significant differences (P value) were determined using a two-tailed, unpaired Student’s t-test assuming a normal distribution. P values are indicated in the corresponding figures. When no statistically significant differences were observed, the corresponding data were not displayed. IC50 and EC50 were calculated using a nonlinear regression curve (log agonist vs. normalized response-variable response). Tumor volumes for individual mice in each treatment group are plotted, and mean tumor volumes are presented for each group as mean ± SD using a scatter plot. Differences in tumor growth were determined by one-way ANOVA adjusted by the Tukey’s correction for multiple comparisons.
We generated two VHH - scFv tandem bispecific antibodies, LiTE and Albu-LiTCo. The LiTE (44) was obtained by fusing the anti-mouse/human EGFR VHH (EGa1) to the anti-mouse CD3ϵ scFv (2C11) through a flexible G4S linker (Figure 1A; Supplementary Figures 1A, B). The LiTCo was generated by fusing the anti-mouse/human PD-L1 VHH (Nb6P) to the anti-mouse 4-1BB scFv (1D8) (10, 22) through a G4S linker (Figure 1A; Supplementary Figures 1C, D). To improve its pharmacokinetic properties, the LiTCo was fused to the N-terminus of an albumin variant with enhanced affinity for human FcRn (Albu-LiTCo) (22, 50). Both antibodies were efficiently expressed in biologically active form by transfected human embryonic kidney HEK293 cells. Western blot analysis under reducing conditions of cell culture supernatants (SNs) showed bands with migration patterns consistent with their estimated molecular weights (Supplementary Figures 2A, C). To assess target specificity, ELISA binding assays were performed. SN of LiTE-lipofected HEK293 cells specifically bound to mouse and human EGFR (Supplementary Figure 2B), while Albu-LiTCo-lipofected SN bound to mouse and human PD-L1, as well as mouse 4-1BB (Supplementary Figures 2D, E). One-step chromatography yielded ∼90% pure LiTE and Albu-LiTCo with post-purification yields of approximately 0.5 mg/L HEK293 SN. SEC analysis revealed major monomeric peaks of 47 kDa (LiTE) and 110 kDa (Albu-LiTCo), consistent with their theoretical molecular weights (Supplementary Figures 3B, D). ELISA showed dose-dependent binding of purified LiTE to EGFR (Supplementary Figure 4A) and purified Albu-LiTCo to PD-L1 and 4-1BB (Supplementary Figures 4B, C). Their ability to specifically recognize the antigens in a cellular context was analyzed by flow cytometry. LiTE bound to mouse CD3 on the T cell line 2B4 (Supplementary Figure 5A) and to human EGFR-expressing mouse CT26 cells (CT26EGFR) (Supplementary Figure 5C), but not to wild-type CT26 cells or human Jurkat T cells. As for Albu-LiTCo, it bound specifically to genetically engineered HEK293 cells to express mouse 4-1BB on their cell surface (HEK2934-1BB) (Supplementary Figure 5B) and to CHO cells expressing human PD-L1 (CHOPD-L1) (Supplementary Figure 5D). No binding to wild-type HEK293 or CHO cells was detected.
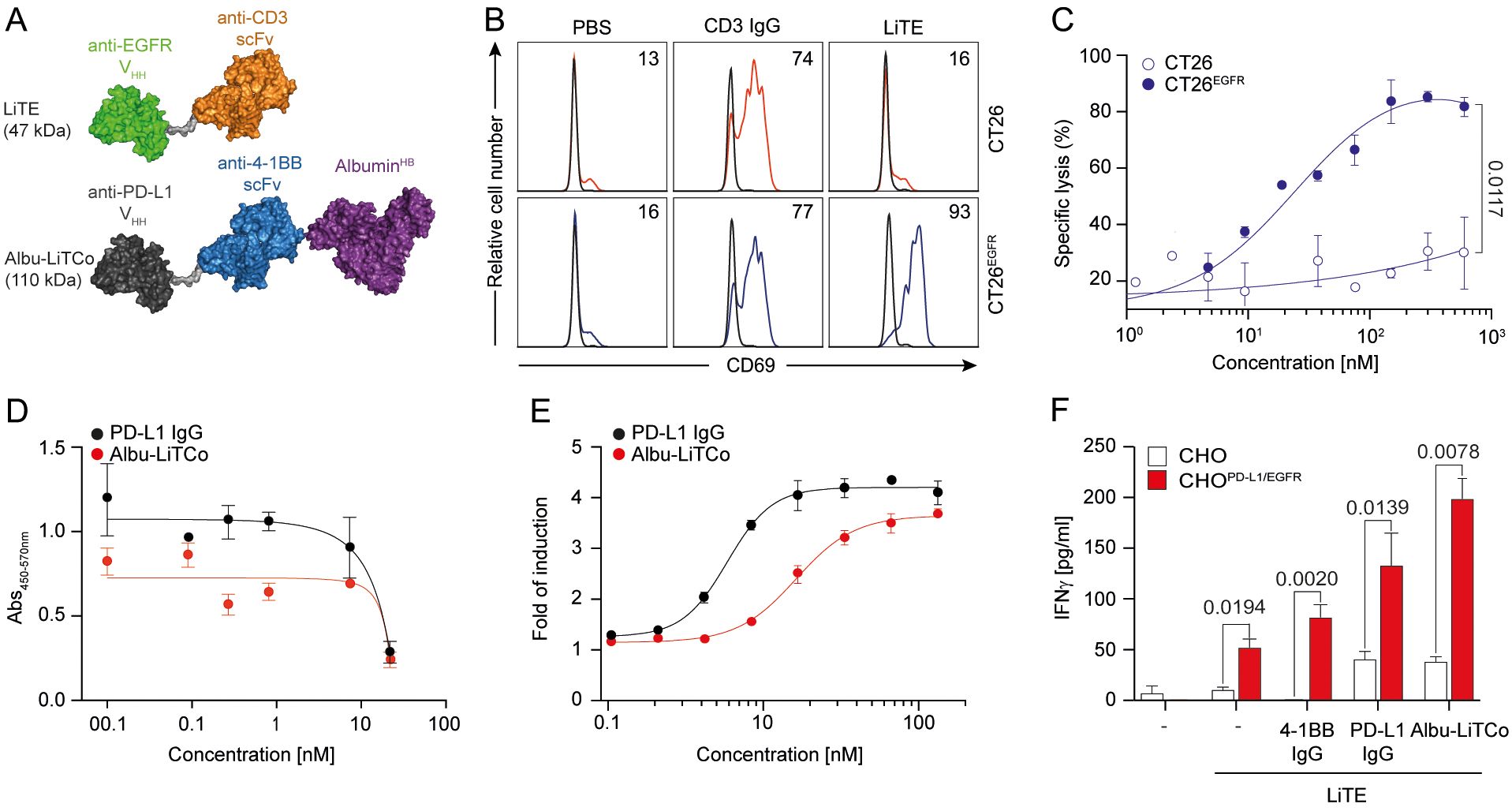
Figure 1. Functional characterization and mechanisms of action of LiTE and Albu-LiTCo antibodies. Schematic representation of EGFR-targeted LiTE and Albu-LiTCo antibodies (A). LiTE-mediated EGFR-dependent activation of tumor cells (B). CT26 and CT26EGFR were cocultured with mouse splenocytes (E:T ratio 5:1) in the presence of LiTE or anti-CD3 IgG at 3.34 nM equimolar concentration. Mouse CD69 expression was measured by flow cytometry after 24 hours (B). LiTE-mediated EGFR-dependent cytotoxicity of tumor cells (C). Luciferase-expressing CT26 and CT26EGFR cells were cocultured with splenocytes (E:T ratio 5:1) and different concentrations of LiTE. Specific lysis was measured by bioluminescence at 48 hours. Percent specific lysis was calculated relative to control. Data are mean ± SD (n = 3). Significance was calculated by unpaired Student’s t test. Competition ELISA for PD-L1/PD-1 interaction inhibition by anti-PD-L1 IgG or Albu-LiTCo (D). Data are mean ± SD (n = 3). One representative experiment from two independent experiments is shown. LogIC50 is shown. Blocking activity of Albu-LiTCo in a cell-based bioassay (E). JurkatPD-1 cells were cocultured with CHOPD-L1 cells and increasing concentrations of anti-PD-L1 IgG or Albu-LiTCo. Luminescence was measured after 6 hours. Data are expressed as fold induction relative to unstimulated JurkatPD-1 cells. Representative dose-concentration curves are shown as mean ± SD (n = 3). IC50 is shown. Agonistic activity of Albu-LiTCo (F). Wild type CHO cells or CHOPD-L1/EGFR were cocultured with LiTE-activated mouse CD8a+ T cells (3.34 nM) in the presence of Albu-LiTCo, anti-4-1BB IgG or anti-PD-L1 IgG at 6.67 nM equimolar dose. IFNγ secretion was determined after 72 h Negative controls (–) consisted of CD8+ T cells cultured with either CHO or CHOPD-L1/EGFR, and in the absence (- control in the left) or presence (- control in the right) of soluble LiTE. Data are mean ± SD (n = 3). One representative experiment of three independent experiments is shown. Significance was calculated by unpaired Student’s t test.
