
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol., 15 January 2025
Sec. Cytokines and Soluble Mediators in Immunity
Volume 15 - 2024 | https://doi.org/10.3389/fimmu.2024.1493306
This article is part of the Research TopicUnraveling the Molecular Mechanisms of Cytokine Signaling in Regulating Inflammatory DiseasesView all 16 articles
 Audrey Cambon1
Audrey Cambon1 Christophe Guervilly2,3
Christophe Guervilly2,3 Clémence Delteil4Nicola Potere5Richard Bachelier1Edwige Tellier1Evelyne Abdili1,6Marine Leprince7Marco Giani5Ildo Polidoro8Valentina Albanese8Paolo Ferrante8Laurence Coffin9Michael Schiffrin9Laurent Arnaud6Romaric Lacroix1,6Sandrine Roque7Jean-Marie Forel2,3
Clémence Delteil4Nicola Potere5Richard Bachelier1Edwige Tellier1Evelyne Abdili1,6Marine Leprince7Marco Giani5Ildo Polidoro8Valentina Albanese8Paolo Ferrante8Laurence Coffin9Michael Schiffrin9Laurent Arnaud6Romaric Lacroix1,6Sandrine Roque7Jean-Marie Forel2,3 Sami Hraiech2,3
Sami Hraiech2,3 Laurent Daniel10
Laurent Daniel10 Laurent Papazian11Françoise Dignat-George1,6
Laurent Papazian11Françoise Dignat-George1,6 Gilles Kaplanski1,7*
Gilles Kaplanski1,7*Rationale: COVID-19-associated acute-respiratory distress syndrome (C-ARDS) results from a direct viral injury associated with host excessive innate immune response mainly affecting the lungs. However, cytokine profile in the lung compartment of C-ARDS patients has not been widely studied, nor compared to non-COVID related ARDS (NC-ARDS).
Objectives: To evaluate caspase-1 activation, IL-1 signature, and other inflammatory cytokine pathways associated with tissue damage using post-mortem lung tissues, bronchoalveolar lavage fluids (BALF), and serum across the spectrum of COVID-19 severity.
Methods: Histological features were described and activated-caspase-1 labeling was performed in 40 post-mortem biopsies. Inflammatory cytokines were quantified in BALF and serum from 19 steroid-treated-C-ARDSand compared to 19 NC-ARDS. Cytokine concentrations were also measured in serum from 128 COVID-19 patients at different severity stages.
Measurements and main results: Typical “diffuse alveolar damage” in lung biopsies were associated with activated caspase-1 expression and vascular lesions. Soluble Caspase-1p20, IL-1β, IL-1Ra, IL-6 and at lower level IFNγ and CXCL-10, were highly elevated in BALF from steroid-treated-C-ARDS as well as in NC-ARDS. IL-1β appeared concentrated in BALF, whereas circulating IL-6 and IL-1Ra concentrations were comparable to those in BALF and correlated with severity. TNFα, TNFR1 and CXCL8 however, were significantly higher in NC-ARDS compared to C-ARDS, treated by steroid.
Conclusions: In the lungs of C-ARDS, both caspase-1 activation with a predominant IL-1β/IL-6 signature and IFNγ -associated chemokines are elevated despite steroid treatment. These pathways may be specifically targeted in ARDS to improve response to treatment and to limit alveolar and vascular lung damage.
Caspase-1 activation and a predominant IL-1/IL-6 signature associated with IFNγ-induced chemokines remain highly detectable in the BALF of steroid-resistant COVID-19-ARDS, arguing for new multi-targeted therapeutics in COVID-19-ARDS. Lung biopsies from deceased COVID-19 patients show Diffuse Alveolar Damage and vasculopathy associated with activated-caspase-1. In the lungs of C-ARDS and NC-ARDS, a predominant caspase-1-induced IL-1β/IL-6 signature and IFNγ -induced chemokines persist.
Coronavirus-disease-2019 (COVID-19), caused by severe-acute-respiratory-syndrome-coronavirus-2 (SARS-CoV-2) is associated with dysregulation of host immune responses (1). In severe forms, COVID-19 causes bilateral pneumonia, that rapidly progresses to acute-respiratory-distress-syndrome (ARDS) and death (1). Immunothrombosis is one of the main complications arising in severe forms of COVID-19, due to the combined hyperactivation of the immune and coagulation systems (2–5). Multiple immune pathways have been shown to play a role in the immunopathogenesis of COVID-19. Type I interferons (IFN) promote the activation of antiviral effector mechanisms and induce the production of pro-inflammatory cytokines (6, 7). Conversely, the impaired type III-IFN response observed in severe cases of COVID-19 may promote innate immunity with hyperinflammation and immunothrombosis (2, 8–10). Increased circulating levels of pro-inflammatory cytokines have been detected in patients with severe COVID-19, as well as increased activation of nuclear-factor-kappa-light chain enhancer of activated B cells (NF-kB) (11). Inflammasomes markers such as the NACHT, LRR, pyrin domain (PYD) domains-containing protein 3 (NLRP3) and the absent-in-melanoma-2 (AIM2) are also activated during COVID-19 (12). Danger-associated molecular patterns (DAMPs) and pathogen-associated molecular patterns (PAMPS) released upon tissue injury are detected by NLRP3, a cytoplasmic protein which is in turn, activated into a macromolecular complex called NLRP3-inflammasome (13, 14). The activated NLRP3-inflammasome engages through the PYD domain of ASC (apoptosis-associated speck-like protein containing a caspase recruitment domain [CARD]) pro-caspase-1, which then undergoes autocatalytic activation to caspase-1. Activated caspase-1 (Casp1p20) is then responsible for transforming pro-IL-1β and pro-IL-18 into biologically active IL-1β and IL-18, two major pro-inflammatory and pro-coagulant cytokines of the IL-1 family. Casp1p20 also cleaves gasdermin-D, which forms cytoplasmic membrane pores and triggers pyroptosis (15). During COVID-19, strong activation of the NLRP3-inflammasome and caspase-1 is induced with subsequent release of IL-1β and IL-18, although these cytokines remain difficult to detect in patient’s blood (14, 16). Although controversial, caspase-1-induced pyroptotic cell death also appears to occur following SARS-CoV-2 infection of mononuclear cells in vitro (15, 17–19). Furthermore, in humanized mice expressing the human ACE2 receptor, SARS-CoV-2 infects macrophages by binding of anti-spike IgG to CD16, resulting in NLRP3-inflammasome activation, IL-1β and IL-18 production, and pyroptosis (19–21). Both pyroptosis and necrosis play an important role in hyperinflammatory syndromes, mainly through a significant release of DAMPs, notably IL-1α, which maintains an upward inflammatory loop (20, 22–24). IL-1α induces inflammasome activation and IL-1β production and, like IL-1β, is mediated through the IL-1 receptor. Despite their central role in amplifying the innate immune response, the role of DAMPs in COVID-19 is poorly understood to date. Moreover, the specific contribution of NLRP3 inflammasome activation and IL-1 signaling to COVID-19 immunopathogenesis remains unclear. In the present study of three prospective cohorts of COVID-19-patients, we investigated caspase-1 activation and IL-1 signature using post-mortem lung tissue, bronchoalveolar lavage fluid (BALF), and serum across the spectrum of COVID-19 severity. We also investigated the hypothetical role of other important inflammatory pathways and DAMPs in COVID-19 immunopathogenesis.
This prospective multicenter study was approved by the Medical Ethics Committee of Aix-Marseille-University (CPP # 1123 HPS1) and by the Assistance Publique de Marseille digital data protection delegate (RGPD2020-47). Patients were divided into three different cohorts (Supplementary Figure S1). All patients underwent a nasopharyngeal swab to confirm SARS-CoV-2 infection with RT-PCR.
Cohort A, consisted of post-mortem lung samples from 40 patients admitted to the public hospitals of Marseille (France) and “Santo Spirito” hospital in Pescara (Italy) with ARDS and SARS-CoV-2 infection who died of respiratory failure. For comparison, we used lung samples from 10 patients with ARDS and a negative RT-PCR SARS-CoV-2, who died from other causes than COVID-19: drowning, thoracic traumatism, epiglottitis, bladder perforation, aortic dissection, congenital cardiopathy and unexplained death.
Cohort B, consisted of 38 patients enrolled between July 1st, 2019 and April 23rd, 2021 in two tertiary university extracorporeal membrane oxygenation (ECMO) centers in Marseille (Hôpital Nord) and Paris (Pitié-Salpétrière). We included intubated and mechanically ventilated (IMV) adults with severe ARDS receiving veno-venous-ECMO for less than 24 hours. 19 patients were RT-PCR confirmed SARS-CoV-2 infected (C-ARDS) and 19 patients were COVID-19 free, with negative RT-PCR for SARS-CoV-2 (NC-ARDS). All C-ARDS patients received dexamethasone 6 mg intravenously for 10 days as standard treatment according to the RECOVERY protocol (17) and anticoagulation. All ARDS-patients underwent BAL and blood sampling within 48 hours of ECMO cannulation and baseline characteristics as well as clinical outcomes were recorded. BALF and serum were collected and stored at -80°C prior to analysis. BALF from 12 patients diagnosed with lung cancer were used as controls. We chose to use these controls because these patients were in diagnostic phasis. All patients had early-stage lung cancer and early-stage lung cancer is a disease with very low systemic and bronchoalveolar inflammation. Of these, 9 patients had non-small-cell lung cancer and 3 patients had small-cell lung cancer.
Cohort C, consisted of 128 patients with COVID-19 pneumonia confirmed by positive RT-PCRand lung computed tomodensitometry, admitted to public hospitals of Marseille, between March 20th, 2020 and April 14th, 2020. Patients were classified according to their clinical manifestations and severity, like in the protocol for novel coronavirus pneumonia (25). 37 patients were hospitalized in intensive care unit with critical COVID-19 and required invasive mechanical ventilation (IMV), were categorized as IMV-COVID-19 patients, (n=37); 21 patients were hospitalized in medical units with severe COVID-19 and required supplemental O2 >6L/min to achieve peripheral oxygen saturation (SpO2) ≥95% (severe COVID-19, n=21), 70 patients were hospitalized in medical units with COVID-19 not meeting the criteria for severe or IMV-COVID-19 and therefore categorized as moderate COVID-19 (n=70). Baseline clinical data and clinical outcomes were recorded for all patients. Serum was collected within 48 hours of hospitalization and stored at -80°C. Serum was also collected from 11 healthy volunteers COVID-19 free, with a negative RT-PCR for SARS-CoV2.
The autopsies were performed by experienced forensic pathologists in accordance with published recommendations (26). The organs were studied both in situ and individually on the dissection table. Lungs were biopsied transparietally (pleura and lung parenchyma samples), in the posterior region and inferior lobar. Pathologic examination was performed on each organ fixed in 10% buffered formalin. Microscopic examinations of the lungs were performed on the central and peripheral areas of each inferior lobe, using 4µm sections stained with hematoxylin, eosin and saffron (HES). The most representative lung samples were analyzed by immunohistochemistry. Slides were incubated overnight at 4°C with a rabbit polyclonal antibody (Ab) anti-activated-caspase-1 (1:100; Merck, St-Louis, USA), followed by an secondary rabbit antibody and HRP-DAB. Images were acquired with the NanoZoomer S360 (Hamamatsu, Massy, France) using a 10×objective (×100 magnification) for HES and for immunohistochemistry.
