 Shay Tzaban1Ori Stern1Elad Zisman1Galit Eisenberg1,2Shiri Klein1,2
Shay Tzaban1Ori Stern1Elad Zisman1Galit Eisenberg1,2Shiri Klein1,2 Shoshana Frankenburg1
Shoshana Frankenburg1 Michal Lotem1,2,3*
Michal Lotem1,2,3*- 1The Lautenberg Center for Immunology and Cancer Research, The Faculty of Medicine, Hebrew University of Jerusalem, Jerusalem, Israel
- 2Center for Melanoma and Cancer Immunotherapy, Sharett Institute of Oncology, Jerusalem, Israel
- 3Hadassah Cancer Research Institute, Hadassah Hebrew University Medical Center, Jerusalem, Israel
Alternative splicing (AS) is a mechanism that generates translational diversity within a genome. Equally important is the dynamic adaptability of the splicing machinery, which can give preference to one isoform over others encoded by a single gene. These isoform preferences change in response to the cell’s state and function. Particularly significant is the impact of physiological alternative splicing in T lymphocytes, where specific isoforms can enhance or reduce the cells’ reactivity to stimuli. This process makes splicing isoforms defining features of cell states, exemplified by CD45 splice isoforms, which characterize the transition from naïve to memory states. Two developments have accelerated the use of AS dynamics for therapeutic interventions: advancements in long-read RNA sequencing and progress in nucleic acid chemical modifications. Improved oligonucleotide stability has enabled their use in directing splicing to specific sites or modifying sequences to enhance or silence particular splicing events. This review highlights immune regulatory splicing patterns with potential significance for enhancing anticancer immunotherapy.
Introduction
Biologists have long been puzzling how the human genome, which bears considerable similarity to lower eukaryotes, is responsible for the complex, sophisticated organisms it creates. Following Sharp and Roberts’ description of RNA splicing, Gilbert, in 1978, hypothesized that alternative splicing (AS) might be the missing layer that leads to the immense protein diversity despite the only 23000-gene human genome.
RNA splicing is a “cut and paste” process, removing introns and rejoining exons from the primary gene transcript, the pre-mRNA. The process relies on the biochemical uniqueness of RNA, which DNA lacks, of extensive flexibility and intrinsic catalytic activity. Small nuclear RNAs that assemble sequentially are directed to conserved sequences in the 5’(GT) and 3’(AG) splice sites on the primary transcript in an orderly manner. Together, the small RNAs and numerous proteins form the spliceosome. An adenosine in the intronic segment performs a nucleophilic attack on the 5′ end, cleaving the 5′ nucleotide (generally the “G” in a GT); a loop is then formed and removed. Following, the exon upstream of the removed intron is ligated to the 5′ end catalyzed by the spliceosomal RNAs and the ribonuclear proteins (RNPs).
Alternative splicing produces variants that differ from the constitutive RNA transcript. It occurs parallel to the transcription process and produces several isoforms from one gene. Each isoform may lack an exon or part of an exon from either side of the constitutive exon. This pattern, called ‘cassette-type alternative exon’ or ‘exon skipping’, is the most common. Intron retention, uncommon in humans, occurs mainly in untranslated regions. See Figure 1 for common splicing patterns.

Figure 1. Alternative splicing patterns.
AS involves 95% of the genes (1). With deep RNA sequencing becoming a more common read-out in experimental systems and longer RNA reads being produced, it is now clear that the pattern of RNA splicing is dynamically regulated and constantly changes (2).
The ratio between a constitutive transcript and its alternatively spliced isoforms depends on splice site recognition, its occupancy by spliceosomal RNPs, and regulatory RNA binding proteins (RBPs) (3). These RBPs bind or complement cis-sequences on the premature transcript on the intron or the exon. Sequences that promote spliceosome assembly at a splice site are called enhancers, and their RBPs are usually serine arginine-rich proteins. Sequences that reduce splice site recognition (silencers) attract heterologous nuclear RNPs (hnRNPs) (4). It is thought that enhancers usually act to generate constitutive mRNA, while the silencers yield AS isoforms. However, how the basic mechanism of AS varies from cell to cell and what determines it remains to be further elucidated.
Here, we focus on the role of AS in the T-cell immune response, particularly on anticancer immunity. This review aims to draw attention to new therapeutic opportunities in the functional distinctions between a constitutive protein/receptor and its splice isoforms. The motivation to unveil the intriguing mechanisms by which AS amplifies and regulates immune functions relies on reports from our group and others that RNA transcripts of the same immune gene can act in different directions or magnitudes in the immune context (5, 6). Thus, AS is an essential layer of immune regulation and a potential therapeutic target.
Alternative splicing is a mechanism of dynamic adaptability
Splicing event regulation
Although the prime outcome of AS is the fold increase in functionally distinct proteins compared to the number of genes, AS also plays a significant role in the most fundamental biological processes: evolution, differentiation, and adaptation. AS is a source of evolutionary development, a determinant of organ, tissue, and cell characteristics, and part of cellular adaptation to a changing environment (2).
The concerted manner by which protein production is shifted from one isoform to another yields the regulatory characteristics of AS. Its preferences differ among tissues and developmental states and respond to extracellular signals in a dynamic manner that precedes or synchronizes with gene transcription. In parallel to the dependency of intracellular processes on transcriptional activation, cellular events emerge from the shift in protein isoform ratios. How splicing events are concerted and what network cascades occur is a field of active research emphasizing health disorders and malignancies (7). The regulation of splicing events depends on both cis-acting regulatory sequences, located in introns or exons, and trans-acting splicing factor proteins that can strengthen or weaken the spliceosome’s recognition of the splice sites (8). These regulatory proteins belong to families of RNA-binding proteins, such as arginine–serine-rich (SR), heterogeneous nuclear ribonucleoprotein (hnRNP), and RNA-binding motif (RBM) proteins (9). They recognize specific regulatory sequences and enhance or inhibit the recognition of neighboring splice sites by the core splicing machinery (7). The expression level of the regulatory proteins is tissue- and state-specific (10), and they are subjected to regulatory splicing themselves (11).
From the evolutionary point of view, alternative splicing varies significantly among species. The insertion of multiple introns that separate exons has derived from ancestral genes and predated AS in eukaryote development. The option to skip exons was enabled by DNA mutations that may have resulted in splice sites with weaker binding affinity for spliceosomal components such as U1 small nuclear ribonucleoprotein (snRNP) (12). While the emergence of alternative splice sites contributed to protein diversity and is partly unique to a species, particularly when changing the reading frame, comparative genomics indicates that sequences that regulate RNA binding proteins are conserved and shared (13, 14).
Over 200,000 identified isoforms are reported in genome databases, and the majority of them lack functional annotation. Some well-studied examples show how events of retention or exclusion of specific domains may change protein cell localization, membrane anchorage, shedding of ectodomains, mRNA stability, and translational efficiency. The molecular alterations that emerge from AS may occur without any change in the level of the general gene’s transcript or before a change (15). Furthermore, the translational changes in reading frames may produce diverse translation outputs (13) and even insert poison exons, resulting in nonsense-mediated mRNA decay (NMD) and diminished protein levels (16). We can conclude that alternative splicing is timed and regulated in a manner that is not necessarily dependent on active simultaneous gene transcription.
Alternative splicing in T lymphocytes
T-cell states are associated with preferential expression of specific splicing isoforms. A unique characteristic of T lymphocytes is that they transform within minutes from a stationary naïve or inactive state to intense activity. In their effector state, T cells must adapt to synthesize large amounts of cytokines, migrate, proliferate, lyse target cells, and address accelerated metabolic needs. Already in 2006, it was found that memory T-cells respond to antigenic stimuli faster than naïve cells by omitting exons 4, 5, and 6 from the extracellular part of the membrane phosphatase CD45. CD45 is expressed in T and B cells and, in its constitutive, full-exon inclusive state, is referred to as CD45RA. The CD45RO variant shows variable exclusion of exons 4, 5, and 6. CD45 dephosphorylates both inhibitory and costimulatory tyrosines of the Src-family kinases (17). Oberdoerffer et al. showed that the transition from the RA to the RO form depends on the activity of the splicing factor hnRNPLL (heterogeneous ribonucleoprotein L-like) (18). HnRNAPLL was suggested to be a master regulator in activated T cells, affecting not only CD45.
Before and in parallel to CD45, specific gene isoforms impacting T-cell function were discovered. Interesting events recorded in activated T-cells included the short isoform of CD28, which induces faster activation (19), splice variants of CD44 and CTLA4, which correlated with a higher risk of autoimmune disease (20, 21), and MALT1A, a paraprotease that integrates TCR activation with the downstream IKK/NF-κB pathway. Reminiscent of CD45, naïve T-cells express MALT1B, a splice isoform missing exon 7, while activated T-cells express MALT1A, which includes exon 7 and is associated with rapid NF-κB signaling and improved lymphocyte function (22, 23).
A landscape view of AS in immune cells was offered by Lynch et al. in 2004, preceding a complete landscape of the comprehensive gene involvement in this phenomenon (22). Although the list of spliced genes described to regulate lymphocyte activation was restricted, the diverse array of functions governed by splicing suggested the substantial ubiquity of this process (23).
In the last few years, analyses have focused on gene families and activation cascades as it becomes clear that AS affects most genes. An example is the production of anti-apoptotic splice isoforms of members of the BCL2 gene family in activated T-cells. Adding costimulation via CD28 increased the ratio of anti-apoptotic splice variants and augmented T-cell proliferation. Interestingly, the genes that displayed significant changes in their splice isoform ratios did not have the highest expression levels (24).
The concept of AS-induced effector transition is not limited to activated T lymphocytes but also plays a critical role in B cell affinity maturation. In these processes, poly-pyrimidine tract binding proteins PTBP1 and PTB3 are splicing factors that drive the appropriate expression of gene sets required to adapt B lymphocytes to antibody-producing cells (25).
Splicing events that generate soluble isoforms of immune receptors
A prevalent splicing pattern observed in immune receptors gives rise to soluble isoforms that lack membrane anchorage and are secreted into the extracellular space. These soluble receptors may regulate signaling cascades, which differ from those initiated by their parental receptor (Table 1). Most prominently, the soluble receptors can function as a decoy of their corresponding ligands and compete with their constitutive, membrane-bound forms (26–35). The ratio between the membranal and the soluble isoforms of a receptor can remain fixed (26, 36). However, it might change depending on the cell’s metabolic, functional, or differentiation state (37, 38). Diverting the pre-mRNA splicing towards the soluble isoforms results in reduced expression of the membrane-bound receptor and can even negate its cellular effect. In a different context, the soluble receptors may have agonistic effects (28, 39) and initiate reverse signaling by binding to other receptors (40–42). For example, glucocorticoid-induced tumor necrosis factor receptor (GITR) ligand that is expressed on plasmacytoid dendritic cells prompts a reverse signal that initiates noncanonical NF-kappaB-dependent induction of indoleamine 2,3-dioxygenase upon binding to soluble GITR. This leads to the tryptophan catabolism immunoregulatory pathway (43). In addition, soluble isoforms have been documented to bind with their ligand to distinct membranal partners, activating trans-signaling pathways (44, 45). Furthermore, some soluble receptors stabilize their ligand configuration or alter their biodistribution (31, 45–47). Another typical example of important alternative splicing of immune cells is the removal of the hydrophobic transmembranal segment of the B-cell receptor to form a secreted immunoglobulin (48). Interestingly, soluble receptors may exert different functions (i.e., agonistic and antagonistic) depending on their concentration (45, 46, 49) (Figure 2).
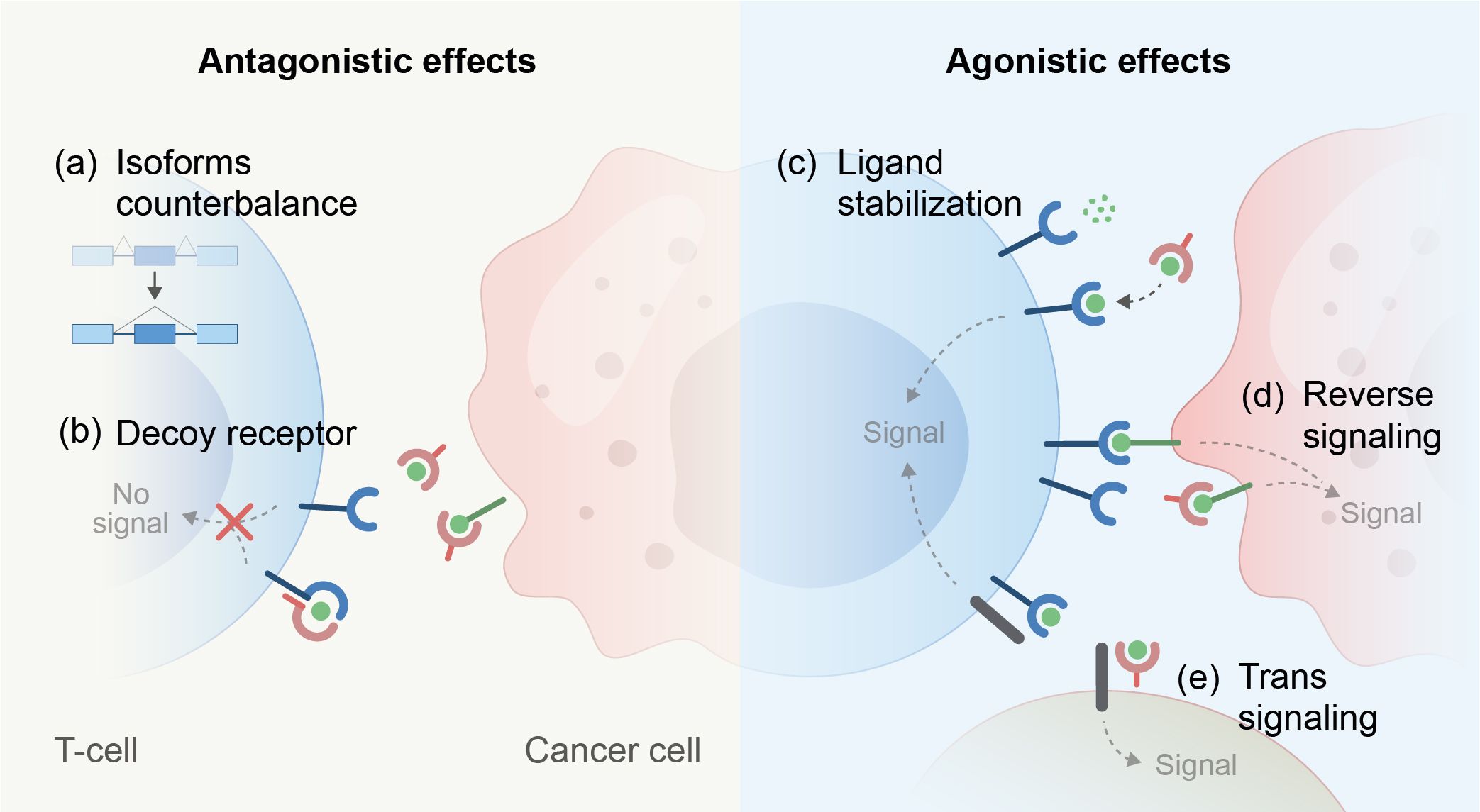
Figure 2. Mechanisms that lead to antagonistic or agonistic effects of soluble ectodomains derived from immune regulatory receptors. (A–E, mechanisms depicted).
