
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 20 September 2024
Sec. Autoimmune and Autoinflammatory Disorders: Autoinflammatory Disorders
Volume 15 - 2024 | https://doi.org/10.3389/fimmu.2024.1458118
This article is part of the Research Topic Basic, clinical, and translational studies of Yao syndrome and other NOD2 related diseases View all 8 articles
 Hafsa Nomani1Song Wu2Ashmia Saif1Frank Hwang1Jane Metzger1Brianne Navetta-Modrov1
Hafsa Nomani1Song Wu2Ashmia Saif1Frank Hwang1Jane Metzger1Brianne Navetta-Modrov1 Peter D. Gorevic1
Peter D. Gorevic1 Ivona Aksentijevich3
Ivona Aksentijevich3 Qingping Yao1*
Qingping Yao1*Objective: Yao syndrome (YAOS) is formerly called nucleotide-binding oligomerization domain containing 2 (NOD2)-associated autoinflammatory disease.We report a large cohort of YAOS.
Methods: We conducted a retrospective analysis of a cohort of adult patients with systemic autoinflammatory diseases (SAIDs). All patients underwent testing for a periodic fever syndrome gene panel.
Results: A total of 194 patients carried NOD2 variants, 152 patients were diagnosed with YAOS, and 42 had mixed autoinflammatory diseases with combined variants in NOD2 and other SAID-associated genes. Demographic, clinical and molecular data were summaried. In sub-group analysis of the 194 patients, individual patients were often identified to carry two or more variants that usually included IVS8 + 158/R702W, IVS8 + 158/L1007fs, IVS8 + 158/V955I, IVS8 + 158/other, or NOD2/variants in other SAID genes. Ninety-nine patients carried single variants. Taken together, these variants contribute to the disease in combination or individually.
Conclusion: This largest cohort has provided comprehensive clinical and genotyping data in YAOS. Variants in the NOD2 gene can give rise to a spectrum from inflammatory bowel disease to autoinflammatory disease.This report further raises awareness of the underdiagnosed disease in the medical community.
Systemic autoinflammatory diseases (SAIDs) primarily derive from abnormal innate immune responses (1). Nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs) are intracellular sensors to pathogen-associated molecular pattern molecules (PAMPs) and play a critical role in innate immune responses (2). Genetic variations in NLRs have been associated with a number of SAIDs. One example is nucleotide-binding oligomerization domain containing 2 (NOD2), a cytosolic sensor to peptidoglycans or muramyl dipeptide (MDP) from bacterial walls. NOD2 contributes to the defense against microorganisms and the regulation of inflammatory processes, primarily in the gut (3). NOD2 genetic variations are associated with diseases such as Crohn’s disease (CD, OMIM 266600), Blau syndrome (BS, OMIM 186580), and Yao syndrome (YAOS, OMIM 617321) (4).
YAOS is formerly designated as NOD2-associated autoinflammatory disease and phenotypically characterized by recurrent fever, dermatitis, arthralgias, gastrointestinal and sicca-like symptoms, distal leg and eyelid swelling (5). Since our initial report in 2011, this disease has been increasingly recognized and reported in North America, Europe, and Asia (6–10). The disease appears more common than initially thought, particularly in the adult patient population. Patients often went through a long diagnostic odyssey before the final disease diagnosis, and approximately 15% of patients filed for disability due to physical and vocational impairments, resulting in a significant burden of disease (5). Due to its association with specific NOD2 gene mutations, molecular testing is necessary for diagnosis. Because of overlapping clinical manifestations between YAOS and other SAIDs, such as recurrent fevers, an autoinflammatory disease gene panel is usually performed using Next Generation and targeted DNA sequencing for a differential diagnosis. Over the last decade, the clinical phenotype and genotype of YAOS have expanded rapidly (11). Herein, we report the largest single-site cohort of YAOS patients and provide an update on clinical phenotypes and related aspects.
We have established a large cohort of adult patients with SAIDs at the Center of Autoinflammatory Diseases in Stony Brook University Hospital since February 2016. These patients were referred from across the U.S. and managed by subspecialists in our group, with clinical, biochemical, and genetic data entered into Electronic Medical Records (EMRs). Specifically, the EMRs of SAID patients with NOD2 mutations were reviewed. Systemic autoimmune diseases, vasculitis, inflammatory bowel disease (IBD), infectious and malignant diseases were excluded after detailed evaluations. All patients underwent molecular testing for an autoinflammatory disease gene panel, including MEFV, TNFRSF1A, NLRP3, MVK, NLRP12, and NOD2. Approximately 20% of patients also had larger scale genetic testing for an autoimmune and autoinflammatory gene panel at the Molecular Diagnostics Laboratory at Invitae in California. According to our published criteria, YAOS was diagnosed based on characteristic clinical phenotype and a specific NOD2 genotype, with the exclusion of related diseases (5). Patients with NOD2 and other variants in two or more different SAID genes were also included to delineate the NOD2 genetic landscape.
Descriptive statistics was used for analysis. Median+/-IQR were reported for continuous variables; category percentages were reported for categorical variables. All analyses were performed using SAS 9.4 (SAS Institute Inc., Cary, NC).
There were 194 patients in this study cohort, including 152 patients carrying genetic variants only in NOD2, and 42 patients with combined genetic variants in NOD2 and other SAID genes. The demographic data and clinical manifestations and their frequencies from the 152 patients with NOD2 variants alone are summarized in Table 1. The clinical features of the disease were graphically featured previously (5, 12) and are exemplified with three additional representative cases in Figure 1.