Soluble Caspase-1 p20 (sCasp1p20) and human IL-1 receptor antagonist (IL-Ra) concentrations were evaluated by QUANTIKINE ELISA assays (Bio-Techne Minneapolis, USA), IL-1α by ProQuantum Human IL-1α immunoassay (Thermo Fisher Scientific, Waltham, USA), HMGB1 by ELISA kit (Bio-Techne), IL-33 by ELISA kit (Abcam, Cambridge, UK), IL-1β by human IL-1β High-Sensitivity ELISA kit (Thermo Fisher Scientific) and soluble sNLRP3, C-X-C motif chemokine ligand (CXCL)10, IL-6, and IL-18 using specific ELISA assays (BD Biosciences, San Jose, USA). IFNγ concentration was quantified by Luminex kit (Thermo Fisher Scientific), and tumor necrosis α (TNFα), soluble tumor necrosis factor receptor-1 (sTNFR-1) and CXCL8 by Luminex kits from Merck (St-Louis, USA).
GraphPad-Prism V.9.2.0 software (GraphPad Software Inc., San Diego, USA) was used for statistical analysis. Values are presented as median with interquartile range (25%-75% percentile) for the indicated number of dosages. Comparisons between groups were performed using Mann-Whitney test for quantitative variables. Before carrying out these statistical tests, we performed a one-factor ANOVA test to highlight any significant differences between the groups. The ANOVA test with a p<0.05 value prompted us to perform a Mann-Whitney test, comparing the groups 2 by 2.
Associations between continuous variables were analysed using Spearman-correlation-test. Statistical significance was defined as p<0.05.
We first examined pathologic findings and in situ caspase-1 activation in C-ARDS-related lung injury using postmortem lung samples from 40 deaths (Table 1). Typical features of alveolar injury (Figure 1A, left panel) were observed in 72.5% of cases among which 23 had diffuse alveolar damage (DAD) (Figure 1Bc). Evidence of vasculopathy was present in 34 cases distributed as follows: 8/34 with thrombo-embolisms (Figure 1Bb), 8/34 with thrombotic microangiopathy (Figure 1Bc), 6/34 with endothelitis (Figure 1Bd) and 26/34 with intimal lesions (Figures 1Bd, Be). In 20/40 of the cases, interstitial inflammation was present, mainly consisting in the presence of mononuclear cells in inflammatory lesions. We then performed immunohistochemistry on lung sections using specific anti-Casp1p20 Ab (Figure 1C). Caspase-1 activation was detected in 14 of 29 (48.2%) lung biopsies presenting with alveolar lesions, in 10/23 (43.4%) of those presenting DAD (Figure 1C, left panel), but in only in 1/10 (10%) among controls. Moreover, Casp1p20 labelling was mostly associated with endoalveolar macrophages (15/20) rather than with interstitial inflammatory cells (4/20) or pneumocytes (1/20). Casp1p20 labelling associated with vascular lesions in 90% of cases (18/20), with alveolar lesions in 70% (14/20) and with interstitial lesions in 45% (9/20).
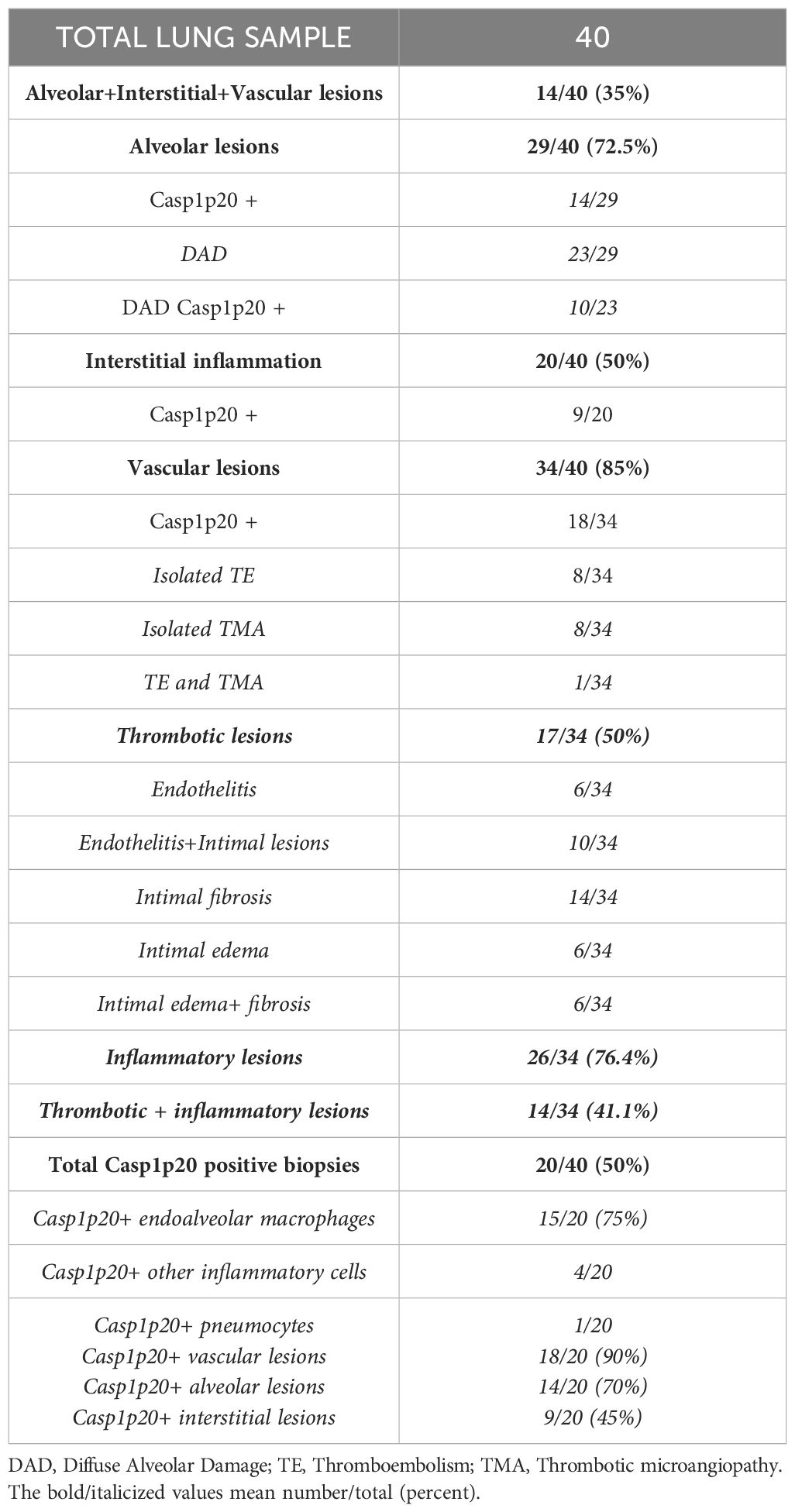
Table 1. Histological analysis of lung sections from 40 patients who died of COVID-19 ARDS: frequency of alveolar, interstitial, vascular lesions and Casp1p20 expression.
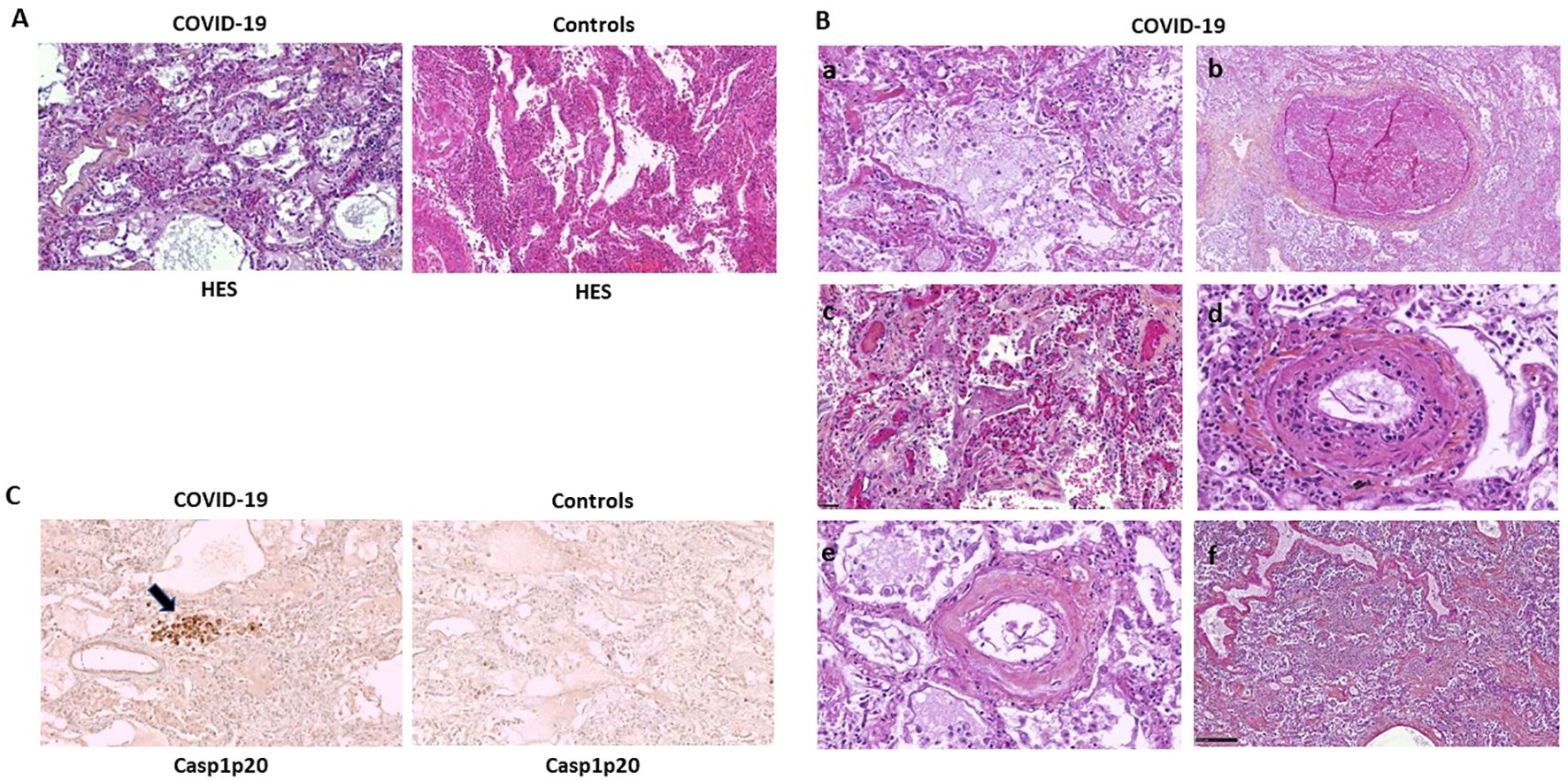
Figure 1. Caspase-1 activation is mostly associated with vascular lesions in C-ARDS lung samples. Paraffine-embedded lung sections (4µm) from patients who died of COVID-19 (A, left panel and B) or controls (A, right panel) stained by hematoxylin-eosin-saffron (HES) coloration (original magnification x100). Microscopic aspect of lesions such as Diffuse Alveolar Damage (DAD) with desquamation of alveolar epithelium (A, left panel and Ba), thrombosis (Bb), intra-alveolar fibrin deposits (Bc), endothelitis (Bd and Be) and interstitial infiltration by mononuclear inflammatory cells (Bf). Activated Caspase-1 (Casp1p20) expression in lung sections of patients who died of COVID-19 (C, left panel) or controls (C, right panel), was analyzed by anti-Casp1p20 immunostaining of macrophages and desquamative pneumocytes (original magnification x100). Scale bar = 250µm.