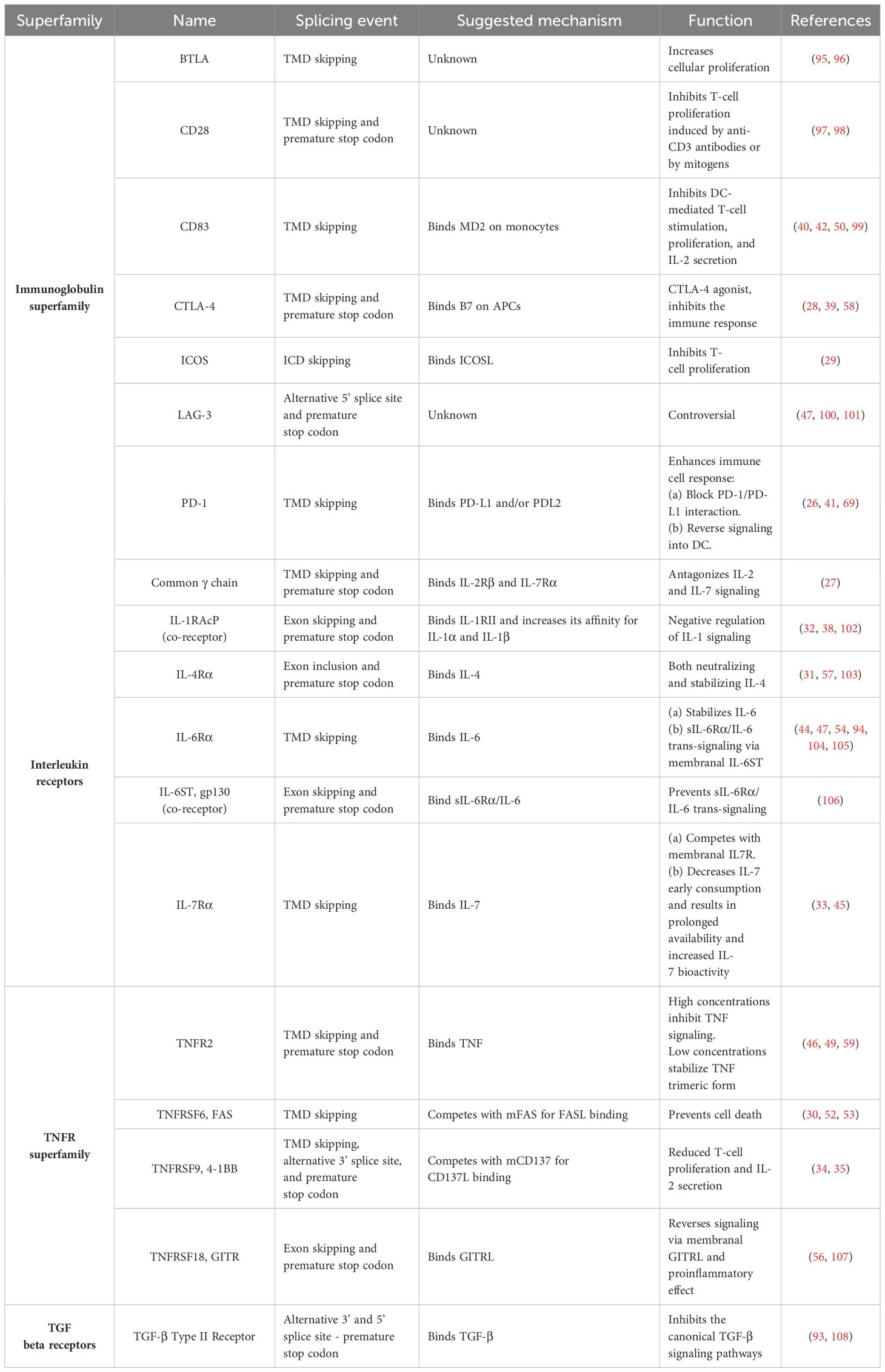
Table 1. Soluble T-cell immune receptors due to alternative splicing.
The common splicing patterns that lead to the generation of soluble isoforms of membranal receptors include (1) Transmembrane domain (TMD) skipping: In the process of alternative splicing, skipping of the transmembrane encoding exon results in the creation of a soluble product encompassing both the intracellular and extracellular domains (26, 33, 50–55); (2) Alternative terminator: a shortened soluble isoform is encoded by a sequence that includes a mutually exclusive exon containing an alternative polyadenylation site, or by alternative splicing that results in frameshift and premature stop codon (48, 56, 57). As a result, the translated proteins include only the extracellular domain, lose their membrane anchorage and become soluble (27, 58, 59).
It should be noted that alternative splicing is not the sole mechanism that creates soluble receptors. These segments can also be made by proteolytic cleavage of extracellular domains by proteases in the extracellular matrix (Figure 3). However, unlike AS, the intracellular domain (ICD) of a cleaved receptor remains anchored and theoretically may retain its effect. The impact of a truncated signaling domain is diverse or unknown. Typical examples of receptors that utilize both mechanisms to produce their soluble formats are cytokine receptors, including the TNF and TNFR superfamily (59, 60). In addition, some immune receptor genes lack the transmembrane domain and are, therefore, constitutively expressed as soluble receptors with linked intracellular and extracellular domains. They mainly function as decoy receptors (61). For example, decoy receptor 3 (DcR3, TNFRSF6B) is a secreted TNFR superfamily member that lacks a transmembrane domain. DcR3 can interrupt FAS-FASL interaction by binding FASL and inhibiting FASL-induced apoptosis (62).
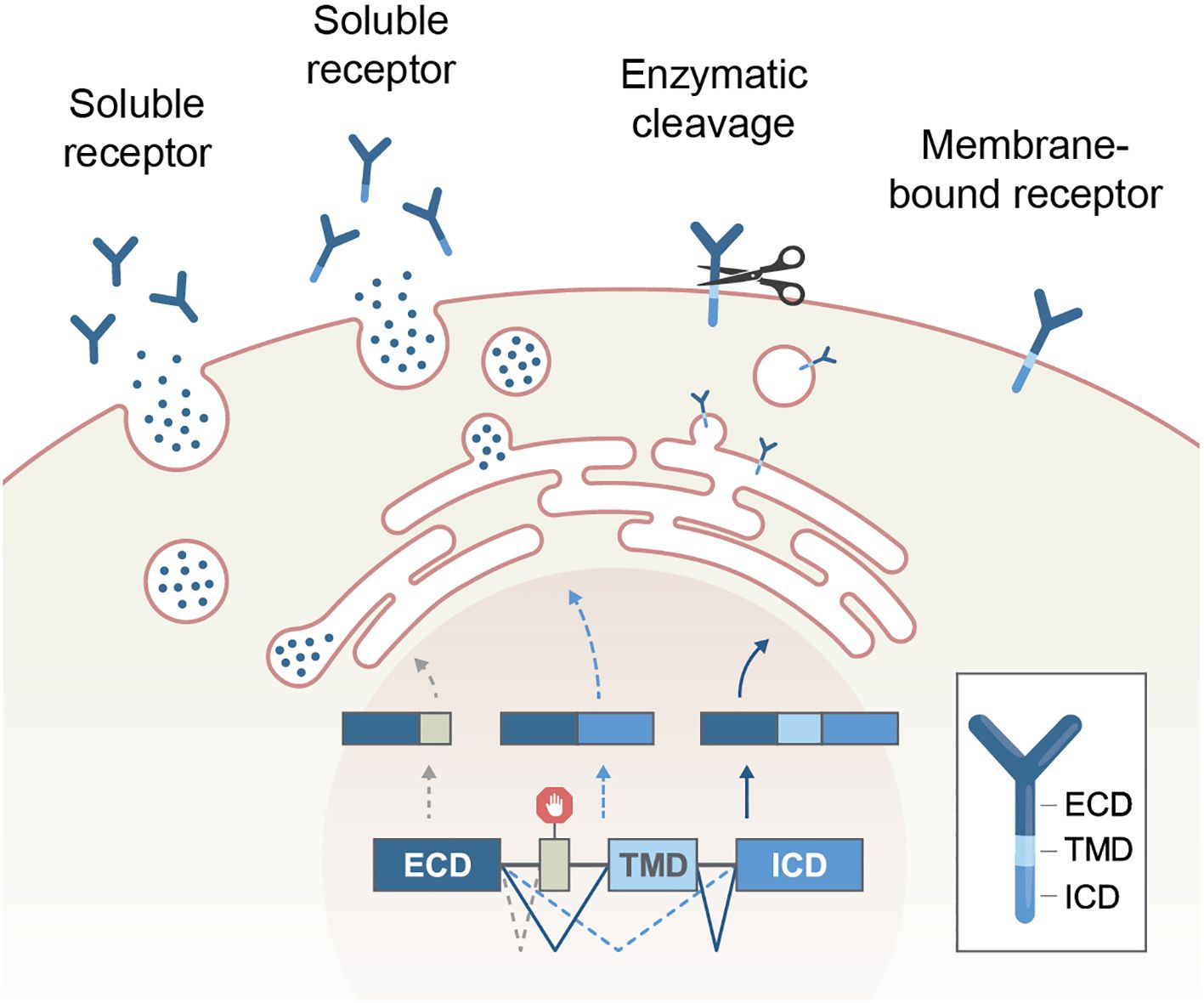
Figure 3. Production of soluble receptors by alternative splicing or enzymatic cleavage. Of note, the intracellular part of a receptor remains only in the cleavage process. ECD, extracellular domain, TMD, transmembrane domain, ICD, intracellular domain.
Alternative splicing of the immunoglobulin superfamily
The immunoglobulin (Ig) superfamily is a large group of proteins with a common Ig domain. The Ig superfamily is critical in the immune response networks (63). Some Ig superfamily receptors can be translated to a soluble form by an alternative splicing process (64, 65). Among these are CTLA4 (39, 58), CD83 (42, 66), and LAG3 (66). Here, we will focus on two specific examples: The programmed cell death receptor PD-1 and the B cell receptors (BCRs) that convert, after splicing, into immunoglobulins (antibodies).
PD-1
Following stimulation, PD-1 is expressed on T-cells in a membrane-bound form (mPD-1). When it binds to its ligand (PD-L1), mPD-1 inhibits the effector functions of T- cells, promotes apoptosis, and restricts proliferation (67, 68). PD-1 exon 3, which encodes the transmembrane domain, can be skipped by alternative splicing, generating a soluble receptor form (sPD-1). The ratio between the two isoforms is consistent during T-cell activation (36). sPD-1 can act as a decoy receptor and compete with the PD-1 receptor on the interaction with the ligands PD-L1 and PD-L2, and block the interaction of PD-L1 with B7-1 (69). The shedded PD-1 ectodomain exerts a similar effect, suppressing the PD-1 inhibitory function (70, 71). It has been speculated that sPD-1 has a reverse signaling effect when binding to PD-L1 on dendritic cells (41).
Immunoglobulins can be membrane-bound or secreted as antibodies. Naive B cells express membrane-bound receptors, usually from the IgM class. Following stimulation, the B cell receptors undergo alternative splicing via an alternative terminator mechanism. As a result, the carboxy terminus no longer contains the hydrophobic transmembrane domain but, instead, has a hydrophilic secretory tail. The secreted antibodies play a crucial independent role during the immune response (48).
Alternative splicing of the TNF-receptor superfamily
The TNFRSF comprises trimeric receptors made of three homologous molecules that initiate signaling pathways involved in inflammation, proliferation, differentiation, cell migration, and induction of cell death (72). Like the Ig superfamily, TNFRSF members share similar splicing patterns that result in soluble isoforms.
FAS (CD95, TNFRSF6) is one of the best-known members of the TNF receptors superfamily. It is abundantly expressed in many tissues, including the gastrointestinal tract, the respiratory system, and lymphoid tissues (73–75). FAS is mainly known for its pro-apoptotic pathway activation following FAS ligand (FASL) binding (76). However, it also has other functions, e.g., it takes part in the differentiation of naïve T cells to memory cells (77). FAS is robustly expressed on T-cells and has an apoptosis-inducing role during T-cell development (78) and activation (79–81). A specific alternative splicing event is the skipping of exon 6, which encodes the transmembrane domain of FAS, resulting in a soluble form of the FAS receptor (52, 53). The FAS exon 6 skipping mechanism has been studied extensively. It has been shown that many splicing factors can regulate this event, among them TIA-1 (82), PTB (82), HuR (83), hnRNP A1 (84), SRSF4 (85), SRSF7 (86), and SRSF6 (87). Similarly to PD-1, the soluble FAS receptor competes with membrane-bound FAS for FASL ligation, thereby limiting FAS signaling (30, 52). Bajgain et al. described the ability of secreted FAS extracellular domain to enhance CAR T-cell antitumor activity against a FAS-ligand-expressing tumor (88).
TGFβ (transforming growth factor beta receptor) TGFβ is of special interest because it controls immunity via a rich network of cells and mediators, with the end result being immune evasion of the cancer tissue. The biological functions of TGFβ are mostly mediated by the monomeric, soluble form of the protein. The monomer is cleaved by proteases in the Golgi complex and later released from glycoproteins that ligate it in a non-covalent manner (89). TGFβ enhances the expansion of regulatory T cells (Tregs), the inhibition of NK and effector T cells, and the induction of immune suppressive cytokines including IL-4 and IL-10. Active TGFβ exerts its effect via receptors that activate SMAD transcription factors, a family with hundreds of regulatory elements. Tumors exploit TGFβ to induce a supportive stroma that weakens the immune response by acting as a mechanical barrier and expressing inhibitory membranal ligands, such as PD-L1 (90, 91). In a series of patients with gynecological cancers who received immune checkpoint inhibitors, a high TGFβ expression score correlated with treatment failure and reduced survival (92). The type II receptor for TGFβ has a splicing variant which lacks the transmembrane domain, and exert a higher binding affinity to the three sub-types of TGFβ. By doing so, it competes with the natural ligands and reduces fibrotic pathology (93).
Type I cytokine receptors
In addition to the Ig and TNFR superfamilies, members of other immune receptor families can generate soluble forms through alternative splicing, including type I cytokine receptors IL6Rα (54, 94), IL-4Rα (57), and IL-7Rα (33). Another example is the common γ chain IL-2, 7, and 15 cytokine receptors, for which skipping exon 6 encoding the transmembrane domain results in a frameshift and a premature stop codon. The resultant proteins contain only the extracellular domain. The soluble IL-2 and IL-7 receptors were reported to impair T-cell signaling and function (27).
Pathology of splicing and alternative splicing
Splicing is an imperative regulator of most cellular functions. Therefore, disrupted splicing regulation can lead to different pathologies, depending on the involved tissue and the protein products of the aberrant transcript. The most investigated pathologies that result from erroneous splicing events include neurodegenerative disorders and cancers. The first arise from germline mutations, while the latter arise from somatic genome aberrations. However, splicing-related mutations can cause many other disorders, such as dilated cardiomyopathy and Marfan syndrome (109, 110).
It remains a mystery why germline splicing-related mutations primarily affect the brain. One theory holds that alternative splicing is crucial in determining the neural cell state (19) and that neural tissue is rich in tissue-specific splicing events. However, not all splicing-related mutations in neural cells lead to a change in alternative splicing. An example of this is Duchenne muscular dystrophy (DMD), where a deletion of an exon leads to the production of a truncated protein via the process of nonsense-mediated decay (NMD) rather than a new isoform of the original protein (111).
Some argue that 15-50% of pathological mutations affect gene splicing (9–11). Nevertheless, these diseases are not regarded as splicing-related disorders since mutations that do not change the coding sequence are typically misclassified as allelic variations (112–114). In addition, the wide use of exome sequencing, which filters out most intronic parts, introduces an inherent bias underscoring splicing mutations (115–118).
Dis-regulated splicing leading to neurodegenerative disorders
Neurodegenerative diseases are a group of disorders caused by the gradual loss of neuronal cell function or structure. Strikingly, splicing-related mutations are one of the leading causes of many neurodegenerative diseases (119). The most investigated neuronal disorder instigated by splicing is spinal muscular atrophy (SMA). Nonetheless, most neural pathologies, such as early-onset Parkinson’s (119–121), Alzheimer’s disease (122), familial dysautonomia (123), and Amyotrophic Lateral Sclerosis (ALS), could evolve from splicing mutations (124, 125).
Given the significant number of neurodegenerative disorders caused by mutations impacting RNA splicing, it is not surprising that there have been numerous efforts to investigate the use of splicing-editing techniques as a treatment option for these conditions. For example, antisense oligonucleotides (ASOs) are being widely researched for their potential use in treating SMA, DMD, and ALS (126). The use of ASOs to manipulate alternative splicing is further discussed elsewhere in this review. The use of CRISPR-Cas9 to affect alternative splicing has been suggested for Duchenne muscular dystrophy (127), and spliceosome-mediated RNA trans-splicing (SMaRT) (128) has been tested in Huntington’s disease (129), DMD (130), and Alzheimer’s disease (131).