The functionality of purified LiTE was further assessed by analyzing T cell activation in cocultures of mouse splenocytes with CT26 or CT26EGFR cells at an effector-to-target (E:T) ratios of 5:1. CD69 was expressed after 24 hours of coculture with CT26EGFR cells, but not with wild-type CT26 cells, as determined by flow cytometry (Figure 1B). Purified LiTE directed T cells to lyse luciferase-expressing CT26EGFR tumor cells in a dose-dependent manner when in vitro cocultured with murine splenocytes at a E:T ratio of 5:1 [median effective concentration (EC50), 15.4 nM] (Figure 1C), indicating that EGFR cross-linking is required for LiTE-induced cytotoxicity. No appreciable lysis of EGFR-negative CT26 tumor cells was detected at any concentration. Cytokine release (IFNγ and IL-2) was only observed when splenocytes were cocultured with CT26EGFR cells (Supplementary Figure 6). Together, purified LiTE was highly monomeric and mediated robust T cell activation that was conditioned on both EGFR and mouse CD3 binding.
The blockade of the PD-L1/PD-1 pathway by Albu-LiTCo was evaluated by competition ELISA and compared to the bivalent anti-PD-L1 mAb atezolizumab. Albu-LiTCo effectively inhibited the PD-1/PD-L1 interaction with a half maximal inhibitory concentration (IC50) of 52.68 nM versus 10.20 nM for atezolizumab (Figure 1D). In cell-based assays using an NFAT-luciferase reporter Jurkat cell line expressing human PD-1, Albu-LiTCo efficiently blocked the PD-1/PD-L1 interaction, resulting in dose-dependent NFAT activation and light emission with an IC50 of 1.627 nM, slightly higher than that observed with atezolizumab (0.586 nM) (Figure 1E).
We studied the 4-1BB agonistic activity of Albu-LiTCo in co-cultures of isolated mouse CD8a+ T cells preactivated with LiTE at 0.175 μg/ml (3.34 nM) in the presence of wild-type CHO cells and CHO cells expressing human PD-L1 and human EGFR (CHOPD-L1/EGFR) (Figure 1F). Albu-LiTCo significantly increased IFNγ secretion of LiTE-activated T cells in co-culture with CHOPD-L1/EGFR cells (P=0.0078) (Figure 1F), but not in co-culture with wild-type CHO cells. LiTE alone also increased IFNγ secretion following EGFR-specific crosslinking but at lower levels (p=0.0194) (Figure 1F). Addition of anti-4-1BB or anti-PD-L1 IgG antibodies also increased IFNγ levels when T CD8a+ cells were co-cultured with CHOPD-L1/EGFR cells in the presence of LiTE (P=0.0020 and P=0.0139, respectively), but the effect was milder than that observed with Albu-LiTCo. IFNγ secretion was negligible in the absence of LiTE-mediated EGFR-specific crosslinking. Activation of CD8a+ T cells with soluble anti-mouse CD3 mAb (0.5 µg/ml) in the presence of CHOPD-L1 cells (Supplementary Figure 7A) or plastic-immobilized PD-L1 (Supplementary Figure 7B) also promoted significantly increased IFNγ levels (P = 0.0026 and P = 0.0067, respectively).
The mRNA-encoding bispecific antibodies contain a 5’ type I cap (5’ Cap: Cap1) and N1-methylpseudouridine modified nucleosides (N1-me-Ψ). The mRNA template includes a 5’ UTR, Kozak sequence, 3’ UTR and poly(A) tail, as well as the LiTE or Albu-LiTCo coding sequences. We first tested whether HEK293 cells transfected with mRNA expressed functional versions of both bispecific antibodies. Bands of the expected molecular weight were detected by Western blot in SN of mRNA-transfected HEK293 cells (Figure 2A). In addition, specific binding to hEGFR by LiTERNA (Figure 2B) and to mPD-L1 and m4-1BB by Albu-LiTCoRNA (Figures 2C, D) was demonstrated by ELISA. Correspondingly, flow cytometry analysis showed binding of LiTERNA to mouse CD3 on 2B4 T cells and to human EGFR on CT26EGFR cells (Figures 2E, G), as well as binding of Albu-LiTCoRNA to stably transfected cells expressing m4-1BB or hPD-L1 (Figures 2F, H). No binding was observed on untransfected cells. LiTERNA (3.3 µg/ml) elicited a strong cytotoxic response in cocultures with murine splenocytes and CT26EGFR cells at an E:T ratio of 5:1 (P=0.019) (Figure 3A), with an efficacy comparable to that of the corresponding recombinant purified LiTE at the highest concentration used (Figure 1C). In addition, CD8a+ T cells preactivated with LiTERNA (3.3 µg/ml) (Figure 3B) or purified LiTE (3.34 nM) (Figure 3C) in the presence of purified Albu-LiTCo (6.67 nM) or Albu-LiTCoRNA (3.9 µg/ml), respectively, showed a significant increase in IFNγ levels when cocultured with CHOPDL1/EGFR cells but not with CHO cells (Figures 3B, C).
