
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 06 January 2025
Sec. Primary Immunodeficiencies
Volume 15 - 2024 | https://doi.org/10.3389/fimmu.2024.1447995
 Sara Posadas-Cantera1,2
Sara Posadas-Cantera1,2 Noriko Mitsuiki1
Noriko Mitsuiki1 Florian Emmerich3
Florian Emmerich3 Virginia Patiño4
Virginia Patiño4 Hanns-Martin Lorenz5
Hanns-Martin Lorenz5 Olaf Neth6Ingunn Dybedal7
Olaf Neth6Ingunn Dybedal7 Kjetil Taskén8
Kjetil Taskén8 Alejandro A. Schäffer9
Alejandro A. Schäffer9 Bodo Grimbacher1,10,11,12*
Bodo Grimbacher1,10,11,12* Laura Gámez-Díaz1,11*
Laura Gámez-Díaz1,11*Introduction: Human Cytotoxic-T-lymphocyte-antigen-4 (CTLA-4) insufficiency caused by heterozygous germline mutations in CTLA4 is a complex immune dysregulation and immunodeficiency syndrome presenting with reduced penetrance and variable disease expressivity, suggesting the presence of disease modifiers that trigger the disease onset and severity. Various genetic and non-genetic potential triggers have been analyzed in CTLA-4 insufficiency cohorts, however, none of them have revealed a clear association to the disease. Multiple HLA haplotypes have been positively or negatively associated with various autoimmune diseases and inborn errors of immunity (IEI) due to the relevance of MHC in the strength of the T cell responses.
Methods: In this exploratory study, we investigated the association of disease onset, severity and clinical manifestations of CTLA-4 insufficiency with specific HLA class I (A, B and C) and class II (DRB1 and DQB1) alleles in forty-three individuals harboring heterozygous mutations in CTLA4. Twenty-six out of the 43 recruited individuals presented moderate or severe clinical symptoms whereas 17 were completely healthy. HLA frequency analysis, odds ratio analysis and genetic linkage analysis were used.
Results: The principal statistical analyses showed no positive association between the HLA genotypes analyzed with the disease onset or the disease severity. We found potential risk associations of HLA-DQB1*05:01 and HLA-DRB1*01:02 with respiratory tract involvement and HLA-C*05:01 with affection of the neurological system in the CTLA-4-insufficient patients. Additionally, we found a potential protective association of HLA-DRB1*01:01 with gastrointestinal symptoms.
Discussion: Even though, our findings suggest that HLA-A, -B, -C, DRB1, and DQB1 do not contribute to the onset or severity of disease in CTLA-4 insufficiency, certain HLA-alleles may influence the manifestation of specific symptoms. We advocate for further investigation of specific class I and class II HLA alleles as potential disease modifiers in larger clinical cohorts of CTLA-4 insufficiency.
The complete activation of T lymphocytes requires at least two signals. The first signal involves the recognition of the major histocompatibility complex (MHC)-bound peptide on antigen presenting cells (APC) by the T-cell receptor (TCR) on T lymphocytes, determining the strength of the T-cell response (1). The second signal involves the binding of B7 costimulatory molecules on APCs with the CD28 receptor expressed on T lymphocytes. Following T-cell activation, intracellular vesicles containing cytotoxic-T-lymphocyte-antigen-4 (CTLA-4) are mobilized to the cell membrane to outcompete CD28 for binding to B7 costimulatory molecules on APCs. After binding, CTLA-4:B7 complexes are internalized into T lymphocytes via transendocytosis for further lysosomal degradation (2). Hence, CTLA-4 is considered a negative immune regulator essential for controlling T-cell activation and maintaining immune homeostasis.
