 Liting Yue1†
Liting Yue1† Jisong Li
Jisong Li Siyuan Song
Siyuan Song Xiaoqin Zhang
Xiaoqin Zhang Yi Wang
Yi Wang- 1Center of Critical Care Medicine, Sichuan Provincial People’s Hospital, University of Electronic Science and Technology of China, Chengdu, Sichuan, China
- 2Department of Gastrointestinal Surgery, Sichuan Provincial People’s Hospital, University of Electronic Science and Technology of China, Chengdu, China
- 3Department of Neuroscience, Baylor College of Medicine, Houston, TX, United States
- 4Clinical Immunology Translational Medicine Key Laboratory of Sichuan Province, Sichuan Provincial People’s Hospital, Chengdu, China
As an effective treatment for diabetes, islet transplantation has garnered significant attention and research in recent years. However, immune rejection and the toxicity of immunosuppressive drugs remain critical factors influencing the success of islet transplantation. While immunosuppressants are essential in reducing immune rejection reactions and can significantly improve the survival rate of islet transplants, improper use of these drugs can markedly increase mortality rates following transplantation. Additionally, the current availability of islet organ donations fails to meet the demand for organ transplants, making xenotransplantation a crucial method for addressing organ shortages. This review will cover the following three aspects: 1) the immune responses occurring during allogeneic islet transplantation, including three stages: inflammation and IBMIR, allogeneic immune response, and autoimmune recurrence; 2) commonly used immunosuppressants in allogeneic islet transplantation, including calcineurin inhibitors (Cyclosporine A, Tacrolimus), mycophenolate mofetil, glucocorticoids, and Bortezomib; and 3) early and late immune responses in xenogeneic islet transplantation and the immune effects of triple therapy (ECDI-fixed donor spleen cells (ECDI-SP) + anti-CD20 + Sirolimus) on xenotransplantation.
1 Introduction
Diabetes is a chronic metabolic disease characterized by high blood glucose levels, affecting over 500 million people worldwide. Type 1 diabetes (T1D) results from an autoimmune response that destroys the insulin-producing β-cells in the body, resulting in the inability to produce insulin to regulate blood glucose levels (1). Since the discovery of insulin in 1922, insulin therapy has been used to treat patients with T1D. This disease requires minute-to-minute regulation of blood glucose levels, and measures such as exogenous insulin supplementation and continuous glucose monitoring (CGM) can have a certain delay in detecting and controlling blood glucose levels, which insulin injections cannot achieve (2, 3). Only by transplanting insulin-producing cells from donors can we precisely measure and deliver the appropriate doses of insulin (4). Additionally, although intensive insulin therapy can improve glycated hemoglobin levels, it does not prevent diabetic complications (5). When patients face severe metabolic complications, failure of exogenous insulin treatment, or when insulin use fails to prevent acute complications, islet transplantation becomes a necessary treatment measure (6). The transplantation of pancreatic tissue, whether whole pancreas or islets, is a clinical option for the treatment of labile type 1 diabetes. Pancreas transplantation is usually performed as a multi-organ transplant procedure; most of these (72%) are combined pancreatorenal procedures. Therefore, it is particularly suitable for patients with type 1 diabetes combined with end-stage renal disease. Open surgery is required to transplant the entire pancreas into the abdominal cavity of the recipient and connect the blood vessels and digestive tract. The operation is complicated and traumatic, and the recovery time is long. Whole organ pancreas transplants restore euglycemia almost immediately following transplantation, and long-term graft survival rates are excellent. Despite the need for immunosuppression, recipient morbidity and mortality decreased significantly, as did the risk of complications associated with poor glycemic control and a better quality of life (7, 8). Islet transplantation refers to the isolation, purification and transplantation of islets from the pancreas of the donor into the recipient (detailed procedures are described below). Islet transplantation is suitable for type 1 diabetes patients who have experienced severe hypoglycemic events. Following Edmonton protocol, the islets are injected directly into the recipient’s liver portal vein under the ultrasound observation, and the operation is less traumatic, the anesthesia time is shorter, the invasion is less, and the recovery time is fast. Although many patients experience significant improvements in blood sugar control after transplantation, exogenous insulin may still be required, and long-term success rates are relatively low.
As an alternative therapy, islet transplantation can sustainably reverse T1D. Successful islet transplantation eliminates the need for stringent blood glucose monitoring and prevents the progression of diabetic complications. However, a significant challenge faced by islet transplantation is the immune response of the body to the foreign islets. When donor islets are exposed to the recipient’s immune system, the implants can trigger a rapid immune response (9, 10). Therefore, the survival rate of islets after isolation and transplantation becomes a major issue. Immunosuppressive therapy is currently the most popular immunomodulation method to ensure the survival of islet grafts. Clinical islet transplantation began in the 1970s (11), but due to various reasons, its clinical efficacy was not ideal. It was not until 1999 that Shapiro et al. (12) proposed and established a standard set, including donor selection, islet equivalent transplantation, and post-operative immunosuppressive regimens. They used a large number of isolated islet cells for transplantation and implemented a new regimen post-operatively, using a steroid-free regimen and reduced doses of calcineurin inhibitors (sirolimus, low-dose tacrolimus, and daclizumab), known as the “Edmonton Protocol” (12). Once this protocol was promoted, clinical results improved significantly, marking an important milestone in clinical IT. With the promotion of the Edmonton clinical protocol and the continuous improvement of islet cell isolation techniques, the survival rate of islet transplantation has significantly improved but is still relatively low compared to other organs. Moreover, it is known that the traditional methods of using immunosuppressive drugs during and after islet transplantation can cause many side effects, such as mouth ulcers, peripheral edema, anemia, weight loss, and paroxysmal diarrhea (9, 13). Therefore, to improve the survival rate after islet transplantation, many issues must be addressed, including islet viability, effective implantation, and the application of immunosuppressants that lead to islet damage (14). Therefore, the purpose of this article is to summarize the immune responses and mechanisms of action of immunosuppressants that occur after islet transplantation to better guide islet transplantation and improve islet survival rates.
2 Immune response in allogeneic islet Transplantation
2.1 Inflammatory response
Clinical islet transplantation requires four steps: perfusion of the donor pancreas, digestion of the pancreas to separate the islets from the exocrine tissue, purification of the islets, and transplantation via the portal vein infusion of islet into the recipient (15). When the prepared islets are infused into the patient’s body through the portal vein, it triggers an inflammatory response. Early inflammatory response leads to the early loss of islet viability, posing a significant challenge to the long-term survival rate of islet transplantation. This early inflammatory reaction significantly affects islet viability, with estimates indicating that up to 50% of transplanted islets may be lost during this initial phase (16). Post-pancreas transplantation, ischemia-reperfusion creates an inflammatory environment, where the Instant Blood-Mediated Inflammatory Reaction (IBMIR) plays a crucial role. Injecting purified islets into the recipient’s portal vein promotes an innate immune-dependent inflammatory response, known as IBMIR.
IBMIR is initiated by the intense activation of the coagulation cascade, where the negatively charged surface of the islets activates the intrinsic coagulation pathway (17), and the tissue factor (TF) expressed by the islets induces the extrinsic coagulation pathway (18). Simultaneously, islets secrete inflammatory factors such as IL-8 and MCP-1, which have chemotactic and pro-inflammatory effects on macrophages and neutrophils (19, 20). Activated platelets can adhere by binding to the extracellular matrix (ECM) and collagen on the surface of islets. Additionally, due to the rapid transient expression of p-selectin on the membranes of activated platelet alpha granules and vascular endothelial Weibel-Palade bodies, the p-selectin lectin-like domain present on neutrophils and monocytes binds with sialyl Lewis x and p-selectin glycoprotein ligand-1, mediating the rolling of neutrophils and monocytes on endothelial cells and their adhesion to platelets (21, 22). On the other hand, vascular endothelial cells secrete IL-6 and IL-8, promoting the aggregation of neutrophils and macrophages (19). Complement activation is triggered by natural immune antibodies IgG and IgM. When isolated islets are exposed to blood, the complement system is rapidly activated, leading to the lysis of islet cells. Simultaneously, the production of anaphylatoxins C3a and C5a further induces the aggregation of macrophages and neutrophils, promoting the release of cytokines such as IL-1, IL-6, IL-8, and TNF-a by monocytes (23). Granulocytes appear 8 hours after islet transplantation, with extensive infiltration into the grafts after 12 hours. Neutrophils are the main members of the granulocyte family and the first line of defense in innate immunity. They contain various cytokines that, when activated, are released and cause damage to islets; neutrophils significantly contribute to the activation and recruitment of macrophages at acute inflammation sites. Once activated, they produce various chemokines to attract monocytes and macrophages. Additionally, neutrophil infiltration leads to the release of cytokines such as TNF-a and macrophage inflammatory protein-1a by T cells and macrophages, which can expand IBMIR and induce subsequent adaptive immunity, triggering and enhancing cellular rejection (23, 24) (Figure 1). Aggregated macrophages continuously secrete cytokines such as IL-6 and IL-8 to sustain the inflammatory response and release pro-inflammatory factors such as IL-1b, IFN-g, and TNF-a. The IL1b secreted by macrophages and neutrophils binds to IL-1b receptors on the surface of islet cells, activating IL-1 receptor-associated kinases and TNF receptor-associated factor 6, leading to the phosphorylation and degradation of IkB, releasing NF-kB, which then enters the nucleus to regulate the transcription of multiple genes, including IL-1, IL-6, TNF-a, and iNOS. TNF-a produced by macrophages and islet cells binds to TNF receptors, activating the NF-kB and MAPK pathways and inducing apoptosis. Apoptosis is mediated by caspase-3 activation through the MAPK pathway or by activating effector caspases, including FADD-mediated caspase-3 activation. IFN-g produced by macrophages binds to IFN-g receptors on islet cells, activating JAK1 and JAK2. Activated JAK2 then activates Signal Transducer and Activator of Transcription 1 (STAT1). STAT1 is then transferred to the nucleus for gene regulation, ultimately leading to islet cell apoptosis. The pro-apoptotic effect of STAT1 may be partially mediated by the activation of caspase-2, caspase-3, and caspase-7 (25). Under the combined action of cytokines IL-1b, TNF-a, and IFN-g, the overexpression of iNOS in b-cells and macrophages leads to excessive synthesis of NO. Subsequently, NO loses electrons and combines with superoxide radicals to form highly reactive peroxynitrite (ONOO-). The cytotoxicity of ONOO subsequently induces islet cell apoptosis. On the other hand, macrophages play an antigen-presenting role, promoting the activation of T cells into CD8+ T cells and CD4+ T cells. Activated T cells produce cytokines such as IFN-g, TNF-a, and lymphotoxin, thereby inducing b-cell apoptosis. (Figure 1). Lisa Özmen et al. (26) exposed human islets to ABO-compatible blood and found that administering Melaglavin dose-dependently eliminated IBMIR. In the absence of or at concentrations below 0.4 μmol/l of Melaglavin, the integrity of islets exposed to blood was lost. However, at concentrations of 1-10 μmol/l, Melaglavin inhibited coagulation and complement activation, leading to reduced platelet and leukocyte activation and consumption. This protective effect indicates that thrombin plays a crucial role in IBMIR and suggests that thrombin inhibition could improve the outcomes of clinical islet transplantation (26). L. Moberg et al. perfused human islets with fresh ABO-compatible blood for 30 minutes. In control samples (containing either only islets or blood with non-inhibitory anti-TF [4503]), coagulation occurred within 15 minutes. However, blood containing inhibitory anti-TF [4509] inhibited coagulation throughout the observation period. The study found that IBMIR is initiated by TF and consistently occurs during clinical islet transplantation, even in the absence of clinical symptoms like portal vein thrombosis. Inhibiting this process may increase the success rate of clinical islet transplantation and reduce the number of donors required per patient (18).
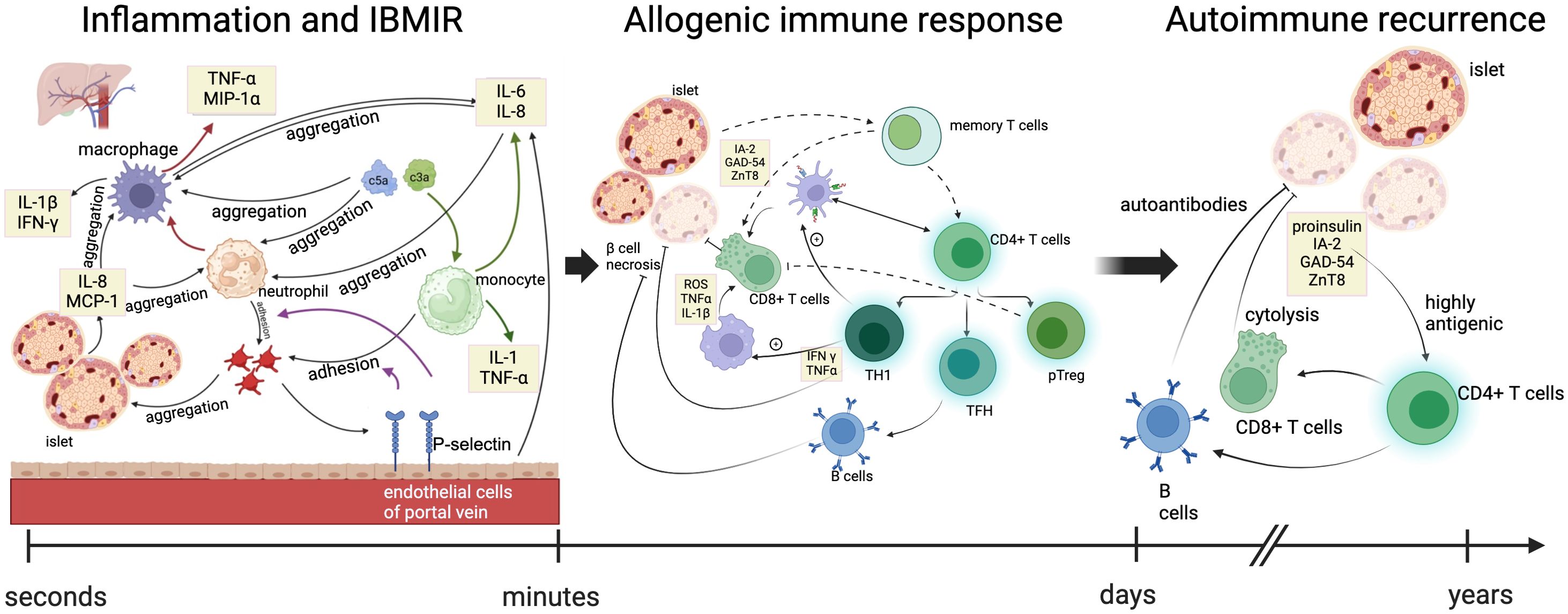
Figure 1. Immune mechanisms at three stages of allogeneic islet transplantation. The immune responses occurring during allogeneic islet transplantation included three stages: inflammation and IBMIR, allogeneic immune response, and autoimmune recurrence. In the early stages of transplantation, islets secrete pro-inflammatory factors and activate the complement system, promoting the recruitment of platelets, neutrophils, and monocyte macrophages to the graft. Vascular endothelial cells secrete cytokines and release P-selectin, promoting the adhesion between monocytes/neutrophils and platelets. Accumulated monocyte macrophages and neutrophils further enhance the recruitment of macrophages and the secretion of cytokines. Green arrows indicate the process by which the complement system promotes cytokine secretion by monocytes. Red arrows indicate the process by which neutrophils enhance cytokine secretion by macrophages. Purple arrows represent the process by which P-selectin promotes the adhesion of monocytes and neutrophils to platelets. Within days after transplantation, the release of inflammatory signals leads to increased cytokine production, with neutrophils signaling macrophages and dendritic cells to the site of islet phagocytosis, presenting antigens on their surface and recruiting adaptive immune cells. The infiltration of helper and cytotoxic T cells further damages the islets, recruiting B cells that produce antibodies against the allogeneic islets and differentiating T cells into memory T cells, ultimately leading to the rejection of the overall allogeneic transplant. TNFα, tumor necrosis factor alpha; MIP1α, macrophage inflammatory protein-1 alpha; IFN-γ, interferon; MCP-1, monocyte chemoattractant protein-1; GAD65, glutamic acid decarboxylase 65; IA-2, insulinoma-associated protein 2; ZnT8, transporter 8. ROS, reactive oxygen species; TH1, T helper 1 cells; TFH, follicular helper T cells; pTreg, peripheral regulatory T cells.
IBMIR, characteristic of innate inflammatory responses and thrombotic pathway, is driven by the activation of the coagulation cascade, with negatively charged islet surfaces activating the intrinsic coagulation pathway, and tissue factor (TF) expressed by the islets triggering the extrinsic pathway.
The innate immune system is the body’s rapid response to an initial infection or injury. In IBMIR, the following components are mainly involved: Neutrophils: They are the first cells to arrive at the transplant site, release inflammatory mediators and oxygen free radicals, mediate local tissue damage and remove pathogens. Monocytes and macrophages: Monocytes are recruited and converted into macrophages, which further release cytokines (such as TNF-α and IL-1) that intensify the inflammatory response and enhance recruitment of immune cells. Cytokines released by neutrophils and macrophages in IBMIR not only promote local inflammatory responses, but may also affect T cell activation and subsequent adaptive immune responses (27).The activation of innate immune cells can lead to apoptosis or necrosis of the transplanted islet cells, thus reducing the survival rate of the grafts. The inflammatory response triggered by IBMIR may cause more immune cells to aggregate, forming positive feedback and further aggravating the damage. There is a close interaction between IBMIR and congenital leukocyte response, which together affect the success rate of islet transplantation.