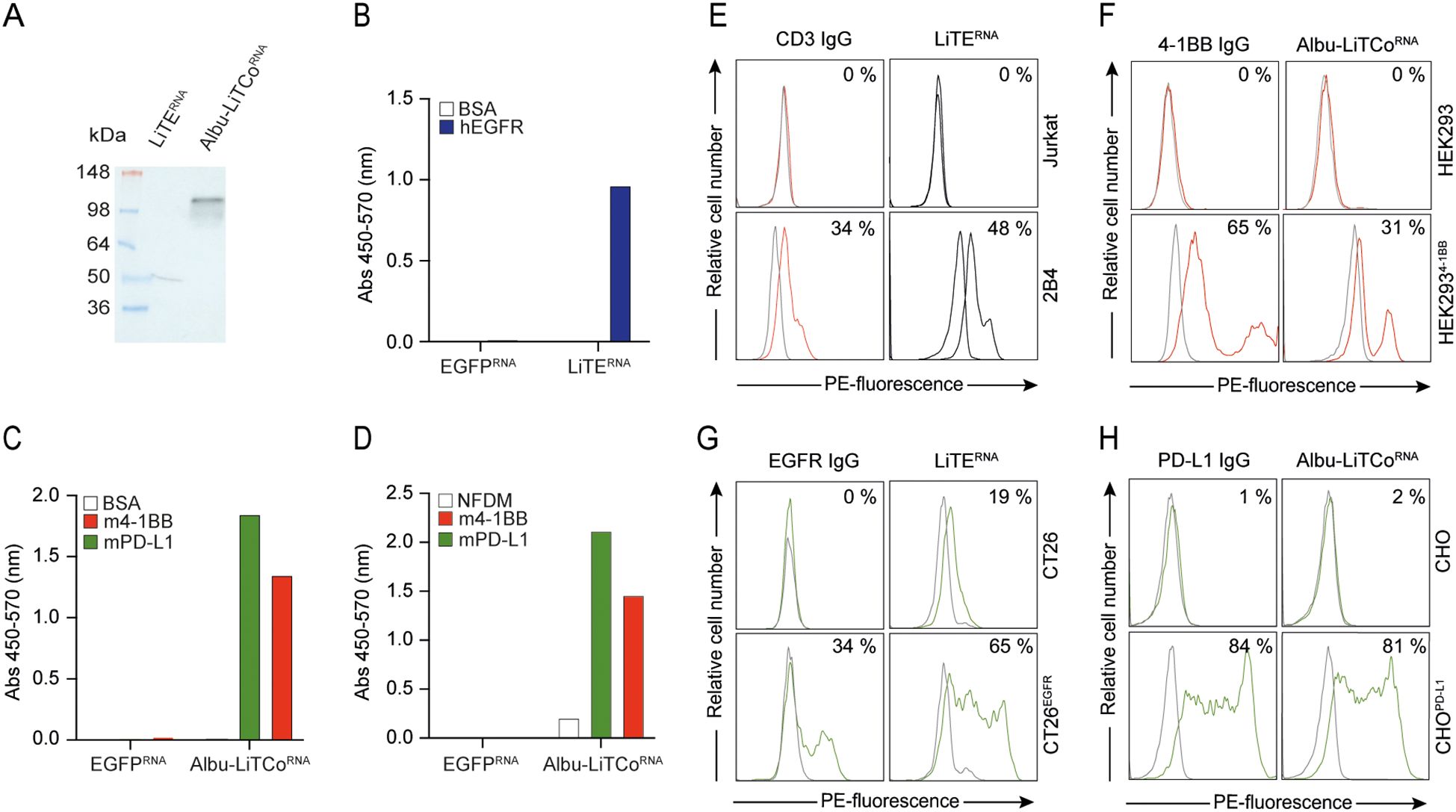
Figure 2. Functional characterization of LiTERNA and Albu-LiTCoRNA. Western blot detection using a HRP-conjugated anti-VHH mAb in the conditioned media of LiTERNA and Albu-LiTCoRNA transfected HEK293 cells. Molecular mass (kDa) is indicated (A). Specific binding of LiTERNA (B) and Albu-LiTCoRNA (C, D) to plastic immobilized specific antigens (hEGFR and m4-1BB, mPD-L1, respectively) demonstrated by ELISA using an HRP-conjugated anti-VHH mAb cocktail (B, C) or HRP-conjugated anti-human serum albumin (HRP-HSA) mAb (D). Data are represented as a mean ± SD (n = 3). BSA, bovine serum albumin; NFDM, non-fat dry milk. Binding of LiTERNA and Albu-LiTCoRNA to cell surface expressed antigens mCD3 (E), m4-1BB (F), hEGFR (G) and hPDL-1 (H) by flow cytometry. The y-axis shows the relative cell number, and the x-axis represents the R-phycoerythrin (PE)-fluorescence, expressed on a linear scale. The anti-CD3 IgG, anti-4-1BB IgG, anti-EGFR IgG and anti-PD-L1 IgG were used as controls. One representative experiment out of two independent experiments is shown. The number indicates the percentage of positive cells (%).
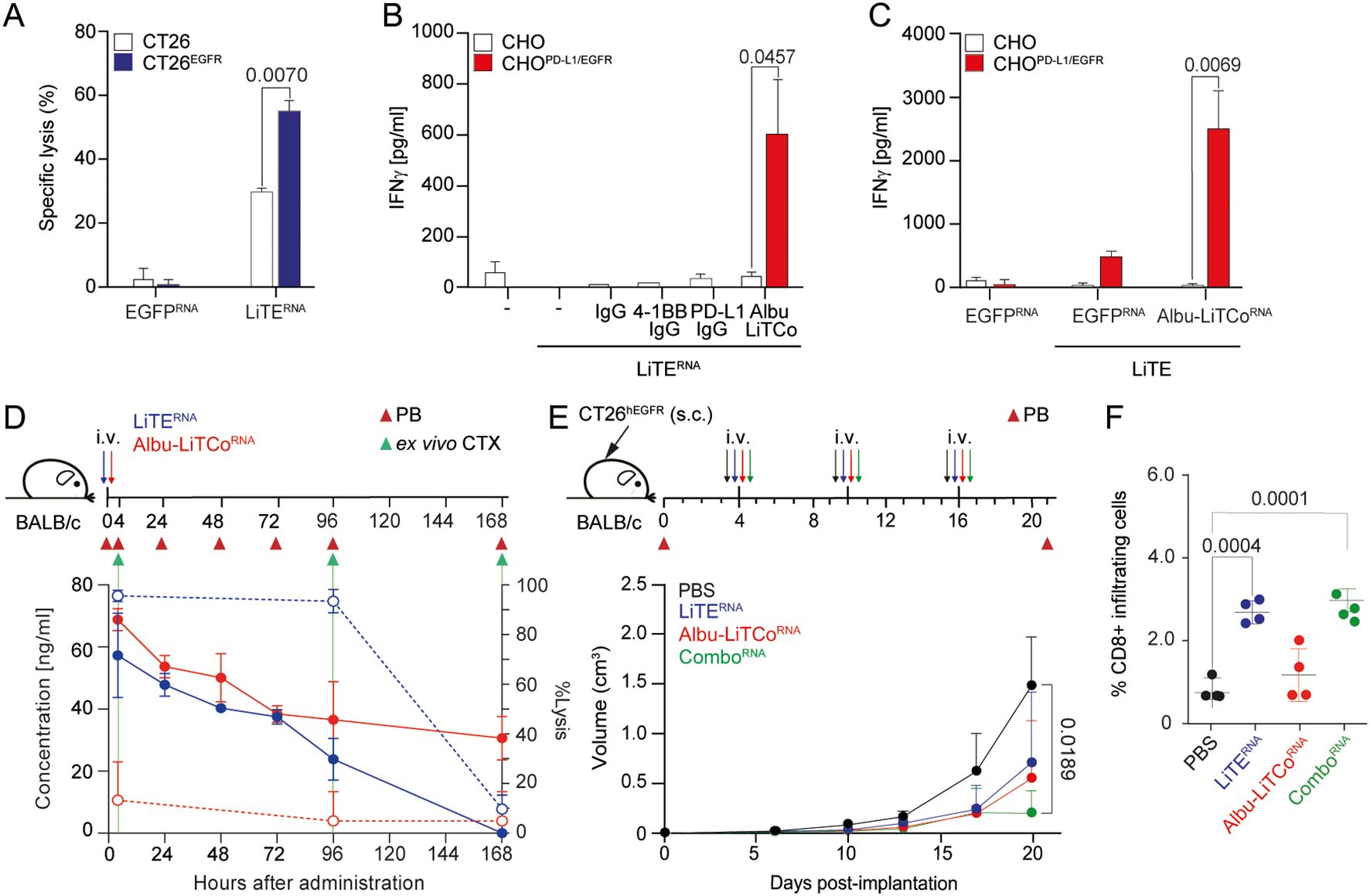
Figure 3. Effects of LiTERNA and Albu-LiTCoRNA on tumor cell cytotoxicity, agonistic costimulatory activity and tumor growth inhibition. EGFR-dependent cytotoxicity by LiTERNA (A). Luciferase-expressing CT26 and CT26EGFR cells were co-cultured with mouse splenocytes at an E:T ratio of 5:1. Specific tumor cell lysis was measured by bioluminescence. The percentage of specific lysis was calculated relative to the same number of tumor cells cultured with splenocytes. Data are expressed as mean ± SD (n = 3). Significance was calculated by unpaired Student’s t test. CHO or CHOPD-L1/EGFR cells were plated with mouse CD8a+ T cells activated with LiTERNA in the presence of Albu-LiTCo (B) or with LiTE in the presence of Albu-LiTCoRNA (C). IFNγ secretion was determined after 72 hours. Data are expressed as mean ± SD (n = 3). Significance was calculated by unpaired Student’s t test. Anti-4-1BB IgG and anti-PD-L1 IgG (B) or EGFPRNA (C) were used as controls. Pharmacokinetic profile of LiTERNA (solid blue line) and Albu-LiTCoRNA (solid red line) expressed as ng/mL (left Y-axis) after a single intravenous (i.v.) administration of mRNA-LNP in BALB/c mice (D). Ex vivo specific tumor lysis of CT26EGFR cells mediated by LiTE-containing mouse serum (dashed blue line) and Albu-LiTCo-containing mouse serum (dashed red line) expressed as % of Lysis (right Y-axis) (D). Target cells modified for the expression of luciferase were co-cultured with splenocytes at the effector/target (E/T) ratio of 5:1 in the presence of mouse serum obtained at 4, 96 and 168 hours post-mRNA-LNP administration. After 48 hours, the percentage of specific tumor lysis was measured by bioluminescence (right Y-axis). Data are presented as mean ± SD (n = 3). BALB/c mice were subcutaneously (s.c.) inoculated with CT26EGFR cells, randomized into n=5/group with similar mean tumor sizes and SDs and treated with 3 i.v. injections of LiTERNA, Albu-LiTCoRNA (10 µg/mouse) as monotherapy or combined (ComboRNA) (E). Average tumor volume growth of mice in each group is shown. Data are presented as mean ± SD. Significance was determined by one-way ANOVA with Tukey’s correction test for multiple comparisons. Quantitative analysis of intratumoral CD8+ T cells in mouse tumor tissue (n = 4/group) (F). Data were calculated as percentage of CD8+ versus total cell number and presented as mean ± SD. Significance was determined by one-way ANOVA with Tukey’s test correction for multiple comparisons.
