 Franklin R. Toapanta1*Jingping Hu1Kari Ann Shirey2Paula J. Bernal1Myron M. Levine1,3Thomas C. Darton4†Claire S. Waddington4†Andrew J. Pollard4Marcelo B. Sztein1,2,3*
Franklin R. Toapanta1*Jingping Hu1Kari Ann Shirey2Paula J. Bernal1Myron M. Levine1,3Thomas C. Darton4†Claire S. Waddington4†Andrew J. Pollard4Marcelo B. Sztein1,2,3*- 1Department of Medicine and Center for Vaccine Development and Global Health, University of Maryland School of Medicine, Baltimore, MD, United States
- 2Department of Microbiology and Immunology, University of Maryland School of Medicine, Baltimore, MD, United States
- 3Department of Pediatrics and Center for Vaccine Development and Global Health, University of Maryland School of Medicine, Baltimore, MD, United States
- 4Oxford Vaccine Group, Department of Paediatrics, University of Oxford and the National Institute for Health and Care Research (NIHR) Oxford Biomedical Research Centre, Oxford, United Kingdom
An oral Controlled Human Infection Model (CHIM) with wild-type S. Typhi was re-established allowing us to explore the development of immunity. In this model, ~55% of volunteers who received the challenge reached typhoid diagnosis criteria (TD), while ~45% did not (NoTD). Intestinal macrophages are one of the first lines of defense against enteric pathogens. Most organs have self-renewing macrophages derived from tissue-resident progenitor cells seeded during the embryonic stage; however, the gut lacks these progenitors, and all intestinal macrophages are derived from circulating monocytes. After infecting gut-associated lymphoid tissues underlying microfold (M) cells, S. Typhi causes a primary bacteremia seeding organs of the reticuloendothelial system. Following days of incubation, a second bacteremia and clinical disease ensue. S. Typhi likely interacts with circulating monocytes or their progenitors in the bone marrow. We assessed changes in circulating monocytes after CHIM. The timepoints studied included 0 hours (pre-challenge) and days 1, 2, 4, 7, 9, 14, 21 and 28 after challenge. TD participants provided extra samples at the time of typhoid diagnosis, and 48-96 hours later (referred as ToD). We report changes in Classical Monocytes -CM-, Intermediate Monocytes -IM- and Non-classical Monocytes -NCM-. Changes in monocyte activation markers were identified only in TD participants and during ToD. CM and IM upregulated molecules related to interaction with bacterial antigens (TLR4, TLR5, CD36 and CD206). Of importance, CM and IM showed enhanced binding of S. Typhi. Upregulation of inflammatory molecules like TNF-α were detected, but mechanisms involved in limiting inflammation were also activated (CD163 and CD354 downregulation). CM upregulated molecules to interact/modulate cells of the adaptive immunity, including T cells (HLA-DR, CD274 and CD86) and B cells (CD257). Both CM and IM showed potential to migrate to the gut as integrin α4β7 was upregulated. Unsupervised analysis revealed 7 dynamic cell clusters. Five of these belonged to CM showing that this is the main population activated during ToD. Overall, we provide new insights into the changes that diverse circulating monocyte subsets undergo after typhoid diagnosis, which might be important to control this disease since these cells will ultimately become intestinal macrophages once they reach the gut.
Introduction
Salmonella enterica serovar Typhi (S. Typhi) is a human restricted pathogen and the agent responsible for typhoid fever. This pathogen continues to be a major public health problem, particularly in developing countries, where young children are disproportionately affected (1–5). The innate immune system is one of the first lines of defense against invading pathogens. Among innate cells, circulating monocytes, derived from bone marrow precursors, show great plasticity and can differentiate into various cell types (e.g., macrophages and dendritic cells -DC-) once they reach their homing organs and are exposed to the appropriate cytokine environment (6–11). During steady state, tissue-resident macrophages from most organs (e.g., brain, liver, lungs) are renewed by tissue-resident progenitor cells that are seeded before birth (12, 13). During acute infections, the pool of macrophages in these tissues increase due to the influx of circulating monocytes that differentiate into macrophages (monocyte-derived macrophages) (14–19). Importantly, the gut lacks tissue-resident macrophage-progenitors; therefore, all macrophages in the gut are monocyte-derived, even those present during steady state (20–24). Animal studies have shown the importance of local macrophages in controlling bacterial growth (25–27); therefore, it is important to determine whether circulating monocytes, which will contribute to the pool of tissue macrophages are affected by S. Typhi infection.
S. Typhi is transmitted by the fecal oral route (3, 28). Following ingestion, the bacteria enter the host via the intestinal mucosa. After infecting gut-associated lymphoid tissues (GALT) underlying microfold (M) cells, S. Typhi uses the GALT lymphatic vessels to reach the great veins (blood) and disseminate systemically seeding organs of the reticuloendothelial system (e.g., liver, spleen, bone marrow). This primary bacteremia (silent) occurs shortly after ingestion of the bacterial inoculum and individuals do not show any clinical symptoms. After a moderately long incubation time (~8-14 days), some individuals will develop a febrile illness. This is accompanied by a low-level bacteremia (1-10 CFU/ml of blood) (29) (secondary bacteremia) (30). The advent of optimized culture-PCR assays has allowed detection of the primary bacteriemia in some volunteers in challenge studies with wild-type S. Typhi (30, 31). Classic blood culture tests usually detect S. Typhi during the secondary bacteremia (29, 32). S. Typhi or bacterial products/metabolites likely encounter bone marrow monocyte precursors and/or blood monocytes during the primary and/or secondary bacteremia. Whether these encounters alter the phenotype of the monocytes remains unknown. This interaction can have important consequences for the host since all gut macrophages, as well as a portion of macrophages in other tissues, are derived from monocytes in circulation.
In humans, the blood monocyte compartment is composed of various subsets identified by the distinct expression of CD14 and CD16. These subsets include Classical Monocytes (CM; CD14+ CD16-), Intermediate Monocytes (IM; CD14+ CD16+) and Non-classical Monocytes (NCM; CD14dim CD16+). CM make up ∼85% of the circulating monocyte pool, whereas the remaining ∼15% consists of IM and NCM (33, 34). Mouse and human studies showed that IM and NCM represent diverse stages in the developmental sequence of monocytes (12, 35–38). CM have a short lifespan in blood (~1-2 days) and more than 95% of these cells are rapidly recruited to tissues to support tissue-resident macrophages (38). CMs that remain in circulation transform into IM, which have a lifespan of ~4 days. IM have similar functional characteristics than CM. Circulating IM eventually become NCM, which represent the final stage in the development of circulating monocytes and are considered luminal blood macrophages (39). These cells have a lifespan of ~7-8 days (38) and have been proposed to act as custodians of the vasculature by patrolling endothelial cell integrity in an LFA-1-dependent fashion (40).
In recent years, studies have tried to understand the role that the diverse monocyte subsets play in health and disease. Reports have shown changes in the frequencies of these subsets during bacterial and viral infections; for example, sepsis patients showed an expansion of the IM and NCM subsets (41–43). Interestingly, during steady state, IM have the highest expression of surface markers for antigen processing and presentation, such as HLA-DR (34, 44). Expression of this marker is significantly reduced in sepsis patients, when compared with healthy controls (42, 43, 45). The degree of recovery in surface expression of this marker is associated with mortality as survivors show an increase HLA-DR expression while non-survivors fail to recover expression of this marker (46–48). Of note, HLA-DR expression changes in sepsis were observed in all monocyte subsets. Whether similar changes are induced by S. Typhi infection during primary or secondary bacteremia remain unknown. In this study, we used specimens from a controlled human infection model of wild-type (wt) S. Typhi that was reestablished by the Oxford Vaccine Group at the University of Oxford. In this outpatient model, challenge with 103 CFU of S. Typhi (Quailes strain) resulted in ~55% of participants reaching typhoid endpoints (referred herein as typhoid diagnosis -TD-) (49). Specimens from volunteers who reached TD, or not, (TD and NoTD, respectively) were used to study the various monocyte subsets. Importantly, in a previous study from our group, we started characterizing some aspects of the monocyte response to wt S. Typhi infection. We showed that monocytes of TD volunteers during time of disease had an activated phenotype (e.g., increase expression of CD38 and CD40), upregulated molecules involved in migration to GALT (e.g., integrin α4β7) and had enhanced ability to bind S. Typhi (50). In the current study, we extended our studies and evaluated in depth the characteristics of various circulating monocytes subsets. In these subsets we assessed expression of markers involved in the interaction with bacteria (TLR-4, TLR-5, TREM-1, CD36, CD163) as well as markers involved in antigen presentation and/or interaction with cells of the adaptive immune response (HLA-DR, CD86, CD274, CD257). Moreover, we assessed the ability of the monocyte subsets to interact with S. Typhi and migrate to the gut as determined by expression of integrin α4β7. The results showed that a multiplicity of changes in the monocyte subsets are elicited following S. Typhi infection.
Materials and methods
Participants, clinical trial description and ethics statement
Samples used in this study were collected as part of a clinical study to establish a human model of typhoid fever (Oxford Vaccine Group, University of Oxford, United Kingdom). The details of the clinical outcomes have already been published (49). In brief, healthy adults (18-60 years-old) with no previous history of typhoid vaccination or residence (>6 months) in endemic areas were enrolled. Volunteers ingested a S. Typhi (Quailes strain) inoculum (103 or 104 CFU) as previously described (49). Typhoid diagnosis included meeting clinical (temperature ≥38°C sustained for ≥ 12 hours) and/or microbiological (blood culture confirmed S. Typhi bacteremia) endpoints. Participants were monitored daily for at least 14 days. The duration and severity of all solicited and unsolicited symptoms experienced were recorded daily as well as oral temperature readings (twice per day). All participants received antibiotic treatment (ciprofloxacin, 500 mg twice daily, 14 days) at typhoid diagnosis, or at day 14 post challenge (or if felt clinically necessary prior to day 14 or typhoid diagnosis). Subsequent visits were performed at days 21 and 28 after challenge. In the current report we describe the results from a subset of volunteers challenged with 103 CFU of S. Typhi. In this subset (n=10), 5 volunteers were diagnosed with typhoid disease (TD) and 5 were not (NoTD). Specimens for this study were selected based on the availability of critical time points to perform a comprehensive evaluation of monocytes subsets. This clinical study was performed by the Oxford Vaccine Group at the Centre for Clinical Vaccinology and Tropical Medicine of the University of Oxford (UK) (49). All volunteers provided a written informed consent at the time of enrollment and the procedures were approved by the Oxfordshire Research Ethics Committee A (10/H0604/53). This clinical study was registered on the UK Clinical Research Network (identifier UKCRN ID 9297).
