 Cong Xie1,2†
Cong Xie1,2† Jingyan Yang3†
Jingyan Yang3† Aman Gul2,4,5†Yifan Li2Rui Zhang6Maimaititusun Yalikun2Xiaotong Lv7Yuhan Lin1,8
Aman Gul2,4,5†Yifan Li2Rui Zhang6Maimaititusun Yalikun2Xiaotong Lv7Yuhan Lin1,8 Qingli Luo2*Huijuan Gao1*
Qingli Luo2*Huijuan Gao1*- 1Department of Endocrinology and Clinical Immunology, Yuquan Hospital, School of Clinical Medicine, Tsinghua University, Beijing, China
- 2Department of Integrative Medicine, Huashan Hospital Affiliated to Fudan University, Fudan Institutes of Integrative Medicine, Fudan University Shanghai Medical College, Shanghai, China
- 3The Third Affiliated Hospital, Beijing University of Chinese Medicine, Beijing, China
- 4Department of Respiratory Medicine, Uyghur Medicines Hospital of Xinjiang Uyghur Autonomous Region, Urumqi, China
- 5College of Life Science and Technology, Xinjiang University, Urumqi, China
- 6Department of Pulmonary and Critical Care Medicine, Shenzhen Hospital of Guangzhou University of Chinese Medicine (Futian), Shenzhen, China
- 7Department of Cardiology, The Second Affiliated Hospital of Tianjin University of Traditional Chinese Medicine, Tianjin, China
- 8Dongzhimen Hospital, Beijing University of Chinese Medicine, Beijing, China
In the present review, we focused on recent translational and clinical discoveries in asthma immunology, facilitating phenotyping and stratified or personalized interventions for patients with this condition. The immune processes behind chronic inflammation in asthma exhibit marked heterogeneity, with diverse phenotypes defining discernible features and endotypes illuminating the underlying molecular mechanisms. In particular, two primary endotypes of asthma have been identified: “type 2-high,” characterized by increased eosinophil levels in the airways and sputum of patients, and “type 2-low,” distinguished by increased neutrophils or a pauci-granulocytic profile. Our review encompasses significant advances in both innate and adaptive immunities, with emphasis on the key cellular and molecular mediators, and delves into innovative biological and targeted therapies for all the asthma endotypes. Recognizing that the immunopathology of asthma is dynamic and continuous, exhibiting spatial and temporal variabilities, is the central theme of this review. This complexity is underscored through the innumerable interactions involved, rather than being driven by a single predominant factor. Integrated efforts to improve our understanding of the pathophysiological characteristics of asthma indicate a trend toward an approach based on disease biology, encompassing the combined examination of the clinical, cellular, and molecular dimensions of the disease to more accurately correlate clinical traits with specific disease mechanisms.
1 Introduction
Asthma is a familiar, chronic, noncommunicable lung disease affecting approximately 300 million people worldwide (1), including 45.7 million adults in China (2). The prevalence of asthma considerably varies across different countries and regions; the prevalence is higher in urban areas and individuals with some risk factors, including allergies, smoking, and air pollution exposure. Although asthma incidence appears to be stabilizing after decades of rapid growth in many developed countries, its prevalence is increasing rapidly in low- and middle-income countries. This increase in prevalence may be owing to the worsening of fossil fuel pollution and the adoption of Westernized lifestyles. Furthermore, the absence of accurate diagnosis and standardized treatments in these developing countries increases the asthma burden on patients, their families, and society as a whole (3).
An expiratory airflow limitation is the primary feature of asthma; however, this limitation is generally reversible but related to airway lumen diameter narrowing. The narrowing occurs because of chronic inflammation in the walls of the airway, which is marked by the infiltration and activation of different immune cells, including eosinophils, neutrophils, lymphocytes, dendritic cells (DCs), innate lymphoid cells (ILCs), and mast cells, inducing processes such as bronchial hyperresponsiveness, mucus hypersecretion, and airway remodeling.
The clinical traits of asthma, i.e., dyspnea, coughing, wheezing, chest tightness, loss of lung function, exacerbation tendency, and asthma severity, suggest that the disease encompasses distinct underlying mechanisms, in which structural and immune cells interact to manifest the pathogenetic features of asthma. However, the relative contribution of these features may differ among patients with asthma, coupled with remarkable differences in genetic variations and environmental exposure; this results in significant heterogeneity in clinical manifestations and inflammatory biomarker expression.
Asthma has different clinical characteristics (“phenotypes”) and underlying causative mechanisms (“endotypes”) (4). Historically, clinicians have categorized asthma into two phenotypes: intrinsic (nonallergic) and extrinsic (allergic) (5). The primary difference between these two phenotypes is that allergic asthma generally occurs during childhood, whereas nonallergic asthma usually begins in adulthood. Allergic asthma typically manifests as acute episodes with increased airway responsiveness after allergen stimulation; it is more responsive to inhaled corticosteroids (ICSs) compared with nonallergic asthma (6). More recently, various clinical parameters, including onset age, condition severity and duration, frequency of acute exacerbation, impairment in respiratory function, level of symptom control, biomarkers, and treatment response, including potential hormone resistance, have been utilized to classify the phenotypes of asthma.
Some of the most common phenotypes, including allergic asthma, nonallergic asthma, adult-onset (late-onset) asthma, asthma with persistent airflow limitation, and obesity-associated asthma, have been listed in the updated 2023 and 2024 Global Initiative for Asthma (GINA) guidelines (7). Simultaneously, researchers in the field of basic medical sciences, particularly immunologists using murine models of allergic asthma and/or inflammation, have confirmed the pivotal role of the elements of the T helper (TH2) immune pathway in exacerbating inflammation and airway hyperreactivity (8, 9). TH2 cells are involved in the generation of cytokines that induce the different essential characteristics of asthma, including tissue eosinophilia (interleukin [IL]-5), bronchial hyperresponsiveness (IL-13), and goblet cell metaplasia (IL-4 and IL-13) (10). Recent studies have extended this understanding and suggested that apart from TH2 cells, other innate immune cells, including mast cells, basophils, group 2 ILCs, IL-4- and/or IL-13-activated macrophages (“M2”), and a small portion of IL-4-secreting natural killer (NK)/NKT cells, also contribute to TH2 cell induced cytokine production in asthma; as a result, the terminology has gradually shifted from “TH2 cell-high” to “type 2-high” asthma (11). However, this “type 2-high” profile, primarily characterized by eosinophilia, is only observed in roughly 50% of patients with asthma (12). The remaining patients, categorized as “type 2-low” asthma, without eosinophilia, exhibit distinct immune features, including airway neutrophilia, obesity-associated systemic inflammation, or minimal immune activation signs in some cases (13).
In patients with asthma, there is specific chronic inflammation in the lower airway mucosa. Although the major cellular components associated with this inflammation type have been ascertained, the interplay between the inflammatory cells in different spatial and temporal dimensions remains unclear (14); furthermore, it is not known how this inflammation translates into asthma symptoms. Similar to other atopic diseases, asthma pathogenesis involves several factors, including genetic predisposition, the airway initiation of specific IgE (sIgE) to respiratory allergens, and an overactive immune system that produces excessive amounts of inflammatory mediators. To date, the acute inflammatory changes observed in asthma have garnered considerable attention; in this chronic condition, inflammation persists for many years in most patients. Superimposed on this chronic inflammatory state are acute inflammatory episodes, which correspond to exacerbations of asthma.
Moving from patients to animal or cellular models and back represents an iterative process by which we can elucidate the intricate pathophysiology of asthma. Herein, we focus on the underlying immunological aspects of asthma in the context of recent insights into its extraordinary heterogeneity by summarizing the findings from human studies on particular pathways along with rigorous basic experimentation that has collected a surplus of molecular details.
2 Pro-inflammatory and anti-inflammatory arms of the immune landscape in asthma
Under the guidance of locally released chemokines, many inflammatory cells are recruited to the lungs from the bloodstream; these cells exert functional properties for asthma development. Furthermore, airway structural cells, including epithelial cells, fibroblasts, and airway smooth muscle cells (SMCs), are essential inflammatory mediator sources that actively participate in the inflammatory process. In individuals with asthma, both innate (mast cells, DCs, eosinophils, neutrophils, basophils, ILCs, monocytes, and macrophages) and adaptive (T and B lymphocytes) immunities are involved in the inflammatory cell profile (15).
A landmark study conducted in the mid-1980s reported the classical CD4+T lymphocyte subsets (TH1 and TH2 cells) (16); since then, it is well-known that TH2 cells orchestrate eosinophilic airway inflammation by producing abundant amounts of IL-4, IL-5, and IL-13 (17). IL-4 is required for allergic sensitization and IgE class switching, IL-5 is warranted for eosinophil survival, IL-13 exerts multifunctional effects in the lungs, including a vital role in controlling mucus production, goblet cell metaplasia, bronchial hyperresponsiveness, and airway remodeling (18). In contrast, TH1 cells release IL-2, interferon (IFN)-γ, and tumor necrosis factor (TNF)-α, possibly conferring a protective role in asthma because they can directly antagonize pathologic TH2 responses to control eosinophilic inflammation (19). To support this, IL-12, a pro-TH1 cell cytokine, administration in mice suppresses antigen-induced airway hyperresponsiveness and inflammation by producing IFN-γ via TH1 cells (20, 21). However, recent studies on the phenotype of type 2-low asthma have demonstrated the dominance of IFN-γ+TH1 cells in severe disease forms, which is potentially associated with corticosteroid refractoriness (22, 23).
In addition to TH2 cells, TH17 cells and their produced cytokine IL-17A are prominent and extensively studied in the context of asthma, particularly in severe, steroid-resistant cases (24). These TH17-derived cytokines, including IL-17 and IL-22, are related to increased neutrophil recruitment in the airways (25). To this end, neutrophil extracellular traps and cytoplasts further promote TH17 polarization and neutrophilic inflammation in severe asthma (26). However, the precise roles of TH17 cells and IL-17 in mouse asthma models remain unknown primarily because IL-17 may play dual regulatory roles: it plays a protective role in the challenge stage but worsens asthma under other conditions (27, 28). In chronic asthma models, IL-17A induces the proliferation of fibroblasts (29), inhibits the anti-inflammatory effects of regulatory T cells (Tregs) (30), and directly contracts bronchial SMCs (31).
Increasing evidence in animals indicates that a major hallmark of several autoimmune disorders, including asthma, is functional defects in Tregs (32). In a broader perspective, as a diverse population, Tregs comprise CD4+CD25+ forkhead box (Fox)p3+ natural and inducible Tregs, IL-10-producing Tr1 cells, transforming growth factor (TGF)-β-producing TH3 cells, and other minor subsets with suppressive functions, including CD4−CD8− T and γδT cells (33). In children with asthma, both CD4+CD25hi Tregs and Foxp3 mRNA expression decrease in the peripheral blood and bronchoalveolar lavage fluid (BALF); this phenomenon can be reversed following treatment with inhaled glucocorticoids (34). Recently, a study has revealed that numerical and functional deficiencies in Tregs may increase the asthma risk in children and young adults; however, the association between Tregs and the risk or severity of asthma in the elderly may be weaker (35).
The discovery of a distinct cohort of IL-9-secreting CD4+T cells, called TH9 cells, has enhanced the intricacies of T cell subsets. These cells are produced in response to IL-4 and TGF-β (36). These TH9 cells facilitate the binding of the transcription factors PU.1 and interferon-regulatory factor (IRF)-4 to the Il9 promoter (37). Furthermore, IL-25 (i.e., IL-17E) enhances IL-9 secretion from TH9 cells (38). IL-9 promotes allergic responses, including IgE production and eosinophilia (39). In allergic inflammation experimental models, mast cell accumulation is IL-9-dependent (40); however, lung-infiltrating mast cells and protease expression in mast cells were significantly decreased in mice with PU.1 deficiency (41). Subsequent studies have demonstrated that the deletion of a regulatory region in the Il9 locus, which is vital for initiating the IL-9 expression and TH9 cell maturation, effectively alleviates allergic lung inflammation (42, 43). Several clinical trials involving a humanized anti-IL-9 monoclonal antibody (mAb), MEDI-528, have been successfully completed in individuals with asthma, demonstrating some degree of efficacy (44, 45).
At present, studies suggest that alveolar macrophages possess comprehensive immunoregulatory capabilities in asthma, beyond those of a pathogenic barrier to lung tissues (46). Based on the stimulation type, surface markers, pattern of secreted cytokines, and functional characteristics, two main polarized macrophage subpopulations have been identified: “M1” macrophages, which are classically activated, and “M2” macrophages, which are alternatively activated (47). Although controversial, some studies have demonstrated that M2 macrophages express TH2-associated cytokines (IL-4 and IL-13) and TGF-β, which participate in type 2 inflammation and airway remodeling in allergic asthma (48). However, M2 macrophages release high levels of IL-10 and TGF-β, playing roles in inflammation resolution, wound repair, and homeostasis maintenance, further complicating the precise function of M2 macrophages (49).
Despite the well-known heterogeneity of asthma, an imbalanced immune microenvironment is a prerequisite for its development. This imbalance encompasses the dynamic interplay of T cells and macrophages, beginning from the initial stages and continuing until disease progression. Instead of attributing the disease solely to specific subsets, alterations in the interactions and functions between different subgroups may play significant roles (50). Such interactions and functions may be referred to as “pro-inflammatory/anti-inflammatory balance regulatory networks” (Figure 1). Notably, the classical paradigm of TH2-skewed immune responses remains relevant; however, emerging evidence suggests that it is considerably more sophisticated in vivo than previously envisaged, involving extremely uneven cell subpopulations and different cytokine expression patterns that dynamically fine-tune themselves based on different spatiotemporal cues.
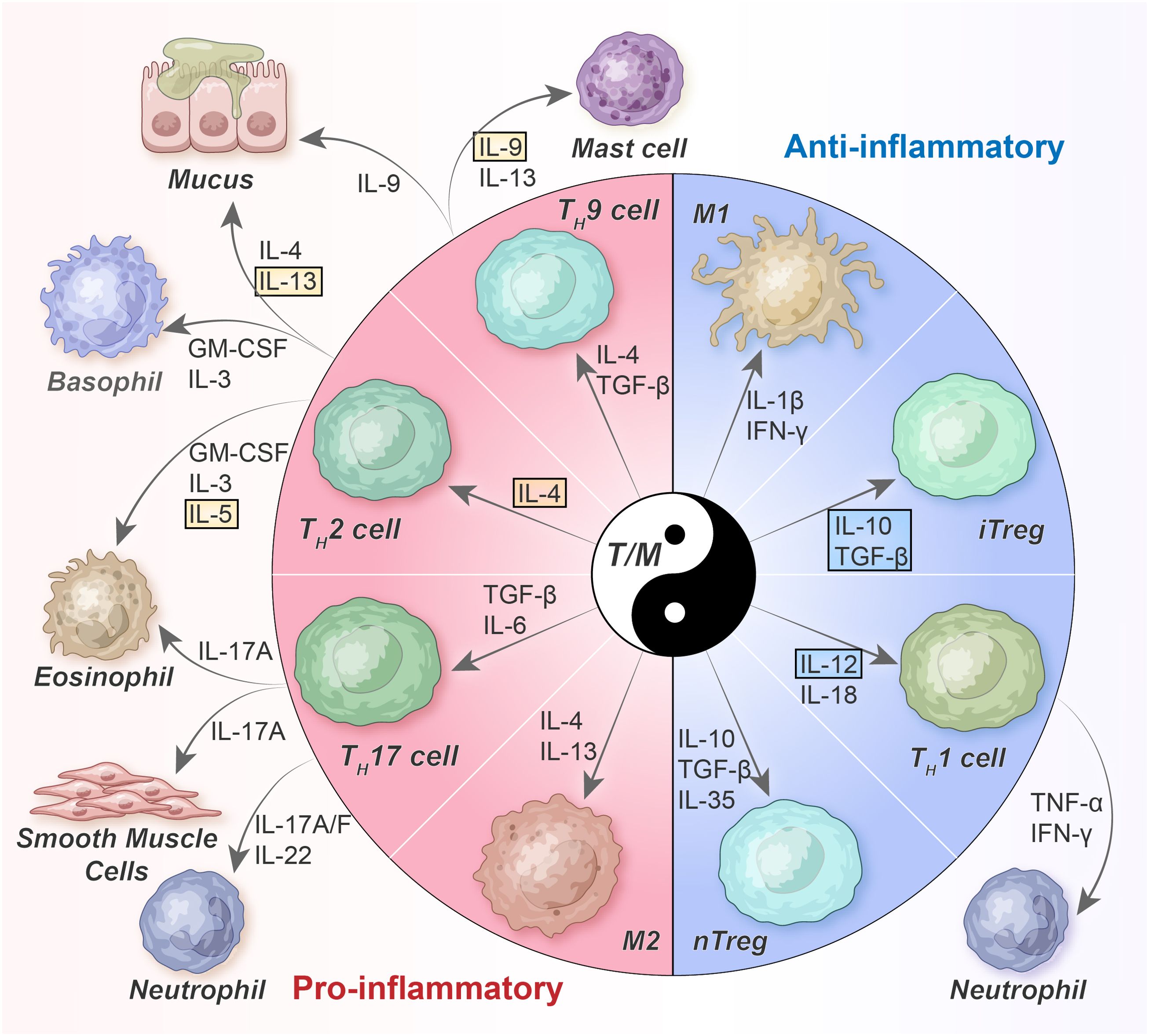
Figure 1. Balanced T-cell/macrophage networks and their cytokine milieu in asthma. TH2 cells orchestrate allergic inflammation by secreting TH2 cytokines such as IL-4, IL-5, IL-9, and IL-13. However, TH1 cells, which are differentiated because of IL-12 and IL-18, inhibit TH2 cells by producing IFN-γ. TH17 cells, affected by IL-6 and TGF-β, follow a distinct differentiation pathway. In general, Tregs mitigate the activity of other TH cells by secreting TGF-β and IL-10; however, their functionality may be compromised under asthma conditions. nTreg, naturally occurring Tregs; iTreg, inducible Tregs; GM-CSF, granulocyte-macrophage colony stimulating factor; TGF, transforming growth factor; TNF, tumor necrosis factor; IFN, interferon.
3 Mechanisms leading to asthma
3.1 Step 1. Dysregulated epithelial barrier and early innate immune response
3.1.1 Epithelial injury, activation, and derived signals
Bronchial epithelial cells, which are strategically positioned at the host and environment interface, play vital roles in preserving respiratory mucosal integrity and stability because of mechanical-physical barriers, ciliary clearance, and immunoregulatory functions; they serve as the first line of defense against pathogens and airborne allergens (51). Early investigation of airway asthma pathology has revealed that epithelial cells are damaged, disintegrated, and dysfunctional. As a result, the “epithelial barrier hypothesis” has been established, suggesting that various allergic and autoimmune diseases have similar triggering mechanisms (52).
Different asthma phenotypes exhibit airway epithelial abnormalities, primarily manifesting as increased epithelial leakage, decreased inflammatory thresholds, ciliated cell shedding, detached columnar cells (Creola bodies), and impaired intercellular adhesion (53). Under homeostatic conditions, an impermeable epithelial barrier is formed; this barrier is maintained primarily by tight junctions (TJs) at the apical end of columnar cells. This barrier is further reinforced via different adhesion mechanisms in the basal and basolateral surfaces of epithelial cells, including adherens junctions (AJs) and (hemi)desmosomes (54). The bronchial biopsies of patients with asthma have revealed that zonula occludens-1 (ZO-1) and occludin, which are TJ proteins, are irregular stained, suggesting functional defects in epithelial connections (55). Compared with control subjects, the cultured airway epithelial cells of patients with asthma also suggest TJ protein degradations (55); in contrast, E-cadherin and α-catenin, AJ proteins, expression is decreased (56).
The vulnerability of the airway epithelium to environmental irritants and the dysfunctional repair mechanisms after such injuries are vital for asthma development (57). Aeroallergens such as house dust mites (HDMs), pollens, and fungi, microbes such as viruses and bacteria, and environmental pollutants such as cigarette smoke, particulate matter (PM)2.5, and diesel exhaust, directly impede the integrity of TJ barriers of the airway epithelium (58). Furthermore, insufficient antioxidant and antiviral mechanisms in asthmatic airways may increase the susceptibility of epithelial cells to oxidative and virus-induced damage.
Genome-wide association studies (GWAS) have confirmed that the genetic predisposition for asthma development is partially associated with barrier dysfunction; single-nucleotide polymorphisms (SNPs) have been identified in multiple genes, including protocadherin-1 (PCDH1), cadherin-related family member-3 (CDHR3), and orosomucoid-like protein isoform-3 (ORMDL3) (53). Furthermore, experimental mouse models that simulate three asthma phenotypes, i.e., type 2high-eosinophilic, type 2low-neutrophilic, and mixed granulocytic, were employed to differentiate the effects of phenotypes on the disruption of the epithelial barrier by focusing on TJ proteins and mucins. Many TJ proteins, including ZO-1 and claudin-18, were decreased in the asthma phenotypes; however, the degree of reduction was different. In contrast, claudin-4 is only overexpressed in neutrophilic asthma. Moreover, phenotype-specific discrepancies are found in mucins: MUC5AC and MUC5B are overexpressed in the asthma phenotypes; however, it is more pronounced in neutrophilic and mixed asthma (59).
