 May Y. Choi
May Y. Choi Karen H. Costenbader
Karen H. Costenbader Marvin J. Fritzler
Marvin J. Fritzler- 1Department of Medicine, Cumming School of Medicine, University of Calgary, Calgary, AB, Canada
- 2McCaig Institute for Bone and Joint Health, Calgary, AB, Canada
- 3Department of Medicine, Div of Rheumatology, Inflammation and Immunity, Brigham and Women’s Hospital, Boston, MA, United States
- 4Medicine, Harvard Medical School, Boston, MA, United States
Introduction: Despite progress in our understanding of disease pathogenesis for systemic autoimmune rheumatic diseases (SARD), these diseases are still associated with high morbidity, disability, and mortality. Much of the strongest evidence to date implicating environmental factors in the development of autoimmunity has been based on well-established, large, longitudinal prospective cohort studies.
Methods: Herein, we review the current state of knowledge on known environmental factors associated with the development of SARD and potential areas for future research.
Results: The risk attributable to any particular environmental factor ranges from 10-200%, but exposures are likely synergistic in altering the immune system in a complex interplay of epigenetics, hormonal factors, and the microbiome leading to systemic inflammation and eventual organ damage. To reduce or forestall the progression of autoimmunity, a better understanding of disease pathogenesis is still needed.
Conclusion: Owing to the complexity and multifactorial nature of autoimmune disease, machine learning, a type of artificial intelligence, is increasingly utilized as an approach to analyzing large datasets. Future studies that identify patients who are at high risk of developing autoimmune diseases for prevention trials are needed.
Introduction
Environmental factors operating on the background of hormonal factors and genetic vulnerability may be accelerating factors included in a long-held paradigm that helps explain the etiology of systemic autoimmune rheumatic disease (SARD), including systemic lupus erythematosus (SLE), rheumatoid arthritis (RA), systemic sclerosis (SSc), Sjögren’s disease (SjD), idiopathic inflammatory myopathies (IIM) and others (1). On the backdrop of an increasing prevalence of SARD and other autoimmune diseases (2–6), potential accelerating factors include several environmental and socioeconomic factors that include alterations of foods, increasing exposure to xenobiotics due to water and air pollution, heat and other extreme weather events (i.e., climate change), biodiversity loss, ultraviolet (UV) light exposure, pandemics and infections, and socioeconomic factors such as changes in personal lifestyles and psychological stress.
Extensive research over the past three to four decades has elucidated the environmental factors associated with SLE (7) and other SARD. In general, the environmental factors can be classified as airborne, waterborne, workplace/occupational, social, and behavioral (8). While it has not been possible to identify a universal environmental “pathogen” for all SARD, there is compelling evidence that some environmental exposures clearly serve as risk factors for disease onset. The central importance of identifying these factors is that many of these factors are actionable and modifiable through intervention and remediation. Expanding the use of machine learning (ML), a form of artificial intelligence (AI), to analyze large datasets including environmental exposures may lead to the identification of other modifiable environmental risk factors, and allow the development of new disease-specific remediation programs (2).
Environmental factors and autoimmunity
The development of SARD has been associated with several lifestyle behaviors. For instance, cigarette smoke (9–11), obesity (12), alcohol use (moderate consumption being protective) (10, 13–15), poor nutrition and intake of ultra-processed foods (16), psychosocial factors (e.g., major depression (17), sleep deprivation (18), child abuse, personal trauma, post-traumatic stress disorder [PTSD]) (19, 20), and reproductive factors (21–23) have been associated with SLE development. Environmental exposures such as air pollution (24), occupational hazards (25), residential proximity to hazardous waste sites or pesticide exposure (26, 27), UV light (28–33), vitamin D deficiency (34), and exposure to viruses (35, 36) have also been linked to increased SLE risk. Similar lifestyle factors have been reported for increased risk of developing RA (moderate alcohol consumption decreases RA risk), SSc, IIM, other SARD, and autoinflammatory conditions (Tables 1, 2).
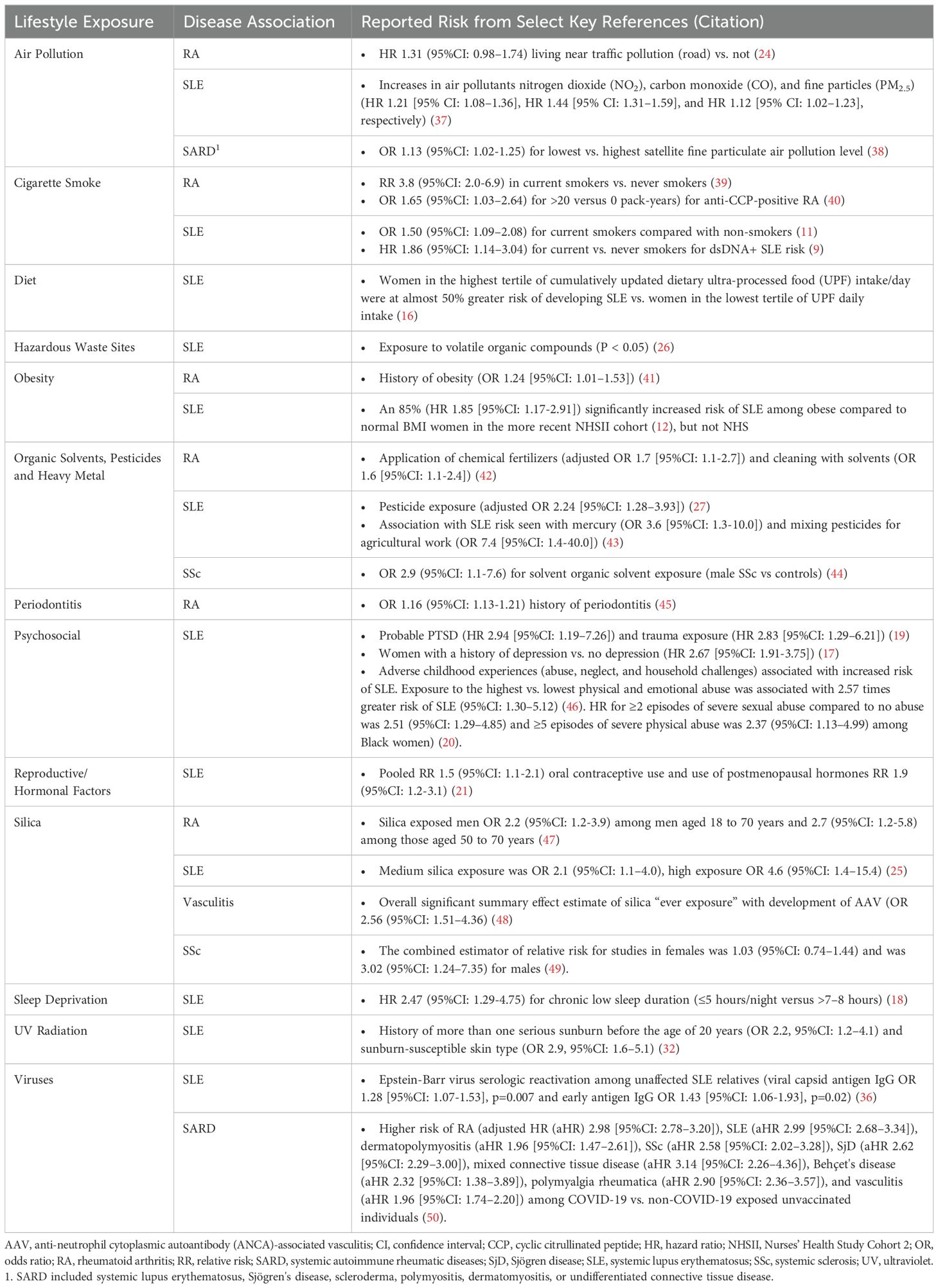
Table 1. Environmental factors that increase risk for systemic autoimmune rheumatic diseases.
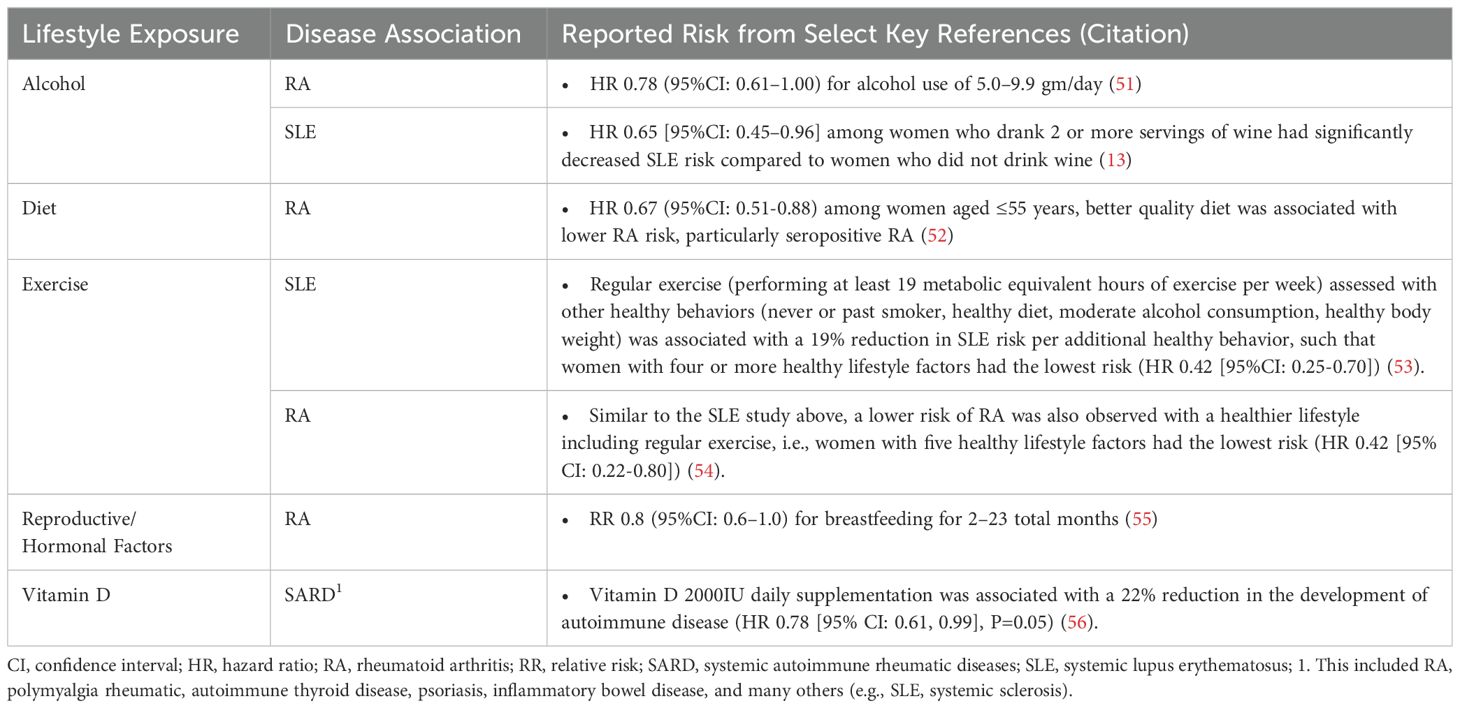
Table 2. Environmental factors that decrease risk for systemic autoimmune rheumatic diseases.
Precisely how and the extent to which these lifestyle factors contribute to individual risk of autoimmune disease likely varies (57, 58). This has been particularly well-studied using large cohort studies including cohorts enrolled in the Nurses’ Health Study (NHS) and Black Women’s Health Study (BWHS). In SLE, each factor independently increases the risk of disease development by 10-200%, but they likely interact with each other and with genetic risk, potentially synergistically, to accelerate brewing autoimmunity in SLE [reviewed in (57–60)]. Using SLE as an example below, we discuss several potential biologic pathways involving epigenomics, the microbiome, and immune dysregulation that lead to inflammation and organ damage, mechanisms that may also apply to the development of other SARD (Figure 1).