Isolation of peripheral blood mononuclear cells (PBMC)
Blood specimens were collected from S. Typhi challenged volunteers and PBMC isolated by density gradient centrifugation and cryopreserved as previously described (49). Blood specimens were collected before challenge and at various time points thereafter. The evaluated time points differed slightly between TD and NoTD volunteers. These included 0 hours (pre-challenge) and days 1 (24h), 2, 4, 7, 9, 14, 21 and 28 after challenge in all subjects. In TD volunteers extra samples were collected at the time of typhoid diagnosis (9-14 days after challenge with 103 CFU of wt S. Typhi (49),) as well as 48 and 96 hours later.
Cell surface staining for flow-cytometry
Cryopreserved PBMC were thawed and after resting for 2-3 hours stained for flow cytometry analyses. In specimens in which TNF-α was assessed, Golgi apparatus inhibitors (Brefeldin A and Monensin) were added immediately after thawing the cells. Following 3 hour of incubation (37°C, 5% CO2) the cells were stained for flow cytometry. PBMC were cultured in complete media [RMPI (Gibco, NY, USA) supplemented with 10% fetal bovine serum (FBS) (Gemini Bioproducts, West Sacramento, CA), 2 mM L-glutamine (Gibco, Grand Island, NY, USA), 1x non-essential amino acids (Gibco, Grand Island, NY, USA), 10 mM HEPES (Gibco, Grand Island, NY, USA), 2.5 mM Sodium pyruvate, (Lonza, Walkersville, MD, USA), 100 U/ml Penicillin, 100 ug/ml streptomycin (Sigma-Aldrich, St. Louis, MO, USA), 50 μg/ml Gentamicin (Gibco, Grand Island, NY, USA)]. Cells were stained for flow cytometry in V-shaped 96-well plates using methods previously described (50, 51). Briefly, 1x106 cells were plated and stained for viability (20 min on ice) using 100 μl of Alexa Fluor 700-succinimidyl ester dye (0.4 μg/ml) (Invitrogen, USA). After 2 washes with flow cytometry staining (FC) buffer (4% FCS, 1X phosphate buffered saline (PBS) and 0.02 Sodium Azide), S. Typhi ISP 1820 labeled with Pacific Blue dye (S. Typhi-PB) were added. Bacteria were labeled as previously described (52) and 1 µl of labeled bacteria (O.D. = 0.4) was diluted in 100 µl of FC buffer and added to the cells for 30 min (RT). Cells were then washed twice with FC buffer, followed by blocking with a mixture of mouse IgG (5 μg in 25 µl of FC buffer) (Meridian Life Sciences, Memphis, TN, USA) and human IgG (5 μg in 25 µl of FC buffer) (Sigma, St Louis, MO) for 10 minutes on ice. Cells were then stained with various antibody cocktails prepared in FC buffer. After washing cells twice with FC buffer, stained cells were fixed with 1% PFA in PBS. In experiments that involve the measurement of intracellular targets, following surface staining, the cells were fixed with IC Fix buffer (Invitrogen) (30 min RT) and following washes (2x) with 1X Permeabilization Buffer (Invitrogen), cells were stained. The intracellular antibody cocktail was prepared in 1X Permeabilization Buffer. Monoclonal antibodies (mAbs) against the following molecules conjugated to the stated fluorochromes were used: CD257-FITC (clone 1D6; eBioscience), CD354-PE (clone TREM-26; BioLegend), CD16-PE-CF594 (clone 3G8; Beckton Dickinson -BD-), CD86-PerCP-Cy5.5 (clone 2331; BD), CD284-PE-Cy7 (clone Y1/82A; eBioscience), HLA-DR-BV570 (clone L243; eBioscience), CD163-BV605 (clone GHI/61; BioLegend), CD14-QDot655 (clone TuK; Invitrogen), CD274-BV711 (clone 29E.2A3; BioLegend), CD285-Alexa Fluor 647 (clone 624915; BD); CD3-Alexa Fluor 700 (clone UCHT1;BD), CD19-Alexa Fluor 700 (clone HIB19; BioLegend), CD36-APC-H7 (clone 5-271; BioLegend), and Integrin α4β7-Alexa Fluor 647 (clone ACT-1; in-house labeled). Antibodies to the next intracellular targets were used: COX-2-PE (clone L128; BD) and TNF-α-BV711 (clone MAb11; BioLegend). Samples were collected in a custom LSRII flow cytometer (BD, USA). Samples were analyzed using FlowJo (Tree Star, San Francisco, CA).
Gene expression
For gene expression analyses, monocytes were enriched using a negative selection kit (StemCell Technologies, Vancouver, Canada). Enriched monocytes (1x105) cells were then plated in 1mL of serum-free media (DMEM, 1% L-glutamine, 1% Penicillin/Streptomycin) supplemented with 5% human AB serum. Cells were incubated for 24h (37°C in 5% CO2) and then total RNA was isolated using TriPure according to manufacturer’s protocol. Transcripts for PTGS2 and MRC1 were assessed by qRT-PCR (ABI Prism 7900 Sequence Detection System and software; Applied Biosystems) as previously described (Shirey MI 2010). The following primer sets were used in these studies: HPRT forward (5’-GTTATGGCGACCCGCAG-3’); HPRT reverse (5’-ACCCTTTCCAAATCCTCAGC-3’); PTGS2 (COX-2) forward: (5’- CCCATGTCAAAACCGAGGTG -3’); PTGS2 (COX-2) reverse (5’- CCGGTGTTGAGCAGTTTTCTC -3’); MRC1 forward (5’- TCCTGGTTTTTGCCTCTGTC -3’); MRC1 reverse (5’- GCACTGGGACTCACTGCAT -3’).
Statistical methods
Differences in the frequencies (percentages) of monocyte subsets at day 0 were analyzed using parametric t-tests (2-sided, paired). Comparisons of marker expression (percentages or mean fluorescence intensity -MFI-) at day 0 between the diverse monocyte subsets were also performed using parametric t-tests (2-sided, paired). In this manuscript, as all TD volunteers had a slightly different timeframe until typhoid diagnosis, we decided to create an artificial timepoint encompassing the average of 48h-TD, 96h-TD and the time point immediately before, for each volunteer. Therefore, this artificial timepoint included the day at which the criteria for typhoid diagnosis were reached. We named this combined timepoint, Time-of-Disease (ToD) to differentiate from time of diagnosis. Changes in expression of markers in TD volunteers following challenge compared to day 0 (pre-challenge) with the average MFIs, percentages or transcript fold-changes at ToD were done using parametric t-tests (2-sided, paired). In TD volunteers, the more profound changes in marker expression were noted during ToD. Comparisons of marker expressions at steady state (day 0) between TD and NoTD volunteers were also assessed using parametric t-tests (2-sided, unpaired). Comparisons of the cell clusters rendered by UMAP, X-shift and Cluster Explorer, between Day 0 and 48h-TD, were done using unpaired, parametric, 2-sided t-tests. All statistical analyses were performed in GraphPad Prism software (GraphPad Software Inc., USA). P values <0.05, without adjustment for multiple comparisons, were considered statistically significant.
Results
Monocyte subset frequencies after S. Typhi challenge
In humans, CD14 and CD16 define three monocyte subsets in circulation, which include Classical (CM; CD14+ CD16-), Intermediate (IM; CD14+ CD6+) and Non-Classical (NCM; CD14- CD16+) Monocytes (Figure 1A; Supplementary Figure 1A). Consistent with the literature, in the volunteers participating in this study, CM comprised around 80% of the monocytes, while IM and NCM each encompassed 5-10% of the monocyte subsets (Figure 1B). Before wt S. Typhi challenge (Day 0), TD and NoTD volunteers showed similar percentages of monocyte subsets (Supplementary Figures 1B–D). Following challenge, TD volunteers were typhoid diagnosis criteria between days 9 and 14. Of note, every volunteer showed a slightly different time frame in which they reached diagnosis criteria. TD volunteers provided additional blood samples at 48- and 96-hours post-typhoid diagnosis (48h-TD and 96h-TD, respectively). We used these two time points plus the one immediately previous, to align the data. In NoTD volunteers, days 9 and 14 were aligned with the Pre-TD and the data point immediately after 96h-TD, respectively. These alignments were used to visualize the TD and NoTD data simultaneously, as shown in Figures 1C–E, which display the percentage of monocytes subsets. IM of TD volunteers showed a clear increase in their percentage between Pre-TD, 48h-TD and 96h-TD timepoints (Figure 1D) while the other two subsets showed limited variations (Figures 1C, E). NoTD volunteers showed minimal changes in the percentages of any monocyte subset. To determine whether the increase of IM in TD volunteers was statistically significant, we compared pre-challenge data (Day 0) to a data point that averaged 48h-TD, 96h-TD and the time point immediately before. This artificial timepoint encompassed the day at which the criteria for typhoid diagnosis were reached. This combined timepoint was named Time-of-Disease (ToD) to differentiate it from time of diagnosis (examples are shown in Supplementary Figures 1E–H). We observed significant increases in the frequencies of IM during ToD (Figure 1F). In contrast, no differences in the percentages of monocytes subsets were observed in NoTD volunteers (Supplementary Figure 1I).