Heterozygous germline mutations in CTLA4 have been identified as the cause of an inborn error of immunity (IEI) known since 2015 as CTLA-4 insufficiency (3, 4). This syndrome is characterized by immunodeficiency and immune dysregulation, leading to clinical symptoms such as hypogammaglobulinemia, recurrent infections, autoimmune cytopenia, hepatosplenomegaly, tissue lymphoid infiltrations, and malignancies (3–6). However, CTLA-4 insufficiency presents with incomplete penetrance and reduced disease expressivity as about 30% of CTLA4 mutation carriers exhibit no clinical symptoms despite having impaired in vitro CTLA-4 function (3, 7). These observations suggest the presence of disease modifiers that trigger disease onset or influence disease severity. To date, several genetic and non-genetic disease modifiers in the context of CTLA-4 insufficiency have been studied. Among non-genetic factors, past infections by Epstein Barr virus (EBV), cytomegalovirus, herpes simplex virus type 1 and 2 and Toxoplasma gondii have been ruled out (8). Regarding genetic factors, a gain-of-function mutation in JAK3 and a loss-of-function mutation in CLEC7A have been proposed as disease modifiers as they seem to contribute to the full disease expressivity in patients with heterozygous mutations in CTLA4 (9, 10). However, these two rare variants in JAK3 and CLEC7A have been identified in only one patient each.
The MHC system is required to deliver the first signal of T cell activation; MHC is also known as the human leukocyte antigen (HLA) system in humans and contains the most polymorphic gene cluster of the entire human genome. Based on the structure and function of the HLA gene products, HLA components are classified into three main groups, class I, class II and class III (11). HLA class I, which includes HLA-A, HLA-B and HLA-C isotypes, presents endogenous peptides to the TCR of CD8+ T cells. In contrast, the TCR of CD4+ T cells recognize exogenous peptides bound to HLA class II that includes HLA-DR, DP, and DQ isotypes. HLA class III encodes, among others, components of the complement system, heat shock proteins and tumor necrosis factor family proteins (12, 13). Specific HLA haplotypes have been commonly associated with disease predisposition or protection in various autoimmune diseases. For instance, several HLA-DRB1 alleles have been associated with susceptibility to rheumatoid arthritis (RA) in different populations, HLA-DQA1 and HLA-DQB1 alleles have been strongly associated with type-1 diabetes and celiac disease, and specific HLA-DRB3*03:01 allele with Crohn’s disease (14, 15). In addition, susceptibility to common variable immunodeficiency (CVID) and deficiency of IgA (sIgAD), both IEI, has also been found to be positively associated with HLA-DQ and HLA-DRB1 alleles (16, 17). These associations are not surprising, given that the specificity of the HLA-peptide–TCR (T-cell receptor) interaction is essential for maintaining an efficient and appropriate adaptive immune response, not only to counteract infection and malignancy, but also to preserve self- tolerance, preventing the generation of autoimmune diseases. The specific underlying mechanisms behind these associations are largely unknown.
We hypothesized that specific HLA alleles may influence disease manifestation, disease onset and disease severity in CTLA-4 insufficiency. In this exploratory study, we analyzed the A, B and C HLA class I alleles, and the DRB1 and DQB1 class II HLA alleles in 43 individuals from seven different families carrying heterozygous mutations in CTLA4. Our results indicate that the investigated HLA alleles are unlikely to be disease modifiers in patients with CTLA-4 insufficiency.
Between 2018 and 2022, we enrolled 43 individuals from seven different families, each harboring heterozygous mutations in CTLA4. These families are identified here as families A, B, C, D, E, F and G. Families A, B and E are from Germany and were recruited at the Center for Chronic Immunodeficiencies, Medical Center - University of Freiburg. Family C was referred from the Department of Immunology and Histocompatibility, Centre for Primary Immunodeficiencies, “Aghia Sophia” Children’s Hospital, Athens, Greece, whereas Family D was referred from the Immunology Team, Montevideo, Uruguay. Families F and G attend the Paediatric Infectious Diseases, Rheumatology and Immunology Unit, Hospital Virgen del Rocio, Instituto de Biomedicina de Sevilla, Spain, and the Department of Hematology, Oslo University Hospital, Norway, respectively. All participating individuals donated blood samples for DNA extraction after signing an informed written consent under local ethics board–approved protocols from each of the above-mentioned institutions. This project was approved by the local ethics committee of University of Freiburg, protocol number 466/18.
Genomic DNA samples were extracted from either whole blood or peripheral blood mononuclear cells following standard protocols. HLA typing encompassing HLA-A, -B, -C, -DRB1, and -DQB1 was performed applying routine methods either through Sanger-Sequencing (ProTrans, Hockenheim, Germany) or next generation sequencing (NGS) using NGSgoR sequencing kits (GenDx, the Netherlands). HLA-typing analysis was performed by aligning the nucleotide sequences to the official HLA-reference database (https://www.ebi.ac.uk/ipd/imgt/hla/), which is used as the reference for research and clinical diagnostics worldwide.
