 Van-Thanh Duong
Van-Thanh Duong Dongjun Lee
Dongjun Lee Yun Hak Kim
Yun Hak Kim Sae-Ock Oh
Sae-Ock Oh
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 14 October 2024
Sec. Primary Immunodeficiencies
Volume 15 - 2024 | https://doi.org/10.3389/fimmu.2024.1460882
UNC13 family (also known as Munc13) proteins are evolutionarily conserved proteins involved in the rapid and regulated secretion of vesicles, including synaptic vesicles and cytotoxic granules. Fast and regulated secretion at the neuronal and immunological synapses requires multiple steps, from the biogenesis of vesicles to membrane fusion, and a complex array of proteins for each step. Defects at these steps can lead to various genetic disorders. Recent studies have shown multiple roles of UNC13D in the secretion of cytotoxic granules by immune cells. Here, the molecular structure and detailed roles of UNC13D in the biogenesis, tethering, and priming of cytotoxic vesicles and in endoplasmic reticulum are summarized. Moreover, its association with immune diseases, including familial hemophagocytic lymphohistiocytosis type 3, macrophage activation syndrome, juvenile idiopathic arthritis, and autoimmune lymphoproliferative syndrome, is reviewed. Finally, the therapeutic application of CRISPR/Cas9-based gene therapy for genetic diseases is introduced.
Fast and regulated secretion of vesicles is critical for neurotransmission at the neuronal synapse and for defense at the immunological synapse (1–4). This requires multiple steps, from the biogenesis of vesicles to membrane fusion, and a complex array of proteins for each step. The UNC13 family (also known as Munc13) proteins are evolutionarily conserved proteins responsible for rapid and regulated secretion of vesicles, including synaptic vesicles and cytotoxic granules (5–8). Their expression and roles in various tissues, including the nervous and immune systems, have been documented previously (5, 6, 9, 10). In addition, their pathological roles have been observed in genetic diseases, such as familial hemophagocytic lymphohistiocytosis, and various types of cancers (11–13). UNC13 family proteins have been observed in primitive animals, even before the emergence of neurons (14, 15). Four isoforms of the UNC13 family have been identified in mammals; these include UNC13A, UNC13B, UNC13C, and UNC13D (16). UNC13A and UNC13C are specifically expressed in nervous tissues. UNC13B is transcribed into two principal isoforms via alternative splicing—ubUNC13B is ubiquitously expressed, whereas bUNC13B is restricted to the brain (9, 17). Northern blotting and quantitative reverse transcription-PCR analysis revealed that UNC13D expression is widespread, with strong expression in hematopoietic cells, including CD4 and CD8 T lymphocytes, CD19 B lymphocytes, natural killer (NK) cells, and CD14 monocytes. Regarding non-hematopoietic cells, UNC13D exhibits strong signals in the lungs, liver, pancreas, and placenta, whereas only modest signals are observed in the brain, heart, skeletal muscle, and kidney (6). A broad review of the UNC13 family members has been published previously (5, 16). The present study focuses on recent progress in research on the role of UNC13D in the immune system.
Structural characteristics of the UNC13 family members have been described previously (8, 9). Although the core domains of UNC13 members are conserved in UNC13D and brain-specific angiogenesis inhibitor 1 associated protein 3, they exhibit structural differences from the cognate UNC13 proteins because they lack three specific domains—an N-terminal C2 domain (C2A), a calmodulin-binding domain, and a diacylglycerol-binding C1 domain (Figure 1). The calmodulin-binding domain is located between the C2A and C1 domains (not marked in Figure 1 for simplicity). In addition to these three characteristic differences, UNC13D has fewer disordered regions than the other family members, which suggests that it is more rigid than other members (Figure 1).
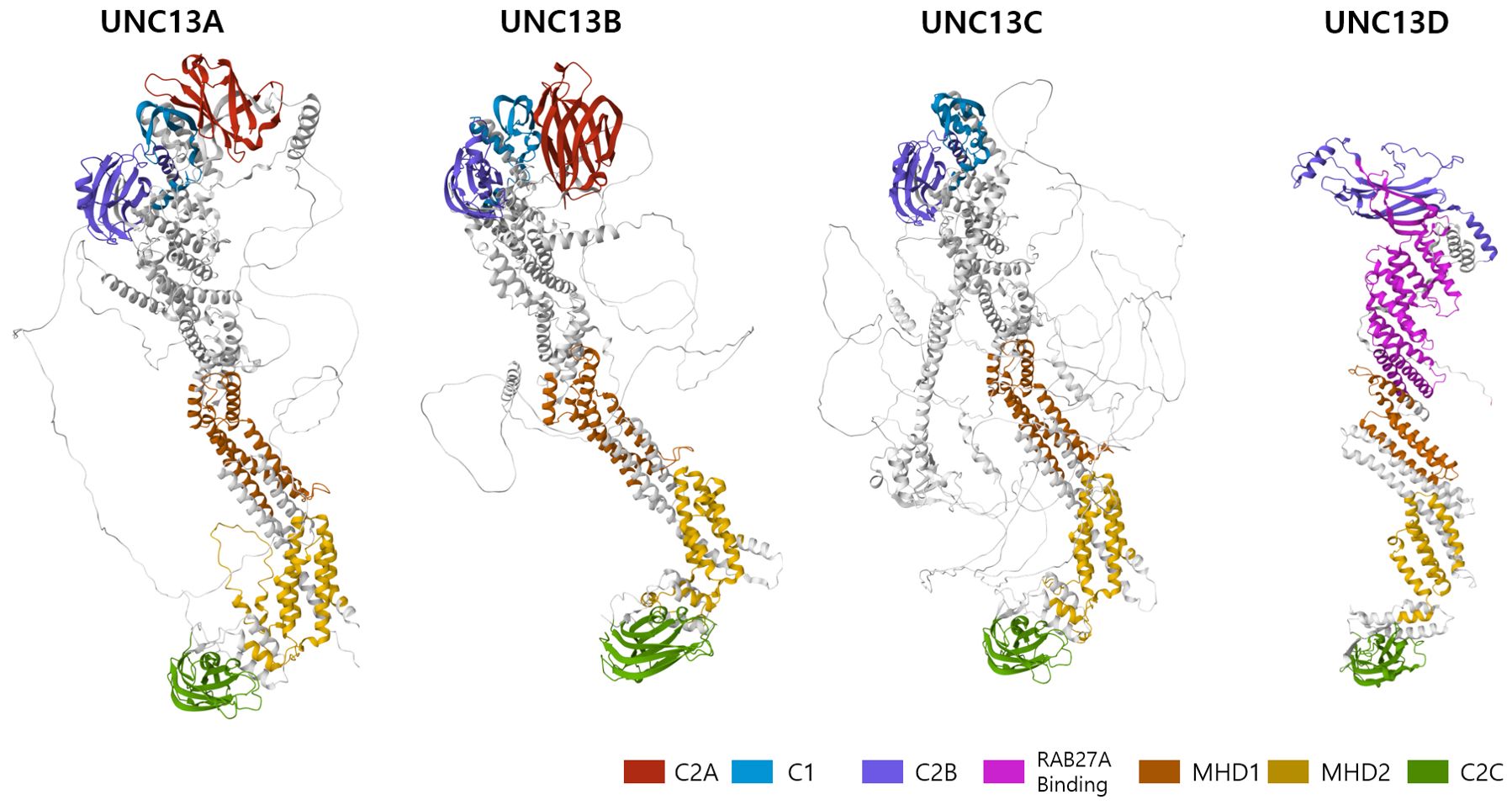
Figure 1. 3D structure of the human UNC13 family members predicted by AlphaFold (https://alphafoldserver.com/). Critical domains are indicated by color code.
Structurally, UNC13D contains two C2 domains separated by long sequences that contain two regions known as Munc13-homology domains (MHD1 and MHD2) and RAB27A binding domain (Figure 1) (8). These domains display distinctive topologies based on the circular permutation of their β strands, wherein the N- and C-termini remain either at the top or bottom of the C2-β sandwich. Type I topology is found in the first C2 domain (C2A), which is comparable to cPLA2 and PLCδ1. Type II topology is found in the second C2 domain (C2B), similar to that encountered in the C2A and C2B domains of synaptotagmin 1 as well as in rabphilin (18, 19). In addition, the MHD-containing region, which separates the two C2 domains, is entirely made up of α helices, some of which have specific lengths and distinct heptad repeats, indicating structures generating coils. UNC13D and its partners may interact through this hypothesized coil-forming structure (6). Furthermore, amino acids 240–543, which constitute the C-terminal end of the C2B domain, are necessary for binding to Rab27a (Figure 1). In particular, residue 280FQLIHK285 is crucial for binding (20).
Unlike other UNC13 proteins, UNC13D does not have the diacylglycerol-binding C1 domain that is essential for neuronal priming. However, the two C2 domains shared by UNC13D and all other UNC13 homologues have been implicated in vesicle priming and exocytosis (8). UNC13D is a versatile protein that associates with numerous molecules and performs diverse functions. Apart from Rab27a, a well-known interacting partner of UNC13D, other Rab GTPases, such as Rab7, Rab11, and Rab37, interact for vesicle trafficking. Several studies have reported that UNC13D is associated with Cd63, Stk24, Ccm3, Doc2 alpha, and S100A10, which regulate exocytosis (Table 1).
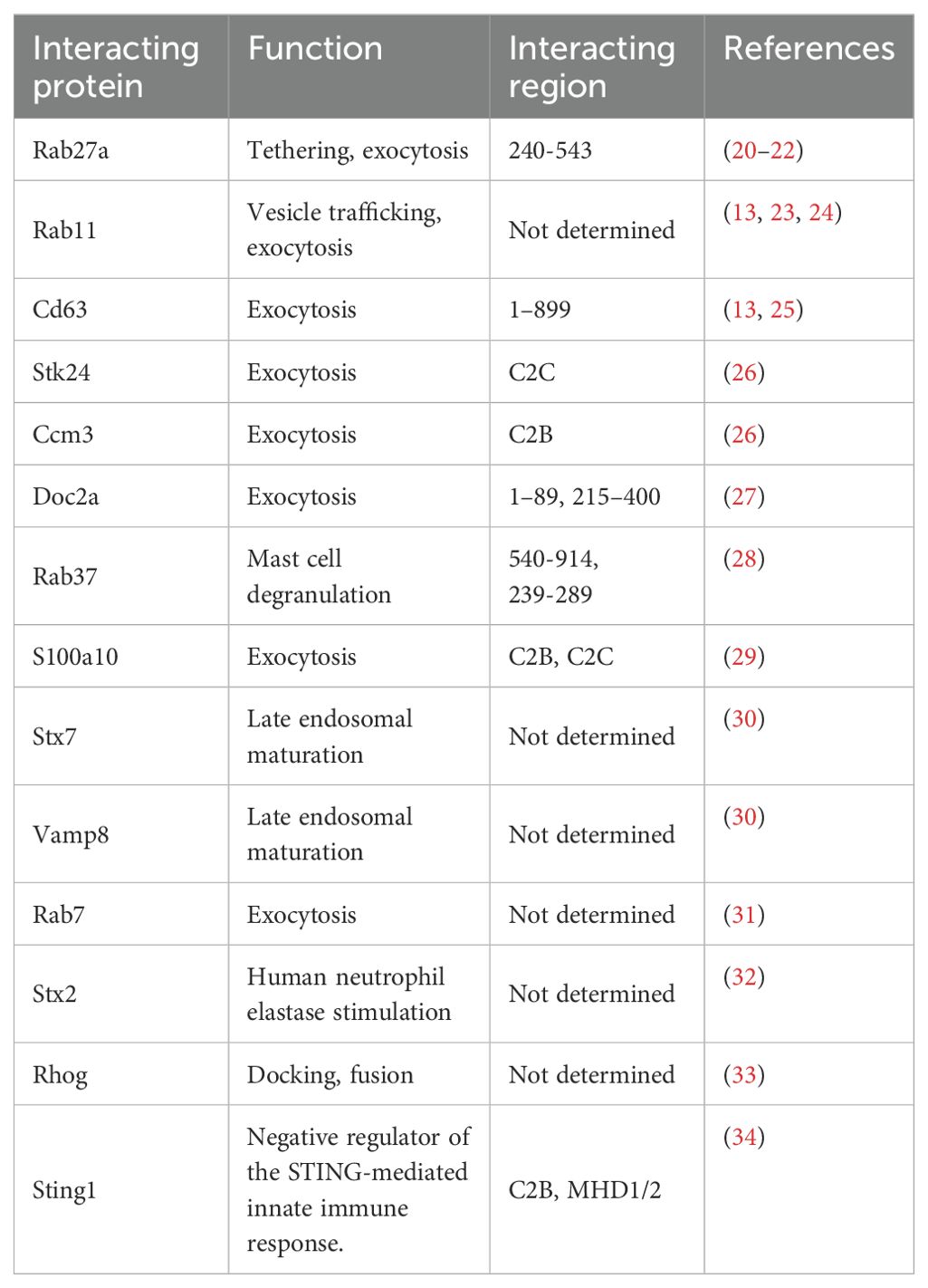
Table 1. UNC13D and its binding proteins.
