
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Immunol. , 26 August 2024
Sec. Primary Immunodeficiencies
Volume 15 - 2024 | https://doi.org/10.3389/fimmu.2024.1451212
 Halyn Orellana1
Halyn Orellana1 Jia Yan1
Jia Yan1 Alex Paul2Mari Tokita1
Alex Paul2Mari Tokita1 Yan Ding3
Yan Ding3 Rajarshi Ghosh1Katie L. Lewis1Joie Davis1
Rajarshi Ghosh1Katie L. Lewis1Joie Davis1 Leila Jamal1
Leila Jamal1 Colleen Jodarski1
Colleen Jodarski1 Morgan Similuk1Nermina Saucier2
Morgan Similuk1Nermina Saucier2 Zhanyang Zhu3Yihe Wang3
Zhanyang Zhu3Yihe Wang3 Sitao Wu3Jason Ruggieri3 NIAID Centralized Sequencing Program Working Group
Sitao Wu3Jason Ruggieri3 NIAID Centralized Sequencing Program Working Group Helen C. Su1Gulbu Uzel1Shareef Nahas3
Helen C. Su1Gulbu Uzel1Shareef Nahas3 Megan Cooper2
Megan Cooper2 Magdalena A. Walkiewicz1*
Magdalena A. Walkiewicz1*Gonadal and gonosomal mosaicism describe phenomena in which a seemingly healthy individual carries a genetic variant in a subset of their gonadal tissue or gonadal and somatic tissue(s), respectively, with risk of transmitting the variant to their offspring. In families with one or more affected offspring, occurrence of the same apparently de novo variants can be an indicator of mosaicism in either parent. Panel-based deep sequencing has the capacity to detect low-level mosaic variants with coverage exceeding the typical limit of detection provided by current, readily available sequencing techniques. In this study, we report three families with more than one affected offspring with either confirmed or apparent parental gonosomal or gonadal mosaicism for PIK3CD pathogenic variants. Data from targeted deep sequencing was suggestive of low-level maternal gonosomal mosaicism in Family 1. Through this approach we did not detect pathogenic variants in PIK3CD from parental samples in Family 2 and Family 3. We conclude that mosaicism was likely confined to the maternal gonads in Family 2. Subsequent long-read genome sequencing in Family 3 showed that the paternal chromosome harbored the pathogenic variant in PIK3CD in both affected children, consistent with paternal gonadal mosaicism. Detection of parental mosaic variants enables accurate risk assessment, informs reproductive decision-making, and provides helpful context to inform clinical management in families with PIK3CD pathogenic variants.
Mosaicism, the presence of multiple genetically distinct cell populations within an individual, is an underrecognized mechanism of pathogenesis in patients with genetic diseases, including inborn errors of immunity (IEI). Parental mosaicism, either gonadal, in which a mosaic variant is present in a subset of gonadal tissue but not somatic tissue, or gonosomal, in which a mosaic variant is present in subsets of both gonadal and somatic tissue, confers risk of transmitting the mutant allele to the offspring(s), which leads to the development of genetic diseases. When unrecognized, parental mosaicism can be mistaken for a de novo change and confound recurrence risk estimates. In some clinical situations, e.g., bone marrow donor considerations, it can also influence the clinical management of the affected individual. Nevertheless, testing for mosaicism can be challenging, and current readily available sequencing techniques such as exome with on average 100X depth of coverage, genome with on average 30X depth of coverage, and Sanger sequencing may not provide the depth of coverage needed to detect all clinically relevant mosaic variants. However, in recent years, techniques with higher sequencing depth allowing for a lower limit of detection have proven invaluable in understanding mosaicism in many different areas, particularly for investigating disorders with dominant inheritance that may result from mosaic variants, including IEI.
