
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Immunol. , 15 November 2024
Sec. Autoimmune and Autoinflammatory Disorders : Autoimmune Disorders
Volume 15 - 2024 | https://doi.org/10.3389/fimmu.2024.1450048
 Ksymena Leśniak1*
Ksymena Leśniak1* Rafał Płoski2†
Rafał Płoski2† Małgorzata Rydzanicz2
Małgorzata Rydzanicz2 Aleksandra Rymarz1Arkadiusz Lubas1Tomasz Syryło3
Aleksandra Rymarz1Arkadiusz Lubas1Tomasz Syryło3 Stanisław Niemczyk1
Stanisław Niemczyk1Cryoglobulinemia is a rare disease characterized by the presence of cryoglobulins in the blood serum. It is usually caused by autoimmune, lymphoproliferative, or infectious factors. The pathogenesis of cryoglobulinemia is not well understood, therefore, genetic testing is very important. We present the case of two adult sisters with different clinical phenotypes of non-infectious cryoglobulinemic vasculitis associated with a rare genetic variant [(Hg38) 1:160323529 C>G, NP_004362.2:p.(Gly203Ala)]. One of the sisters suffered from essential mixed cryoglobulinemia, while the other suffered from cryoglobulinemia associated with systemic connective tissue disease. In both cases, genetic tests revealed a variant in the COPA gene, encoding coatomer subunit alpha. Mutations in the COPA gene are associated with COPA syndrome, an autoimmune interstitial lung, joint, and kidney monogenic disease, found mainly in children. Only 15 pathogenic COPA variants have been reported thus far which suggests that the full spectrum of disease manifestations remains unknown. Ours is the first report of the association of the COPA gene with non-infectious cryoglobulinemic vasculitis in adults. This unexpected finding may direct research into the pathogenesis of cryoglobulinemia and new treatment strategies for this rare disease.
Cryoglobulinemia is a unique disease at a crossroads between autoimmune and lymphoproliferative disorders and is characterized by the presence of cryoglobulins in the blood serum. Cryoglobulins are immunoglobulins that precipitate in vitro at <37°C and dissolve when heated. Once in the blood system, these molecules can cause organ damage through the occlusion of blood vessels or through vasculitis (1, 2). Cryoglobulins are produced via monoclonal or polyclonal expansion of B lymphocytes as a consequence of lymphoproliferative disorders or persistent immune stimulation caused by chronic infections or autoimmune diseases (3–5). There are three subtypes of cryoglobulins based on their clonality and the type of immunoglobulins they contain (6). Type I is closely related to hematological diseases such as multiple myeloma, Waldenstrom’s macroglobulinemia, and chronic lymphocytic leukemia (7). Types II and III [referred to as mixed cryoglobulinemia, (MC)] occur in infections [mainly Hepatitis C Virus (HCV)], autoimmune diseases [mainly systemic lupus erythematosus (SLE) and Sjögren’s syndrome (SS)] and, less frequently, cancers (type B lymphomas) (8–10). Mixed cryoglobulinemia, which is not related to the above diseases, is called “essential MC” (1).
Mixed cryoglobulinemia belongs to the group of systemic small vessel vasculitis (cryoglobulinemic vasculitis) and, due to its monoclonal component, is also a monoclonal gammopathy of clinical significance (MGCS) (11, 12). The clinical symptoms of MC range from mild (the classic triad of symptoms: purpura, arthralgia, and fatigue), through more serious neurological (peripheral neuropathy), nephrological (chronic glomerulonephritis), and hepatological manifestations (chronic hepatitis), to life-threatening systemic manifestations such as vasculitis or, less frequently, the development of cancer (11, 13–15). The proposed classification criteria for cryoglobulinemic vasculitis are based on the clinical symptoms of organ involvement and the detection of cryoglobulins in serum, but also on serological tests indicating decreased complement component 4 (C4) and increased rheumatoid factor (RF) levels (16).
Treatment of this rare disease poses a challenge and depends on the etiopathogenesis and clinical manifestations. There are three treatment strategies for cryoglobulinemia: antiviral treatment, conventional immunosuppression, and biological treatment. Rituximab, a biological drug that depletes B lymphocytes, is an essential component of modern immunosuppressive regimens in cryoglobulinemia (17, 18).
Despite years of research and observation, many aspects of cryoglobulinemia are still unclear, especially regarding the pathogenesis of this rare disease.
We present the case reports of two adult sisters with non-infectious mixed cryoglobulinemia, in whom exome sequencing (ES) revealed a variant in the gene encoding coatomer subunit alpha (COPA) which was absent in the remaining family members, including the third healthy sister (Figure 1).
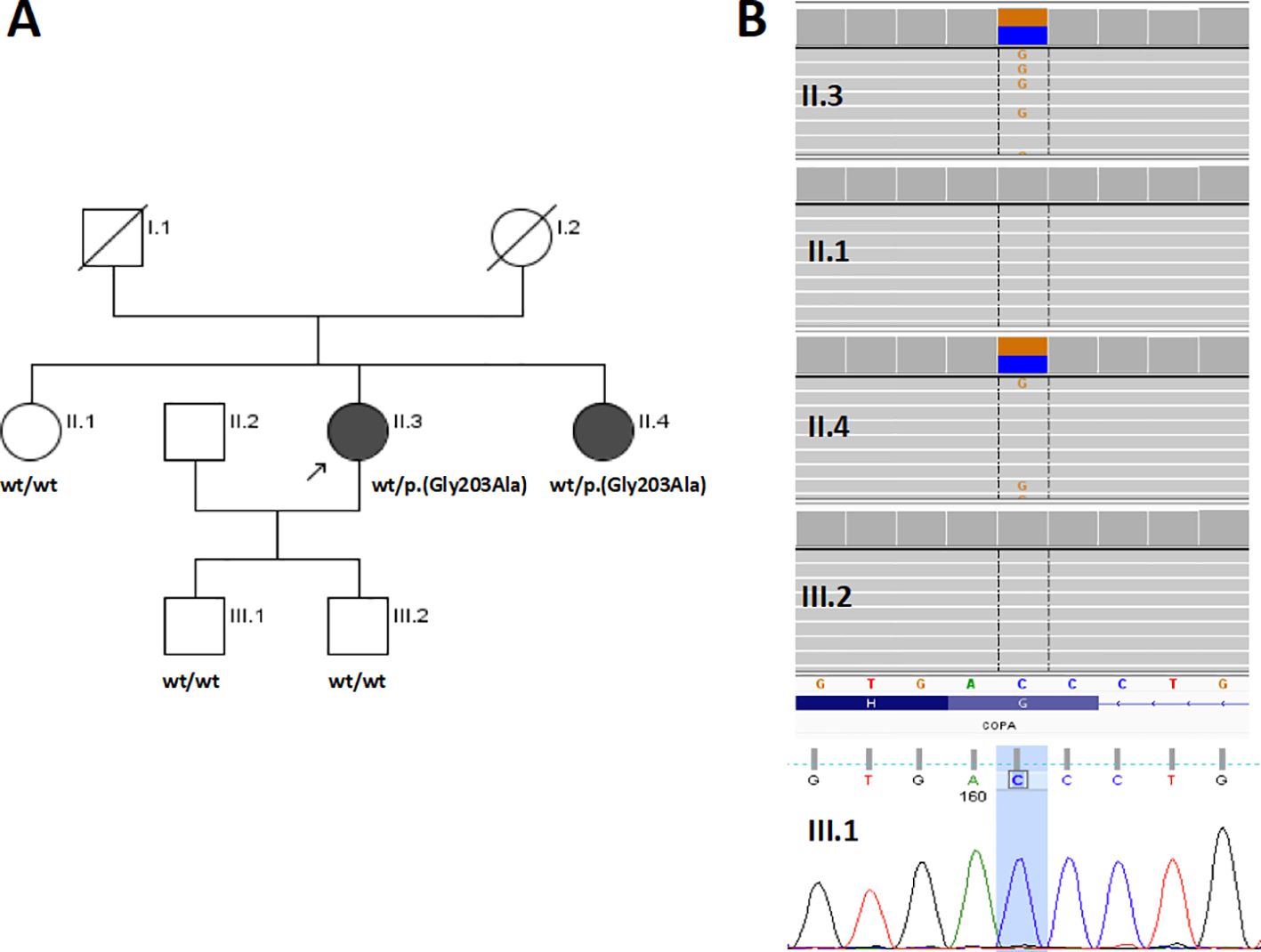
Figure 1. Genetic study of a Polish family with COPA syndrome. (A) Pedigree of the examined family with phenotype/genotype information; circles represent females, squares indicate males, filled symbols indicate an affected individual. The proband is marked with a black arrow; wt = wild-type. (B) The COPA p.(Gly203Ala) variant validation and family study results. For the amplicon deep sequencing (II.1, II.3, II.4, and III.2), an Integrative Genomic Viewer screen shot is presented. For the Sanger sequencing (III.1), a part of the chromatogram is presented.
