 Mohammed Yosri
Mohammed Yosri Mohamed Dokhan
Mohamed Dokhan Elizabeth Aboagye2
Elizabeth Aboagye2 Mouhamad Al Moussawy
Mouhamad Al Moussawy Hossam A. Abdelsamed
Hossam A. Abdelsamed- 1The Regional Center for Mycology and Biotechnology, Al-Azhar University, Cairo, Egypt
- 2Immunology Center of Georgia (IMMCG), Medical College of Georgia (MCG), Augusta University, Augusta, GA, United States
- 3Starzl Transplantation Institute, School of Medicine, University of Pittsburgh, Pittsburgh, PA, United States
- 4Department of Physiology, Augusta University, Augusta, GA, United States
The immune system is endowed with the capacity to distinguish between self and non-self, so-called immune tolerance or “consciousness of the immune system.” This type of awareness is designed to achieve host protection by eliminating cells expressing a wide range of non-self antigens including microbial-derived peptides. Such a successful immune response is associated with the secretion of a whole spectrum of soluble mediators, e.g., cytokines and chemokines, which not only contribute to the clearance of infected host cells but also activate T cells that are not specific to the original cognate antigen. This kind of non-specific T-cell activation is called “bystander activation.” Although it is well-established that this phenomenon is cytokine-dependent, there is evidence in the literature showing the involvement of peptide/MHC recognition depending on the type of T-cell subset (naive vs. memory). Here, we will summarize our current understanding of the mechanism(s) of bystander T-cell activation as well as its biological significance in a wide range of diseases including microbial infections, cancer, auto- and alloimmunity, and chronic inflammatory diseases such as atherosclerosis.
1 Introduction
The Oxford Language Dictionary defines “bystander” as a person who is physically present during an ongoing specific event but not directly involved in it. Nature is full of bystander examples that might be of symbiotic benefits or deleterious consequences. For instance, during photosynthesis, green-leaved plants release oxygen into the atmosphere as a by-product of the process (1). Consequently, eukaryotes can perform cellular respiration using released oxygen to produce their cellular energy. This interconnected chain of events represents a clear example of a bystander effect where eukaryotic cells, not necessarily participating directly in photosynthesis, get affected beneficially from using their oxygen by-product.
Similarly, cytokines as by-products of functional immune responses (2, 3) can activate a wide range of immune cells including T cells but not necessarily those specific to the initial non-self antigen. In other words, antigen non-specific T cells that happen to be present during an ongoing immune response can be activated by cytokines as bystanders, a phenomenon known as “bystander T cell activation” (Figure 1A) (4–6). However, the high cost of this process can lead to “epitope spreading,” where collateral damage of the host cells results in the release of new antigens and the activation of T cells within the microenvironment leading to autoimmunity (Figure 1B) (7–9).
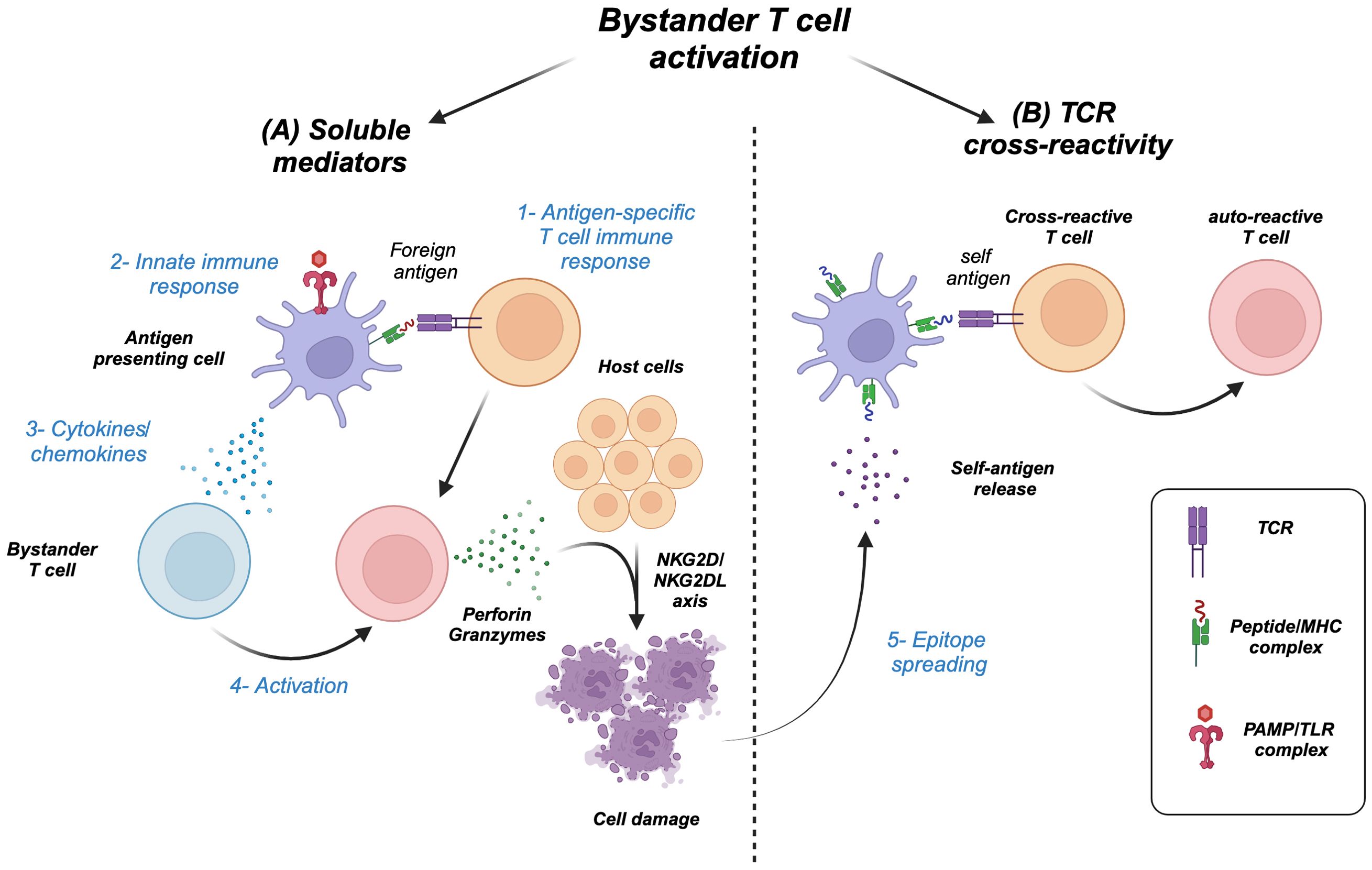
Figure 1. Types of T cell bystander activation. (A) 1) Antigen-specific immune response and/or 2) innate stimulation of antigen-presenting cells results in 3) a wave of cytokines/chemokines that 4) activate bystander T cells, which can cause host cell damage through effector molecules (granzymes/perforin) and NK receptor–ligand interaction (NKG2D/NKG2DL). (B) 5) The damage can release new self-antigens, a process called epitope spreading, which in turn activates cross-reactive T cells including initial antigen-specific T cells to become autoreactive.
Although it is widely accepted that bystander memory T cell activation is cytokine-dependent (10–12), there is evidence supporting the role of the self-peptide/MHC complex along with cytokines in the activation of naive T cells (5, 13–17). In this review, we will shed light on the mechanisms of bystander T-cell activation (Figure 1). Furthermore, we will discuss the biological relevance of this process in autoimmunity, cancer, transplantation, microbial infection, and atherosclerosis.
2 Mechanisms of bystander T cell activation
The communication between different cell types in the body is mainly governed by at least two processes: 1) receptor–ligand interaction and (2) soluble mediators (18). Likewise, T cells can be activated through their T-cell receptor (TCR) via interaction with the peptide/MHC complex, presented by APCs (receptor–ligand complex) (19, 20) and/or soluble mediators including cytokines and microbial products (21–34), where both types of communication can contribute to the bystander activation of T cells.
Bystander T-cell activation is defined as the activation of T cells that are non-specific to the original antigen (35). In other words, during viral infections, there could be activation of T cells not necessarily specific to viral antigens (35). For instance, several studies demonstrated the activation of antigen non-specific T cells during mouse lymphocytic choriomeningitis virus (LCMV) infection (35–37). Additionally, LCMV-specific cytotoxic T cells (CTLs) undergo activation and proliferation following infection with unrelated viruses including murine cytomegalovirus (MCMV) and vaccinia virus (VV) (38).
2.1 Bystander T cell activation via soluble mediators versus cross-reactivity
Bystander T-cell activation can actually happen through two mechanisms: first, via activation of cross-reactive TCRs (Figure 1B) (39–42), and second, via activation through soluble mediators including cytokines (21–25, 27, 28, 30–33, 43) because of the strong immune response to the original foreign antigen (Figure 1A). Although the phenomenon of bystander T-cell activation is widely associated with stimulation via soluble mediators including cytokines, several follow-up studies demonstrated the involvement of peptide–MHC/TCR interaction in the process (15, 16, 44, 45). These studies served as an impetus for us to further understand the mechanisms regulating this process as well as the differential requirement for each mechanism that can be dependent on the T-cell subtype (naive vs. memory). Furthermore, we would like to clarify the concept of cross-reactivity and its relation to bystander T-cell activation. Cross-reactivity is simply the ability of a single TCR to recognize multiple peptide–MHC complexes, which was first hinted as a hypothesis by Matzinger and Bevan (46). Indeed, it is plausible for the immune system to develop such a feature of TCR degeneracy or so-called heterologous immunity to recognize a vast number of microbial epitopes, yet molecular mimicry could be the high cost of such capacity where an overlap between microbial and self-epitopes results in an immune response against self and the development of autoimmune diseases. A classic example of such a phenomenon is rheumatic carditis, where cardiac myosin is cross-reactive with a virulence factor related to group A streptococcus (GAS), Streptococcus pyogenes, called the M protein (47–49). One can speculate that the host-immune response against the bacteria damages the heart tissue by exposing new epitopes including myosin. Consequently, the initial T-cell immune response becomes cross-reactive against the self-proteins causing autoimmunity (Figure 1B). Additionally, we will discuss in the next sections, many types of autoimmune diseases that are characterized by the presence of bystander-activated antigen non-specific T cells in the inflamed/damaged organ, hinting to their activation either via the proinflammatory microenvironment and/or cross-reactivity. Yet, it is challenging to dissect bystander T-cell activation via cytokines versus cross-reactivity, as TCRs will have some degree of responsiveness to self, i.e., tonic signaling. In the following sections, we will discuss the mechanisms regulating bystander T-cell activation and their effect on naive and memory CD8 T cells.
2.1.1 The role of microbial products and cytokines in naive and memory T-cell activation
The secretion of cytokines in a given microenvironment and the role of pathogen-associated microbial patterns (PAMPs) are two intermingled processes, where activation via PAMPs results in the secretion of cytokines. Consequently, pathogen recognition receptors (PRRs) are upregulated. PAMPs are known to bind directly to T cells through PRRs including Toll-like receptors (TLRs), acting as costimulatory signal, activating them to upregulate activation markers and secretion of cytokines (50–52). Indeed, TLR2 engagement in T cells enhances their effector functions and survival (26, 29), while TLR7 activation in T cells contributes to the pathogenesis of multiple sclerosis in a preclinical animal model (34). In vitro infection of splenocytes with Burkholderia pseudomallei bacteria results in rapid expression of IFNγ in CD8 CD44hi (activated/memory) compared to naive CD8 CD44lo cells (30). Furthermore, the Sprent lab showed that memory T cells were responsive to TLR ligands including poly I:C (TLR3 ligand) and LPS (TLR4 ligand), which can induce type I interferon and consequently can synthetize memory CD8 T cells to IL-15 and IL-2 by upregulation of IL-2/IL-15Rβ (CD122) (5, 53). To further examine the role of PAMPs on T-cell bystander activation in vivo, poly I:C was injected into naive mice mimicking viral infections, which resulted in the proliferation of polyclonal memory CD8 T cells in a TCR-independent manner (5).
Several studies examine the effect of the proinflammatory cytokines such as IL-12 and IL-18 as well as the common gamma chain cytokine IL-15 on bystander T-cell activation (Figure 2B) (21, 23–25, 28, 31, 43). For example, the combination of both IL-12 and IL-18 induced the proliferation and IFNγ expression from memory CD8 T cells (23, 28, 32). Furthermore, IL-12 augments the induction of IFNγ from T cells by enhancing the expression of IL-18R (54). Additionally, IL-15 alone in the absence of TCR stimulation can activate memory CD8 T cells to proliferate and acquire effector functions, while prolonged exposure enhances their cytotoxicity i.e., upregulation of perforin, granzymes, CD107a, granulysin, and NK receptors such as NKG2D (21, 24, 25, 31, 43). NKG2D can mediate direct cell killing in the absence of TCR stimulation (4, 55), recognizing its ligands including stress-induced proteins MICA and MICB in humans and H60 and Mult1 in mice (56). This binding results in the activation of downstream signaling through DAP10/12. The killing of target cells can result in the release of self-antigens that can activate bystander T cells amplifying an autoimmune response through epitope spreading (Figures 1A, 3A, 4A). For instance, in celiac disease, an autoimmune disease, intestinal epithelial cells express IL-15, which upregulates NKG2D on T cells infiltrating the intestine, where they can target the epithelial causing damage and possibly the release of self-antigens activating T cells and amplifying the bystander T-cell response (57). IL-15 can also play an important role in the migration of bystander memory CD8 T cells to the site of inflammation. During acute hepatitis A, IL-15 upregulates CCR5 chemokine receptor enhancing their requirement for liver parenchyma (58) (Figure 3A). The synergy between cytokines plays an important role in bystander T-cell activation. For instance, both IL-23 and IL-1β can act synergistically to induce the expression of IFNγ, IL-22, and GM-CSF in effector/memory CD4 T cells (59–61). Another example of cytokine synergy and bystander activation is depicted in IL-2 and IL-33. Indeed, IL-2 upregulates IL-33R (ST2) in Th2 CD4 T cells making them express IL-13 (61). Furthermore, the IL-1 family member proinflammatory cytokine IL-18 can induce the expression of IFNγ from naive and memory CD4 T cells and IL-17 from Th17 CD4 T cells in an antigen-independent manner (59, 61–63). In response to IL-18, IL-18Rα+ memory CD4 T cells express a wide spectrum of cytokines including IL-5, IL-6, IL-13, IL-22, GM-CSF, TNFα, and IFNγ (64). The upregulation of IL-18Rα can be attributed to IL-12, which acts synergistically with IL-18 and induces Th1 and Th17 to express IFNγ in a TCR-independent manner (59, 61). Finally, high doses of IL-2 override the requirement of TCR stimulation and synthesize naive CD4 T cells to become responsive to IL-12 and IL-18 and acquire a Th1 phenotype and express IFNγ (62).