In addition to its essential role as a physical barrier, the airway epithelium also modulates the initial innate immune response (Figure 2). Epithelial cells express several pattern recognition receptors (PRRs), rapidly detecting and responding to pathogen-associated molecular patterns (PAMPs) found in microorganisms as well as damage-associated molecular patterns (DAMPs) released due to tissue injury, cellular stress and cell death (60).
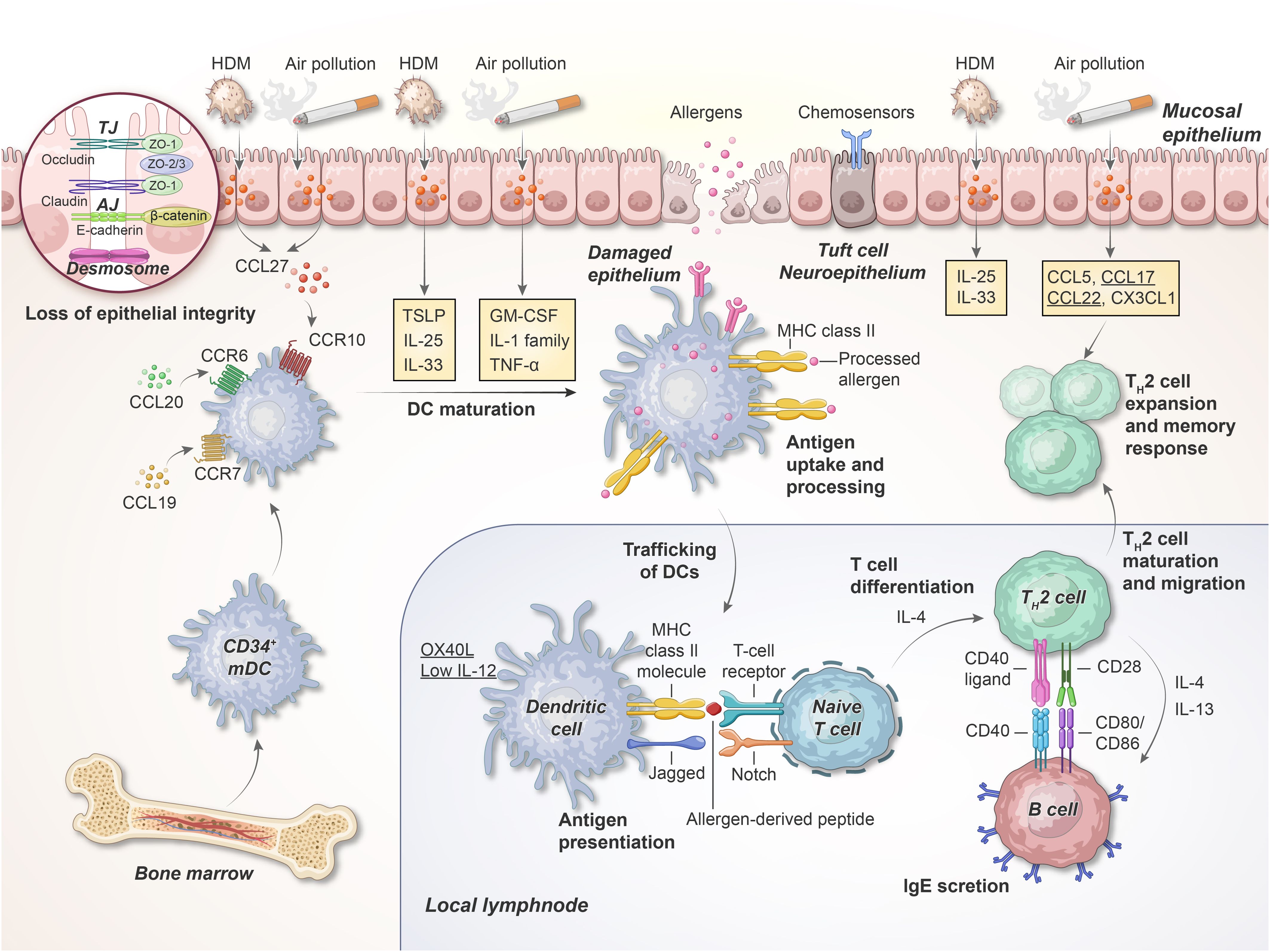
Figure 2. Early phase of allergen sensitizations in the airway. Inhaled allergens and air pollutants with protease activity can cleave epithelial TJs and trigger the PRRs on the epithelial cells; this results in the production of cytokines, including IL-1, IL-25, IL-33, TSLP, GM-CSF, and TNF-α. When these cytokines are released, DCs migrate toward the T cell region of the adjacent lymph nodes. Here, DCs interact with naive T cells via the TCR, MHC class II molecules, and co-stimulatory molecules, thereby facilitating TH cell differentiation. HDM, house dust mite; TJ, tight junction; AJ, adherens junction; ZO, zonula occludens; TSLP, thymic stromal lymphopoietin; mDC, myeloid DCs, MHC, major histocompatibility complex. This illustration was adapted from Holgate, 2012 (75).
Because of PRR activation on epithelial cells, large amounts of cytokines, chemokines, and antimicrobial peptides are secreted, attracting and activating innate and adaptive immune cells. During bacterial pathogen invasion, bacterial cell wall components can be sensed via various PRRs on airway epithelial cells, including toll-like receptor (TLR)-2, which recognizes elements such as lipoteichoic acid in gram-positive bacteria, TLR4, which recognizes lipopolysaccharide (LPS) in gram-negative bacteria, and NOD1 and NOD2, which recognize peptidoglycans. As a result, nuclear factor kappa B (NF-κB) is activated, initiating immune responses and eventually regulating bacterial clearance (61).
During viral infection, TLR3, TLR7/8, retinoic acid-inducible gene I (RIG-I), melanoma differentiation-associated protein-5 (MDA5), and laboratory of genetic and physiology-2 (LGP2) can recognize nucleic acid patterns (62). Furthermore, the activation of PRR-dependent epithelial cells results in the production of endogenous danger signals (i.e., DAMPs), including free adenosine triphosphate (ATP), uric acid, and high-motility group box 1 (HMGB1) protein, and activation of DCs with granulocyte-macrophage colony-stimulating factor (GM-CSF), IL-1α, and IL-33 (63).
The contemporary viewpoint suggests that tissue disturbances are the primary factor responsible for type 2 immunity rather than direct antigen recognition. In genetically susceptible individuals, with impaired epithelial barrier function, airways are vulnerable to viral infections and inhaled allergens in early life. This triggers immature DCs, guiding TH2 cell responses and sensitization to local allergens (64). Airway exposure to allergens, pollutants, or pathogens results in the release of epithelial-derived cytokines such as IL-33 and IL-25 and thymic stromal lymphopoietin (TSLP), a member of the IL-2 cytokine family. These cytokines, commonly called “alarmins,” exert pleiotropic properties; however, they synergistically activate DCs, ILC2s, memory TH2 cells, eosinophils, and mast cells, acting upstream in a sustained type 2 immune response cascade (65).
In mice, IL-33 and IL-25 activate OX40-ligand (OX40L, or CD252) expression on ILC2s, thereby activating ILC2 proliferation and cytokine production (66); in contrast, TSLP can prime DCs to improve type 2 immunity by activating T and B cells. If either one or a combination of these “alarmins” are neutralized, the development of the salient characteristics of asthma, including eosinophilia, airway hyperactivity, and peribronchial collagen deposition, may be inhibited, contingent upon the model allergen used (67–70). Moreover, clinical research suggests that after allergen inhalation for 24 h, the expression level of IL-33, IL-25, and TSLP is increased in the airway epithelium of patients with allergic asthma, correlating with the degree of airway obstruction (71). Large-scale GWAS have confirmed that SNPs in IL33 (located at 9p24.1), IL1RL1 (encoding the IL-33 receptor; also called suppression of tumorigenicity 2 [ST2], located at the 2q12.1 locus), and TSLP (5q22.1 locus) are positively associated with the risk of developing asthma; furthermore, epigenome analysis has revealed that they frequently exhibit an active chromatin state (72). Therefore, epithelial-derived alarmins may function as “early signals” in patients with different asthma phenotypes and can serve as potential therapeutic targets for allergic airway inflammation. At present, a mAb directed against TSLP (tezepelumab), has been approved for treating severe asthma in step 5 of the GINA guidelines (73). Biologic agents targeting IL-33 such as itepekimab are undergoing clinical trials (74) (Table 1).
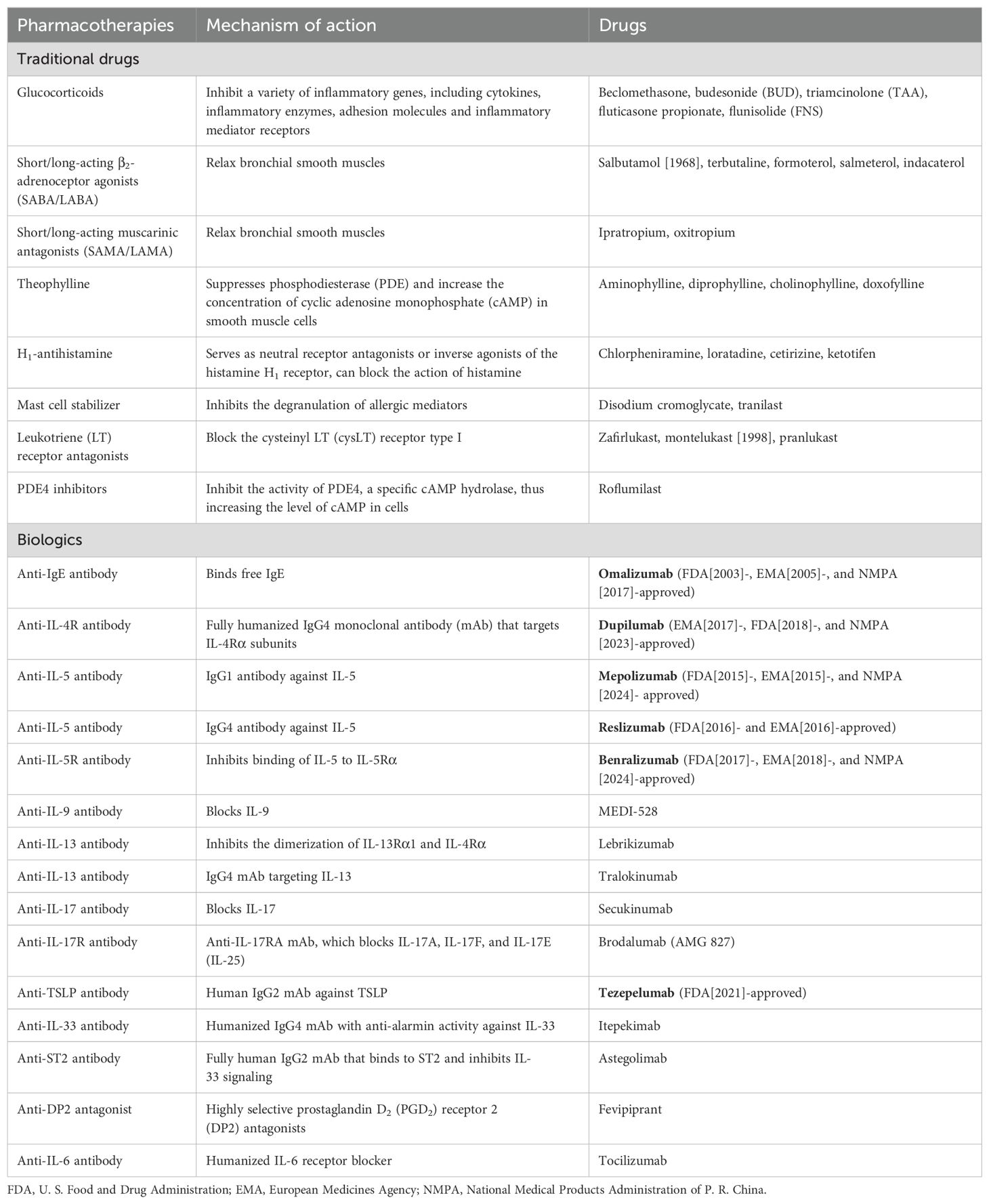
Table 1. Classic and novel treatments developed for asthma.
3.1.2 DCs deliver immunogenic messages to naive T cells
The interaction between specialized antigen-presenting airway DCs and T cells facilitates allergen sensitization. Allergen processing into small peptides and selectively presenting these processed peptides to the T cell receptors (TCRs) of naive T cells via major histocompatibility complex (MHC) class II molecules (i.e., the “first signal” or antigen-specific signal) are the underlying mechanisms (75).
Effective allergen signaling warrants co-stimulatory interplay between DCs and T cells that occurs in local lymphoid collections; this results in the differentiation of T cells into TH2-type T cells (i.e., the “second signal” or co-stimulatory signal) (76). Specifically, the activation of epithelial cells can result in the release of chemoattractants (C-C motif chemokine ligand [CCL]-20, CCL19 and CCL27, the ligands for CCR6, CCR7, and CCR10, respectively) that attract immature DCs that then differentiate and activate inflammation and adaptive immunity. A subset of conventional DCs (cDC2s) that depend on IRF4 for their development is responsible for initiating TH2 responses in mouse lungs and other organs (77). Several epithelial-derived cytokines, including IL-1α (78), GM-CSF (78), IL-33 (79), TSLP (80), and CSF1 (81), can directly target CD11b+CD172a (SIRPα)+ cDC2s, involved in the differentiation of TH2 cells. However, TH2 responses are not induced by lung-resident CD103+XCR1+ cDC1s (IRF8 and the basic leucine zipper transcriptional factor ATF-like 3 [Batf3]-dependent) and monocyte-derived DCs (moDCs). In fact, they may confer protection against asthma development by producing IL-12, a TH1-associated cytokine, thereby inhibiting TH2 responses (82). Moreover, plasmacytoid DCs (pDCs) play a tolerogenic role in allergic lung inflammation, inducing Foxp3+ Tregs in response to inhaled antigens, at least partially by upregulating programmed death-ligand 1 (PD-L1, or CD274), a T cell inhibitory ligand (83).
T cell differentiation is driven by the migration of allergen-loaded cDC2s to the regional lymph nodes from the lung tissues; this may be regulated by ILC2-derived IL-13 and type I IFNs (84, 85). Furthermore, epithelial cell-derived cytokines and chemokines, including IL-25, IL-33, CCL17 (thymus- and activation-regulated chemokine, TARC), and CCL22 (macrophage-derived chemokine, MDC), affect the activation of DCs, maturation of TH2 cells, and their mucosal migration. In asthma models challenged with allergen, CD11b+ DCs may be an important source of CCL17 and CCL22 (by activating their CCR4 receptors), TH2 cell-attracting inflammatory chemokines, and eosinophil-selective chemokines (i.e., the “third signal” or DC-secreted cytokines) (86). In clinical settings, activated DCs considerably increase in the airways of individuals suffering from asthma and ongoing inflammation (87); the high-affinity receptor FcϵRI is expressed in their lung cDC2s express (88, 89), with increased levels of CD86 and OX40L (90).
Indeed, because DCs can perceive danger signals, process antigens, and migrate to draining lymph nodes, they occupy the intersection between innate and adaptive immunities in the lungs.
3.2 Step 2. Adaptive immune reaction: features of type 2-high and type 2-low inflammation
Because tolerance or immune regulation fails in step 1, adaptive immune inflammation develops in the lung; this comprises CD4+ TH2, TH1, TH17, ILC2, and IgE-producing B cells (Figure 3). TH2 cytokines such as IL-4, IL-5, and IL-13 primarily drive allergic inflammation. However, pro-inflammatory cytokines, including TNF-α and IL-1β, augment inflammatory responses and play a role in more severe diseases. Some cytokines, including IL-10 and IL-12, exert anti-inflammatory properties and appear to be insufficient among individuals with asthma.
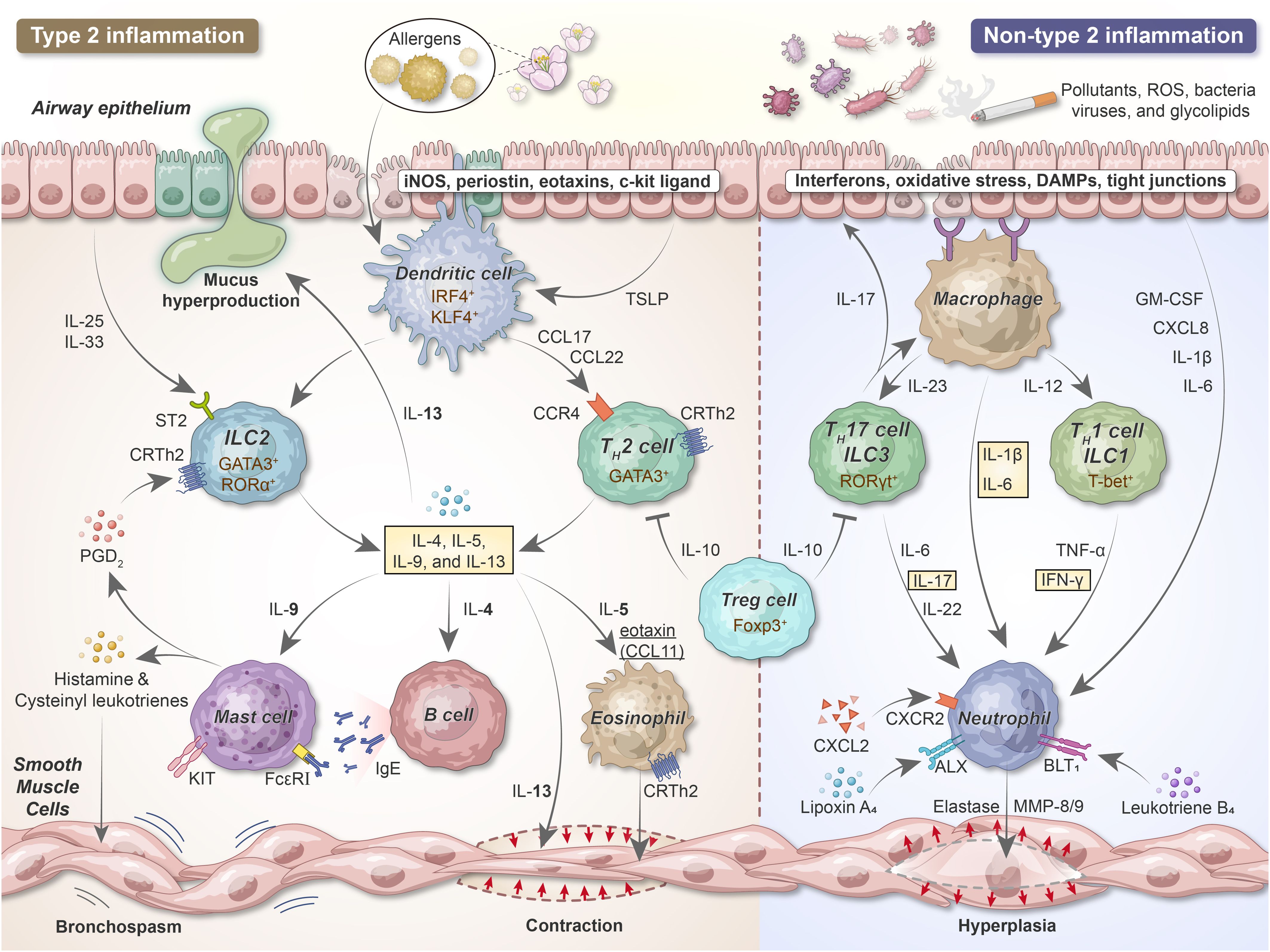
Figure 3. The development of airway inflammation and bronchial hyperresponsiveness in acute asthma. After sensitization, epithelial cells release alarmins (TSLP, IL-25, and IL-33) that activate DCs and ILCs. Upon the uptake, processing, and presentation of antigens to naive T cells, DCs promote naive T cell differentiation into TH2 lymphocytes. ILC2s and TH2 secrete IL-4, IL-5, IL-9, and IL-13, exerting vital roles in type 2 inflammation. Pollutants, cigarette smoke, viruses, and bacteria can damage and stimulate the airway epithelium, thereby releasing IL-1β, IL-6, and chemokines such as IL-8 acting as neutrophil chemoattractants. DCs and macrophages recruit neutrophils and release pro-inflammatory cytokines via TH1/ILC1 and TH17/ILC3 cells. iNOS, inducible nitric oxide synthase; IRF4, interferon regulatory factor 4; KLF4, Kruppel-like factor 4; ST2, suppressor of tumorigenicity 2; GATA, GATA-binding protein; ROR, RAR-related orphan receptor; Foxp3, forkhead box protein 3; T-bet, T-box expressed in T cells; PGD2, prostaglandin D2; MMP, matrix metalloproteinase.
