 Maria Giovanna Desimio1†
Maria Giovanna Desimio1† Daniela Angela Covino
Daniela Angela Covino Caterina Cancrini
Caterina Cancrini Margherita Doria
Margherita Doria- 1Research Unit of Primary Immunodeficiency, Bambino Gesù Children’s Hospital, IRCCS, Rome, Italy
- 2Department of Systems Medicine, University of Rome “Tor Vergata”, Rome, Italy
The Epstein–Barr virus (EBV) is usually acquired during infancy as an asymptomatic infection and persists throughout life in a latent state under the control of the host immune system. However, EBV is associated with various malignant diseases that preferentially develop in immunodeficient individuals. Accumulating evidence suggests an important role for NK cells, though the mechanisms by which EBV evades or triggers NK cell responses are poorly understood. Here, we generated EBV-immortalized lymphoblastoid cell lines stably expressing an inducible form of the BZLF1 early lytic viral protein (LCL-Z) to challenge primary NK cells with EBV+ targets in either the latent or lytic phase of infection. We show that entry into the lytic phase results in drastic downregulation of HLA-E but not HLA-A, -B, and -C molecules and in increased expression of ligands for the activating NKG2D receptor, with MICB being upregulated at the cell membrane and released in a soluble form while ULBP2 and ULBP4 accumulate intracellularly. Furthermore, LCL-Z cells are killed by NK cells in an NKG2D-dependent manner and to a much higher extent during the lytic phase, but HLA-class I molecules constrain killing throughout the viral life cycle; unexpectedly, the antibody-mediated block of the inhibitory NKG2A receptor results in reduced lysis of lytic LCL-Z cells that are nearly devoid of the cognate HLA-E ligand. Accordingly, we show that NKG2A+ NK cell subsets, specifically CD56bright and NKG2A+KIR+CD56dim cells, are those that preferentially respond against cells with lytic EBV replication. Overall, these results shed light on NK/EBV+ cell interactions providing new information for improving NK cell-based immunotherapies to treat EBV-induced diseases.
1 Introduction
Over 95% of the world’s population is infected by the Epstein–Barr virus (EBV), an oncogenic γ-herpesvirus that is transmitted via saliva to naïve B cells in oral lymphoid tissues (1). EBV establishes a latent infection through stepwise restriction of viral protein and non-coding RNA (ncRNA) expression (from type III to 0 latency programs), hence persisting for life in memory B cells in which it may occasionally switch to a lytic replication phase, allowing virus reseeding and transmission to a different host (1, 2). EBV transition from latency to lytic replication is under the control of cellular factors, including signaling pathways, epigenetic regulation, and stress conditions, and of viral gene products, with the immediate-early BZLF1 protein having a key role in the transactivation of most lytic genes that allow the virus to replicate (3). Primary EBV infection normally occurs during infancy without causing symptoms, yet it may lead to infectious mononucleosis (IM), particularly when deferred into puberty; additionally, mostly in individuals with an immunodeficiency caused by co-infections, inborn error of immunity (IEI), or immunosuppressive therapies, EBV is associated with various lymphoproliferative disorders (LPDs) and malignancies affecting B cells or other atypical viral targets, including Burkitt’s lymphoma (BL), Hodgkin’s lymphoma (HL), NK or T cell lymphoma, nasopharyngeal carcinoma (NPC), and gastric carcinoma (GC), overall being responsible for approximately 1.5% of all human tumors (4). EBV-related pathologies arise from impaired or dysregulated immune responses against EBV, from the activity of various viral proteins and ncRNAs that promote tumorigenic transformation and immune evasion, and, possibly, from viral and host factors not yet identified (1, 4–6).
In healthy individuals, EBV-specific CD8 T cells are crucial for viral load suppression during primary infection, then persist as long-lasting memory cells (5). Before development of virus-specific T-cell responses, EBV is challenged by the encounter with innate immune cells, such as natural killer (NK) cells, which are abundant at the site of virus entry and function as the first line of defense against pathogens (7). NK cells are cytotoxic lymphocytes armed with the antigen-independent capacity to kill infected, tumor-transformed, and, in general, “stressed” cells via secretion of lytic granules; such innate response is controlled via the balance between opposite signaling pathways: on the one hand, a negative regulation is exerted by inhibitory receptors, including inhibitory killer immunoglobulin receptors (iKIRs), NKG2A, and LILRB-1, upon engagement with ligands belonging to the HLA-class I protein family; on the other hand, activating receptors, which comprise NKG2D, DNAM-1, natural cytotoxicity receptors (NCRs: NKp30, NKp44, and NKp46), NKG2C, and activating KIRs (aKIRs), mediate NK cell activation following recognition of ligands that are either virally encoded or upregulated on infected/stressed cells (8–10). NK cell-mediated killing also occurs via antibody-dependent cellular cytotoxicity (ADCC) triggered by the CD16 low-affinity Fc receptor or via apoptosis induced by death receptor ligands (TRAIL and CD95-L). Moreover, activated NK cells secrete cytokines, particularly IFN-γ and TNF, and chemokines that attract and stimulate T cells and innate cells, thus promoting broad immune responses (8, 10). Most receptors are expressed in NK cells in a random manner, generating a remarkable phenotypic and functional NK cell heterogeneity (11). In peripheral blood, two NK cell populations can be distinguished: a minor subset (~10%) consists of CD56brightCD16−/+ cells (referred to as CD56bright), which are efficient cytokine producers and commonly considered as immature precursors of the most abundant highly cytotoxic CD56dimCD16+ subset (referred to as CD56dim) (12). Phenotypically, CD56bright cells are characterized by the expression of NKG2A and various activating receptors, while the CD56dim subset comprises a heterogeneous population of NK cells with different degrees of differentiation, which implies the gradual loss of NKG2A and sequential acquisition of iKIRs and the CD57 maturation marker (13, 14). Additionally, among CD56dim cells, a population of NKG2C+ cells with features of adaptive immunity, also referred to as Memory-Like, has been identified in HCMV-seropositive individuals (15).
A large body of clinical and experimental evidence supports the important role of NK cells in the control of EBV during primary infection and beyond, as we recently reviewed (16). First, discovery of six isolated NK cell deficiencies as well as impaired NK cell development or function in more than 50 different IEIs illustrated the critical role of these cells in anti-viral defense, particularly in the prevention of severe infection with EBV and other herpesviruses (17). Second, reduced NK cell frequency and/or cytotoxicity were associated with EBV+ LPDs and tumors by comparison with equivalent EBV− diseases, pointing towards a key role of NK cells in restraining EBV-related pathogenesis (18–22). Furthermore, studies on acute EBV infection in IM patients and in a humanized mouse model of IM showed the expansion of early differentiated CD56dimNKG2A+ NK cells with effective anti-EBV activity before initiation of virus specific T-cell responses (23, 24); additionally, NK cell depletion experiments in mice demonstrated that NK cells not only suppressed EBV viremia and prevented EBV-induced malignancies, but also reduced lymphocytosis and the consequent IM symptoms by cytotoxic clearance of activated CD8 T cells (23). Finally, studies in EBV-infected individuals pointed at the anti-viral potential of distinct NK cell subsets, specifically mature CD56dimNKG2A+CD57+ cells (25) and tonsillar CD56brightIFNhigh cells (7).
The knowledge on NK cell biology has greatly increased in recent years, leading to the development of novel immunotherapies such as adoptive transfer of potentiated NK cells, which offer exciting promise for the treatment of EBV-associated diseases. However, for successful clinical applications, current understanding of the mechanisms that regulate the capacity of NK cells to kill EBV+ cell targets should be improved. Indeed, several experimental studies have shown the capacity of various EBV proteins and ncRNAs to modulate the expression of ligands for both inhibitory and activating NK receptors, though results are often controversial and little is known on the consequences for NK cell-mediated killing [reviewed in Ref (16).]. As for HLA-class I molecules, these are highly expressed on latently EBV-infected cells through the activity of viral factors such as LMP1 and then they are downregulated by various lytic proteins acting at different levels (from gene transcription to protein trafficking) once the virus has entered the productive replication program; hence, the interaction of HLA-class I on EBV+ cells with cognate iKIRs inhibiting NK cell activation could be either efficient or impaired depending on the latent or lytic phase, respectively. This model is in line with NK cell-mediated killing being more efficient against targets with lytic EBV replication reported in studies in vitro and in a mouse model (23, 24, 26), yet is at odds with the lack of KIR expression on those NK cell subsets that react against EBV in vivo during IM and upon in vitro challenge with EBV-infected targets (24). Another controversial matter concerns EBV modulation of ligands for NK cell activating receptors, particularly those recognized by NKG2D (MICA/MICB and ULBP1–6 family of proteins, referred to as NKG2DLs) and DNAM-1 (CD112 and CD155 proteins, referred to as DNAM-1Ls). Expression of cell-surface NKG2DLs, which is normally highly restricted but can be induced in response to infection, transformation, or other cellular stress (27, 28), was described on latently EBV-infected cells in some reports (24, 29, 30), even though there is no consensus on the type of ligand expressed (i.e., MICA, MICB, ULBP1, or ULBP4) and, in other studies, none of the NKG2DLs was found (26, 31); a few studies also analyzed NKG2DLs after entry in the EBV lytic phase, showing disparate results ranging from further MICA up-modulation (24), de novo induced ULBP1 expression (26), or complete absence of NKG2DLs (31). Analogously, contrasting evidence was provided for DNAM-1Ls that, depending on the report, were either absent in all phases of the EBV life cycle (31) or induced during lytic replication (24, 26). Furthermore, experimental studies demonstrated the capacity of individual EBV proteins or ncRNAs to either repress (e.g., mir-BART2/7, LMP2, and EBNA1) or induce (e.g., LMP1 and BZLF1) specific NKG2DL expression, without an apparent separation of inhibitory and stimulatory activities relative to the viral replication phases [reviewed in Ref (16).]. Overall, a growing number of disparate lines of evidence suggest that EBV has evolved strategies to modulate the expression of ligands for NK cell receptors but further investigation is clearly needed to understand which receptor/ligand axes effectively mediate the capacity of NK cells to control EBV infection throughout the viral life cycle.
Here, we set up EBV-immortalized lymphoblastoid cell lines (LCLs) with stable expression of an inducible form of BZLF1 to challenge primary NK cells with targets in either the latent or lytic phase of EBV infection, and investigated killing efficiency, NK cell receptor/ligand interactions, and which subset of NK cells was most responsive.
2 Materials and methods
2.1 Cells
Primary NK cells and EBV-immortalized LCLs were maintained in complete RPMI 1640 medium (cRPMI) and 293T cells were maintained in complete Dulbecco’s modified Eagle’s medium (cDMEM) both supplemented with 10% fetal bovine serum, 2 mM L-glutamine, and 100 units/mL penicillin–streptomycin (all from Euroclone). PBMCs of healthy subjects were obtained by separation on Ficoll gradients of buffy coats from a donor bank. Ethical committee approval and written informed consent from all participants were obtained, in accordance with the Declaration of Helsinki. Primary NK cells were isolated from PBMCs by negative selection with the EasySep NK-cell Enrichment Kit (Stem Cell Technologies) according to the manufacturer’s protocol. The purity (~95%) of isolated NK (CD3-CD56+CD16−/+) was assessed by immunolabeling and flow cytometry analysis. LCLs were generated by infecting PBMCs with the laboratory strain B95-8 (ATCC) of EBV according to standard techniques, and maintained at ~0.5 × 106/mL by passaging every 4 days and adding fresh cRPMI once a week. Stably transduced LCL-Z cells were maintained in cRPMI supplemented with 1 μg/mL of puromycin (Sigma-Aldrich/Merck) and, to induce EBV lytic replication, 200 nM of 4-hydroxytamoxifen (HT) diluted in cRPMI from a 25 mM stock in methanol (Sigma-Aldrich/Merck).