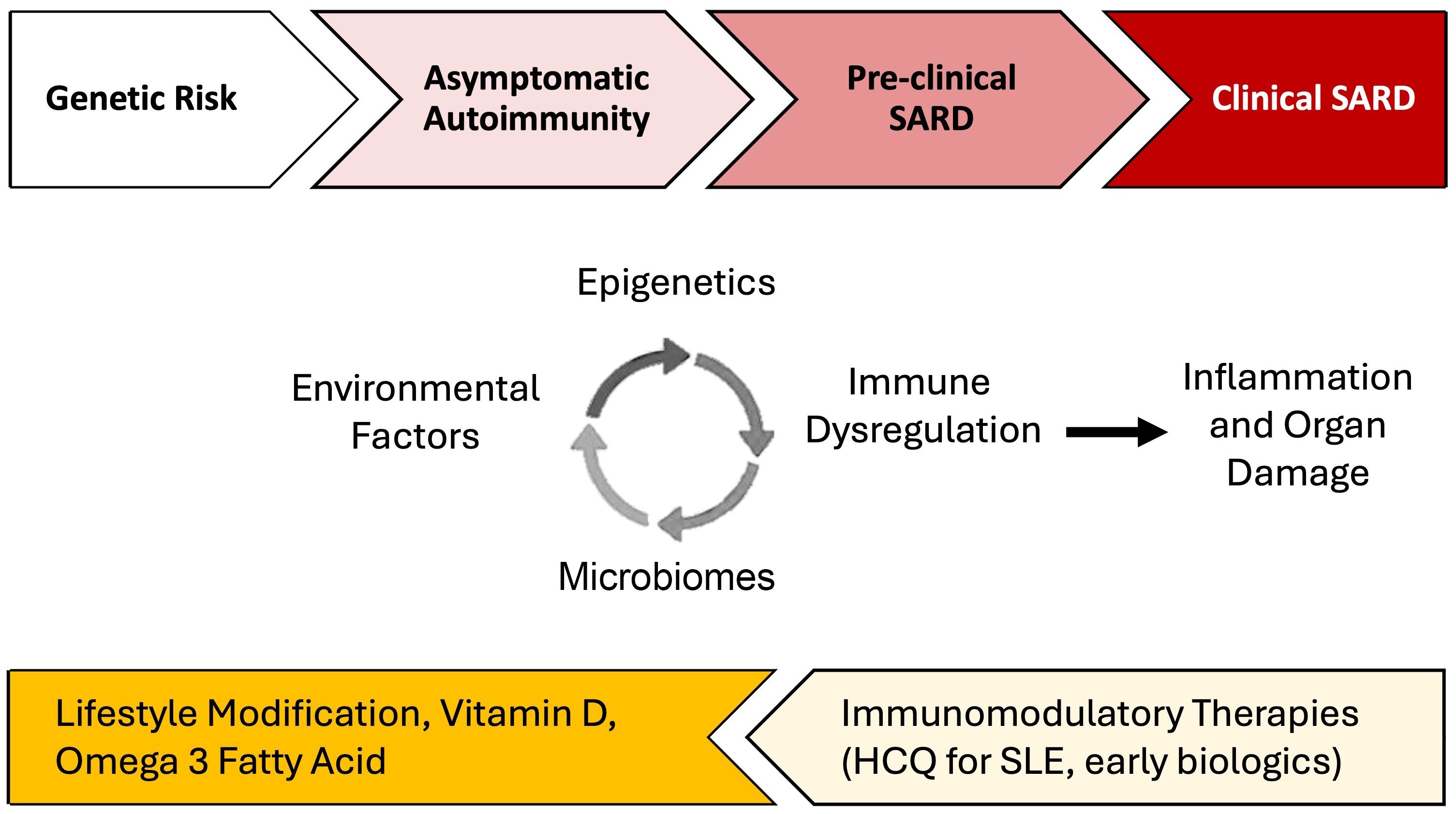
Figure 1. Environmental factor-associated pathogenesis and personalized preventative vs. treatment interventions for systemic autoimmune rheumatic diseases (SARD). Among individuals genetically predisposed to SARD development, unhealthy lifestyle behaviors and other environmental factors can trigger dysregulation in the microbiome, epigenetic changes, and immune dysregulation which, together, drive inflammation. In turn, inflammation can drive further derangements in the microbiome, cause distinct epigenetic changes, and lead to additional immune dysregulation. During the periods of asymptomatic autoimmunity and pre-clinical SARD, this positive feedback leads to a process wherein inflammation becomes chronic and self-sustaining, ultimately driving autoimmunity and eventually leading to organ damage and clinical disease. Effective lifestyle interventions, supplementation, and early introduction of immunomodulatory therapies may help prevent disease progression. There may be a potential role for treatments such as hydroxychloroquine for pre-SLE [SMILE trial underway (61)] and Abatacept, a T-cell co-stimulation inhibitor, for pre-RA (62, 63).
Common pathways of pathogenesis: immune dysregulation, epigenomics, the microbiome
Immune dysregulation
Inflammation is an adaptive response to stressors that involves multiple physiological processes that include the innate and adaptive immune systems. In turn, inflammation regulates – and is regulated by – several highly interconnected systems including the epigenome and microbiome (64). Unhealthy lifestyle behaviors (i.e., smoking, sedentary lifestyle, and consumption of ultra-processed foods) promote systemic inflammation leading to chronic inflammatory diseases, including SARD. Before developing overt clinical manifestations, individuals developing SARD have a period of asymptomatic autoimmunity and inflammation of variable intensity and duration, characterized by increasing oxidative stress, loss of immune tolerance, autoantibody formation, immune complex deposition and complement activation, epigenetic modifications, and upregulation and/or downregulation of cytokine expression [reviewed in (65)].
In SLE, both obesity and exposure to the toxic components of cigarette smoke induce oxidative stress (66). This, in turn, raises intracellular levels of reactive oxygen species that damage DNA producing immunogenic DNA adducts that can lead to the production of ‘pathogenic’ anti-double-stranded DNA antibodies (dsDNA) (67–69). In the NHS and NHSII cohorts, smokers were at higher risk of developing anti-dsDNA positive SLE compared to never-smokers (hazard ratio [HR] 1.86 [95% confidence interval (CI): 1.14-13.04]), while there were no significant associations between smoking status or pack-years and overall SLE or anti-dsDNA negative SLE (9). In addition to elevated oxidative stress, the byproducts of smoking could also augment native autoreactive B cells (11) and induce pulmonary antinuclear antibody (ANA) as demonstrated in the lungs of exposed mice (70). Smoking may also influence specific genes in the pathogenesis of SLE (57). An individual with a high SLE genetic risk score or GRS (score based on 86 single-nucleotide polymorphisms and 10 classic HLA alleles previously associated with SLE) and a status of current/recent smoking was strongly associated with SLE risk (odds ratio [OR] 1.5, p=0.0003 versus more distant past/never smoking) and even stronger in the presence of anti-dsDNA antibodies. Not surprisingly, smoking also affects circulating cytokines. Elevated SARD-related cytokines including the B-cell lymphocyte stimulator (BlyS) (70), tumor necrosis factor-alpha (TNF-α), and interleukin (IL)-6 (71, 72), but lower IL-10 (an anti-inflammatory cytokine) have been detected in smokers (73). These cytokines affect the function of T cells and CD4+ regulatory T cells, which are important in maintaining self-tolerance. Similarly, adipose tissue, in particular visceral fat, secretes pro-inflammatory adipocyte-derived cytokines and exhibits higher levels of C-reactive protein (CRP), TNF-α receptor 2, and IL-6 than non-obese individuals (74).
The association between SLE risk and diet is less clear in humans (75–77) compared to other autoimmune diseases such as RA [reviewed in (78)]. There is evidence from SLE-prone mice models that low dietary fiber intake and a Western-type diet (i.e., high in sugar, fat, refined grains, and red meat) are associated with increased autoantibody production (79, 80). In the BWHS, a diet high in carbohydrates and low in fats was associated with an increased risk of developing SLE in African American women (HR 1.88 [95%CI: 1.06-3.35]) (75). Consumption of ultra-processed foods, in particular sugar and artificially sweetened beverages, has been associated with an increased risk of developing SLE among women (16). Low to moderate alcohol consumption (approximately 1/2 drink a day), on the other hand, has been shown to reduce the risk of SLE development among women (10, 13–15). Alcohol (e.g., ethanol) and antioxidants may counteract the changes induced by smoking and obesity, i.e., inhibiting key enzymes in DNA synthesis and suppressing TNF-α, IL-6, IL-8, and interferon (IFN)-γ that lower systemic inflammation (81, 82).
Several studies have reported an association between lack of sleep and SLE risk in humans (18, 83, 84). In the NHS and NHSII cohorts, chronic low sleep duration (</=5 hours/night versus the recommended >7-8 hours) was associated with increased SLE risk (adjusted HR 2.47 [95% CI: 1.29, 4.75]), with stronger effects among those with body pain and depression. In sleep-deprived individuals, increased levels of IL-6 and TNF-α have been reported (85–89). In SLE-prone mice, sleep deprivation was associated with accelerated production of autoantibodies and earlier disease onset (90). Sleep disturbances arising in individuals who have had childhood or adult trauma, PTSD, or occupational stress from working night or rotating shifts, may also explain why these factors have also been linked to SLE onset (17, 19, 20, 43, 91, 92). In the NHSII, PTSD, a condition arising after exposure to trauma and marked by severe psychological stress, was associated with increased SLE risk (HR 2.94 [95% CI: 1.19-7.26], p<0.05) compared to women with no trauma, even after adjusting for other SLE risk factors smoking, body mass index (BMI), and oral contraceptive use (19). In the NHS and NHSII, women with a history of depression had a higher risk of SLE (HR 2.67 [95:CI: 1.91-3.75] p<0.001) compared to women with no depression (17). Systemic inflammation, denoted by elevated TNF, IL-6, and CRP levels, has been repeatedly reported in individuals with emotional stress and distress (91, 93–102).
There is also evidence that sex hormones are important in SLE development (21, 22), a disease, like some other SARD, that predominantly affects females. In SLE, a population-based nested case-control study using the UK’s General Practice Research Database demonstrated that there was a dose-response in oral contraceptive pill (ethinyl estradiol) and SLE risk (adjusted rate ratio [aRR] 1.42, 1.63, and 2.92 for < or =30 microgram, 31-49 microgram, and 50 microgram, respectively) (22). They also reported that the rate was particularly increased among females who recently started taking oral contraceptive pills (aRR 2.52 [95%CI: 1.14-5.57]) compared with longer-term current users. Estrogen prevents B cell receptor-mediated apoptosis and upregulates several genes that contribute to B cell activation and survival (cd22, shp-1, bcl-2, and vcam-1) (103).
Chemical and physical exposures have also been historically linked to SLE onset, including crystalline silica dust (25, 33, 104, 105), heavy metals such as mercury (43), air pollution and other respiratory particulates (38, 106), residential proximity to hazardous waste sites (26), agricultural pesticides (27, 43, 107), and organic solvents (42, 44). Proposed mechanisms of pathogenesis include stimulation of cellular necrosis and release of intracellular antigens resulting in systemic inflammation and IFN upregulation. These environmental exposures have also been described as important risk factors in the development of RA (42), SSc (44), and vasculitis (48). A comprehensive review of the literature (~1980-2010) on environmental factors and SARD development concluded that among these chemical factors, crystalline silica exposure, solvent exposure, and smoking had the strongest level of evidence (108). Since then, however, multiple studies have been published. The evidence for metal exposure and SARD development including mercury at that time was felt to be insufficient, although there is renewed interest in mercury-induced autoimmunity in more recent studies (109, 110). Mercury exposure has been associated with autoimmune features that are more consistent with pre- or sub-clinical autoimmunity in humans, and in animal studies, acts independently of type I IFN to induce milder disease (111).
UVB radiation can exacerbate pre-existing SLE, however, whether it contributes to SLE disease onset or pathogenesis is less clear. While UVB radiation can up-regulate Th2 cells and down-regulate Th1 cells, induce IL-10 production, increase type I IFN expression, and prolong T cell activation to increase SLE risk (29–31), another subset of UV radiation, UVA, is used as a phototherapy modality to treat cutaneous forms of lupus (112). UVB also has an important role in vitamin D3 synthesis in the skin, which has been hypothesized to lower SLE risk (28, 113). Vitamin D deficiency is reportedly common among SLE patients (34) and is important in the regulatory pathways of numerous genes involved in inflammation and immunity including IL-2 inhibition, antibody production, and lymphocyte proliferation (114, 115). We will later discuss a large, randomized, double-blind, placebo-controlled clinical trial called the vitamin D and omega 3 trial (VITAL) trial, where vitamin D 2000 IU daily supplementation was associated with a 22% reduction in the development of autoimmune disease (HR 0.78 [95% CI: 0.61, 0.99], p=0.05) (56).