LiTERNA and Albu-LiTCoRNA were formulated with a polymer/lipid-based nanoparticle (LNP) to ensure efficient translation in the liver following intravenous (i.v.) administration (40). Following a single dose of 10 µg of mRNA-LNP, translated proteins peaked within 4 hours (approximately 100 ng/ml). Protein levels remained at 40 ng/ml for 72 hours in the case of LiTERNA and 30 ng/ml for up to 168 hours in the case of Albu-LiTCoRNA (Figure 3D). As previously reported, the engineered human albuminHB fused to the LiTCo molecule significantly enhanced its PK properties (22). In addition, in vivo production of the bispecific antibodies promoted significantly longer circulating half-lives (LiTE; terminal t1/2, 18.9 ± 0.018 hours; Albu-LiTCo; terminal t1/2, 49.62 ± 0.050 hours) compared to purified protein (Supplementary Figure 8) (22). Purified LiTE exhibited rapid clearance from the circulation, with a terminal half-life of just 0.59 ± 0.001 hours, whereas the Albu-LiTCo demonstrated a significantly extended circulatory half-life of 22.15 ± 0.017 hours (Supplementary Figure 8). To confirm the cytotoxicity potential of in vivo produced LiTE, serum from mice that had received 10 µg of mRNA-LNP LiTERNA or Albu-LiTCoRNA was added to cocultures of mouse splenocytes with either EGFR-positive (CT26EGFR) or EGFR-negative (CT26) cells at an E:T ratio of 5:1. LiTE-containing mouse serum collected at 4 hours and 4 days after LiTERNA-LNP i.v. administration mediated EGFR-specific cytotoxicity of CT26EGFR tumor cells ex vivo, indicating full functionality of the antibody produced in vivo (dashed blue line) (Figure 3D). A correlation between serum LiTE levels and specific cytotoxic activity was observed. No significant lysis of EGFR-expressing cells was detected in cocultures with mouse serum collected on day 7 (Figure 3D). No cytotoxicity was observed in cocultures with Albu-LiTCo mouse serum at any time point (dashed red line) (Figure 3D). Lysis of EGFR-negative CT26 tumor cells was negligible in the presence of serum containing either LiTE- or Albu-LiTCo (Supplementary Figure 9).
After demonstrating that pharmacologically active levels of both bispecific antibodies could be achieved by i.v. mRNA-LNP administration, we investigated their antitumor activity in an immunocompetent mouse model subcutaneously xenografted with CT26EGFR colorectal tumors (10, 22), which constitutively expresses low levels of PD-L1 that increase in response to soluble IFNγ (Supplementary Figure 10). Mice were treated once a week with i.v. injection of 10 μg LiTE mRNA-LNP (LiTERNA), Albu-LiTCo mRNA-LNP (Albu-LiTCoRNA), the combination (ComboRNA) or PBS for 3 weeks. Both LiTERNA and Albu-LiTCoRNA, when administered as monotherapy, resulted in a delayed tumor growth that was not statistically significant. However, the therapeutic effect of ComboRNA was superior to both monotherapies, leading to a significant reduction in tumor growth (P = 0.0189) (Figure 3E). In addition, the ComboRNA achieved at least one complete regression out of five mice bearing CT26EGFR tumors (Supplementary Figure 11D). Neither treatment resulted in the development of splenomegaly or hepatomegaly (Supplementary Figures 12A, B), and IFNγ levels evaluated 96 hours after the second injection were comparable to those found in PBS-treated mice (Supplementary Figure 12C). To quantify lymphocyte infiltration immunohistochemical (IHC) staining of mouse CD8 was performed on tumor xenografts. CD8+ T cells increased significantly in tumors treated with LiTERNA and ComboRNA (P = 0.004 and P = 0.001, respectively) (Figure 3F; Supplementary Figure 12D).