First, we examined the activation of the NLRP3-inflammasome signaling pathway. sNLRP3 was significantly increased in BALF from C-ARDS and NC-ARDS patients compared to controls (p<0.0001, respectively; Figure 2A). Similar results were obtained when sCasp1p20 was measured in BALF (Figure 2B). sNLRP3 was also significantly increased in the serum of C-ARDS and NC-ARDS patients compared to controls (p<0.05 and p<0.0001 respectively; Figure 2A), as was sCasp1p20 in the serum of C-ARDS and NC-ARDS patients compared to controls (p<0.001 and p<0.0001, respectively; Figure 2B). However, no significant difference was found, when comparing sNLRP3 and sCasp1p20 levels in BALF from C-ARDS versus NC-ARDS patients (p=0.66 and p=0.16, respectively; Figures 2A, B). Importantly, sNLRP3 levels were significantly higher in BALF from C-ARDS patients compared to serum from the same patients (p<0.001; Figure 2A), while this was not true for sCasp1p20 (p>0.05, Figure 2B).
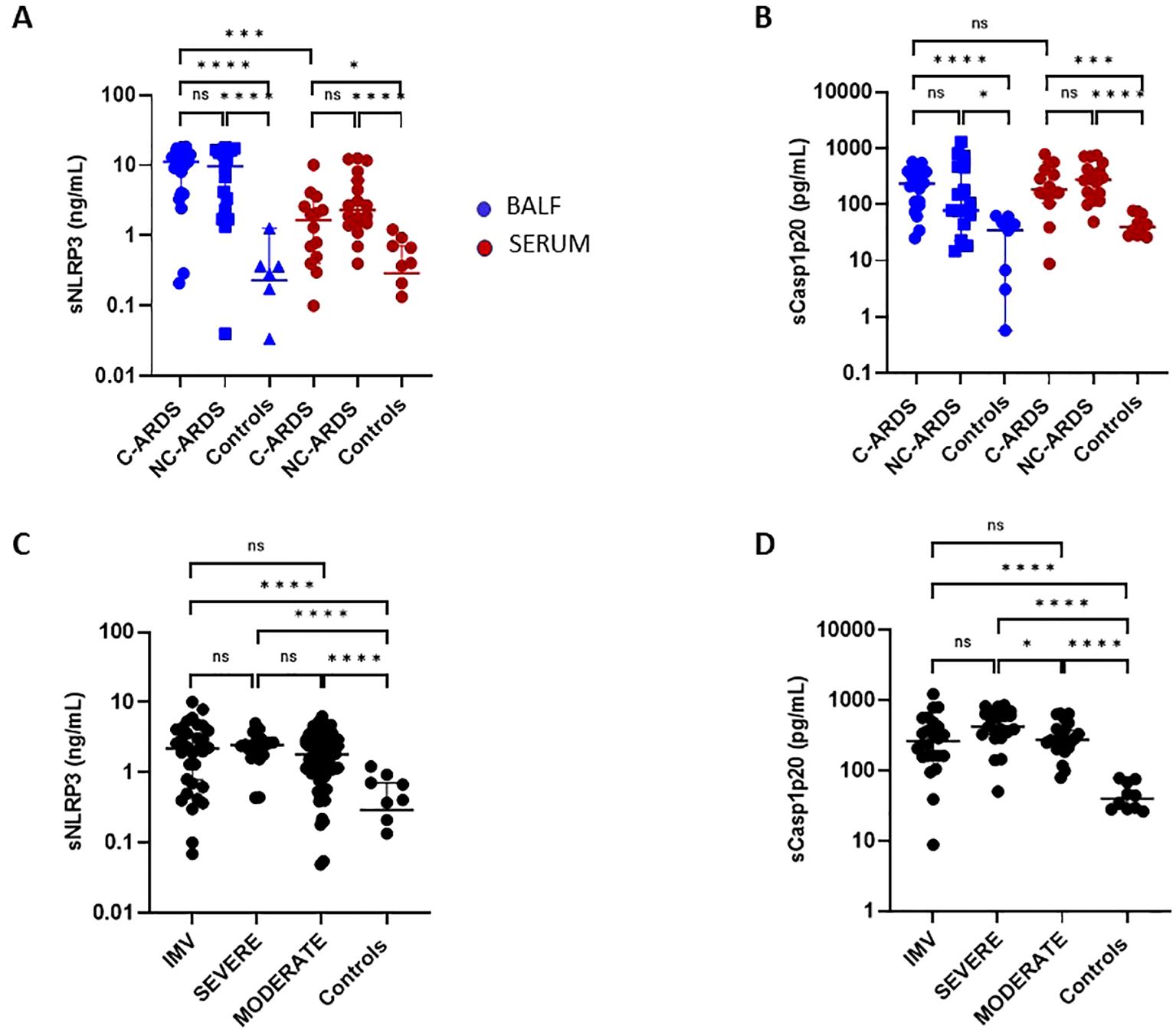
Figure 2. Elevated sNLRP3 and sCasp1p20 concentrations in C-ARDS and in NC-ARDS. sNLRP3 (A) and sCasp1p20 (B) proteins concentrations were measured by ELISA in the BALF (blue) from patients with COVID-19 ARDS (C-ARDS, n=19), non-COVID-19 ARDS (NC-ARDS, n=19) or with lung cancer as respective controls (Controls, n=8). Concentrations of the same proteins were measured in the serum (red) from patients with C-ARDS (n=14), NC-ARDS (n=19) or healthy donors as respective controls (n=12). sNLRP3 (C) proteins concentrations were measured in the serum from COVID-19 patients (C: n=122) and sCasp1p20 (D) proteins concentrations were measured in the serum from COVID-19 patients (D: n=69) and from healthy donors as respective controls (n=10). COVID patients were classified in IMV forms (C: n=32 and D: 24 respectively), severe forms (C n= 20 and D: n=21 respectively) and moderate forms (C: n= 70 and D: 24 respectively) and compared to controls. Numbers of patients tested for cytokines assays could varied in each cohort, according to technical difficulties. Each dot represents the value from a single individual (*p<0.05, ***p<0.001, ****p<0.0001). ns, no statistically significant.
We then assessed circulating sNLRP3 and sCasp1p20 levels in relation to disease severity (Figures 2C, D). Overall, serum sNLRP3 levels were significantly increased in COVID-19 patients compared to controls at all disease stages (Figure 2C; Table 2). Similar results were obtained for sCasp1p20 levels (Figure 2D; Table 2). When stratified, sCasp1p20 levels were significantly higher in patients with severe versus moderate COVID-19 (p<0.01; Figure 2D).
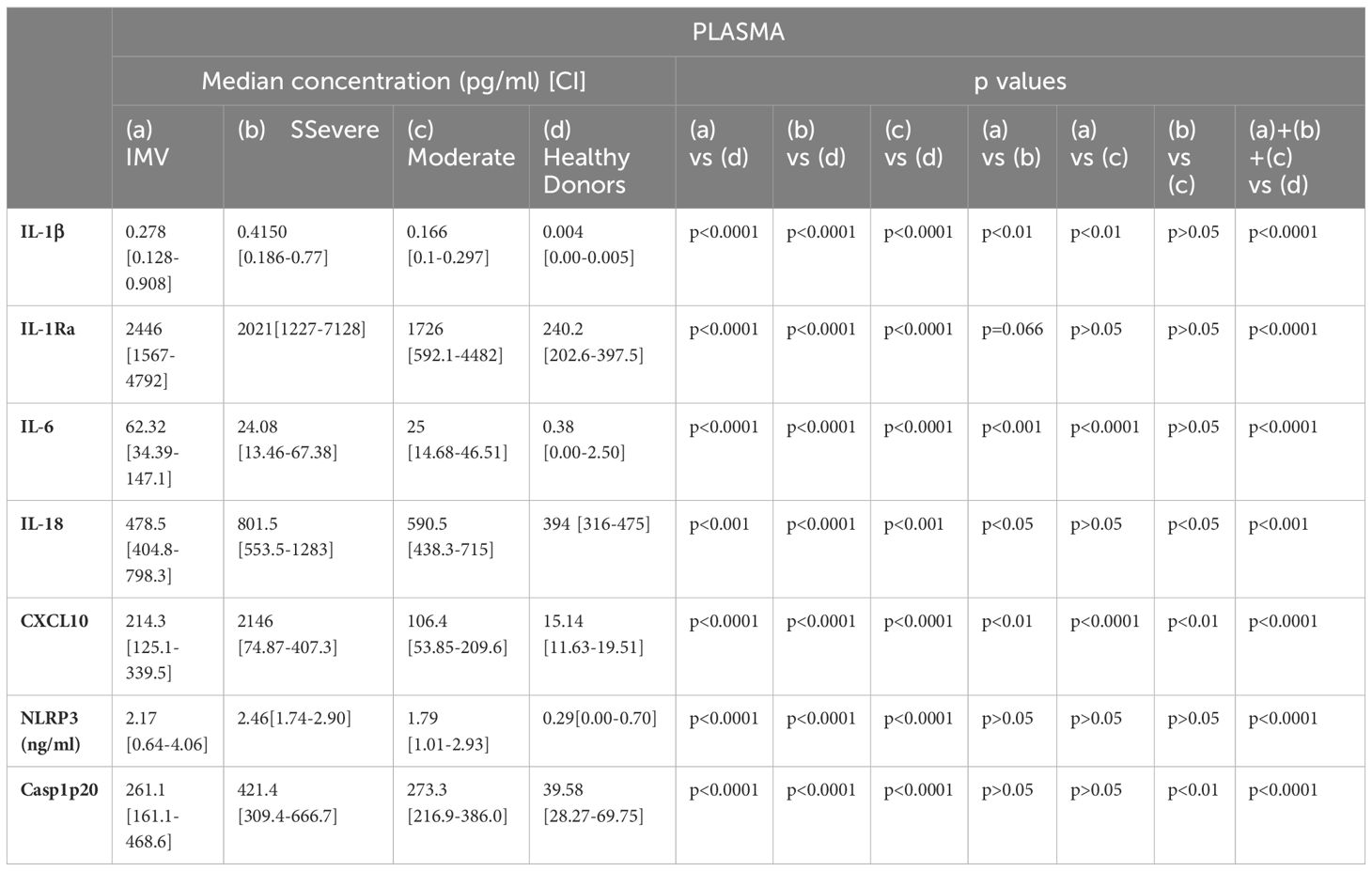
Table 2. Associations of cytokines and pro-inflammatory proteins with COVID-19 severity.
To assess the consequences of NLRP3-inflammasome and caspase-1 activation in COVID-19 patients, we measured the levels of IL-1β, IL-1Ra and IL-6 in both BALF and serum of C-ARDS or NC-ARDS patients and in control subjects. Despite inter-individual variability, IL-1β (p<0.0001 and p<0.0001, respectively; Figure 3A), IL-1Ra (p<0.0001 and p<0.0001, respectively; Figure 3B) and IL6 (p<0.001 and p<0.001 respectively; Figure 3C) concentrations were significantly increased in BALF from C-ARDS and NC-ARDS patients compared to controls. In serum, IL-1β was detectable at very low concentrations but was significantly higher in C-ARDS and NC-ARDS patients compared to controls (p<0.0001 and p<0.0001, respectively; Figure 3A). In contrast, the concentrations of IL-1Ra (p<0.0001 and p<0.0001, respectively; Figure 3B) and IL-6 concentrations (p<0.0001 and p<0.0001, respectively; Figures 3C) were well detectable in the serum and significantly higher in C-ARDS and NC-ARDS than in controls. We also evaluated whether cytokine concentrations in BALF and serum differed between C-ARDS and NC-ARDS. We found no significant differences in IL-1β, IL-Ra and IL-6 concentrations in subjects with C-ARDS versus NC-ARDS in either BALF or serum (Figures 3A–C). Interestingly, IL-1β levels, from subjects with C-ARDS, were significantly higher in BALF than in serum (p<0.0001; Figure 3A), unlike IL-1Ra and IL-6 (p>0.05 and p>0.05; respectively; Figures 3B, C).