According to the latest reports, heterozygous mutations in the COPA gene (chr.1 q23.2) result in COPA syndrome, a rare autoinflammatory disease inherited in an autosomal dominant manner with incomplete penetrance and variable expression (19).
The classic COPA syndrome phenotype mainly includes interstitial lung disease, most often in the form of intra-alveolar hemorrhage, arthritis, glomerulonephritis, and high titers of circulating autoantibodies (19, 20). Most symptoms appear in early childhood, 64% by the age of 5. Thus far, only a few cases have been reported with symptoms occurring over the age of 50 years (21–23).
According to The Human Gene Mutation Database (HGMD), only 15 different high-confidence pathogenic COPA variants have been reported in the literature thus far, raising the possibility that the full spectrum of the disease phenotype is not known.
A 58-year-old patient with hypothyroidism in the course of Hashimoto’s disease and bipolar disorder was admitted to the Department of Nephrology in Warsaw (in 2012) for the diagnosis of purpura. A medical interview revealed pain in the upper abdomen and in the joints of the hands and lower limbs, and then a small-spotted rash on the lower legs appeared a few weeks before the admission (Figure 2). The patient’s family history included a sister diagnosed with cryoglobulinemia and Sjögren’s syndrome.
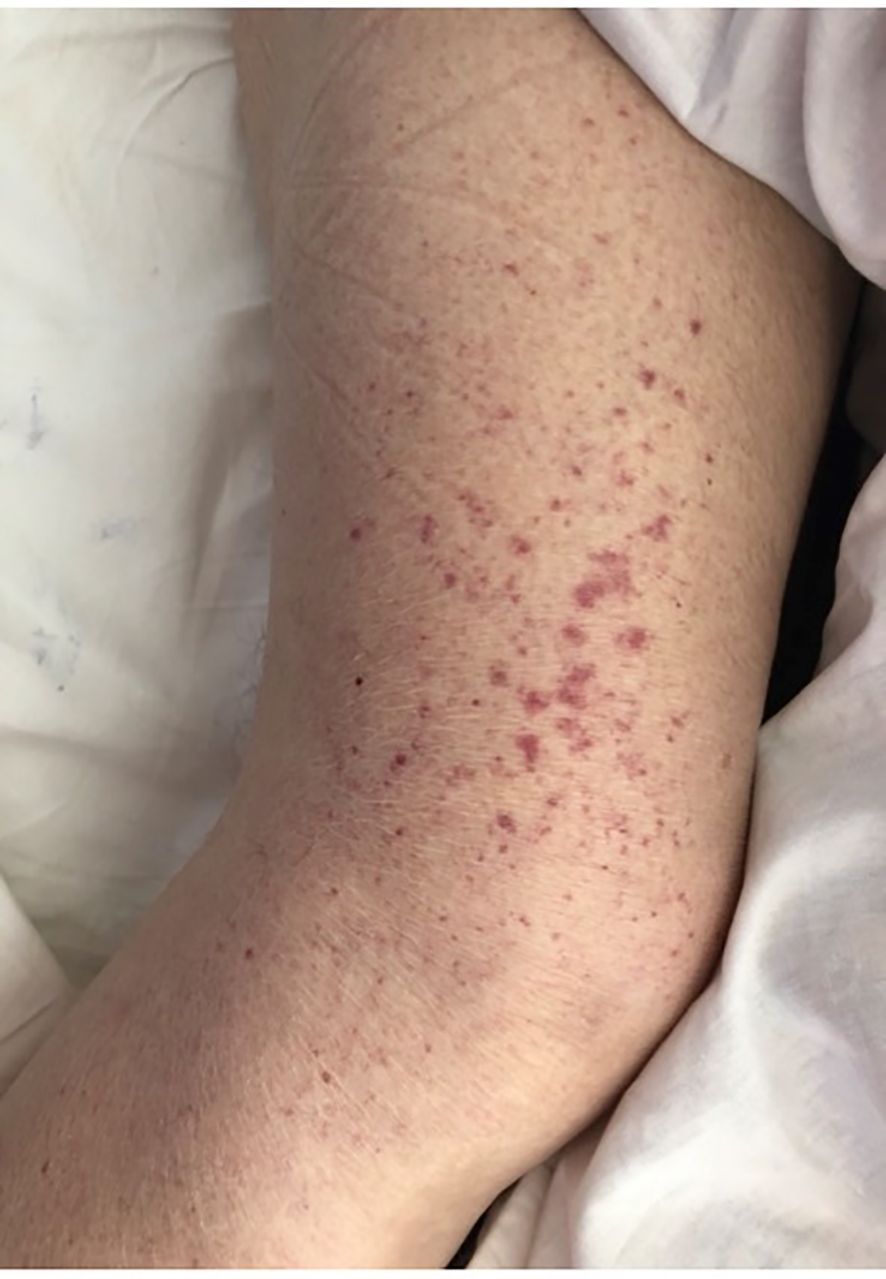
Figure 2. Cryoglobulinemic vasculitis presents as a small-spotted rash on the lower legs in our index case.
The physical examination revealed no abnormalities apart from purpura on the skin of the lower legs. Laboratory tests performed during hospitalization revealed increased inflammatory markers: erythrocyte sedimentation rate (ESR) of 52 mm/h, C-reactive protein (CRP) level of 3.7mg/dl; elevated rheumatoid factor (RF 64 IU/ml), and significantly reduced complement C4 (0.03 g/l). Kidney function was normal, creatinine was in the range of 0.7mg/dl, and a general urine test showed no abnormalities.