To destroy the target cells, immune cells (e.g., NK and CD8 T cells) secrete cytotoxic granules into them (7). The multistep generation of cytotoxic granules containing perforin, granzymes, and granulysin has been documented using electron microscopy and kinetic tracer uptake experiments (35–37). This cytotoxic process is triggered by the recognition of target cells via immune receptors and plasma membrane (PM) proteins (38). Cytotoxic T lymphocytes (CTLs) are activated through the interaction of T-cell receptors (TCR) with MHC class I molecules and the recognition of antigenic peptides derived from the target cells. NK cell cytotoxicity is initiated by the ligation of activating receptors and is regulated by the recognition of self MHC class I molecules. Cytotoxic lymphocytes employ two distinct pathways to perform their effector functions. The first step involves the binding of the Fas ligand on CTLs and NK cells to its receptor on the target cell, leading to oligomerization of the receptor and apoptosis of the target cells (39). The cytotoxic lytic granule pathway, also known as the second pathway, is a rapid and efficient exocytic transport pathway for vesicular structures that carry lytic molecules such as perforin and granzymes (4). The early stage in cytotoxic granule generation involves merging of lysosomes with ‘exocytic vesicles’ (Figure 2) (20, 24, 40). In cytotoxic T cells, the exocytic vesicles originate from the fusion of late endosomes, containing Rab27a and Rab7, with recycling endosomes, containing Rab11 and UNC13D. During the fusion of the late and recycling endosomes, UNC13D, localized in the recycling endosome, plays a critical role by interacting with Rab27a, localized in the late endosome. Similarly, in NK cells, CD16, NKG2D, and 2B4 recruit UNC13D to the cytotoxic granules (41). In addition, UNC13D localizes to Rab7+ and Rab27b+ vesicles and merges with Rab11+ endosomes to generate UNC13D+/Rab7+/Rab11+ endosomal vesicles in mast cells (31). Nevertheless, the mechanism underlying UNC13D localization needs to be further explored.
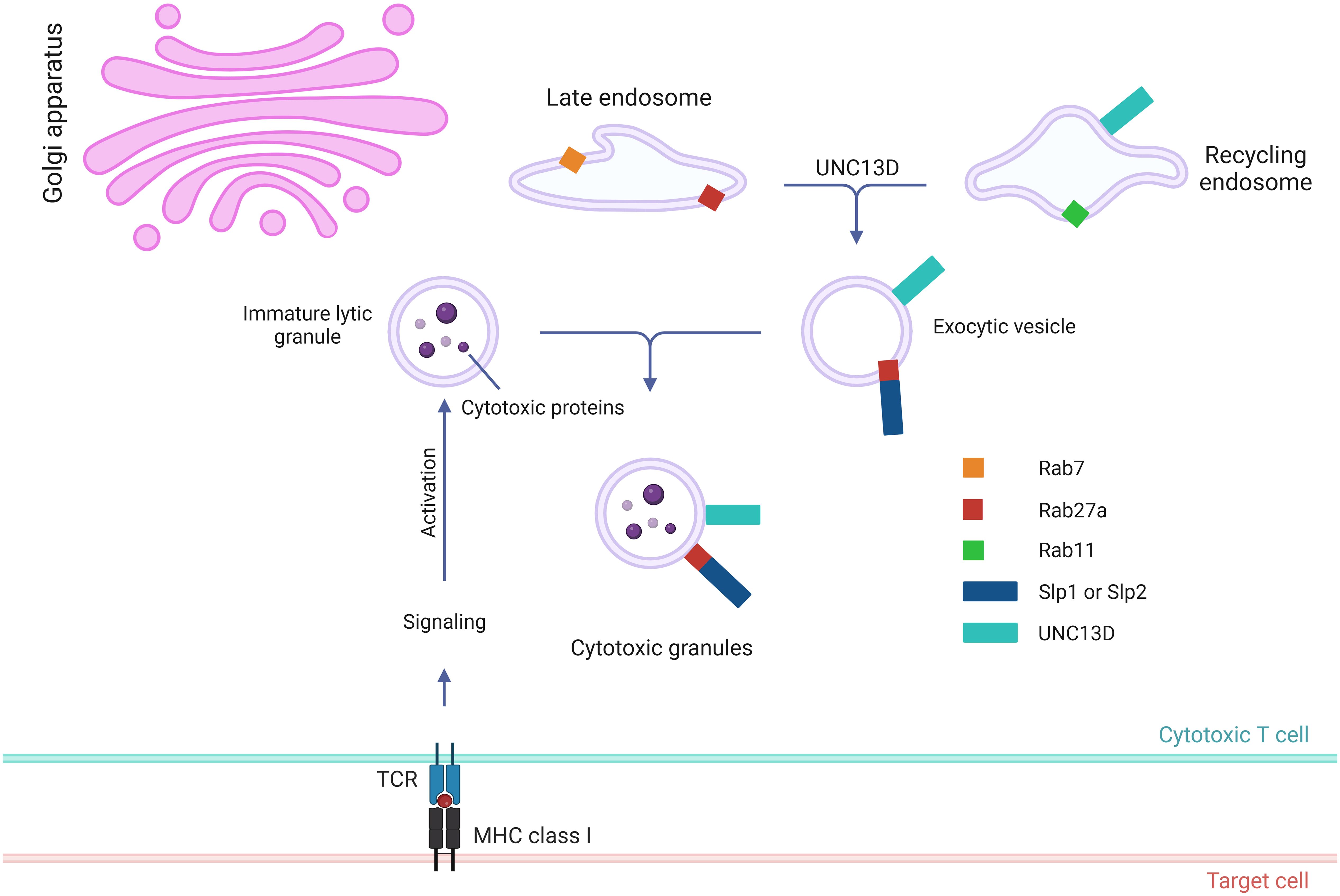
Figure 2. Role of UNC13D in the biogenesis of cytotoxic granules. Stepwise genesis of cytotoxic granules from late and recycling endosomes via exocytic vesicle is presented. Cytotoxic proteins, such as granzymes, perforin, and granulysin, are synthesized in the endoplasmic reticulum before being transported to the Golgi apparatus. UNC13D is responsible for coordinating the subsequent interaction between recycling endosomes via Rab11 and late endosomes via Rab7 to generate exocytic vesicles that are specific to the regulated secretory pathway. These vesicles contain effector proteins essential for exocytosis, such as UNC13D, Rab27a, and synaptotagmin-like proteins (Slps).
After the generation of cytotoxic granules, which are meant to fuse with the PM, they need to be transported around the immunological synapse to create a readily releasable pool for the secretion (Figure 3). To maintain this pool, the cytotoxic granules must be loosely docked (tethered) to increase their dwelling time (42) near the PM. During this tethering step, prior to tight docking by soluble NSF attachment protein receptor (SNARE) proteins, Rab27a proteins and their effector molecules play critical roles (43). The interaction between Rab27a and Slp2a, a synaptotagmin-like protein, is critical for localizing cytotoxic granules near the PM via C2 domains (44, 45). The finding that UNC13D has C2 domains that can associate with PM phospholipids and interact with Rab27a suggests that UNC13D regulates the tethering of cytotoxic granules to the PM. The UNC13D mutant, which cannot bind to Rab27a, severely impairs the stalling behavior upon the initiation of immune receptor signaling (20). In addition, UNC13D restricts cytotoxic granules to the PM after the lipopolysaccharide stimulation in neutrophils (46). Because the binding of C2 domains of UNC13D to PM phospholipids requires Ca2+ (47) and no significant defects are observed in the tethering step in patients with familial hemophagocytic lymphohistiocytosis type 3 (FHL3) having UNC13D mutations (6), further studies are required to elucidate the role of UNC13D in the tethering step.
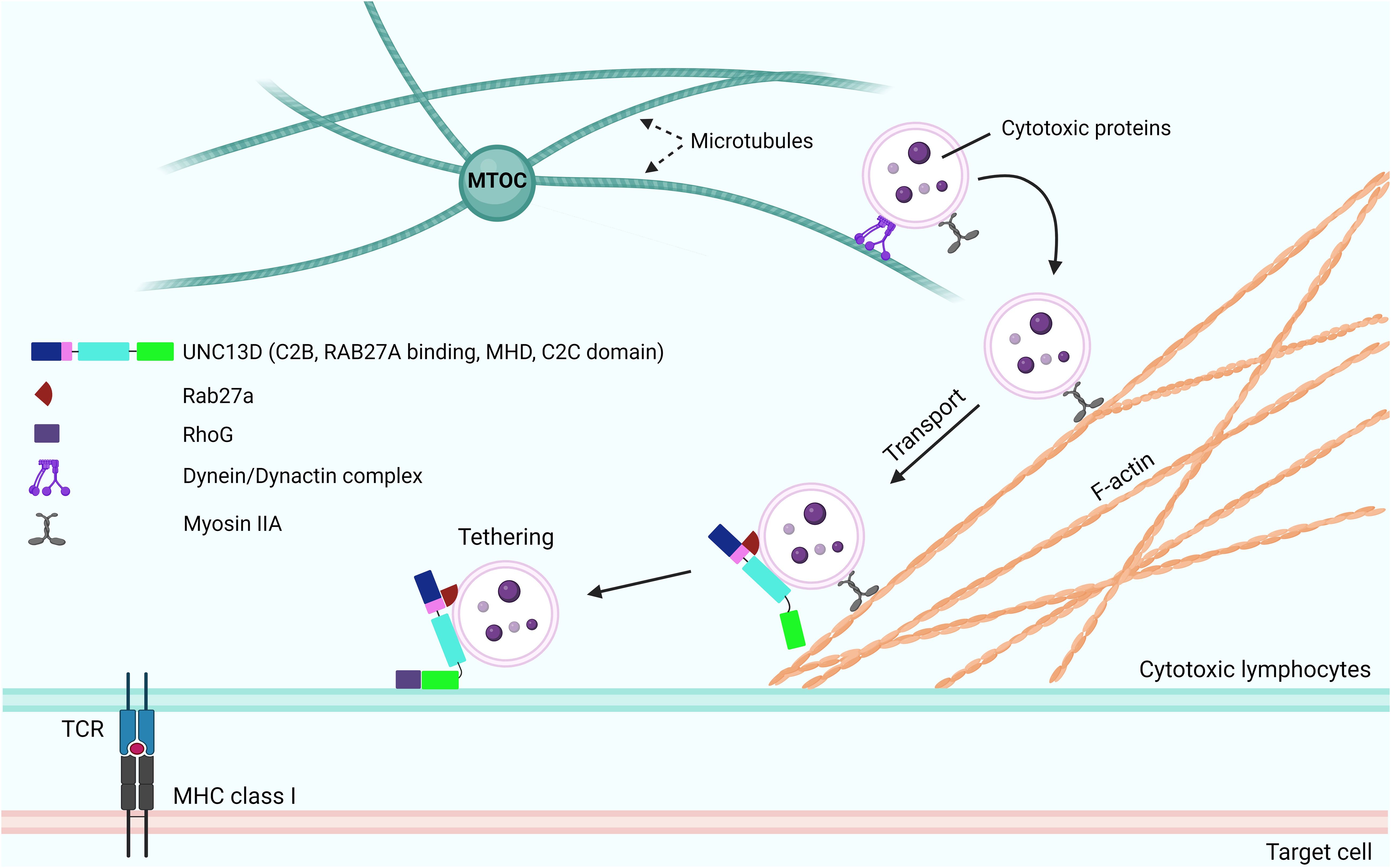
Figure 3. Role of UNC13D in the tethering and docking of cytotoxic granules. Following the activation of receptors of cytotoxic lymphocytes and initiation of signaling cascades, the cytotoxic granules are transported toward the MTOC along microtubules in a dynein-dynactin complex-dependent manner. The MTOC and granules polarize toward the contact area of the target cell, where the granules switch from microtubules to an F-actin network, at the immunological synapse. Subsequently, they move through the cortical filamentous actin meshwork because of the activity of the actin motor protein myosin IIA. Through the involvement of Rab27a and RhoG, the cytotoxic granules can transport and attach to the plasma membrane. While UNC13D interacts with Rab27a and associates with lytic granules through its MHD domains, the tethering of docked granules to the plasma membrane is orchestrated by UNC13D, which interacts with RhoG through its phospholipid-binding C2C domain. MTOC, microtubule organizing center; F-actin, filamentous actin; MHDs, Munc-homology domains; RhoG, Ras Homolog Family Member G.