Viral triggers, particularly Epstein-Barr Virus (EBV), have also been associated with SLE development (35). In a recent study of 436 unaffected SLE patient relatives who were followed for 6.3 ± 3.9 years and evaluated for interim transitioning to SLE, increased serological reactivation of EBV was associated with higher risk of transitioning to SLE (viral capsid antigen IgG OR 1.28 [95%CI: 1.07-1.53], p=0.007 and expression of EBV early antigen IgG (OR 1.43 [95%CI: 1.06-1.93], p=0.02) (36). Proposed mechanisms include molecular mimicry and the release of EBV-encoded small RNAs from infected cells resulting in the induction of type-1 IFN and proinflammatory cytokines via activating toll-like receptor (TLR)-3 signaling (116). The interest in triggering of autoimmune conditions by viral infections was renewed during the coronavirus disease 2019 (COVID-19) pandemic when there were outbreaks of pediatric inflammatory multisystemic syndrome [PIMS also referred to as multisystem inflammatory syndrome in children (MIS-C)] that reportedly followed severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection in children. These reports included cases of Kawasaki-like disease, Kawasaki disease shock syndrome, toxic shock syndrome, myocarditis and macrophage activation syndrome (117–119). In adults, SARS-CoV-2 infection has also been linked to a higher risk of developing a diverse spectrum of new-onset autoimmune diseases as highlighted by two large retrospective studies (50, 120). Chang et al. used data from the TriNetX network and propensity score matching (two cohorts [COVID-19 and non-COVID-19] of 887,455 SARS-CoV-2 unvaccinated individuals) to identify the incidence of autoimmune conditions during the study period (1 January 2020 to 31 December 2021) (50). Unlike EBV, there was a wider spectrum of SARD seen including higher risk of RA (adjusted hazard ratio (aHR) 2.98 [95%CI: 2.78–3.20]), SLE (aHR 2.99 [95%CI: 2.68–3.34]), dermato/polymyositis (aHR 1.96 [95%CI: 1.47–2.61]), SSc (aHR 2.58 [95%CI: 2.02–3.28]), SjD (aHR 2.62 [95%CI: 2.29–3.00]), and other autoimmune diseases. Future studies that elucidate how viruses, such as SARS-CoV-2, increase the risk of SARD development may help implement preventive measures and early treatment in individuals who have had these infections to prevent morbidity and mortality.
A key pathway involved in both anti-viral response and the pathogenesis of SLE and other SARD including IIM and SSc is the type I IFN pathway (121). Approximately 50-70% of adult and pediatric SLE patients have an upregulated IFN signature, a cluster of IFN-stimulated genes, that correlates with disease activity and severity (122). A recent study demonstrated that type-1 IFN inhibits the aryl hydrocarbon receptor (AHR) pathway. Suppressed AHR signaling promotes T cell production of CXC ligand 13 (CXCL13), a chemokine that regulates B cell recruitment and lymphoid aggregation in inflamed tissues (123). AHR is important for sensing changes in the cellular milieu provided by the environment, diet, commensal flora, and host metabolism (124). In response to these environmental ligands, AHR has a protective role against inflammation by downregulating pro-inflammatory pathways (124). In the gut, AHR is expressed in epithelial cells and immune cells in the lamina propria to also stabilize the gut epithelial barrier (124). In the central nervous system, AHR is upregulated in astrocytes and microglia in response to ligands that cross the blood-brain barrier (124). Lower AHR expression has been described as a potential mechanism of pathogenesis for several autoimmune conditions including inflammatory bowel disease (125), multiple sclerosis (126), and psoriasis (127). In SLE, deficits in the AHR-driven immunoregulation exacerbated by the type-1 IFN may explain how alterations in the environment lead to the development of autoimmunity and uncontrolled inflammation. Moreover, polycyclic aromatic hydrocarbons, smoking, air pollution, and other environmental exposures cause DNA methylation changes in the AHR repressor genes, potentially linking these exposures to the development of autoimmunity (128–130). Future studies are warranted to elucidate the pathways by which regulation of the AHR pathway is related to lymphocyte activation status in the pathogenesis of autoimmunity.
Epigenetic changes
The currently accepted etiologic model for SARD implicates an interaction of inherited genetic factors and environmental exposures over time. DNA methylation (DNAm), an epigenetic change controlling gene expression, is influenced by both genetics and environmental exposures and therefore, may provide a critical link between them [reviewed in (131–133)]. For instance, UV light exposure, infections, silica, heavy metals and pesticide exposures, cigarette smoking, and air pollution are all thought to inhibit DNAm by oxidative stress, which could promote SARD onset specifically or non-specifically (134). In addition to DNAm, cigarette smoking is linked to the activation of enzymes that regulate other types of epigenetic modifications (i.e., post-translational modifications of histones via methylation, acetylation, phosphorylation, ubiquitination, and regulation of non-coding RNA sequences) to mediate the expression of multiple inflammatory genes, thereby participating in the onset development of autoinflammatory diseases (135).
DNAm occurs when a methyl group is added to a cytosine base in a cytosine-phosphate-guanine dinucleotide (CpG) which, in general, silences nearby gene expression. By comparison, demethylation activates gene expression. These changes, mainly demethylation and in particular IFN gene hypomethylation, have been observed in various cell subsets, including CD4 T cells in patients affected by SLE (136–145). Upregulation of type I IFN in SLE is thought to induce an “IFN epigenomic signature”, activating latent enhancers and “bookmarking” chromatin, reprogramming genes to be hyper-responsive, amplifying the inflammatory cascade (146–148). Emerging data reveal that some of these epigenetic changes are correlated with SLE disease manifestations (malar and discoid rash, dsDNA autoantibodies, lupus nephritis) and disease severity (137, 139, 144, 149), and are highly specific to SLE such that they distinguish individuals with existing SLE from controls and other SARD (141, 150). Well-designed epidemiologic studies are still needed to determine whether other epigenetic changes precede the development of SARD and whether such changes could be modified to abrogate disease.
Microbiome influences
There is mounting evidence that imbalances in the microbiota contribute to metabolic and immune regulatory dysfunction, which may contribute to the pathogenesis of chronic inflammatory diseases such as SARD [reviewed in (151)]. Several independent reported studies of 16S rRNA libraries have identified characteristic patterns of gut dysbiosis in SLE, in which there is an inverse relationship between disease activity and overall biodiversity of the intestinal microbiota (152–154). In studies of 61 female SLE patients, there was an eight-fold increase in Ruminococcus gnavus abundance compared to the healthy subjects, and most patients with high R. gnavus abundance had active nephritis (152). Increases in R. gnavus abundance have also been observed in other diseases including allergies and spondyloarthropathies with inflammatory bowel disease (155–157). Importantly, many strains of R. gnavus express a VH3 B cell repertoire (BCR) targeted B cell superantigen, particularly relevant to SLE given the importance of B cell activation in disease pathogenesis (158).
Evidence suggests that SLE patients may suffer chronic microbial translocation through impaired gut barrier integrity contributing to immunologic dysregulation (159). Oral microbiome studies confirm that SLE patients have a distinct microbiome signature compared to healthy controls, with evidence of translocation of bacteria, e.g., Veillonella species, from the oral cavity to the intestine (160, 161).
In healthy adults, the microbiome, even at the level of strains, is relatively stable over many years (162). However, the microbiome can be altered by diet, sleep, exercise, stress, medications (antibiotics and non-antibiotics), and the environment (163). Perturbations in the gut microbiome composition have been suggested to trigger SLE onset or disease flares and vice versa (164). In-depth studies examining the impact of lifestyle and environmental factors on changes to the microbiome and subsequent risk of autoimmune diseases are needed.
Other host barriers should also be considered as potential targets for prevention including the oral cavity and lung mucosa as these have been identified as sites of pathogenic autoreactive immune responses that contribute to autoimmune disease. The initiation of RA by inflammation characterized by an aberrant Th-17-dominated immune response, neutrophil activation, antigen citrullination, and anti-cyclic citrullinated peptide (CCP) production is exacerbated by microbial dysbiosis, the presence of oral pathobionts (e.g., Porphyromonas gingivalis and Aggregatibacter actinomycetemcomitans), and periodontitis has been described (45, 165–167).
The lung mucosa is another site of protein citrullination leading to RA development, promoted by microbial infection or dysbiosis and the inhalation of pollutants such as tobacco smoke or other pollutants (168, 169). This anti-CCP production and translocation into the systemic circulation has been proposed to accelerate the development of RA with interstitial lung disease for individuals who are genetically predisposed (e.g., gain-of-function MUC5B promoter variant reducing mucociliary function in small airways responsible for clearing inhaled particles in the lungs (170)). It is difficult to be certain that microbiome alterations observed in recent studies of SARD patients are not due to established and treated disease. Additional studies of the microbiome before disease onset are warranted.
Mitigation of environmental factors
Traditional cohort studies
Our current understanding of lifestyle factors and autoimmune diseases has largely depended on large observational epidemiological studies (53, 54, 171). Many of these studies used self-reported data including the use of validated and standardized questionnaires. These studies also relied on the retention of subjects in the long term to enable repeated measurement of lifestyle behaviors. Nevertheless, these studies have filled important knowledge gaps in our understanding of the link between environmental exposures and autoimmunity.
In the NHS and NHSII cohorts, our group demonstrated that adherence to multiple healthy behaviors (healthy diet (highest 40th percentile of the Alternative Healthy Eating Index), regular exercise (performing at least 19 metabolic equivalent hours of exercise per week), never or past smoker, moderate alcohol consumption (drinking ≥5 gm/day alcohol), and maintaining a healthy body weight (body mass index <25 kg/m2) was associated with a 19% reduction in SLE risk per additional healthy behavior, such that women with four or more healthy lifestyle factors had the lowest risk (HR 0.42 [95%CI: 0.25-0.70]) (53). An even greater reduction per healthy behavior (22%) was observed for the risk of anti-dsDNA-positive SLE. Overall, the population-attributable risk, or the proportion of the risk in this population that could be attributed to these five modifiable lifestyle risk factors was 47.7% [95%CI: 23.1-66.6%]. Using the same cohorts and similar modeling, a lower risk of RA was also observed with a healthier lifestyle, i.e., women with five healthy lifestyle factors had the lowest risk (HR 0.42 [95%CI: 0.22-0.80]) (54). Therefore, a significant proportion of the risks of both SLE and RA may be preventable by adhering to healthy lifestyles.
Intervention and prevention trials
There is a scarcity of clinical trials examining lifestyle and environmental interventions and prevention strategies to reduce the risk of autoimmune disease development. One of the challenges in designing a strong and well-powered prevention study is identifying which at-risk individuals to study. Our group has previously developed SLE risk prediction models having 76% accuracy by combining family history, genetic factors, and lifestyle, medical and behavioral exposures that classify a woman’s risk of SLE in the next two years (172). There is also a rapidly growing panel of potential biomarkers of SLE risk or early disease including anti-dense fine speckled 70 (DFS70) as a rule-out SARD test (173), anti-C1q antibodies as a rule-in test (174), cytokines and chemokines (175, 176), IFN signature (177), as well as markers of complement activation (178). Therefore, identifying individuals for screening, risk-stratifying, assessing biomarkers, and testing intervention and prevention strategies before clinical disease onset has recently become possible (65, 179).
In a pivotal randomized, double-blind, placebo-controlled vitamin D and omega 3 trial (VITAL) trial with a two-by-two factorial design (n=25 871 participants followed for a median of 5.3 years), vitamin D (2000IU/day) supplementation for five years [with or without omega 3 fatty acid (1000 mg/day)] had a significant reduction in the risk of confirmed autoimmune disease of 22% (HR 0.78 [95% CI: 0.61, 0.99], p=0.05) (56). This included RA, polymyalgia rheumatica, autoimmune thyroid disease, psoriasis, inflammatory bowel disease, and others (e.g., SLE, SSc). Individuals who received an omega-3 fatty acid supplementation (with or without vitamin D supplementation) had a reduced rate of incident autoimmune disease by 15% but this was not statistically significant. However, the two-year post-intervention observation study where participants were no longer provided with any supplements but were invited to continue being observed while off assigned supplements, demonstrated that the protective effects of the 5.3 years of randomized exposure to 2000 IU/day of vitamin D dissipated, but the randomized supplementation with 1,000 mg/day of omega-3 fatty acids for the 5.3 years was seen to have a sustained effect in reducing autoimmune disease incidence (180). The results suggest that vitamin D supplementation of 2000 IU/day should be given continuously for long-term prevention of autoimmune disease, while the beneficial effects of omega-3 fatty acids may be more sustained.
The only SLE-specific prevention trial to date is the “Study of Anti-Malarials in Incomplete Lupus Erythematosus (SMILE)” (61), which was set to determine whether SLE progression can be abrogated by using hydroxychloroquine (HCQ) among patients with a positive ANA test and at least one (but not three or more) additional clinical or laboratory criterion from the 2012 Systemic Lupus Inception Collaborating Clinics (SLICC) classification criteria (181). This highly anticipated, multicenter, randomized, double-blind, placebo-controlled, 24-month trial is expected to be completed soon.