In this study, we combine for the first time two mRNA-encoded bispecific antibodies with complementary mechanisms of action and demonstrate their potential to induce effective antitumor responses with a favorable toxicity profile. We designed and characterized two VHH-scFv tandem bispecific antibodies. The anti-EGFR x anti-CD3 LiTE specifically activates and redirects polyclonal mouse T cells to kill EGFR-positive cancer cells, while the Albu-LiTCo combines PD-L1/PD-1 axis blockade with PD-L1-dependent 4-1BB costimulation and enhances EGFR-specific LiTE T cell-mediated activation in vitro.
A disadvantage of Fc-free, fragment-based bispecific antibodies administered as purified proteins is their short half-life and the need for frequent dosing or continuous infusion to achieve sustained therapeutic concentrations, which severely limit their clinical potential (51, 52). In this context, mRNA-based delivery of therapeutic antibodies may be a promising approach (53–55) as it allows in vivo expression, avoiding the complex manufacturing process of proteins and potentially increasing serum half-life, and achieves remarkable therapeutic effects (40, 56–58). Here, we implemented a polymer/lipid-based formulation for the systemic delivery of mRNA-encoded bispecific antibodies (40). In vitro-translated LiTERNA and Albu-LiTCoRNA were shown to have comparable biological functionalities to purified proteins, and after i.v. administration of mRNA-LNP, pharmacokinetic analysis showed that this approach resulted in sustained in vivo production of both bispecific antibodies. Given the favorable toxicity profile of Albu-LiTCo, we employed a HLE strategy to prolong its circulation time, thereby increasing the probability of reaching the tumor and loco-regional lymph nodes, to block the PD-1/PD-L1 axis and provide PD-L1-specific 4-1BB costimulation to induce the proliferation and survival of antigen primed tumor-reactive T cells. Albu-LiTCo was fused to albuminHB, a human variant sequence with high avidity for FcRn (22, 37), with a half-life of 49.62 hours after mRNA-LNP administration. For inducing polyclonal T cell engagement, we designed an albumin-free LiTE molecule with a half-life of 0.59 hours when administered as a purified protein and of 18.9 hours after mRNA-LNP delivery, ensuring a shorter systemic exposure cycle compared to Albu-LiTCo and minimizing on-target off-tumor toxicity. In fact, although the anti-EGFR VHH recognizes mouse EGFR, no significant adverse effects were observed during treatment for 3 weeks with LiTE as mRNA monotherapy or combo with Albu-LiTCo. The use of Albu-LiTCoRNA as monotherapy results in limited antitumor responses in vivo. PD-L1 has been shown to be upregulated on tumor, stromal and immune cells upon IFNγ release (59–61). TCE-mediated T cell activation could lead to an increase in PD-1 expression, thereby reducing treatment efficacy (35, 61, 62). Co-administration of TCE with ICIs has shown improved tumor control and synergistic effects in numerous preclinical studies (17, 62–65). Consistent with this, our study demonstrated that ComboRNA treatment induced a significantly enhanced antitumor response even at low serum levels of both bispecific antibodies (in the ng/ml range). Furthermore, our results showed that LiTERNA was able to increase intratumoral CD8+ T cell infiltration both as monotherapy and in combination, but the co-administration of Albu-LiTCoRNA effectively improved T cell functionality. In addition, the ComboRNA demonstrated effective tumor suppression without associated toxic effects (30, 66, 67).
In conclusion, our results suggest that combined in vivo delivery of two mRNAs encoding an EGFR-specific TCE and a PD-L1-dependent 4-1BB agonist is a safe and feasible strategy to induce antitumor responses. The tumor model used was designed to evaluate the biological activity and potential efficacy of the mRNA-based strategy in a controlled setting, rather than to replicate the clinical context of advanced tumors. Future studies will focus on optimizing the dosing regimen and evaluating the long-term efficacy of the treatment.
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
All animal procedures were performed in accordance with European Union Directive 86/609/EEC and Recommendation 2007/526/EC, enforced in Spanish law under RD 1201/2005. Animal protocols were performed in strict adherence to the guidelines stated in the International Guiding Principles for Biomedical Research Involving Animals, established by the Council for International Organizations of Medical Sciences (CIOMS) and were approved by the Ethics Committee of Animal Experimentation of the Instituto Investigación Sanitaria Puerta de Hierro-Segovia de Arana (Hospital Universitario Puerta de Hierro Majadahonda, Madrid, Spain). Procedures were also approved by the Animal Welfare Division of the Environmental Affairs Council of the Government of Madrid (076/19). The study was conducted in accordance with the local legislation and institutional requirements.
OH: Formal analysis, Investigation, Methodology, Writing – original draft, Writing – review & editing. RN: Formal analysis, Investigation, Methodology, Writing – original draft, Writing – review & editing. SF: Formal analysis, Methodology, Writing – review & editing. LR-P: Investigation, Methodology, Writing – review & editing. AT-G: Investigation, Methodology, Writing – review & editing. LD-A: Investigation, Methodology, Writing – review & editing. MG-R: Investigation, Methodology, Writing – review & editing. NS-P: Methodology, Writing – review & editing. LV: Methodology, Writing – review & editing. CS: Funding acquisition, Writing – review & editing. KH: Writing – review & editing. LS: Funding acquisition, Writing – original draft, Writing – review & editing. MC: Conceptualization, Formal analysis, Funding acquisition, Investigation, Methodology, Writing – original draft, Writing – review & editing. LÁ-V: Conceptualization, Funding acquisition, Writing – original draft, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. CS reports grants from the Instituto de Salud Carlos III (ISCIII) (PI23/00565) and co-funded by the European Union, and from Gobierno de Navarra, Departamento de Salud (GN21-2022) and Fundación Intheos. LS reports grants from ICSIII (PI19/00132, PI22/00085). MC reports grants from Centro para el Desarrollo Tecnológico y la Innovación (CDTI) (IDI-20210744 and ABS-20220276), Ministerio de Ciencia e Innovación MCIN/AEI/10.13039/501100011033/(CPP2022-009762 and CPP2022-009765), and Gobierno de Navarra (PEN-0011-1411-2023-000002). LA-V reports grants from MCIN/AEI/10.13039/501100011033 (PID2023-148429OB-I00, PID2020-117323RB-100, PDC2021-121711-100, CPP2022-009762 and CPP2022-009765), the ISCIII (DTS20/00089), the Comunidad de Madrid (S2022/BMD-7225 - NEXT-GEN-CART-MAD-CM), Fundación CRIS contra el Cáncer (FCRIS-2021-0090 and FCRIS-2023-0070), Asociación Española contra el Cáncer (AECC) (PROYE19084ALVA and PRYGN234844ALVA), Fundación ‘‘La Caixa’’ (HR21-00761-IL7R-LungCan) and Fundación FERO (BBASELGAFERO2024-01). OH was supported by an industrial PhD fellowship from the Comunidad de Madrid (IND2020/BMD-17668). RN was supported by a Torres-Quevedo grant from the MCIN/AEI/10.13039/501100011033 (PTQ- 2019-010541). LR-P was supported by a predoctoral fellowship from the Chair of Immunology, Universidad Francisco de Vitoria/Merck. LD-A was supported by a Rio Hortega grant from the ISCIII (CM20/00004). MG-R was supported by an industrial PhD fellowship from the Comunidad de Madrid (IND2022/BMD-23732).