Specifically, a recent study investigated the incidence of parental gonosomal mosaicism in 92 families with IEIs with de novo germline variants and in 36 families with IEIs with moderate-to-high suspicion of gene mosaicism (1). Parental gonosomal mosaicism was detected in approximately 7% of families with de novo mutations, and gonadal, somatic, gonosomal, or revertant mosaicism was detected in approximately 64% of families with moderate-to-high suspicion of gene mosaicism (1). Amplicon-based deep sequencing (ADS) showed low-level parental mosaicism in families with IEIs, including activated PI3K delta syndrome 1 (APDS1), autoimmune lymphoproliferative syndrome, Wiskott-Aldrich syndrome, and X-linked agammaglobulinemia (1, 2). This study identified a familial heterozygous c.3061G>A (p.Glu1021Lys) pathogenic variant in PIK3CD, defects in which cause APDS1, by Sanger sequencing in two siblings with APDS1. ADS analysis of parental tissue samples in this family, including a paternal sperm sample, did not detect the pathogenic variant in either parent, suggesting maternal gonadal mosaicism in this family (1). Two additional cases of mosaicism for PIK3CD have been reported. The first study used next generation sequencing (NGS) based methods on a parent-child trio to elucidate the origin of a homozygous c.3061G>A (p.Glu1021Lys) pathogenic variant in PIK3CD in a DNA sample extracted from blood in an affected child. Sanger sequencing did not detect the pathogenic variant in DNA extracted from either parental blood sample. However, ultra-deep sequencing at 2.5 million X depth of coverage of DNA extracted from peripheral leukocytes detected low level mosaicism (1.64% variant allele fraction) for the c.3061G>A (p.Glu1021Lys) PIK3CD pathogenic variant in in the apparently healthy mother, consistent with her having gonosomal mosaicism. Further analysis showed segmental uniparental disomy accounting for the presence of the homozygous PIK3CD c.3061G>A (p.Glu1021Lys) variant in the child (3). The second study identified a familial heterozygous c.3061G>A (p.Glu1021Lys) pathogenic variant in PIK3CD by Sanger sequencing of three half-siblings who shared the same father (4). Sanger sequencing did not detect the pathogenic variant in DNA extracted from paternal or maternal blood samples. However, Sanger sequencing detected the familial PIK3CD variant in a paternal DNA sample extracted from semen, which was consistent with paternal gonadal mosaicism, though these results don’t exclude possible low level gonosomal mosaicism.
Delineating parental origin of mosaic findings can inform recurrence risk estimation (5, 6). In families with one or more affected offspring, probabilistic modeling of mosaicism estimates recurrence risk to be higher in cases of maternal gonadal mosaicism compared with cases with paternal origin (5). Further, recurrence risk for additional offspring is estimated to be higher in instances of gonosomal mosaicism compared with mosaicism confined to gonadal tissue for both maternal and paternal transmission (5).
Accordingly, the use of high depth sequencing for detection and diagnosis of low-level mosaicism may be considered for the management and recurrence risk estimation for families with IEIs with multiple affected children and negative parental testing. High coverage NGS-based technologies afford high sensitivity for detecting variants that comprise as little as 1% of the original sample, allowing higher detection of mosaicism. Further, long read sequencing allows phasing of variants when the tissue carrying the variant is unavailable. Here, we present a study demonstrating application of these technologies to elucidate the origin of mosaicism in apparently unaffected parents in three unrelated families with familial heterozygous PIK3CD pathogenic variants.
In Family 1, we identified a heterozygous c.1002C>G (p.Asn334Lys) pathogenic variant in PIK3CD in three half-siblings (II.1, II.2, and II.3) who share the same mother (I.2) (Figure 1A). Patient II.1 is an 18-year-old male who showed symptoms of APDS1 starting in infancy, including sinopulmonary infections, splenomegaly, and thrombocytopenia. Half-siblings, patients II.2 and II.3 displayed symptoms from birth; II.2, a 1-year-old female, had cytopenia and leukopenia, while II.3 had cytopenia (Supplementary Table 1). Their mother has asthma and allergic rhinitis but is otherwise relatively healthy. A complete list of patient symptoms can be found in Supplementary Table 1. Through exome sequencing of genomic DNA samples extracted from whole blood from the proband (II.1) and the half-sibling (II.2), we detected the PIK3CD variant in 40/80 reads (50% variant allele fraction (VAF)) and 121/228 reads (53% VAF), respectively (Figure 1B). We confirmed both results by Sanger sequencing (Figure 1B). The proband’s second half-sibling (II.3) was tested at an outside laboratory and reported to be heterozygous for the familial pathogenic variant. We also performed exome sequencing on a maternal DNA sample extracted from saliva and did not detect the familial PIK3CD pathogenic variant at 43 read depth at this locus. We then performed panel-based deep sequencing on the maternal DNA sample extracted from saliva and isolated monocytes. Through this approach, we detected the familial PIK3CD variant in 29/11,811 reads (0.25% VAF) in the maternal DNA sample from saliva but did not detect the variant in an additional DNA sample from isolated monocytes. Preliminary analyses of over 500 samples that underwent the same targeted deep sequencing show a mean depth coverage of 8915X at the variant locus in Family 1. For the C>G variant, the mean number of reads across all samples supporting the alternate G allele was 0.42 +/- 1.41, with a maximum of 29 reads, which was significantly above the background. Our results are consistent with low-level gonosomal mosaicism in the mother.