The tests for antinuclear antibodies (ANA) and antineutrophil cytoplasmic antibodies (ANCA) and those against double-stranded DNA (ds-DNA) and soluble nuclear antigens (Ro, La, Sm) were negative. Infection with hepatitis B Virus (HBV), HCV, and human immunodeficiency virus (HIV) was excluded. A chest x-ray and abdominal ultrasound showed no significant abnormalities. The abdominal pain was transient and did not recur. No changes in the gastric or duodenal mucosa were reported in gastroscopy. A skin biopsy was performed, which showed signs of leukocytoclastic small vessel vasculitis. Antinuclear antibodies were negative and the patient did not present symptoms of dryness typical of Sjögren’s syndrome, therefore the diagnostics were not further investigated. Since cryoglobulinemia was considered in the differential diagnosis, a test was performed which confirmed the presence of cryoglobulins. After immunochemical analysis which revealed the presence of polyclonal IgM and polyclonal IgG cryoglobulins, a mild form of mixed cryoglobulinemia type III was diagnosed. Immunosuppressive treatment was implemented including glucocorticosteroids (prednisone 55mg/day) and methotrexate (15mg/week). Within a few months, despite the treatment, the purpura recurred with a tendency to form confluent changes. Additionally, sensory disturbances appeared in the lower limbs, accompanied by a significant increase in the concentration of cryoglobulins (4.3 g/L). Simultaneously, a monoclonal component of the IgM class with kappa light chains appeared among the cryoglobulins. Another serological diagnosis for systemic connective tissue disease was performed but its results were negative. A bone marrow biopsy excluded multiple myeloma and other hematological malignancies. Increased inflammation markers and a decrease in complement C4 persisted. HCV RNA was unreactive. To diagnose any sensory disorders, an MRI of the head, electroneurography (ENG), and a neurological consultation were performed showing evidence of vascular brain damage (diffused, small lesions typical of cerebral vasculitis in MRI) and axonal sensory-motor polyneuropathy, most likely in the course of cryoglobulinemic vasculitis. The patient was diagnosed with essential mixed cryoglobulinemia type II. Due to the lack of effectiveness of the current treatment and the signs of disease progression with the involvement of the nervous system, it was decided to intensify the treatment and change the therapy to monthly intravenous cyclophosphamide (CYC) infusions. During the treatment, despite the administration of 5 g of CYC, no response with a decrease in cryoglobulins was obtained and the purpura recurred. Moreover, abdominal pain, nausea, musculoskeletal pain, very severe pain in the lower limbs with a burning sensation and numbness in the feet, increasing swelling of the lower legs, bruising of the three toes of the left foot, and a low-grade fever occurred. Cryoglobulins were still present in the blood serum. Due to the risk of necrosis of the toes, the patient qualified for therapeutic plasma exchange (TPE) procedures. A total of 5 TPE procedures were performed without complications and then another CYC pulse therapy was administered. The treatment resulted in the improvement of both the general condition and the blood supply to the left toes, however, disease remission was not achieved. Due to the lack of effectiveness of the current therapy, it was possible to use rituximab in the Polish conditions. It was decided to administer rituximab (RTX, 1.0 g on days 1 and 15) and prednisone was continued with a gradual reduction of the dose. A transient clinical remission of the disease was achieved without cryoglobulin negativity. The lymphocyte CD19 population was regularly determined by flow cytometry, but these results did not significantly influence therapeutic decisions.
One year after RTX administration, inflammatory state parameters and the cryoglobulin concentration increased and symptoms of kidney involvement appeared in the form of hematuria. Therefore, it was decided to start treatment with mycophenolate mofetil (MMF) and prednisone administration was continued. The clinical remission with cryoglobulin negativity was achieved but lasted only 8 months. The disease then relapsed, requiring the administration of a second course of rituximab. Prednisone was continued at a low dose, and after a few months, MMF treatment was added which resulted in a year-long remission of the disease with negative cryoglobulins.
Due to the high risk of recurrence of cryoglobulinemia symptoms and associated complications and the confirmed high effectiveness and safety of the previously used biological treatment, the patient received booster doses of rituximab (after 4 and 6 years of the disease). Despite complex immunosuppressive therapy, the patient’s cryoglobulinemic vasculitis could not be sufficiently controlled. The disease kept recurring and in its final stage, 8 years after diagnosis, an ischemic stroke occurred. Repeated emergency TPE procedures were planned, but they were not carried out. The patient died in 2019 as a result of COVID-19-related severe respiratory failure (before the availability of vaccinations).
A 50-year-old patient with a history of Sjögren’s syndrome and Hashimoto’s disease for approximately 10 years was admitted to the Rheumatology Department in Cracow (2007) due to recurrent purpura on the lower limbs for 2 years with accompanying pain in the limbs. Physical examination revealed abnormalities including purpura on the skin of the lower legs and thighs, which was confluent in some places, discoloration in sites where the changes disappeared, sensory disturbances in the distal parts of the lower limbs, and persistent dryness of the eyes and mouth. Moreover, abnormal test results included leukopenia in the blood count, accelerated ESR (50mm/h) with normal CRP, and a decrease in both complement components, especially C4 (C3: 0.768 g/L; C4: 0.019 g/L). Serological tests revealed the presence of rheumatoid factor with a negative anti-cyclic citrullinated peptide antibodies (anti-CCP) result and the presence of antinuclear antibodies with a homogeneous + granular type (titer of:20408) and mitochondrial type (titer of:5120). Tests for dsDNA, nRNP, Sm, SS-A (Ro), SS-B (La), Scl-70, PM-Scl, Jo-1 and ANCA were negative. However, cryoglobulins were detected in blood serum. Hepatitis B, hepatitis C, and HIV infections were excluded. TSH level was normal. A chest x-ray revealed fibrosis in the lower parts of the lungs. The abdominal ultrasound examination did not show any abnormalities. Cryoglobulinemia with the involvement of the skin and peripheral nervous system secondary to systemic connective tissue disease (CTD) was diagnosed and treatment with methylprednisolone was initiated at the initial dose of 16mg/day which resulted in clinical remission. After a year, anti-Ro-52 (+++) antibodies were detected and after 13 years anti-SCL 70 (+) antibodies appeared. Hydroxychloroquine 250mg/day was added to the glucocorticoids. Due to the history of cryoglobulinemia and persistent C4 hypocomplementemia, the patient was subjected to strict surveillance, considering the higher risk of developing lymphoma.
After 11 years, in the absence of any respiratory symptoms, a follow-up CT scan of the chest revealed a left lung tumor (Figure 3A). The positron emission tomography-computed tomography (PET-CT) [on 18F-fluorodeoxyglucose-positron emission tomography (FDG-PET/CT)] revealed a polycyclic nodular lesion of the left lung with dimensions of approx. 25x17mm, with increased glucose metabolism (Figure 3B). It was decided to remove segment 10 of the left lung. Histopathological examinations of sections from this lesion demonstrated granulomas composed of epithelioid and giant cells accompanied by an abundant inflammatory component of lymphoid cells, foci of necrosis in some areas, fibrosis of the perivascular parenchyma, and areas of destruction of the bronchiolar wall. First, tuberculosis and other bacterial and fungal infections were ruled out. Due to the lack of typical clinical features, sarcoidosis was also ruled out. The test for ANCA was negative and cryoglobulins were still detected in the blood serum. Based on subsequent chest CT scans (which found ground glass zones, subpleural densities, small nodules, and small fibrosis in the lungs), interstitial lung disease closely resembling nonspecific interstitial pneumonia (NSIP) in the course of Sjögren’s syndrome was diagnosed and therefore, oral MMF was added to previous glucocorticoids in the treatment. The treatment provided stabilization which was visible in lung imaging tests, however, the patient developed symptoms of bronchial asthma. In the pulmonary function testing (PFT), an obstructive ventilatory defect was found and the patient required inhalant drugs.