2.2 Allogeneic immune response
The allogeneic immune response, which is adaptive immunity, occurs later but leads to long-term functional reduction of β-cells, resulting in a significant portion of islets losing their insulin independence. Analysis of pancreatic sections from T1D patients reveals significant immune infiltration within individual islets, confirming the crucial role of CD4 and CD8 T cells in β-cell destruction (14, 28). Despite high levels of systemic inflammation markers in T2D patients, their islets do not exhibit similar T cell infiltration, in stark contrast to the pancreatic sections of T1D patients, making islet autoantibodies a differential diagnostic marker between T1D and T2D (4). The presence or development of alloreactivity (against human leukocyte antigens, HLA) and its impact on allogeneic graft survival is well-defined in the solid organ transplantation literature. Donor-specific antibodies (DSA) binding to endothelial cells or islets (which constitutively express Class I HLA and aberrantly upregulate Class II HLA) can activate the classical complement pathway. Even in the absence of complement, some DSAs can promote antibody-dependent cellular cytotoxicity, where innate immune cells bind to Fc fragments, triggering the release of cytolytic enzymes by neutrophils and NK cells. C4d is a degradation product of the classical complement pathway, covalently bonded to the endothelium, serving as a marker for antibody-mediated immunity (4). Transplanting allogeneic islets or pancreas to T1D recipients expressing major and minor histocompatibility antigens on endogenous islets and pancreas can elicit complex adaptive B cell and T cell responses, leading to classical allogeneic graft rejection.
Key effector immune cells include cytotoxic T cells (CD8+ T cells), macrophages, plasma cells, and CD4+ T helper cells. In human T1D, existing evidence from single-islet studies from the Network for Pancreatic Organ Donors with Diabetes suggests that β-cell destruction is largely mediated by direct contact between CD8 T cells and β-cells, as well as CD4 T cell-mediated M1 macrophage polarization (29–31).
CD8+ T cells eliminate cells presenting non-self antigens by inducing apoptosis through the release of cytotoxic molecules (such as granzymes and perforin) or through cell-surface interactions (such as the binding of Fas ligand (also known as CD95L) on T cells to Fas receptors on the target cells) (32). Activated CD8+ T cells infiltrating the graft also induce macrophage activation, particularly through the expression of pro-inflammatory cytokines such as IFN-γ (33).
Macrophages typically exhibit pro-inflammatory characteristics and display M1 polarization during acute rejection, producing pro-inflammatory cytokines that lead to direct cellular damage and coordinate pro-inflammatory immune responses (34). Their primary function is phagocytosis, recognizing damaged allogeneic graft tissue through pattern recognition receptors such as Toll-like receptors. As antigen-presenting cells, macrophages can present allogeneic antigens on MHC class II molecules, thereby promoting the adaptive immune response (35).
Plasma cells are another type of effector immune cell derived from B cells and form the cornerstone of humoral immunity. They enable the body to combat foreign invaders not only by neutralizing pathogens but also by performing various effector functions, including regulating hypersensitivity reactions, activating the complement cascade, and modulating the mucosal microbiome. However, their activity can be problematic in solid organ transplantation (36). In transplantation, plasma cells can produce donor-specific antibodies (DSAs), which lead to acute and chronic rejection by activating the complement system, resulting in vascular injury and graft loss. The impact of DSAs has been extensively evaluated in various solid organ transplants (37–39).
CD4+ T helper cells play a critical role in immune rejection. They coordinate the activation of other immune cells, such as B cells and cytotoxic T cells, to enhance the immune response against allogeneic material. These CD4+ T cells are capable of producing and releasing various cytokines, including interferon-gamma (IFN-γ) and interleukin-2 (IL-2). Additionally, CD4+ T cells actively interact with B cells, promoting antibody production and thereby strengthening humoral immunity (40, 41). CD4 T cells can provide “help” to B cells and stimulate antibody production (as described above), as well as promote effector CD8 T cell responses and stimulate resident macrophages in the islets (42, 43).
Auto-reactive CD4 T cells interact with dendritic cells presenting islet antigens (44) and can differentiate into T helper 1 (TH1) cells, follicular helper T cells (TFH), peripheral regulatory T cells (pTreg), or anergic cells. TFH cells help B cells produce high-affinity islet-specific antibodies (29). TH1 cells activate dendritic cells and enhance antigen presentation to islet-specific CD8 T cells (45), thereby inducing the proliferation of effector CD8 T cells (45). TH1 cells migrate to the pancreas (46), secrete pro-inflammatory cytokines interferon-γ (IFNγ) and TNFα, and induce β-cell death (47). TH1-derived IFNγ and TNFα stimulate M1 macrophages in the islets to produce reactive oxygen species (ROS), TNFα, and IL-1β (48), further amplifying the cycle of β-cell death (30)The resulting inflammation leads to increased infiltration of CD8 T cells, which directly kill β-cells via perforin and granzyme B (49), while natural and peripheral regulatory T cells (nTreg and pTreg) attempt to suppress this response through TGFβ and IL-10 (50).
2.3 Autoimmune recurrence
Patients with T1D and concurrent autoantibodies have a lower success rate for islet transplantation due to the presence of memory CD4+ and CD8+ T cells, which rapidly reactivate to target islet antigens (IA-2, GAD-54, and ZnT8) and destroy the transplanted islets (42, 43). Patients with T1D who have long-term β-cell transplants still have the ability to destroy islets. Reviewing the case of David Sutherland’s identical twin transplant surgery, where the pancreas of an unaffected twin was transplanted into the twin with long-term T1D without immunosuppression, resulted in the loss of transplanted β-cell function and pancreatitis (51, 52). This is because most individuals’ immune systems develop the ability to distinguish self from non-self. In T1D, the loss of the ability to recognize insulin-producing islet β-cells as self leads to an autoimmune response, which destroys β-cells in the natural pancreas (53, 54). This autoimmune response is primarily mediated by T cells, which are the main effector cells in the β-cell destruction process. Moreover, there is ample evidence that isolated allogeneic islet transplants may cause autoimmune recurrence in a small but significant proportion of patients. In the autoimmune process, when islets or pancreas are transplanted into recipients with T1D, donor β-cells express β-cell-specific antigens that are attacked by T cells and B cells (55–58). These include insulin (proinsulin), glutamic acid decarboxylase 65 (GAD65), insulinoma-associated protein 2 (IA2), and zinc transporter 8 (ZnT8), which are highly antigenic to both B cells and T cells in humans (59). This explains why islet autoantibodies sometimes rise sharply within weeks after transplantation. This increase usually occurs without any signs of allogeneic immunity (60). Therefore, transplanting islets or pancreas into T1D recipients is a renewed challenge to the autoreactive memory response and may lead to the recurrence of autoimmune function post-transplant.
In summary, when allogeneic islets are transplanted into T1D patients, a comprehensive immune response is elicited against the foreign tissue. Besides the classic rejection of the allogeneic graft, the outcomes of islet or pancreatic transplantation may be severely impacted by early intense inflammatory responses and the reactivation of autoimmunity. In simple terms, the three stages of immune response experienced are inflammation and IBMIR, allogeneic immune response, and autoimmune recurrence. Within days after transplantation, the release of inflammatory signals leads to increased cytokine production, with neutrophils signaling macrophages and dendritic cells to the site of islet phagocytosis, presenting antigens on their surface and recruiting adaptive immune cells. The infiltration of helper and cytotoxic T cells further damages the islets, recruiting B cells that produce antibodies against the allogeneic islets and differentiating T cells into memory T cells, ultimately leading to the rejection of the overall allogeneic transplant. The entire process of allogeneic transplant rejection may be amplified in T1D patients because they have effectively primed T cells specifically targeting β-cells.
3 Immunosuppressants in allogeneic islet transplantation
A major issue in islet transplantation is transplant rejection. To prevent this complication, immunosuppressive drugs such as cyclosporine, tacrolimus, mycophenolate mofetil, and corticosteroids must be used (61). However, immunosuppressants have severe side effects, including inducing diabetes, nephrotoxicity, and carcinogenic effects (62–65).
3.1 Calcineurin inhibitors
There are many types of calcineurin inhibitors (CNIs), such as the commonly used cyclosporine and rapamycin. The potent immunosuppressive properties of cyclosporine were discovered in 1976. Cyclosporine blocks the clonal expansion of resting T cells by inhibiting the transcription of genes encoding IL-2 and the high-affinity IL-2 receptor, which is crucial for T cell activation (66).
Tacrolimus (FK506) was the first macrolide antibiotic explored for its effective immunosuppressive properties in 1987 (67, 68). The mechanism of toxicity of tacrolimus is as follows: tacrolimus binds to the immunophilin FK506-binding protein 12 (FKBP12) to form a complex that binds and inhibits the mammalian target of rapamycin (mTOR) kinase, thereby exerting immunosuppressive activity (69, 70). This kinase is a key regulator of cell metabolism, growth, and proliferation. Importantly, inhibition of mTOR by tacrolimus causes cell cycle arrest in the mid-to-late G1 phase, thus potentially inhibiting tumor cell growth and, importantly, its immunosuppressive function by inhibiting T cell and B cell proliferation (71). However, FKBP12 and mTOR are ubiquitously expressed. Therefore, there is a possibility of “off-target” effects on cells other than tumor and immune regulatory cells.
mTOR kinase exists in two distinct complexes: mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2). They have different substrates and are regulated differently (Figure 2). Although they share some core components, such as mTOR, mLST8, and DEPTOR, they also contain other unique proteins. For example, a unique component of mTORC1 is RAPTOR (regulatory associated protein of mTOR), which acts as a bridge to bind mTOR to its downstream effectors (72, 73). An important component of mTORC2 is the protein Rictor (rapamycin-insensitive companion of mTOR), which is necessary for the formation of the mTORC2 complex and its kinase activity (74, 75). Importantly, mTORC1 is highly sensitive to inhibition by rapamycin, whereas mTORC2 was initially thought to be resistant to rapamycin (74, 75), but in fact, it is sensitive to long-term rapamycin treatment in some cell types (76–78). Therefore, both complexes may play a role in the immunosuppressive and toxic effects of rapamycin. Consistent with its role as a key regulator of cell metabolism, proliferation, and growth, mTORC1 activity is regulated by nutrients, growth factors, and cellular energy levels (Figure 2). The best-characterized targets of mTORC1 are eIF4E-binding protein (4E-BP) and S6 kinase protein (S6K), both of which play important roles in the regulation of protein synthesis.
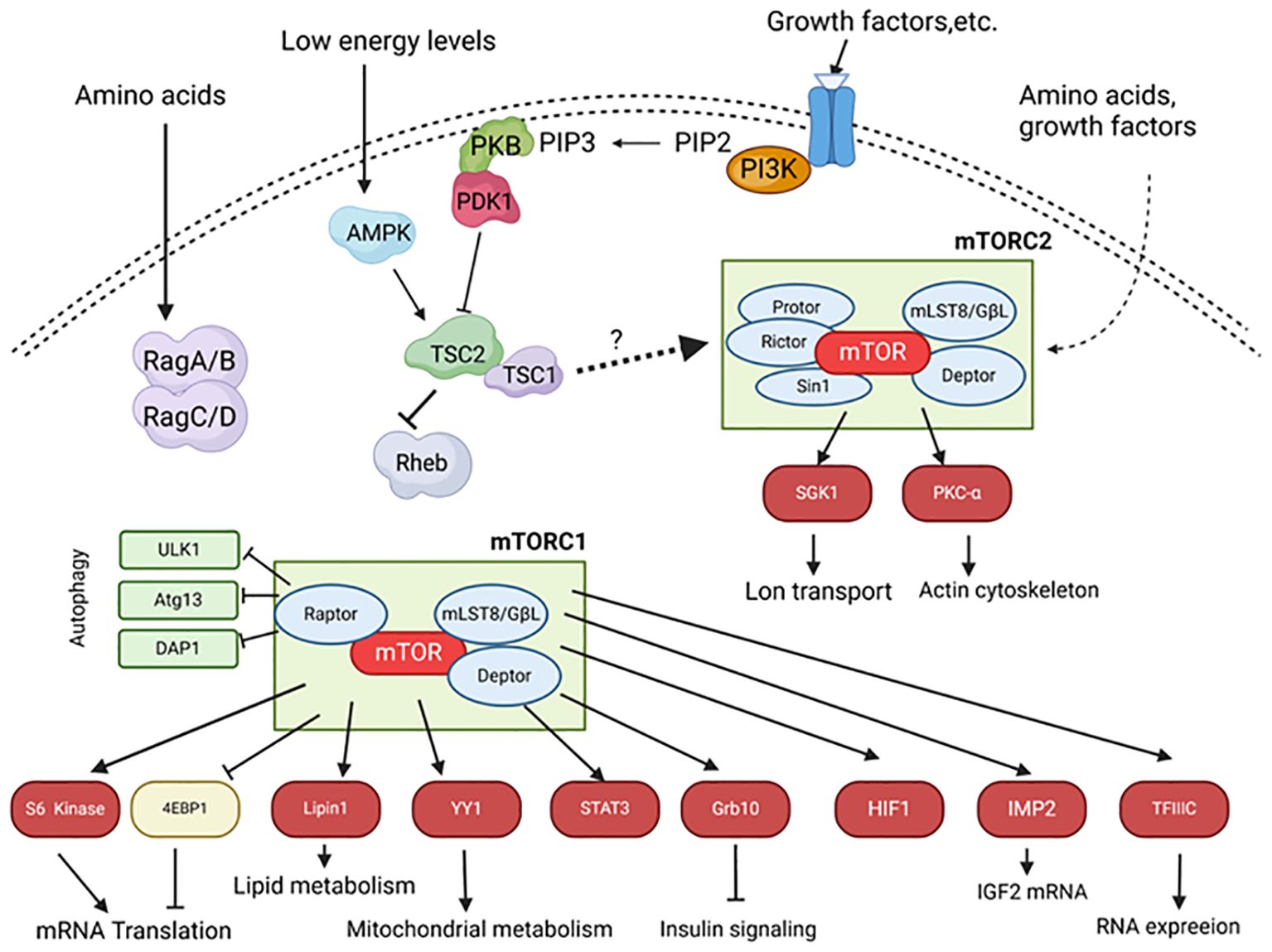
Figure 2. The mTOR signaling pathway in islets. Upon stimulation by insulin and other growth factors, phosphoinositide 3-kinase (PI3K) converts phosphatidylinositol 4,5-bisphosphate (PIP2) into phosphatidylinositol 3,4,5-trisphosphate (PIP3), which localizes PKB to the membrane and activates it through PDK1 and mTORC2. Activated PKB phosphorylates and inhibits TSC1/2. Rheb, a small GTPase inhibited by TSC2, positively regulates mTORC1 activity. mTORC1 phosphorylates S6 kinase 1/2 and 4EBP1, leading to increased mRNA translation. Amino acids activate mTORC1 through Rag A/B and C/D. Under low energy conditions, the ratio of AMP to ATP increases, activating AMP-activated kinase (AMPK), which phosphorylates and activates the TSC1/2 complex, thereby inhibiting mTORC1. mTORC2 activity is primarily mediated through unknown pathways. mTORC2 phosphorylates and activates PKB, serum- and glucocorticoid-induced kinase 1 (SGK1), and PKC. Arrows indicate stimulatory effects; block ends indicate inhibitory effects; solid lines represent direct effects, and dashed lines represent indirect effects. Atg13, Autophagy-related protein 13; DAP1, Death-associated protein 1; Deptor, DEP domain-containing mTOR-interacting protein; 4EBP, eIF4E-binding protein; GbL-G, Protein Gβ-like; HIF1, Hypoxia-inducible factor 1; IMP2, Insulin-like growth factor 2 mRNA-binding protein; mLST8, Mammalian lethal with Sec13 protein 8; PDK1, Phosphoinositide-dependent protein kinase 1; Protor, Protein observed with Rictor; Raptor, Regulatory associated protein of mTOR; Rictor, Rapamycin-insensitive companion of mTOR; Sin1, Stress-activated protein kinase-interacting protein 1; TFIIIC, Transcription factor 3C; ULK1, Unc-51 like kinase 1.
The role of mTORC1 in B cell function is as follows. An important aspect of maintaining glucose homeostasis is the maintenance of pancreatic B cell mass and the ability of B cell mass to increase in insulin-resistant states such as obesity. The increase in B cell mass is due to increased neogenesis (progenitor cell generation) and proliferation (hyperplasia), hypertrophy, and reduced apoptosis. There is substantial evidence indicating that rapamycin significantly reduces the proliferation of B cells and progenitor cells, thereby affecting the maintenance of B cell mass. The most compelling evidence for the role of mTORC1 in regulating B cell mass comes from in vivo transgenic mouse models (79). Overactivation of mTORC1 by selectively overexpressing Rheb (80) or deleting TSC1 (81) or TSC2 (81, 82) in B cells leads to increased B cell size and mass, along with improved insulin secretion and glucose tolerance. These effects may be partially mediated by S6K, as mice lacking S6K1 or rpS6 exhibit hypoinsulinemia and glucose intolerance with reduced B cell size (83, 84). Additionally, transgenic mice overexpressing constitutively active S6K exhibit improved glucose tolerance and enhanced insulin secretion with increased B cell size (85). Although these studies strongly suggest the critical role of mTORC1, manipulation of mTOR upstream regulators (e.g., Rheb) may affect pathways beyond mTORC1, so causality cannot be definitively established. There is extensive work investigating the role of mTOR in regulating cell proliferation in certain cell types, but little is known about the exact mechanisms by which mTORC1 signaling regulates B cell cycle progression. However, it is known that mTORC1 can regulate the synthesis and stability of cyclins D2 and D3 in B cells (86). These cyclins form complexes with cyclin-dependent kinase 4, controlling cell cycle progression. In rat islets treated with rapamycin, reduced levels of cyclins D1 and D2 were observed, accompanied by decreased β-cell proliferation (87).
mTORC1 also appears to play a role in insulin secretion by pancreatic B cells. Knockdown of TSC1 in mice results in significantly increased insulin production, independent of B cell mass (81). Additionally, long-term rapamycin treatment inhibits glucose-stimulated insulin secretion (GSIS) in cloned B cell lines as well as rodent and human islets. However, it is unclear whether this effect is mediated by mTORC1 or mTORC2. The control of insulin secretion in B cells involves many complex signaling pathways, and the mechanism by which rapamycin regulates insulin secretion remains unknown. One proposed mechanism is that inhibition of mTORC1 reduces mitochondrial function, particularly the activity of α-ketoglutarate dehydrogenase. This leads to reduced carbohydrate metabolism, thereby decreasing mitochondrial ATP production (88), which is known to regulate insulin secretion in B cells (89). Another explanation is that rapamycin promotes autophagy, a process primarily controlled by mTORC1 rather than mTORC2, or intracellular degradation of cytoplasmic proteins involved in insulin production, leading to inhibition of insulin secretion (89).