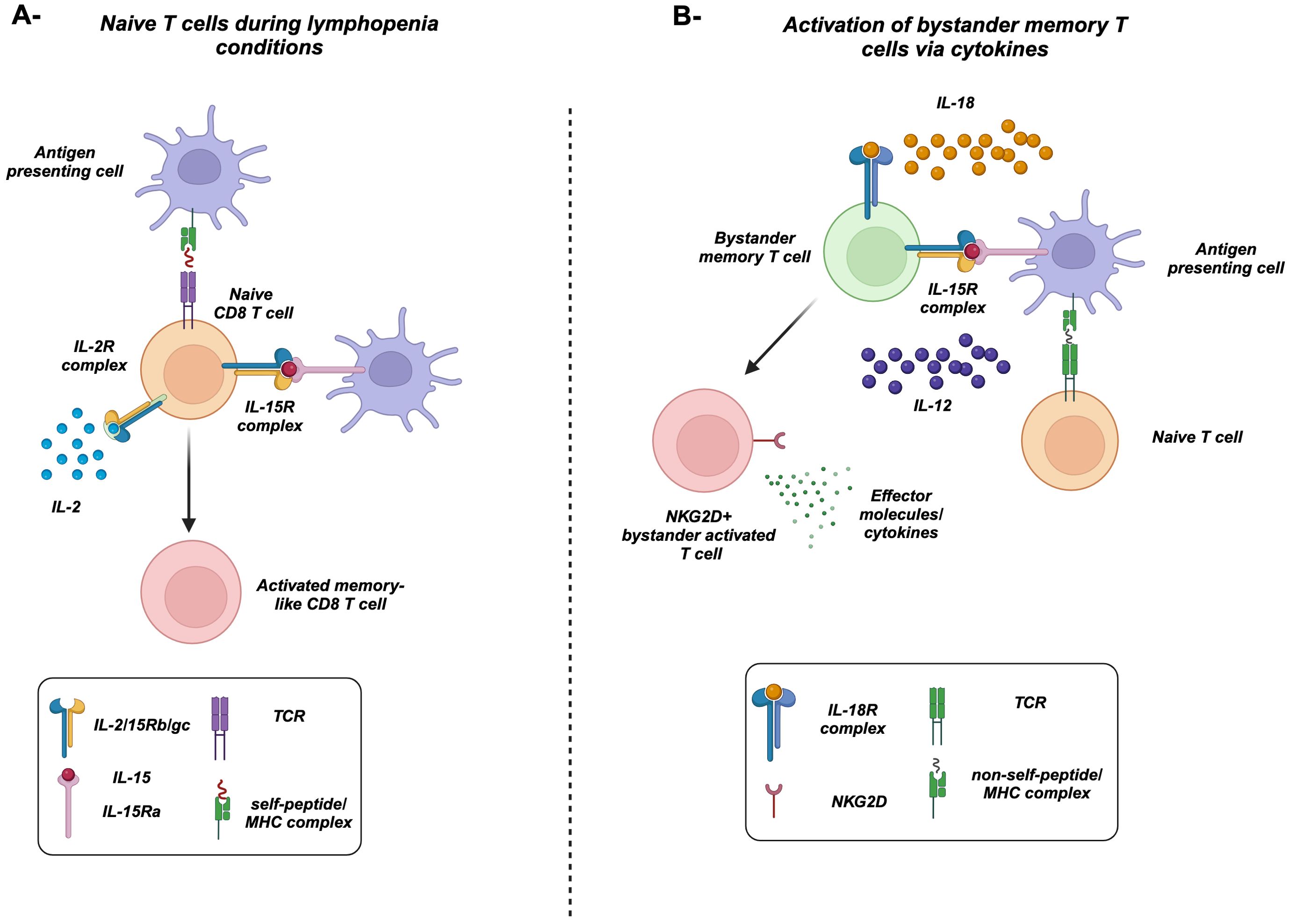
Figure 2. Bystander-activated memory T cells versus homeostatic proliferation of naive T cells. (A) Under lymphopenia conditions, naive CD8 T cells, recognizing a self-antigen presented by MHC-I, respond to the increased levels of IL-2 and cross-presented IL-15. They acquire a proliferative effector/memory-like phenotype. (B) Antigen-specific naive T cells recognize its specific antigen presented on MHC. In the meantime, IL-15 cross-presented by activated antigen-presenting cells, along with IL-12 and IL-18, activates bystander memory T cells in a TCR-independent manner. Activated memory cells upregulate the natural killer group 2D receptors (NKG2D) and produce high levels of effector molecules/cytokines including IFNγ along with granzymes and perforin.
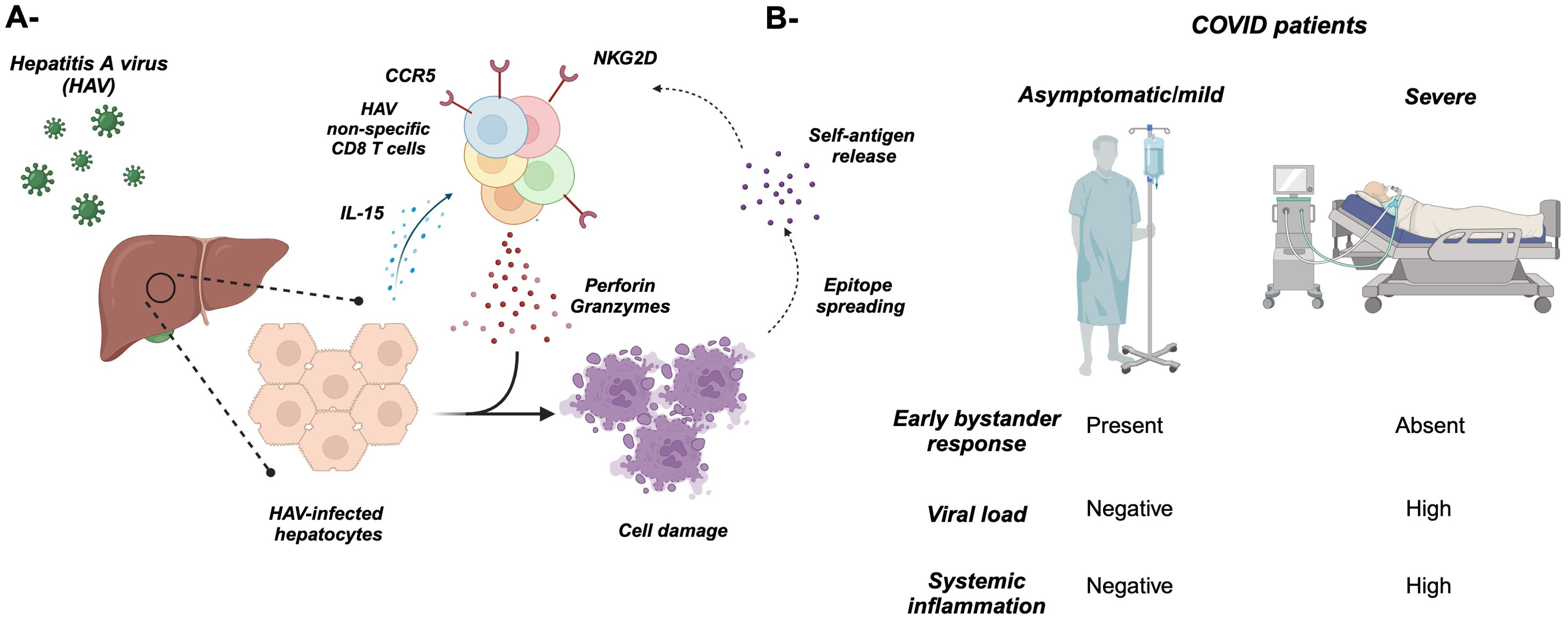
Figure 3. Bystander T cell activation during microbial infections. (A) Infection of hepatocytes with hepatitis A virus (HAV) results in the release of IL-15, which activates non-specific “bystander” CD8 T cells including EBV- and CMV-specific (depicted in different colors). These activated T cells cause damage to the hepatocytes through NKG2D and cytotoxic cytokines/molecules including perforin, granzymes, and IFNγ. The overall hepatocyte damage might result in the release of new self-antigens that activate T cells and cause autoimmunity through “epitope spreading” (dotted arrows). (B) Asymptomatic/mild COVD-19 patients are characterized by an early burst of bystander T-cell activation, which might protect them from disease progression to severe clinical presentation.
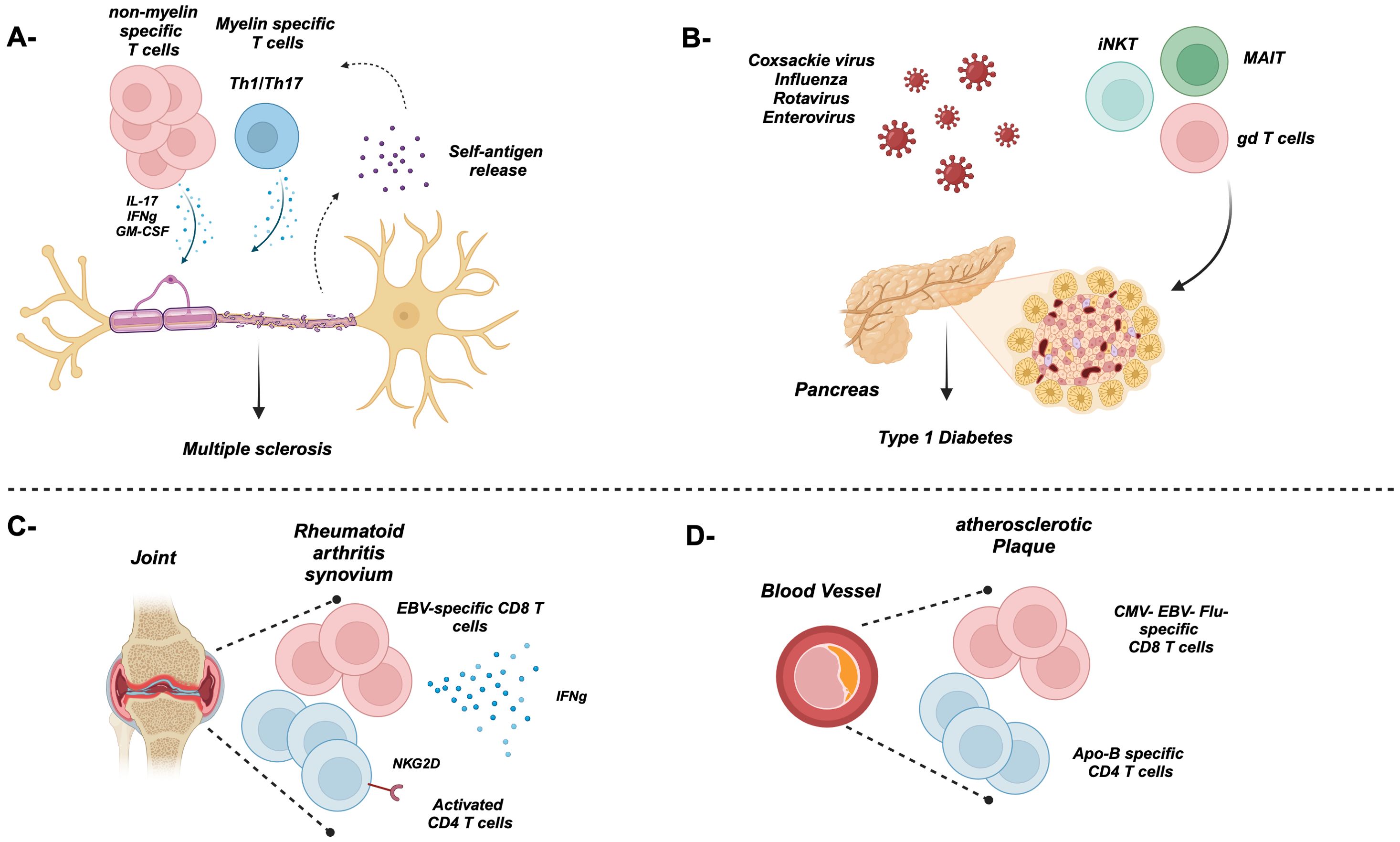
Figure 4. Bystander T cell activation during autoimmune diseases. (A) Myelin- and non-myelin-specific T cells contribute to the damage of the neuron’s myelin sheath during multiple sclerosis through proinflammatory cytokines. The release of new self-antigens might exacerbate the disease through the activation of T cells non-specific to the original antigen. (B) The Coxsackie virus as well as others can trigger and contribute to the damage of insulin-secreting beta cells in several ways. Furthermore, innate-like bystander T cells such as iNKT, MAIT, and gd T cells also play a pathogenic role in T1D. (C) Majority of T cells in the synovium are antigen non-specific including EBV- and CMV-specific, causing damage to the synovium through the secretion of proinflammatory cytokines. (D) An atherosclerotic plaque lining a blood vessel is rich in ApoB-specific and non-specific T cells.
Lymphopenic conditions following chemo- and radiotherapy represent another source of cytokines, where these conditions result in a significant decrease in the pool of T cells. Consequently, there is an increase in the bioavailability of homeostatic cytokines including IL-7 and IL-15, which results in the expansion of T cells, a process called lymphopenia-induced homeostatic proliferation (LIP) (17, 65–69). Regarding naive CD8 T cells, they can actually proliferate and acquire a memory-like phenotype and functions in response to lymphopenia in a self-peptide/MHC-dependent manner, independent of non-self antigens (Figure 2A) (15, 16, 44, 45, 70–74). Under steady-state conditions, naive CD8 T cells can also acquire a memory-like phenotype in the presence of IL-15 and the transcription factor EOMES (75–77). These antigen-inexperienced cells are termed “virtual memory” T cells (TVM) (10, 75, 77, 78), which accumulate during aging (79–81). Later, it was demonstrated that naive T cells that undergo LIP are phenotypically very similar to TVM (77, 82). In summary, these studies demonstrated the importance of cytokines along with tonic TCR stimulation in the activation of naive CD8 T cells. Hence, we speculate that naive T cells can also undergo another type of bystander activation that is different from memory CD8 T cells, i.e., dependent on both cytokines and the self-peptide/MHC complex.
One can speculate that this type of differential requirement could be attributed to the epigenetic programming specific to each cell population making memory T cells poised for rapid recall of effector responses compared to naive CD8 T cells. In fact, human memory CD8 T cells maintain permissively an open unmethylated state of promoter regions associated with effector molecules and cytokines (e.g., IFNγ and Prf1) during homeostatic proliferation compared to naive CD8 T cells (83, 84). Furthermore, naive CD8 T cells were not able to express IFNγ following classical in-vitro TCR/CD28 stimulation even in the presence of common gamma-chain cytokines (85). However, they upregulate the activation marker CD38 and express IFNγ in the presence of anti-CD3/CD28 antibodies and the proinflammatory cytokine IL-12 (85).