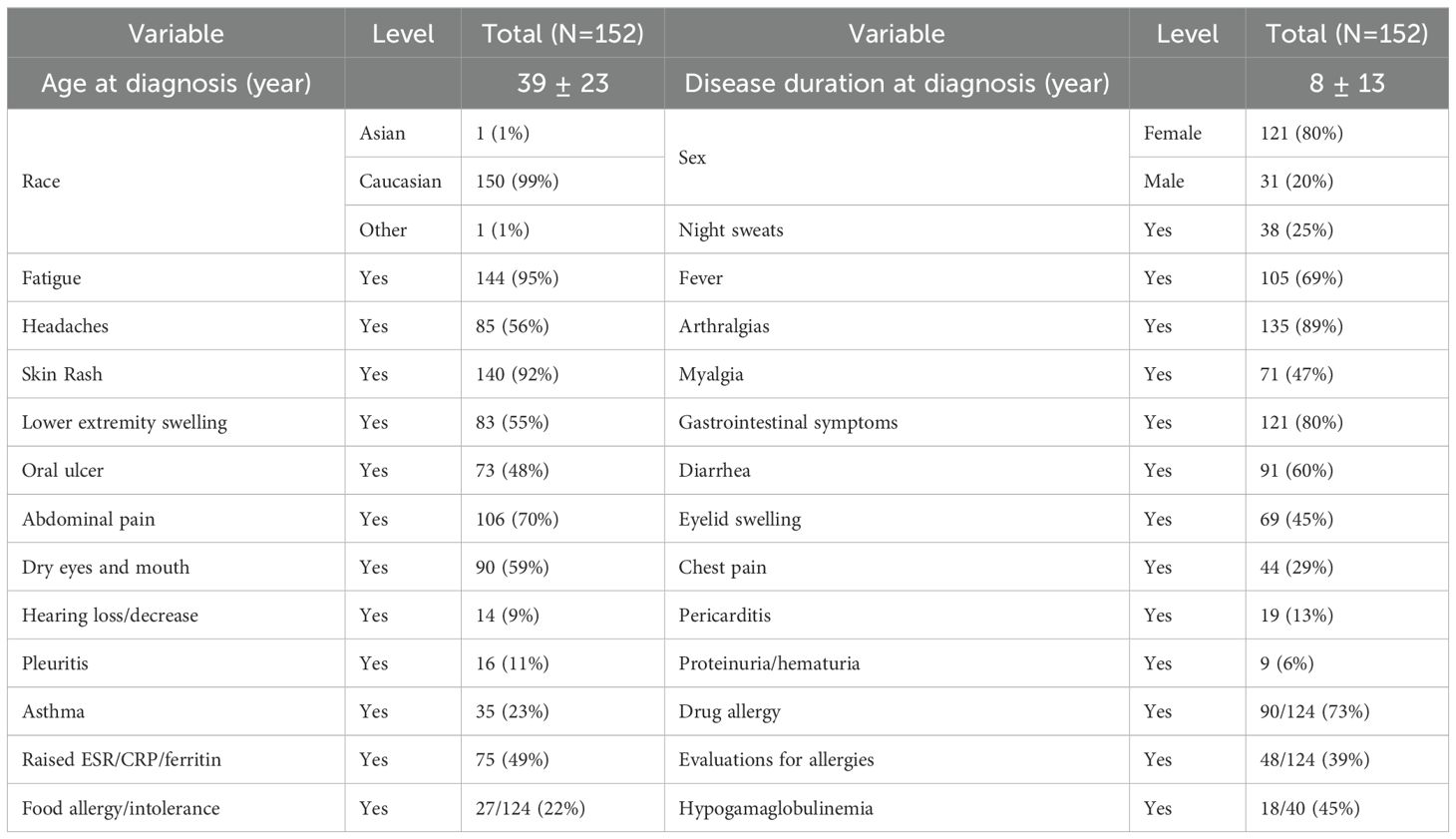
Table 1. Demographic, clinical and laboratory data in YAOS patients.
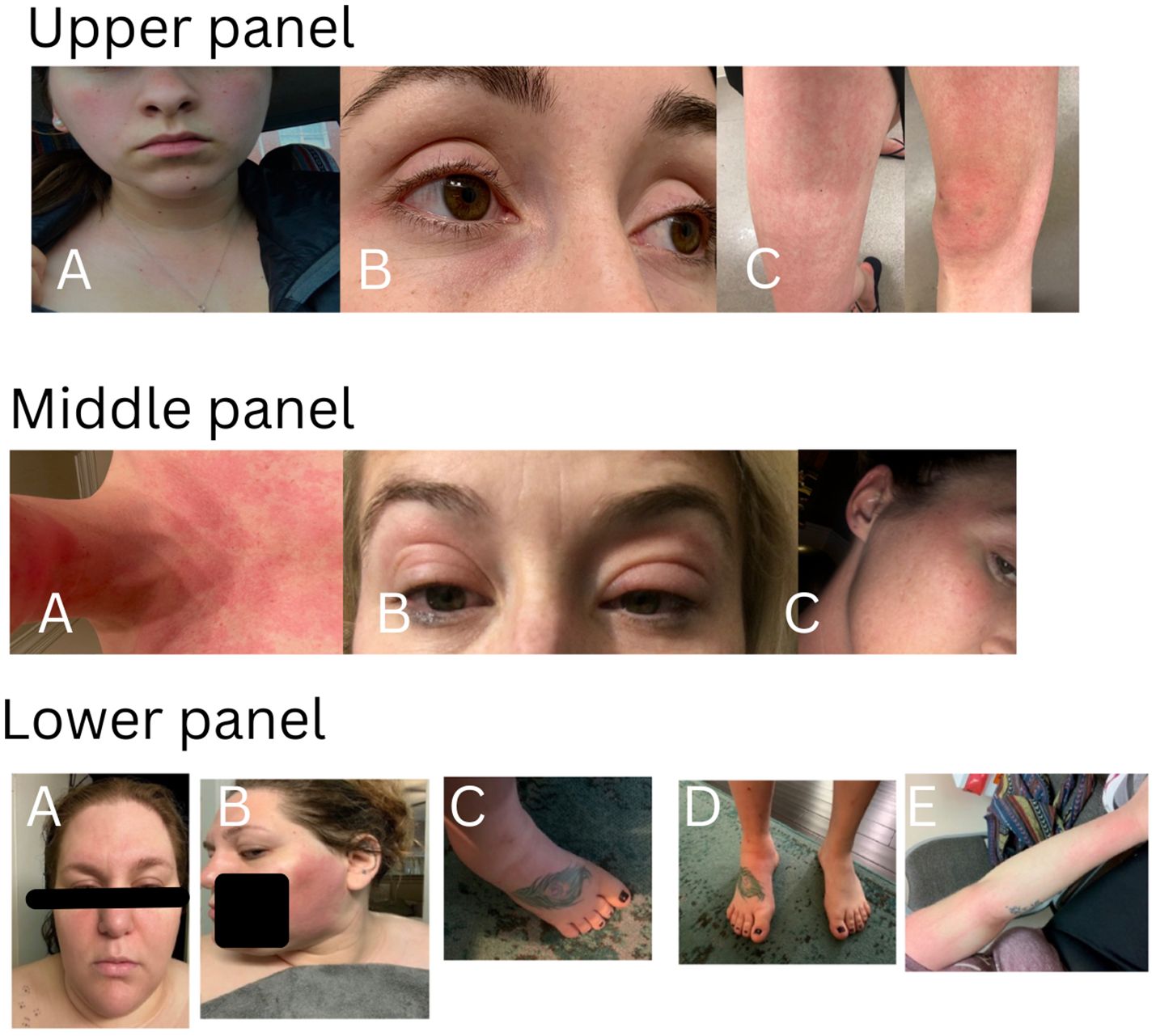
Figure 1. Cutaneous presentations in three representative patients. Upper panel: patchy erythema on cheeks, anterior neck and upper chest (A), eyelid redness and mild puffiness (B), erythematous rash on the thigh and knee with swelling (C). This patient carries heterozygous NOD2 IVS8 + 158 only and had a good response to IL-1 inhibitor therapy. Middle panel: diffuse redness on the chin, anterior neck and chest (A), bilateral eyelid swelling with discoloration (B), redness on the right upper cheek, temple and external ear (C). This patient carries heterozygous NOD2 V955I only and had a good response to IL-1 inhibitor therapy. Lower panel: erythematous patches on the face, forehead and bilateral eyelid swelling with discoloration(A), large patchy erythema on the left cheek (B), swollen ankle and foot (C, D), erythematous patches on the arm (E). This patient carries heterozygous and compound NOD2 IVS8 + 158/1007fs/V955I and had a good response to IL-1 inhibitor therapy.