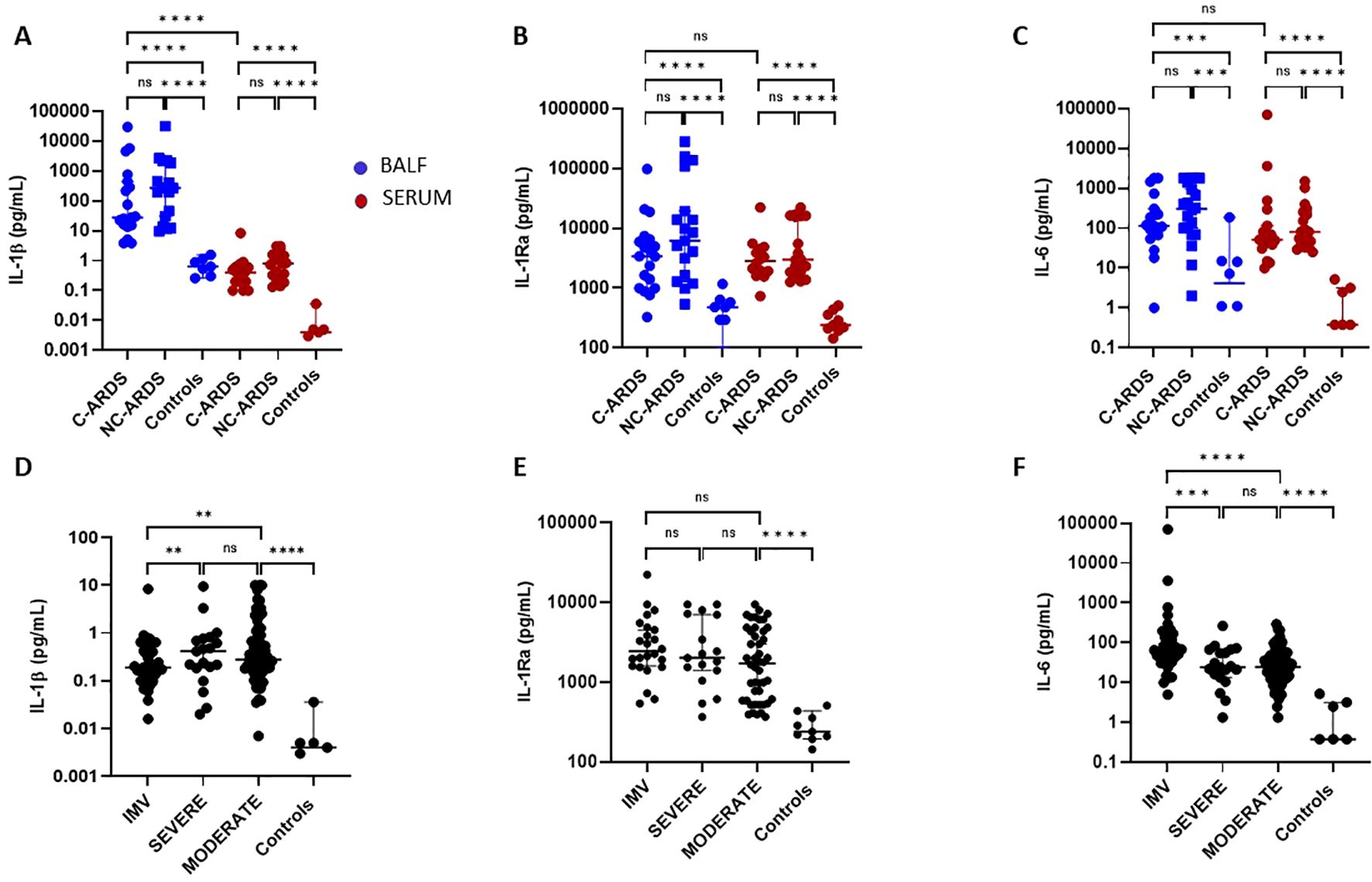
Figure 3. Activation of the IL-1β/IL-6 pathway in C-ARDS and in NC-ARDS. IL-1β (A), IL-1Ra (B), and IL-6 (C) concentrations were measured in the BALF (blue) from patients with C-ARDS (n=19), NC-ARDS (n=19) or with lung cancer as specific controls (Controls, n=8). Concentrations of the same proteins were measured in serum (red) from patients with C-ARDS (n=19), NC-ARDS (n=19) or healthy donors as specific controls (n=9). IL-1β (D), IL-1Ra (E), and IL-6 (F) concentrations were measured in the serum from COVID-19 patients (n=128) and healthy donors as respective controls (n=8). COVID patients were classified in IMV (D: n=37, E: n=24 and F: n=37 respectively), severe (D: n= 21, E: n=17 and F: n=21 respectively) and moderate forms (D: n =70, E: n= 46 and F: n=70 respectively) and compared to controls. Numbers of patients tested for cytokines assays could varied in each cohort, according to technical difficulties. Each dot represents the value from a single individual (ns: p>0.05, **p<0.01, ***p<0.001, ****p<0.0001).
We then assessed serum IL-1β, IL-1Ra and IL-6 concentrations in COVID-19 patients according to disease severity. Serum IL-1β was increased in all COVID-19 patients compared to healthy subjects (Table 2, Figure 3D) as was IL-1Ra. (Table 2, Figure 3E), and IL-6 (Table 2, Figure 3F). Intriguingly). Interestingly, circulating IL-1β levels were lower in patients with IMV-COVID-19 compared to severe or moderate COVID-19 (Table 2, Figure 3D), while no difference was observed for IL-1Ra (p>0.05; Figure 3E). Moreover, circulating IL-6 levels were significantly higher in patients with IMV-COVID compared to severe or moderate COVID-19 (Table 2, Figure 3F).
Since production of IL-18/IFNγ can also be affected by NLRP3-inflammasome and caspase-1 activation, we assessed IL-18, IFNγ, and CXCL10 levels in BALF and serum of patients with C-ARDS or NC-ARDS and of control subjects. In BALF, IL-18 was elevated in both C-ARDS and NC-ARDS patients compared to the controls (p<0.05 and p<0.01, respectively; Figure 4A) but detectable at very low concentrations. IL-18 was considerably higher in serum than in BALF from C-ARDS patients (p<0.0001, Figure 4A) and in serum of C-ARDS and NC-ARDS patients than in controls (p<0.001 and p<0.01 respectively; Figure 4A).
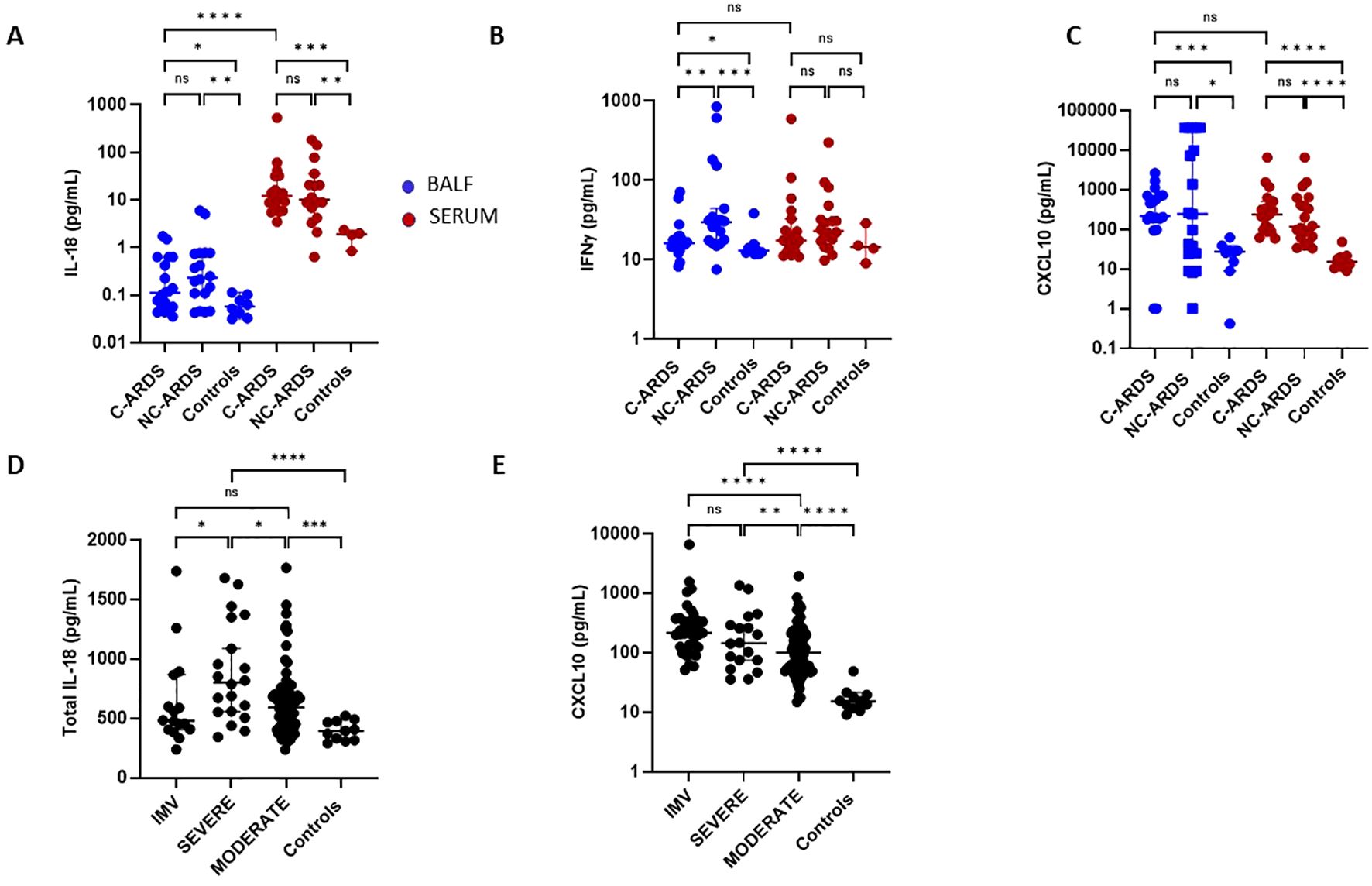
Figure 4. Activation of the IL-18/IFNγ pathway in C-ARDS and in NC-ARDS. IL-18 (A), IFNγ (B) and CXCL10 (C) concentrations were measured in the BALF (blue) from patients with C-ARDS (n=19), NC-ARDS (n=18) and with lung cancer as specific controls (n=8). Concentrations of the same proteins were measured in serum (red) from patients with C-ARDS, (n=19), NC-ARDS, (n=19) and healthy donors as controls (A: n=4, B: n= 4 and C: n=11 respectively). IL-18 (D) and CXCL10 (E) concentrations were measured in serum from COVID-19 patients (n= 127) and from healthy donors as respective controls (n=11). COVID patients were classified in IMV (D: n=16 and E: n= 37, respectively), severe (n=20) and moderate forms (n= 70) and compared to controls. Numbers of patients tested for cytokines assays could varied in each cohort, according to technical difficulties. Each dot represents the value from a single individual (*p<0.05, **p<0.01, ***p<0.001, ****p<0.0001). ns, no statistically significant.