Dis-regulated splicing causing cancer and immune evasion
Splicing-related mutations in cancer can be grouped into three categories: 1-those affecting the core spliceosome complex, resulting in new isoforms; 2-those impacting splicing factors, affecting the expression levels of multiple isoforms; and 3-those affecting splicing recognition sites, altering the expression level of a single gene or creating new isoforms (Table 2; Figure 4).
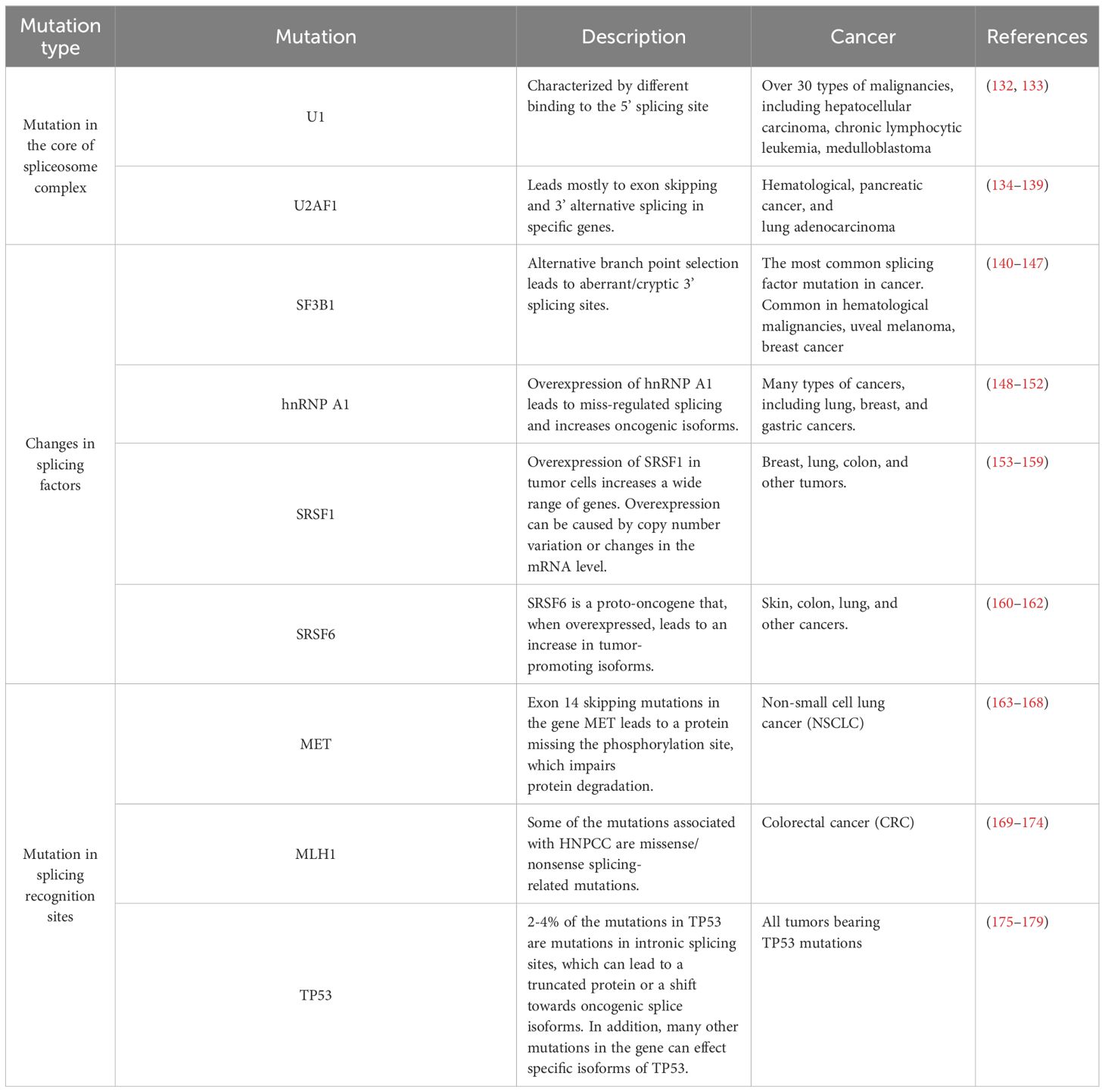
Table 2. Specific mutations associated with splicing dis-regulation in human cancers.
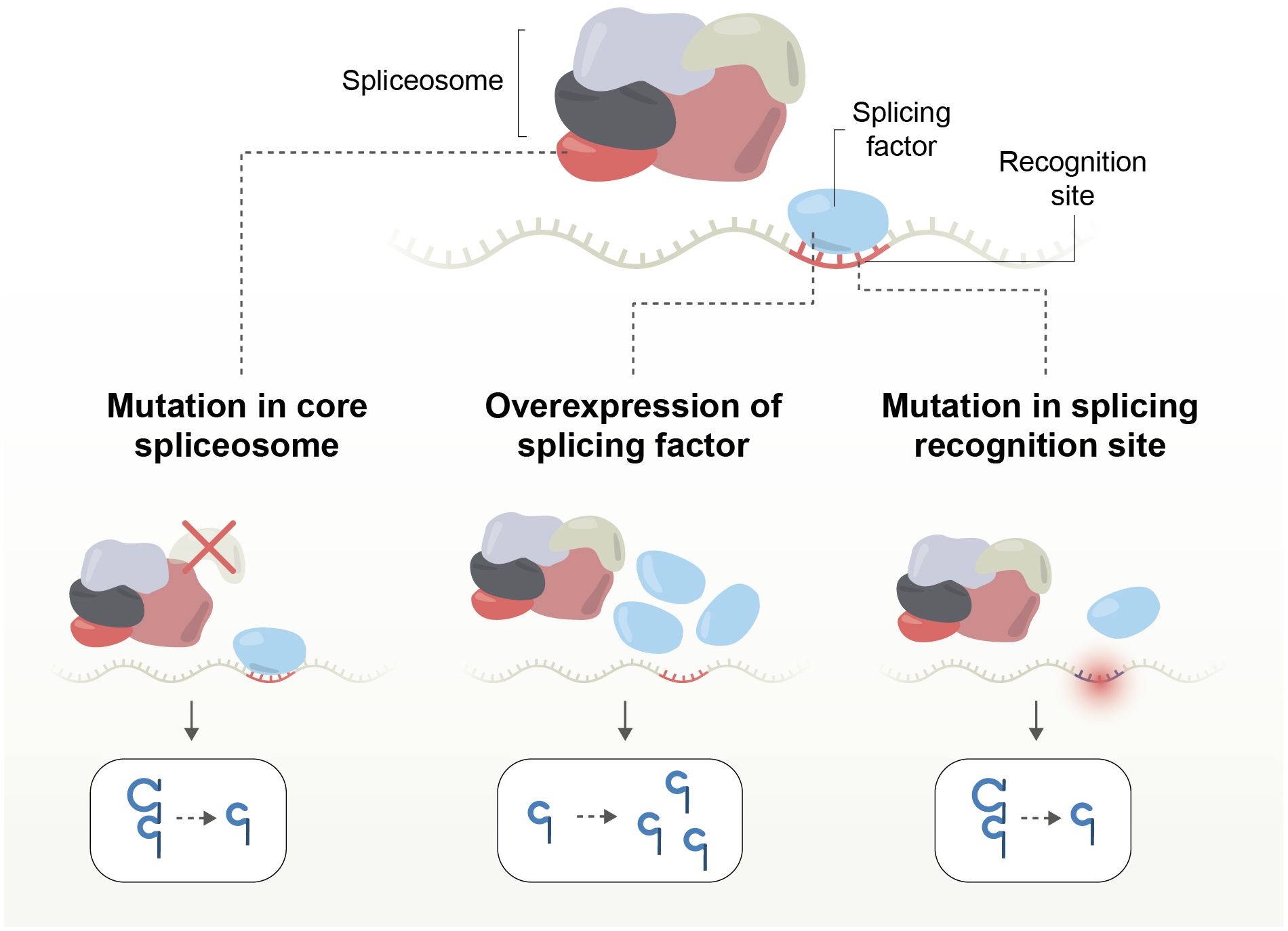
Figure 4. Mechanisms of splicing disruption by mutations affecting the core spliceosome complex; splicing factors, or splicing recognition sites, altering the expression level of a single gene or creating new isoforms.
Splicing factors mutations are particularly prevalent in myeloid neoplasms; for example, SF3B1, that increases anti-apoptotic isoforms, enhances tumor proliferation and progression, and is associated with poor survival of patients (134, 140, 141, 180, 181). U2AF1 is another splicing factor mutated in myeloid malignancies that drives altered splicing preferences. Intronic mutations are more frequent than exonic, and a third of somatic mutations in the exon-intron boundary are associated with splicing changes. If a mutation occurs in a 5’ or 3’ splicing site, there is a greater than 50% chance of it leading to a splicing shift (182).
Splicing can be employed as a cancer treatment approach in various forms: using single-stranded oligonucleotides to change the splicing of specific genes and switch between oncogenic and tumor-suppressing forms, as has been demonstrated for the BCL gene (67); regulating specific splicing factors through drugs that directly impact them, such as blocking SF3B1 (68); or by attacking the pathway which the mutant splicing factor exploits. Thus, tumors with driver mutations in SF3B1 or U2AF1 may be vulnerable to NMD inhibition (68–72). Some widely used therapies, such as camptothecin and cisplatin, have been found to impact RNA splicing, potentially contributing to their efficacy (73–75).
As discussed in the following paragraph, recent attention has focused on the generation of neo-antigens by including erroneous transcripts. However, altered splicing and the emergence of usually unexpressed isoforms independently impact tumor immunogenicity. These effects often hinder the anticancer immune response. For example, HLA tumor-enriched alternative splicing events occur in 10-30% of lung and breast cancers, affecting MHC expression. When HLA expression is inconsistent, the ability of tumor epitopes to be presented and recognized is diminished or completely lost (183). In ovarian cancer, certain splicing factors, such as BUD31, SF3B4, and CTNNBL1, may indirectly support immune evasion (184). This immune escape may involve increased PD-L1 expression and primary resistance to PD-1 inhibitors. Such mechanisms are seen in clear renal cell carcinoma, where an exon-including splicing event in the chromatin remodeling gene PBRM1 contributes to immune evasion (185).
Generation of cancer neo-antigens by mutations in splicing factors
While reports indicate that altered splicing isoforms contribute to tumor immune evasion, splicing alterations are now attracting significant interest as a source of cancer antigenicity. This interest stems from the potential of AS to drive isoforms that include retained intronic sequences. These intronic transcripts, in turn, may form neoantigens—peptide sequences that have not had the opportunity to tolerize the immune system. Such newly transcribed sequences hold the potential for generating protective immunity and improving clinical responses to immune checkpoint inhibitors (61–64).
Several pharmacological compounds have been used that either degrade splicing factors, disrupt spliceosome assembly, or inhibit nonsense-mediated decay (186, 187). One example is indisulam, an anticancer sulfonamide that generates aberrant transcripts. Interestingly, indisulam does not directly inhibit cancer growth; instead, it triggers a T-cell response against cryptic sequences from abnormal RNA, which impedes tumor progression. Other splicing-disruptive compounds, such as pladienolide B and H3B-8800, are currently being evaluated in experimental systems and clinical trials for myeloid neoplasms. Predicting the effect of splice manipulation on the tumor microenvironment is challenging, but as will be discussed, induction of soluble ectodomains from immune-modulatory receptors may interfere with immune checkpoint inhibitors. Soluble PD1, for instance, may saturate PD-1 blocking antibodies and reduce their availability to rescue exhausted antitumor T cells (188).
RNA sequencing for splicing analysis
The technology developed to sequence RNA and obtain long transcript reads that capture added or missing nucleotides was crucial to assessing AS in health and disease.
Bulk RNA sequencing (RNA-seq) is mainly performed using two methodologies (Figure 5). The first is short-read sequencing, which can sequence RNA molecules in reads of up to 301 base pairs (bp), for which the Illumina platform is commonly used. The second is long-read sequencing, known as “third-generation sequencing.” This method can sequence up to 26,000 bp RNA molecules in the NanoporeTM platform (189). Long-read sequencing has the advantage of identifying full-length transcripts derived from each gene. However, this sequencing method had an accuracy of only 90% and is, therefore, error-prone. Erroneous sequencing interferes with the alignment of the reads to a reference genome and thus can miss sutured exons and their splice junction, an important feature required to determine the splicing pattern (190). However, recently, Nanopore announced that its sequencing accuracy has increased to 99.9%.
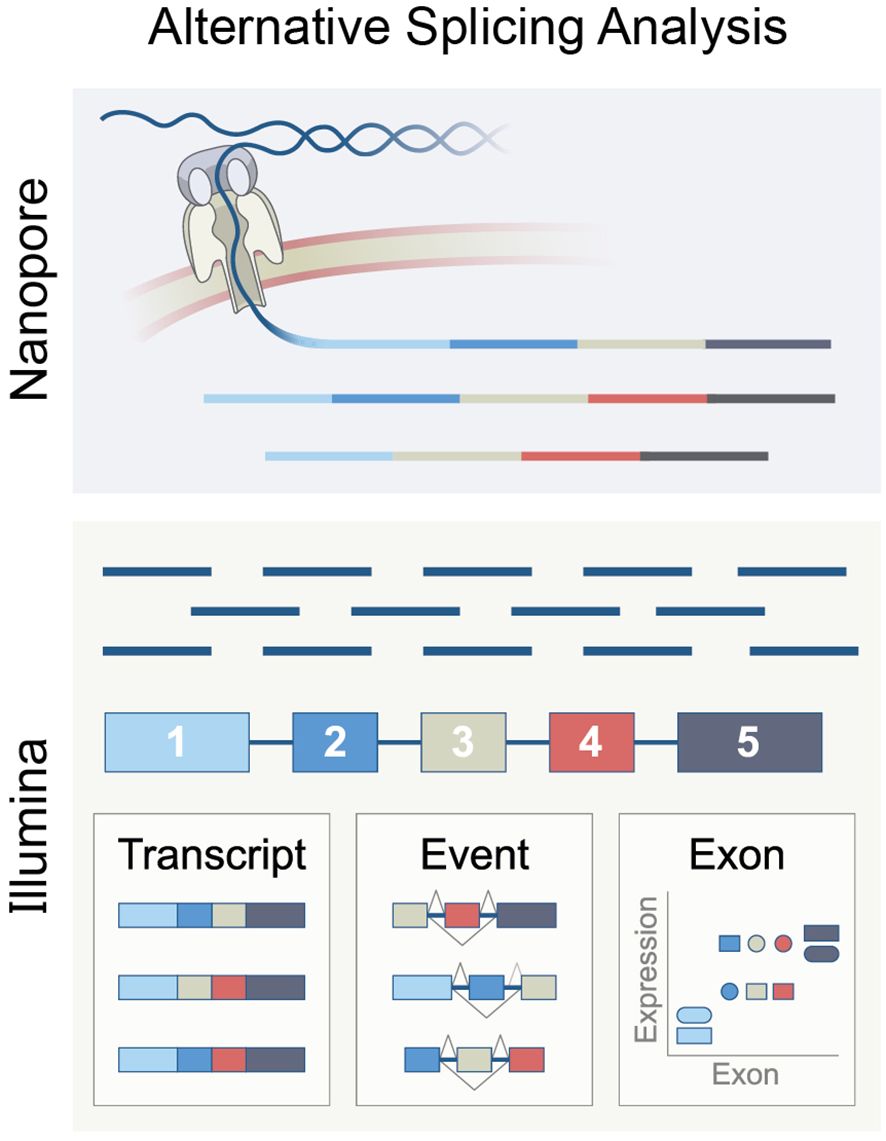
Figure 5. The principle of RNA splicing analysis using Nanopore long-reads or Ilumina short-reads, representing methods based on exon, isoform, or event.