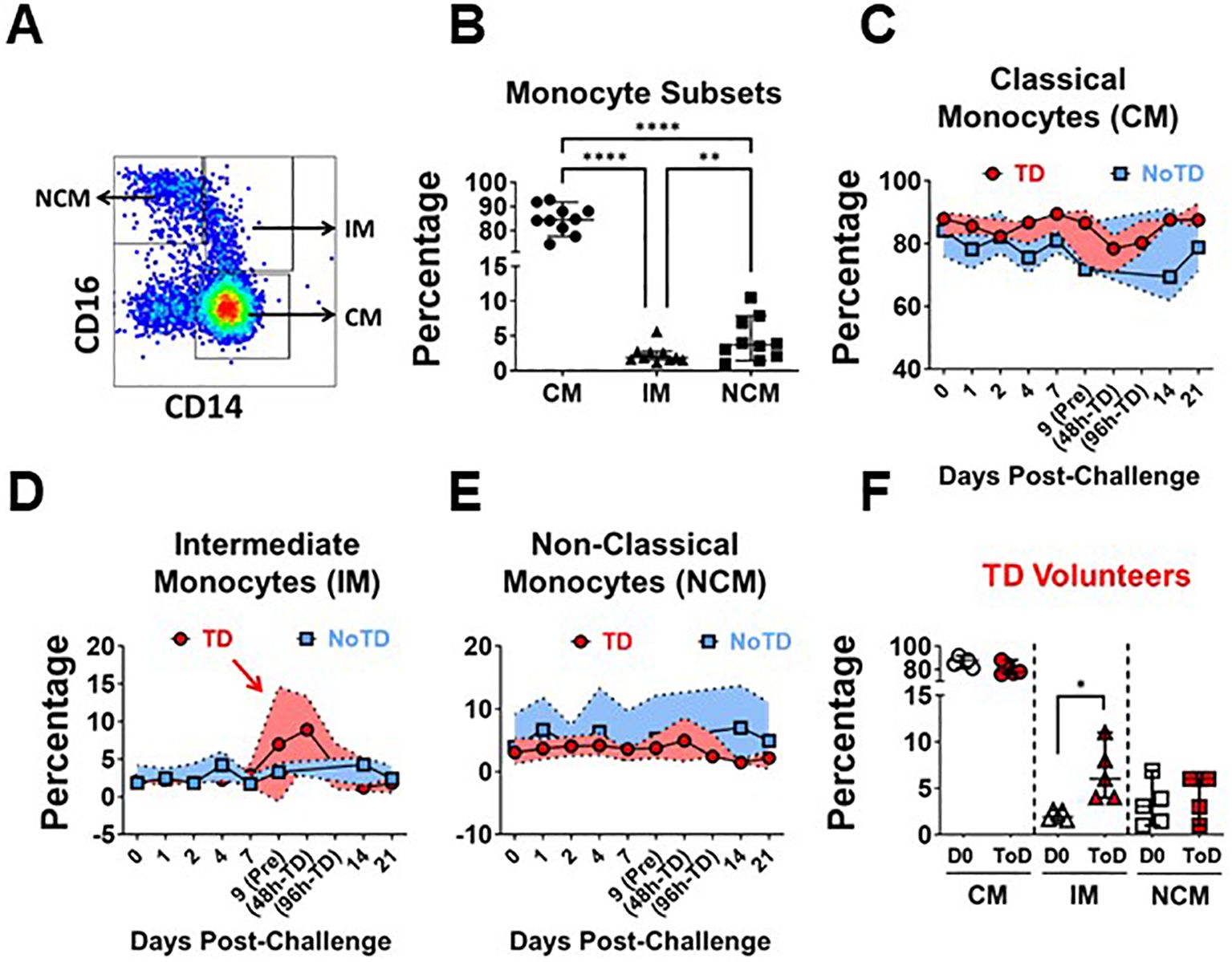
Figure 1. Frequency (percentage) of monocyte subsets before and after wt S. Typhi challenge. (A) displays the three circulating monocyte subsets identified by CD14 and CD16. (B) shows the percentage distribution of these subsets in the volunteers studied. (C–E) show the percentages of the populations overtime for CM, IM and NCM, respectively, in TD (red) and NoTD (blue) volunteers. (F) shows the comparison of the percentage of the monocytes subsets between Day 0 (pre-challenge) vs. Time-of-Disease (ToD). CM, Classical Monocytes; IM, Intermediate Monocytes; NCM, Non-classical Monocytes; ToD, Time of disease. Plots (B, F) show the Median and 95CI. (C–E) show median and interquartile range. The p values were calculated using paired t-tests (2-sided). *p<0.05, **p<0.005, ****p<0.0001.
Changes in pattern recognition receptors by monocyte subsets
Studies have shown differential gene transcription and protein expression profiles between the monocyte subsets (38, 39, 41, 53–55); however, little is known about changes induced by S. Typhi infection. Importantly, during S. Typhi hematic dissemination, particularly during the second bacteremia with coincides with febrile illness, the bacteria are likely to encounter blood monocytes. Therefore, we assessed receptors that play a role in the interaction with bacteria, including CD284 (TLR4), CD285 (TLR5) and CD36 (scavenger receptor). Expression of these markers was assessed in all monocyte subsets before (Day 0; Supplementary Figures 2A–C) and after challenge. Following wt S. Typhi challenge, changes in the expression of these markers among TD volunteers were predominant during ToD. CM showed a significant upregulation of CD284, CD285 and CD36. IM showed a significant upregulation of CD285 and NCM an upregulation of CD284 (Figures 2A–C). Upregulation of these molecules suggested that blood monocytes, particularly CM, could have an enhanced ability to bind S. Typhi during the disease days. To determine whether this was the case, we labeled S. Typhi (killed) with a fluorescent dye and exposed monocytes to these bacteria. CM and IM showed enhanced binding capacity for the S. Typhi during ToD, while NCM did not (Figures 2D, E). Supplementary Figure 2D shows differences in avidity for S. Typhi by the monocyte subsets at baseline. CD206 mediates non-opsonic phagocytic uptake of a wide variety of microbes including yeast, fungi, protozoa, and bacteria. This receptor has been shown to bind several bacterial molecules such as LPS and polysaccharides (56). CD206 was assessed by qRT-PCR (MRC1 gene) in enriched monocytes from TD volunteers and we observed enhanced MRC1 expression during ToD (Figure 2F). The results suggested that circulating monocytes of TD volunteers, particularly CM, were more prone to interact with S. Typhi during ToD through modulation of the expression of various receptors.
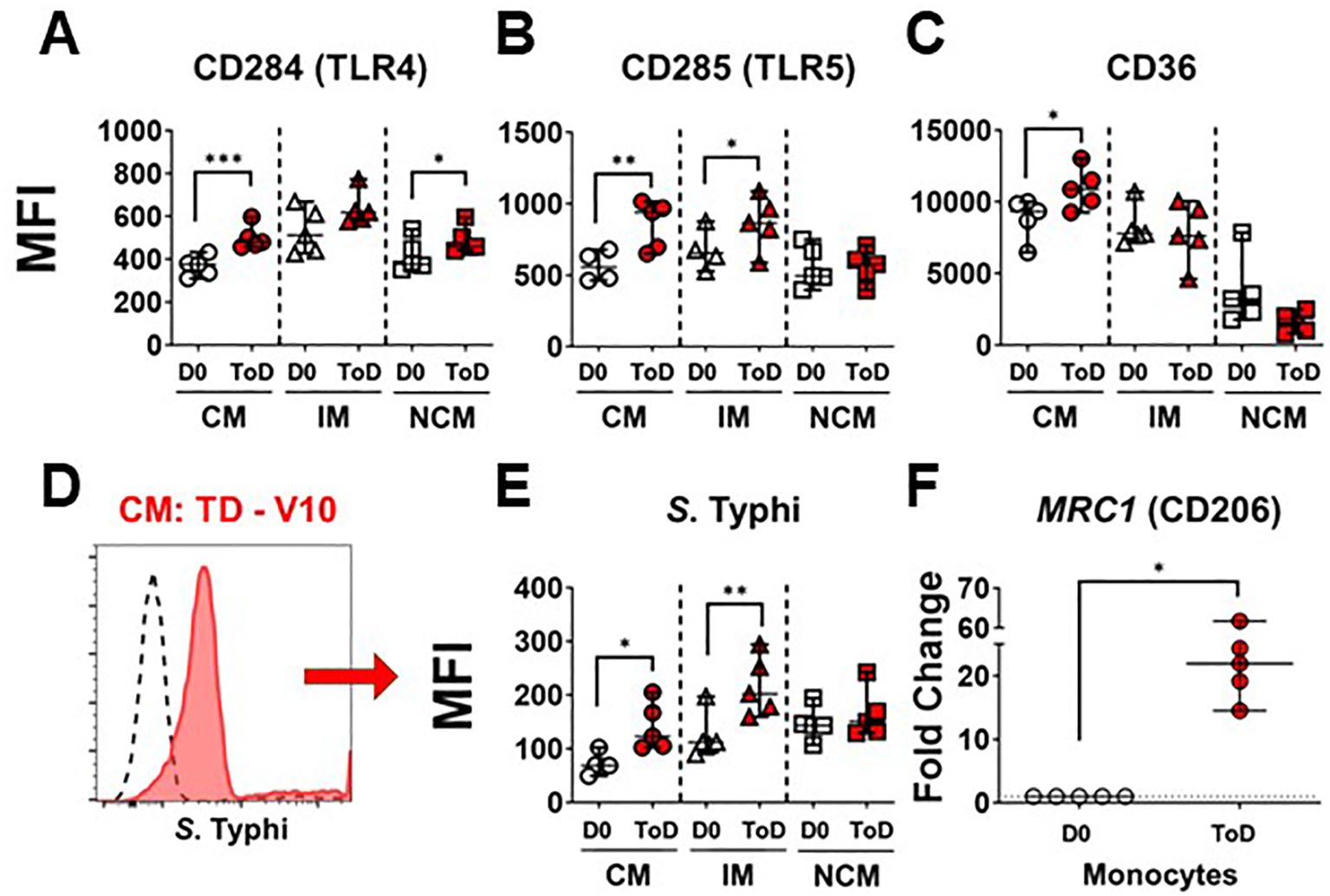
Figure 2. Expression of PRRs in circulating monocyte subsets after wt S. Typhi challenge in TD volunteers. (A–C) show changes in expression (MFI) of CD284 (TLR4), CD285 (TLR5) and CD36, respectively, after challenge in TD volunteers. The data compares D0 vs. ToD in the monocyte subsets. (D) displays an overlay of CM binding S. Typhi at baseline (dotted line histogram) and ToD (ref filled histogram) as an example. (E) displays compiled data of S. Typhi binding by the diverse monocyte subsets in TD volunteers. (F) shows the fold change of MRC1 during ToD by enriched monocytes from TD volunteers. MFI, Median Fluorescence Intensity; CM, Classical Monocytes; IM, Intermediate Monocytes; NCM, Non-classical Monocytes; ToD, Time of disease. All panels show the median and 95 CI. The p values were calculated using paired t-tests (2-sided). *p<0.05, **p<0.005, ***p<0.001.
Inflammation and regulatory markers in monocyte subsets
The enhanced avidity for S. Typhi by CM and IM during ToD in TD volunteers prompted us to investigate whether these cells also exhibited upregulation of inflammatory markers by assessing expression of TNF-α production following wt challenge. The percentage of CM and IM producing this cytokine increased significantly during ToD (Figure 3A; Supplementary Figure 3F). It is worth noting that at steady state IM produced significatively more TNF-α than CM or NCM (Supplementary Figure 3A). We also investigated expression of COX-2, another key inflammatory marker. Contrary to the expected, after wt S. Typhi challenge, the expression of COX-2 did not change in any of the monocyte subsets in TD volunteers during ToD (Figure 3B). We confirmed these results by qRT-PCR in enriched monocytes from TD volunteers and showed that PTGS2 (COX-2) transcripts did not increase during ToD (Supplementary Figure 3E). Basal levels of COX-2 expression in the diverse monocyte subsets are shown in Supplementary Figure 3B.