2.1.2 The role of the peptide/MHC complex in naive and memory T-cell activation
As we have discussed earlier, naive CD8 T cells can proliferate and upregulate T-cell activation markers under lymphopenic conditions (Figure 2A), which suggests the importance of cytokines in LIP (86). However, they still require an interaction with the self-peptide MHC complex, where adoptive transfer of FACS-purified P14 naive CD8 T cells (LCMV antigen-specific CD8 T cells) into irradiated MHC-I-deficient mice failed to induce such proliferation and activation compared to WT mice (15). Furthermore, Cho et al. demonstrated that naive CD8 T cells failed to undergo proliferation upon transfer into irradiated MHC-I KO and TAP-1 KO mice compared to WT mice (13). These findings suggest the requirement for a self-peptide/MHC-I complex to drive such proliferation and activation of naive T cells, hinting toward a link between the self-peptide/MHC complex and responsiveness to cytokines. Indeed, Stoklasek et al. demonstrated that adoptive transfer of naive OT-1 CD8 T cells into irradiated MHC-I-deficient mice failed to undergo proliferation even after IL-15/IL15Rα treatment, suggesting the need for MHC-I interaction for an adequate IL-15 response in naive CD8 T cells (87). Along the same lines, LCMV acute infection results in the upregulation of CD122 (IL-2/IL-15Rβ) in CD8 T cells (88). Thus far, the abovementioned observations give us a clue toward the role of the self-peptide/MHC–TCR axis possibly by sensitizing naive cells to be responsive to common gamma chain cytokines. Although it is clear that cytokines play an important role in memory T-cell bystander activation, Goplen et al. demonstrated the activation of the downstream TCR signaling following bystander activation of memory T cells (14). In fact, the proinflammatory cytokines, such as IL-12, TNFα, and IL-15 that typically drive bystander memory T-cell activation, lead to the phosphorylation of CD3ε chain that propagated downstream the signaling pathway of TCR including ZAP70 and its downstream messengers LAT. Hence, this study suggested that proinflammatory cytokines make use of TCR/CD3 signalosome to facilitate bystander effector/memory CD8 T-cell responses. To elucidate the role of MHC in the bystander activation of memory CD8 T cells, the authors adoptively transfer VSV-specific CD8 memory T cells to congenic MHC class I-sufficient or -deficient hosts followed by infection with unrelated infection (Listeria monocytogenes—Lm) showing a reduction in IFNγ expression in MHC-I-deficient mice compared to WT mice (14). These data suggest the importance of MHC-I in the bystander activation of memory CD8 T cells. The obvious question is “Can these cells protect against unrelated infection?” A direct experiment will be the adoptive transfer of antigen-specific memory T cells to naive mice followed by infection with an unrelated pathogen. For instance, Soudja et al. showed that the adoptive transfer of LCMV polyclonal memory CD8 T cells or OT-1 memory CD8 T cells from mice infected with Lm-Ova decreases the bacterial load of Lm in WT mice (89). Similarly, Berg et al. observed protection against Lm using vaccinia virus expressing Ova (VacV-Ova) as a primary infection (23).
3 Biological significance of bystander T cell activation
While bystander T-cell activation is widely described in humans and multiple mouse models (10, 12), the function of bystander T cells could vary from one disease to another, i.e., protective (23, 43, 89) or pathogenic (90–94). Furthermore, other studies did not observe a significant contribution of the process toward pathogen clearance (35, 95, 96). This might be attributed to the masking effect of other cell types including γδ T cells, ILCs, and unconventional memory T cells contributing to the ongoing immune response (10). The lack of a consensus agreement on the function of bystander T cells could result from the usage of different preclinical experimental mouse infection models. In the next sections, we will discuss the role of bystander T cell activation not only during immune responses against pathogens but also in cancer, atherosclerosis, autoimmunity, and transplantation.
3.1 Bystander T cell activation during microbial infections
The occurrence of bystander T-cell activation has been well-documented in human infectious diseases (4). For instance, in patients with primary human immunodeficiency virus (HIV) and hepatitis B virus (HBV) infections, a robust T-cell activation was detected not only in antigen-specific CD8 T cells but also in EBV, CMV, and Flu-specific CD8 T cells (97, 98). Furthermore, HIV patients with interrupted antiretroviral treatment are characterized by activation of antigen-specific and non-specific CTLs (99).
These observations have been attributed to the common gamma chain cytokine IL-15 in the expansion and activation of bystander T cells (98, 100). Yet, the remaining question still remain “What role do these bystander T cells play during infection?” Is it protective or pathogenic? In the case of HBV, bystander-activated CD8 T cells might not contribute to HBV clearance but are implicated in precipitating the immunopathology associated with viral hepatitis. Those cells contribute to the hepatic parenchymal injury, characteristic of chronic viral hepatitis (101). Along the same lines, acute hepatitis A viral (HAV) infection is associated with severe hepatic parenchymal injury that was thought to be precipitated by HAV-specific CD8 T cells (Figure 3A) (102). However, in chimpanzees, HAV-specific CD8 T cells fail to produce significant levels of IFNγ or CD107a post-stimulation with HAV-specific peptides, demonstrating the hypofunction of viral-specific CTLs (103). To further understand the role of CD8 T cells during HAV infection, Kim et al. (55) assessed the specificity of activated CD8 T cells during acute HAV infection. They found that a substantial proportion of activated CD38+ HLA-DR+ CD8 T cells in the periphery of acutely HAV-infected patients were unrelated to HAV but specific against Flu, EBV, CMV, vaccinia virus, and respiratory syncytial virus (55), echoing earlier observations in HIV and HBV patients. Additionally, activation of bystander CMV- and EBV-specific CD8 T cells has been associated with acute pediatric hepatitis E virus (HEV) (104). Furthermore, there was an increase in the frequency of polyclonal Th1 CD4 T cells and CD8 T cells in the peripheral blood (104).
These observations pose the question “What is the mechanism behind the expansion and activation of non-cognate antigen-specific “bystander” T cells? Increasing levels of IL-15 and IL-18 have been observed in the sera of acutely HAV- and HEV-infected patients, suggesting a role of these cytokines in the antigen-independent activation of unrelated virus-specific CD8 T cells in these patients (55, 104). Furthermore, infecting human hepatoma cell lines (HepG2) with HAV resulted in the production of high levels of IL-15, which has been shown to drive HAV-unrelated CD8 T-cell activation (upregulation of NKG2D) in the absence of cognate antigenic stimulation (55). To evaluate the role of these bystander activated CD8 T cells in inducing liver tissue damage, the authors showed that IL-15-activated CD8 T cells isolated from healthy controls and intrahepatic CD8 T cells isolated from the livers of acute hepatitis A (AHA) patients were capable of lysing K562 target cells which do not express MHC class I and also lysing liver-derived Huh-7 cells (55). Furthermore, CMV-specific CD8 T cells isolated from the periphery of AHA patients but not healthy adults were able to lyse K562 cells (55). These results suggested that the hepatic injury associated with AHA infection is TCR independent and can be induced by IL-15-activated HAV-unrelated CD8 T cells in an NKG2D-dependent mechanism. Indeed, the group was able to infer a positive correlation between the proportion of activated HAV-unrelated virus-specific CD8 T cells and liver tissue damage during acute hepatitis A infection.
Another example showing the contribution of bystander T-cell activation to viral infection is the zoonotic viral hemorrhagic fever caused by the Lassa virus which is endemic to western Africa. Using a bone marrow chimera mouse model followed by Lassa viral infection, the authors demonstrated the activation of polyclonal CD8 T cells as well as OT-1 cells which is specific to ovalbumin (105). Along the same lines, in asymptomatic or mild symptomatic coronavirus disease 2019 (COVID-19) patients, there is a burst of COVID-19 non-specific CD8 T cells compared to severe symptomatic patients (Figure 3B). This early expansion of bystander CD8 T cells could play a role in the prevention of disease progression. Harnessing this non-specific response early during COVID-19 could be a better strategy to avoid disease progression (106). This work provided an up-close mechanistic insight on the role of bystander-activated T cells in the immunopathology of a disease.
3.2 Role of bystander T cells in cancer
It is widely accepted that tumor-infiltrating T cells (TILs) are heterogeneous in their specificity, i.e., a hybrid of both tumor-specific and non-specific T cells, where the proportion of tumor antigen-specific CD8 T cells (CTLs) among the TILs varies depending on the tumor type (107–110). For instance, melanoma tumors might harbor 50%–80% tumor-reactive CTLs out of their TILs. On the other hand, TILs of cold tumors such as serous ovarian cancer and microsatellite-stable colorectal cancers lack a tumor-reactive T-cell repertoire (107, 109, 110). The TILs of such tumors harbor some viral-specific T cells such as EBV-reactive CD8 T cells (110). Similarly, glioblastoma, renal cell carcinoma, endometrial carcinoma, head and neck, thyroid, and breast cancers house EBV, CMV, and Flu-specific memory CD8 T cells (108).
3.2.1 Bystander T cell recruitment
Recruitment of these bystander CD8 T cells to the tumor microenvironment is mostly driven by chemokines and inflammatory cues enriched in the tumoral niche and independent of a cognate antigen (111, 112). The chemokines can be secreted from tumor cells such as CCL5 secreted by the cancer cells. Next, CXCL9 produced from IFNγ-stimulated antigen-presenting cells recruits T cells expressing CXCR3. CXCL10 is also involved in the process of recruiting CXCR3+ T cells, which are mostly bystander in nature (113). However, tumor-specific T cells can also express both chemokine receptors (CXCR3 and CCR5), which suggests that the recruitment of bystander T cells is simply a collateral phenomenon accompanied by recruitment of tumor-specific T cells (114). In both scenarios, bystander T cells infiltrate and accumulate in the tumor (111, 112).
3.2.2 Approaches to harness bystander T cells
The detection of non-exhausted viral-specific T cells in various tumors and their correlation with better tumor control (115, 116) encouraged the idea of harnessing their cytotoxicity especially that a lot of tumoral antigens are either self or modified self-antigens that fail to mount a strong TCR stimulation (117–119).
3.2.2.1 Vaccination and infection mouse models
One way to fulfill this aim is by establishing OT-1 chimeras through infecting wild-type naive mice that harbor congenic OT-1 CD8 T cells with VSV-expressing OVA. This approach results in mounting OT-1 memory CD8 T-cell response 30+ days post-infection (108). Injecting VSV viral peptide reactivated the OT-1 memory T cells, which is associated with delayed B16 melanoma growth. Furthermore, treating tumor-bearing mice with systemic anti-PDL-1 antibodies along with VSV-OVA peptide intratumorally resulted in the eradication of B16 tumors in 34% of the mice (108). These data suggested that activation of bystander memory viral-specific T cells enhances antitumor immunity. This effect is even enhanced with PD-L1 blockade. Indeed, using PD-1KO mouse models or blocking the PD-1/PD-L1 axis enhances the functionality of bystander T cells but reduces the longevity of T cells (120). Along the same lines, intratumoral treatment of melanoma with heat-inactivated Flu virus resulted in reduced tumor growth and increased CD8 T-cell infiltration. Such treatment was shown to sensitize “cold” non-immunogenic tumors to checkpoint immunotherapy, rendering them “hot” and responsive tumors (121). Even at a transcriptomic level, Caushi et al. showed the enrichment of virus-specific CD8 T cells in tumor tissues of lung cancer patients who received immune checkpoint blockade. Those viral-specific CD8 T cells expressed effector genes higher than tumor-specific CD8 T cells (122). These results suggested that the concurrence of infection and cancer can lead to the activation of pre-existing viral-specific CD8 T cells in the tumor microenvironment in a TCR-independent manner.
3.2.2.2 Cytokines and chimeric antigen receptor T cell-based approaches
Another way to activate bystander T cells is through cytokine-based immunotherapy and chimeric antigen receptor (CAR) T cells. Indeed, injecting mice-bearing tumors (B16, 3LL, or Renca) with anti-CD40/IL-2 resulted in vigorous expansion of memory CD44hi NKG2D+ CD8 T cells and significant protective anti-tumoral effect (123). Recently, it has been demonstrated that CTLs can kill tumors that downregulate MHC-I through the NKG2D–NKG2DL axis (124). Furthermore, the IL-15-based superagonist (ALT-803) results in an antigen-independent enrichment of innate-like CD8 T cells expressing NKG2D with an antitumor activity (125). In vivo, ALT-803 can eliminate myelomas from the bone marrow providing survival benefit for the treated mice (125). These studies suggest that the anti-tumoral effect can be induced by cytokine-based immunotherapy and is mediated by the expanded NKG2D+ memory CD8 T cells in an MHC-independent manner. Additionally, CAR T cells can be designed in a way specific to both CD3 chain and ubiquitous tumor antigen such as EphA2 (126), which is commonly expressed by glioblastoma, lung, breast, and prostate cancers (127, 128). Hence, these “engager” cells are not only directed toward a tumor but also can activate bystander T cells (126). Along the same lines, using the scRNA-Seq approach, Kaminiski demonstrated for the first time the transcriptome signature of CARneg CD8 bystander T cells following CAR therapy for B-cell leukemia enrich for NK-like markers (CD160, KLRD1, and KIR3DL2) and chemokines and chemokine receptors (CCR9 and CCL5) and less of naive signature (129).
The obvious question here is “Is there any role for bystander CD4 T cells?” Can CD4 and CD8 cooperate to enhance tumor killing? Lee et al. discussed nicely the role of bystander CD4 T cells in infection, autoimmunity, and cancer (130). Furthermore, Joncker et al. co-injected TCR transgenic CD4 and CD8 T cells that can recognize HY male antigen into female mice bearing fibrosarcoma tumors. Following priming of the mice with male cells, there were accumulation and expansion of both Tg CD4 and CD8 T cells, which were considered bystander, into the tumor and tumor-draining lymph nodes (131). Furthermore, Schietinger et al. showed the cooperation between CD4 and CD8 T cells as a requirement for bystander killing of tumor cells using transgenic mouse models (132). Taken together, the aforementioned studies suggest an active contribution of the bystander CD4 and CD8 T cells among TILs to the anti-tumor immunity in mice and humans.
3.3 Bystander T cells and auto- and alloimmunity
Auto- and alloimmune responses are considered two faces for the same coin. In autoimmune disorders, immune responses are dysregulated resulting in the persistence and activation of self-reactive T cells causing disease sequalae (133). However, the initial events and propagation of autoimmunity are still not very well understood. In the case of alloimmunity, graft rejection results from the recognition of non-self antigens associated with the donor organ (134). Hence, both immune responses resulted from the break in self-tolerance, but the cause behind them is different. One is through missing self and the development of autoimmunity, while in the case of graft rejection, it is the recognition of non-self antigens within the donor graft.
3.3.1 Bystander T cells in autoimmune diseases
During a vigorous immune response mediated by an infection, vaccination, or even alloantigens, bystander activation of low-affinity self-reactive CD8 T cells that escaped thymic negative selection seems to be a plausible hypothesis for autoimmunity that has been assessed and studied for the past few decades. For instance, this can be explained by the onset or recurrence of autoimmune disorders following vaccination or solid organ transplantation (135–140). The contribution of bystander activation to autoimmune disease pathogenesis stems from early seminal work on a herpetic stromal keratitis (HSK) preclinical mouse model, an autoimmune disorder resulting from corneal infection with HSV-1. HSV-1-infected OVA transgenic RAG−/− or SCID mice whose TCR is reactive against the OVA peptide only resulted in HSK. These results suggested that autoimmunity does not require a TCR-mediated activation and demonstrated that this activation of T cells is beyond antigen specificity (141, 142).
