 Kana Masuda-Kuroki
Kana Masuda-Kuroki Shahrzad Alimohammadi
Shahrzad Alimohammadi Samantha Lowry
Samantha Lowry Anna Di Nardo
Anna Di Nardo
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 26 September 2024
Sec. Inflammation
Volume 15 - 2024 | https://doi.org/10.3389/fimmu.2024.1469829
This article is part of the Research Topic Lipid-Immune Interplay in Chronic Inflammatory-Based Disorders View all 5 articles
Background: Psoriasis is an inflammatory skin condition where immune cells play a significant role. The importance of the cross-talk between keratinocytes and immune cells in the pathogenesis of psoriasis has recently been reaffirmed. Recent studies have found that several S1PR functional antagonists, other than S1PR2, are effective in improving psoriasis. This study aims to investigate the role of S1PR2 in psoriasis, that has not been investigated before.
Methods: Spatial transcriptomics, RT-qPCR, and flow cytometry were used to map the immune cell landscape and its association with metabolic pathways in an imiquimod (IMQ)-induced psoriasis-like inflammation in S1pr2fl/fl K14-Cre mice that could not sense sphingosine-1-phosphate (S1P) in the epidermis through the S1PR2 receptor.
Results: Our analysis suggests that S1PR2 in keratinocytes plays a major role in psoriasis-like inflammation compared to other S1PRs. It acts as a down-regulator, inhibiting the recruitment of Th17 cells into the skin. In IMQ-induced psoriasis skin, both S1pr2-/- and S1pr2fl/fl K14-Cre mice showed higher expressions of proinflammatory cytokines such as TNF-α, IL-17A, and IL-1β together with higher expressions of MyD88/NF-κB pathway compared to the wild-type mice. Remarkably, in IMQ-treated mice, the deletion of S1pr2 in keratinocytes only resulted in a larger population of Th17 cells in skin-draining lymph nodes. Other S1PR modulators did not improve the worsening of psoriasis-like inflammation caused by S1PR2 deficiency in keratinocytes.
Conclusion: This study reaches two main conclusions: signals from keratinocytes play a central role in creating an immune environment that promotes the development of psoriasis, and stimulating S1PR2, instead of suppressing it, represents a potential therapeutic approach for psoriasis.
Psoriasis is a chronic inflammatory skin disease characterized by scaly and elevated erythema, which involves numerous cell types in its inflammatory cascades. Recent studies on the pathogenesis of psoriasis have focused on immune cells and revealed that activated dendritic cells (DCs) promote helper T cell differentiation into Th1 and Th17 cells, which secrete cytokines stimulating epidermal keratinocytes to form hyperproliferation (1). On the other hand, keratinocytes respond to stimuli by secreting antimicrobial peptides, cytokines, and chemokines, further promoting the activation of T cells, DCs, and neutrophils to circulate an inflammatory loop to evoke a chronic inflammation (2, 3). Therefore, keratinocytes are considered a key factor in the inflammatory loop not only as an executor but also as a trigger for immune responses in psoriasis (4).
Keratinocytes constitute the outermost layer of the skin and are surrounded by an intercellular lipid matrix composed of ceramides, cholesterol, and fatty acids (5). Sphingosine 1-phosphate (S1P), one of the bioactive sphingolipid mediators, is metabolically generated from ceramide (6). S1P regulates cellular activities, including cell proliferation and immune responses, by binding to the G-protein-coupled receptor S1PR1-5 (7).
Recently, several groups have found that the levels of sphingosine, S1P, and ceramide in psoriasis lesional skin are significantly higher than in healthy skin in humans (8, 9). Moreover, lipidomic profiling has revealed that the skin lesions in imiquimod (IMQ)-induced psoriasis mouse models have higher ceramides than those in the control mouse skin (10). In addition, some research groups treated psoriasis mice with S1P or S1PR modulators except for S1PR2 (11, 12). These studies confirmed that Sphingolipids and their signaling, except S1PR2, promote the pathogenesis of psoriasis. However, it has not been directly confirmed whether direct inhibition or promotion of S1PR2 alters psoriasis inflammation. Thus, it was still unclear whether S1PR2 has any role in psoriasis. Since previous studies found that S1PR2 is involved in proinflammatory cytokine production in the epidermis as a response to various external stimuli (13) and has a role in regulating keratinocyte proliferation (14), we hypothesized that S1PR2 plays a vital role in the pathogenesis of psoriasis.
This report demonstrates the involvement of S1PR2 in keratinocytes in regulating psoriasis-like inflammation. This study investigates induced psoriasis inflammation in S1PR2-knockout mice, keratinocyte-specific S1PR2 conditional knockout mice, and S1PR2-blocked human keratinocytes. Our study found that the S1PR2 deficiency in keratinocytes activates the myeloid differentiation primary response gene 88 (MyD88)/nuclear factor-kappa B (NF-κB) pathway, leading to Th17 recruiting into the skin.
Here, we present compelling evidence that keratinocytes play a central role in initiating psoriasis inflammation and regulating other cells through S1PR2 signaling. This pathway may offer a novel treatment for psoriasis.
Balb/c mice, that is S1pr2+/+ were purchased from Jackson Lab (Bar Harbor, ME). S1pr2-/- mice were provided by Dr. Chun’s laboratory (15). S1pr2fl/fl mice were gifted from Dr. Hla’s laboratory at Boston Children’s Hospital (MA, USA). K14-Cre-transgenic mice were gifted from Dr. Gallo’s laboratory at the University of California San Diego (CA, USA). Epidermis-specific S1PR2 knockout mice (S1pr2fl/fl K14-Cre) were generated by crossing S1pr2fl/fl mice with K14-Cre-transgenic mice as described previously (16). All mice were housed in the laboratory animal facility at the University of California San Diego in individual ventilated plastic cages and were provided food and water ad libitum under temperature-controlled conditions with a 12 h light/dark cycles. All experiments were performed with 3 times with at least 3 mice in each group each time. For a total of at least 9 mice were used for each group in the experiment. All animal protocols were reviewed and approved by University of California San Diego (IACUC approval number: s10288).
Mice at 8 weeks old of S1pr2+/+, S1pr2-/-, S1pr2fl/fl, and S1pr2fl/fl K14-Cre were depilated on the back skin 1 day before the treatment. After depilation, mice received a daily topical dose of 62.5 mg of commercially available IMQ cream (5%) (Catalog No. 51672414506, Taro Pharmaceutical Industries, Hawthorne, NY) for 7 days on the depilated back skin as described previously (17). Over 7 days, one group of mice received 10 μg of FTY720 dissolved in 80μL of ethanol 1 hour before administration of IMQ. FTY720 dosage and administration timing were per previous papers (18, 19). Skin samples and axillary lymph node (LN)s were collected for further analysis 24 hours after the last IMQ application. The severity of erythema and scale was evaluated using an objective scoring system from 0 to 4 (0, none; 1, slight; 2, moderate; 3, marked; and 4, very marked) as described previously (20). Axillary LNs were measured using Kynup Digital Caliper (Deqing Liang Feng Electronic & Technology Co., Ltd., Zhejiang, China), and weighed by Mettler Toledo Analytical Balance MS104TS/00 (Catalog No. 30133522, Mettler Toledo, Columbus, OH).
Primary normal human epidermal keratinocytes NHEKs (Catalog No. C0015C, Thermo Fisher Scientific, Waltham, MA) were cultured in EpiLife Medium with 60 mM calcium (Catalog No. MEPI500CA, Thermo Fisher Scientific, Waltham, MA) supplemented with Human Keratinocyte Growth Supplement (Catalog No. S0015, Thermo Fisher Scientific, Waltham, MA) at 37 °C in 5% CO2. Subconfluent cells were seeded in 6-well plates and grown to 80% confluence before treatment.
NHEKs cultured in 6-well plates were incubated with phosphate-buffered saline (PBS) or 10 μM of JTE013 (Catalog No. 10009458, Cayman Chemical Company, Ann Arbor, MI) at 37°C in 5% CO2 for 2 hours (13), then incubated with PBS for 24 hours or 100 μM of IMQ (Catalog No. 99011-78-6, InvivoGen, San Diego, CA) for 2, 5, or 24 hours at 37°C in 5% CO2 (21, 22).
Total RNAs from mouse dorsal epidermis and NHEKs were obtained by Quick RNA Miniprep Kit (Catalog No. R1055, Zymo Research, Irvine, CA). cDNA conversion from RNA and real-time RT-qPCR were performed according to the previous report (16). The primers and the probes used for real-time RT-qPCR are listed in Supplementary Tables S1, S2, respectively. The volume of RNA and cDNA was measured by NanoDrop 2000 Spectrophotometer (Catalog No. ND2000CLAPTOP, Thermo Scientific, Waltham, MA). The expression of target genes was normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) expression and analyzed by the 2– ΔΔct method.
Formalin-fixed and paraffin-embedded (FFPE) mouse skin samples were sectioned, and HE stained at the University of California San Diego Biorepository and Tissue and Technology Shared Resource. For immunofluorescence staining of FFPE sections, unstained sections were deparaffinized, rehydrated, and allowed to reach boiling temperature and maintained at a sub-boiling temperature for 10 minutes with antigen retrieval solution (Catalog No. 00495558, Invitrogen, Waltham, MA). After antigen retrieval, immunofluorescent staining was performed using the primary and fluorochrome-conjugated secondary antibodies listed in Supplementary Table S3. Nuclei were counterstained with 4’,6-diamidino-2-phenylindole (DAPI) and slides were mounted with Prolong Gold Anti-Fade Reagent with DAPI (Catalog No. 8961, Cell Signaling Technology, Danvers, MA).