It is not completely clear how the activity of mTORC2 is regulated, but there is evidence that it can be stimulated by amino acids and growth factors (90, 91). Downstream targets of mTORC2 include protein kinase C (PKC)-α (85–87) and protein kinase B (PKB) (92), two serine/threonine kinases that play roles in the regulation of key cellular processes such as apoptosis, proliferation, motility, and differentiation, as well as serum- and glucocorticoid-induced kinase 1 (93), which plays a role in the control of ion transport (94) (Figure 2).
In mice, B cell-specific deletion of the Rictor gene (an important component of mTORC2) is associated with reduced plasma insulin levels due to decreased insulin secretion from islets, leading to hyperglycemia (95). This is related to reduced B cell mass and proliferation but does not increase B cell apoptosis. Research by Adam D. Barlow et al. has demonstrated that knocking down Rictor in rat islets using small interfering RNA results in increased B cell apoptosis and reduced GSIS (76). These studies specifically demonstrate that mTORC2 activity plays a dominant role in B cell survival and function. Importantly, prolonged rapamycin treatment (24 hours) of MIN6 cells, rat islets, or human islets leads to dissociation of mTORC2, thereby inhibiting its expression. This precedes the toxic effects of rapamycin on function and activity, occurring simultaneously with reduced PKB phosphorylation and downstream signaling. Interestingly, the expression of constitutively active PKB in MIN6 cells and rat islets can mitigate the harmful effects of rapamycin on GSIS and cell viability (76). Overall, this suggests that rapamycin B cell toxicity is primarily mediated through inhibition of mTORC2 and its subsequent impact on PKB signaling. However, this is based on in vitro experiments with B cells and needs to be further confirmed in vivo.
Extensive research indicates that PKB, as a key downstream effector of mTORC2, plays an important role in B cell survival and function. These studies further reveal the potential role of mTORC2 in B cell homeostasis. For instance, transgenic mice expressing constitutively active PKB in B cells show a significant increase in B cell mass due to increased B cell number and size (96, 97). This is manifested by significantly elevated plasma insulin levels, improved glucose tolerance, and resistance to streptozotocin-induced diabetes. In INS-1 cells, rat B cell lines, and primary rat B cells, expression of constitutively active PKB has also been shown to protect against lipotoxicity (98), cytokine-induced cytotoxicity (99), and AMPK-mediated cytotoxicity (100). Conversely, studies in transgenic mice lacking PKB show significantly elevated blood glucose levels, reduced insulin levels, and impaired glucose tolerance.
Rapamycin is a key immunosuppressant, particularly in islet cell and kidney transplantation. However, extensive in vitro and in vivo evidence strongly suggests that rapamycin has harmful effects on pancreatic B cells and peripheral insulin sensitivity. This toxicity is mainly because rapamycin inhibits mTOR, which is part of complex signaling pathways controlling many important cellular functions (including mRNA translation, cell proliferation, cell growth, differentiation, protein synthesis, angiogenesis, and apoptosis) through mTORC1 and mTORC2 (71). In summary, rapamycin-induced B cell toxicity and insulin resistance are likely mediated primarily through mTORC2 rather than mTORC1 (76, 95).
In addition to the above mechanisms, although rapamycin is structurally unrelated to cyclosporine, it shares many intracellular pathways that inhibit calcineurin and subsequently block IL-2 production. It acts by limiting the dephosphorylation and translocation of nuclear factor of activated T cells (NFAT). NFATs play a critical role in T cell activation. When T cells are stimulated by antigens, intracellular calcium levels rise rapidly, activating calcineurin. Activated calcineurin dephosphorylates NFATs, exposing nuclear localization signals and causing NFATs to translocate from the cytoplasm to the nucleus. In the nucleus, NFATs bind to specific DNA sequences, regulating the transcription of related genes and participating in T cell proliferation, differentiation, and cytokine production. Calcineurin signaling is essential for insulin secretion and β-cell proliferation (101), and specific inactivation of calcineurin in β-cells is associated with age-related hyperglycemia (102). Apoptosis of islet cells related to calcineurin inhibition is also thought to occur through the limitation of cAMP response element-binding protein (CREB), which reduces the expression of insulin receptor substrate-2 (IRS-2), limits Akt phosphorylation, and affects insulin secretion (103, 104). CNIs also reduce the expression of cell surface glucose transporter 4 (GLUT4) and decrease insulin-stimulated glucose uptake in adipocytes (105), potentially leading to peripheral insulin resistance.
Moreover, rapamycin promotes reduced mitochondrial Ca2+ uptake, which has been shown to impair respiration and ATP production, leading to impaired glucose-stimulated insulin secretion (GSIS) (106). CNIs, particularly rapamycin, enhance the deleterious effects of glucolipotoxicity on β-cells, inducing the expression of forkhead box protein O1 (FoxO1), thereby limiting proliferation (107), and reducing insulin content and secretion (108). Rapamycin causes reversible graft dysfunction, characterized by amyloid deposition and macrophage infiltration in transplanted islets (101), with no clear evidence of β-cell death. Ultrastructural examination of the grafts shows reduced insulin granules, accompanied by increased transcripts associated with extracellular matrix deposition and inflammation. Heparin is primarily used to reduce IBMIR-mediated cell destruction of islets, promoting the fibrillation of human islet amyloid polypeptide (IAPP) and has been shown to simultaneously promote amyloid deposition and reduce β-cell apoptosis (109). Rapamycin exerts its antifibrotic function by inhibiting JAK2/STAT3 signaling activation through targeting JAK2, thereby inhibiting M2 macrophage polarization (110). After transplanting human islets into NSG mice, rapamycin inhibits β-cell function by activating islet-resident macrophages through inhibition of the NFAT pathway and by stimulating macrophages to produce IL-1β through increased amyloid deposition in the transplanted islets (111). Heparinase treatment significantly reduces amyloid deposition and subsequent β-cell toxicity (112).
As two types of CNIs, cyclosporine and rapamycin have similar mechanisms of action, but rapamycin has been shown to be 10-100 times more potent than cyclosporine in inhibiting mixed lymphocyte cultures and the generation of cytotoxic T cells in vitro (66).
3.2 Mycophenolate Mofetil
In 1993, Mycophenolate Mofetil (MMF), the 2-4 morpholinoethyl ester of the biologically active compound mycophenolic acid, was introduced as a new immunosuppressant (113). MMF reversibly inhibits inosine monophosphate dehydrogenase (IMPDH), a key enzyme in the de novo synthesis of the purine nucleotides in DNA (i.e., guanine and adenine) (114). Lymphocytes play a crucial role in graft rejection, and without IMPDH, they cannot produce sufficient amounts of purines (115). Consequently, MMF can prevent the proliferation of T cells and B cells, thereby inhibiting antibody production. Additionally, by lowering intracellular GTP levels in lymphocytes, MMF inhibits glycosylation and the expression of certain adhesion molecules, thus reducing lymphocyte migration to the graft (116). However, its effect on T cell proliferation has garnered more attention due to the critical role of T cells in the allogeneic response (117). MMF is considered a safe drug, with the most commonly reported side effects being mild and primarily involving the gastrointestinal system (diarrhea, abdominal pain, nausea, and vomiting) (118–120). Its main advantage is the lack of nephrotoxicity and diabetogenic effects, making MMF an important drug in kidney and islet transplantation.
3.3 Glucocorticoids
T1D is secondary to the initial autoimmunity of islets, resulting from the inflammatory destruction of β-cells (74). Inflammatory macrophages are key in maintaining islet injury (121). Pro-inflammatory cytokines, partly derived from macrophages and damaged β-cells, further inhibit β-cell function by inducing nitric oxide production (122, 123). As T1D progresses, pro-inflammatory cytokines inhibit β-cell regeneration, stimulate peripheral insulin resistance, and maintain insulin inflammation (124). Glucocorticoids (GC) are used clinically for their powerful anti-inflammatory and immunosuppressive effects (125), but high doses of glucocorticoids promote peripheral insulin resistance and inhibit β-cell function (62, 63, 126), thus discouraging their use in T1D treatment and transplantation protocols (12). However, the general notion that GC’s effects on β-cells are purely harmful has been increasingly challenged (127–131). It has now been demonstrated that selective GC regeneration within β-cells can prevent inflammatory β-cell destruction, suggesting that GC-targeting therapy with 11β-hydroxysteroid dehydrogenase type 1 (11β-HSD1) may improve the course of T1D and islet transplantation aggravated by high-dose hormones.
3.4 Bortezomib
In addition to the health risks posed by infections and cancer due to broad immunosuppression, numerous studies report that widely used immunosuppressants such as glucocorticoids or calcineurin inhibitors are cytotoxic to islet β-cells (71, 132). Thus, there has been a need to develop tolerance-promoting regimens that can retain the viability and function of islets post-transplantation. Bortezomib, a selective inhibitor of the 26S proteasome, has been FDA-approved for treating relapsed multiple myeloma (133, 134). Bortezomib’s mechanism of action involves inhibiting the proteasomal degradation of IκB, thereby inhibiting the activation of nuclear factor κB (NF-κB) (135, 136). Since NF-κB is a key transcription factor involved in the expression of various genes related to immune responses, numerous studies have demonstrated the immunosuppressive effects of bortezomib. It selectively depletes alloreactive T cells in vitro and reduces the secretion of T helper 1 (Th1) cell cytokines (137). Additionally, bortezomib can modulate the function of dendritic cells (DCs): treatment with bortezomib induces a skewed phenotypic maturation of DCs in response to lipopolysaccharides (LPS) and other endogenous stimuli while reducing cytokine production (138). Other studies have also reported that bortezomib can prevent graft-versus-host disease (GVHD) and allograft rejection in mouse models of allogeneic stem cell and cardiac transplantation (139, 140). Furthermore, bortezomib can inhibit the activation of rapamycin-resistant memory T cells without affecting the viability of regulatory T cells (Tregs) in non-human primates (141). Overall, these immunomodulatory effects suggest that bortezomib has the potential to be a promising immunosuppressant candidate in islet transplantation.
So-Hee Hong et al. (142) conducted a related study using BALB/c spleen cells to pre-sensitize C57 BL/6 mice, administering low-dose bortezomib (0.1 mg/kg) for 4 consecutive days to observe its immunosuppressive effects in vivo. Since NF-κB is the primary transcription factor for DC maturation, DC maturation status was detected by measuring the expression levels of MHC class II molecules and other co-stimulatory molecules in CD11c+ DCs. The conclusions suggested that low-dose bortezomib only reduced the expression of MHC class II molecules without affecting other co-stimulatory molecules expressed on DCs. Unlike other studies showing bortezomib’s inhibitory effect on alloreactive T cells with high-dose treatment, short-term low-dose bortezomib treatment did not significantly affect the percentage of splenic effector memory cells (CD4+CD44 and CD8+CD44) and the number of T cells producing allogeneic antigen-specific interferon-γ. Based on these results, it was speculated that low-dose bortezomib might inhibit DCs in vivo by altering their MHC class II expression.
Additionally, some studies suggest that high-dose rapamycin treatment impairs β-cell regeneration and reduces islet engraftment, adversely affecting islet transplantation (143, 144). Therefore, So-Hee Hong et al. (142) developed a new combination therapy based on low-dose bortezomib and rapamycin, which is highly tolerable and minimally cytotoxic to β-cells, as a potential alternative and tolerance-promoting immunosuppressive regimen in allogeneic islet transplantation. They tested the efficacy of low-dose bortezomib alone or in combination with rapamycin in an islet transplantation model. Low-dose (0.1 mg/kg) bortezomib treatment groups showed longer graft survival rates compared to control groups (0.05 mg/kg group: P=0.1, 0.1 mg/kg group: P=0.0036). Low-dose (1 mg/kg) rapamycin was added to the same transplantation environment. Compared to the control group, the 0.05 mg/kg bortezomib + rapamycin group (P=0.0011) and the 0.1 mg/kg bortezomib + rapamycin group showed significantly prolonged islet graft survival (P=0.001). Although not statistically significant, the combination of rapamycin and bortezomib increased graft survival compared to the bortezomib-only treatment group. In the 0.1 mg/kg bortezomib plus rapamycin treatment group, 4 out of 6 mice maintained normoglycemia for 100 days, while 2 out of 6 mice in the 0.1 mg/kg bortezomib-only treatment group maintained normoglycemia for 100 days. Additionally, the mean graft survival period increased from 24 days to 58 days after adding rapamycin to the 0.05 mg/kg bortezomib treatment group. To determine whether low-dose bortezomib + rapamycin treatment induces immune tolerance, grafts were removed from recipient mice that maintained normoglycemia for over 100 days, and a second graft (islets from BALB/c donors) was transplanted into the contralateral kidney. Interestingly, mice with the second graft maintained normoglycemia for 50 days without any immunosuppression. To determine whether this tolerance was systemic, BALB/c and C3H (third-party) as well as C57 BL/6 (control) skin grafts were transplanted into the flank of the second transplant recipients. The C3H skin grafts were rejected on day 14 post-transplant (DPT). The rejection of BALB/c skin grafts was somewhat delayed but ultimately rejected on DPT 18. Unexpectedly, the rejection of BALB/c skin appeared to result in the rejection of the second islet graft, as blood glucose levels returned to hyperglycemia 20 days after the skin rejection reaction. Thus, it was concluded that this combination therapy induced tolerance to islet-specific antigens, and its inhibitory effect was insufficient to prevent strong skin graft rejection. Many studies have shown that Th1 cells are major participants in graft rejection in various transplant models, with interferon-γ playing a key role by activating cytotoxic CD8+ T cells (145, 146). So-Hee Hong et al. investigated whether bortezomib alone or in combination with rapamycin could reduce Th1 and interferon-γ-producing cells. Splenocytes from allogeneic islet transplant mice that maintained normoglycemia for over 60 days were stimulated in vitro with irradiated BALB/c splenocytes, followed by ELISPOT analysis. The combination therapy group showed almost no detectable interferon-γ-producing cells. Although a reduction in interferon-γ-producing cells was also observed in the bortezomib-only group, it was not as pronounced as in the combination therapy group. Moreover, no significant changes were observed in other cytokine-producing cells in the combination therapy group. MLR assays were used to detect BALB/c-specific T cell responses in recipient mice treated with bortezomib + rapamycin. The results indicated that the mice’s T cells had a lower proliferative response to BALB/c antigens but not to third-party C3H antigens. Therefore, these results suggest that low-dose, short-term combination therapy with bortezomib and rapamycin significantly increases graft survival and induces tolerance to islet antigens while inducing severe BALB/c-specific T cell hyporesponsiveness, increased Tregs, and reduced inflammatory cytokines (142).
4 Xenotransplantation
The increasing number of patients in need of organ transplants has made xenotransplantation of islets a potential future treatment option for diabetic patients due to the shortage of organ donors. According to recent advances in preclinical studies on non-human primates, porcine islets may be the ideal choice among various animal organs and tissues for xenotransplantation (147), mainly due to the biochemical compatibility of porcine and human insulin and the potential to obtain a large number of donor pigs through relatively short turnover breeding strategies. Additionally, another theoretical advantage of porcine islets is their potential resistance to autoimmune recurrence against human β-cells (148). The main barrier to interspecies transplantation is the preformed xenogeneic antibodies that cause hyperacute rejection. Hyperacute rejection (HAR) is a rapidly occurring rejection in islet transplantation and other organ transplants, usually occurring within minutes to hours after transplantation. This condition arises from the interaction between pre-existing antibodies from humans or non-human primates (NHP) and the antigens present in the graft (149).Among these antibodies, the most common are IgMs and IgGs that identify galactose-α1,3-galactose (α-Gal) residues, which are attached to glycoproteins and glycolipids by the α1,3 galactosyltransferase (α1,3GT) found in non-primate genomes. Humans and apes do not have α-Gal epitopes (150). Furthermore, approximately 70–90% of these antibodies specifically target α-Gal epitopes (151). As a result, when an organ from a pig is transplanted into a human or a non-human primate (NHP), the existing anti-Gal antibodies attach to the α-Gal epitopes found on the graft’s vascular endothelium. This interaction triggers the production of complement component 3b (C3b), activates the complement system (152), and leads to the formation of a membrane attacking complex (MAC).These responses result in the lysis of endothelial cells, damage to the vasculature, and ultimately, rejection of the graft (153, 154). Additionally, the disruption of endothelial vascular integrity leads to interstitial hemorrhage, tissue ischemia, and necrosis (155, 156). Additionally, the failure of the graft is exacerbated by thrombotic occlusion of capillaries, fibrinoid necrosis in arterial walls, and the accumulation of neutrophils (157). The histopathological characteristics of hyperacute rejection (HAR) include compromised vascular integrity, edema, thrombi rich in fibrin and platelets, as well as interstitial hemorrhage accompanied by extensive deposition of immunoglobulins and terminal complement products on the walls of vessels (157). In order to reduce the occurrence of hyperacute rejection, the following measures can be taken: first, immunosuppressants mentioned in this paper are the main measures; knocking out the a1,3GT gene in pigs (GTKO pigs) (158).With the identification of carbohydrate xenoantigens (159) and advances in genetic engineering, it is possible to eliminate these xenoantigens (160) to prevent hyperacute rejection. However, T-cell-mediated xenogeneic immune responses are very intense and more challenging to control compared to immune responses against allogeneic antigens (161).
The xenogeneic T-cell response to porcine islets can be triggered through both direct and indirect antigen presentation (162). Once activated, T cells can mediate graft destruction through direct cytotoxicity (163) or by differentiating into cytokine-producing helper T cells that assist B cells in class switching and antibody production, or by activating innate cells such as macrophages and NK cells involved in xenotransplant rejection (164, 165). Th1 and Th2 cytokines, such as IFN-γ and IL-4, play significant roles in this process (166–168). It has been experimentally demonstrated that the infusion of carbodiimide-fixed donor splenocytes (ECDI-SP) can exert effective immunoregulatory effects through the silent clearance of apoptotic cells, effectively inducing donor-specific tolerance (169–172).
4.1 Early acute inflammatory response
Islet xenotransplantation represents a promising therapeutic alternative for treating type 1 diabetes. However, shortly after transplanting donor islets into the recipient, a robust innate immune response is triggered, including an IBMIR, which adversely affects the functionality of the islet transplant (153).