3.3.1.1 Multiple sclerosis
Multiple sclerosis (MS) is essentially caused by autoimmune-mediated demyelination of neurons of the central nervous system. Disease relapse or exacerbation has been associated with recent microbial infections (143). Although thought to be mainly driven by activated myelin protein self-reactive T cells such as myelin oligodendritic glycoprotein (MOG)-specific T cells that are cross-reactive to microbial epitopes, more recent studies evaluating the pathogenesis of the disease in murine model known as experimental autoimmune encephalitis (EAE) demonstrated that bystander activation of autoreactive T cells stands as an alternative mechanism for MS pathogenesis. For instance, majority of the CD4 T cells infiltrating the central nervous system (CNS) are not specific to MOG (60), where they are mainly expressing cytokines related to Th17 cells including IL-17A, IFNγ, and GM-CSF (Figure 4A). Suppression of these cells can be mediated by Tregs. Indeed, Kim et al. showed that engineered myelin basic protein (MBP)-specific Tregs had the capacity to suppress not only antigen-specific T cells but also non-specific ones (144). Furthermore, other studies showed that Tregs can suppress T cells in an antigen-independent manner (145, 146). The obvious question is “How can these cells get activated in a TCR-independent fashion?” Indeed, the IL-1 family member IL-1β induces CD4 T cells to express GM-CSF and the transcription factor Bhlh40. Furthermore, human CD4 T cells isolated from an MS patient expressed IL-1R1 as well as TLR2 and TLR4, which contributed also to the production of IL-6, IL-17A, IFNγ, and GM-CSF (147). Nogai et al. showed that, in a CD4 T-cell model, treating transgenic mice that only recognize the MBP Ac1-11 peptide with LPS resulted in the development of EAE (148). Intriguingly, CD4 bystander T cells can also play a neuroprotective role as demonstrated by other labs (149). This kind of protection is mainly mediated by IL-4, suggesting that Th2-mediated immune response could be protective against EAE (149). Furthermore, the IL-12 family member IL-27 can induce the upregulation of PD-L1 in CD4 T cells ameliorating EAE (150). The damage of host cells as result of the ongoing immune response can result in the release of new self-antigens, a phenomenon called epitope (determinant) spreading. The recognition of self-antigens results in autoimmunity (151). For instance, in relapsing–remitting experimental autoimmune encephalomyelitis (R-EAE), a model for MS, epitope spreading plays a crucial role in disease exacerbation, where T cells are initially activated by specific immunodominant epitopes, such as PLP (139–151) or MBP (84–104). However, damage to the CNS exposes additional myelin epitopes that were not originally targeted (intermolecular epitope spreading). On the other hand, intramolecular epitope spreading occurs with the same protein. In this scenario, an autoantibody can bind to the same protein but at a different protein region compared to initial binding (152). Furthermore, there are wide varieties of pancreatic tissue antigen GAD-65 and proinsulin in type 1 diabetes patients compared to healthy controls supporting intramolecular epitope spreading (153).
3.3.1.2 Type 1 diabetes
Type 1 diabetes (T1D) is characterized by self-reactive T cells that damage insulin-secreting beta cells in the pancreatic islet of Langerhans (154). Although the initial trigger of the disease is not completely understood, viral infections mediated by the Coxsackie virus as well as other viruses (Figure 4B) can contribute to beta-cell destruction via a) direct infection of beta cells, b) inflammatory cytokines, and c) molecular mimicry where viral epitopes overlap with autoantigens (155–158). Majority of the T cells in the islets are “bystanders” non-specific to islet antigens and mainly play a protective role by reducing the accessibility of the antigen-specific T cells to the autoantigens (159). Furthermore, several studies demonstrated the pathogenic role of innate-like T cells including MAIT, γδ T cells, and iNKT cells in T1D (160–162) (Figure 4B).
3.3.1.3 Rheumatoid arthritis
Bystander activation of CD8 T cells has been proposed in a wide spectrum of autoimmune diseases including rheumatoid arthritis (RA) and celiac disease (57, 163–165). Indeed, EBV- and CMV-specific activated CD8 T cells have been detected in the synovial fluid of rheumatoid arthritis patients (166) (Figure 4C). The activation of these unrelated CD8 T cells is thought to be mediated by a chronic inflammatory environment enriched in the synovial milieu. Indeed, TLR2 activation initiates RA through T cell IFNγ secretion in the absence of a specific antigen (167). Additionally, bacterial lipopolysaccharides can activate osteoclast contributing to the proinflammatory environment (168). Although it has been demonstrated that CD8 T cells expressing TLR4 correlates with disease severity and expression of effector cytokines/molecules (169), other reports suggested their regulatory function (170, 171).
In regard to CD4 T cells, NKG2D+ CD4 T cells are enriched in the peripheral blood and synovium of RA patients (163). The NKG2D ligand MICA/B is upregulated in the synovium in response to proinflammatory cytokines including TNFα, which hints a role of the NKG2D/NKG2DL axis in joint damage (163). RA can develop in another preclinical autoimmune disease model (Sjogren syndrome), suggesting a mechanism of bystander T cell activation via cross-reactivity; however, the authors did not rule out this possibility (172).
3.3.1.4 Atherosclerosis
Over the last few years, the importance of autoimmunity has been widely accepted in the pathogenesis of atherosclerosis, a common pathology underlying cardiovascular diseases (CVDs) characterized by arterial wall plaque formation rich in lipids and immune cells (173). Indeed, atherosclerosis patients harbor circulating autoantibodies against apolipoprotein B containing lipoproteins (ApoB) such as LDL, which indicates loss of self-tolerance (174, 175). This actually served as an impetus for several labs to shift their efforts to understand the mechanisms underlying autoimmunity during atherosclerosis. For instance, two studies by Wang et al. and Depuydt et al. used a combination of scRNA-Seq and TCR-Seq to demonstrate the breakdown of peripheral tolerance in atherosclerosis (176, 177). In the mouse study of Wang et al., the highest degree of plaque-specific T-cell expansion was observed in the cytotoxic CD8 T-cell compartment expressing CD69 and Fosb, both indicating TCR signaling. Similar observations were documented in human subjects with coronary artery disease (CAD) (178). Additionally, the majority of CD8 T-cell clones within aortic plaques shared TCR patterns with those seen in aorta draining lymph nodes (RLNs) of an Apoe−/− mouse atherosclerosis model (176). Other studies revealed the presence of ApoB-specific CD4 T cells in mice and patients with atherosclerosis (Figure 4D).
Although the abovementioned studies strongly suggest the autoimmune nature of atherosclerosis, the trigger of autoimmunity is not completely understood. However, few recent observations in the literature might explain the phenomenon. For instance, episodes of infections are correlated with CAD complications (179, 180). This process might involve CD8 cells found in atherosclerotic plaques (176, 177, 181–188). A small percentage of these cells have shown cross-reactivity to influenza virus, Epstein–Barr virus (EBV), and cytomegalovirus (CMV) in the plaque (189). Along the same lines, de Jong et al. found that ~2% of TCR sequences in the plaques were specific to viral epitopes, where the T cells exhibit signs of activation (190). To investigate if this process happens through molecular mimicry, the authors eluted peptides from MHC-I molecules followed by mass spectrometry. They observed several peptides that could be cross-reactive; however, it has not been tested if these peptides can stimulate T cells from the plaque. An alternative explanation could be the activation of the plaque T cells in an antigen-independent manner, where cytokines secreted during inflammation can activate T cells in the plaque non-specifically.
3.3.1.5 Systemic lupus erythematosus
Systemic lupus erythematosus (SLE) is a complex autoimmune disease characterized by a defect in the clearance of apoptotic cells, which results in the breakdown of self-tolerance and the production of autoantibodies against dsDNA leading to system organ damage including the kidneys (191, 192). Activated bystander CD4 T cells expressing NKG2D secrete IL-10 and TGF-β, suggesting an immunoregulatory role in SLE (193). Furthermore, in vitro, these cells can inhibit T cell proliferation (193). On the contrary, Dermatophagoides pteronyssinus group 2 (Der p2), a main allergen for house dust mite, causes the upregulation of proinflammatory cytokines from PBMCs of allergic SLE patients. This stimulation can activate B cells for autoantibody production in a bystander manner (194).
3.3.2 Bystander T cells in transplantation
In transplantation, the role of bystander T-cell activation has not been widely discussed; however, several studies reported the significant effect of TLR agonists and microbial infections on graft rejection in preclinical mouse models (195–200). For instance, Welsh et al. showed that despite treatment of thymectomized mice with the tolerance induction regimen (co-stimulation blockade, e.g., anti-CD154 and donor-specific leukocyte), which classically induces skin graft survival for more than 100 days, acute LCMV infection precipitates graft rejection (200). Furthermore, TLR agonist treatment results in rapid skin allograft rejection despite the co-stimulation blockade treatment (197, 198). In a human cardiac allograft vasculopathy (CAV) study, the authors suggest the infiltration of bystander T cells into the graft (201).
3.4 Therapeutic implications of bystander T-cell activation
As we have discussed earlier in the previous sections, bystander T cell activation can lead to a pathological versus protective effect depending on the disease context. For instance, acute viral hepatitis infection is associated with liver parenchymal cell damage mainly mediated by bystander T cells (Figure 3A). Conversely, an early burst of bystander T-cell activation is associated with asymptomatic/mild COVID-19 symptoms but absent in patients with severe symptoms (Figure 3B). Hence, it seems plausible to first define the role of bystander T cells in a specific disease setting and then decide to harness or inhibit these cells. Although both disease settings are mediated by viral infections, it is not clear why bystander cells are protective in one scenario versus the other. One can speculate that the inflammatory environment and tissue type discrepancy could play a role.
Since bystander T cells are activated in an antigen-independent manner and lack antigen specificity, it is difficult to target them. However, the NK receptor NKG2D could be one of the surface proteins to target. Indeed, blocking NKG2D in HBV preclinical mouse model reduces liver injury (Figure 5) (202). On the contrary, during Listeria monocytogenes infection, blocking NKG2D exacerbates the disease, suggesting a protective role of NKG2D+ cells specifically CD8 T cells (11, 43). In the case of COVID-19 patients, the phenomenon of early bystander burst T-cell activation can be exploited to avoid disease progression and induce early viral control (106). However, it is not clear why a cohort of patients develop this early bystander activation while others do not.
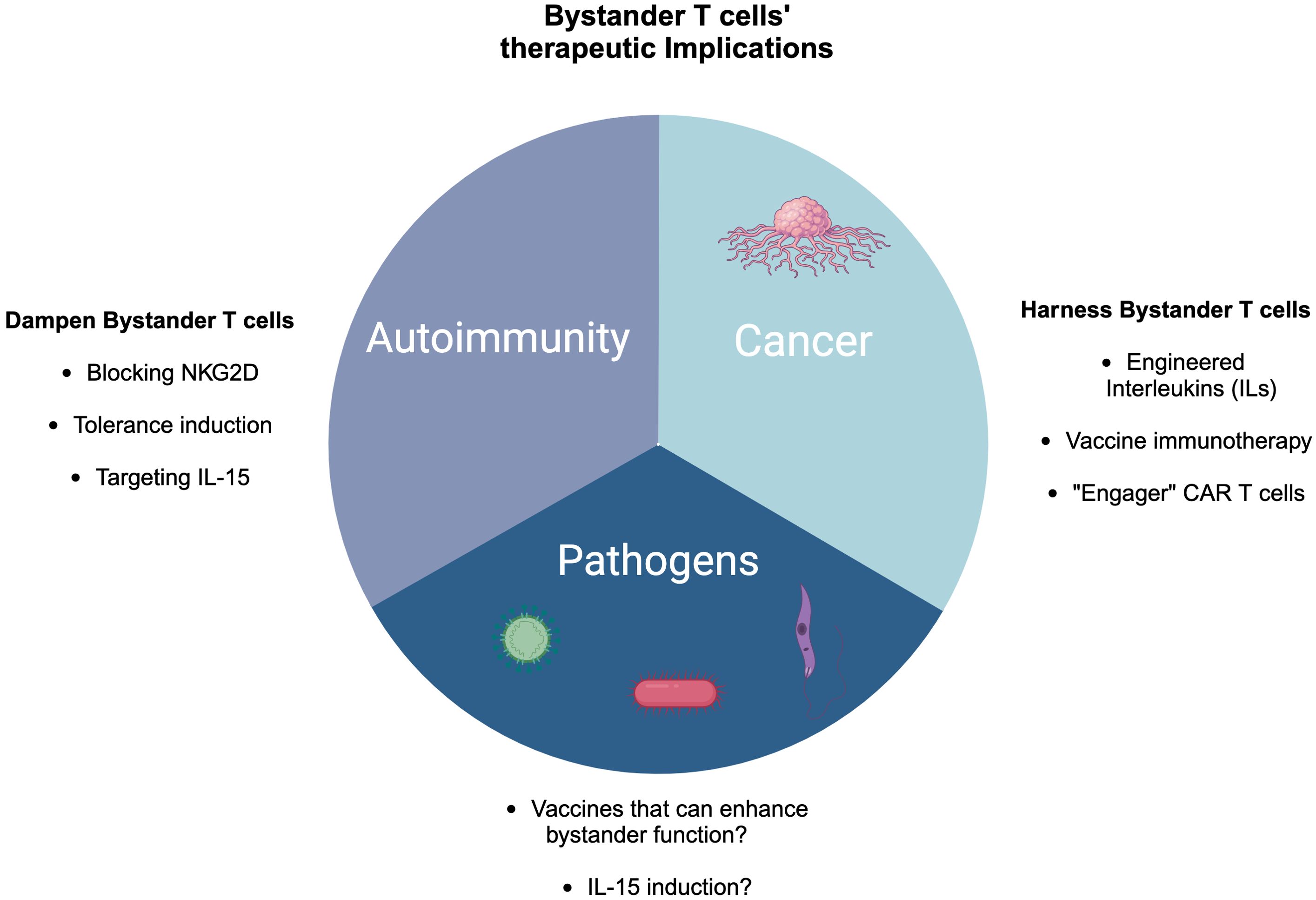
Figure 5. Therapeutic implications of bystander T cells in cancer, pathogen control, and autoimmunity. In cancer, there are several ways to harness bystander T cells including engineered cytokines, vaccine immunotherapy, and engager CAR T cells. In the case of autoimmunity, blocking NKG2D, targeting IL-15, and tolerance induction might be potential approaches to dampen bystander T cells, while in pathogen control, IL-15 induction and developing vaccines could be novel strategies.
The heterogeneity of TILs along with the expression of NKG2D can be exploited in various ways to harness anti-tumor immunity. For instance, cytokine-induced killer cell therapy can be one approach where a cocktail of cytokines including IL-2 and IFNγ can be used to upregulate NK cell receptors such as NKG2D (203, 204). Similarly, engineered interleukins (ILs) such as IL-15 and a combination of IL-2 and anti-CD40 can enhance expansion and induce cancer cell killing via NKG2D in a TCR-independent manner (114, 123, 125, 205, 206) (Figure 5).