IFNγ levels in BALF were significantly higher in both C-ARDS and NC-ARDS than in controls (p<0.05 and p<0.001, respectively; Figure 4B). Conversely, serum INFγ concentrations from C-ARDS and NC-ARDS patients were not different from controls (p>0.05 respectively; Figure 4B). CXCL10 was increased in BALF from C-ARDS and NC-ARDS compared to controls (p<0.001 and p<0.05, respectively; Figure 4C) and in serum from C-ARDS and NC-ARDS compared to controls (p<0.0001 and p<0.0001, respectively; Figure 4C). However, no significant differences of CXCL10 concentrations were observed when comparing BALF from C-ARDS versus NC-ARDS (p=0.98; Figure 4C).
When stratifying according to disease severity, serum IL-18 was overall increased in all COVID-19 subgroups compared to controls (Table 2, Figure 4D). Circulating IL-18 was slightly higher in severe compared to moderate COVID-19 (p<0.05; Figure 4D), but lower in IMV-COVID-19 compared to severe COVID-19 (p<0.05; Figure 4D). Serum CXCL10 was increased in all COVID-19 patients compared to controls, (Table 2, Figure 4E). Plasma). Serum levels of CXCL10 appeared to increase with the severity of COVID-19 (Table 2, Figure 4E).
To define predictive biomarkers of severity, we then explored the correlation between serum cytokine levels and widely used scores reflective of patient clinical status. Circulating IL-1Ra and IL-6 positively correlated with SOFA score (p<0.01, respectively; Supplementary Figures S2A, B), whereas sNLRP3 concentrations positively correlated with SAPS2 score (p<0.05; Supplementary Figure S2C).
To evaluate the pathogenic contribution of DAMPs in C-ARDS, we measured IL-1α and HMGB1 levels in BALF of C-ARDS or NC-ARDS patients and of control subjects. Both IL-1α and HMGB1 levels were detectable and significantly higher in BALF from C-ARDS and NC-ARDS patients than from controls (for IL-1α, p<0.0001 and p<0.001, respectively; Figure 5A and for HMGB1, p<0.001 and p<0.0001, respectively; Figure 5B). However, no difference was observed between patients with C-ARDS and with NC-ARDS concerning IL-1α or HMGB1 concentrations in BALF (p=0.20 and p=0.3, respectively; Figures 5A, B).

Figure 5. Elevated DAMPS concentrations in BALF of C-ARDS, and TNFα, sTNFR-1, CXCL8 concentrations in C-ARDS and NC-ARDS. IL-1α (A) and HMGB1 (B) concentrations were measured in the BALF from patients with C-ARDS (n=19), NC-ARDS (n=18) and with lung cancer as respective controls (n=11). TNFα (C), TNFR-1 (D), CXCL8 (E) concentrations were measured in the BALF (blue) from patients with C-ARDS (n=19), NC-ARDS, (n=19) and with lung cancer as controls (n=8). Concentrations of the same proteins were measured in serum (red) from patients C-ARDS, NC-ARDS and healthy donors as controls (n=10). Numbers of patients tested for cytokines assays could varied in each cohort, according to technical difficulties. Each dot represents the value from a single individual (*p<0.05, **p<0.01, ***p<0.001, ****p<0.0001). ns, no statistically significant.
To determine the contribution of other inflammatory cytokines to the immunopathogenesis of C-ARDS, we measured TNFα, TNF-R1 and CXCL8 in BALF and serum of patients with C-ARDS, NC-ARDS and of controls. Surprisingly, concentrations of TNFα, sTNFR-1 and CXCL8 in BALF of C-ARDS were significantly lower than in NC-ARDS (Table 3, Figures 5C–E), but significantly higher than in controls for sTNFR-1 and CXCL8 (Table 3, Figures 5D, E). Similarly, serum levels of TNFα, sTNFR-1 and CXCL8 in C-ARDS were significantly lower than in NC-ARDS (Table 3, Figures 5C–E) and serum levels of sTNFR-1 was higher than in controls (p<0.0001; Figure 5D).
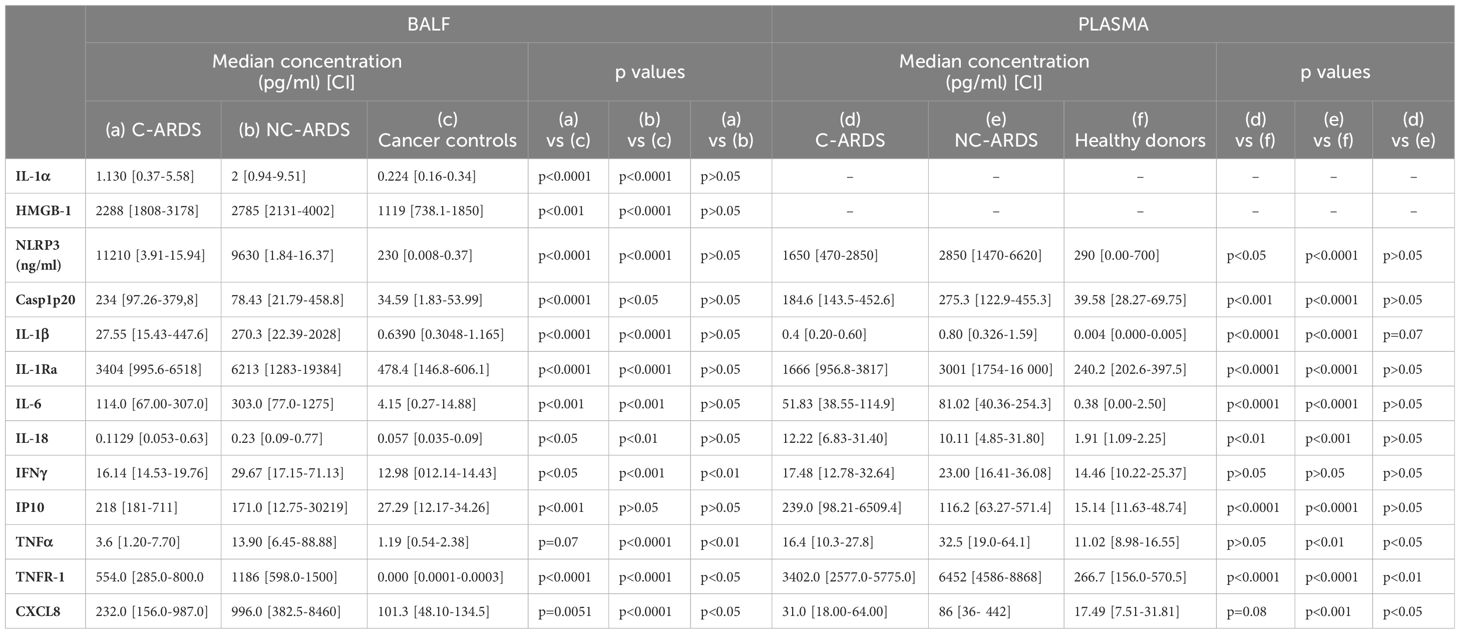
Table 3. Cytokine concentrations in BALF and plasma from COVID-19/non-COVID-19 ARDS patients and respective controls.
In this prospective multicenter study, we used several approaches such as histopathological and immunohistochemical analyses on post-mortem tissues from subjects with fatal C-ARDS, and immune profiling of BALF and serum from patients across the COVID-19 severity spectrum to provide information on NLRP3-inflammasome activation and the involvement of IL-1 signaling in COVID-19 patients.
Consistent with previous reports (27–30), DAD was present in almost 60% of subjects with fatal COVID-19-ARDS, and interstitial mononuclear cell inflammation in half of them. Inflammatory vascular lesions, including endothelitis, were detected in the vast majority (>80%) of subjects died of COVID-19 disease. In the same time, pulmonary thromboembolism and thrombotic microangiopathy were also frequent (>40%), confirming the idea that vasculopathy, coagulopathy and immunothrombosis contribute centrally to the pathogenesis of refractory ARDS in patients with COVID-19 (2, 29, 31–34).
Expression and localization of Casp1p20, an indicator of NLRP3-inflammasome activation, were assessed in the lungs of patients with refractory C-ARDS. Interestingly, intense Casp1p20 expression was also detected in abundance (>90%) at sites of vascular injury and vessel thrombosis, indicating a spatial association between NLRP3-inflammasome activation and COVID-19-associated vasculopathy.
In agreement with autopsy findings, increased levels of sNLRP3 and sCasp1p20 were observed in both BALF and serum from C-ARDS, with sNLRP3 concentrations considerably higher in BALF than in serum. The lung is thus a major site of inflammasome assembly and activation during C-ARDS. In addition, serum levels of sNLRP3 and sCasp1p20 were globally increased in all COVID-19 patients, with Casp1p20 concentrations higher in severely ill compared to moderate ill patients. This suggests the involvement of caspase-1 in hyperinflammatory responses. Collectively, these findings are consistent with activation of the NLRP3-inflammasome pathway in both lungs and blood and also corroborate previous observations showing elevated Casp1p20 levels in COVID-19 patients and inflammasome activation in patient-derived PBMCs and in CD14+ macrophages resident in COVID-19 lung tissue (17, 29, 35, 36). NLRP3-inflammasome activation can be directly triggered by multiple SARS-CoV-2-derived proteins, including spike protein and ORF3a, which act as PAMPs (19).
Activation of the NLRP3-inflammasome and subsequent cleavage of caspase-1 are responsible for the processing of IL-1β and IL-18 precursors into biologically active cytokines. In turn, IL-1β induces IL-6 synthesis, as well as the production of IL-1Ra, a natural endogenous inhibitor of IL-1. IL-18, in combination with IL-12, is a major inducer of IFNγ production (37). Due to its extremely short half-life, IL-β is usually detected at very low concentrations in the bloodstream, even in disorders typically driven by IL-1β. As expected, we observed low serum IL-β concentrations in COVID-19 patients, which were, however, significantly higher than controls. Interestingly, we found high concentrations of IL-1β (reaching >10,000pg/ml in some individuals) in BALF, indicating intense activation of the pulmonary NLRP3-inflammasome and IL-1β production during COVID-19 disease. These observations corroborate and extend data from single-cell RNA transcriptomic analyses suggesting increased IL-1β in BALF of smaller cohorts of COVID-19 patients (38–40). In contrast to IL-1β, IL-1Ra and IL-6 are generally secreted at higher concentrations and appeared elevated in both BALF and serum of COVID-19 patients. In our study, serum IL-6 correlates with the severity of COVID-19, as previously shown by others (41, 42). Our results, showing abundant expression of activated caspase-1 associated with pulmonary vascular injury and thrombosed vessels, as well as elevated levels of NLRP3, caspase-1, IL-1β and IL-6 in the pulmonary microenvironment, confirm the role of the NLRP3-inflammasome pathway in COVID-19-associated immunothrombosis, as we previously suggested (14).
IL-18 was increased in BALF but especially in the serum of C-ARDS patients compared to controls, showing a positive association with disease severity as previously described (17, 43). However, serum IL-18 levels remained lower than those observed in other hyperinflammatory syndromes (44). We found low but detectable concentrations of IFNγ in C-ARDS patients, which in BALF appeared higher in C-ARDS and NC-ARDS than in controls. However, an IFNγ-induced signature is probably present in COVID-19, since CXCL10 (in common with CXCL9), an important IFNγ-induced chemokine, is highly detectable, as previously observed in several IFNγ-mediated syndromes (45). CXCL10 levels were elevated in BALF and serum of C-ARDS patients and correlated with disease severity. Several other reports have shown an elevated CXCL10 signature during COVID-19, including in BALF (39, 40, 46) and CXCL10 is a good circulating marker of disease severity (43, 47), consistent with the fact that a Th-1 signature can develop over time during severe COVID-19 (43, 48).