2.2 Generation of LCL-Z cell lines
A lentivirus was generated by a standard protocol based on three DNA vectors including the p8272 plasmid, a kind gift of Paul J. Farrell (Imperial College Faculty of Medicine, London) that codes for the early lytic EBV protein BZLF1 fused to the HT-responsive mutated estrogen receptor hormone-binding domain and for the puromycin resistance enzyme (32), the packaging (psPAX2), and envelope-encoding (pVSV-G) plasmids, which were transfected into 293T cells using Lipofectamine™ 3000 (Thermo Fisher Scientific) according to the manufacturer’s instructions. Forty-eight hours after transfection, the lentivirus-containing medium was harvested, frozen in aliquots and titrated by serial dilution in 293T cell cultures followed by flow cytometry analysis of BZLF1+ cells after 2 days. Finally, LCLs were subjected to spin infection [2,000g for 60 min at room temperature in the presence 8 μg/mL polybrene (Sigma-Aldrich/Merck)] with lentivirus at a multiplicity of infection (m.o.i.) of 25, then cultivated for 24 h before replacing the medium with fresh cRPMI. Three days later, 1 μg/mL of puromycin was added to cultures to select stably transduced cell lines, referred to as LCL-Z cells, expressing the inducible BZLF1-HT protein.
2.3 Measurement of EBV replication by qPCR
To measure EBV replication, total DNA was extracted from LCL-Z cells treated or not with 200 nM HT for 24, 48, and 72 h using the QIAmp DNA Blood Kit (Quiagen) and analyzed by qPCR using SensiFAST SYBR Green PCR master mix (Bioline) using primers specific for the viral BALF5 gene and the PIK3R1 control gene (BALF5/F, 5′-CGGAGTTGTTATCAAAGAGGC-3′; BALF5/R, 5′-CGAGAAAGACGGAGATGGC-3′; PIK3R1/F, 5′-TTATCAAGCTCGTGGAAGCC-3′; PIK2R1/R, 5′-TGTAAACGGCTGCTGGAAT-3′). The cycling conditions were as follows: 95°C for 2 min, followed by 40 cycles of 95°C for 5 s and 60°C for 30 s. Quantitative PCR was performed using the Applied Biosystems 7300 Real-Time PCR System. The increase of EBV replication initiated by HT-activated BZLF1 over spontaneous replication was evaluated by dividing BALF5 DNA amounts by those measured in non-treated control samples (hence set to 1).
2.4 Flow cytometry
In all experiments except the cytotoxicity assays, cell viability was assessed by using the LIVE/DEAD fixable NEAR-IR dead cell stain kit according to the manufacturer’s protocol (Thermo Fisher Scientific). To label cell-surface molecules, cells were incubated for 20 min at 4°C with specific mouse monoclonal antibodies (mAbs). To detect intracellular molecules, cells were fixed with 1% paraformaldehyde (PFA) for 10 min, then permeabilized with Permeabilizing Solution 2 (BD Pharmingen) for 10 min before incubation at room temperature for 30 min with specific mAbs. For unconjugated mAbs, a further incubation with AF647-coniugated goat anti-mouse IgG (GAM) (Thermo Fisher Scientific) was performed. Immunolabeled cells resuspended in 1% PFA were acquired on a Cytoflex (Beckman Coulter). Positive cell gating was set using fluorescence minus one control (FMO). The specific mean fluorescence intensity (MFI) for each labeled marker was calculated by subtrac8ng the value obtained with isotype control antibody. Data analyses were performed using Kaluza (Beckman Coulter).
The following mAbs were used: CD3/AF700 (UCHT1), CD56/PerCpCy5.5 (B159), and CD16/BV510 (3G8) from BD Pharmingen; NKG2A/FITC and NKG2C/PE from Miltenyi Biotec; HLA-E (3D12) and CD57/PE-Cy7 (TB01) from eBioscience; CD16/AF700 (3G8), NKG2D/BV785 (1D11), KIR2DL1/S1/S3/S5/APC (HP-MA4), KIR2DL2/L3/S2/APC (DX27), KIR3DL1/APC (DX9), CD155 (SKII.4), CD112 (TX313), and CD48/APC (BJ40) from BioLegend; MICA (AMO1) and ULBP3 (CUMO3) from BamOmaB; MICB (MAB1599), ULBP1 (MAB1380), and ULBP2/5/6 (MAB1298) from R&D Systems; and ULBP4 (6E6) and BZLF1/AF488 (BZ1) from Santa Cruz Biotechnology; HLA-A, -B, and -C (W6/32) were kindly provided by Patrizio Giacomini (Istituto Regina Elena, Rome, Italy).
2.5 Enzyme-linked immunosorbent assay
LCL-Z cells were plated at 2.5 × 106/mL in cRPMI alone or containing 200 nM HT and exposed 25 μM MMPI-III (Calbiochem/Merck) or equivalent amounts of its solvent (DMSO). After 48 h, culture medium was collected and analyzed by enzyme-linked immunosorbent assay (ELISA) to measure the concentration of sMICA and sMICB (from R&D Systems) according to the manufacturer’s instructions and sULBP2 following a previously described procedure (33).
2.6 NK cell lysis assay
Flow cytometry-based cytotoxicity assays were performed using LCL-Z cells treated with HT for 24, 48, and 72 h as targets and purified NK cells pre-activated for 18 h with 500 IU/mL IL-2 (Peprotech) as effectors. Briefly, LCL-Z cells were labeled with Cell Proliferation Dye eFluor™ 450 (eBioscience, Thermo Fisher Scientific) according to the manufacturer’s instructions, washed twice, and then seeded in cRPMI either alone (spontaneous lysis control) or with effector cells at an effector-to-target cell (E:T) ratio of 2:1 for 4 h. Cells in the co-culture were collected and labeled with 7-aminoactinomycin D (7-AAD; Sigma-Aldrich/Merck) for 20 min, fixed/permeabilized, stained for intracellular BZLF1, and analyzed by flow cytometry. The percentage of specific lysis of target cells (gated as eFluor450+) was calculated as follows: 100 × (%7-AAD+ target cells in sample − basal %7-AAD+ target cells)/(100 − basal %7-AAD+ target cells). In blocking experiments, prior to co-culture with targets, NK cells were incubated for 30 min at room temperature with saturating amounts of blocking antibodies anti-NKG2D (1D11; R&D Systems) and anti-DNAM-1 (11A8; BioLegend), both used at 1 μg/106 cells, anti-NKG2A (Z199; Beckman Coulter) used at 3 μg/106 cells, or equivalent amounts of purified IgG as control. To block HLA class I molecules, saturating amounts of anti-HLA-class I IgM clone A6136 (1:20 dilution of the A6136 hybridoma supernatant; kindly provided by Lorenzo Moretta, Bambino Gesù Children’s Hospital, Rome, Italy) were added to the medium of the 4-h co-cultures.
2.7 Degranulation of NK cell subsets
Effectors and targets were prepared as for the lysis assay; specifically, IL-2-stimulated NK cells were seeded either alone (no target control) or together with targets (eFluor450-labeled LCL-Z cells pre-exposed or not to HT for 48 h) at an E:T ratio of 2:1 in cRPMI. The medium was supplemented with a 1:200 dilution of either anti-CD107a/BV605 or control mouse IgG1/BV605 (BD Biosciences). Cells were cultivated for 6 h, with the addition of Golgi Stop (1:1500; BD Biosciences) and Brefeldin A (10 μg/mL; Sigma-Aldrich/Merck) after the first hour, then subjected to staining for cell-surface NK markers (i.e., CD16, CD56, NKG2A/C/D, CD57, NKG2A, and KIR2DL1/2DL2/3DL1) and analyzed by flow cytometry to identify maturation subsets and their degranulation efficiency (i.e., %CD107a+) within gated (eFluor450-) NK cells.
2.8 Statistical analysis
All experiments have been performed independently at least three times. GraphPad Prism 6.0 software was used to perform all statistical analyses. A value of p < 0.05 was considered statistically significant.
3 Results
3.1 Activation of inducible BZLF1 efficiently initiates lytic EBV replication in transduced LCLs
Very useful for experimental studies is the capacity of EBV, specifically the B95-8 prototype strain, to transform in vitro primary B cells into LCLs that express the full latency program (type III: six EBNA proteins, LMP1, LMP2, and various ncRNAs) and grow indefinitely in culture. Here, to study NK cell interactions with EBV-infected cells in both latent and lytic phases of the virus life cycle, we generated healthy donor-derived LCLs with constitutive expression of the immediate-early lytic EBV protein BZLF1 that is kept inactive by fusion to an HT-responsive portion of the estrogen receptor, herein referred to as LCL-Z cells. As measured by flow cytometry in a 72-h time course, the low frequency of BZLF1+ cells that spontaneously enter the lytic phase in LCL-Z cultures rapidly rises and increases over time upon the addition of HT (Figures 1A, B), in line with BZLF1 functioning as the key switch between latent and lytic replication. As expected, non-transduced LCL cultures do not respond to HT treatment and present a very low percentage of BZLF1+ cells analogously to untreated LCL-Z cultures (Figure 1B). The increment of lytic EBV replication was confirmed by measuring viral DNA that increased in LCL-Z cells upon HT exposure for 24, 48, and 72 h by 5-, 10-, and 15-folds, respectively, as compared to untreated cells (Figure 1C).