Allele-specific frequencies were calculated in cases and controls irrespective of their pedigree status. All HLA alleles were coded as either present or absent.
We used FASTLINK v. 4.1P (18–20) to compute logarithm of odds (LOD) scores for the observed data and we used the related simulation package FASTSLINK v. 3.0 (21) to estimate the power to detect linkage. For simplicity, we show single-marker analyses with four different parameter settings, while noting that other parameter settings gave qualitatively similar results. We modeled the modifier locus as dominant because there are parent-child affected pairs. We assumed a penetrance of 0.9 (90%) for the modifier disease associated allele. In three out of four parameter combinations, we allowed a phenocopy rate of 0.01, meaning that the modifier would not be active if there is a phenocopy. We varied the frequency of the modifier allele among (0.01, 0.10, 0.20, 0.50). The genetic linkage analysis differs fundamentally from the HLA allele frequency analysis in that the LOD scores for each family are computed separately for each family and added. Thus, the disease-associated marker allele(s) can be different in each family and one can still detect genetic linkage. We assumed that the frequency of each HLA allele is equal for each locus. For the power calculations we used a 5-allele marker placed at 5 centiMorgans from the putative modifier locus. Using 100 replicates for each parameter setting, we estimated the probability of observing a LOD score above the thresholds of 1, 2, or 3.
The CHAI-morbidity score is a disease assessment score that was developed based on the data of the first 133 CTLA-4 insufficiency patients analyzed (7) and an additional 73 unpublished patients. The CHAI morbidity score quantifies organ involvement by integrating specific laboratory values including flow cytometry results, imaging data, and physiological functional results into a score and is described in detail in (22).