IBMIR is triggered by the xenogeneic contact between blood and islets, involving the activation of coagulation and complement systems, as well as complex interactions between leukocytes and platelets, which significantly impact the function and survival of xenografts, thereby adversely affecting the outcomes of islet xenotransplantation (17, 173) (Figure 3). Therefore, the following section explains the mechanisms of IBMIR components.
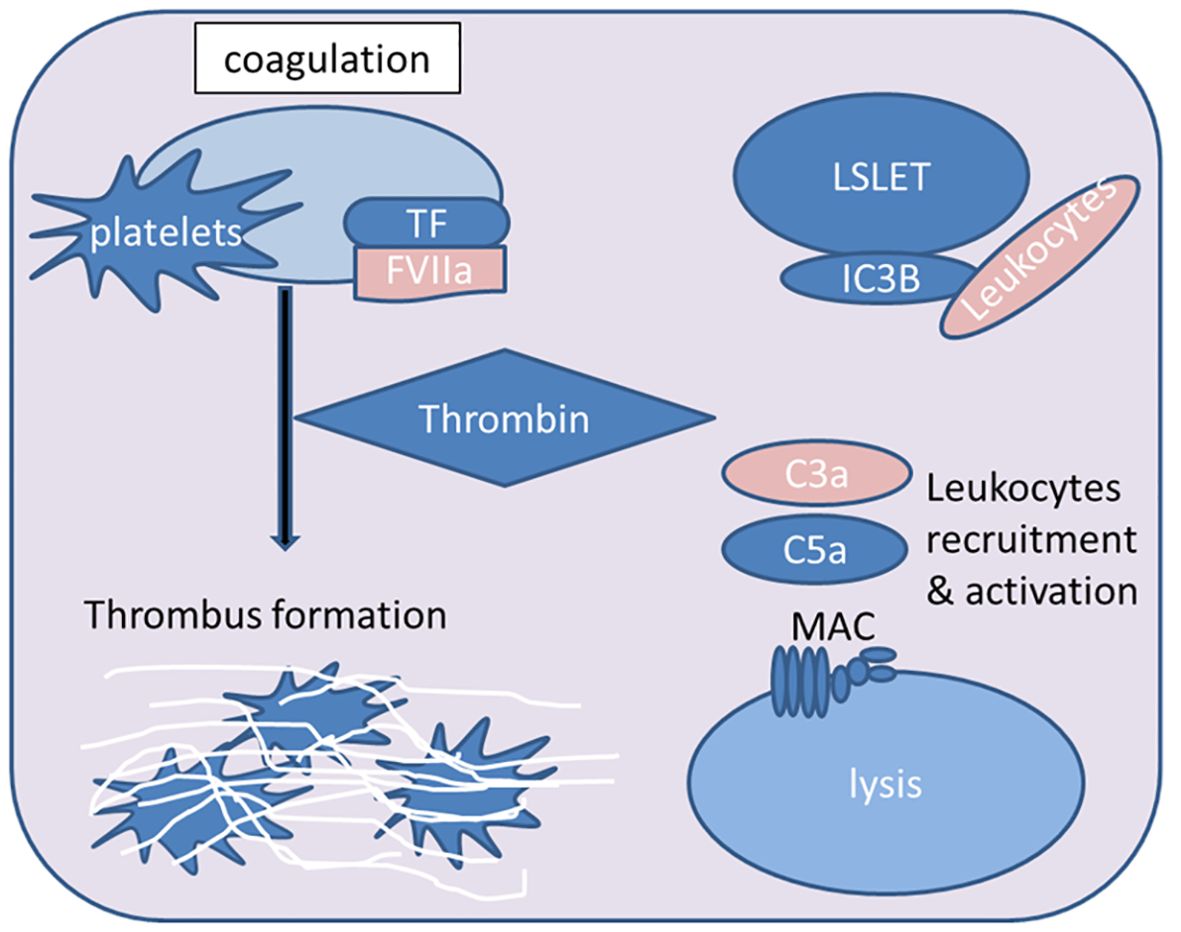
Figure 3. Overview of key steps in the IBMIR process during islet xenotransplantation. The contact between xenogeneic blood and islets triggers the activation of the extrinsic coagulation pathway mediated by tissue factor (TF). Consequently, downstream effector thrombin is produced, leading to fibrin deposition and thrombosis. The attachment of platelets to the islets further supports the pro-coagulant effect. Activated complement fragments (iC3b) deposit on the islet surface, and the anaphylatoxins C3a and C5a activate and attract leukocytes. The formation of the membrane attack complex (MAC) mediates islet lysis (FVIIa, activated coagulation factor VII; MAC, membrane attack complex).
Research for IBMIR found that platelet-independent complement activation was observed 30 minutes after porcine islets were exposed to plasma, and the formation of membrane attack complexes could be observed in porcine islet tissue pathology sections 60 minutes later, with up to 40% of islets losing their function (174). Complement system activation occurs through three different pathways (known as the classical pathway, lectin pathway, and alternative pathway), depending on the nature of the initial trigger. Regardless of the activation pathway, all pathways converge at the cleavage of C3 by C3 convertase. C3 convertase cleaves the central component C3 into the anaphylatoxins C3a and C3b (175), with the primary function of C3b and its cleavage product iC3b being opsonization for phagocytosis. Additionally, iC3b can bind to complement receptors CR3 and CR4, leading to immune cell adhesion and activation (176, 177). Since complement activation is associated with the proteolytic cleavage of its components, proteases represent another “non-traditional” pathway of complement activation (178, 179).
The classical pathway (CP) is triggered by antigen-antibody complexes recognized by C1q. A major process in this pathway is the production of CP C3 convertase C4b2b, generated by the cleavage of C4 into C4a and C4b, followed by the splitting of C2 into C2a and C2b (180). Activation of the lectin pathway (LP) is initiated by the binding of mannose-binding lectin (MBL) or ficolins to pathogen surfaces, involving the participation of MBL-associated serine proteases MASP-1 and MASP-2, which is significantly similar to CP activation (181).
The spontaneous hydrolysis of C3 to C3(H2O) accounts for the constitutive and continuous low-level activation of the alternative pathway (AP) (182). The generated C3b assembles the APC3 convertase C3bBb together with factor B and factor D (183). The APC3 convertase complex is stabilized by the binding of properdin (184–186).
In all three pathways, the cleavage of C3 to produce C3b is a major component of C5 convertase, which cleaves C5 into the anaphylatoxins C5a and C5b (187). C5b participates in the formation of the membrane attack complex (MAC) by recruiting complement components C6, C7, C8, and C9, with the primary function of mediating the lysis of pathogens or target cells (188).
On the other hand, C3a and C5a anaphylatoxins, by interacting with G-protein-coupled C3a and C5a receptors, are highly effective chemoattractants, promoting the recruitment of inflammatory cells to sites of injury or infection. Furthermore, C3a and C5a can activate immune cells, upregulating the expression and release of inflammatory cytokines and mediators (175, 189).
The coagulation cascade is involved in both hemostasis and thrombosis (190). The tissue factor of the so-called extrinsic pathway is a core participant in coagulation (191), involved in the pathology of thrombosis, including cardiovascular diseases (192, 193) and biomaterial-related processes (194). Inflammatory stimuli or endothelial cell activation produce the extrinsic factor X complex composed of TF and activated coagulation factor VII (FVIIa) (195). The extrinsic factor X complex, in turn, promotes the activation of factor X (FX), which, together with activated FVa and Ca2+, forms the prothrombinase complex that mediates the conversion of prothrombin to thrombin (196). Thrombin can activate platelets, cleave prothrombin into thrombin, leading to the formation of insoluble thrombin (197).
Coagulation and thrombosis are involved in acute reactions to both allogeneic and xenogeneic islet transplantation (18) (198). Notably, exposure of human or porcine islets to human blood results in rapid activation of coagulation, evidenced by upregulated TF levels (199) and significant thrombin generation (26). Moreover, allogeneic islet transplantation is associated with thrombotic manifestations, such as fibrin deposition and the localization of transplanted islets within thrombi (198). Therefore, endogenous antithrombotic agents are significant as potential beneficial modulators of IBMIR. The fine-tuning of the coagulation cascade (200) is mediated by antithrombin III (ATIII), which inactivates thrombin, FXa, and FIXa (201); activated protein C (APC), which, along with protein S, blocks FVa and FVIIIa (202); tissue factor pathway inhibitor (TFPI); and thrombomodulin (TM). TFPI binds and inhibits FXa or the TF/FVIIa complex (203). TM’s anticoagulant activity is mediated by binding to thrombin. The TM-thrombin complex further promotes the generation of APC (204). However, thrombin bound to TM can cleave and activate thrombin-activatable fibrinolysis inhibitor (TAFI) (205), conferring procoagulant properties by blocking fibrinolysis. In the context of xenogeneic islet transplantation, transgenic pigs overexpressing hemostasis-regulating molecules have been generated. For this purpose, the expression of hTFPI protected the xenografts, promoting the achievement of normoglycemia after xenotransplantation. Porcine TM has been shown to be a poor cofactor for human thrombin, resulting in the loss of its protective function and increased coagulation (206). Thus, transgenic overexpression of hTM in pigs can avoid the thrombotic manifestations observed after xenotransplantation of porcine islets (207).
The contact of host blood with transplanted islets rapidly triggers a series of thrombo-inflammatory responses, including upregulation of TF expression (199) and thrombin generation (26). Additionally, the induction of TAFI further propagates the procoagulant effect (208). Intravascular coagulation is induced (209), forming thrombi that capture the islets (198). Concurrently, activation of CP and AP of the complement system occurs, generating anaphylatoxins that lead to the recruitment of inflammatory cells to the graft. Moreover, active complement fragments deposit on the graft, promoting complement-dependent islet lysis (210). Platelets and leukocytes infiltrate the transplantation site and adhere to the islet surface (26, 211). Consequently, the integrity of the islet grafts is compromised, leading to substantial early loss of transplanted islets (212, 213). The acute destruction of a significant proportion of transplanted islets by IBMIR is the primary reason why a high number of islets are required for effective islet transplantation (214). Interestingly, the degree of islet damage increases with the decreasing compatibility between donor and recipient species. Therefore, in the case of xenogeneic islet transplantation, IBMIR becomes more relevant because the recipient cannot control IBMIR induced by xenogeneic islet transplantation due to incompatibility between regulators and effectors, respectively, for the IBMIR of xenografts and recipient cells (215). Furthermore, regulatory proteins are considerably lacking in porcine islet preparations (216). Thus, developing effective treatment regimens targeting the regulatory parameters of IBMIR is imperative (173) (Figure 3).
In further studies targeting IBMIR, Bennet et al. cultured isolated islets in whole blood in the presence of soluble CR1 (sCR1). They demonstrated that sCR1 treatment blocked complement activation associated with IBMIR and protected the islets from damage. Simultaneous inhibition with sCR1 and heparin eliminated the adverse effects of IBMIR by reducing the activation of coagulation, complement, and leukocytes. Interestingly, in vivo experiments confirmed the protective effect of sCR1, as the use of this inhibitor supported islet integrity, which could be evaluated by the reduction in insulin release shortly after transplantation (198).
Notably, isolated islets can act as a source of procoagulant factors. TF, the main trigger of coagulation in vivo, has been found in isolated islets (18, 199), and its knockout (217, 218) or inhibition with specific antibodies (219) has been shown to be beneficial in blocking IBMIR. Interestingly, nicotinamide (a vitamin B derivative) has been used to reduce the expression levels of TF and coagulation, thereby improving IBMIR (20), and leading to improved islet function after transplantation (220).
Islet xenografts can be considered as foreign biological surfaces, and exposure to recipient blood triggers a strong innate immune response. Therefore, an emerging strategy to eliminate the adverse effects of IBMIR is to coat the surface of isolated islets with inhibitory molecules, thereby locally inhibiting the coagulation and complement systems at the transplant site. A 14-patient Phase 1/2a study in New Zealand showed that neonatal porcine islets encapsulated with alginate-poly-L-ornithine-alginate (APA) were safe and reduced unawareness of hypoglycemia in patients with type 1 diabetes (221). Strategies such as donor-specific hematopoietic progenitor cell transplantation (mixed chimerism) and concomitant donor-specific thymus transplantation showed great promise for improving immune tolerance (221, 222).
4.2 Early acute rejection
Studies have shown that during early acute rejection of porcine islet xenografts, the rejecting host graft exhibits direct and indirect anti-donor T cell IL-17 responses and produces strong anti-pig antibodies with severe B cell infiltration (148). IL-17 produced by the early donor stimulus dominates the early acute rejection response rather than IFN-γ production. Treatment with porcine ECDI-SP inhibits the host anti-pig IL-17 response, and when combined with transient B cell depletion (such as anti-CD20 monoclonal antibody) and short-course sirolimus, this triple therapy significantly and durably suppresses the host anti-pig IL-17 response and significantly prolongs the survival time of porcine islet xenografts (223). During early acute rejection, B cells may help induce the differentiation of IL-17-producing T cells and the production of xenogeneic antibodies by plasma cells. Studies have shown that B cell antigens presented by B1 B cells can effectively promote Th17 differentiation (224–226). Conversely, Th17 cells are effective B cell helper cells that can induce B cell proliferation in vitro and trigger their class switching in vivo (227). It can be imagined that the induced xenogeneic IL-17 response feeds back to promote B cell proliferation and differentiation, establishing a positive feedback loop between B cells and Th17 cells, effectively promoting early acute rejection of islet xenografts.
4.3 Late rejection
In the context of late rejection initially protected by porcine ECDI-SP + anti-CD20 + sirolimus triple therapy, it was found that late rejection appeared to be entirely cell-mediated, as xenogeneic antibodies could not be detected after the rejection of the islet xenografts. Secondly, the phenomenon of late rejection seemed to always be associated with highly aggressive B cell infiltration in the graft. Thirdly, indirect xenogeneic IFN-γ responses appeared before the late rejection after B cell reconstitution (148). It can be imagined that newly emerged B cells directly acquire xenogeneic antigens in the graft and induce indirect anti-donor IFN-γ responses. Graft-infiltrating B cells may also directly initiate cytotoxic T lymphocytes within the graft, leading to the in situ destruction of the graft (228).
5 Strategies for treating the immune response to xenogeneic islet transplantation
5.1 Islet encapsulation
Islet encapsulation is an advanced method of islet transplantation, where isolated islets from humans or pigs can be transplanted without the need for toxic immunosuppression. This proves particularly beneficial for porcine islet xenotransplantation. Encapsulating islets with a semipermeable barrier allows for the exchange of nutrients and hormones, including insulin, while maintaining immune isolation, thus overcoming one of the major obstacles of xenotransplantation. Although clinical trials of porcine islets have achieved some success in New Zealand and Argentina, more research may be needed to develop optimal encapsulation methods and materials before this technology is ready for larger clinical trials in the United States. Key factors influencing encapsulation technology include:
1. Capsule size and material: Traditionally, smaller capsules are believed to be more effective due to easier material exchange through the capsule (229). However, recent studies suggest that spherical materials with diameters ≥1.5 mm exhibit significantly better biocompatibility compared to smaller or differently shaped counterparts (230). An in vivo study demonstrated that 1.5 mm alginate-encapsulated rat islets could restore blood glucose control in streptozotocin-induced diabetic C57 BL/6 mice for up to 180 days. This indicates that biocompatibility might be more crucial than material exchange efficiency in terms of effectiveness. Alginate consists of linear binary copolymers of β-D-mannuronic acid and α-L-guluronic acid. The length and sequence of mannuronic and guluronic acid chains in alginate hydrogels, as well as the mannuronic to guluronic acid ratio (M
ratio), determine alginate’s mechanical strength, elasticity, durability, permeability, and swelling properties. The use of multivalent cations (Ca²+, Ba²+) and polycations (poly-L-lysine or poly-L-ornithine) during alginate synthesis alters its properties (231). For example, alginate-poly-L-ornithine capsules provide high biocompatibility, better stability, and improved mechanical strength but induce excessive pericapsular cell overgrowth and macrophage activation, leading to capsule fibrosis. Multivalent cations like barium can avoid such fibrosis but reduce molar selectivity.
2. Transplantation site: Since the early 1970s, the liver infused via the portal vein has been widely accepted as the optimal site for islet transplantation in rodents. This principle, due to the prevalence and importance of rodent studies, has been extended to most animal models and nearly become the preferred site for microencapsulated islet transplantation (232). However, subsequent studies have identified several reasons why the liver is not the best site, including: (1) interaction with blood flow causing IBMIR, reducing islet mass by up to 50%; (2) the possibility of thrombosis during infusion; (3) relatively lower oxygen tension compared to the pancreas (233, 234). Intraperitoneal transplantation is also common, mainly due to its low volume limitation on grafts. However, this site has several drawbacks, including lack of close contact with blood flow, uncertain distribution of encapsulated islets, and the tendency for capsules to stack in the pelvic cavity of bipedal animals, making them difficult to retrieve. Subcapsular kidney transplantation is often used in animal models, considering the large number of encapsulated islets used for clinical transplantation and the lack of aggregation at this site. The subcutaneous space can be used for large numbers of encapsulated islets; however, this site is notoriously poor for blood access. Prevascularized subcutaneous spaces seem promising for both device and device-free methods (235), though this area requires two surgeries for prevascularization and actual transplantation. The omental pouch can also be used without two surgeries, potentially making it an ideal site for encapsulated islet implantation. In fact, studies using immunocompetent diabetic rat models have shown long-term function of encapsulated islets in the omental pouch (236). Researchers have also explored potential sites such as the gastric submucosa, peritoneal space, spleen, bone, and muscle. Animal models have identified specific advantages of several alternative sites, such as low blood contact to reduce IBMIR or the ability to biopsy the site after islet delivery, but to date, these positive results have been offset by equally compelling negative factors such as insufficient oxygen supply, surgical difficulty, or the need for more islets to correct blood glucose imbalance. Ongoing research may eventually yield a better site for islet infusion, though the liver remains the best choice for clinical islet transplantation despite its recognized limitations.
Porcine and microencapsulated islets have both been used clinically without significant side effects. However, compared to allogeneic naked islet transplantation, this method’s effectiveness is still suboptimal. Improving islet quality, enhancing capsule biocompatibility, and determining suitable implantation sites are crucial for the implementation of this therapy (237). Further research should make this method as effective as allogeneic naked islet transplantation, representing a real breakthrough in overcoming donor shortages and avoiding or mitigating the side effects associated with immunosuppressive drugs.