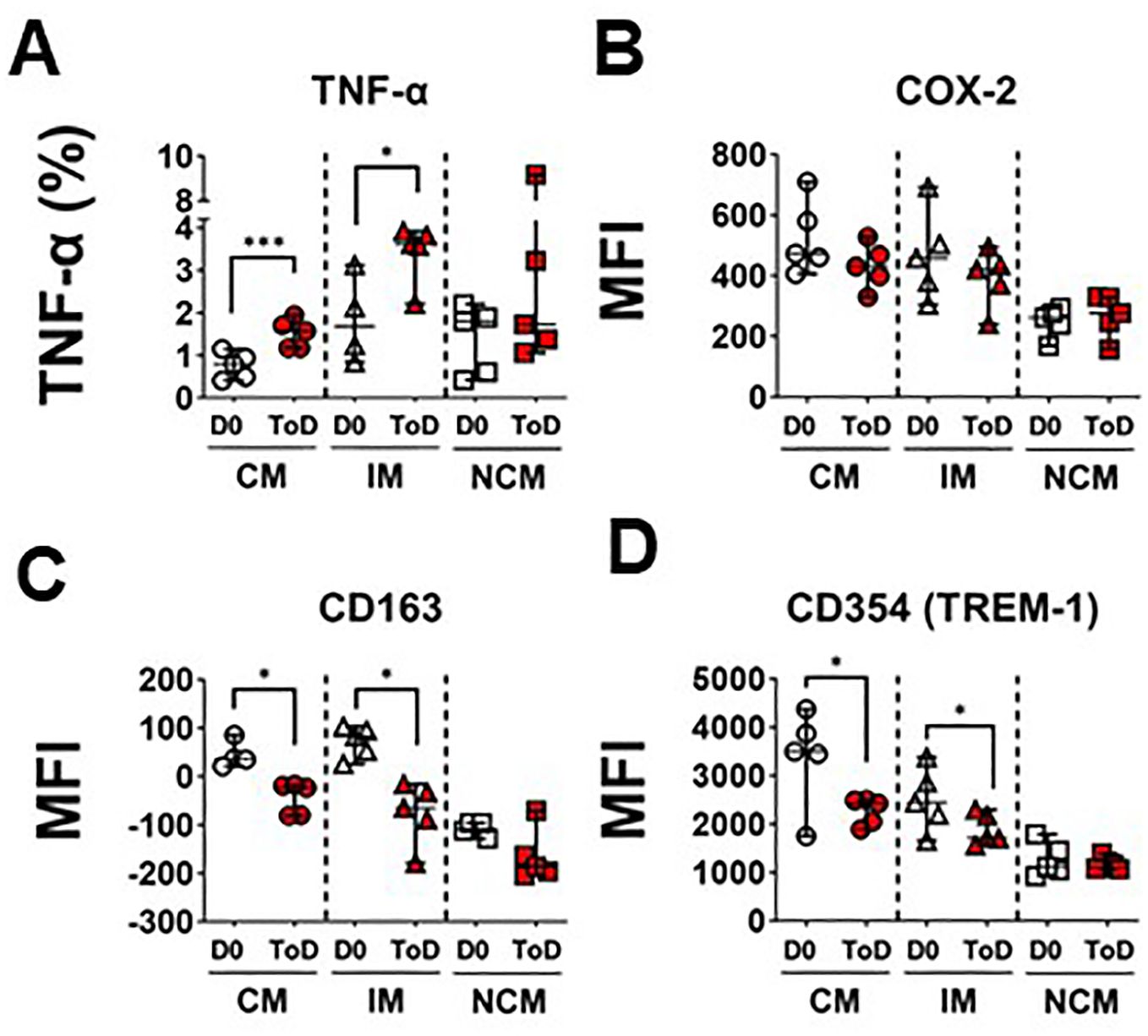
Figure 3. Markers of inflammation and their regulation in circulating monocyte subsets after wt S. Typhi challenge in TD volunteers. (A) displays changes in the percentage of cells producing TNF-α at baseline (D0) and ToD in circulating monocyte subsets in TD volunteers. (B) displays expression of COX-2 (MFI) at day 0 and during ToD. (C) shows changes in the expression of CD163 between D0 and ToD in TD volunteers. (D) shows changes in the expression of CD354 (TREM-1) between D0 and ToD in TD volunteers. MFI, Median Fluorescence Intensity; CM, Classical Monocytes; IM, Intermediate Monocytes; NCM, Non-classical Monocytes; ToD, Time of disease. All panels, except (A) show the median and 95 CI. The p values were calculated using paired t-tests (2-sided). *p<0.05, ***p<0.001.
We also investigated whether mechanisms that monocytes use to limit dysregulated inflammation were activated. For this, we assessed surface expression of CD163 after challenge. In monocytes, CD163 promotes bacteria-induced pro-inflammatory cytokine production; however, extracellular TLR activation leads to shedding of CD163 as soluble CD163 (sCD163). Shedding of this molecule may be a mechanism to decrease acute and severe monocyte activation and inflammation. Interestingly, following wt S. Typhi challenge, surface expression of CD163 was reduced on CM and IM monocytes during ToD (Figure 3C). Moreover, since the mechanism of CD163 cleavage from the surface is mediated by TLR activation, we investigated whether there was downregulation of receptors involved in amplification of the TLR signal, such CD354 (TREM-1). After wt S. Typhi challenge, the expression of CD354 was significantly reduced in CM and IM during ToD (Figure 3D). Steady state expression of CD163 and CD354 is shown in Supplementary Figures 3C, D, respectively. Taken together, the data suggest that in TD volunteers during ToD S. Typhi induced expression of inflammatory markers in CM and IM, but inflammation was controlled, at least in part, by mechanisms involved in dampening the inflammatory response.
Interaction with cells of the adaptive immune response and homing to GALT
TD volunteers showed induction of an inflammatory response by CM and IM during ToD, albeit controlled, as suggested by the reduction in the expression of CD163 and CD354. This also suggested that monocytes were being activated and likely upregulated molecules involved in their interaction with cells of the adaptive immune response. Therefore, we assessed expression of proteins involved in interaction with T cells including HLA-DR, CD274 (PD-L1) and CD86 (Figure 4). Upregulation of these proteins is associated with activation of antigen presenting cells, including monocyte/macrophages. Even though CD274 (PD-L1) is associated with regulation of T cell function, this protein is highly upregulated during the acute infection phase and therefore considered an activation marker (57). Following wt challenge, all the monocyte subsets in TD volunteers upregulated these molecules, except for CD86 by IM. (Figures 4A–C). Baseline expression levels of these proteins in the monocyte subsets are shown in Supplementary Figure 4A–C. At baseline (D0) IM expressed the highest levels of HLA-DR, CD274 and CD86, followed by NCM. Conversely, CM expressed the lowest levels of these receptors. We also assessed expression of CD257 (BAFF), which allows monocyte interaction with B cells. After challenge, this molecule was significantly upregulated by CM during ToD (Figure 4D). Baseline expression of CD257 is shown in Supplementary Figure 4D. At baseline, IM monocytes showed the highest expression of this marker, followed by NCM and CM. Taken together, these data suggest that monocytes, particularly CM were activated and ready to interact with T and B cells.
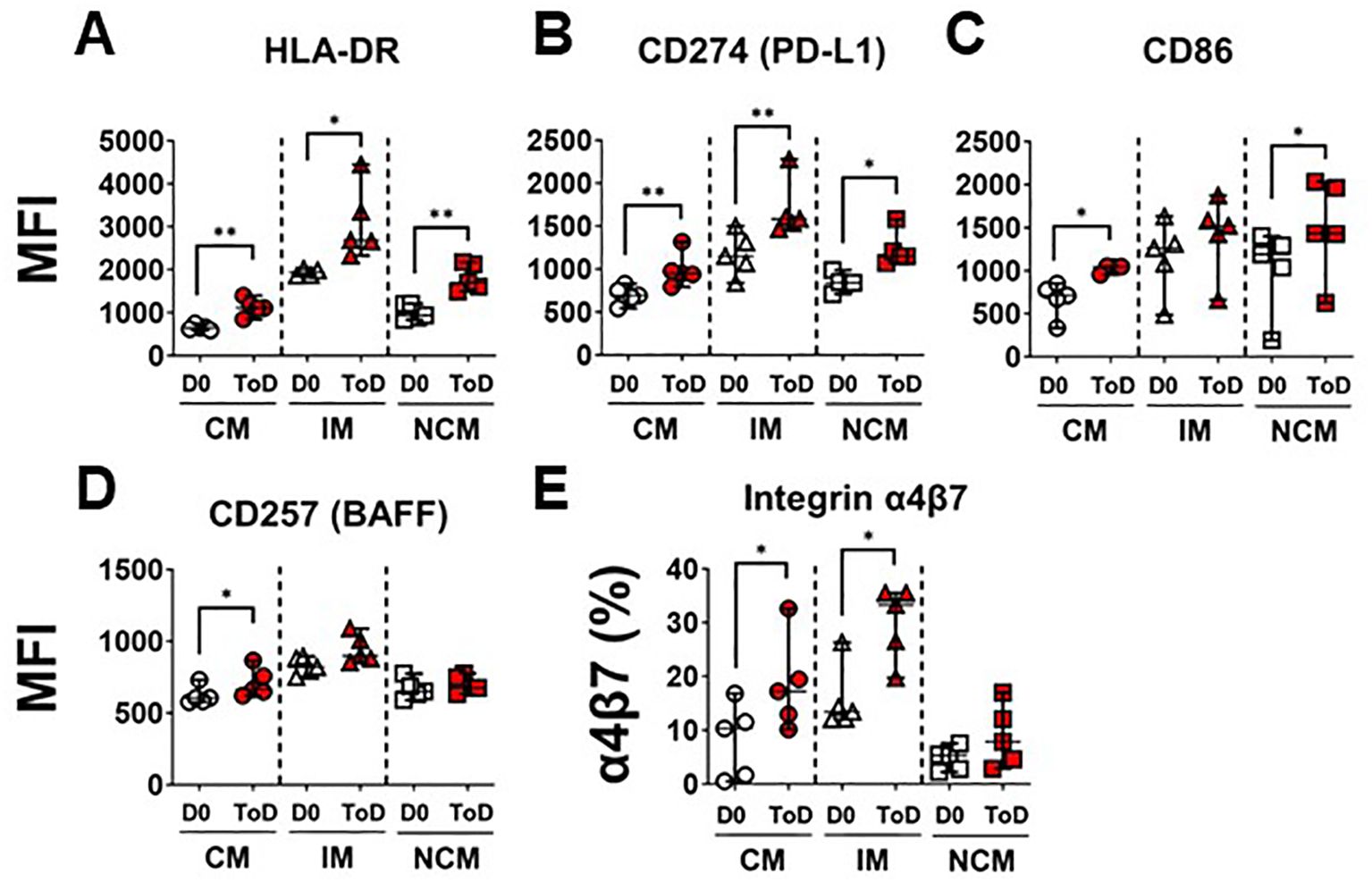
Figure 4. Monocyte receptors for interactions with T and B cells. (A–C) display changes in expression (MFI) of monocyte receptors that interact with T cells, including HLA-DR (A), CD274 (B) and CD86 (C). (D) shows changes in the expression (MFI) of CD257, which monocytes use to interact with B cells. (E) display changes in expression (percentage) integrin α4β7. Changes are shown between D0 and ToD in TD volunteers. MFI, Median Fluorescence Intensity; CM, Classical Monocytes; IM, Intermediate Monocytes; NCM, Non-classical Monocytes; ToD, Time of disease. All panels, except (A), show the median and 95 CI. The p values were calculated using paired t-tests (2-sided). *p<0.05, **p<0.005.