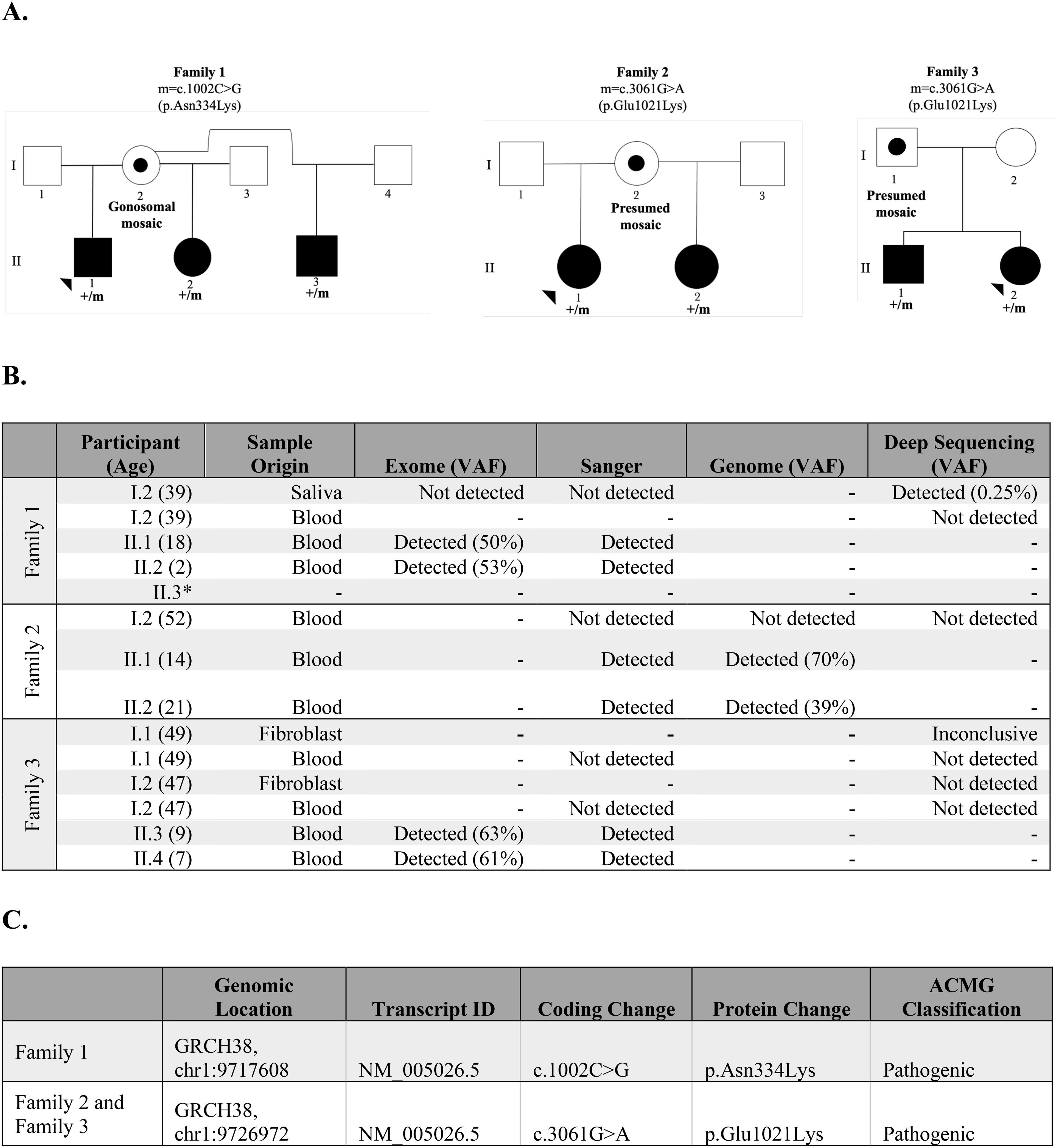
Figure 1. Summary of findings of families 1, 2, and 3. (A) Pedigree of the families, including the coding change and the specific amino acid substitution. Black solid symbols represent affected subjects, the open symbol represents an unaffected subject, the dotted symbol represents a subject carrying a postzygotic mutation, squares represent male subjects, and circles represent female subjects. (B) Detection of PIK3CD pathogenic variant in exome, Sanger, genome, or targeted deep sequencing in families 1, 2, and 3. (C) Genomic information of pathogenic PIK3CD variants in families 1, 2, and 3.