Inflammasome-dependent caspase-1 activation can also trigger pyroptosis with subsequent release of intracellular contents (49, 50). Increased levels of intracellular proteins such as NLRP3 and Casp1p20 observed in BALF and serum from C-ARDS patients suggest widespread cell death resulting from direct viral cytotoxicity or inflammatory damage. Consequently, we also found detectable levels of DAMPs such as IL-1α and HMGB1 in BAL of C-ARDS patients. These intranuclear proteins are released after cell death. They probably originate from injured cells of respiratory tractus, although the nature of these cells (bronchial epithelial, alveolar or endothelial…) remains uncertain. The specific contribution of DAMPs to COVID-19 immunopathogenesis remains incompletely understood, but the role of DAMPS is investigated in the review by Land et al. When viral load is too high, for example, in the respiratory tract, authors suppose a “forcing” of many virus-infected host cells to decide to commit “suicidal” regulated cell death (e.g., necroptosis, pyroptosis) associated with release of large amounts of DAMPs. Ironically, although the aim of this “suicidal” cell death is to save and restore organismal homeostasis, the intrinsic release of excessive amounts of DAMPs leads to those dysregulated hyperinflammatory responses—as typically involved in the pathogenesis of acute respiratory distress syndrome and systemic inflammatory response syndrome in respiratory viral infections (21). DAMPS could be released as a direct consequence of SARS-CoV-2 cytotoxicity, triggering cell necrosis, or indirectly because of the excessive inflammatory response, leading to necroptosis or pyroptosis associated with the release of large amounts of DAMPs. The excessive emission of DAMPS thus released into the extracellular environment could promote dysregulated hyperinflammatory responses, and pro-inflammatory programs, such as the activation of inflammasomes, notably the NLRP3 inflammasome (21, 51–53). In response to NLRP3 inflammasome activation, Caspase-1 is cleaved, resulting in pyroptosis. Moreover, IL-1ß and IL-18 released after cleavage by activated Caspase-1 belong to DAMPS. DAMPS could therefore participate in a positive feedback loop of the innate immune response induced by SARS-CoV-2 infection. The authors defined different classes of activating DAMPS in the event of cell lysis, and suppressor DAMPS or compensatory SAMPS in the initiation of controlled defense responses favoring, depending on their balance, either inflammation or resolution of inflammation in the event of infection (21). Two studies have reported elevated circulating concentrations of HMGB1 and calprotectin (S100A8/A9, a marker of neutrophil activation) in serum from COVID-19 patients (54, 55). In a study by Renieris et al., mice injected with serum from COVID-19 patients showed elevated pulmonary expression of pro-inflammatory molecules, including TNFα, IL-6 and CXCL10 resembling human COVID-19. Treatment of these mice with anakinra (which blocks both IL-1α and IL-1β signaling) or with antibodies selectively targeting IL-1α significantly attenuated COVID-19-like pulmonary immunopathology, potentially identifying IL-1α as a mediator of inflammation and tissue-specific injury (54, 56). Taken together, these observations may help to explain the limited efficacy of the anti-IL-1β monoclonal antibody, canakinumab, compared to anakinra in COVID-19 (57–61). Aware of the limitations of these observations, we plan to pursue this work by characterizing and specifically targeting DAMPS and eventually SAMPS in BALF and serum from ARDS patients.
We found that NLRP3-inflammasome activation and concentrations of IL-1α, IL-β, IL-6, IL-1Ra or CXCL10 were broadly similar in C-ARDS and NC-ARDS. However, concentrations of TNFα, sTNF-R1 and CXCL8 both in BALF and serum, were significantly higher in NC-ARDS than in C-ARDS, suggesting that inflammatory pathways involving NF-kB could be probably more up-regulated in NC-ARDS than in C-ARDS. A more likely explanation of these results may be due to the different use of steroids, which are well-known inhibitors of NF-kB-induced gene transcription. Glucocorticoids act by modulating the gene expression of several proteins involved in the inflammatory response. The reduced transcriptional activity of NF-κB is key to the anti-inflammatory effects of glucocorticoids. As expression of many of the pro-inflammatory cytokines implicated in COVID-19 is NF-κB-dependent, including IL-6, TNFα, and CXCL10 among others (62). Indeed, all C-ARDS subjects included in this study received steroids as a part of standard and recommended treatment, whereas NC-ARDS subjects were steroids free. It should be noted that all C-ARDS patients included in this study were in fact refractory to this steroid therapy, experiencing severe clinical deterioration despite well-conducted treatment, and that in these patients, steroids demonstrated almost a poor biological inhibitory effect on IL-1α, IL-β, IL-6, IL-1Ra or CXCL10 levels (62). The deterioration of COVID-19 patients is inextricably linked to immunopathological phenomena rather than viral load. The aggressive inflammatory responses in COVID-19 result in damage to the airways, termed ARDS, which may lead to respiratory failure and death. Corticosteroid treatment reduced mortality, but subsequent meta-analyses have again highlighted the importance of timing when considering corticosteroid administration, as corticosteroid use was associated with increased viral load. NLRP3 inflammasome activation is strongly correlated with COVID-19 severity and part of dexamethasone’s clinical effect in COVID-19 may be via NLRP3 inhibition (63). But our results suggest that these inflammatory cytokines,IL-1α, IL-β, IL-6, IL-1Ra or CXCL10, may be poorly targeted by steroids and may require to be specifically targeted in C-ARDS patients with poor response to steroids (64–66). Thus, in pursuit of this work, we aim to assess and analyze some of the anti-inflammatory cytokines, dependent from NLRP3 inflammasome pathway or from NF-κB pathway, to show how the balance between pro- and anti-inflammatory cytokines is affected in C-ARDS, NC-ARDS and control pulmonary vasculopathies, before and after steroid treatment. Furthermore, data showing similar IL-1/IL-6 cytokine profiles in C-ARDS and NC-ARDS patients suggest that these cytokine pathways may be highly activated in all types of ARDS, leading to the conclusion that ARDS should be considered as an hyperinflammatory state with DAD and severe prognosis in which novel anti-inflammatory strategies targeting these cytokines merit further mechanistic investigation (30). Specific NLRP3 inhibitors are currently undergoing clinical trials for the treatment of COVID-19 (63).
This study is limited in terms of power and clinical significance. Our control patients represented an imperfect control, since they were patients who might have incipient bronchopulmonary cancer. Bronchopulmonary cancer develops in inflammatory tumor microenvironment, which may involve the NLRP3 pathways. Tumor cell-intrinsic mechanisms of NLRP3 activation in the tumor microenvironment has been described by Tengesdal. et al (67). Aberrant activation of NLRP3 within the tumor microenvironment results in increased IL-1β and IL-18 secretion. Dysregulation of these cytokines induce tumor promoting mechanisms, such as angiogenesis, immunosuppression and metastasis. NLRP3 activation therefore represents a key immune checkpoint within the tumor microenvironnement, acting as a master switch for inflammation-mediated tumor progression. Nevertheless, according to our results from controls, ELISA protein expression levels of the inflammatory cytokines from the IL-1 family, and dependent from NLRP3 pathway, were significantly lower in BAL and in serum than in the other groups studied. Moreover, NLRP3 activation in cancer remains controversial. The role of the NLRP3 inflammasome therefore appears to be secondary in this inflammatory tumor microenvironment, after Transforming growth factor β (TGF-β). The article by Zhao et al. characterizes the tumor microenvironment in relation to carcinogenesis, and the predominant role of TGF-ß/SMAD4 signaling in cancer. TGF-β signaling pathway plays important roles in many biological processes, including cancer initiation and progression (68). Another predominantly inflammatory pathway, is interferon persistent signaling. Mathew et al. described the persistent IFN signaling as a potent immunoregulatory effect, promoting carcinogenesis. In cancer cells and immune cells, chronic IFN-I signaling is linked with cancer resistance in humans. IFN signaling thus appears to be involved in the carcinogenesis process, and argue to using combined JAK inhibition and PD-1 immunotherapy for non-small cell lung cancer patients (69). It seemed ethically difficult to propose another group controls totally asymptomatic.
Limitations of our study lie in the small number of patients in the study groups, and the missing data, resulting from mainly technical reasons, like insufficient quantity of samples or poor preservation of samples.
Finally, these results are therefore not generalizable but raise the hypothesis of an IL-1/IL-6 and IFNγ/IL-18 cytokine signature in ARDS BAL, in response to activation of the NLRP3 inflammasome in lung cells. These results are preliminary data to support clinical trials for new NLRP3 inhibitors, anti-IL-1 and anti-IL-6 therapies or JAK ½ inhibitors in COVID-ARDS and in ARDS.
As a conclusion, the results of this study support a role for the NLRP3-inflammasome activation and IL-1 in the immunopathogenesis of COVID-19. The presence of increased plasma levels of sNLRP3, sCasp1p20 and IL-1 cytokines (IL-1β, IL-18) was demonstrated in hospitalized patients across the spectrum of COVID-19 severity. Upregulation of this pro-inflammatory IL-1β/IL-6 pathway was also observed in both BALF and plasma obtained from critically ill patients with steroid-refractory-C-ARDS, but predominantly in BALF, better reflecting the inflammatory alveolar microevironment. Consistent with NLRP3-inflammasome activation, caspase-1 activation was detected on post-mortem lung tissues and mainly registered with lung-residing macrophages, localizing predominantly at the areas of alveolar injury and foremost-injured vasculature and thrombosis, hence supporting NLRP3-inflammasome activation as a putative mechanism contributing to immunothrombosis in COVID-19. IL-18/IFNγ-induced pathways also appeared to play a role in patients with steroid-resistant C-ARDS and may be insufficiently targeted by steroid therapy alone, arguing for new multi-target strategies in COVID-ARDS.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
This prospective multicenter study was approved by the Medical Ethics Committee of Aix-Marseille-University (CPP # 1123 HPS1) and by the Assistance Publique de Marseille digital data protection delegate (RGPD2020-47). The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
AC: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. CG: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – review & editing. CD: Formal analysis, Methodology, Writing – review & editing. NP: Methodology, Project administration, Resources, Supervision, Writing – review & editing. RB: Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Validation, Writing – review & editing. ET: Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Visualization, Writing – review & editing. EA: Data curation, Formal analysis, Investigation, Methodology, Writing – review & editing. ML: Data curation, Investigation, Software, Writing – review & editing. MG: Data curation, Methodology, Writing – review & editing. IP: Data curation, Investigation, Methodology, Writing – review & editing. VA: Data curation, Investigation, Methodology, Writing – review & editing. PF: Data curation, Investigation, Methodology, Writing – review & editing. LC: Formal analysis, Resources, Writing – review & editing. MS: Formal Analysis, Methodology, Resources, Writing – review & editing. LA: Data curation, Formal analysis, Investigation, Methodology, Validation, Writing – review & editing. RL: Data curation, Methodology, Supervision, Validation, Visualization, Writing – review & editing. SR: Formal analysis, Methodology, Writing – review & editing. J-MF: Data curation, Formal analysis, Methodology, Writing – review & editing. SH: Data curation, Formal Analysis, Investigation, Methodology, Writing – review & editing. LD: Writing – review & editing. LP: Writing – review & editing. FD-G: Conceptualization, Resources, Writing – review & editing. GK: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing.
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
To AB2 Bio EPFL Innovation Park Building B, Lausanne, Switzerland.