As indicated above, CM leave circulation and migrate to various tissues to become terminally differentiated cells. Pathogens that initially infect and gain access through the gut mucosal, like S. Typhi, are expected to elicit migration of activated immune cells to gut associated lymphoid tissues (GALT). Therefore, we assessed expression of integrin α4β7 by the monocyte subsets. Integrin α4β7 binds to the Mucosal vascular Addressin Cell Adhesion Molecule-1 (MAdCAM-1), which is expressed on Peyer patches, mesenteric lymph nodes, and lamina propria venules; therefore, facilitating the migration of immune cells to GALT. Following challenge, CM and IM of TD volunteers increased expression of this homing molecule suggesting that these cells are primed to migrate to the GALT (Figure 4E; Supplementary Figure 4F). Baseline expression of integrin α4β7 is shown in Supplementary Figure 4E. At baseline, IM had the highest frequency of cells expressing integrin α4β7.
To further explore the immunophenotypic characteristics of circulating monocytes, we performed unsupervised analyses in concatenated samples from Day 0 and 48h-TD from four TD volunteers. Timepoint 48-TD was selected because this showed the most significant changes among TD volunteers. This analysis included the 3 monocytes subsets (monocyte population). Samples from one volunteer where not included as one marker was omitted by mistake during staining and therefore could not be concatenated appropriately (Figure 5). Cells clusters rendered by a dimensionality reduction analysis (UMAP) were assessed using X-shift (Figure 5A). Seven cell clusters were identified (Figure 5B) and the marker expression composition assessed using Cluster Explorer. NCM and IM were represented in clusters 1 and 5, respectively, and all other clusters were part of the CM population (Figure 5C). Concatenated samples were deconvoluted to determine the clusters present on the UMAP at day 0 and 48h-TD for each volunteer. Figure 5D shows data from one representative volunteer highlighting prominent changes. For example, cluster 7 was present at day 0, but not at 48h-TD. On the other hand, clusters 3 and 4 were not present at day 0 but predominated at 48h-TD. The frequency (percentage) of clusters at day 0 and 48h-TD for each volunteer was compared. Clusters 3, 4 and 5 increased significantly in frequency at TD-48h, while clusters 2 and 7 decreased (Figures 5E, F). Clusters 3 and 4 were part of the CM population, appeared at 48h-TD, showed an activated phenotype (HLA-DR high and CD163 low) and high capacity to migrate to GALT (high integrin α4β7 expression). Of these clusters, clusters 3 and 6 (conventional monocytes) showed high binding for S. Typhi. Clusters 1 and 6 did not show changes between day 0 and 48-TD (Supplementary Figure 6H). Overall, these data show that monocyte responses to S. Typhi infection are very dynamic and involve CM and IM. The data suggest that CM are the main population involved in these responses and that defined CM-subsets are likely to be the dominant, but this aspect needs to be better defined in future studies. IM were involved in responses to S. Typhi during ToD to a lower extent than CM.
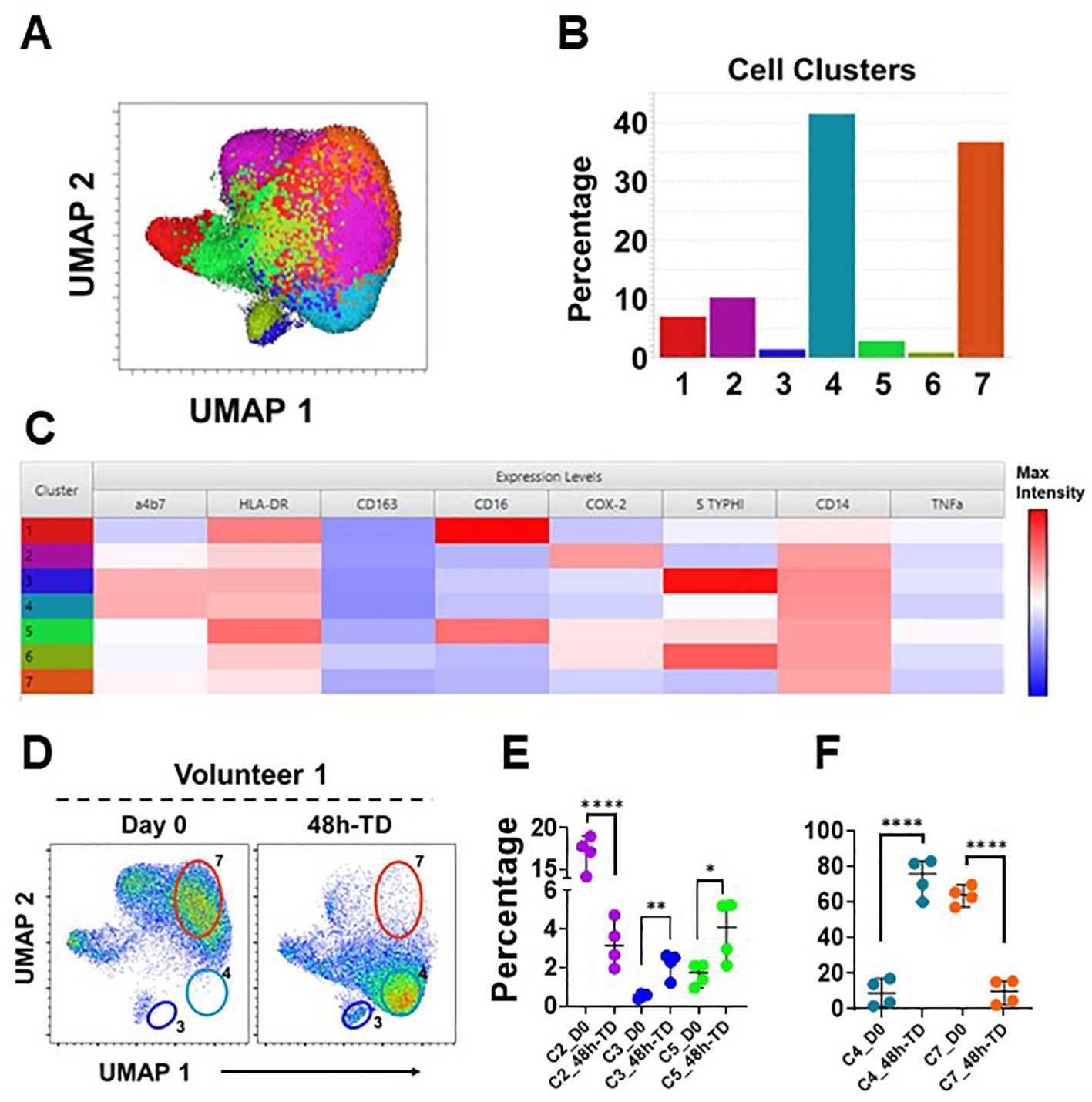
Figure 5. Unsupervised analysis. (A) shows a UMAP representation of monocytes (Day 0 and 48h-TD concatenated from 4 TD volunteers) overlayed with the seven cell clusters identified by X-Shift. (B) displays the size (percentage) of the clusters and (C) shows their composition and expression intensity of the markers in each cluster. (D) shows the UMAP deconvoluted on a representative volunteer at day 0 (left) and 48h-TD (right). The position of some clusters (3, 4 and 7) is shown to highlight the differences between these timepoints. (E) shows the changes in size (percentage) of clusters 2, 3 and 5 between Day 0 and 48h-TD. (F) shows similar changes, but in clusters 4 and 7. (E, F) show the median and 95 CI. The p values were calculated using unpaired t-tests (2-sided). *p<0.05, **p<0.005, ****p<0.0001.
Discussion
S. Typhi is a human restricted pathogen and the agent responsible for typhoid fever. These bacteria enter the host via the intestinal mucosa and after infecting local cells (gut macrophages and other phagocytes) disseminate causing a systemic disease. Circulating monocytes show great plasticity and can differentiate into various cell types (e.g., macrophages and dendritic cells -DC-) once they reach the tissues. Various monocytes subsets have been described in circulation including CM, IM and NCM. The re-establishment of a S. Typhi model has allowed to study in detail several arms of the immune system in those volunteers that reached typhoid diagnosis (TD) criteria. Importantly, typhoid fever reaches the blood seeding reticuloendothelial organs (primary bacteremia) and after ~8-14 days incubation, causes a secondary bacteremia that coincides with febrile illness. Bone marrow is part of the reticuloendothelial system and a site that houses monocyte precursors (mature monocytes remain in bone marrow for 2-3 days before being released into circulation). Therefore, S. Typhi can encounter monocytes either in bone marrow or in circulation. Unlike other tissues, which have a population of self-renewing tissue macrophages, all gut macrophages are derived from circulating monocytes. Therefore, we investigated whether volunteers that develop TD showed significant changes in circulating monocytes and whether there were differences within the diverse subsets. Typhoid fever causes low-level bacteremia (1-10 CFU/ml of blood) and therefore the number of monocytes that encounter the bacteria might be limited. Despite this, it is quite remarkable the profound changes noted in circulating monocytes, suggesting that the bacteria in blood are only partially responsible for these changes. It is likely that bacterial products or metabolites (derived from infected cells or other immune cells) could be involved in amplifying the monocyte activation/response.
We observed that monocyte markers involved in interaction with bacterial antigens (CD284, CD285, CD36, MRC1) were elevated in TD volunteers during ToD, particularly in the CM and IM subsets (Figures 2A–C, F). Moreover, this was associated with an enhanced ability of monocytes to bind S. Typhi (Figure 2E). The enhanced ability of monocytes to bind S. Typhi during ToD agrees with a previous report from our group (50). In the current study, we extended the assessments to include monocyte subsets and showed that CM and IM are the main subpopulations binding S. Typhi. We expected an increased expression of CD284 and CD285, as these receptors recognize LPS and flagellin, both present in S. Typhi. Less clear was the role of CD36, a scavenger receptor. CD36 facilitates innate immune responses against gram-positive bacteria such as S. aureus and S. pneumoniae. However, the role of CD36 in the defense against Gram negative bacteria has been less evident (58–61). It was recently shown that the absence of this receptor significantly increased mice susceptibility to acute intrapulmonary infection by K. pneumonia, suggesting a role for CD36 in controlling infections to Gram-negative bacteria (62). Our data provides evidence that CD36 also plays a role during S. Typhi infection, another Gram-negative pathogen (Figure 2C).