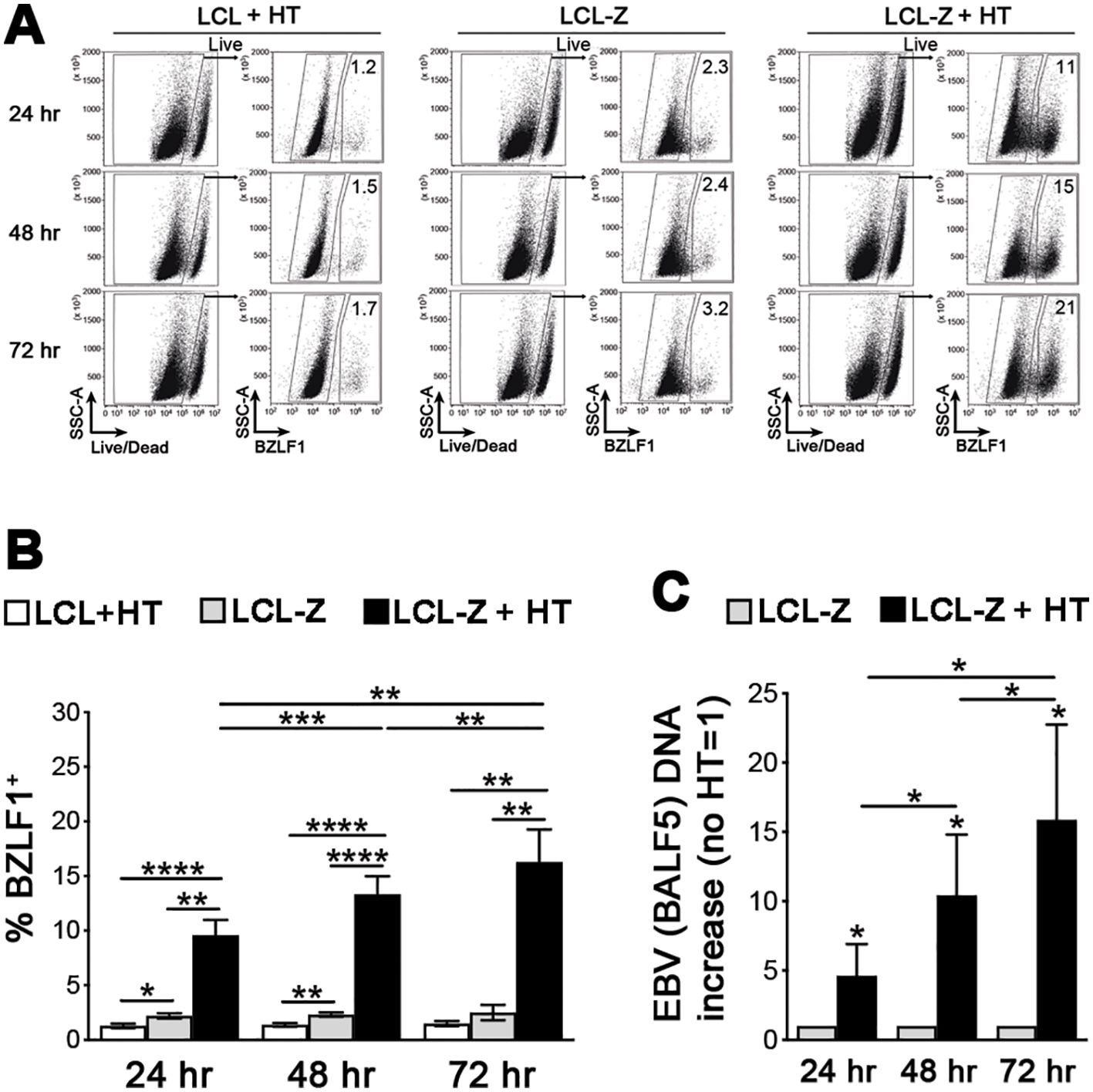
Figure 1. BZLF1 activation triggers efficient EBV lytic replication in LCL-Z cells. (A) Representative flow cytometry analysis of BZLF1 expression in LCL-Z cells after culture with or without 400 nM HT for 24, 48, and 72 h; original non-transduced cells treated with HT (LCL + HT) are also shown. (B) Bar graphs with dot plots show the mean ± SEM percentage of BZLF1+ cells in LCLs derived from six different donors that were stably transduced with BZLF1 and exposed or not to HT for 24, 48, and 72 h (LCL-Z and LCL-Z + HT, respectively); in parallel, non-transduced LCL-Z cells were exposed to HT and analyzed. (C) LCL-Z cells treated or not with HT for the indicated time were subjected to qPCR to quantify EBV viral DNA. Each bar represents the mean and standard deviation for the viral DNA level (e.g., BALF5 polymerase gene) after normalization, calculated by dividing for the DNA level in the untreated sample for six independent experiments. *p < 0.05, **p < 0.01, ***p<0.001, ****p<0.0001 by paired t-test.
3.2 The switch to lytic EBV replication induces the expression of MICB and downregulates HLA class I molecules, particularly HLA-E
Then, we used the inducible LCL-Z cell system to analyze several ligands for NK cells during EBV latency and upon switch to lytic replication, with a special focus on those molecules that have been controversially implicated in EBV-NK cell interaction in previous studies. Specifically, expression of NKG2DLs (MICA/B and ULBP1–4), DNAM-1Ls (CD112 and CD155), HLA-A, -B, and -C, HLA-E, and CD48 was measured by flow cytometry in both BZLF1− and BZLF1+ LCL-Z cells after exposure to HT. Figure 2A shows representative results illustrating that, among NKG2DLs, MICB and, to a lesser extent, ULBP1, ULBP2, and ULBP4 were expressed on BZLF1− cells in LCL-Z as well as in control non-transduced LCL cultures, while DNAM-1Ls were absent. This pattern was maintained in BZLF1+ LCL-Z cells with the exception of MICB expression that was significantly increased in terms of positive cells (18 ± 4 vs. 24 ± 5% MICB+, mean ± SEM) as well as MFI (1,400 ± 277 vs. 1,880 ± 300 MICB MFI) and of a trend towards downregulation of ULBP2 by comparing with BZLF1− counterparts in six independent cultures (Figures 2A, B). As to HLA molecules, HLA-A, -B, and -C levels were modestly reduced in BZLF1+ as compared with BZLF1− cells in LCL-Z cultures (75% ± 6% vs. 70% ± 6% HLA-A, -B, and -C+ and 26,670 ± 9,990 vs. 17,550 ± 5,170 HLA-A, -B, and -C MFI), whereas HLA-E was drastically downregulated in BZLF1+(35% ± 4% vs. 8% ± 1% HLA-E+ and 3,540 ± 1,160 vs. 374 ± 89 HLA-E MFI). A partial loss of HLA-E was also evident in BZLF1− cells of HT-induced LCL-Z cultures (as compared to BZLF1− LCLs, Figure 2A), suggesting that HLA-E expression was being affected by EBV starting from the very early phases of latency exit, when BZLF1 amounts are too low for positive cell gating by flow-cytometry. On the other hand, CD48 was expressed by all cells to a level that did not vary between BZLF1+ and BZLF1− cells (Figures 2A, B). Results are shown at 48 h post-treatment with HT, but phenotypic changes start earlier (24 h) and are maintained at 72 h (data not shown).
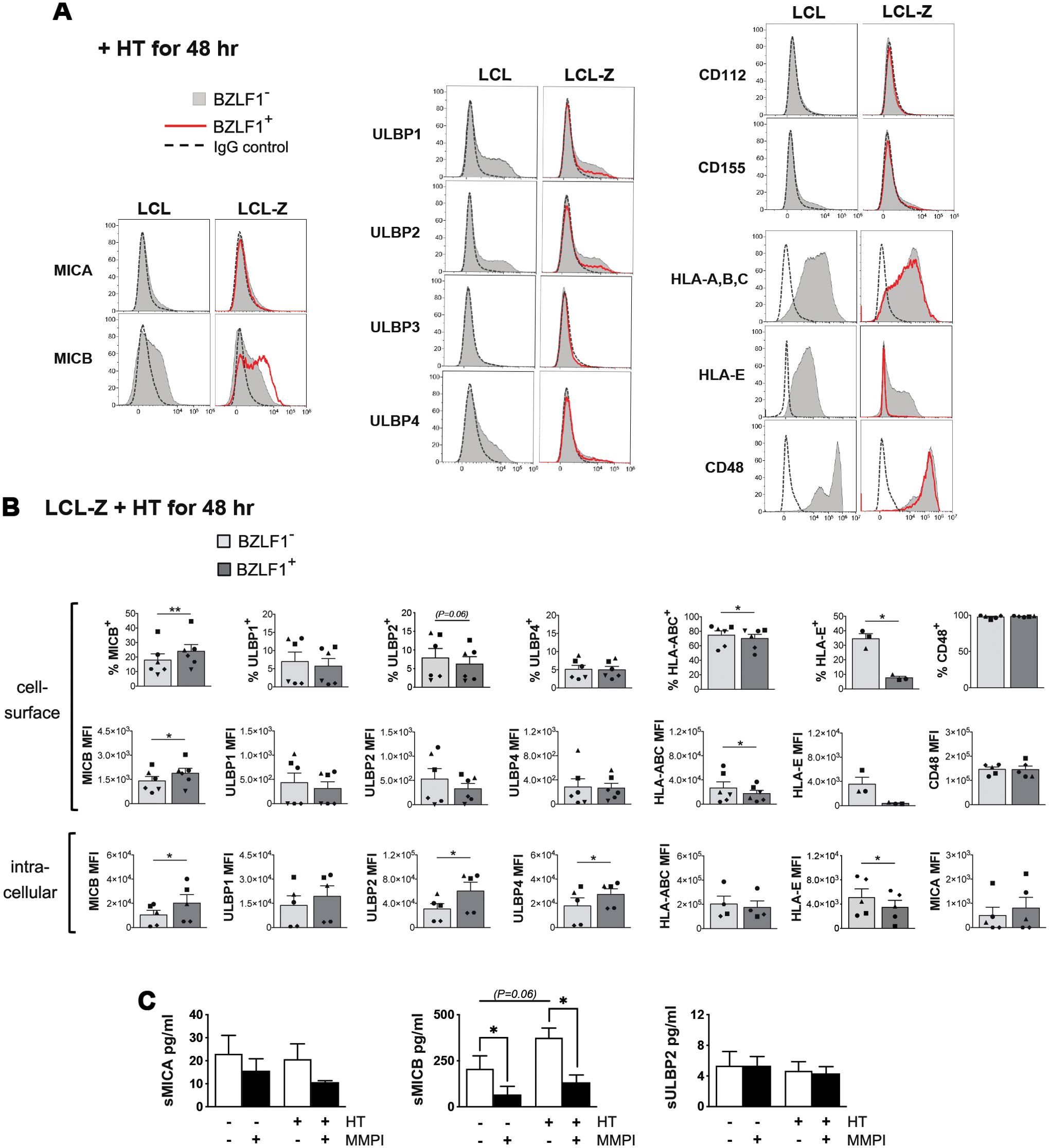
Figure 2. Expression of ligands for NK receptors during the latent and lytic phases of the EBV life cycle. (A) Flow cytometry histograms comparing the levels of NKG2DLs (MICA, MICB, and ULBP1–4), DNAM-1Ls (CD112 and CD155), HLA-A, -B, and -C, HLA-E, and CD48 on gated BZLF1+ (red line) and BZLF1− (filled gray) cells in LCL and LCL-Z cultures treated with HT for 48 h are shown for a representative experiment. Staining with control Ig isotype is depicted as a dashed line. (B) Bar graphs with dot plots depict the mean ± SEM percentage of positive cells and mean fluorescence intensity (MFI) at the cell surface (top panels) and MFI obtained by intracellular staining (bottom panels) for NK receptor ligands expressed in BZLF1− and BZLF1+ LCL-Z cells exposed to HT for 48 (h) Five or six different LCL-Z cells (each represented with a distinctive symbol) were analyzed for each molecule. Comparisons between BZLF1− and BZLF1+ cells were performed by paired t-test. (C) Soluble NKG2DLs (sMICA, sMICB, and sULBP2) were measured in the medium of LCL-Z cells cultivated for 48 h without treatments or in the presence of HT and/or MMPI. Bars show mean ± SEM from three independent experiments and statistics was performed using paired t-test. Significant differences are indicated (*p < 0.05, **p < 0.01) as well as nearly significant p-values.