Pie charts were generated using GraphPad Prism version 9.0.0, Odds ratios and 95% confidence intervals were calculated using the R package epitools and visualized with the ggplot2 R package.
We collected data from seven families, including a total of 43 individuals carrying heterozygous mutations in CTLA4 (Figure 1A). Six of these families were from European countries, and one family was from South America. Eighteen out of the 43 recruited individuals were male (41.8%) while 25 were female (58.2%). At recruitment, the average age of our carrier cohort was 45.8 years, with an age range from 15 to 91 years (Tables 1, 2).
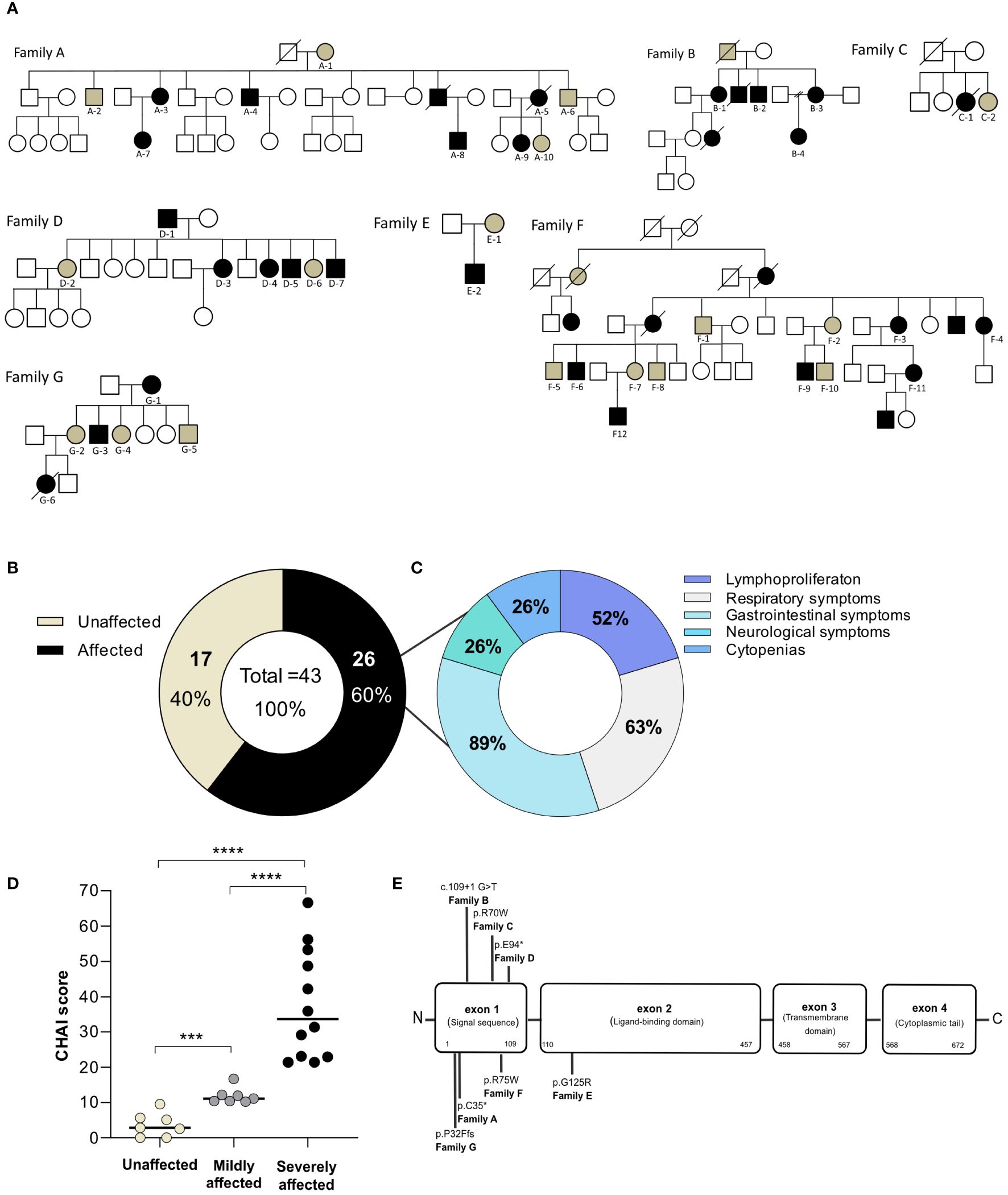
Figure 1. Characteristics of the study population. (A) Family pedigrees depicting CTLA4 mutation carriers and recruited individuals. Squares indicate male subjects and circles indicate female subjects. Filled symbols indicate subjects with an identified mutation in CTLA4. Black symbols designate affected patients whereas brown symbols designate unaffected CTLA4 mutation carriers. Recruited individuals are designated with an ID below their corresponding symbol. Crossed-out symbols indicate deceased subjects. (B) Classification of CTLA4 mutation carriers into unaffected (brown) and affected groups (black) based on the requirement of medical attention. (C) Clinical symptoms of the affected CTLA4 mutation carriers clustered in five main groups according to the organ affected. (D) CHAI score classified the CTLA4 mutation carriers into unaffected <10 points (light brown), mildly affected between 10 to 20 points (gray) and severely affected >20 points (black) (n=19). Mann Whitney test. p-value >0.0001. (E) Schematic representation of the type and distribution of the CTLA4 mutations identified in our cohort. *p<0.05; **p<0.01; ***p<0.001, ****p<0.0001.