Diverse asthma phenotypes are primarily driven by the complex interaction between type 1 and type 2 immune pathways (Table 2). In the early 1990s, some years after studies on the immune system in mice helped develop the TH1/TH2 T-lymphocyte-focused paradigm, the concept that airway inflammation in atopic asthma is associated with activated TH2 cells was first recognized (91). In another study, GATA3 was identified as a master transcription factor for TH2 cell development and cytokine production (92). Likewise, T-bet (93) and retinoic acid-related orphan receptor γt (RORγt) (94) are vital for differentiating TH1 and TH17 cells, respectively. In the last decade, solid data regarding the mechanisms underlying lineage development and the molecules associated with ILC subset functions have been obtained, wherein ILC1s, ILC2s, and ILC3s share and mirror the characteristics of CD4+ TH1, TH2, and TH17 cells (95). ILC1s, similar to TH1 and NK cells, generate IFN-γ upon IL-12 and IL-15 stimulation and depend on T-bet for their development. ILC2s depend on GATA3 and RORα and generate type 2 cytokines such as IL-5, IL-13, and IL-9, but little IL-4. ILC3 development requires RORγt; they respond to IL-23 and IL-1β, thereby producing IL-17 and IL-22.
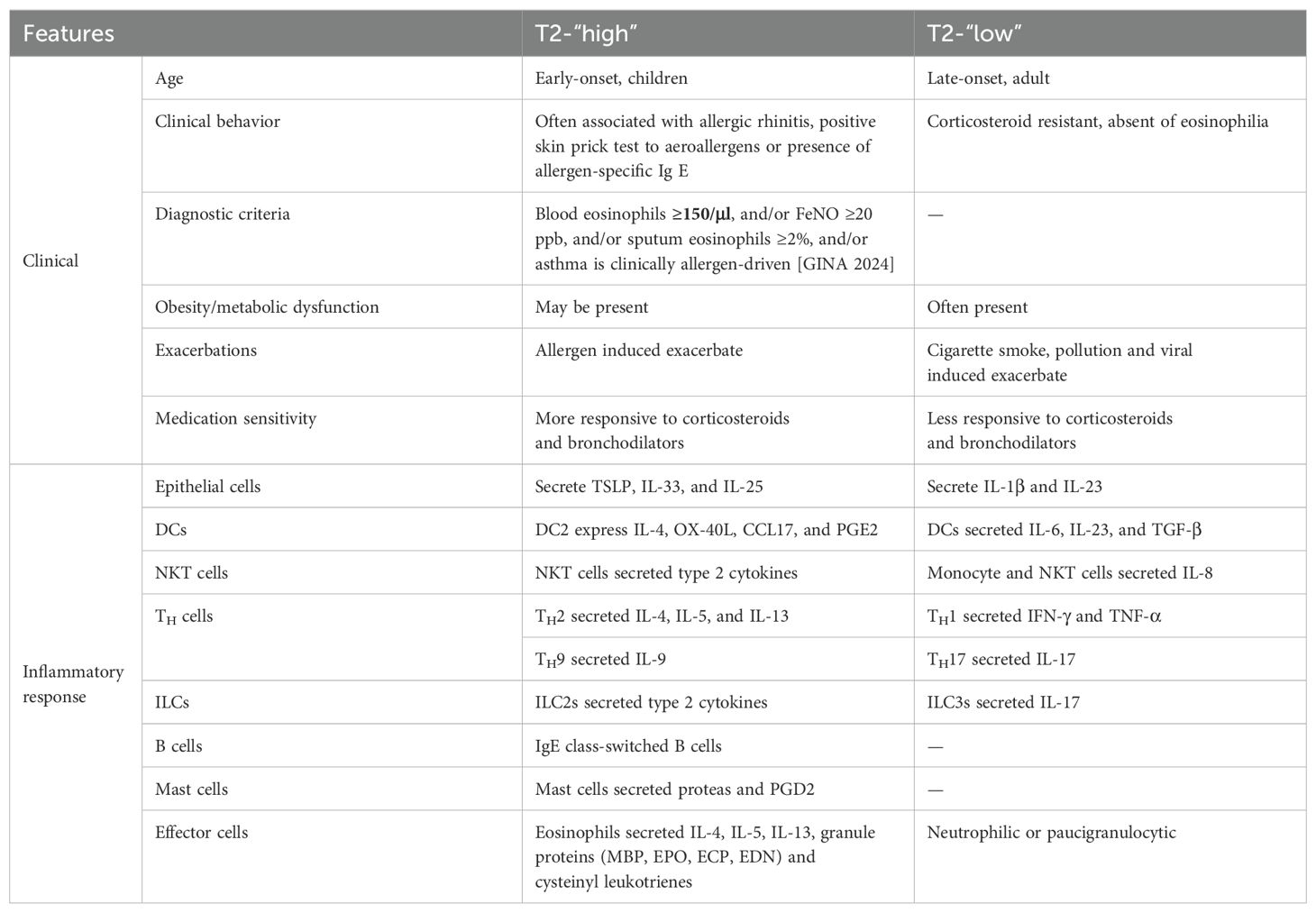
Table 2. Asthma phenotypes and cellular mechanism of type 2-high and type 2-low inflammation.
The ability of both CD4+ T cells and ILCs to rapidly release various cytokines in response to environmental stimuli such as tissue damage, pathogen invasion, or cellular stress is a fundamental characteristic. Accumulating evidence indicates that the plasticity and maintenance of the subsets of TH cells and ILCs are regulated by a delicate balance between their transcription factors, which are activated by differentiation-oriented cytokines; furthermore, they are affected by epigenetic modifications due to tissue microenvironment alterations (96). With an in-depth understanding of these cell types, we can gain invaluable insight into the mechanisms underlying the development of type 2-high and type 2-low asthma phenotypes (97).
3.2.1 Type 2 inflammation
Immunologists use mice to set up an ovalbumin (OVA)-induced allergic asthma model; this provides a window for elucidating the pathophysiology of the type 2-high asthma endotype and developing novel therapeutics (98). This is a pure TH2 response in which sensitization is achieved by intraperitoneally injecting the model antigen alum (adjuvant)-emulsified OVA and challenging with aerosolized OVA; this promotes IL-4, IL-5, IL-9, and IL-13 production and OVA-specific IgE and IgG1 synthesis.
Using this model, it was discovered that IL-4, via IL-4Rα, can promote IgE class switch recombination of B cells and plasma cell differentiation, worsen bronchial hyperreactivity, and induce the expression of adhesion molecules such as ICAM-1 (CD54) and VCAM-1 (CD106), priming the vascular endothelium for eosinophils extravasation (99). Uniquely, IL-4 is critically involved in the differentiation of naive TH cells into TH2 cells.
IL-5 is an essential cytokine for eosinophil development, maturation, activation, proliferation, and survival (100). However, it may not exert chemotactic effects on eosinophils. CCR3 is selectively activated by eotaxin-1 (CCL11), eotaxin-2 (CCL24), and eotaxin-3 (CCL26) (101), combined with the expression of some adhesion molecules, including VCAM-1 (which is upregulated by IL-4 and IL-13) (102), facilitating the recruitment of eosinophils from the bloodstream to the lung mucosa and interstitium.
IL-13 and IL-4 have similar biological properties because both cytokines can function via the IL-4Rα chain and phosphorylate STAT6, a downstream transcription factor (103). However, the key difference is that IL-13 majorly participates in the effector phase of type 2 immune responses, thereby affecting the development of several traditional pathophysiological characteristics of asthma owing to the effects it exerts on lung structural cells, including epithelial cells (mucus-secreting goblet cell differentiation and proliferation), SMCs (smooth muscle hypertrophy induction and enhanced contractility), fibroblasts (extracellular matrix [ECM] production), and endothelial cells (vascular remodeling) (8, 104). Unlike IL-4, IL-13 plays no role in the differentiation of T cells because IL-13R is not expressed in immature T cells. This disparity may be because canonical TH2 cells produce high levels of IL-13 at the effector site of the lungs, whereas IL-4 is primarily produced by T follicular helper (TFH) cells in the lymph nodes (105). Furthermore, in airway epithelial cells, IL-13 augments the expression of inducible nitric oxide synthase (iNOS); iNOS is primarily involved in generating fractional exhaled nitric oxide (FeNO), a diagnostic biomarker to examine type 2 inflammation in the respiratory tract (106).
IL-9 is released by a subset of CD4+ T cells (TH9 cells), potentially by classical TH2 cells, as well as by ILC2s. IL-9 drives mast cell survival, bronchial hyperresponsiveness, mucus cell metaplasia, and airway wall remodeling in mouse models (107). Therefore, IL-4, IL-5, IL-9, and IL-13, serving as classical type 2 cytokines, share common characteristics; however, each cytokine exhibits an exclusive functional profile.
The discovery that eosinophil-rich responses could be induced in mice lacking T and B cells has piqued our interest over the past few years that ILC2s as an important player in the pathogenesis of asthma (108). ILC2s are significantly increased in the blood and in the bronchoalveolar of asthmatic patients (109). Both ILC2s and TH2 cells belong to the lymphoid lineage and generate similar cytokine patterns. Their functions considerably overlap in asthma, although there are some detailed differences. In the presence of IL-25 or IL-33, ILC2s can directly control the key features of type 2 asthma, including eosinophilia, bronchial hyperreactivity, and goblet cell hyperplasia, by producing IL-5, IL-9, and IL-13 (110). Furthermore, ILCs function as antigen-presenting cells that use MHC class II molecules to present antigenic epitopes and express OX40L to support CD4+ T lymphocyte activity (111, 112). Moreover, in local tissue settings, ILC2s interact with both DCs and TH2 cells in complex bidirectional crosstalk, modulating the intensity of type 2 responses to properly respond to perceived environmental threats (84, 113).
Cellular activation and inflammatory mediator release are representative characteristics of type 2-high asthma, which can be observed via mast cell degranulation and eosinophil vacuolation. The interplay between IgE and high-affinity FcϵRI on granulocytes, including mast cells and basophils, results in the initiation of cell activation and degranulation, releasing multiple preformed and newly synthesized mediators, cytokines, chemokines, and growth factors. The ready-made mediators stored in cytoplasmic granules include biogenic amines (e.g., histamine and serotonin), neutral proteases (e.g., tryptase, chymase, and carboxypeptidase A), proteoglycans (e.g., heparin and chondroitin sulfate), and some cytokines (e.g., TNF-α) and growth factors (e.g., vascular endothelial growth factor A [VEGFA]) (114). TH2-dependent trypsin-expression cell (MCT) are the main type of mast cells that contribute to mild-to-moderate allergic asthma (115). However, in more severe asthma forms, mast cells containing both trypsin and chymase (MCTC) become dominant; compared with MCT, MCTC relies more on stem-cell factors (SCF, i.e., the KIT ligand) for survival (116).
Lipid-derived mediators can be secreted by FcϵRI aggregation-activated mast cells. They are responsible for arachidonic acid metabolism via the cyclooxygenase (COX) and lipoxygenase (LOX) pathways, releasing prostaglandins (PGs, particularly PGD2), leukotriene B4 (LTB4), and cysteinyl leukotrienes (CysLTs, including LTC4, LTD4, and LTE4) (117). Histamines, PGs, and CysLTs as potent bronchoconstrictors and can lead to bronchospasm, vasodilation, plasma leakage, mucus production, and elevated cellular recruitment in the lungs. PGD2 acts on type 1 PGD2 receptor (DP1) to contract the airway smooth muscle and CRTH2, a TH2 cell-expressed chemokine receptor, i.e., DP2, to chemoattract TH2 cells, ILC2s, and eosinophils (118). Several anti-allergic drugs targeting the abovementioned mediators, including antihistamines, leukotriene modifiers, mast cell membrane stabilizers, and DP2-receptor antagonists, have been developed to decrease type 2 inflammation in the airways. These drugs are majorly used as adjunctive therapies for patients who inadequately respond to regular treatment strategies for allergic asthma, particularly those with allergic rhinitis and allergic skin diseases.
In recent years, non-steroidal anti-inflammatory-drug (NSAID)-exacerbated respiratory disease (NERD) has attracted increasing attention (119). As a prominent severe type 2-high asthma phenotype that appears in adulthood, it is characterized by increased production of CysLTs, high prevalence of coexisting chronic rhinosinusitis with nasal polyps (CRSwNP), and hypersensitivity to aspirin, emphasizing that dysregulation of arachidonic acid metabolism likely exacerbates the worsening of upper and lower airway symptoms of asthma.
3.2.2 Non-type 2 inflammation
Asthma is generally linked to increased eosinophils and TH2 cytokines; however, some patients present with a predominantly neutrophilic disease, also called “non-type 2” or “type 2-low” asthma; this asthma phenotype lacks TH2 cytokine signatures but may exhibit severe glucocorticoid resistance (13). Other features associated with severe neutrophilic asthma include advanced age, impaired lung function, decreased reversibility of bronchodilator responsiveness, microbial infections, tobacco consumption, and obesity (120). Although the molecular mechanism underlying airway neutrophilic inflammation remains unelucidated, it primarily involves the IFN-γ-mediated type 1 and IL-17-mediated type 3 immune pathways (121).
In general, PRRs (such as TLRs) activation in response to microbial infections leads to a type 1 immune response; it involves immune cells that can release IFN-γ, including CD4+ TH1 cells, type 1 ILCs (ILC1s), NK cells, and CD8+ cytotoxic T (TC1) cells (122). The development of naïve T cells in the TH1 or TC1 direction can be induced by intracellular microbes that interact with the TLRs on DCs in the presence of IL-12 and IL-18 derived from DCs and IFN-γ derived from NK cells or ILC1s. The BALF cells isolated from patients with severe type 2-low asthma and lung tissues obtained from corresponding mouse models showed increased levels of IFN-γ and decreased expression of secretory leukocyte protease inhibitor (SLPI), which correlated with high airway resistance and steroid insensitivity (123). At present, the pathogenic roles of type 1 immunity in asthma remain controversial. IFN-γ signaling may be absent in the airway epithelial cells of patients with asthma (124); this abnormality increases their vulnerability to viral infections and worsens asthma (125). In contrast, other studies have demonstrated that severe asthma episodes are associated with high IFN-γ and IL-17A expression in the airways (126, 127). Despite receiving high-dose corticosteroid treatment, CD4+ T cells with IFN-γ+ (TH1 cells) were higher in the BALF of patients with severe asthma than in that of patients with mild or moderate asthma (123, 126).
IL-17 cytokine family members, including IL-17A and IL-17F, mediate the type 3 immune process. Extracellular bacteria and fungi induce IL-1β and IL-23 production by myeloid DCs (mDCs); as a result, primitive CD161+ T cells differentiate into CD4+ TH17 or CD8+ TC17 cells and trigger ILC3s to generate cytokines (122). Furthermore, IL-17A, IL-17F, and IL-22 regulate neutrophilic influx into tissues by inducing airway epithelial and stromal cells to generate cytokines such as G-CSF, GM-CSF, and IL-6, as well as chemokines such as CXCL1, CXCL6, and CXCL8 (IL-8); this promotes neutrophil activation and migration (128). Other cells, including γδT cells, NKT cells, and granulocytes, are also known to secrete IL-17 cytokines (129). In some individuals with moderate-to-severe asthma, these cytokines are increased in the blood, sputum, and bronchial biopsies, correlating with increased disease severity (130). In mice and humans, TH17-related cytokines play a role in airway remodeling by inducing mucous cell metaplasia, promoting fibroblast and SMC proliferation, and directly contracting bronchial SMCs, thereby narrowing the airway (31, 131). Unfortunately, despite the relatively large amount of evidence via observational studies, a randomized clinical trial of brodalumab, an IL-17 receptor-neutralizing antibody, in patients with poorly controlled moderate-to-severe asthma failed to exhibit significant benefits in all-comers (132) (Table 1).
3.2.3 Phenotype overlap and systemic inflammation
Owing to a strong relationship between heterogeneity and asthma, the dichotomous viewpoint of asthma categorization may be oversimplified; it only appears at the extremes of the continuous spectrum (129). Based on asthma endotype complexity, two primary inflammatory signatures involving TH2 or TH17 cells and their respective cytokines have been categorized as discrete subpopulations; however, recent evidence suggests the simultaneous occurrence of type 2 and non-type 2 immune responses in some patients (133). Most TH cells display a degree of plasticity that depends on environmental factors and may be redirected toward other effector CD4+ cells. Interestingly, a study suggested that the TH2 and TH17 inflammatory pathways are mutually regulated in patients with asthma; the suppression of the TH2 pathway promotes a TH17 response; therefore, the dual blockade of TH2 and TH17 functionalities may be rewarding for asthma treatment (134).
Historically, IL-17 was considered to be derived from conventional TH17 cells; however, a novel CD4+ TH2 memory or effector cell subset that collectively expresses GATA3 and RORγt and produces TH2 and TH17 cytokines has now been identified; this subset persists as the predominant IL-17-producing T cell population during the chronic phase of asthma (135). In individuals with severe and corticoid-resistant asthma, dual-positive TH2/TH17 cells (i.e., IL-17-producing TH2 cells) were significantly increased in the peripheral blood and BALF (136, 137). These dual-positive TH2/TH17 cells are characterized by higher levels of IL-4 production and increased expression of MEK1 (mitogen-activated protein-extracellular signal-regulated kinase [ERK] kinase 1), mediating resistance to dexamethasone-induced cell death (136). This can partially explain why a TH2/TH17predominant endotype causes more severe asthma compared with the traditional TH2predominat and TH2low endotype (136). Additional research has demonstrated that IL-1β, IL-6, anti-IFN-γ, and IL-21, which is a cytokine environment that stimulates T cells in asthma to differentiate into the same biphenotypic cells, promote dual-positive TH2/TH17 cell differentiation, worsening asthma (137). Similarly, under specific alterations in the inflammatory microenvironment, TH17 cells repolarize toward the TH2 profile, adjusting their cytokine expression to a TH2-like pattern (138).
In a recent cross-sectional study in humans, phenotype distribution was examined in patients with mild-to-severe asthma (139). Phenotype overlap is extremely common in patients with asthma (73.4%), encompassing combinations of type 2-related, non-type 2-related, and mixed type 2/non-type 2 inflammation. The last group is particularly concerning; it accounts for approximately 50% of the total number of patients and exhibits the worst clinical outcomes. The average age or onset age is younger in the type 2 group, intermediate in the mixed one, and older in the non-type 2 group and might reflect immune inflammatory status evolution, beginning from a canonical type 2 signature and progressively transforming into a more complex mixed type 2/non-type 2 signature. As individuals age, those who develop pure type 2 asthma in the early years may encounter various environmental stimuli throughout their lives, possibly triggering different pathways and altering the original type 2 predominance (140).
Asthma is being increasingly acknowledged as a systemic disease, in which inflammation is not limited to the airways but remarkably cross-communicates with other distant organs by releasing inflammatory mediators (141). In patients with asthma, increased IL-6 and IL-1β levels are markers for systemic inflammation and are associated with reduced forced expiratory volume in 1 second (FEV1) and elevated acute exacerbations (142, 143). The analysis of the clinical traits of patients with asthma suggested that an imbalanced diet with excessive calorie intake and the resulting metabolically related inflammation can affect both innate and adaptive defense mechanisms in the respiratory tract (144). The burden of obesity-associated inflammation may increase the risk of developing asthma. White adipose tissue secretes pro-inflammatory cytokines such as IL-1β, IL-6, TNF-α, and leptin, which may negatively impact lower airway function by increasing airway hyperreactivity (145). Interestingly, increased plasma IL-6 levels and systemic inflammation are observed in both type 2-high and type 2-low asthma; however, they are not associated with upstream type 2 inflammation, with no data for the increase in IL-6 expression in the sputum (143).
3.3 Step 3. Transition from airway inflammation to structural changes
3.3.1 Chronicity of inflammation
Inflammation persists when humans are continuously or repeatedly exposed to viruses, bacteria, allergens, air pollutants, tobacco smoke, and/or oxidative stress; furthermore, there are many innate immune cells in the bronchial mucosa epithelium, including eosinophils, neutrophils, basophils, and monocyte–macrophage lineage cells, as well as blood-derived adaptive immune cells, including TH2 cells, other T cell types, and B cells. Chronic inflammation and airway remodeling simultaneously occur because of the continuous cycle of epithelial damage and repair; this results in disease chronicity, which is a characteristic of asthma. In general, the inflammatory process of asthma is primarily confined to the conducting airways. However, with disease progression to a more chronic state, inflammatory cells infiltrate the proximal of the trachea and larynx and distal of the smaller airways, periodically involving neighboring alveoli. The inflammatory responses in the small airways may primarily occur outside the airway smooth muscle, whereas submucosal inflammation is predominant in the large airways.
