 Li-juan Ding
Li-juan Ding Xin Jiang
Xin Jiang Te Li2
Te Li2 Shudong Wang
Shudong Wang- 1Department of Radiation Oncology, The First Hospital of Jilin University, Changchun, Jilin, China
- 2Department of Geriatrics, The First Hospital of Jilin University, Changchun, Jilin, China
- 3Department of Cardiology, The First Hospital of Jilin University, Changchun, Jilin, China
Protein post-translational modifications (PTMs) represent a crucial aspect of cellular regulation, occurring after protein synthesis from mRNA. These modifications, which include phosphorylation, ubiquitination, acetylation, methylation, glycosylation, Sumoylation, and palmitoylation, play pivotal roles in modulating protein function. PTMs influence protein localization, stability, and interactions, thereby orchestrating a variety of cellular processes in response to internal and external stimuli. Dysregulation of PTMs is linked to a spectrum of diseases, such as cancer, inflammatory diseases, and neurodegenerative disorders. UFMylation, a type of PTMs, has recently gained prominence for its regulatory role in numerous cellular processes, including protein stability, response to cellular stress, and key signaling pathways influencing cellular functions. This review highlights the crucial function of UFMylation in the development and progression of tumors, underscoring its potential as a therapeutic target. Moreover, we discuss the pivotal role of UFMylation in tumorigenesis and malignant progression, and explore its impact on cancer immunotherapy. The article aims to provide a comprehensive overview of biological functions of UFMylation and propose how targeting UFMylation could enhance the effectiveness of cancer immunotherapy strategies.
Introduction
Protein post-translation modification (PTM), a kind of chemical modification of a protein, occurs after proteins are translated from messenger RNA (mRNA) (1, 2). PTMs play an important role in regulation of protein function via controlling its localization, stability and interactions with other proteins, leading to regulation of various cellular processes upon different internal and external signals (3). Dysregulation of PTMs leads to various diseases, including cancer, inflammatory diseases and neurodegenerative disorders (4). Better understanding the mechanisms of PTMs could help us identify potential therapeutic targets for diverse diseases. There are more than 400 types of PTMs, which affect protein functions (5). The common types of PTMs include phosphorylation (addition of a phosphate group) (6), ubiquitination (addition of ubiquitin molecules) (7, 8), acetylation (addition of an acetyl group) (9), methylation (addition of a methyl group) (10), glycosylation (addition of carbohydrate group) (11), Sumoylation (addition of small ubiquitin-like modifier) (12, 13), palmitoylation (addition of palmitic acid) (14).
In recent years, UFMylation has been emerged to regulate various cellular processes, such as protein stability, cellular stress responses to different internal and external signals, and cellular signaling pathways that regulate cellular biological functions (15, 16). UFMylation belongs to a PTM and involves the covalent attachment of ubiquitin-fold modifier 1 (UFM1) to target proteins (17). UFMylation typically involves three important enzymatic steps, activation, conjugation and ligation (18, 19). UFM1 is activated by an E1 activating enzyme to initiate UFMylation, which prepares it for transfer to the next enzyme. Then, the activated UFM1 is transferred to an E2 conjugating enzyme, which acts as an intermediary carrier in the UFMylation process. Lastly, an E3 ligase enzyme promoted the transfer of UFM1 from the E2 enzyme to the target protein, which completes the UFMylation process and influencing the function of the target proteins (20). There are similarities between UFMylation and ubiquitination. Both UFMylation and ubiquitination are PTMs where small proteins (UFM1 and ubiquitin) are covalently attached to target protein. Both processes involve a series of enzymatic steps by E1, E2 and E3 enzymes. Both UFMylation and ubiquitination are reversible processes because specific proteases can remove UFM1 or ubiquitin from target proteins. Moreover, both modifications can alter the function, localization, interaction and stability of proteins. Evidence has shown that UFMylation is involved in numerous cellular functions, such as DNA damage response, endoplasmic reticulum (ER) stress response, and the regulation of the immune system (21). Hence, in this review, we discuss the role of UFMylation in tumorigenesis and progression. Moreover, we describe the functions of UFMylation in cancer immunotherapy. Targeting UFMylation could be useful for improving cancer immunotherapy.
UFM1 conjugation system
UFM1 is a novel ubiquitin-like molecule (UBL) with evolutionary presence across multiple species. Like ubiquitin, UFM1 undergoes a sequential three-step enzymatic process for covalent attachment to its substrates. This process involves the UFM1-activating enzyme (E1), which is known as UBA5 (ubiquitin-like modifier-activating enzyme 5), the UFM1-conjugating enzyme 1 (E2, UFC1), and the UFM1-specific ligase 1 (E3, UFL1) (21). Specifically, UBA5 is a key enzyme involved in the UFMylation pathway, which functions similarly to an E1 enzyme in the ubiquitin system, catalyzing the initial step of activating the ubiquitin-like protein UFM1. UBA5 first activates UFM1 by forming a high-energy thioester bond between the C-terminal glycine of UFM1 and the cysteine residue in the active site of UBA5. This step is ATP-dependent, requiring energy to activate UFM1 for subsequent conjugation. After activation, UFM1 is transferred from UBA5 to the UFC1, which is crucial for the next step in attaching UFM1 to target proteins (22). UFL1 acts as a ligase, specifically an E3 ligase in the UFMylation process, to catalyze the final step of attaching UFM1 to target proteins. De-UFMylation is carried out by specific enzymes UFSPs (UFM1 specific peptidases). The proteases UFSP1 and UFSP2 involve in the maturation of the UFM1 precursor and de‐UFMylation process. These enzymes reverse the attachment of UFM1 to target proteins, thereby regulating the functions and interactions of these proteins. DDRGK1 (DDRGK domain containing 1) and CDK5RAP3 involve in regulation of the UFMylation system. UFBP1 (UFM1-specific binding protein 1) interacts with UFM1 and the E3 ligase UFL1, helping to facilitate the conjugation of UFM1 to substrate proteins (Figure 1).
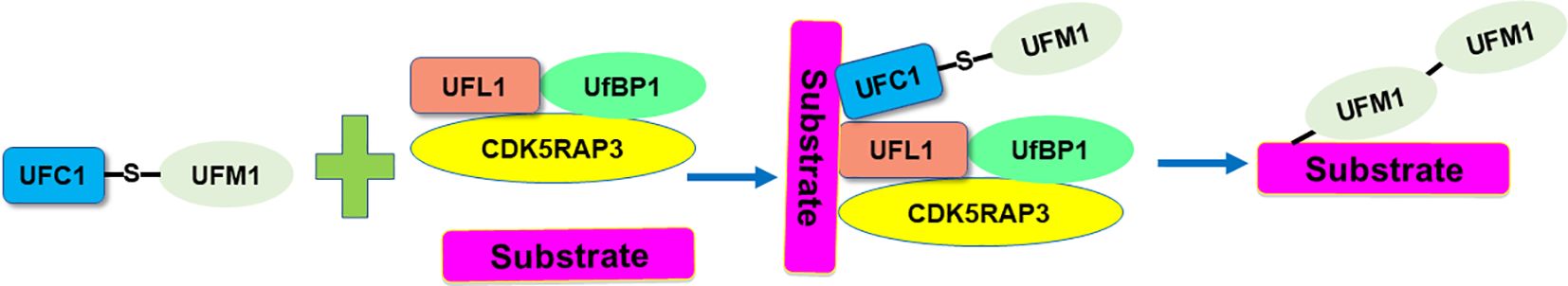
Figure 1. Illustration of the UFMylation process.
RCAD/UFL1 plays roles in various cellular processes, including ER signaling, the UPR, and neurodegeneration (22, 23). One study reveals that RCAD/UFL1 is critical for embryonic development, HSC survival, and erythroid differentiation. Deletion of RCAD/UFL1 significantly impairs hematopoietic development, leading to severe anemia, cytopenia, and eventual lethality. Depletion of RCAD/UFL1 induces elevated endoplasmic reticulum stress and activates the UPR in bone marrow cells. Additionally, the absence of RCAD/UFL1 disrupts autophagic degradation, increases mitochondrial mass and ROS (reactive oxygen species), and triggers a DNA damage response, p53 activation, and heightened cell death in HSCs (hematopoietic stem cell). This study supports the essential role of RCAD/UFL1 in murine hematopoiesis and development via maintaining cellular homeostasis (24). The UFMylation pathway that is facilitated by UBA5 influences various cellular processes including protein stability, cellular stress responses, and cellular signaling pathways. Dysregulation of UFMylation pathway can contribute to disease mechanisms, particularly in neurodegenerative diseases and cancer. In the following paragraphs, we will describe the mechanisms by which aberrant UFMylation contributes to tumorigenesis in various cancer types.
UFMylation in various cancer types
Liu et al. reported UFMylation kept p53 stability via antagonizing its ubiquitination (25). It is clear that p53, which is known as the “guardian of the genome”, is a crucial protein known primarily for its important role in preventing cancer (26, 27). Tumor suppressor p53 has been reported to regulate cell cycle, DNA repair, apoptosis and response to stress (28). Mutations of p53 have been observed in many cancer types and involves in tumorigenesis, suggesting that understanding how p53 works and how its activity is controlled or altered in cancer cells is critical for developing new therapeutic strategies (29). Evidence has suggested that p53 can be regulated by PTMs (30). Liu and coworkers found that UFM1 can covalently modify p53, which stabilizes the protein by reducing its ubiquitination and subsequent proteasomal degradation. Mechanistically, UFL1 competes with MDM2 (mouse double minute 2 homolog) for binding to p53, thereby stabilizing it. Knockdown of UFL1 or DDRGK1 leads to decreased p53 stability, enhanced cell growth, and tumor formation in vivo (25). Moreover, both UFL1 and DDRGK1 are downregulated in a significant proportion of renal cell carcinomas, where their expression levels positively associated with p53 levels. These findings underscore UFMylation as a vital PTM that maintains p53 stability and tumor-suppressive activity, suggesting that targeting UFMylation could be a viable therapeutic strategy in cancer treatment. In the following sections, we will describe the various role of UFMylation in different cancer types (25).
Breast cancer
Breast cancer is one of the most common cancers among women worldwide (31). The most common forms of breast cancer are ductal carcinoma in situ and invasive ductal carcinoma (32). Triple-negative breast cancer (TNBC) is a subtype of breast cancer characterized by the absence of three common receptors: estrogen receptors (ER), progesterone receptors (PR), and human epidermal growth factor receptor 2 (HER2) (33, 34). TNBC accounts for about 10-20% of all breast cancers. TNBC does not respond to hormonal therapy (such as tamoxifen or aromatase inhibitors) or therapies that target HER2 receptors, such as Herceptin (trastuzumab) (35, 36). Hence, TNBC tends to be more aggressive and has a higher probability of metastasis, particularly in the first few years after diagnosis. Factors that can increase the risk of breast cancer include age, genetic mutations (such as BRCA1 and BRCA2), family history of breast cancer, radiation exposure, obesity, etc. (37). Common treatments for breast cancer include surgery, radiation therapy, chemotherapy, hormone therapy, targeted therapy and immunotherapy (38–40). Understanding breast tumorigenesis is essential for prevention, early detection, and effective treatment strategies, which significantly improve the prognosis (41).