A similar HCQ prevention trial in RA (“Strategy to Prevent the Onset of Clinically-Apparent Rheumatoid Arthritis” or STOP-RA) was halted early due to the futility of the treatment (182). In the interim analysis it was observed that in individuals who were anti-CCP positive but without inflammatory arthritis at baseline, one year of HCQ was not superior to placebo in preventing or delaying the development of inflammatory arthritis, and the classification of individuals as having RA at 3 years (probabilities of RA development were 34% in the HCQ arm and 36% in the placebo; p=0.844). Therefore, in RA, HCQ did not help prevent or delay the onset of clinical disease compared to placebo. The study did suggest however that anti-CCP at levels of ≥40 units will be an important enrolment criterion in future RA prevention studies. Therefore, as we strive towards a future of prevention over cure in any SARD, a better and more standardized approach to identifying the timing of intervention and which patients are at the highest risk is urgently needed to ensure the success of prevention trials.
Other RA prevention trials such as the “TREAT Early Arthralgia to Reverse or Limit Impending Exacerbation to Rheumatoid arthritis” (TREAT EARLIER) trial examining one year of methotrexate also did not meet its endpoint of development of clinical arthritis among individuals with arthralgia clinically suspected of progressing to RA and magnetic resonance imaging (MRI)-detected subclinical joint inflammation (183). The T-cell co-stimulation inhibitor abatacept has shown greater promise in delaying RA development in two different at-risk populations. In the “Abatacept inhibits inflammation and onset of rheumatoid arthritis in individuals at high risk” or ARIAA trial, abatacept treatment for six months among RA-at-risk individuals (anti-CCP positive and showing MRI signs of inflammation) reduced subclinical joint inflammation and delays the development of RA (62). In the “Arthritis Prevention In the Pre-clinical Phase of RA with Abatacept” (APIPPRA) trial, at-risk individuals were defined as individuals with arthralgia, anti-CCP plus rheumatoid factor (RF) positive or high anti-CCP titers ≥3 x upper limit of normal plus RF negative, without synovitis at baseline (63). In this randomized, double-blind, multicenter, parallel, placebo-controlled, phase 2b clinical trial, 52 weeks of abatacept treatment reduced RA development over two years compared to placebo. However, by 24 months, the effect of abatacept treatment on symptom burden and subclinical inflammation as determined by ultrasound was not sustained. Therefore, longer treatment with abatacept beyond 12 months might be required. These studies again highlight the need for criteria that identify at-risk individuals from patients with early RA and the most appropriate time to target preventative interventions (184).
Future technologies for research on environmental exposures and SARD
In the last decade, there has been an exponential uptake of AI technologies to study diseases including SARD [reviewed in (185–187)]. Much of this is due to greater access to a variety of data sources, e.g., images, efficient data collection tools, and supercomputer and analytic methods to rapidly compute. ML is a type of AI that refers to utilizing computers to perform specific tasks by learning from the data rather than being explicitly programmed with instructions such as traditional statistical tests. Within ML, different algorithms are generally categorized into supervised, unsupervised, reinforcement, and deep learning.
In the study of SARD, ML has proven useful in developing prediction models for diagnosis and disease outcomes and in elucidating pathogenesis [reviewed in (185)]. As SARD are highly complex, multifactorial, and heterogeneous diseases, ML is an ideal approach because it can reveal patterns and interactions between variables in large and complex datasets more accurately and efficiently than traditional statistical methods. As we enter an era of ‘multi-omics’, information on our patients is becoming increasingly ‘layered’ and challenging to interpret and ML holds promise for new insights and interpretations.
Utilizing ML, we recently demonstrated that there are four unique SLE clusters defined by longitudinal autoantibody profiles alone (188). While these clusters are predictive of disease activity, treatment requirements, complications, and mortality, it also points to autoantibodies as being a fundamental underlying mechanism of immune dysregulation and disease pathogenesis of SLE. This approach can be adopted to study pathogenesis for other SARD and inform more personalized monitoring and treatment plans. The focus of current SLE ML models is on the identification of patients with established disease or the prediction of specific SLE manifestations, e.g., nephritis, neuropsychiatric disease. This includes a validated diagnostic algorithm called the SLE Risk Probability Index (SLERPI) where a SLERPI score of greater than 7 was highly accurate (94.2%) and sensitive for detecting early disease (93.8%) and severe manifestations including kidney (97.9%) and neuropsychiatric involvement (91.8%) (189). Future studies to develop ML models that predict the development of new-onset SLE utilizing datasets that include environmental exposures are needed.
Conclusions
Our examination of risk and protective factors for SARD development, including adherence to multiple healthy lifestyle behaviors, has helped our understanding of the pathogenesis of autoimmunity that involves immune dysregulation, epigenetics, and an altered microbiome. Multiple environmental exposures, including social and behavioral factors throughout our lifespan are likely synergistic and interactive with each other and with genetic factors, influencing the immune system in a complex interplay of epigenetic, hormonal, and microbiome influences, leading to systemic inflammation and eventual organ damage in some. While a major focus has been placed on identifying new targets for disease treatment, shifting the care paradigm to disease prevention is an attractive proposition, especially as our ability to identify high-risk individuals improves. In the few prevention trials that have been conducted, the importance of identifying patients at the highest risk and the likelihood of benefiting from preventative treatment has been highlighted, and thus far, biomarkers have played a critical role in risk stratification. Given the complexity and vast clinical heterogeneity of SARD, ML approaches will become increasingly relied upon to study SARD pathogenesis and prevention.
Author contributions
MC: Conceptualization, Formal analysis, Investigation, Validation, Visualization, Writing – original draft, Writing – review & editing. KC: Conceptualization, Investigation, Project administration, Resources, Supervision, Validation, Writing – original draft, Writing – review & editing. MF: Conceptualization, Supervision, Validation, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. KC is supported by NIH K24 AR066109 and R01 AR057327. MC is supported by CIHR, McCaig Institute for Bone and Joint Health, Lupus Foundation of America, Lupus Canada, and the Arthritis Society.
Conflict of interest
MC has received consulting fees from AstraZeneca, Mallinckrodt Pharmaceuticals, MitogenDx, Werfen, Celltrion, Organon, and GlaxoSmithKline. MF is the Medical Director of Mitogen Diagnostics Corp and a consultant to Werfen.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
aHR, adjusted hazards ratio; AI, artificial intelligence; ANA, antinuclear antibody; BWHS, Black Women’s Health Study; CI, confidence interval; COVID-19; coronavirus disease 2019; BlyS, B-cell lymphocyte stimulator; CCP, cyclic citrullinated peptide; CRP, C-reactive protein; dsDNA, anti-double-stranded DNA; DNAm, DNA methylation; EBV, Epstein-Barr virus; GRS, genetic risk score; HCQ, hydroxychloroquine; HLA, human lymphocyte antigen; HR, hazard ratio; ML, machine learning; IFN, interferon; IL, interleukin; IIM, idiopathic inflammatory myopathies; IU, international units; NHS, Nurses’ Health Study; OR, odds ratio; NHSII, PTSD, post-traumatic stress disorder; RA, rheumatoid arthritis; rRNA, ribosomal RNA; SARD, systemic autoimmune rheumatic diseases; SARS-CoV2, severe acute respiratory syndrome coronavirus 2; SjD, Sjögren disease; SLE, systemic lupus erythematosus; SSc, systemic sclerosis; TNF, tumor necrosis factor; UV, ultraviolet; VH3 BCR, VH3 B Cell Repertoire.
References
1. Pollard KM. Environment, autoantibodies, and autoimmunity. Front Immunol. (2015) 6:60. doi: 10.3389/fimmu.2015.00060
2. Miller FW. The increasing prevalence of autoimmunity and autoimmune diseases: an urgent call to action for improved understanding, diagnosis, treatment, and prevention. Curr Opin Immunol. (2023) 80:102266. doi: 10.1016/j.coi.2022.102266
3. Heidari H, Lawrence DA. Climate stressors and physiological dysregulations: mechanistic connections to pathologies. Int J Environ Res Public Health. (2023) 21. doi: 10.3390/ijerph21010028
4. Skevaki C, Nadeau KC, Rothenberg ME, Alahmad B, Mmbaga BT, Masenga GG, et al. Impact of climate change on immune responses and barrier defense. J Allergy Clin Immunol. (2024) 153(5):1194–205. doi: 10.1016/j.jaci.2024.01.016
5. Lee AS, Aguilera J, Efobi JA, Jung YS, Seastedt H, Shah MM, et al. Climate change and public health: The effects of global warming on the risk of allergies and autoimmune diseases: The effects of global warming on the risk of allergies and autoimmune diseases. EMBO Rep. (2023) 24:e56821. doi: 10.15252/embr.202356821
6. Pollard KM, Christy JM, Cauvi DM, Kono DH. Environmental xenobiotic exposure and autoimmunity. Curr Opin Toxicol. (2018) 10:15–22. doi: 10.1016/j.cotox.2017.11.009
7. Cardelli C, Zucchi D, Elefante E, Signorini V, Menchini M, Stagnaro C, et al. Environment and systemic lupus erythematosus. Clin Exp Rheumatol. (2024) 42:1104–14. doi: 10.55563/clinexprheumatol/17vmqc
8. Silman AJ, Hochberg MC. Occupational and environmental influences on scleroderma. Rheum Dis Clin North Am. (1996) 22:737–49. doi: 10.1016/S0889-857X(05)70298-2
9. Barbhaiya M, Tedeschi SK, Lu B, Malspeis S, Kreps D, Sparks JA, et al. Cigarette smoking and the risk of systemic lupus erythematosus, overall and by anti-double stranded DNA antibody subtype, in the Nurses' Health Study cohorts. Ann Rheum Dis. (2018) 77:196–202. doi: 10.1136/annrheumdis-2017-211675
10. Cozier YC, Barbhaiya M, Castro-Webb N, Conte C, Tedeschi SK, Leatherwood C, et al. Relationship of cigarette smoking and alcohol consumption to incidence of systemic lupus erythematosus in a prospective cohort study of black women. Arthritis Care Res (Hoboken). (2019) 71:671–7. doi: 10.1002/acr.23703
11. Costenbader KH, Kim DJ, Peerzada J, Lockman S, Nobles-Knight D, Petri M, et al. Cigarette smoking and the risk of systemic lupus erythematosus: a meta-analysis. Arthritis Rheumatism. (2004) 50:849–57. doi: 10.1002/art.20049
12. Tedeschi SK, Barbhaiya M, Malspeis S, Lu B, Sparks JA, Karlson EW, et al. Obesity and the risk of systemic lupus erythematosus among women in the Nurses' Health Studies. Semin Arthritis Rheumatol. (2017) 47:376–83. doi: 10.1016/j.semarthrit.2017.05.011
13. Barbhaiya M, Lu B, Sparks JA, Malspeis S, Chang SC, Karlson EW, et al. Influence of alcohol consumption on the risk of systemic lupus erythematosus among women in the nurses' Health study cohorts. Arthritis Care Res (Hoboken). (2017) 69:384–92. doi: 10.1002/acr.22945
14. Bengtsson AA, Rylander L, Hagmar L, Nived O, Sturfelt G. Risk factors for developing systemic lupus erythematosus: a case-control study in southern Sweden. Rheumatol (Oxford). (2002) 41:563–71. doi: 10.1093/rheumatology/41.5.563
15. Wang J, Pan HF, Ye DQ, Su H, Li XP. Moderate alcohol drinking might be protective for systemic lupus erythematosus: a systematic review and meta-analysis. Clin Rheumatol. (2008) 27:1557–63. doi: 10.1007/s10067-008-1004-z
16. Rossato S, Oakes EG, Barbhaiya M, Sparks JA, Malspeis S, Willett WC, et al. Ultra-processed food intake and risk of systemic lupus erythematosus among women followed in the nurses’ health study cohorts. Arthritis Care Res. (2024), 1–11. doi: 10.1002/acr.25395