Authors LC and MS were employed by the company AB2 Bio. Author NP has received a training fellowship from the International Society on Thrombosis and Haemostasis ISTH, and a research grant from the International Network of VENous Thromboembolism Clinical Research Networks INVENT, outside of the present work. Author CG reported personal consulting fees from Xenios FMC outside the submitted work.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2024.1493306/full#supplementary-material
Supplementary Figure 1 | Flowchart of the study design including 3 cohorts of COVID-19 patients.
Supplementary Figure 2 | Circulating IL-6, IL-1Ra and sNLRP3 concentrations correlated with clinical severity in C-ARDS. Correlations between circulating IL-1Ra (A), IL-6 (B) and SOFA, or between soluble sNLRP3 and SAPS2 (C) in C-ARDS patients. SOFA, Sequential-Organ-Failure-Assessment; SAPS2, Simplified-Acute-Physiology-Score-2. Spearman R correlation coefficients are indicated.
COVID, Coronavirus Disease-2019; DAMPS, Danger-associated molecular patterns; PAMPS, Pathogen-associated molecular patterns; BALF, Bronchoalveolar lavage fluids; IL-, Interleukin; IFN-γ, interferon-gamma; CXCL-, Chemokines; HMGB1, High–mobility group box 1; Il-1Ra, Interleukin- Receptor antagonist; TNFR-1, Tumor necrosis factor receptor 1; ARDS, Acute-respiratory-distress-syndrome; NF-kB, Nuclear factor kappa-light-chain; NACTH, NAIP, CIITA, HET-E et TEP1 NTPases implicated in apoptosis and MHC transcription activation”; LRR, Leucine-Rich Repeat; PYD, Pyrin Domain; NLRP3, NOD-like receptor family, pyrin domain containing 3; AIM2, Absent-In-Melanoma-2; ASC, Apoptosis-associated speck-like protein; CARD, Caspase Recruitment Domain; CD, Cluster Differenciation; IgG, Immunoglobin G; ECMO, Extracorporeal Membrane Oxygenation; ICU, Intensive Care Unit; IMV, Invasive Mechanical Ventilation; ARDS, Acute-Respiratory-Distress-Syndrome; RT-PCR, Reverse Transcriptase Polymerase Chain Reaction; HES, Hematoxylin, Eosin and Saffron; ELISA, Enzyme-linked Immunosorbent assay; TNF-a, Tumor necrosis factor-a; DAD, Diffuse Alveolar Damage; PBMC, Peripheral Blood Mononuclear Cells; ACE, Angiotensin-Converting Enzyme; ORF3, Open reading frame; RNA, Ribonucleic acid; Ab, antibody; PANapotosis, Pyrosis, Apoptosis, Necroptosis.
1. Jin Y, Yang H, Ji W, Wu W, Chen S, Zhang W, et al. Virology, epidemiology, pathogenesis, and control of COVID-19. Viruses. (2020) 12:372. doi: 10.3390/v12040372
2. Bonaventura A, Vecchié A, Dagna L, Martinod K, Dixon DL, Van Tassell BW, et al. Endothelial dysfunction and immunothrombosis as key pathogenic mechanisms in COVID-19. Nat Rev Immunol. (2021) 21:319−29. doi: 10.1038/s41577-021-00536-9
3. Vallier L, Bouriche T, Bonifay A, Judicone C, Bez J, Franco C, et al. Increasing the sensitivity of the human microvesicle tissue factor activity assay. Thromb Res. (2019) 182:64−74. doi: 10.1016/j.thromres.2019.07.011
4. Choi D, Waksman O, Shaik A, Mar P, Chen Q, Cho DJ, et al. Association of blood viscosity with mortality among patients hospitalized with COVID-19. J Am Coll Cardiol. (2022) 80:316−28. doi: 10.1016/j.jacc.2022.04.060
5. Ranucci M, Ballotta A, Dedda UD, Baryshnikova E, Poli MD, Resta M, et al. The procoagulant pattern of patients with COVID-19 acute respiratory distress syndrome. J Thromb Haemostasis. (2022) 18:1747. doi: 10.1111/jth.14854
6. McNab F, Mayer-Barber K, Sher A, Wack A, O’Garra A. Type I interferons in infectious disease. Nat Rev Immunol. (2015) 15:87−103. doi: 10.1038/nri3787
7. Hadjadj J, Yatim N, Barnabei L, Corneau A, Boussier J, Smith N, et al. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science. (2020) 369:718−24. doi: 10.1126/science.abc6027
8. Mizurini DM, Hottz ED, Bozza PT, Monteiro RQ. Fundamentals in covid-19-associated thrombosis: molecular and cellular aspects. Front Cardiovasc Med 17 déc. (2021) 8:785738. doi: 10.3389/fcvm.2021.785738
9. Tang L, Yin Z, Hu Y, Mei H. Controlling cytokine storm is vital in COVID-19. Front Immunol 30 nov. (2020) 11:570993. doi: 10.3389/fimmu.2020.570993
10. Zhang F, Mears JR, Shakib L, Beynor JI, Shanaj S, Korsunsky I, et al. IFN-γ and TNF-α drive a CXCL10+ CCL2+ macrophage phenotype expanded in severe COVID-19 lungs and inflammatory diseases with tissue inflammation. Genome Med 20 avr. (2021) 13:64. doi: 10.1186/s13073-021-00881-3
11. Su C, Rousseau S, Emad A. Identification of transcriptional regulatory network associated with response of host epithelial cells to SARS-CoV-2. Sci Rep 14 déc. (2021) 11:23928. doi: 10.1038/s41598-021-03309-5
12. Vora SM, Lieberman J, Wu H. Inflammasome activation at the crux of severe COVID-19. Nat Rev Immunol. (2021) 21:694−703. doi: 10.1038/s41577-021-00588-x
13. Jo EK, Kim JK, Shin DM, Sasakawa C. Molecular mechanisms regulating NLRP3 inflammasome activation. Cell Mol Immunol. (2016) 13:148−59. doi: 10.1038/cmi.2015.95
14. Potere N, Del Buono MG, Caricchio R, Cremer PC, Vecchié A, Porreca E, et al. Interleukin-1 and the NLRP3 inflammasome in COVID-19: Pathogenetic and therapeutic implications. eBioMedicine. (2022) 85:104299. doi: 10.1016/j.ebiom.2022.104299
15. Silva CMS, Wanderley CWS, Veras FP, Gonçalves AV, Lima MHF, Toller-Kawahisa JE, et al. Gasdermin-D activation by SARS-CoV-2 triggers NET and mediate COVID-19 immunopathology. Crit Care. (2022) 26:206. doi: 10.1186/s13054-022-04062-5
16. Bertoni A, Penco F, Mollica H, Bocca P, Prigione I, Corcione A, et al. Spontaneous NLRP3 inflammasome-driven IL-1-β secretion is induced in severe COVID-19 patients and responds to anakinra treatment. J Allergy Clin Immunol. (2022) 150:796−805. doi: 10.1016/j.jaci.2022.05.029
17. Rodrigues TS, de Sá KSG, Ishimoto AY, Becerra A, Oliveira S, Almeida L, et al. Inflammasomes are activated in response to SARS-CoV-2 infection and are associated with COVID-19 severity in patients. J Exp Med. (2020) 218:e20201707. doi: 10.1084/jem.20201707
18. Ferreira AC, Soares VC, de Azevedo-Quintanilha IG, Dias S da SG, Fintelman-Rodrigues N, Sacramento CQ, et al. SARS-CoV-2 engages inflammasome and pyroptosis in human primary monocytes. Cell Death Discovery 1 mars. (2021) 7:43. doi: 10.1038/s41420-021-00428-w
19. Junqueira C, Crespo Â, Ranjbar S, Ingber J, Parry B, Ravid S, et al. SARS-CoV-2 infects blood monocytes to activate NLRP3 and AIM2 inflammasomes, pyroptosis and cytokine release. medRxiv [Preprint]. (2021) 8:2021.03.06.21252796. doi: 10.1101/2021.03.06.21252796
20. Cicco S, Cicco G, Racanelli V, Vacca A. Neutrophil extracellular traps (NETs) and damage-associated molecular patterns (DAMPs): two potential targets for COVID-19 treatment. Mediators Inflamm. (2020) 2020:7527953. doi: 10.1155/2020/7527953
21. Land WG. Role of DAMPs in respiratory virus-induced acute respiratory distress syndrome—with a preliminary reference to SARS-CoV-2 pneumonia. Genes Immun. (2021) 22:141−60. doi: 10.1038/s41435-021-00140-w
22. Karki R, Sharma BR, Tuladhar S, Williams EP, Zalduondo L, Samir P, et al. Synergism of TNF-α and IFN-γ Triggers inflammatory cell death, tissue damage, and mortality in SARS-coV-2 infection and cytokine shock syndromes. Cell. (2021) 184:149–168.e17. doi: 10.1016/j.cell.2020.11.025
23. Xu H, Akinyemi IA, Chitre SA, Loeb JC, Lednicky JA, McIntosh MT, et al. SARS-CoV-2 viroporin encoded by ORF3a triggers the NLRP3 inflammatory pathway. Virology. (2022) 568:13−22. doi: 10.1016/j.virol.2022.01.003
24. Shen X, He L, Cai W. Role of lipopolysaccharides in the inflammation and pyroptosis of alveolar epithelial cells in acute lung injury and acute respiratory distress syndrome. J Inflammation Res. (2024) 17:5855−69. doi: 10.2147/JIR.S479051
25. Wei PF. Diagnosis and treatment protocol for novel coronavirus pneumonia (Trial version 7). Chin Med J (Engl). (2020) 133(9):1087–95. doi: 10.1097/CM9.0000000000000819
26. Hanley B, Lucas SB, Youd E, Swift B, Osborn M. Autopsy in suspected COVID-19 cases. J Clin Pathol. (2020) 73:239−42. doi: 10.1136/jclinpath-2020-206522
27. Delorey TM, Ziegler CGK, Heimberg G, Normand R, Yang Y, Segerstolpe Å, et al. COVID-19 tissue atlases reveal SARS-CoV-2 pathology and cellular targets. Nature. (2021) 595:107−13. doi: 10.1038/s41586-021-03570-8
28. Wichmann D, Sperhake JP, Lütgehetmann M, Steurer S, Edler C, Heinemann A, et al. Autopsy findings and venous thromboembolism in patients with COVID-19: A prospective cohort study. Ann Intern Med. (2020) 173(4):268–77. doi: 10.7326/M20-2003
29. Barton LM, Duval EJ, Stroberg E, Ghosh S, Mukhopadhyay S. COVID-19 autopsies, Oklahoma, USA. Am J Clin Pathol. (2020) 153(6):725–33. doi: 10.1093/ajcp/aqaa062
30. Serdaroglu E, Kesici S, Bayrakci B, Kale G. Diffuse alveolar damage correlation with clinical diagnosis of pediatric acute respiratory distress syndrome. J Pediatr Intensive Care. (2021) 10:052−7. doi: 10.1055/s-0040-1714127
31. Xu G, Qi F, Li H, Yang Q, Wang H, Wang X, et al. The differential immune responses to COVID-19 in peripheral and lung revealed by single-cell RNA sequencing. Cell Discovery. (2020) 6:73. doi: 10.1038/s41421-020-00225-2
32. Flaumenhaft R, Enjyoji K, Schmaier AA. Vasculopathy in COVID-19. Blood. (2022) 140:222−35. doi: 10.1182/blood.2021012250
33. Hottz ED, Martins-Gonçalves R, Palhinha L, Azevedo-Quintanilha IG, de Campos MM, Sacramento CQ, et al. Platelet-monocyte interaction amplifies thromboinflammation through tissue factor signaling in COVID-19. Blood Adv. (2022) 6:5085−99. doi: 10.1182/bloodadvances.2021006680