In Family 2, we identified a heterozygous c.3061G>A (p.Glu1021Lys) pathogenic variant in PIK3CD in two half-siblings (II.1 and II.2) who share the same mother (I.2) (Figure 1C). Patient II.1 and patient II.2 are 14- and 21-year-old females, respectively, diagnosed with APDS1 and developed symptoms of APDS1 during infancy, starting with otitis media (Supplementary Table 1).Their mother has Crohn’s disease. Through genome sequencing of genomic DNA extracted from whole blood from the proband (II.1) and half-sibling (II.2), we detected the PIK3CD variant in 21/30 reads (70% VAF) and 14/36 (39% VAF), respectively (Figure 1B). We confirmed both results by Sanger sequencing. We also performed genome sequencing on a maternal DNA sample extracted from blood and did not detect the familial PIK3CD pathogenic variant. We performed deep sequencing at an average depth of 9,677 X coverage at the PIK3CD locus to detect possible low-level mosaicism in her blood sample. Through this approach, we did not detect the c.3061G>A (p.Glu1021Lys) pathogenic variant. This finding suggests the presence of gonadal mosaicism in the proband’s mother, which could not be experimentally confirmed because of the non-availability of gonadal tissue for testing. However, we cannot rule out the highly low probability of recurrence of a de novo variant.
In Family 3, we identified a familial heterozygous c.3061G>A (p.Glu1021Lys) pathogenic variant in PIK3CD in two affected siblings (II.1 and II.2) born to non-consanguineous parents (I.1 and I.2) of reportedly diverse genomic ancestry (Figure 1C). Patient II.1 is a 9-year-old male who showed symptoms of APDS1 starting at age four, primarily sinopulmonary infections. Patient II.2 is a 7-year-old female that displayed symptoms of APDS1 at one year of age, initially presenting with thrombocytopenia (Supplementary Table 1). Their mother and father are both apparently unaffected with no major health issues. Through exome sequencing of genomic DNA extracted from whole blood from the proband (II.1) and the sibling (II.2), we detected the PIK3CD variant in 27/43 reads (63% VAF) and 19/31 reads (61% VAF), respectively (Figure 1B). We confirmed these results through Sanger sequencing. We also performed Sanger sequencing on the parental DNA samples extracted from whole blood and we did not detect the familial pathogenic variant in either sample. Additionally, previously completed exome sequencing at an outside lab did not detect the variant in either parent. We performed deep sequencing on parental genomic DNA samples extracted from both blood and fibroblasts. However, at an average depth of 9,677 X coverage at the PIK3CD locus, we did not detect the familial pathogenic variant in blood-derived DNA samples from either parent. Deep sequencing of the DNA extracted from fibroblast samples did not detect the variant in the mother and results for the father were inconclusive. We performed long-read genome sequencing on DNA samples extracted from blood from all four members of Family 3 to determine the parental chromosome of origin harboring the c.3061G>A (p.Glu1021Lys) pathogenic variant in the two affected siblings. Haplotype analyses of chromosomal regions surrounding the pathogenic allele showed that the pathogenic variant in PIK3CD resides on the paternal allele in both affected children (Figure 2) (Supplementary Methods). Together, these findings are best explained by gonadal mosaicism in the father.