Since pathogen-derived peptides are widely expressed by tumors, such as glioblastoma, expressing CMV antigens, while melanoma is rich in bacterial antigens, harnessing viral-specific bystander T cells within the tumor microenvironment via vaccination-based immunotherapies could be an alternative strategy (Figure 5). For example, active influenza vaccine enhanced lung cancer outcomes in both mouse models and human patients (121). Additionally, the study revealed that intratumoral vaccination with heat-inactivated influenza virus notably reduced skin melanoma and improved survival rates (105). CAR T cells with dual TCR (one for the tumor antigen and virus-specific receptor) could even be a better strategy to enhance the efficacy of tumor killing (114, 207) (Figure 5).
3.5 Future directions and concluding remarks
Antigen specificity is the hallmark of the adaptive immune response (208); but, view less the existence of innate memory has been recently described during alloimmune responses (209, 210). However, antigen non-specific T cells can proliferate, get activated, and acquire effector functions in an inflammatory milieu. The latter is referred to as bystander activation. Bystander activation has been always been seen as an alternative TCR-independent activation pathway of memory T cells. Regarding naive T cells, they are responsive to cytokines and require a self-peptide–MHC engagement to induce their activation and proliferation. This phenomenon called homeostatic proliferation, which occurs in lymphopenic conditions such as irradiation-induced bone marrow ablation, HIV infection, and other lymphopenia-inducing processes, could be the naive T-cell counterpart version of bystander activation. The differential requirement of cytokines versus both tonic TCR signaling and cytokines could be attributed to epigenetic programs acquired during the development of both T-cell subsets (naive vs. memory). Although the mechanisms underlying bystander T-cell activation are well documented, the function can vary depending on the human disease setting or the preclinical animal model as we have discussed earlier in several examples such as microbial infection, cancer, autoimmunity, and transplantation.
To exploit the function of bystander T cells in the context of autoimmunity, we can draw parallels from tumor immunity and microbial infections. However, the disease setting is different. For instance, in cancer, several studies as discussed above seek approaches to harness the effector functions of bystander T cells in the tumor microenvironment (Figure 5). Indeed, bystander T cells do not exhibit an exhaustion signature compared to tumor-specific T cells in melanoma patients (116). However, in autoimmunity, strategies should be developed to dampen the cytotoxicity of autoreactive T cells or induce tolerance. Similar to microbial infections, anti-NKG2D could be one approach to inhibit bystander T cells in the context of autoimmunity (Figure 5). However, the approach of one-size-fits-all cannot be applied to autoimmunity since some autoimmune diseases are characterized by immunoregulatory bystander T cells such as in the case of SLE where activated bystander CD4 T cells expressing NKG2D secrete IL-10 and TGF-β (193). Furthermore, bystander CD4 T cells in MS can play a neuroprotective role as well (149). Taken together, utilizing bystander T cells in cell therapy represents a potential opportunity as a future perspective where harnessing tumor immunity is a great example. However, in the case of other diseases such as autoimmunity, the approach should be tailored based on the disease context, the tissue environment, and other factors. Finally, bystander T cells could play an important role in designing novel vaccines, enhancing their efficacy.
Author contributions
MY: Writing – original draft, Writing – review & editing. MD: Writing – original draft, Writing – review & editing. MM: Writing – original draft, Writing – review & editing. HA: Writing – original draft, Writing – review & editing. EA: Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. IMMCG, Medical College of Georgia, Augusta University start up funds were used for publication fees.
Acknowledgments
All figures were created by Biorender Software.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Ruben S. Photosynthesis and phosphorylation. J Am Chem Soc (JACS). (1943) 65:279–82. doi: 10.1021/ja01242a041
2. Banyer JL, Hamilton NH, Ramshaw IA, Ramsay AJ. Cytokines in innate and adaptive immunity. Rev Immunogenet. (2000) 2:359–73.
3. Imanishi J. Expression of cytokines in bacterial and viral infections and their biochemical aspects. J Biochem. (2000) 127:525–30. doi: 10.1093/oxfordjournals.jbchem.a022636
4. Kim TS, Shin EC. The activation of bystander CD8(+) T cells and their roles in viral infection. Exp Mol Med. (2019) 51:1–9. doi: 10.1038/s12276-019-0316-1
5. Tough DF, Borrow P, Sprent J. Induction of bystander T cell proliferation by viruses and type I interferon in vivo. Science. (1996) 272:1947–50. doi: 10.1126/science.272.5270.1947
6. Tough DF, Sprent J. Viruses and T cell turnover: evidence for bystander proliferation. Immunol Rev. (1996) 150:129–42. doi: 10.1111/j.1600-065X.1996.tb00699.x
7. Lehmann PV, Forsthuber T, Miller A, Sercarz EE. Spreading of T-cell autoimmunity to cryptic determinants of an autoantigen. Nature. (1992) 358:155–7. doi: 10.1038/358155a0
8. Ross A, Wu J, Carlock C, Glass W 2nd, Lou YH. Inter-molecular epitope spreading does not lead to extension of autoimmunity beyond target tissue in autoimmune glomerulonephritis. PloS One. (2018) 13:e0202988. doi: 10.1371/journal.pone.0202988
9. Scofield RH, Kaufman KM, Baber U, James JA, Harley JB, Kurien BT. Immunization of mice with human 60-kd Ro peptides results in epitope spreading if the peptides are highly homologous between human and mouse. Arthritis Rheum. (1999) 42:1017–24. doi: 10.1002/1529-0131(199905)42:5<1017::AID-ANR22>3.0.CO;2-7
10. Paprckova D, Salyova E, Michalik J, Stepanek O. Bystander activation in memory and antigen-inexperienced memory-like CD8 T cells. Curr Opin Immunol. (2023) 82:102299. doi: 10.1016/j.coi.2023.102299
11. Whiteside SK, Snook JP, Williams MA, Weis JJ. Bystander T cells: A balancing act of friends and foes. Trends Immunol. (2018) 39:1021–35. doi: 10.1016/j.it.2018.10.003
12. Lee H, Jeong S, Shin EC. Significance of bystander T cell activation in microbial infection. Nat Immunol. (2022) 23:13–22. doi: 10.1038/s41590-021-00985-3
13. Cho JH, Boyman O, Kim HO, Hahm B, Rubinstein MP, Ramsey C, et al. An intense form of homeostatic proliferation of naive CD8+ cells driven by IL-2. J Exp Med. (2007) 204:1787–801. doi: 10.1084/jem.20070740
14. Goplen NP, Saxena V, Knudson KM, Schrum AG, Gil D, Daniels MA, et al. IL-12 signals through the TCR to support CD8 innate immune responses. J Immunol. (2016) 197:2434–43. doi: 10.4049/jimmunol.1600037
15. Murali-Krishna K, Ahmed R. Cutting edge: naive T cells masquerading as memory cells. J Immunol. (2000) 165:1733–7. doi: 10.4049/jimmunol.165.4.1733
16. Goldrath AW, Bogatzki LY, Bevan MJ. Naive T cells transiently acquire a memory-like phenotype during homeostasis-driven proliferation. J Exp Med. (2000) 192:557–64. doi: 10.1084/jem.192.4.557
17. Goldrath AW, Sivakumar PV, Glaccum M, Kennedy MK, Bevan MJ, Benoist C, et al. Cytokine requirements for acute and Basal homeostatic proliferation of naive and memory CD8+ T cells. J Exp Med. (2002) 195:1515–22. doi: 10.1084/jem.20020033
18. Armingol E, Officer A, Harismendy O, Lewis NE. Deciphering cell-cell interactions and communication from gene expression. Nat Rev Genet. (2021) 22:71–88. doi: 10.1038/s41576-020-00292-x
19. Norcross MA. A synaptic basis for T-lymphocyte activation. Ann Immunol (Paris). (1984) 135D:113–34. doi: 10.1016/S0769-2625(84)81105-8
20. Shah K, Al-Haidari A, Sun J, Kazi JU. T cell receptor (TCR) signaling in health and disease. Signal Transduct Target Ther. (2021) 6:412. doi: 10.1038/s41392-021-00823-w
21. Balin SJ, Pellegrini M, Klechevsky E, Won ST, Weiss DI, Choi AW, et al. Human antimicrobial cytotoxic T lymphocytes, defined by NK receptors and antimicrobial proteins, kill intracellular bacteria. Sci Immunol. (2018) 3. doi: 10.1126/sciimmunol.aat7668
22. Beadling C, Slifka MK. Differential regulation of virus-specific T-cell effector functions following activation by peptide or innate cytokines. Blood. (2005) 105:1179–86. doi: 10.1182/blood-2004-07-2833
23. Berg RE, Crossley E, Murray S, Forman J. Memory CD8+ T cells provide innate immune protection against Listeria monocytogenes in the absence of cognate antigen. J Exp Med. (2003) 198:1583–93. doi: 10.1084/jem.20031051
24. Correia MP, Cardoso EM, Pereira CF, Neves R, Uhrberg M, Arosa FA. Hepatocytes and IL-15: a favorable microenvironment for T cell survival and CD8+ T cell differentiation. J Immunol. (2009) 182:6149–59. doi: 10.4049/jimmunol.0802470
25. Correia MP, Costa AV, Uhrberg M, Cardoso EM, Arosa FA. IL-15 induces CD8+ T cells to acquire functional NK receptors capable of modulating cytotoxicity and cytokine secretion. Immunobiology. (2011) 216:604–12. doi: 10.1016/j.imbio.2010.09.012
26. Cottalorda A, Verschelde C, Marcais A, Tomkowiak M, Musette P, Uematsu S, et al. TLR2 engagement on CD8 T cells lowers the threshold for optimal antigen-induced T cell activation. Eur J Immunol. (2006) 36:1684–93. doi: 10.1002/eji.200636181
27. Ge C, Monk IR, Pizzolla A, Wang N, Bedford JG, Stinear TP, et al. Bystander activation of pulmonary trm cells attenuates the severity of bacterial pneumonia by enhancing neutrophil recruitment. Cell Rep. (2019) 29:4236–4244 e3. doi: 10.1016/j.celrep.2019.11.103
28. Kambayashi T, Assarsson E, Lukacher AE, Ljunggren HG, Jensen PE. Memory CD8+ T cells provide an early source of IFN-gamma. J Immunol. (2003) 170:2399–408. doi: 10.4049/jimmunol.170.5.2399
29. Komai-Koma M, Jones L, Ogg GS, Xu D, Liew FY. TLR2 is expressed on activated T cells as a costimulatory receptor. Proc Natl Acad Sci U.S.A. (2004) 101:3029–34.
30. Lertmemongkolchai G, Cai G, Hunter CA, Bancroft GJ. Bystander activation of CD8+ T cells contributes to the rapid production of IFN-gamma in response to bacterial pathogens. J Immunol. (2001) 166:1097–105. doi: 10.4049/jimmunol.166.2.1097
31. Liu K, Catalfamo M, Li Y, Henkart PA, Weng NP. IL-15 mimics T cell receptor crosslinking in the induction of cellular proliferation, gene expression, and cytotoxicity in CD8+ memory T cells. Proc Natl Acad Sci U.S.A. (2002) 99:6192–7.
32. Raue HP, Beadling C, Haun J, Slifka MK. Cytokine-mediated programmed proliferation of virus-specific CD8(+) memory T cells. Immunity. (2013) 38:131–9.
33. Raue HP, Brien JD, Hammarlund E, Slifka MK. Activation of virus-specific CD8+ T cells by lipopolysaccharide-induced IL-12 and IL-18. J Immunol. (2004) 173:6873–81. doi: 10.4049/jimmunol.173.11.6873
34. Ye J, Wang Y, Liu X, Li L, Opejin A, Hsueh EC, et al. TLR7 signaling regulates th17 cells and autoimmunity: novel potential for autoimmune therapy. J Immunol. (2017) 199:941–54. doi: 10.4049/jimmunol.1601890
35. Ehl S, Hombach J, Aichele P, Hengartner H, Zinkernagel RM, et al. Bystander activation of cytotoxic T cells: studies on the mechanism and evaluation of in vivo significance in a transgenic mouse model. J Exp Med. (1997) 185:1241–51. doi: 10.1084/jem.185.7.1241
36. Murali-Krishna K, Altman JD, Suresh M, Sourdive DJ, Zajac AJ, Miller JD, et al. Counting antigen-specific CD8 T cells: a reevaluation of bystander activation during viral infection. Immunity. (1998) 8:177–87. doi: 10.1016/S1074-7613(00)80470-7
37. Zarozinski CC, Welsh RM. Minimal bystander activation of CD8 T cells during the virus-induced polyclonal T cell response. J Exp Med. (1997) 185:1629–39. doi: 10.1084/jem.185.9.1629
38. Yang HY, Dundon PL, Nahill SR, Welsh RM. Virus-induced polyclonal cytotoxic T lymphocyte stimulation. J Immunol. (1989) 142:1710–8. doi: 10.4049/jimmunol.142.5.1710
39. Brehm MA, Pinto AK, Daniels KA, Schneck JP, Welsh RM, Selin LK. T cell immunodominance and maintenance of memory regulated by unexpectedly cross-reactive pathogens. Nat Immunol. (2002) 3:627–34. doi: 10.1038/ni806
40. Chen HD, Fraire AE, Joris I, Brehm MA, Welsh RM, Selin LK. Memory CD8+ T cells in heterologous antiviral immunity and immunopathology in the lung. Nat Immunol. (2001) 2:1067–76. doi: 10.1038/ni727
41. Rehermann B, Shin EC. Private aspects of heterologous immunity. J Exp Med. (2005) 201:667–70. doi: 10.1084/jem.20050220
42. Welsh RM, Selin LK. No one is naive: the significance of heterologous T-cell immunity. Nat Rev Immunol. (2002) 2:417–26. doi: 10.1038/nri820
43. Chu T, Tyznik AJ, Roepke S, Berkley AM, Woodward-Davis A, Pattacini L, et al. Bystander-activated memory CD8 T cells control early pathogen load in an innate-like, NKG2D-dependent manner. Cell Rep. (2013) 3:701–8. doi: 10.1016/j.celrep.2013.02.020
44. Bender J, Mitchell T, Kappler J, Marrack P. CD4+ T cell division in irradiated mice requires peptides distinct from those responsible for thymic selection. J Exp Med. (1999) 190:367–74. doi: 10.1084/jem.190.3.367
45. Goldrath AW, Bevan MJ. Low-affinity ligands for the TCR drive proliferation of mature CD8+ T cells in lymphopenic hosts. Immunity. (1999) 11:183–90. doi: 10.1016/S1074-7613(00)80093-X
46. Matzinger P, Bevan MJ. Hypothesis: why do so many lymphocytes respond to major histocompatibility antigens? Cell Immunol. (1977) 29:1–5.