Yoo et al. discovered that the UFMylation of the nuclear receptor coactivator ASC1 (activating signal cointegrator 1) is critical for the activation of estrogen receptor alpha (ERα) in response to 17β-estradiol (E2). In the absence of E2, ASC1 remains non-modified due to its association with the UFM1-specific protease UfSP2. Upon E2 exposure, ERα displaces UfSP2 from ASC1, leading to its UFMylation. This modification enhances the interactions between ASC1 and other coactivators such as p300 and SRC1 at ERα-responsive gene promoters (42). Manipulating ASC1 levels or its UFMylation status profoundly influences ERα-driven tumor progression. Specifically, ASC1 overexpression or reduced UfSP2 expression promotes tumor development, an effect mitigated by tamoxifen. Conversely, expression of a UFMylation-deficient ASC1 variant or suppression of the UFM1-activating enzyme UBA5 inhibits tumor growth. These findings highlight the pivotal role of ASC1 UFMylation in modulating breast cancer development via ERα transactivation, presenting novel insights into the regulatory mechanisms of hormone-responsive breast cancer (42).
Yoo et al. further found that ERα is modified by UFM1, and this UFMylation substantially enhances ERα stability and transactivation capabilities. Specifically, suppressing UFSP2 significantly increases ERα stability by reducing its ubiquitination. Conversely, ERα stability is compromised by inhibiting UFMylation through the silencing of UBA5 (43). Furthermore, this group identified Lys171 and Lys180 on ERα as primary UFM1 acceptor sites, and show that substituting these lysine residues with arginine (2KR mutation) drastically diminishes ERα stability. Furthermore, this 2KR mutation impedes 17β-estradiol-induced transactivity of ERα and downregulates the expression of key downstream targets such as pS2, cyclin D1, and c-Myc, indicating the necessity of UFMylation for transcriptional function of ERα. The 2KR mutation also inhibits anchorage-independent colony growth in MCF7 cells. Importantly, components of the UFM1 conjugation machinery, including UBA5, UFC1, UFL1, and UFBP1, are markedly elevated in ERα-positive breast cancer cell lines and tissues. These results underscore the vital function of ERα UFMylation in enhancing its stability and transcriptional activity, thereby promoting breast cancer progression (43).
Glioblastoma
Glioblastoma, which is often called glioblastoma multiforme (GBM), is one of the most aggressive types of primary brain cancer in adults (44). Glioblastoma is highly malignant due to its rapid growth and tendency to invade surrounding brain tissue. Glioblastomas have high level of cellular diversity because they contain a mix of cell types, making them difficult to treat (45). Symptoms of glioblastoma can vary depending on the location of the tumor in the brain but typically include headaches, nausea, seizures, cognitive impairments, and neurological deficits such as weakness or sensory changes (46). MRI (magnetic resonance imaging) and CT (computed tomography) scans are used to diagnose glioblastomas, which can detect the size and location of the tumor. A biopsy is used to confirm glioblastoma diagnosis (47). Clinically, treatment for glioblastoma involves a combination of surgery, radiation therapy, immunotherapy and chemotherapy (48–50). In general, after surgery removes tumor as much as possible, radiation therapy in combination with chemotherapy kill any remaining cancer cells (51). Prognosis of patients with glioblastoma remains poor with median survival times ranging from 12 to 18 months. Understanding the genetic and molecular mechanism underlying glioblastoma is critical to develop more effective treatments that target specific and important pathways in glioblastoma oncogenesis and resistance to current chemotherapy, such as temozolomide (52, 53).
One study used genome-wide CRISPR-Cas9 approach explored genetic vulnerabilities in glioblastoma stem cells (GSCs). Moreover, this group explored the mechanisms of temozolomide sensitivity in GSCs (54). Utilizing CRISPR-Cas9 in patient-derived GSCs, the entire coding genome was investigated to identify critical pathways that drive growth and underpin the gene-essential circuitry of GBM stemness and cell proliferation. This group highlighted the significance of SOCS3 (suppressor of cytokine signaling 3), USP8 (ubiquitin specific peptidase 8), DOT1L (disruptor of telomeric silencing 1-like), which belong to the SOX transcription factor family. Moreover, protein ufmylation is critical in promoting GSC growth. Specifically, all members of the ufmylation pathway, including UBA5, UFC1, UFL1, and UFSP2, were crucial, as single-gene knockouts significantly reduced cell fitness. This highlights each step in this pathway as a potential therapeutic target (54). Treatment of 14 patient-derived GSC cultures with DKM 2-93, which is an inhibitor of UBA5, demonstrated strong anti-proliferative effects. Additionally, depletion of UFC1 and UFSP2 promoted CHOP expression, an ER-stress response marker, suggesting a connection to ER homeostasis maintenance. The identification of SEL1L and HRD1 as GBM-specific fitness genes further underscores the potential of targeting proteostasis networks in therapeutic strategies for GBM. These findings point to the UFMylation pathway as a critical component for GBM stem cell maintenance and a promising target for developing new GBM treatments. Furthermore, this study uncovered mechanisms of temozolomide resistance, suggesting potential avenues for combination therapy. This genome-wide functional approach enhanced the understanding of GBM growth dynamics and drug resistance (54).
Renal cancer
Renal cancer, which is often known as kidney cancer, is one of the common cancers in human. The most common type of renal cancer is renal cell carcinoma (RCC), which makes up about 90% of all kidney cancers (55). Within RCC, there are several subtypes, including clear cell, papillary, and chromophobe renal cell carcinomas. Several risk factors for renal cancer development have been reported, including smoking, obesity, high blood pressure, family history of renal cancer, and genetic conditions (56). Diagnostic approaches of renal cancer include ultrasound, CT scans, MRI and sometimes biopsy (57, 58). Treatments for renal cancer include surgery, cryoablation, radiation therapy, targeted therapy and immunotherapy (59). Patients with localized renal cancers often have a high cure rate with surgery, while patients with advanced and metastatic renal cancers often have a poor prognosis (60). Advancements in the understanding of molecular biology of renal cancer could develop novel treatments and extend patient survival. Wang et al. reported that UFMylation is activated in renal cancer (61). Our group investigated the relationship between UFMylation, autophagy, and the UPS in renal cancer. UFMylation levels remained unchanged with either activation or inhibition of autophagy, as evidenced by consistent LC3 conversion rates in renal cancer tissues compared to adjacent non-cancerous tissues. This suggests that the increase in UFMylation observed in renal cancer is independent of autophagy. Inhibition of the UPS with MG132 led to increased UFMylation in renal cancer cells, indicating a potential association between UFMylation and UPS activity. Additionally, UFMylation levels did not correlate with VHL gene mutations, which is frequently mutated in renal cancer and serves as a key E3 ligase in the UPS (62). Moreover, treatment with sunitinib, targeting multiple tyrosine kinases, significantly reduced UFMylation levels, while upregulation of active UFM1 was found to partially counteract the anti-tumor effects of sunitinib. These findings suggest that UFMylation might serve as a novel molecular target for the treatment of renal cancer (61).
Colon cancer
Colon cancer is one of the most common types of cancer in both men and women. Numerous factors that can increase the risk of colon cancer include older age, family history of colon cancer or polyps, inflammatory intestinal conditions, such as ulcerative colitis or Crohn’s disease, a diet low in fiber and high in fat and calories, diabetes, obesity, smoking, and alcohol (63, 64). Colonoscopy and biopsy are helpful for colon cancer diagnosis. Treatments for colon cancer have surgery, radiation therapy, chemotherapy and targeted therapy (65). Colon cancer can be highly treatable if it is caught early via awareness, colonoscopy screen (66). Zhou et al. performed genomic profiling of the UFMylation family genes and discovered a tumor suppressor UFSP2 in colon cancer (67). This study utilized TCGA data cohort and analyzed the genomic alterations of eight UFMylation family genes, including UFSP1, UFSP2, UFM1, UFL1, UFC1, DDRGK1, UBA5 and CDK5RAP3. Moreover, 55 recurrent and focal SCNA events were uncovered in UFMylation family genes in 33 cancer types. UFSP2 gene was often deleted in 14 cancer types. The frequency for copy number loss of UFSP2, UFM1 and UFL1 was 31%, 31% and 18%, respectively. The frequency for copy number gain for UFC1, UFSP1 and DDRGK1 was 34%, 34% and 30%. Notably, UFSP2 copy number was heterozygous loss in cancer tissues and tumor cells, suggesting a possible haploinsufficiency of the UFSP2 gene. 11% of TCGA samples displayed high-level copy number alterations in at least one UFMylation gene (67). These alterations typically manifested in a mutually exclusive pattern of amplifications and deletions, suggesting that UFMylation genes may have overlapping functions. UFMylation genes generally displayed less than 5% somatic mutations and less than 0.1% transcript fusions. Additionally, RNA sequencing data analysis from the TCGA samples revealed ubiquitous expression of UFMylation genes across various cancer types. Moreover, UFSP2 mutation is frequently observed in colon cancer and uterine corpus endometrial carcinoma. This pattern, along with high recurrent copy number loss, indicate that UFSP2 may perform tumor suppressive function (67). Previous research supports this assertion, showing that knockdown of UFSP2 enhances breast cancer cell growth and tumor formation. Furthermore, significantly reduced levels of UFSP2 mRNA and protein levels were observed in multiple cancer tissues. Knockdown of UFSP2 expression was observed to significantly enhance the growth rates of colon cancer cells in vitro and in vivo. Additionally, an increase in total UFMylation levels was detected after UFSP2 depletion in cells and xenograft tumors, linking UFSP2 genomic alterations to the functional role of UFMylation in colon cancer (67). GSEA analysis showed that the loss of UFSP2 predominantly affected pathways related to DNA replication, the cell cycle, the spliceosome, the ribosome, and mismatch repair. By knocking down UFSP2 in colon cancer cell lines, there was an increase in the expression of key marker genes, including PCNA and MCM2 for DNA replication, CDK4 and CCND1 for cell cycle regulation, RPL26 for ribosome protein synthesis. These results further substantiate the tumor suppressive properties of UFSP2 in colon cancer (67).
Pancreatic cancer
Pancreatic cancer has a worse prognosis in human cancer types. This is due to pancreas deep location in the body. In addition, pancreatic cancer often has no symptoms until it has reached an advanced stage, making it difficult to detect early (68). Risk factors for pancreatic cancer have smoking, chronic pancreatitis, diabetes, family history of pancreatic cancer, obesity, etc (69). Treatment options for pancreatic cancer have surgery, radiation therapy, chemotherapy, and targeted therapy (70–72). Because pancreatic cancer has a lower 5-year survival rate, it is important to discover early detection and innovative treatment approaches (73). To achieve this goal, exploration of molecular mechanisms of pancreatic oncogenesis is necessary. One group identified that UBA5 could be a target for pancreatic cancer via chemoproteomic screening of covalent ligands (74). Another group identified that the UFMylation of RPL10 (ribosomal protein L10) contributed to pancreatic adenocarcinoma development (75). The ufmylation of RPL10 was found in both pancreatic cancer cells and tissues. RPL10 UFMylation enhances cell proliferation and promoted cancer cell stemness, primarily through upregulated expression of KLF4 (kruppel-like transcription factor 4). Additionally, mutagenesis of RPL10 UFMylation sites further solidified the link between RPL10 UFMylation and these cellular behaviors, including proliferation and stemness. Overall, UFMylation of RPL10 is a crucial process that promotes the stemness of pancreatic cancer cells, contributing to the progression of pancreatic cancer (75).