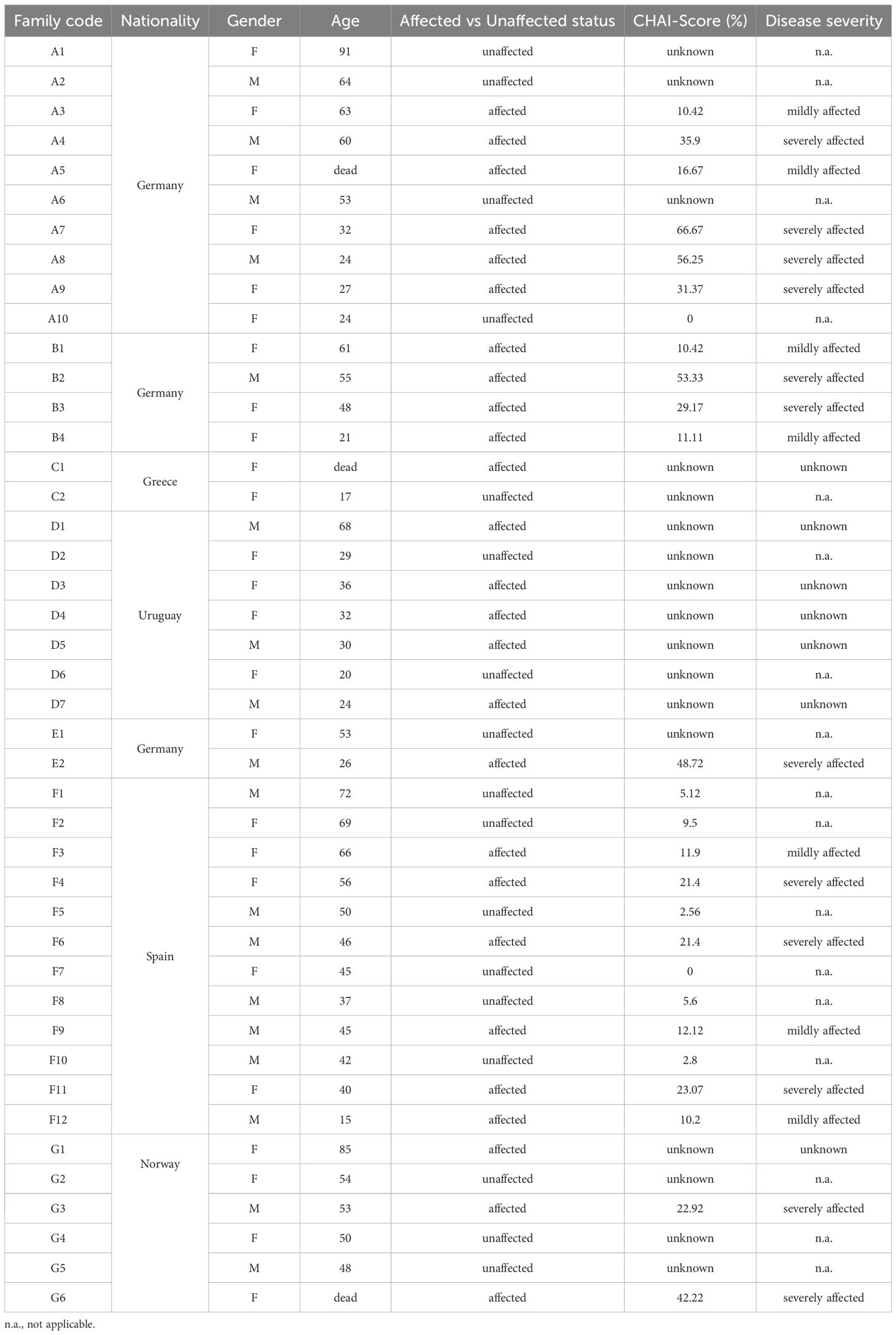
Table 1. Baseline characteristics of the study population.
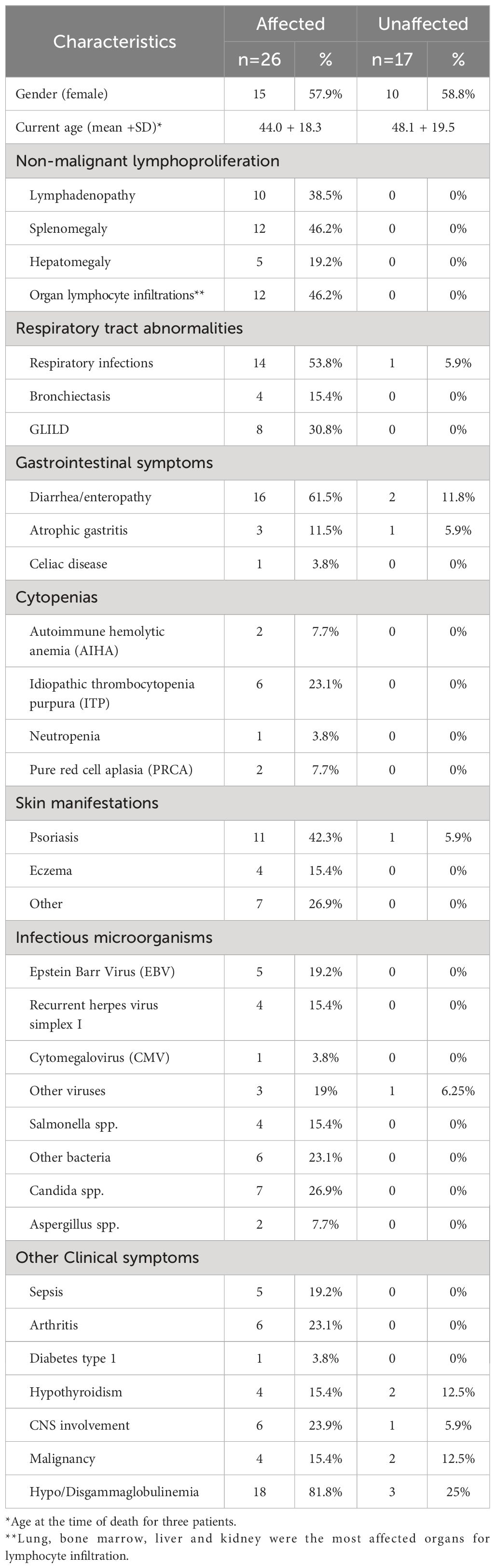
Table 2. Clinical characteristics of the study population.