17. Roberts AL, Kubzansky LD, Malspeis S, Feldman CH, Costenbader KH. Association of depression with risk of incident systemic lupus erythematosus in women assessed across 2 decades. JAMA Psychiatry. (2018) 75:1225–33. doi: 10.1001/jamapsychiatry.2018.2462
18. Choi MY, Malspeis S, Sparks JA, Cui J, Yoshida K, Costenbader KH. Association of sleep deprivation and the risk of developing systemic lupus erythematosus among women. Arthritis Care Res (Hoboken). (2023) 75:1206–12. doi: 10.1002/acr.25017
19. Roberts AL, Malspeis S, Kubzansky LD, Feldman CH, Chang SC, Koenen KC, et al. Association of trauma and posttraumatic stress disorder with incident systemic lupus erythematosus in a longitudinal cohort of women. Arthritis Rheumatol. (2017) 69:2162–9. doi: 10.1002/art.40222
20. Cozier YC, Barbhaiya M, Castro-Webb N, Conte C, Tedeschi S, Leatherwood C, et al. Association of child abuse and systemic lupus erythematosus in Black women during adulthood. Arthritis Care Res. (2021) 73:833–40. doi: 10.1002/acr.24188
21. Costenbader KH, Feskanich D, Stampfer MJ, Karlson EW. Reproductive and menopausal factors and risk of systemic lupus erythematosus in women. Arthritis Rheumatol. (2007) 56:1251–62. doi: 10.1002/art.22510
22. Bernier MO, Mikaeloff Y, Hudson M, Suissa S. Combined oral contraceptive use and the risk of systemic lupus erythematosus. Arthritis Rheumatol. (2009) 61:476–81. doi: 10.1002/art.24398
23. Lateef A, Petri M. Hormone replacement and contraceptive therapy in autoimmune diseases. J Autoimmun. (2012) 38:J170–6. doi: 10.1016/j.jaut.2011.11.002
24. Hart JE, Laden F, Puett RC, Costenbader KH, Karlson EW. Exposure to traffic pollution and increased risk of rheumatoid arthritis. Environ Health Perspectives. (2009) 117:1065–9. doi: 10.1289/ehp.0800503
25. Parks CG, Cooper GS, Nylander-French LA, Sanderson WT, Dement JM, Cohen PL, et al. Occupational exposure to crystalline silica and risk of systemic lupus erythematosus: a population-based, case-control study in the southeastern United States. Arthritis Rheumatol. (2002) 46:1840–50. doi: 10.1002/art.10368
26. Karlson EW, Watts J, Signorovitch J, Bonetti M, Wright E, Cooper GS, et al. Effect of glutathione S-transferase polymorphisms and proximity to hazardous waste sites on time to systemic lupus erythematosus diagnosis: results from the Roxbury lupus project. Arthritis Rheumatol. (2007) 56:244–54. doi: 10.1002/art.22308
27. Williams JN, Chang SC, Sinnette C, Malspeis S, Parks CG, Karlson EW, et al. Pesticide exposure and risk of systemic lupus erythematosus in an urban population of predominantly African-American women. Lupus. (2018) 27:2129–34. doi: 10.1177/0961203318805844
28. Barbhaiya M, Costenbader KH. Ultraviolet radiation and systemic lupus erythematosus. Lupus. (2014) 23:588–95. doi: 10.1177/0961203314530488
29. Sontheimer C, Liggitt D, Elkon KB. Ultraviolet B irradiation causes stimulator of interferon genes-dependent production of protective type I interferon in mouse skin by recruited inflammatory monocytes. Arthritis Rheumatol. (2017) 69:826–36. doi: 10.1002/art.39987
30. Yin Q, Xu X, Lin Y, Lv J, Zhao L, He R. Ultraviolet B irradiation induces skin accumulation of plasmacytoid dendritic cells: a possible role for chemerin. Autoimmunity. (2014) 47:185–92. doi: 10.3109/08916934.2013.866105
31. Wolf SJ, Estadt SN, Theros J, Moore T, Ellis J, Liu J, et al. Ultraviolet light induces increased T cell activation in lupus-prone mice via type I IFN-dependent inhibition of T regulatory cells. J Autoimmun. (2019) 103:102291. doi: 10.1016/j.jaut.2019.06.002
32. Bengtsson A, Rylander L, Hagmar L, Nived O, Sturfelt G. Risk factors for developing systemic lupus erythematosus: a case–control study in southern Sweden. Rheumatology. (2002) 41:563–71. doi: 10.1093/rheumatology/41.5.563
33. Cooper GS, Wither J, Bernatsky S, Claudio JO, Clarke A, Rioux JD, et al. Occupational and environmental exposures and risk of systemic lupus erythematosus: silica, sunlight, solvents. Rheumatology. (2010) 49:2172–80. doi: 10.1093/rheumatology/keq214
34. Ritterhouse LL, Crowe SR, Niewold TB, Kamen DL, Macwana SR, Roberts VC, et al. Vitamin D deficiency is associated with an increased autoimmune response in healthy individuals and in patients with systemic lupus erythematosus. Ann rheumatic diseases. (2011) 70:1569–74. doi: 10.1136/ard.2010.148494
35. Jog NR, James JA. Epstein barr virus and autoimmune responses in systemic lupus erythematosus. Front Immunol. (2020) 11:623944. doi: 10.3389/fimmu.2020.623944
36. Jog NR, Young KA, Munroe ME, Harmon MT, Guthridge JM, Kelly JA, et al. Association of Epstein-Barr virus serological reactivation with transitioning to systemic lupus erythematosus in at-risk individuals. Ann Rheumatic Diseases. (2019) 78:1235–41. doi: 10.1136/annrheumdis-2019-215361
37. Jung C-R, Chung W-T, Chen W-T, Lee R-Y, Hwang B-F. Long-term exposure to traffic-related air pollution and systemic lupus erythematosus in Taiwan: A cohort study. Sci Total Environment. (2019) 668:342–9. doi: 10.1016/j.scitotenv.2019.03.018
38. Bernatsky S, Smargiassi A, Barnabe C, Svenson LW, Brand A, Martin RV, et al. Fine particulate air pollution and systemic autoimmune rheumatic disease in two Canadian provinces. Environ Res. (2016) 146:85–91. doi: 10.1016/j.envres.2015.12.021
39. Heliovaara M, Aho K, Aromaa A, Knekt P, Reunanen A. Smoking and risk of rheumatoid arthritis. J Rheumatol. (1993) 20:1830–5.
40. Pedersen M, Jacobsen S, Klarlund M, Pedersen BV, Wiik A, Wohlfahrt J, et al. Environmental risk factors differ between rheumatoid arthritis with and without auto-antibodies against cyclic citrullinated peptides. Arthritis Res Ther. (2006) 8:1–15. doi: 10.1186/ar2022
41. Crowson CS, Matteson EL, Davis JM III, Gabriel SE. Contribution of obesity to the rise in incidence of rheumatoid arthritis. Arthritis Care Res. (2013) 65:71–7. doi: 10.1002/acr.21660
42. Parks CG, Hoppin JA, De Roos AJ, Costenbader KH, Alavanja MC, Sandler DP. Rheumatoid arthritis in agricultural health study spouses: associations with pesticides and other farm exposures. Environ Health perspectives. (2016) 124:1728–34. doi: 10.1289/EHP129
43. Cooper GS, Parks CG, Treadwell EL, St Clair EW, Gilkeson GS, Dooley MA. Occupational risk factors for the development of systemic lupus erythematosus. J Rheumatol. (2004) 31:1928–33.
44. Nietert PJ, Sutherland SE, Silver RM, Pandey JP, Knapp RG, Hoel DG, et al. Is occupational organic solvent exposure a risk factor for scleroderma? Arthritis Rheumatism: Off J Am Coll Rheumatol. (1998) 41:1111–8.
45. Chen H-H, Huang N, Chen Y-M, Chen T-J, Chou P, Lee Y-L, et al. Association between a history of periodontitis and the risk of rheumatoid arthritis: a nationwide, population-based, case–control study. Ann Rheumatic Diseases. (2013) 72:1206–11. doi: 10.1136/annrheumdis-2012-201593
46. Feldman CH, Malspeis S, Leatherwood C, Kubzansky L, Costenbader KH, Roberts AL. Association of childhood abuse with incident systemic lupus erythematosus in adulthood in a longitudinal cohort of women. J Rheumatol. (2019) 46:1589–96. doi: 10.3899/jrheum.190009
47. Stolt P, Källberg H, Lundberg I, Sjögren B, Klareskog L, Alfredsson L. Silica exposure is associated with increased risk of developing rheumatoid arthritis: results from the Swedish EIRA study. Ann rheumatic diseases. (2005) 64:582–6. doi: 10.1136/ard.2004.022053
48. Gómez-Puerta JA, Gedmintas L, Costenbader KH. The association between silica exposure and development of ANCA-associated vasculitis: systematic review and meta-analysis. Autoimmun Rev. (2013) 12:1129–35. doi: 10.1016/j.autrev.2013.06.016
49. McCormic ZD, Khuder SS, Aryal BK, Ames AL, Khuder SA. Occupational silica exposure as a risk factor for scleroderma: a meta-analysis. Int Arch Occup Environ Health. (2010) 83:763–9. doi: 10.1007/s00420-009-0505-7
50. Chang R, Chen TY, Wang SI, Hung YM, Chen HY, Wei CC. Risk of autoimmune diseases in patients with COVID-19: A retrospective cohort study. EClinicalMedicine. (2023) 56. doi: 10.1016/j.eclinm.2022.101783
51. Lu B, Solomon DH, Costenbader KH, Karlson EW. Alcohol consumption and risk of incident rheumatoid arthritis in women: a prospective study. Arthritis Rheumatol. (2014) 66:1998–2005. doi: 10.1002/art.38634
52. Hu Y, Sparks JA, Malspeis S, Costenbader KH, Hu FB, Karlson EW, et al. Long-term dietary quality and risk of developing rheumatoid arthritis in women. Ann rheumatic diseases. (2017) 76:1357–64. doi: 10.1136/annrheumdis-2016-210431
53. Choi MY, Hahn J, Malspeis S, Stevens EF, Karlson EW, Sparks JA, et al. Association of a combination of healthy lifestyle behaviors with reduced risk of incident systemic lupus erythematosus. Arthritis Rheumatol. (2022) 74:274–83. doi: 10.1002/art.41935
54. Hahn J, Malspeis S, Choi MY, Stevens E, Karlson EW, Lu B, et al. Association of healthy lifestyle behaviors and the risk of developing rheumatoid arthritis among women. Arthritis Care Res (Hoboken). (2023) 75:272–6. doi: 10.1002/acr.24862
55. Karlson EW, Mandl LA, Hankinson SE, Grodstein F. Do breast-feeding and other reproductive factors influence future risk of rheumatoid arthritis?: Results from the Nurses' Health Study. Arthritis Rheumatism. (2004) 50:3458–67. doi: 10.1002/art.20621
56. Hahn J, Cook NR, Alexander EK, Friedman S, Walter J, Bubes V, et al. Vitamin D and marine omega 3 fatty acid supplementation and incident autoimmune disease: VITAL randomized controlled trial. BMJ. (2022) 376:e066452. doi: 10.1136/bmj-2021-066452
57. Cui J, Raychaudhuri S, Karlson EW, Speyer C, Malspeis S, Guan H, et al. Interactions between genome-wide genetic factors and smoking influencing risk of systemic lupus erythematosus. Arthritis Rheumatol. (2020) 72:1863–71. doi: 10.1002/art.41414
58. Young KA, Munroe ME, Guthridge JM, Kamen DL, Niewold TB, Gilkeson GS, et al. Combined role of vitamin D status and CYP24A1 in the transition to systemic lupus erythematosus. Ann Rheum Dis. (2017) 76:153–8. doi: 10.1136/annrheumdis-2016-209157
59. Barbhaiya M, Costenbader KH. Environmental exposures and the development of systemic lupus erythematosus. Curr Opin Rheumatol. (2016) 28:497–505. doi: 10.1097/BOR.0000000000000318
60. Woo JM, Parks CG, Jacobsen S, Costenbader KH, Bernatsky S. The role of environmental exposures and gene-environment interactions in the etiology of systemic lupus erythematous. J Intern Med. (2022). doi: 10.1111/joim.13448
61. Olsen NJ, James JA, Arriens C, Ishimori ML, Wallace DJ, Kamen DL, et al. Study of Anti-Malarials in Incomplete Lupus Erythematosus (SMILE): study protocol for a randomized controlled trial. Trials. (2018) 19:694. doi: 10.1186/s13063-018-3076-7
62. Rech J, Tascilar K, Hagen M, Kleyer A, Manger B, Schoenau V, et al. Abatacept inhibits inflammation and onset of rheumatoid arthritis in individuals at high risk (ARIAA): a randomised, international, multicentre, double-blind, placebo-controlled trial. Lancet. (2024) 403(10429):850–9. doi: 10.1016/S0140-6736(23)02650-8
63. Cope AP, Jasenecova M, Vasconcelos JC, Filer A, Raza K, Qureshi S, et al. Abatacept in individuals at high risk of rheumatoid arthritis (APIPPRA): a randomised, double-blind, multicentre, parallel, placebo-controlled, phase 2b clinical trial. Lancet. (2024) 403(10429):838–49. doi: 10.1016/S0140-6736(23)02649-1
64. Vodovotz Y, Barnard N, Hu FB, Jakicic J, Lianov L, Loveland D, et al. Prioritized research for the prevention, treatment, and reversal of chronic disease: recommendations from the lifestyle medicine research summit. Front Med (Lausanne). (2020) 7:585744. doi: 10.3389/fmed.2020.585744
65. Choi MY, Costenbader KH. Understanding the concept of pre-clinical autoimmunity: prediction and prevention of systemic lupus erythematosus: identifying risk factors and developing strategies against disease development. Front Immunol. (2022) 13:890522. doi: 10.3389/fimmu.2022.890522
66. Manna P, Jain SK. Obesity, oxidative stress, adipose tissue dysfunction, and the associated health risks: causes and therapeutic strategies. Metab Syndr Relat Disord. (2015) 13:423–44. doi: 10.1089/met.2015.0095
67. Włodarczyk M, Nowicka G. Obesity DNA. Damage, and development of obesity-related diseases. Int J Mol Sci. (2019) 20(5):1146. doi: 10.3390/ijms20051146
68. Petruzzelli S, Celi A, Pulerà N, Baliva F, Viegi G, Carrozzi L, et al. Serum antibodies to benzo (a) pyrene diol epoxide-DNA adducts in the general population: effects of air pollution, tobacco smoking, and family history of lung diseases. Cancer Res. (1998) 58:4122–6.