Oral squamous cell carcinoma
Oral squamous cell carcinoma (OSCC) is the most common type of oral cancer. Several risk factors have contributed to OSCC, including tobacco use, heavy alcohol consumption, HPV (human papillomavirus) exposure, chronic dental irritation and dietary factors (76, 77). A biopsy is important for OSCC diagnosis after a clinical examination (78). Imaging test can be used to determine the extent of cancer and tumor invasion, such as X-rays, CT scans, MRI and PET scans (79). Treatments of OSCC have surgery, radiation therapy, chemotherapy or a combination, which are dependent on the tumor stage and location (80, 81). Recently, UFM1 was reported to enhance OSCC progression via regulating immune infiltration (82). Higher expression of UFM1 was found in OSCC and its overexpression was linked to shorter overall survival, indicating that UFM1 could serve as an adverse prognostic factor in OSCC. Furthermore, UFM1 expression could predict poor outcomes in OSCC patients. Functionally, reducing UFM1 expression suppressed the cell proliferation, attenuated cell migration, and invasion in OSCC. Additionally, UFM1 was linked to various immune cells, including Th17 cells, T helper cells, and cytotoxic cells, as well as with processes of ubiquitination. These findings highlight the potential of UFM1 as a biomarker for OSCC prognosis and a target for therapeutic intervention (82).
Hepatocellular carcinoma
Hepatocellular carcinoma (HCC) is the most common type of primary liver cancer. HCC is often associated with chronic liver disease and cirrhosis. Risk factors of HCC development have chronic hepatitis B or C infection, cirrhosis, alcohol consumption, aflatoxin exposure, nonalcoholic fatty liver disease (NAFLD), etc (83). Besides imaging tests such as ultrasound, CT scans and MRIs, and a biopsy, alpha-fetoprotein (AFP) test in blood may aid in diagnosis (84). Treatments for HCC include surgical resection, liver transplantation, chemoembolization, radiofrequency ablation, targeted therapy drugs and immunotherapy (85–87). Chen et al. reported that lncRNA B3GALT5-AS1 regulated the miR-934/UFM1 axis and inhibited tumor progression in HCC (88). A reduction in B3GALT5-AS1 levels was seen in both HCC cell lines and tissues. Upregulation of B3GALT5-AS1 inhibited the malignant properties of HCC cells. The effects of B3GALT5-AS1 overexpression can be reversed by miR-934 mimics, establishing miR-934 as a downstream effector. Additionally, the impact of miR-934 inhibition was mitigated by UFM1 downregulation, highlighting a link between miR-934 and UFM1. B3GALT5-AS1 suppresses the PI3K(phosphoinositide 3-kinases)/AKT signaling pathway via UFM1. Collectively, B3GALT5-AS1 acts as a potent suppressor of HCC by modulating miR-934 and UFM1, pointing to its potential as a HCC therapeutic target (88). Moreover, loss of UFL1/HFBP1 in hepatocytes activated mTOR pathway and facilitated liver damage and liver carcinogenesis via using hepatocyte-specific Ufl1Δ/Δhep and Ufbp1Δ/Δhep mice (89). High-fat diet (HFD) was used to induce fatty liver disease, and diethylnitrosamine (DEN) was used to trigger liver cancer. Results demonstrated that deletion of Ufl1 or Ufbp1 in hepatocytes led to early signs of liver damage such as hepatocyte apoptosis and mild steatosis by 2 months, progressing to severe hepatocellular ballooning, extensive fibrosis, and steatohepatitis by 6-8 months. Notably, over 50% of the knockout mice developed spontaneous HCC by 14 months. These mice also showed increased susceptibility to HFD-induced fatty liver and DEN-induced HCC. The Ufl1/Ufbp1 complex was found to bind with and inhibit the mTOR/GβL complex, thus reducing mTORC1 activity. Loss of Ufl1 or Ufbp1 dissociated this complex, leading to enhanced mTOR pathway and HCC development. Ufl1 and Ufbp1 play critical roles in preventing liver fibrosis, steatohepatitis, and HCC by acting as inhibitors of the mTOR pathway, highlighting their potential as therapeutic targets for liver diseases (89).
Gastric cancer
Gastric cancer is one of the common cancer types in the world, which can spread throughout the stomach and to other organs, particularly the esophagus, lungs, lymph nodes, and liver (90). The most common type of gastric cancer is adenocarcinoma, which accounts for about 90% of all gastric cancer cases. Several factors contribute to gastric cancer development, such as helicobacter pylori infection, diet, smoking, family history, chronic gastritis, stomach polyps (91). Gastric cancer is often asymptomatic in the early stages, which can delay diagnosis. Diagnosis typically involves endoscopy, a biopsy and CT scans (92, 93). Treatments for gastric cancer include surgery, chemotherapy, radiation therapy, targeted therapy and immunotherapy (94). The association between the UFM1 expression and the outcomes of gastric cancer patients who had surgery was determined by Lin and coworkers. Lin et al. utilized public databases to explore the relationship between UFM1 and treatment outcomes by single-sample gene set enrichment analysis (ssGSEA). Moreover, the expression of UFM1 was determined in cancerous and paracancerous gastric cancer tissues. UFM1 expression was decreased and interacted with CDK5RAP3. Moreover, CDK5RAP3 and UFM1 expression was inversely associated with one key oncogenic pathway activation, AKT. Low expression of UFM1 and CDK5RAP3 was associated with poor prognosis, which was independent predictor to predict overall survival of gastric cancer patients. Moreover, in combination of UFM1, CDK5RAP3 and TNM (tumor, node, metastasis) stages increased the accuracy of prognosis prediction in gastric cancer patients (95).
Lin et al. further found that UFM1 inhibited the expression of PDK1 via negative regulation of PI3K/AKT signaling, leading to suppression of invasive ability in gastric cancer (96). UFM1 expression was decreased at both protein level and mRNA level in gastric cancer tissues, which was associated with low 5-year survival rate in patients with gastric cancer. Upregulation of UFM1 inhibited migration and invasion abilities in gastric cancer cells, while downregulation of UFM1 exhibited the opposite functions. In vivo study further identified the tumor suppressive function of UFM1 in nude mouse model. Mechanistically, UFM1 enhanced the PDK1 ubiquitination level, leading to reduction of the PDK1 protein level, thereby reducing the AKT phosphorylation at Ser473 (96). Another study reported that upregulation of UFBP1 expression correlates with increased progression-free survival in gastric cancer after platinum-based chemotherapy. UFBP1 enhances the sensitivity of gastric cancer cells to cisplatin, whereas its knockdown reduces this sensitivity. Proteomic analysis indicated a significant reduction in aldo-keto reductase 1Cs (AKR1Cs) protein levels due to UFBP1 overexpression. Additionally, UFBP1 modulates ROS production in response to cisplatin. Mechanistic insights showed that UFBP1 decreases AKR1Cs expression and Nrf2 transcriptional activity, promoting Nrf2 degradation via K48-linked polyubiquitination. Further cell and mouse experiments confirmed that UFBP1 amplifies cisplatin sensitivity through the Nrf2/AKR1C axis. These findings identify UFBP1 as a potential prognostic biomarker for gastric cancer and provide a mechanistic basis for personalized chemotherapy approaches (97).
UFMylation in immunotherapy
Cancer immunotherapy is a type of cancer treatment that harnesses the immune system to fight cancer (98). Although immune cells can recognize and destroy cancer cells, cancer often finds ways to evade immune detection. Immunotherapy aims to boost the immune ability to combat cancer more effectively (99). There are several types of immunotherapy, including checkpoint inhibitors, CAR (chimeric antigen receptors) T-cell therapy, cancer vaccines, monoclonal antibodies (100). Common checkpoint proteins in immunotherapy include PD-1 (programmed death-1), PD-L1 (programmed death ligand-1) and CTLA-4 (cytotoxic T-lymphocyte associated protein 4), which block the immune cells to attack cancer cells (101, 102). CAR T-cell therapy involves modifying T-cells via engineering to produce special receptors on their surface, leading to the better destroying cancer cells (103). Cancer vaccine induces the immune system to target tumor cells, which can help prevent the cancer from growing or recurrence (104, 105). Immunotherapy can be a powerful treatment option for some types of cancer. However, it fails to work for everyone, and figuring out why and how to improve immunotherapy is a major area of research in the field of oncology. Recently, UFMylation has been reported to involve in immunotherapy in cancer (106, 107). Therefore, we describe the role of UFMylation in regulation of immunotherapy in the following paragraphs.
UFMylation of pirin
Pancreatic ductal adenocarcinoma (PDAC) is the most common type of pancreatic cancer, accounting for about 90% of all pancreatic cancers. PDAC is particularly aggressive with late detection and poor prognosis (37). Treatment options for PDAC depend on the disease stage at diagnosis and may include surgery, chemotherapy, radiation therapy, immunotherapy, or a combination of these approaches (72, 108). One study identified a potent approach to induce ferroptosis in PDAC and stimulate antitumor immune response. Utilizing patient-derived organoid models and a KPC mouse model, which is known as LSL-KrasG12D/+, LSL-Trp53R172H/+, Pdx-1-Cre, this study showed that downregulation of macrophage-capping protein (MCP) reduced UFMylation of pirin (PIR), suggesting that PIR is a potential UFM1 substrate. This inhibition led to decreased transcription of GPX4 (glutathione peroxidase 4), a ferroptosis biomarker, and enhanced the cytoplasmic release of HMGB1 (high mobility group box 1). The reduced GPX4 levels initiate ferroptosis, while the released HMGB1 promotes pro-inflammatory M1-like macrophage polarization. Consequently, therapeutic targeting of MCP (monocyte chemoattractant protein 1) not only triggered ferroptosis but also activated antitumor, pro-inflammatory responses, offering a dual antitumor effect. Moreover, a nanosystem for specifically silencing MCP was developed, which provided a novel approach for PDAC treatment (109).
PD-1 UFMylation
PD-1 and PD-L1 are key proteins involved in the immune system “checkpoint”, which manage immune responses to avoid attacking the body tissues. In the context of cancer, however, PD-1 and PD-L1 proteins are often exploited by tumor cells to evade immune destruction (110, 111). PD-1 is found on the surface of T-cells and acts as an immune checkpoint to keep the immune system in check. When PD-1 binds with its ligands, PD-L1 or PD-L2, it sends an inhibitory signal to T-cells, reducing their activity to attack the tumor cells (112). PD-L1 is mainly expressed on the surface of cancer cells. Targeting the interaction between PD-1 and PD-L1 is a promising strategy for cancer immunotherapy via using checkpoint inhibitors, such as anti-PD-1 antibodies (pembrolizumab and nivolumab) and anti-PD-L1 antibodies (atezolizumab) (113, 114). Recently, one study demonstrated that ablation of UFL1 in T cells inhibited PD-1 UFMylation to increase anti-tumor immunity (106). This study investigated the physiological role of UFMylation in T cells by examining mice with a conditional knockout (cKO) of Ufl1, focusing on tumor immunity. Ufl1 cKO mice demonstrate superior tumor control. Single-cell RNA sequencing analysis of these mice reveals an increase in tumor-infiltrating cytotoxic CD8+ T cells. Moreover, UFL1 regulated the UFMylation of PD-1, which in turn inhibited PD-1 ubiquitination and degradation (106). Additionally, AMPK-mediated phosphorylation of UFL1 at Thr536 impaired PD-1 UFMylation, leading to PD-1 degradation and enhanced CD8+ T cell activation. Consequently, the ablation of UFL1 in T cells diminishes PD-1 stability, promoting a robust anti-tumor immune response. This improved tumor immunity is particularly evident in the enhanced response of Ufl1 cKO mice to anti-CTLA-4 immunotherapy. This study not only clarify the significant role of UFMylation in T-cell function but also position UFL1 as a promising target for cancer therapy (106).