Figure 3. (A) A CT scan of the chest revealed a left lung tumor (blue arrow). (B) FDG-PET/CT examination revealed a polycyclic nodular lesion of the left lung with dimensions of approx. 25x17mm, with a maximum standardized uptake value (SUVmax) of 11.6.
Finally, 17 years after the diagnosis of cryoglobulinemia, the symptoms of the disease have not recurred, the patient feels well, and she is under the care of a rheumatologist and pneumonologist.
Genomic DNA was extracted from the whole blood of all study participants using standard methods. In the index case (proband), MONO ES was performed using a Twist Human Core Exome 2.0 + Comp Spike-in + Twist mtDNA Panel (Twist Bioscience, South San Francisco, CA, USA). The enriched library was pair-end sequenced (2x100 bp) on a NovaSeq 6000 (Illumina, San Diego, CA, USA) to a mean coverage of 120x, GE10 (coverage greater or equal 10) = 98%, GE20 = 97.7%. The raw data analysis and variants prioritization were performed as previously described (24). In silico pathogenicity prediction was performed based on the in-house developed platform GeneBe (https://zgm.wum.edu.pl/cloud2/) and the American College of Medical Genetics and Genomics (ACMG) guidelines (25, 26). Conservation scores were provided by Varsome (27).
Segregation of the variant considered causative was further examined in all available members of the proband’s family, including two sisters (unaffected II.1 and affected II.4) and two of the proband’s unaffected sons (III.1 and III.2) (Figure 1). Variant validation and family study were performed by amplicon deep sequencing in II.1, II.3, II.4, and III.2 using a Nextera XT Kit (Illumina) sequenced as described above for ES, and for III.1 by direct Sanger sequencing with a BigDye Terminator v3.1 Kit (Applied Biosystems, Foster City, USA) on an ABI 3500Xl Genetic Analyzer (Applied Biosystems).
The study protocol was approved by the local ethics committee (Military Institute of Medicine Bioethics Committee: approval number 37/WIM/2018). Written informed consent was obtained from all participants or their legal guardians in accordance with the Declaration of Helsinki.
In the index case, a novel [(Hg38) 1:160323529 C>G, NM_004371.4:c.608G>C, NP_004362.2:p.(Gly203Ala)] heterozygous missense variant located in exon 8 of the COPA gene was identified and prioritized as disease-causing. The p.(Gly203Ala) has 0 frequency in the gnomAD v4.0.0 database (accessed 11 March 2024) and the in-house database of Polish individuals composed of >11,000 exomes (accessed 11 March 2024). The variant was predicted to be pathogenic by AlphaMissense (0.95), CADD (32), Eigen_PC (0,73), FATHMM_MKL (0,99), IST_S2 (1,0), PrimateAI (0,91), and Polyphen (0,99); according to an automatically implemented ACMG classification, the variant was scored as VUS (Variant of Uncertain Significance, 4 points, PM2 Moderate, PP2 Supporting, PP3 Supporting) (25). However, when the location in a mutation hot spot region was taken into account (PM1 moderate, see Discussion) the classification changed to Likely Pathogenic (6 points). The c.608G variant is evolutionarily conserved (phyloP100 score: 7.281) and is located in a region moderately constrained for coding variation (CCRS=82.2). The family study revealed the presence of p.(Gly203Ala) in the index case’s affected sister (II.4) and its absence in index case’s unaffected sister (II.1) and both unaffected sons (III.1, III.2) (Figures 1A, B).
COPA syndrome, also known as an autoimmune interstitial lung, joint, and kidney disease (MIM # 616414), is a rare autosomal dominant disorder caused by heterozygous missense variants located in exons 8 and 9 of the COPA gene and highly conserved WD40 domain of the COPA protein (28, 29). However, incomplete penetrance with unaffected carriers is also reported (19, 30).
In our study, we present a novel COPA p.(Gly203Ala) missense variant in a Polish family with two affected sisters presenting with symptoms of cryoglobulinemic vasculitis. While the pathogenicity of the variant cannot be proven, it has a very low population frequency (absent from available database) and is predicted to be so by a number of in silico tools including the powerful recently-developed AI-based AlphaMissense (31). Furthermore, all pathogenic COPA variants reported thus far in the literature cluster in exons 8 and 9 between amino acids 230-285 (19, 21, 23, 32–42) with only a single exception for position 75 (41). This clustering is also apparent when ClinVar-reported pathogenic/likely pathogenic COPA variants are analyzed (6 variants reported in amino acid regions 230-245, visualized at https://gnomad.broadinstitute.org/gene/COPA accessed 10 05 2024). The location of the variant we report in the proximity of the abovementioned domain (exon 8, amino acid pos. 203) is another argument supporting its pathogenicity, especially when considering that the COPA gene consists of 33 exons, encoding 1225 aa. It is noteworthy that after inclusion of this information, the ACMG classification of COPA p.(Gly203Ala) changed to Likely Pathogenic.
The presented cases are of particular importance because they concern a rare form of non-infectious cryoglobulinemia that could not be explained by known causes.
In recent years, various studies of the pathogenesis of cryoglobulinemia have been conducted, but they mostly focused on patients with HCV infection. For example, Zignego et al. provided the results of a multicenter genome-wide association study (GWAS) focusing on the analysis of HCV-related cryoglobulinemia at the genome level which revealed a significant association between cryoglobulinemic vasculitis and the presence of single nucleotide polymorphisms (SNPs) in the vicinity of the NOTCH4 and MHC class II genes on chromosome 6 (43). In turn, Gragnani et al. demonstrated a linkage between two polymorphisms that was previously confirmed in the HCV-related MC (NOTCH4 rs2071286 and HLA-II rs9461776) in patients with HCV-related lymphoproliferative disorders (LPDs) at risk of non-Hodgkin’s lymphoma (NHL) (44).
Therefore, it was surprising that the index case patient and her affected sister were carriers of a mutation typical for Mendelian syndrome of immune dysregulation.
In recent decades, many important discoveries have been made in the field of genetically determined diseases related to the dysregulation of the immune system. Thus far, over 400 different phenotypes of single-gene inborn errors of immunity (IEIs) have been described, which include not only infections but also allergies, autoimmunity, autoinflammation, and malignancy.
One of these is coatomer subunit alpha (COPA) syndrome, classified by the International Union of Immunologic Societies (IUIS) expert committee on inborn errors of immunity under the “auto-inflammatory, other” category (45). COPA syndrome can be defined as a complex disorder of the immune system, both at the innate (autoinflammatory) and adaptive (autoimmune) levels (19, 38, 46).COPA codes for the coatomer subunit alpha protein, a part of the coat protein complex I (COPI), which, as a component of the cell’s vesicular transport mechanism, is responsible for the retrograde movement of proteins from the Golgi apparatus back to the endoplasmic reticulum (ER) (19). The presence of a COPA mutation is associated with ER stress, which results in the release of pro-inflammatory cytokines and an expansion of T-helper type 17 (TH17) cells (19). In turn, alterations in T lymphocyte populations promote the dysregulation of the immune system and favor autoimmune diseases (47). Recent research indicates a key role of the stimulator of interferon genes (STING), a master regulator of innate immunity, in the pathogenesis of COPA syndrome (38). In patients with the COPA mutation, the accumulation of activated STING in the cell results in increased type I interferon (IFN) signaling (21, 48). The production of IFN type I is increased, similar to another well-described type I interferonopathy STING-associated vasculopathy with onset in infancy (SAVI) (49).