To further investigate the impact on NK cell ligands of lytic EBV replication, HT-exposed LCL-Z were labeled after permeabilization for NKG2DL or HLA molecules to measure their intracellular expression levels (Figure 2B, lower panels). Of note, the intracellular levels of MICB as well as of ULBP2 and ULBP4 were increased in BZLF1+ cells as compared with BZLF1− cells, while expression of other ligands was very low/undetectable in both cell populations; moreover, a significant reduction of HLA-E but not HLA-A, -B, and -C levels within BZLF1+ cells was found (Figure 2B). Finally, since NKG2DLs can be regulated via proteolytic shedding at the cell membrane, the presence of soluble MICA, MICB, and ULBP2 (referred to as sMICA, sMICB, and sULBP2) was analyzed by ELISA using the cell culture medium of LCL-Z exposed or not to HT for 48 h, with or without a broad-spectrum matrix metalloproteinase inhibitor (MMPI). Results showed that, while sMICA and sULBP2 were barely detectable in all conditions, sMICB accumulated in an MMP-dependent manner in LCL-Z cultures and to a higher extent in cultures with lytic EBV replication (Figure 2C).
Overall, these data demonstrate that NKG2DLs, but not DNAM-1Ls, are modulated during EBV infection; specifically, cell-surface MICB is expressed and can be released in a soluble form during latency and to a higher extent in cells with lytic EBV replication, whereas other NKG2DLs are very low or absent at the cell membrane in all phases of the viral life cycle, though ULBP2 and ULBP4 accumulate within cells in the lytic stage. In addition, all cell-surface HLA molecules are downregulated in cells with lytic EBV replication, with a much stronger effect on HLA-E that nearly disappears as compared with a modest reduction of HLA-A, -B, and -C molecules.
3.3 EBV entry into the lytic phase sensitizes LCLs to NK cell-mediated killing
Next, we used HT-stimulated LCL-Z to assess the susceptibility to NK cell-mediated killing of infected cells in both latent and lytic phases of the EBV life cycle. To this end, NK cells purified from healthy donors and activated with IL-2 served as effectors (E) in a 5-h flow cytometry-based cytotoxicity assay in which LCL-Z treated with HT for 24, 48, and 72 h were used as targets (T) at an E:T ratio of 2:1 (the gating strategy is shown in Figure 3A). We found that LCL-Z were rather poorly killed by NK cells during the latent phase at each post-stimulation time point, with a specific lysis of BZLF1− cells corresponding to 11.5% ± 3.3%, 6.1% ± 0.4%, and 7.2% ± 1.4% (mean ± SEM; Figure 3B). Conversely, the EBV switch to lytic replication significantly enhanced NK cell-mediated clearance, resulting in 34.5% ± 4.4%, 37.9% ± 6.1%, and 28.5% ± 6.6% lysis of BZLF1+ cells (Figure 3B). In these experiments, LCL-Z targets were killed by allogeneic NK cells, but analogous results can be obtained in autologous settings (Supplementary Figure S1).
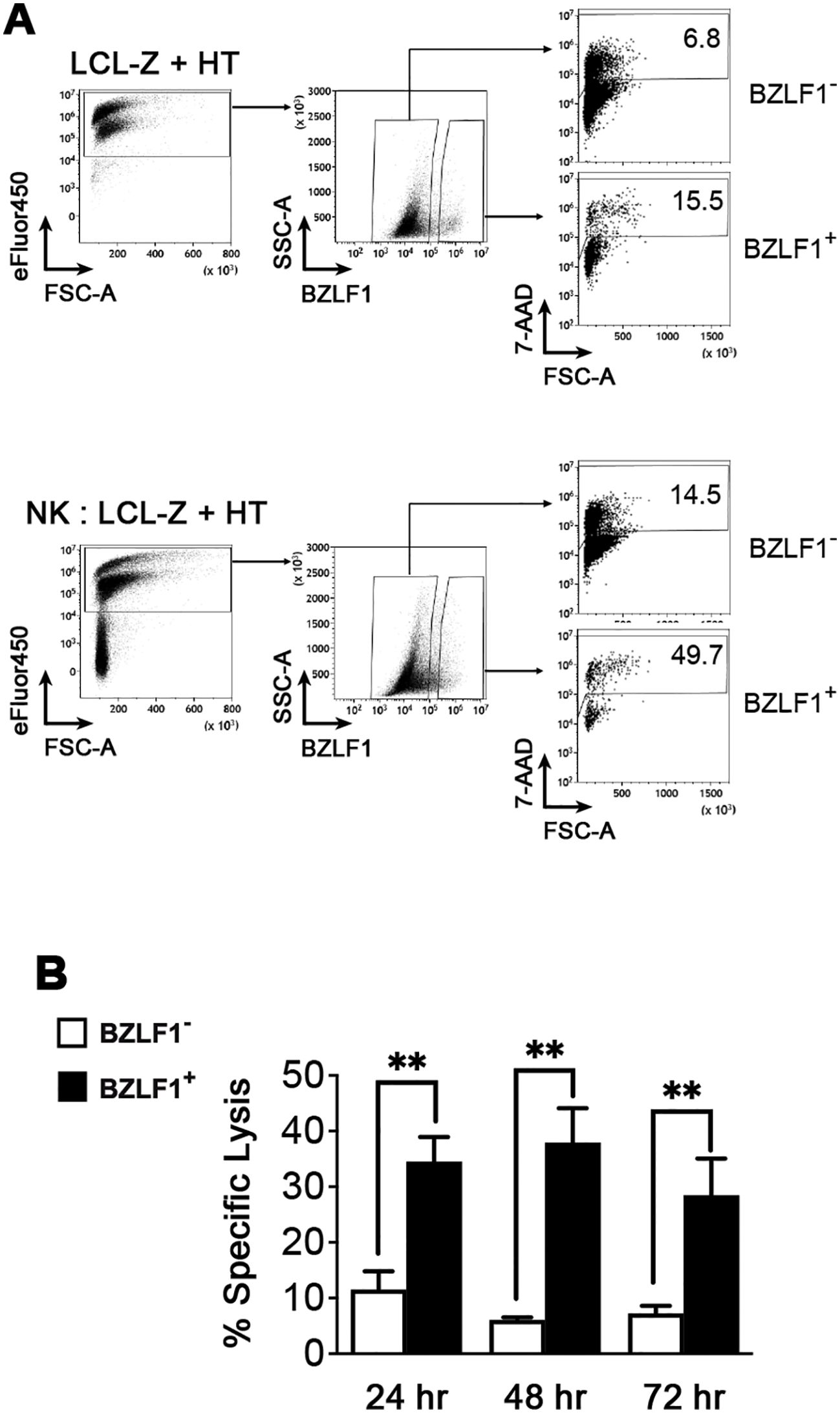
Figure 3. Sensitivity of LCLs with latent or lytic EBV infection to NK cell-mediated killing. A killing assay was set up in which LCL-Z targets stimulated with HT for 48 h were labeled with eFluor450 and then cultivated for 5 h with primary NK cells pre-activated with IL-2 at an E:T ratio of 2:1. The cultures were analyzed by flow cytometry to measure the frequency of dead (7-AAD+) cells among eFluor450+ targets gated as BZLF1− and BZLF1+. (A) The gating strategy and percentages of dead cells are shown for a representative sample of LCL-Z + HT targets cultivated separately (top panels) or together with NK (bottom panels). (B) The percentage of NK cell-mediated specific lysis (mean ± SEM) was measured in eight different LCL-Z cell lines. **p < 0.01 by paired t-test.
3.4 MICB upregulation on LCLs translates into NKG2D-mediated killing by NK cells
To examine the receptor/ligand pairs that mediate NK cell recognition and killing of LCLs with both latent and lytic EBV infection, lysis assays were performed in the presence of blocking antibodies directed against NKG2D or DNAM-1; in addition, pan-HLA class I specific antibody (IgM clone A6136) was added or not during the lysis assay. Blocking NKG2D but not DNAM-1 resulted in decreased NK cell-mediated killing of both BZLF1− and BZLF1+ cells (Figure 4), consistent with MICB expression and the absence of DNAM-1Ls on these cell targets described herein (Figures 2A, B). On the other hand, the addition of anti-HLA class I antibody resulted in a higher cytotoxicity against not only BZLF1− cells but also BZLF1+ cells, which display reduced HLA-A, -B, and -C and nearly absent HLA-E on their membrane (Figures 2A, B), indicating that residual expression of class I molecules on cells undergoing lytic EBV replication is sufficient for engaging inhibitory NK receptors, hence constraining activation of NK cell cytotoxicity (Figure 4). Finally, antibody-mediated blocking of NKG2A, an inhibitory receptor engaged by HLA-E that has been implicated in anti-EBV NK cell responses in previous in vivo and in vitro studies (24, 34, 35), had no effect on BZLF1− cell killing but significantly reduced the lysis of BZLF1+ cells (Figure 4), suggesting a positive role for NKG2A in the capacity of NK cells to clear LCLs undergoing lytic EBV replication.