5.2 Immunosuppressants for xenotransplantation
In 1994, CG Groth reported the first xenotransplantation of porcine fetal islet-like cell clusters in a T1D patient. This study demonstrated the feasibility of porcine islet transplantation but did not show improvement in the patient’s condition (238). Over the following decades, porcine islet xenotransplantation has been more thoroughly explored in preclinical trials with non-human primates (239, 240). Humoral rejection is the main obstacle to the success of xenotransplantation. The α1,3Gal epitope, present on the surface of almost all animals except humans and some primates, is the primary antigen causing hyperacute rejection in pig-to-human and pig-to-non-human primate islet xenotransplantation. The University of Pittsburgh and Revivicor, Inc. designed Gal knockout (GTKO) pigs that do not express Gal (158). This proved to be a significant milestone in the development of xenotransplantation. Although other xenogeneic antigens were later discovered, Gal remains the most relevant, and GTKO pigs are considered a potential choice for eventual clinical translation. However, Gal knockout does not prevent islet rejection, and other genetic manipulations have been explored. In 2009, the Pittsburgh islet team first demonstrated long-term function (up to one year) of islet grafts in streptozotocin-induced diabetic non-human primates transplanted with porcine islets genetically modified to express human complement regulatory protein (hCD46). hCD46 expressed on porcine islets limited antibody-mediated rejection, allowing for the reduction of immunosuppression to maintain sufficient islet mass for long-term normal function. However, it did not reduce the initial islet loss associated with IBMIR as expected (241). This led to the further development of multigene pig islet donors capable of providing multifaceted protection to enhance islet transplantation. Five years later, the same group achieved similar success, long-term transplantation of islets from multigene pigs for the first time. A pig with four modified genes, (i) GTKO, and (ii) hCD46, (iii) human tissue factor pathway inhibitor (hTFPI) for antithrombotic and anti-inflammatory effects, and (iv) CTLA4-Ig to inhibit cellular immune responses, demonstrated improved success rates in retaining islet mass early postoperatively and maintaining islet implantation and function for up to one year during transplantation (242). This study also provided preliminary insights into glucose metabolism in pigs expressing human genes regulated by the insulin promoter, demonstrating that multiple islet-targeting transgenes inserted into pigs were not harmful to islet function and opened the door to further experiments and genetic manipulation for islet xenotransplantation (243). Multigene donor pigs have been shown to be a reproducibly effective source of islets for pig-to-non-human primate xenotransplantation (209). CG Park and colleagues at Seoul National University in Korea are conducting ongoing research of great significance for islet xenotransplantation, successfully maintaining normal blood glucose levels in diabetic primates within 600 days post porcine islet transplantation (244). A common feature of these successful long-term porcine-to-non-human primate islet studies is the use of CD154 monoclonal antibody (mAb)-based immunosuppression to prevent rejection. Although there is evidence that anti-CD154 mAb is effective and safe in pig-to-non-human primate islet transplantation models (245), it is associated with thromboembolic complications in humans and is not clinically translatable.
Despite promising data on the use of anti-CD40 antibody (co-stimulatory blockade) in organ xenotransplantation (246, 247), the islet xenotransplantation community is still searching for a clinically translatable immunosuppressant that can successfully prevent rejection without causing excessive side effects. New techniques for targeted genomic editing, particularly clustered regularly interspaced short palindromic repeats (CRISPR)-associated protein-9 nuclease (Cas9), offer hope that further genetic manipulation of porcine islets can improve compatibility between host and donor, thus allowing successful control of rejection with previously unfeasible immunosuppression. The field of islet xenotransplantation is steadily advancing and may soon approach clinical-grade experience and technology to begin clinical trials (248).
6 Conclusion and perspectives
6.1 Affirming islet transplantation
Islet transplantation holds great promise for the treatment of T1DM, as it offers the potential to restore euglycemia in a reliable manner, protects against hypoglycemia and glycemic liability in a way that exogenous insulin administration has thus far been unable to achieve. It has reduced many complications of diabetes and greatly improved patient healing.
6.2 Role and limitations of immunosuppressants
Limited islet survival after implantation hinders the success of IT due to innate immune attack through IBMIR, recurrent autoimmune islet destruction, or alloimmune rejection. The need for lifelong immunosuppressive therapy and the attendant risks of infection, cancer, and nephrotoxicity pose their own unique additional challenges, making this treatment unattractive to all but those at risk of severe brittle hypoglycemia. Optimizing new blood vessel formation by better controlling angiogenesis, suppressing inflammation, and reducing oxidative stress can all further improve outcomes.
6.3 Challenges and difficulties
The number of islets available for transplantation is a major limitation for both autoislet and alloislet approaches to β-cell replacement therapy. Therefore, the establishment of an unlimited source of islet tissue for transplantation has been a long-sought-after goal.
6.4 Future research directions
Stem cells: Significant progress has been made in the science and application of pluripotent stem cells, which are now entering early-stage pilot clinical trials. The possibility that cell transplantation can be accomplished with less need for immunosuppressants remains a real possibility, and progress is being made in immunomodulatory control through Treg infusion, MSC co-transplantation, and other innovative approaches.
Porcine islet xenotransplantation: Porcine islets have the advantage of targeting normal insulin similar to that present in humans, as well as the physiological ability to handle the heavy demands of insulin secretion. Importantly, porcine IAPP contains amino acid substitutions in the region corresponding to residues 20 to 29 that prevent the formation of fibrils (249, 250). Disadvantages include the larger immunologic barrier of xenogeneic than allogeneic tissue that presents an additional risk for hyperacute rejection and requires more intensive immunosuppression (239, 240),
Islet encapsulation technology: Islet encapsulation provides a barrier to protect transplanted islets, mainly by preventing excessive fibrosis, promoting local vascularization, and preventing future chronic immunosuppressive rejection. The latest data from NOD mice appear to confirm that agarose microencapsulated islets protect against autoimmune reactions (251). A recent paper published in PNAS shows that large encapsulated islets placed in the omentum protect grafts from immune attack and improve glucose metabolic control (252). These data demonstrate the potential of this technique as a safe method for successful islet transplantation.
Author contributions
LY: Writing – original draft. JL: Writing – original draft. MY: Writing – original draft. SS: Writing – review & editing, Validation. XZ: Writing – review & editing, Supervision, Funding acquisition. YW: Writing – review & editing, Validation, Supervision, Funding acquisition, Conceptualization.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This study was supported by the National Natural Science Foundation of China (81802504), Department of Sichuan Science and Technology of China (2024YFH0234).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
2. Batra L, Shrestha P, Zhao H, Woodward KB, Togay A, Tan M, et al. Localized immunomodulation with PD-L1 results in sustained survival and function of allogeneic islets without chronic immunosuppression. J Immunol. (2020) 204:2840–51. doi: 10.4049/jimmunol.2000055
3. DeWitt DE, Hirsch IB. Outpatient insulin therapy in type 1 and type 2 diabetes mellitus: scientific review. Jama. (2003) 289:2254–64. doi: 10.1001/jama.289.17.2254
4. Chen QD, Liu L, Zhao XH, Liang JB, Li SW. Challenges and opportunities in the islet transplantation microenvironment: a comprehensive summary of inflammatory cytokine, immune cells, and vascular endothelial cells. Front Immunol. (2023) 14:1293762. doi: 10.3389/fimmu.2023.1293762
5. Pambianco G, Costacou T, Ellis D, Becker DJ, Klein R, Orchard TJ. The 30-year natural history of type 1 diabetes complications: the Pittsburgh Epidemiology of Diabetes Complications Study experience. Diabetes. (2006) 55:1463–9. doi: 10.2337/db05-1423
6. Kandaswamy R, Skeans MA, Gustafson SK, Carrico RJ, Prentice MA, Israni AK, et al. Pancreas. Am J Transplant. (2016) 16 (Suppl 2):47–68. doi: 10.1111/ajt.13667
7. Sutherland DE, Gruessner RW, Dunn DL, Matas AJ, Humar A, Kandaswamy R, et al. Lessons learned from more than 1,000 pancreas transplants at a single institution. Ann Surg. (2001) 233:463–501. doi: 10.1097/00000658-200104000-00003
8. Meloche RM. Transplantation for the treatment of type 1 diabetes. World J Gastroenterol. (2007) 13:6347–55. doi: 10.3748/wjg.v13.i47.6347
9. Gibly RF, Graham JG, Luo X, Lowe WL Jr., Hering BJ, Shea LD. Advancing islet transplantation: from engraftment to the immune response. Diabetologia. (2011) 54:2494–505. doi: 10.1007/s00125-011-2243-0
10. Kanak MA, Takita M, Kunnathodi F, Lawrence MC, Levy MF, Naziruddin B. Inflammatory response in islet transplantation. Int J Endocrinol. (2014) 2014:451035. doi: 10.1155/2014/451035
11. Sutherland DE, Gores PF, Farney AC, Wahoff DC, Matas AJ, Dunn DL, et al. Evolution of kidney, pancreas, and islet transplantation for patients with diabetes at the University of Minnesota. Am J Surg. (1993) 166:456–91. doi: 10.1016/S0002-9610(05)81142-0
12. Shapiro AM, Lakey JR, Ryan EA, Korbutt GS, Toth E, Warnock GL, et al. Islet transplantation in seven patients with type 1 diabetes mellitus using a glucocorticoid-free immunosuppressive regimen. N Engl J Med. (2000) 343:230–8. doi: 10.1056/NEJM200007273430401
13. Hirshberg B, Rother KI, Digon BJ 3rd, Lee J, Gaglia JL, Hines K, et al. Benefits and risks of solitary islet transplantation for type 1 diabetes using steroid-sparing immunosuppression: the National Institutes of Health experience. Diabetes Care. (2003) 26:3288–95. doi: 10.2337/diacare.26.12.3288
14. Rother KI, Harlan DM. Challenges facing islet transplantation for the treatment of type 1 diabetes mellitus. J Clin Invest. (2004) 114:877–83. doi: 10.1172/JCI200423235
15. Shapiro AM, Pokrywczynska M, Ricordi C. Clinical pancreatic islet transplantation. Nat Rev Endocrinol. (2017) 13:268–77. doi: 10.1038/nrendo.2016.178
16. Ricordi C. Islet transplantation: a brave new world. Diabetes. (2003) 52:1595–603. doi: 10.2337/diabetes.52.7.1595
17. Bennet W, Groth CG, Larsson R, Nilsson B, Korsgren O. Isolated human islets trigger an instant blood mediated inflammatory reaction: implications for intraportal islet transplantation as a treatment for patients with type 1 diabetes. Ups J Med Sci. (2000) 105:125–33. doi: 10.1517/03009734000000059
18. Moberg L, Johansson H, Lukinius A, Berne C, Foss A, Källen R, et al. Production of tissue factor by pancreatic islet cells as a trigger of detrimental thrombotic reactions in clinical islet transplantation. Lancet. (2002) 360:2039–45. doi: 10.1016/S0140-6736(02)12020-4
19. Delaune V, Berney T, Lacotte S, Toso C. Intraportal islet transplantation: the impact of the liver microenvironment. Transpl Int. (2017) 30:227–38. doi: 10.1111/tri.2017.30.issue-3
20. Moberg L, Olsson A, Berne C, Felldin M, Foss A, Källen R, et al. Nicotinamide inhibits tissue factor expression in isolated human pancreatic islets: implications for clinical islet transplantation. Transplantation. (2003) 76:1285–8. doi: 10.1097/01.TP.0000098905.86445.0F
21. Langer HF, Chavakis T. Leukocyte-endothelial interactions in inflammation. J Cell Mol Med. (2009) 13:1211–20. doi: 10.1111/j.1582-4934.2009.00811.x
22. Purdy M, Obi A, Myers D, Wakefield T. P- and E- selectin in venous thrombosis and non-venous pathologies. J Thromb Haemost. (2022) 20:1056–66. doi: 10.1111/jth.15689
23. Yan LL, Ye LP, Chen YH, He SQ, Zhang CY, Mao XL, et al. The influence of microenvironment on survival of intraportal transplanted islets. Front Immunol. (2022) 13:849580. doi: 10.3389/fimmu.2022.849580
24. Vivot K, Benahmed MA, Seyfritz E, Bietiger W, Elbayed K, Ruhland E, et al. A metabolomic approach ((1)H HRMAS NMR spectroscopy) supported by histology to study early post-transplantation responses in islet-transplanted livers. Int J Biol Sci. (2016) 12:1168–80. doi: 10.7150/ijbs.15189
25. Sironi JJ, Ouchi T. STAT1-induced apoptosis is mediated by caspases 2, 3, and 7. J Biol Chem. (2004) 279:4066–74. doi: 10.1074/jbc.M307774200
26. Ozmen L, Ekdahl KN, Elgue G, Larsson R, Korsgren O, Nilsson B. Inhibition of thrombin abrogates the instant blood-mediated inflammatory reaction triggered by isolated human islets: possible application of the thrombin inhibitor melagatran in clinical islet transplantation. Diabetes. (2002) 51:1779–84. doi: 10.2337/diabetes.51.6.1779
27. Chen D, Zhang N, Fu S, Schröppel B, Guo Q, Garin A, et al. CD4+ CD25+ regulatory T-cells inhibit the islet innate immune response and promote islet engraftment. Diabetes. (2006) 55:1011–21. doi: 10.2337/diabetes.55.04.06.db05-1048
28. Marfil-Garza BA, Hefler J, Verhoeff K, Lam A, Dajani K, Anderson B, et al. Pancreas and islet transplantation: comparative outcome analysis of a single-centre cohort over 20-years. Ann Surg. (2023) 277:672–80. doi: 10.1097/SLA.0000000000005783
29. Rodriguez-Calvo T, Ekwall O, Amirian N, Zapardiel-Gonzalo J, von Herrath MG. Increased immune cell infiltration of the exocrine pancreas: a possible contribution to the pathogenesis of type 1 diabetes. Diabetes. (2014) 63:3880–90. doi: 10.2337/db14-0549
30. Coppieters KT, Dotta F, Amirian N, Campbell PD, Kay TW, Atkinson MA, et al. Demonstration of islet-autoreactive CD8 T cells in insulitic lesions from recent onset and long-term type 1 diabetes patients. J Exp Med. (2012) 209:51–60. doi: 10.1084/jem.20111187
31. Babon JA, DeNicola ME, Blodgett DM, Crèvecoeur I, Buttrick TS, Maehr R, et al. Analysis of self-antigen specificity of islet-infiltrating T cells from human donors with type 1 diabetes. Nat Med. (2016) 22:1482–7. doi: 10.1038/nm.4203
32. Duneton C, Winterberg PD, Ford ML. Activation and regulation of alloreactive T cell immunity in solid organ transplantation. Nat Rev Nephrol. (2022) 18:663–76. doi: 10.1038/s41581-022-00600-0
33. Halloran PF, Einecke G, Sikosana MLN, Madill-Thomsen K. The biology and molecular basis of organ transplant rejection. Handb Exp Pharmacol. (2022) 272:1–26. doi: 10.1007/164_2021_557
34. Li J, Li C, Zhuang Q, Peng B, Zhu Y, Ye Q, et al. The evolving roles of macrophages in organ transplantation. J Immunol Res. (2019) 2019:5763430. doi: 10.1155/2019/5763430
35. Ronca V, Wootton G, Milani C, Cain O. The immunological basis of liver allograft rejection. Front Immunol. (2020) 11:2155. doi: 10.3389/fimmu.2020.02155
36. Agarwal D, Allman D, Naji A. Novel therapeutic opportunities afforded by plasma cell biology in transplantation. Am J Transplant. (2020) 20:1984–91. doi: 10.1111/ajt.15813
37. Ho EK, Vlad G, Vasilescu ER, de la Torre L, Colovai AI, Burke E, et al. Pre- and posttransplantation allosensitization in heart allograft recipients: major impact of de novo alloantibody production on allograft survival. Hum Immunol. (2011) 72:5–10. doi: 10.1016/j.humimm.2010.10.013
38. Loupy A, Hill GS, Jordan SC. The impact of donor-specific anti-HLA antibodies on late kidney allograft failure. Nat Rev Nephrol. (2012) 8:348–57. doi: 10.1038/nrneph.2012.81
39. Morrell MR, Pilewski JM, Gries CJ, Pipeling MR, Crespo MM, Ensor CR, et al. De novo donor-specific HLA antibodies are associated with early and high-grade bronchiolitis obliterans syndrome and death after lung transplantation. J Heart Lung Transplant. (2014) 33:1288–94. doi: 10.1016/j.healun.2014.07.018
40. Le Moine A, Goldman M. Non-classical pathways of cell-mediated allograft rejection: new challenges for tolerance induction? Am J Transplant. (2003) 3:101–6. doi: 10.1034/j.1600-6143.2002.00026.x
41. Plenter RJ, Grazia TJ, Doan AN, Gill RG, Pietra BA. CD4 T cells mediate cardiac xenograft rejection via host MHC Class II. J Heart Lung Transplant. (2012) 31:1018–24. doi: 10.1016/j.healun.2012.05.018
42. Reffet S, Thivolet C. Immunology of pancreatic islet transplantation. Diabetes Metab. (2006) 32:523–6. doi: 10.1016/S1262-3636(06)72805-1
43. Regnell SE, Lernmark Å. Early prediction of autoimmune (type 1) diabetes. Diabetologia. (2017) 60:1370–81. doi: 10.1007/s00125-017-4308-1
44. Sun JK, Keenan HA, Cavallerano JD, Asztalos BF, Schaefer EJ, Sell DR, et al. Protection from retinopathy and other complications in patients with type 1 diabetes of extreme duration: the joslin 50-year medalist study. Diabetes Care. (2011) 34:968–74. doi: 10.2337/dc10-1675
45. Soeldner JS, Tuttleman M, Srikanta S, Ganda OP, Eisenbarth GS. Insulin-dependent diabetes mellitus and autoimmunity: islet-cell autoantibodies, insulin autoantibodies, and beta-cell failure. N Engl J Med. (1985) 313:893–4. doi: 10.1056/NEJM198510033131417
46. Eisenbarth GS. Type I diabetes mellitus. A chronic autoimmune disease. N Engl J Med. (1986) 314:1360–8. doi: 10.1056/NEJM198605223142106
47. Zhang L, Gianani R, Nakayama M, Liu E, Kobayashi M, Baschal E, et al. Type 1 diabetes: chronic progressive autoimmune disease. Novartis Found Symp. (2008) 292:85–94. doi: 10.1002/9780470697405.ch7
48. Sutherland DE, Goetz FC, Sibley RK. Recurrence of disease in pancreas transplants. Diabetes. (1989) 38 (Suppl 1):85–7. doi: 10.2337/diab.38.1.s85
49. Sarikonda G, Pettus J, Phatak S, Sachithanantham S, Miller JF, Wesley JD, et al. CD8 T-cell reactivity to islet antigens is unique to type 1 while CD4 T-cell reactivity exists in both type 1 and type 2 diabetes. J Autoimmun. (2014) 50:77–82. doi: 10.1016/j.jaut.2013.12.003
50. Rodriguez-Calvo T, Suwandi JS, Amirian N, Zapardiel-Gonzalo J, Anquetil F, Sabouri S, et al. Heterogeneity and lobularity of pancreatic pathology in type 1 diabetes during the prediabetic phase. J Histochem Cytochem. (2015) 63:626–36. doi: 10.1369/0022155415576543
51. Sibley RK, Sutherland DE, Goetz F, Michael AF. Recurrent diabetes mellitus in the pancreas iso- and allograft. A light and electron microscopic and immunohistochemical analysis of four cases. Lab Invest. (1985) 53:132–44.