For immunofluorescence staining of NHEKs, cells were cultured on the cover glass in 10-cm petri dish for 24 hours and fixed with 4% paraformaldehyde. Fixed cells were stained with primary and fluorochrome-conjugated secondary antibody listed in Supplementary Table S3. For negative control, fixed cells were stained only with fluorochrome-conjugated secondary antibody. Nuclei were counterstained with DAPI with Prolong Gold Anti-Fade Reagent with DAPI (Catalog No. 8961, Cell Signaling Technology, Danvers, MA). Fluorescence images were obtained with a fluorescence microscope EVOS M5000 (Catalog No. AMF5000, Invitrogen, Waltham, MA).
Axillary LNs were extracted from mice, and LN cells were isolated for flow cytometry analysis. Surface staining was performed for 20 minutes at 4°C with fluorochrome-conjugated antibodies. Fc receptors were blocked with BD Pharmingen Purified Rat Anti-Mouse CD16/CD32 (Mouse BD Fc Block) (Catalog No. AB_394656, BD Biosciences, Franklin Lakes, NJ) diluted 1:100 before staining. For intracellular staining, the cells were fixed and permeabilized using BioLegend Cyto-Fast Fix/Perm Buffer Set (Catalog No. 426803, BioLegend, San Diego, CA), then stained with fluorochrome-conjugated antibodies at room temperature for 30 minutes according to the manufacturer’s instruction. Fixable Viability Dye eFluor 506 (Catalog No. 65-0866-14, Invitrogen, Waltham, MA) were used for viability dye. Fluorescein isothiocyanate (FITC)-conjugated anti-mouse IL-17A monoclonal antibody (mAb) (Catalog No. 506907), APC-conjugated anti-mouse CD4 mAb (Catalog No. 100515), and BV605-conjugated anti-mouse CD8 mAb (Catalog No. 100744) were purchased from BioLegend (San Diego, CA). Samples were measured in flow cytometer, and data were analyzed with FlowJo software (FlowJo, Ashland, OR).
ELISA kit (Catalog No. MBS2708370, MyBioSource, San Diego, CA) were used to determine NF-κB protein expression in mouse skin. Proteins were detected from supernatants of tissue homogenate according to the manufacturer’s instructions. Optical density (OD) value was detected with a micro-plate reader SpectraMax iD3 (Catalog No. ID3-STD, Molecular Devices, San Jose, CA). ELISA measurements were normalized to each sample’s total weight or volume as described previously (23).
FFPE mouse skin samples that passed the RNA quality control (DV200 > 30%) were used for spatial transcriptomic library construction and sequencing. The tissues were prepared according to the Visium CytAssist Spatial Gene Expression for FFPE-Tissue Preparation Guide (CG000518, 10× Genomics, Pleasanton, CA, USA). FFPE mouse skin samples were sectioned into 10 μm, then deparaffinization, probe hybridization and probe ligation were performed. The tissue was permeabilized and the mRNA was reverse transcribed into cDNA with barcode containing slide location information on a designated area of 11 mm × 11 mm square. cDNA was then amplified and processed to obtain cDNA libraries according to the manufacturer’s protocol. To verify the size of PCR-enriched fragments, the template size distribution was checked using Agilent D1000 ScreenTape (Agilent Technologies, Santa Clara, CA). Sequencing was performed at the IGM Genomics Center at the University of California, San Diego. Libraries were sequenced using NovaSeq X Plus 10B (Illumina, San Diego, CA) with a read length of 150 – 1000 bp. Partek flow software version 11 (Partek Incorporated, Chesterfield, MO, USA) was used to process H&E images and FASTQ files and perform spatial gene expression analysis and pathway enrichment analysis. The sequencing results were guaranteed to be accurate as follows; Number of reads: IMQ-treated S1pr2fl/fl, 577,665,006; IMQ-treated S1pr2fl/fl K14-Cre, 496,579,139; Number of spots under tissue: IMQ-treated S1pr2fl/fl, 1,213; IMQ-treated S1pr2fl/fl K14-Cre, 975; Mean reads per spot: 903,639; 1,010,791; Median genes per spot: 1,758; 1,046.
In all experiments, all samples were performed in triplicates, and values were expressed as mean ± SD. Two-way ANOVA and Tukey’s multiple comparison test determined statistical significances of differences for multiple group analysis. Student t-test was performed to compare two groups. P < 0.05 was considered significant. Analyses were performed through the GraphPad Prism software version 7 (GraphPad Software, Boston, MA).
To investigate whether S1PR2 has any effects on psoriasis inflammation, we induced psoriasis-like skin inflammation with 5% IMQ cream on the depilated dorsal skin of S1pr2-/- mice, which has no S1PR2 in their whole body and that of S1pr2+/+ mice, S1pr2 wild type control. After IMQ application, S1pr2-/- mice had much more scales on their dorsal skin than S1pr2+/+ mice, though the severity of erythema was not different between S1pr2-/- mice and S1pr2+/+ mice (Figures 1A, D). None developed systemic symptoms other than skin manifestations. No difference in body shape and size of the mice was observed between the S1pr2+/+ mice and S1pr2-/- mice. Histological analyses with hematoxylin and eosin (H&E) staining of S1pr2-/- mice showed more parakeratosis in the stratum corneum, and more inflammatory cell infiltration in the lower dermis than that of S1pr2+/+ mice after IMQ application (Figure 1B). In addition, immunofluorescence staining revealed that S1pr2-/- mice had more neutrophils in the lower dermis than S1pr2+/+ mice after IMQ application (Figure 1C). We then quantified the mRNA expression levels of proinflammatory cytokines in the epidermis. We found that S1pr2-/- mice epidermis had significantly higher mRNA expressions of Tnf-α, Il-17a, and Il-1β than S1pr2+/+ mice after IMQ application (Figure 1E). These results demonstrate the involvement of S1PR2 in psoriasis pathogenesis and suggest that deletion of S1PR2 amplifies psoriasis inflammation.
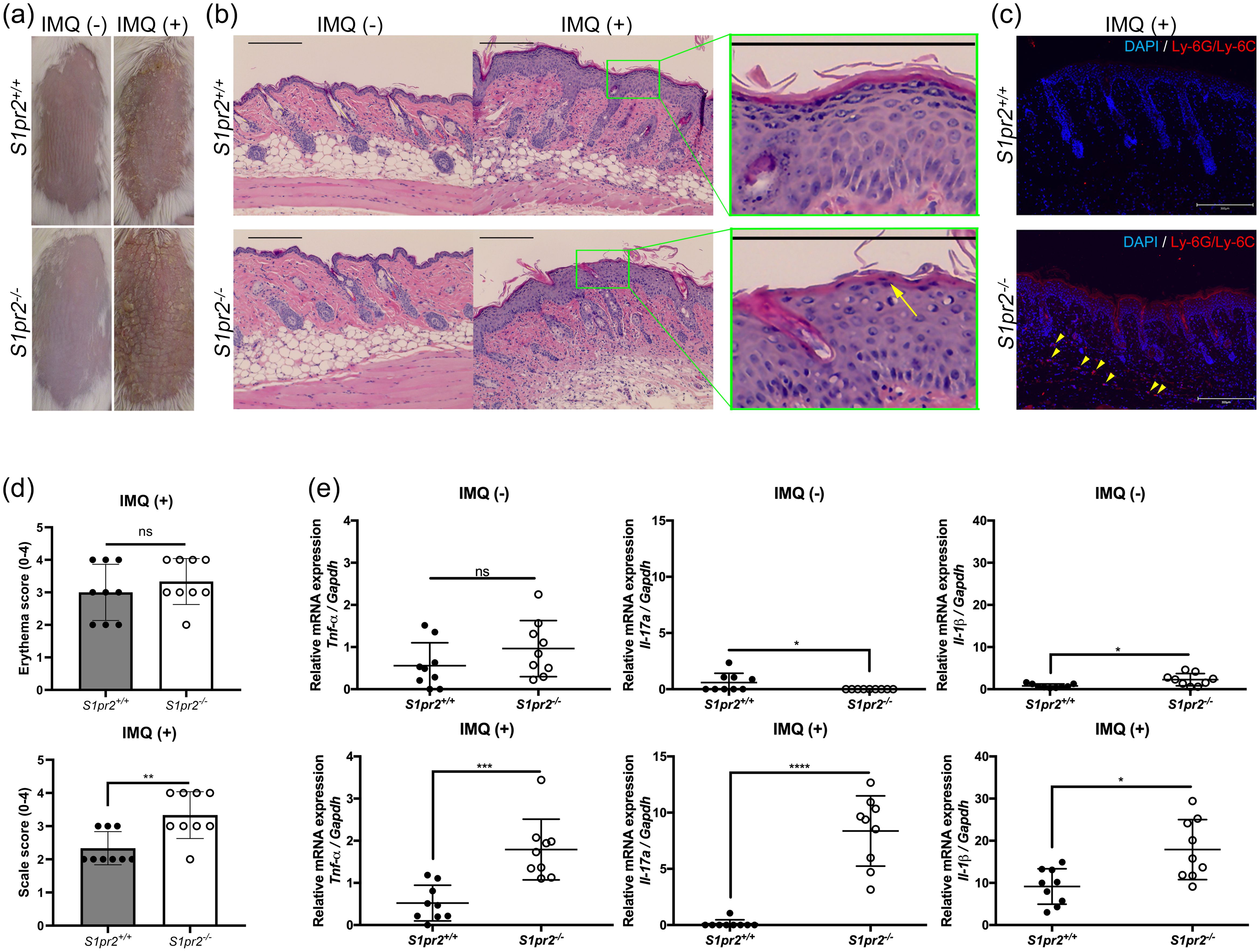
Figure 1. S1PR2 contributes psoriasis-like inflammation in the murine skin. (A) Representative clinical manifestations and (B) H&E staining images of dorsal skin from S1pr2+/+ and S1pr2-/-mice. Arrow shows parakeratosis. (C) Ly-6G/Ly-6C (red) and nuclei (blue) immunofluorescence images of S1pr2+/+ and S1pr2-/- mouse skin 1 week after IMQ application. Arrowhead shows Ly-6G/Ly-6C positive cells. (D) Severity score of erythema (upper panel) and scaling (lower panel). (E) mRNA expressions of Tnf-α, Il-17a, and Il-1β of Balb/c and S1pr2-/- mouse epidermis was measured with RT-qPCR and normalized to Gapdh mRNA expression level. Data shown are the mean ± SD and represent three independent experiments with similar results. Scale bar = 300 μm. ****p < 0.0001, ***p <0.0005, **p < 0.005, *p < 0.05. S1PR2, sphingosine 1-phosphate receptor 2; IMQ, imiquimod; ns, not significant; Gapdh, glyceraldehyde-3-phosphate.