47. Cunningham MW. Pathogenesis of group A streptococcal infections. Clin Microbiol Rev. (2000) 13:470–511. doi: 10.1128/CMR.13.3.470
48. Galvin JE, Hemric ME, Ward K, Cunningham MW. Cytotoxic mAb from rheumatic carditis recognizes heart valves and laminin. J Clin Invest. (2000) 106:217–24. doi: 10.1172/JCI7132
49. Krisher K, Cunningham MW. Myosin: a link between streptococci and heart. Science. (1985) 227:413–5. doi: 10.1126/science.2578225
50. Seyda M, Elkhal A, Quante M, Falk CS, Tullius SG. T cells going innate. Trends Immunol. (2016) 37:546–56. doi: 10.1016/j.it.2016.06.004
51. Reynolds JM, Dong C. Toll-like receptor regulation of effector T lymphocyte function. Trends Immunol. (2013) 34:511–9. doi: 10.1016/j.it.2013.06.003
52. Rahman AH, Taylor DK, Turka LA. The contribution of direct TLR signaling to T cell responses. Immunol Res. (2009) 45:25–36. doi: 10.1007/s12026-009-8113-x
53. Zhang X, Sun S, Hwang I, Tough DF, Sprent J. Potent and selective stimulation of memory-phenotype CD8+ T cells in vivo by IL-15. Immunity. (1998) 8:591–9. doi: 10.1016/S1074-7613(00)80564-6
54. Yoshimoto T, Takeda K, Tanaka T, Ohkusu K, Kashiwamura S, Okamura H, et al. IL-12 up-regulates IL-18 receptor expression on T cells, Th1 cells, and B cells: synergism with IL-18 for IFN-gamma production. J Immunol. (1998) 161:3400–7. doi: 10.4049/jimmunol.161.7.3400
55. Kim J, Chang DY, Lee HW, Lee H, Kim JH, Sung PS, et al. Innate-like cytotoxic function of bystander-activated CD8(+) T cells is associated with liver injury in acute hepatitis A. Immunity. (2018) 48:161–173 e5. doi: 10.1016/j.immuni.2017.11.025
56. Raulet DH. Roles of the NKG2D immunoreceptor and its ligands. Nat Rev Immunol. (2003) 3:781–90. doi: 10.1038/nri1199
57. Meresse B, Chen Z, Ciszewski C, Tretiakova M, Bhagat G, Krausz TN, et al. Coordinated induction by IL15 of a TCR-independent NKG2D signaling pathway converts CTL into lymphokine-activated killer cells in celiac disease. Immunity. (2004) 21:357–66. doi: 10.1016/j.immuni.2004.06.020
58. Seo IH, Eun HS, Kim JK, Lee H, Jeong S, Choi SJ, et al. IL-15 enhances CCR5-mediated migration of memory CD8(+) T cells by upregulating CCR5 expression in the absence of TCR stimulation. Cell Rep. (2021) 36:109438. doi: 10.1016/j.celrep.2021.109438
59. Lee YK, Landuyt AE, Lobionda S, Sittipo P, Zhao Q, Maynard CL. TCR-independent functions of Th17 cells mediated by the synergistic actions of cytokines of the IL-12 and IL-1 families. PloS One. (2017) 12:e0186351. doi: 10.1371/journal.pone.0186351
60. Lee HG, Lee JU, Kim DH, Lim S, Kang I, Choi JM. Pathogenic function of bystander-activated memory-like CD4(+) T cells in autoimmune encephalomyelitis. Nat Commun. (2019) 10:709. doi: 10.1038/s41467-019-08482-w
61. Guo L, Wei G, Zhu J, Liao W, Leonard WJ, Zhao K, et al. IL-1 family members and STAT activators induce cytokine production by Th2, Th17, and Th1 cells. Proc Natl Acad Sci U.S.A. (2009) 106:13463–8.
62. Chakir H, Lam DK, Lemay AM, Webb JR. Bystander polarization” of CD4+ T cells: activation with high-dose IL-2 renders naive T cells responsive to IL-12 and/or IL-18 in the absence of TCR ligation. Eur J Immunol. (2003) 33:1788–98. doi: 10.1002/eji.200323398
63. Munk RB, Sugiyama K, Ghosh P, Sasaki CY, Rezanka L, Banerjee K, et al. Antigen-independent IFN-gamma production by human naive CD4 T cells activated by IL-12 plus IL-18. PloS One. (2011) 6:e18553.
64. Holmkvist P, Roepstorff K, Uronen-Hansson H, Sanden C, Gudjonsson S, Patschan O, et al. A major population of mucosal memory CD4+ T cells, coexpressing IL-18Ralpha and DR3, display innate lymphocyte functionality. Mucosal Immunol. (2015) 8:545–58. doi: 10.1038/mi.2014.87
65. Bosco N, Agenes F, Ceredig R. Effects of increasing IL-7 availability on lymphocytes during and after lymphopenia-induced proliferation. J Immunol. (2005) 175:162–70. doi: 10.4049/jimmunol.175.1.162
66. Bourgeois C, Stockinger B. T cell homeostasis in steady state and lymphopenic conditions. Immunol Lett. (2006) 107:89–92. doi: 10.1016/j.imlet.2006.08.001
67. Kimura MY, Pobezinsky LA, Guinter TI, Thomas J, Adams A, Park JH, et al. IL-7 signaling must be intermittent, not continuous, during CD8(+) T cell homeostasis to promote cell survival instead of cell death. Nat Immunol. (2013) 14:143–51. doi: 10.1038/ni.2494
68. Martin CE, Spasova DS, Frimpong-Boateng K, Kim HO, Lee M, Kim KS, et al. Interleukin-7 availability is maintained by a hematopoietic cytokine sink comprising innate lymphoid cells and T cells. Immunity. (2017) 47:171–182 e4. doi: 10.1016/j.immuni.2017.07.005
69. Tan JT, Dudl E, LeRoy E, Murray R, Sprent J, Weinberg KI, et al. IL-7 is critical for homeostatic proliferation and survival of naive T cells. Proc Natl Acad Sci U.S.A. (2001) 98:8732–7.
70. Ernst B, Lee DS, Chang JM, Sprent J, Surh CD. The peptide ligands mediating positive selection in the thymus control T cell survival and homeostatic proliferation in the periphery. Immunity. (1999) 11:173–81. doi: 10.1016/S1074-7613(00)80092-8
71. Murali-Krishna K, Lau LL, Sambhara S, Lemonnier F, Altman J, Ahmed R. Persistence of memory CD8 T cells in MHC class I-deficient mice. Science. (1999) 286:1377–81. doi: 10.1126/science.286.5443.1377
72. Tanchot C, Lemonnier FA, Perarnau B, Freitas AA, Rocha B. Differential requirements for survival and proliferation of CD8 naive or memory T cells. Science. (1997) 276:2057–62. doi: 10.1126/science.276.5321.2057
73. Viret C, Wong FS, Janeway CA Jr. Designing and maintaining the mature TCR repertoire: the continuum of self-peptide:self-MHC complex recognition. Immunity. (1999) 10:559–68. doi: 10.1016/S1074-7613(00)80055-2
74. Sprent J, Surh CD. Normal T cell homeostasis: the conversion of naive cells into memory-phenotype cells. Nat Immunol. (2011) 12:478–84. doi: 10.1038/ni.2018
75. Lauvau G, Goriely S. Memory CD8+ T cells: orchestrators and key players of innate immunity? PloS Pathog. (2016) 12:e1005722.
76. Sosinowski T, White JT, Cross EW, Haluszczak C, Marrack P, Gapin L, et al. CD8alpha+ dendritic cell trans presentation of IL-15 to naive CD8+ T cells produces antigen-inexperienced T cells in the periphery with memory phenotype and function. J Immunol. (2013) 190:1936–47. doi: 10.4049/jimmunol.1203149
77. White JT, Cross EW, Kedl RM. Antigen-inexperienced memory CD8(+) T cells: where they come from and why we need them. Nat Rev Immunol. (2017) 17:391–400. doi: 10.1038/nri.2017.34
78. Akue AD, Lee JY, Jameson SC. Derivation and maintenance of virtual memory CD8 T cells. J Immunol. (2012) 188:2516–23. doi: 10.4049/jimmunol.1102213
79. Chiu BC, Martin BE, Stolberg VR, Chensue SW. Cutting edge: Central memory CD8 T cells in aged mice are virtual memory cells. J Immunol. (2013) 191:5793–6. doi: 10.4049/jimmunol.1302509
80. Renkema KR, Li G, Wu A, Smithey MJ, Nikolich-Zugich J. Two separate defects affecting true naive or virtual memory T cell precursors combine to reduce naive T cell responses with aging. J Immunol. (2014) 192:151–9. doi: 10.4049/jimmunol.1301453
81. Rudd BD, Venturi V, Li G, Samadder P, Ertelt JM, Way SS, et al. Nonrandom attrition of the naive CD8+ T-cell pool with aging governed by T-cell receptor:pMHC interactions. Proc Natl Acad Sci U.S.A. (2011) 108:13694–9.
82. Haluszczak C, Akue AD, Hamilton SE, Johnson LD, Pujanauski L, Teodorovic L, et al. The antigen-specific CD8+ T cell repertoire in unimmunized mice includes memory phenotype cells bearing markers of homeostatic expansion. J Exp Med. (2009) 206:435–48. doi: 10.1084/jem.20081829
83. Abdelsamed HA, Moustaki A, Fan Y, Dogra P, Ghoneim HE, Zebley CC, et al. Human memory CD8 T cell effector potential is epigenetically preserved during in vivo homeostasis. J Exp Med. (2017) 214:1593–606. doi: 10.1084/jem.20161760
84. Akondy RS, Fitch M, Edupuganti S, Yang S, Kissick HT, Li KW, et al. Origin and differentiation of human memory CD8 T cells after vaccination. Nature. (2017) 552:362–7. doi: 10.1038/nature24633
85. Zebley CC, Abdelsamed HA, Ghoneim HE, Alli S, Brown C, Haydar D, et al. Proinflammatory cytokines promote TET2-mediated DNA demethylation during CD8 T cell effector differentiation. Cell Rep. (2021) 37:109796. doi: 10.1016/j.celrep.2021.109796
86. Kawabe T, Yi J, Sprent J. Homeostasis of naive and memory T lymphocytes. Cold Spring Harb Perspect Biol. (2021) 13. doi: 10.1101/cshperspect.a037879
87. Stoklasek TA, Colpitts SL, Smilowitz HM, Lefrancois L. MHC class I and TCR avidity control the CD8 T cell response to IL-15/IL-15Ralpha complex. J Immunol. (2010) 185:6857–65. doi: 10.4049/jimmunol.1001601
88. Mathews DV, Dong Y, Higginbotham LB, Kim SC, Breeden CP, Stobert EA, et al. CD122 signaling in CD8+ memory T cells drives costimulation-independent rejection. J Clin Invest. (2018) 128:4557–72. doi: 10.1172/JCI95914
89. Soudja SM, Ruiz AL, Marie JC, Lauvau G. Inflammatory monocytes activate memory CD8(+) T and innate NK lymphocytes independent of cognate antigen during microbial pathogen invasion. Immunity. (2012) 37:549–62. doi: 10.1016/j.immuni.2012.05.029
90. Crosby EJ, Clark M, Novais FO, Wherry EJ, Scott P. Lymphocytic choriomeningitis virus expands a population of NKG2D+CD8+ T cells that exacerbates disease in mice coinfected with leishmania major. J Immunol. (2015) 195:3301–10. doi: 10.4049/jimmunol.1500855
91. Crosby EJ, Goldschmidt MH, Wherry EJ, Scott P. Engagement of NKG2D on bystander memory CD8 T cells promotes increased immunopathology following Leishmania major infection. PloS Pathog. (2014) 10:e1003970. doi: 10.1371/journal.ppat.1003970
92. Whiteside SK, Snook JP, Ma Y, Sonderegger FL, Fisher C, Petersen C, et al. IL-10 deficiency reveals a role for TLR2-dependent bystander activation of T cells in lyme arthritis. J Immunol. (2018) 200:1457–70. doi: 10.4049/jimmunol.1701248
93. Jones RE, Kay T, Keller T, Bourdette D. Nonmyelin-specific T cells accelerate development of central nervous system APC and increase susceptibility to experimental autoimmune encephalomyelitis. J Immunol. (2003) 170:831–7. doi: 10.4049/jimmunol.170.2.831
94. Grifoni A, Weiskopf D, Ramirez SI, Mateus J, Dan JM, Moderbacher CR, et al. Targets of T cell responses to SARS-coV-2 coronavirus in humans with COVID-19 disease and unexposed individuals. Cell. (2020) 181:1489–1501 e15. doi: 10.1016/j.cell.2020.05.015
95. Lee JY, Hamilton SE, Akue AD, Hogquist KA, Jameson SC, et al. Virtual memory CD8 T cells display unique functional properties. Proc Natl Acad Sci U.S.A. (2013) 110:13498–503.
96. Osborn JF, Mooster JL, Hobbs SJ, Munks MW, Barry C, Harty JT, et al. Enzymatic synthesis of core 2 O-glycans governs the tissue-trafficking potential of memory CD8(+) T cells. Sci Immunol. (2017) 2. doi: 10.1126/sciimmunol.aan6049
97. Doisne JM, Urrutia A, Lacabaratz-Porret C, Goujard C, Meyer L, Chaix ML, et al. CD8+ T cells specific for EBV, cytomegalovirus, and influenza virus are activated during primary HIV infection. J Immunol. (2004) 173:2410–8. doi: 10.4049/jimmunol.173.4.2410
98. Sandalova E, Laccabue D, Boni C, Tan AT, Fink K, Ooi EE, et al. Contribution of herpesvirus specific CD8 T cells to anti-viral T cell response in humans. PloS Pathog. (2010) 6:e1001051. doi: 10.1371/journal.ppat.1001051
99. Bastidas S, Graw F, Smith MZ, Kuster H, Gunthard HF, Oxenius A. CD8+ T cells are activated in an antigen-independent manner in HIV-infected individuals. J Immunol. (2014) 192:1732–44. doi: 10.4049/jimmunol.1302027
100. Younes SA, Freeman ML, Mudd JC, Shive CL, Reynaldi A, Panigrahi S, et al. IL-15 promotes activation and expansion of CD8+ T cells in HIV-1 infection. J Clin Invest. (2016) 126:2745–56. doi: 10.1172/JCI85996
101. Shin EC, Sung PS, Park SH. Immune responses and immunopathology in acute and chronic viral hepatitis. Nat Rev Immunol. (2016) 16:509–23. doi: 10.1038/nri.2016.69
102. Wang M, Feng Z. Mechanisms of hepatocellular injury in hepatitis A. Viruses. (2021) 13. doi: 10.3390/v13050861
103. Zhou Y, Callendret B, Xu D, Brasky KM, Feng Z, Hensley LL, et al. Dominance of the CD4(+) T helper cell response during acute resolving hepatitis A virus infection. J Exp Med. (2012) 209:1481–92. doi: 10.1084/jem.20111906
104. Morita A, Imagawa K, Iwasaki T, Yaita K, Sakai A, Takada H. Activation of bystander CD8(+) T cells in a pediatric patient with acute hepatitis E. Immunol Med. (2024) p:1–7. doi: 10.1080/25785826.2024.2378542
105. Port JR, Wozniak DM, Oestereich L, Pallasch E, Becker-Ziaja B, Muller J, et al. Severe human lassa fever is characterized by nonspecific T-cell activation and lymphocyte homing to inflamed tissues. J Virol. (2020) 94. doi: 10.1128/JVI.01367-20
106. Bergamaschi L, Mescia F, Turner L, Hanson AL, Kotagiri P, Dunmore BJ, et al. Longitudinal analysis reveals that delayed bystander CD8+ T cell activation and early immune pathology distinguish severe COVID-19 from mild disease. Immunity. (2021) 54:1257–1275 e8. doi: 10.1016/j.immuni.2021.05.010
107. Kvistborg P, Shu CJ, Heemskerk B, Fankhauser M, Thrue CA, Toebes M, et al. TIL therapy broadens the tumor-reactive CD8(+) T cell compartment in melanoma patients. Oncoimmunology. (2012) 1:409–18.