52. Sutherland DE, Sibley R, Xu XZ, Michael A, Srikanta AM, Taub F, et al. Twin-to-twin pancreas transplantation: reversal and reenactment of the pathogenesis of type I diabetes. Trans Assoc Am Physicians. (1984) 97:80–7.
53. Baxter AG. Immunogenetics and the cause of autoimmune disease. Autoimmunity. (1997) 25:177–89. doi: 10.3109/08916939709008024
54. Bach JF, Chatenoud L. Tolerance to islet autoantigens in type 1 diabetes. Annu Rev Immunol. (2001) 19:131–61. doi: 10.1146/annurev.immunol.19.1.131
55. Thivolet C, Abou-Amara S, Martin X, Lefrancois N, Petruzzo P, McGregor B, et al. Serological markers of recurrent beta cell destruction in diabetic patients undergoing pancreatic transplantation. Transplantation. (2000) 69:99–103. doi: 10.1097/00007890-200001150-00018
56. Pugliese A, Reijonen HK, Nepom J, Burke GW 3rd. Recurrence of autoimmunity in pancreas transplant patients: research update. Diabetes Manag (Lond). (2011) 1:229–38. doi: 10.2217/dmt.10.21
57. Laughlin E, Burke G, Pugliese A, Falk B, Nepom G. Recurrence of autoreactive antigen-specific CD4+ T cells in autoimmune diabetes after pancreas transplantation. Clin Immunol. (2008) 128:23–30. doi: 10.1016/j.clim.2008.03.459
58. Vendrame F, Pileggi A, Laughlin E, Allende G, Martin-Pagola A, Molano RD, et al. Recurrence of type 1 diabetes after simultaneous pancreas-kidney transplantation, despite immunosuppression, is associated with autoantibodies and pathogenic autoreactive CD4 T-cells. Diabetes. (2010) 59:947–57. doi: 10.2337/db09-0498
59. Di Lorenzo TP, Peakman M, Roep BO. Translational mini-review series on type 1 diabetes: Systematic analysis of T cell epitopes in autoimmune diabetes. Clin Exp Immunol. (2007) 148:1–16. doi: 10.1111/j.1365-2249.2006.03244.x
60. Braghi S, Bonifacio E, Secchi A, Di Carlo V, Pozza G, Bosi E. Modulation of humoral islet autoimmunity by pancreas allotransplantation influences allograft outcome in patients with type 1 diabetes. Diabetes. (2000) 49:218–24. doi: 10.2337/diabetes.49.2.218
61. Alejandro R, Lehmann R, Ricordi C, Kenyon NS, Angelico MC, Burke G, et al. Long-term function (6 years) of islet allografts in type 1 diabetes. Diabetes. (1997) 46:1983–9. doi: 10.2337/diab.46.12.1983
62. Delaunay F, Khan A, Cintra A, Davani B, Ling ZC, Andersson A, et al. Pancreatic beta cells are important targets for the diabetogenic effects of glucocorticoids. J Clin Invest. (1997) 100:2094–8. doi: 10.1172/JCI119743
63. Drachenberg CB, Klassen DK, Weir MR, Wiland A, Fink JC, Bartlett ST, et al. Islet cell damage associated with tacrolimus and cyclosporine: morphological features in pancreas allograft biopsies and clinical correlation. Transplantation. (1999) 68:396–402. doi: 10.1097/00007890-199908150-00012
64. Kahan BD, Koch SM. Current immunosuppressant regimens: considerations for critical care. Curr Opin Crit Care. (2001) 7:242–50. doi: 10.1097/00075198-200108000-00006
65. Yale JF, Roy RD, Grose M, Seemayer TA, Murphy GF, Marliss EB. Effects of cyclosporine on glucose tolerance in the rat. Diabetes. (1985) 34:1309–13. doi: 10.2337/diab.34.12.1309
66. Nanji SA, Shapiro AM. Islet transplantation in patients with diabetes mellitus: choice of immunosuppression. BioDrugs. (2004) 18:315–28. doi: 10.2165/00063030-200418050-00004
67. Ochiai T, Nagata M, Nakajima K, Suzuki T, Sakamoto K, Enomoto K, et al. Studies of the effects of FK506 on renal allografting in the beagle dog. Transplantation. (1987) 44:729–33. doi: 10.1097/00007890-198712000-00001
68. Ochiai T, Nakajima K, Nagata M, Suzuki T, Asano T, Uematsu T, et al. Effect of a new immunosuppressive agent, FK 506, on heterotopic cardiac allotransplantation in the rat. Transplant Proc. (1987) 19:1284–6.
69. Sabatini DM, Erdjument-Bromage H, Lui M, Tempst P, Snyder SH. RAFT1: a mammalian protein that binds to FKBP12 in a rapamycin-dependent fashion and is homologous to yeast TORs. Cell. (1994) 78:35–43. doi: 10.1016/0092-8674(94)90570-3
70. Thomson AW, Turnquist HR, Raimondi G. Immunoregulatory functions of mTOR inhibition. Nat Rev Immunol. (2009) 9:324–37. doi: 10.1038/nri2546
71. Barlow AD, Nicholson ML, Herbert TP. Evidence for rapamycin toxicity in pancreatic β-cells and a review of the underlying molecular mechanisms. Diabetes. (2013) 62:2674–82. doi: 10.2337/db13-0106
72. Hara K, Maruki Y, Long X, Yoshino K, Oshiro N, Hidayat S, et al. Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell. (2002) 110:177–89. doi: 10.1016/S0092-8674(02)00833-4
73. Kim DH, Sarbassov DD, Ali SM, King JE, Latek RR, Erdjument-Bromage H, et al. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell. (2002) 110:163–75. doi: 10.1016/S0092-8674(02)00808-5
74. Jacinto E, Loewith R, Schmidt A, Lin S, Rüegg MA, Hall A, et al. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat Cell Biol. (2004) 6:1122–8. doi: 10.1038/ncb1183
75. Sarbassov DD, Ali SM, Kim DH, Guertin DA, Latek RR, Erdjument-Bromage H, et al. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr Biol. (2004) 14:1296–302. doi: 10.1016/j.cub.2004.06.054
76. Barlow AD, Xie J, Moore CE, Campbell SC, Shaw JA, Nicholson ML, et al. Rapamycin toxicity in MIN6 cells and rat and human islets is mediated by the inhibition of mTOR complex 2 (mTORC2). Diabetologia. (2012) 55:1355–65. doi: 10.1007/s00125-012-2475-7
77. Zeng Z, Sarbassov dos D, Samudio IJ, Yee KW, Munsell MF, Ellen Jackson C, et al. Rapamycin derivatives reduce mTORC2 signaling and inhibit AKT activation in AML. Blood. (2007) 109:3509–12. doi: 10.1182/blood-2006-06-030833
78. Sarbassov DD, Ali SM, Sengupta S, Sheen JH, Hsu PP, Bagley AF, et al. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell. (2006) 22:159–68. doi: 10.1016/j.molcel.2006.03.029
79. Xie J, Herbert TP. The role of mammalian target of rapamycin (mTOR) in the regulation of pancreatic β-cell mass: implications in the development of type-2 diabetes. Cell Mol Life Sci. (2012) 69:1289–304. doi: 10.1007/s00018-011-0874-4
80. Hamada S, Hara K, Hamada T, Yasuda H, Moriyama H, Nakayama R, et al. Upregulation of the mammalian target of rapamycin complex 1 pathway by Ras homolog enriched in brain in pancreatic beta-cells leads to increased beta-cell mass and prevention of hyperglycemia. Diabetes. (2009) 58:1321–32. doi: 10.2337/db08-0519
81. Mori H, Inoki K, Opland D, Münzberg H, Villanueva EC, Faouzi M, et al. Critical roles for the TSC-mTOR pathway in β-cell function. Am J Physiol Endocrinol Metab. (2009) 297:E1013–22. doi: 10.1152/ajpendo.00262.2009
82. Shigeyama Y, Kobayashi T, Kido Y, Hashimoto N, Asahara S, Matsuda T, et al. Biphasic response of pancreatic beta-cell mass to ablation of tuberous sclerosis complex 2 in mice. Mol Cell Biol. (2008) 28:2971–9. doi: 10.1128/MCB.01695-07
83. Pende M, Kozma SC, Jaquet M, Oorschot V, Burcelin R, Le Marchand-Brustel Y, et al. Hypoinsulinaemia, glucose intolerance and diminished beta-cell size in S6K1-deficient mice. Nature. (2000) 408:994–7. doi: 10.1038/35050135
84. Ruvinsky I, Sharon N, Lerer T, Cohen H, Stolovich-Rain M, Nir T, et al. Ribosomal protein S6 phosphorylation is a determinant of cell size and glucose homeostasis. Genes Dev. (2005) 19:2199–211. doi: 10.1101/gad.351605
85. Elghazi L, Balcazar N, Blandino-Rosano M, Cras-Méneur C, Fatrai S, Gould AP, et al. Decreased IRS signaling impairs beta-cell cycle progression and survival in transgenic mice overexpressing S6K in beta-cells. Diabetes. (2010) 59:2390–9. doi: 10.2337/db09-0851
86. Balcazar N, Sathyamurthy A, Elghazi L, Gould A, Weiss A, Shiojima I, et al. mTORC1 activation regulates beta-cell mass and proliferation by modulation of cyclin D2 synthesis and stability. J Biol Chem. (2009) 284:7832–42. doi: 10.1074/jbc.M807458200
87. Niclauss N, Bosco D, Morel P, Giovannoni L, Berney T, Parnaud G. Rapamycin impairs proliferation of transplanted islet β cells. Transplantation. (2011) 91:714–22. doi: 10.1097/TP.0b013e31820c10c8
88. Shimodahira M, Fujimoto S, Mukai E, Nakamura Y, Nishi Y, Sasaki M, et al. Rapamycin impairs metabolism-secretion coupling in rat pancreatic islets by suppressing carbohydrate metabolism. J Endocrinol. (2010) 204:37–46. doi: 10.1677/JOE-09-0216
89. Maechler P, Wollheim CB. Mitochondrial function in normal and diabetic beta-cells. Nature. (2001) 414:807–12. doi: 10.1038/414807a
90. Gan X, Wang J, Su B, Wu D. Evidence for direct activation of mTORC2 kinase activity by phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem. (2011) 286:10998–1002. doi: 10.1074/jbc.M110.195016
91. Tato I, Bartrons R, Ventura F, Rosa JL. Amino acids activate mammalian target of rapamycin complex 2 (mTORC2) via PI3K/Akt signaling. J Biol Chem. (2011) 286:6128–42. doi: 10.1074/jbc.M110.166991
92. Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. (2005) 307:1098–101. doi: 10.1126/science.1106148
93. García-Martínez JM, Alessi DR. mTOR complex 2 (mTORC2) controls hydrophobic motif phosphorylation and activation of serum- and glucocorticoid-induced protein kinase 1 (SGK1). Biochem J. (2008) 416:375–85. doi: 10.1042/BJ20081668
94. Diakov A, Korbmacher C. A novel pathway of epithelial sodium channel activation involves a serum- and glucocorticoid-inducible kinase consensus motif in the C terminus of the channel's alpha-subunit. J Biol Chem. (2004) 279:38134–42. doi: 10.1074/jbc.M403260200
95. Gu Y, Lindner J, Kumar A, Yuan W, Magnuson MA. Rictor/mTORC2 is essential for maintaining a balance between beta-cell proliferation and cell size. Diabetes. (2011) 60:827–37. doi: 10.2337/db10-1194
96. Bernal-Mizrachi E, Wen W, Stahlhut S, Welling CM, Permutt MA. Islet beta cell expression of constitutively active Akt1/PKB alpha induces striking hypertrophy, hyperplasia, and hyperinsulinemia. J Clin Invest. (2001) 108:1631–8. doi: 10.1172/JCI200113785
97. Tuttle RL, Gill NS, Pugh W, Lee JP, Koeberlein B, Furth EE, et al. Regulation of pancreatic beta-cell growth and survival by the serine/threonine protein kinase Akt1/PKBalpha. Nat Med. (2001) 7:1133–7. doi: 10.1038/nm1001-1133
98. Wrede CE, Dickson LM, Lingohr MK, Briaud I, Rhodes CJ. Protein kinase B/Akt prevents fatty acid-induced apoptosis in pancreatic beta-cells (INS-1). J Biol Chem. (2002) 277:49676–84. doi: 10.1074/jbc.M208756200
99. Collier JJ, Fueger PT, Hohmeier HE, Newgard CB. Pro- and antiapoptotic proteins regulate apoptosis but do not protect against cytokine-mediated cytotoxicity in rat islets and beta-cell lines. Diabetes. (2006) 55:1398–406. doi: 10.2337/db05-1000
100. Cai Y, Wang Q, Ling Z, Pipeleers D, McDermott P, Pende M, et al. Akt activation protects pancreatic beta cells from AMPK-mediated death through stimulation of mTOR. Biochem Pharmacol. (2008) 75:1981–93. doi: 10.1016/j.bcp.2008.02.019
101. Xu C, Niu YJ, Liu XJ, Teng YQ, Li CF, Wang HY, et al. Tacrolimus reversibly reduces insulin secretion, induces insulin resistance, and causes islet cell damage in rats. Int J Clin Pharmacol Ther. (2014) 52:620–7. doi: 10.5414/CP202090
102. Dai C, Walker JT, Shostak A, Padgett A, Spears E, Wisniewski S, et al. Tacrolimus- and sirolimus-induced human β cell dysfunction is reversible and preventable. JCI Insight. (2020) 5(1):e130770. doi: 10.1172/jci.insight.130770
103. Pereira MJ, Palming J, Rizell M, Aureliano M, Carvalho E, Svensson MK, et al. and tacrolimus reduce the amount of GLUT4 at the cell surface in human adipocytes: increased endocytosis as a potential mechanism for the diabetogenic effects of immunosuppressive agents. J Clin Endocrinol Metab. (2014) 99:E1885–94. doi: 10.1210/jc.2014-1266
104. Lombardi A, Trimarco B, Iaccarino G, Santulli G. Impaired mitochondrial calcium uptake caused by tacrolimus underlies beta-cell failure. Cell Commun Signal. (2017) 15:47. doi: 10.1186/s12964-017-0203-0
105. Okamoto H, Hribal ML, Lin HV, Bennett WR, Ward A, Accili D. Role of the forkhead protein FoxO1 in beta cell compensation to insulin resistance. J Clin Invest. (2006) 116:775–82. doi: 10.1172/JCI24967
106. Triñanes J, Rodriguez-Rodriguez AE, Brito-Casillas Y, Wagner A, De Vries APJ, Cuesto G, et al. Deciphering tacrolimus-induced toxicity in pancreatic β Cells. Am J Transplant. (2017) 17:2829–40. doi: 10.1111/ajt.14323
107. Ekberg H, Tedesco-Silva H, Demirbas A, Vítko S, Nashan B, Gürkan A, et al. Reduced exposure to calcineurin inhibitors in renal transplantation. N Engl J Med. (2007) 357:2562–75. doi: 10.1056/NEJMoa067411
108. Vincenti F, Friman S, Scheuermann E, Rostaing L, Jenssen T, Campistol JM, et al. Results of an international, randomized trial comparing glucose metabolism disorders and outcome with cyclosporine versus tacrolimus. Am J Transplant. (2007) 7:1506–14. doi: 10.1111/j.1600-6143.2007.01749.x
109. Kale A, Rogers NM. No time to die-how islets meet their demise in transplantation. Cells. (2023) 12(5):796. doi: 10.3390/cells12050796
110. Liu B, Jiang Q, Chen R, Gao S, Xia Q, Zhu J, et al. Tacrolimus ameliorates bleomycin-induced pulmonary fibrosis by inhibiting M2 macrophage polarization via JAK2/STAT3 signaling. Int Immunopharmacol. (2022) 113:109424. doi: 10.1016/j.intimp.2022.109424
111. Duan K, Liu J, Zhang J, Chu T, Liu H, Lou F, et al. Advancements in innate immune regulation strategies in islet transplantation. Front Immunol. (2023) 14:1341314. doi: 10.3389/fimmu.2023.1341314
112. Henderson JR, Moss MC. A morphometric study of the endocrine and exocrine capillaries of the pancreas. Q J Exp Physiol. (1985) 70:347–56. doi: 10.1113/expphysiol.1985.sp002920
113. Allison AC, Eugui EM. The design and development of an immunosuppressive drug, mycophenolate mofetil. Springer Semin Immunopathol. (1993) 14:353–80. doi: 10.1007/BF00192309
114. Allison AC, Eugui EM. Preferential suppression of lymphocyte proliferation by mycophenolic acid and predicted long-term effects of mycophenolate mofetil in transplantation. Transplant Proc. (1994) 26:3205–10.
115. Allison AC, Hovi T, Watts RW, Webster AD. The role of de novo purine synthesis in lymphocyte transformation. Ciba Found Symp. (1977) 48):207–24.