Based on the clinical status information provided by the treating physicians or extracted from the literature (7, 22, 23), the 43 recruited individuals were classified as affected or unaffected CTLA4 mutation carriers (Table 1). The affected group included all CTLA4 mutation carriers experiencing moderate or severe clinical symptoms, whereas the unaffected group included all those CTLA4 mutation carriers not requiring specialized medical treatment up to the time of this study (7, 23). Out of the 43 CTLA4 mutation carriers, 26 were classified as affected and 17 were classified as unaffected (Figure 1B and Table 1). Gender distribution was conserved in the affected (11 male and 15 female) and the unaffected groups (7 male and 10 female). The average age was also similar in the affected (44 years old) and unaffected groups (48.1 years old) (Table 2). At the time of the study, 40 patients were alive, while three affected CTLA4 mutation carriers had passed away at the ages of 20 (code A5), 25 (code C1), and 37 (code G6) years.
Clinical manifestations of the 26 affected CTLA4 mutation carriers were clustered into five main different groups: i) non-malignant lymphoproliferation, ii) respiratory tract illnesses, iii) gastrointestinal symptoms, iv) neurological clinical manifestations, v) cytopenias (Figure 1C). Non-malignant lymphoproliferation included lymphadenopathy, splenomegaly, hepatomegaly, and lymphocyte organ infiltrations. Respiratory tract conditions included respiratory infections, bronchiectasis and Granulomatous and Lymphocytic Interstitial Lung Diseases (GLILD). Gastrointestinal symptoms included diarrhea, enteropathy and atrophic gastritis. The cytopenias group involved autoimmune hemolytic anemia (AIHA), idiopathic thrombocytopenia purpura (ITP), neutropenia and pure red cell aplasia (PRCA). Clinical manifestations are summarized in Table 1. Based on the CHAI morbidity score calculated in 19 affected individuals, twelve patients were classified as severely affected (CHAI score >20%), and seven as mildly affected (score between 10% and 20%) (Figure 1D and Table 1).
Sequencing of CTLA4 confirmed, two nonsense mutations, three missense mutations, one insertion and one frameshift mutation. Specifically, Family A and Family D had nonsense mutations (c.105C>A; p.C35*, and c.280G>T; E94*, respectively), whereas Family C, E and F had missense mutations (c.208C>T; p.R70W, c.373G>A; p.G125R, and c.223C>T; p.R75W, respectively). Family B had one insertion (c.109 + 1G>T), and one frameshift mutation was found in Family G (c.94_101delinsTTCTCTTCATCA; p.P32Ffs*) (Figure 1E). Functional testing of CTLA-4-mediated transendocytosis in regulatory T cells from all members of Families A (n=10), B (n=4), C (n=2) and F (n=12) reported elsewhere, showed reduced transendocytosis of CD80 regardless the affected or unaffected status of the individual (3, 7).
HLA genotyping of HLA-A,-B,-C was performed in 38 CTLA4 mutation carriers due to sample availability whereas 43 CTLA4 mutation carriers were sequenced for -DRB1 and -DQB1 alleles (Table 2). The genotype information included a two-to-four digit HLA allele. For each clinical manifestation group (Figure 1C), allele-specific overall frequencies were calculated in cases and controls irrespective of the pedigree status. We predicted the haplotypes of G-1, G-2, and G-4 based on allele frequencies. The genotype of HLA-DRB1 of G-1, G-2, and G-4 presented up to two-digits as DRB1*13s. In addition, the most likely genotype formed a haplotype with DQB1*06:04, which has been reported as DRB1*13:02 in the allele frequency net database (AFND) (24). The results of genotypes and serotypes are shown in Table 3.