Since YAOS has been previously described in detail, herein we update and focus on certain clinical manifestations that were not previously reported or highlighted. Recurrent fever is common and can last a few hours to weeks. Dermatitis is known to be a common feature primarily manifesting as patchy erythema on the face, chest, or limbs. In addition, urticaria and livedo reticularis were seen in a minority of patients. Eczema-like patches can occur near the earlobe and adjacent areas. Individual patients may present with mixed cutaneous findings of the rashes described above. Generally, rashes are more related to heat than cold and more often occur in summertime or after having a hot shower. Histopathological findings vary, but spongiotic dermatitis and perivascular lymphocytic infiltrate are most common (6). Patients may present with brief episodes of red external ears without pain or thickening distinct from the cartilaginous involvement of relapsing polychondritis. Sicca symptoms are common without typical findings of primary Sjögren syndrome. Eyelid swelling with or without discoloration can occur often in the morning and can be unilateral or bilateral. Patients may have respiratory symptoms, such as chronic sinusitis, asthma-like symptoms, or intermittent episodes of unexplained shortness of breath. Chest pain, pericarditis and pleuritis occurred in 29%, 13%, and 11% of patients, respectively. Several patients developed pleuritis or pericarditis so severe that decortication surgery or placement of a pericardial window was required. Several patients had abnormal cardiac findings including tachycardia and ventricular hypertrophy, dilation, and mild pulmonary hypertension. Gastrointestinal symptoms often include cyclic abdominal pain, bloating and/or nonbloody diarrhea in the majority, whereas other patients may have intermittent episodes of nausea, vomiting or constipation. Extensive gastrointestinal workups did not show convincing evidence of IBD. Oligo- or polyarthralgias are common and usually intermittent without causing articular deformities or destruction. Distal leg swelling of unclear cause, which often involves the ankle and foot, is a characteristic finding, and may be unilateral or bilateral, descriptively resembling lymphedema in some patients. Patients (approximately 16%) may present with episodic superficial (cervical, axillary, or inguinal) and/or deep (intra-abdominal) lymphadenopathy, reactive hyperplasia without granulomatous change mostly. Neurologically, complaints of headaches are common, and several patients were diagnosed with meningitis or ventriculitis. Patient-reported drug allergies and food allergy/intolerance were 73% and 22%, respectively in YAOS (Table 1), including cutaneous flushing or allergic phenomena, and even anaphylactic reactions. Forty-one percent of patients underwent evaluations for atopic disorders by allergists/immunologists, and idiopathic chronic urticaria, or mast cell activation syndrome was often suspected. Hypogammaglobinemia can also occur (13). Among the 152 patients with NOD2 variants only in the current study, 40 were tested for Immunoglobulin (Ig) quantitation, and 45% (18/40) had low Ig of different degrees.
Mild leukocytosis and anemia could occur. Approximately 50% of patients had documented elevated acute phase reactants such as ESR and/or CRP. Patients may have detectable antinuclear antibodies (ANAs) of low titers, but serologic testing for systemic autoimmune diseases is negative for anti-extractable nuclear antigen (ENA), anti-double stranded DNA (dsDNA) antibodies, Rheumatoid factor and Cyclic citrullinated peptide (CCP) antibodies among others.
In summary, YAOS is a systemic inflammatory disease, primarily affecting cutaneous, musculoskeletal, lymphoreticular, cardiopulmonary and gastrointestinal system with rare involvement of internal solid organs. The characteristic clinical phenotypes are recurrent fever, dermatitis, GI symptoms, eyelid swelling, and distal leg swelling to constitute a constellation with other uncharacteristic symptoms. With these in mind, a proper diagnostic workup for autoinflammatory process may be initiated, including genetic testing.
A mutational spectrum of NOD2 has been identified to increase susceptibility to the disease, including 24 gene variants (Table 2). Most patients carry variants within the Leucine Rich Repeat (LRR) and adjacent region, and some variants reside in the central NACHT domain encoded by exon 4 (R702W, R703C) (Figure 2). Of the 24 variants, 19 are rare (MAF<1%) and 5 are classified as low-frequency or common. Most variants have been previously reported to increase susceptibility to YAOS (11, 12) (7). For example, NOD2 IVS8 + 158, V955I, R702W, and 1007fs are main variants in YAOS. In our sub-group analysis of the 194 patients with positive NOD2 variants (Figure 3), 95 (49%) patients carried two or more variants that usually included IVS8 + 158/R702W, IVS8 + 158/L1007fs, IVS8 + 158/V955I, IVS8 + 158/N852S, or IVS8 + 158/other (approximately 27%), or NOD2 with other SAID gene variants (22%). These combinations often consisted of common and low frequency NOD2 variants. Twenty-three percent of patients carried NOD2 IVS8 + 158 only, and 17.0% carried V955I only. Thirty-seven (19%) carried 19 rare NOD2 variants, including 6 single variants and 13 in combination with other variants. Of the 37 patients, 21 carried single rare NOD2 variants and 16 also carried another NOD2 or other SAID gene variants. The variant combinations were usually low-penetrance/rare variants in NOD2 or other SAID genes such as MEFV: E148Q or NLRP3: Q705K. (Figure 3). Genotyping combinations in YAOS can be common and low frequency NOD2 variants or low frequency or common NOD2 variants with rare NOD2 variants. Taken together, these data underpin the contributions of these NOD2 variants to the disease in combination or individually.
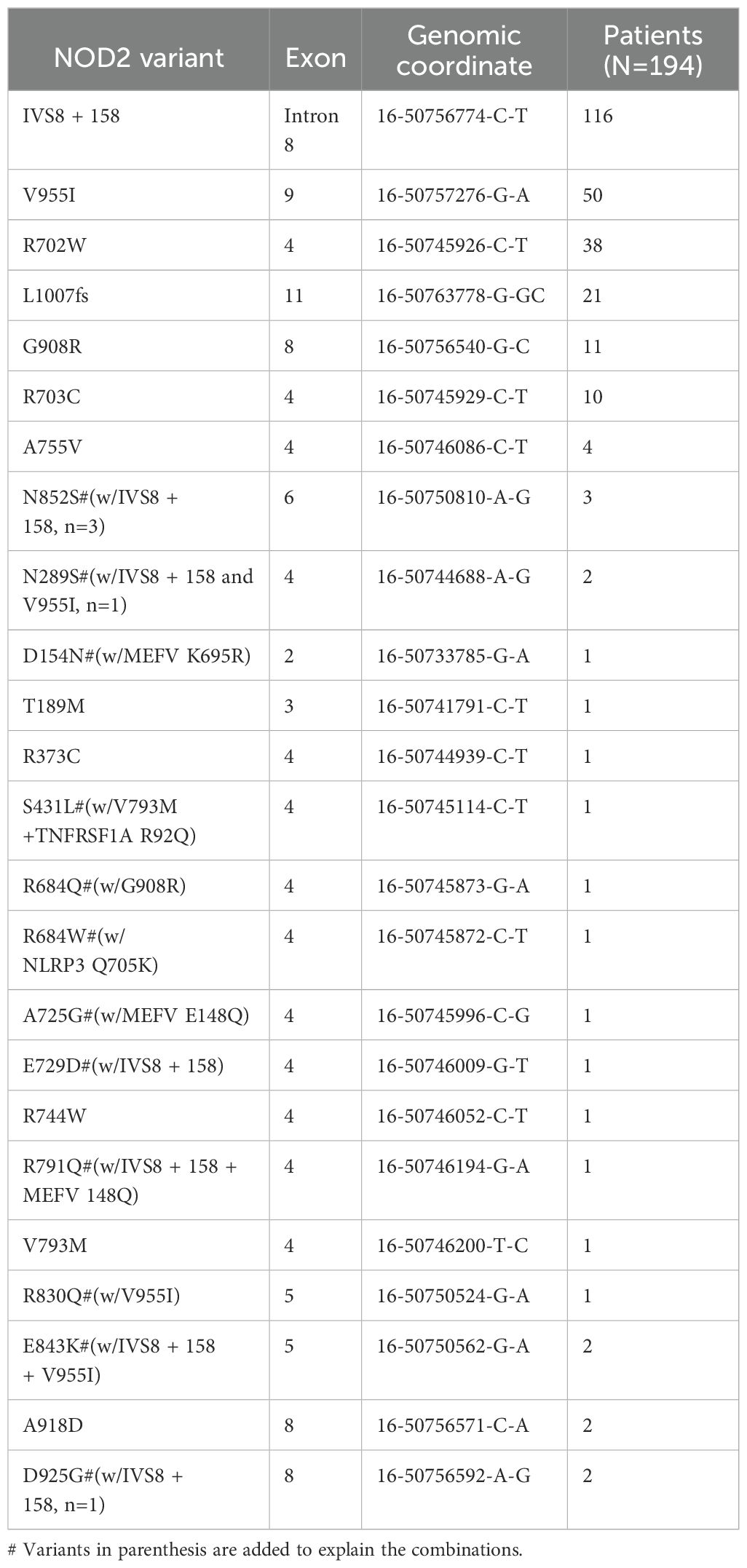
Table 2. Genotyping results in Yao syndrome.