Another important molecule for monocytes/macrophages is CD206. CD206 recognizes and binds a wide range of ligands, including peptide hormones, lysosomal hydrolases, mannose, fucose, and collagen, along with allergens and microbial products including CpG DNA. CD206 also mediates the non-opsonic phagocytic uptake by macrophages of a wide variety of microbes including yeast, fungi, protozoa, and bacteria. In the case of bacterial infections, it is known that CD206 binds LPS and polysaccharides (56). Moreover, soluble CD206 (sCD206) has been identified in the ascites fluid from cirrhosis patients with spontaneous bacterial peritonitis and is considered a marker of macrophage activation in these cases (63). To our knowledge, no data regarding the role of this molecule in S. Typhi infection has been reported. Herein we show that enriched monocytes from TD volunteers increased expression of MRC1 (CD206), probably the result of interactions with antigens from the bacteria, further suggesting activation of these cells.
TD volunteers showed evidence that S. Typhi induced inflammation, as the percentage of CM and IM producing TNF-α was upregulated during ToD. However, it appears that factors involved in controlling inflammation were also at play since CD163 was downregulated during ToD. CD163 is a member of the scavenger receptor cysteine-rich (SRCR) superfamily class B, is exclusively expressed by monocytes and macrophages and is able to bind both Gram-positive and Gram-negative organisms (64). Extracellular TLR activation leads to shedding of CD163 as soluble CD163 (sCD163). Because surface CD163 on macrophages functions as an innate immune receptor for bacteria (64), its shedding may be a mechanism to decrease acute and severe monocyte activation and inflammation. Several observations support this contention. For example, physiological doses of LPS acutely stimulate CD163 shedding within 1 hour, followed by increased expression on the surface of monocytes and macrophages 24 hours later (65–67). Moreover, levels of CD163 on monocytes are inversely correlated with the levels of sCD163 found in-vitro in tissue culture medium and in-vivo in plasma (65). Therefore, the decrease in CD163 expression by CM and IM (Figure 3C) in TD volunteers during ToD most likely reflects activation of mechanisms to control the inflammation, possibly triggered by the engagement of TLR4/5 by S. Typhi. Furthermore, since the mechanism of CD163 cleavage from the surface is mediated by TLR activation, we assessed whether receptors involved in amplification of TLR signals (e.g., CD354), were downregulated. We showed that CM and IM exhibited downregulation of this marker, providing further evidence that mechanisms involved in controlling inflammation were activated. The lack of increase in COX-2 expression (Figure 3B; Supplementary Figure 3E) might also be a consequence of these regulatory mechanisms.
The increased expression of HLA-DR, CD274 and CD86 by TD volunteers during ToD confirmed that these cells, particularly CM and IM, were activated and possibly ready to interact with T cells. Additionally, CM and IM showed increased expression of integrin α4β7 suggesting that these cells were migrating to gut lymphoid tissues. Integrin α4β7 upregulation is also in agreement with our previous studies (50). Additionally, these findings suggest that interaction with the bacteria or bacterial products triggered monocyte activation during the systemic dissemination of S. Typhi and before reaching the tissues. Interestingly, little is known about the role of CD257 in Salmonella infection. CD257 is a homotrimer that is found either on the cell surface as a type II transmembrane protein or is released in a soluble form after cleavage (68, 69). CD257 is required for B cell survival, maturation, activation and homeostasis and is found on neutrophils, monocytes, macrophages and dendritic cells (68, 70, 71). Monocytes are one of the major cell types expressing and secreting CD257 during infection and inflammation. A recent report showed that CD257 is important for protection from Salmonella infection in a mouse model (72). We showed that CM exhibited increased CD257 expression on their surface during ToD, suggesting a role for this molecule in Salmonella infections in humans. Future research should address this issue in more detail, including assessment of this cytokine in sera, as this molecule is ultimately released from the cells and is only transiently expressed on the surface of monocytes and other innate cells.
Unsupervised analysis showed that the CM were the main subset responding during TD, as 5 of the 7 cell clusters identified belonged to this subset. Significant changes were identified at the 48h-TD timepoint. Of note, some clusters were present only at day 0, but were either not present or their presence was greatly reduced at the 48hTD time point (e.g., Cluster 7). Other cell clusters behaved in the opposite way, such as cluster 4, which was present only at 48h-TD (Figure 5). Of importance is the identification of small clusters within CM, such as cell cluster 3, whose cells showed an activated phenotype, increased capacity to migrate to GALT and enhanced binding of S. Typhi. Future studies will be directed to investigate the role that these cells play in typhoid disease.
The data collected in this study, despite that involved only 5 TD and 5 noTD volunteers, provided consistent, significant results. Additionally, the data from TD volunteers reproduced aspects we had previously reported in the overall monocyte population (e.g., upregulation of integrin α4β7); therefore, despite the limited number of volunteers the results are consistent across studies (50). The data in the current study suggest that CM are the main monocyte population being activated by S. Typhi during ToD. Importantly, CM are likely the most important population since they are not only the largest monocyte subset, but also most of them appear to have the capacity to migrate to the gut tissues to differentiate into macrophages or DCs. Importantly, IM showed upregulation of most of the markers assessed, including integrin α4β7, which might reflect their transition status towards NCM. Alternatively, it might suggest that during acute infection, IM might be malleable and able to revert to CM or perform similar functions to CM. Of note, in TD volunteers, no marker showed changes compared to day 0, before day 9. It is also important to note that at baseline, each monocyte sub-population expressed diverse degrees of the molecules assessed (Supplementary Figures 2-4), which is in line with previous reports from other groups and reflect their differentiation status (38, 39, 53, 54, 73). Also, at baseline TD and NoTD volunteers showed similar expression levels of the molecules assessed (Supplementary Figure 5). Importantly, the findings in TD volunteers agree with previously published data from our group (50). In this manuscript, we have expanded the observations to include diverse monocyte subsets. One limitation of our study is its descriptive nature. Despite the fact that the human challenge model allows a detailed characterization of the immune response, it does not allow experimental immunological approaches such as depletion of cell populations before infection, cells/genes knock out/in, tissue harvesting, etc. that are common in animal models. To address this issue, we and other groups are developing systems that mimic human tissues to better characterize immunity to S. Typhi and other pathogens at the local level and in lymphoid tissues (74–79).
Finally, NoTD volunteers did not show any changes in the expression of the molecules assessed at any of the timepoints assessed (Supplementary Tables 1-3). Supplementary Figure 1I shows one example of the lack of changes in the markers assessed in NoTD volunteers. It is also important to note that in our previous study we reported that monocytes of NoTD volunteers showed enhanced binding of S. Typhi 24h after infection (Day 1). In the current study we did not detect this phenomenon. The size of the inoculum could be partially responsible for these differences, as in the previous manuscript we reported findings in volunteers that received 104 CFU; while in the current one from subjects that received 103 CFU. A lower inoculum could result in less activation of the mechanisms that lead to enhanced avidity for the bacteria in NoTD volunteers. Another possibility could involve differences in timing of the enhanced S. Typhi binding. If the S. Typhi binding peak in NoTD volunteers that received 103 CFU took place earlier or later than 24h (e.g., 12h or 36h) the current blood sampling could not have captured this phenomenon. Future studies will investigate this aspect in more detail. In sum, we provide new insights into the changes that diverse circulating monocyte subsets undergo after typhoid diagnosis (TD volunteers during ToD), which might be important to control this disease since circulating monocytes, particularly CM, will ultimately become intestinal or tissue macrophages once they reach the gut or other target tissues.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
Ethics statement
The studies involving humans were approved by Oxfordshire Research Ethics Committee A (10/H0604/53). The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
Author contributions
FT: Writing – original draft, Writing – review & editing, Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision. JH: Data curation, Formal analysis, Methodology, Writing – original draft, Writing – review & editing. KS: Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Resources, Writing – original draft, Writing – review & editing. PB: Data curation, Formal analysis, Methodology, Writing – original draft, Writing – review & editing. ML: Resources, Writing – original draft, Writing – review & editing. TD: Resources, Writing – original draft, Writing – review & editing. CW: Resources, Writing – original draft, Writing – review & editing. AP: Resources, Writing – original draft, Writing – review & editing. MS: Conceptualization, Funding acquisition, Investigation, Methodology, Resources, Supervision, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported, in part, by NIAID, NIH, DHHS federal research grants R01 AI036525, U19 AI082655 and U19AI181108 (Cooperative Center for Human Immunology (CCHI)) to MS. FT and KS were supported in part by a Pilot Project funded by the UMB CCHI. This work was also supported in part by funds through the National Cancer Institute Support Grant (CCSG) P30 CA134274. Partial funding for open access was provided by the University of Maryland Health Sciences and Human Services Library's Open Access Fund.