Psoriasis can be triggered by damaging stimuli such as trauma or scratching (24). Keratinocytes, which constitute the outermost layer of the skin, receive these stimuli first and directly. To assess whether the deletion of S1PR2 only in keratinocytes affects psoriasis inflammation, we induced psoriasis-like skin inflammation on the depilated dorsal skin of S1pr2fl/fl K14-Cre mice, that lack of S1PR2 only in keratinocytes, and that of S1pr2fl/fl mice. Consistent with the results of S1pr2-/- mice, S1pr2fl/fl K14-Cre mice showed more scales on their dorsal skin than S1pr2fl/fl mice after IMQ application, though the severity of erythema was not different between S1pr2fl/fl K14-Cre mice and S1pr2fl/fl mice (Figures 2A, D). None developed systemic symptoms other than skin manifestations. No difference in body shape and size of the mice was observed between the S1pr2fl/fl mice and S1pr2fl/fl K14-Cre mice. H&E staining of S1pr2fl/fl K14-Cre mice presented more parakeratosis in the stratum corneum and more inflammatory cell infiltration in the lower dermis than that of S1pr2fl/fl mice (Figure 2B). Immunofluorescence staining analysis showed that S1pr2fl/fl K14-Cre mice had more neutrophils in the lower dermis than S1pr2fl/fl mice (Figure 2C). We investigated proinflammatory cytokine profiles and found that S1pr2fl/fl K14-Cre mice epidermis had significantly higher mRNA expressions of TNF-α, Il-17A, and IL-1β than that of S1pr2fl/fl mice after IMQ application (Figure 2E).

Figure 2. S1PR2 in keratinocytes regulates psoriasis-like inflammation in the murine skin. (A) Representative clinical manifestations and (B) H&E staining images of dorsal skin from S1pr2fl/fl and S1pr2fl/fl K14-Cre mice. (C) Ly-6G/Ly-6C (red) and nuclei (blue) immunofluorescence images of S1pr2fl/fl and S1pr2fl/fl K14-Cre mouse skin 1 week after IMQ application. Arrowhead shows Ly-6G/Ly-6C positive cells. (D) Severity score of erythema (upper panel) and scaling (lower panel). (E) mRNA expressions of Tnf-α, Il-17a, and Il-1β of S1pr2fl/fl and S1pr2fl/fl K14-Cre mouse epidermis were measured with RT-qPCR and normalized to Gapdh mRNA expression level. Data shown are the mean ± SD and are representative of three independent experiments with similar results. Scale bar = 300 μm. ****p < 0.0001, ***p <0.0005, **p < 0.005, *p < 0.05. ns, not significant.
Several studies on mice revealed that the S1PR modulator fingolimod (FTY720) binds S1PR1, 3, 4, and 5, and selective S1PR1 modulator Syl930 ameliorates psoriasis inflammation (12, 25). We, therefore, investigated whether all other S1PRs could regulate inflammation in psoriasis exacerbated by S1PR2 deficiency in keratinocytes. We treated IMQ-induced psoriasis skin of S1pr2fl/fl and S1pr2fl/fl K14-Cre mice with a topical application of FTY720 and compared cytokine profiles with control and IMQ-only groups (Supplementary Figure S1A). With FTY720 and IMQ, S1pr2fl/fl K14-Cre mice still had more scaly erythema than S1pr2fl/fl mice (Supplementary Figures S1B, C), and S1pr2fl/fl K14-Cre mice epidermis had significantly higher mRNA expressions of TNF-α and IL-1β than that of S1pr2fl/fl mice. These results suggest that S1PR2 in keratinocytes plays a crucial role in regulating inflammation in psoriasis and that S1PR2 is more effective in controlling psoriasis than other S1PRs.
IL-17A plays a critically important role in psoriasis inflammation, and Th17 cells are thought to be the main source of IL-17A in psoriasis (26). To investigate whether S1PR2 deletion in keratinocytes affects Th17 cell increase in psoriasis-like skin, we examined the distribution of CD4+ IL-17A+ and CD8+ IL-17A+ T cells in the skin-draining LNs from S1pr2fl/fl K14-Cre and S1pr2fl/fl mice after IMQ application. In terms of size and weight of LNs, S1pr2fl/fl K14-Cre mice had larger and heavier LNs than S1pr2fl/fl mice (Figures 3A, B). In addition, we observed a larger number of CD4+IL-17+ T cells in the skin-draining LNs of S1pr2fl/fl K14-Cre mice than that of S1pr2fl/fl mice after IMQ administration, while no change was observed in the population of CD8+IL-17+ T cells between S1pr2fl/fl K14-Cre and S1pr2fl/fl mice (Figures 3C–G). We also examined the distribution of Cd4+Il-17a+ and Cd8a+Il-17a+ areas in the skin by spatial sequencing to investigate the population of Th17 cells and Tc17 cells in the skin. Contrary to the LN results, a lower percentage of Cd4+Il-17a+ areas and a lower percentage of Cd8a+Il-17a+ areas were observed in the IMQ-treated S1pr2fl/fl K14-Cre mouse skin, compared to S1pr2fl/fl mouse skin (Supplementary Figures S2A, B). Next, we examined Cd2+Cd3+Il-17a+ areas to investigate the distribution of γδ T cells and observed a smaller number in the IMQ-treated S1pr2fl/fl K14-Cre mouse skin (Supplementary Figure S2C). It has been reported that IL-17A production by CD8+ T cells is upregulated in psoriatic skin (27), and the results of this study suggest that S1PR2 deficiency increases IL-17A production from CD8+ cells in the psoriatic skin region. These results suggest that deleting S1pr2 in mouse keratinocytes affects skin-resident cells and the recruitment of IL-17-producing cells during psoriasis-like inflammation.
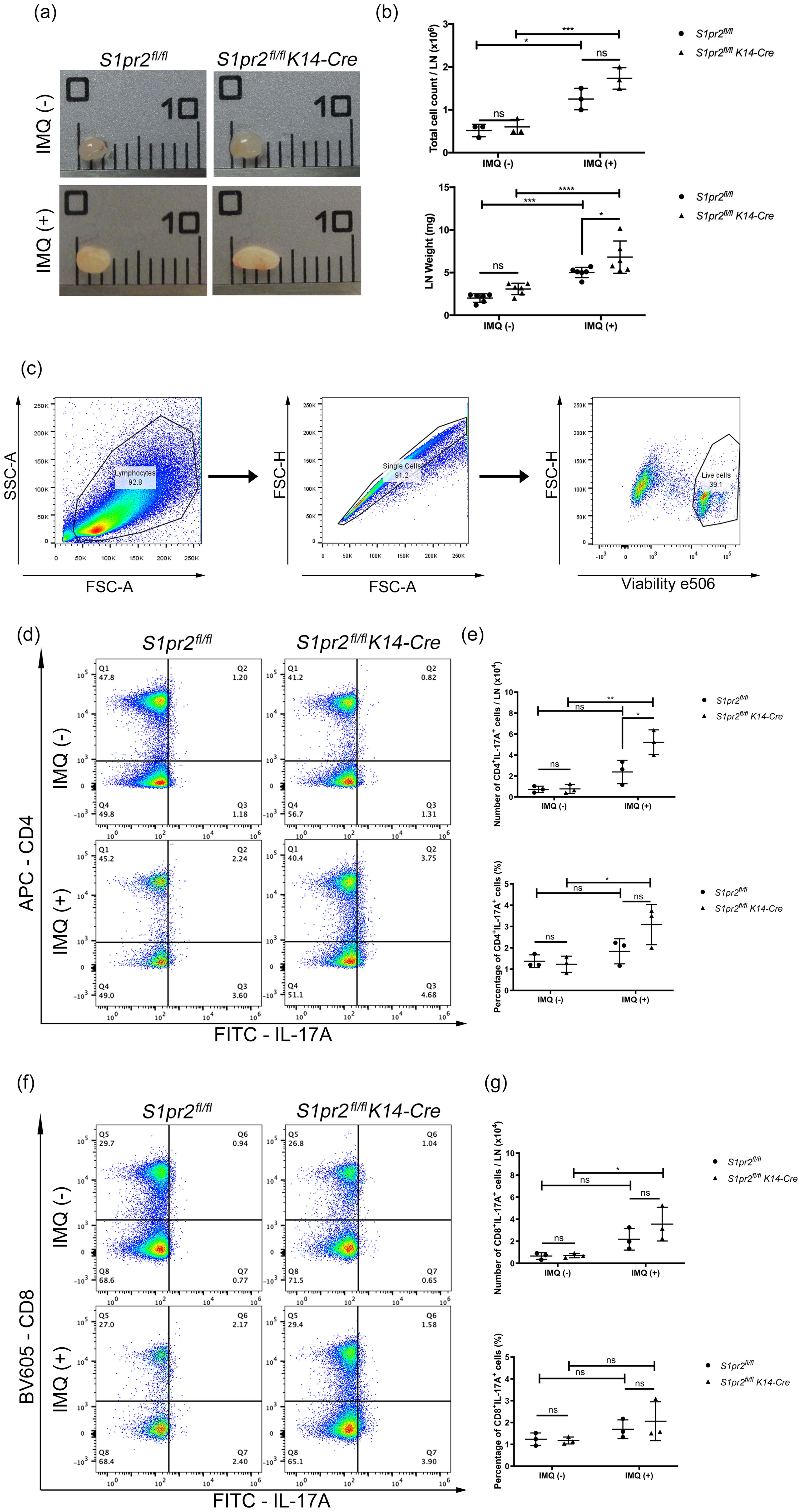
Figure 3. S1PR2 deletion in keratinocytes increases Th17 cell population in skin-draining lymph nodes. (A) Representative images, (B) total cell count and weight of skin-draining lymph nodes of IMQ-treated S1pr2fl/fl and S1pr2fl/fl K14-Cre mice. In each group, a total of six axillary LNs were obtained from the right and left armpit from three mice. Caliper grid line distance is 1 mm. (C, D, F) Flow cytometry analysis of skin-draining LN cells of IMQ-treated S1pr2fl/fl and S1pr2fl/fl K14-Cre mice. The number in each quadrant represents the percentage of the cells. (E, G) Bar graph shows the population of CD4+IL-17A+ cells and CD8+IL-17A+ cells among live cells as measured by flow cytometry analysis (C, D, F). LN, lymph node.