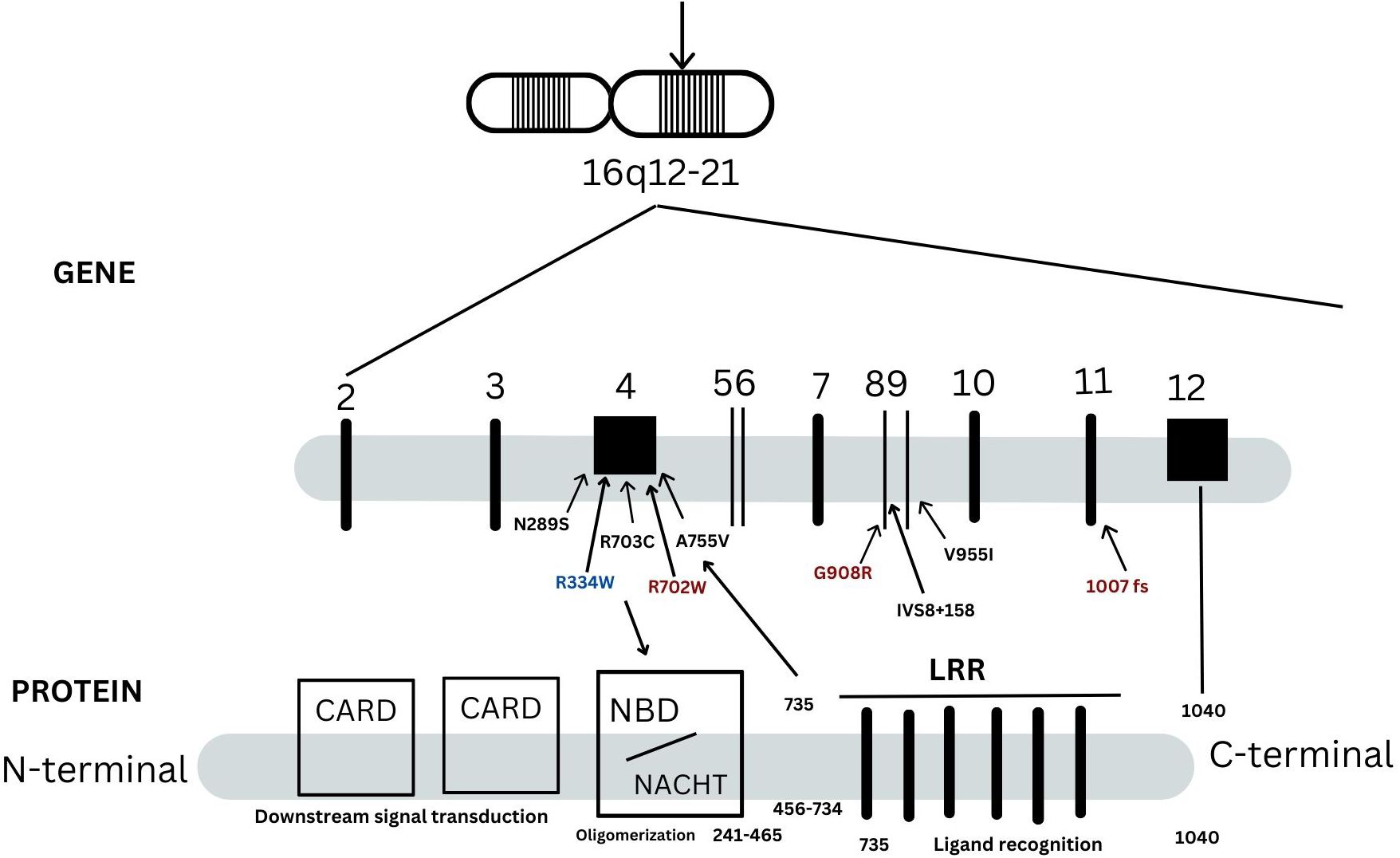
Figure 2. A schematic representation of NOD2 gene and protein. There are 12 exons in the NOD2 gene as indicated in vertical lines. NOD2 protein is composed of 1040 amino acids and is divided into three regions: the leucin-rich repeats (LRRs), nucleotide-binding domain (NBD) and caspase recruitment domains (CARDs). They are responsible for bacterial recognition, NOD2 self-oligomerization and down steam signal transduction, respectively. Three main variants in red, 1007fs, Q908R, R702W, are identified in -40% patients with Crohn’s disease. These three NOD2 variants, IVS8 + 158, V955I or other variants are linked to Yao syndrome (YAOS). Most YAOS-associated variants are within the region encoding LRRs and are detected often in combination in individual patients. In a minority of YAOS patients, variants in exon 4(non-Blau syndrome associated variants) and between the exon 4 and the region encoding the LRRs are identified; these are usually rare variants and were detected singly or in combination with the LRR variants. Blau syndrome-associated variants are within exon 4 and are of high penetrance.