69. Mooney LA, Perera FP, Van Bennekum AM, Blaner WS, Karkoszka J, Covey L, et al. Gender differences in autoantibodies to oxidative DNA base damage in cigarette smokers. Cancer Epidemiol Prev Biomarkers. (2001) 10:641–8.
70. Morissette MC, Gao Y, Shen P, Thayaparan D, Bérubé JC, Paré PD, et al. Role of BAFF in pulmonary autoantibody responses induced by chronic cigarette smoke exposure in mice. Physiol Rep. (2016) 4(24):e1. doi: 10.14814/phy2.13057
71. Bermudez EA, Rifai N, Buring JE, Manson JE, Ridker PM. Relation between markers of systemic vascular inflammation and smoking in women. Am J Cardiol. (2002) 89:1117–9. doi: 10.1016/S0002-9149(02)02284-1
72. Tracy RP, Psaty BM, Macy E, Bovill EG, Cushman M, Cornell ES, et al. Lifetime smoking exposure affects the association of C-reactive protein with cardiovascular disease risk factors and subclinical disease in healthy elderly subjects. Arteriosclerosis thrombosis Vasc Biol. (1997) 17:2167–76. doi: 10.1161/01.ATV.17.10.2167
73. Hahn J, Leatherwood C, Malspeis S, Liu X, Lu B, Roberts AL, et al. Associations between smoking and systemic lupus erythematosus-related cytokines and chemokines among US female nurses. Arthritis Care Res. (2021) 73:1583–9. doi: 10.1002/acr.24370
74. Panagiotakos DB, Pitsavos C, Yannakoulia M, Chrysohoou C, Stefanadis C. The implication of obesity and central fat on markers of chronic inflammation: The ATTICA study. Atherosclerosis. (2005) 183:308–15. doi: 10.1016/j.atherosclerosis.2005.03.010
75. Castro-Webb N, Cozier YC, Barbhaiya M, Ruiz-Narvaez EA, Li S, Costenbader KH, et al. Association of macronutrients and dietary patterns with risk of systemic lupus erythematosus in the Black Women's Health Study. Am J Clin Nutr. (2021) 114:1486–94. doi: 10.1093/ajcn/nqab224
76. Tedeschi SK, Barbhaiya M, Sparks JA, Karlson EW, Kubzansky LD, Roberts AL, et al. Dietary patterns and risk of systemic lupus erythematosus in women. Lupus. (2020) 29:67–73. doi: 10.1177/0961203319888791
77. Barbhaiya M, Tedeschi S, Sparks JA, Leatherwood C, Karlson EW, Willett WC, et al. Association of dietary quality with risk of incident systemic lupus erythematosus in the nurses' Health studies. Arthritis Care Res. (2020) 73(9):1250–8. doi: 10.1002/acr.24443
78. Tedeschi SK, Costenbader KH. Is there a role for diet in the therapy of rheumatoid arthritis? Curr Rheumatol Rep. (2016) 18(5):23. doi: 10.1007/s11926-016-0575-y
79. Schafer AL, Eichhorst A, Hentze C, Kraemer AN, Amend A, Sprenger DTL, et al. Low dietary fiber intake links development of obesity and lupus pathogenesis. Front Immunol. (2021) 12:696810. doi: 10.3389/fimmu.2021.696810
80. Pan Y, Ke H, Yan Z, Geng Y, Asner N, Palani S, et al. The western-type diet induces anti-HMGB1 autoimmunity in Apoe(-/-) mice. Atherosclerosis. (2016) 251:31–8. doi: 10.1016/j.atherosclerosis.2016.05.027
81. Waldschmidt TJ, Cook RT, Kovacs EJ. Alcohol and inflammation and immune responses: summary of the 2006 Alcohol and Immunology Research Interest Group (AIRIG) meeting. Alcohol. (2008) 42:137–42. doi: 10.1016/j.alcohol.2007.11.003
82. Wirleitner B, Schroecksnadel K, Winkler C, Schennach H, Fuchs D. Resveratrol suppresses interferon-gamma-induced biochemical pathways in human peripheral blood mononuclear cells in vitro. Immunol Lett. (2005) 100:159–63. doi: 10.1016/j.imlet.2005.03.008
83. Young KA, Munroe ME, Harley JB, Guthridge JM, Kamen DL, Gilkensen GS, et al. Less than 7 hours of sleep per night is associated with transitioning to systemic lupus erythematosus. Lupus. (2018) 27:1524–31. doi: 10.1177/0961203318778368
84. Hsiao YH, Chen YT, Tseng CM, Wu LA, Lin WC, Su VY, et al. Sleep disorders and increased risk of autoimmune diseases in individuals without sleep apnea. Sleep. (2015) 38:581–6. doi: 10.5665/sleep.4574
85. Irwin MR, Wang M, Campomayor CO, Collado-Hidalgo A, Cole S. Sleep deprivation and activation of morning levels of cellular and genomic markers of inflammation. Arch Intern Med. (2006) 166:1756–62. doi: 10.1001/archinte.166.16.1756
86. Vgontzas AN, Zoumakis E, Bixler EO, Lin HM, Follett H, Kales A, et al. Adverse effects of modest sleep restriction on sleepiness, performance, and inflammatory cytokines. J Clin Endocrinol Metab. (2004) 89:2119–26. doi: 10.1210/jc.2003-031562
87. Bollinger T, Bollinger A, Skrum L, Dimitrov S, Lange T, Solbach W. Sleep-dependent activity of T cells and regulatory T cells. Clin Exp Immunol. (2009) 155:231–8. doi: 10.1111/j.1365-2249.2008.03822.x
88. Clinton JM, Davis CJ, Zielinski MR, Jewett KA, Krueger JM. Biochemical regulation of sleep and sleep biomarkers. J Clin Sleep Med. (2011) 7:S38–42. doi: 10.5664/JCSM.1360
89. Wilder-Smith A, Mustafa FB, Earnest A, Gen L, Macary PA. Impact of partial sleep deprivation on immune markers. Sleep Med. (2013) 14:1031–4. doi: 10.1016/j.sleep.2013.07.001
90. Palma BD, Gabriel A Jr., Colugnati FA, Tufik S. Effects of sleep deprivation on the development of autoimmune disease in an experimental model of systemic lupus erythematosus. Am J Physiol Regul Integr Comp Physiol. (2006) 291:R1527–32. doi: 10.1152/ajpregu.00186.2006
91. Calcagni E, Elenkov I. Stress system activity, innate and T helper cytokines, and susceptibility to immune-related diseases. Ann N Y Acad Sci. (2006) 1069:62–76. doi: 10.1196/annals.1351.006
92. Case SM, Feldman CH, Guan H, Stevens E, Kubzansky LD, Koenen KC, et al. Posttraumatic stress disorder (PTSD) and risk of systemic lupus erythematosus (SLE) among medicaid recipients. Arthritis Care Res (Hoboken). (2021) 75(1):174–9. doi: 10.1002/acr.24758
93. Sumner JA, Chen Q, Roberts AL, Winning A, Rimm EB, Gilsanz P, et al. Posttraumatic stress disorder onset and inflammatory and endothelial function biomarkers in women. Brain Behav Immun. (2018) 69:203–9. doi: 10.1016/j.bbi.2017.11.013
94. Passos IC, Vasconcelos-Moreno MP, Costa LG, Kunz M, Brietzke E, Quevedo J, et al. Inflammatory markers in post-traumatic stress disorder: a systematic review, meta-analysis, and meta-regression. Lancet Psychiatry. (2015) 2:1002–12. doi: 10.1016/S2215-0366(15)00309-0
95. Gill J, Vythilingam M, Page GG. Low cortisol, high DHEA, and high levels of stimulated TNF-alpha, and IL-6 in women with PTSD. J Trauma Stress. (2008) 21:530–9. doi: 10.1002/jts.20372
96. Pace TW, Wingenfeld K, Schmidt I, Meinlschmidt G, Hellhammer DH, Heim CM. Increased peripheral NF-kappaB pathway activity in women with childhood abuse-related posttraumatic stress disorder. Brain Behav Immun. (2012) 26:13–7. doi: 10.1016/j.bbi.2011.07.232
97. Lindqvist D, Wolkowitz OM, Mellon S, Yehuda R, Flory JD, Henn-Haase C, et al. Proinflammatory milieu in combat-related PTSD is independent of depression and early life stress. Brain Behav Immun. (2014) 42:81–8. doi: 10.1016/j.bbi.2014.06.003
98. Slopen N, Kubzansky LD, McLaughlin KA, Koenen KC. Childhood adversity and inflammatory processes in youth: a prospective study. Psychoneuroendocrinology. (2013) 38:188–200. doi: 10.1016/j.psyneuen.2012.05.013
99. Gola H, Engler H, Sommershof A, Adenauer H, Kolassa S, Schedlowski M, et al. Posttraumatic stress disorder is associated with an enhanced spontaneous production of pro-inflammatory cytokines by peripheral blood mononuclear cells. BMC Psychiatry. (2013) 13:40. doi: 10.1186/1471-244X-13-40
100. Hartwell KJ, Moran-Santa Maria MM, Twal WO, Shaftman S, DeSantis SM, McRae-Clark AL, et al. Association of elevated cytokines with childhood adversity in a sample of healthy adults. J Psychiatr Res. (2013) 47:604–10. doi: 10.1016/j.jpsychires.2013.01.008
101. Danese A, Moffitt TE, Pariante CM, Ambler A, Poulton R, Caspi A. Elevated inflammation levels in depressed adults with a history of childhood maltreatment. Arch Gen Psychiatry. (2008) 65:409–15. doi: 10.1001/archpsyc.65.4.409
102. Stewart JC, Rand KL, Muldoon MF, Kamarck TW. A prospective evaluation of the directionality of the depression-inflammation relationship. Brain Behav Immun. (2009) 23:936–44. doi: 10.1016/j.bbi.2009.04.011
103. Grimaldi CM, Cleary J, Dagtas AS, Moussai D, Diamond B. Estrogen alters thresholds for B cell apoptosis and activation. J Clin Invest. (2002) 109:1625–33. doi: 10.1172/JCI0214873
104. Boudigaard SH, Schlunssen V, Vestergaard JM, Sondergaard K, Toren K, Peters S, et al. Occupational exposure to respirable crystalline silica and risk of autoimmune rheumatic diseases: a nationwide cohort study. Int J Epidemiol. (2021) 50:1213–26. doi: 10.1093/ije/dyaa287
105. Finckh A, Cooper GS, Chibnik LB, Costenbader KH, Watts J, Pankey H, et al. Occupational silica and solvent exposures and risk of systemic lupus erythematosus in urban women. Arthritis Rheumatol. (2006) 54:3648–54. doi: 10.1002/art.22210
106. Bernatsky S, Smargiassi A, Johnson M, Kaplan GG, Barnabe C, Svenson L, et al. Fine particulate air pollution, nitrogen dioxide, and systemic autoimmune rheumatic disease in Calgary, Alberta. Environ Res. (2015) 140:474–8. doi: 10.1016/j.envres.2015.05.007
107. Parks CG, Walitt BT, Pettinger M, Chen JC, de Roos AJ, Hunt J, et al. Insecticide use and risk of rheumatoid arthritis and systemic lupus erythematosus in the Women's Health Initiative Observational Study. Arthritis Care Res (Hoboken). (2011) 63:184–94. doi: 10.1002/acr.20335
108. Miller FW, Alfredsson L, Costenbader KH, Kamen DL, Nelson LM, Norris JM, et al. Epidemiology of environmental exposures and human autoimmune diseases: findings from a National Institute of Environmental Health Sciences Expert Panel Workshop. J Autoimmun. (2012) 39:259–71. doi: 10.1016/j.jaut.2012.05.002
109. Crowe W, Allsopp PJ, Watson GE, Magee PJ, Strain JJ, Armstrong DJ, et al. Mercury as an environmental stimulus in the development of autoimmunity - A systematic review. Autoimmun Rev. (2017) 16:72–80. doi: 10.1016/j.autrev.2016.09.020
110. Pollard KM, Cauvi DM, Toomey CB, Hultman P, Kono DH. Mercury-induced inflammation and autoimmunity. Biochim Biophys Acta Gen Subj. (2019) 1863:129299. doi: 10.1016/j.bbagen.2019.02.001
111. Pollard KM, Escalante GM, Huang H, Haraldsson KM, Hultman P, Christy JM, et al. Induction of systemic autoimmunity by a xenobiotic requires endosomal TLR trafficking and signaling from the late endosome and endolysosome but not type I IFN. J Immunol. (2017) 199:3739–47. doi: 10.4049/jimmunol.1700332
112. McGrath H. Ultraviolet-A1 irradiation decreases clinical disease activity and autoantibodies in patients with systemic lupus erythematosus. Clin Exp Rheumatol. (1994) 12:129–35.