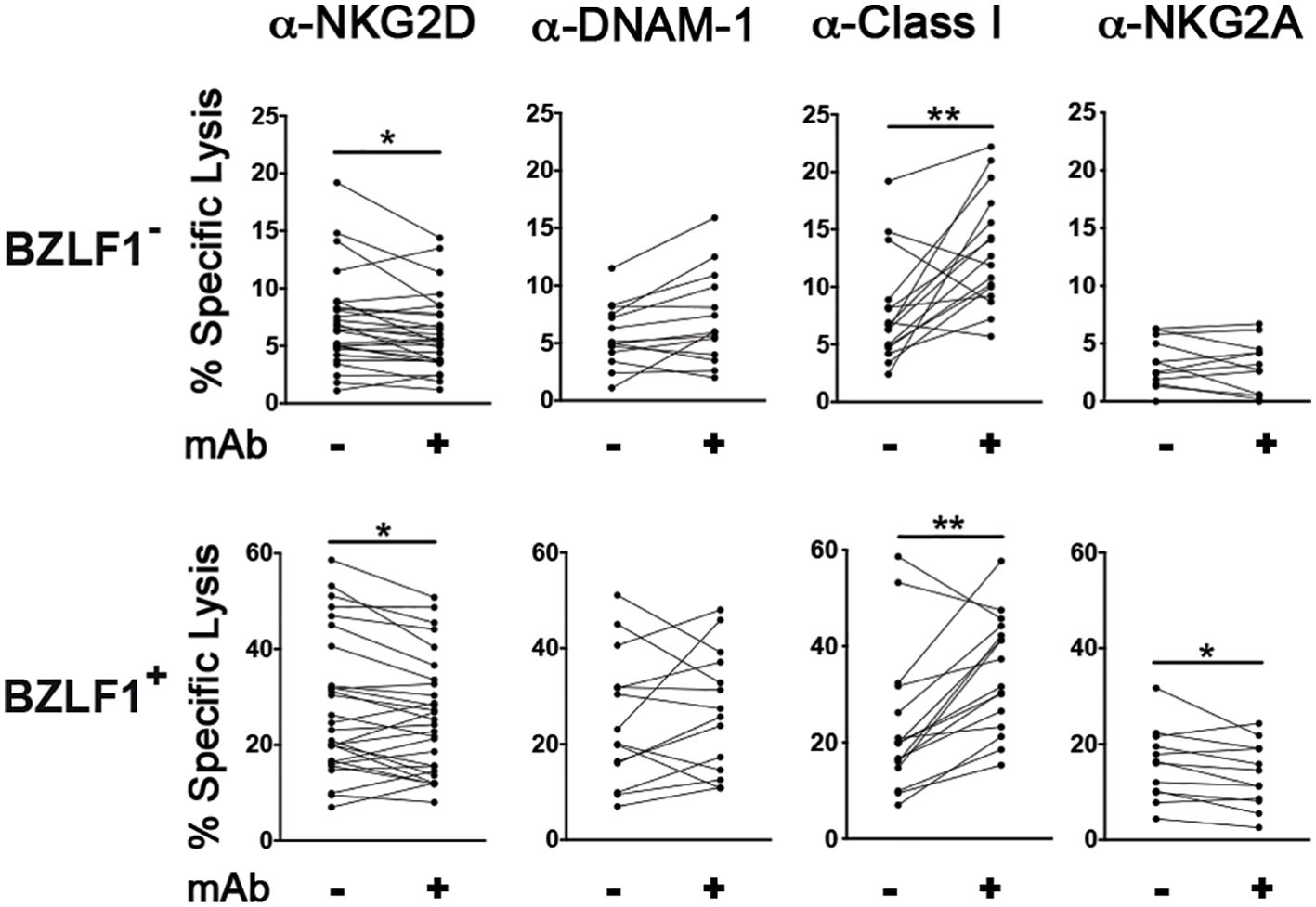
Figure 4. Effect of blocking antibodies on NK cell-mediated killing of LCL targets with latent or lytic EBV infection. Multiple HT-treated LCL-Z cell lines were employed as targets in NK lysis assays described in Figure 1 in which antibodies blocking NKG2D (n = 28), DNAM-1 (n = 14), Class I molecules (n = 16), or NKG2A (n = 11) were used. The percentage of specific lysis was measured for both BZLF1− and BZLF1+ targets. Statistics was carried out using paired Wilcoxon (panels with anti-HLA-Class I blocking antibody) and t-test for non-parametric and parametric distributions, respectively (*p < 0.05, **p < 0.01).
3.5 CD56bright and NKG2A+KIR+CD56dim NK cell subsets efficiently respond against LCLs with lytic EBV replication
The vast majority of IL-2-activated NK cells express NKG2D and hence have the potential to recognize and kill MICB+ LCL targets, whereas NKG2A is expressed by CD56bright cells and subsets of CD56dim cells corresponding to nearly half of the NK cell population (36); specifically, 44% ± 8% of NK cell samples tested in the present study were NKG2A+. Therefore, we thought to investigate which subset of NK cells reacted against LCLs with and without EBV reactivation by comparing the degranulation activity (in terms of CD107a expression) of IL-2-stimulated NK cells that have been cultivated for 5 h in medium alone (no targets) or with autologous LCL-Z pre-exposed or not to HT, and then gated into various maturation subsets by flow cytometry. The gating strategy separated CD56bright cells and five subsets of CD56dim cells further dissected in NKG2A+KIR− (referred to as Early-Differentiated, ED), NKG2A+KIR+, NKG2A−KIR−, NKG2A−KIR+CD57+ (referred to as Mature, M), and NKG2A−KIR+CD57+NKG2C+ (Memory-Like, ML). Conventionally, CD56bright cells comprise both CD16− and CD16+ cells while CD56dim cells are all CD16+; however, herein CD16− cells were also included in the CD56dim gate because we observed a partial loss of CD16 expression in CD56dim cells after exposure to LCL-Z targets, likely consequent to CD16 shedding by activated NK cells (37), while other markers were largely maintained (Supplementary Figure S2). By analyzing the entire population of IL-2-stimulated NK cells (CD56bright + CD56dim cells), their basal degranulation modestly increased upon co-culture with LCL-Z cells but it was significantly higher if LCL-Z were previously exposed to HT (Figure 5A), in accord with killing data (Figure 3). Moreover, we found a general trend towards the higher frequency of CD107a+ in all gated subsets of NK cells challenged with LCLs as compared with NK cells cultured without targets (Figure 5B); of note, HT-exposed LCL-Z targets induced a statistically significant increment of CD56bright and NKG2A+KIR+ cells within the CD107a+ population, indicating that these NK subsets are those that preferably respond against cells with lytic EBV replication. Results also showed a higher degranulation of the more differentiated M and ML subsets upon challenge with LCL-Z + HT targets, although not in one tested donor who also had the lowest overall degranulation activity (Figure 5A, diamond symbol) despite the unvarying NK cell phenotype and maturation pattern (Supplementary Figure S3).
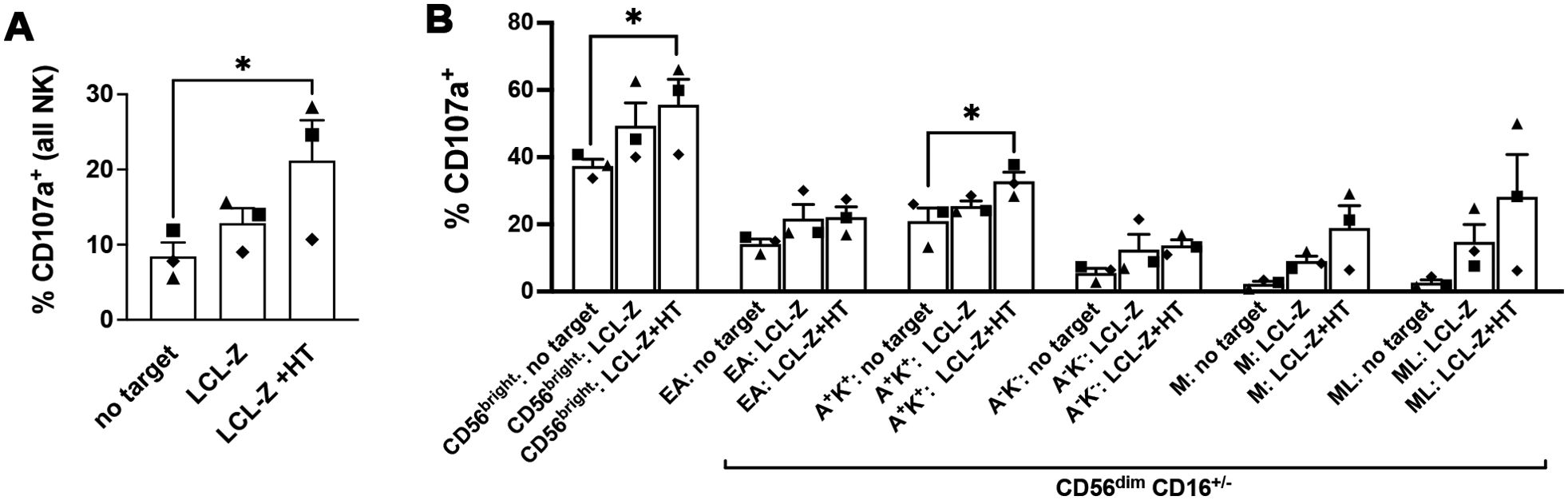
Figure 5. Frequencies of NK cells responding to LCLs with and without lytic EBV activation. Primary IL-2-stimulated NK cells were cultivated for 5 h alone or in the presence of autologous LCL-Z cells pre-treated or not with HT for 48 h, then analyzed by flow cytometry to measure expression of the CD107a degranulation marker. (A) Frequencies of CD107a+ NK cells are reported for the three culture conditions (no targets, with LCL-Z, or HT-treated LCL-Z). (B) Analysis of the distribution of gated CD107a+ NK cells into maturation subsets including CD56bright and five CD56dim subsets identified as NKG2A+KIR− (Early-Differentiated, ED), NKG2A+KIR+ (A+K+), NKG2A−KIR− (A-K-), NKG2A−KIR+CD57+ (Mature, M), and NKG2A−KIR+CD57+NKG2C+ (Memory-Like, ML). KIR labeling was performed combining three different antibodies recognizing KIR2DL1, KIR2DL2, and KIR3DL1. Bars show mean ± SEM of results obtained with three independent donors (each represented with a distinctive symbol). Statistics was performed using Friedman with Dunn’s multiple comparisons test (*p < 0.05).
4 Discussion
EBV is usually acquired in young age and persists throughout life in a state of latency without causing harm, yet various EBV-related LPD and malignant diseases can be incurred, particularly in patients with primary or secondary immunodeficiency. For EBV, there is no preventing/therapeutic vaccine or effective drug available, at present, and EBV-induced diseases are treated with conventional strategies, such as surgery and chemotherapy, which are often burdened by complications. On the other hand, rapidly evolving immunotherapies have recently offered novel opportunities, with some strategies already having proved their efficacy in EBV-related malignancies, including adoptive transfer of EBV-specific T cells and use of antibodies targeting B cells or blocking immune checkpoint signaling (38–41). Another promising approach to treat EBV+ malignancies rely on NK cell-based therapies that include modified NK cells with engineered receptors and activation/expansion of NK cell populations for adoptive transfer; of note, NK cells can outperform T cells in adoptive cell therapy because they have a general lower risk of graft-versus-host disease or other adverse events, and display effector functions that are not sensitive to escape mutations in T-cell antigens, which often occur in EBV (42, 43). Importantly, accumulating evidence has demonstrated that the induction of EBV-induced malignancies is not an exclusive prerogative of viral latency proteins as lytic factors also contribute to tumorigenesis (6, 44, 45); hence, therapeutic targeting of both latent and lytic phases may be essential.