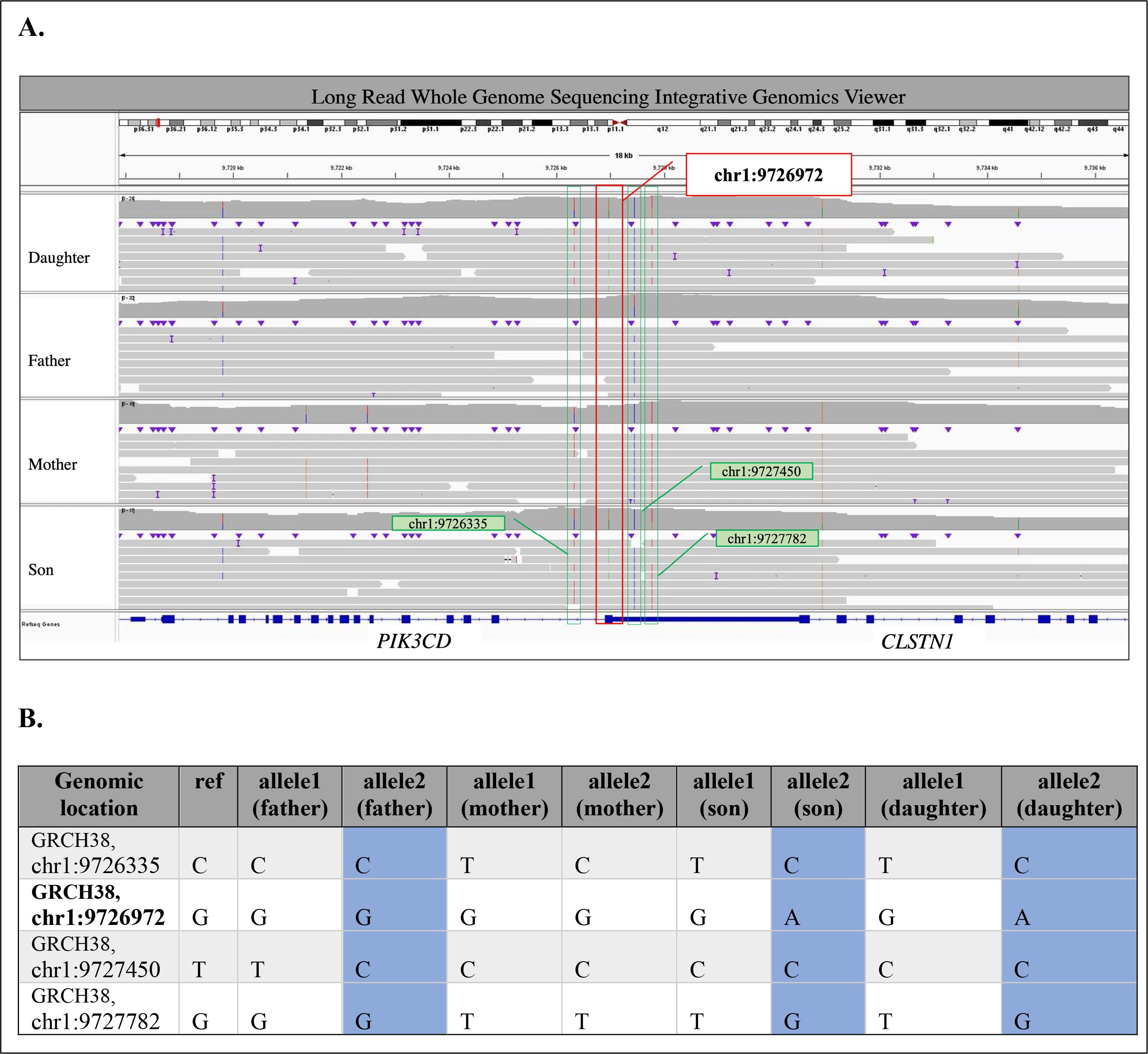
Figure 2. Summary of Long Read Whole Genome Sequencing analysis in Family 3. (A) Variants near the pathogenic c.3061G>A (p.Glu1021Lys) PIK3CD variant (chr1:9726972G>A; highlighted in red box) in Integrative Genomics Viewer (https://www.igv.org) for Daughter, Father, Mother, and Son from top to bottom panel. Variants surrounding pathogenic variant are highlighted in green boxes. (B) Phased variants near the pathogenic c.3061G>A (p.Glu1021Lys) PIK3CD variant (chr1:9726972G>A; in bold). Shared paternal haplotype is highlighted in blue.