OH, RN and MC are all employees of Leadartis. SF formerly worked at Leadartis. LA-V and LS are co-founders of Leadartis.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2024.1494206/full#supplementary-material
1. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. (2012) 12:252–64. doi: 10.1038/nrc3239
2. Sprooten J, De Wijngaert P, Vanmeerbeerk I, Martin S, Vangheluwe P, Schlenner S, et al. Necroptosis in immuno-oncology and cancer immunotherapy. Cells. (2020) 9:1823. doi: 10.3390/cells9081823
3. Warmuth S, Gunde T, Snell D, Brock M, Weinert C, Simonin A, et al. Engineering of a trispecific tumor-targeted immunotherapy incorporating 4-1BB co-stimulation and PD-L1 blockade. Oncoimmunology. (2021) 10:2004661. doi: 10.1080/2162402X.2021.2004661
4. Qiao Y, Qiu Y, Ding J, Luo N, Wang H, Ling X, et al. Cancer immune therapy with PD-1-dependent CD137 co-stimulation provides localized tumour killing without systemic toxicity. Nat Commun. (2021) 12:6360. doi: 10.1038/s41467-021-26645-6
5. Geuijen C, Tacken P, Wang L-C, Klooster R, van Loo PF, Zhou J, et al. A human CD137×PD-L1 bispecific antibody promotes anti-tumor immunity via context-dependent T cell costimulation and checkpoint blockade. Nat Commun. (2021) 12:4445. doi: 10.1038/s41467-021-24767-5
6. Melero I, Hervas-Stubbs S, Glennie M, Pardoll DM, Chen L. Immunostimulatory monoclonal antibodies for cancer therapy. Nat Rev Cancer. (2007) 7:95–106. doi: 10.1038/nrc2051
7. Chester C, Sanmamed MF, Wang J, Melero I. Immunotherapy targeting 4-1BB: mechanistic rationale, clinical results, and future strategies. Blood. (2018) 131:49–57. doi: 10.1182/blood-2017-06-741041
8. Melero I, Sanmamed MF, Glez-Vaz J, Luri-Rey C, Wang J, Chen L. CD137 (4-1BB)-based cancer immunotherapy on its 25th anniversary. Cancer Discovery. (2023) 13:552–69. doi: 10.1158/2159-8290.CD-22-1029
9. Melero I, Shuford WW, Newby SA, Aruffo A, Ledbetter JA, Hellström KE, et al. Monoclonal antibodies against the 4-1BB T-cell activation molecule eradicate established tumors. Nat Med. (1997) 3:682–5. doi: 10.1038/nm0697-682
10. Compte M, Harwood SL, Muñoz IG, Navarro R, Zonca M, Perez-Chacon G, et al. A tumor-targeted trimeric 4-1BB-agonistic antibody induces potent anti-tumor immunity without systemic toxicity. Nat Commun. (2018) 9:4809. doi: 10.1038/s41467-018-07195-w
11. Li F, Ravetch JV. Antitumor activities of agonistic anti-TNFR antibodies require differential FcγRIIB coengagement in vivo. Proc Natl Acad Sci U.S.A. (2013) 110:19501–6. doi: 10.1073/pnas.1319502110
12. Xu Y, Szalai AJ, Zhou T, Zinn KR, Chaudhuri TR, Li X, et al. Fc gamma Rs modulate cytotoxicity of anti-Fas antibodies: implications for agonistic antibody-based therapeutics. J Immunol. (2003) 171:562–8. doi: 10.4049/jimmunol.171.2.562
13. Segal NH, Logan TF, Hodi FS, McDermott D, Melero I, Hamid O, et al. Results from an integrated safety analysis of urelumab, an agonist anti-CD137 monoclonal antibody. Clin Cancer Res. (2017) 23:1929–36. doi: 10.1158/1078-0432.CCR-16-1272
14. Segal NH, He AR, Doi T, Levy R, Bhatia S, Pishvaian MJ, et al. Phase I study of single-agent utomilumab (PF-05082566), a 4-1BB/CD137 agonist, in patients with advanced cancer. Clin Cancer Res. (2018) 24:1816–23. doi: 10.1158/1078-0432.CCR-17-1922
15. Qi X, Li F, Wu Y, Cheng C, Han P, Wang J, et al. Optimization of 4-1BB antibody for cancer immunotherapy by balancing agonistic strength with FcγR affinity. Nat Commun. (2019) 10:2141. doi: 10.1038/s41467-019-10088-1
16. Xu Y, Szalai AJ, Zhou T, Zinn KR, Chaudhuri TR, Li X, et al. FcγRs modulate cytotoxicity of anti-fas antibodies: implications for agonistic antibody-based therapeutics. J Immunol. (2003) 171:562–8. doi: 10.4049/jimmunol.171.2.562
17. Claus C, Ferrara C, Xu W, Sam J, Lang S, Uhlenbrock F, et al. Tumor-targeted 4-1BB agonists for combination with T cell bispecific antibodies as off-the-shelf therapy. Sci Transl Med. (2019) 11:eaav5989. doi: 10.1126/scitranslmed.aav5989
18. Hinner MJ, Aiba RSB, Jaquin TJ, Berger S, Dürr MC, Schlosser C, et al. Tumor-localized costimulatory T-cell engagement by the 4-1BB/HER2 bispecific antibody-anticalin fusion PRS-343. Clin Cancer Res. (2019) 25:5878–89. doi: 10.1158/1078-0432.CCR-18-3654
19. Kamata-Sakurai M, Narita Y, Hori Y, Nemoto T, Uchikawa R, Honda M, et al. Antibody to CD137 activated by extracellular adenosine triphosphate is tumor selective and broadly effective in vivo without systemic immune activation. Cancer Discovery. (2021) 11:158–75. doi: 10.1158/2159-8290.CD-20-0328
20. Compte M, Harwood SL, Erce-Llamazares A, Tapia-Galisteo A, Romero E, Ferrer I, et al. An fc-free EGFR-specific 4-1BB-agonistic trimerbody displays broad antitumor activity in humanized murine cancer models without toxicity. Clin Cancer Res. (2021) 27:3167–77. doi: 10.1158/1078-0432.CCR-20-4625
21. Navarro R, Frago S, Hangiu O, Erce-Llamazares A, Lázaro-Gorines R, Morcillo MA, et al. Pharmacokinetics and safety of LEAD-452, an EGFR-specific 4-1BB-agonistic trimerbody in non-human primates. Toxicol Appl Pharmacol. (2024) 487:116961. doi: 10.1016/j.taap.2024.116961
22. Hangiu O, Compte M, Dinesen A, Navarro R, Tapia-Galisteo A, Mandrup OA, et al. Tumor targeted 4-1BB agonist antibody-albumin fusions with high affinity to FcRn induce anti-tumor immunity without toxicity. iScience. (2022) 25:104958. doi: 10.1016/j.isci.2022.104958
23. Qu QX, Zhu XY, Du WW, Wang HB, Shen Y, Zhu YB, et al. 4-1BB agonism combined with PD-L1 blockade increases the number of tissue-resident CD8+ T cells and facilitates tumor abrogation. Front Immunol. (2020) 11:577. doi: 10.3389/fimmu.2020.00577