Recently, Kalinichenko et al. identified RhoG as a new binding partner of UNC13D (33). They reported that the protein-protein interaction between RhoG and UNC13D is crucial for docking UNC13D to the PM, as it enables membrane fusion. Mechanistically, UNC13D lacks a C1 lipid-binding domain and cannot bind to lipids alone. In contrast, RhoG can bind to phospholipids in an activation-dependent manner; thus, the assistance of RhoG is required to bind to membrane lipids.
Following the tethering process, SNARE proteins play critical roles in the tight docking of cytotoxic granules to the PM. The priming event involves the formation of a substantial trans-protein complex consisting of SNARE proteins on vesicles (v-SNAREs) and on the PM (t-SNAREs) (Figure 4) (48). The UNC13 family members play a role in priming synaptic vesicles in presynaptic neurons (10, 49), whereas UNC13D has a similar function in CTLs and NK cells (4). Experiments with v- and t-SNARE liposomes revealed that UNC13D facilitates the formation of trans-SNARE complexes and that the fusion of liposomes is Ca2+-dependent. Boswell et al. reported that the C2B domain of UNC13D is crucial for Ca2+-dependent binding to t-SNAREs, whereas the C2C domain is responsible for Ca2+-dependent interactions with phospholipids (47). At the immunological synapse, the interactions between t-SNARE proteins (SNAP23 and STX11) and v-SNARE proteins (VAMP7) can cause the trans-core SNARE complex to overcome the barrier of negative repulsion between two lipid membranes and water molecules. Additionally, the MHD domains of the UNC13 family proteins are associated with priming functions (50). The phenotype observed in patients with FHL3 also supports the role of UNC13D in this step. Mutations in UNC13D result in the formation of cells in which granules can dock at the PM but are unable to release their contents, which indicates that UNC13D plays a vital role in priming cytotoxic lytic granules before fusion can occur (6, 20).
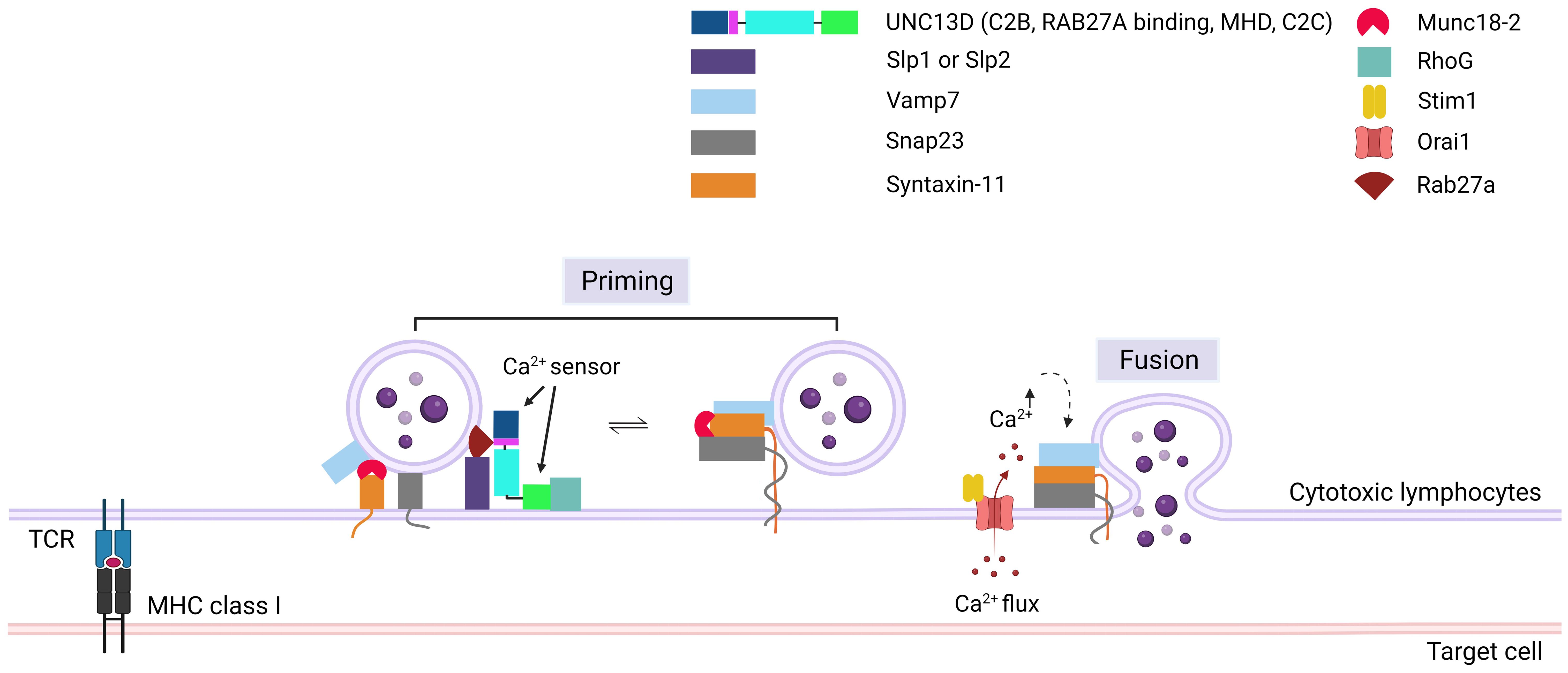
Figure 4. Role of UNC13D in cytotoxic granule priming. The formation of trans-SNARE complex is presented. Docking of mature cytotoxic granules at the PM is achieved through the interaction of Rab27a with Slp1 or Slp2. Moreover, cytotoxic granules interact with a docking complex formed by Munc18-2 and Stx11 in the closed conformation. The docked granules are primed by UNC13D following calcium flux, possibly through the UNC13D-mediated conversion of syntaxin-11 to an open conformation by displacing Munc18-2. Additionally, UNC13D is involved in the formation of an initial bridge connecting the granule membrane to the plasma membrane by interacting with RhoG via its C2C domain. Finally, a complex known as SNARE is established by the interaction of plasma membrane Snap23 and syntaxin-11 with granule membrane Vamp7, triggering the fusion of vesicles with the plasma membrane, resulting in the release of granule content into the synaptic cleft at the immunological synapse. Orai1, Calcium release-activated calcium channel protein 1; Slp1/2, synaptotagmin-like proteins1/2; SNARE, soluble N-ethylmaleimide-sensitive factor accessory protein receptor; RhoG, Ras Homolog Family Member G; Stim1, Stromal interaction molecule 1; Snap23, Synaptosomal-associated protein 23; Vamp7, Vesicle-associated membrane protein 7.
During the formation of the trans-SNARE complex, the inhibition of STX11 expression by MUNC18-2 may be released by UNC13D (51). MUNC18-2 (STXBP2) maintains an unsuitable (closed) state for STX to form the trans-SNARE complex. The interaction of UNC13D with MUNC18-2 allows the transition of STX11 to an open state, facilitating the formation of the trans-SNARE complex. This interaction is consistent with the finding that the MHD domains of UNC13A interact with STX1/STXBP1 in neurons, enabling STXBP1 to recruit VAMP2 (52–55). In addition, MUNC18-2 has been co-immunoprecipitated with UNC13D in platelets (56).
The cyclic guanosine monophosphate (GMP)-adenosine monophosphate (cGAMP) synthase (cGAS)-stimulator of interferon genes (STING) pathway is associated with aberrant exogenously or endogenously derived cytosolic DNA (57). Binding of cGAS to aberrant DNA results in the generation of cGAMP and activation of STING, which is located in the ER. During STING activation, the STING dimer is oligomerized and transported to the Golgi apparatus via the ER-Golgi intermediate compartment (ERGIC). Finally, the IRF3 and NF-κB pathways are activated to promote inflammatory reactions. UNC13D inhibits STING signaling by attenuating its oligomerization in the ER (34). A recent study showed that UNC13D co-precipitates with STING and co-localizes in the ER. Overexpression of UNC13D in HeLa cells inhibits the translocation of STING to ERGIC (34). Accordingly, patients with FHL3 having low UNC13D levels show hyperinflammatory responses (6).
UNC13D is composed of 32 exons and 31 introns (6). To date, at least 200 distinct mutations in the UNC13D gene have been documented as a contributing factor to FHL3, in which approximately 36.6%, 15.6% and 47.6% exhibited homozygous, heterozygous, compound heterozygous mutations, respectively (58). A systemic study of 322 patients showed that mutations were identified in 53.4% of exons and 46.6% of introns. Patients with severe features demonstrated a 1.6-fold increase in the prevalence of exonic mutations compared to intronic mutations, whereas the distribution of exonic and intronic mutations was nearly balanced in patients with mild features (58). Missense and splice mutations were the most frequent in severe and mild feature patients, respectively.
Among intronic mutations, the variant c.118-308C>T and the 253-kb inversion along with c.117 + 143A>G, c.118-307G>A, c.754-1G>C, c.1596 + 1G>C are notable pathogenic variants of UNC13D (59–64) (Table 2). Among exonic mutations, at least six pathogenic mutations were identified by cell-based functional assays (Table 2). Three of them (E379K, L403P, R414C) were located in the RAB27A interaction domain, one (G863D) in the MHD2 domain, one (A918) in the C2C domain (Figure 1), one (T76M) in the N-terminal.

Table 2. Notable pathogenic mutations of UNC13D .
It is important to explore how the expression of UNC13D is regulated in different lymphocyte subsets, given the essential role this protein plays in lymphocyte cytotoxicity (6). In 2000, Koch and his team reported a ubiquitous UNC13D expression pattern; however, a more in-depth analysis has not been performed since that initial finding (9). Remarkably, mutations occurring in an evolutionarily conserved region of UNC13D intron 1 have been frequently associated with the development of FHL3 (60–63). This sequence serves as an overall enhancer and regulates the expression of an alternative UNC13D isoform that has a unique N-terminus (60, 90). Using isoform specific antibodies, Galgano et al. (91) revealed that the alternative UNC13D isoform, which possesses a unique N-terminus, is preferentially expressed in human lymphocytes and platelets, whereas the conventional isoform is mainly expressed in monocytes and neutrophils. The distinct N-terminal regions of the two isoforms did not alter the localization or trafficking of UNC13D to the immunological synapse in cytotoxic T cells. Additionally, the ectopic expression of both isoforms proficiently restored exocytosis in T cells lacking UNC13D, which were derived from patients with FHL3. The research established that the conventional and alternative isoforms of UNC13D show different expression profiles in hematopoietic cell subsets, while exhibiting similar localization and contribution in T cell exocytosis (91). Although similar localization and functional contribution of both N-terminals, there might be some difference in susceptibility to bacterial infection and clinical manifestations in splice 1 mutation patients. In FHL3 patients, c.118-308C>T intronic homozygous mutation reduced conventional and alternative UNC13D transcript expression and affected cytotoxic exocytosis (90). However, in recurrent macrophage activation syndrome (MAS) and systemic juvenile idiopathic arthritis patients, c.117 + 143A>G mutation did not affect UNC13D expression in other immune cell types (64).
Hemophagocytic lymphohistiocytosis (HLH) is a life-threatening hyperinflammatory syndrome caused by the defective functions of CTLs, NK cells, and macrophages, resulting in cytokine storms and immune cell-mediated damage across multiple organ systems (92, 93). Dysregulated mechanisms responsible for impaired granule-mediated cytotoxicity result in prolonged contact between lymphocytes and their target cells. In addition, the inability of the immune system to eliminate the target cells, antigen-presenting cells, and cytotoxic cells results in failure to terminate the immune response (93–95). Consequently, a continuous increase is observed in cytokine levels, T-cell proliferation, and macrophage activation, ultimately leading to multi-organ failure (96, 97). HLH is typically divided into primary and secondary forms, based on the presence or absence of genetic defects. Familial HLH (FHL), a term for primary HLH, is inherited as an autosomal recessive condition, whereas secondary HLH is caused by infections, autoimmune disease, or malignancies (98, 99).