The finding of a mutation in the COPA gene in sisters with cryoglobulinemia, which is a lymphoproliferative disorder, may therefore indicate a relationship between intracellular disorders and the inappropriate activation of B cells, which requires further research.
It appears that COPA gene mutations do not show a genotype-phenotype correlation for the typical symptoms of COPA syndrome, although the strength of any conclusive statement is limited due to the recent identification of this disease and a limited number of patients. COPA syndrome is an ultra-rare disease with a limited number of cases reported thus far. It is diagnosed mainly in children, hence it is rarely included in a differential diagnosis in the adult population (50). The most characteristic symptom of COPA syndrome is lung involvement; however, the symptoms of the disease may change over time in individual patients along with the appearance of autoantibodies and affect other organs and systems, sometimes leading to their failure and the need for transplantation. Pulmonary involvement in young children is often manifested by intra-alveolar bleeding, while later, interstitial lung disease (ILD) involving cysts/nonspecific interstitial pneumonia (NSIP) (on imaging) and follicular bronchiolitis, lymphocytic interstitial pneumonia, and/or interstitial fibrosis (on lung biopsy) can be observed (19, 21, 51, 52). More than 90% of patients with COPA syndrome have joint pain or arthritis, and approximately 40% of them, mainly in the second decade of life, show symptoms of kidney involvement (glomerulonephritis on biopsy). Laboratory tests reveal increased inflammatory state parameters and the presence of autoantibodies in the blood serum (ANA, ANCA, and/or RF/CCP) (19, 20, 28). Although the term “COPA syndrome” is typically reserved for cases with pulmonary involvement with or without joint or renal involvement, the COPA syndrome phenotype is widening (23). Moreover, in patients with COPA syndrome, especially in later childhood or adolescence, symptoms such as intra-alveolar hemorrhage or glomerulonephritis suggest SLE or ANCA-vasculitis, which may pose diagnostic difficulties (22, 53). The sisters with a mutation in the COPA gene in this case report did not present with “typical COPA syndrome” but it is well-known that a monogenic disease may have different phenotypes (54, 55). First of all, our patients were adults, but it should be emphasized that the diagnosis of a rare disease in both suggested a genetic basis. There are a few reports in the available literature that describe sisters suffering from cryoglobulinemia and brothers suffering from cryoglobulinemia and macroglobulinemia, however, the data comes from a time when genetic tests were not accessible (56–58).
Some typical features of the COPA syndrome, including interstitial lung disease and positive autoimmune serologies, were described in the sister of our index case (Table 1). However, a histopathological examination of the tumor in the left lung revealed granulomas, which have not been previously reported in COPA syndrome (19, 37, 52). In contrast to ANCA-positive vasculitis, granulomas are not a typical manifestation of lung involvement in cryoglobulinemia (1). In turn, the clinical picture of the index case was dominated by symptoms of cryoglobulinemic small vessel vasculitis, mainly involving the skin and the peripheral/central nervous system, which are organs less frequently affected in COPA syndrome (Table 1) (19, 34). Symptoms of vasculitis in the course of cryoglobulinemia were reported in both sisters. According to current knowledge, cryoglobulinemic vasculitis can also cause pulmonary alveolar hemorrhage and glomerulonephritis, which are a typical presentation of COPA syndrome (14, 59). The assessment of cryoglobulins is included in the differential diagnosis of renal-pulmonary syndrome in adults so this may be why we have not found any information in the literature about cryoglobulins in COPA syndrome predominantly diagnosed in young children.
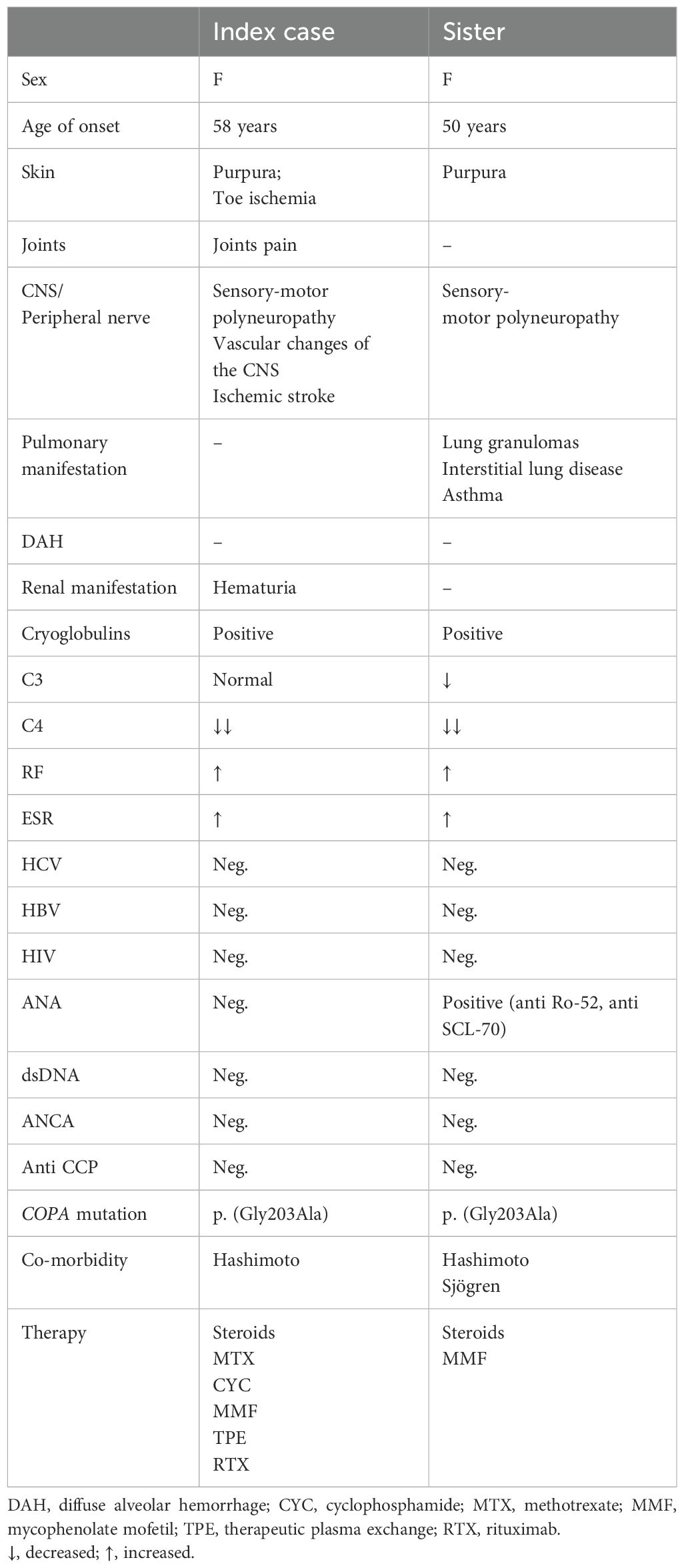
Table 1. Clinical and serological characteristics of the index case and her sister with cryoglobulinemia.