Since Illumina sequencing is well-established and widely used, most splicing analysis tools are designed for short reads. Analyzing bulk RNA-seq from Illumina data can be done in three ways. The first is determining the exon expression level and comparing its expression in varying biological settings or states. This method is called “exon-based”. The second method aims to deduce isoform expression from the short reads sequencing. This method is called “isoform-based”. The third approach, called “event-based,” computes the relative inclusion of an exon between two exons. This approach utilizes reads of splice junctions that overlap at least two exons.
A comparison of the main computational tools based on these three methods concluded that the event-based and exon-based tools while having a relatively low overlap, seem to work the best. It is suggested that concurrent use of the two methods yields the optimal splicing map of a given cellular population (191).
Another critical parameter to consider when performing splicing analysis is the quality of the RNA-seq data. In this regard, two features need to be accounted for: the depth of the sequencing and the length of the reads. Mehmood et al. (191) have noted that a depth between 40-60 million reads per sample will be sufficient for a robust splicing analysis. When considering reading length, 100 bp reads were the threshold for thoroughly detecting splicing junctions (192). It is also advised to sequence the data using paired-end sequencing to increase the read length.
Extracting splicing data from single-cell RNA-seq is even more complex. In general, to apply splicing analysis tools, the samples must be produced to capture the full transcript. However, most single-cell RNA-seq technologies are based on a 3’ or 5’ capturing of the RNA molecule. As a result, while preparing the sequencing libraries, only the transcript’s end is included; thus, there are limited options for splicing analysis (193). The main exception to these technologies is Smart-seq sequencing, which captures reads from all over the transcript. This technology enables splicing analysis with the limitation of read depth and length. This was demonstrated with the single-cell splicing analysis tool ‘Expedition.’ In their study, Yan Song et al. (194) used Smart-seq2 sequencing with a mean of 25 million reads per cell and a 100 bp read length. In comparison, 10XGenomics™ recommends a sequencing depth of 20,000-50,000 reads per cell and a read length of 28 bp (195); this is shallow sequencing compared to Yan Song’s analysis. To overcome these problems, a new single-cell long-read RNA sequencing technology based on Pacific Bioscience’s sequencing, called MAS-ISO-seq, was recently introduced. This technology is still new and needs further investigation. Furthermore, a joint project of Nanopore and 10x Genomics produced long-reads in single-cell RNA sequencing (196).
Splicing modification using antisense oligonucleotides
During the 70s, evidence accumulated for the promising ability of small RNA molecules to control translation processes (197–199). Paterson et al. were the first to generate a translation-inhibiting system based on a complementary mRNA-DNA hybrid, resulting in reversibly arrested β globin translation (199). In 1978, Paul Zamecnik and Mary Stephenson used synthetic DNA against RSV (200). Their 13-nucleotide product hybridized with the viral mRNA and prevented viral replication (201). Later, it was shown that the mechanism of action of the short, single-stranded oligonucleotides included RNase-H1 assembly to the DNA-mRNA hybrid, cleavage, and mRNA degradation (202). Despite the promising results, the research in this field was paused for a decade, mainly due to technical issues related to nucleotide synthesis (203, 204), skepticism about the ability of nucleic acids to enter target cells, and restricted knowledge of the human genome (205). The progress in these aspects re-ignited the research in nucleic acids-based manipulation. The chemically modified, short, single-stranded antisense oligonucleotides (ASOs) improved durability, cellular uptake, delivery, and post-transcriptional effects.
Chemical modifications
The advancement of chemical modifications of nucleic acids marked a significant milestone in the clinical application of this compound class. A key outcome was the development of splice-switching antisense oligonucleotides (SSOs), designed to modify alternative splicing patterns and enhance exon skipping. Specifically, chemical modifications that reduce RNaseH activity form a stable DNA-mRNA hybrid, preventing subsequent RNA degradation (206). These SSOs can be directed towards splice-site sequences, hindering and redirecting the spliceosome to an alternative splice site in the subsequent exon (207, 208) (Figure 6).
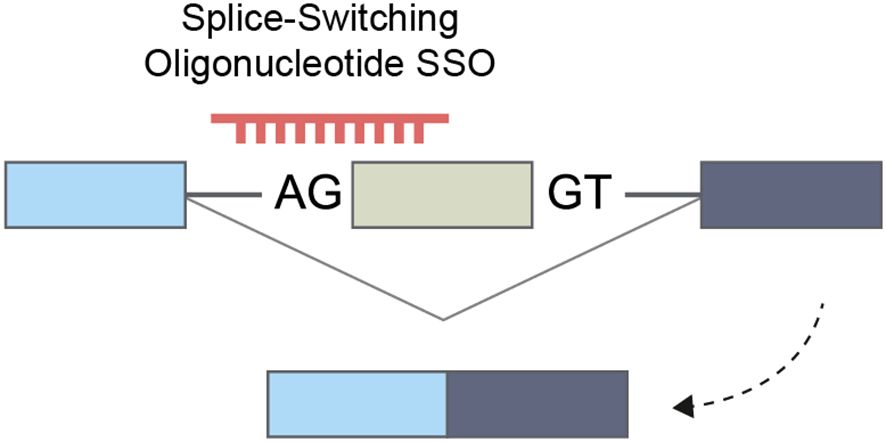
Figure 6. Splice-switching oligonucleotide that enhances exon skipping and increases the expression of an alternative isoform.
In addition to splicing alterations via complementation to splice sites, SSO can modify splicing by targeting splicing enhancers (ESE, ISE) or silencers (ESS, ISS) (209, 210). These interventions may interrupt splicing by inhibiting linkage to splicing factors, leading to exon exclusion or inclusion. In addition to whole exon skipping, the pre-mRNA splicing modulation can result in intron retention, alternative 5′ and 3′ splice sites, alternative promoter, or alternative polyadenylation sites (209).
Finding SSO-targetable splicing motifs is not trivial. A systematic scan of the exon of interest is necessary to spot the precise sequence, which the SSO should complement to alter the wild-type splicing pattern.
Two types of chemical modification are currently used for FDA-approved drugs: 2′-O-methoxyethyl (2′-MOE) nucleosides with phosphorothioate (PS) backbone and phosphorodiamidate morpholino oligomers (PMO) with a N, N-dimethylamino phosphorodiamidate backbone (Figure 7).
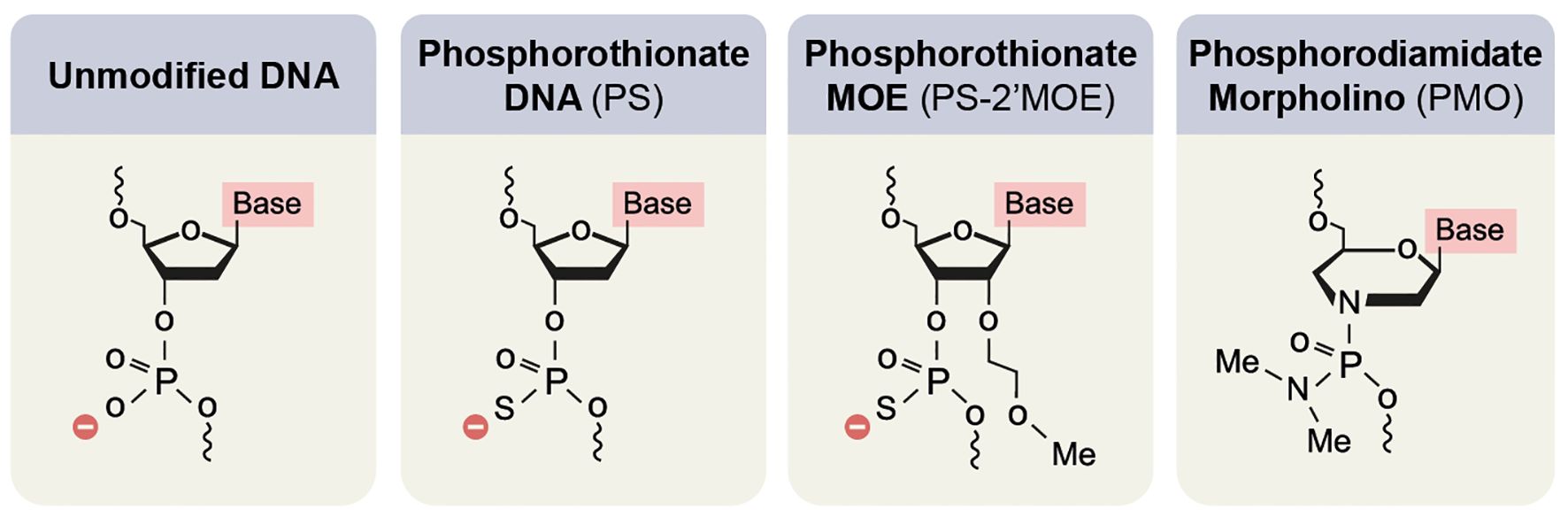
Figure 7. Main chemical modifications of nucleic acids to improve the clinical applicability of oligonucleotides.
2’-MOE belongs to a group of modifications in the 2’O of the furanose ring of the nucleic acid. Other prevalent modifications are 2′-O-methyl (2′-OMe), locked nucleic acid (LNA), and SSOs containing 2′-constrained ethyl (2′-cEt). Alongside the RNaseH1 resistance, the 2’O modifications increase the SSO affinity (211). High affinity is attributed to higher potency, longer half-life, and less immune-provoking properties (212, 213). 2’MOE modifications are usually accompanied by switching Oxygen in the backbone to Sulfur (PS). This switch decreases the SSO affinity but improves the resistance to nuclease activity (214, 215) and molecular binding to proteins – resulting in reduced kidney clearing (216) and improved uptake by target cells (217–219).
In PMO (220), a morpholino ring replaces the furanose ring. In addition, the negatively charged backbone is replaced by a N, N-dimethylamino phosphorodiamidate backbone. As a result of these changes, the SSOs have higher in vivo tolerance but faster kidney clearance, which requires a higher dosage (221, 222).
Although these are the main modifications currently used for SSO drugs, recent publications have shown how additional chemical modifications can further improve splicing modulation. For example, Langner et al. synthesized a hybrid that combines PMO modification with a PS backbone, which exhibits higher efficiency than 2’-MOE modification with the same backbone (223).
SSO-based drugs and clinical trials
Eighteen RNA-targeted oligonucleotide drugs have been approved, including five SSOs (206). The most advanced examples of clinical use of SSOs are in the field of genetic neuromuscular diseases.
The first SSO that the FDA approved is used for spinal muscular atrophy (SMA) treatment. The drug nusinersen, approved by the FDA in 2016, is an SSO with a 2′-MOE modification and a PS backbone. Nusinersen targets the splicing silencer located in SMN2 intron 7 pre-mRNA, and by blocking the binding of hnRNPA1 and A2, it promotes higher exon 7 inclusion, increasing the SMN2 protein synthesis (224, 225). The treatment results in prolonged survival and a dramatic improvement in motor development.
Other approved SSO drugs are used for the treatment of Duchenne muscular dystrophy (DMD). This severe, progressive muscle-wasting disease causes difficulty in movement and breathing and, eventually, early death. It is caused by mutations in the DMD gene, leading to impaired dystrophin protein production (226). In recent years, the FDA has approved four drugs based on a mechanism of SSO with PMO modification. The first drug approved, eteplirsen, was approved in 2016 and causes mutated exon 51 skipping (227). Three additional drugs that work in a similar mechanism have been approved in recent years for different mutations that lead to DMD: golodirsen, which causes exon 53 skipping, was approved in 2019 (228); viltolarsen, approved in 2020, also causes exon 53 skipping (229); and casimersen, approved in 2021, induces exon 45 skipping (230). To date, DMD is the only disease for which even modest, consistent clinical benefit has been shown using PMOs. Thus, PMO SSOs have demonstrated minimal and doubtful applicability in mammalian systems (227, 231).
Considering the achievements of SSOs in DMD and SMA, several groups have recently published promising data demonstrating the potential of ASO in other diseases. For example, Yang et al. (232) show the use of SSO to prevent a splicing pattern that arises from an alternative 3’ splice site between SYNGAP1 exon 10 and exon 11. This splicing pattern leads to nonsense-mediated decay (NMD). Mutations in this gene are a common cause of autism and intellectual disability. Using SSO with 2′-MOE modification increased the expression of the active protein in an in vitro system. Promising results for the use of SSO can also be seen in the treatment of Dravet syndrome (233), Huntington’s disease (234), and fragile X syndrome (235). Similarly, in cystic fibrosis, Oren et al. (236) and Michaels et al. (237) demonstrate the use of SSO that leads to mutated exon 23 skipping, increasing the expression of the CFTR protein.
SSO and cancer treatment
The use of SSO in cancer treatment is still in its early stages. There is currently no approved drug, but there are ongoing research studies. The primary approach for anticancer SSO is modulating the alternative splicing of oncogenes toward NMD, nonfunctional dominant negative isoforms, or isoforms with the opposite function.
For example, Dewaele et al. (238) used PMO-modified SSO for MDM4 exon 6 skipping, resulting in nonsense-mediated decay and rescue of MDM4’s target - the tumor suppressor protein p53. The SSO administration reduces diffuse large B cell lymphoma growth both in vitro and in vivo.
Using SSOs for translatable alternative splicing isoforms was shown in the human epidermal growth factor receptor 2 (HER2) case. HER2 is an oncogene and established therapeutic target in a large subset of women with breast cancer (239). Wan et al. (240) and Pankratova et al. (241) used SSOs to skip HER2 exons 15 and 19, respectively. The manipulations resulted in the upregulation of Δ15HER2, a HER2 inhibitor isoform, and Δ19HER2, a dominant negative isoform, leading to apoptosis and inhibition of proliferation.
Khurshid et al. (242) recently proposed using SSO for patients with rhabdomyosarcoma (RMS). In their article, the group describes the modification of the insulin receptor splicing pattern by targeting the binding site of the splicing factor CELF1. This prevents the skipping of exon 11, leading to an increase in the expression of the receptor in its full form (IR-B). The use of SSO in an RMS cell line system led to a decrease in proliferation, migration, and angiogenesis.
Manipulating cancer-associated metabolic programs using SSO was demonstrated by Wang et al. (243). The group found that elements in exon 10 of the pyruvate kinase M (PKM) gene influence the choice between the inclusion of exon 10 and exon 9. Exon 10 inclusion, the M2 isoform, is common in cancer tumors and is associated with their ability to switch to aerobic glycolysis (Warburg effect). The group demonstrated the possibility of using SSO for splicing modulation in favor of exon 9 inclusion and showed that the manipulation could lead to apoptosis of glioblastoma cell lines. Recently, the group showed a similar effect in a hepatocellular carcinoma mouse system (244). In summary, the use of SSOs to manipulate the immune system is still in its early stages. While there is significant progress in understanding the immune system at the molecular landscape, many complexities regarding the manipulation of T cells are yet to be unraveled.
Author contributions
ST: Conceptualization, Investigation, Methodology, Writing – original draft, Writing – review & editing. OS: Formal analysis, Investigation, Writing – original draft, Writing – review & editing. EZ: Data curation, Investigation, Methodology, Writing – original draft, Writing – review & editing. GE: Conceptualization, Supervision, Writing – review & editing. SK: Conceptualization, Investigation, Writing – review & editing. SF: Writing – review & editing. ML: Conceptualization, Project administration, Supervision, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. The preparation of this review, part of the work performed by the Lotem lab in Jerusalem and reported here, as well as publication expenses, are supported by Dr. Miriam and Sheldon G. Adelson Medical Research Foundation and the European Union’s Horizon 2021 Research and Innovation Programme under grant agreement no. 101057250.