Cytotoxicity is a complex process that involves the generation of lytic granules containing perforin and granzyme B. These granules are then transported and docked to the membrane, followed by exocytosis at the immunological synaptic interphase. Ultimately, this process leads to target cell lysis (4). To date, four genetic abnormalities—PRF1 encoding perforin (FHL2) (100), UNC13D encoding Munc13-4 (FHL3) (6), STX11 encoding syntaxin-11 (FHL4) (101), and STXBP2 encoding Munc18-2 (FHL5)—have been detected in FHL (102, 103). Perforin acts as a cytolytic effector molecule for CTLs and NK cells, creating pore-like structures in the membranes of target cells. In contrast, UNC13D, syntaxin-11, and Munc18–2 play roles in the intracellular trafficking and membrane fusion of cytolytic granules. Thus, the cytolytic activity of NK cells and CTLs is inherently impaired in patients with FHL (104). UNC13D deficiency was initially reported in 2003 and is the second most prevalent genetically determined variant of FHL, referred to as FHL3. It accounts for 20–40% of the FHL incidences, depending on geographic distribution and ethnicity (85, 105). More than 100 mutations have been identified in the UNC13D gene, distributed across 32 exons, and the most frequent molecular alterations are those affecting mRNA splicing (71, 72, 83).
MAS is a type of HLH associated with rheumatological diseases such as systemic juvenile idiopathic arthritis (sJIA) (106). The clinical and laboratory characteristics of MAS and HLH exhibit striking similarities (107). In the last few years, an association has been observed between MAS in patients with juvenile idiopathic arthritis (JIA) and UNC13D mutations, particularly polymorphic variants in critical regulatory regions (64, 108, 109). Specifically, a novel functional intronic alteration within UNC13D, c.117 + 143A>G, was found to be related to MAS and JIA (64). Yang et al. identified a homozygous missense mutation in UNC13D (c.2588G>A, p. G863D), a hotspot mutation in the Chinese population, associated with recurrent MAS (110). This relationship may partially explain the increased risk of MAS development in specific patients. Studies on patients with UNC13D mutations experiencing juvenile polymyositis and MAS provide important insights into the pathogenesis of familial HLH (110).
JIA is the most common pediatric rheumatological disease (111) and is classified into the following subtypes: oligoarticular, polyarticular, enthesitis-related, psoriatic, and systemic JIA. HLH is the most prevalent in patients with sJIA, who are at the risk of MAS development (112). Several research groups have consistently reported a decrease in perforin levels in both CTLs and NK cells, along with impaired NK cell function in patients with sJIA (113, 114). Hazen et al. reported that a child suffering from JIA had compound heterozygous mutations in UNC13D; one allele contained a known mutation in the splice donor site of intron 9 (753 + 3 [G>A]), whereas the other allele carried a novel missense mutation in exon 18 (1579 [C>T] R527W), which reduced the cytotoxic function of NK cells. These findings suggest that sJIA may be considered a form of haemophagocytic syndrome (115). Following this, variations in UNC13D were observed in three of eighteen individuals with s-JIA/MAS, two of whom had biallelic mutations (109). Recently, exome sequencing analysis revealed two heterozygous UNC13D variants (p.R727Q and p.A59T) in an individual diagnosed with JIA (116).
ALPS is a well-known disease resulting from genetic defects in components of the FAS pathway, including FAS/TNFRSF6, FASL/TNFSF6, and CASP10 (58). Sequencing analysis identified four rare missense variants of UNC13D, including p.Cys112Ser, p.Val781Ile, and a haplotype containing both p.Ile848Leu and p.Ala995Pro, in three patients with ALPS. Additionally, a homozygous missense variation (c.2588G > A, p.G863D) was found in the UNC13D gene by Gu et al. (117). Such variations ultimately lead to excessive activation of T cells and macrophages, one of which is associated with the impaired development of Treg cells and severe ALPS-like symptoms (117). PRF1 and UNC13D can serve as risk factors for the development of ALPS in patients who do not exhibit any defects in the FAS pathway (118, 119). These findings suggest that rare loss-of-function variations in UND13D are potential risk factors for ALPS development (120).
For patients who are suspected to have FHL3, initial step involves the detection of its protein and an assessment of its functional properties. Although western blotting was a prevalent method for detection; however, it is important to note that SDS-PAGE can alter the secondary structure of proteins, which could lead to variations in the detection results. It has been documented that the expression of UNC13D is markedly greater in platelets than in peripheral blood mononuclear cells (PBMCs) and in the populations of CD8, CD4, CD14, and CD19 (121). The detection of UNC13D protein in platelets using western blotting or flow cytometry is an effective method for screening individuals diagnosed with FHL3. However, in the acute phase of disease that required platelet transfusion, it was challenging to identify FHL3 using western blot analysis. Alternatively, the recently introduced flow cytometric analysis of intraplatelet UNC13D protein expression has revealed the existence of bimodal populations of platelets, which include both normal and those lacking UNC13D (121). Although the flow cytometry tends to be more sensitive, but the results may vary depending on the antibody selection (122).
To provide a comprehensive genetic evaluation of FHL3 patients, sequence analysis can be chosen either by direct Sanger sequencing or by Next-Generation Sequencing (123, 124). However, the evaluation of pathogenicity of each genetic variant is required. FHL3 exhibited a range of clinical manifestations that were both diverse and complex. In some cases, patients showed atypical symptoms, resulting in challenges in achieving an accurate diagnosis. Zhao et al. suggests that the expression of UNC13D protein may not fully represent the pathogenic implications of UNC13D variants (122).
Recently in vitro cell-based functional assays for the evaluation of UNC13D variants were introduced because western blotting, flow cytometry and sequencing techniques could not directly indicate the functional status of UNC13D and immunotherapies can distort the cytotoxicity assay (89). In this assay, after knocking out endogenous UNC13D, its variant was introduced into cells and cytotoxic activity of CD8+ T and NK cells and their degranulation capacity were measured. These functional assays can distinguish pathogenic from nonpathogenic mutations. Interestingly, late-onset manifestation of mutation was also functionally evaluated.
Patients carrying mutations in the UNC13D gene, whether homozygous or compound heterozygous, that result in abnormal, truncated, or missing UNC13D variants, suffer from familial hemophagocytic lymphohistiocytosis type 3. Individuals diagnosed with FHL3 often develop acute HLH within their first year of life, and commonly manifest with fever, hepatosplenomegaly, cytopenia, and distinctive laboratory features, frequently presenting with neurological manifestations (125–127). So, the first phase of treatment focuses on managing hyper-inflammation and etoposide-based chemotherapy is administered. Alternatively, immunotherapy utilizing antithymocyte globulin (128–131) is employed as a first-line therapy, although more favorable outcomes have been observed with the monoclonal anti-CD52 antibody alemtuzumab (132–135). Combinatorial immunochemotherapy is employed to regulate the cytokine storm and inhibit cellular proliferation; however, it frequently leads to significant side effects after prolonged treatment (92).
For the replacement of dysfunctional immune system (Figure 5), the second phase of treatment usually involves hematopoietic stem cell (HSC) transplantation after myeloablative/reductive conditioning; however, the mortality rate remains high (92, 136–139). The incomplete remission of disease activity, conditioning-related toxicities, and graft rejection during the transplantation are major risks that pose significant challenges (140, 141). The long-term survival rate of patients with FHL is approximately 50% (137, 142), making it imperative for clinical advancements using new methods to improve treatment outcomes and reduce side effects.
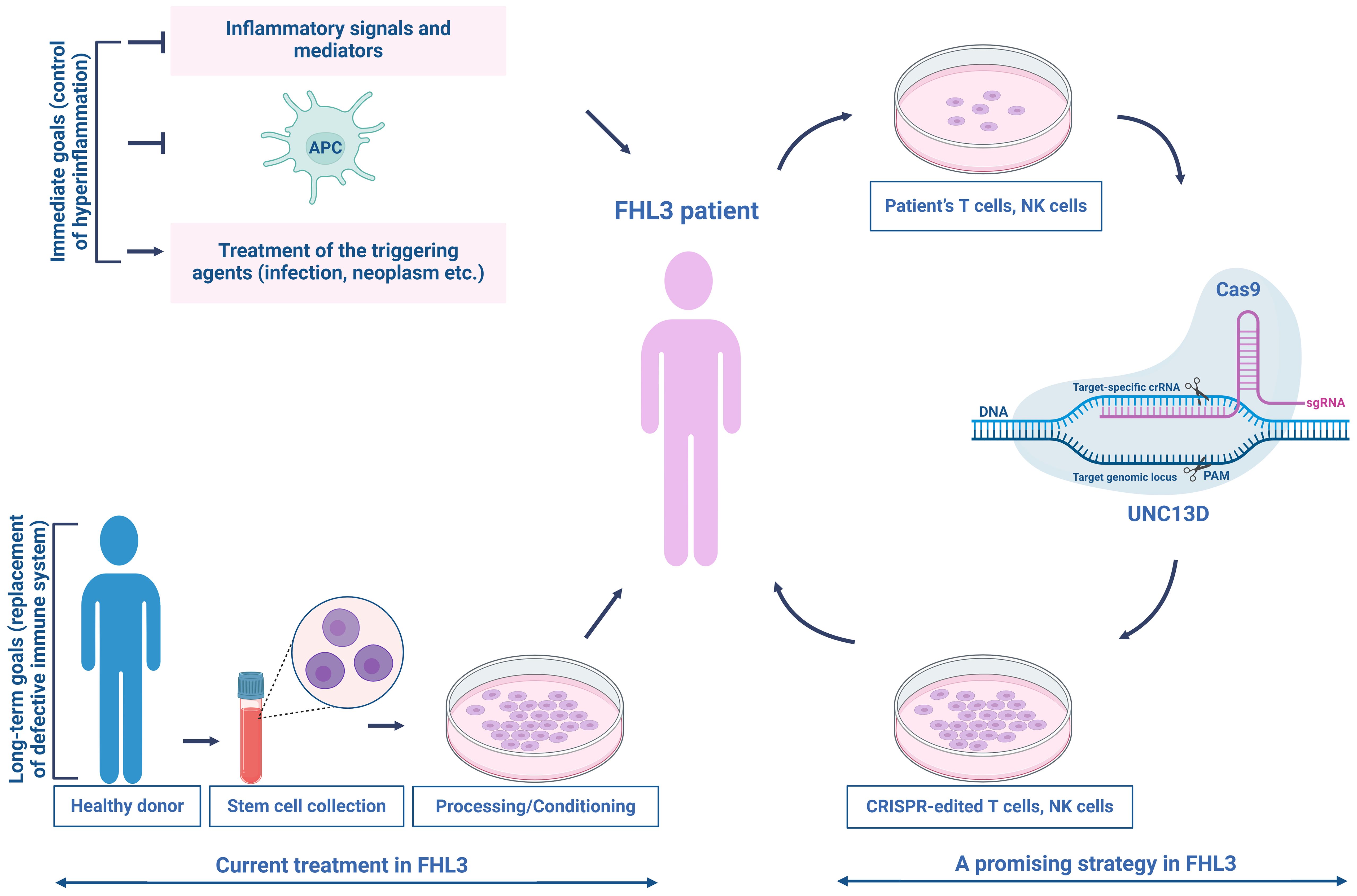
Figure 5. Current treatment and a promising therapeutic strategy for patients with FHL3. The current treatment for FHL3 is typically divided into two phases—controlling hyperinflammation and replacing the defective immune system. In the first phase, the standard of care consists of etoposide-based chemotherapy (HLH-94 treatment protocol) to control severe inflammation, eradicate over-stimulated antigen-presenting cells, and manage the triggering agent (infection, neoplasm, etc.). The second phase commonly involves HSC transplantation after myeloablative/reductive conditioning. Targeted genome editing could offer a solution to the problems associated with current treatments, restoring NK/T cell cytotoxic function. This promising strategy establishes a beginning for the clinical integration of autologous NK/T cell therapy in patients with FHL3.
Recently, gene therapy is extensively studied for the functional recovery for the UNC13D defects. As the second most common type of primary HLH, FHL3 is a potential candidate for gene therapy. A feasible strategy is to employ autologous, repaired T cells as a transitional step toward transplant therapy to enhance the health of the patient and minimize the potential risks associated with HSC transplantation (HSCT). The first gene therapy study on FHL3 was published by Soheili et al. (143), which involved the transduction of activated peripheral blood mononuclear cells from three FHL3 patients. This was achieved using either a vesicular stomatitis virus G protein or a measles H/F pseudotyped LV vector encoding a complementary DNA product from human UNC13D. Transplanting EBV-induced tumors into NSG mice, followed by the infusion of transduced T cells from patient with FHL3, resulted in a noticeable reduction in tumor size (143). Soheili et al. successfully transduced HSC derived from UNC13D null (Jinx) mice, a model for FHL3, demonstrating that gene therapy improved the in vitro metrics and viral clearance (144). In 2019, Dettmer et al. used a gamma retrovirus-based gene therapy construct to transduce cells from patient with FHL3 cells, resulting in improved degranulation capacity and cytotoxicity in transduced cells (145). Previous research showed that introducing the UNC13D gene into Jinx HSCs using lentiviral transfer successfully restored T-cell function in the FHL3 (Jinx) disease model, with 15% of degranulation-competent cells being sufficient to trigger a degranulation response comparable to that of the wild type (146). Despite the advancements in lentiviral and retroviral gene therapy, concerns still remain regarding insertional mutagenesis (147–149) and the regulation of UNC13D expression (9, 30, 150, 151).