PD-L1 UFMylation
Zhou et al. reported that dysregulation of PD-L1 by UFMylation disrupted tumor immune evasion (107). In this study, PD-L1 was identified as a target of UFMylation. Moreover, UFMylation contributes to the destabilization of PD-L1 by promoting its ubiquitination. Disrupting PD-L1 UFMylation through the depletion of UFL1 or UFM1, or by defective UFMylation of PD-L1 itself, stabilized PD-L1 in various human and murine cancer cell lines, which impair antitumor immunity both in vitro and in vivo. Additionally, a reduction in UFL1 expression was observed across multiple cancers. Moreover, lower UFL1 levels were associated negatively with anti-PD-1 therapy response in melanoma patients. Notably, a covalent inhibitor of UFSP2 that enhanced UFMylation activity was developed, potentially augmenting the efficacy of PD-1 blockade therapies. This work revealed a new regulatory mechanism of PD-L1, suggesting UFMylation as a novel therapeutic target in oncology (107).
PLAC8 UFMylation
Placenta specific 8 (PLAC8) has been revealed to regulate cell growth in tumorigenesis (115). Mao et al. reported that PLCA8 inhibited cell apoptosis via activation of the PI3K/AKT/NF-κB pathway in breast cancer (116). PLAC8/MAPK (mitogen-activated protein kinase) axis regulated tamoxifen sensitivity in breast cancer, which was abrogated by curcumin-mediated protein stability change (117). Chen et al. reported that PLAC8 enhanced Adriamycin resistance by reduction of autophagy in breast cancer (118). PLAC8 was increased in TNBC and underwent UFM1-mediated modification, which enhances its stability and influences cellular proliferation. Importantly, PLAC8 modulated the immune response by regulating PD-L1 ubiquitination levels. Clinical data from breast cancer patients further revealed that PLAC8 expression was elevated in TNBC compared to non-TNBC and was associated positively with PD-L1 expression. These findings introduce a new PLAC8-regulated pathway in TNBC, offering new insights for clinical diagnosis and opening potential avenues for immunotherapeutic interventions in breast cancer subtype (119).
UFMylation regulates RIG-I
RIG-I, an RNA-binding protein, initiates the antiviral innate immune response by activating downstream signaling through the adaptor protein MAVS, leading to the production of type I and III interferons (IFNs). This signaling cascade is localized at endoplasmic reticulum (ER)–mitochondrial contact sites. UFL1 as a component recruited to these contact sites following RIG-I activation. UFL1 and the UFMylation process are critical for IFN induction after RIG-I activation. Post RNA virus infection, UFL1 associates with the membrane-targeting protein 14–3-3ϵ, facilitating its recruitment to activated RIG-I, thus enhancing downstream immune signaling. Significantly, there was an increase in UFM1 conjugation of 14–3-3ϵ post RIG-I activation. Furthermore, the absence of UFMylation disrupts the interaction between 14–3-3ϵ and RIG-I, consequently impeding the linkage to MAVS (mitochondrial antiviral signaling protein) and the subsequent IFN-inducing signal transduction. These findings establish UFMylation as a crucial regulatory mechanism in RIG-I-mediated signaling and as a pivotal posttranslational modulator of IFN induction, which highlight potential targets for modulating antiviral immune responses (120).
Inhibitors for UFMylation
Metformin
Metformin is a widely used medication primarily for treating type 2 diabetes (121). It is recognized for its effectiveness, safety, and cost-efficiency, making it one of the most commonly prescribed drugs for managing diabetes around the world (122). Metformin works by improving the sensitivity of body tissues to insulin, thereby facilitating better cellular uptake of glucose, which lowers blood sugar levels (123). In addition, metformin has other potential health benefits and uses in prediabetes (124, 125), polycystic ovary syndrome (PCOS) (126, 127), aging (128, 129) and extending life span (130). Moreover, metformin has attracted interest for its potential in longevity via influencing fundamental aging factors (131). Metformin has been implicated to play a critical role in cancer treatment and prevention (132, 133). For example, metformin inhibited cell proliferation and glycolysis via regulation of ADAMTS12 in gastric cancer (134). Metformin reduced tumor cell stemness induced by paclitaxel via regulating FOXO3a in non-small-cell lung cancer (135). Metformin displayed antineoplastic functions via modulation of TGF-β and p38/ERK/MAPK signaling pathways in PTEN-deficient endometrial cancer (136).
Ferroptosis is a type of programmed cell death, which is distinct from other forms of cell death like apoptosis, necrosis, and autophagy (137). The process is heavily dependent on iron, which contributes to the production of ROS and lipid peroxidation (138). Targeting ferroptosis has been identified for an effective strategy for cancer treatment (139). Metformin has been reported to target apoptosis, necroptosis and ferroptosis in breast cancer cells (140). Moreover, metformin targeted miR-324-3p and GPX4, leading to induction of ferroptosis in breast cancer (141). One study showed that metformin inhibited autophagy via influencing lncRNA H19 and resulted in induction of ferroptosis in breast cancer (142). Another study showed that metformin induced ferroptosis and increased sorafenib sensitivity via regulation of ATF4 (activating transcription factor 4) and STAT3 (signal transducer and activator of transcription 3) in hepatocellular carcinoma cells (143). Metformin regulated the Nrf2/HO-1 signaling pathway and facilitated ferroptosis in lung cancer (144). Recently, Yang et al. reported that metformin mediated ferroptosis via suppression of UFMylation of SLC7A11 in breast cancer (145). Metformin promoted ferroptosis independently of AMPK to inhibit tumor growth. Metformin elevated levels of Fe2+ and lipid reactive oxygen species within cells. Metformin disrupted the stability of the protein SLC7A11, a key regulator of ferroptosis, by impeding its UFMylation. Additionally, when combined with sulfasalazine, metformin synergistically enhanced ferroptosis and suppressed the growth of breast cancer cells (145). Hence, metformin inhibited UFMylation of SLC7A11 in breast cancer.
UBA5 inhibitor
One group developed a selective micromolar inhibitor of UBA5, compound 8.5, to block UFM1 protein conjugation. This group introduced a new organometallic inhibitor that features a core scaffold incorporating adenosine and zinc(II)cyclen. The inhibitor targets UBA5 selectively and noncompetitively, distinguishing itself from other E1 enzymes and a broad spectrum of human kinases. In vitro data showed that this inhibitor of UBA5 selectively hindered the proliferation of cancer cells, particularly effective at concentrations above 50μM in cells with elevated levels of UBA5. This inhibitor could offer potential insights into therapeutic interventions via regulation of UFM1 pathway (146). Another group used chemoproteomic screening of covalent ligands and found that UBA5 might be a potential therapy target for pancreatic cancer. Moreover, this research led to the discovery of DKM 2-93, a covalent ligand that reduced cell survival and tumor growth in vivo in pancreatic cancer. DKM 2-93 achieved its therapeutic effects by covalently modifying the catalytic cysteine of UBA5, thereby inhibiting its function in activating the UFM1 for protein UFMylation. UBA5 could be a potent target for pancreatic cancer therapy and DKM 2-93 might be a relatively selective inhibitor of UBA5 (74). Fang et al. reported the properties of usenamine A, a natural compound derived from the lichen Usnea longissimi, known for its inhibitory effects on UBA5. In vitro studies demonstrated that usenamine A effectively inhibits cell proliferation, induces G2/M phase arrest, autophagy, and endoplasmic reticulum stress in breast cancer cells. These findings suggest that usenamine A holds potential as a therapeutic agent to inhibit the breast tumorigenesis and progression in part via regulation of UBA5 (147).
Conclusions and perspectives
In conclusion, UFMylation plays an essential role in the development and progression of tumors (Table 1). Moreover, UFMylation is critically involved in regulation of cancer immunotherapy (Table 2). Utilizing UFMylation as a target may enhance the efficacy of cancer treatment and immunotherapy (Figure 2). As UFMylation emerges as a critical PTM with substantial implications in cellular regulation and disease, future research directions are poised to expand our understanding and harness this pathway therapeutically. UFMylation of PTIP (Pax2 transactivation domain interacting protein) was reported to confer chemoresistance in BRCA1-deficine cells (148). Further detailed studies are needed to delineate the precise molecular mechanisms by which UFMylation influences cellular processes such as protein stability, signal transduction, and stress responses. Advanced techniques such as cryo-electron microscopy and single-molecule fluorescence could provide deeper insights into the structural and functional dynamics of UFMylation at the molecular level. Although a link between UFMylation and diseases like cancer, neurodegeneration, and inflammatory disorders has been established, future research should focus on identifying specific UFMylation-related pathways that are dysregulated in these conditions. This involves comprehensive proteomic studies and genetic screening to map out the UFMylation substrates and their pathophysiological roles in various diseases. In addition, there is a compelling need to develop specific inhibitors or enhancers of UFMylation enzymes as potential therapeutic agents. Drug discovery efforts should be intensified to identify small molecules or biologics that can modulate the UFMylation pathway with high specificity and low toxicity. Given its role in critical cellular functions, UFMylation could serve as a biomarker for disease progression or therapeutic response, particularly in cancer and immune-related conditions. Research should be directed towards validating UFMylation-related proteins or modifications as diagnostic, prognostic, or predictive biomarkers. Combining UFMylation studies with other omics approaches (genomics, transcriptomics, metabolomics) can yield comprehensive insights into how UFMylation interacts with other cellular pathways and influences complex biological systems. Such integrative studies would be crucial for constructing a holistic model of cellular regulation and disease manifestation. Finally, the translation of bench research to bedside application will require rigorous clinical trials to evaluate the safety and efficacy of UFMylation-targeted therapies. In summary, research on UFMylation can advance from basic science to clinical applications, offering new strategies for treating a variety of diseases and enhancing our understanding of cellular homeostasis.