Acknowledgments
Figures were created with HandMed by Dr. Yitzchak Yadegari.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Nilsen TW, Graveley BR. Expansion of the eukaryotic proteome by alternative splicing. Nature. (2010) 463:457–63. doi: 10.1038/nature08909
2. Baralle FE, Giudice J. Alternative splicing as a regulator of development and tissue identity. Nat Rev Mol Cell Biol. (2017) 18:437–51. doi: 10.1038/nrm.2017.27
3. Barash Y, Calarco JA, Gao W, Pan Q, Wang X, Shai O, et al. Deciphering the splicing code. Nature. (2010) 465:53–9. doi: 10.1038/nature09000
4. Holmes ME, Hertel KJ. Interdependent regulation of alternative splicing by SR and hnRNP proteins. bioRxiv. (2024). doi: 10.1101/2024.08.19.608666
5. Hajaj E, Zisman E, Tzaban S, Merims S, Cohen J, Klein S, et al. Alternative splicing of the inhibitory immune checkpoint receptor SLAMF6 generates a dominant positive form, boosting T-cell effector functions. Cancer Immunol Res. (2021) 9:637–50. doi: 10.1158/2326-6066.CIR-20-0800
6. Hanawa H, Ma Y, Mikolajczak SA, Charles ML, Yoshida T, Yoshida R, et al. A novel costimulatory signaling in human T lymphocytes by a splice variant of CD28. Blood. (2002) 99:2138–45. doi: 10.1182/blood.V99.6.2138
7. Bonnal SC, Lopez-Oreja I, Valcarcel J. Roles and mechanisms of alternative splicing in cancer - implications for care. Nat Rev Clin Oncol. (2020) 17:457–74. doi: 10.1038/s41571-020-0350-x
8. Lee Y, Rio DC. Mechanisms and regulation of alternative pre-mRNA splicing. Annu Rev Biochem. (2015) 84:291–323. doi: 10.1146/annurev-biochem-060614-034316
9. Fu XD, Ares M Jr. Context-dependent control of alternative splicing by RNA-binding proteins. Nat Rev Genet. (2014) 15:689–701. doi: 10.1038/nrg3778
10. Badr E, ElHefnawi M, Heath LS. Computational identification of tissue-specific splicing regulatory elements in human genes from RNA-seq data. PloS One. (2016) 11:e0166978. doi: 10.1371/journal.pone.0166978
11. Lareau LF, Brenner SE. Regulation of splicing factors by alternative splicing and NMD is conserved between kingdoms yet evolutionarily flexible. Mol Biol Evol. (2015) 32:1072–9. doi: 10.1093/molbev/msv002
12. Carmel I, Tal S, Vig I, Ast G. Comparative analysis detects dependencies among the 5’ splice-site positions. RNA. (2004) 10:828–40. doi: 10.1261/rna.5196404
13. Magen A, Ast G. The importance of being divisible by three in alternative splicing. Nucleic Acids Res. (2005) 33:5574–82. doi: 10.1093/nar/gki858
14. Brooks AN, Yang L, Duff MO, Hansen KD, Park JW, Dudoit S, et al. Conservation of an RNA regulatory map between Drosophila and mammals. Genome Res. (2011) 21:193–202. doi: 10.1101/gr.108662.110
15. Dillman AA, Hauser DN, Gibbs JR, Nalls MA, McCoy MK, Rudenko IN, et al. mRNA expression, splicing and editing in the embryonic and adult mouse cerebral cortex. Nat Neurosci. (2013) 16:499–506. doi: 10.1038/nn.3332
16. de Oliveira Freitas MaChado C, Schafranek M, Bruggemann M, Hernandez Canas MC, Keller M, Di Liddo A, et al. Poison cassette exon splicing of SRSF6 regulates nuclear speckle dispersal and the response to hypoxia. Nucleic Acids Res. (2023) 51:870–90. doi: 10.1093/nar/gkac1225
17. Salmond RJ, McNeill L, Holmes N, Alexander DR. CD4+ T cell hyper-responsiveness in CD45 transgenic mice is independent of isoform. Int Immunol. (2008) 20:819–27. doi: 10.1093/intimm/dxn040
18. Oberdoerffer S, Moita LF, Neems D, Freitas RP, Hacohen N, Rao A. Regulation of CD45 alternative splicing by heterogeneous ribonucleoprotein, hnRNPLL. Science. (2008) 321:686–91. doi: 10.1126/science.1157610
19. Zhang X, Chen MH, Wu X, Kodani A, Fan J, Doan R, et al. Cell-type-specific alternative splicing governs cell fate in the developing cerebral cortex. Cell. (2016) 166:1147–62 e15. doi: 10.1016/j.cell.2016.07.025
20. Latini A, Novelli L, Ceccarelli F, Barbati C, Perricone C, De Benedittis G, et al. mRNA expression analysis confirms CD44 splicing impairment in systemic lupus erythematosus patients. Lupus. (2021) 30:1086–93. doi: 10.1177/09612033211004725
21. AlFadhli S. Overexpression and secretion of the soluble CTLA-4 splice variant in various autoimmune diseases and in cases with overlapping autoimmunity. Genet Test Mol Biomarkers. (2013) 17:336–41. doi: 10.1089/gtmb.2012.0391
22. Ruefli-Brasse AA, French DM, Dixit VM. Regulation of NF-kappaB-dependent lymphocyte activation and development by paracaspase. Science. (2003) 302:1581–4. doi: 10.1126/science.1090769
23. Meininger I, Griesbach RA, Hu D, Gehring T, Seeholzer T, Bertossi A, et al. Alternative splicing of MALT1 controls signalling and activation of CD4(+) T cells. Nat Commun. (2016) 7:11292. doi: 10.1038/ncomms11292
24. Blake D, Radens CM, Ferretti MB, Gazzara MR, Lynch KW. Alternative splicing of apoptosis genes promotes human T cell survival. Elife. (2022) 11. doi: 10.7554/eLife.80953.sa2
25. Monzon-Casanova E, Screen M, Diaz-Munoz MD, Coulson RMR, Bell SE, Lamers G, et al. The RNA-binding protein PTBP1 is necessary for B cell selection in germinal centers. Nat Immunol. (2018) 19:267–78. doi: 10.1038/s41590-017-0035-5
26. Khan M, Zhao Z, Arooj S, Fu Y, Liao G. Soluble PD-1: predictive, prognostic, and therapeutic value for cancer immunotherapy. Front Immunol. (2020) 11:587460. doi: 10.3389/fimmu.2020.587460
27. Hong C, Luckey MA, Ligons DL, Waickman AT, Park JY, Kim GY, et al. Activated T cells secrete an alternatively spliced form of common gamma-chain that inhibits cytokine signaling and exacerbates inflammation. Immunity. (2014) 40:910–23. doi: 10.1016/j.immuni.2014.04.020
28. Ward FJ, Dahal LN, Wijesekera SK, Abdul-Jawad SK, Kaewarpai T, Xu H, et al. The soluble isoform of CTLA-4 as a regulator of T-cell responses. Eur J Immunol. (2013) 43:1274–85. doi: 10.1002/eji.201242529
29. Li X, Li J, Zheng Y, Lee SJ, Zhou J, Giobbie-Hurder A, et al. Granulocyte-macrophage colony-stimulating factor influence on soluble and membrane-bound ICOS in combination with immune checkpoint blockade. Cancer Immunol Res. (2023) 11:1100–13. doi: 10.1158/2326-6066.CIR-22-0702
30. Papoff G, Cascino I, Eramo A, Starace G, Lynch DH, Ruberti G. An N-terminal domain shared by Fas/Apo-1 (CD95) soluble variants prevents cell death in vitro. J Immunol. (1996) 156:4622–30. doi: 10.4049/jimmunol.156.12.4622
31. Sato TA, Widmer MB, Finkelman FD, Madani H, Jacobs CA, Grabstein KH, et al. Recombinant soluble murine IL-4 receptor can inhibit or enhance IgE responses in vivo. J Immunol. (1993) 150:2717–23. doi: 10.4049/jimmunol.150.7.2717
32. Smith DE, Hanna R, Della F, Moore H, Chen H, Farese AM, et al. The soluble form of IL-1 receptor accessory protein enhances the ability of soluble type II IL-1 receptor to inhibit IL-1 action. Immunity. (2003) 18:87–96. doi: 10.1016/S1074-7613(02)00514-9
33. Crawley AM, Faucher S, Angel JB. Soluble IL-7R alpha (sCD127) inhibits IL-7 activity and is increased in HIV infection. J Immunol. (2010) 184:4679–87. doi: 10.4049/jimmunol.0903758
34. Luu K, Shao Z, Schwarz H. The relevance of soluble CD137 in the regulation of immune responses and for immunotherapeutic intervention. J Leukoc Biol. (2020) 107:731–8. doi: 10.1002/JLB.2MR1119-224R
35. Thum E, Shao Z, Schwarz H. CD137, implications in immunity and potential for therapy. Front Biosci (Landmark Ed). (2009) 14:4173–88. doi: 10.2741/3521
36. Nielsen C, Ohm-Laursen L, Barington T, Husby S, Lillevang ST. Alternative splice variants of the human PD-1 gene. Cell Immunol. (2005) 235:109–16. doi: 10.1016/j.cellimm.2005.07.007
37. Blake D, Lynch KW. The three as: Alternative splicing, alternative polyadenylation and their impact on apoptosis in immune function. Immunol Rev. (2021) 304:30–50. doi: 10.1111/imr.v304.1
38. Jensen LE, Whitehead AS. Expression of alternatively spliced interleukin-1 receptor accessory protein mRNAs is differentially regulated during inflammation and apoptosis. Cell Signal. (2003) 15:793–802. doi: 10.1016/S0898-6568(03)00039-1
39. Oaks MK, Hallett KM. Cutting edge: a soluble form of CTLA-4 in patients with autoimmune thyroid disease. J Immunol. (2000) 164:5015–8. doi: 10.4049/jimmunol.164.10.5015
40. Grosche L, Knippertz I, Konig C, Royzman D, Wild AB, Zinser E, et al. The CD83 molecule - an important immune checkpoint. Front Immunol. (2020) 11:721. doi: 10.3389/fimmu.2020.00721
41. Kuipers H, Muskens F, Willart M, Hijdra D, van Assema FB, Coyle AJ, et al. Contribution of the PD-1 ligands/PD-1 signaling pathway to dendritic cell-mediated CD4+ T cell activation. Eur J Immunol. (2006) 36:2472–82. doi: 10.1002/eji.200635978
42. Horvatinovich JM, Grogan EW, Norris M, Steinkasserer A, Lemos H, Mellor AL, et al. Soluble CD83 inhibits T cell activation by binding to the TLR4/MD-2 complex on CD14(+) monocytes. J Immunol. (2017) 198:2286–301. doi: 10.4049/jimmunol.1600802
43. Grohmann U, Volpi C, Fallarino F, Bozza S, Bianchi R, Vacca C, et al. Reverse signaling through GITR ligand enables dexamethasone to activate IDO in allergy. Nat Med. (2007) 13:579–86. doi: 10.1038/nm1563
44. Vardam TD, Zhou L, Appenheimer MM, Chen Q, Wang WC, Baumann H, et al. Regulation of a lymphocyte-endothelial-IL-6 trans-signaling axis by fever-range thermal stress: hot spot of immune surveillance. Cytokine. (2007) 39:84–96. doi: 10.1016/j.cyto.2007.07.184
45. Lundstrom W, Highfill S, Walsh ST, Beq S, Morse E, Kockum I, et al. Soluble IL7Ralpha potentiates IL-7 bioactivity and promotes autoimmunity. Proc Natl Acad Sci United States America. (2013) 110:E1761–70. doi: 10.1073/pnas.1222303110
46. Aderka D, Engelmann H, Maor Y, Brakebusch C, Wallach D. Stabilization of the bioactivity of tumor necrosis factor by its soluble receptors. J Exp Med. (1992) 175:323–9. doi: 10.1084/jem.175.2.323
47. Peters M, Jacobs S, Ehlers M, Vollmer P, Mullberg J, Wolf E, et al. The function of the soluble interleukin 6 (IL-6) receptor in vivo: sensitization of human soluble IL-6 receptor transgenic mice towards IL-6 and prolongation of the plasma half-life of IL-6. J Exp Med. (1996) 183:1399–406. doi: 10.1084/jem.183.4.1399
48. Murphy KM, Weaver C. Janeway’s Immunobiology. New York and London: Garland Science/Taylor & Francis Group L (2017) p. 196–7.
49. Kefaloyianni E. Soluble forms of cytokine and growth factor receptors: mechanisms of generation and modes of action in the regulation of local and systemic inflammation. FEBS letters. (2022) 596:589–606. doi: 10.1002/1873-3468.14305
50. Dudziak D, Nimmerjahn F, Bornkamm GW, Laux G. Alternative splicing generates putative soluble CD83 proteins that inhibit T cell proliferation. J Immunol. (2005) 174:6672–6. doi: 10.4049/jimmunol.174.11.6672
51. Goodwin RG, Friend D, Ziegler SF, Jerzy R, Falk BA, Gimpel S, et al. Cloning of the human and murine interleukin-7 receptors: demonstration of a soluble form and homology to a new receptor superfamily. Cell. (1990) 60:941–51. doi: 10.1016/0092-8674(90)90342-C
52. Cheng J, Zhou T, Liu C, Shapiro JP, Brauer MJ, Kiefer MC, et al. Protection from Fas-mediated apoptosis by a soluble form of the Fas molecule. Science. (1994) 263:1759–62. doi: 10.1126/science.7510905
53. Cascino I, Fiucci G, Papoff G, Ruberti G. Three functional soluble forms of the human apoptosis-inducing Fas molecule are produced by alternative splicing. J Immunol. (1995) 154:2706–13. doi: 10.4049/jimmunol.154.6.2706
54. Horiuchi S, Koyanagi Y, Zhou Y, Miyamoto H, Tanaka Y, Waki M, et al. Soluble interleukin-6 receptors released from T cell or granulocyte/macrophage cell lines and human peripheral blood mononuclear cells are generated through an alternative splicing mechanism. Eur J Immunol. (1994) 24:1945–8. doi: 10.1002/eji.1830240837
55. Monaghan SF, Banerjee D, Chung CS, Lomas-Neira J, Cygan KJ, Rhine CL, et al. Changes in the process of alternative RNA splicing results in soluble B and T lymphocyte attenuator with biological and clinical implications in critical illness. Mol Med. (2018) 24:32. doi: 10.1186/s10020-018-0036-3
56. Nocentini G, Ronchetti S, Bartoli A, Spinicelli S, Delfino D, Brunetti L, et al. Identification of three novel mRNA splice variants of GITR. Cell Death Differ. (2000) 7:408–10. doi: 10.1038/sj.cdd.4400670
57. Kruse S, Forster J, Kuehr J, Deichmann KA. Characterization of the membrane-bound and a soluble form of human IL-4 receptor alpha produced by alternative splicing. Int Immunol. (1999) 11:1965–70. doi: 10.1093/intimm/11.12.1965
58. Magistrelli G, Jeannin P, Herbault N, Benoit De Coignac A, Gauchat JF, Bonnefoy JY, et al. A soluble form of CTLA-4 generated by alternative splicing is expressed by nonstimulated human T cells. Eur J Immunol. (1999) 29:3596–602. doi: 10.1002/(SICI)1521-4141(199911)29:11<3596::AID-IMMU3596>3.0.CO;2-Y
59. Lainez B, Fernandez-Real JM, Romero X, Esplugues E, Canete JD, Ricart W, et al. Identification and characterization of a novel spliced variant that encodes human soluble tumor necrosis factor receptor 2. Int Immunol. (2004) 16:169–77. doi: 10.1093/intimm/dxh014
60. Philippe C, Roux-Lombard P, Fouqueray B, Perez J, Dayer JM, Baud L. Membrane expression and shedding of tumour necrosis factor receptors during activation of human blood monocytes: regulation by desferrioxamine. Immunology. (1993) 80:300–5.