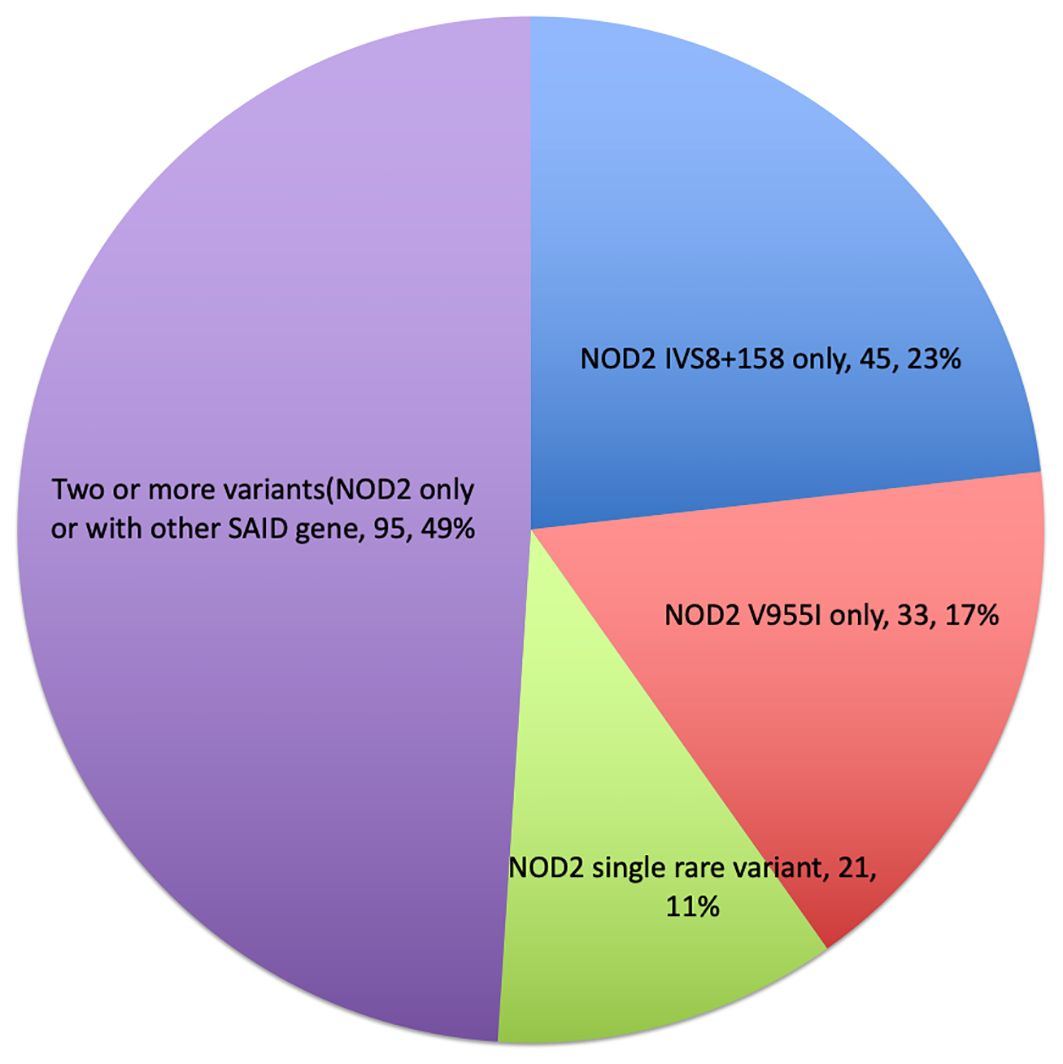
Figure 3. Genotyping distribution in disease (piechart).
YAOS is an increasingly recognized autoinflammatory disease associated with NOD2 mutations. In this largest patient cohort, we delineated the genotyping architecture of NOD2 and updated clinical phenotypes (Table 1, Figure 2). This study provides a more detailed genetic map and clinical data useful for prompt recognition and diagnosis.
First, the intronic variant NOD2 IVS8 + 158 (rs5743289) has been identified in approximately 60% of our adult patients, of whom 94% were heterozygote carriers. However, a significant portion of these patients also carrried concurrent one or two low- frequency or rare NOD2 variants in the form of compound heterozygous variants. For example, NOD2 IVS8 + 158 can coincide with R702W (missense mutation), 1007fs (frameshift mutation), V955I (missense mutation), N852S(missense mutation), or other NOD2 variants. In the current study, 26% (50) of patients carried heterozygous NOD2 V955I, of whom 33 had V955I only and 17 were concurrent with other NOD2 variants.
Based on our studies and the literature, low-frequency and common variants of NOD2 can give rise to a spectrum from IBD to SAID. Apart from YAOS, NOD2 mutations are well known to be associated with CD and BS. Up to 40% of patients with CD carry three main NOD2 variants, R702W, G908R, 1007fs, located in exons 4, 8 and 11, respectively (14). CD, therefore, is a major differential diagnosis for YAOS given the shared genotypes in both diseases. In a recent study, supervised machine learning was used to classify IBD patients by subtype using whole exome sequecing (WES) data; NOD2 was found to be the top gene for discriminating CD and ulcerative colitis, regardless of gene panel used (15). In another study of 1,183 pediatric patients with IBD using WES, NOD2 variants were found to contribute to early-onset CD in which the landscape of NOD2 variants was defined (16). Compared to the genetic map in the pediatric patients, the NOD2 genetic architecture as determined by targeted gene panel sequencing in YAOS appears somewhat similar (Figure 2). However, biallelic rare and low-frequency NOD2 variants were identified more often in pediatric patients (8%), either as homozygote or compound heterozygous variants, R702W, 1007fs, and G908R. In our cohort, only one 28-year-old female carried homozygous NOD2 G908R without IBD. Altogether, these data indicate that homozygotes and compound heterozygotes of these three variants cause early-onset disease, likely due to their double gene dose effect. In addition, BS is an autosomal dominant disease characterized by the triad of granulomatous dermatitis, uveitis and inflammatory arthritis (17). BS primarily occurs in children, and adult-onset disease is extremely rare. Both dermatitis and arthritis are symmetric and persistent, and fever and gastrointestinal symptoms are unusual in the American and European populations (18, 19). In contrast, approximately 50% (26/50) of Japanese patients with BS were reported to have fever, while GI symptoms were nearly absent (20). BS is caused by the high-penetrance NOD2 variants in exon 4, such as R334W, R334Q and a dozen other rare NOD2 variants (20, 21). Of note, these variants were not identified in our current cohort. Taken together, these data support that the NOD2 genotypes have varying degrees of penetrance and they can contribute to different phenotypes. In fact, the same genotype can be associated with different phenotypes due to contribution of other genetic, developmental, and environmental variables (22, 23).
Human genetic disorders have been traditionally classified as monogenic (Mendelian) or complex (polygenic). In Mendelian diseases (Huntington disease), there were slight but no predominant differences in sex ratios (24, 25), as primary mutations are highly penetrant and major determinants in the pathogenesis of diseases. Similarly, a female to male ratio (24/26) was not predominant for BS (20). By contrast, female/male ratio was 52/48 for CD patients without NOD2 variants and 62/38 for patients with one NOD2 variant (14). In YAOS, women account for 80% of the patient population. Both CD (up to 40% with NOD2 variants) and YAOS are associated with NOD2 variants of incomplete penetrance and only a small proportion of individuals with such variants develop disease. These diseases or disease status cannot be explained by the monogenic disease model. To supplement the traditional dichotomous classification, we recently proposed and defined the Genetically Transitional Disease (GTD) concept (26). GTD refers to a disease status between monogenic and polygenic, where a gene mutation is necessary but not sufficient to cause disease (26). This concept has been now applied to certain rheumatic diseases (27), including autoinflammatory diseases. Human genetic disorders generally result from interactions between candidate genes, genetic background, and environment among others (28). Genetic background refers to all other related genes that may interact with the gene of interest to potentially influence specific phenotype in concert with environment, such as diet (23, 29). GTD highlights the pervasive influence of genetic background together with environment. Unlike monogenic diseases, genetic background (including X chromosome and maternally transmitted mitochondrial DNA) and sex hormones could play more important roles in YAOS and CD with low-penetrance NOD2 variants, possibly resulting in female predominance (30).
Notably, in our cohort of patients, approximately 50% of patients carried two or more NOD2 variants, or NOD2 with other SAID gene variants, including MEFV, NLRP3 and NLRP12. We recently reported that patients with combined NOD2 and other gene variants may present with mixed autoinflammatory diseases, but YAOS was enriched. Such combinations may contribute to the etiological complexity of disease due to the genetic background in relation to SAID genes (30).