Microscopically, inflammation can affect all layers of the airwall in patients with chronic asthma. The epithelium does not exhibit the normal pseudostratified appearance and may be stripped, with only the basal layer remaining. The basal cells are hyperplastic, with squamous metaplasia. Furthermore, goblet cell hyperplasia is observed. Characteristically, the basement membrane of the epithelium is thickened and hyalinized. Moreover, mucous glands in the bronchial submucosa are hyperplastic. In addition, the submucosa is edematous and contains a mixed inflammatory infiltrate, with different numbers of eosinophils. Bronchial smooth muscle hyperplasia and hypertrophy are the most typical features of status asthmaticus. In all bronchial tube wall layers, eosinophil, monocyte, lymphocyte, and plasma cell infiltration is observed. In some cases of fatal asthma, mucus plugs comprising mucin glycoproteins and plasma proteins may block the airways; furthermore, Charcot–Leyden crystals, the disintegrating product of eosinophils, are often observed in the tube walls and mucus plugs (146).
Of note, several reinforcing feedback loops promote the perpetuation of chronic airway inflammation among patients with asthma (Figure 4). For example, in type 2-high asthma, TH2 cells and ILC2s are significant IL-4, IL-5, and IL-13 sources; furthermore, infiltrating mast cells, eosinophils, and basophils play vital roles in producing type 2 cytokines. Collectively, these cells maintain the environment needed for the survival of type 2 cytokines, thereby supporting chronic disease development. IL-4 neutralizes the suppressive function of Tregs and inhibits their generation, allowing the differentiation of IL-4-producing TH2 cells or ILC2s (147, 148). The co-engagement of sIgE bound to FcϵRI receptors on mast cells and basophils results in the massive release of IL-4, a major mediator of B cell class switching and IgE synthesis. IL-5 mediates eosinophil differentiation and function, whereas eosinophils, in turn, secrete large amounts of IL-5. Alarmins that activate TH2 cells and ILC2s are released by damaged airway epithelial cells, leading to cytokine (IL-4 and IL-13) production; this significantly increase the expression of histone deacetylases (HDACs) and silent information regulator genes (SIRTs), whose activities are inversely correlated with the integrity of the epithelial barrier (149).
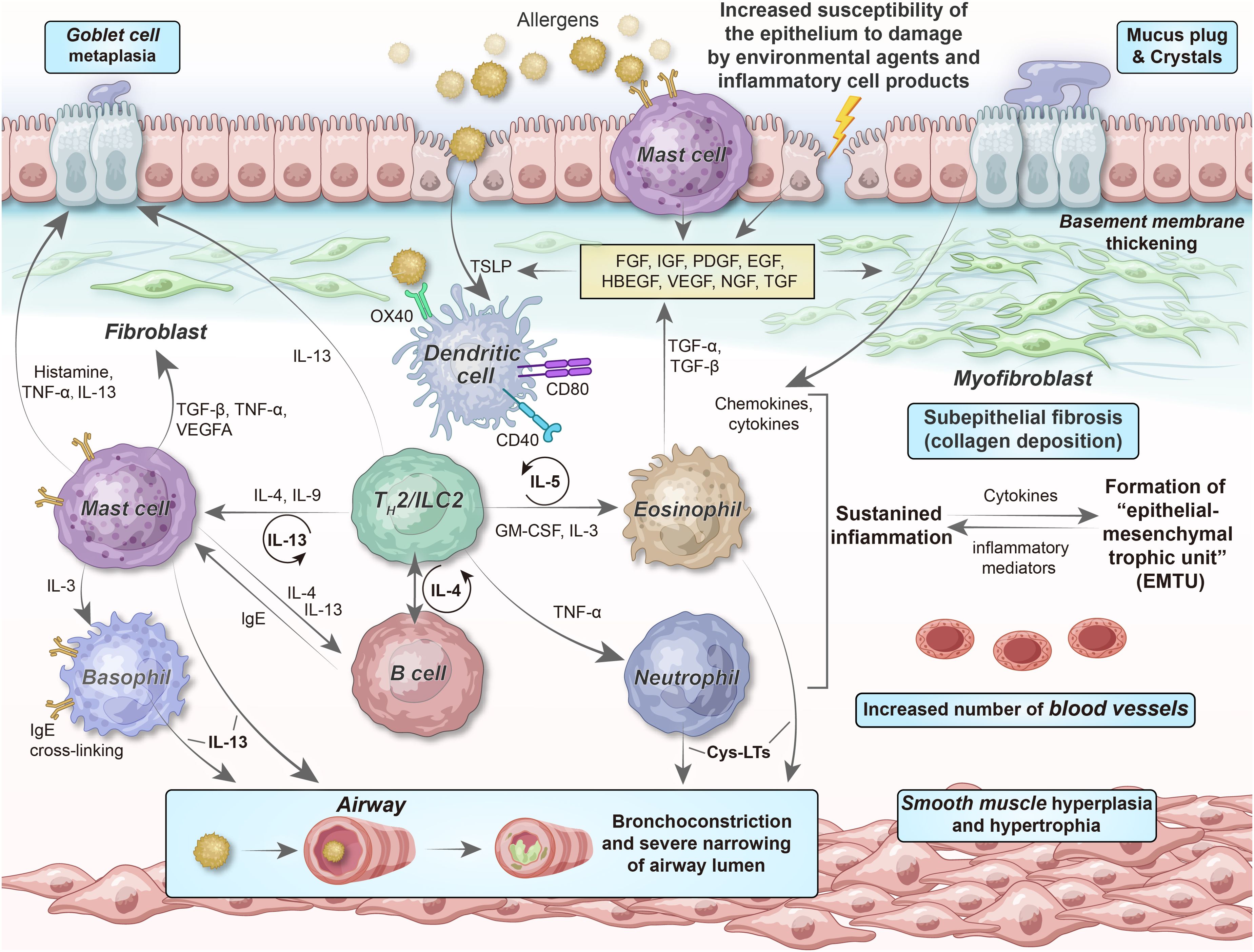
Figure 4. Relationship of epithelial–mesenchymal communication to airway inflammation and remodeling in chronic persistent asthma. The chronicity of inflammation is associated with repeated damage–repair responses that contribute to establishing the EMTU, which, in turn, provides a continuous tissue environment (“soil”) for TH2 cell/ILC2-related inflammation (“seed”). The interdependency among chronic inflammation, altered immunity, and structural changes, involving epithelial cells, (myo)fibroblasts, SMCs, and their secretory ECM, microvasculature, and neural networks, can describe why chronic airway inflammation persists even if obvious environmental stimuli are absent, as well as the reason for the failure to obtain complete therapeutic responses to anti-inflammatory drugs at the more severe and chronic end of the asthma spectrum. FGF, fibroblast growth factor; IGF, insulin-like growth factor; PDGF, platelet-derived growth factor; EGF, epidermal growth factor; HBEGF, heparin-binding EGF-like growth factor; VEGF, vascular endothelial growth factor; NGF, nerve growth factor; TGF, transforming growth factor; Cys-LTs, cysteinyl leukotrienes.
To break the abovementioned vicious cycle and inhibit continuous asthma progression, several anti-inflammatory drugs have been developed (Table 1). However, the redundancy of cytokine sources and overlapping effects of inflammatory mediators increases the robustness of the pro-inflammatory network, making treatment with a single targeted therapy (e.g., cytokine-specific monoclonal antibodies) challenging. To date, the inhaled administration of glucocorticoids remains the most effective treatment modality for asthma; they modulate (mostly downregulate) the expression of approximately 200 genes to exert anti-inflammatory effects.
Recently, the concept of “trained innate immunity” has garnered attention to explain the pathological mechanisms underlying chronic inflammation in asthma (150). Immunologic memory, long considered a specific feature of adaptive immunity, primarily depends on antigen receptor gene rearrangements and lymphocyte clone production. However, emerging literature suggests that the innate immune system can exhibit memory features (151). Abnormal innate immune memory frequently exacerbates the inflammatory responses observed in asthma. Furthermore, repeated exposure to different non-specific stimuli can result in conditionally trained immunity in innate immune cells, including airway epithelial cells, DCs, ILC2s, mast cells, monocytes, macrophages, and NK cells. Depending on the upregulation of pro-inflammatory factors such as IL-1, IL-6, TNF-α, and CCL17 or anti-inflammatory mediators such as IL-10, these cells develop into a pro-inflammatory or anti-inflammatory state. Signaling pathways impinging on transcription factors and an intricate interconnection between epigenetic modifications and metabolic reprogramming may help maintain this state, possibly favoring (pro-inflammatory state) or preventing (anti-inflammatory state) asthma development or progression (152).
3.3.2 Bronchial epithelial–mesenchymal transition and lung tissue remodeling
Airway remodeling, an outstanding feature of chronic asthma, is characterized by aberrant epithelial repair and fibroblast accumulation, contributing to ECM deposition, which results in fixed bronchial obstruction (153). EMT is a dynamic approach in which epithelial cells acquire mesenchymal characteristics and lose their epithelial phenotype; it plays a vital role in normal development, tissue remodeling, fibrosis, and cancer progression. Based on its functional significance, EMT can be classified into three types: type I EMT presents during embryonic development, type II EMT participates in wound healing and organ fibrosis, and type III EMT is related to the metastasis of malignant tumors and transformation of tumor phenotypes. Among these types, type II EMT participates in asthmatic airway remodeling (154).
In general, epithelial injury and ciliopathies; mucosal edema; goblet cell hyperplasia; basal membrane thickening; increased blood vessel supply; increased mass of subepithelial fibroblasts, myofibroblasts, and airway SMCs; and ECM protein deposition are the pathological changes that occur during airway remodeling. These events result in excessive airway reactivity, with mucus formation and plugging extending into the small airways, resulting in airway trapping and overinflation; this ultimately results in decreased lung function in patients with chronic asthma (155).
As a hallmark of airway remodeling, subepithelial fibrosis is proportional to disease severity and duration (156). Through EMT, airway epithelial cells lose apical–basal polarity, undergo cytoskeletal changes, and lose intercellular adhesion and TJs; furthermore, the expression level of epithelial markers, including E-cadherin, is decreased and that of interstitial markers, including α-smooth muscle actin (α-SMA) and vimentin, is increased (157, 158); this results in airway epithelial cell differentiation into myofibroblasts, thereby exacerbating the degree of subepithelial fibrosis (159). Among the signaling pathways involved in EMT, the TGF-β1/Smad, Wnt/β-catenin, and Sonic Hedgehog signaling pathways have been extensively studied and occupy a central position (160).
The prevailing view is that inflammation is the primary driver and amplifier of most airway remodeling processes. Various cytokines, chemokines, and growth factors released from inflammatory and tissue cells in the airways form a complex signaling milieu, driving structural changes in the lung tissue. In type 2-high asthma, activated eosinophils release cytotoxic granular proteins, including eosinophil cationic protein (ECP), major binding protein (MBP), eosinophil peroxidase (EPO), and eosinophil-derived neurotoxin (EDN), LTC4, and platelet-activating factor (PAF), leading to airway constriction, mucus secretion, and increased blood permeability (161). Furthermore, eosinophils are a major source of TGF-β, inducing subepithelial fibrosis, airway smooth muscle hypertrophy, and goblet cell proliferation (162). Epithelial damage and delayed repair stimulate the production of several growth factors, including epidermal growth factor (EGF), TGF-β, and VEGF, which driving airway fibrosis and SMC, neuronal, and capillary proliferation, resembling a chronic wound scenario (163). SMCs are the primary structural cells present in the bronchial airways. During asthmatic airway inflammation, airway SMCs undergo continuous proliferation and hypertrophy, along with deposition of ECM and differentiation of goblet cells (164). Based on these structural changes, SMCs also participate in inflammatory and remodeling processes via the expression of cell adhesion molecules (CAMs), cytokine receptors, chemokine receptors, and TLRs (165). Furthermore, by releasing cytokines such as IL-4, IL-9, and IL-13, TH2 cells and ILC2s promote subepithelial fibrosis, epithelial goblet cell metaplasia, and SMC proliferation (166). In another striking animal study, the researchers reported that when airway inflammation levels were almost similar, mice without TH17 cells had lesser airway remodeling than controls. Therefore, TH17 cells induce airway remodeling in a TH2 response-independent manner (30).
Although airway remodeling is frequently associated with inflammation, this perspective is being challenged and should not be assumed to occur downstream from a single (or central) mechanism. First, airway remodeling can occur in early disease stages or preschool-aged children; however, modeling is less pronounced in adult-onset asthmatic airways (167), suggesting that it is simply not a consequence of inflammation. Second, more severe tissue remodeling, characterized by the excessive deposition of connective tissues with gradual lung function loss, is rare in moderate-to-severe asthma and only develops over time in patients receiving insufficient therapy (54). Lastly, anti-inflammatory treatment may partially reverse airway remodeling and airflow obstruction; however, it generally requires long-term rehabilitation (168).
A new hypothesis about persistent asthma has emerged in which epithelial damage in individuals with asthma triggers abnormal communication between the epithelium and basal myofibroblast sheaths, which is mediated via growth factors, thereby driving airway remodeling; this is called activation of the “epithelial–mesenchymal trophic unit (EMTU)” (169). In contrast, the cytokine milieu generated by the EMTU endows a favorable environment for sustained chronic inflammation. This leads to several functionally significant alterations in the architecture of the affected tissue, including considerable airway wall thickening (including the epithelium, lamina reticularis, submucosa, and smooth muscles), ECM protein deposition (including periostin (170), fibronectin (171), tenascin-C, osteopontin, and “repair-type” collagens I, III, V, and VI), and goblet cell hyperplasia; this is linked to increased mucus production. In individuals who have such airway wall thickening, bronchoconstriction severely narrows the airway lumen compared with that occurring in normal thickness airway walls.
Irrespective of the underlying mechanism, repeated bronchoconstriction, in and of itself, upregulates pro-fibrogenic cytokines and deposits subepithelial collagen (172). Therefore, in addition to chronic inflammation and tissue injury response, changes in mechanical stress may also lead to airway wall remodeling via the action of epithelial-inducible proteins such as YKL-40 (encoded by the gene CHI3L1; i.e., chitinase-3-like protein 1) (173), resistin-like molecule-β (RELMβ) (174), and members of the plasminogen activator system (175).
A groundbreaking endoscopic treatment has highlighted the importance of airway remodeling and mechanical transduction as drivers of asthma pathogenesis. Bronchial thermoplasty (BT) is a non-pharmacological intervention in which therapeutic radio-frequency energy is applied in a controlled manner to the bronchial walls to heat the tissues; BT can decrease the frequency and severity of asthmatic attacks (Evidence B) (176). This temperature-controlled radio-frequency energy is locally delivered to the proximal airways to produce clinical effects and alleviate patient symptoms by decreasing the mass of smooth muscles. However, the other effects of BT on airway remodeling and how it provides clinical benefits remain unclear, potentially involving the downregulation of cytokines such as TGF-β and RANTES (CCL5) (177), inhibition of RBM (reticular basement membrane) thickness and ECM deposition (178), modulation of innervation and vascularization, and improvement of the regenerative capacity of the airway epithelium (179). Nevertheless, evidence regarding its effectiveness and long-term safety is limited, and, at present, it can only be leveraged in research trials or within the scope of national registries as an additional treatment for some adult patients with moderate-to-severe asthma.
3.4 Step 4. Turning point: disease exacerbation or remission
3.4.1 Allergen-induced exacerbation of type 2-high asthma
As previously discussed, in stable type 2-high asthma, the airway epithelium releases TSLP, IL-25, and IL-33, differentiating DC-activated TH2 cells. Therefore, IL-5 and GM-CSF are secreted by these TH2 cells, which along with epithelial-derived eotaxins, monocyte chemotactic proteins (MCPs), and RANTES regulate eosinophil production, maturation, recruitment, and activation. Ultimately, the local degranulation of lung eosinophils damages the airway epithelium. Meanwhile, IL-9 and IL-13, as TH2 cytokines, induce goblet cell metaplasia (62).
Several other factors, including indoor allergens such as dust mites, pet fur, and cockroaches; outdoor allergens such as catkins, pollen, fungi, and cold air; and dietary allergies such as aspirin and some high-protein fish, shrimp, crabs, and eggs, can aggravate inflammatory responses during the acute worsening of type 2-high asthma. The PRRs in the airway epithelium can detect allergens and other environmental stimuli. PM2.5 extracts can acutely exacerbate allergic lung inflammation in an inflammasome-dependent manner via the TLR2/NF-κB/NLRP3 pathway (180). Overlaid on the general type 2-high inflammatory response, epithelial cells produce TARC (CCL17), a major chemokine for recruiting TH17 cells; TARC enhances the pro-inflammatory effects of TH2 cells by releasing IL-17 and secreting IL-8, leading to neutrophil recruitment (Figure 5). Evidence suggests that TARC levels are significantly elevated in the BALF of individuals with allergen-challenged asthma exacerbation. The combinations of IL-4, IL-13, and TNF-α elicit TARC release from airway SMCs, thereby promoting the force generation of smooth muscles and airway stenosis (181).
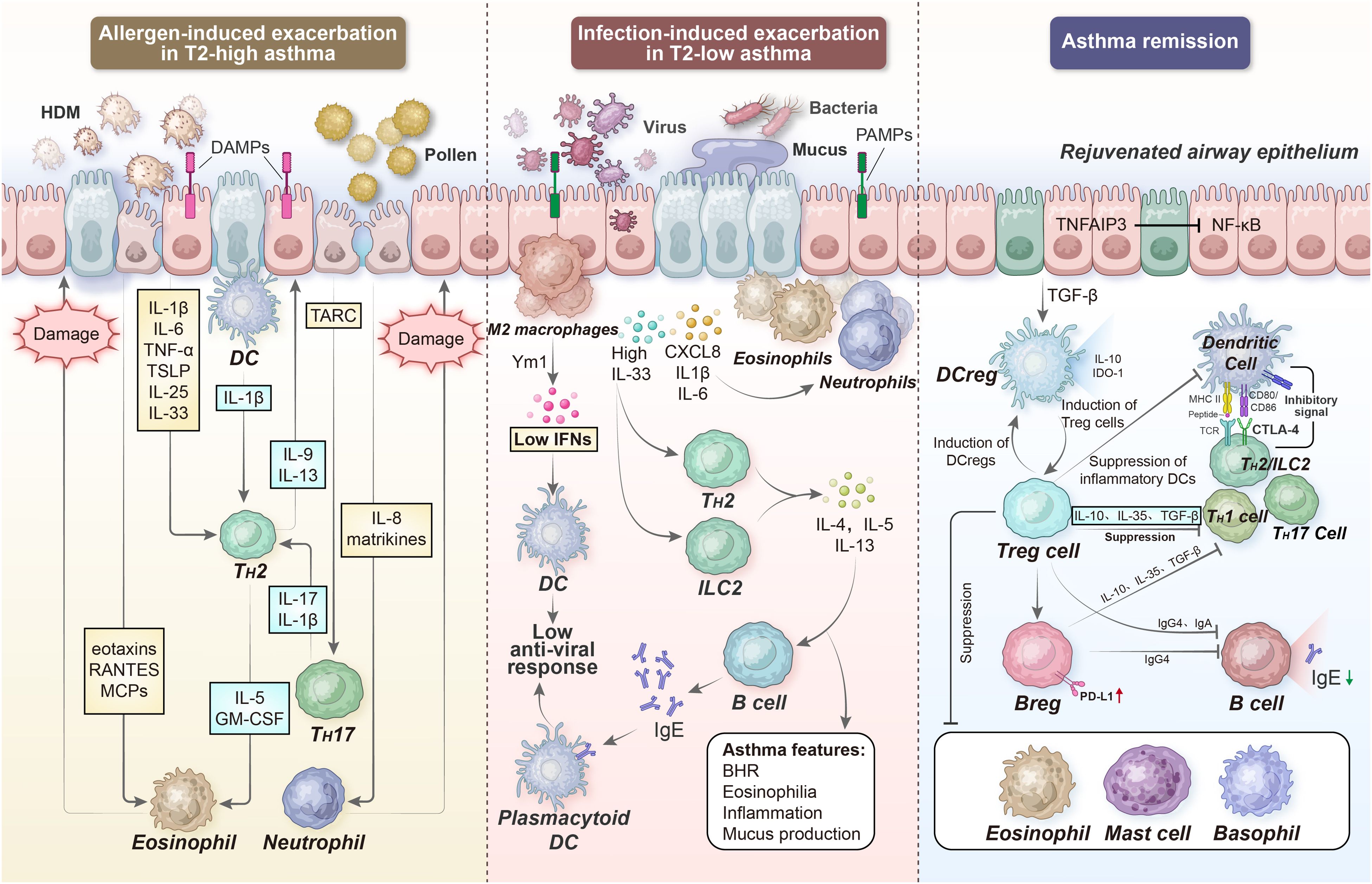
Figure 5. Acute exacerbation and remission of asthma. During allergen- and infection-induced asthma exacerbation, the PRRs in the airway epithelium can detect many other additional factors. Subsequently, epithelial cells in the airway release TSLP, IL-25, and IL-33 to support DC-activated TH2 cell differentiation; on the other hand, they secrete TARC and IL-8 to recruit TH17 cells and neutrophils, respectively. In turn, the local degranulation of lung eosinophils and neutrophils damages the airway epithelium, further impairing barrier integrity and enhancing ongoing inflammation. However, several regulatory immune cell types, including DCregs, Tregs, and Bregs can limit or resolve inflammation, partially via IL-10-, TGF-β-, and IL-35-dependent mechanisms. DAMPs, damage-associated molecular patterns; PAMPs, pathogen-associated molecular patterns; TARC, thymus activation regulated chemokine; RANTES, regulated upon activation normal T cell expressed and secreted; Ym1, chitinase-like protein 3; BHR, bronchial hyperresponsiveness; TNFAIP3, tumor necrosis factor alpha-induced protein 3; IDO-1, indoleamine 2,3-dioxygenase 1, CTLA-4, cytotoxic T-lymphocyte-associated protein 4.