61. Levine SJ. Molecular mechanisms of soluble cytokine receptor generation. J Biol Chem. (2008) 283:14177–81. doi: 10.1074/jbc.R700052200
62. Sung HH, Juang JH, Lin YC, Kuo CH, Hung JT, Chen A, et al. Transgenic expression of decoy receptor 3 protects islets from spontaneous and chemical-induced autoimmune destruction in nonobese diabetic mice. J Exp Med. (2004) 199:1143–51. doi: 10.1084/jem.20031939
63. Odales J, Guzman Valle J, Martinez-Cortes F, Manoutcharian K. Immunogenic properties of immunoglobulin superfamily members within complex biological networks. Cell Immunol. (2020) 358:104235. doi: 10.1016/j.cellimm.2020.104235
64. Gu D, Ao X, Yang Y, Chen Z, Xu X. Soluble immune checkpoints in cancer: production, function and biological significance. J Immunother Cancer. (2018) 6:132. doi: 10.1186/s40425-018-0449-0
65. Chakrabarti R, Kapse B, Mukherjee G. Soluble immune checkpoint molecules: Serum markers for cancer diagnosis and prognosis. Cancer Rep (Hoboken). (2019) 2:e1160. doi: 10.1002/cnr2.1160
66. Triebel F. LAG-3: a regulator of T-cell and DC responses and its use in therapeutic vaccination. Trends Immunol. (2003) 24:619–22. doi: 10.1016/j.it.2003.10.001
67. Sharpe AH, Pauken KE. The diverse functions of the PD1 inhibitory pathway. Nat Rev. (2018) 18:153–67. doi: 10.1038/nri.2017.108
68. Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol. (2008) 26:677–704. doi: 10.1146/annurev.immunol.26.021607.090331
69. Song MY, Park SH, Nam HJ, Choi DH, Sung YC. Enhancement of vaccine-induced primary and memory CD8(+) T-cell responses by soluble PD-1. J Immunother. (2011) 34:297–306. doi: 10.1097/CJI.0b013e318210ed0e
70. He L, Zhang G, He Y, Zhu H, Zhang H, Feng Z. Blockade of B7-H1 with sPD-1 improves immunity against murine hepatocarcinoma. Anticancer Res. (2005) 25:3309–13.
71. He YF, Zhang GM, Wang XH, Zhang H, Yuan Y, Li D, et al. Blocking programmed death-1 ligand-PD-1 interactions by local gene therapy results in enhancement of antitumor effect of secondary lymphoid tissue chemokine. J Immunol. (2004) 173:4919–28. doi: 10.4049/jimmunol.173.8.4919
72. Dostert C, Grusdat M, Letellier E, Brenner D. The TNF family of ligands and receptors: communication modules in the immune system and beyond. Physiol Rev. (2019) 99:115–60. doi: 10.1152/physrev.00045.2017
73. Fine A, Anderson NL, Rothstein TL, Williams MC, Gochuico BR. Fas expression in pulmonary alveolar type II cells. Am J Physiol. (1997) 273:L64–71. doi: 10.1152/ajplung.1997.273.1.L64
74. Pinkoski MJ, Brunner T, Green DR, Lin T. Fas and Fas ligand in gut and liver. Am J Physiol Gastrointest Liver Physiol. (2000) 278:G354–66. doi: 10.1152/ajpgi.2000.278.3.G354
75. Krammer PH. CD95’s deadly mission in the immune system. Nature. (2000) 407:789–95. doi: 10.1038/35037728
76. Golstein P, Griffiths GM. An early history of T cell-mediated cytotoxicity. Nat Rev. (2018) 18:527–35. doi: 10.1038/s41577-018-0009-3
77. Klebanoff CA, Scott CD, Leonardi AJ, Yamamoto TN, Cruz AC, Ouyang C, et al. Memory T cell-driven differentiation of naive cells impairs adoptive immunotherapy. J Clin Invest. (2016) 126:318–34. doi: 10.1172/JCI81217
78. Sprent J, Kishimoto H. The thymus and negative selection. Immunol Rev. (2002) 185:126–35. doi: 10.1034/j.1600-065X.2002.18512.x
79. Alderson MR, Tough TW, Davis-Smith T, Braddy S, Falk B, Schooley KA, et al. Fas ligand mediates activation-induced cell death in human T lymphocytes. J Exp Med. (1995) 181:71–7. doi: 10.1084/jem.181.1.71
80. Kunkele A, Johnson AJ, Rolczynski LS, Chang CA, Hoglund V, Kelly-Spratt KS, et al. Functional Tuning of CARs Reveals Signaling Threshold above Which CD8+ CTL Antitumor Potency Is Attenuated due to Cell Fas-FasL-Dependent AICD. Cancer Immunol Res. (2015) 3:368–79. doi: 10.1158/2326-6066.CIR-14-0200
81. Horton BL, Williams JB, Cabanov A, Spranger S, Gajewski TF. Intratumoral CD8(+) T-cell apoptosis is a major component of T-cell dysfunction and impedes antitumor immunity. Cancer Immunol Res. (2018) 6:14–24. doi: 10.1158/2326-6066.CIR-17-0249
82. Izquierdo JM, Majos N, Bonnal S, Martinez C, Castelo R, Guigo R, et al. Regulation of Fas alternative splicing by antagonistic effects of TIA-1 and PTB on exon definition. Mol Cell. (2005) 19:475–84. doi: 10.1016/j.molcel.2005.06.015
83. Izquierdo JM, Hu antigen R. (HuR) functions as an alternative pre-mRNA splicing regulator of Fas apoptosis-promoting receptor on exon definition. J Biol Chem. (2008) 283:19077–84. doi: 10.1074/jbc.M800017200
84. Oh H, Lee E, Jang HN, Lee J, Moon H, Sheng Z, et al. hnRNP A1 contacts exon 5 to promote exon 6 inclusion of apoptotic Fas gene. Apoptosis. (2013) 18:825–35. doi: 10.1007/s10495-013-0824-8
85. Jang HN, Liu Y, Choi N, Oh J, Ha J, Zheng X, et al. Binding of SRSF4 to a novel enhancer modulates splicing of exon 6 of Fas pre-mRNA. Biochem Biophys Res Commun. (2018) 506:703–8. doi: 10.1016/j.bbrc.2018.10.123
86. Tejedor JR, Papasaikas P, Valcarcel J. Genome-wide identification of Fas/CD95 alternative splicing regulators reveals links with iron homeostasis. Mol Cell. (2015) 57:23–38. doi: 10.1016/j.molcel.2014.10.029
87. Choi N, Jang HN, Oh J, Ha J, Park H, Zheng X, et al. SRSF6 regulates the alternative splicing of the apoptotic fas gene by targeting a novel RNA sequence. Cancers (Basel). (2022) 14(8):1990. doi: 10.3390/cancers14081990
88. Bajgain P, Torres Chavez AG, Balasubramanian K, Fleckenstein L, Lulla P, Heslop HE, et al. Secreted fas decoys enhance the antitumor activity of engineered and bystander T cells in fas ligand-expressing solid tumors. Cancer Immunol Res. (2022) 10:1370–85. doi: 10.1158/2326-6066.CIR-22-0115
89. Batlle E, Massague J. Transforming growth factor-beta signaling in immunity and cancer. Immunity. (2019) 50:924–40. doi: 10.1016/j.immuni.2019.03.024
90. Chakravarthy A, Khan L, Bensler NP, Bose P, De Carvalho DD. TGF-beta-associated extracellular matrix genes link cancer-associated fibroblasts to immune evasion and immunotherapy failure. Nat Commun. (2018) 9:4692. doi: 10.1038/s41467-018-06654-8
91. Tauriello DVF, Palomo-Ponce S, Stork D, Berenguer-Llergo A, Badia-Ramentol J, Iglesias M, et al. TGFbeta drives immune evasion in genetically reconstituted colon cancer metastasis. Nature. (2018) 554:538–43. doi: 10.1038/nature25492
92. Ni Y, Soliman A, Joehlin-Price A, Rose PG, Vlad A, Edwards RP, et al. High TGF-beta signature predicts immunotherapy resistance in gynecologic cancer patients treated with immune checkpoint inhibition. NPJ Precis Oncol. (2021) 5:101. doi: 10.1038/s41698-021-00242-8
93. Bertolio MS, La Colla A, Carrea A, Romo A, Canziani G, Echarte SM, et al. A novel splice variant of human TGF-beta type II receptor encodes a soluble protein and its fc-tagged version prevents liver fibrosis in vivo. Front Cell Dev Biol. (2021) 9:690397. doi: 10.3389/fcell.2021.690397
94. Lust JA, Donovan KA, Kline MP, Greipp PR, Kyle RA, Maihle NJ. Isolation of an mRNA encoding a soluble form of the human interleukin-6 receptor. Cytokine. (1992) 4:96–100. doi: 10.1016/1043-4666(92)90043-Q
95. Garapati VP, Lefranc MP. IMGT Colliers de Perles and IgSF domain standardization for T cell costimulatory activatory (CD28, ICOS) and inhibitory (CTLA4, PDCD1 and BTLA) receptors. Dev Comp Immunol. (2007) 31:1050–72. doi: 10.1016/j.dci.2007.01.008
96. Andrzejczak A, Karabon L. BTLA biology in cancer: from bench discoveries to clinical potentials. biomark Res. (2024) 12:8. doi: 10.1186/s40364-024-00556-2
97. Magistrelli G, Jeannin P, Elson G, Gauchat JF, Nguyen TN, Bonnefoy JY, et al. Identification of three alternatively spliced variants of human CD28 mRNA. Biochem Biophys Res Commun. (1999) 259:34–7. doi: 10.1006/bbrc.1999.0725
98. Hebbar M, Jeannin P, Magistrelli G, Hatron PY, Hachulla E, Devulder B, et al. Detection of circulating soluble CD28 in patients with systemic lupus erythematosus, primary Sjogren’s syndrome and systemic sclerosis. Clin Exp Immunol. (2004) 136:388–92. doi: 10.1111/j.1365-2249.2004.02427.x
99. Lechmann M, Krooshoop DJ, Dudziak D, Kremmer E, Kuhnt C, Figdor CG, et al. The extracellular domain of CD83 inhibits dendritic cell-mediated T cell stimulation and binds to a ligand on dendritic cells. J Exp Med. (2001) 194:1813–21. doi: 10.1084/jem.194.12.1813
100. Burnell SEA, Capitani L, MacLachlan BJ, Mason GH, Gallimore AM, Godkin A. Seven mysteries of LAG-3: a multi-faceted immune receptor of increasing complexity. Immunother Adv. (2022) 2:ltab025. doi: 10.1093/immadv/ltab025
101. Gorgulho J, Roderburg C, Beier F, Bokemeyer C, Brummendorf TH, Loosen SH, et al. Soluble lymphocyte activation gene-3 (sLAG3) and CD4/CD8 ratio dynamics as predictive biomarkers in patients undergoing immune checkpoint blockade for solid Malignancies. Br J cancer. (2024) 130:1013–22. doi: 10.1038/s41416-023-02558-7
102. Frenay J, Bellaye PS, Oudot A, Helbling A, Petitot C, Ferrand C, et al. IL-1RAP, a key therapeutic target in cancer. Int J Mol Sci. (2022) 23(23):14918. doi: 10.3390/ijms232314918
103. Blum H, Wolf M, Enssle K, Rollinghoff M, Gessner A. Two distinct stimulus-dependent pathways lead to production of soluble murine interleukin-4 receptor. J Immunol. (1996) 157:1846–53. doi: 10.4049/jimmunol.157.5.1846
104. Schumertl T, Lokau J, Rose-John S, Garbers C. Function and proteolytic generation of the soluble interleukin-6 receptor in health and disease. Biochim Biophys Acta Mol Cell Res. (2022) 1869:119143. doi: 10.1016/j.bbamcr.2021.119143
105. Hurst SM, Wilkinson TS, McLoughlin RM, Jones S, Horiuchi S, Yamamoto N, et al. Il-6 and its soluble receptor orchestrate a temporal switch in the pattern of leukocyte recruitment seen during acute inflammation. Immunity. (2001) 14:705–14. doi: 10.1016/S1074-7613(01)00151-0
106. Wolf J, Waetzig GH, Chalaris A, Reinheimer TM, Wege H, Rose-John S, et al. Different soluble forms of the interleukin-6 family signal transducer gp130 fine-tune the blockade of interleukin-6 trans-signaling. J Biol Chem. (2016) 291:16186–96. doi: 10.1074/jbc.M116.718551
107. Tian J, Zhang B, Rui K, Wang S. The role of GITR/GITRL interaction in autoimmune diseases. Front Immunol. (2020) 11:588682. doi: 10.3389/fimmu.2020.588682
108. Romo A, Rodriguez TM, Yu G, Dewey RA. Chimeric TbetaRII-SE/Fc overexpression by a lentiviral vector exerts strong antitumoral activity on colorectal cancer-derived cell lines in vitro and on xenografts. Cancer Gene Ther. (2024) 31:174–85. doi: 10.1038/s41417-023-00694-z
109. Fenix AM, Miyaoka Y, Bertero A, Blue SM, Spindler MJ, Tan KKB, et al. Gain-of-function cardiomyopathic mutations in RBM20 rewire splicing regulation and re-distribute ribonucleoprotein granules within processing bodies. Nat Commun. (2021) 12:6324. doi: 10.1038/s41467-021-26623-y
110. Wells QS, Becker JR, Su YR, Mosley JD, Weeke P, D’Aoust L, et al. Whole exome sequencing identifies a causal RBM20 mutation in a large pedigree with familial dilated cardiomyopathy. Circ Cardiovasc Genet. (2013) 6:317–26. doi: 10.1161/CIRCGENETICS.113.000011
111. Aartsma-Rus A, Van Deutekom JC, Fokkema IF, Van Ommen GJ, Den Dunnen JT. Entries in the Leiden Duchenne muscular dystrophy mutation database: an overview of mutation types and paradoxical cases that confirm the reading-frame rule. Muscle Nerve. (2006) 34:135–44. doi: 10.1002/mus.20586
112. Pagani F, Stuani C, Tzetis M, Kanavakis E, Efthymiadou A, Doudounakis S, et al. New type of disease causing mutations: the example of the composite exonic regulatory elements of splicing in CFTR exon 12. Hum Mol Genet. (2003) 12:1111–20. doi: 10.1093/hmg/ddg131
113. Cartegni L, Chew SL, Krainer AR. Listening to silence and understanding nonsense: exonic mutations that affect splicing. Nat Rev Genet. (2002) 3:285–98. doi: 10.1038/nrg775
114. Pagani F, Baralle FE. Genomic variants in exons and introns: identifying the splicing spoilers. Nat Rev Genet. (2004) 5:389–96. doi: 10.1038/nrg1327
115. Bergsma AJ, van der Wal E, Broeders M, van der Ploeg AT, Pim Pijnappel WWM. Alternative splicing in genetic diseases: improved diagnosis and novel treatment options. Int Rev Cell Mol Biol. (2018) 335:85–141. doi: 10.1016/bs.ircmb.2017.07.008
116. Chou J, Ohsumi TK, Geha RS. Use of whole exome and genome sequencing in the identification of genetic causes of primary immunodeficiencies. Curr Opin Allergy Clin Immunol. (2012) 12:623–8. doi: 10.1097/ACI.0b013e3283588ca6
117. Ars E, Kruyer H, Gaona A, Serra E, Lazaro C, Estivill X. Prenatal diagnosis of sporadic neurofibromatosis type 1 (NF1) by RNA and DNA analysis of a splicing mutation. Prenat Diagn. (1999) 19:739–42. doi: 10.1002/(SICI)1097-0223(199908)19:8<739::AID-PD626>3.0.CO;2-A
118. Baralle D, Baralle M. Splicing in action: assessing disease causing sequence changes. J Med Genet. (2005) 42:737–48. doi: 10.1136/jmg.2004.029538