24. Jeong S, Park E, Kim H-D, Sung E, Kim H, Jeon J, et al. Novel anti-4-1BB×PD-L1 bispecific antibody augments anti-tumor immunity through tumor-directed T-cell activation and checkpoint blockade. J Immunother Cancer. (2021) 9:e00242. doi: 10.1136/jitc-2021-002428
25. Muik A, Garralda E, Altintas I, Gieseke F, Geva R, Ben-Ami E, et al. Preclinical characterization and phase I trial results of a bispecific antibody targeting PD-L1 and 4-1BB (GEN1046) in patients with advanced refractory solid tumors. Cancer Discovery. (2022) 12:1248–65. doi: 10.1158/2159-8290.CD-21-1345
26. Goebeler M-E, Bargou RC. T cell-engaging therapies - BiTEs and beyond. Nat Rev Clin Oncol. (2020) 17:418–34. doi: 10.1038/s41571-020-0347-5
27. Wang P, Sun LL, Clark R, Hristopoulos M, Chiu CPC, Dillon M, et al. Novel anti-LY6G6D/CD3 T-cell-dependent bispecific antibody for the treatment of colorectal cancer. Mol Cancer Ther. (2022) 21:974–85. doi: 10.1158/1535-7163.MCT-21-0599
28. Iizuka A, Nonomura C, Ashizawa T, Kondou R, Ohshima K, Sugino T, et al. A T-cell-engaging B7-H4/CD3-bispecific fab-scFv antibody targets human breast cancer. Clin Cancer Res. (2019) 25:2925–34. doi: 10.1158/1078-0432.CCR-17-3123
29. Dang K, Castello G, Clarke SC, Li Y, Balasubramani A, Boudreau A, et al. Attenuating CD3 affinity in a PSMAxCD3 bispecific antibody enables killing of prostate tumor cells with reduced cytokine release. J Immunother Cancer. (2021) 9:e002488. doi: 10.1136/jitc-2021-002488
30. Ellerman D. Bispecific T-cell engagers: Towards understanding variables influencing the in vitro potency and tumor selectivity and their modulation to enhance their efficacy and safety. Methods. (2019) 154:102–17. doi: 10.1016/j.ymeth.2018.10.026
31. Brinkmann U, Kontermann RE. The making of bispecific antibodies. MAbs. (2017) 9:182–212. doi: 10.1080/19420862.2016.1268307
32. Blanco B, Compte M, Lykkemark S, Sanz L, Alvarez-Vallina L. T cell-redirecting strategies to “STAb” Tumors: beyond CARs and bispecific antibodies. Trends Immunol. (2019) 40:243–57. doi: 10.1016/j.it.2019.01.008
33. Przepiorka D, Ko C-W, Deisseroth A, Yancey CL, Candau-Chacon R, Chiu H-J, et al. FDA approval: blinatumomab. Clin Cancer Res. (2015) 21:4035–9. doi: 10.1158/1078-0432.CCR-15-0612
34. Cech P, Skórka K, Dziki L, Giannopoulos K. T-cell engagers-the structure and functional principle and application in hematological Malignancies. Cancers (Basel). (2024) 16:1580. doi: 10.3390/cancers16081580
35. Middelburg J, Kemper K, Engelberts P, Labrijn AF, Schuurman J, van Hall T. Overcoming challenges for CD3-bispecific antibody therapy in solid tumors. Cancers (Basel). (2021) 13:287. doi: 10.3390/cancers13020287
36. Paz-Ares L, Champiat S, Lai WV, Izumi H, Govindan R, Boyer M, et al. Tarlatamab, a first-in-class DLL3-targeted bispecific T-cell engager, in recurrent small-cell lung cancer: an open-label, phase I study. J Clin Oncol. (2023) 41:2893–903. doi: 10.1200/JCO.22.02823
37. Mandrup OA, Ong SC, Lykkemark S, Dinesen A, Rudnik-Jansen I, Dagnæs-Hansen NF, et al. Programmable half-life and anti-tumour effects of bispecific T-cell engager-albumin fusions with tuned FcRn affinity. Commun Biol. (2021) 4:310. doi: 10.1038/s42003-021-01790-2
38. Sahin U, Karikó K, Türeci Ö. mRNA-based therapeutics–developing a new class of drugs. Nat Rev Drug Discovery. (2014) 13:759–80. doi: 10.1038/nrd4278
39. Van Hoecke L, Roose K. How mRNA therapeutics are entering the monoclonal antibody field. J Transl Med. (2019) 17:54. doi: 10.1186/s12967-019-1804-8
40. Stadler CR, Bähr-Mahmud H, Celik L, Hebich B, Roth AS, Roth RP, et al. Elimination of large tumors in mice by mRNA-encoded bispecific antibodies. Nat Med. (2017) 23:815–7. doi: 10.1038/nm.4356
41. Stadler CR, Ellinghaus U, Fischer L, Bähr-Mahmud H, Rao M, Lindemann C, et al. Preclinical efficacy and pharmacokinetics of an RNA-encoded T cell-engaging bispecific antibody targeting human claudin 6. Sci Transl Med. (2024) 16:eadl2720. doi: 10.1126/scitranslmed.adl2720
42. Pozzi C, Cuomo A, Spadoni I, Magni E, Silvola A, Conte A, et al. The EGFR-specific antibody cetuximab combined with chemotherapy triggers immunogenic cell death. Nat Med. (2016) 22:624–31. doi: 10.1038/nm.4078
43. Navarro R, Tapia-Galisteo A, Martín-García L, Tarín C, Corbacho C, Gómez-López G, et al. TGF-β-induced IGFBP-3 is a key paracrine factor from activated pericytes that promotes colorectal cancer cell migration and invasion. Mol Oncol. (2020) 14:2609–28. doi: 10.1002/1878-0261.12779
44. Mølgaard K, Harwood SL, Compte M, Merino N, Bonet J, Alvarez-Cienfuegos A, et al. Bispecific light T-cell engagers for gene-based immunotherapy of epidermal growth factor receptor (EGFR)-positive Malignancies. Cancer Immunol Immunother. (2018) 67:1251–60. doi: 10.1007/s00262-018-2181-5
45. Gilliland LK, Norris NA, Marquardt H, Tsu TT, Hayden MS, Neubauer MG, et al. Rapid and reliable cloning of antibody variable regions and generation of recombinant single chain antibody fragments. Tissue Antigens. (1996) 47:1–20. doi: 10.1111/j.1399-0039.1996.tb02509.x
46. Silva-Pilipich N, Blanco E, Lozano T, Martisova E, Igea A, Herrador-Cañete G, et al. Local delivery of optimized nanobodies targeting the PD-1/PD-L1 axis with a self-amplifying RNA viral vector induces potent antitumor responses. Cancer Lett. (2023) 561:216139. doi: 10.1016/j.canlet.2023.216139
47. Bern M, Nilsen J, Ferrarese M, Sand KMK, Gjølberg TT, Lode HE, et al. An engineered human albumin enhances half-life and transmucosal delivery when fused to protein-based biologics. Sci Transl Med. (2020) 12:287. doi: 10.1126/scitranslmed.abb0580