34. Paul O, Tao JQ, West E, Litzky L, Feldman M, Montone K, et al. Pulmonary vascular inflammation with fatal coronavirus disease 2019 (COVID-19): possible role for the NLRP3 inflammasome. Respir Res. (2022) 23:25. doi: 10.1186/s12931-022-01944-8
35. Aymonnier K, Ng J, Fredenburgh LE, Zambrano-Vera K, Münzer P, Gutch S, et al. Inflammasome activation in neutrophils of patients with severe COVID-19. Blood Adv. (2022) 6:2001−13. doi: 10.1182/bloodadvances.2021005949
36. Courjon J, Dufies O, Robert A, Bailly L, Torre C, Chirio D, et al. Heterogeneous NLRP3 inflammasome signature in circulating myeloid cells as a biomarker of COVID-19 severity. Blood Adv. (2021) 5:1523−34. doi: 10.1182/bloodadvances.2020003918
37. Kaplanski G. Interleukin-18: Biological properties and role in disease pathogenesis. Immunol Rev. (2018) 281:138−53. doi: 10.1111/imr.2018.281.issue-1
38. Xu Z, Shi L, Wang Y, Zhang J, Huang L, Zhang C, et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir Med. (2020) 8:420−2. doi: 10.1016/S2213-2600(20)30076-X
39. Liao M, Liu Y, Yuan J, Wen Y, Xu G, Zhao J, et al. Single-cell landscape of bronchoalveolar immune cells in patients with COVID-19. Nat Med. (2020) 26:842−4. doi: 10.1038/s41591-020-0901-9
40. Zhou Z, Ren L, Zhang L, Zhong J, Xiao Y, Jia Z, et al. Heightened innate immune responses in the respiratory tract of COVID-19 patients. Cell Host Microbe. (2020) 27:883–890.e2. doi: 10.1016/j.chom.2020.04.017
41. Bivona G, Agnello L, Ciaccio M. Biomarkers for prognosis and treatment response in COVID-19 patients. Ann Lab Med. (2021) 41:540−8. doi: 10.3343/alm.2021.41.6.540
42. Broman N, Rantasärkkä K, Feuth T, Valtonen M, Waris M, Hohenthal U, et al. IL-6 and other biomarkers as predictors of severity in COVID-19. Ann Med. (2021) 53:410−2. doi: 10.1080/07853890.2020.1840621
43. Lucas C, Wong P, Klein J, Castro TBR, Silva J, Sundaram M, et al. Longitudinal analyses reveal immunological misfiring in severe COVID-19. Nature. (2020) 584:463–9. doi: 10.1038/s41586-020-2588-y
44. Kessel C, Hedrich CM, Foell D. Innately adaptive or truly autoimmune: is there something unique about systemic juvenile idiopathic arthritis? Arthritis Rheumatol. (2020) 72:210−9. doi: 10.1002/art.41107
45. Prencipe G, Bracaglia C, Caiello I, Pascarella A, Francalanci P, Pardeo M, et al. The interferon-gamma pathway is selectively up-regulated in the liver of patients with secondary hemophagocytic lymphohistiocytosis. PloS One. (2019) 14:e0226043. doi: 10.1371/journal.pone.0226043
46. Zaid Y, Doré É, Dubuc I, Archambault AS, Flamand O, Laviolette M, et al. Chemokines and eicosanoids fuel the hyperinflammation within the lungs of patients with severe COVID-19. J Allergy Clin Immunol août. (2021) 148:368–380.e3. doi: 10.1016/j.jaci.2021.05.032
47. Diorio C, Henrickson SE, Vella LA, McNerney KO, Chase J, Burudpakdee C, et al. Multisystem inflammatory syndrome in children and COVID-19 are distinct presentations of SARS-CoV-2. J Clin Invest. (2020) 130:5967−75. doi: 10.1172/JCI140970
48. Basheer M, Saad E, Kananeh M, Asad L, Khayat O, Badarne A, et al. Cytokine patterns in COVID-19 patients: which cytokines predict mortality and which protect against? Curr Issues Mol Biol. (2022) 44(10):4735–47. doi: 10.3390/cimb44100323
49. Kelley N, Jeltema D, Duan Y, He Y. The NLRP3 inflammasome: an overview of mechanisms of activation and regulation. Int J Mol Sci 6 juill. (2019) 20:3328. doi: 10.3390/ijms20133328
50. Pan P, Shen M, Yu Z, Ge W, Chen K, Tian M, et al. SARS-CoV-2 N protein promotes NLRP3 inflammasome activation to induce hyperinflammation. Nat Commun. (2021) 12:4664. doi: 10.1038/s41467-021-25015-6
51. Silva-Lagos LA, Pillay J, van Meurs M, Smink A, van der Voort PHJ, de Vos P. DAMPening COVID-19 severity by attenuating danger signals. Front Immunol. (2021) 12:720192. doi: 10.3389/fimmu.2021.720192/full
52. Fan X, Song JW, Wang SY, Cao WJ, Wang XW, Zhou MJ, et al. Changes of damage associated molecular patterns in COVID-19 patients. Infect Dis Immun. (2021) 1:20−7. doi: 10.1097/01.ID9.0000733572.40970.6c
53. Parthasarathy U, Martinelli R, EH V, Best K, Therien AG. The impact of DAMP-mediated inflammation in severe COVID-19 and related disorders. Biochem Pharmacol janv. (2022) 195:114847. doi: 10.1016/j.bcp.2021.114847
54. Renieris G, Karakike E, Gkavogianni T, Droggiti DE, Stylianakis E, Andriopoulou T, et al. IL-1 mediates tissue-specific inflammation and severe respiratory failure in COVID-19. J Innate Immun. (2022) 14:643−56. doi: 10.1159/000524560
55. Shi H, Zuo Y, Yalavarthi S, Gockman K, Zuo M, Madison JA, et al. Neutrophil calprotectin identifies severe pulmonary disease in COVID-19. J Leukoc Biol. (2021) 109:67−72. doi: 10.1002/JLB.3COVCRA0720-359R
56. Silvin A, Chapuis N, Dunsmore G, Goubet AG, Dubuisson A, Derosa L, et al. Elevated calprotectin and abnormal myeloid cell subsets discriminate severe from mild COVID-19. Cell 17 sept. (2020) 182:1401–1418.e18. doi: 10.1016/j.cell.2020.08.002
57. Kyriazopoulou E, Panagopoulos P, Metallidis S, Dalekos GN, Poulakou G, Gatselis N, et al. An open label trial of anakinra to prevent respiratory failure in COVID-19. Elife. 8 mars. (2021) 10:e66125. doi: 10.7554/eLife.66125
58. Caricchio R, Abbate A, Gordeev I, Meng J, Hsue PY, Neogi T, et al. Effect of canakinumab vs placebo on survival without invasive mechanical ventilation in patients hospitalized with severe COVID-19: A randomized clinical trial. JAMA. (2021) 326(3):230–9. doi: 10.1001/jama.2021.9508
59. Kyriazopoulou E, Poulakou G, Milionis H, Metallidis S, Adamis G, Tsiakos K, et al. Early treatment of COVID-19 with anakinra guided by soluble urokinase plasminogen receptor plasma levels: a double-blind, randomized controlled phase 3 trial. Nat Med. (2021) 27(10):1752–60. doi: 10.1038/s41591-021-01499-z
60. Cauchois R, Koubi M, Delarbre D, Manet C, Carvelli J, Blasco VB, et al. Early IL-1 receptor blockade in severe inflammatory respiratory failure complicating COVID-19. Proc Natl Acad Sci U.S.A. (2020) 117:18951−3. doi: 10.1073/pnas.2009017117
61. Audemard-Verger A, Le Gouge A, Pestre V, Courjon J, Langlois V, Vareil MO, et al. Efficacy and safety of anakinra in adults presenting deteriorating respiratory symptoms from COVID-19: A randomized controlled trial. PloS One. (2022) 17:e0269065. doi: 10.1371/journal.pone.0269065
62. De Bosscher K, Vanden Berghe W, Haegeman G. The interplay between the glucocorticoid receptor and nuclear factor-kappaB or activator protein-1: molecular mechanisms for gene repression. Endocr Rev. (2003) 24(4):488–522. doi: 10.1210/er.2002-0006
63. Hooftman A, O’Neill LAJ. Can NLRP3 inhibitors improve on dexamethasone for the treatment of COVID-19? Curr Res Pharmacol Drug Discovery. (2021) 2:100048. doi: 10.1016/j.crphar.2021.100048
64. Forsyth CB, Zhang L, Bhushan A, Swanson B, Zhang L, Mamede JI, et al. The SARS-coV-2 S1 spike protein promotes MAPK and NF-kB activation in human lung cells and inflammatory cytokine production in human lung and intestinal epithelial cells. Microorganisms. (2022) 10:1996. doi: 10.3390/microorganisms10101996
65. Olajide OA, Iwuanyanwu VU, Lepiarz-Raba I, Al-Hindawi AA. Induction of exaggerated cytokine production in human peripheral blood mononuclear cells by a recombinant SARS-coV-2 spike glycoprotein S1 and its inhibition by dexamethasone. Inflammation. (2021) 44:1865−77. doi: 10.1007/s10753-021-01464-5
66. Colás-Algora N, Muñoz-Pinillos P, Cacho-Navas C, Avendaño-Ortiz J, de Rivas G, et al. Simultaneous targeting of il-1-signaling and il-6-trans-signaling preserves human pulmonary endothelial barrier function during a cytokine storm-brief report. Arterioscler Thromb Vasc Biol. 43(11):2213–22. doi: 10.1161/ATVBAHA.123.319695.
67. Tengesdal IW, Dinarello CA, Marchetti C. NLRP3 and cancer: pathogenesis and therapeutic opportunities. Pharmacol Ther nov. (2023) 251:108545. doi: 10.1016/j.pharmthera.2023.108545
68. Zhao M, Mishra L, Deng CX. The role of TGF-β/SMAD4 signaling in cancer. Int J Biol Sci. (2018) 14:111−23. doi: 10.7150/ijbs.23230
Keywords: acute respiratory distress syndrome, vasculopathy, caspase-1, cytokines, bronchoalveolar fluid, COVID-19
Citation: Cambon A, Guervilly C, Delteil C, Potere N, Bachelier R, Tellier E, Abdili E, Leprince M, Giani M, Polidoro I, Albanese V, Ferrante P, Coffin L, Schiffrin M, Arnaud L, Lacroix R, Roque S, Forel J-M, Hraiech S, Daniel L, Papazian L, Dignat-George F and Kaplanski G (2025) Caspase-1 activation, IL-1/IL-6 signature and IFNγ-induced chemokines in lungs of COVID-19 patients. Front. Immunol. 15:1493306. doi: 10.3389/fimmu.2024.1493306
Received: 08 September 2024; Accepted: 11 December 2024;
Published: 15 January 2025.
Edited by:
Jian Zheng, University of Louisville, United StatesReviewed by:
Chiara Colarusso, University of Salerno, ItalyCopyright © 2025 Cambon, Guervilly, Delteil, Potere, Bachelier, Tellier, Abdili, Leprince, Giani, Polidoro, Albanese, Ferrante, Coffin, Schiffrin, Arnaud, Lacroix, Roque, Forel, Hraiech, Daniel, Papazian, Dignat-George and Kaplanski. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gilles Kaplanski, Z2lsbGVzLmthcGxhbnNraUBhcC1obS5mcg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.