Recent advancements in targeted genome editing have shown great promise in addressing these challenges. Dettmer et al. (145) employed a CRISPR/Cas9-based strategy to delete an intronic cryptic splice donor site within UNC13D, achieving a high success rate (Figure 5). Jinx mice receiving UNC13D-edited HSCs exhibited notable chimerism without any detectable off-target effects. These mice were completely protected against lymphocytic choriomeningitis virus-induced HLH and showed no signs of clonal T-cell expansion or leukemogenesis (152). Moreover, repaired long-lived T memory stem (TSCM) cells can provide extended protection, as genetically modified T cells can survive for over ten years (153, 154). TSCM cells, similar to naïve T (TN) cells, have a low differentiation ability compared to other memory T-cell subsets and are suggested to occupy the highest position in the T-cell hierarchy within a progressive model of T-cell differentiation (155). Following allogeneic HSCT, only TN and TSCM cells can regenerate the entire spectrum of T-memory cell subsets, including TSCM cells (156). In FHL, many children have genetic loss-of-function mutations that disrupt the functions of proteins necessary for the normal cytotoxic activities of T and NK cells (93). Recently, the efficacy of precisely repaired autologous T cells in addressing FHL has been highlighted. Adeno-associated virus-templated CRISPR-Cas9 was employed to successfully restore the function of memory T cells lacking perforin-1 in mice. These repaired cells were subsequently used to treat life-threatening hyperinflammation induced by EBV in a mouse model of FHL. Additionally, T cells derived from pediatric patients with FHL2 and FHL3 were effectively expanded and repaired, resulting in the recovery of CD8 + T-cell cytotoxicity while maintaining a TSCM cell-like phenotype (157).
Autologous gene-modified T cells have been utilized in cancer treatment of numerous patients, demonstrating their safety and lack of malignant transformation. Accordingly, the potential impact of unwanted on-and off-target events may be less critical for autologous T cells (157, 158). However, to apply precise gene editing in clinical practice, a comprehensive evaluation of the potential off-target sites is required (Figure 5).
Although notable progress has been made in understanding the role of UNC13D in the nervous system, its function in the immune system remains poorly characterized. Recent cancer studies have highlighted its importance (12, 13). The regulation of vesicle trafficking and its contribution to tumor progression need further exploration. In the immune system, the role of UNC13D in the priming of cytotoxic granules is relatively well established. However, its involvement in other steps, such as the biogenesis and tethering of cytotoxic granules, is not well understood. In particular, the association of its biological role in the immune system with the progression of immunological diseases needs to be characterized. For therapeutic development involving UNC13D, lipid nanoparticles for the delivery of therapeutic genes must be examined because of their potential in gene therapy (159, 160).
VD: Conceptualization, Investigation, Visualization, Data curation, Writing – original draft. DL: Conceptualization, Supervision, Writing – review & editing, Validation. YK: Conceptualization, Supervision, Validation, Writing – review & editing. SO: Conceptualization, Funding acquisition, Investigation, Supervision, Visualization, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by a 2-year research grant from Pusan National University.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Rettig J, Neher E. Emerging roles of presynaptic proteins in Ca-triggered exocytosis. Science. (2002) 298:781–5. doi: 10.1126/science.1075375
2. Sudhof TC. The synaptic vesicle cycle. Annu Rev Neurosci. (2004) 27:509–47. doi: 10.1146/annurev.neuro.26.041002.131412
3. Wojcik SM, Brose N. Regulation of membrane fusion in synaptic excitation-secretion coupling: speed and accuracy matter. Neuron. (2007) 55:11–24. doi: 10.1016/j.neuron.2007.06.013
4. de Saint Basile G, Ménasché G, Fischer A. Molecular mechanisms of biogenesis and exocytosis of cytotoxic granules. Nat Rev Immunol. (2010) 10:568–79. doi: 10.1038/nri2803
5. Dittman JS. Unc13: a multifunctional synaptic marvel. Curr Opin Neurobiol. (2019) 57:17–25. doi: 10.1016/j.conb.2018.12.011
6. Feldmann J, Callebaut I, Raposo G, Certain S, Bacq D, Dumont C, et al. Munc13-4 is essential for cytolytic granules fusion and is mutated in a form of familial hemophagocytic lymphohistiocytosis (FHL3). Cell. (2003) 115:461–73. doi: 10.1016/S0092-8674(03)00855-9
7. van der Sluijs P, Zibouche M, van Kerkhof P. Late steps in secretory lysosome exocytosis in cytotoxic lymphocytes. Front Immunol. (2013) 4:359. doi: 10.3389/fimmu.2013.00359
8. Dudenhöffer-Pfeifer M, Schirra C, Pattu V, Halimani M, Maier-Peuschel M, Marshall MR, et al. Different Munc13 isoforms function as priming factors in lytic granule release from murine cytotoxic T lymphocytes. Traffic. (2013) 14:798–809. doi: 10.1111/tra.2013.14.issue-7
9. Koch H, Hofmann K, Brose N. Definition of Munc13-homology-domains and characterization of a novel ubiquitously expressed Munc13 isoform. Biochem J. (2000) 349:247–53. doi: 10.1042/bj3490247
10. Augustin I, Betz A, Herrmann C, Jo T, Brose N. Differential expression of two novel Munc13 proteins in rat brain. Biochem J. (1999) 337:363–71. doi: 10.1042/bj3370363
11. Duong VT, Ha M, Kim J, Kim JY, Park S, Reshma KM, et al. Recycling machinery of integrin coupled with focal adhesion turnover via RAB11-UNC13D-FAK axis for migration of pancreatic cancer cells. J Transl Med. (2024) 22:800. doi: 10.1186/s12967-024-05630-9
12. Lofstedt A, Ahlm C, Tesi B, Bergdahl IA, Nordenskjold M, Bryceson YT, et al. Haploinsufficiency of UNC13D increases the risk of lymphoma. Cancer. (2019) 125:1848–54. doi: 10.1002/cncr.32011
13. Messenger SW, Woo SS, Sun Z, Martin TFJ. A Ca(2+)-stimulated exosome release pathway in cancer cells is regulated by Munc13-4. J Cell Biol. (2018) 217:2877–90. doi: 10.1083/jcb.201710132
14. Varoqueaux F, Fasshauer D. Getting nervous: an evolutionary overhaul for communication. Annu Rev Genet. (2017) 51:455–76. doi: 10.1146/annurev-genet-120116-024648
15. Burkhardt P, Sprecher SG. Evolutionary origin of synapses and neurons - Bridging the gap. Bioessays. (2017) 39. doi: 10.1002/bies.201700024
16. Ansari U, Chen V, Sedighi R, Syed B, Muttalib Z, Ansari K, et al. Role of the UNC13 family in human diseases: A literature review. AIMS Neurosci. (2023) 10:388–400. doi: 10.3934/Neuroscience.2023029
17. Brose N, Hofmann K, Hata Y, Sudhof TC. Mammalian homologues of Caenorhabditis elegans unc-13 gene define novel family of C2-domain proteins. J Biol Chem. (1995) 270:25273–80. doi: 10.1074/jbc.270.42.25273
18. Nalefski EA, Falke JJ. The C2 domain calcium-binding motif: Structural and functional diversity. Protein Sci. (1996) 5:2375–90. doi: 10.1002/pro.5560051201
19. Rizo J, Sudhof TC. C2-domains, structure and function of a universal Ca2+-binding domain. J Biol Chem. (1998) 273:15879–82. doi: 10.1074/jbc.273.26.15879
20. Elstak ED, Neeft M, Nehme NT, Voortman J, Cheung M, Goodarzifard M, et al. The munc13-4-rab27 complex is specifically required for tethering secretory lysosomes at the plasma membrane. Blood. (2011) 118:1570–8. doi: 10.1182/blood-2011-02-339523
21. Neeft M, Wieffer M, de Jong AS, Negroiu G, Metz CHG, van Loon A, et al. Munc13-4 is an effector of Rab27a and controls secretion of lysosomes in hematopoietic cells. Mol Biol Cell. (2005) 16:731–41. doi: 10.1091/mbc.e04-10-0923
22. Shirakawa R, Higashi T, Tabuchi A, Yoshioka A, Nishioka H, Fukuda M, et al. Munc13-4 is a GTP-Rab27-binding protein regulating dense core granule secretion in platelets. J Biol Chem. (2004) 279:10730–7. doi: 10.1074/jbc.M309426200
23. Johnson JL, He J, Ramadass M, Pestonjamasp K, Kiosses WB, Zhang JZ, et al. Munc13-4 is a Rab11-binding protein that regulates Rab11-positive vesicle trafficking and docking at the plasma membrane. J Biol Chem. (2016) 291:3423–38. doi: 10.1074/jbc.M115.705871
24. Ménager MM, Ménasché G, Romao M, Knapnougel P, Ho CH, Garfa M, et al. Secretory cytotoxic granule maturation and exocytosis require the effector protein hMunc13-4. Nat Immunol. (2007) 8:257–67. doi: 10.1038/ni1431
25. Elstak ED, te Loo M, Tesselaar K, van Kerkhof P, Loeffen J, Grivas D, et al. A novel Dutch mutation in UNC13D reveals an essential role of the C2B domain in munc13-4 function. Pediatr Blood Cancer. (2012) 58:598–605. doi: 10.1002/pbc.23253
26. Zhang Y, Tang WW, Zhang HF, Niu XF, Xu YK, Zhang JS, et al. A network of interactions enables CCM3 and STK24 to coordinate UNC13D-driven vesicle exocytosis in neutrophils. Dev Cell. (2013) 27:215–26. doi: 10.1016/j.devcel.2013.09.021
27. Higashio H, Nishimura N, Ishizaki H, Miyoshi J, Orita S, Sakane A, et al. Doc2α and Munc13-4 regulate Ca-dependent secretory lysosome exocytosis in mast cells. J Immunol. (2008) 180:4774–84. doi: 10.4049/jimmunol.180.7.4774
28. Higashio H, Satoh Y, Saino T. Mast cell degranulation is negatively regulated by the Munc13-4-binding small-guanosine triphosphatase Rab37. Sci Rep. (2016) 6. doi: 10.1038/srep22539
29. Chehab T, Santos NC, Holthenrich A, Koerdt SN, Disse J, Schuberth C, et al. A novel Munc13-4/S100A10/annexin A2 complex promotes Weibel-Palade body exocytosis in endothelial cells. Mol Biol Cell. (2017) 28:1688–700. doi: 10.1091/mbc.e17-02-0128
30. Chicka MC, Ren Q, Richards D, Hellman LM, Zhang J, Fried MG, et al. Role of Munc13-4 as a Ca2+-dependent tether during platelet secretion. Biochem J. (2016) 473:627–39. doi: 10.1042/BJ20151150
31. Woo SS, James DJ, Martin TFJ. Munc13-4 functions as a Ca sensor for homotypic secretory granule fusion to generate endosomal exocytic vacuoles. Mol Biol Cell. (2017) 28:792–808. doi: 10.1091/mbc.e16-08-0617
32. Xu R, Zhou J, Zhou XD, Li Q, Perelman JM, Kolosov VP. Munc13-4 mediates human neutrophil elastase-induced airway mucin5AC hypersecretion by interacting with syntaxin2. Mol Med Rep. (2018) 18:1015–24. doi: 10.3892/mmr.2018.9015
33. Kalinichenko A, Casoni GP, Dupré L, Trotta L, Huemer J, Galgano D, et al. RhoG deficiency abrogates cytotoxicity of human lymphocytes and causes hemophagocytic lymphohistiocytosis. Blood. (2021) 137:2033–45. doi: 10.1182/blood.2020008738