The potential involvement of skin barrier dysfunction in the pathogenesis of psoriasis has been previously discussed (28). Some reports showed an increase of tight junction proteins and filaggrin in psoriasis lesions (29, 30), while others showed a decrease (31, 32). Thus, a consensus has yet to be reached. Since our previous studies have shown that S1PR2 is essential for maintaining and recovering epidermal skin barrier function (13, 16), we investigated whether the skin barrier is altered in psoriasis-like inflammation lacking epidermal S1PR2. RT- qPCR analysis showed no specific trend in mRNA expression levels of the skin barrier proteins between the control and the IMQ-treated group in either S1pr2fl/fl or S1pr2fl/fl K14-Cre mice (Supplementary Figure S3). Our results suggest that impairment of the skin barrier function is not involved in the pathogenesis of psoriasis.
In a mouse model of psoriasis, IMQ acts as a TLR7 ligand to induce the expression of cytokines required for activation of the Th17 pathway and inflammatory factors necessary for psoriasis development (33). To explore how S1PR2 affects IMQ-induced psoriasis-like inflammation, we examined mRNA expressions of the TLR7 signaling pathway in the epidermis of S1PR2-deficient mice. In the epidermis of IMQ-treated S1pr2-/- mice, mRNA levels of Myd88, Irf7, Nfkb1, and Rela were higher than in wild-type mice, but no significant difference was observed in the mRNA level of Tlr7 (Figure 4A). Similar to that, the epidermis of IMQ-treated S1pr2 fl/fl K14-Cre mice had higher mRNA levels of Myd88, Traf6, Irf7, Nfkb1, and Rela compared to S1pr2 fl/fl mice (Figure 4B). In addition, the epidermis of IMQ-treated S1pr2 fl/fl K14-Cre mice had higher protein levels of NF-κB (Figure 4C). This result shows that the deletion of S1PR2 causes an excessive increase in the MyD88/NF-κB pathway, not solely due to TLR7 stimulation.
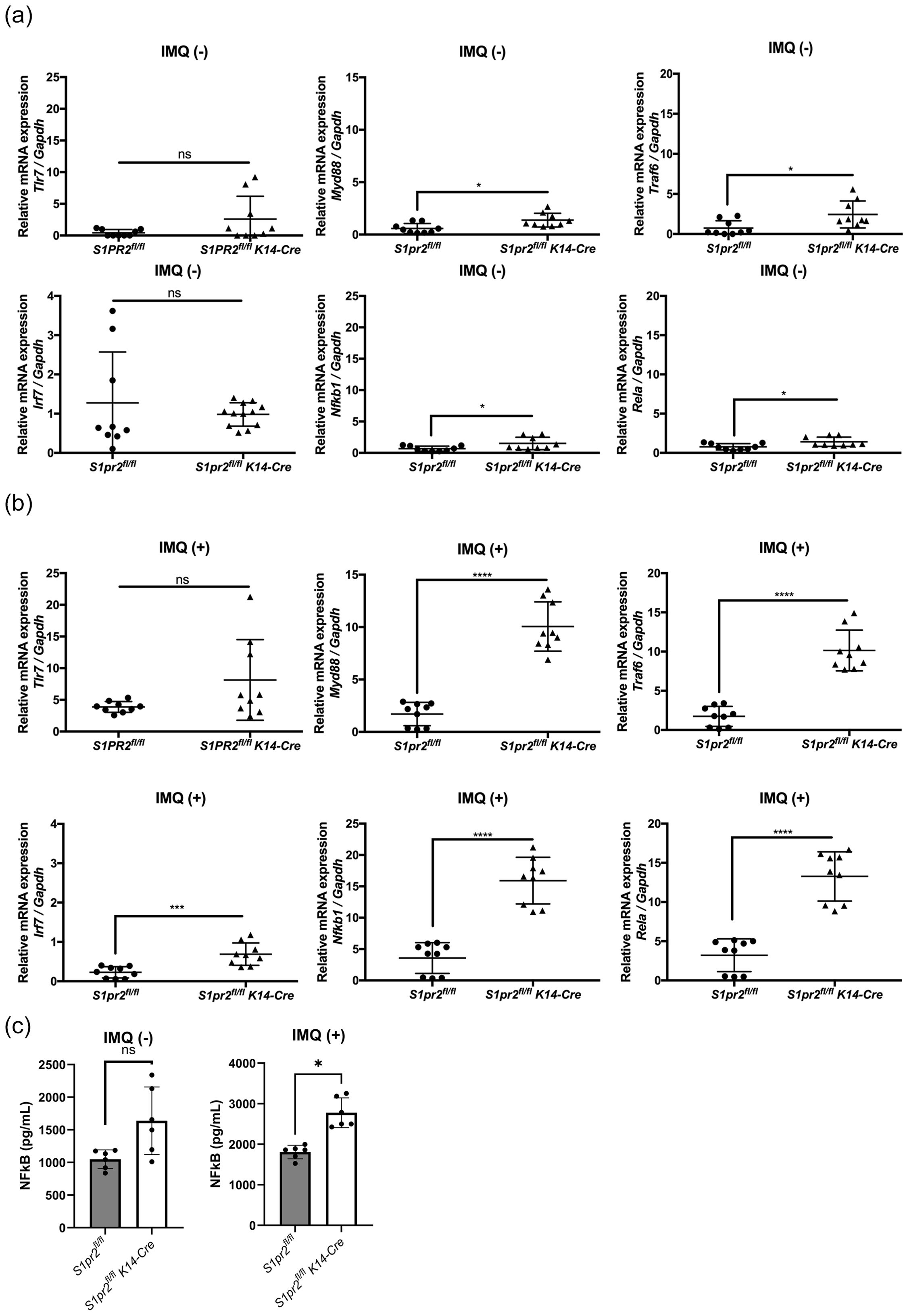
Figure 4. S1PR2 deletion increases MyD88/NF-κB pathway molecules but not affects TLR7 expression. mRNA expressions of Tlr7, Myd88, Traf6, Irf7, Nfkb1, and Rela of (A) Balb/c and S1pr2-/- mouse epidermis, and (B) S1pr2fl/fl and S1pr2fl/fl K14-Cre mouse epidermis were measured with RT-qPCR and normalized to Gapdh mRNA expression level. (C) The protein level of NF-κB was measured by enzyme-linked immunosorbent assay. Data shown are the mean ± SD (n = 3) and are representative of three independent experiments with similar results. ****p < 0.0001, ***p <0.0005, **p < 0.005, *p < 0.05. ns, not significant.
To confirm histology and qPCR analysis, we performed spatial transcriptomics of whole skin tissue from IMQ-treated S1pr2 fl/fl and S1pr2 fl/fl K14-Cre mouse. Graph-based clustering analysis and Uniform Manifold Approximation and Projection for Dimension Reduction (UMAP) showed that epidermis and dermis were classified in separate clusters for IMQ-treated S1pr2 fl/fl and S1pr2 fl/fl K14-Cre mouse, although subcutaneous tissue was classified as a same cluster in both IMQ-treated S1pr2 fl/fl and S1pr2 fl/fl K14-Cre mouse (Figure 5A). In addition, the spatial gene expression analysis showed an upregulation of Tnf, Il-17a, Il-17c, Il-17f, and Il-1β in the epidermis of IMQ-treated S1pr2 fl/fl K14-Cre mouse, consistent with the findings from the pathway enrichment analysis (Figures 5B, C). In addition, gene expression levels of Il-12 cytokine families such as Il-12a, Il-12b, Il-23a, and Il-27 were upregulated in the epidermis of IMQ-treated S1pr2 fl/fl K14-Cre mouse (Figures 5B, C). These data support the fact that the IL-23/Th17 axis is recognized as a key link in the immunopathogenesis of psoriasis, and S1P has been reported to affect IL-23-mediated signaling (34). Moreover, the spatial gene expression analysis and pathway enrichment analysis revealed that the MyD88/NF-κB pathway is upregulated in IMQ-treated S1pr2 fl/fl K14-Cre mouse epidermis, while TLR domain-containing adapter inducing IFN-β (TRIF) and TRAF3, a MyD88-independent signaling pathway, is downregulated (Figure 5C, lower panel). These results support the histology and qPCR findings that S1PR2 deletion in keratinocytes induces MyD88/NF-κB pathway increase in the epidermis, resulting in proinflammatory cytokine increase after IMQ stimulation. Since angiogenesis is an exacerbating factor of psoriatic inflammation (35), we also investigated the gene expression levels of vascular endothelial growth factor (VEGF), hypoxia-inducible factor (HIF), the murine IL-8 homolog CXCL1, and angiopoietin that are considered to be the main players in angiogenesis (Supplementary Figure S4). The spatial gene expression analysis and pathway enrichment analysis showed that S1PR2 deletion did not alter these expressions or even reduce them. This is consistent with the fact that S1PR2 does not change the severity of erythema, suggesting that epidermal S1PR2 does not affect angiogenesis.