Two-hit hypothesis has been demonstrated in the development of tumors (31). We assumed that a two-hit-like theory, i.e., two or more germline mutational events in the same gene (intragenic) or different genes (extragenic) are required for the development of some SAIDs (27). Approximately 27% patients with YAOS carry two or more low-penetrance NOD2 variants, while 19% carry one rare NOD2 variant along with one or more low-penetrance variants in NOD2 or other SAID genes (Figure 3). In CD, NOD2 gene-dosage effects have been noted. For example, individuals carrying any one of the CD associated risk alleles (R702W, G908R, L1007fs) have two to four fold increased risk for developing CD (32), whereas carriers of two or more of the same NOD2 variants have a 15–40 fold increased risk for developing CD (14). Individuals with two or more rare and low-frequency NOD2 variants (homozygotes or compound heterozygotes) have been found to develop early-onset CD (16). Together, these data support our postulate that the two-hit like theory could be operational in the NOD2-associated diseases. We believe that this theory is compatible with intragenic or extragenic epistasis or gene to gene interactions as in our study (30). Based on this theory, some YAOS patients with monoallelic NOD2 variants may be further examined to search for another one or more germline or somatic mutational events.
NOD2 is highly expressed in monocytes, macrophages, dendritic, and paneth cells (3). NOD2 binds MDP and this ligation causes self-oligomerization leading to activation of downstream signal transduction pathways, including NF-kB (33). High-impact BS-associted mutations are gain-of-function and result in constitutive protein activation (34). BS-associated mutations can also result in loss of NOD2 cross-regulatory function (35). In contrast, CD-associated NOD2 variants are considered loss-of-function with respect to sensing and they impair the NOD2 signaling pathway activation, causing abnormal production of cytokines (36).
NOD2 IVS8 + 158 is an intronic missense mutation, aka c.2798 + 158 C>T or JW1 (37). Non-coding variants have been increasingly reported to be associated with diseases, and recommendations on clinical interpretations of such variants have been proposed (38). In general, intronic mutations are known to cause disease through alterations in splicing sites, splicing regulatory elements, or disruption of transcription regulatory motifs and non-coding RNA genes (39). Our prior study did not find an alteration in the splicing site of NOD2 IVS8 + 158, but NOD2 transcript level and basal p38 mitogen-activated protein kinase (MAPK) activity were significantly elevated in peripheral blood mononuclear cells (PBMCs) from YAOS patients (40). We also demonstrated the presence of dysregulated NOD2 pathway signaling and cytokine profiles in YAOS (40). PBMCs in cocultures with MDP produced higher levels of IL-6 and MAPK in YAOS patients with the NOD2 IVS8 + 158 variant, suggesting gain-of-function. Cytokines and NF-kB activation were significantly lower, favoring loss-of-function in patients with compound NOD2 IVS8 + 158/R702W perhaps due to their additive influences, profoundly compromising the NOD2 activation pathway. Using RNA sequencing and transcriptomic analysis, NOD2-mediated signaling pathway was found to be hyperactive with overproduction of pro-inflammatory cytokines such as IL-1 and IL-6 in YAOS patients with NOD2 variant Q902K (41). YAOS is considered to be a GTD and may result from a complex interaction of genetic variants in NOD2 and genetic background from other innate immune sensor genes, in combination with environmental triggers. For example, gastrointestinal surgeries may trigger or exacerbate disease (5) and we have reported that COVID-19 infection or vaccinations can elicit disease expression or exacerbation (42).
In summary, BS-associated NOD2 variants are highly penetrant, whereas CD- and YAOS-associated NOD2 variants are of low penetrance, and both diseases share NOD2 variants, such as R702W, L1007fs and G908R. Functional study of YAOS has been limited to date. Further study will be needed to refine molecular mechanisms of YAOS.
In the current article, we briefly discuss therapeutic options. Short courses of glucocorticoids are effective for flares and sulfasalazine was effective in 40%. Hydroxychloroquine was tried in many patients with minimal or temporary effects. Methotrexate was ineffective overall. Biologics are second choice if there is failure to improve with or intolerance of sulfasalazine. Unlike BS or CD, IL-1 antagonists are generally effective for YAOS (Table 3). In a Greek study of 167 patients with SAIDs, YAOS was diagnosed in 12 patients (7% of the entire cohort), largely substantiating the clinical phenotype, genotype and therapeutic response as we previously reported (10). For non-responders, a Janus kinase (JAK) inhibitor or IL-6 inhibitor may be tried. Based on our experience, sulfasalazine or hydroxychloroquine combined with an IL-1 inhibitor or JAK inhibitor could be used to maximize effectiveness for some cases. While anti-TNFα is a major therapy for BS (43), it produced mixed or temporary improvement for the treatment of YAOS. Further studies are warranted to identify or develop more effective new drugs to treat YAOS.
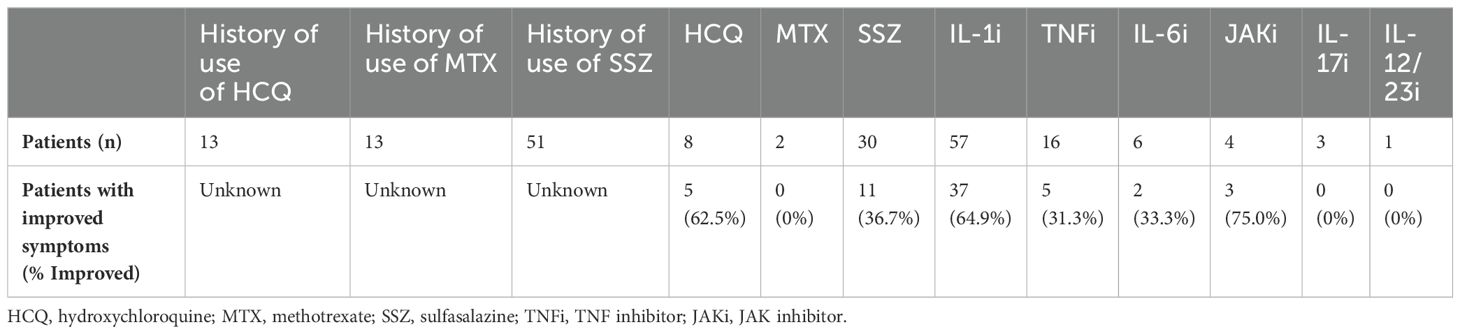
Table 3. Therapeutic data for YAOS patients.
Genetic counseling using the GTD concept may be appropriate and supplemental due to the identification of the NOD2 variants. This will help patients understand the medical, psychological, and familial implications of genetic contributions to their disease. Approximately 15% of YAOS patients report a positive family history (6). Therefore, we recommend that family members and relatives be genotyped only if they experience similar autoinflammatory symptoms. Prognostically, most patients have recurrent disease flares of varying frequencies, and some may eventually develop chronic and persistent disease. To date, none of our patients have developed CD over a decade long follow up. A minority may develop chronic pain syndrome or fibromyalgia, consequently affecting physical and/or mental function. Some patients in the current study were initially diagnosed with Still’s disease or Periodic Fever, Aphthous Stomatitis, Pharyngitis, Adenitis (PFAPA), in which diagnostic criteria are clinical, as these criteria were established before periodic fever gene testing was available. In those clinical scenarios, we suggest testing for a periodic fever syndrome gene panel, including NOD2 whole gene sequencing (44). Very rarely were male patients complicated by Whipple’s disease, especially after a long-term immunosuppression (33).