Abnormal contraction of the bronchial smooth muscle is a key pathological process that induces airway hyperresponsiveness in asthma. The inflammatory cytokines generated during asthma exacerbation, primarily IL-13, can not only directly act on IL-13 and IL-4 receptors on airway SMCs (182), thereby enhancing agonist-evoked excitatory effects by upregulating pro-inflammatory mediators such as IL-1β and TNF-α (183), but also modulate G protein-coupled receptor (GPCR, e.g., muscarinic receptor)-related signaling pathways and/or inhibit cAMP production, thereby altering calcium homeostasis in airway SMCs (184). Newly synthesized mediators released from airway SMCs promote immune cell recruitment and activation, thereby sharpening an ongoing inflammatory response. Persistent airway inflammation helps enhance the strength generated by airway smooth muscles, possibly increasing the number and size of airway SMCs. Understanding the regulation of airway smooth muscle function and combining this information with the underlying immunologic processes that drive asthma pathogenesis may become an important breakthrough in addressing the treatment mysteries of asthma (185).
In a very recent study, using a human allergen-induced asthma exacerbation model, single-cell RNA sequencing (scRNA-seq) was utilized to compare the lower airway mucosa of patients with asthma and allergic controls without asthma (186). The airway epithelium of patients with asthma was highly dynamic, with the upregulation of the genes involved in mucus metaplasia, matrix remodeling, and glycolysis; however, antioxidant and growth factor pathways observed in controls were not induced. In particular, following allergen exposure, IL9-expressing pathogenic TH2 cells were observed in the asthmatic airways, along with the enrichment of cDC2s and CCR2-expressing monocyte-derived cells, which upregulated the expression of inflammatory mediators and metalloproteinases. Moreover, a unique TH2-mononuclear phagocyte-basal cell interactome was discovered in patients with asthma, characterized by type 2 programming of immune and structural cells and the involvement of additional signals, including the TNF family and cellular metabolism pathways.
3.4.2 Infection-induced exacerbation of type 2-low asthma
Across all age groups, respiratory tract infections are closely associated with wheezing illnesses, possibly affecting asthma development and severity. Airway infection caused by viruses, chlamydia, or mycoplasma may play a vital role in asthma pathogenesis: repeated respiratory infections during early childhood (particularly respiratory syncytial virus [RSV]) encompass the strongest predictors of future asthma risk (187); on the other hand, to date, viral infections (primarily rhinovirus [RV]) are the most common reason for the acute worsening of already established adult asthma; this results in the acute aggravation of disease symptoms, warranting increased medication use and emergency department visits and hospitalization and intensive care measures in some cases (188).
In most patients with asthma, relatively mild respiratory viruses such as RV, RSV, adenovirus, human metapneumovirus, influenza viruses, and parainfluenza viruses lead to acute exacerbation. After infection, host cells such as airway epithelial cells induce inflammatory responses to counteract the viruses. This cellular response may facilitate the acute exacerbation of asthma.
Cellular responses to viruses are initiated by TLR-3, TLR-7, and TLR-8, RIG-I, and MDA5 detecting a single-stranded RNA (or else a double-stranded RNA during replication) (189). The activation of these receptors drives a vigorous innate immune response via the induction of primary interferons (e.g., IFN-β), which results in the activation of NKT cells and alternative activation of macrophages (M2), thereby maintaining airway pathology through the production of IL-13 (190). In the lungs, anti-viral immunity heavily depends on type I (IFN-α and IFN-β) and type III (IL-29 [IFN-λ1], IL-28A [IFN-λ2], IL-28B [IFN-λ3], and IFN-λ4) IFN production, cytokine (such as IL-1, IFN-γ, TNF, IL-6, IL-12, and IL-18) secretion, and chemokine (such as CCL3, CCL5, CXCL8, and CXCL10) release (191) (Figure 5).
Many studies have suggested that IFN production is either delayed (192) or deficient (124, 193) in patients with asthma, resulting in the absence of adequate clearance rates for their immune responses to viral infections (194). This defect is associated with epithelial barrier and TJ disruption. Recently, studies have demonstrated that the exogenous dosing of IFN-β decreases viral exacerbations in mild-to-moderate asthma by restoring impaired antiviral innate immunity (195, 196). Additional factors that affect viral infection severity and asthma exacerbation risk include the enhanced expression of viral receptors (such as ICAM-1, low density lipoprotein receptor [LDLR], and CDHR3) (197, 198), high body mass index of patients (199), excessive increase in blood eosinophils (patients with type 2-high asthma) (199), reproducible changes in the DNA methylome (200, 201), and disturbances in the airway microbiota (202, 203).
Compared with viral respiratory infections, bacterial infections have a smaller and less important effect. In patients with acute asthma exacerbation, only a small proportion have isolated bacteria from their sputum (204).Airway bacterial infections may occur secondary to viral-induced epithelial barrier disruption, resulting in increased inflammation and risk of asthma exacerbation (205). However, there is limited evidence linking bacterial infections to acute asthma exacerbation. In a large-scale study, the colonization levels of dominant bacterial pathogens were not statistically significantly different in children with recurring and acute wheezing and those without a wheezing history (206).
Notably, the long-term oral administration of low-dose azithromycin, a macrolide antibiotic, can significantly decrease the acute exacerbation of eosinophilic and non-eosinophilic asthma, improve the quality of life of patients with severe asthma, and decrease the risk of lower respiratory tract infections (207, 208). Azithromycin exerts antibacterial, anti-inflammatory, and immunomodulatory properties and is cost-effective. At present, guidelines recommend using this antibiotic for refractory asthma. Nevertheless, potential issues such as antimicrobial resistance and side effects such as cardiac toxicity and gastrointestinal adverse reactions may restrict the widespread use of this antibiotic (209, 210).
Infection-induced asthma is not always a type 2-low inflammation. Allergic bronchopulmonary aspergillosis (ABPA) is a type 2-high inflammatory lung disease caused by an allergy to Aspergillus fumigatus, commonly presenting with treatment-resistant asthma and recurrent pulmonary shadows, which may be accompanied by bronchiectasis (211). This disease is not rare, but it is often misdiagnosed or overlooked clinically. Classical immunology suggests that the usual response of the human host to fungal clearance is a TH1 CD4+ T-cell response, which mediates the phagocytic function of macrophages and neutrophils. In contrast, the immune reaction in ABPA is predominantly mediated by TH2 cells, which not only cannot eradicate the fungi but also cause a massive influx and degranulation of mast cells and eosinophils, releasing various inflammatory mediators and cytokines, including IL-4, IL-5, IL-13, as well as total and A.fumigatus-specific IgE. Persistent inflammation will lead to airway mucus plugging, bronchiectasis, and pulmonary fibrosis. If not controlled, it can culminate in end-organ damage and clinical manifestations, which warrants our vigilance.
3.4.3 Suppression and resolution of airway inflammation
Although asthma cannot be completely cured, it is possible to further pursue more realistic and achievable goals. Remission is defined as the long-term absence of disease signs and symptoms, accompanied or not by the normalization of the underlying pathology. Clinically remitted asthma requires stable lung function and the endorsement of patients or clinicians, in addition to at least 12 months without significant asthma symptoms and exacerbations; on the other hand, complete remission (cure) warrants the normalization of the underlying pathology, including resolution of airway inflammation (212). Both remission types may be achieved with or without treatment. Based on the clinical settings and research populations, clinicians and researchers can flexibly adjust the achievable and measurable definitions of asthma remission.
The introduction of ICS in the 1980s revolutionized the treatment of asthma, and to this day it remains the cornerstone for gaining optimal asthma control. ICS is generally effective in mitigating symptoms of mild-to-moderate asthma, enhancing lung function, and preventing exacerbations. The emergence of combination therapy with ICS, long-acting β2-agonists (LABA), and long-acting muscarinic antagonists (LAMA) has further improved asthma management. However, some studies have indicated that patients with inactive asthma, even when in complete remission, still exhibit some degree of persistent subclinical airway inflammation, hyperresponsiveness, and remodeling (213–215), possibly determining the future risk of recurrence (216, 217). Furthermore, many cross-sectional studies have examined the inflammatory markers associated with clinical and complete asthma remission in different samples such as blood, sputum, BALF, or endobronchial biopsies and eosinophils, neutrophils, mast cells, IgE, FeNO, iNOS, histamine, ECP, and EPO (214, 218–222). Most of these studies have confirmed that these markers are higher in subjects with asthma remission than in healthy controls and lower than in those with persistent asthma (223), although some studies have found no significant differences between the groups.
Further defining the clinical features and pathophysiology of asthma remission may be beneficial; however, future studies to explore its phenotype and underlying mechanisms are vital (212). Here, we focus on the immunological aspects of the natural ablation of asthma inflammation, accentuating the role of allergen immunotolerance and regulatory immune cells.
In healthy individuals, an overt immune response is not provoked during exposure to innocuous environmental antigens, if not combined with tissue damage or danger signals. Mechanistically, the clonal anergy state of T cells can be induced by isolated TCR stimulation in the absence of co-stimulatory signals. When the antigen (allergen) dose is extremely low, lymphocytes cannot be effectively activated, resulting in the unresponsiveness of the immune system (low-zone tolerance); in contrast, if the antigen over occupied TCRs, it may lead to the apoptosis of effector T cells or the induction of Treg cells that suppress immune responses, thereby also presenting an unresponsive state (high-zone tolerance). This provides a theoretical basis for the later proposed “hygiene hypothesis” and “diet-microbiome hypothesis” (224). The former suggests that in the urbanized regions of highly industrialized countries, owing to improvements in sanitation and public health and decreased exposure to infectious sources during early life, the adaptive immune system of children may not be fully exercised and developed, increasing the risk of asthma, allergic diseases, and autoimmune conditions. In contrast, rural lifestyles and large families, combined with unhygienic contact with older siblings, livestock, and soil, confer some level of protection and tolerance against allergens (225). The latter offers another explanation, i.e., a decrease in dietary fiber intake and an increase in fat intake and changes in eating habits and dietary structures will modify the composition of gut microbiota, leading to a loss of symbiotic functions of non-pathogenic bacteria and an overall reduction in microbiome diversity, as well as an increase in allergic reactions (226).
Prior exposure to some environmental factors and microbial contents, including farm dust, butyrate, LPS, and N-glycolylneuraminic acid, can suppress the responses of the airway epithelium to allergens because they induce the expression of TNF-α induced protein-3 (TNFAIP3, also called A20), a negative regulator of NF-κB activation (227). This effect inhibits the production of IL-33, GM-CSF, and the DC chemokine CCL20 by epithelial cells in response to allergen inhalation, and promotes the production of tolerance-inducing cytokines such as TGF-β; this induces tolerogenic or regulatory DCs (DCregs) that promote Treg development. In particular, the dampening of immunologic responsiveness and maintenance of allergen tolerance contribute to regulatory immune cells, notably Tregs and regulatory B cells (Bregs) (228) (Figure 5). Tregs exert a broad suppressive effect on several immune cells; this effect is achieved by secreting cytokines such as IL-10 and TGF-β and other inhibitory factors such as indoleamine 2,3-dioxygenase (IDO-1), as well as by providing co-inhibitory molecules such as cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), programmed cell death protein 1 (PD-1, or CD279), and PD-L1 (229). Tregs directly suppress mast cells, eosinophils, basophils, ILC2s, and inflammatory DCs involved in programming effector T cell subsets (TH1, TH2, and TH17 cells). Furthermore, In Treg-derived cytokines, primarily IL-10, IL-35, and TGF-β, induce Bregs and produce IgG4 and IgA antibodies via B cells. In addition, Bregs produce anti-inflammatory cytokines, thereby suppressing effector T-cell responses (230).
A key event in generating normal immune responses to allergens is redirecting allergen-specific effector T cells toward a regulatory phenotype; this phenomenon accounts for some degree of the clinical efficacy of allergen-specific immunotherapy (AIT) (231). In instances where an immunologically proven allergen-driven mechanism of asthma, AIT represents the sole etiologic remedy for allergic manifestations (232, 233). It involves repeatedly administering high doses of causative allergens, generally via subcutaneous injection (SCIT) or sublingual application (SLIT), to induce a permanent state of tolerance and long-term benefits after discontinuing the treatment (234). AIT can be safely used to treat adolescents and adults with mild-to-moderate and well-controlled allergic asthma, not only significantly controlling symptoms and decreasing acute attacks but also decreasing the need for ICS dosage. Furthermore, AIT can prevent the further development of rhinitis into asthmatic symptoms in children (235). However, caution should be exercised when using AIT to treat uncontrolled and severe asthma (236).
4 Challenges ahead and future directions
Despite being in close contact with harmful chemicals and pathogenic microorganisms in the external environment, the human lungs can maintain efficient gas exchange. This relies on the protective role of the immune system of the lung mucosa against various harmful factors. The dynamic regulation of lung structure and immune cells is an essential guarantee for maintaining lung immune homeostasis. The disruption of this immune homeostasis may result in asthma development. As a heterogeneous condition, asthma presents with chronic airway inflammation, with the involvement of various cells and cellular components and association with airway hyperresponsiveness. If bronchial asthma is not promptly diagnosed and treated, it can lead to irreversible airway narrowing and tissue remodeling with disease progression.
In the last few years, a significant paradigm shift has been observed in our perception of asthma pathobiology, largely because of an improved understanding of its heterogeneity and different endotypes. Initially, asthma was considered a unique TH2 cell-mediated disease, a dogma largely developed from mouse asthma models that drove the development of several type 2-oriented monoclonal antibodies. As present, these biologics are successfully used in clinical settings, primarily to decrease the frequency of exacerbations in patients receiving conventional therapy. However, this immune process is absent in 50% of patients; this condition is termed “type 2-low asthma,” whose existence and definition remain uncertain, encompassing various asthma subtypes such as neutrophilic, mixed granulocytic, or paucigranulocytic forms and is characterized by normal eosinophils and low type 2 inflammation marker expression.
Various aspects of innate or adaptive immunity responses to allergens, environmental triggers, or viruses are involved in developing allergen sensitization, asthma symptoms, exacerbations, and treatment responses. Extensive crosstalk exists between airway epithelial and immune cells during disease initiation and persistence, indicating that epithelial barrier restoration in asthma requires greater attention. With an increase in the complexity and severity of disease manifestations, the complexity of the accompanying immunopathology also increases, with the possible involvement of additional adaptive immunity elements and structural changes in the airways. While there has been significant advancement in the understanding of some of the current immunological advances in asthma, additional studies are warranted to ascribe the mechanisms underlying asthma inception and identify additional biomarkers that facilitate targeted interventions, by prioritizing the development of tools for the rapid, accurate, and low-cost diagnosis of the endotypes and sub-endotypes of asthma.
Clinicians have begun to realize that the one-size-fits-all “stepwise approach to therapy” cannot entirely meet the optimal diagnostic and therapeutic needs of all patients, particularly those with severe or refractory asthma. The concept of “treatable traits (TTs)” has been proposed as a way to address the diverse pathophysiological factors involved in severe asthma and overcome the limitations of existing treatment strategies (237). This notion of personalized medicine, which advocates multidisciplinary teamwork and is based on multidimensional assessment, represents a shift toward precision medicine. By offering greater flexibility and comprehensiveness in treatment, this concept can significantly improve health-related quality of life and asthma control while decreasing acute exacerbations (238, 239).
Immunological advances have always resonated with the progress of clinical asthma research, mutually complementing each other. However, new curative or even preventive treatments that can control symptoms in patients with asthma are warranted in the future, to alleviate the significant burden it places on society. For example, motivated by the marked success of an adoptive cellular immunotherapy based on the chimeric antigen receptor (CAR) for treating various malignant tumor types, a research group developed a cytokine-anchored CAR-T (CCAR-T) cell system using chimeric IL-5/CD28/CD3ζ receptors and revealed the targeted killing effect of IL-5-anchored CCAR-T cells on eosinophils in vivo and in vitro, as well as their protective effect on allergic airway inflammation, significantly surpassing the quintessential therapeutic window of current mAb-based treatments in clinical settings (240). This research group innovatively employed the CCAR-T cell system to treat severe eosinophilic asthma, which may be a milestone achievement in future research on various intractable allergic diseases.
A unified and innovative approach is required to address the challenges posed by asthma. Contemporary cutting-edge methods, including but not restricted to single-cell sequencing, phenomics, genetic lineage tracing, tissue imaging systems, and organoid technology, which can achieve obtain highly multiplexed information with subcellular spatial resolution, and their in-depth computational analysis may help better define asthma in the forthcoming years. It is imperative to underscore that the key to the successful development of personalized and phenotype-specific asthma treatments lies in continuously collaborating with clinical experts and immunologists and integrating bench and bedside approaches.
Author contributions
CX: Conceptualization, Visualization, Writing – original draft. JY: Writing – original draft. AG: Writing – original draft. YFL: Writing – original draft. RZ: Writing – original draft. MY: Writing – original draft. XL: Writing – original draft. YHL: Writing – original draft. QL: Funding acquisition, Supervision, Writing – review & editing. HG: Conceptualization, Funding acquisition, Supervision, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by the National Natural Science Foundation of China (NSFC, no. 82104812 to HG and no. 82004318 to QL), the Young Elite Scientists Sponsorship Program by China Association for Science and Technology (to HG), and the Capital’s Funds for Health Improvement and Research (CFH, no. 2024-2-4131 to HG). The opinions, results, and conclusions reported in this paper are those of the authors and are independent from the funding sources, and all of the funders had no involvement in the writing, analysis, or interpretation of results, or the decision to submit for publication.