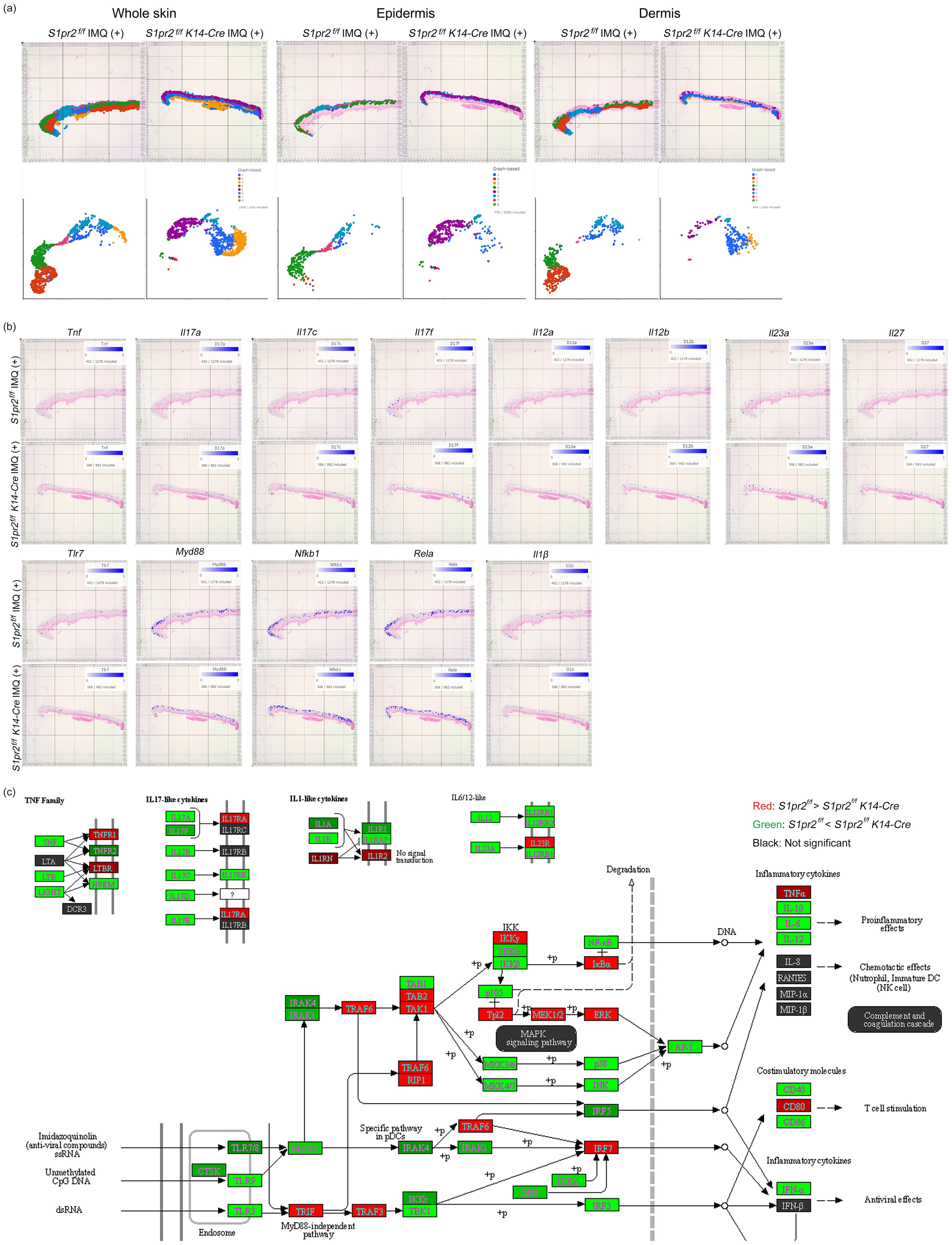
Figure 5. Spatial transcriptomics reveals that S1PR2 deletion in keratinocytes results in psoriasis-like inflammation in the epidermis. (A) Graph-based clustering (upper lane) and UMAPs (lower lane) of S1pr2 fl/fl and S1pr2 fl/fl K14-Cre mouse epidermis after IMQ administration. (B) Spatial gene expression analysis of Tnf, Il-17a, Il-17c, Il-17f, Il-12a, Il-12b, Il-23a, Il-27, Tlr7, Myd88, Nfkb1, Rela, and Il-1β in S1pr2 fl/fl and S1pr2 fl/fl K14-Cre mouse epidermis after IMQ administration. (C) Representatives of pathway enrichment analysis of the mRNA levels by spatial transcriptomics of S1pr2 fl/fl and S1pr2 fl/fl K14-Cre mouse epidermis after IMQ administration. Genes upregulated and downregulated in S1pr2 fl/fl K14-Cre mouse epidermis are highlighted in green and red, respectively. Fold change > 2 and adjusted P value < 0.05.
It was previously stated that NHEKs express TLRs 1, 2, 3, 4, 5, 6, and 9 but not TLR7 and TLR8 (36–38). However, a recent study found that human keratinocytes express TLR7 after calcium-induced differentiation (21). Since the expression of TLR7 is an essential factor in whether the IMQ is an appropriate reagent to induce psoriasis-like inflammation in NHEKs, we performed immunofluorescence staining on NHEKs to confirm that they express TLR7. According to the immunofluorescence staining analysis, we found that NHEKs expressed TLR7 and concluded that NHEKs could be stimulated with IMQ to induce a psoriatic condition similar to that of keratinocytes in a psoriasis mouse model (Figure 6A).
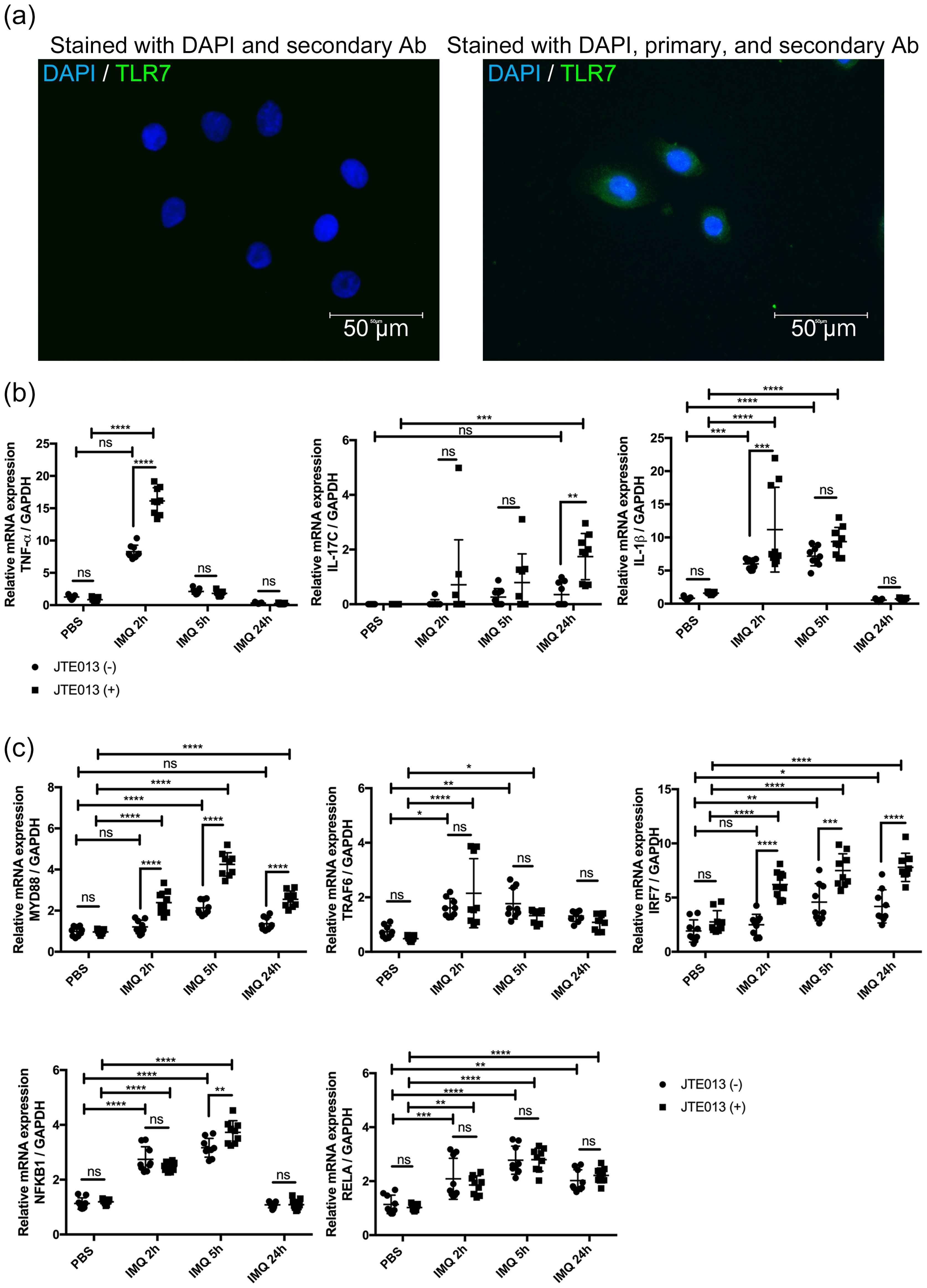
Figure 6. S1PR2 blockade in NHEK increases psoriasis-related inflammation. (A) Representative immunofluorescence images of TLR7 (green) and nuclei (blue) of NHEKs. Left panel shows NHEKs stained with DAPI and secondary antibody. Right panel shows NHEKs stained with DAPI, primary, and secondary antibody (B) mRNA expressions of TNF-α, Il-17C, IL-1β, and (C) mRNA expressions of MYD88, TRAF6, IRF7, NFKB1, and RELA of NHEKs treated with PBS for 24 hours or with IMQ for 2, 5, and 24 hours. Before IMQ treatment, S1PR2 was inhibited by incubating NHEKs with 10 μM JTE013 for 2 hours. Each gene expression was measured with RT-qPCR and normalized to GAPDH mRNA expression level. Data shown are the mean ± SD (n = 3) and are representative of three independent experiments with similar results. ****p < 0.0001, ***p <0.0005, **p < 0.005. ns, not significant.