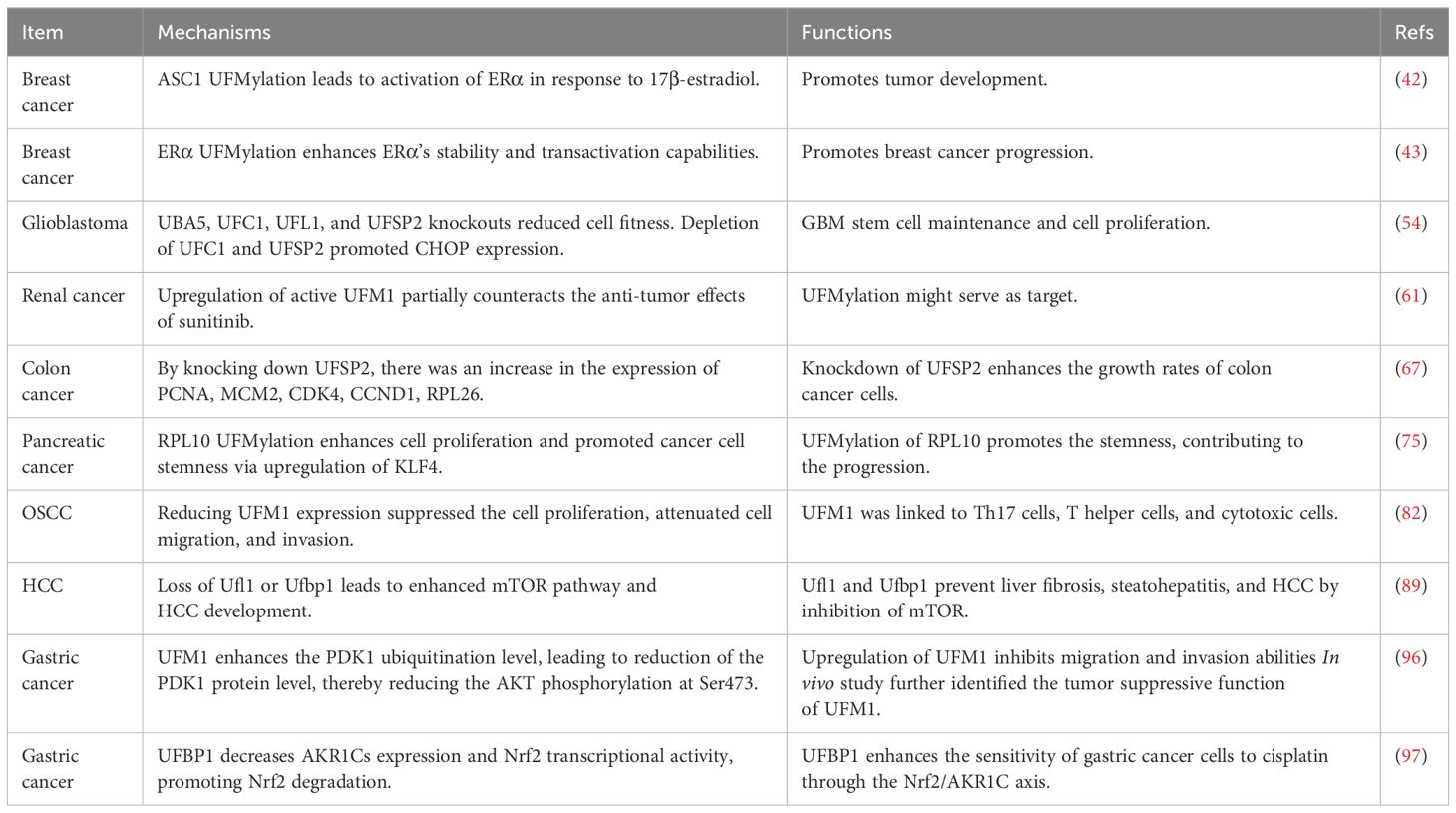
Table 1. Role of UFMylation in tumorigenesis.
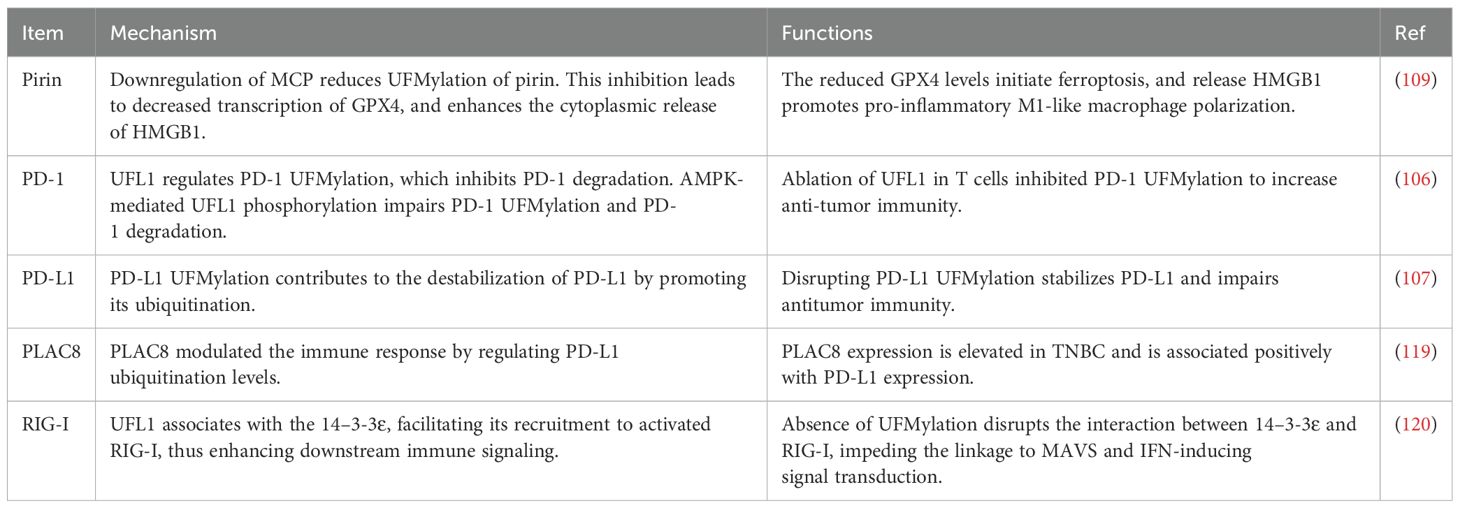
Table 2. Role of UFMylation in immunotherapy.
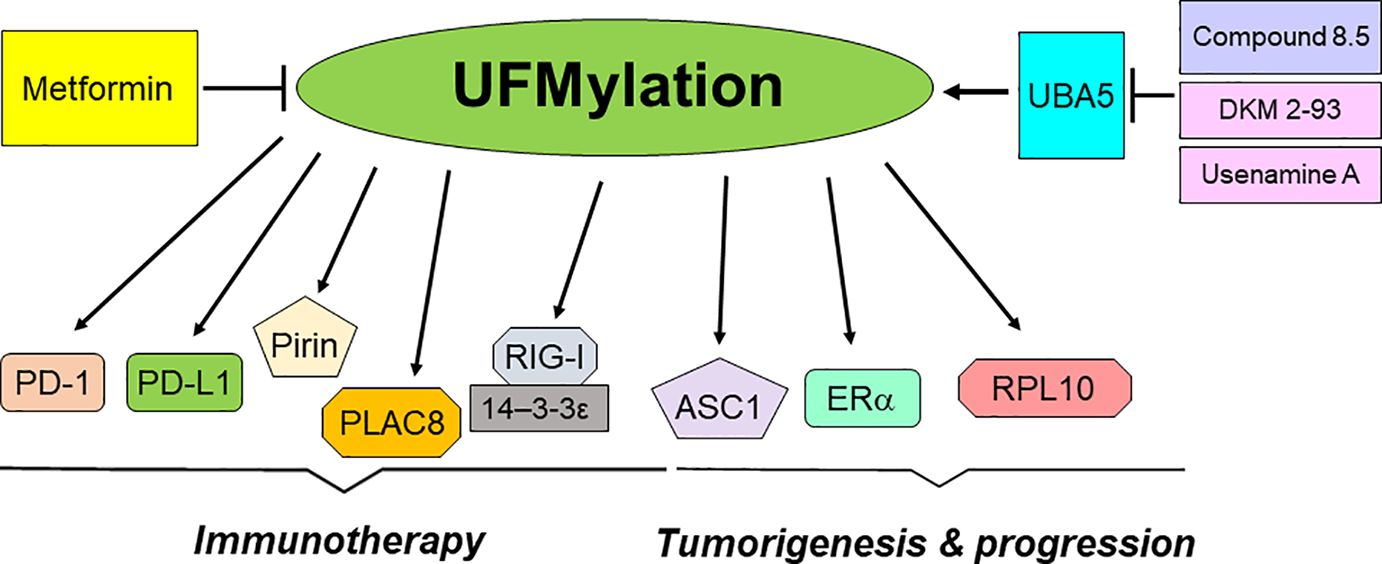
Figure 2. UFMylation in regulation of tumorigenesis and immunotherapy.
Author contributions
L-JD: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Software, Writing – original draft. XJ: Data curation, Formal analysis, Investigation, Methodology, Software, Writing – original draft. TL: Data curation, Formal analysis, Investigation, Methodology, Project administration, Software, Writing – review & editing. SW: Conceptualization, Funding acquisition, Project administration, Supervision, Validation, Visualization, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the National Natural Science Foundation of China (82070399).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Walsh G, Jefferis R. Post-translational modifications in the context of therapeutic proteins. Nat Biotechnol. (2006) 24:1241–52. doi: 10.1038/nbt1252
2. Li W, Li F, Zhang X, Lin HK, Xu C. Insights into the post-translational modification and its emerging role in shaping the tumor microenvironment. Signal Transduct Target Ther. (2021) 6(1):422. doi: 10.1038/s41392-021-00825-8
3. Kitamura N, Galligan JJ. A global view of the human post-translational modification landscape. Biochem J. (2023) 480:1241–65. doi: 10.1042/BCJ20220251
4. Hogg SJ, Beavis PA, Dawson MA, Johnstone RW. Targeting the epigenetic regulation of antitumour immunity. Nat Rev Drug Discovery. (2020) 19:776–800. doi: 10.1038/s41573-020-0077-5
5. Ramazi S, Zahiri J. Posttranslational modifications in proteins: resources, tools and prediction methods. Database (Oxford). (2021) 2021:baab012. doi: 10.1093/database/baab012
6. Ubersax JA, Ferrell JE Jr. Mechanisms of specificity in protein phosphorylation. Nat Rev Mol Cell Biol. (2007) 8:530–41. doi: 10.1038/nrm2203
7. Beck DB, Werner A, Kastner DL, Aksentijevich I. Disorders of ubiquitylation: unchained inflammation. Nat Rev Rheumatol. (2022) 18:435–47. doi: 10.1038/s41584-022-00778-4
8. Wang W, Liu W, Chen Q, Yuan Y, Wang P. Targeting CSC-related transcription factors by E3 ubiquitin ligases for cancer therapy. Semin Cancer Biol. (2022) 87:84–97. doi: 10.1016/j.semcancer.2022.11.002
9. Shvedunova M, Akhtar A. Modulation of cellular processes by histone and non-histone protein acetylation. Nat Rev Mol Cell Biol. (2022) 23:329–49. doi: 10.1038/s41580-021-00441-y
10. Greer EL, Shi Y. Histone methylation: a dynamic mark in health, disease and inheritance. Nat Rev Genet. (2012) 13:343–57. doi: 10.1038/nrg3173
11. Schjoldager KT, Narimatsu Y, Joshi HJ, Clausen H. Global view of human protein glycosylation pathways and functions. Nat Rev Mol Cell Biol. (2020) 21:729–49. doi: 10.1038/s41580-020-00294-x
12. Geoffroy MC, Hay RT. An additional role for SUMO in ubiquitin-mediated proteolysis. Nat Rev Mol Cell Biol. (2009) 10(8):564–8. doi: 10.1038/nrm2707
13. Vertegaal ACO. Signalling mechanisms and cellular functions of SUMO. Nat Rev Mol Cell Biol. (2022) 23:715–31. doi: 10.1038/s41580-022-00500-y
14. SM F, Abrami L, Linder ME, Bamji SX, Dickinson BC, van der Goot FG. Mechanisms and functions of protein S-acylation. Nat Rev Mol Cell Biol. (2024) 25(6):488–509. doi: 10.1038/s41580-024-00700-8
15. Wang X, Xu X, Wang Z. The post-translational role of UFMylation in physiology and disease. Cells. (2023) 12(21):2543. doi: 10.3390/cells12212543
16. Wei Y, Xu X. UFMylation: A unique & Fashionable modification for life. Genomics Proteomics Bioinf. (2016) 14:140–6. doi: 10.1016/j.gpb.2016.04.001
17. Millrine D, Peter JJ, Kulathu Y. A guide to UFMylation, an emerging posttranslational modification. FEBS J. (2023) 290:5040–56. doi: 10.1111/febs.16730
18. Zhou X, Mahdizadeh SJ, Le Gallo M, Eriksson LA, Chevet E, Lafont E. UFMylation: a ubiquitin-like modification. Trends Biochem Sci. (2024) 49:52–67. doi: 10.1016/j.tibs.2023.10.004
19. Gerakis Y, Quintero M, Li H, Hetz C. The UFMylation system in proteostasis and beyond. Trends Cell Biol. (2019) 29:974–86. doi: 10.1016/j.tcb.2019.09.005
20. Banerjee S, Kumar M, Wiener R. Decrypting UFMylation: how proteins are modified with UFM1. Biomolecules. (2020) 10(10):1442. doi: 10.3390/biom10101442
21. Jing Y, Mao Z, Chen F. UFMylation system: an emerging player in tumorigenesis. Cancers (Basel). (2022) 14(14):3501. doi: 10.3390/cancers14143501
22. Jiang Q, Wang Y, Xiang M, Hua J, Zhou T, Chen F, et al. UFL1, a UFMylation E3 ligase, plays a crucial role in multiple cellular stress responses. Front Endocrinol (Lausanne). (2023) 14:1123124. doi: 10.3389/fendo.2023.1123124
23. Xie Z, Fang Z, Pan Z. Ufl1/RCAD, a Ufm1 E3 ligase, has an intricate connection with ER stress. Int J Biol Macromol. (2019) 135:760–7. doi: 10.1016/j.ijbiomac.2019.05.170
24. Zhang M, Zhu X, Zhang Y, Cai Y, Chen J, Sivaprakasam S, et al. RCAD/Ufl1, a Ufm1 E3 ligase, is essential for hematopoietic stem cell function and murine hematopoiesis. Cell Death Differ. (2015) 22:1922–34. doi: 10.1038/cdd.2015.51
25. Liu J, Guan D, Dong M, Yang J, Wei H, Liang Q, et al. UFMylation maintains tumour suppressor p53 stability by antagonizing its ubiquitination. Nat Cell Biol. (2020) 22:1056–63. doi: 10.1038/s41556-020-0559-z
26. Hassin O, Oren M. Drugging p53 in cancer: one protein, many targets. Nat Rev Drug Discovery. (2023) 22:127–44. doi: 10.1038/s41573-022-00571-8
27. Tuval A, Strandgren C, Heldin A, Palomar-Siles M, Wiman KG. Pharmacological reactivation of p53 in the era of precision anticancer medicine. Nat Rev Clin Oncol. (2024) 21:106–20. doi: 10.1038/s41571-023-00842-2
28. Peuget S, Zhou X, Selivanova G. Translating p53-based therapies for cancer into the clinic. Nat Rev Cancer. (2024) 24:192–215. doi: 10.1038/s41568-023-00658-3
29. Huang Y, Che X, Wang PW, Qu X. p53/MDM2 signaling pathway in aging, senescence and tumorigenesis. Semin Cancer Biol. (2024) 101:44–57. doi: 10.1016/j.semcancer.2024.05.001
30. Bode AM, Dong Z. Post-translational modification of p53 in tumorigenesis. Nat Rev Cancer. (2004) 4:793–805. doi: 10.1038/nrc1455
31. Bray F, Laversanne M, Sung H, Ferlay J, Siegel RL, Soerjomataram I, et al. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. (2024) 74:229–63. doi: 10.3322/caac.21834
32. Harbeck N, Penault-Llorca F, Cortes J, Gnant M, Houssami N, Poortmans P, et al. Breast cancer. Nat Rev Dis Primers. (2019) 5:66. doi: 10.1038/s41572-019-0111-2
33. Marra A, Chandarlapaty S, Modi S. Management of patients with advanced-stage HER2-positive breast cancer: current evidence and future perspectives. Nat Rev Clin Oncol. (2024) 21:185–202. doi: 10.1038/s41571-023-00849-9
34. Li YQ, Sun FZ, Li CX, Mo HN, Zhou YT, Lv D, et al. RARRES2 regulates lipid metabolic reprogramming to mediate the development of brain metastasis in triple negative breast cancer. Mil Med Res. (2023) 10:34. doi: 10.1186/s40779-023-00470-y
35. Bianchini G, De Angelis C, Licata L, Gianni L. Treatment landscape of triple-negative breast cancer - expanded options, evolving needs. Nat Rev Clin Oncol. (2022) 19:91–113. doi: 10.1038/s41571-021-00565-2
36. Sirhan Z, Thyagarajan A, Sahu RP. The efficacy of tucatinib-based therapeutic approaches for HER2-positive breast cancer. Mil Med Res. (2022) 9:39. doi: 10.1186/s40779-022-00401-3
37. Siegel RL, Giaquinto AN, Jemal A. Cancer statistics, 2024. CA Cancer J Clin. (2024) 74:12–49. doi: 10.3322/caac.21820
38. Will M, Liang J, Metcalfe C, Chandarlapaty S. Therapeutic resistance to anti-oestrogen therapy in breast cancer. Nat Rev Cancer. (2023) 23:673–85. doi: 10.1038/s41568-023-00604-3