113. Costenbader KH, Feskanich D, Holmes M, Karlson EW, Benito-Garcia E. Vitamin D intake and risks of systemic lupus erythematosus and rheumatoid arthritis in women. Ann Rheum Dis. (2008) 67:530–5. doi: 10.1136/ard.2007.072736
114. Cutolo M, Otsa K, Paolino S, Yprus M, Veldi T, Seriolo B. Vitamin D involvement in rheumatoid arthritis and systemic lupus erythaematosus. Ann Rheum Dis. (2009) 68:446–7. doi: 10.1136/ard.2008.093476
115. Iruretagoyena M, Hirigoyen D, Naves R, Burgos PI. Immune response modulation by vitamin D: role in systemic lupus erythematosus. Front Immunol. (2015) 6:513. doi: 10.3389/fimmu.2015.00513
116. Iwakiri D, Zhou L, Samanta M, Matsumoto M, Ebihara T, Seya T, et al. Epstein-Barr virus (EBV)-encoded small RNA is released from EBV-infected cells and activates signaling from Toll-like receptor 3. J Exp Med. (2009) 206:2091–9. doi: 10.1084/jem.20081761
117. Verdoni L, Mazza A, Gervasoni A, Martelli L, Ruggeri M, Ciuffreda M, et al. An outbreak of severe Kawasaki-like disease at the Italian epicentre of the SARS-CoV-2 epidemic: an observational cohort study. Lancet. (2020) 395:1771–8. doi: 10.1016/S0140-6736(20)31103-X
118. Riphagen S, Gomez X, Gonzalez-Martinez C, Wilkinson N, Theocharis P. Hyperinflammatory shock in children during COVID-19 pandemic. Lancet. (2020) 395:1607–8. doi: 10.1016/S0140-6736(20)31094-1
119. Belhadjer Z, Méot M, Bajolle F, Khraiche D, Legendre A, Abakka S, et al. Acute heart failure in multisystem inflammatory syndrome in children in the context of global SARS-CoV-2 pandemic. Circulation. (2020) 142:429–36. doi: 10.1161/CIRCULATIONAHA.120.048360
120. Tesch F, Ehm F, Vivirito A, Wende D, Batram M, Loser F, et al. Incident autoimmune diseases in association with SARS-CoV-2 infection: a matched cohort study. Clin Rheumatol. (2023) 42:2905–14. doi: 10.1007/s10067-023-06670-0
121. Banchereau J, Pascual V. Type I interferon in systemic lupus erythematosus and other autoimmune diseases. Immunity. (2006) 25:383–92. doi: 10.1016/j.immuni.2006.08.010
122. Wahadat MJ, Bodewes ILA, Maria NI, van Helden-Meeuwsen CG, van Dijk-Hummelman A, Steenwijk EC, et al. Type I IFN signature in childhood-onset systemic lupus erythematosus: a conspiracy of DNA- and RNA-sensing receptors? Arthritis Res Ther. (2018) 20:4. doi: 10.1186/s13075-017-1501-z
123. Law C, Wacleche VS, Cao Y, Pillai A, Sowerby J, Hancock B, et al. Interferon subverts an AHR–JUN axis to promote CXCL13+ T cells in lupus. Nature. (2024) 631, 857–866. doi: 10.1038/s41586-024-07627-2
124. Rothhammer V, Quintana FJ. The aryl hydrocarbon receptor: an environmental sensor integrating immune responses in health and disease. Nat Rev Immunol. (2019) 19:184–97. doi: 10.1038/s41577-019-0125-8
125. Zhao Y, Ma T, Chen W, Chen Y, Li M, Ren L, et al. MicroRNA-124 promotes intestinal inflammation by targeting aryl hydrocarbon receptor in Crohn’s disease. J Crohn's Colitis. (2016) 10:703–12. doi: 10.1093/ecco-jcc/jjw010
126. Quintana FJ, Basso AS, Iglesias AH, Korn T, Farez MF, Bettelli E, et al. Control of Treg and TH17 cell differentiation by the aryl hydrocarbon receptor. Nature. (2008) 453:65–71. doi: 10.1038/nature06880
127. Lebwohl MG, Stein Gold L, Strober B, Papp KA, Armstrong AW, Bagel J, et al. Phase 3 trials of tapinarof cream for plaque psoriasis. New Engl J Med. (2021) 385:2219–29. doi: 10.1056/NEJMoa2103629
128. Andersen AM, Lei MK, Beach SRH, Philibert RA, Sinha S, Colgan JD. Cigarette and cannabis smoking effects on GPR15+ Helper T cell levels in peripheral blood: relationships with epigenetic biomarkers. Genes (Basel). (2020) 11. doi: 10.3390/genes11020149
129. Talbot J, Peres RS, Pinto LG, Oliveira RDR, Lima KA, Donate PB, et al. Smoking-induced aggravation of experimental arthritis is dependent of aryl hydrocarbon receptor activation in Th17 cells. Arthritis Res Ther. (2018) 20:119. doi: 10.1186/s13075-018-1609-9
130. Tantoh DM, Wu MC, Chuang CC, Chen PH, Tyan YS, Nfor ON, et al. AHRR cg05575921 methylation in relation to smoking and PM(2.5) exposure among Taiwanese men and women. Clin Epigenet. (2020) 12:117. doi: 10.1186/s13148-020-00908-3
131. Somers EC, Richardson BC. Environmental exposures, epigenetic changes and the risk of lupus. Lupus. (2014) 23:568–76. doi: 10.1177/0961203313499419
132. Teruel M, Sawalha AH. Epigenetic variability in systemic lupus erythematosus: what we learned from genome-wide DNA methylation studies. Curr Rheumatol Rep. (2017) 19:32. doi: 10.1007/s11926-017-0657-5
133. Richardson B, Scheinbart L, Strahler J, Gross L, Hanash S, Johnson M. Evidence for impaired T cell DNA methylation in systemic lupus erythematosus and rheumatoid arthritis. Arthritis Rheumatol. (1990) 33:1665–73. doi: 10.1002/art.1780331109
134. Gorelik GJ, Yarlagadda S, Patel DR, Richardson BC. Protein kinase Cdelta oxidation contributes to ERK inactivation in lupus T cells. Arthritis Rheumatol. (2012) 64:2964–74. doi: 10.1002/art.34503
135. Zong D, Liu X, Li J, Ouyang R, Chen P. The role of cigarette smoke-induced epigenetic alterations in inflammation. Epigenet Chromatin. (2019) 12:65. doi: 10.1186/s13072-019-0311-8
136. Lanata CM, Chung SA, Criswell LA. DNA methylation 101: what is important to know about DNA methylation and its role in SLE risk and disease heterogeneity. Lupus Sci Med. (2018) 5:e000285. doi: 10.1136/lupus-2018-000285
137. Chung SA, Nititham J, Elboudwarej E, Quach HL, Taylor KE, Barcellos LF, et al. Genome-wide assessment of differential DNA methylation associated with autoantibody production in systemic lupus erythematosus. PloS One. (2015) 10:e0129813. doi: 10.1371/journal.pone.0129813
138. Mok A, Solomon O, Nayak RR, Coit P, Quach HL, Nititham J, et al. Genome-wide profiling identifies associations between lupus nephritis and differential methylation of genes regulating tissue hypoxia and type 1 interferon responses. Lupus Sci Med. (2016) 3:e000183. doi: 10.1136/lupus-2016-000183
139. Coit P, Renauer P, Jeffries MA, Merrill JT, McCune WJ, Maksimowicz-McKinnon K, et al. Renal involvement in lupus is characterized by unique DNA methylation changes in naive CD4+ T cells. J Autoimmun. (2015) 61:29–35. doi: 10.1016/j.jaut.2015.05.003
140. Absher DM, Li X, Waite LL, Gibson A, Roberts K, Edberg J, et al. Genome-wide DNA methylation analysis of systemic lupus erythematosus reveals persistent hypomethylation of interferon genes and compositional changes to CD4+ T-cell populations. PloS Genet. (2013) 9:e1003678. doi: 10.1371/journal.pgen.1003678
141. Zhao M, Zhou Y, Zhu B, Wan M, Jiang T, Tan Q, et al. IFI44L promoter methylation as a blood biomarker for systemic lupus erythematosus. Ann Rheum Dis. (2016) 75:1998–2006. doi: 10.1136/annrheumdis-2015-208410
142. Miller S, Tsou PS, Coit P, Gensterblum-Miller E, Renauer P, Rohraff DM, et al. Hypomethylation of STAT1 and HLA-DRB1 is associated with type-I interferon-dependent HLA-DRB1 expression in lupus CD8+ T cells. Ann Rheum Dis. (2019) 78:519–28. doi: 10.1136/annrheumdis-2018-214323
143. Coit P, Ortiz-Fernandez L, Lewis EE, McCune WJ, Maksimowicz-McKinnon K, Sawalha AH. A longitudinal and transancestral analysis of DNA methylation patterns and disease activity in lupus patients. JCI Insight. (2020) 5. doi: 10.1172/jci.insight.143654
144. Lanata CM, Paranjpe I, Nititham J, Taylor KE, GianFrancesco M, Paranjpe M, et al. A phenotypic and genomics approach in a multi-ethnic cohort to subtype systemic lupus erythematosus. Nat Commun. (2019) 10:3902. doi: 10.1038/s41467-019-11845-y
145. Joseph S, George NI, Green-Knox B, Treadwell EL, Word B, Yim S, et al. Epigenome-wide association study of peripheral blood mononuclear cells in systemic lupus erythematosus: Identifying DNA methylation signatures associated with interferon-related genes based on ethnicity and SLEDAI. J Autoimmun. (2019) 96:147–57. doi: 10.1016/j.jaut.2018.09.007
146. Barrat FJ, Crow MK, Ivashkiv LB. Interferon target-gene expression and epigenomic signatures in health and disease. Nat Immunol. (2019) 20:1574–83. doi: 10.1038/s41590-019-0466-2
147. Tsou PS, Coit P, Kilian NC, Sawalha AH. EZH2 modulates the DNA methylome and controls T cell adhesion through junctional adhesion molecule A in lupus patients. Arthritis Rheumatol. (2018) 70:98–108. doi: 10.1002/art.40338
148. Park SH, Kang K, Giannopoulou E, Qiao Y, Kang K, Kim G, et al. Type I interferons and the cytokine TNF cooperatively reprogram the macrophage epigenome to promote inflammatory activation. Nat Immunol. (2017) 18:1104–16. doi: 10.1038/ni.3818
149. Renauer P, Coit P, Jeffries MA, Merrill JT, McCune WJ, Maksimowicz-McKinnon K, et al. DNA methylation patterns in naive CD4+ T cells identify epigenetic susceptibility loci for malar rash and discoid rash in systemic lupus erythematosus. Lupus Sci Med. (2015) 2:e000101. doi: 10.1136/lupus-2015-000101
150. Breitbach ME, Ramaker RC, Roberts K, Kimberly RP, Absher D. Population-specific patterns of epigenetic defects in the B cell lineage in patients with systemic lupus erythematosus. Arthritis Rheumatol. (2020) 72:282–91. doi: 10.1002/art.41083
151. Silverman GJ, Azzouz DF, Alekseyenko AV. Systemic Lupus Erythematosus and dysbiosis in the microbiome: cause or effect or both? Curr Opin Immunol. (2019) 61:80–5. doi: 10.1016/j.coi.2019.08.007
152. Azzouz D, Omarbekova A, Heguy A, Schwudke D, Gisch N, Rovin BH, et al. Lupus nephritis is linked to disease-activity associated expansions and immunity to a gut commensal. Ann Rheum Dis. (2019) 78:947–56. doi: 10.1136/annrheumdis-2018-214856
153. He Z, Shao T, Li H, Xie Z, Wen C. Alterations of the gut microbiome in Chinese patients with systemic lupus erythematosus. Gut Pathog. (2016) 8:64. doi: 10.1186/s13099-016-0146-9
154. Hevia A, Milani C, Lopez P, Cuervo A, Arboleya S, Duranti S, et al. Intestinal dysbiosis associated with systemic lupus erythematosus. mBio. (2014) 5:e01548–14. doi: 10.1128/mBio.01548-14