The presence of gonadal or gonosomal mosaicism is strongly suggested when a dominantly inherited disease recurs in offspring of apparently healthy parents; such cases have been reported in families with PIK3CD pathogenic variants (1, 3, 4). Here, we report a case series of three families with either confirmed or apparent gonosomal or gonadal mosaicism for pathogenic PIK3CD variants. These families had multiple affected children with the same pathogenic PIK3CD variant. Parents in each family were unaffected and did not have any apparent symptoms attributed to APDS1. Data from targeted deep sequencing following exome and Sanger sequencing supported maternal gonosomal mosaicism in Family 1. Through this approach we did not detect the pathogenic variant in maternal blood samples for Family 2, suggesting that mosaicism was likely confined to her gonads. Data from long-read genome sequencing following inconclusive targeted deep sequencing supported paternal gonadal mosaicism in Family 3. Although, we did not measure the level of mosaicism in the gametes, we hypothesize that the level of mosaicism is likely higher in the gonads than what we observed in peripheral tissues, based on our findings in these three families. Additional studies are needed to determine whether higher mosaicism in the gonads could account for the 100% incidence of affected children in each family we examined.
Our data from Families 1 and 2 are consistent with theoretical calculations of increased germline mosaicism in maternal samples (5). In families with more than one affected child and apparently unaffected parents, the likelihood of maternal mosaicism for the PIK3CD variant is increased (1, 3). Furthermore, data from Family 1 supports theoretical increased recurrence risk estimates due to the presence of maternal gonosomal mosaicism, as gonosomal mosaicism and maternal origin are both major determinants of elevated recurrence risk compared with gonadal mosaicism and paternal origin, respectively. (5). This is because an apparently mosaic parent with multiple affected offspring may have a larger fraction of variants in their germline cells than is typical, making transmission of a mutant gamete more likely.
In Family 3, deep sequencing data could not conclusively identify the parental origin of the pathogenic PIK3CD variant in the affected siblings. Long-read sequencing on parental blood samples with haplotype analysis was used to delineate the parent of origin of the transmitted allele in the offspring (7). By phasing the surrounding variant regions in the affected siblings with the parental chromosome that had shared single nucleotide variants (SNVs) we determined which parent transmitted the risk haplotype. We captured unique SNVs from each parent, as they were each from diverse genomic ancestries. This analysis demonstrated the utility of long-read sequencing to investigate parent of origin following inconclusive deep sequencing.
Detection of mosaicism has serious implications in hematopoietic stem cell transplantation for families with affected offspring and potential donor parents; however, the results of this study do not impact the clinical management of the affected patients themselves. Detection of low-level mosaicism through targeted deep sequencing of a parent prior to transplantation could reduce the likelihood of bone marrow rejection or failure, as mosaic parents are not suitable donors due to potential hematopoietic phenotypes conferred by the mosaic variant.
Additionally, clarifying that an apparently de novo variant is actually due to parental gonadal or gonosomal mosaicism is important for providing accurate recurrence risk estimates. Genetic counselors often rely on an empirical understanding of recurrence risk. Elucidating low level parental mosaicism in families with affected children with apparently de novo variants can provide families with more accurate recurrence risk assessment during counseling sessions. Recurrence calculations can help guide family planning discussions and direct the use of resources such as preimplantation and prenatal genetic diagnostic testing (8).
Although prior studies showed mosaicism that was stable over time in families with IEIs (1, 9), individuals with mosaicism may show later disease onset and/or milder or subclinical phenotypes than individuals with non-mosaic variants in the same genes (10). The level of mosaicism in PIK3CD at which APDS1 develops is unknown at this time and it may depend on the variant type and the cells types in which it is expressed. A prior report of a mother with gonosomal mosaicism for PIK3CD at 37 years of age with1.64% VAF had no apparent clinical phenotype (3). Similarly, the mother we identified in Family 1 at the time of evaluation at 39 years of age, also had no apparent clinical phenotype with 0.25% VAF. At this time, we have no data to determine her clinical course in the future. Based on prior publications suggesting stability of PIK3CD mosaicism over time, we hypothesize that that the level of mosaicism will remain stable and not lead to subsequent phenotypic manifestation of disease in the unaffected parents; nevertheless, this hypothesis lacks experimental confirmation (1, 3). Given our limited understanding of the dynamics of mosaicism and subsequent disease progression, future studies characterizing its cellular and clinical spectrum throughout life are warranted.
This report should be interpreted in light of several limitations. First, the limited availability of tissues for testing did not allow for a wide view of the landscape of mosaicism across multiple cell lineages in these families. Further, the depth of deep sequencing of 9,677X may limit detection of lower levels of mosaicism than what we have detected in this study. Finally, data on the positive and negative predictive values for deep sequencing require additional longitudinal studies that follow phenotypic features of participants with varying VAFs. The extent of baseline mosaicism of IEI genes in healthy individuals across the lifespan and the clinical impact of mosaic changes remains to be addressed.