108. Rosato PC, Wijeyesinghe S, Stolley JM, Nelson CE, Davis RL, Manlove LS, et al. Virus-specific memory T cells populate tumors and can be repurposed for tumor immunotherapy. Nat Commun. (2019) 10:567. doi: 10.1038/s41467-019-08534-1
109. Scheper W, Kelderman S, Fanchi LF, Linnemann C, Bendle G, de Rooij MAJ, et al. Low and variable tumor reactivity of the intratumoral TCR repertoire in human cancers. Nat Med. (2019) 25:89–94. doi: 10.1038/s41591-018-0266-5
110. Simoni Y, Becht E, Fehlings M, Loh CY, Koo SL, Teng KWW, et al. Bystander CD8(+) T cells are abundant and phenotypically distinct in human tumour infiltrates. Nature. (2018) 557:575–9. doi: 10.1038/s41586-018-0130-2
111. Kohli K, Pillarisetty VG, Kim TS. Key chemokines direct migration of immune cells in solid tumors. Cancer Gene Ther. (2022) 29:10–21. doi: 10.1038/s41417-021-00303-x
112. Dangaj D, Bruand M, Grimm AJ, Ronet C, Barras D, Duttagupta PA, et al. Cooperation between constitutive and inducible chemokines enables T cell engraftment and immune attack in solid tumors. Cancer Cell. (2019) 35:885–900 e10. doi: 10.1016/j.ccell.2019.05.004
113. Maurice NJ, McElrath MJ, Andersen-Nissen E, Frahm N, Prlic M. CXCR3 enables recruitment and site-specific bystander activation of memory CD8(+) T cells. Nat Commun. (2019) 10:4987. doi: 10.1038/s41467-019-12980-2
114. Meier SL, Satpathy AT, Wells DK. Bystander T cells in cancer immunology and therapy. Nat Cancer. (2022) 3:143–55. doi: 10.1038/s43018-022-00335-8
115. Danahy DB, Berton RR, Badovinac VP. Cutting edge: antitumor immunity by pathogen-specific CD8 T cells in the absence of cognate antigen recognition. J Immunol. (2020) 204:1431–5. doi: 10.4049/jimmunol.1901172
116. Oliveira G, Stromhaug K, Klaeger S, Kula T, Frederick DT, Le PM, et al. Phenotype, specificity and avidity of antitumour CD8(+) T cells in melanoma. Nature. (2021) 596:119–25. doi: 10.1038/s41586-021-03704-y
117. Cole DK, Pumphrey NJ, Boulter JM, Sami M, Bell JI, Gostick E, et al. Human TCR-binding affinity is governed by MHC class restriction. J Immunol. (2007) 178:5727–34. doi: 10.4049/jimmunol.178.9.5727
118. Hay ZLZ, Knapp JR, Magallon RE, O'Connor BP, Slansky JE. Low TCR binding strength results in increased progenitor-like CD8+ Tumor-infiltrating lymphocytes. Cancer Immunol Res. (2023) 11:570–82. doi: 10.1158/2326-6066.CIR-22-0761
119. Wei P, Jordan KR, Buhrman JD, Lei J, Deng H, Marrack P, et al. Structures suggest an approach for converting weak self-peptide tumor antigens into superagonists for CD8 T cells in cancer. Proc Natl Acad Sci U.S.A. (2021) 118. doi: 10.1073/pnas.2100588118
120. Le CT, Vick LV, Collins C, Dunai C, Sheng MK, Khuat LT, et al. Regulation of human and mouse bystander T cell activation responses by PD-1. JCI Insight. (2023) 8. doi: 10.1172/jci.insight.173287
121. Newman JH, Chesson CB, Herzog NL, Bommareddy PK, Aspromonte SM, Pepe R, et al. Intratumoral injection of the seasonal flu shot converts immunologically cold tumors to hot and serves as an immunotherapy for cancer. Proc Natl Acad Sci U.S.A. (2020) 117:1119–28.
122. Caushi JX, Zhang J, Ji Z, Vaghasia A, Zhang B, Hsiue EH, et al. Transcriptional programs of neoantigen-specific TIL in anti-PD-1-treated lung cancers. Nature. (2021) 596:126–32. doi: 10.1038/s41586-021-03752-4
123. Tietze JK, Wilkins DE, Sckisel GD, Bouchlaka MN, Alderson KL, Weiss JM, et al. Delineation of antigen-specific and antigen-nonspecific CD8(+) memory T-cell responses after cytokine-based cancer immunotherapy. Blood. (2012) 119:3073–83. doi: 10.1182/blood-2011-07-369736
124. Lerner EC, Woroniecka KI, D'Anniballe VM, Wilkinson DS, Mohan AA, Lorrey SJ, et al. CD8(+) T cells maintain killing of MHC-I-negative tumor cells through the NKG2D-NKG2DL axis. Nat Cancer. (2023) 4:1258–72. doi: 10.1038/s43018-023-00600-4
125. Wong HC, Jeng EK, Rhode PR. The IL-15-based superagonist ALT-803 promotes the antigen-independent conversion of memory CD8(+) T cells into innate-like effector cells with antitumor activity. Oncoimmunology. (2013) 2:e26442.
126. Iwahori K, Kakarla S, Velasquez MP, Yu F, Yi Z, Gerken C, et al. Engager T cells: a new class of antigen-specific T cells that redirect bystander T cells. Mol Ther. (2015) 23:171–8. doi: 10.1038/mt.2014.156
127. Biao-xue R, Xi-guang C, Shuan-ying Y, Wei L, Zong-juan M. EphA2-dependent molecular targeting therapy for Malignant tumors. Curr Cancer Drug Targets. (2011) 11:1082–97. doi: 10.2174/156800911798073050
128. Wykosky J, Debinski W. The EphA2 receptor and ephrinA1 ligand in solid tumors: function and therapeutic targeting. Mol Cancer Res. (2008) 6:1795–806. doi: 10.1158/1541-7786.MCR-08-0244
129. Kaminski J, Fleming RA, Alvarez-Calderon F, Winschel MB, McGuckin C, Ho EE, et al. B-cell-directed CAR T-cell therapy activates CD8+ cytotoxic CARneg bystander T cells in patients and nonhuman primates. Blood. (2024) 144:46–60. doi: 10.1182/blood.2023022717
130. Lee HG, Cho MJ, Choi JM. Bystander CD4(+) T cells: crossroads between innate and adaptive immunity. Exp Mol Med. (2020) 52:1255–63. doi: 10.1038/s12276-020-00486-7
131. Joncker NT, Marloie MA, Chernysheva A, Lonchay C, Cuff S, Klijanienko J, et al. Antigen-independent accumulation of activated effector/memory T lymphocytes into human and murine tumors. Int J Cancer. (2006) 118:1205–14. doi: 10.1002/ijc.v118:5
132. Schietinger A, Philip M, Liu RB, Schreiber K, Schreiber H. Bystander killing of cancer requires the cooperation of CD4(+) and CD8(+) T cells during the effector phase. J Exp Med. (2010) 207:2469–77. doi: 10.1084/jem.20092450
133. Kronenberg M, Rudensky A. Regulation of immunity by self-reactive T cells. Nature. (2005) 435:598–604. doi: 10.1038/nature03725
134. Lakkis FG, Sayegh MH. Memory T cells: a hurdle to immunologic tolerance. J Am Soc Nephrol. (2003) 14:2402–10. doi: 10.1097/01.ASN.0000085020.78117.70
135. Hashimoto E, Shimada M, Noguchi S, Taniai M, Tokushige K, Hayashi N. Disease recurrence after living liver transplantation for primary biliary cirrhosis: a clinical and histological follow-up study. Liver Transpl. (2001) 7:588–95. doi: 10.1053/jlts.2001.25357
136. Hubscher SG, Elias E, Buckels JA, Mayer AD, McMaster P, Neuberger JM. Primary biliary cirrhosis. Histological evidence of disease recurrence after liver transplantation. J Hepatol. (1993) 18:173–84. doi: 10.1016/S0168-8278(05)80244-2
137. Liermann Garcia RF, Evangelista Garcia C, McMaster P, Neuberger J. Transplantation for primary biliary cirrhosis: retrospective analysis of 400 patients in a single center. Hepatology. (2001) 33:22–7. doi: 10.1053/jhep.2001.20894
138. Neuberger J, Portmann B, Calne R, Williams R. Recurrence of autoimmune chronic active hepatitis following orthotopic liver grafting. Transplantation. (1984) 37:363–5. doi: 10.1097/00007890-198404000-00009
139. Sylvestre PB, Batts KP, Burgart LJ, Poterucha JJ, Wiesner RH. Recurrence of primary biliary cirrhosis after liver transplantation: Histologic estimate of incidence and natural history. Liver Transpl. (2003) 9:1086–93. doi: 10.1053/jlts.2003.50213
140. Vadala M, Poddighe D, Laurino C, Palmieri B. Vaccination and autoimmune diseases: is prevention of adverse health effects on the horizon? EPMA J. (2017) 8:295–311. doi: 10.1007/s13167-017-0101-y
141. Banerjee K, Biswas PS, Kumaraguru U, Schoenberger SP, Rouse BT. Protective and pathological roles of virus-specific and bystander CD8+ T cells in herpetic stromal keratitis. J Immunol. (2004) 173:7575–83. doi: 10.4049/jimmunol.173.12.7575
142. Gangappa S, Babu JS, Thomas J, Daheshia M, Rouse BT. Virus-induced immunoinflammatory lesions in the absence of viral antigen recognition. J Immunol. (1998) 161:4289–300. doi: 10.4049/jimmunol.161.8.4289
143. Marrodan M, Alessandro L, Farez MF, Correale J. The role of infections in multiple sclerosis. Mult Scler. (2019) 25:891–901. doi: 10.1177/1352458518823940
144. Kim YC, Zhang AH, Yoon J, Culp WE, Lees JR, Wucherpfennig KW, et al. Engineered MBP-specific human Tregs ameliorate MOG-induced EAE through IL-2-triggered inhibition of effector T cells. J Autoimmun. (2018) 92:77–86. doi: 10.1016/j.jaut.2018.05.003
145. Karim M, Feng G, Wood KJ, Bushell AR. CD25+CD4+ regulatory T cells generated by exposure to a model protein antigen prevent allograft rejection: antigen-specific reactivation in vivo is critical for bystander regulation. Blood. (2005) 105:4871–7. doi: 10.1182/blood-2004-10-3888
146. Thornton AM, Shevach EM. Suppressor effector function of CD4+CD25+ immunoregulatory T cells is antigen nonspecific. J Immunol. (2000) 164:183–90. doi: 10.4049/jimmunol.164.1.183
147. Ferreira TB, Hygino J, Wing AC, Kasahara TM, Sacramento PM, Camargo S, et al. Different interleukin-17-secreting Toll-like receptor(+) T-cell subsets are associated with disease activity in multiple sclerosis. Immunology. (2018) 154:239–52. doi: 10.1111/imm.2018.154.issue-2
148. Nogai A, Siffrin V, Bonhagen K, Pfueller CF, Hohnstein T, Volkmer-Engert R, et al. Lipopolysaccharide injection induces relapses of experimental autoimmune encephalomyelitis in nontransgenic mice via bystander activation of autoreactive CD4+ cells. J Immunol. (2005) 175:959–66. doi: 10.4049/jimmunol.175.2.959
149. Walsh JT, Hendrix S, Boato F, Smirnov I, Zheng J, Lukens JR, et al. MHCII-independent CD4+ T cells protect injured CNS neurons via IL-4. J Clin Invest. (2015) 125:699–714. doi: 10.1172/JCI76210
150. Hirahara K, Ghoreschi K, Yang XP, Takahashi H, Laurence A, Vahedi G, et al. Interleukin-27 priming of T cells controls IL-17 production in trans via induction of the ligand PD-L1. Immunity. (2012) 36:1017–30. doi: 10.1016/j.immuni.2012.03.024
151. Vanderlugt CJ, Miller SD. Epitope spreading. Curr Opin Immunol. (1996) 8:831–6. doi: 10.1016/S0952-7915(96)80012-4
152. Poole BD, Schneider RI, Guthridge JM, Velte CA, Reichlin M, Harley JB, et al. Early targets of nuclear RNP humoral autoimmunity in human systemic lupus erythematosus. Arthritis Rheum. (2009) 60:848–59. doi: 10.1002/art.24306
153. Ott PA, Dittrich MT, Herzog BA, Guerkov R, Gottlieb PA, Putnam AL, et al. T cells recognize multiple GAD65 and proinsulin epitopes in human type 1 diabetes, suggesting determinant spreading. J Clin Immunol. (2004) 24:327–39. doi: 10.1023/B:JOCI.0000029120.77824.41
154. Dwyer AJ, Shaheen ZR, Fife BT. Antigen-specific T cell responses in autoimmune diabetes. Front Immunol. (2024) 15:1440045. doi: 10.3389/fimmu.2024.1440045
155. Jun HS, Yoon JW. The role of viruses in type I diabetes: two distinct cellular and molecular pathogenic mechanisms of virus-induced diabetes in animals. Diabetologia. (2001) 44:271–85. doi: 10.1007/s001250051614
156. Isaacs SR, Foskett DB, Maxwell AJ, Ward EJ, Faulkner CL, Luo JYX, et al. Viruses and type 1 diabetes: from enteroviruses to the virome. Microorganisms. (2021) 9. doi: 10.20944/preprints202106.0574.v1
157. Lloyd RE, Tamhankar M, Lernmark A. Enteroviruses and type 1 diabetes: multiple mechanisms and factors? Annu Rev Med. (2022) 73:483–99.