155. Chua H-H, Chou H-C, Tung Y-L, Chiang B-L, Liao C-C, Liu H-H, et al. Intestinal dysbiosis featuring abundance of Ruminococcus gnavus associates with allergic diseases in infants. Gastroenterology. (2018) 154:154–67. doi: 10.1053/j.gastro.2017.09.006
156. Breban M, Tap J, Leboime A, Said-Nahal R, Langella P, Chiocchia G, et al. Faecal microbiota study reveals specific dysbiosis in spondyloarthritis. Ann rheumatic diseases. (2017) 76:1614–22. doi: 10.1136/annrheumdis-2016-211064
157. Hall AB, Yassour M, Sauk J, Garner A, Jiang X, Arthur T, et al. A novel Ruminococcus gnavus clade enriched in inflammatory bowel disease patients. Genome Med. (2017) 9:1–12. doi: 10.1186/s13073-017-0490-5
158. Bunker JJ, Drees C, Watson AR, Plunkett CH, Nagler CR, Schneewind O, et al. B cell superantigens in the human intestinal microbiota. Sci Transl Med. (2019) 11(507):eaau9356. doi: 10.1126/scitranslmed.aau9356
159. Shi L, Zhang Z, Yu AM, Wang W, Wei Z, Akhter E, et al. The SLE transcriptome exhibits evidence of chronic endotoxin exposure and has widespread dysregulation of non-coding and coding RNAs. PloS One. (2014) 9:e93846. doi: 10.1371/journal.pone.0093846
160. Li BZ, Zhou HY, Guo B, Chen WJ, Tao JH, Cao NW, et al. Dysbiosis of oral microbiota is associated with systemic lupus erythematosus. Arch Oral Biol. (2020) 113:104708. doi: 10.1016/j.archoralbio.2020.104708
161. Correa JD, Calderaro DC, Ferreira GA, Mendonca SM, Fernandes GR, Xiao E, et al. Subgingival microbiota dysbiosis in systemic lupus erythematosus: association with periodontal status. Microbiome. (2017) 5:34. doi: 10.1186/s40168-017-0252-z
162. Faith JJ, Guruge JL, Charbonneau M, Subramanian S, Seedorf H, Goodman AL, et al. The long-term stability of the human gut microbiota. Science. (2013) 341:1237439. doi: 10.1126/science.1237439
163. Maier L, Pruteanu M, Kuhn M, Zeller G, Telzerow A, Anderson EE, et al. Extensive impact of non-antibiotic drugs on human gut bacteria. Nature. (2018) 555:623–8. doi: 10.1038/nature25979
164. Azzouz DF, Chen Z, Izmirly PM, Chen LA, Li Z, Zhang C, et al. Longitudinal gut microbiome analyses and blooms of pathogenic strains during lupus disease flares. Ann rheumatic diseases. (2023) 82:1315–27. doi: 10.1136/ard-2023-223929
165. Mikuls TR, Payne JB, Yu F, Thiele GM, Reynolds RJ, Cannon GW, et al. Periodontitis and Porphyromonas gingivalis in patients with rheumatoid arthritis. Arthritis Rheumatol. (2014) 66:1090–100. doi: 10.1002/art.38348
166. Mikuls TR, Thiele GM, Deane KD, Payne JB, O'Dell JR, Yu F, et al. Porphyromonas gingivalis and disease-related autoantibodies in individuals at increased risk of rheumatoid arthritis. Arthritis Rheumatism. (2012) 64:3522–30. doi: 10.1002/art.34595
167. Konig MF, Abusleme L, Reinholdt J, Palmer RJ, Teles RP, Sampson K, et al. Aggregatibacter actinomycetemcomitans–induced hypercitrullination links periodontal infection to autoimmunity in rheumatoid arthritis. Sci Trans Med. (2016) 8:369ra176–369ra176. doi: 10.1126/scitranslmed.aaj1921
168. Scher JU, Joshua V, Artacho A, Abdollahi-Roodsaz S, Öckinger J, Kullberg S, et al. The lung microbiota in early rheumatoid arthritis and autoimmunity. Microbiome. (2016) 4:1–10. doi: 10.1186/s40168-016-0206-x
169. Reynisdottir G, Karimi R, Joshua V, Olsen H, Hensvold AH, Harju A, et al. Structural changes and antibody enrichment in the lungs are early features of anti–citrullinated protein antibody–positive rheumatoid arthritis. Arthritis Rheumatol. (2014) 66:31–9. doi: 10.1002/art.38201
170. Juge P-A, Lee JS, Ebstein E, Furukawa H, Dobrinskikh E, Gazal S, et al. MUC5B promoter variant and rheumatoid arthritis with interstitial lung disease. New Engl J Med. (2018) 379:2209–19. doi: 10.1056/NEJMoa1801562
171. Rodriguez Huerta MD, Trujillo-Martin MM, Rua-Figueroa I, Cuellar-Pompa L, Quiros-Lopez R, Serrano-Aguilar P, et al. Healthy lifestyle habits for patients with systemic lupus erythematosus: A systemic review. Semin Arthritis Rheumatol. (2016) 45:463–70. doi: 10.1016/j.semarthrit.2015.09.003
172. Cui J, Malspeis S, Choi MY, Lu B, Sparks JA, Yoshida K, et al. Risk prediction models for incident systemic lupus erythematosus among women in the Nurses' health study cohorts using genetics, family history, and lifestyle and environmental factors. Semin Arthritis Rheumatol. (2023) 58:152143. doi: 10.1016/j.semarthrit.2022.152143
173. Choi MY, Clarke AE, St Pierre Y, Hanly JG, Urowitz MB, Romero-Diaz J, et al. The prevalence and determinants of anti-DFS70 autoantibodies in an international inception cohort of systemic lupus erythematosus patients. Lupus. (2017) 26:1051–9. doi: 10.1177/0961203317692437
174. Olsen NJ, McAloose C, Carter J, Han BK, Raman I, Li QZ, et al. Clinical and immunologic profiles in incomplete lupus erythematosus and improvement with hydroxychloroquine treatment. Autoimmune Dis. (2016) 2016:8791629. doi: 10.1155/2016/8791629
175. Lu R, Munroe ME, Guthridge JM, Bean KM, Fife DA, Chen H, et al. Dysregulation of innate and adaptive serum mediators precedes systemic lupus erythematosus classification and improves prognostic accuracy of autoantibodies. J Autoimmun. (2016) 74:182–93. doi: 10.1016/j.jaut.2016.06.001
176. Munroe ME, Lu R, Zhao YD, Fife DA, Robertson JM, Guthridge JM, et al. Altered type II interferon precedes autoantibody accrual and elevated type I interferon activity prior to systemic lupus erythematosus classification. Ann Rheum Dis. (2016) 75:2014–21. doi: 10.1136/annrheumdis-2015-208140
177. Niewold TB, Hua J, Lehman TJA, Harley JB, Crow MK. High serum IFN-α activity is a heritable risk factor for systemic lupus erythematosus. Genes Immunity. (2007) 8:492–502. doi: 10.1038/sj.gene.6364408
178. Ramsey-Goldman R, Alexander RV, Massarotti EM, Wallace DJ, Narain S, Arriens C, et al. Complement activation in patients with probable systemic lupus erythematosus and ability to predict progression to american college of rheumatology-classified systemic lupus erythematosus. Arthritis Rheumatol. (2020) 72(1):78–88. doi: 10.1002/art.41093
179. Deane KD. Targeting environmental risks to prevent rheumatic disease. Rheum Dis Clin North Am. (2022) 48:931–43. doi: 10.1016/j.rdc.2022.06.011
180. Costenbader KH, Cook NR, Lee IM, Hahn J, Walter J, Bubes V, et al. Vitamin D and marine n-3 fatty acids for autoimmune disease prevention: outcomes two years after completion of a double-blind, placebo-controlled trial. Arthritis Rheumatol. (2024) 76(6):973–83. doi: 10.1002/art.42811
181. Petri M, Orbai AM, Alarcon GS, Gordon C, Merrill JT, Fortin PR, et al. Derivation and validation of the Systemic Lupus International Collaborating Clinics classification criteria for systemic lupus erythematosus. Arthritis Rheumatol. (2012) 64:2677–86. doi: 10.1002/art.34473
182. Deane KD, Striebich C, Feser M, Demoruelle K, Moss L, Bemis E, et al. Hydroxychloroquine does not prevent the future development of rheumatoid arthritis in a population with baseline high levels of antibodies to citrullinated protein antigens and absence of inflammatory arthritis: interim analysis of the StopRA trial. Arthritis Rheumatol. (2022) 74:3180–2.
183. Krijbolder DI, Verstappen M, van Dijk BT, Dakkak YJ, Burgers LE, Boer AC, et al. Intervention with methotrexate in patients with arthralgia at risk of rheumatoid arthritis to reduce the development of persistent arthritis and its disease burden (TREAT EARLIER): a randomised, double-blind, placebo-controlled, proof-of-concept trial. Lancet. (2022) 400:283–94. doi: 10.1016/S0140-6736(22)01193-X
184. Finckh A, Courvoisier D, Lamacchia C. Recherche clinique en rhumatismes i. Measuring ACPA in the general population or primary care: is it useful? RMD Open. (2020) 6(1). doi: 10.1136/rmdopen-2019-001085
185. Zhan K, Buhler KA, Chen IY, Fritzler MJ, Choi MY. Systemic lupus in the era of machine learning medicine. Lupus Sci Med. (2024) 11. doi: 10.1136/lupus-2023-001140
186. Filipp FV. Opportunities for artificial intelligence in advancing precision medicine. Curr Genet Med Rep. (2019) 7:208–13. doi: 10.1007/s40142-019-00177-4
187. Foulquier N, Redou P, Saraux A. How health information technologies and artificial intelligence may help rheumatologists in routine practice. Rheumatol Ther. (2019) 6:135–8. doi: 10.1007/s40744-019-0154-6
188. Choi MY, Chen I, Clarke AE, Fritzler MJ, Buhler KA, Urowitz M, et al. Machine learning identifies clusters of longitudinal autoantibody profiles predictive of systemic lupus erythematosus disease outcomes. Ann rheumatic diseases. (2023) 82:927–36. doi: 10.1136/ard-2022-223808
189. Adamichou C, Genitsaridi I, Nikolopoulos D, Nikoloudaki M, Repa A, Bortoluzzi A, et al. Lupus or not? SLE Risk Probability Index (SLERPI): a simple, clinician-friendly machine learning-based model to assist the diagnosis of systemic lupus erythematosus. Ann Rheum Dis. (2021) 80:758–66. doi: 10.1136/annrheumdis-2020-219069
Keywords: autoimmunity, autoimmune diseases, environment, autoantibodies, epigenetics, microbiome, machine learning, artificial intelligence
Citation: Choi MY, Costenbader KH and Fritzler MJ (2024) Environment and systemic autoimmune rheumatic diseases: an overview and future directions. Front. Immunol. 15:1456145. doi: 10.3389/fimmu.2024.1456145
Received: 28 June 2024; Accepted: 16 August 2024;
Published: 10 September 2024.
Edited by:
Frederick Miller, National Institute of Environmental Health Sciences (NIH), United StatesReviewed by:
Shepherd Schurman, National Institutes of Health (NIH), United StatesKenneth Michael Pollard, The Scripps Research Institute, United States
Adam Schiffenbauer, National Institutes of Health (NIH), United States
Copyright © 2024 Choi, Costenbader and Fritzler. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: May Y. Choi, bWF5LmNob2lAdWNhbGdhcnkuY2E=