In summary, as we describe in this study, detection of mosaic variants in PIK3CD in seemingly healthy parents improves risk assessment and clinical management for families with apparently de novo variants in more than one offspring.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: dbGaP accession ID phs001899.v3.p1.
The studies involving humans were approved by the National Institutes of Health (NIH) Institutional Review Board (NCT03206099). The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
HO: Data curation, Formal Analysis, Investigation, Visualization, Writing – original draft. JY: Conceptualization, Data curation, Formal Analysis, Investigation, Supervision, Visualization, Writing – review & editing. AP: Conceptualization, Data curation, Formal Analysis, Methodology, Validation, Writing – review & editing. MT: Conceptualization, Methodology, Writing – review & editing. YD: Data curation, Formal Analysis, Methodology, Visualization, Writing – review & editing. RG: Formal Analysis, Methodology, Writing – review & editing. KL: Project administration, Writing – review & editing. JD: Data curation, Writing – review & editing. LJ: Data curation, Writing – review & editing. CJ: Data curation, Writing – review & editing. MS: Data curation, Writing – review & editing. NS: Project administration, Writing – review & editing. ZZ: Data curation, Formal Analysis, Methodology, Visualization, Writing – review & editing. YW: Data curation, Formal Analysis, Methodology, Visualization, Writing – review & editing. SW: Data curation, Formal Analysis, Methodology, Visualization, Writing – review & editing. JR: Project administration, Writing – review & editing. HS: Data curation, Investigation, Writing – review & editing. GU: Conceptualization, Data curation, Investigation, Writing – review & editing. SN: Data curation, Formal Analysis, Investigation, Methodology, Validation, Writing – review & editing. MC: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Resources, Validation, Writing – review & editing. MW: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Supervision, Writing – review & editing.
The members of the NIAID Centralized Sequencing Program working group are Bryce A. Seifert, Nadjalisse Reynolds-Lallement, Rachel Gore Moses, Michael Kamen, Sarah Bannon, Sophie Byers, Ekaterina Damskey, Sruthi Srinivasan, Adrienne Borges, and Nicole Gentile.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This research was funded by the Division of Intramural Research, National Institutes of Allergy and Infectious Diseases, and 1ZIAAI001059 to HS. Work was supported by NIH grant R21AI168957 and The Center for Pediatric Immunology at Washington University and St. Louis Children’s Hospital (to MC).
The authors would like to thank the participants and their families for their valuable contributions to this research. We also acknowledge the sequencing services and professional partnership from the Center for Inherited Disease Research, Human Genome Sequencing Center. Special thanks to Julie Niemela, Karen Frank, and Sergio Rosenzweig, from the NIH Clinical Center Department of Laboratory Medicine; to Justin Lack and Wenjia Cao, from the NIAID Collaborative Bioinformatics Resource team; and the Genome Research Integration Team, including Daniel Veltri, Sandhya Xirasagar, Andrew Oler, Eric Karlins, Xi Cheng, Jason Barnett, Nikita Vlasenko, Ke Huang, Robert Gilmore, Satishkumar Ranganathan, Emily Tran, Alvin Yun, Joseph Mackey, Svetlana Yazhuk, Smilee Samuel, Younjie Fan, Conrad Shyu, Jenica Abrudan, Cyrus Afrasiabi, Zhiwen Li, Lingwen Zhang, Menolin Sharma, Krishnaveni Kaladi, Giovanni Borjas, Amjad Elhassan, Laurel Myers, and Joanne Berghout; to Jose Jaison. This research was supported by the Intramural Research Program at NIH at the NIAID Division of Intramural Research.