Acknowledgments
We are indebted to the volunteers who participated in the studies. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Allergy and Infectious Diseases or the National Institutes of Health or other granting institutions. We would like to acknowledge Ms. Regina Harley from the CVD Flow and Mass Cytometry Core. Flow cytometry experiments were performed at the Flow Cytometry and Mass Cytometry Core Facility, a shared service of the University of Maryland School of Medicine Center for Innovative Biomedical Resources (CIBR), Baltimore, Maryland.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2024.1454857/full#supplementary-material
References
1. Lozano R, Naghavi M, Foreman K, Lim S, Shibuya K, Aboyans V, et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet. (2012) 380:2095–128. doi: 10.1016/S0140-6736(12)61728-0
2. Mead PS, Slutsker L, Dietz V, McCaig LF, Bresee JS, Shapiro C, et al. Food-related illness and death in the United States. Emerg Infect Dis. (1999) 5:607–25. doi: 10.3201/eid0505.990502
3. Meiring JE, Khanam F, Basnyat B, Charles RC, Crump JA, Debellut F, et al. Typhoid fever. Nat Rev Dis Primers. (2023) 9:71. doi: 10.1038/s41572-023-00480-z
4. Chowdhury AR, Mukherjee D, Chatterjee R, Chakravortty D. Defying the odds: Determinants of the antimicrobial response of Salmonella Typhi and their interplay. Mol Microbiol. (2024) 121:213–29. doi: 10.1111/mmi.15209
5. Carey ME, McCann NS, Gibani MM. Typhoid fever control in the 21st century: where are we now? Curr Opin Infect Dis. (2022) 35:424–30. doi: 10.1097/QCO.0000000000000879
6. van Furth R, Cohn ZA. THE ORIGIN AND KINETICS OF MONONUCLEAR PHAGOCYTES. J Exp Med. (1968) 128:415–35. doi: 10.1084/jem.128.3.415
7. Peters JH, Ruppert J, Gieseler RK, Najar HM, Xu H. Differentiation of human monocytes into CD14 negative accessory cells: do dendritic cells derive from the monocytic lineage? Pathobiology. (1991) 59:122–6. doi: 10.1159/000163628
8. Romani N, Gruner S, Brang D, Kampgen E, Lenz A, Trockenbacher B, et al. Proliferating dendritic cell progenitors in human blood. J Exp Med. (1994) 180:83–93. doi: 10.1084/jem.180.1.83
9. Sallusto F, Lanzavecchia A. Efficient presentation of soluble antigen by cultured human dendritic cells is maintained by granulocyte/macrophage colony-stimulating factor plus interleukin 4 and downregulated by tumor necrosis factor alpha. J Exp Med. (1994) 179:1109–18. doi: 10.1084/jem.179.4.1109
10. Rigamonti A, Villar J, Segura E. Monocyte differentiation within tissues: a renewed outlook. Trends Immunol. (2023) 44:999–1013. doi: 10.1016/j.it.2023.10.005
11. Pang J, Koh TJ. Proliferation of monocytes and macrophages in homeostasis, infection, injury, and disease. J Leukoc Biol. (2023) 114:532–46. doi: 10.1093/jleuko/qiad093
12. Yona S, Kim KW, Wolf Y, Mildner A, Varol D, Breker M, et al. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity. (2013) 38:79–91. doi: 10.1016/j.immuni.2012.12.001
13. Hashimoto D, Chow A, Noizat C, Teo P, Beasley MB, Leboeuf M, et al. Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity. (2013) 38:792–804. doi: 10.1016/j.immuni.2013.04.004
14. Serbina NV, Pamer EG. Monocyte emigration from bone marrow during bacterial infection requires signals mediated by chemokine receptor CCR2. Nat Immunol. (2006) 7:311–7. doi: 10.1038/ni1309
15. Liao CT, Andrews R, Wallace LE, Khan MW, Kift-Morgan A, Topley N, et al. Peritoneal macrophage heterogeneity is associated with different peritoneal dialysis outcomes. Kidney Int. (2017) 91:1088–103. doi: 10.1016/j.kint.2016.10.030
16. Nahrendorf M, Swirski FK, Aikawa E, Stangenberg L, Wurdinger T, Figueiredo JL, et al. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J Exp Med. (2007) 204:3037–47. doi: 10.1084/jem.20070885
17. Zigmond E, Samia-Grinberg S, Pasmanik-Chor M, Brazowski E, Shibolet O, Halpern Z, et al. Infiltrating monocyte-derived macrophages and resident kupffer cells display different ontogeny and functions in acute liver injury. J Immunol. (2014) 193:344–53. doi: 10.4049/jimmunol.1400574
18. Avraham-Davidi I, Yona S, Grunewald M, Landsman L, Cochain C, Silvestre JS, et al. On-site education of VEGF-recruited monocytes improves their performance as angiogenic and arteriogenic accessory cells. J Exp Med. (2013) 210:2611–25. doi: 10.1084/jem.20120690
19. Arnold L, Henry A, Poron F, Baba-Amer Y, van Rooijen N, Plonquet A, et al. Inflammatory monocytes recruited after skeletal muscle injury switch into antiinflammatory macrophages to support myogenesis. J Exp Med. (2007) 204:1057–69. doi: 10.1084/jem.20070075
20. Bain CC, Bravo-Blas A, Scott CL, Perdiguero EG, Geissmann F, Henri S, et al. Constant replenishment from circulating monocytes maintains the macrophage pool in the intestine of adult mice. Nat Immunol. (2014) 15:929–37. doi: 10.1038/ni.2967
21. Bain CC, Schridde A. Origin, differentiation, and function of intestinal macrophages. Front Immunol. (2018) 9:2733. doi: 10.3389/fimmu.2018.02733
22. Bernardo D, Marin AC, Fernandez-Tome S, Montalban-Arques A, Carrasco A, Tristan E, et al. Human intestinal pro-inflammatory CD11c(high)CCR2(+)CX3CR1(+) macrophages, but not their tolerogenic CD11c(-)CCR2(-)CX3CR1(-) counterparts, are expanded in inflammatory bowel disease. Mucosal Immunol. (2018) 11:1114–26. doi: 10.1038/s41385-018-0030-7
23. Muller PA, Matheis F, Mucida D. Gut macrophages: key players in intestinal immunity and tissue physiology. Curr Opin Immunol. (2020) 62:54–61. doi: 10.1016/j.coi.2019.11.011
24. Yip JLK, Balasuriya GK, Spencer SJ, Hill-Yardin EL. The role of intestinal macrophages in gastrointestinal homeostasis: heterogeneity and implications in disease. Cell Mol Gastroenterol Hepatol. (2021) 12:1701–18. doi: 10.1016/j.jcmgh.2021.08.021
25. Harrington KA, Hormaeche CE. Expression of the innate resistance gene Ity in mouse Kupffer cells infected with Salmonella typhimurium in vitro. Microb Pathog. (1986) 1:269–74. doi: 10.1016/0882-4010(86)90051-3
26. Maskell DJ, Hormaeche CE, Harrington KA, Joysey HS, Liew FY. The initial suppression of bacterial growth in a salmonella infection is mediated by a localized rather than a systemic response. Microb Pathog. (1987) 2:295–305. doi: 10.1016/0882-4010(87)90127-6
27. Mastroeni P, Harrison JA, Robinson JH, Clare S, Khan S, Maskell DJ, et al. Interleukin-12 is required for control of the growth of attenuated aromatic-compound-dependent salmonellae in BALB/c mice: role of gamma interferon and macrophage activation. Infect Immun. (1998) 66:4767–76. doi: 10.1128/IAI.66.10.4767-4776.1998
28. Manesh A, Meltzer E, Jin C, Britto C, Deodhar D, Radha S, et al. Typhoid and paratyphoid fever: a clinical seminar. J Travel Med. (2021) 28:1–13. doi: 10.1093/jtm/taab012
29. Watson KC. Laboratory and clinical investigation of recovery of Salmonella typhi from blood. J Clin Microbiol. (1978) 7:122–6. doi: 10.1128/jcm.7.2.122-126.1978
30. Darton TC, Zhou L, Blohmke CJ, Jones C, Waddington CS, Baker S, et al. Blood culture-PCR to optimise typhoid fever diagnosis after controlled human infection identifies frequent asymptomatic cases and evidence of primary bacteraemia. J Infect. (2017) 74:358–66. doi: 10.1016/j.jinf.2017.01.006
31. Zhou L, Pollard AJ. A novel method of selective removal of human DNA improves PCR sensitivity for detection of Salmonella Typhi in blood samples. BMC Infect Dis. (2012) 12:164. doi: 10.1186/1471-2334-12-164
32. Coleman W, Buxton BH. The bacteriology of the blood in convalescence from typhoid fever. With a theory of the pathogenesis of the disease. J Med Res. (1909) 21:83–93.
33. Passlick B, Flieger D, Ziegler-Heitbrock HW. Identification and characterization of a novel monocyte subpopulation in human peripheral blood. Blood. (1989) 74:2527–34. doi: 10.1182/blood.V74.7.2527.2527
34. Wong KL, Tai JJ, Wong WC, Han H, Sem X, Yeap WH, et al. Gene expression profiling reveals the defining features of the classical, intermediate, and nonclassical human monocyte subsets. Blood. (2011) 118:e16–31. doi: 10.1182/blood-2010-12-326355
35. Sunderkotter C, Nikolic T, Dillon MJ, Van Rooijen N, Stehling M, Drevets DA, et al. Subpopulations of mouse blood monocytes differ in maturation stage and inflammatory response. J Immunol. (2004) 172:4410–7. doi: 10.4049/jimmunol.172.7.4410
36. Yrlid U, Jenkins CD, MacPherson GG. Relationships between distinct blood monocyte subsets and migrating intestinal lymph dendritic cells in vivo under steady-state conditions. J Immunol. (2006) 176:4155–62. doi: 10.4049/jimmunol.176.7.4155
37. Thomas GD, Hanna RN, Vasudevan NT, Hamers AA, Romanoski CE, McArdle S, et al. Deleting an nr4a1 super-enhancer subdomain ablates ly6C(low) monocytes while preserving macrophage gene function. Immunity. (2016) 45:975–87. doi: 10.1016/j.immuni.2016.10.011
38. Patel AA, Zhang Y, Fullerton JN, Boelen L, Rongvaux A, Maini AA, et al. The fate and lifespan of human monocyte subsets in steady state and systemic inflammation. J Exp Med. (2017) 214:1913–23. doi: 10.1084/jem.20170355
39. Ginhoux F, Jung S. Monocytes and macrophages: developmental pathways and tissue homeostasis. Nat Rev Immunol. (2014) 14:392–404. doi: 10.1038/nri3671
40. Auffray C, Fogg D, Garfa M, Elain G, Join-Lambert O, Kayal S, et al. Monitoring of blood vessels and tissues by a population of monocytes with patrolling behavior. Science. (2007) 317:666–70. doi: 10.1126/science.1142883
41. Wong KL, Yeap WH, Tai JJ, Ong SM, Dang TM, Wong SC. The three human monocyte subsets: implications for health and disease. Immunol Res. (2012) 53:41–57. doi: 10.1007/s12026-012-8297-3
42. Poehlmann H, Schefold JC, Zuckermann-Becker H, Volk HD, Meisel C. Phenotype changes and impaired function of dendritic cell subsets in patients with sepsis: a prospective observational analysis. Crit Care. (2009) 13:R119. doi: 10.1186/cc7969