116. Allison AC, Eugui EM. Mycophenolate mofetil and its mechanisms of action. Immunopharmacology. (2000) 47:85–118. doi: 10.1016/S0162-3109(00)00188-0
117. Rosenberg AS, Singer A. Cellular basis of skin allograft rejection: an in vivo model of immune-mediated tissue destruction. Annu Rev Immunol. (1992) 10:333–58. doi: 10.1146/annurev.iy.10.040192.002001
118. Mycophenolate mofetil for the treatment of a first acute renal allograft rejection: three-year follow-up. The Mycophenolate Mofetil Acute Renal Rejection Study Group. Transplantation. (2001) 71:1091–7. doi: 10.1097/00007890-200104270-00014
119. Mathew TH. A blinded, long-term, randomized multicenter study of mycophenolate mofetil in cadaveric renal transplantation: results at three years. Tricontinental Mycophenolate Mofetil Renal Transplantation Study Group. Transplantation. (1998) 65:1450–4. doi: 10.1097/00007890-199806150-00007
120. Pescovitz MD, Conti D, Dunn J, Gonwa T, Halloran P, Sollinger H, et al. Intravenous mycophenolate mofetil: safety, tolerability, and pharmacokinetics. Clin Transplant. (2000) 14:179–88. doi: 10.1034/j.1399-0012.2000.140301.x
121. Hutchings P, Rosen H, O'Reilly L, Simpson E, Gordon S, Cooke A. Transfer of diabetes in mice prevented by blockade of adhesion-promoting receptor on macrophages. Nature. (1990) 348:639–42. doi: 10.1038/348639a0
122. Corbett JA, Mikhael A, Shimizu J, Frederick K, Misko TP, McDaniel ML, et al. Nitric oxide production in islets from nonobese diabetic mice: aminoguanidine-sensitive and -resistant stages in the immunological diabetic process. Proc Natl Acad Sci U.S.A. (1993) 90:8992–5.
123. Flodström M, Tyrberg B, Eizirik DL, Sandler S. Reduced sensitivity of inducible nitric oxide synthase-deficient mice to multiple low-dose streptozotocin-induced diabetes. Diabetes. (1999) 48:706–13. doi: 10.2337/diabetes.48.4.706
124. Eizirik DL, Colli ML, Ortis F. The role of inflammation in insulitis and beta-cell loss in type 1 diabetes. Nat Rev Endocrinol. (2009) 5:219–26. doi: 10.1038/nrendo.2009.21
125. Silverman MN, Sternberg EM. Glucocorticoid regulation of inflammation and its functional correlates: from HPA axis to glucocorticoid receptor dysfunction. Ann N Y Acad Sci. (2012) 1261:55–63. doi: 10.1111/j.1749-6632.2012.06633.x
126. Lambillotte C, Gilon P, Henquin JC. Direct glucocorticoid inhibition of insulin secretion. An in vitro study of dexamethasone effects in mouse islets. J Clin Invest. (1997) 99:414–23. doi: 10.1172/JCI119175
127. Buchwald P, Bocca N, Marzorati S, Hochhaus G, Bodor N, Stabler C, et al. Feasibility of localized immunosuppression: 1. Exploratory studies with glucocorticoids in a biohybrid device designed for cell transplantation. Pharmazie. (2010) 65:421–8.
128. Hult M, Ortsäter H, Schuster G, Graedler F, Beckers J, Adamski J, et al. Short-term glucocorticoid treatment increases insulin secretion in islets derived from lean mice through multiple pathways and mechanisms. Mol Cell Endocrinol. (2009) 301:109–16. doi: 10.1016/j.mce.2008.09.038
129. Lund T, Fosby B, Korsgren O, Scholz H, Foss A. Glucocorticoids reduce pro-inflammatory cytokines and tissue factor in vitro and improve function of transplanted human islets in vivo. Transpl Int. (2008) 21:669–78. doi: 10.1111/tri.2008.21.issue-7
130. Rafacho A, Marroquí L, Taboga SR, Abrantes JL, Silveira LR, Boschero AC, et al. Glucocorticoids in vivo induce both insulin hypersecretion and enhanced glucose sensitivity of stimulus-secretion coupling in isolated rat islets. Endocrinology. (2010) 151:85–95. doi: 10.1210/en.2009-0704
131. Turban S, Liu X, Ramage L, Webster SP, Walker BR, Dunbar DR, et al. Optimal elevation of β-cell 11β-hydroxysteroid dehydrogenase type 1 is a compensatory mechanism that prevents high-fat diet-induced β-cell failure. Diabetes. (2012) 61:642–52. doi: 10.2337/db11-1054
132. Balamurugan AN, Bottino R, Giannoukakis N, Smetanka C. Prospective and challenges of islet transplantation for the therapy of autoimmune diabetes. Pancreas. (2006) 32:231–43. doi: 10.1097/01.mpa.0000203961.16630.2f
133. Colson K, Doss DS, Swift R, Tariman J, Thomas TE. Bortezomib, a newly approved proteasome inhibitor for the treatment of multiple myeloma: nursing implications. Clin J Oncol Nurs. (2004) 8:473–80. doi: 10.1188/04.CJON.473-480
134. Field-Smith A, Morgan GJ, Davies FE. Bortezomib (Velcadetrade mark) in the treatment of multiple myeloma. Ther Clin Risk Manag. (2006) 2:271–9. doi: 10.2147/tcrm.2006.2.3.271
135. Brignole C, Marimpietri D, Pastorino F, Nico B, Di Paolo D, Cioni M, et al. Effect of bortezomib on human neuroblastoma cell growth, apoptosis, and angiogenesis. J Natl Cancer Inst. (2006) 98:1142–57. doi: 10.1093/jnci/djj309
136. Vodanovic-Jankovic S, Hari P, Jacobs P, Komorowski R, Drobyski WR. NF-kappaB as a target for the prevention of graft-versus-host disease: comparative efficacy of bortezomib and PS-1145. Blood. (2006) 107:827–34. doi: 10.1182/blood-2005-05-1820
137. Blanco B, Pérez-Simón JA, Sánchez-Abarca LI, Carvajal-Vergara X, Mateos J, Vidriales B, et al. Bortezomib induces selective depletion of alloreactive T lymphocytes and decreases the production of Th1 cytokines. Blood. (2006) 107:3575–83. doi: 10.1182/blood-2005-05-2118
138. Nencioni A, Schwarzenberg K, Brauer KM, Schmidt SM, Ballestrero A, Grünebach F, et al. Proteasome inhibitor bortezomib modulates TLR4-induced dendritic cell activation. Blood. (2006) 108:551–8. doi: 10.1182/blood-2005-08-3494
139. Sun K, Welniak LA, Panoskaltsis-Mortari A, O'Shaughnessy MJ, Liu H, Barao I, et al. Inhibition of acute graft-versus-host disease with retention of graft-versus-tumor effects by the proteasome inhibitor bortezomib. Proc Natl Acad Sci U.S.A. (2004) 101:8120–5. doi: 10.1073/pnas.0401563101
140. Sun K, Wilkins DE, Anver MR, Sayers TJ, Panoskaltsis-Mortari A, Blazar BR, et al. Differential effects of proteasome inhibition by bortezomib on murine acute graft-versus-host disease (GVHD): delayed administration of bortezomib results in increased GVHD-dependent gastrointestinal toxicity. Blood. (2005) 106:3293–9. doi: 10.1182/blood-2004-11-4526
141. Kim JS, Lee JI, Shin JY, Kim SY, Shin JS, Lim JH, et al. Bortezomib can suppress activation of rapamycin-resistant memory T cells without affecting regulatory T-cell viability in non-human primates. Transplantation. (2009) 88:1349–59. doi: 10.1097/TP.0b013e3181bd7b3a
142. Hong SH, Kim K, Shin JS, Chung H, Park CG. A combination regimen of low-dose bortezomib and rapamycin prolonged the graft survival in a murine allogeneic islet transplantation model. Immunol Lett. (2019) 216:21–7. doi: 10.1016/j.imlet.2019.10.005
143. Berney T, Secchi A. Rapamycin in islet transplantation: friend or foe? Transpl Int. (2009) 22:153–61. doi: 10.1111/j.1432-2277.2008.00743.x
144. Zhang N, Su D, Qu S, Tse T, Bottino R, Balamurugan AN, et al. Sirolimus is associated with reduced islet engraftment and impaired beta-cell function. Diabetes. (2006) 55:2429–36. doi: 10.2337/db06-0173
145. Diamond AS, Gill RG. An essential contribution by IFN-gamma to CD8+ T cell-mediated rejection of pancreatic islet allografts. J Immunol. (2000) 165:247–55. doi: 10.4049/jimmunol.165.1.247
146. Simeonovic CJ, Townsend MJ, Morris CF, Hapel AJ, Fung MC, Mann DA, et al. Immune mechanisms associated with the rejection of fetal murine proislet allografts and pig proislet xenografts: comparison of intragraft cytokine mRNA profiles. Transplantation. (1999) 67:963–71. doi: 10.1097/00007890-199904150-00006
147. Kim HJ, Byun N, Kwon O, Park CG. Cross-sensitization between xeno- and allo-antigens on subsequent allogeneic and xenogeneic pancreatic islet transplantation in a murine model. Biochem Biophys Res Commun. (2016) 480:474–8. doi: 10.1016/j.bbrc.2016.10.076
148. Kang HK, Wang S, Dangi A, Zhang X, Singh A, Zhang L, et al. Differential role of B cells and IL-17 versus IFN-γ During early and late rejection of pig islet xenografts in mice. Transplantation. (2017) 101:1801–10. doi: 10.1097/TP.0000000000001489
149. Cooper DK, Human PA, Lexer G, Rose AG, Rees J, Keraan M, et al. Effects of cyclosporine and antibody adsorption on pig cardiac xenograft survival in the baboon. J Heart Transplant. (1988) 7:238–46.
150. Cooper DKC, Ekser B, Tector AJ. Immunobiological barriers to xenotransplantation. Int J Surg. (2015) 23:211–6. doi: 10.1016/j.ijsu.2015.06.068
151. Galili U. Natural anti-carbohydrate antibodies contributing to evolutionary survival of primates in viral epidemics? Glycobiology. (2016) 26:1140–50. doi: 10.1093/glycob/cww088
152. Zeyland J, Lipiński D, Słomski R. The current state of xenotransplantation. J Appl Genet. (2015) 56:211–8. doi: 10.1007/s13353-014-0261-6
153. Yang YG, Sykes M. Xenotransplantation: current status and a perspective on the future. Nat Rev Immunol. (2007) 7:519–31. doi: 10.1038/nri2099
154. Kobayashi T, Cooper DK. Anti-Gal, alpha-Gal epitopes, and xenotransplantation. Subcell Biochem. (1999) 32:229–57. doi: 10.1007/978-1-4615-4771-6_10
155. Platt JL, Fischel RJ, Matas AJ, Reif SA, Bolman RM, Bach FH. Immunopathology of hyperacute xenograft rejection in a swine-to-primate model. Transplantation. (1991) 52:214–20. doi: 10.1097/00007890-199108000-00006
156. Schuurman HJ, Cheng J, Lam T. Pathology of xenograft rejection: a commentary. Xenotransplantation. (2003) 10:293–9. doi: 10.1034/j.1399-3089.2003.02092.x
157. Ierino FL, Sandrin MS. Spectrum of the early xenograft response: from hyperacute rejection to delayed xenograft injury. Crit Rev Immunol. (2007) 27:153–66. doi: 10.1615/CritRevImmunol.v27.i2
158. Phelps CJ, Koike C, Vaught TD, Boone J, Wells KD, Chen SH, et al. Production of alpha 1,3-galactosyltransferase-deficient pigs. Science. (2003) 299:411–4. doi: 10.1126/science.1078942
159. Park JY, Park MR, Kwon DN, Kang MH, Oh M, Han JW, et al. Alpha 1,3-galactosyltransferase deficiency in pigs increases sialyltransferase activities that potentially raise non-gal xenoantigenicity. J Biomed Biotechnol. (2011) 2011:560850. doi: 10.1155/2011/560850
160. Chen Y, Stewart JM, Gunthart M, Hawthorne WJ, Salvaris EJ, O'Connell PJ, et al. Xenoantibody response to porcine islet cell transplantation using GTKO, CD55, CD59, and fucosyltransferase multiple transgenic donors. Xenotransplantation. (2014) 21:244–53. doi: 10.1111/xen.12091
161. Hering BJ, Walawalkar N. Pig-to-nonhuman primate islet xenotransplantation. Transpl Immunol. (2009) 21:81–6. doi: 10.1016/j.trim.2009.05.001
162. Griesemer A, Yamada K, Sykes M. Xenotransplantation: immunological hurdles and progress toward tolerance. Immunol Rev. (2014) 258:241–58. doi: 10.1111/imr.12152
163. Yi S, Feng X, Wang Y, Kay TW, Wang Y, O'Connell PJ. CD4+ cells play a major role in xenogeneic human anti-pig cytotoxicity through the Fas/Fas ligand lytic pathway. Transplantation. (1999) 67:435–43. doi: 10.1097/00007890-199902150-00017
164. Lin ML, Zhan Y, Nutt SL, Brady J, Wojtasiak M, Brooks AG, et al. NK cells promote peritoneal xenograft rejection through an IFN-gamma-dependent mechanism. Xenotransplantation. (2006) 13:536–46. doi: 10.1111/j.1399-3089.2006.00348.x
165. Yi S, Feng X, Hawthorne WJ, Patel AT, Walters SN, O'Connell PJ. CD4+ T cells initiate pancreatic islet xenograft rejection via an interferon-gamma-dependent recruitment of macrophages and natural killer cells. Transplantation. (2002) 73:437–46. doi: 10.1097/00007890-200202150-00019
166. Lee RS, Yamada K, Womer KL, Pillsbury EP, Allison KS, Marolewski AE, et al. Blockade of CD28-B7, but not CD40-CD154, prevents costimulation of allogeneic porcine and xenogeneic human anti-porcine T cell responses. J Immunol. (2000) 164:3434–44. doi: 10.4049/jimmunol.164.6.3434
167. Mulley WR, Wee JL, Christiansen D, Milland J, Ierino FL, Sandrin MS. Lentiviral expression of CTLA4Ig inhibits primed xenogeneic lymphocyte proliferation and cytokine responses. Xenotransplantation. (2006) 13:248–52. doi: 10.1111/j.1399-3089.2006.00297.x
168. Singh NP, Guo L, Mhoyan A, Shirwan H. Predominant expression of Th2 cytokines and interferon-gamma in xenogeneic cardiac grafts undergoing acute vascular rejection. Transplantation. (2003) 75:586–90. doi: 10.1097/01.TP.0000052594.83318.68
169. Getts DR, Turley DM, Smith CE, Harp CT, McCarthy D, Feeney EM, et al. Tolerance induced by apoptotic antigen-coupled leukocytes is induced by PD-L1+ and IL-10-producing splenic macrophages and maintained by T regulatory cells. J Immunol. (2011) 187:2405–17. doi: 10.4049/jimmunol.1004175
170. Kheradmand T, Wang S, Bryant J, Tasch JJ, Lerret N, Pothoven KL, et al. Ethylenecarbodiimide-fixed donor splenocyte infusions differentially target direct and indirect pathways of allorecognition for induction of transplant tolerance. J Immunol. (2012) 189:804–12. doi: 10.4049/jimmunol.1103705
171. Martin AJ, McCarthy D, Waltenbaugh C, Goings G, Luo X, Miller SD. Ethylenecarbodiimide-treated splenocytes carrying male CD4 epitopes confer histocompatibility Y chromosome antigen transplant protection by inhibiting CD154 upregulation. J Immunol. (2010) 185:3326–36. doi: 10.4049/jimmunol.1000802
172. Turley DM, Miller SD. Peripheral tolerance induction using ethylenecarbodiimide-fixed APCs uses both direct and indirect mechanisms of antigen presentation for prevention of experimental autoimmune encephalomyelitis. J Immunol. (2007) 178:2212–20. doi: 10.4049/jimmunol.178.4.2212
173. Nilsson B, Ekdahl KN, Korsgren O. Control of instant blood-mediated inflammatory reaction to improve islets of Langerhans engraftment. Curr Opin Organ Transplant. (2011) 16:620–6. doi: 10.1097/MOT.0b013e32834c2393
174. Liuwantara D, Chew YV, Favaloro EJ, Hawkes JM, Burns HL, O'Connell PJ, et al. Characterizing the mechanistic pathways of the instant blood-mediated inflammatory reaction in xenogeneic neonatal islet cell transplantation. Transplant Direct. (2016) 2:e77. doi: 10.1097/TXD.0000000000000590
175. Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: a key system for immune surveillance and homeostasis. Nat Immunol. (2010) 11:785–97. doi: 10.1038/ni.1923
176. Ehlers MR. CR3: a general purpose adhesion-recognition receptor essential for innate immunity. Microbes Infect. (2000) 2:289–94. doi: 10.1016/S1286-4579(00)00299-9
177. Lachmann PJ. The amplification loop of the complement pathways. Adv Immunol. (2009) 104:115–49. doi: 10.1016/S0065-2776(08)04004-2
178. Amara U, Flierl MA, Rittirsch D, Klos A, Chen H, Acker B, et al. Molecular intercommunication between the complement and coagulation systems. J Immunol. (2010) 185:5628–36. doi: 10.4049/jimmunol.0903678
179. Oikonomopoulou K, Ricklin D, Ward PA, Lambris JD. Interactions between coagulation and complement–their role in inflammation. Semin Immunopathol. (2012) 34:151–65. doi: 10.1007/s00281-011-0280-x
180. Gaboriaud C, Ling WL, Thielens NM, Bally I, Rossi V. Deciphering the fine details of c1 assembly and activation mechanisms: "mission impossible"? Front Immunol. (2014) 5:565. doi: 10.3389/fimmu.2014.00565
181. Gál P, Barna L, Kocsis A, Závodszky P. Serine proteases of the classical and lectin pathways: similarities and differences. Immunobiology. (2007) 212:267–77. doi: 10.1016/j.imbio.2006.11.002
182. Thurman JM, Holers VM. The central role of the alternative complement pathway in human disease. J Immunol. (2006) 176:1305–10. doi: 10.4049/jimmunol.176.3.1305
183. Ricklin D. Manipulating the mediator: modulation of the alternative complement pathway C3 convertase in health, disease and therapy. Immunobiology. (2012) 217:1057–66. doi: 10.1016/j.imbio.2012.07.016
184. Alcorlo M, Tortajada A, Rodríguez de Córdoba S, Llorca O. Structural basis for the stabilization of the complement alternative pathway C3 convertase by properdin. Proc Natl Acad Sci U.S.A. (2013) 110:13504–9. doi: 10.1073/pnas.1309618110
185. Hourcade DE. The role of properdin in the assembly of the alternative pathway C3 convertases of complement. J Biol Chem. (2006) 281:2128–32. doi: 10.1074/jbc.M508928200
186. Lesher AM, Nilsson B, Song WC. Properdin in complement activation and tissue injury. Mol Immunol. (2013) 56:191–8. doi: 10.1016/j.molimm.2013.06.002
187. Rawal N, Pangburn MK. Structure/function of C5 convertases of complement. Int Immunopharmacol. (2001) 1:415–22. doi: 10.1016/S1567-5769(00)00039-4
188. Bubeck D. The making of a macromolecular machine: assembly of the membrane attack complex. Biochemistry. (2014) 53:1908–15. doi: 10.1021/bi500157z
189. Klos A, Tenner AJ, Johswich KO, Ager RR, Reis ES, Köhl J. The role of the anaphylatoxins in health and disease. Mol Immunol. (2009) 46:2753–66. doi: 10.1016/j.molimm.2009.04.027
190. Williams JC, Mackman N. Tissue factor in health and disease. Front Biosci (Elite Ed). (2012) 4:358–72. doi: 10.2741/e383
191. Owens AP 3rd, Mackman N. Tissue factor and thrombosis: The clot starts here. Thromb Haemost. (2010) 104:432–9. doi: 10.1160/TH09-11-0771
192. Saha D, S S, Sergeeva EG, Ionova ZI, Gorbach AV. Tissue factor and atherothrombosis. Curr Pharm Des. (2015) 21:1152–7. doi: 10.2174/1381612820666141013154946
193. Samad F, Ruf W. Inflammation, obesity, and thrombosis. Blood. (2013) 122:3415–22. doi: 10.1182/blood-2013-05-427708
194. Kourtzelis I, Markiewski MM, Doumas M, Rafail S, Kambas K, Mitroulis I, et al. Complement anaphylatoxin C5a contributes to hemodialysis-associated thrombosis. Blood. (2010) 116:631–9. doi: 10.1182/blood-2010-01-264051
195. Edgington TS, Mackman N, Brand K, Ruf W. The structural biology of expression and function of tissue factor. Thromb Haemost. (1991) 66:67–79.