119. Nik S, Bowman TV. Splicing and neurodegeneration: Insights and mechanisms. Wiley Interdiscip Rev RNA. (2019) 10:e1532. doi: 10.1002/wrna.2019.10.issue-4
120. Samaranch L, Lorenzo-Betancor O, Arbelo JM, Ferrer I, Lorenzo E, Irigoyen J, et al. PINK1-linked parkinsonism is associated with Lewy body pathology. Brain. (2010) 133:1128–42. doi: 10.1093/brain/awq051
121. Fu RH, Liu SP, Huang SJ, Chen HJ, Chen PR, Lin YH, et al. Aberrant alternative splicing events in Parkinson’s disease. Cell Transplant. (2013) 22:653–61. doi: 10.3727/096368912X655154
122. Tollervey JR, Wang Z, Hortobagyi T, Witten JT, Zarnack K, Kayikci M, et al. Analysis of alternative splicing associated with aging and neurodegeneration in the human brain. Genome Res. (2011) 21:1572–82. doi: 10.1101/gr.122226.111
123. Anderson SL, Coli R, Daly IW, Kichula EA, Rork MJ, Volpi SA, et al. Familial dysautonomia is caused by mutations of the IKAP gene. Am J Hum Genet. (2001) 68:753–8. doi: 10.1086/318808
124. Licatalosi DD, Darnell RB. Splicing regulation in neurologic disease. Neuron. (2006) 52:93–101. doi: 10.1016/j.neuron.2006.09.017
125. Li D, McIntosh CS, Mastaglia FL, Wilton SD, Aung-Htut MT. Neurodegenerative diseases: a hotbed for splicing defects and the potential therapies. Transl Neurodegener. (2021) 10:16. doi: 10.1186/s40035-021-00240-7
126. Holm A, Hansen SN, Klitgaard H, Kauppinen S. Clinical advances of RNA therapeutics for treatment of neurological and neuromuscular diseases. RNA Biol. (2022) 19:594–608. doi: 10.1080/15476286.2022.2066334
127. Amoasii L, Long C, Li H, Mireault AA, Shelton JM, Sanchez-Ortiz E, et al. Single-cut genome editing restores dystrophin expression in a new mouse model of muscular dystrophy. Sci Transl Med. (2017) 9(418):eaan8081. doi: 10.1126/scitranslmed.aan8081
128. Puttaraju M, Jamison SF, Mansfield SG, Garcia-Blanco MA, Mitchell LG. Spliceosome-mediated RNA trans-splicing as a tool for gene therapy. Nat Biotechnol. (1999) 17:246–52. doi: 10.1038/6986
129. Rindt H, Yen PF, Thebeau CN, Peterson TS, Weisman GA, Lorson CL. Replacement of huntingtin exon 1 by trans-splicing. Cell Mol Life Sci. (2012) 69:4191–204. doi: 10.1007/s00018-012-1083-5
130. Lorain S, Peccate C, Le Hir M, Griffith G, Philippi S, Precigout G, et al. Dystrophin rescue by trans-splicing: a strategy for DMD genotypes not eligible for exon skipping approaches. Nucleic Acids Res. (2013) 41:8391–402. doi: 10.1093/nar/gkt621
131. Avale ME, Rodriguez-Martin T, Gallo JM. Trans-splicing correction of tau isoform imbalance in a mouse model of tau mis-splicing. Hum Mol Genet. (2013) 22:2603–11. doi: 10.1093/hmg/ddt108
132. Shuai S, Suzuki H, Diaz-Navarro A, Nadeu F, Kumar SA, Gutierrez-Fernandez A, et al. The U1 spliceosomal RNA is recurrently mutated in multiple cancers. Nature. (2019) 574:712–6. doi: 10.1038/s41586-019-1651-z
133. Suzuki H, Kumar SA, Shuai S, Diaz-Navarro A, Gutierrez-Fernandez A, De Antonellis P, et al. Recurrent noncoding U1 snRNA mutations drive cryptic splicing in SHH medulloblastoma. Nature. (2019) 574:707–11. doi: 10.1038/s41586-019-1650-0
134. Yoshida K, Sanada M, Shiraishi Y, Nowak D, Nagata Y, Yamamoto R, et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature. (2011) 478:64–9. doi: 10.1038/nature10496
135. Graubert TA, Shen D, Ding L, Okeyo-Owuor T, Lunn CL, Shao J, et al. Recurrent mutations in the U2AF1 splicing factor in myelodysplastic syndromes. Nat Genet. (2011) 44:53–7. doi: 10.1038/ng.1031
136. Corrigendum. Comprehensive molecular profiling of lung carcinoma. Can Genome Atlas Res Netw. Nat. (2014) 511(7511):543–50. doi: 10.1038/nature13385
137. Bailey P, Chang DK, Nones K, Johns AL, Patch AM, Gingras MC, et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature. (2016) 531:47–52. doi: 10.1038/nature16965
138. Brooks AN, Choi PS, de Waal L, Sharifnia T, Imielinski M, Saksena G, et al. A pan-cancer analysis of transcriptome changes associated with somatic mutations in U2AF1 reveals commonly altered splicing events. PloS One. (2014) 9:e87361. doi: 10.1371/journal.pone.0087361
139. Przychodzen B, Jerez A, Guinta K, Sekeres MA, Padgett R, Maciejewski JP, et al. Patterns of missplicing due to somatic U2AF1 mutations in myeloid neoplasms. Blood. (2013) 122:999–1006. doi: 10.1182/blood-2013-01-480970
140. Lopez-Canovas JL, Del Rio-Moreno M, Garcia-Fernandez H, Jimenez-Vacas JM, Moreno-Montilla MT, Sanchez-Frias ME, et al. Splicing factor SF3B1 is overexpressed and implicated in the aggressiveness and survival of hepatocellular carcinoma. Cancer Lett. (2021) 496:72–83. doi: 10.1016/j.canlet.2020.10.010
141. Papaemmanuil E, Cazzola M, Boultwood J, Malcovati L, Vyas P, Bowen D, et al. Somatic SF3B1 mutation in myelodysplasia with ring sideroblasts. New Engl J Med. (2011) 365:1384–95. doi: 10.1056/NEJMoa1103283
142. Furney SJ, Pedersen M, Gentien D, Dumont AG, Rapinat A, Desjardins L, et al. SF3B1 mutations are associated with alternative splicing in uveal melanoma. Cancer Discovery. (2013) 3:1122–9. doi: 10.1158/2159-8290.CD-13-0330
143. Maguire SL, Leonidou A, Wai P, Marchio C, Ng CK, Sapino A, et al. SF3B1 mutations constitute a novel therapeutic target in breast cancer. J pathol. (2015) 235:571–80. doi: 10.1002/path.2015.235.issue-4
144. Gentien D, Kosmider O, Nguyen-Khac F, Albaud B, Rapinat A, Dumont AG, et al. A common alternative splicing signature is associated with SF3B1 mutations in Malignancies from different cell lineages. Leukemia. (2014) 28:1355–7. doi: 10.1038/leu.2014.28
145. Obeng EA, Chappell RJ, Seiler M, Chen MC, Campagna DR, Schmidt PJ, et al. Physiologic expression of sf3b1(K700E) causes impaired erythropoiesis, aberrant splicing, and sensitivity to therapeutic spliceosome modulation. Cancer Cell. (2016) 30:404–17. doi: 10.1016/j.ccell.2016.08.006
146. Zhou Z, Gong Q, Wang Y, Li M, Wang L, Ding H, et al. The biological function and clinical significance of SF3B1 mutations in cancer. biomark Res. (2020) 8:38. doi: 10.1186/s40364-020-00220-5
147. Alsafadi S, Houy A, Battistella A, Popova T, Wassef M, Henry E, et al. Cancer-associated SF3B1 mutations affect alternative splicing by promoting alternative branchpoint usage. Nat Commun. (2016) 7:10615. doi: 10.1038/ncomms10615
148. Sun G, Zhou H, Chen K, Zeng J, Zhang Y, Yan L, et al. Retraction Note: HnRNP A1 - mediated alternative splicing of CCDC50 contributes to cancer progression of clear cell renal cell carcinoma via ZNF395. J Exp Clin Cancer Res. (2023) 42:43. doi: 10.1186/s13046-023-02613-4
149. Cogoi S, Rapozzi V, Cauci S, Xodo LE. Critical role of hnRNP A1 in activating KRAS transcription in pancreatic cancer cells: A molecular mechanism involving G4 DNA. Biochim Biophys Acta Gen Subj. (2017) 1861:1389–98. doi: 10.1016/j.bbagen.2016.11.031
150. Chen Y, Liu J, Wang W, Xiang L, Wang J, Liu S, et al. High expression of hnRNPA1 promotes cell invasion by inducing EMT in gastric cancer. Oncol Rep. (2018) 39:1693–701. doi: 10.3892/or.2018.6273
151. Ryu HG, Jung Y, Lee N, Seo JY, Kim SW, Lee KH, et al. HNRNP A1 promotes lung cancer cell proliferation by modulating VRK1 translation. Int J Mol Sci. (2021) 22(11):5506. doi: 10.3390/ijms22115506
152. Loh TJ, Moon H, Cho S, Jang H, Liu YC, Tai H, et al. CD44 alternative splicing and hnRNP A1 expression are associated with the metastasis of breast cancer. Oncol Rep. (2015) 34:1231–8. doi: 10.3892/or.2015.4110
153. Lv Y, Zhang W, Zhao J, Sun B, Qi Y, Ji H, et al. SRSF1 inhibits autophagy through regulating Bcl-x splicing and interacting with PIK3C3 in lung cancer. Signal Transduct Target Ther. (2021) 6:108. doi: 10.1038/s41392-021-00495-6
154. Karni R, de StanChina E, Lowe SW, Sinha R, Mu D, Krainer AR. The gene encoding the splicing factor SF2/ASF is a proto-oncogene. Nat Struct Mol Biol. (2007) 14:185–93. doi: 10.1038/nsmb1209
155. Comiskey DF Jr., Montes M, Khurshid S, Singh RK, Chandler DS. SRSF2 regulation of MDM2 reveals splicing as a therapeutic vulnerability of the p53 pathway. Mol Cancer Res. (2020) 18:194–203. doi: 10.1158/1541-7786.MCR-19-0541
156. Lei S, Zhang B, Huang L, Zheng Z, Xie S, Shen L, et al. SRSF1 promotes the inclusion of exon 3 of SRA1 and the invasion of hepatocellular carcinoma cells by interacting with exon 3 of SRA1pre-mRNA. Cell Death Discovery. (2021) 7:117. doi: 10.1038/s41420-021-00498-w
157. Liu H, Gong Z, Li K, Zhang Q, Xu Z, Xu Y. SRPK1/2 and PP1alpha exert opposite functions by modulating SRSF1-guided MKNK2 alternative splicing in colon adenocarcinoma. J Exp Clin Cancer Res. (2021) 40:75. doi: 10.1186/s13046-021-01877-y
158. Jiang L, Huang J, Higgs BW, Hu Z, Xiao Z, Yao X, et al. Genomic landscape survey identifies SRSF1 as a key oncodriver in small cell lung cancer. PloS Genet. (2016) 12:e1005895. doi: 10.1371/journal.pgen.1005895
159. Anczukow O, Akerman M, Clery A, Wu J, Shen C, Shirole NH, et al. SRSF1-regulated alternative splicing in breast cancer. Mol Cell. (2015) 60:105–17. doi: 10.1016/j.molcel.2015.09.005
160. Jensen MA, Wilkinson JE, Krainer AR. Splicing factor SRSF6 promotes hyperplasia of sensitized skin. Nat Struct Mol Biol. (2014) 21:189–97. doi: 10.1038/nsmb.2756
161. Wan L, Yu W, Shen E, Sun W, Liu Y, Kong J, et al. SRSF6-regulated alternative splicing that promotes tumour progression offers a therapy target for colorectal cancer. Gut. (2019) 68:118–29. doi: 10.1136/gutjnl-2017-314983
162. Cohen-Eliav M, Golan-Gerstl R, Siegfried Z, Andersen CL, Thorsen K, Orntoft TF, et al. The splicing factor SRSF6 is amplified and is an oncoprotein in lung and colon cancers. J pathol. (2013) 229:630–9. doi: 10.1002/path.4129
163. Kong-Beltran M, Seshagiri S, Zha J, Zhu W, Bhawe K, Mendoza N, et al. Somatic mutations lead to an oncogenic deletion of met in lung cancer. Cancer Res. (2006) 66:283–9. doi: 10.1158/0008-5472.CAN-05-2749
164. Onozato R, Kosaka T, Kuwano H, Sekido Y, Yatabe Y, Mitsudomi T. Activation of MET by gene amplification or by splice mutations deleting the juxtamembrane domain in primary resected lung cancers. J Thorac Oncol. (2009) 4:5–11. doi: 10.1097/JTO.0b013e3181913e0e
165. Schrock AB, Frampton GM, Suh J, Chalmers ZR, Rosenzweig M, Erlich RL, et al. Characterization of 298 patients with lung cancer harboring MET exon 14 skipping alterations. J Thorac Oncol. (2016) 11:1493–502. doi: 10.1016/j.jtho.2016.06.004
166. Peschard P, Fournier TM, Lamorte L, Naujokas MA, Band H, Langdon WY, et al. Mutation of the c-Cbl TKB domain binding site on the Met receptor tyrosine kinase converts it into a transforming protein. Mol Cell. (2001) 8:995–1004. doi: 10.1016/S1097-2765(01)00378-1
167. Awad MM, Lee JK, Madison R, Classon A, Kmak J, Frampton GM, et al. Characterization of 1,387 NSCLCs with MET exon 14 (METex14) skipping alterations (SA) and potential acquired resistance (AR) mechanisms. J Clin Oncol. (2020) 38. doi: 10.1200/JCO.2020.38.15_suppl.9511
168. Heist RS, Shim HS, Gingipally S, Mino-Kenudson M, Le L, Gainor JF, et al. MET exon 14 skipping in non-small cell lung cancer. Oncologist. (2016) 21. doi: 10.1634/theoncologist.2015-0510
169. Gorlov IP, Gorlova OY, Frazier ML, Amos CI. Missense mutations in hMLH1 and hMSH2 are associated with exonic splicing enhancers. Am J Hum Genet. (2003) 73:1157–61. doi: 10.1086/378819
170. Liu T, Tannergard P, Hackman P, Rubio C, Kressner U, Lindmark G, et al. Missense mutations in hMLH1 associated with colorectal cancer. Hum Genet. (1999) 105:437–41. doi: 10.1007/s004399900160
171. Wijnen J, Khan PM, Vasen H, Menko F, van der Klift H, van den Broek M, et al. Majority of hMLH1 mutations responsible for hereditary nonpolyposis colorectal cancer cluster at the exonic region 15-16. Am J Hum Genet. (1996) 58:300–7.
172. Stella A, Wagner A, Shito K, Lipkin SM, Watson P, Guanti G, et al. A nonsense mutation in MLH1 causes exon skipping in three unrelated HNPCC families. Cancer Res. (2001) 61:7020–4.
173. Clarke LA, Veiga I, Isidro G, Jordan P, Ramos JS, Castedo S, et al. Pathological exon skipping in an HNPCC proband with MLH1 splice acceptor site mutation. Genes Chromosomes Cancer. (2000) 29:367–70. doi: 10.1002/1098-2264(2000)9999:9999<::AID-GCC1051>3.0.CO;2-V
174. Tanko Q, Franklin B, Lynch H, Knezetic J. A hMLH1 genomic mutation and associated novel mRNA defects in a hereditary non-polyposis colorectal cancer family. Mutat Res. (2002) 503:37–42. doi: 10.1016/S0027-5107(02)00031-3
175. Smeby J, Sveen A, Eilertsen IA, Danielsen SA, Hoff AM, Eide PW, et al. Transcriptional and functional consequences of TP53 splice mutations in colorectal cancer. Oncogenesis. (2019) 8:35. doi: 10.1038/s41389-019-0141-3
176. Bodner SM, Minna JD, Jensen SM, D’Amico D, Carbone D, Mitsudomi T, et al. Expression of mutant p53 proteins in lung cancer correlates with the class of p53 gene mutation. Oncogene. (1992) 7:743–9.