34. Song P, Yang WW, Lou KF, Dong H, Zhang H, Wang BM, et al. UNC13D inhibits STING signaling by attenuating its oligomerization on the endoplasmic reticulum. EMBO Rep. (2022) 23. doi: 10.15252/embr.202255099
35. Peters PJ, Geuze HJ, Vanderdonk HA, Slot JW, Griffith JM, Stam NJ, et al. Molecules relevant for T-cell-target cell-interaction are present in cytolytic granules of human lymphocytes-T. Eur J Immunol. (1989) 19:1469–75. doi: 10.1002/eji.1830190819
36. Raposo G, Tenza D, Mecheri S, Peronet R, Bonnerot C, Desaymard C. Accumulation of major histocompatibility complex class II molecules in mast cell secretory granules and their release upon degranulation. Mol Biol Cell. (1997) 8:2631–45. doi: 10.1091/mbc.8.12.2631
37. Raposo G, Tenza D, Murphy DM, Berson JF, Marks MS. Distinct protein sorting and localization to premelanosomes, melanosomes, and lysosomes in pigmented melanocytic cells. J Cell Biol. (2001) 152:809–23. doi: 10.1083/jcb.152.4.809
38. Smith-Garvin JE, Koretzky GA, Jordan MS. T cell activation. Annu Rev Immunol. (2009) 27:591–619. doi: 10.1146/annurev.immunol.021908.132706
39. Jenkins MR, Griffiths GM. The synapse and cytolytic machinery of cytotoxic T cells. Curr Opin Immunol. (2010) 22:308–13. doi: 10.1016/j.coi.2010.02.008
40. Sanchez-Ruiz Y, Valitutti S, Dupre L. Stepwise maturation of lytic granules during differentiation and activation of human CD8 T lymphocytes. PloS One. (2011) 6. doi: 10.1371/journal.pone.0027057
41. Wood SM, Meeths M, Chiang SCC, Bechensteen AG, Boelens JJ, Heilmann C, et al. Different NK cell-activating receptors preferentially recruit Rab27a or Munc13-4 to perforin-containing granules for cytotoxicity. Blood. (2009) 114:4117–27. doi: 10.1182/blood-2009-06-225359
42. Qu B, Pattu V, Junker C, Schwarz EC, Bhat SS, Kummerow C, et al. Docking of lytic granules at the immunological synapse in human CTL requires Vti1b-dependent pairing with CD3 endosomes. J Immunol. (2011) 186:6894–904. doi: 10.4049/jimmunol.1003471
43. Phatarpekar PV, Billadeau DD. Molecular regulation of the plasma membrane-proximal cellular steps involved in NK cell cytolytic function. J Cell Sci. (2020) 133. doi: 10.1242/jcs.240424
44. Holt O, Kanno E, Bossi G, Booth S, Daniele T, Santoro A, et al. Slp1 and slp2-a localize to the plasma membrane of CTL and contribute to secretion from the immunological synapse. Traffic. (2008) 9:446–57. doi: 10.1111/j.1600-0854.2008.00714.x
45. Ménasché G, Ménager MM, Lefebvre JM, Deutsch E, Athman R, Lambert N, et al. A newly identified isoform of Slp2a associates with Rab27a in cytotoxic T cells and participates to cytotoxic granule secretion. Blood. (2008) 112:5052–62. doi: 10.1182/blood-2008-02-141069
46. Johnson JL, Hong H, Monfregola J, Kiosses WB, Catz SD. Munc13-4 restricts motility of Rab27a-expressing vesicles to facilitate lipopolysaccharide-induced priming of exocytosis in neutrophils. J Biol Chem. (2011) 286:5647–56. doi: 10.1074/jbc.M110.184762
47. Boswell KL, James DJ, Esquibel JM, Bruinsma S, Shirakawa R, Horiuchi H, et al. Munc13-4 reconstitutes calcium-dependent SNARE-mediated membrane fusion. J Cell Biol. (2012) 197:301–12. doi: 10.1083/jcb.201109132
48. Tang BL. A unique SNARE machinery for exocytosis of cytotoxic granules and platelets granules. Mol Membr Biol. (2015) 32:120–6. doi: 10.3109/09687688.2015.1079934
49. Augustin I, Rosenmund C, Sudhof TC, Brose N. Munc13-1 is essential for fusion competence of glutamatergic synaptic vesicles. Nature. (1999) 400:457–61. doi: 10.1038/22768
50. Guan R, Dai H, Rizo J. Binding of the Munc13-1 MUN domain to membrane-anchored SNARE complexes. Biochemistry. (2008) 47:1474–81. doi: 10.1021/bi702345m
51. Benavides N, Spessott WA, Sanmillan ML, Vargas M, Livingston MS, Erickson N, et al. STXBP2-R190C variant in a patient with neonatal hemophagocytic lymphohistiocytosis (HLH) and G6PD deficiency reveals a critical role of STXBP2 domain 2 on granule exocytosis. Front Immunol. (2020) 11:545414. doi: 10.3389/fimmu.2020.545414
52. Lai Y, Choi UB, Leitz J, Rhee HJ, Lee C, Altas B, et al. Molecular mechanisms of synaptic vesicle priming by Munc13 and Munc18. Neuron. (2017) 95:591–607 e10. doi: 10.1016/j.neuron.2017.07.004
53. Ma C, Su L, Seven AB, Xu Y, Rizo J. Reconstitution of the vital functions of Munc18 and Munc13 in neurotransmitter release. Science. (2013) 339:421–5. doi: 10.1126/science.1230473
54. Wang S, Li Y, Gong J, Ye S, Yang X, Zhang R, et al. Munc18 and Munc13 serve as a functional template to orchestrate neuronal SNARE complex assembly. Nat Commun. (2019) 10:69. doi: 10.1038/s41467-018-08028-6
55. Ma L, Rebane AA, Yang G, Xi Z, Kang Y, Gao Y, et al. Munc18-1-regulated stage-wise SNARE assembly underlying synaptic exocytosis. Elife. (2015) 4. doi: 10.7554/eLife.09580
56. Al Hawas R, Ren Q, Ye S, Karim ZA, Filipovich AH, Whiteheart SW. Munc18b/STXBP2 is required for platelet secretion. Blood. (2012) 120:2493–500. doi: 10.1182/blood-2012-05-430629
57. Decout A, Katz JD, Venkatraman S, Ablasser A. The cGAS-STING pathway as a therapeutic target in inflammatory diseases. Nat Rev Immunol. (2021) 21:548–69. doi: 10.1038/s41577-021-00524-z
58. Amirifar P, Ranjouri MR, Abolhassani H, Moeini Shad T, Almasi-Hashiani A, Azizi G, et al. Clinical, immunological and genetic findings in patients with UNC13D deficiency (FHL3): A systematic review. Pediatr Allergy Immunol. (2021) 32:186–97. doi: 10.1111/pai.13323
59. Yamamoto K, Ishii E, Sako M, Ohga S, Furuno K, Suzuki N, et al. Identification of novel MUNC13-4 mutations in familial haemophagocytic lymphohistiocytosis and functional analysis of MUNC13-4-deficient cytotoxic T lymphocytes. J Med Genet. (2004) 41:763–7. doi: 10.1136/jmg.2004.021121
60. Meeths M, Chiang SC, Wood SM, Entesarian M, Schlums H, Bang B, et al. Familial hemophagocytic lymphohistiocytosis type 3 (FHL3) caused by deep intronic mutation and inversion in UNC13D. Blood. (2011) 118:5783–93. doi: 10.1182/blood-2011-07-369090
61. Entesarian M, Chiang SC, Schlums H, Meeths M, Chan MY, Mya SN, et al. Novel deep intronic and missense UNC13D mutations in familial haemophagocytic lymphohistiocytosis type 3. Br J Haematol. (2013) 162:415–8. doi: 10.1111/bjh.2013.162.issue-3
62. Seo JY, Song JS, Lee KO, Won HH, Kim JW, Kim SH, et al. Founder effects in two predominant intronic mutations of UNC13D, c.118-308C>T and c.754-1G>C underlie the unusual predominance of type 3 familial hemophagocytic lymphohistiocytosis (FHL3) in Korea. Ann Hematol. (2013) 92:357–64. doi: 10.1007/s00277-012-1628-6
63. Qian Y, Johnson JA, Connor JA, Valencia CA, Barasa N, Schubert J, et al. The 253-kb inversion and deep intronic mutations in UNC13D are present in North American patients with familial hemophagocytic lymphohistiocytosis 3. Pediatr Blood Cancer. (2014) 61:1034–40. doi: 10.1002/pbc.24955
64. Schulert GS, Zhang M, Husami A, Fall N, Brunner H, Zhang K, et al. Brief report: novel UNC13D intronic variant disrupting an NF-kappaB enhancer in a patient with recurrent macrophage activation syndrome and systemic juvenile idiopathic arthritis. Arthritis Rheumatol. (2018) 70:963–70. doi: 10.1002/art.40438
65. Zhang K, Chandrakasan S, Chapman H, Valencia CA, Husami A, Kissell D, et al. Synergistic defects of different molecules in the cytotoxic pathway lead to clinical familial hemophagocytic lymphohistiocytosis. Blood. (2014) 124:1331–4. doi: 10.1182/blood-2014-05-573105
66. Nakamura L, Bertling A, Brodde MF, Zur Stadt U, Schulz AS, Ammann S, et al. First characterization of platelet secretion defect in patients with familial hemophagocytic lymphohistiocytosis type 3 (FHL-3). Blood. (2015) 125:412–4. doi: 10.1182/blood-2014-07-587568
67. Xiong HY, Alipanahi B, Lee LJ, Bretschneider H, Merico D, Yuen RK, et al. RNA splicing. The human splicing code reveals new insights into the genetic determinants of disease. Science. (2015) 347:1254806. doi: 10.1126/science.1254806
68. Rego S, Dagan-Rosenfeld O, Zhou W, Sailani MR, Limcaoco P, Colbert E, et al. High-frequency actionable pathogenic exome variants in an average-risk cohort. Cold Spring Harb Mol Case Stud. (2018) 4. doi: 10.1101/mcs.a003178
69. Gadoury-Levesque V, Dong L, Su R, Chen J, Zhang K, Risma KA, et al. Frequency and spectrum of disease-causing variants in 1892 patients with suspected genetic HLH disorders. Blood Adv. (2020) 4:2578–94. doi: 10.1182/bloodadvances.2020001605
70. Santoro A, Cannella S, Bossi G, Gallo F, Trizzino A, Pende D, et al. Novel Munc13-4 mutations in children and young adult patients with haemophagocytic lymphohistiocytosis. J Med Genet. (2006) 43:953–60. doi: 10.1136/jmg.2006.041863
71. Santoro A, Cannella S, Trizzino A, Bruno G, De Fusco C, Notarangelo LD, et al. Mutations affecting mRNA splicing are the most common molecular defect in patients with familial hemophagocytic lymphohistiocytosis type 3. Haematol the Hematol J. (2008) 93:1086–90. doi: 10.3324/haematol.12622
72. Sieni E, Cetica V, Santoro A, Beutel K, Mastrodicasa E, Meeths M, et al. Genotype-phenotype study of familial haemophagocytic lymphohistiocytosis type 3. J Med Genet. (2011) 48:343–52. doi: 10.1136/jmg.2010.085456
73. Alsina L, Colobran R, de Sevilla MF, Catala A, Vinas L, Ricart S, et al. Novel and atypical splicing mutation in a compound heterozygous UNC13D defect presenting in Familial Hemophagocytic Lymphohistiocytosis triggered by EBV infection. Clin Immunol. (2014) 153:292–7. doi: 10.1016/j.clim.2014.04.019
74. Manno EC, Salfa I, Palma P, Bertaina A, Lombardi A, Moretta F, et al. Familial hemophagocytic lymphohistiocytosis type 3 diagnosed at school age: a case report. J Pediatr Hematol Oncol. (2014) 36:e128–30. doi: 10.1097/MPH.0b013e318292bc7c
75. Capalbo A, Valero RA, Jimenez-Almazan J, Pardo PM, Fabiani M, Jimenez D, et al. Optimizing clinical exome design and parallel gene-testing for recessive genetic conditions in preconception carrier screening: Translational research genomic data from 14,125 exomes. PloS Genet. (2019) 15:e1008409. doi: 10.1371/journal.pgen.1008409
76. Gray PE, Shadur B, Russell S, Mitchell R, Gallagher K, Thia K, et al. Neonatal cytomegalovirus palatal ulceration and bocavirus pneumonitis associated with a defect of lymphocyte cytotoxicity caused by mutations in UNC13D. J Pediatr Infect Dis Soc. (2019) 8:73–6. doi: 10.1093/jpids/pix112
77. Gokce M, Balta G, Unal S, Oguz K, Cetin M, Gumruk F. Spinal cord involvement in a child with familial hemophagocytic lymphohistiocytosis. J Pediatr Neurosci. (2012) 7:194–6. doi: 10.4103/1817-1745.106477