Acknowledgments
The authors express the gratitude to Dr. Huahua Zhong (National Center for Neurological Disorders, Huashan Hospital Affiliated to Fudan University, Shanghai, China), Dr. Jinrong Lin (Institute of Sports Medicine, Fudan University, Shanghai, China; Present address: Botnar Research Centre, Nuffield Department of Orthopaedics, Rheumatology, and Musculoskeletal Sciences, University of Oxford, Oxford, UK) and Dr. Yong Wu (Department of Nephrology, Hunan Provincial Key Laboratory of Kidney Disease and Blood Purification, the 2nd Xiangya Hospital of Central South University, Changsha, China) for their honest-to-goodness assistance with language editing and figure artwork.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Porsbjerg C, Melen E, Lehtimaki L, Shaw D. Asthma. Lancet. (2023) 401:858–73. doi: 10.1016/S0140-6736(22)02125-0
2. Huang K, Yang T, Xu J, Yang L, Zhao J, Zhang X, et al. Prevalence, risk factors, and management of asthma in China: A national cross-sectional study. Lancet. (2019) 394:407–18. doi: 10.1016/S0140-6736(19)31147-X
3. Meghji J, Mortimer K, Agusti A, Allwood BW, Asher I, Bateman ED, et al. Improving lung health in low-income and middle-income countries: from challenges to solutions. Lancet. (2021) 397:928–40. doi: 10.1016/S0140-6736(21)00458-X
4. Wenzel SE. Asthma phenotypes: the evolution from clinical to molecular approaches. Nat Med. (2012) 18:716–25. doi: 10.1038/nm.2678
5. Walker C, Bode E, Boer L, Hansel TT, Blaser K, Virchow JC Jr. Allergic and nonallergic asthmatics have distinct patterns of T-cell activation and cytokine production in peripheral blood and bronchoalveolar lavage. Am Rev Respir Dis. (1992) 146:109–15. doi: 10.1164/ajrccm/146.1.109
6. Green RH, Brightling CE, Woltmann G, Parker D, Wardlaw AJ, Pavord ID. Analysis of induced sputum in adults with asthma: identification of subgroup with isolated sputum neutrophilia and poor response to inhaled corticosteroids. Thorax. (2002) 57:875–9. doi: 10.1136/thorax.57.10.875
7. Venkatesan P. 2023 Gina report for asthma. Lancet Respir Med. (2023) 11:589. doi: 10.1016/S2213-2600(23)00230-8
8. Wills-Karp M, Luyimbazi J, Xu X, Schofield B, Neben TY, Karp CL, et al. Interleukin-13: central mediator of allergic asthma. Science. (1998) 282:2258–61. doi: 10.1126/science.282.5397.2258
9. Grunig G, Warnock M, Wakil AE, Venkayya R, Brombacher F, Rennick DM, et al. Requirement for il-13 independently of il-4 in experimental asthma. Science. (1998) 282:2261–3. doi: 10.1126/science.282.5397.2261
10. Lambrecht BN, Hammad H, Fahy JV. The cytokines of asthma. Immunity. (2019) 50:975–91. doi: 10.1016/j.immuni.2019.03.018
11. Hammad H, Lambrecht BN. The basic immunology of asthma. Cell. (2021) 184:1469–85. doi: 10.1016/j.cell.2021.02.016
12. Fahy JV. Type 2 inflammation in asthma–present in most, absent in many. Nat Rev Immunol. (2015) 15:57–65. doi: 10.1038/nri3786
13. Sze E, Bhalla A, Nair P. Mechanisms and therapeutic strategies for non-T2 asthma. Allergy. (2020) 75:311–25. doi: 10.1111/all.13985
14. Luo W, Hu J, Xu W, Dong J. Distinct spatial and temporal roles for th1, th2, and th17 cells in asthma. Front Immunol. (2022) 13:974066. doi: 10.3389/fimmu.2022.974066
15. Barnes PJ. Cellular and molecular mechanisms of asthma and copd. Clin Sci (Lond). (2017) 131:1541–58. doi: 10.1042/CS20160487
16. Mosmann TR, Cherwinski H, Bond MW, Giedlin MA, Coffman RL. Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. J Immunol. (1986) 136:2348–57. doi: 10.4049/jimmunol.136.7.2348
17. Harker JA, Lloyd CM. T helper 2 cells in asthma. J Exp Med. (2023) 220:e20221094. doi: 10.1084/jem.20221094
18. Finkelman FD, Hogan SP, Hershey GK, Rothenberg ME, Wills-Karp M. Importance of cytokines in murine allergic airway disease and human asthma. J Immunol. (2010) 184:1663–74. doi: 10.4049/jimmunol.0902185
19. Szabo SJ, Sullivan BM, Peng SL, Glimcher LH. Molecular mechanisms regulating th1 immune responses. Annu Rev Immunol. (2003) 21:713–58. doi: 10.1146/annurev.immunol.21.120601.140942
20. Gavett SH, O'Hearn DJ, Li X, Huang SK, Finkelman FD, Wills-Karp M. Interleukin 12 inhibits antigen-induced airway hyperresponsiveness, inflammation, and th2 cytokine expression in mice. J Exp Med. (1995) 182:1527–36. doi: 10.1084/jem.182.5.1527
21. Cohn L, Homer RJ, Niu N, Bottomly K. T helper 1 cells and interferon gamma regulate allergic airway inflammation and mucus production. J Exp Med. (1999) 190:1309–18. doi: 10.1084/jem.190.9.1309
22. Wisniewski JA, Muehling LM, Eccles JD, Capaldo BJ, Agrawal R, Shirley DA, et al. T(H)1 signatures are present in the lower airways of children with severe asthma, regardless of allergic status. J Allergy Clin Immunol. (2018) 141:2048–60.e13. doi: 10.1016/j.jaci.2017.08.020
23. Britt RD Jr., Thompson MA, Sasse S, Pabelick CM, Gerber AN, Prakash YS. Th1 cytokines tnf-alpha and ifn-gamma promote corticosteroid resistance in developing human airway smooth muscle. Am J Physiol Lung Cell Mol Physiol. (2019) 316:L71–81. doi: 10.1152/ajplung.00547.2017
24. Newcomb DC, Peebles RS Jr. Th17-mediated inflammation in asthma. Curr Opin Immunol. (2013) 25:755–60. doi: 10.1016/j.coi.2013.08.002
25. Bullone M, Carriero V, Bertolini F, Folino A, Mannelli A, Di Stefano A, et al. Elevated serum ige, oral corticosteroid dependence and il-17/22 expression in highly neutrophilic asthma. Eur Respir J. (2019) 54:1900068. doi: 10.1183/13993003.00068-2019
26. Krishnamoorthy N, Douda DN, Bruggemann TR, Ricklefs I, Duvall MG, Abdulnour RE, et al. Neutrophil cytoplasts induce T(H)17 differentiation and skew inflammation toward neutrophilia in severe asthma. Sci Immunol. (2018) 3:eaao4747. doi: 10.1126/sciimmunol.aao4747
27. Schnyder-Candrian S, Togbe D, Couillin I, Mercier I, Brombacher F, Quesniaux V, et al. Interleukin-17 is a negative regulator of established allergic asthma. J Exp Med. (2006) 203:2715–25. doi: 10.1084/jem.20061401
28. Wakashin H, Hirose K, Maezawa Y, Kagami S, Suto A, Watanabe N, et al. Il-23 and th17 cells enhance th2-cell-mediated eosinophilic airway inflammation in mice. Am J Respir Crit Care Med. (2008) 178:1023–32. doi: 10.1164/rccm.200801-086OC
29. Bellini A, Marini MA, Bianchetti L, Barczyk M, Schmidt M, Mattoli S. Interleukin (Il)-4, il-13, and il-17a differentially affect the profibrotic and proinflammatory functions of fibrocytes from asthmatic patients. Mucosal Immunol. (2012) 5:140–9. doi: 10.1038/mi.2011.60
30. Zhao J, Lloyd CM, Noble A. Th17 responses in chronic allergic airway inflammation abrogate regulatory T-cell-mediated tolerance and contribute to airway remodeling. Mucosal Immunol. (2013) 6:335–46. doi: 10.1038/mi.2012.76
31. Kudo M, Melton AC, Chen C, Engler MB, Huang KE, Ren X, et al. Il-17a produced by alphabeta T cells drives airway hyper-responsiveness in mice and enhances mouse and human airway smooth muscle contraction. Nat Med. (2012) 18:547–54. doi: 10.1038/nm.2684
32. Lin W, Truong N, Grossman WJ, Haribhai D, Williams CB, Wang J, et al. Allergic dysregulation and hyperimmunoglobulinemia E in foxp3 mutant mice. J Allergy Clin Immunol. (2005) 116:1106–15. doi: 10.1016/j.jaci.2005.08.046
33. Tang Q, Bluestone JA. The foxp3+ Regulatory T cell: A jack of all trades, master of regulation. Nat Immunol. (2008) 9:239–44. doi: 10.1038/ni1572
34. Hartl D, Koller B, Mehlhorn AT, Reinhardt D, Nicolai T, Schendel DJ, et al. Quantitative and functional impairment of pulmonary cd4+Cd25hi regulatory T cells in pediatric asthma. J Allergy Clin Immunol. (2007) 119:1258–66. doi: 10.1016/j.jaci.2007.02.023
35. Birmingham JM, Chesnova B, Wisnivesky JP, Calatroni A, Federman J, Bunyavanich S, et al. The effect of age on T-regulatory cell number and function in patients with asthma. Allergy Asthma Immunol Res. (2021) 13:646–54. doi: 10.4168/aair.2021.13.4.646
36. Veldhoen M, Uyttenhove C, van Snick J, Helmby H, Westendorf A, Buer J, et al. Transforming growth factor-beta 'Reprograms' the differentiation of T helper 2 cells and promotes an interleukin 9-producing subset. Nat Immunol. (2008) 9:1341–6. doi: 10.1038/ni.1659
37. Kaplan MH. The transcription factor network in th9 cells. Semin Immunopathol. (2017) 39:11–20. doi: 10.1007/s00281-016-0600-2
38. Angkasekwinai P, Chang SH, Thapa M, Watarai H, Dong C. Regulation of il-9 expression by il-25 signaling. Nat Immunol. (2010) 11:250–6. doi: 10.1038/ni.1846
39. Koch S, Sopel N, Finotto S. Th9 and other il-9-producing cells in allergic asthma. Semin Immunopathol. (2017) 39:55–68. doi: 10.1007/s00281-016-0601-1
40. Pajulas A, Fu Y, Cheung CCL, Chu M, Cannon A, Alakhras N, et al. Interleukin-9 promotes mast cell progenitor proliferation and ccr2-dependent mast cell migration in allergic airway inflammation. Mucosal Immunol. (2023) 16:432–45. doi: 10.1016/j.mucimm.2023.05.002
41. Sehra S, Yao W, Nguyen ET, Glosson-Byers NL, Akhtar N, Zhou B, et al. Th9 cells are required for tissue mast cell accumulation during allergic inflammation. J Allergy Clin Immunol. (2015) 136:433–40.e1. doi: 10.1016/j.jaci.2015.01.021
42. Lloyd CM, Harker JA. Epigenetic control of interleukin-9 in asthma. N Engl J Med. (2018) 379:87–9. doi: 10.1056/NEJMcibr1803610
43. Schwartz DM, Farley TK, Richoz N, Yao C, Shih HY, Petermann F, et al. Retinoic acid receptor alpha represses a th9 transcriptional and epigenomic program to reduce allergic pathology. Immunity. (2019) 50:106–20.e10. doi: 10.1016/j.immuni.2018.12.014
44. Parker JM, Oh CK, LaForce C, Miller SD, Pearlman DS, Le C, et al. Safety profile and clinical activity of multiple subcutaneous doses of medi-528, a humanized anti-interleukin-9 monoclonal antibody, in two randomized phase 2a studies in subjects with asthma. BMC Pulm Med. (2011) 11:14. doi: 10.1186/1471-2466-11-14
45. Oh CK, Leigh R, McLaurin KK, Kim K, Hultquist M, Molfino NA. A randomized, controlled trial to evaluate the effect of an anti-interleukin-9 monoclonal antibody in adults with uncontrolled asthma. Respir Res. (2013) 14:93. doi: 10.1186/1465-9921-14-93
46. Britt RD Jr., Ruwanpathirana A, Ford ML, Lewis BW. Macrophages orchestrate airway inflammation, remodeling, and resolution in asthma. Int J Mol Sci. (2023) 24:10451. doi: 10.3390/ijms241310451
47. Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. (2014) 41:14–20. doi: 10.1016/j.immuni.2014.06.008
48. Girodet PO, Nguyen D, Mancini JD, Hundal M, Zhou X, Israel E, et al. Alternative macrophage activation is increased in asthma. Am J Respir Cell Mol Biol. (2016) 55:467–75. doi: 10.1165/rcmb.2015-0295OC
49. Saradna A, Do DC, Kumar S, Fu QL, Gao P. Macrophage polarization and allergic asthma. Transl Res. (2018) 191:1–14. doi: 10.1016/j.trsl.2017.09.002
50. Liu X, Fang L, Guo TB, Mei H, Zhang JZ. Drug targets in the cytokine universe for autoimmune disease. Trends Immunol. (2013) 34:120–8. doi: 10.1016/j.it.2012.10.003
51. Hewitt RJ, Lloyd CM. Regulation of immune responses by the airway epithelial cell landscape. Nat Rev Immunol. (2021) 21:347–62. doi: 10.1038/s41577-020-00477-9
52. Akdis CA. Does the epithelial barrier hypothesis explain the increase in allergy, autoimmunity and other chronic conditions? Nat Rev Immunol. (2021) 21:739–51. doi: 10.1038/s41577-021-00538-7
53. Hellings PW, Steelant B. Epithelial barriers in allergy and asthma. J Allergy Clin Immunol. (2020) 145:1499–509. doi: 10.1016/j.jaci.2020.04.010
54. Komlosi ZI, van de Veen W, Kovacs N, Szucs G, Sokolowska M, O'Mahony L, et al. Cellular and molecular mechanisms of allergic asthma. Mol Aspects Med. (2022) 85:100995. doi: 10.1016/j.mam.2021.100995
55. Xiao C, Puddicombe SM, Field S, Haywood J, Broughton-Head V, Puxeddu I, et al. Defective epithelial barrier function in asthma. J Allergy Clin Immunol. (2011) 128:549–56.e1-12. doi: 10.1016/j.jaci.2011.05.038
56. de Boer WI, Sharma HS, Baelemans SM, Hoogsteden HC, Lambrecht BN, Braunstahl GJ. Altered expression of epithelial junctional proteins in atopic asthma: possible role in inflammation. Can J Physiol Pharmacol. (2008) 86:105–12. doi: 10.1139/y08-004
57. Raby KL, Michaeloudes C, Tonkin J, Chung KF, Bhavsar PK. Mechanisms of airway epithelial injury and abnormal repair in asthma and copd. Front Immunol. (2023) 14:1201658. doi: 10.3389/fimmu.2023.1201658
58. Celebi Sozener Z, Cevhertas L, Nadeau K, Akdis M, Akdis CA. Environmental factors in epithelial barrier dysfunction. J Allergy Clin Immunol. (2020) 145:1517–28. doi: 10.1016/j.jaci.2020.04.024
59. Tan HT, Hagner S, Ruchti F, Radzikowska U, Tan G, Altunbulakli C, et al. Tight junction, mucin, and inflammasome-related molecules are differentially expressed in eosinophilic, mixed, and neutrophilic experimental asthma in mice. Allergy. (2019) 74:294–307. doi: 10.1111/all.13619
60. Heijink IH, Kuchibhotla VNS, Roffel MP, Maes T, Knight DA, Sayers I, et al. Epithelial cell dysfunction, a major driver of asthma development. Allergy. (2020) 75:1902–17. doi: 10.1111/all.14421
61. Uehara A, Fujimoto Y, Fukase K, Takada H. Various human epithelial cells express functional toll-like receptors, nod1 and nod2 to produce anti-microbial peptides, but not proinflammatory cytokines. Mol Immunol. (2007) 44:3100–11. doi: 10.1016/j.molimm.2007.02.007
62. Frey A, Lunding LP, Ehlers JC, Weckmann M, Zissler UM, Wegmann M. More than just a barrier: the immune functions of the airway epithelium in asthma pathogenesis. Front Immunol. (2020) 11:761. doi: 10.3389/fimmu.2020.00761
63. Hammad H, Lambrecht BN. Barrier epithelial cells and the control of type 2 immunity. Immunity. (2015) 43:29–40. doi: 10.1016/j.immuni.2015.07.007
64. Hammad H, Lambrecht BN. Dendritic cells and epithelial cells: linking innate and adaptive immunity in asthma. Nat Rev Immunol. (2008) 8:193–204. doi: 10.1038/nri2275
65. Gauvreau GM, Bergeron C, Boulet LP, Cockcroft DW, Cote A, Davis BE, et al. Sounding the alarmins-the role of alarmin cytokines in asthma. Allergy. (2023) 78:402–17. doi: 10.1111/all.15609
66. Halim TYF, Rana BMJ, Walker JA, Kerscher B, Knolle MD, Jolin HE, et al. Tissue-restricted adaptive type 2 immunity is orchestrated by expression of the costimulatory molecule ox40l on group 2 innate lymphoid cells. Immunity. (2018) 48:1195–207.e6. doi: 10.1016/j.immuni.2018.05.003
67. Chu DK, Llop-Guevara A, Walker TD, Flader K, Goncharova S, Boudreau JE, et al. Il-33, but not thymic stromal lymphopoietin or il-25, is central to mite and peanut allergic sensitization. J Allergy Clin Immunol. (2013) 131:187–200.e1-8. doi: 10.1016/j.jaci.2012.08.002
68. Gregory LG, Jones CP, Walker SA, Sawant D, Gowers KH, Campbell GA, et al. Il-25 drives remodelling in allergic airways disease induced by house dust mite. Thorax. (2013) 68:82–90. doi: 10.1136/thoraxjnl-2012-202003
69. Iijima K, Kobayashi T, Hara K, Kephart GM, Ziegler SF, McKenzie AN, et al. Il-33 and thymic stromal lymphopoietin mediate immune pathology in response to chronic airborne allergen exposure. J Immunol. (2014) 193:1549–59. doi: 10.4049/jimmunol.1302984
70. Van Dyken SJ, Nussbaum JC, Lee J, Molofsky AB, Liang HE, Pollack JL, et al. A tissue checkpoint regulates type 2 immunity. Nat Immunol. (2016) 17:1381–7. doi: 10.1038/ni.3582
71. Wang W, Li Y, Lv Z, Chen Y, Li Y, Huang K, et al. Bronchial allergen challenge of patients with atopic asthma triggers an alarmin (Il-33, tslp, and il-25) response in the airways epithelium and submucosa. J Immunol. (2018) 201:2221–31. doi: 10.4049/jimmunol.1800709
72. Demenais F, Margaritte-Jeannin P, Barnes KC, Cookson WOC, Altmuller J, Ang W, et al. Multiancestry association study identifies new asthma risk loci that colocalize with immune-cell enhancer marks. Nat Genet. (2018) 50:42–53. doi: 10.1038/s41588-017-0014-7
73. Corren J, Menzies-Gow A, Chupp G, Israel E, Korn S, Cook B, et al. Efficacy of tezepelumab in severe, uncontrolled asthma: pooled analysis of the pathway and navigator clinical trials. Am J Respir Crit Care Med. (2023) 208:13–24. doi: 10.1164/rccm.202210-2005OC
74. Wechsler ME, Ruddy MK, Pavord ID, Israel E, Rabe KF, Ford LB, et al. Efficacy and safety of itepekimab in patients with moderate-to-severe asthma. N Engl J Med. (2021) 385:1656–68. doi: 10.1056/NEJMoa2024257
75. Holgate ST. Innate and adaptive immune responses in asthma. Nat Med. (2012) 18:673–83. doi: 10.1038/nm.2731
76. Edner NM, Carlesso G, Rush JS, Walker LSK. Targeting co-stimulatory molecules in autoimmune disease. Nat Rev Drug Discovery. (2020) 19:860–83. doi: 10.1038/s41573-020-0081-9
77. Deckers J, Sichien D, Plantinga M, Van Moorleghem J, Vanheerswynghels M, Hoste E, et al. Epicutaneous sensitization to house dust mite allergen requires interferon regulatory factor 4-dependent dermal dendritic cells. J Allergy Clin Immunol. (2017) 140:1364–77.e2. doi: 10.1016/j.jaci.2016.12.970
78. Willart MA, Deswarte K, Pouliot P, Braun H, Beyaert R, Lambrecht BN, et al. Interleukin-1alpha controls allergic sensitization to inhaled house dust mite via the epithelial release of gm-csf and il-33. J Exp Med. (2012) 209:1505–17. doi: 10.1084/jem.20112691
79. Rank MA, Kobayashi T, Kozaki H, Bartemes KR, Squillace DL, Kita H. Il-33-activated dendritic cells induce an atypical th2-type response. J Allergy Clin Immunol. (2009) 123:1047–54. doi: 10.1016/j.jaci.2009.02.026
80. Melum GR, Farkas L, Scheel C, Van Dieren B, Gran E, Liu YJ, et al. A thymic stromal lymphopoietin-responsive dendritic cell subset mediates allergic responses in the upper airway mucosa. J Allergy Clin Immunol. (2014) 134:613–21.e7. doi: 10.1016/j.jaci.2014.05.010
81. Moon HG, Kim SJ, Lee MK, Kang H, Choi HS, Harijith A, et al. Colony-stimulating factor 1 and its receptor are new potential therapeutic targets for allergic asthma. Allergy. (2020) 75:357–69. doi: 10.1111/all.14011
82. Conejero L, Khouili SC, Martinez-Cano S, Izquierdo HM, Brandi P, Sancho D. Lung cd103+ Dendritic cells restrain allergic airway inflammation through il-12 production. JCI Insight. (2017) 2:e90420. doi: 10.1172/jci.insight.90420
83. Kool M, van Nimwegen M, Willart MA, Muskens F, Boon L, Smit JJ, et al. An anti-inflammatory role for plasmacytoid dendritic cells in allergic airway inflammation. J Immunol. (2009) 183:1074–82. doi: 10.4049/jimmunol.0900471