39. Swain SM, Shastry M, Hamilton E. Targeting HER2-positive breast cancer: advances and future directions. Nat Rev Drug Discovery. (2023) 22:101–26. doi: 10.1038/s41573-022-00579-0
40. Ponde NF, Zardavas D, Piccart M. Progress in adjuvant systemic therapy for breast cancer. Nat Rev Clin Oncol. (2019) 16:27–44. doi: 10.1038/s41571-018-0089-9
41. Britt KL, Cuzick J, Phillips KA. Key steps for effective breast cancer prevention. Nat Rev Cancer. (2020) 20:417–36. doi: 10.1038/s41568-020-0266-x
42. Yoo HM, Kang SH, Kim JY, Lee JE, Seong MW, Lee SW, et al. Modification of ASC1 by UFM1 is crucial for ERalpha transactivation and breast cancer development. Mol Cell. (2014) 56:261–74. doi: 10.1016/j.molcel.2014.08.007
43. Yoo HM, Park JH, Kim JY, Chung CH. Modification of ERalpha by UFM1 increases its stability and transactivity for breast cancer development. Mol Cells. (2022) 45:425–34. doi: 10.14348/molcells.2022.0029
44. Miller KD, Ostrom QT, Kruchko C, Patil N, Tihan T, Cioffi G, et al. Brain and other central nervous system tumor statistics, 2021. CA Cancer J Clin. (2021) 71:381–406. doi: 10.3322/caac.21693
45. Rabah N, Ait Mohand FE, Kravchenko-Balasha N. Understanding glioblastoma signaling, heterogeneity, invasiveness, and drug delivery barriers. Int J Mol Sci. (2023) 24(18):14256. doi: 10.3390/ijms241814256
46. Shahcheraghi SH, Alimardani M, Lotfi M, Lotfi M, Uversky VN, Guetchueng ST, et al. Advances in glioblastoma multiforme: Integrating therapy and pathology perspectives. Pathol Res Pract. (2024) 257:155285. doi: 10.1016/j.prp.2024.155285
47. He Y, Dong XH, Zhu Q, Xu YL, Chen ML, Liu Z. Ultrasound-triggered microbubble destruction enhances the radiosensitivity of glioblastoma by inhibiting PGRMC1-mediated autophagy in vitro and in vivo. Mil Med Res. (2022) 9:9. doi: 10.1186/s40779-022-00369-0
48. Noorani I, Mischel PS, Swanton C. Leveraging extrachromosomal DNA to fine-tune trials of targeted therapy for glioblastoma: opportunities and challenges. Nat Rev Clin Oncol. (2022) 19:733–43. doi: 10.1038/s41571-022-00679-1
49. Lim M, Xia Y, Bettegowda C, Weller M. Current state of immunotherapy for glioblastoma. Nat Rev Clin Oncol. (2018) 15:422–42. doi: 10.1038/s41571-018-0003-5
50. Guo QL, Dai XL, Yin MY, Cheng HW, Qian HS, Wang H, et al. Nanosensitizers for sonodynamic therapy for glioblastoma multiforme: current progress and future perspectives. Mil Med Res. (2022) 9:26. doi: 10.1186/s40779-022-00386-z
51. Mao M, Wu Y, He Q. Recent advances in targeted drug delivery for the treatment of glioblastoma. Nanoscale. (2024) 16:8689–707. doi: 10.1039/d4nr01056f
52. Obrador E, Moreno-Murciano P, Oriol-Caballo M, Lopez-Blanch R, Pineda B, Gutierrez-Arroyo JL, et al. Glioblastoma therapy: past, present and future. Int J Mol Sci. (2024) 25(5):2529. doi: 10.3390/ijms25052529
53. Sharma S, Chepurna O, Sun T. Drug resistance in glioblastoma: from chemo- to immunotherapy. Cancer Drug Resist. (2023) 6:688–708. doi: 10.20517/cdr.2023.82
54. MacLeod G, Bozek DA, Rajakulendran N, Monteiro V, Ahmadi M, Steinhart Z, et al. Genome-wide CRISPR-cas9 screens expose genetic vulnerabilities and mechanisms of temozolomide sensitivity in glioblastoma stem cells. Cell Rep. (2019) 27:971–986 e9. doi: 10.1016/j.celrep.2019.03.047
55. Gansler T, Fedewa S, Amin MB, Lin CC, Jemal A. Trends in reporting histological subtyping of renal cell carcinoma: association with cancer center type. Hum Pathol. (2018) 74:99–108. doi: 10.1016/j.humpath.2018.01.010
56. Hsieh JJ, Purdue MP, Signoretti S, Swanton C, Albiges L, Schmidinger M, et al. Renal cell carcinoma. Nat Rev Dis Primers. (2017) 3:17009. doi: 10.1038/nrdp.2017.9
57. Zieren RC, Zondervan PJ, Pienta KJ, Bex A, de Reijke TM, Bins AD. Diagnostic liquid biopsy biomarkers in renal cell cancer. Nat Rev Urol. (2024) 21:133–57. doi: 10.1038/s41585-023-00818-y
58. Usher-Smith J, Simmons RK, Rossi SH, Stewart GD. Current evidence on screening for renal cancer. Nat Rev Urol. (2020) 17:637–42. doi: 10.1038/s41585-020-0363-3
59. Siva S, Kothari G, Muacevic A, Louie AV, Slotman BJ, Teh BS, et al. Radiotherapy for renal cell carcinoma: renaissance of an overlooked approach. Nat Rev Urol. (2017) 14:549–63. doi: 10.1038/nrurol.2017.87
60. Ginzburg S, Tomaszewski JJ, Kutikov A. Focal ablation therapy for renal cancer in the era of active surveillance and minimally invasive partial nephrectomy. Nat Rev Urol. (2017) 14:669–82. doi: 10.1038/nrurol.2017.143
61. Wang S, Jia M, Su M, Hu X, Li J, Xu Y, et al. Ufmylation is activated in renal cancer and is not associated with von hippel-lindau mutation. DNA Cell Biol. (2020) 39:654–60. doi: 10.1089/dna.2019.5225
62. Liao C, Hu L, Zhang Q. Von Hippel-Lindau protein signalling in clear cell renal cell carcinoma. Nat Rev Urol. (2024) 39(4):654–60. doi: 10.1038/s41585-024-00876-w
63. Siegel RL, Wagle NS, Cercek A, Smith RA, Jemal A. Colorectal cancer statistics, 2023. CA Cancer J Clin. (2023) 73:233–54. doi: 10.3322/caac.21772
64. O'Keefe SJ. Diet, microorganisms and their metabolites, and colon cancer. Nat Rev Gastroenterol Hepatol. (2016) 13:691–706. doi: 10.1038/nrgastro.2016.165
65. Gangadhar T, Schilsky RL. Molecular markers to individualize adjuvant therapy for colon cancer. Nat Rev Clin Oncol. (2010) 7:318–25. doi: 10.1038/nrclinonc.2010.62
66. Ricciardiello L, Ahnen DJ, Lynch PM. Chemoprevention of hereditary colon cancers: time for new strategies. Nat Rev Gastroenterol Hepatol. (2016) 13:352–61. doi: 10.1038/nrgastro.2016.56
67. Zhou J, Ma X, Xu L, Liang Q, Mao J, Liu J, et al. Genomic profiling of the UFMylation family genes identifies UFSP2 as a potential tumour suppressor in colon cancer. Clin Transl Med. (2021) 11:e642. doi: 10.1002/ctm2.642
68. Collisson EA, Bailey P, Chang DK, Biankin AV. Molecular subtypes of pancreatic cancer. Nat Rev Gastroenterol Hepatol. (2019) 16:207–20. doi: 10.1038/s41575-019-0109-y
69. Klein AP. Pancreatic cancer epidemiology: understanding the role of lifestyle and inherited risk factors. Nat Rev Gastroenterol Hepatol. (2021) 18:493–502. doi: 10.1038/s41575-021-00457-x
70. Stoop TF, Theijse RT, Seelen LWF, Groot Koerkamp B, van Eijck CHJ, Wolfgang CL, et al. Preoperative chemotherapy, radiotherapy and surgical decision-making in patients with borderline resectable and locally advanced pancreatic cancer. Nat Rev Gastroenterol Hepatol. (2024) 21:101–24. doi: 10.1038/s41575-023-00856-2
71. Hu ZI, O'Reilly EM. Therapeutic developments in pancreatic cancer. Nat Rev Gastroenterol Hepatol. (2024) 21:7–24. doi: 10.1038/s41575-023-00840-w
72. Huang X, Zhang G, Tang TY, Gao X, Liang TB. Personalized pancreatic cancer therapy: from the perspective of mRNA vaccine. Mil Med Res. (2022) 9:53. doi: 10.1186/s40779-022-00416-w
73. Singhi AD, Wood LD. Early detection of pancreatic cancer using DNA-based molecular approaches. Nat Rev Gastroenterol Hepatol. (2021) 18(7):457–68. doi: 10.1038/s41575-021-00470-0
74. Roberts AM, Miyamoto DK, Huffman TR, Bateman LA, Ives AN, Akopian D, et al. Chemoproteomic screening of covalent ligands reveals UBA5 as a novel pancreatic cancer target. ACS Chem Biol. (2017) 12:899–904. doi: 10.1021/acschembio.7b00020
75. Wang K, Chen S, Wu Y, Wang Y, Lu Y, Sun Y, et al. The ufmylation modification of ribosomal protein L10 in the development of pancreatic adenocarcinoma. Cell Death Dis. (2023) 14:350. doi: 10.1038/s41419-023-05877-y
76. Wu N, Li Y, Ma X, Huang Z, Chen Z, Chen W, et al. High incidence of HPV infection in minors with oral squamous cell carcinoma. Diagn Pathol. (2024) 19:51. doi: 10.1186/s13000-024-01470-9