In conclusion, our study of the largest YAOS cohort has updated the clinical phenotype and genotype in the disease. The study provides more comprehensive data and deeper understanding of the contribution of NOD2 genetic variants to adult-onset inflammatory disease.
Given that the Two-hit like theory is postulated to be a potential mechanism of the gene variants in YAOS, functional study of compound NOD2 variants and combined variants with other related SAID genes might be needed in the future to unveil novel immunobiological mechanisms underlying the disease.
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
The studies involving humans were approved by Stony Brook University institutional review board. The studies were conducted in accordance with the local legislation and institutional requirements. The ethics committee/institutional review board waived the requirement of written informed consent for participation from the participants or the participants’ legal guardians/next of kin because This is a retrospective study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
HN: Data curation, Investigation, Methodology, Resources, Writing – review & editing. SW: Data curation, Formal analysis, Investigation, Methodology, Validation, Writing – review & editing. AS: Data curation, Investigation, Methodology, Resources, Writing – review & editing. FH: Investigation, Resources, Writing – review & editing. JM: Resources, Writing – review & editing. BN-M: Resources, Writing – review & editing, Investigation. PG: Writing – review & editing, Resources. IA: Investigation, Writing – review & editing, Data curation, Methodology. QY: Resources, Writing – review & editing, Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Project administration, Supervision, Validation, Writing – original draft.
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
The authors are thankful to Dr. Baozhong Xin, Molecular Laboratory, DDC, Middlefield, Ohio, USA for performing the genetic testing. We are grateful to Mrs. Lyn Hastings, Communication Specialist, the Department of Medicine, Stony Brook University, USA for making the composite figures.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. The author declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Kastner DL, Aksentijevich I, Goldbach-Mansky R. Autoinflammatory disease reloaded: a clinical perspective. Cell. (2010) 140:784–90. doi: 10.1016/j.cell.2010.03.002
2. Chou WC, Jha S, Linhoff MW, Ting JP. The NLR gene family: from discovery to present day. Nat Rev Immunol. (2023) 23:635–54. doi: 10.1038/s41577-023-00849-x
3. Yao Q. Nucleotide-binding oligomerization domain containing 2: Structure, function, and diseases. Semin Arthritis Rheumatol. (2013) 43:125–30. doi: 10.1016/j.semarthrit.2012.12.005
4. Yao Q, Li E, Shen B. Autoinflammatory disease with focus on NOD2-associated disease in the era of genomic medicine. Autoimmunity Autoimmunity. (2019) 52:48–56. doi: 10.1080/08916934.2019.1613382
5. Yao Q, Shen B. A systematic analysis of treatment and outcomes of NOD2-associated autoinflammatory disease. Am J Med. (2017) 130:365 e13– e18. doi: 10.1016/j.amjmed.2016.09.028
6. Yao Q, Su LC, Tomecki KJ, Zhou L, Jayakar B, Shen B. Dermatitis as a characteristic phenotype of a new autoinflammatory disease associated with NOD2 mutations. J Am Acad Dermatol. (2013) 68:624–31. doi: 10.1016/j.jaad.2012.09.025
7. Navetta-Modrov B, Nomani H, Yun M, Yang J, Salvemini J, Aroniadis O, et al. A novel nucleotide-binding oligomerization domain 2 genetic marker for Yao syndrome. J Am Acad Dermatol. (2023) 89:166–8. doi: 10.1016/j.jaad.2023.02.029
8. Trueb B, Zhuang L, Keller I, von Kockritz L, Kuchen S, Dufour JF, et al. Coincidence of NOD2-associated autoinflammatory disease (Yao syndrome) and HCV infection with fatal consequences: interaction between genes and environment. J Clin Rheumatol. (2021) 27:S592–4. doi: 10.1097/RHU.0000000000000963
9. Zhang J, Huang X, Shen M. Expanding clinical characteristics and genotypic profiling of yao syndrome in chinese patients. Front Immunol. (2024) 15. doi: 10.3389/fimmu.2024.1444542
10. Karamanakos A, Vougiouka O, Sapountzi E, Venetsanopoulou AI, Tektonidou MG, Germenis AE, et al. The expanding clinical spectrum of autoinflammatory diseases with NOD2 variants: a case series and literature review. Front Immunol. (2024) 15:1342668. doi: 10.3389/fimmu.2024.1342668
11. Yao Q, Kontzias A. Expansion of phenotypic and genotypic spectrum in yao syndrome: A case series. J Clin Rheumatol. (2022) 28:e156–60. doi: 10.1097/RHU.0000000000001655
12. Yao Q, Shen M, McDonald C, Lacbawan F, Moran R, Shen B. NOD2-associated autoinflammatory disease: a large cohort study. Rheumatol (Oxford). (2015) 54:1904–12. doi: 10.1093/rheumatology/kev207
13. Yao Q, Shen M, Fernandez J. NOD2-associated autoinflammatory disease and immune deficiency. J Allergy Clin Immunol Pract. (2016) 4:780–2. doi: 10.1016/j.jaip.2016.02.016
14. Lesage S, Zouali H, Cezard JP, Colombel JF, Belaiche J, Almer S, et al. CARD15/NOD2 mutational analysis and genotype-phenotype correlation in 612 patients with inflammatory bowel disease. Am J Hum Genet. (2002) 70:845–57. doi: 10.1086/339432
15. Stafford IS, Ashton JJ, Mossotto E, Cheng G, Beattie RM, Ennis S. Supervised machine learning classifies inflammatory bowel disease patients by subtype using whole exome sequencing data. J Crohns Colitis. (2023) 17:1672–80. doi: 10.1093/ecco-jcc/jjad084
16. Horowitz JE, Warner N, Staples J, Crowley E, Gosalia N, Murchie R, et al. Mutation spectrum of NOD2 reveals recessive inheritance as a main driver of Early Onset Crohn’s Disease. Sci Rep. (2021) 11:5595. doi: 10.1038/s41598-021-84938-8
17. Rose CD, Arostegui JI, Martin TM, Espada G, Scalzi L, Yague J, et al. NOD2-associated pediatric granulomatous arthritis, an expanding phenotype: study of an international registry and a national cohort in Spain. Arthritis Rheumatol. (2009) 60:1797–803. doi: 10.1002/art.24533
18. Rose CD, Martin TM, Wouters CH. Blau syndrome revisited. Curr Opin Rheumatol. (2011) 23:411–8. doi: 10.1097/BOR.0b013e328349c430
19. Punzi L, Furlan A, Podswiadek M, Gava A, Valente M, De Marchi M, et al. Clinical and genetic aspects of Blau syndrome: a 25-year follow-up of one family and a literature review. Autoimmun Rev. (2009) 8:228–32. doi: 10.1016/j.autrev.2008.07.034