177. Surget S, Khoury MP, Bourdon JC. Uncovering the role of p53 splice variants in human Malignancy: a clinical perspective. Onco Targets Ther. (2013) 7:57–68. doi: 10.2147/OTT.S53876
178. Bourdon JC. p53 and its isoforms in cancer. Br J cancer. (2007) 97:277–82. doi: 10.1038/sj.bjc.6603886
179. Leroy B, Anderson M, Soussi T. TP53 mutations in human cancer: database reassessment and prospects for the next decade. Hum Mutat. (2014) 35:672–88. doi: 10.1002/humu.2014.35.issue-6
180. Bamopoulos SA, Batcha AMN, Jurinovic V, Rothenberg-Thurley M, Janke H, Ksienzyk B, et al. Clinical presentation and differential splicing of SRSF2, U2AF1 and SF3B1 mutations in patients with acute myeloid leukemia. Leukemia. (2020) 34:2621–34. doi: 10.1038/s41375-020-0839-4
181. Martin M, Masshofer L, Temming P, Rahmann S, Metz C, Bornfeld N, et al. Exome sequencing identifies recurrent somatic mutations in EIF1AX and SF3B1 in uveal melanoma with disomy 3. Nat Genet. (2013) 45:933–6. doi: 10.1038/ng.2674
182. Group PTC, Calabrese C, Davidson NR, Demircioglu D, Fonseca NA, He Y, et al. Genomic basis for RNA alterations in cancer. Nature. (2020) 578:129–36. doi: 10.1038/s41586-020-1970-0
183. Puttick C, Jones TP, Leung MM, Galvez-Cancino F, Liu J, Varas-Godoy M, et al. MHC Hammer reveals genetic and non-genetic HLA disruption in cancer evolution. Nat Genet. (2024) 56:2121–31. doi: 10.1038/s41588-024-01883-8
184. Wei L, Li Y, Chen J, Wang Y, Wu J, Yang H, et al. Alternative splicing in ovarian cancer. Cell Commun Signal. (2024) 22:507. doi: 10.1186/s12964-024-01880-8
185. Cho N, Kim SY, Lee SG, Park C, Choi S, Kim EM, et al. Alternative splicing of PBRM1 mediates resistance to PD-1 blockade therapy in renal cancer. EMBO J. (2024) 43(22):5421–44. doi: 10.1038/s44318-024-00262-7
186. Lu SX, De Neef E, Thomas JD, Sabio E, Rousseau B, Gigoux M, et al. Pharmacologic modulation of RNA splicing enhances anti-tumor immunity. Cell. (2021) 184:4032–47 e31. doi: 10.1016/j.cell.2021.05.038
187. Carvalho T, Martins S, Rino J, Marinho S, Carmo-Fonseca M. Pharmacological inhibition of the spliceosome subunit SF3b triggers exon junction complex-independent nonsense-mediated decay. J Cell sci. (2017) 130:1519–31. doi: 10.1242/jcs.202200
188. Mahoney KM, Shukla SA, Patsoukis N, Chaudhri A, Browne EP, Arazi A, et al. A secreted PD-L1 splice variant that covalently dimerizes and mediates immunosuppression. Cancer Immunol Immunother. (2019) 68:421–32. doi: 10.1007/s00262-018-2282-1
189. Jain M, Abu-Shumays R, Olsen HE, Akeson M. Advances in nanopore direct RNA sequencing. Nat Methods. (2022) 19:1160–4. doi: 10.1038/s41592-022-01633-w
190. Krizanovic K, Echchiki A, Roux J, Sikic M. Evaluation of tools for long read RNA-seq splice-aware alignment. Bioinformatics. (2018) 34:748–54. doi: 10.1093/bioinformatics/btx668
191. Mehmood A, Laiho A, Venalainen MS, McGlinchey AJ, Wang N, Elo LL. Systematic evaluation of differential splicing tools for RNA-seq studies. Brief Bioinform. (2020) 21:2052–65. doi: 10.1093/bib/bbz126
192. Chhangawala S, Rudy G, Mason CE, Rosenfeld JA. The impact of read length on quantification of differentially expressed genes and splice junction detection. Genome Biol. (2015) 16:131. doi: 10.1186/s13059-015-0697-y
193. Ziegenhain C, Vieth B, Parekh S, Reinius B, Guillaumet-Adkins A, Smets M, et al. Comparative analysis of single-cell RNA sequencing methods. Mol Cell. (2017) 65:631–43 e4. doi: 10.1016/j.molcel.2017.01.023
194. Song Y, Botvinnik OB, Lovci MT, Kakaradov B, Liu P, Xu JL, et al. Single-cell alternative splicing analysis with expedition reveals splicing dynamics during neuron differentiation. Mol Cell. (2017) 67:148–61 e5. doi: 10.1016/j.molcel.2017.06.003
195. Illumina. Available online at: https://emea.support.illumina.com/bulletins/2020/04/maximum-read-length-for-illumina-sequencing-platforms.html.
196. Available online at: https://nanoporetech.com/resource-centre/application-note/isoform-detection-single-cell-and-spatial-resolution.
197. Heywood SM, Kennedy DS. Purification of myosin translational control RNA and its interaction with myosin messenger RNA. Biochemistry. (1976) 15:3314–9. doi: 10.1021/bi00660a023
198. Bester AJ, Kennedy DS, Heywood SM. Two classes of translational control RNA: their role in the regulation of protein synthesis. Proc Natl Acad Sci U S A. (1975) 72:1523–7. doi: 10.1073/pnas.72.4.1523
199. Paterson BM, Roberts BE, Kuff EL. Structural gene identification and mapping by DNA-mRNA hybrid-arrested cell-free translation. Proc Natl Acad Sci U S A. (1977) 74:4370–4. doi: 10.1073/pnas.74.10.4370
200. Zamecnik PC, Stephenson ML. Inhibition of Rous sarcoma virus replication and cell transformation by a specific oligodeoxynucleotide. Proc Natl Acad Sci U S A. (1978) 75:280–4. doi: 10.1073/pnas.75.1.280
201. Stephenson ML, Zamecnik PC. Inhibition of Rous sarcoma viral RNA translation by a specific oligodeoxyribonucleotide. Proc Natl Acad Sci U S A. (1978) 75:285–8. doi: 10.1073/pnas.75.1.285
202. Donis-Keller H. Site specific enzymatic cleavage of RNA. Nucleic Acids Res. (1979) 7:179–92. doi: 10.1093/nar/7.1.179
203. Sinha ND, Biernat J, McManus J, Koster H. Polymer support oligonucleotide synthesis XVIII: use of beta-cyanoethyl-N,N-dialkylamino-/N-morpholino phosphoramidite of deoxynucleosides for the synthesis of DNA fragments simplifying deprotection and isolation of the final product. Nucleic Acids Res. (1984) 12:4539–57. doi: 10.1093/nar/12.11.4539
204. Huang Y, Knouse KW, Qiu S, Hao W, Padial NM, Vantourout JC, et al. A P(V) platform for oligonucleotide synthesis. Science. (2021) 373(6560):1265–70. doi: 10.1126/science.abi9727
205. Tamm I, Dorken B, Hartmann G. Antisense therapy in oncology: new hope for an old idea? Lancet. (2001) 358:489–97. doi: 10.1016/S0140-6736(01)05629-X
206. Martin Egli MM. Chemistry, structure and function of approved oligonucleotide therapeutics. Nucleic Acids Res. (2023) 51:2529–73. doi: 10.1093/nar/gkad067
207. Mourich DV, Oda SK, Schnell FJ, Crumley SL, Hauck LL, Moentenich CA, et al. Alternative splice forms of CTLA-4 induced by antisense mediated splice-switching influences autoimmune diabetes susceptibility in NOD mice. Nucleic Acid Ther. (2014) 24:114–26. doi: 10.1089/nat.2013.0449
208. Graziewicz MA, Tarrant TK, Buckley B, Roberts J, Fulton L, Hansen H, et al. An endogenous TNF-alpha antagonist induced by splice-switching oligonucleotides reduces inflammation in hepatitis and arthritis mouse models. Mol Ther. (2008) 16:1316–22. doi: 10.1038/mt.2008.85
209. Bauman J, Jearawiriyapaisarn N, Kole R. Therapeutic potential of splice-switching oligonucleotides. Oligonucleotides. (2009) 19:1–13. doi: 10.1089/oli.2008.0161
210. Mendell JR, Goemans N, Lowes LP, Alfano LN, Berry K, Shao J, et al. Longitudinal effect of eteplirsen versus historical control on ambulation in Duchenne muscular dystrophy. Ann Neurol. (2016) 79:257–71. doi: 10.1002/ana.24555
211. Crooke ST, Vickers TA, Lima WF, Wu H-J. Antisense Drug Technology: Principles, Strategies, and Applications. Crooke ST, editor. Boca Raton, Fla., United States: CRC Press (2008) p. 3–46.
212. Swayze EE, Bhat B. Antisense Drug Technology: Principles, Strategies, and Applications. Crooke ST, editor. Boca Raton, Fla., United States: CRC Press (2008) p. 143–82.
213. Henry S. Antisense Drug Technology-Principles, Strategies, and Applications. Crooke ST, editor. Boca Raton, Fla., United States: CRC Press (2008) p. 327–64.
214. Gleave ME, Monia BP. Antisense therapy for cancer. Nat Rev Cancer. (2005) 5:468–79. doi: 10.1038/nrc1631
215. Eckstein F. Phosphorothioates, essential components of therapeutic oligonucleotides. Nucleic Acid Ther. (2014) 24:374–87. doi: 10.1089/nat.2014.0506
216. Sands H, Gorey-Feret LJ, Cocuzza AJ, Hobbs FW, Chidester D, Trainor GL. Biodistribution and metabolism of internally 3H-labeled oligonucleotides. I. Comparison of a phosphodiester and a phosphorothioate. Mol Pharmacol. (1994) 45:932–43.
217. Bennett CF, Swayze EE. RNA targeting therapeutics: molecular mechanisms of antisense oligonucleotides as a therapeutic platform. Annu Rev Pharmacol Toxicol. (2010) 50:259–93. doi: 10.1146/annurev.pharmtox.010909.105654
218. Wang S, Allen N, Vickers TA, Revenko AS, Sun H, Liang XH, et al. Cellular uptake mediated by epidermal growth factor receptor facilitates the intracellular activity of phosphorothioate-modified antisense oligonucleotides. Nucleic Acids Res. (2018) 46:3579–94. doi: 10.1093/nar/gky145
219. Brown DA, Kang SH, Gryaznov SM, DeDionisio L, Heidenreich O, Sullivan S, et al. Effect of phosphorothioate modification of oligodeoxynucleotides on specific protein binding. J Biol Chem. (1994) 269:26801–5. doi: 10.1016/S0021-9258(18)47090-1
220. Iversen PL. Antisense Drug Technology. Crooke ST, editor. Boca Raton, FL: CRC Press (2008) p. 565–82.
221. Summerton J. Morpholino antisense oligomers: the case for an RNase H-independent structural type. Biochim Biophys Acta. (1999) 1489:141–58. doi: 10.1016/S0167-4781(99)00150-5
222. Geary RS. Antisense oligonucleotide pharmacokinetics and metabolism. Expert Opin Drug Metab Toxicol. (2009) 5:381–91. doi: 10.1517/17425250902877680
223. Langner HK, Jastrzebska K, Caruthers MH. Synthesis and characterization of thiophosphoramidate morpholino oligonucleotides and chimeras. J Am Chem Soc. (2020) 142:16240–53. doi: 10.1021/jacs.0c04335
224. Finkel RS, Mercuri E, Darras BT, Connolly AM, Kuntz NL, Kirschner J, et al. Nusinersen versus sham control in infantile-onset spinal muscular atrophy. N Engl J Med. (2017) 377:1723–32. doi: 10.1056/NEJMoa1702752
225. De Vivo DC, Hwu W-L, Reyna SP, Farwell W, Gheuens S, Sun P, et al. Interim efficacy and safety results from the phase 2 NURTURE study evaluating nusinersen in presymptomatic infants with spinal muscular atrophy. Neurology. (2017) 2017:S46.003.
226. Duan D, Goemans N, Takeda S, Mercuri E, Aartsma-Rus A. Duchenne muscular dystrophy. Nat Rev Dis Primers. (2021) 7:13. doi: 10.1038/s41572-021-00248-3
227. Lim KR, Maruyama R, Yokota T. Eteplirsen in the treatment of Duchenne muscular dystrophy. Drug Des Devel Ther. (2017) 11:533–45. doi: 10.2147/DDDT.S97635
229. Dhillon S. Viltolarsen: first approval. Drugs. (2020) 80:1027–31. doi: 10.1007/s40265-020-01339-3
231. Crooke ST, Liang XH, Baker BF, Crooke RM. Antisense technology: A review. J Biol Chem. (2021) 296:100416. doi: 10.1016/j.jbc.2021.100416
232. Yang R, Feng X, Arias-Cavieres A, Mitchell RM, Polo A, Hu K, et al. Upregulation of SYNGAP1 expression in mice and human neurons by redirecting alternative splicing. Neuron. (2023) 111:1637–50 e5. doi: 10.1016/j.neuron.2023.02.021
233. Yuan Y, Lopez-Santiago L, Denomme N, Chen C, O’Malley HA, Hodges SL, et al. ASO restores excitability, GABA signalling and sodium current density in a model of Dravet syndrome. Brain. (2024) 147(4):1231–46. doi: 10.1093/brain/awad349
234. Kim H, Lenoir S, Helfricht A, Jung T, Karneva ZK, Lee Y, et al. A pathogenic proteolysis-resistant huntingtin isoform induced by an antisense oligonucleotide maintains huntingtin function. JCI Insight. (2022) 7(17):e154108. doi: 10.1172/jci.insight.154108
235. Shah S, Sharp KJ, Raju Ponny S, Lee J, Watts JK, Berry-Kravis E, et al. Antisense oligonucleotide rescue of CGG expansion-dependent FMR1 mis-splicing in fragile X syndrome restores FMRP. Proc Natl Acad Sci U S A. (2023) 120:e2302534120. doi: 10.1073/pnas.2302534120
236. Oren YS, Avizur-BarChad O, Ozeri-Galai E, Elgrabli R, Schirelman MR, Blinder T, et al. Antisense oligonucleotide splicing modulation as a novel Cystic Fibrosis therapeutic approach for the W1282X nonsense mutation. J cystic fibrosis: Off J Eur Cystic Fibrosis Society. (2022) 21:630–6. doi: 10.1016/j.jcf.2021.12.012
237. Michaels WE, Pena-Rasgado C, Kotaria R, Bridges RJ, Hastings ML. Open reading frame correction using splice-switching antisense oligonucleotides for the treatment of cystic fibrosis. Proc Natl Acad Sci U S A. (2022) 119(3):e2114886119. doi: 10.1073/pnas.2114886119
238. Dewaele M, Tabaglio T, Willekens K, Bezzi M, Teo SX, Low DH, et al. Antisense oligonucleotide-mediated MDM4 exon 6 skipping impairs tumor growth. J Clin Invest. (2016) 126:68–84. doi: 10.1172/JCI82534
239. Oh DY, Bang YJ. HER2-targeted therapies - a role beyond breast cancer. Nat Rev Clin Oncol. (2020) 17:33–48. doi: 10.1038/s41571-019-0268-3
240. Wan J, Sazani P, Kole R. Modification of HER2 pre-mRNA alternative splicing and its effects on breast cancer cells. Int J Cancer. (2009) 124:772–7. doi: 10.1002/ijc.v124:4
241. Pankratova S, Nielsen BN, Shiraishi T, Nielsen PE. PNA-mediated modulation and redirection of Her-2 pre-mRNA splicing: specific skipping of erbB-2 exon 19 coding for the ATP catalytic domain. Int J Oncol. (2010) 36:29–38.
242. Khurshid S, Montes M, Comiskey DF Jr., Shane B, Matsa E, Jung F, et al. Splice-switching of the insulin receptor pre-mRNA alleviates tumorigenic hallmarks in rhabdomyosarcoma. NPJ Precis Oncol. (2022) 6:1. doi: 10.1038/s41698-021-00245-5
Keywords: alternative splicing, cancer, immunotherapy, T lymphocytes, immune receptors
Citation: Tzaban S, Stern O, Zisman E, Eisenberg G, Klein S, Frankenburg S and Lotem M (2025) Alternative splicing of modulatory immune receptors in T lymphocytes: a newly identified and targetable mechanism for anticancer immunotherapy. Front. Immunol. 15:1490035. doi: 10.3389/fimmu.2024.1490035
Received: 02 September 2024; Accepted: 25 November 2024;
Published: 07 January 2025.
Edited by:
Zhihao Wang, Wuhan University, ChinaReviewed by:
Hao Wu, Exelixis Inc., United StatesShengjian Huang, Chengdu Chipscreen Pharmaceutical Ltd, China
Copyright © 2025 Tzaban, Stern, Zisman, Eisenberg, Klein, Frankenburg and Lotem. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michal Lotem, bWxvdGVtQGhhZGFzc2FoLm9yZy5pbA==