78. Bienemann K, Daschkey S, Sorensen J, Schwabe D, Klingebiel T, Honscheid A, et al. A novel homozygous mutation in UNC13D presenting as Epstein-Barr-virus-associated lymphoproliferative disease at 9 years of age. Leuk Lymphoma. (2016) 57:2949–51. doi: 10.1080/10428194.2016.1177724
79. Gray PE, Shadur B, Russell S, Mitchell R, Buckley M, Gallagher K, et al. Late-onset non-HLH presentations of growth arrest, inflammatory arachnoiditis, and severe infectious mononucleosis, in siblings with hypomorphic defects in UNC13D. Front Immunol. (2017) 8:944. doi: 10.3389/fimmu.2017.00944
80. Hori M, Yasumi T, Shimodera S, Shibata H, Hiejima E, Oda H, et al. A CD57(+) CTL degranulation assay effectively identifies familial hemophagocytic lymphohistiocytosis type 3 patients. J Clin Immunol. (2017) 37:92–9. doi: 10.1007/s10875-016-0357-3
81. Shabrish S, Kelkar M, Yadav RM, Bargir UA, Gupta M, Dalvi A, et al. The spectrum of clinical, immunological, and molecular findings in familial hemophagocytic lymphohistiocytosis: experience from India. Front Immunol. (2021) 12:612583. doi: 10.3389/fimmu.2021.612583
82. Zur Stadt U, Beutel K, Kolberg S, Schneppenheim R, Kabisch H, Janka G, et al. Mutation spectrum in children with primary hemophagocytic lymphohistiocytosis: molecular and functional analyses of PRF1, UNC13D, STX11, and RAB27A. Hum Mutat. (2006) 27:62–8. doi: 10.1002/humu.20274
83. Rudd E, Bryceson YT, Zheng C, Edner J, Wood SM, Ramme K, et al. Spectrum, and clinical and functional implications of mutations in familial haemophagocytic lymphohistiocytosis. J Med Genet. (2008) 45:134–41. doi: 10.1136/jmg.2007.054288
84. Rohr J, Beutel K, Maul-Pavicic A, Vraetz T, Thiel J, Warnatz K, et al. Atypical familial hemophagocytic lymphohistiocytosis due to mutations in UNC13D and STXBP2 overlaps with primary immunodeficiency diseases. Haematologica. (2010) 95:2080–7. doi: 10.3324/haematol.2010.029389
85. Cetica V, Sieni E, Pende D, Danesino C, De Fusco C, Locatelli F, et al. Genetic predisposition to hemophagocytic lymphohistiocytosis: Report on 500 patients from the Italian registry. J Allergy Clin Immunol. (2016) 137:188–+. doi: 10.1016/j.jaci.2015.06.048
86. Meng L, Pammi M, Saronwala A, Magoulas P, Ghazi AR, Vetrini F, et al. Use of exome sequencing for infants in intensive care units: ascertainment of severe single-gene disorders and effect on medical management. JAMA Pediatr. (2017) 171:e173438. doi: 10.1001/jamapediatrics.2017.3438
87. Li H, Benson LA, Henderson LA, Solomon IH, Kennedy AL, Soldatos A, et al. Central nervous system-restricted familial hemophagocytic lymphohistiocytosis responds to hematopoietic cell transplantation. Blood Adv. (2019) 3:503–7. doi: 10.1182/bloodadvances.2018027417
88. Jin Z, Wang Y, Wang J, Zhang J, Wu L, Gao Z, et al. Primary hemophagocytic lymphohistiocytosis in adults: the utility of family surveys in a single-center study from China. Orphanet J Rare Dis. (2018) 13:17. doi: 10.1186/s13023-017-0753-7
89. Noori T, Rudd-Schmidt JA, Kane A, Frith K, Gray PE, Hu H, et al. A cell-based functional assay that accurately links genotype to phenotype in familial HLH. Blood. (2023) 141:2330–42. doi: 10.1182/blood.2022018398
90. Cichocki F, Schlums H, Li H, Stache V, Holmes T, Lenvik TR, et al. Transcriptional regulation of Munc13-4 expression in cytotoxic lymphocytes is disrupted by an intronic mutation associated with a primary immunodeficiency. J Exp Med. (2014) 211:1079–91. doi: 10.1084/jem.20131131
91. Galgano D, Soheili T, Voss M, Torralba-Raga L, Tesi B, Cichocki F, et al. Alternative UNC13D promoter encodes a functional Munc13-4 isoform predominantly expressed in lymphocytes and platelets. Front Immunol. (2020) 11:1154. doi: 10.3389/fimmu.2020.01154
92. Maakaroun NR, Moanna A, Jacob JT, Albrecht H. Viral infections associated with haemophagocytic syndrome. Rev Med Virol. (2010) 20:93–105. doi: 10.1002/rmv.v20:2
93. Al-Samkari H, Berliner N. Hemophagocytic lymphohistiocytosis. Annu Rev Pathol. (2018) 13:27–49. doi: 10.1146/annurev-pathol-020117-043625
94. Menasche G, Feldmann J, Fischer A, de Saint Basile G. Primary hemophagocytic syndromes point to a direct link between lymphocyte cytotoxicity and homeostasis. Immunol Rev. (2005) 203:165–79. doi: 10.1111/j.0105-2896.2005.00224.x
95. Terrell CE, Jordan MB. Perforin deficiency impairs a critical immunoregulatory loop involving murine CD8(+) T cells and dendritic cells. Blood. (2013) 121:5184–91. doi: 10.1182/blood-2013-04-495309
96. Ammann S, Lehmberg K, U. zur Stadt G, Rensing-Ehl A, Klemann C, Heeg M, et al. Primary and secondary hemophagocytic lymphohistiocytosis have different patterns of T-cell activation, differentiation and repertoire. Eur J Immunol. (2017) 47:364–73. doi: 10.1002/eji.201646686
97. Chaturvedi V, Marsh RA, Zoref-Lorenz A, Owsley E, Chaturvedi V, Nguyen TC, et al. T-cell activation profiles distinguish hemophagocytic lymphohistiocytosis and early sepsis. Blood. (2021) 137:2337–46. doi: 10.1182/blood.2020009499
98. Chen X, Wang F, Zhang Y, Teng W, Wang M, Nie D, et al. Genetic variant spectrum in 265 Chinese patients with hemophagocytic lymphohistiocytosis: Molecular analyses of PRF1, UNC13D, STX11, STXBP2, SH2D1A, and XIAP. Clin Genet. (2018) 94:200–12. doi: 10.1111/cge.2018.94.issue-2
99. Weisfeld-Adams JD, Frank Y, Havalad V, Hojsak JM, Posada R, Kaicker SM, et al. Diagnostic challenges in a child with familial hemophagocytic lymphohistiocytosis type 3 (FHLH3) presenting with fulminant neurological disease. Childs Nervous System. (2009) 25:153–9. doi: 10.1007/s00381-008-0744-z
100. Stepp SE, Dufourcq-Lagelouse R, Le Deist F, Bhawan S, Certain S, Mathew PA, et al. Perforin gene defects in familial hemophagocytic lymphohistiocytosis. Science. (1999) 286:1957–9. doi: 10.1126/science.286.5446.1957
101. U. zur Stadt S, Diler AS, Henter JI, Kabisch H, Schneppenheim R, Nürnberg P, et al. Hennies, Linkage of familial hemophagocytic lymphohistiocytosis (FHL) type-4 to chromosome 6q24 and identification of mutations in syntaxin 11. Hum Mol Genet. (2005) 14:827–34. doi: 10.1093/hmg/ddi076
102. Côte M, Ménager MM, Burgess A, Mahlaoui N, Picard C, Schaffner C, et al. Munc18-2 deficiency causes familial hemophagocytic lymphohistiocytosis type 5 and impairs cytotoxic granule exocytosis in patient NK cells. J Clin Invest. (2009) 119:3765–73. doi: 10.1172/JCI40732
103. U. zur Stadt J, Seifert W, Koch F, Grieve S, Pagel J, Strauss J, et al. Familial hemophagocytic lymphohistiocytosis type 5 (FHL-5) is caused by mutations in Munc18-2 and impaired binding to Syntaxin 11. Am J Hum Genet. (2009) 85:482–92. doi: 10.1016/j.ajhg.2009.09.005
104. Sieni E, Cetica V, Heckmann Y, Coniglio ML, Da Ros M, Ciambotti B, et al. Familial hemophagocytic lymphonistiocytosis: When rare diseases shed light on immune system functioning. Front Immunol. (2014) 5. doi: 10.3389/fimmu.2014.00167
105. Nagai K, Yamamoto K, Fujiwara H, An J, Ochi T, Suemori K, et al. Subtypes of familial hemophagocytic lymphohistiocytosis in Japan based on genetic and functional analyses of cytotoxic T lymphocytes. PloS One. (2010) 5. doi: 10.1371/journal.pone.0014173
106. Gioia C, Paroli M, Izzo R, Di Sanzo L, Rossi E, Pignatelli P, et al. Pathogenesis of hemophagocytic lymphohistiocytosis/macrophage activation syndrome: A case report and review of the literature. Int J Mol Sci. (2024) 25. doi: 10.3390/ijms25115921
107. Grom AA, Mellins ED. Macrophage activation syndrome: advances towards understanding pathogenesis. Curr Opin Rheumatol. (2010) 22:561–6. doi: 10.1097/01.bor.0000381996.69261.71
108. Kaufman KM, Linghu B, Szustakowski JD, Husami A, Yang F, Zhang K, et al. Whole-exome sequencing reveals overlap between macrophage activation syndrome in systemic juvenile idiopathic arthritis and familial hemophagocytic lymphohistiocytosis. Arthritis Rheumatol. (2014) 66:3486–95. doi: 10.1002/art.v66.12
109. Zhang K, Biroschak J, Glass DN, Thompson SD, Finkel T, Passo MH, et al. Macrophage activation syndrome in patients with systemic juvenile idiopathic arthritis is associated with MUNC13-4 polymorphisms. Arthritis Rheum. (2008) 58:2892–6. doi: 10.1002/art.23734
110. Yang X, Yao H, Zhao Q, Zhao Q, Yan X, Zhang Z, et al. UNC13D mutation in a patient with juvenile polymyositis with recurrent macrophage activation syndrome. Rheumatol (Oxford). (2021) 60:e404–6. doi: 10.1093/rheumatology/keab391
111. Zaripova LN, Midgley A, Christmas SE, Beresford MW, Baildam EM, Oldershaw RA. Juvenile idiopathic arthritis: from aetiopathogenesis to therapeutic approaches. Pediatr Rheumatol Online J. (2021) 19:135. doi: 10.1186/s12969-021-00629-8
112. Atteritano M, David A, Bagnato G, Beninati C, Frisina A, Iaria C, et al. Haemophagocytic syndrome in rheumatic patients. A systematic review. Eur Rev Med Pharmacol Sci. (2012) 16:1414–24.
113. Villanueva J, Lee S, Giannini EH, Graham TB, Passo MH, Filipovich A, et al. Natural killer cell dysfunction is a distinguishing feature of systemic onset juvenile rheumatoid arthritis and macrophage activation syndrome. Arthritis Res Ther. (2005) 7:R30–7. doi: 10.1186/ar1453
114. Wulffraat NM, Rijkers GT, Elst E, Brooimans R, Kuis W. Reduced perforin expression in systemic juvenile idiopathic arthritis is restored by autologous stem-cell transplantation. Rheumatol (Oxford). (2003) 42:375–9. doi: 10.1093/rheumatology/keg074
115. Hazen MM, Woodward AL, Hofmann I, Degar BA, Grom A, Filipovich AH, et al. Mutations of the hemophagocytic lymphohistiocytosis-associated gene UNC13D in a patient with systemic juvenile idiopathic arthritis. Arthritis Rheum. (2008) 58:567–70. doi: 10.1002/art.23199
116. Batlle-Maso L, Mensa-Vilaro A, Solis-Moruno M, Marques-Bonet T, Arostegui JI, Casals F. Genetic diagnosis of autoinflammatory disease patients using clinical exome sequencing. Eur J Med Genet. (2020) 63:103920. doi: 10.1016/j.ejmg.2020.103920
117. Gu H, Ma J, Chen Z, Wang J, Zhang R, Wu R. Synergistic defects of novo FAS and homozygous UNC13D leading to autoimmune lymphoproliferative syndrome-like disease: A 10-year-old Chinese boy case report. Gene. (2018) 672:45–9. doi: 10.1016/j.gene.2018.05.097
118. Tan C, Ozgul RK, Cagdas Ayvaz D, Tezcan I, Sanal O. Polymorphisms in FAS and CASP8 genes may contribute to the development of ALPS phenotype: a study in 25 patients with probable ALPS. Turk J Pediatr. (2015) 57:141–5.