20. Matsuda T, Kambe N, Ueki Y, Kanazawa N, Izawa K, Honda Y, et al. Clinical characteristics and treatment of 50 cases of Blau syndrome in Japan confirmed by genetic analysis of the NOD2 mutation. Ann Rheum Dis. (2020) 79:1492–9. doi: 10.1136/annrheumdis-2020-217320
21. Wu D, Shen M. Two Chinese pedigrees of Blau syndrome with thirteen affected members. Clin Rheumatol. (2018) 37:265–70. doi: 10.1007/s10067-017-3758-7
22. Vogt G, Huber M, Thiemann M, van den Boogaart G, Schmitz OJ, Schubart CD. Production of different phenotypes from the same genotype in the same environment by developmental variation. J Exp Biol. (2008) 211:510–23. doi: 10.1242/jeb.008755
23. Kammenga JE. The background puzzle: how identical mutations in the same gene lead to different disease symptoms. FEBS J. (2017) 284:3362–73. doi: 10.1111/febs.2017.284.issue-20
24. Pham Nguyen TP, Bravo L, Gonzalez-Alegre P, Willis AW. Geographic barriers drive disparities in specialty center access for older adults with huntington's disease. J Huntingtons Dis. (2022) 11:81–9. doi: 10.3233/JHD-210489
25. Keogh RH, Szczesniak R, Taylor-Robinson D, Bilton D. Up-to-date and projected estimates of survival for people with cystic fibrosis using baseline characteristics: A longitudinal study using UK patient registry data. J Cyst Fibros. (2018) 17:218–27. doi: 10.1016/j.jcf.2017.11.019
26. Yao Q, Gorevic P, Shen B, Gibson G. Genetically transitional disease: a new concept in genomic medicine. Trends Genet. (2023) 39:98–108. doi: 10.1016/j.tig.2022.11.002
27. Niewold TB, Aksentijevich I, Gorevic PD, Gibson G, Yao Q. Genetically transitional disease: conceptual understanding and applicability to rheumatic disease. Nat Rev Rheumatol. (2024) 20:301–10. doi: 10.1038/s41584-024-01086-9
28. Monk D, Mackay DJG, Eggermann T, Maher ER, Riccio A. Genomic imprinting disorders: lessons on how genome, epigenome and environment interact. Nat Rev Genet. (2019) 20:235–48. doi: 10.1038/s41576-018-0092-0
29. Mullis MN, Matsui T, Schell R, Foree R, Ehrenreich IM. The complex underpinnings of genetic background effects. Nat Commun. (2018) 9:3548. doi: 10.1038/s41467-018-06023-5
30. Nomani H, Deng Z, Navetta-Modrov B, Yang J, Yun M, Aroniadis O, et al. Implications of combined NOD2 and other gene mutations in autoinflammatory diseases. Front Immunol. (2023) 14:1265404. doi: 10.3389/fimmu.2023.1265404
31. Hino O, Kobayashi T, Mourning Dr, Alfred G. Knudson: the two-hit hypothesis, tumor suppressor genes, and the tuberous sclerosis complex. Cancer Sci. (2017) 108:5–11. doi: 10.1111/cas.2017.108.issue-1
32. Economou M, Trikalinos TA, Loizou KT, Tsianos EV, Ioannidis JP. Differential effects of NOD2 variants on Crohn's disease risk and phenotype in diverse populations: a metaanalysis. Am J Gastroenterol. (2004) 99:2393–404. doi: 10.1111/j.1572-0241.2004.40304.x
33. Williamson KA, Yun M, Koster MJ, Arment C, Patnaik A, Chang TW, et al. Susceptibility of nucleotide-binding oligomerization domain 2 mutations to Whipple's disease. Rheumatol (Oxford). (2024) 63:1291–6. doi: 10.1093/rheumatology/kead372
34. Chamaillard M, Philpott D, Girardin SE, Zouali H, Lesage S, Chareyre F, et al. Gene-environment interaction modulated by allelic heterogeneity in inflammatory diseases. Proc Natl Acad Sci U S A. (2003) 100:3455–60. doi: 10.1073/pnas.0530276100
35. Mao L, Dhar A, Meng G, Fuss I, Montgomery-Recht K, Yang Z, et al. Blau syndrome NOD2 mutations result in loss of NOD2 cross-regulatory function. Front Immunol. (2022) 13:988862. doi: 10.3389/fimmu.2022.988862
36. Beynon V, Cotofana S, Brand S, Lohse P, Mair A, Wagner S, et al. NOD2/CARD15 genotype influences MDP-induced cytokine release and basal IL-12p40 levels in primary isolated peripheral blood monocytes. Inflammation Bowel Dis. (2008) 14:1033–40. doi: 10.1002/ibd.20441
37. Sugimura K, Taylor KD, Lin YC, Hang T, Wang D, Tang YM, et al. A novel NOD2/CARD15 haplotype conferring risk for Crohn disease in Ashkenazi Jews. Am J Hum Genet. (2003) 72:509–18. doi: 10.1086/367848
38. Ellingford JM, Ahn JW, Bagnall RD, Baralle D, Barton S, Campbell C, et al. Recommendations for clinical interpretation of variants found in non-coding regions of the genome. Genome Med. (2022) 14:73. doi: 10.1186/s13073-022-01073-3
39. Vaz-Drago R, Custodio N, Carmo-Fonseca M. Deep intronic mutations and human disease. Hum Genet. (2017) 136:1093–111. doi: 10.1007/s00439-017-1809-4
40. McDonald C, Shen M, Johnson EE, Kabi A, Yao Q. Alterations in nucleotide-binding oligomerization domain-2 expression, pathway activation, and cytokine production in Yao syndrome. Autoimmunity. (2018) 51:53–61. doi: 10.1080/08916934.2018.1442442
41. Zhang J, Luo Y, Wu B, Huang X, Zhao M, Wu N, et al. Identifying functional dysregulation of NOD2 variant Q902K in patients with Yao syndrome. Arthritis Res Ther. (2024) 26:58. doi: 10.1186/s13075-024-03286-w
42. Rivera EG, Patnaik A, Salvemini J, Jain S, Lee K, Lozeau D, et al. SARS-CoV-2/COVID-19 and its relationship with NOD2 and ubiquitination. Clin Immunol. (2022) 238:109027. doi: 10.1016/j.clim.2022.109027
43. Matsuda T, Kambe N, Takimoto-Ito R, Ueki Y, Nakamizo S, Saito MK, et al. Potential benefits of TNF targeting therapy in blau syndrome, a NOD2-associated systemic autoinflammatory granulomatosis. Front Immunol. (2022) 13:895765. doi: 10.3389/fimmu.2022.895765
Keywords: autoinflammatory disease, Blau syndrome, phenotype, genetically transitional disease, genotype, inflammatory bowel disease, NOD2, Yao syndrome
Citation: Nomani H, Wu S, Saif A, Hwang F, Metzger J, Navetta-Modrov B, Gorevic PD, Aksentijevich I and Yao Q (2024) Comprehensive clinical phenotype, genotype and therapy in Yao syndrome. Front. Immunol. 15:1458118. doi: 10.3389/fimmu.2024.1458118
Received: 01 July 2024; Accepted: 06 September 2024;
Published: 20 September 2024.
Edited by:
Emanuele Bizzi, ASST Fatebenefratelli Sacco, ItalyReviewed by:
Katerina Laskari, National and Kapodistrian University of Athens, GreeceCopyright © 2024 Nomani, Wu, Saif, Hwang, Metzger, Navetta-Modrov, Gorevic, Aksentijevich and Yao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qingping Yao, cWluZ3BpbmcueWFvQHN0b255YnJvb2ttZWRpY2luZS5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.