43. Skrzeczynska J, Kobylarz K, Hartwich Z, Zembala M, Pryjma J. CD14+CD16+ monocytes in the course of sepsis in neonates and small children: monitoring and functional studies. Scand J Immunol. (2002) 55:629–38. doi: 10.1046/j.1365-3083.2002.01092.x
44. Zawada AM, Rogacev KS, Rotter B, Winter P, Marell RR, Fliser D, et al. SuperSAGE evidence for CD14++CD16+ monocytes as a third monocyte subset. Blood. (2011) 118:e50–61. doi: 10.1182/blood-2011-01-326827
45. Kim OY, Monsel A, Bertrand M, Coriat P, Cavaillon JM, Adib-Conquy M. Differential down-regulation of HLA-DR on monocyte subpopulations during systemic inflammation. Crit Care. (2010) 14:R61. doi: 10.1186/cc8959
46. Monneret G, Lepape A, Voirin N, Bohe J, Venet F, Debard AL, et al. Persisting low monocyte human leukocyte antigen-DR expression predicts mortality in septic shock. Intensive Care Med. (2006) 32:1175–83. doi: 10.1007/s00134-006-0204-8
47. Genel F, Atlihan F, Ozsu E, Ozbek E. Monocyte HLA-DR expression as predictor of poor outcome in neonates with late onset neonatal sepsis. J Infect. (2010) 60:224–8. doi: 10.1016/j.jinf.2009.12.004
48. Caille V, Chiche JD, Nciri N, Berton C, Gibot S, Boval B, et al. Histocompatibility leukocyte antigen-D related expression is specifically altered and predicts mortality in septic shock but not in other causes of shock. Shock. (2004) 22:521–6. doi: 10.1097/01.shk.0000143410.63698.57
49. Waddington CS, Darton TC, Jones C, Haworth K, Peters A, John T, et al. An outpatient, ambulant-design, controlled human infection model using escalating doses of salmonella typhi challenge delivered in sodium bicarbonate solution. Clin Infect Diseases. (2014) 58:1230–40. doi: 10.1093/cid/ciu078
50. Toapanta FR, Bernal PJ, Fresnay S, Darton TC, Jones C, Waddington CS, et al. Oral wild-type salmonella typhi challenge induces activation of circulating monocytes and dendritic cells in individuals who develop typhoid disease. PloS Negl Trop diseases. (2015) 9:e0003837. doi: 10.1371/journal.pntd.0003837
51. Toapanta FR, Ross TM. Impaired immune responses in the lungs of aged mice following influenza infection. Respir Res. (2009) 10:112. doi: 10.1186/1465-9921-10-112
52. Toapanta FR, Bernal PJ, Sztein MB. Diverse phosphorylation patterns of B cell receptor-associated signaling in naive and memory human B cells revealed by phosphoflow, a powerful technique to study signaling at the single cell level. Front Cell Infect Microbiol. (2012) 2:128. doi: 10.3389/fcimb.2012.00128
53. Boyette LB, Macedo C, Hadi K, Elinoff BD, Walters JT, Ramaswami B, et al. Phenotype, function, and differentiation potential of human monocyte subsets. PloS One. (2017) 12:e0176460. doi: 10.1371/journal.pone.0176460
54. Hamers AAJ, Dinh HQ, Thomas GD, Marcovecchio P, Blatchley A, Nakao CS, et al. Human monocyte heterogeneity as revealed by high-dimensional mass cytometry. Arterioscler Thromb Vasc Biol. (2019) 39:25–36. doi: 10.1161/ATVBAHA.118.311022
55. Yang J, Zhang L, Yu C, Yang XF, Wang H. Monocyte and macrophage differentiation: circulation inflammatory monocyte as biomarker for inflammatory diseases. biomark Res. (2014) 2:1. doi: 10.1186/2050-7771-2-1
56. Zamze S, Martinez-Pomares L, Jones H, Taylor PR, Stillion RJ, Gordon S, et al. Recognition of bacterial capsular polysaccharides and lipopolysaccharides by the macrophage mannose receptor. J Biol Chem. (2002) 277:41613–23. doi: 10.1074/jbc.M207057200
57. Li J, Tang Z, Xie M, Hang C, Yu Y, Li C. Association between elevation of plasma biomarkers and monocyte dysfunction and their combination in predicting sepsis: An observational single-centre cohort study. Innate Immun. (2020) 26:514–27. doi: 10.1177/1753425920926602
58. Hoebe K, Georgel P, Rutschmann S, Du X, Mudd S, Crozat K, et al. CD36 is a sensor of diacylglycerides. Nature. (2005) 433:523–7. doi: 10.1038/nature03253
59. Sharif O, Matt U, Saluzzo S, Lakovits K, Haslinger I, Furtner T, et al. The scavenger receptor CD36 downmodulates the early inflammatory response while enhancing bacterial phagocytosis during pneumococcal pneumonia. J Immunol. (2013) 190:5640–8. doi: 10.4049/jimmunol.1202270
60. Stuart LM, Deng J, Silver JM, Takahashi K, Tseng AA, Hennessy EJ, et al. Response to Staphylococcus aureus requires CD36-mediated phagocytosis triggered by the COOH-terminal cytoplasmic domain. J Cell Biol. (2005) 170:477–85. doi: 10.1083/jcb.200501113
61. Jimenez-Dalmaroni MJ, Xiao N, Corper AL, Verdino P, Ainge GD, Larsen DS, et al. Soluble CD36 ectodomain binds negatively charged diacylglycerol ligands and acts as a co-receptor for TLR2. PloS One. (2009) 4:e7411. doi: 10.1371/journal.pone.0007411
62. Olonisakin TF, Li H, Xiong Z, Kochman EJ, Yu M, Qu Y, et al. CD36 provides host protection against klebsiella pneumoniae intrapulmonary infection by enhancing lipopolysaccharide responsiveness and macrophage phagocytosis. J Infect Dis. (2016) 214:1865–75. doi: 10.1093/infdis/jiw451
63. Stengel S, Quickert S, Lutz P, Ibidapo-Obe O, Steube A, Kose-Vogel N, et al. Peritoneal level of CD206 associates with mortality and an inflammatory macrophage phenotype in patients with decompensated cirrhosis and spontaneous bacterial peritonitis. Gastroenterology. (2020) 158:1745–61. doi: 10.1053/j.gastro.2020.01.029
64. Fabriek BO, van Bruggen R, Deng DM, Ligtenberg AJ, Nazmi K, Schornagel K, et al. The macrophage scavenger receptor CD163 functions as an innate immune sensor for bacteria. Blood. (2009) 113:887–92. doi: 10.1182/blood-2008-07-167064
65. Weaver LK, Hintz-Goldstein KA, Pioli PA, Wardwell K, Qureshi N, Vogel SN, et al. Pivotal advance: activation of cell surface Toll-like receptors causes shedding of the hemoglobin scavenger receptor CD163. J Leukoc Biol. (2006) 80:26–35. doi: 10.1189/jlb.1205756
66. Brenchley JM, Price DA, Schacker TW, Asher TE, Silvestri G, Rao S, et al. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat Med. (2006) 12:1365–71. doi: 10.1038/nm1511
67. Ancuta P, Kamat A, Kunstman KJ, Kim EY, Autissier P, Wurcel A, et al. Microbial translocation is associated with increased monocyte activation and dementia in AIDS patients. PloS One. (2008) 3:e2516. doi: 10.1371/journal.pone.0002516
68. Schneider P, MacKay F, Steiner V, Hofmann K, Bodmer JL, Holler N, et al. BAFF, a novel ligand of the tumor necrosis factor family, stimulates B cell growth. J Exp Med. (1999) 189:1747–56. doi: 10.1084/jem.189.11.1747
69. Moore PA, Belvedere O, Orr A, Pieri K, LaFleur DW, Feng P, et al. BLyS: member of the tumor necrosis factor family and B lymphocyte stimulator. Science. (1999) 285:260–3. doi: 10.1126/science.285.5425.260
70. Shu HB, Hu WH, Johnson H. TALL-1 is a novel member of the TNF family that is down-regulated by mitogens. J Leukoc Biol. (1999) 65:680–3. doi: 10.1002/jlb.65.5.680
71. Nardelli B, Belvedere O, Roschke V, Moore PA, Olsen HS, Migone TS, et al. Synthesis and release of B-lymphocyte stimulator from myeloid cells. Blood. (2001) 97:198–204. doi: 10.1182/blood.V97.1.198
72. Kuley R, Draves KE, Fuller DH, Giltiay NV, Clark EA, Giordano D. B cell activating factor (BAFF) from neutrophils and dendritic cells is required for protective B cell responses against Salmonella typhimurium infection. PloS One. (2021) 16:e0259158. doi: 10.1371/journal.pone.0259158
73. Buechler C, Ritter M, Orso E, Langmann T, Klucken J, Schmitz G. Regulation of scavenger receptor CD163 expression in human monocytes and macrophages by pro- and antiinflammatory stimuli. J Leukoc Biol. (2000) 67:97–103. doi: 10.1002/jlb.67.1.97
74. Salerno-Goncalves R, Fasano A, Sztein MB. Engineering of a multicellular organotypic model of the human intestinal mucosa. Gastroenterology. (2011) 141:e18–20. doi: 10.1053/j.gastro.2011.04.062
75. Sztein MB, Bafford AC, Salerno-Goncalves R. Salmonella enterica serovar Typhi exposure elicits ex vivo cell-type-specific epigenetic changes in human gut cells. Sci Rep. (2020) 10:13581. doi: 10.1038/s41598-020-70492-2
76. Nickerson KP, Llanos-Chea A, Ingano L, Serena G, Miranda-Ribera A, Perlman M, et al. A versatile human intestinal organoid-derived epithelial monolayer model for the study of enteric pathogens. Microbiol spectrum. (2021) 9:e0000321. doi: 10.1128/Spectrum.00003-21
77. Salerno-Goncalves R, Chen H, Bafford AC, Izquierdo M, Hormazabal JC, Lagos R, et al. Early host immune responses in a human organoid-derived gallbladder monolayer to Salmonella Typhi strains from patients with acute and chronic infections: a comparative analysis. Front Immunol. (2024) 15:1334762. doi: 10.3389/fimmu.2024.1334762
78. Mohn KG, Brokstad KA, Islam S, Oftung F, Tondel C, Aarstad HJ, et al. Early induction of cross-reactive CD8+ T-cell responses in tonsils after live-attenuated influenza vaccination in children. J Infect Dis. (2020) 221:1528–37. doi: 10.1093/infdis/jiz583
Keywords: S. Typhi, human oral challenge, CHIM, classical monocytes, intermediate monocytes, non-classical monocytes
Citation: Toapanta FR, Hu J, Shirey KA, Bernal PJ, Levine MM, Darton TC, Waddington CS, Pollard AJ and Sztein MB (2024) Changes in monocyte subsets in volunteers who received an oral wild-type Salmonella Typhi challenge and reached typhoid diagnosis criteria. Front. Immunol. 15:1454857. doi: 10.3389/fimmu.2024.1454857
Received: 25 June 2024; Accepted: 08 August 2024;
Published: 27 August 2024.
Edited by:
Farha Naz, University of Virginia, United StatesReviewed by:
Roland Lang, University Hospital Erlangen, GermanyPietro Mastroeni, University of Cambridge, United Kingdom
Copyright © 2024 Toapanta, Hu, Shirey, Bernal, Levine, Darton, Waddington, Pollard and Sztein. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Franklin R. Toapanta, ZnRvYXBhbnRhQHNvbS51bWFyeWxhbmQuZWR1; Marcelo B. Sztein, TXN6dGVpbkBzb20udW1hcnlsYW5kLmVkdQ==
†Present address: Thomas C. Darton, School of Medicine and Population Health, University of Sheffield, Sheffield, United Kingdom Claire S. Waddington, Department of Medicine, Imperial College London, London, United Kingdom