We next blocked S1PR2 on NHEKs using JTE013 and stimulated them with IMQ to investigate whether S1PR2 in human keratinocytes affects in the same way as in the murine model in psoriasis inflammation. Consistent with mouse model results, S1PR2 blockade of NHEKs significantly increased mRNA expressions of TNF-α at 2 hours, IL-1β at 5 hours, and IL-17C at 24 hours after IMQ administration (Figure 6B). Furthermore, in S1PR2 blocked NHEKs, mRNA expressions of MyD88 and IRF7 were observed at any time after IMQ administration, and that of NFKB1 was observed 5 hours after IMQ administration (Figure 6C). Altogether our in vivo and in vitro data indicate a critical role for keratinocyte S1PR2 in psoriasis-like inflammation through regulating MyD88/NF-κB pathway.
S1P is one of the sphingolipids produced via the degradation of ceramide and the phosphorylation of sphingosine acts as a signaling molecule by binding receptors S1PRs (S1PR1-5) (7). Each S1PR is known to have a different function on cellular responses, such as cell proliferation, differentiation, apoptosis, and immune responses (39). Among the five S1PRs, S1PR1 has been primarily studied for its association with psoriasis (35, 40). In addition, fingolimod, an S1PR1 functional antagonist that binds to S1PR3-5, has been reported to inhibit the migration of T cells from the LN to the skin and alleviate IMQ-induced psoriasis (12). However, the role of S1PR2 in psoriasis has yet to be investigated. Our study demonstrated that S1PR2 deletion increases psoriasis inflammation. Namely, our data indicated that S1PR2 acts as an immunosuppressor in psoriasis pathogenesis.
Since the discovery that T cells play a major role in the pathogenesis of psoriasis, research on the pathogenesis and treatment of psoriasis has primarily targeted immune cells (41–45). However, the importance of crosstalk between keratinocytes and immune cells in psoriasis has received increasing attention recently (4, 46). In psoriasis lesions, external stimulation activates keratinocytes to produce proinflammatory cytokines such as TNF-α and IL-1β, stimulating dendritic cells to induce differentiation of helper T cells (47–49). One of the helper T cells subsets the Th17 cell, produces IL-17A, and stimulates the keratinocytes to produce proinflammatory cytokines such as IL-1β, IL-17C, and IL-8, which induces the recruitment of neutrophils (2, 50–52). IL-1β secreted from keratinocytes acts on keratinocytes to promote cytokine production via the MyD88/NF-κB pathway (52, 53). This positive feedback forms a loop to maintain the inflammatory phase of psoriasis. Our data demonstrated that S1PR2 in keratinocytes interacts with the MyD88/NF-κB pathway and regulates this feedback loop.
In studies other than the skin and keratinocytes, S1PR2 has been reported to have pro-inflammatory functions. For example, vascular endothelial cell inflammation is suppressed in S1PR2-deficient mice (54), and osteoclast formation is inhibited in S1PR2-deficient mice (55). On the other hand, it has been reported that S1PR2 acts as a suppressive mediator in tumors; S1PR2 signaling confers tumor suppressive activity in diffuse large B-cell lymphoma (56), and lower S1PR2 values are associated with a worse prognosis in cervical squamous cell carcinoma (57).
Several studies have reported that S1PR2 ameliorates atopic dermatitis in skin diseases by promoting the Th2 cell-attracting capacity of dendritic cells (58, 59). However, another study has shown that S1PR2 deficiency increases skin inflammation by causing hyperpermeability and exacerbating mechanical stress damage and bacterial penetration (13). Our data showed that the absence of S1PR2 in keratinocytes worsened skin inflammation in psoriasis. This suggests that S1PR2 is anti-inflammatory in keratinocytes, in contrast to its effects on other cell types.
In addition, although several studies reported that FTY720, a functional antagonist of S1PR1, 3, 4, and 5, ameliorated psoriasis, the study in mice found that topical FTY720 does not alter psoriasis inflammation (19). This was hypothesized to be due to S1PR2’s function restraining hyperplasia in the skin (14, 60). Our data showed that the exacerbation of psoriasis caused by S1PR2 deficiency was not ameliorated by the administration of FTY720. This supports the notion that S1PR2 has anti-inflammatory properties in keratinocytes, and psoriasis worsens when absent. Furthermore, the study suggests that the absence of S1PR2 signaling cannot be compensated for by modulating other S1PRs. Since our previous study revealed that S1PR2 has a crucial role in skin barrier maintenance (13), we investigated the relationship between skin barrier function and psoriasis inflammation, which has been discussed in recent years (28). Of note, the association between skin barrier function and psoriasis inflammation is not consistent, with some clinical studies showing increased expression of tight junction proteins in the psoriasis skin (29, 61), while some clinical studies have shown decreased tight junction proteins and filaggrin expression in psoriasis (31, 32). In addition, a study that analyzed filaggrin and tight junction proteins in human psoriasis, ex vivo models, and in vitro models found that tight junction changes were observed in the early stage of psoriasis but not in developed psoriasis (30). This study shows that S1PR2 in keratinocytes inhibits psoriasis inflammation due to the MyD88/NF-κB pathway but not due to the skin barrier impairment. Since our data did not show any correlation between skin barrier and psoriasis inflammation, we support the idea that skin barrier changes are not responsible for psoriasis cytokine production.
MyD88 is an adaptor protein that signals downstream of TLRs and IL-1 receptor family members and activates the transcription factor NF-κB (62, 63). Thus, MyD88 is a central molecule that regulates immune responses in many cell types. However, MyD88 does not elicit the same response in different cell types. MyD88-mediated TLR signaling induces the secretion of Th1 and Th2 cytokines in mast cells (64). On the other hand, MyD88 in keratinocytes regulates Th1 and Th17 but not Th2 responses in allergic reactions (65). In addition, developing atopic and contact dermatitis requires MyD88 signaling in dendritic cells but not in keratinocytes (65–67). In our previous study, S1PR2 deletion in keratinocytes increased mRNA expressions of Il-1β and Cxcl1 of the contact dermatitis mouse model but did not change other cytokines and clinical manifestations (16). The difference between psoriasis and contact dermatitis is due to the specific regulation of the MyD88/NF-κB pathway by S1PR2 in keratinocytes. Thus, the mechanism we found applies only to psoriasis inflammation.
A possible mechanism for S1PR2 to suppress the MyD88/NF-κB pathway is its balance with the MyD88-independent TLR signaling pathway. Our spatial transcriptomic analysis revealed that in the presence of S1PR2, MyD88-independent TLR signaling pathways such as TRIF and TRAF3 are activated when stimulated with IMQ, although the MyD88/NF-κB pathway is downregulated. However, further studies are required to determine the cause of this imbalanced signaling pathway.
In addition, this study’s limitation is that it did not focus on immune cells such as DCs or macrophages and analyzed specifically the Th17 cell population. Since DCs and macrophages are also key players in the immune response closely associated with psoriasis pathogenesis, these cells should be investigated in the future. Moreover, it is necessary to study the spleen, a tissue where immune cells are assembled, in order to further investigate the effects on systemic inflammation.
Our study is groundbreaking because it is the first to show that a signal generated by a lipid in keratinocytes can directly generate psoriatic changes in the skin. We followed the effects of the S1P signal generated by the keratinocytes and how it reverberates throughout the skin.
In summary, we demonstrated that S1PR2 is involved in regulating psoriasis inflammation and, in particular, that S1PR2 in keratinocytes plays an essential role in this regulation. This study emphasizes the significance of keratinocytes in psoriasis research and suggests targeting S1PR2 as a novel treatment approach.
The datasets generated and analyzed for this study can be found at https://www.ncbi.nlm.nih.gov/bioproject/PRJNA1161425, hosted by National Center for Biotechnology Information (NCBI), accession number PRJNA1161425; GEO: GSE277246.
The animal study was approved by Institutional Animal Care and Use Committee approval number: s10288. The study was conducted in accordance with the local legislation and institutional requirements.
KM-K: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Visualization, Writing – original draft, Writing – review & editing. SA: Writing – review & editing. SL: Investigation, Methodology, Writing – review & editing. AD: Conceptualization, Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This research was funded by the NIH National Institute of Allergy & Infectious Disease (NIAID), grant number 2R01AI093957 and the National Eczema Association (NEA), grant number ECRG133.
We thank Mr. George Bertini for his support in the spatial transcriptomic analysis and Dr. George Sen for his thoughtful suggestions.
The authors declare that the research was conducted without any commercial or financial relationships that could potentially create a conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2024.1469829/full#supplementary-material
DC, dendritic cell; FFPE, Formalin-fixed and paraffin-embedded; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; IMQ, imiquimod; MyD88, myeloid differentiation primary response gene 88; NF-κB, nuclear factor-kappa B; NHEK, normal human epidermal keratinocyte; PBS, phosphate-buffered saline; RT-qPCR, Reverse transcription-quantitative polymerase chain reaction; S1P, Sphingosine 1-phosphate; S1PR, Sphingosine 1-phosphate receptor; TNF, Tumor necrosis factor; TRAF, Tumor necrosis factor receptor–associated factor; Th17 cells, T helper 17 cells; UMAP, Uniform Manifold Approximation and Projection for Dimension Reduction.