ZZ, YW, SW, JR, SN, and YD are employed by Infinity-Biologix LLC D/B/A SAMPLED.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be constructed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2024.1451212/full#supplementary-material
1. Mensa-Vilaró A, Bravo García-Morato M, de la Calle-Martin O, Franco-Jarava C, Martínez-Saavedra MT, González-Granado LI, et al. Unexpected relevant role of gene mosaicism in patients with primary immunodeficiency diseases. J Allergy Clin Immunol. (2019) 143:359–68. doi: 10.1016/j.jaci.2018.09.009
2. Rivière JG, Franco-Jarava C, Martínez-Gallo M, Aguiló-Cucurull A, Blasco-Pérez L, Paramonov I, et al. Uncovering low-level maternal gonosomal mosaicism in X-linked agammaglobulinemia: Implications for genetic counseling. Front Immunol. (2020) 11. doi: 10.3389/fimmu.2020.00046
3. Wang Y, Chen X, Yang Q, Tang W, Jia Y, Zhou L, et al. E1021K homozygous mutation in PIK3CD leads to activated PI3K-delta syndrome 1. J Clin Immunol. (2020) 40:378–87. doi: 10.1007/s10875-020-00749-y
4. Segundo GRS, Takano OA, Moraes LSL, Nadaf MISV, Fernandes SJ, Ochs HD, et al. Paternal gonadal mosaicism as cause of a puzzling inheritance pattern of activated PI3-kinase delta syndrome. Ann Allergy Asthma Immunol Off Publ Am Coll Allergy Asthma Immunol. (2017) 119:564–6. doi: 10.1016/j.anai.2017.09.054
5. Campbell IM, Stewart JR, James RA, Lupski JR, Stankiewicz P, Olofsson P, et al. Parent of origin, mosaicism, and recurrence risk: Probabilistic modeling explains the broken symmetry of transmission genetics. Am J Hum Genet. (2014) 95:345–59. doi: 10.1016/j.ajhg.2014.08.010
6. Campbell IM, Yuan B, Robberecht C, Pfundt R, Szafranski P, McEntagart ME, et al. Parental somatic mosaicism is underrecognized and influences recurrence risk of genomic disorders. Am J Hum Genet. (2014) 95:173–82. doi: 10.1016/j.ajhg.2014.07.003
7. Noyes MD, Harvey WT, Porubsky D, Sulovari A, Li R, Rose NR, et al. Familial long-read sequencing increases yield of de novo mutations. Am J Hum Genet. (2022) 109:631–46. doi: 10.1016/j.ajhg.2022.02.014
8. Xu N, Shi W, Cao X, Zhou X, Jin L, Huang HF, et al. Parental mosaicism detection and preimplantation genetic testing in families with multiple transmissions of de novo mutations. J Med Genet. (2023) 60:910–7. doi: 10.1136/jmg-2022-108920
9. Rowczenio DM, Gomes SM, Aróstegui JI, Mensa-Vilaro A, Omoyinmi E, Trojer H, et al. Late-onset cryopyrin-associated periodic syndromes caused by somatic NLRP3 mosaicism-UK single center experience. Front Immunol. (2017) 8. doi: 10.3389/fimmu.2017.01410
10. Truty R, Rojahn S, Ouyang K, Kautzer C, Kennemer M, Pineda-Alvarez D, et al. Patterns of mosaicism for sequence and copy-number variants discovered through clinical deep sequencing of disease-related genes in one million individuals. Am J Hum Genet. (2023) 110:551–64. doi: 10.1016/j.ajhg.2023.02.013
Keywords: gonadal mosaicism, gonosomal mosaicism, PIK3CD, inborn errors of immunity, primary immunodeficiency diseases
Citation: Orellana H, Yan J, Paul A, Tokita M, Ding Y, Ghosh R, Lewis KL, Davis J, Jamal L, Jodarski C, Similuk M, Saucier N, Zhu Z, Wang Y, Wu S, Ruggieri J, NIAID Centralized Sequencing Program Working Group, Su HC, Uzel G, Nahas S, Cooper M and Walkiewicz MA (2024) Case report: Deep sequencing and long-read genome sequencing refine prior genetic analyses in families with apparent gonadal mosaicism in PIK3CD-related activated PI3K delta syndrome. Front. Immunol. 15:1451212. doi: 10.3389/fimmu.2024.1451212
Received: 18 June 2024; Accepted: 29 July 2024;
Published: 26 August 2024.
Edited by:
Sudhir Gupta, University of California, Irvine, United StatesReviewed by:
Elena Wen-Yuan Hsieh, University of Colorado Anschutz Medical Campus, United StatesCopyright © 2024 Orellana, Yan, Paul, Tokita, Ding, Ghosh, Lewis, Davis, Jamal, Jodarski, Similuk, Saucier, Zhu, Wang, Wu, Ruggieri, NIAID Centralized Sequencing Program Working Group, Su, Uzel, Nahas, Cooper and Walkiewicz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Magdalena A. Walkiewicz, bWFnZGFsZW5hLndhbGtpZXdpY3pAbmloLmdvdg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.