196. Walker RK, Krishnaswamy S. The activation of prothrombin by the prothrombinase complex. The contribution of the substrate-membrane interaction to catalysis. J Biol Chem. (1994) 269:27441–50. doi: 10.1016/S0021-9258(18)47005-6
197. Mosesson MW. Fibrinogen and fibrin structure and functions. J Thromb Haemost. (2005) 3:1894–904. doi: 10.1111/j.1538-7836.2005.01365.x
198. Bennet W, Sundberg B, Lundgren T, Tibell A, Groth CG, Richards A, et al. Damage to porcine islets of Langerhans after exposure to human blood in vitro, or after intraportal transplantation to cynomologus monkeys: protective effects of sCR1 and heparin. Transplantation. (2000) 69:711–9. doi: 10.1097/00007890-200003150-00007
199. Johansson H, Lukinius A, Moberg L, Lundgren T, Berne C, Foss A, et al. Tissue factor produced by the endocrine cells of the islets of Langerhans is associated with a negative outcome of clinical islet transplantation. Diabetes. (2005) 54:1755–62. doi: 10.2337/diabetes.54.6.1755
200. Davie EW, Fujikawa K, Kisiel W. The coagulation cascade: initiation, maintenance, and regulation. Biochemistry. (1991) 30:10363–70. doi: 10.1021/bi00107a001
201. Hirsh J. Current anticoagulant therapy–unmet clinical needs. Thromb Res. (2003) 109 Suppl 1:S1–8. doi: 10.1016/S0049-3848(03)00250-0
202. Bouwens EA, Stavenuiter F, Mosnier LO. Mechanisms of anticoagulant and cytoprotective actions of the protein C pathway. J Thromb Haemost. (2013) 11 Suppl 1:242–53. doi: 10.1111/jth.12247
203. Wood JP, Ellery PE, Maroney SA, Mast AE. Biology of tissue factor pathway inhibitor. Blood. (2014) 123:2934–43. doi: 10.1182/blood-2013-11-512764
205. Mosnier LO, Meijers JC, Bouma BN. Regulation of fibrinolysis in plasma by TAFI and protein C is dependent on the concentration of thrombomodulin. Thromb Haemost. (2001) 85:5–11.
206. Roussel JC, Moran CJ, Salvaris EJ, Nandurkar HH, d'Apice AJ, Cowan PJ. Pig thrombomodulin binds human thrombin but is a poor cofactor for activation of human protein C and TAFI. Am J Transplant. (2008) 8:1101–12. doi: 10.1111/j.1600-6143.2008.02210.x
207. Wuensch A, Baehr A, Bongoni AK, Kemter E, Blutke A, Baars W, et al. Regulatory sequences of the porcine THBD gene facilitate endothelial-specific expression of bioactive human thrombomodulin in single- and multitransgenic pigs. Transplantation. (2014) 97:138–47. doi: 10.1097/TP.0b013e3182a95cbc
208. Wang S, Zhao Z, Cong Z, Suo G. Thrombin-activatable fibrinolysis inhibitor is activated in an instant blood-mediated inflammatory reaction after intraportal islet transplant. Exp Clin Transplant. (2014) 12:62–6. doi: 10.6002/ect
209. Hawthorne WJ, Salvaris EJ, Phillips P, Hawkes J, Liuwantara D, Burns H, et al. Control of IBMIR in neonatal porcine islet xenotransplantation in baboons. Am J Transplant. (2014) 14:1300–9. doi: 10.1111/ajt.12722
210. Tjernberg J, Ekdahl KN, Lambris JD, Korsgren O, Nilsson B. Acute antibody-mediated complement activation mediates lysis of pancreatic islets cells and may cause tissue loss in clinical islet transplantation. Transplantation. (2008) 85:1193–9. doi: 10.1097/TP.0b013e31816b22f3
211. Goto M, Johansson H, Maeda A, Elgue G, Korsgren O, Nilsson B. Low molecular weight dextran sulfate prevents the instant blood-mediated inflammatory reaction induced by adult porcine islets. Transplantation. (2004) 77:741–7. doi: 10.1097/01.TP.0000114872.26990.4F
212. Biarnés M, Montolio M, Nacher V, Raurell M, Soler J, Montanya E. Beta-cell death and mass in syngeneically transplanted islets exposed to short- and long-term hyperglycemia. Diabetes. (2002) 51:66–72. doi: 10.2337/diabetes.51.1.66
213. van der Windt DJ, Bottino R, Casu A, Campanile N, Cooper DK. Rapid loss of intraportally transplanted islets: an overview of pathophysiology and preventive strategies. Xenotransplantation. (2007) 14:288–97. doi: 10.1111/j.1399-3089.2007.00419.x
214. Harlan DM, Kenyon NS, Korsgren O, Roep BO. Current advances and travails in islet transplantation. Diabetes. (2009) 58:2175–84. doi: 10.2337/db09-0476
215. van der Windt DJ, Bottino R, Kumar G, Wijkstrom M, Hara H, Ezzelarab M, et al. Clinical islet xenotransplantation: how close are we? Diabetes. (2012) 61:3046–55. doi: 10.2337/db12-0033
216. Tai JH, Sun H, Liu W, Melling CW, Hasilo C, White DJ. Isolating human islets of Langerhans causes loss of decay accelerating factor (CD55) on beta-cells. Cell Transplant. (2008) 17:1349–59. doi: 10.3727/096368908787648092
217. Ji M, Yi S, Smith-Hurst H, Phillips P, Wu J, Hawthorne W, et al. The importance of tissue factor expression by porcine NICC in triggering IBMIR in the xenograft setting. Transplantation. (2011) 91:841–6. doi: 10.1097/TP.0b013e3182106091
218. Ma X, Ye B, Gao F, Liang Q, Dong Q, Liu Y, et al. Tissue factor knockdown in porcine islets: an effective approach to suppressing the instant blood-mediated inflammatory reaction. Cell Transplant. (2012) 21:61–71. doi: 10.3727/096368911X580563
219. Berman DM, Cabrera O, Kenyon NM, Miller J, Tam SH, Khandekar VS, et al. Interference with tissue factor prolongs intrahepatic islet allograft survival in a nonhuman primate marginal mass model. Transplantation. (2007) 84:308–15. doi: 10.1097/01.tp.0000275401.80187.1e
220. Jung DY, Park JB, Joo SY, Joh JW, Kwon CH, Kwon GY, et al. Effect of nicotinamide on early graft failure following intraportal islet transplantation. Exp Mol Med. (2009) 41:782–92. doi: 10.3858/emm.2009.41.11.084
221. Matsumoto S, Tan P, Baker J, Durbin K, Tomiya M, Azuma K, et al. Clinical porcine islet xenotransplantation under comprehensive regulation. Transplant Proc. (2014) 46:1992–5. doi: 10.1016/j.transproceed.2014.06.008
222. Yamada K, Sykes M, Sachs DH. Tolerance in xenotransplantation. Curr Opin Organ Transplant. (2017) 22:522–8. doi: 10.1097/MOT.0000000000000466
223. Lee FT, Dangi A, Shah S, Burnette M, Yang YG, Kirk AD, et al. Rejection of xenogeneic porcine islets in humanized mice is characterized by graft-infiltrating Th17 cells and activated B cells. Am J Transplant. (2020) 20:1538–50. doi: 10.1111/ajt.15763
224. Weber MS, Prod'homme T, Patarroyo JC, Molnarfi N, Karnezis T, Lehmann-Horn K, et al. B-cell activation influences T-cell polarization and outcome of anti-CD20 B-cell depletion in central nervous system autoimmunity. Ann Neurol. (2010) 68:369–83. doi: 10.1002/ana.22081
225. Zhong X, Gao W, Degauque N, Bai C, Lu Y, Kenny J, et al. Reciprocal generation of Th1/Th17 and T(reg) cells by B1 and B2 B cells. Eur J Immunol. (2007) 37:2400–4. doi: 10.1002/eji.200737296
226. Zhong X, Lau S, Bai C, Degauque N, Holodick NE, Steven SJ, et al. A novel subpopulation of B-1 cells is enriched with autoreactivity in normal and lupus-prone mice. Arthritis Rheum. (2009) 60:3734–43. doi: 10.1002/art.25015
227. Mitsdoerffer M, Lee Y, Jäger A, Kim HJ, Korn T, Kolls JK, et al. Proinflammatory T helper type 17 cells are effective B-cell helpers. Proc Natl Acad Sci U.S.A. (2010) 107:14292–7. doi: 10.1073/pnas.1009234107
228. Graham KL, Krishnamurthy B, Fynch S, Ayala-Perez R, Slattery RM, Santamaria P, et al. Intra-islet proliferation of cytotoxic T lymphocytes contributes to insulitis progression. Eur J Immunol. (2012) 42:1717–22. doi: 10.1002/eji.201242435
229. Calafiore R, Basta G. Clinical application of microencapsulated islets: actual prospectives on progress and challenges. Adv Drug Delivery Rev. (2014) 67-68:84–92. doi: 10.1016/j.addr.2013.09.020
230. Veiseh O, Doloff JC, Ma M, Vegas AJ, Tam HH, Bader AR, et al. Size- and shape-dependent foreign body immune response to materials implanted in rodents and non-human primates. Nat Mater. (2015) 14:643–51. doi: 10.1038/nmat4290
231. Krishnan R, Alexander M, Robles L, Foster CE 3rd, Lakey JR. Islet and stem cell encapsulation for clinical transplantation. Rev Diabetes Stud. (2014) 11:84–101. doi: 10.1900/RDS.2014.11.84
232. Bottino R, Knoll MF, Knoll CA, Bertera S, Trucco MM. The future of islet transplantation is now. Front Med (Lausanne). (2018) 5:202. doi: 10.3389/fmed.2018.00202
233. Merani S, Toso C, Emamaullee J, Shapiro AM. Optimal implantation site for pancreatic islet transplantation. Br J Surg. (2008) 95:1449–61. doi: 10.1002/bjs.6391
234. Van Der Windt DJ, Echeverri GJ, Ijzermans JNM, Cooper DKC. The choice of anatomical site for islet transplantation. Cell Transplant. (2008) 17:1005–14. doi: 10.3727/096368908786991515
235. Pepper AR, Gala-Lopez B, Pawlick R, Merani S, Kin T, Shapiro AM. A prevascularized subcutaneous device-less site for islet and cellular transplantation. Nat Biotechnol. (2015) 33:518–23. doi: 10.1038/nbt.3211
236. Pareta R, McQuilling JP, Sittadjody S, Jenkins R, Bowden S, Orlando G, et al. Long-term function of islets encapsulated in a redesigned alginate microcapsule construct in omentum pouches of immune-competent diabetic rats. Pancreas. (2014) 43:605–13. doi: 10.1097/MPA.0000000000000107
237. Shimoda M, Matsumoto S. Microencapsulation in clinical islet xenotransplantation. Methods Mol Biol. (2017) 1479:335–45. doi: 10.1007/978-1-4939-6364-5_25
238. Groth CG, Korsgren O, Tibell A, Tollemar J, Möller E, Bolinder J, et al. Transplantation of porcine fetal pancreas to diabetic patients. Lancet. (1994) 344:1402–4. doi: 10.1016/S0140-6736(94)90570-3
239. Cardona K, Korbutt GS, Milas Z, Lyon J, Cano J, Jiang W, et al. Long-term survival of neonatal porcine islets in nonhuman primates by targeting costimulation pathways. Nat Med. (2006) 12:304–6. doi: 10.1038/nm1375
240. Hering BJ, Wijkstrom M, Graham ML, Hårdstedt M, Aasheim TC, Jie T, et al. Prolonged diabetes reversal after intraportal xenotransplantation of wild-type porcine islets in immunosuppressed nonhuman primates. Nat Med. (2006) 12:301–3. doi: 10.1038/nm1369
241. van der Windt DJ, Bottino R, Casu A, Campanile N, Smetanka C, He J, et al. Long-term controlled normoglycemia in diabetic non-human primates after transplantation with hCD46 transgenic porcine islets. Am J Transplant. (2009) 9:2716–26. doi: 10.1111/j.1600-6143.2009.02850.x
242. Bottino R, Wijkstrom M, van der Windt DJ, Hara H, Ezzelarab M, Murase N, et al. Pig-to-monkey islet xenotransplantation using multi-transgenic pigs. Am J Transplant. (2014) 14:2275–87. doi: 10.1111/ajt.12868
243. Wijkstrom M, Bottino R, Iwase H, Hara H, Ekser B, van der Windt D, et al. Glucose metabolism in pigs expressing human genes under an insulin promoter. Xenotransplantation. (2015) 22:70–9. doi: 10.1111/xen.12145
244. Shin JS, Kim JM, Kim JS, Min BH, Kim YH, Kim HJ, et al. Long-term control of diabetes in immunosuppressed nonhuman primates (NHP) by the transplantation of adult porcine islets. Am J Transplant. (2015) 15:2837–50. doi: 10.1111/ajt.13345
245. Bottino R, Knoll MF, Graeme-Wilson J, Klein EC, Ayares D, Trucco M, et al. Safe use of anti-CD154 monoclonal antibody in pig islet xenotransplantation in monkeys. Xenotransplantation. (2017) 24(1). doi: 10.1111/xen.12283
246. Iwase H, Hara H, Ezzelarab M, Li T, Zhang Z, Gao B, et al. Immunological and physiological observations in baboons with life-supporting genetically engineered pig kidney grafts. Xenotransplantation. (2017) 24(2). doi: 10.1111/xen.12293
247. Mohiuddin MM, Singh AK, Corcoran PC, Thomas Iii ML, Clark T, Lewis BG, et al. Chimeric 2C10R4 anti-CD40 antibody therapy is critical for long-term survival of GTKO.hCD46.hTBM pig-to-primate cardiac xenograft. Nat Commun. (2016) 7:11138. doi: 10.1038/ncomms11138
248. Ellis CE, Korbutt GS. Justifying clinical trials for porcine islet xenotransplantation. Xenotransplantation. (2015) 22:336–44. doi: 10.1111/xen.12196
249. Potter KJ, Abedini A, Marek P, Klimek AM, Butterworth S, Driscoll M, et al. Islet amyloid deposition limits the viability of human islet grafts but not porcine islet grafts. Proc Natl Acad Sci U.S.A. (2010) 107:4305–10. doi: 10.1073/pnas.0909024107
250. Zhang X, Cheng B, Gong H, Li C, Chen H, Zheng L, et al. Porcine islet amyloid polypeptide fragments are refractory to amyloid formation. FEBS Lett. (2011) 585:71–7. doi: 10.1016/j.febslet.2010.11.050
251. Kobayashi T, Aomatsu Y, Iwata H, Kin T, Kanehiro H, Hisanga M, et al. Survival of microencapsulated islets at 400 days posttransplantation in the omental pouch of NOD mice. Cell Transplant. (2006) 15:359–65. doi: 10.3727/000000006783981954
Keywords: islet transplantation, immune response, immunosuppressants, xenotransplantation, allogenic and xenogenic islet transplantation
Citation: Yue L, Li J, Yao M, Song S, Zhang X and Wang Y (2024) Cutting edge of immune response and immunosuppressants in allogeneic and xenogeneic islet transplantation. Front. Immunol. 15:1455691. doi: 10.3389/fimmu.2024.1455691
Received: 27 June 2024; Accepted: 27 August 2024;
Published: 13 September 2024.
Edited by:
Shiva Pathak, Stanford University, United StatesReviewed by:
Raphael Meier, University of Maryland, United StatesNaoaki Sakata, Fukuoka University, Japan
Copyright © 2024 Yue, Li, Yao, Song, Zhang and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yi Wang, d195aTIwMjJAMTYzLmNvbQ==; Xiaoqin Zhang, ZG9jenhxQDE2My5jb20=; Siyuan Song, c2kteXVhbi5zb25nQGJjbS5lZHU=
†These authors have contributed equally to this work