84. Halim TY, Steer CA, Matha L, Gold MJ, Martinez-Gonzalez I, McNagny KM, et al. Group 2 innate lymphoid cells are critical for the initiation of adaptive T helper 2 cell-mediated allergic lung inflammation. Immunity. (2014) 40:425–35. doi: 10.1016/j.immuni.2014.01.011
85. Webb LM, Lundie RJ, Borger JG, Brown SL, Connor LM, Cartwright AN, et al. Type I interferon is required for T helper (Th) 2 induction by dendritic cells. EMBO J. (2017) 36:2404–18. doi: 10.15252/embj.201695345
86. Beaty SR, Rose CE Jr., Sung SS. Diverse and potent chemokine production by lung cd11bhigh dendritic cells in homeostasis and in allergic lung inflammation. J Immunol. (2007) 178:1882–95. doi: 10.4049/jimmunol.178.3.1882
87. El-Gammal A, Oliveria JP, Howie K, Watson R, Mitchell P, Chen R, et al. Allergen-induced changes in bone marrow and airway dendritic cells in subjects with asthma. Am J Respir Crit Care Med. (2016) 194:169–77. doi: 10.1164/rccm.201508-1623OC
88. Dutertre CA, Becht E, Irac SE, Khalilnezhad A, Narang V, Khalilnezhad S, et al. Single-cell analysis of human mononuclear phagocytes reveals subset-defining markers and identifies circulating inflammatory dendritic cells. Immunity. (2019) 51:573–89.e8. doi: 10.1016/j.immuni.2019.08.008
89. Naessens T, Morias Y, Hamrud E, Gehrmann U, Budida R, Mattsson J, et al. Human lung conventional dendritic cells orchestrate lymphoid neogenesis during chronic obstructive pulmonary disease. Am J Respir Crit Care Med. (2020) 202:535–48. doi: 10.1164/rccm.201906-1123OC
90. Vroman H, Tindemans I, Lukkes M, van Nimwegen M, de Boer GM, Tramper-Stranders GA, et al. Type ii conventional dendritic cells of asthmatic patients with frequent exacerbations have an altered phenotype and frequency. Eur Respir J. (2020) 55:1900859. doi: 10.1183/13993003.00859-2019
91. Robinson DS, Hamid Q, Ying S, Tsicopoulos A, Barkans J, Bentley AM, et al. Predominant th2-like bronchoalveolar T-lymphocyte population in atopic asthma. N Engl J Med. (1992) 326:298–304. doi: 10.1056/NEJM199201303260504
92. Zheng W, Flavell RA. The transcription factor gata-3 is necessary and sufficient for th2 cytokine gene expression in cd4 T cells. Cell. (1997) 89:587–96. doi: 10.1016/s0092-8674(00)80240-8
93. Szabo SJ, Kim ST, Costa GL, Zhang X, Fathman CG, Glimcher LH. A novel transcription factor, T-bet, directs th1 lineage commitment. Cell. (2000) 100:655–69. doi: 10.1016/s0092-8674(00)80702-3
94. Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, et al. The orphan nuclear receptor rorgammat directs the differentiation program of proinflammatory il-17+ T helper cells. Cell. (2006) 126:1121–33. doi: 10.1016/j.cell.2006.07.035
95. Vivier E, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, et al. Innate lymphoid cells: 10 years on. Cell. (2018) 174:1054–66. doi: 10.1016/j.cell.2018.07.017
96. Korchagina AA, Shein SA, Koroleva E, Tumanov AV. Transcriptional control of ilc identity. Front Immunol. (2023) 14:1146077. doi: 10.3389/fimmu.2023.1146077
97. ASamoah K, Chung KF, Zounemat Kermani N, Bodinier B, Dahlen SE, Djukanovic R, et al. Proteomic signatures of eosinophilic and neutrophilic asthma from serum and sputum. EBioMedicine. (2023) 99:104936. doi: 10.1016/j.ebiom.2023.104936
98. Woodrow JS, Sheats MK, Cooper B, Bayless R. Asthma: the use of animal models and their translational utility. Cells. (2023) 12:1091. doi: 10.3390/cells12071091
99. Corry DB, Folkesson HG, Warnock ML, Erle DJ, Matthay MA, Wiener-Kronish JP, et al. Interleukin 4, but not interleukin 5 or eosinophils, is required in a murine model of acute airway hyperreactivity. J Exp Med. (1996) 183:109–17. doi: 10.1084/jem.183.1.109
100. Pelaia C, Paoletti G, Puggioni F, Racca F, Pelaia G, Canonica GW, et al. Interleukin-5 in the pathophysiology of severe asthma. Front Physiol. (2019) 10:1514. doi: 10.3389/fphys.2019.01514
101. Ying S, Robinson DS, Meng Q, Rottman J, Kennedy R, Ringler DJ, et al. Enhanced expression of eotaxin and ccr3 mrna and protein in atopic asthma. Association with airway hyperresponsiveness and predominant co-localization of eotaxin mrna to bronchial epithelial and endothelial cells. Eur J Immunol. (1997) 27:3507–16. doi: 10.1002/eji.1830271252
102. Nagata M, Sedgwick JB, Vrtis R, Busse WW. Endothelial cells upregulate eosinophil superoxide generation via vcam-1 expression. Clin Exp Allergy. (1999) 29:550–61. doi: 10.1046/j.1365-2222.1999.00506.x
103. Kuperman D, Schofield B, Wills-Karp M, Grusby MJ. Signal transducer and activator of transcription factor 6 (Stat6)-deficient mice are protected from antigen-induced airway hyperresponsiveness and mucus production. J Exp Med. (1998) 187:939–48. doi: 10.1084/jem.187.6.939
104. Zhu Z, Homer RJ, Wang Z, Chen Q, Geba GP, Wang J, et al. Pulmonary expression of interleukin-13 causes inflammation, mucus hypersecretion, subepithelial fibrosis, physiologic abnormalities, and eotaxin production. J Clin Invest. (1999) 103:779–88. doi: 10.1172/JCI5909
105. Dell'Aringa M, Reinhardt RL. Notch signaling represents an important checkpoint between follicular T-helper and canonical T-helper 2 cell fate. Mucosal Immunol. (2018) 11:1079–91. doi: 10.1038/s41385-018-0012-9
106. Chibana K, Trudeau JB, Mustovich AT, Hu H, Zhao J, Balzar S, et al. Il-13 induced increases in nitrite levels are primarily driven by increases in inducible nitric oxide synthase as compared with effects on arginases in human primary bronchial epithelial cells. Clin Exp Allergy. (2008) 38:936–46. doi: 10.1111/j.1365-2222.2008.02969.x
107. Kearley J, Erjefalt JS, Andersson C, Benjamin E, Jones CP, Robichaud A, et al. Il-9 governs allergen-induced mast cell numbers in the lung and chronic remodeling of the airways. Am J Respir Crit Care Med. (2011) 183:865–75. doi: 10.1164/rccm.200909-1462OC
108. Thio CL, Chang YJ. The modulation of pulmonary group 2 innate lymphoid cell function in asthma: from inflammatory mediators to environmental and metabolic factors. Exp Mol Med. (2023) 55:1872–84. doi: 10.1038/s12276-023-01021-0
109. Winkler C, Hochdorfer T, Israelsson E, Hasselberg A, Cavallin A, Thorn K, et al. Activation of group 2 innate lymphoid cells after allergen challenge in asthmatic patients. J Allergy Clin Immunol. (2019) 144:61–9.e7. doi: 10.1016/j.jaci.2019.01.027
110. Klein Wolterink RG, Kleinjan A, van Nimwegen M, Bergen I, de Bruijn M, Levani Y, et al. Pulmonary innate lymphoid cells are major producers of il-5 and il-13 in murine models of allergic asthma. Eur J Immunol. (2012) 42:1106–16. doi: 10.1002/eji.201142018
111. Oliphant CJ, Hwang YY, Walker JA, Salimi M, Wong SH, Brewer JM, et al. Mhcii-mediated dialog between group 2 innate lymphoid cells and cd4(+) T cells potentiates type 2 immunity and promotes parasitic helminth expulsion. Immunity. (2014) 41:283–95. doi: 10.1016/j.immuni.2014.06.016
112. Drake LY, Iijima K, Kita H. Group 2 innate lymphoid cells and cd4+ T cells cooperate to mediate type 2 immune response in mice. Allergy. (2014) 69:1300–7. doi: 10.1111/all.12446
113. Halim TY, Hwang YY, Scanlon ST, Zaghouani H, Garbi N, Fallon PG, et al. Group 2 innate lymphoid cells license dendritic cells to potentiate memory th2 cell responses. Nat Immunol. (2016) 17:57–64. doi: 10.1038/ni.3294
114. Galli SJ, Tsai M, Piliponsky AM. The development of allergic inflammation. Nature. (2008) 454:445–54. doi: 10.1038/nature07204
115. Fajt ML, Wenzel SE. Mast cells, their subtypes, and relation to asthma phenotypes. Ann Am Thorac Soc. (2013) 10 Suppl:S158–64. doi: 10.1513/AnnalsATS.201303-064AW
116. Balzar S, Fajt ML, Comhair SA, Erzurum SC, Bleecker E, Busse WW, et al. Mast cell phenotype, location, and activation in severe asthma. Data from the severe asthma research program. Am J Respir Crit Care Med. (2011) 183:299–309. doi: 10.1164/rccm.201002-0295OC
117. Elieh Ali Komi D, Bjermer L. Mast cell-mediated orchestration of the immune responses in human allergic asthma: current insights. Clin Rev Allergy Immunol. (2019) 56:234–47. doi: 10.1007/s12016-018-8720-1
118. Fajt ML, Gelhaus SL, Freeman B, Uvalle CE, Trudeau JB, Holguin F, et al. Prostaglandin D(2) pathway upregulation: relation to asthma severity, control, and th2 inflammation. J Allergy Clin Immunol. (2013) 131:1504–12. doi: 10.1016/j.jaci.2013.01.035
119. Woo SD, Luu QQ, Park HS. Nsaid-exacerbated respiratory disease (Nerd): from pathogenesis to improved care. Front Pharmacol. (2020) 11:1147. doi: 10.3389/fphar.2020.01147
120. Carr TF, Kraft M. Use of biomarkers to identify phenotypes and endotypes of severeasthma. Ann Allergy Asthma Immunol. (2018) 121:414–20. doi: 10.1016/j.anai.2018.07.029
121. Fitzpatrick AM, Chipps BE, Holguin F, Woodruff PG. T2-"Low" Asthma: overview and management strategies. J Allergy Clin Immunol Pract. (2020) 8:452–63. doi: 10.1016/j.jaip.2019.11.006
122. Annunziato F, Romagnani C, Romagnani S. The 3 major types of innate and adaptive cell-mediated effector immunity. J Allergy Clin Immunol. (2015) 135:626–35. doi: 10.1016/j.jaci.2014.11.001
123. Raundhal M, Morse C, Khare A, Oriss TB, Milosevic J, Trudeau J, et al. High ifn-gamma and low slpi mark severe asthma in mice and humans. J Clin Invest. (2015) 125:3037–50. doi: 10.1172/JCI80911
124. Wark PA, Johnston SL, Bucchieri F, Powell R, Puddicombe S, Laza-Stanca V, et al. Asthmatic bronchial epithelial cells have a deficient innate immune response to infection with rhinovirus. J Exp Med. (2005) 201:937–47. doi: 10.1084/jem.20041901
125. Edwards MR, Regamey N, Vareille M, Kieninger E, Gupta A, Shoemark A, et al. Impaired innate interferon induction in severe therapy resistant atopic asthmatic children. Mucosal Immunol. (2013) 6:797–806. doi: 10.1038/mi.2012.118
126. Oriss TB, Raundhal M, Morse C, Huff RE, Das S, Hannum R, et al. Irf5 distinguishes severe asthma in humans and drives th1 phenotype and airway hyperreactivity in mice. JCI Insight. (2017) 2:e91019. doi: 10.1172/jci.insight.91019
127. Bhakta NR, Christenson SA, Nerella S, Solberg OD, Nguyen CP, Choy DF, et al. Ifn-stimulated gene expression, type 2 inflammation, and endoplasmic reticulum stress in asthma. Am J Respir Crit Care Med. (2018) 197:313–24. doi: 10.1164/rccm.201706-1070OC
128. Veldhoen M. Interleukin 17 is a chief orchestrator of immunity. Nat Immunol. (2017) 18:612–21. doi: 10.1038/ni.3742
129. Lambrecht BN, Hammad H. The immunology of asthma. Nat Immunol. (2015) 16:45–56. doi: 10.1038/ni.3049
130. Al-Ramli W, Prefontaine D, Chouiali F, Martin JG, Olivenstein R, Lemiere C, et al. T(H)17-associated cytokines (Il-17a and il-17f) in severe asthma. J Allergy Clin Immunol. (2009) 123:1185–7. doi: 10.1016/j.jaci.2009.02.024
131. Chang Y, Al-Alwan L, Risse PA, Halayko AJ, Martin JG, Baglole CJ, et al. Th17-associated cytokines promote human airway smooth muscle cell proliferation. FASEB J. (2012) 26:5152–60. doi: 10.1096/fj.12-208033
132. Busse WW, Holgate S, Kerwin E, Chon Y, Feng J, Lin J, et al. Randomized, double-blind, placebo-controlled study of brodalumab, a human anti-il-17 receptor monoclonal antibody, in moderate to severe asthma. Am J Respir Crit Care Med. (2013) 188:1294–302. doi: 10.1164/rccm.201212-2318OC
133. Seys SF, Grabowski M, Adriaensen W, Decraene A, Dilissen E, Vanoirbeek JA, et al. Sputum cytokine mapping reveals an 'Il-5, il-17a, il-25-high' Pattern associated with poorly controlled asthma. Clin Exp Allergy. (2013) 43:1009–17. doi: 10.1111/cea.12125
134. Choy DF, Hart KM, Borthwick LA, Shikotra A, Nagarkar DR, Siddiqui S, et al. Th2 and th17 inflammatory pathways are reciprocally regulated in asthma. Sci Transl Med. (2015) 7:301ra129. doi: 10.1126/scitranslmed.aab3142
135. Wang YH, Voo KS, Liu B, Chen CY, Uygungil B, Spoede W, et al. A novel subset of cd4(+) T(H)2 memory/effector cells that produce inflammatory il-17 cytokine and promote the exacerbation of chronic allergic asthma. J Exp Med. (2010) 207:2479–91. doi: 10.1084/jem.20101376
136. Irvin C, Zafar I, Good J, Rollins D, Christianson C, Gorska MM, et al. Increased frequency of dual-positive th2/th17 cells in bronchoalveolar lavage fluid characterizes a population of patients with severe asthma. J Allergy Clin Immunol. (2014) 134:1175–86.e7. doi: 10.1016/j.jaci.2014.05.038
137. Sun W, Yuan Y, Qiu L, Zeng Q, Jia J, Xiang X, et al. Increased proportion of dual-positive th2-th17 cells promotes a more severe subtype of asthma. Can Respir J. (2021) 2021:9999122. doi: 10.1155/2021/9999122
138. Tortola L, Jacobs A, Pohlmeier L, Obermair FJ, Ampenberger F, Bodenmiller B, et al. High-dimensional T helper cell profiling reveals a broad diversity of stably committed effector states and uncovers interlineage relationships. Immunity. (2020) 53:597–613.e6. doi: 10.1016/j.immuni.2020.07.001
139. Han YY, Zhang X, Wang J, Wang G, Oliver BG, Zhang HP, et al. Multidimensional assessment of asthma identifies clinically relevant phenotype overlap: A cross-sectional study. J Allergy Clin Immunol Pract. (2021) 9:349–62.e18. doi: 10.1016/j.jaip.2020.07.048
140. Ricciardolo FLM, Guida G, Bertolini F, Di Stefano A, Carriero V. Phenotype overlap in the natural history of asthma. Eur Respir Rev. (2023) 32:220201. doi: 10.1183/16000617.0201-2022
141. Pite H, Aguiar L, Morello J, Monteiro EC, Alves AC, Bourbon M, et al. Metabolic dysfunction and asthma: current perspectives. J Asthma Allergy. (2020) 13:237–47. doi: 10.2147/JAA.S208823
142. Morjaria JB, Babu KS, Vijayanand P, Chauhan AJ, Davies DE, Holgate ST. Sputum il-6 concentrations in severe asthma and its relationship with fev1. Thorax. (2011) 66:537. doi: 10.1136/thx.2010.136523
143. Peters MC, McGrath KW, Hawkins GA, Hastie AT, Levy BD, Israel E, et al. Plasma interleukin-6 concentrations, metabolic dysfunction, and asthma severity: A cross-sectional analysis of two cohorts. Lancet Respir Med. (2016) 4:574–84. doi: 10.1016/S2213-2600(16)30048-0
144. Cottrell L, Neal WA, Ice C, Perez MK, Piedimonte G. Metabolic abnormalities in children with asthma. Am J Respir Crit Care Med. (2011) 183:441–8. doi: 10.1164/rccm.201004-0603OC
145. McGinley B, Punjabi NM. Obesity, metabolic abnormalities, and asthma: establishing causal links. Am J Respir Crit Care Med. (2011) 183:424–5. doi: 10.1164/rccm.201009-1525ED
146. Persson EK, Verstraete K, Heyndrickx I, Gevaert E, Aegerter H, Percier JM, et al. Protein crystallization promotes type 2 immunity and is reversible by antibody treatment. Science. (2019) 364:eaaw4295. doi: 10.1126/science.aaw4295
147. Mantel PY, Kuipers H, Boyman O, Rhyner C, Ouaked N, Ruckert B, et al. Gata3-driven th2 responses inhibit tgf-beta1-induced foxp3 expression and the formation of regulatory T cells. PLoS Biol. (2007) 5:e329. doi: 10.1371/journal.pbio.0050329
148. Noval Rivas M, Burton OT, Oettgen HC, Chatila T. Il-4 production by group 2 innate lymphoid cells promotes food allergy by blocking regulatory T-cell function. J Allergy Clin Immunol. (2016) 138:801–11.e9. doi: 10.1016/j.jaci.2016.02.030
149. Steelant B, Wawrzyniak P, Martens K, Jonckheere AC, Pugin B, Schrijvers R, et al. Blocking histone deacetylase activity as a novel target for epithelial barrier defects in patients with allergic rhinitis. J Allergy Clin Immunol. (2019) 144:1242–53.e7. doi: 10.1016/j.jaci.2019.04.027
150. Wegmann M. Trained immunity in allergic asthma. J Allergy Clin Immunol. (2023) 151:1471–3. doi: 10.1016/j.jaci.2023.02.023
151. Netea MG, Dominguez-Andres J, Barreiro LB, Chavakis T, Divangahi M, Fuchs E, et al. Defining trained immunity and its role in health and disease. Nat Rev Immunol. (2020) 20:375–88. doi: 10.1038/s41577-020-0285-6
152. Fanucchi S, Dominguez-Andres J, Joosten LAB, Netea MG, Mhlanga MM. The intersection of epigenetics and metabolism in trained immunity. Immunity. (2021) 54:32–43. doi: 10.1016/j.immuni.2020.10.011
153. Varricchi G, Brightling CE, Grainge C, Lambrecht BN, Chanez P. Airway remodelling in asthma and the epithelium: on the edge of a new era. Eur Respir J. (2024) 63:2301619. doi: 10.1183/13993003.01619-2023
154. Rout-Pitt N, Farrow N, Parsons D, Donnelley M. Epithelial mesenchymal transition (Emt): A universal process in lung diseases with implications for cystic fibrosis pathophysiology. Respir Res. (2018) 19:136. doi: 10.1186/s12931-018-0834-8
155. Hopp RJ, Wilson MC, Pasha MA. Small airway disease in pediatric asthma: the who, what, when, where, why, and how to remediate. A review and commentary. Clin Rev Allergy Immunol. (2022) 62:145–59. doi: 10.1007/s12016-020-08818-1
156. Sun Y, Shi Z, Liu B, Li X, Li G, Yang F, et al. Ykl-40 mediates airway remodeling in asthma via activating fak and mapk signaling pathway. Cell Cycle. (2020) 19:1378–90. doi: 10.1080/15384101.2020.1750811
157. Sun Z, Ji N, Ma Q, Zhu R, Chen Z, Wang Z, et al. Epithelial-mesenchymal transition in asthma airway remodeling is regulated by the il-33/cd146 axis. Front Immunol. (2020) 11:1598. doi: 10.3389/fimmu.2020.01598
158. Song Y, Wang Z, Jiang J, Piao Y, Li L, Xu C, et al. Dek-targeting aptamer dta-64 attenuates bronchial emt-mediated airway remodelling by suppressing tgf-beta1/smad, mapk and pi3k signalling pathway in asthma. J Cell Mol Med. (2020) 24:13739–50. doi: 10.1111/jcmm.15942
159. Yang N, Zhang H, Cai X, Shang Y. Epigallocatechin-3-gallate inhibits inflammation and epithelial−Mesenchymal transition through the pi3k/akt pathway via upregulation of pten in asthma. Int J Mol Med. (2018) 41:818–28. doi: 10.3892/ijmm.2017.3292
160. Pain M, Bermudez O, Lacoste P, Royer PJ, Botturi K, Tissot A, et al. Tissue remodelling in chronic bronchial diseases: from the epithelial to mesenchymal phenotype. Eur Respir Rev. (2014) 23:118–30. doi: 10.1183/09059180.00004413
161. Canas JA, Sastre B, Rodrigo-Munoz JM, Fernandez-Nieto M, Barranco P, Quirce S, et al. Eosinophil-derived exosomes contribute to asthma remodelling by activating structural lung cells. Clin Exp Allergy. (2018) 48:1173–85. doi: 10.1111/cea.13122
162. Kay AB, Phipps S, Robinson DS. A role for eosinophils in airway remodelling in asthma. Trends Immunol. (2004) 25:477–82. doi: 10.1016/j.it.2004.07.006