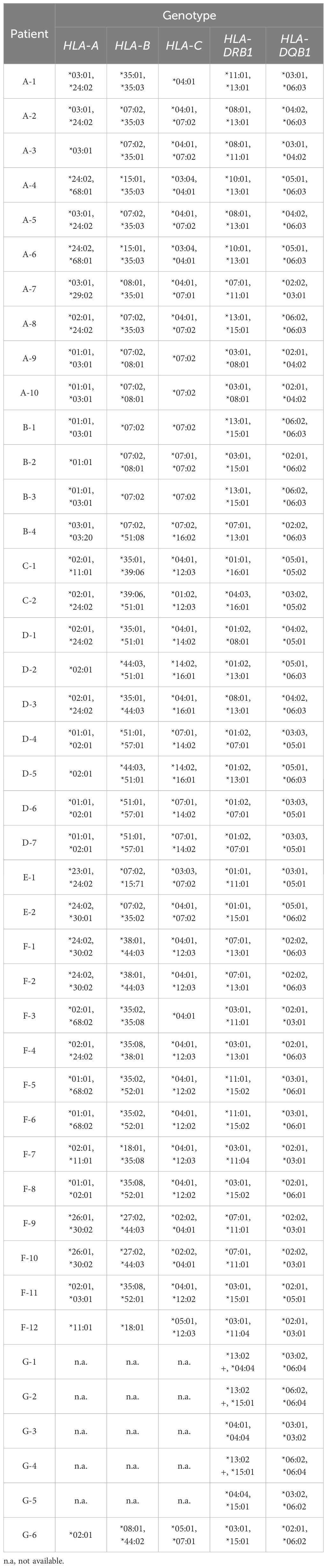
Table 3. HLA haplotype associations.
The odds ratio (OR) is widely used to quantify the association between an outcome and exposure to a factor. OR >1 indicates higher odds in the exposed individuals, implying increased risk of the outcome, while an OR <1 suggests lower odds, indicating protection. The 95% confidence interval (95% CI) estimates the precision of the OR and is often used as an indicator of statistical significance when it does not overlap the null value (OR= 1) (25). To further assess the potential association of single HLA allele frequencies with disease onset, severity or symptoms in CTLA-4 insufficiency, OR were derived from contingency tables, 95% CI were obtained with the adjusted Wald method for small sample size. Contingency tables containing zero values were adjusted using the Haldane and Anscombe correction (26). P-values were calculated using Fisher’s exact test without adjustment for multiple testing due to the exploratory nature of the study. However, Bonferroni-adjusted p-values are displayed in the association tables (Table 4; Supplementary Tables 1-6).
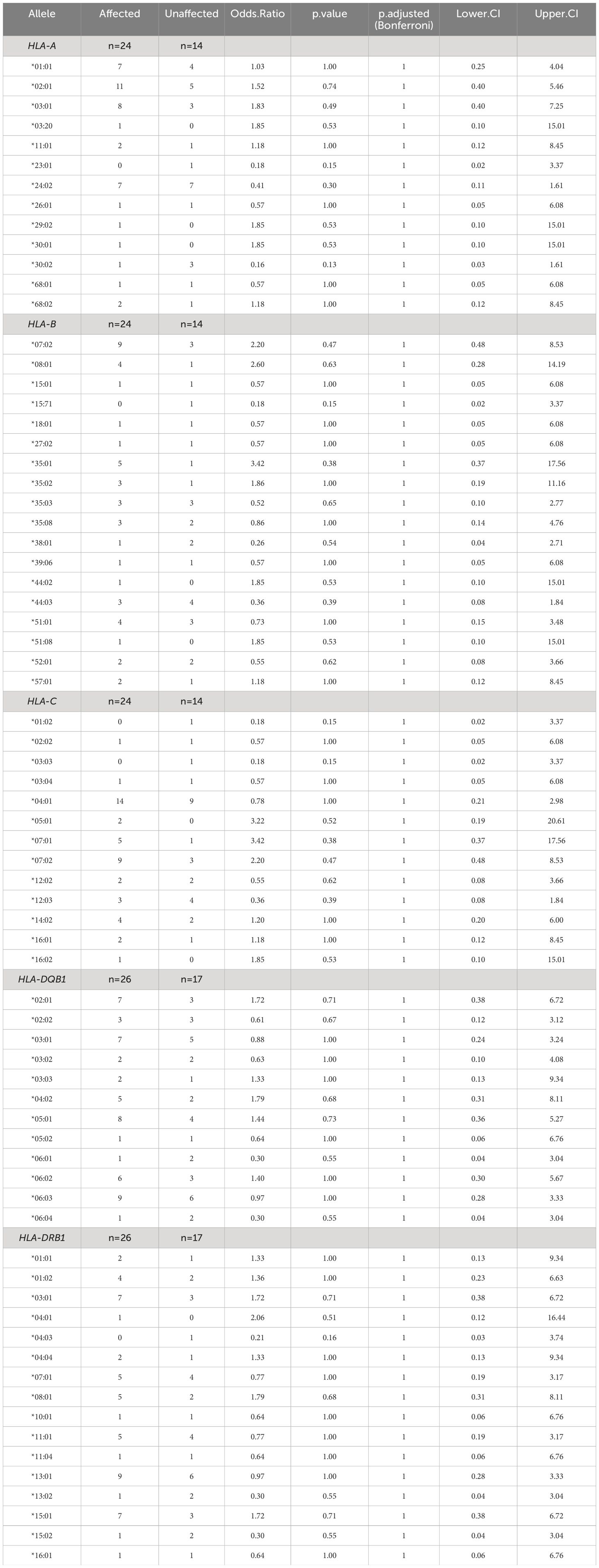
Table 4. Association of HLA-I and HLA-II alleles with affected status of CTLA4 mutation carriers.
No statistically significant difference in the odds of the outcome (affected status) between exposed and unexposed groups was observed for each HLA allele, with p-values > 0.05 (Table 4). To facilitate visualization and interpretation, OR and 95% CI were expressed on a logarithmic scale, which is symmetric around one (Figure 2). Positive values indicate positive associations (>0), and negative values indicate negative associations (<0). Similarly, OR analyses for disease severity showed no significant association with HLA alleles (Supplementary Figure 1, Supplementary Table 1). However, potential positive associations (raw