Vasculitis may be the leading symptom or accompanying feature in many monogenic autoinflammatory diseases (60–66). Furthermore, COPA syndrome, along with other autoinflammatory diseases such as SAVI, CANDLE (chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature), and Aicardi–Goutières syndrome can be classified as monogenic diseases with vasculitis (60, 61). If we examine the histopathological results of the lungs of patients with COPA syndrome, we can find signs of vasculitis in most patients with necrosis of the capillary walls, and neutrophils are often identified along the capillaries, which could suggest an immunological origin of pulmonary hemorrhages (20).
Currently, there is no specific drug for COPA syndrome, and its symptoms are treated with immunosuppression regimens used in immunological disorders since these features dominate the clinical picture of COPA syndrome. Based on recent reports demonstrating an increased expression of type 1 interferon (IFN)-stimulated genes (ISGs) in patients with a mutation in the COPA gene, it has been assumed that drugs blocking type I interferon signaling (i.e., Janus kinase inhibitors such as baricitinib and ruxolitinib) may be effective (48). Indeed, these drugs have been used in patients with autoinflammatory interferonopathies including COPA syndrome (67–69).
Thus far, reports show short-term results, and greater clinical experience is required for definitive recommendations. Considering the multidrug resistance in the course of cryoglobulinemia in our index case patient, we should perhaps use interferon pathway inhibitors as the next line of treatment, however, genetic tests were performed at the end of the patient’s life, precluding a possible trial.
The finding of a mutation in the COPA gene in sisters with non-infectious mixed cryoglobulinemia is a valuable contribution to research on the pathogenesis of this rare disease. Moreover, the results of the genetic tests of the presented cases allow us to expand the phenotype of COPA syndrome in adults to include cryoglobulinemic vasculitis.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The studies involving humans were approved by Military Institute of Medicine Bioethics Committee: approval number 37/WIM/2018. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
KL: Conceptualization, Data curation, Investigation, Resources, Writing – original draft. RP: Conceptualization, Writing – original draft, Investigation, Supervision, Writing – review & editing. MR: Data curation, Investigation, Writing – original draft. AR: Supervision, Writing – review & editing. AL: Supervision, Writing – review & editing. TS: Writing – review & editing. SN: Conceptualization, Project administration, Supervision, Writing – review & editing.
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
2. Sansonno D, Tucci FA, Ghebrehiwet B, Lauletta G, Peerschke EI, Conteduca V, et al. Role of the receptor for the globular domain of C1q protein in the pathogenesis of hepatitis C virus-related cryoglobulin vascular damage. J Immunol. (2009) 183:6013–20. doi: 10.4049/jimmunol.0902038
3. Cacoub P, Poynard T, Ghillani P, Charlotte F, Olivi M, Piette JC, et al. Extrahepatic manifestations of chronic hepatitis C. MULTIVIRC Group Multidepartment Virus C. Arthritis Rheumatol. (1999) 42:2204–12. doi: 10.1002/15290131(199910)42:10<2204::AID-ANR24>3.0.CO;2-D
4. Visentini M, Conti V, Cristofoletti C, Lazzeri C, Marrapodi R, Russo G, et al. Clonal expansion and functional exhaustion of monoclonal marginal zone B cells in mixed cryoglobulinemia: the yin and yang of HCV-driven lymphoproliferation and autoimmunity. Autoimmun Rev. (2013) 12:430–5. doi: 10.1016/j.autrev.2012.08.016
5. Ramos-Casals M, De Vita S, Tzioufas AG. Hepatitis C virus, Sjoügren’s syndrome and B-cell lymphoma: linking infection, autoimmunity and cancer. Autoimmun Rev. (2005) 4:8–15. doi: 10.1016/j.autrev.2004.04.004
6. Brouet JC, Clauvel JP, Danon F, Klein M, Seligmann M. Biologic and clinical significance of cryoglobulins: a report of 86 cases. Am J Med. (1974) 57:775–88. doi: 10.1016/0002-9343(74)90852-3
7. Trejo O, Ramos-Casals M, López-Guillermo A, Carrasco MG, Yagüe J, Cervera R, et al. Hematologic Malignancies in patients with cryoglobulinaemia: association with autoimmune and chronic viral diseases. Semin Arthritis Rheumatol. (2003) 33:19–28. doi: 10.1053/sarh.2003.50020
8. Ferri C, Greco F, Longombardo G, Palla P, Moretti A, Marzo E, et al. Association between hepatitis C virus and mixed cryoglobulinemia. Clin Exp Rheumatol. (1991) 9:621–4.
9. Tzioufas AG, Boumba DS, Skopouli FN, Moutsopoulos HM. Mixed monoclonal cryoglobulinaemia and monoclonal rheumatoid factor cross-reactive idiotypes as predictive factors for the development of lymphoma in primary Sjogren’s syndrome. Arthritis Rheumatol. (1996) 39:767–72. doi: 10.1002/art.1780390508
10. Saadoun D, Sellam J, Ghillani-Dalbin P, Crecel R, Piette JC, Cacoub P, et al. Increased risks of lymphoma and death among patients with non-hepatitis C virus-related mixed cryoglobulinemia. Arch Intern Med. (2006) 166:2101–8. doi: 10.1001/archinte.166.19.2101
11. Jennette JC, Falk RJ, Bacon PA, Basu N, Cid MC, Ferrario F, et al. 2012 revised international chapel hill consensus conference nomenclature of vasculitides. Arthritis Rheum. (2013) 65:1–11. doi: 10.1002/art.37715
12. Fermand JP, Bridoux F, Dispenzieri A, Jaccard A, Kyle RA, Leung N, et al. Monoclonal gammopathy of clinical significance: a novel concept with therapeutic implications. Blood. (2018) 132:1478–85. doi: 10.1182/blood-2018-04-839480
13. Leśniak K, Rymarz A, Lubas A, Niemczyk S. Noninfectious, severe cryoglobulinemic vasculitis with renal involvement - safety and efficacy of long-term treatment with rituximab. Int J Nephrol Renovasc Dis. (2021) 14:267–77. doi: 10.2147/IJNRD.S315388
14. Ferri C, Sebastiani M, Giuggioli D, Cazzato M, Longombardo G, Antonelli A, et al. Mixed cryoglobulinemia: demographic, clinical, and serologic features and survival in 231 patients. Semin Arthritis Rheumatol. (2004) 33:355–74. doi: 10.1016/j.semarthrit.2003.10.001