77. Wang J, Gao B. Mechanisms and potential clinical implications of oral microbiome in oral squamous cell carcinoma. Curr Oncol. (2023) 31:168–82. doi: 10.3390/curroncol31010011
78. Gupta S, Singh B, Abhishek R, Gupta S, Sachan M. The emerging role of liquid biopsy in oral squamous cell carcinoma detection: advantages and challenges. Expert Rev Mol Diagn. (2024) 24:311–31. doi: 10.1080/14737159.2024.2340997
79. Ghaderi H, Roshan-Zamir M, Jafarinia M, Kruger E. Oral squamous cell carcinoma: focus on biomarkers for screening. J Dent (Shiraz). (2024) 25:1–16. doi: 10.30476/dentjods.2023.96159.1924
80. Fu J, Alhaskawi A, Dong Y, Jin F, Chen J, Zou X, et al. Improving oral squamous cell carcinoma diagnosis and treatment with fluorescence molecular imaging. Photodiagnosis Photodyn Ther. (2023) 44:103760. doi: 10.1016/j.pdpdt.2023.103760
81. Alsaeedi SM, Aggarwal S. The holistic review on occurrence, biology, diagnosis, and treatment of oral squamous cell carcinoma. Cureus. (2022) 14:e30226. doi: 10.7759/cureus.30226
82. Ke D, Guo HH, Jiang N, Shi RS, Fan TY. Inhibition of UFM1 expression suppresses cancer progression and is linked to the dismal prognosis and immune infiltration in oral squamous cell carcinoma. Aging (Albany NY). (2023) 15:13059–76. doi: 10.18632/aging.205219
83. Yang JD, Hainaut P, Gores GJ, Amadou A, Plymoth A, Roberts LR. A global view of hepatocellular carcinoma: trends, risk, prevention and management. Nat Rev Gastroenterol Hepatol. (2019) 16:589–604. doi: 10.1038/s41575-019-0186-y
84. Llovet JM, Kelley RK, Villanueva A, Singal AG, Pikarsky E, Roayaie S, et al. Hepatocellular carcinoma. Nat Rev Dis Primers. (2021) 7:6. doi: 10.1038/s41572-020-00240-3
85. Llovet JM, Pinyol R, Yarchoan M, Singal AG, Marron TU, Schwartz M, et al. Adjuvant and neoadjuvant immunotherapies in hepatocellular carcinoma. Nat Rev Clin Oncol. (2024) 21:294–311. doi: 10.1038/s41571-024-00868-0
86. Huang DQ, Singal AG, Kanwal F, Lampertico P, Buti M, Sirlin CB, et al. Hepatocellular carcinoma surveillance - utilization, barriers and the impact of changing aetiology. Nat Rev Gastroenterol Hepatol. (2023) 20:797–809. doi: 10.1038/s41575-023-00818-8
87. Lu Y, Gao Y, Yang H, Hu Y, Li X. Nanomedicine-boosting icaritin-based immunotherapy of advanced hepatocellular carcinoma. Mil Med Res. (2022) 9:69. doi: 10.1186/s40779-022-00433-9
88. Chen E, Zhou B, Bian S, Ni W, Chen Z. The lncRNA B3GALT5-AS1 Functions as an HCC Suppressor by Regulating the miR-934/UFM1 Axis. J Oncol. (2021) 2021:1776432. doi: 10.1155/2021/1776432
89. Chen F, Sheng L, Zhou T, Yan L, Loveless R, Li H, et al. Loss of Ufl1/Ufbp1 in hepatocytes promotes liver pathological damage and carcinogenesis through activating mTOR signaling. J Exp Clin Cancer Res. (2023) 42:110. doi: 10.1186/s13046-023-02681-6
90. Wang W, Yang YJ, Zhang RH, Deng JY, Sun Z, Seeruttun SR, et al. Standardizing the classification of gastric cancer patients with limited and adequate number of retrieved lymph nodes: an externally validated approach using real-world data. Mil Med Res. (2022) 9:15. doi: 10.1186/s40779-022-00375-2
91. Thrift AP, Wenker TN, El-Serag HB. Global burden of gastric cancer: epidemiological trends, risk factors, screening and prevention. Nat Rev Clin Oncol. (2023) 20:338–49. doi: 10.1038/s41571-023-00747-0
92. Nobel T, Sihag S. Advances in diagnostic, staging, and restaging evaluation of esophageal and gastric cancer. Surg Oncol Clin N Am. (2024) 33:467–85. doi: 10.1016/j.soc.2024.02.002
93. Burz C, Pop V, Silaghi C, Lupan I, Samasca G. Prognosis and treatment of gastric cancer: A 2024 update. Cancers (Basel). (2024) 16(9):1708. doi: 10.3390/cancers16091708
94. Alsina M, Arrazubi V, Diez M, Tabernero J. Current developments in gastric cancer: from molecular profiling to treatment strategy. Nat Rev Gastroenterol Hepatol. (2023) 20:155–70. doi: 10.1038/s41575-022-00703-w
95. Lin M, Lian NZ, Cao LL, Huang CM, Zheng CH, Li P, et al. Down-regulated expression of CDK5RAP3 and UFM1 suggests a poor prognosis in gastric cancer patients. Front Oncol. (2022) 12:927751. doi: 10.3389/fonc.2022.927751
96. Lin JX, Xie XS, Weng XF, Qiu SL, Yoon C, Lian NZ, et al. UFM1 suppresses invasive activities of gastric cancer cells by attenuating the expres7sion of PDK1 through PI3K/AKT signaling. J Exp Clin Cancer Res. (2019) 38:410. doi: 10.1186/s13046-019-1416-4
97. Hu Z, Wang X, Li D, Cao L, Cui H, Xu G. UFBP1, a key component in ufmylation, enhances drug sensitivity by promoting proteasomal degradation of oxidative stress-response transcription factor Nrf2. Oncogene. (2021) 40:647–62. doi: 10.1038/s41388-020-01551-1
98. Fenis A, Demaria O, Gauthier L, Vivier E, Narni-Mancinelli E. New immune cell engagers for cancer immunotherapy. Nat Rev Immunol. (2024) 24(7):471–86. doi: 10.1038/s41577-023-00982-7
99. Schenkel JM, Pauken KE. Localization, tissue biology and T cell state - implications for cancer immunotherapy. Nat Rev Immunol. (2023) 23:807–23. doi: 10.1038/s41577-023-00884-8
100. Tang L, Huang ZP, Mei H, Hu Y. Insights gained from single-cell analysis of chimeric antigen receptor T-cell immunotherapy in cancer. Mil Med Res. (2023) 10:52. doi: 10.1186/s40779-023-00486-4
101. Mehrotra S, Kupani M, Kaur J, Kaur J, Pandey RK. Immunotherapy guided precision medicine in solid tumors. Adv Protein Chem Struct Biol. (2024) 140:249–92. doi: 10.1016/bs.apcsb.2024.02.004
102. Ma GL, Lin WF. Immune checkpoint inhibition mediated with liposomal nanomedicine for cancer therapy. Mil Med Res. (2023) 10:20. doi: 10.1186/s40779-023-00455-x
103. Ramapriyan R, Vykunta VS, Vandecandelaere G, Richardson LGK, Sun J, Curry WT, et al. Altered cancer metabolism and implications for next-generation CAR T-cell therapies. Pharmacol Ther. (2024) 259:108667. doi: 10.1016/j.pharmthera.2024.108667
104. Grant M, Ni Lee L, Chinnakannan S, Tong O, Kwok J, Cianci N, et al. Unlocking cancer vaccine potential: What are the key factors? Hum Vaccin Immunother. (2024) 20:2331486. doi: 10.1080/21645515.2024.2331486
105. Liu N, Xiao X, Zhang Z, Mao C, Wan M, Shen J. Advances in cancer vaccine research. ACS Biomater Sci Eng. (2023) 9:5999–6023. doi: 10.1021/acsbiomaterials.3c01154
106. He C, Xing X, Chen HY, Gao M, Shi J, Xiang B, et al. UFL1 ablation in T cells suppresses PD-1 UFMylation to enhance anti-tumor immunity. Mol Cell. (2024) 84:1120–1138 e8. doi: 10.1016/j.molcel.2024.01.024
107. Zhou J, Ma X, He X, Chen B, Yuan J, Jin Z, et al. Dysregulation of PD-L1 by UFMylation imparts tumor immune evasion and identified as a potential therapeutic target. Proc Natl Acad Sci U.S.A. (2023) 120:e2215732120. doi: 10.1073/pnas.2215732120
108. Khayat S, Choudhary K, Claude Nshimiyimana J, Gurav J, Hneini A, Nazir A, et al. Pancreatic cancer: from early detection to personalized treatment approaches. Ann Med Surg (Lond). (2024) 86:2866–72. doi: 10.1097/MS9.0000000000002011
109. Li G, Liao C, Chen J, Wang Z, Zhu S, Lai J, et al. Targeting the MCP-GPX4/HMGB1 axis for effectively triggering immunogenic ferroptosis in pancreatic ductal adenocarcinoma. Adv Sci (Weinh). (2024) 11(21):e2308208. doi: 10.1002/advs.202308208
110. Lin X, Kang K, Chen P, Zeng Z, Li G, Xiong W, et al. Regulatory mechanisms of PD-1/PD-L1 in cancers. Mol Cancer. (2024) 23:108. doi: 10.1186/s12943-024-02023-w
111. Hou B, Chen T, Zhang H, Li J, Wang P, Shang G. The E3 ubiquitin ligases regulate PD-1/PD-L1 protein levels in tumor microenvironment to improve immunotherapy. Front Immunol. (2023) 14:1123244. doi: 10.3389/fimmu.2023.1123244
112. Javed SA, Najmi A, Ahsan W, Zoghebi K. Targeting PD-1/PD-L-1 immune checkpoint inhibition for cancer immunotherapy: success and challenges. Front Immunol. (2024) 15:1383456. doi: 10.3389/fimmu.2024.1383456