In the present study, to investigate the interaction of NK cells with EBV throughout its life cycle, we generated multiple inducible LCL-Z cell lines that, in the absence of stimuli, typically resemble EBV-associated B-cell lymphomas with type III latency, but then, upon HT-mediated activation of recombinant BZLF1 protein, they rapidly enter into the lytic replication phase. The lytic EBV replication, for which no permissive cell system is currently available, is usually investigated in those few cells among LCL or BL cell cultures in which the virus is spontaneously reactivated or, more often, after treatment with inducing agents such as phorbol esters, calcium ionophores, histone deacetylase inhibitors, or Ig crosslinkers. Conversely, in the LCL-Z cell system, the switch to the lytic replication phase occurs efficiently with a physiologic, i.e., BZLF1-initiated, modality, hence avoiding the use of EBV-reactivating drugs that profoundly alter gene expression of the host cell. By analyzing LCL-Z cells with and without lytic reactivation, we demonstrated that, among NKG2DLs and DNAM-1Ls, only MICB is significantly expressed at the cellular membrane during latency and to a higher level in cells that entered the lytic cycle, which translates into killing by NK cells in a manner that is dependent on the cognate NKG2D receptor. In addition, we detected increased intracellular levels of MICB as well as of ULBP2 and ULBP4 within lytic cells and accumulation in LCL-Z cultures of sMICB that was higher in cultures with lytic EBV replication. Apparently, infection with EBV results in up-modulation of NKG2DLs, particularly MICB, which is a common cellular defense against viruses mediated by activation of stress response pathways (46) while, analogously to other viruses, EBV has evolved the capacity to contrast cell-surface NKG2DL expression in order to avoid immune recognition, specifically retaining MICB, ULBP2, and ULBP4 into intracellular compartments and inducing the release of sMICB from the cell membrane through the activity of viral factors that are currently unknown. Our results are in line with some previous studies showing that latently infected LCLs express MICB along with, whenever tested, low levels of MICA and ULBP4, and they are susceptible to NKG2D-dependent killing by NK cells as well as EBV-specific CD8 T-cell clones or γδ T cells (29, 30, 47). Also in accordance with our data, it was shown that the LMP2A latency protein of EBV, which activates signaling pathways to promote the proliferation of latently infected cells, induces MICA and MICB expression at the transcriptional level, while, on the other hand, reducing the cell-surface expression of both MIC proteins and ULBP4 through mechanisms that have not been identified as yet (30, 48). Additional lines of evidence indicating that EBV has developed means to contrast NKG2DL expression was provided by studies on viral ncRNAs showing that miR-BART2-5p and miR-BART7 impair translation of MICB and MICA mRNA, respectively (29, 49). These EBV activities have important functional consequences for NK cell responses, since inhibition of miR-BART2-5p in latent LCLs resulted in MICB upregulation and enhanced killing by NK cells (29), whereas overexpression of miR-BART7 in NPC lowered rates of NK cell-mediated lysis (49). Finally, studies in patients with mutated MAGT1 (referred to as “X-linked MAGT1 deficiency with increased susceptibility to EBV-infection and N-linked glycosylation defect”, XMEN) clearly pointed at a key role of the NKG2D/NKG2DL axis in the control of EBV-related diseases, which are the main cause of severe morbidity in these patients (50); specifically, because of the absence of MAGT1-mediated glycosylation, NKG2D is strongly downregulated on patients’ NK and CD8 T cells that, as a consequence, are poorly cytotoxic in vitro against autologous NKG2DL+ (i.e., positively stained by means of a recombinant NKG2D-Fc molecule) LCLs, unless NKG2D expression is previously restored. It should be mentioned, though, that some previous lines of evidence are discordant with our results; in particular, one study showed that cells with lytic EBV replication do not express various tested NKG2DLs (i.e., MICA, MICB, and ULBP2) or DNAM-1Ls (CD122 and CD155) and are killed by NK cells in a manner that is independent of NKG2D but requires DNAM-1, implying the engagement with a hypothetical DNAM-1L yet to be identified (31). We suggest that discrepancies are linked to differences in the experimental system employed, consisting in challenging an NK cell line (NKL) with lytic BL cells (AKBM treated with anti-IgG) or spontaneously reactivated LCLs.
To evade CD8 T-cell responses, EBV has developed a complex strategy targeting the antigen presentation machinery (APM) and HLA class I molecules that, on the other hand, may activate NK cells through the lack of inhibitory KIR/HLA class I interactions according to the “missing self” model of NK cell lysis (8). Specifically, by means of EBV activation in BL cell lines, transduction of individual EBV genes into EBV− tumor cell lines, or EBV infection of primary B cells, it has been demonstrated that expression of AMP and HLA class I is stimulated by the latent LMP1 protein while the opposite effect is exerted by various lytic proteins and ncRNAs, with cell-surface HLA-A, -B, and -C molecules being drastically downregulated following entry into the lytic phase [reviewed in Ref (51).]. In the present study, we found that LCL and unstimulated LCL-Z cells express high HLA-A, -B, and -C levels in agreement with previous reports (24, 52), which reflects the nature of activated blasts and expression of LMP1; once entered into the lytic phase, HLA-A, -B, and -C levels were reduced both at the cell surface and intracellularly in a statistically significant manner, yet to a very modest extent. In accord with these results, antibody-mediated block of HLA molecules resulted in higher NK cell-mediated killing of both BZLF1− and BZLF1+ targets, indicating that LCLs are protected from cytolysis by the interaction between HLA ligands with inhibitory KIRs on effector cells throughout the viral life cycle. Therefore, partial reduction of HLA-A, -B, and -C during lytic EBV replication is important for the virus to escape CD8 T-cell recognition, particularly via the downregulation of specific HLA-B molecules that present immunodominant EBV peptide during primary B-cell infection (53), but apparently is not sufficient to trigger NK cell activation. It should be noted, though, that NK cell inhibition via KIR/HLA-A, -B, and -C interactions shall be confirmed with EBV+ targets other than LCLs, for instance, EBV-associated tumors in which HLA class I molecules can be downregulated also by cellular oncogenic factors, as shown for c-Myc in the context of NPC (54).
Moreover, our results show an important role for the NKG2A/HLA-E interaction in the capacity of NK cells to kill LCL targets. In line with some previous studies in BL cells (26, 55), we found that latent LCLs efficiently express HLA-E, non-classical HLA class I molecules that present peptides derived from leader sequences of HLA-A, -B, -C, and -G molecules; then, upon EBV switch to lytic replication, HLA-E is drastically downregulated. HLA-E/peptide complexes normally suppress NK cell function by binding to CD94 in the heterodimeric NKG2A/CD94 receptor, which results in NKG2A-mediated inhibitory signaling; this important immune checkpoint can be targeted by blocking antibodies that are currently in clinical development to treat HLA-E+ tumors (56). Unexpectedly, we found that blocking of NKG2A via a specific antibody had no effect on the capacity of NK cells to kill latent LCLs, notwithstanding their HLA-E expression, and clearance of lytic BZLF1+ cells that are nearly devoid of HLA-E was rather decreased. This apparent discrepancy, however, can be explained by considering some peculiar aspects of HLA-E expression in EBV-infected cells as well as of NKG2A biology. First, peptides derived from both latent and lytic EBV proteins have been shown to associate with HLA-E molecules and prevent NKG2A triggering, thus unleashing the cytotoxic responses of NKG2A+ NK cells (57, 58), a phenomenon that may account for the lack of NKG2A-mediated inhibition of LCL killing in our cell system. Second, while KIR-mediated inhibition of NK cells follows a linear relationship with the levels of HLA-A, -B, and -C on targets, high HLA-E levels are required for NKG2A to inhibit activated NK cells (59). Moreover, inhibitory signals delivered by KIRs and NKG2A are needed for NK cells to become fully functional through a process referred to as “education” (also “arming” or “licensing”) in which NKG2A has the strongest impact (60). Accordingly, it was reported that tumors with low HLA-E levels are killed more efficiently by NKG2A+ than NKG2A− NK cells and independently of KIR/HLA-A, -B, and -C interactions (61), indicating that better education by NKG2A can prevail on its inhibitory signaling, at least when HLA-E is poorly expressed on target cells. Therefore, we suggest that NKG2A cannot inhibit NK cell activation against LCLs due to the association of decoy viral peptides to HLA-E that impair engagement of NKG2A throughout the EBV life cycle as well as to insufficient HLA-E expression during the lytic phase. On the other hand, NKG2A identifies the population of NK cells that react against LCLs with lytic EBV replication, since the use of anti-NKG2A antibody reduced killing of BZLF1+ cells in our lysis assay. We assume that, in the absence of NKG2A/HLA-E association, engagement with anti-NKG2A antibody may trigger receptor-mediated inhibitory signals and, ultimately, inhibit cytotoxicity of NKG2A+ NK cells against BZLF1+ targets. Accordingly, by investigating the phenotype of NK cells that degranulated against LCL-Z cells with or without EBV reactivation, we showed that NKG2A+ NK cell subsets, specifically CD56bright and NKG2A+KIR+CD56dim cells, are those that better respond against cells with lytic EBV replication. Our results are largely in agreement with previous in vivo and in vitro evidence, including recruitment in the tonsils of EBV+ individuals of a CD56bright NKG2A+ NK cell subset with a strong IFN-γ-mediated capacity to suppress EBV replication in vitro (7) and expansion during IM of circulating NKG2A+CD56dim NK cells that are proficiently activated when challenged with lytic BL cells (23, 24). In addition, in vitro experiments using IL-2-activated NK cells as effectors showed that CD56dimNKG2A+ cells are preferentially activated against autologous LCLs, although targets with lytic EBV replication were not tested (34). When in-depth analysis of NKG2A+CD56dim cells reacting in vitro against EBV+ targets has been performed, this showed not only high expression of NKG2D and loss of CD16 in line with our results (34, 35, 62), but also lack of KIR expression, which is at odds with our finding that NKG2A+KIR+CD56dim cells are those that better respond against lytic LCLs. However, we think that the present study differs from others in that it is the first one to investigate the overall distribution into maturation subsets of NK cells that have reacted against autologous LCLs with either latent or lytic EBV replication. Interestingly, the simultaneous presence of NKG2A and KIRs is indicative of highly educated CD56dim cells provided with full responsiveness to target cells (63).
It may seem counterintuitive that EBV has evolved means to impair NKG2A-mediated inhibition of NK cells and elicit the antiviral effector function of NKG2A+ NK cells. However, two distinct viruses, HIV-1 and SARS-CoV-2, also encode for HLA-E binding peptides that abrogate inhibition of NKG2A+ NK cells, so that infected cell targets (CD4 T and epithelial lung cells, respectively) are killed in vitro more efficiently by NKG2A+ NK cells (both CD56bright and NKG2A+ CD56dim) than NKG2A− counterparts (64, 65). It is plausible that viral HLA-E binding peptides correspond to conserved sequences critical for the replication fitness of EBV, HIV-1, and SARS-CoV-2 viruses that, on the other hand, have evolved other immune evasion strategies (e.g., downregulation of ligands for activating receptors) to counterbalance missing-self activation of NKG2A+ NK cells.
This study has limitations because host genetic factors that fine-tune NK cell functionality have not been considered, including HLA polymorphisms that influence HLA-E stability and education of NKG2A+ NK cells (66) and HLA alleles and KIR haplotypes associated with EBV infection and disease (67). Another aspect that deserves to be further investigated is the capacity to clear EBV+ targets of adaptive NK cells that express the inhibitory CD94/NKG2C receptor for HLA-E and display superior effector functions in the context of HCMV infection (15). In general, all our results should be confirmed in clinical samples. In particular, it should be investigated whether MICB is upregulated in various EBV-associated tumors and eventually accumulate in a soluble form in patients’ plasma. Likewise, the expression and function of NKG2D in NK cells of patients with EBV-related disease require investigation since chronic binding to cell-surface or soluble NKG2DLs can lead to receptor “detuning” and overall desensitization of NK cells (68). These studies will shed light on the potential of the NKG2D/MICB axis as a therapeutic target in EBV-driven malignancies. At present, several bioengineered molecules harnessing the NKG2D pathway against cancer have been developed such as antibodies that block MIC protein shedding, the extracellular NKG2D domain fused to immune activating components, and the NKG2D-based chimeric antigen receptor (CAR.NKG2D) [reviewed in Ref (69).]. Moreover, we speculate that the development of allogenic HLA mismatched NK cells with the NKG2A+KIR+ phenotype and enhanced expression of NKG2D may harbor great potential for adoptive cell therapies in patients with EBV+ malignancies. Finally, the efficacy of NK cell-based therapies may be further increased by the combination with the “lytic induction” approach (70), which implies reactivation of EBV with histone deacetylase inhibitors or other epigenetic drugs that are well-known for their potent capacity to induce NKG2DL expression among various biological effects (71).