1. Nestle FO, Di Meglio P, Qin JZ, Nickoloff BJ. Skin immune sentinels in health and disease. Nat Rev Immunol. (2009) 9:679–91. doi: 10.1038/nri2622
2. Harper EG, Guo C, Rizzo H, Lillis JV, Kurtz SE, Skorcheva I, et al. Th17 cytokines stimulate CCL20 expression in keratinocytes in vitro and in vivo: implications for psoriasis pathogenesis. J Invest Dermatol. (2009) 129:2175–83. doi: 10.1038/jid.2009.65
3. Lande R, Botti E, Jandus C, Dojcinovic D, Fanelli G, Conrad C, et al. The antimicrobial peptide LL37 is a T-cell autoantigen in psoriasis. Nat Commun. (2014) 5:5621. doi: 10.1038/ncomms6621
4. Ni X, Lai Y. Keratinocyte: A trigger or an executor of psoriasis? J Leukoc Biol. (2020) 108:485–91. doi: 10.1002/jlb.5mr0120-439r
5. Elias PM. Epidermal lipids, barrier function, and desquamation. J Invest Dermatol. (1983) 80:44s–9s. doi: 10.1038/jid.1983.12
6. Spiegel S, Merrill AH Jr. Sphingolipid metabolism and cell growth regulation. FASEB J. (1996) 10:1388–97. doi: 10.1096/fasebj.10.12.8903509
7. Obinata H, Hla T. Sphingosine 1-phosphate and inflammation. Int Immunol. (2019) 31:617–25. doi: 10.1093/intimm/dxz037
8. Moon SH, Kim JY, Song EH, Shin MK, Cho YH, Kim NI. Altered levels of sphingosine and sphinganine in psoriatic epidermis. Ann Dermatol. (2013) 25:321–6. doi: 10.5021/ad.2013.25.3.321
9. Matwiejuk M, Mysliwiec H, Lukaszuk B, Lewoc M, Malla H, Mysliwiec P, et al. The interplay between bioactive sphingolipids in the psoriatic skin and the severity of the disease. Int J Mol Sci. (2023) 24. doi: 10.3390/ijms241411336
10. Liu P, Hou G, Kuang Y, Li L, Chen C, Yan B, et al. Lipidomic profiling reveals metabolic signatures in psoriatic skin lesions. Clin Immunol. (2023) 246:109212. doi: 10.1016/j.clim.2022.109212
11. Jin J, Xue N, Liu Y, Fu R, Wang M, Ji M, et al. A novel S1P1 modulator IMMH002 ameliorates psoriasis in multiple animal models. Acta Pharm Sin B. (2020) 10:276–88. doi: 10.1016/j.apsb.2019.11.006
12. Okura I, Kamata M, Asano Y, Mitsui A, Shimizu T, Sato S, et al. Fingolimod ameliorates imiquimod-induced psoriasiform dermatitis by sequestrating interleukin-17-producing?d T cells in secondary lymph nodes. J Dermatol Sci. (2021) 102:116–25. doi: 10.1016/j.jdermsci.2021.04.004
13. Igawa S, Ohzono A, Pham P, Wang Z, Nakatsuji T, Dokoshi T, et al. Sphingosine 1-phosphate receptor 2 is central to maintaining epidermal barrier homeostasis. J Invest Dermatol. (2021) 141:1188–1197.e1185. doi: 10.1016/j.jid.2020.09.026
14. Schüppel M, Kürschner U, Kleuser U, Schäfer-Korting M, Kleuser B. Sphingosine 1-phosphate restrains insulin-mediated keratinocyte proliferation via inhibition of Akt through the S1P2 receptor subtype. J Invest Dermatol. (2008) 128:1747–56. doi: 10.1038/sj.jid.5701259
15. Ishii I, Ye X, Friedman B, Kawamura S, Contos JJ, Kingsbury MA, et al. Marked perinatal lethality and cellular signaling deficits in mice null for the two sphingosine 1-phosphate (S1P) receptors, S1P(2)/LP(B2)/EDG-5 and S1P(3)/LP(B3)/EDG-3. J Biol Chem. (2002) 277: 25152–25159. doi: 10.1074/jbc.M200137200
16. Masuda-Kuroki K, Alimohammadi S, Di Nardo A. S. epidermidis rescues allergic contact dermatitis in sphingosine 1-phosphate receptor 2-deficient skin. Int J Mol Sci. (2023) 24. doi: 10.3390/ijms241713190
17. van der Fits L, Mourits S, Voerman JS, Kant M, Boon L, Laman JD, et al. Imiquimod-induced psoriasis-like skin inflammation in mice is mediated via the IL-23/IL-17 axis. J Immunol. (2009) 182:5836–45. doi: 10.4049/jimmunol.0802999
18. Reines I, Kietzmann M, Mischke R, Tschernig T, Lüth A, Kleuser B, et al. Topical application of sphingosine-1-phosphate and FTY720 attenuate allergic contact dermatitis reaction through inhibition of dendritic cell migration. J Invest Dermatol. (2009) 129:1954–62. doi: 10.1038/jid.2008.454
19. Schaper K, Dickhaut J, Japtok L, Kietzmann M, Mischke R, Kleuser B, et al. Sphingosine-1-phosphate exhibits anti-proliferative and anti-inflammatory effects in mouse models of psoriasis. J Dermatol Sci. (2013) 71:29–36. doi: 10.1016/j.jdermsci.2013.03.006
20. Matsuno A, Sumida H, Nakanishi H, Ikeyama Y, Ishii T, Omori I, et al. Keratinocyte proline-rich protein modulates immune and epidermal response in imiquimod-induced psoriatic skin inflammation. Exp Dermatol. (2023) 32:2121–30. doi: 10.1111/exd.14960
21. Li ZJ, Sohn KC, Choi DK, Shi G, Hong D, Lee HE, et al. Roles of TLR7 in activation of NF-kappaB signaling of keratinocytes by imiquimod. PloS One. (2013) 8:e77159. doi: 10.1371/journal.pone.0077159
22. Bochenska K, Moskot M, Gabig-Ciminska M. Use of cytokine mix-, imiquimod-, and serum-induced monoculture and lipopolysaccharide- and interferon gamma-treated co-culture to establish in vitro psoriasis-like inflammation models. Cells. (2021) 10. doi: 10.3390/cells10112985
23. Wang Z, Mascarenhas N, Eckmann L, Miyamoto Y, Sun X, Kawakami T, et al. Skin microbiome promotes mast cell maturation by triggering stem cell factor production in keratinocytes. J Allergy Clin Immunol. (2017) 139:1205–1216.e1206. doi: 10.1016/j.jaci.2016.09.019
24. Eyre RW, Krueger GG. Response to injury of skin involved and uninvolved with psoriasis, and its relation to disease activity: Koebner and ‘reverse’ Koebner reactions. Br J Dermatol. (1982) 106:153–9. doi: 10.1111/j.1365-2133.1982.tb00924.x
25. Ji M, Xue N, Lai F, Zhang X, Zhang S, Wang Y, et al. Validating a selective S1P(1) receptor modulator syl930 for psoriasis treatment. Biol Pharm Bull. (2018) 41:592–6. doi: 10.1248/bpb.b17-00939
26. Martin DA, Towne JE, Kricorian G, Klekotka P, Gudjonsson JE, Krueger JG, et al. The emerging role of IL-17 in the pathogenesis of psoriasis: preclinical and clinical findings. J Invest Dermatol. (2013) 133:17–26. doi: 10.1038/jid.2012.194
27. Kurihara K, Fujiyama T, Phadungsaksawasdi P, Ito T, Tokura Y. Significance of IL-17A-producing CD8(+)CD103(+) skin resident memory T cells in psoriasis lesion and their possible relationship to clinical course. J Dermatol Sci. (2019) 95:21–7. doi: 10.1016/j.jdermsci.2019.06.002
28. Orsmond A, Bereza-Malcolm L, Lynch T, March L, Xue M. Skin barrier dysregulation in psoriasis. Int J Mol Sci. (2021) 22. doi: 10.3390/ijms221910841
29. Peltonen S, Riehokainen J, Pummi K, Peltonen J. Tight junction components occludin, ZO-1, and claudin-1, -4 and -5 in active and healing psoriasis. Br J Dermatol. (2007) 156:466–72. doi: 10.1111/j.1365-2133.2006.07642.x
30. Kirschner N, Poetzl C, von den Driesch P, Wladykowski E, Moll I, Behne MJ, et al. Alteration of tight junction proteins is an early event in psoriasis: putative involvement of proinflammatory cytokines. Am J Pathol. (2009) 175:1095–106. doi: 10.2353/ajpath.2009.080973
31. Visconti B, Paolino G, Carotti S, Pendolino AL, Morini S, Richetta AG, et al. Immunohistochemical expression of VDR is associated with reduced integrity of tight junction complex in psoriatic skin. J Eur Acad Dermatol Venereol. (2015) 29:2038–42. doi: 10.1111/jdv.12736
32. Zhang H, Jia T, Che D, Peng B, Chu Z, Song X, et al. Decreased TET2/5-hmC reduces the integrity of the epidermal barrier via epigenetic dysregulation of filaggrin in psoriatic lesions. J Dermatol Sci. (2024) 113:103–12. doi: 10.1016/j.jdermsci.2024.01.004
33. Lu J, Zhong X, Guo C, Tang L, Yu N, Peng C, et al. TLR7-MyD88-DC-CXCL16 axis results neutrophil activation to elicit inflammatory response in pustular psoriasis. Cell Death Dis. (2023) 14:315. doi: 10.1038/s41419-023-05815-y
34. Baeyens A, Bracero S, Chaluvadi VS, Khodadadi-Jamayran A, Cammer M, Schwab SR. Monocyte-derived S1P in the lymph node regulates immune responses. Nature. (2021) 592:290–5. doi: 10.1038/s41586-021-03227-6
35. Syed SN, Raue R, Weigert A, von Knethen A, Brüne B. Macrophage S1PR1 signaling alters angiogenesis and lymphangiogenesis during skin inflammation. Cells. (2019) 8. doi: 10.3390/cells8080785