15. Ramos-Casals M, Robles A, Brito-Zeron P, Nardi N, Nicolás JM, Forns X, et al. Life-threatening cryoglobulinemia: clinical and immunological characterization of 29 cases. Semin Arthritis Rheumatol. (2006) 36:189–96. doi: 10.1016/j.semarthrit.2006.08.005
16. Quartuccio L, Isola M, Corazza L, Ramos-Casals M, Retamozo S, Ragab GM, et al. Validation of the classification criteria for cryoglobulinaemic vasculitis. Rheumatol (Oxford). (2014) 53:2209–13. doi: 10.1093/rheumatology/keu271
17. Terrier B, Krastinova E, Marie I, Launay D, Lacraz A, Belenotti P, et al. Management of noninfectious mixed cryoglobulinemia vasculitis: data from 242 cases included in the CryoVas survey. Blood. (2012) 119:5996–6004. doi: 10.1182/blood-2011-12-396028
18. De Vita S, Quartuccio L, Isola M, Mazzaro C, Scaini P, Lenzi M, et al. A randomized controlled trial of rituximab for the treatment of severe cryoglobulinemic vasculitis. Arthritis Rheumatol. (2012) 64:843–53. doi: 10.1002/art.34331
19. Watkin LB, Jessen B, Wiszniewski W, Vece TJ, Jan M, Sha Y, et al. COPA mutations impair ER-Golgi transport and cause hereditary autoimmune-mediated lung disease and arthritis. Nat Genet. (2015) 47:654–60. doi: 10.1038/ng.3279
20. Vece TJ, Watkin LB, Nicholas SK, Canter D, Braun MC, Guillerman RP, et al. Copa syndrome: a novel autosomal dominant immune dysregulatory disease. J Clin Immunol. (2016) 36:377–87. doi: 10.1007/s10875-016-0271-8
21. Kato T, Yamamoto M, Honda Y, Orimo T, Sasaki I, Murakami K, et al. Augmentation of stimulator of interferon genes–induced type I interferon production in COPA syndrome. Arthritis Rheumatol. (2021) 73:2105–15. doi: 10.1002/art.41790
22. Cabrera-Pérez JS, Branch J, Reyes A, Michael M, Eldin KW, Silva-Carmona M, et al. A zebra at the rodeo: dyspnea, hematuria, and a family history of arthritis. Arthrit Care Res. (2022) 74:165–70. doi: 10.1002/acr.24368
23. Taveira-DaSilva AM, Markello TC, Kleiner DE, Jones AM, Groden C, Macnamara E. et al: Expanding the phenotype of COPA syndrome: a kindred with typical and atypical features. J Med Genet. (2019) 56:778–82. doi: 10.1136/jmedgenet-2018-105560
24. Rydzanicz M, Zwoliński P, Gasperowicz P, Pollak A, Kostrzewa G, Walczak A, et al. A recurrent de novo variant supports KCNC2 involvement in the pathogenesis of developmental and epileptic encephalopathy. Am J Med Genet A. (2021) 185:3384–9. doi: 10.1002/ajmg.a.62455
25. Stawiński P, Płoski R. Genebe.net: Implementation and validation of an automatic ACMG variant pathogenicity criteria assignment. Clin Genet. (2024) 106(2):119–26. doi: 10.1111/cge.14516
26. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
27. Kopanos C, Tsiolkas V, Kouris A, Chapple CE, Albarca Aguilera M, Meyer R, et al. VarSome: the human genomic variant search engine. Bioinformatics. (2019) 35:1978–80. doi: 10.1093/bioinformatics/bty897
28. Frémond ML, Nathan N. COPA syndrome, 5 years after: Where are we? Joint Bone Spine. (2021) 88:105070. doi: 10.1016/j.jbspin.2020.09.002
29. Zeng J, Hao J, Zhou W, Zhou Z, Miao H. A novel mutation c.841C>T in COPA syndrome of an 11-year-old boy: A case report and short literature review. Front Pediatr. (2021) 9:773112. doi: 10.3389/fped.2021.773112
30. Zheng Y, Du Y, Wu Y, Li F, Gu W, Zhao C. COPA syndrome caused by a novel p.Arg227Cys COPA gene variant. Mol Genet Genomic Med. (2024) 12:e2309. doi: 10.1002/mgg3.2309
31. Cheng J, Novati G, Pan J, Bycroft C, Žemgulytė A, Applebaum T, et al. Accurate proteome-wide missense variant effect prediction with AlphaMissense. Science. (2023) 381:eadg7492. doi: 10.1126/science.adg7492
32. Stray-Pedersen A, Sorte HS, Samarakoon P, Gambin T, Chinn IK, Coban Akdemir ZH, et al. Primary immunodeficiency diseases: Genomic approaches delineate heterogeneous Mendelian disorders. J Allergy Clin Immunol. (2017) 139:232–45. doi: 10.1016/j.jaci.2016.05.042
33. Noorelahi R, Perez G, Otero HJ. Imaging findings of Copa syndrome in a 12-year-old boy. Pediatr Radiol. (2018) 48:279–82. doi: 10.1007/s00247-017-3961-3
34. Jensson BO, Hansdottir S, Arnadottir GA, Sulem G, Kristjansson RP, Oddsson A, et al. COPA syndrome in an Icelandic family caused by a recurrent missense mutation in COPA. BMC Med Genet. (2017) 18:129. doi: 10.1186/s12881-017-0490-8
35. Patwardhan A, Spencer CH. An unprecedented COPA gene mutation in two patients in the same family: comparative clinical analysis of newly reported patients with other known COPA gene mutations. Pediatr Rheumatol Online J. (2019) 17:59. doi: 10.1186/s12969-019-0359-9
36. Lin KZ, Zhang FF, Xiao JH. A pathogenic mutation in the COPA gene causes a family of COPA syndrome. Zhonghua Er Ke Za Zhi. (2020) 58:58–9. doi: 10.3760/cma.j.issn.0578-1310.2020.01.015
37. Prenzel F, Harfst J, Schwerk N, Ahrens F, Rietschel E, Schmitt-Grohé S, et al. Lymphocytic interstitial pneumonia and follicular bronchiolitis in children: A registry-based case series. Pediatr Pulmonol. (2020) 55:909–17. doi: 10.1002/ppul.24680
38. Deng Z, Chong Z, Law CS, Mukai K, Ho FO, Martinu T, et al. A defect in COPI-mediated transport of STING causes immune dysregulation in COPA syndrome. J Exp Med. (2020) 217:e20201045. doi: 10.1084/jem.20201045
39. Thaivalappil SS, Garrod AS, Borowitz SM, Watkin LB, Lawrence MG. Persistent unexplained transaminitis in COPA syndrome. J Clin Immunol. (2021) 41:205–8. doi: 10.1007/s10875-020-00832-4
40. Bader-Meunier B, Bustaffa M, Iskounen T, Carter E, Marsh JA, Baujat G, et al. Rheumatoid factor positive polyarticular juvenile idiopathic arthritis associated with a novel COPA mutation. Rheumatol (Oxford). (2021) 60:e171–3. doi: 10.1093/rheumatology/keaa763
41. Ripen AM, Chear CT, Baharin MF, Nallusamy R, Chan KC, Kassim A, et al. A single-center pilot study in Malaysia on the clinical utility of whole-exome sequencing for inborn errors of immunity. Clin Exp Immunol. (2021) 206:119–28. doi: 10.1111/cei.13626
42. Psarianos P, Kwan JYY, Dell S, Wee WB, Rey-McIntyre K, Chen H, et al. COPA syndrome (Ala239Pro) presenting with isolated follicular bronchiolitis in early childhood: case report. J Clin Immunol. (2021) 41:1660–3. doi: 10.1007/s10875-021-01082-8
43. Zignego AL, Wojcik GL, Cacoub P, Visentini M, Casato M, Mangia A, et al. Genome-wide association study of hepatitis C virus- and cryoglobulin-related vasculitis. Genes Immun. (2014) 15:500–5. doi: 10.1038/gene.2014.41