158. Padgett LE, Broniowska KA, Hansen PA, Corbett JA, Tse HM. The role of reactive oxygen species and proinflammatory cytokines in type 1 diabetes pathogenesis. Ann N Y Acad Sci. (2013) 1281:16–35. doi: 10.1111/j.1749-6632.2012.06826.x
159. Christoffersson G, Chodaczek G, Ratliff SS, Coppieters K, von Herrath MG. Suppression of diabetes by accumulation of non-islet-specific CD8(+) effector T cells in pancreatic islets. Sci Immunol. (2018) 3. doi: 10.1126/sciimmunol.aam6533
160. Simoni Y, Beaudoin L, Bui LC, Michel ML, Coumoul X, et al. NOD mice contain an elevated frequency of iNKT17 cells that exacerbate diabetes. Eur J Immunol. (2011) 41:3574–85. doi: 10.1002/eji.201141751
161. Markle JG, Mortin-Toth S, Wong AS, Geng L, Hayday A, Danska JS. gammadelta T cells are essential effectors of type 1 diabetes in the nonobese diabetic mouse model. J Immunol. (2013) 190:5392–401. doi: 10.4049/jimmunol.1203502
162. Rouxel O, Da Silva J, Beaudoin L, Nel I, Tard C, Cagninacci L, et al. Cytotoxic and regulatory roles of mucosal-associated invariant T cells in type 1 diabetes. Nat Immunol. (2017) 18:1321–31. doi: 10.1038/ni.3854
163. Groh V, Bruhl A, El-Gabalawy H, Nelson JL, Spies T. Stimulation of T cell autoreactivity by anomalous expression of NKG2D and its MIC ligands in rheumatoid arthritis. Proc Natl Acad Sci U.S.A. (2003) 100:9452–7.
164. Horwitz MS, Bradley LM, Harbertson J, Krahl T, Lee J, Sarvetnick N. Diabetes induced by Coxsackie virus: initiation by bystander damage and not molecular mimicry. Nat Med. (1998) 4:781–5. doi: 10.1038/nm0798-781
165. Pane JA, Coulson BS. Lessons from the mouse: potential contribution of bystander lymphocyte activation by viruses to human type 1 diabetes. Diabetologia. (2015) 58:1149–59. doi: 10.1007/s00125-015-3562-3
166. Tan LC, Mowat AG, Fazou C, Rostron T, Roskell H, Dunbar PR, et al. Specificity of T cells in synovial fluid: high frequencies of CD8(+) T cells that are specific for certain viral epitopes. Arthritis Res. (2000) 2:154–64. doi: 10.1186/ar80
167. Sobek V, Birkner N, Falk I, Wurch A, Kirschning CJ, Wagner H, et al. Direct Toll-like receptor 2 mediated co-stimulation of T cells in the mouse system as a basis for chronic inflammatory joint disease. Arthritis Res Ther. (2004) 6:R433–46. doi: 10.1186/ar1212
168. Brennan FM, McInnes IB. Evidence that cytokines play a role in rheumatoid arthritis. J Clin Invest. (2008) 118:3537–45. doi: 10.1172/JCI36389
169. Tripathy A, Khanna S, Padhan P, Smita S, Raghav S, Gupta B. Direct recognition of LPS drive TLR4 expressing CD8(+) T cell activation in patients with rheumatoid arthritis. Sci Rep. (2017) 7:933. doi: 10.1038/s41598-017-01033-7
170. Carvalheiro H, da Silva JA, Souto-Carneiro MM. Potential roles for CD8(+) T cells in rheumatoid arthritis. Autoimmun Rev. (2013) 12:401–9. doi: 10.1016/j.autrev.2012.07.011
171. Petrelli A, van Wijk F. CD8(+) T cells in human autoimmune arthritis: the unusual suspects. Nat Rev Rheumatol. (2016) 12:421–8. doi: 10.1038/nrrheum.2016.74
172. Kobayashi M, Yasui N, Ishimaru N, Arakaki R, Hayashi Y. Development of autoimmune arthritis with aging via bystander T cell activation in the mouse model of Sjogren’s syndrome. Arthritis Rheum. (2004) 50:3974–84. doi: 10.1002/art.v50:12
173. Roy P, Orecchioni M, Ley K. How the immune system shapes atherosclerosis: roles of innate and adaptive immunity. Nat Rev Immunol. (2022) 22:251–65. doi: 10.1038/s41577-021-00584-1
174. Tsimikas S, Brilakis ES, Lennon RJ, Miller ER, Witztum JL, McConnell JP, et al. Relationship of IgG and IgM autoantibodies to oxidized low density lipoprotein with coronary artery disease and cardiovascular events. J Lipid Res. (2007) 48:425–33. doi: 10.1194/jlr.M600361-JLR200
175. Tsimikas S, Palinski W, Witztum JL. Circulating autoantibodies to oxidized LDL correlate with arterial accumulation and depletion of oxidized LDL in LDL receptor-deficient mice. Arterioscler Thromb Vasc Biol. (2001) 21:95–100. doi: 10.1161/01.ATV.21.1.95
176. Wang Z, Zhang X, Lu S, Zhang C, Ma Z, Su R, et al. Pairing of single-cell RNA analysis and T cell antigen receptor profiling indicates breakdown of T cell tolerance checkpoints in atherosclerosis. Nat Cardiovasc Res. (2023) 2:290–306. doi: 10.1038/s44161-023-00218-w
177. Depuydt MAC, Schaftenaar FH, Prange KHM, Boltjes A, Hemme E, Delfos L, et al. Single-cell T cell receptor sequencing of paired human atherosclerotic plaques and blood reveals autoimmune-like features of expanded effector T cells. Nat Cardiovasc Res. (2023) 2:112–25. doi: 10.1038/s44161-022-00208-4
178. Iqneibi S, Saigusa R, Khan A, Oliaeimotlagh M, Armstrong Suthahar SS, Kumar S, et al. Single cell transcriptomics reveals recent CD8T cell receptor signaling in patients with coronary artery disease. Front Immunol. (2023) 14:1239148. doi: 10.3389/fimmu.2023.1239148
179. Campbell LA, Rosenfeld ME. Infection and atherosclerosis development. Arch Med Res. (2015) 46:339–50. doi: 10.1016/j.arcmed.2015.05.006
180. Eberhardt N, Noval MG, Kaur R, Amadori L, Gildea M, Sajja S, et al. SARS-CoV-2 infection triggers pro-atherogenic inflammatory responses in human coronary vessels. Nat Cardiovasc Res. (2023) 2:899–916. doi: 10.1038/s44161-023-00336-5
181. Depuydt MAC, Prange KHM, Slenders L, Ord T, Elbersen D, Boltjes A, et al. Microanatomy of the human atherosclerotic plaque by single-cell transcriptomics. Circ Res. (2020) 127:1437–55. doi: 10.1161/CIRCRESAHA.120.316770
182. Fernandez DM, Rahman AH, Fernandez NF, Chudnovskiy A, Amir ED, Amadori L, et al. Single-cell immune landscape of human atherosclerotic plaques. Nat Med. (2019) 25:1576–88. doi: 10.1038/s41591-019-0590-4
183. Gewaltig J, Kummer M, Koella C, Cathomas G, Biedermann BC. Requirements for CD8 T-cell migration into the human arterial wall. Hum Pathol. (2008) 39:1756–62. doi: 10.1016/j.humpath.2008.04.018
184. Grivel JC, Ivanova O, Pinegina N, Blank PS, Shpektor A, Margolis LB, et al. Activation of T lymphocytes in atherosclerotic plaques. Arterioscler Thromb Vasc Biol. (2011) 31:2929–37. doi: 10.1161/ATVBAHA.111.237081
185. Jonasson L, Holm J, Skalli O, Bondjers G, Hansson GK. Regional accumulations of T cells, macrophages, and smooth muscle cells in the human atherosclerotic plaque. Arteriosclerosis. (1986) 6:131–8. doi: 10.1161/01.ATV.6.2.131
186. Kyaw T, Winship A, Tay C, Kanellakis P, Hosseini H, Cao A, et al. Cytotoxic and proinflammatory CD8+ T lymphocytes promote development of vulnerable atherosclerotic plaques in apoE-deficient mice. Circulation. (2013) 127:1028–39. doi: 10.1161/CIRCULATIONAHA.112.001347
187. Stemme S, Holm J, Hansson GK. T lymphocytes in human atherosclerotic plaques are memory cells expressing CD45RO and the integrin VLA-1. Arterioscler Thromb. (1992) 12:206–11. doi: 10.1161/01.ATV.12.2.206
188. Zernecke A, Winkels H, Cochain C, Williams JW, Wolf D, Soehnlein O, et al. Meta-analysis of leukocyte diversity in atherosclerotic mouse aortas. Circ Res. (2020) 127:402–26. doi: 10.1161/CIRCRESAHA.120.316903
189. Chowdhury RR, D'Addabbo J, Huang X, Veizades S, Sasagawa K, Louis DM, et al. Human coronary plaque T cells are clonal and cross-react to virus and self. Circ Res. (2022) 130:1510–30. doi: 10.1161/CIRCRESAHA.121.320090
190. de Jong MJM, Schaftenaar FH, Depuydt MAC, Lozano Vigario F, Janssen GMC, Peeters J, et al. Virus-associated CD8(+) T-cells are not activated through antigen-mediated interaction inside atherosclerotic lesions. Arterioscler Thromb Vasc Biol. (2024) 44:1302–14. doi: 10.1161/ATVBAHA.123.320539
191. Bakshi J, Segura BT, Wincup C, Rahman A. Unmet needs in the pathogenesis and treatment of systemic lupus erythematosus. Clin Rev Allergy Immunol. (2018) 55:352–67. doi: 10.1007/s12016-017-8640-5
192. Choi J, Kim ST, Craft J. The pathogenesis of systemic lupus erythematosus-an update. Curr Opin Immunol. (2012) 24:651–7. doi: 10.1016/j.coi.2012.10.004
193. Dai Z, Turtle CJ, Booth GC, Riddell SR, Gooley TA, Stevens AM, et al. Normally occurring NKG2D+CD4+ T cells are immunosuppressive and inversely correlated with disease activity in juvenile-onset lupus. J Exp Med. (2009) 206:793–805. doi: 10.1084/jem.20081648
194. Yu SJ, Liao EC, Tsai JJ. Der p 2 can induce bystander activation of B cells derived from patients with systemic lupus erythematosus. Immunobiology. (2014) 219:958–63. doi: 10.1016/j.imbio.2014.07.018
195. Ahmed EB, Wang T, Daniels M, Alegre ML, Chong AS. IL-6 induced by Staphylococcus aureus infection prevents the induction of skin allograft acceptance in mice. Am J Transplant. (2011) 11:936–46. doi: 10.1111/j.1600-6143.2011.03476.x
196. Chen L, Wang T, Zhou P, Ma L, Yin D, Shen J, et al. TLR engagement prevents transplantation tolerance. Am J Transplant. (2006) 6:2282–91. doi: 10.1111/j.1600-6143.2006.01489.x
197. Thornley TB, Brehm MA, Markees TG, Shultz LD, Mordes JP, Welsh RM, et al. TLR agonists abrogate costimulation blockade-induced prolongation of skin allografts. J Immunol. (2006) 176:1561–70. doi: 10.4049/jimmunol.176.3.1561
198. Thornley TB, Phillips NE, Beaudette-Zlatanova BC, Markees TG, Bahl K, Brehm MA, et al. Type 1 IFN mediates cross-talk between innate and adaptive immunity that abrogates transplantation tolerance. J Immunol. (2007) 179:6620–9. doi: 10.4049/jimmunol.179.10.6620
199. Wang T, Chen L, Ahmed E, Ma L, Yin D, Zhou P, et al. Prevention of allograft tolerance by bacterial infection with Listeria monocytogenes. J Immunol. (2008) 180:5991–9. doi: 10.4049/jimmunol.180.9.5991
200. Welsh RM, Markees TG, Woda BA, Daniels KA, Brehm MA, Mordes JP, et al. Virus-induced abrogation of transplantation tolerance induced by donor-specific transfusion and anti-CD154 antibody. J Virol. (2000) 74:2210–8. doi: 10.1128/JVI.74.5.2210-2218.2000
201. Habal MV, Miller AMI, Rao S, Lin S, Obradovic A, Khosravi-Maharlooei M, et al. T cell repertoire analysis suggests a prominent bystander response in human cardiac allograft vasculopathy. Am J Transplant. (2021) 21:1465–76. doi: 10.1111/ajt.16333
202. Vilarinho S, Ogasawara K, Nishimura S, Lanier LL, Baron JL. Blockade of NKG2D on NKT cells prevents hepatitis and the acute immune response to hepatitis B virus. Proc Natl Acad Sci U.S.A. (2007) 104:18187–92.
203. Pievani A, Borleri G, Pende D, Moretta L, Rambaldi A, Golay J. Dual-functional capability of CD3+CD56+ CIK cells, a T-cell subset that acquires NK function and retains TCR-mediated specific cytotoxicity. Blood. (2011) 118:3301–10. doi: 10.1182/blood-2011-02-336321
204. Zhang Y, Schmidt-Wolf IGH. Ten-year update of the international registry on cytokine-induced killer cells in cancer immunotherapy. J Cell Physiol. (2020) 235:9291–303. doi: 10.1002/jcp.v235.12
205. Hu J, Zhu S, Xia X, Zhang L, Kleinerman ES, Li S. CD8+T cell-specific induction of NKG2D receptor by doxorubicin plus interleukin-12 and its contribution to CD8+T cell accumulation in tumors. Mol Cancer. (2014) 13:34. doi: 10.1186/1476-4598-13-34
206. Wrangle JM, Velcheti V, Patel MR, Garrett-Mayer E, Hill EG, Ravenel JG, et al. ALT-803, an IL-15 superagonist, in combination with nivolumab in patients with metastatic non-small cell lung cancer: a non-randomised, open-label, phase 1b trial. Lancet Oncol. (2018) 19:694–704. doi: 10.1016/S1470-2045(18)30148-7
207. Pule MA, Savoldo B, Myers GD, Rossig C, Russell HV, Dotti G, et al. Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblastoma. Nat Med. (2008) 14:1264–70. doi: 10.1038/nm.1882
208. Tippalagama R, Chihab LY, Kearns K, Lewis S, Panda S, Willemsen L, et al. Antigen-specificity measurements are the key to understanding T cell responses. Front Immunol. (2023) 14:1127470. doi: 10.3389/fimmu.2023.1127470
209. Dai H, Lan P, Zhao D, Abou-Daya K, Liu W, Chen W. PIRs mediate innate myeloid cell memory to nonself MHC molecules. Science. (2020) 368:1122–7. doi: 10.1126/science.aax4040
Keywords: T cells, bystander, activation, cytokines, peptide/MHC complex, cross-reactivity
Citation: Yosri M, Dokhan M, Aboagye E, Al Moussawy M and Abdelsamed HA (2024) Mechanisms governing bystander activation of T cells. Front. Immunol. 15:1465889. doi: 10.3389/fimmu.2024.1465889
Received: 17 July 2024; Accepted: 31 October 2024;
Published: 27 November 2024.
Edited by:
Derk Amsen, Sanquin Research, NetherlandsReviewed by:
Govindarajan Thangavelu, University of Minnesota Twin Cities, United StatesJae Ho Cho, Chonnam National University Medical School, Republic of Korea
Anjie Zhen, UCLA Department of Medicine, United States
Copyright © 2024 Yosri, Dokhan, Aboagye, Al Moussawy and Abdelsamed. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mouhamad Al Moussawy, bW1vdXNzYXd5QGdtYWlsLmNvbQ==; Hossam A. Abdelsamed, aGFiZGVsc2FtZWRAYXVndXN0YS5lZHU=