113. Llovet JM, Castet F, Heikenwalder M, Maini MK, Mazzaferro V, Pinato DJ, et al. Immunotherapies for hepatocellular carcinoma. Nat Rev Clin Oncol. (2022) 19:151–72. doi: 10.1038/s41571-021-00573-2
114. Greten TF, Villanueva A, Korangy F, Ruf B, Yarchoan M, Ma L, et al. Biomarkers for immunotherapy of hepatocellular carcinoma. Nat Rev Clin Oncol. (2023) 20:780–98. doi: 10.1038/s41571-023-00816-4
115. Mao M, Cheng Y, Yang J, Chen Y, Xu L, Zhang X, et al. Multifaced roles of PLAC8 in cancer. biomark Res. (2021) 9:73. doi: 10.1186/s40364-021-00329-1
116. Mao M, Chen Y, Jia Y, Yang J, Wei Q, Li Z, et al. PLCA8 suppresses breast cancer apoptosis by activating the PI3k/AKT/NF-kappaB pathway. J Cell Mol Med. (2019) 23:6930–41. doi: 10.1111/jcmm.14578
117. Mao M, Hu D, Yang J, Chen Y, Zhang X, Shen J, et al. Regulation of tamoxifen sensitivity by the PLAC8/MAPK pathway axis is antagonized by curcumin-induced protein stability change. J Mol Med (Berl). (2021) 99:845–58. doi: 10.1007/s00109-021-02047-5
118. Chen Y, Jia Y, Mao M, Gu Y, Xu C, Yang J, et al. PLAC8 promotes adriamycin resistance via blocking autophagy in breast cancer. J Cell Mol Med. (2021) 25:6948–62. doi: 10.1111/jcmm.16706
119. Mao M, Chen Y, Yang J, Cheng Y, Xu L, Ji F, et al. Modification of PLAC8 by UFM1 affects tumorous proliferation and immune response by impacting PD-L1 levels in triple-negative breast cancer. J Immunother Cancer. (2022) 10(12):e005668. doi: 10.1136/jitc-2022-005668
120. Snider DL, Park M, Murphy KA, Beachboard DC, Horner SM. Signaling from the RNA sensor RIG-I is regulated by ufmylation. Proc Natl Acad Sci U.S.A. (2022) 119:e2119531119. doi: 10.1073/pnas.2119531119
121. Chaudhary S, Kulkarni A. Metformin: past, present, and future. Curr Diabetes Rep. (2024) 24:119–30. doi: 10.1007/s11892-024-01539-1
122. Patel D, Ayesha IE, Monson NR, Klair N, Patel U, Saxena A, et al. The effectiveness of metformin in diabetes prevention: A systematic review and meta-analysis. Cureus. (2023) 15:e46108. doi: 10.7759/cureus.46108
123. Hostalek U, Gwilt M, Hildemann S. Therapeutic use of metformin in prediabetes and diabetes prevention. Drugs. (2015) 75:1071–94. doi: 10.1007/s40265-015-0416-8
124. Hosey CM, Halpin K, Yan Y. Considering metformin as a second-line treatment for children and adolescents with prediabetes. J Pediatr Endocrinol Metab. (2022) 35:727–32. doi: 10.1515/jpem-2021-0200
125. Khokhar A, Umpaichitra V, Chin VL, Perez-Colon S. Metformin use in children and adolescents with prediabetes. Pediatr Clin North Am. (2017) 64:1341–53. doi: 10.1016/j.pcl.2017.08.010
126. Nassif DS, Januario BL, Sousa BA, Thabane L, Abbade JF. Effectiveness of metformin to pregnant women with PCOS to reduce spontaneous abortion and gestational diabetes mellitus: a protocol for an overview of reviews. BMJ Open. (2024) 14:e078217. doi: 10.1136/bmjopen-2023-078217
127. Attia GM, Almouteri MM, Alnakhli FT. Role of metformin in polycystic ovary syndrome (PCOS)-related infertility. Cureus. (2023) 15:e44493. doi: 10.7759/cureus.44493
128. Khan J, Pernicova I, Nisar K, Korbonits M. Mechanisms of ageing: growth hormone, dietary restriction, and metformin. Lancet Diabetes Endocrinol. (2023) 11:261–81. doi: 10.1016/S2213-8587(23)00001-3
129. Sunjaya AP, Sunjaya AF. Targeting ageing and preventing organ degeneration with metformin. Diabetes Metab. (2021) 47:101203. doi: 10.1016/j.diabet.2020.09.009
130. Valencia WM, Palacio A, Tamariz L, Florez H. Metformin and ageing: improving ageing outcomes beyond glycaemic control. Diabetologia. (017) 60:1630–8. doi: 10.1007/s00125-017-4349-5
131. Triggle CR, Mohammed I, Bshesh K, Marei I, Ye K, Ding H, et al. Metformin: Is it a drug for all reasons and diseases? Metabolism. (2022) 133:155223. doi: 10.1016/j.metabol.2022.155223
132. Galal MA, Al-Rimawi M, Hajeer A, Dahman H, Alouch S, Aljada A. Metformin: A dual-role player in cancer treatment and prevention. Int J Mol Sci. (2024) 25(7):4083. doi: 10.3390/ijms25074083
133. O’Connor L, Bailey-Whyte M, Bhattacharya M, Butera G, Hardell KNL, Seidenberg AB, et al. Association of metformin use and cancer incidence: a systematic review and meta-analysis. J Natl Cancer Inst. (2024) 116:518–29. doi: 10.1093/jnci/djae021
134. Chen R, Chen J, Chen M, Zhou S, Jiang P. Metformin suppresses proliferation and glycolysis of gastric cancer by modulating ADAMTS12. Genes Environ. (2024) 46:1. doi: 10.1186/s41021-023-00296-z
135. Tang Z, Zhang Y, Yu Z, Luo Z. Metformin suppresses stemness of non-small-cell lung cancer induced by paclitaxel through FOXO3a. Int J Mol Sci. (2023) 24(23):16611. doi: 10.3390/ijms242316611
136. Ruiz-Mitjana A, Vidal-Sabanes M, Navaridas R, Perramon-Guell A, Yeramian A, Nicholson-Sabate N, et al. Metformin exhibits antineoplastic effects on Pten-deficient endometrial cancer by interfering with TGF-beta and p38/ERK MAPK signalling. BioMed Pharmacother. (2023) 168:115817. doi: 10.1016/j.biopha.2023.115817
137. Bell HN, Stockwell BR, Zou W. Ironing out the role of ferroptosis in immunity. Immunity. (2024) 57:941–56. doi: 10.1016/j.immuni.2024.03.019
138. Li K, Fan C, Chen J, Xu X, Lu C, Shao H, et al. Role of oxidative stress-induced ferroptosis in cancer therapy. J Cell Mol Med. (2024) 28:e18399. doi: 10.1111/jcmm.18399
139. Khan A, Huo Y, Guo Y, Shi J, Hou Y. Ferroptosis is an effective strategy for cancer therapy. Med Oncol. (2024) 41:124. doi: 10.1007/s12032-024-02317-5
140. Dias Lopes NM, Marinello PC, Sanches LJ, da Silva Brito WA, Lovo-Martins MI, Pinge-Filho P, et al. Patterns of cell death induced by metformin in human MCF-7 breast cancer cells. Pathol Res Pract. (2020) 216:153199. doi: 10.1016/j.prp.2020.153199
141. Hou Y, Cai S, Yu S, Lin H. Metformin induces ferroptosis by targeting miR-324-3p/GPX4 axis in breast cancer. Acta Biochim Biophys Sin (Shanghai). (2021) 53:333–41. doi: 10.1093/abbs/gmaa180
142. Chen J, Qin C, Zhou Y, Chen Y, Mao M, Yang J. Metformin may induce ferroptosis by inhibiting autophagy via lncRNA H19 in breast cancer. FEBS Open Bio. (2022) 12:146–53. doi: 10.1002/2211-5463.13314
143. Hu Z, Zhao Y, Li L, Jiang J, Li W, Mang Y, et al. Metformin promotes ferroptosis and sensitivity to sorafenib in hepatocellular carcinoma cells via ATF4/STAT3. Mol Biol Rep. (2023) 50:6399–413. doi: 10.1007/s11033-023-08492-4
144. Deng C, Xiong L, Chen Y, Wu K, Wu J. Metformin induces ferroptosis through the Nrf2/HO-1 signaling in lung cancer. BMC Pulm Med. (2023) 23:360. doi: 10.1186/s12890-023-02655-6
145. Yang J, Zhou Y, Xie S, Wang J, Li Z, Chen L, et al. Metformin induces Ferroptosis by inhibiting UFMylation of SLC7A11 in breast cancer. J Exp Clin Cancer Res. (2021) 40:206. doi: 10.1186/s13046-021-02012-7
146. da Silva SR, Paiva SL, Bancerz M, Geletu M, Lewis AM, Chen J, et al. A selective inhibitor of the UFM1-activating enzyme, UBA5. Bioorg Med Chem Lett. (2016) 26:4542–7. doi: 10.1016/j.bmcl.2015.10.015
147. Fang B, Li Z, Qiu Y, Cho N, Yoo HM. Inhibition of UBA5 expression and induction of autophagy in breast cancer cells by usenamine A. Biomolecules. (2021) 11(9):1348. doi: 10.3390/biom11091348
Keywords: Ufmylation, oncogenesis, treatment, immunotherapy, cancer
Citation: Ding L-j, Jiang X, Li T and Wang S (2024) Role of UFMylation in tumorigenesis and cancer immunotherapy. Front. Immunol. 15:1454823. doi: 10.3389/fimmu.2024.1454823
Received: 25 June 2024; Accepted: 06 August 2024;
Published: 23 August 2024.
Edited by:
Zichuan Liu, Tianjin University, ChinaCopyright © 2024 Ding, Jiang, Li and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shudong Wang, c2h1ZG9uZ193YW5nQGpsdS5lZHUuY24=