36. Pivarcsi A, Bodai L, Réthi B, Kenderessy-Szabó A, Koreck A, Széll M, et al. Expression and function of Toll-like receptors 2 and 4 in human keratinocytes. Int Immunol. (2003) 15:721–30. doi: 10.1093/intimm/dxg068
37. Köllisch G, Kalali BN, Voelcker V, Wallich R, Behrendt H, Ring J, et al. Various members of the Toll-like receptor family contribute to the innate immune response of human epidermal keratinocytes. Immunology. (2005) 114:531–41. doi: 10.1111/j.1365-2567.2005.02122.x
38. Lebre MC, van der Aar AM, van Baarsen L, van Capel TM, Schuitemaker JH, Kapsenberg ML, et al. Human keratinocytes express functional Toll-like receptor 3, 4, 5, and 9. J Invest Dermatol. (2007) 127:331–41. doi: 10.1038/sj.jid.5700530
39. Blaho VA, Hla T. An update on the biology of sphingosine 1-phosphate receptors. J Lipid Res. (2014) 55:1596–608. doi: 10.1194/jlr.R046300
40. Bell M, Foley D, Naylor C, Robinson C, Riley J, Epemolu O, et al. Discovery of super soft-drug modulators of sphingosine-1-phosphate receptor 1. Bioorg Med Chem Lett. (2018) 28:3255–9. doi: 10.1016/j.bmcl.2018.07.044
41. Gottlieb SL, Gilleaudeau P, Johnson R, Estes L, Woodworth TG, Gottlieb AB, et al. Response of psoriasis to a lymphocyte-selective toxin (DAB389IL-2) suggests a primary immune, but not keratinocyte, pathogenic basis. Nat Med. (1995) 1:442–7. doi: 10.1038/nm0595-442
42. Yawalkar N, Karlen S, Hunger R, Brand CU, Braathen LR. Expression of interleukin-12 is increased in psoriatic skin. J Invest Dermatol. (1998) 111:1053–7. doi: 10.1046/j.1523-1747.1998.00446.x
43. Lee E, Trepicchio WL, Oestreicher JL, Pittman D, Wang F, Chamian F, et al. Increased expression of interleukin 23 p19 and p40 in lesional skin of patients with psoriasis vulgaris. J Exp Med. (2004) 199:125–30. doi: 10.1084/jem.20030451
44. Lowes MA, Kikuchi T, Fuentes-Duculan J, Cardinale I, Zaba LC, Haider AS, et al. Psoriasis vulgaris lesions contain discrete populations of Th1 and Th17 T cells. J Invest Dermatol. (2008) 128:1207–11. doi: 10.1038/sj.jid.5701213
45. Cai Y, Shen X, Ding C, Qi C, Li K, Li X, et al. Pivotal role of dermal IL-17-producing γδ T cells in skin inflammation. Immunity. (2011) 35:596–610. doi: 10.1016/j.immuni.2011.08.001
46. Kamata M, Tada Y. Crosstalk: keratinocytes and immune cells in psoriasis. Front Immunol. (2023) 14:1286344. doi: 10.3389/fimmu.2023.1286344
47. Kondo T, Ohshima T. The dynamics of inflammatory cytokines in the healing process of mouse skin wound: a preliminary study for possible wound age determination. Int J Legal Med. (1996) 108:231–6. doi: 10.1007/bf01369816
48. Cai Y, Xue F, Fleming C, Yang J, Ding C, Ma Y, et al. Differential developmental requirement and peripheral regulation for dermal Vγ4 and Vγ6T17 cells in health and inflammation. Nat Commun. (2014) 5:3986. doi: 10.1038/ncomms4986
49. Schön MP, Erpenbeck L. The interleukin-23/interleukin-17 axis links adaptive and innate immunity in psoriasis. Front Immunol. (2018) 9:1323. doi: 10.3389/fimmu.2018.01323
50. Teunissen MB, Koomen CW, de Waal Malefyt R, Wierenga EA, Bos JD. Interleukin-17 and interferon-gamma synergize in the enhancement of proinflammatory cytokine production by human keratinocytes. J Invest Dermatol. (1998) 111:645–9. doi: 10.1046/j.1523-1747.1998.00347.x
51. Johansen C, Vinter H, Soegaard-Madsen L, Olsen LR, Steiniche T, Iversen L, et al. Preferential inhibition of the mRNA expression of p38 mitogen-activated protein kinase regulated cytokines in psoriatic skin by anti-TNFα therapy. Br J Dermatol. (2010) 163:1194–204. doi: 10.1111/j.1365-2133.2010.10036.x
52. Cai Y, Xue F, Quan C, Qu M, Liu N, Zhang Y, et al. A critical role of the IL-1β-IL-1R signaling pathway in skin inflammation and psoriasis pathogenesis. J Invest Dermatol. (2019) 139:146–56. doi: 10.1016/j.jid.2018.07.025
53. Swindell WR, Beamer MA, Sarkar MK, Loftus S, Fullmer J, Xing X, et al. RNA-seq analysis of IL-1B and IL-36 responses in epidermal keratinocytes identifies a shared myD88-dependent gene signature. Front Immunol. (2018) 9:80. doi: 10.3389/fimmu.2018.00080
54. Zhang G, Yang L, Kim GS, Ryan K, Lu S, O’Donnell RK, et al. Critical role of sphingosine-1-phosphate receptor 2 (S1PR2) in acute vascular inflammation. Blood. (2013) 122:443–55. doi: 10.1182/blood-2012-11-467191
55. Yu H. Sphingosine-1-phosphate receptor 2 regulates proinflammatory cytokine production and osteoclastogenesis. PloS One. (2016) 11:e0156303. doi: 10.1371/journal.pone.0156303
56. Stelling A, Hashwah H, Bertram K, Manz MG, Tzankov A, Müller A. The tumor suppressive TGF-β/SMAD1/S1PR2 signaling axis is recurrently inactivated in diffuse large B-cell lymphoma. Blood. (2018) 131:2235–46. doi: 10.1182/blood-2017-10-810630
57. Zhang Y, Wang H, Lu J, Lv Q, Yun B, Ge Z, et al. Down-regulation of S1PR2 is correlated with poor prognosis and immune infiltrates in cervical squamous cell carcinoma and endocervical adenocarcinoma. Int J Immunopathol Pharmacol. (2023) 37:3946320231178131. doi: 10.1177/03946320231178131
58. Kang J, Lee JH, Im DS. Topical application of S1P(2) antagonist JTE-013 attenuates 2,4-dinitrochlorobenzene-induced atopic dermatitis in mice. Biomol Ther (Seoul). (2020) 28:537–41. doi: 10.4062/biomolther.2020.036
59. Park SJ, Im DS. Blockage of sphingosine-1-phosphate receptor 2 attenuates 2,4-dinitrochlorobenzene-induced atopic dermatitis in mice. Acta Pharmacol Sin. (2020) 41:1487–96. doi: 10.1038/s41401-020-0412-8
60. Thieme M, Zillikens D, Sadik CD. Sphingosine-1-phosphate modulators in inflammatory skin diseases - lining up for clinical translation. Exp Dermatol. (2017) 26:206–10. doi: 10.1111/exd.13174
61. Sikora M, Chrabąszcz M, Waśkiel-Burnat A, Rakowska A, Olszewska M, Rudnicka L. Claudin-3 - a new intestinal integrity marker in patients with psoriasis: association with disease severity. J Eur Acad Dermatol Venereol. (2019) 33:1907–12. doi: 10.1111/jdv.15700
62. Medzhitov R, Preston-Hurlburt P, Kopp E, Stadlen A, Chen C, Ghosh S, et al. MyD88 is an adaptor protein in the hToll/IL-1 receptor family signaling pathways. Mol Cell. (1998) 2:253–8. doi: 10.1016/s1097-2765(00)80136-7
63. Horng T, Barton GM, Flavell RA, Medzhitov R. The adaptor molecule TIRAP provides signaling specificity for Toll-like receptors. Nature. (2002) 420:329–33. doi: 10.1038/nature01180
64. Di Nardo A, Chang YL, Alimohammadi S, Masuda-Kuroki K, Wang Z, Sriram K, et al. Mast cell tolerance in the skin microenvironment to commensal bacteria is controlled by fibroblasts. Cell Rep. (2023) 42:112453. doi: 10.1016/j.celrep.2023.112453
65. Didovic S, Opitz FV, Holzmann B, Förster I, Weighardt H. Requirement of MyD88 signaling in keratinocytes for Langerhans cell migration and initiation of atopic dermatitis-like symptoms in mice. Eur J Immunol. (2016) 46:981–92. doi: 10.1002/eji.201545710
66. Li C, Maillet I, Mackowiak C, Viala C, Di Padova F, Li M, et al. Experimental atopic dermatitis depends on IL-33R signaling via MyD88 in dendritic cells. Cell Death Dis. (2017) 8:e2735. doi: 10.1038/cddis.2017.90
Keywords: keratinocyte biology, psoriasis, sphingosine 1-phosphate, sphingosine 1-phosphate receptor 2, Th 17 cells
Citation: Masuda-Kuroki K, Alimohammadi S, Lowry S and Di Nardo A (2024) Sphingosine 1-phosphate receptor 2 in keratinocytes plays a key role in reducing inflammation in psoriasis. Front. Immunol. 15:1469829. doi: 10.3389/fimmu.2024.1469829
Received: 24 July 2024; Accepted: 30 August 2024;
Published: 26 September 2024.
Edited by:
Barbara Romano, University of Naples Federico II, ItalyReviewed by:
Anella Saviano, University of Naples Federico II, ItalyCopyright © 2024 Masuda-Kuroki, Alimohammadi, Lowry and Di Nardo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anna Di Nardo, YWRpbmFyZG9AdWNzZC5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.