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
The studies involving humans were approved by Ethics Committee of the Bambino Gesù Children’s Hospital. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
Author contributions
MGD: Formal analysis, Investigation, Visualization, Writing – review & editing. DC: Investigation, Writing – review & editing. CC: Funding acquisition, Supervision, Writing – review & editing. MD: Conceptualization, Formal analysis, Funding acquisition, Methodology, Supervision, Visualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the Italian Ministry of Health with “Current Research funds” to MD and CC.
Acknowledgments
We are grateful to Lorenzo Moretta (Bambino Gesù Children’s Hospital, Rome, Italy), Patrizio Giacomini (Istituto Regina Elena, Rome, Italy), and Paul J. Farrell (Imperial College Faculty of Medicine, London, UK) for providing essential reagents. We thank Angela Vacca and Nicola Tumino (Bambino Gesù Children’s Hospital) for help with Class I antibody-mediated blocking protocol. We also thank Antonio Cappelli and other members of SIMT (Immunotransfusion Service, Bambino Gesù Children’s Hospital) for assistance in obtaining buffy coats.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2024.1467304/full#supplementary-material
References
1. Farrell PJ. Epstein-barr virus and cancer. Annu Rev Pathol. (2019) 14:29–53. doi: 10.1146/annurev-pathmechdis-012418-013023
2. Murata T, Sugimoto A, Inagaki T, Yanagi Y, Watanabe T, Sato Y, et al. Molecular basis of epstein-barr virus latency establishment and lytic reactivation. Viruses. (2021) 13:2344. doi: 10.3390/v13122344
3. Li H, Liu S, Hu J, Luo X, Li N, M Bode A, et al. Epstein-Barr virus lytic reactivation regulation and its pathogenic role in carcinogenesis. Int J Biol Sci. (2016) 12:1309–18. doi: 10.7150/ijbs.16564
4. Shannon-Lowe C, Rickinson A. The global landscape of EBV-associated tumors. Front Oncol. (2019) 9:713. doi: 10.3389/fonc.2019.00713
5. Taylor GS, Long HM, Brooks JM, Rickinson AB, Hislop AD. The immunology of Epstein-Barr virus-induced disease. Annu Rev Immunol. (2015) 33:787–821. doi: 10.1146/annurev-immunol-032414-112326
6. Münz C. Latency and lytic replication in Epstein-Barr virus-associated oncogenesis. Nat Rev Microbiol. (2019) 17:691–700. doi: 10.1038/s41579-019-0249-7
7. Lünemann A, Vanoaica LD, Azzi T, Nadal D, Münz C. A distinct subpopulation of human NK cells restricts B cell transformation by EBV. J Immunol. (2013) 191:4989–95. doi: 10.4049/jimmunol.1301046
8. Abel AM, Yang C, Thakar MS, Malarkannan S. Natural killer cells: development, maturation, and clinical utilization. Front Immunol. (2018) 9:1869. doi: 10.3389/fimmu.2018.01869
9. Sivori S, Vacca P, Del Zotto G, Munari E, Mingari MC, Moretta L. Human NK cells: surface receptors, inhibitory checkpoints, and translational applications. Cell Mol Immunol. (2019) 16:430–41. doi: 10.1038/s41423-019-0206-4
10. Mace EM. Human natural killer cells: Form, function, and development. J Allergy Clin Immunol. (2023) 151:371–85. doi: 10.1016/j.jaci.2022.09.022
11. Horowitz A, Strauss-Albee DM, Leipold M, Kubo J, Nemat-Gorgani N, Dogan OC, et al. Genetic and environmental determinants of human NK cell diversity revealed by mass cytometry. . Sci Transl Med. (2013) 5:208ra145. doi: 10.1126/scitranslmed.3006702
12. Cichocki F, Grzywacz B, Miller JS. Human NK cell development: one road or many? Front Immunol. (2019) 10:2078. doi: 10.3389/fimmu.2019.02078
13. Béziat V, Descours B, Parizot C, Debré P, Vieillard V. NK cell terminal differentiation: correlated stepwise decrease of NKG2A and acquisition of KIRs. PloS One. (2010) 5:e11966. doi: 10.1371/journal.pone.0011966
14. Bjorkstrom NK, Ljunggren HG, Sandberg JK. CD56 negative NK cells: origin, function, and role in chronic viral disease. Trends Immunol. (2010) 31:401–6. doi: 10.1016/j.it.2010.08.003
15. López-Botet M, Vilches C, Redondo-Pachón D, Muntasell A, Pupuleku A, Yélamos J, et al. Dual role of natural killer cells on graft rejection and control of cytomegalovirus infection in renal transplantation. Front Immunol. (2017) 8:166. doi: 10.3389/fimmu.2017.00166
16. Desimio MG, Covino DA, Rivalta B, Cancrini C, Doria M. The role of NK cells in EBV infection and related diseases: current understanding and hints for novel therapies. Cancers (Basel). (2023) 15:1914. doi: 10.3390/cancers15061914
17. Mace EM, Orange JS. Emerging insights into human health and NK cell biology from the study of NK cell deficiencies. Immunol Rev. (2019) 287:202–25. doi: 10.1111/imr.12725
18. Wiesmayr S, Webber SA, Macedo C, Popescu I, Smith L, Luce J, et al. Decreased NKp46 and NKG2D and elevated PD-1 are associated with altered NK-cell function in pediatric transplant patients with PTLD. Eur J Immunol. (2012) 42:541–50. doi: 10.1002/eji.201141832
19. Forconi CS, Cosgrove CP, Saikumar-Lakshmi P, Nixon CE, Foley J, Ong’echa JM, et al. Poorly cytotoxic terminally differentiated CD56-CD16+ NK cells accumulate in Kenyan children with Burkitt lymphomas. Blood Adv. (2018) 2:1101–14. doi: 10.1182/bloodadvances.2017015404
20. Nakid-Cordero C, Choquet S, Gauthier N, Balegroune N, Tarantino N, Morel V, et al. Distinct immunopathological mechanisms of EBV-positive and EBV-negative posttransplant lymphoproliferative disorders. Am J Transplant. (2021) 21:2846–63. doi: 10.1111/ajt.16547
21. Png YT, Yang AZY, Lee MY, Chua MJM, Lim CM. The role of NK cells in EBV infection and EBV-associated NPC. Viruses. (2021) 13:300. doi: 10.3390/v13020300
22. Pánisová E, Lünemann A, Bürgler S, Kotur M, Lazarovici J, Danu A, et al. Reduced frequency of cytotoxic CD56dim CD16+ NK cells leads to impaired antibody-dependent degranulation in EBV-positive classical Hodgkin lymphoma. Cancer Immunol Immunother. (2022) 71:13–24. doi: 10.1007/s00262-021-02956-x
23. Chijioke O, Müller A, Feederle R, Barros MHM, Krieg C, Emmel V, et al. Human natural killer cells prevent infectious mononucleosis features by targeting lytic Epstein-Barr virus infection. Cell Rep. (2013) 5:1489–98. doi: 10.1016/j.celrep.2013.11.041
24. Azzi T, Lünemann A, Murer A, Ueda S, Béziat V, Malmberg K, et al. Role for early-differentiated natural killer cells in infectious mononucleosis. Blood. (2014) 124:2533–43. doi: 10.1182/blood-2014-01-553024
25. Hendricks DW, Balfour HHJ, Dunmire SK, Schmeling DO, Hogquist KA, Lanier LL. Cutting edge: NKG2C(hi)CD57+ NK cells respond specifically to acute infection with cytomegalovirus and not Epstein-Barr virus. J Immunol. (2014) 192:4492–6. doi: 10.4049/jimmunol.1303211
26. Pappworth IY, Wang EC, Rowe M. The switch from latent to productive infection in epstein-barr virus-infected B cells is associated with sensitization to NK cell killing. J Virol. (2007) 81:474–82. doi: 10.1128/JVI.01777-06
27. Lanier LL. NKG2D receptor and its ligands in host defense. Cancer Immunol Res. (2015) 3:575–82. doi: 10.1158/2326-6066.CIR-15-0098
28. Zingoni A, Molfetta R, Fionda C, Soriani A, Paolini R, Cippitelli M, et al. NKG2D and its ligands: “One for all, all for one. Front Immunol. (2018) 9:476. doi: 10.3389/fimmu.2018.00476
29. Nachmani D, Stern-Ginossar N, Sarid R, Mandelboim O. Diverse herpesvirus microRNAs target the stress-induced immune ligand MICB to escape recognition by natural killer cells. Cell Host Microbe. (2009) 5:376–85. doi: 10.1016/j.chom.2009.03.003
30. Rancan C, Schirrmann L, Hüls C, Zeidler R, Moosmann A. Latent membrane protein LMP2A impairs recognition of EBV-infected cells by CD8+ T cells. PloS Pathog. (2015) 11:e1004906. doi: 10.1371/journal.ppat.1004906
31. Williams LR, Quinn LL, Rowe M, Zuo J. Induction of the lytic cycle sensitizes epstein-barr virus-infected B cells to NK cell killing that is counteracted by virus-mediated NK cell evasion mechanisms in the late lytic cycle. J Virol. (2015) 90:947–58. doi: 10.1128/JVI.01932-15
32. McDonald C, Karstegl CE, Kellam P, Farrell PJ. Regulation of the Epstein-Barr virus Zp promoter in B lymphocytes during reactivation from latency. J Gen Virol. (2010) 91:622–9. doi: 10.1099/vir.0.017277-0
33. Waldhauer I, Steinle A. Proteolytic release of soluble UL16-binding protein 2 from tumor cells. Cancer Res. (2006) 66:2520–6. doi: 10.1158/0008-5472
34. Hatton O, Strauss-Albee DM, Zhao NQ, Haggadone MD, Pelpola JS, Krams SM, et al. NKG2A-expressing natural killer cells dominate the response to autologous lymphoblastoid cells infected with epstein-barr virus. Front Immunol. (2016) 7:607. doi: 10.3389/fimmu.2016.00607
35. Djaoud Z, Guethlein LA, Horowitz A, Azzi T, Nemat-Gorgani N, Olive D, et al. Two alternate strategies for innate immunity to Epstein-Barr virus: One using NK cells and the other NK cells and γδ T cells. J Exp Med. (2017) 214:1827–41. doi: 10.1084/jem.20161017
36. Manser AR, Uhrberg M. Age-related changes in natural killer cell repertoires: impact on NK cell function and immune surveillance. Cancer Immunol Immunother. (2016) 65:417–26. doi: 10.1007/s00262-015-1750-0