48. Pardi N, Muramatsu H, Weissman D, Karikó K. In vitro transcription of long RNA containing modified nucleosides. Methods Mol Biol. (2013) 969:29–42. doi: 10.1007/978-1-62703-260-5_2
49. Bankhead P, Loughrey MB, Fernández JA, Dombrowski Y, McArt DG, Dunne PD, et al. QuPath: Open source software for digital pathology image analysis. Sci Rep. (2017) 7:16878. doi: 10.1038/s41598-017-17204-5
50. Mandrup OA, Howard KA. Bioengineered solutions to improve cancer immunotherapies. Ther Delivery. (2021) 12:339–41. doi: 10.4155/tde-2021-0019
51. Li H, Er Saw P, Song E. Challenges and strategies for next-generation bispecific antibody-based antitumor therapeutics. Cell Mol Immunol. (2020) 17:451–61. doi: 10.1038/s41423-020-0417-8
52. Suurs FV, Lub-de Hooge MN, de Vries EGE, de Groot DJA. A review of bispecific antibodies and antibody constructs in oncology and clinical challenges. Pharmacol Ther. (2019) 201:103–19. doi: 10.1016/j.pharmthera.2019.04.006
53. Sanz L, Álvarez-Vallina L. Engineered mRNA and the rise of next-generation antibodies. Antibodies (Basel). (2021) 10:37. doi: 10.3390/antib10040037
54. Zhao Y, Gan L, Ke D, Chen Q, Fu Y. Mechanisms and research advances in mRNA antibody drug-mediated passive immunotherapy. J Transl Med. (2023) 21:693. doi: 10.1186/s12967-023-04553-1
55. Schlake T, Thess A, Thran M, Jordan I. 30334070. Cell Mol Life Sci. (2019) 76:301–28. doi: 10.1007/s00018-018-2935-4
56. Bähr-Mahmud H, Ellinghaus U, Stadler CR, Fischer L, Lindemann C, Chaturvedi A, et al. Preclinical characterization of an mRNA-encoded anti-Claudin 18.2 antibody. Oncoimmunology. (2023) 12:2255041. doi: 10.1080/2162402X.2023.2255041
57. Wu L, Wang W, Tian J, Qi C, Cai Z, Yan W, et al. Intravenous delivery of RNA encoding anti-PD-1 human monoclonal antibody for treating intestinal cancer. J Cancer. (2022) 13:579–88. doi: 10.7150/jca.63991
58. Rybakova Y, Kowalski PS, Huang Y, Gonzalez JT, Heartlein MW, DeRosa F, et al. mRNA delivery for therapeutic anti-HER2 antibody expression in vivo. Mol Ther. (2019) 27:1415–23. doi: 10.1016/j.ymthe.2019.05.012
59. Chen S, Crabill GA, Pritchard TS, McMiller TL, Wei P, Pardoll DM, et al. Mechanisms regulating PD-L1 expression on tumor and immune cells. J Immunother Cancer. (2019) 7:305. doi: 10.1186/s40425-019-0770-2
60. Bacac M, Fauti T, Sam J, Colombetti S, Weinzierl T, Ouaret D, et al. A novel carcinoembryonic antigen T-cell bispecific antibody (CEA TCB) for the treatment of solid tumors. Clin Cancer Res. (2016) 22:3286–97. doi: 10.1158/1078-0432.CCR-15-1696
61. Sam J, Colombetti S, Fauti T, Roller A, Biehl M, Fahrni L, et al. Combination of T-cell bispecific antibodies with PD-L1 checkpoint inhibition elicits superior anti-tumor activity. Front Oncol. (2020) 10:575737. doi: 10.3389/fonc.2020.575737
62. Kobold S, Pantelyushin S, Rataj F, Vom Berg J. Rationale for combining bispecific T cell activating antibodies with checkpoint blockade for cancer therapy. Front Oncol. (2018) 8:285. doi: 10.3389/fonc.2018.00285
63. Belmontes B, Sawant DV, Zhong W, Tan H, Kaul A, Aeffner F, et al. Immunotherapy combinations overcome resistance to bispecific T cell engager treatment in T cell-cold solid tumors. Sci Transl Med. (2021) 13:eabd1524. doi: 10.1126/scitranslmed.abd1524
64. Griessinger CM, Olafsen T, Mascioni A, Jiang ZK, Zamilpa C, Jia F, et al. The PET-tracer 89Zr-df-IAB22M2C enables monitoring of intratumoral CD8 T-cell infiltrates in tumor-bearing humanized mice after T-cell bispecific antibody treatment. Cancer Res. (2020) 80:2903–13. doi: 10.1158/0008-5472.CAN-19-3269
65. Skokos D, Waite JC, Haber L, Crawford A, Hermann A, Ullman E, et al. A class of costimulatory CD28-bispecific antibodies that enhance the antitumor activity of CD3-bispecific antibodies. Sci Transl Med. (2020) 12:eaaw7888. doi: 10.1126/scitranslmed.aaw7888
66. Bacac M, Klein C, Umana P. CEA TCB: A novel head-to-tail 2:1 T cell bispecific antibody for treatment of CEA-positive solid tumors. Oncoimmunology. (2016) 5:e1203498. doi: 10.1080/2162402X.2016.1203498
67. Harwood SL, Alvarez-Cienfuegos A, Nuñez-Prado N, Compte M, Hernández-Pérez S, Merino N, et al. ATTACK, a novel bispecific T cell-recruiting antibody with trivalent EGFR binding and monovalent CD3 binding for cancer immunotherapy. Oncoimmunology. (2017) 7:e1377874. doi: 10.1080/2162402X.2017.1377874
Keywords: T-cell engager, costimulatory antibody, cancer immunotherapy, mRNA encoded bispecific antibodies, combined RNA
Citation: Hangiu O, Navarro R, Frago S, Rubio-Pérez L, Tapia-Galisteo A, Díez-Alonso L, Gómez-Rosel M, Silva-Pilipich N, Vanrell L, Smerdou C, Howard KA, Sanz L, Álvarez-Vallina L and Compte M (2025) Effective cancer immunotherapy combining mRNA-encoded bispecific antibodies that induce polyclonal T cell engagement and PD-L1-dependent 4-1BB costimulation. Front. Immunol. 15:1494206. doi: 10.3389/fimmu.2024.1494206
Received: 10 September 2024; Accepted: 16 December 2024;
Published: 06 January 2025.
Edited by:
Jose A. Garcia-Sanz, Spanish National Research Council (CSIC), SpainReviewed by:
Brigitte Kerfelec, INSERM U1068 Centre de Recherche en Cancérologie de Marseille (CRCM), FranceCopyright © 2025 Hangiu, Navarro, Frago, Rubio-Pérez, Tapia-Galisteo, Díez-Alonso, Gómez-Rosel, Silva-Pilipich, Vanrell, Smerdou, Howard, Sanz, Álvarez-Vallina and Compte. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marta Compte, bWFydGEuY29tcHRlQGxlYWRhcnRpcy5jb20=; Luis Álvarez-Vallina, bGFsdmFyZXp2QGV4dC5jbmlvLmVz
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.