163. Lopez-Guisa JM, Powers C, File D, Cochrane E, Jimenez N, Debley JS. Airway epithelial cells from asthmatic children differentially express proremodeling factors. J Allergy Clin Immunol. (2012) 129:990–7.e6. doi: 10.1016/j.jaci.2011.11.035
164. Gosens R, Grainge C. Bronchoconstriction and airway biology: potential impact and therapeutic opportunities. Chest. (2015) 147:798–803. doi: 10.1378/chest.14-1142
165. Joubert P, Hamid Q. Role of airway smooth muscle in airway remodeling. J Allergy Clin Immunol. (2005) 116:713–6. doi: 10.1016/j.jaci.2005.05.042
166. Boucherat O, Boczkowski J, Jeannotte L, Delacourt C. Cellular and molecular mechanisms of goblet cell metaplasia in the respiratory airways. Exp Lung Res. (2013) 39:207–16. doi: 10.3109/01902148.2013.791733
167. Tan DJ, Walters EH, Perret JL, Lodge CJ, Lowe AJ, Matheson MC, et al. Age-of-asthma onset as a determinant of different asthma phenotypes in adults: A systematic review and meta-analysis of the literature. Expert Rev Respir Med. (2015) 9:109–23. doi: 10.1586/17476348.2015.1000311
168. Boulet LP. Airway remodeling in asthma: update on mechanisms and therapeutic approaches. Curr Opin Pulm Med. (2018) 24:56–62. doi: 10.1097/MCP.0000000000000441
169. Holgate ST, Holloway J, Wilson S, Bucchieri F, Puddicombe S, Davies DE. Epithelial-mesenchymal communication in the pathogenesis of chronic asthma. Proc Am Thorac Soc. (2004) 1:93–8. doi: 10.1513/pats.2306034
170. Baldo DC, Romaldini JG, Pizzichini MMM, Cancado JED, Dellavance A, Stirbulov R. Periostin as an important biomarker of inflammatory phenotype T2 in Brazilian asthma patients. J Bras Pneumol. (2023) 49:e20220040. doi: 10.36416/1806-3756/e20220040
171. Ge Q, Zeng Q, Tjin G, Lau E, Black JL, Oliver BG, et al. Differential deposition of fibronectin by asthmatic bronchial epithelial cells. Am J Physiol Lung Cell Mol Physiol. (2015) 309:L1093–102. doi: 10.1152/ajplung.00019.2015
172. Grainge CL, Lau LC, Ward JA, Dulay V, Lahiff G, Wilson S, et al. Effect of bronchoconstriction on airway remodeling in asthma. N Engl J Med. (2011) 364:2006–15. doi: 10.1056/NEJMoa1014350
173. Park JA, Drazen JM, Tschumperlin DJ. The chitinase-like protein ykl-40 is secreted by airway epithelial cells at base line and in response to compressive mechanical stress. J Biol Chem. (2010) 285:29817–25. doi: 10.1074/jbc.M110.103416
174. Grainge C, Dulay V, Ward J, Sammut D, Davies E, Green B, et al. Resistin-like molecule-beta is induced following bronchoconstriction of asthmatic airways. Respirology. (2012) 17:1094–100. doi: 10.1111/j.1440-1843.2012.02215.x
175. Chu EK, Cheng J, Foley JS, Mecham BH, Owen CA, Haley KJ, et al. Induction of the plasminogen activator system by mechanical stimulation of human bronchial epithelial cells. Am J Respir Cell Mol Biol. (2006) 35:628–38. doi: 10.1165/rcmb.2006-0040OC
176. Chaudhuri R, Rubin A, Sumino K, Lapa ESJR, Niven R, Siddiqui S, et al. Safety and effectiveness of bronchial thermoplasty after 10 years in patients with persistent asthma (Bt10+): A follow-up of three randomised controlled trials. Lancet Respir Med. (2021) 9:457–66. doi: 10.1016/S2213-2600(20)30408-2
177. Denner DR, Doeing DC, Hogarth DK, Dugan K, Naureckas ET, White SR. Airway inflammation after bronchial thermoplasty for severe asthma. Ann Am Thorac Soc. (2015) 12:1302–9. doi: 10.1513/AnnalsATS.201502-082OC
178. Wijsman PC, Goorsenberg AWM, Keijzer N, d'Hooghe JNS, Ten Hacken NHT, Shah PL, et al. Airway wall extracellular matrix changes induced by bronchial thermoplasty in severe asthma. J Allergy Clin Immunol. (2023) 153:435–46.e4. doi: 10.1016/j.jaci.2023.09.035
179. d'Hooghe JNS, Ten Hacken NHT, Weersink EJM, Sterk PJ, Annema JT, Bonta PI. Emerging understanding of the mechanism of action of bronchial thermoplasty in asthma. Pharmacol Ther. (2018) 181:101–7. doi: 10.1016/j.pharmthera.2017.07.015
180. Dai MY, Chen FF, Wang Y, Wang MZ, Lv YX. Liu RY. Particulate matters induce acute exacerbation of allergic airway inflammation via the tlr2/nf-kappab/nlrp3 signaling pathway. Toxicol Lett. (2020) 321:146–54. doi: 10.1016/j.toxlet.2019.12.013
181. Faffe DS, Whitehead T, Moore PE, Baraldo S, Flynt L, Bourgeois K, et al. Il-13 and il-4 promote tarc release in human airway smooth muscle cells: role of il-4 receptor genotype. Am J Physiol Lung Cell Mol Physiol. (2003) 285:L907–14. doi: 10.1152/ajplung.00120.2003
182. Kibe A, Inoue H, Fukuyama S, Machida K, Matsumoto K, Koto H, et al. Differential regulation by glucocorticoid of interleukin-13-induced eosinophilia, hyperresponsiveness, and goblet cell hyperplasia in mouse airways. Am J Respir Crit Care Med. (2003) 167:50–6. doi: 10.1164/rccm.2110084
183. Tliba O, Deshpande D, Chen H, Van Besien C, Kannan M, Panettieri RA Jr., et al. Il-13 enhances agonist-evoked calcium signals and contractile responses in airway smooth muscle. Br J Pharmacol. (2003) 140:1159–62. doi: 10.1038/sj.bjp.0705558
184. Amrani Y, Panettieri RA Jr. Modulation of calcium homeostasis as a mechanism for altering smooth muscle responsiveness in asthma. Curr Opin Allergy Clin Immunol. (2002) 2:39–45. doi: 10.1097/00130832-200202000-00007
185. Adams DC, Hariri LP, Miller AJ, Wang Y, Cho JL, Villiger M, et al. Birefringence microscopy platform for assessing airway smooth muscle structure and function in vivo. Sci Transl Med. (2016) 8:359ra131. doi: 10.1126/scitranslmed.aag1424
186. Alladina J, Smith NP, Kooistra T, Slowikowski K, Kernin IJ, Deguine J, et al. A human model of asthma exacerbation reveals transcriptional programs and cell circuits specific to allergic asthma. Sci Immunol. (2023) 8:eabq6352. doi: 10.1126/sciimmunol.abq6352
187. Rosas-Salazar C, Chirkova T, Gebretsadik T, Chappell JD, Peebles RS Jr., Dupont WD, et al. Respiratory Syncytial Virus Infection during Infancy and Asthma during Childhood in the USA (Inspire): A Population-Based, Prospective Birth Cohort Study. Lancet. (2023) 401:1669–80. doi: 10.1016/S0140-6736(23)00811-5
188. Jartti T, Gern JE. Role of viral infections in the development and exacerbation of asthma in children. J Allergy Clin Immunol. (2017) 140:895–906. doi: 10.1016/j.jaci.2017.08.003
189. Slater L, Bartlett NW, Haas JJ, Zhu J, Message SD, Walton RP, et al. Co-ordinated role of tlr3, rig-I and mda5 in the innate response to rhinovirus in bronchial epithelium. PLoS Pathog. (2010) 6:e1001178. doi: 10.1371/journal.ppat.1001178
190. Kim EY, Battaile JT, Patel AC, You Y, Agapov E, Grayson MH, et al. Persistent activation of an innate immune response translates respiratory viral infection into chronic lung disease. Nat Med. (2008) 14:633–40. doi: 10.1038/nm1770
191. Ortega H, Nickle D, Carter L. Rhinovirus and asthma: challenges and opportunities. Rev Med Virol. (2021) 31:e2193. doi: 10.1002/rmv.2193
192. Veerati PC, Troy NM, Reid AT, Li NF, Nichol KS, Kaur P, et al. Airway epithelial cell immunity is delayed during rhinovirus infection in asthma and copd. Front Immunol. (2020) 11:974. doi: 10.3389/fimmu.2020.00974
193. Zhu J, Message SD, Mallia P, Kebadze T, Contoli M, Ward CK, et al. Bronchial mucosal ifn-alpha/beta and pattern recognition receptor expression in patients with experimental rhinovirus-induced asthma exacerbations. J Allergy Clin Immunol. (2019) 143:114–25.e4. doi: 10.1016/j.jaci.2018.04.003
194. Holt PG, Mok D, Panda D, Renn L, Fabozzi G, deKlerk NH, et al. Developmental regulation of type 1 and type 3 interferon production and risk for infant infections and asthma development. J Allergy Clin Immunol. (2019) 143:1176–82.e5. doi: 10.1016/j.jaci.2018.08.035
195. Djukanovic R, Harrison T, Johnston SL, Gabbay F, Wark P, Thomson NC, et al. The effect of inhaled ifn-beta on worsening of asthma symptoms caused by viral infections. A randomized trial. Am J Respir Crit Care Med. (2014) 190:145–54. doi: 10.1164/rccm.201312-2235OC
196. Watson A, Spalluto CM, McCrae C, Cellura D, Burke H, Cunoosamy D, et al. Dynamics of ifn-beta responses during respiratory viral infection. Insights for therapeutic strategies. Am J Respir Crit Care Med. (2020) 201:83–94. doi: 10.1164/rccm.201901-0214OC
197. Basnet S, Palmenberg AC, Gern JE. Rhinoviruses and their receptors. Chest. (2019) 155:1018–25. doi: 10.1016/j.chest.2018.12.012
198. Bonnelykke K, Sleiman P, Nielsen K, Kreiner-Moller E, Mercader JM, Belgrave D, et al. A genome-wide association study identifies cdhr3 as a susceptibility locus for early childhood asthma with severe exacerbations. Nat Genet. (2014) 46:51–5. doi: 10.1038/ng.2830
199. Denlinger LC, Phillips BR, Ramratnam S, Ross K, Bhakta NR, Cardet JC, et al. Inflammatory and comorbid features of patients with severe asthma and frequent exacerbations. Am J Respir Crit Care Med. (2017) 195:302–13. doi: 10.1164/rccm.201602-0419OC
200. McErlean P, Favoreto S Jr., Costa FF, Shen J, Quraishi J, Biyasheva A, et al. Human rhinovirus infection causes different DNA methylation changes in nasal epithelial cells from healthy and asthmatic subjects. BMC Med Genomics. (2014) 7:37. doi: 10.1186/1755-8794-7-37
201. Lund RJ, Osmala M, Malonzo M, Lukkarinen M, Leino A, Salmi J, et al. Atopic asthma after rhinovirus-induced wheezing is associated with DNA methylation change in the smad3 gene promoter. Allergy. (2018) 73:1735–40. doi: 10.1111/all.13473
202. McCauley K, Durack J, Valladares R, Fadrosh DW, Lin DL, Calatroni A, et al. Distinct nasal airway bacterial microbiotas differentially relate to exacerbation in pediatric patients with asthma. J Allergy Clin Immunol. (2019) 144:1187–97. doi: 10.1016/j.jaci.2019.05.035
203. McCauley KE, Flynn K, Calatroni A, DiMassa V, LaMere B, Fadrosh DW, et al. Seasonal airway microbiome and transcriptome interactions promote childhood asthma exacerbations. J Allergy Clin Immunol. (2022) 150:204–13. doi: 10.1016/j.jaci.2022.01.020
204. Cazzola M, Matera MG, Rossi F. Bronchial hyperresponsiveness and bacterial respiratory infections. Clin Ther. (1991) 13:157–71.
205. Kim CK, Callaway Z, Gern JE. Viral infections and associated factors that promote acute exacerbations of asthma. Allergy Asthma Immunol Res. (2018) 10:12–7. doi: 10.4168/aair.2018.10.1.12
206. Nagayama Y, Tsubaki T, Nakayama S, Sawada K, Taguchi K, Toba T, et al. Bacterial colonization in respiratory secretions from acute and recurrent wheezing infants and children. Pediatr Allergy Immunol. (2007) 18:110–7. doi: 10.1111/j.1399-3038.2006.00492.x
207. Gibson PG, Yang IA, Upham JW, Reynolds PN, Hodge S, James AL, et al. Effect of azithromycin on asthma exacerbations and quality of life in adults with persistent uncontrolled asthma (Amazes): A randomised, double-blind, placebo-controlled trial. Lancet. (2017) 390:659–68. doi: 10.1016/S0140-6736(17)31281-3
208. Brusselle GG, Vanderstichele C, Jordens P, Deman R, Slabbynck H, Ringoet V, et al. Azithromycin for prevention of exacerbations in severe asthma (Azisast): A multicentre randomised double-blind placebo-controlled trial. Thorax. (2013) 68:322–9. doi: 10.1136/thoraxjnl-2012-202698
209. Taylor SL, Leong LEX, Mobegi FM, Choo JM, Wesselingh S, Yang IA, et al. Long-term azithromycin reduces haemophilus influenzae and increases antibiotic resistance in severe asthma. Am J Respir Crit Care Med. (2019) 200:309–17. doi: 10.1164/rccm.201809-1739OC
210. Undela K, Goldsmith L, Kew KM, Ferrara G. Macrolides versus placebo for chronic asthma. Cochrane Database Syst Rev. (2021) 11:CD002997. doi: 10.1002/14651858.CD002997.pub5
211. Agarwal R, Sehgal IS, Dhooria S, Muthu V, Prasad KT, Bal A, et al. Allergic bronchopulmonary aspergillosis. Indian J Med Res. (2020) 151:529–49. doi: 10.4103/ijmr.IJMR_1187_19
212. Carpaij OA, Burgess JK, Kerstjens HAM, Nawijn MC, van den Berge M. A review on the pathophysiology of asthma remission. Pharmacol Ther. (2019) 201:8–24. doi: 10.1016/j.pharmthera.2019.05.002
213. Broekema M, Volbeda F, Timens W, Dijkstra A, Lee NA, Lee JJ, et al. Airway eosinophilia in remission and progression of asthma: accumulation with a fast decline of fev(1). Respir Med. (2010) 104:1254–62. doi: 10.1016/j.rmed.2010.03.030
214. Broekema M, Timens W, Vonk JM, Volbeda F, Lodewijk ME, Hylkema MN, et al. Persisting remodeling and less airway wall eosinophil activation in complete remission of asthma. Am J Respir Crit Care Med. (2011) 183:310–6. doi: 10.1164/rccm.201003-0494OC
215. Mak JC, Ho SP, Ho AS, Law BK, Cheung AH, Ho JC, et al. Sustained elevation of systemic oxidative stress and inflammation in exacerbation and remission of asthma. ISRN Allergy. (2013) 2013:561831. doi: 10.1155/2013/561831
216. Boulet LP, Turcotte H, Brochu A. Persistence of airway obstruction and hyperresponsiveness in subjects with asthma remission. Chest. (1994) 105:1024–31. doi: 10.1378/chest.105.4.1024
217. van den Toorn LM, Overbeek SE, de Jongste JC, Leman K, Hoogsteden HC, Prins JB. Airway inflammation is present during clinical remission of atopic asthma. Am J Respir Crit Care Med. (2001) 164:2107–13. doi: 10.1164/ajrccm.164.11.2006165
218. Kim BS, Lee E, Lee MJ, Kang MJ, Yoon J, Cho HJ, et al. Different functional genes of upper airway microbiome associated with natural course of childhood asthma. Allergy. (2018) 73:644–52. doi: 10.1111/all.13331
219. Waserman S, Nair P, Snider D, Conway M, Jayaram L, McCleary LM, et al. Local and systemic immunological parameters associated with remission of asthma symptoms in children. Allergy Asthma Clin Immunol. (2012) 8:16. doi: 10.1186/1710-1492-8-16
220. van Den Toorn LM, Prins JB, Overbeek SE, Hoogsteden HC, de Jongste JC. Adolescents in clinical remission of atopic asthma have elevated exhaled nitric oxide levels and bronchial hyperresponsiveness. Am J Respir Crit Care Med. (2000) 162:953–7. doi: 10.1164/ajrccm.162.3.9909033
221. Warke TJ, Fitch PS, Brown V, Taylor R, Lyons JD, Ennis M, et al. Outgrown asthma does not mean no airways inflammation. Eur Respir J. (2002) 19:284–7. doi: 10.1183/09031936.02.00882002
222. Boulet LP, Turcott H, Plante S, Chakir J. Airway function, inflammation and regulatory T cell function in subjects in asthma remission. Can Respir J. (2012) 19:19–25. doi: 10.1155/2012/347989
223. Tomiita M, Campos-Alberto E, Shima M, Namiki M, Sugimoto K, Kojima H, et al. Interleukin-10 and interleukin-5 balance in patients with active asthma, those in remission, and healthy controls. Asia Pac Allergy. (2015) 5:210–5. doi: 10.5415/apallergy.2015.5.4.210
224. Wypych TP, Marsland BJ, Ubags NDJ. The impact of diet on immunity and respiratory diseases. Ann Am Thorac Soc. (2017) 14:S339–S47. doi: 10.1513/AnnalsATS.201703-255AW
225. Bach JF. Revisiting the hygiene hypothesis in the context of autoimmunity. Front Immunol. (2020) 11:615192. doi: 10.3389/fimmu.2020.615192
227. Schuijs MJ, Willart MA, Vergote K, Gras D, Deswarte K, Ege MJ, et al. Farm Dust and Endotoxin Protect against Allergy through A20 Induction in Lung Epithelial Cells. Science. (2015) 349:1106–10. doi: 10.1126/science.aac6623
228. Palomares O, Akdis M, Martin-Fontecha M, Akdis CA. Mechanisms of immune regulation in allergic diseases: the role of regulatory T and B cells. Immunol Rev. (2017) 278:219–36. doi: 10.1111/imr.12555
229. Martin-Orozco E, Norte-Munoz M, Martinez-Garcia J. Regulatory T cells in allergy and asthma. Front Pediatr. (2017) 5:117. doi: 10.3389/fped.2017.00117
230. Wang L, Fu Y, Chu Y. Regulatory B cells. Adv Exp Med Biol. (2020) 1254:87–103. doi: 10.1007/978-981-15-3532-1_8
231. Durham SR, Shamji MH. Allergen immunotherapy: past, present and future. Nat Rev Immunol. (2023) 23:317–28. doi: 10.1038/s41577-022-00786-1
232. Pfaar O, Lou H, Zhang Y, Klimek L, Zhang L. Recent developments and highlights in allergen immunotherapy. Allergy. (2018) 73:2274–89. doi: 10.1111/all.13652
233. Kappen JH, Agache I, Jutel M, Pillai P, Corrigan CJ. Allergen immunotherapy for asthma. J Allergy Clin Immunol Pract. (2024) 12:23–30. doi: 10.1016/j.jaip.2023.11.031
234. Durham SR, Penagos M. Sublingual or subcutaneous immunotherapy for allergic rhinitis? J Allergy Clin Immunol. (2016) 137:339–49.e10. doi: 10.1016/j.jaci.2015.12.1298
235. Farraia M, Paciencia I, Castro Mendes F, Cavaleiro Rufo J, Shamji M, Agache I, et al. Allergen immunotherapy for asthma prevention: A systematic review and meta-analysis of randomized and non-randomized controlled studies. Allergy. (2022) 77:1719–35. doi: 10.1111/all.15295
236. Caimmi D, Demoly P. A review of allergen immunotherapy in asthma. Allergy Asthma Proc. (2022) 43:310–3. doi: 10.2500/aap.2022.43.210113
237. Agusti A, Gibson PG, McDonald VM. Treatable traits in airway disease: from theory to practice. J Allergy Clin Immunol Pract. (2023) 11:713–23. doi: 10.1016/j.jaip.2023.01.011
238. McDonald VM, Clark VL, Cordova-Rivera L, Wark PAB, Baines KJ, Gibson PG. Targeting treatable traits in severe asthma: A randomised controlled trial. Eur Respir J. (2020) 55:1901509. doi: 10.1183/13993003.01509-2019
239. Janssen SMJ, van Helvoort HAC, Tjalma TA, Antons JC, Djamin RS, Simons SO, et al. Impact of treatable traits on asthma control and quality of life. J Allergy Clin Immunol Pract. (2023) 11:1823–33.e4. doi: 10.1016/j.jaip.2023.02.034
Keywords: lung, asthma, immunity, inflammation, allergy, TH2 cytokines, immunotherapy
Citation: Xie C, Yang J, Gul A, Li Y, Zhang R, Yalikun M, Lv X, Lin Y, Luo Q and Gao H (2024) Immunologic aspects of asthma: from molecular mechanisms to disease pathophysiology and clinical translation. Front. Immunol. 15:1478624. doi: 10.3389/fimmu.2024.1478624
Received: 10 August 2024; Accepted: 18 September 2024;
Published: 08 October 2024.
Edited by:
Jian Zheng, University of Louisville, United StatesReviewed by:
Gandhi Fernando Pavon, National Institute of Respiratory Diseases (INER), MexicoXin Sun, Air Force Medical University, China
Copyright © 2024 Xie, Yang, Gul, Li, Zhang, Yalikun, Lv, Lin, Luo and Gao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Huijuan Gao, Z2FvaHVpanVhbkBtYWlsLnRzaW5naHVhLmVkdS5jbg==; Qingli Luo, cWluZ3FpbmdsdW8yMDEwQDE2My5jb20=
†These authors have contributed equally to this work and share first authorship