37. Romee R, Foley B, Lenvik T, Wang Y, Zhang B, Ankarlo D, et al. NK cell CD16 surface expression and function is regulated by a disintegrin and metalloprotease-17 (ADAM17). Blood. (2013) 121:3599–608. doi: 10.1182/blood-2012-04-425397
38. McLaughlin LP, Bollard CM, Keller MD. Adoptive T cell therapy for epstein-barr virus complications in patients with primary immunodeficiency disorders. Front Immunol. (2018) 9:556. doi: 10.3389/fimmu.2018.00556
39. Al Hamed R, Bazarbachi AH, Mohty M. Epstein-Barr virus-related post-transplant lymphoproliferative disease (EBV-PTLD) in the setting of allogeneic stem cell transplantation: a comprehensive review from pathogenesis to forthcoming treatment modalities. Bone Marrow Transplant. (2020) 55:25–39. doi: 10.1038/s41409-019-0548-7
40. Smith C, McGrath M, Neller MA, Matthews KK, Crooks P, Le Texier L, et al. Complete response to PD-1 blockade following EBV-specific T-cell therapy in metastatic nasopharyngeal carcinoma. NPJ Precis Oncol. (2021) 5:24–7. doi: 10.1038/s41698-021-00162-7
41. Li W, Duan X, Chen X, Zhan M, Peng H, Meng Y, et al. Immunotherapeutic approaches in EBV-associated nasopharyngeal carcinoma. Front Immunol. (2023) 13:1079515. doi: 10.3389/fimmu.2022.1079515
42. Heipertz EL, Zynda ER, Stav-Noraas TE, Hungler AD, Boucher SE, Kaur N, et al. Current perspectives on “Off-the-shelf” Allogeneic NK and CAR-NK cell therapies. Front Immunol. (2021) 12:732135. doi: 10.3389/fimmu.2021.732135
43. Vivier E, Rebuffet L, Narni-Mancinelli E, Cornen S, Igarashi RY, Fantin VR. Natural killer cell therapies. Nature. (2024) 626:727–36. doi: 10.1038/s41586-023-06945-1
44. Manners O, Murphy JC, Coleman A, Hughes DJ, Whitehouse A. Contribution of the KSHV and EBV lytic cycles to tumourigenesis. Curr Opin Virol. (2018) 32:60–70. doi: 10.1016/j.coviro.2018.08.014
45. Murata T, Okuno Y, Sato Y, Watanabe T, Kimura H. Oncogenesis of CAEBV revealed: Intragenic deletions in the viral genome and leaky expression of lytic genes. Rev Med Virol. (2020) 30:e2095. doi: 10.1002/rmv.2095
46. Cerboni C, Fionda C, Soriani A, Zingoni A, Doria M, Cippitelli M, et al. The DNA damage response: A common pathway in the regulation of NKG2D and DNAM-1 ligand expression in normal, infected, and cancer cells. Front Immunol. (2014) 4:508. doi: 10.3389/fimmu.2013.00508
47. Kong Y, Cao W, Xi X, Ma C, Cui L, He W. The NKG2D ligand ULBP4 binds to TCRgamma9/delta2 and induces cytotoxicity to tumor cells through both TCRgammadelta and NKG2D. Blood. (2009) 114:310–7. doi: 10.1182/blood-2008-12-196287
48. Singh S, Banerjee S. Downregulation of HLA-ABC expression through promoter hypermethylation and downmodulation of MIC-A/B surface expression in LMP2A-positive epithelial carcinoma cell lines. Sci Rep. (2020) 10:5415–0. doi: 10.1038/s41598-020-62081-0
49. Wong T, Chen S, Zhang M, Chan JY, Gao W. Epstein-Barr virus-encoded microRNA BART7 downregulates major histocompatibility complex class I chain-related peptide A and reduces the cytotoxicity of natural killer cells to nasopharyngeal carcinoma. Oncol Lett. (2018) 16:2887–92. doi: 10.3892/ol.2018.9041
50. Chaigne-Delalande B, Li FY, O’Connor GM, Lukacs MJ, Jiang P, Zheng L, et al. Mg2+ regulates cytotoxic functions of NK and CD8 T cells in chronic EBV infection through NKG2D. Science. (2013) 341:186–91. doi: 10.1126/science.1240094
51. Jasinski-Bergner S, Mandelboim O, Seliger B. Molecular mechanisms of human herpes viruses inferring with host immune surveillance. J Immunother Cancer. (2020) 8:e000841. doi: 10.1136/jitc-000841
52. Wiesner M, Zentz C, Mayr C, Wimmer R, Hammerschmidt W, Zeidler R, et al. Conditional immortalization of human B cells by CD40 ligation. PloS One. (2008) 3:e1464. doi: 10.1371/journal.pone.0001464
53. Albanese M, Tagawa T, Bouvet M, Maliqi L, Lutter D, Hoser J, et al. Epstein-Barr virus microRNAs reduce immune surveillance by virus-specific CD8+ T cells. Proc Natl Acad Sci U.S.A. (2016) 113:E6467–75. doi: 10.1073/pnas.1605884113
54. Tudor CS, Dawson CW, Eckhardt J, Niedobitek G, Büttner AC, Seliger B, et al. c-Myc and EBV-LMP1: two opposing regulators of the HLA class I antigen presentation machinery in epithelial cells. Br J Cancer. (2012) 106:1980–8. doi: 10.1038/bjc.2012.197
55. Griffin BD, Gram AM, Mulder A, Van Leeuwen D, Claas FHJ, Wang F, et al. EBV BILF1 evolved to downregulate cell surface display of a wide range of HLA class I molecules through their cytoplasmic tail. J Immunol. (2013) 190:1672–84. doi: 10.4049/jimmunol.1102462
56. Fisher JG, Doyle ADP, Graham LV, Khakoo SI, Blunt MD. Disruption of the NKG2A:HLA-E immune checkpoint axis to enhance NK cell activation against cancer. Vaccines (Basel). (2022) 10:1993. doi: 10.3390/vaccines10121993
57. Ulbrecht M, Modrow S, Srivastava R, Peterson PA, Weiss EH. Interaction of HLA-E with peptides and the peptide transporter in vitro: implications for its function in antigen presentation. J Immunol. (1998) 160:4375–85. doi: 10.4049/jimmunol.160.9.4375
58. Mbiribindi B, Pena JK, Arvedson MP, Moreno Romero C, McCarthy SR, Hatton OL, et al. Epstein-Barr virus peptides derived from latent cycle proteins alter NKG2A + NK cell effector function. Sci Rep. (2020) 10:19973–3. doi: 10.1038/s41598-020-76344-3
59. Cheent KS, Jamil KM, Cassidy S, Liu M, Mbiribindi B, Mulder A, et al. Synergistic inhibition of natural killer cells by the nonsignaling molecule CD94. Proc Natl Acad Sci U.S.A. (2013) 110:16981–6. doi: 10.1073/pnas.1304366110
60. Sim MJW, Stowell J, Sergeant R, Altmann DM, Long EO, Boyton RJ. KIR2DL3 and KIR2DL1 show similar impact on licensing of human NK cells. Eur J Immunol. (2016) 46:185–91. doi: 10.1002/eji.201545757
61. Mahaweni NM, Ehlers FAI, Sarkar S, Janssen JWH, Tilanus MGJ, Bos GMJ, et al. NKG2A Expression Is Not per se Detrimental for the Anti-Multiple Myeloma Activity of Activated Natural Killer Cells in an In Vitro System Mimicking the Tumor Microenvironment. Front Immunol. (2018) 9:1415. doi: 10.3389/fimmu.2018.01415
62. Granzin M, Soltenborn S, Müller S, Kollet J, Berg M, Cerwenka A, et al. Fully automated expansion and activation of clinical-grade natural killer cells for adoptive immunotherapy. Cytotherapy. (2015) 17:621–32. doi: 10.1016/j.jcyt.2015.03.611
63. Björkström NK, Riese P, Heuts F, Andersson S, Fauriat C, Ivarsson MA, et al. Expression patterns of NKG2A, KIR, and CD57 define a process of CD56dim NK-cell differentiation uncoupled from NK-cell education. Blood. (2010) 116:3853–64. doi: 10.1182/blood-2010-04-281675
64. Davis ZB, Cogswell A, Scott H, Mertsching A, Boucau J, Wambua D, et al. A conserved HIV-1-derived peptide presented by HLA-E renders infected T-cells highly susceptible to attack by NKG2A/CD94-bearing natural killer cells. PloS Pathog. (2016) 12:e1005421. doi: 10.1371/journal.ppat.1005421
65. Hammer Q, Dunst J, Christ W, Picarazzi F, Wendorff M, Momayyezi P, et al. SARS-CoV-2 Nsp13 encodes for an HLA-E-stabilizing peptide that abrogates inhibition of NKG2A-expressing NK cells. Cell Rep. (2022) 38:110503. doi: 10.1016/j.celrep.2022.110503
66. Horowitz A, Djaoud Z, Nemat-Gorgani N, Blokhuis J, Hilton HG, Béziat V, et al. Class I HLA haplotypes form two schools that educate NK cells in different ways. Sci Immunol. (2016) 1:eaag1672. doi: 10.1126/sciimmunol.aag1672
67. Palmer WH, Norman PJ. The impact of HLA polymorphism on herpesvirus infection and disease. Immunogenetics. (2023) 75:231–47. doi: 10.1007/s00251-022-01288-z
68. Coudert JD, Scarpellino L, Gros F, Vivier E, Held W. Sustained NKG2D engagement induces cross-tolerance of multiple distinct NK cell activation pathways. Blood. (2008) 111:3571–8. doi: 10.1182/blood-2007-07-100057
69. Maurer S, Zhong X, Prada BD, Mascarenhas J, de Andrade LF. The latest breakthroughs in immunotherapy for acute myeloid leukemia, with a special focus on NKG2D ligands. Int J Mol Sci. (2022) 23:15907. doi: 10.3390/ijms232415907
70. Yiu SPT, Dorothea M, Hui KF, Chiang AKS. Lytic induction therapy against epstein-barr virus-associated Malignancies: past, present, and future. Cancers (Basel). (2020) 12:2142. doi: 10.3390/cancers12082142
Keywords: Epstein-Barr virus (EBV), lymphoblastoid cell line (LCL), latency, lytic replication, natural killer (NK) cells, cytotoxicity, NKG2D, NKG2A
Citation: Desimio MG, Covino DA, Cancrini C and Doria M (2024) Entry into the lytic cycle exposes EBV-infected cells to NK cell killing via upregulation of the MICB ligand for NKG2D and activation of the CD56bright and NKG2A+KIR+CD56dim subsets. Front. Immunol. 15:1467304. doi: 10.3389/fimmu.2024.1467304
Received: 19 July 2024; Accepted: 08 November 2024;
Published: 29 November 2024.
Edited by:
Lucia Mundo, University of Limerick, IrelandReviewed by:
Cristina Cerboni, Sapienza University of Rome, ItalyLisa Kronstad, Midwestern University, United States
Copyright © 2024 Desimio, Covino, Cancrini and Doria. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Margherita Doria, bWFyZ2hlcml0YS5kb3JpYUBvcGJnLm5ldA==; ZG9yaWFAdW5pcm9tYTIuaXQ=
†These authors have contributed equally to this work