119. Boggio E, Arico M, Melensi M, Dianzani I, Ramenghi U, Dianzani U, et al. Mutation of FAS, XIAP, and UNC13D genes in a patient with a complex lymphoproliferative phenotype. Pediatrics. (2013) 132:e1052–8. doi: 10.1542/peds.2012-1838
120. Arico M, Boggio E, Cetica V, Melensi M, Orilieri E, Clemente N, et al. Variations of the UNC13D gene in patients with autoimmune lymphoproliferative syndrome. PloS One. (2013) 8:e68045. doi: 10.1371/journal.pone.0068045
121. Murata Y, Yasumi T, Shirakawa R, Izawa K, Sakai H, Abe J, et al. Rapid diagnosis of FHL3 by flow cytometric detection of intraplatelet Munc13-4 protein. Blood. (2011) 118:1225–30. doi: 10.1182/blood-2011-01-329540
122. Zhao Q, Zhao Q, Tang X, An Y, Zhang Z, Tomomasa D, et al. Atypical familial hemophagocytic lymphohistiocytosis type 3 in children: A report of cases and literature review. Pediatr Allergy Immunol. (2024) 35:e14136. doi: 10.1111/pai.14136
123. Babol-Pokora K, Wolowiec M, Popko K, Jaworowska A, Bryceson YT, Tesi B, et al. Molecular genetics diversity of primary hemophagocytic lymphohistiocytosis among Polish pediatric patients. Arch Immunol Ther Exp (Warsz). (2021) 69:31. doi: 10.1007/s00005-021-00635-4
124. Giri PP, Biswas N, Chakravarty S. Familial hemophagocytic lymphohistiocytosis due to mutation of UNC13D gene. Indian J Hematol Blood Transfus. (2016) 32:344–6. doi: 10.1007/s12288-014-0494-x
125. Heeg M, Ammann S, Klemann C, Panning M, Falcone V, Hengel H, et al. Is an infectious trigger always required for primary hemophagocytic lymphohistiocytosis? Lessons from in utero and neonatal disease. Pediatr Blood Cancer. (2018) 65. doi: 10.1002/pbc.v65.11
126. Bergsten E, Horne A, Hed Myrberg I, Arico M, Astigarraga I, Ishii E, et al. Stem cell transplantation for children with hemophagocytic lymphohistiocytosis: results from the HLH-2004 study. Blood Adv. (2020) 4:3754–66. doi: 10.1182/bloodadvances.2020002101
127. Wegehaupt O, Wustrau K, Lehmberg K, Ehl S. Cell versus cytokine - directed therapies for hemophagocytic lymphohistiocytosis (HLH) in inborn errors of immunity. Front Immunol. (2020) 11:808. doi: 10.3389/fimmu.2020.00808
128. Merli P, Algeri M, Gaspari S, Locatelli F. Novel therapeutic approaches to familial HLH (Emapalumab in FHL). Front Immunol. (2020) 11:608492. doi: 10.3389/fimmu.2020.608492
129. Ehl S, Astigarraga I, von Bahr Greenwood T, Hines M, Horne A, Ishii E, et al. Recommendations for the use of etoposide-based therapy and bone marrow transplantation for the treatment of HLH: consensus statements by the HLH steering committee of the histiocyte society. J Allergy Clin Immunol Pract. (2018) 6:1508–17. doi: 10.1016/j.jaip.2018.05.031
130. Mahlaoui N, Ouachee-Chardin M, de Saint Basile G, Neven B, Picard C, Blanche S, et al. Immunotherapy of familial hemophagocytic lymphohistiocytosis with antithymocyte globulins: a single-center retrospective report of 38 patients. Pediatrics. (2007) 120:e622–8. doi: 10.1542/peds.2006-3164
131. Henter JI, Horne A, Arico M, Egeler RM, Filipovich AH, Imashuku S, et al. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. (2007) 48:124–31. doi: 10.1002/pbc.21039
132. Locatelli F, Jordan MB, Allen C, Cesaro S, Rizzari C, Rao A, et al. Emapalumab in children with primary hemophagocytic lymphohistiocytosis. N Engl J Med. (2020) 382:1811–22. doi: 10.1056/NEJMoa1911326
133. Vallurupalli M, Berliner N. Emapalumab for the treatment of relapsed/refractory hemophagocytic lymphohistiocytosis. Blood. (2019) 134:1783–6. doi: 10.1182/blood.2019002289
134. Moshous D, Briand C, Castelle M, Dupic L, Morelle G, Abou Chahla W, et al. Alemtuzumab as first line treatment in children with familial lymphohistiocytosis. Blood. (2019) 134. doi: 10.1182/blood-2019-124477
135. Jordan MB, Hildeman D, Kappler J, Marrack P. An animal model of hemophagocytic lymphohistiocytosis (HLH): CD8+ T cells and interferon gamma are essential for the disorder. Blood. (2004) 104:735–43. doi: 10.1182/blood-2003-10-3413
136. Trottestam H, Berglof E, Horne A, Onelov E, Beutel K, Lehmberg K, et al. Risk factors for early death in children with haemophagocytic lymphohistiocytosis. Acta Paediatr. (2012) 101:313–8. doi: 10.1111/j.1651-2227.2011.02501.x
137. Bergsten E, Horne A, Arico M, Astigarraga I, Egeler RM, Filipovich AH, et al. Confirmed efficacy of etoposide and dexamethasone in HLH treatment: long-term results of the cooperative HLH-2004 study. Blood. (2017) 130:2728–38. doi: 10.1182/blood-2017-06-788349
138. Jordan MB, Allen CE, Weitzman S, Filipovich AH, McClain KL. How I treat hemophagocytic lymphohistiocytosis. Blood. (2011) 118:4041–52. doi: 10.1182/blood-2011-03-278127
139. Gholam C, Grigoriadou S, Gilmour KC, Gaspar HB. Familial haemophagocytic lymphohistiocytosis: advances in the genetic basis, diagnosis and management. Clin Exp Immunol. (2011) 163:271–83. doi: 10.1111/j.1365-2249.2010.04302.x
140. Marsh RA, Hebert K, Kim S, Dvorak CC, Aquino VM, Baker KS, et al. Comparison of hematopoietic cell transplant conditioning regimens for hemophagocytic lymphohistiocytosis disorders. J Allergy Clin Immunol. (2022) 149:1097–1104 e2. doi: 10.1016/j.jaci.2021.07.031
141. Lehmberg K, Albert MH, Beier R, Beutel K, Gruhn B, Kroger N, et al. Treosulfan-based conditioning regimen for children and adolescents with hemophagocytic lymphohistiocytosis. Haematologica. (2014) 99:180–4. doi: 10.3324/haematol.2013.094730
142. Trottestam H, Horne A, Arico M, Egeler RM, Filipovich AH, Gadner H, et al. Chemoimmunotherapy for hemophagocytic lymphohistiocytosis: long-term results of the HLH-94 treatment protocol. Blood. (2011) 118:4577–84. doi: 10.1182/blood-2011-06-356261
143. Soheili T, Riviere J, Ricciardelli I, Durand A, Verhoeyen E, Derrien AC, et al. Gene-corrected human Munc13-4-deficient CD8+ T cells can efficiently restrict EBV-driven lymphoproliferation in immunodeficient mice. Blood. (2016) 128:2859–62. doi: 10.1182/blood-2016-07-729871
144. Soheili T, Durand A, Sepulveda FE, Riviere J, Lagresle-Peyrou C, Sadek H, et al. Gene transfer into hematopoietic stem cells reduces HLH manifestations in a murine model of Munc13-4 deficiency. Blood Adv. (2017) 1:2781–9. doi: 10.1182/bloodadvances.2017012088
145. Dettmer V, Bloom K, Gross M, Weissert K, Aichele P, Ehl S, et al. Retroviral UNC13D gene transfer restores cytotoxic activity of T cells derived from familial hemophagocytic lymphohistiocytosis type 3 patients in vitro. Hum Gene Ther. (2019) 30:975–84. doi: 10.1089/hum.2019.025
146. Takushi SE, Paik NY, Fedanov A, Prince C, Doering CB, Spencer HT, et al. Lentiviral gene therapy for familial hemophagocytic lymphohistiocytosis type 3, caused by UNC13D genetic defects. Hum Gene Ther. (2020) 31:626–38. doi: 10.1089/hum.2019.329
147. Goyal S, Tisdale J, Schmidt M, Kanter J, Jaroscak J, Whitney D, et al. Acute myeloid leukemia case after gene therapy for sickle cell disease. N Engl J Med. (2022) 386:138–47. doi: 10.1056/NEJMoa2109167
148. Nobles CL, Sherrill-Mix S, Everett JK, Reddy S, Fraietta JA, Porter DL, et al. CD19-targeting CAR T cell immunotherapy outcomes correlate with genomic modification by vector integration. J Clin Invest. (2020) 130:673–85. doi: 10.1172/JCI130144
149. Biasco L, Rothe M, Buning H, Schambach A. Analyzing the genotoxicity of retroviral vectors in hematopoietic cell gene therapy. Mol Ther Methods Clin Dev. (2018) 8:21–30. doi: 10.1016/j.omtm.2017.10.002
150. Ramadass M, Catz SD. Molecular mechanisms regulating secretory organelles and endosomes in neutrophils and their implications for inflammation. Immunol Rev. (2016) 273:249–65. doi: 10.1111/imr.2016.273.issue-1
151. Harper MT, van den Bosch MT, Hers I, Poole AW. Platelet dense granule secretion defects may obscure alpha-granule secretion mechanisms: evidence from Munc13-4-deficient platelets. Blood. (2015) 125:3034–6. doi: 10.1182/blood-2014-12-618439
152. Dettmer-Monaco V, Weissert K, Ammann S, Monaco G, Lei L, Grassel L, et al. Gene editing of hematopoietic stem cells restores T-cell response in familial hemophagocytic lymphohistiocytosis. J Allergy Clin Immunol. (2024) 153:243–255 e14. doi: 10.1016/j.jaci.2023.08.003
153. Oliveira G, Ruggiero E, Stanghellini MT, Cieri N, D’Agostino M, Fronza R, et al. Tracking genetically engineered lymphocytes long-term reveals the dynamics of T cell immunological memory. Sci Transl Med. (2015) 7:317ra198. doi: 10.1126/scitranslmed.aac8265
154. Biasco L, Scala S, Basso Ricci L, Dionisio F, Baricordi C, Calabria A, et al. In vivo tracking of T cells in humans unveils decade-long survival and activity of genetically modified T memory stem cells. Sci Transl Med. (2015) 7:273ra13. doi: 10.1126/scitranslmed.3010314
155. Gattinoni L, Speiser DE, Lichterfeld M, Bonini C. T memory stem cells in health and disease. Nat Med. (2017) 23:18–27. doi: 10.1038/nm.4241
156. Cieri N, Oliveira G, Greco R, Forcato M, Taccioli C, Cianciotti B, et al. Generation of human memory stem T cells after haploidentical T-replete hematopoietic stem cell transplantation. Blood. (2015) 125:2865–74. doi: 10.1182/blood-2014-11-608539
157. Li X, Wirtz T, Weber T, Lebedin M, Lowenstein ED, Sommermann T, et al. Precise CRISPR-Cas9 gene repair in autologous memory T cells to treat familial hemophagocytic lymphohistiocytosis. Sci Immunol. (2024) 9:eadi0042. doi: 10.1126/sciimmunol.adi0042
158. Wagner DL, Koehl U, Chmielewski M, Scheid C, Stripecke R. Review: sustainable clinical development of CAR-T cells - switching from viral transduction towards CRISPR-Cas gene editing. Front Immunol. (2022) 13:865424. doi: 10.3389/fimmu.2022.865424
159. Masarwy R, Stotsky-Oterin L, Elisha A, Hazan-Halevy I, Peer D. Delivery of nucleic acid based genome editing platforms via lipid nanoparticles: Clinical applications. Adv Drug Delivery Rev. (2024) 211:115359. doi: 10.1016/j.addr.2024.115359
Keywords: UNC13D, cytotoxic granule secretion, immune diseases, FHL3, gene therapy
Citation: Duong V-T, Lee D, Kim YH and Oh S-O (2024) Functional role of UNC13D in immune diseases and its therapeutic applications. Front. Immunol. 15:1460882. doi: 10.3389/fimmu.2024.1460882
Received: 11 July 2024; Accepted: 24 September 2024;
Published: 14 October 2024.
Edited by:
Samuel Cern Cher Chiang, Cincinnati Children’s Hospital Medical Center, United StatesReviewed by:
Snehal Shabrish, Advanced Centre for Treatment, Research and Education in Cancer (ACTREC), IndiaCopyright © 2024 Duong, Lee, Kim and Oh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sae-Ock Oh, aGVkZ2Vob2dAcHVzYW4uYWMua3I=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.