44. Gragnani L, Fognani E, De Re V, Libra M, Garozzo A, Caini P, et al. Notch4 and mhc class II polymorphisms are associated with hcv-related benign and Malignant lymphoproliferative diseases. Oncotarget. (2017) 8:71528–35. doi: 10.18632/oncotarget.17655
45. Bousfiha A, Moundir A, Tangye SG, Picard C, Jeddane L, Al-Herz W, et al. The 2022 update of IUIS phenotypical classification for human inborn errors of immunity. J Clin Immunol. (2022) 42:1508–20. doi: 10.1007/s10875-022-01352-z
46. Deng Z, Law CS, Ho FO, Wang KM, Jones KD, Shin JS, et al. A defect in thymic tolerance causes T cell-mediated autoimmunity in a murine model of COPA syndrome. J Immunol. (2020) 204:2360–73. doi: 10.4049/jimmunol.2000028
47. de Jesus AA, Goldbach-Mansky R. Newly recognized Mendelian disorders with rheumatic manifestations. Curr Opin Rheumatol. (2015) 27:511–9. doi: 10.1097/BOR.0000000000000207
48. Volpi S, Tsui J, Mariani M, Pastorino C, Caorsi R, Sacco O, et al. Type I interferon pathway activation in COPA syndrome. Clin Immunol. (2018) 187:33–6. doi: 10.1016/j.clim.2017.10.001
49. Liu Y, Jesus AA, Marrero B, Yang D, Ramsey SE, Sanchez GAM, et al. Activated STING in a vascular and pulmonary syndrome. N Engl J Med. (2014) 371:507–18. doi: 10.1056/NEJMoa1312625
50. Simchoni N, Vogel TP, Shum AK. COPA syndrome from diagnosis to treatment: A clinician’s guide. Rheum Dis Clin North Am. (2023) 49:789–804. doi: 10.1016/j.rdc.2023.06.005
51. Nguyen HN, Salman R, Vogel TP, Silva-Carmona M, DeGuzman M, Guillerman RP. Imaging findings of COPA syndrome. Pediatr Radiol. (2023) 53:844–53. doi: 10.1007/s00247-023-05600-1
52. Tsui JL, Estrada OA, Deng Z, Wang KM, Law CS, Elicker BM, et al. Analysis of pulmonary features and treatment approaches in the COPA syndrome. ERJ Open Res. (2018) 4:00017–2018. doi: 10.1183/23120541.00017-2018
53. Khalifi SBE, Viel S, Lahoche A, Frémond ML, Lopez J, Lombard C, et al. COPA syndrome as a cause of lupus nephritis. Kidney Int Rep. (2019) 4:1187–9. doi: 10.1016/j.ekir.2019.04.014
54. Rice GI, Rodero MP, Crow YJ. Human disease phenotypes associated with mutations in TREX1. J Clin Immunol. (2015) 35:235–43. doi: 10.1007/s10875-015-0147-3
55. Al-Mayouf SM, Sunker A, Abdwani R, Abrawi SA, Almurshedi F, Alhashmi N, et al. Loss-of-function variant in DNASE1L3 causes a familial form of systemic lupus erythematosus. Nat Genet. (2011) 43:1186–8. doi: 10.1038/ng.975
56. Weinberger A, Berliner S, Shoenfeld Y, Pinkhas J. Idiopathic mixed cryoglobulinemia in two sisters. Acta Haematol. (1981) 65:219–20. doi: 10.1159/000207182
57. Dammacco F, Scarpioni L, Antonaci S, Bonomo L. Cryoimmunoglobulinemia in four sisters. Acta Haematol. (1978) 59:215–22. doi: 10.1159/000207764
58. Biro I. Occurrence of cryoglobulinemia and macrocryogelglobulinemia in brothers. Pathol Biol. (1962) 103:1709–12.
59. Retamozo S, Diaz-Lagares C, Bosch X, de Vita S, Ramos-Casals M. Life-threatening cryoglobulinemia. Autoimmune diseases: acute Complex situations. (2011), 133–62. doi: 10.1007/978-0-85729-358-9_10
60. Demir S, Sag E, Dedeoglu F, Ozen S. Vasculitis in systemic autoinflammatory diseases. Front Pediatr. (2018) 6:377. doi: 10.3389/fped.2018.00377
61. Demirkaya E, Arici ZS, Romano M, Berard RA, Aksentijevich I. Current state of precision medicine in primary systemic vasculitides. Front Immunol. (2019) 10:2813. doi: 10.3389/fimmu.2019.02813
62. Aksentijevich I, Masters SL, Ferguson PJ, Dancey P, Frenkel J, van Royen-Kerkhoff A, et al. An autoinflammatory disease with deficiency of the interleukin-1-receptor antagonist. N Engl J Med. (2009) 360:2426–37. doi: 10.1056/NEJMoa0807865
63. Khatibi K, Heit JJ, Telischak NA, Elbers JM, Do HM. Cerebral vascular findings in PAPA syndrome: cerebral arterial vasculopathy or vasculitis and a posterior cerebral artery dissecting aneurysm. J neurointerv Surg. (2016) 8:e29. doi: 10.1136/neurintsurg-2015-011753.rep
64. Kolivras A, Theunis A, Ferster A, Lipsker D, Sass U, Dussart A, et al. Cryopyrin-associated periodic syndrome: an autoinflammatory disease manifested as neutrophilic urticarial dermatosis with additional perieccrine involvement. J Cutan Pathol. (2011) 38:202–8. doi: 10.1111/j.1600-0560.2010.01638.x
65. Munoz J, Rodière M, Jeremiah N, Rieux-Laucat F, Oojageer A, Rice GI, et al. Stimulator of interferon genes-associated vasculopathy with onset in infancy: A mimic of childhood granulomatosis with polyangiitis. JAMA Dermatol. (2015) 151:872–7. doi: 10.1001/jamadermatol.2015.0251
66. Muscari I, Iacoponi F, Cantarini L, Lucherini OM, Simonini G, Brizi MG, et al. The diagnostic evaluation of patients with potential adult-onset autoinflammatory disorders: our experience and review of the literature. Autoimmun Rev. (2012) 12:10–3. doi: 10.1016/j.autrev.2012.07.015
67. Sanchez GAM, Reinhardt A, Ramsey S, Wittkowski H, Hashkes PJ, Berkun Y, et al. JAK1/2 inhibition with baricitinib in the treatment of autoinflammatory interferonopathies. J Clin Invest. (2018) 128:3041–52. doi: 10.1172/JCI98814
68. Krutzke S, Rietschel C, Horneff G. Baricitinib in therapy of COPA syndrome in a 15-year-old girl. Eur J Rheumatol. (2020) 7:S78–81. doi: 10.5152/eurjrheum.2019.18177
Keywords: COPA gen, COPA syndrome, cryoglobulinemia, cryoglobulinemia vasculitis, mixed cryoglobulinemia
Citation: Leśniak K, Płoski R, Rydzanicz M, Rymarz A, Lubas A, Syryło T and Niemczyk S (2024) Non-infectious mixed cryoglobulinemia as a new clinical presentation of mutation in the gene encoding coatomer subunit alpha: a case report of two adult sisters. Front. Immunol. 15:1450048. doi: 10.3389/fimmu.2024.1450048
Received: 16 June 2024; Accepted: 25 October 2024;
Published: 15 November 2024.
Edited by:
Jeffrey J. Pu, Harvard Medical School, United StatesReviewed by:
Marcella Visentini, Sapienza University of Rome, ItalyCopyright © 2024 Leśniak, Płoski, Rydzanicz, Rymarz, Lubas, Syryło and Niemczyk. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ksymena Leśniak, a2xlc25pYWtAd2ltLm1pbC5wbA==
†ORCID: Rafał Płoski, orcid.org/0000-0001-6286-5526
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.