p-values) of respiratory symptoms with HLA-DQB1*05:01 and HLA-DRB1*01:02 showed p-values <0.05 and 95% CI greater than one, indicating a trend that might be confirmed with increased sample size (Supplementary Table 2). HLA-DQB1*05:01 (OR 4.89, 95% CI: 1.14-17.63, p-value 0.04) was present in almost half of the individuals with respiratory involvement (8 out of 17 from five different families), while only 15% (4 out of 26 from four different families) individuals with no respiratory involvement carried this allele (Figure 3). HLA-DRB1*01:02 (OR 10.42, 95% CI: 1.09-51.43, p-value 0.03) was found in 29% (5 out of 17) of the subjects with respiratory manifestations but only in 4% (1 out of 26) of the subjects without respiratory symptoms (Figure 3). Noteworthy, all members carrying this HLA allele were from family D. A third HLA-allele, HLA-C*05:01, showed a positive association with neurological symptoms with a raw p-value of 0.04 and 28.64 odds ratio (95% CI: 1.42-180.9). Only seven patients exhibited neurological symptoms, from whom two, belonging to different families, carried this allele (Supplementary Table 3 and Figure 3). Finally, the HLA-DRB1*01:01 allele demonstrated a protective association with gastrointestinal symptoms (OR 0.1, 95% CI: 0.02-1.66, p-value 0.04, Supplementary Table 4). This allele was found in two affected individuals from two separate families, and one unaffected individual from one of these families. Importantly, none of the 24 individuals who experienced gastrointestinal symptoms carried this allele (Figure 3). While other associations were identified with p-values < 0.05, the confidence intervals overlapped the null value, and the alleles were present in only one individual or a single family. These associations included HLA-C*16:01 with respiratory tract involvement; HLA-A*29:02, HLA-A*30:01, and HLA-B*44:02 with neurological manifestations; HLA-DQB1*06:01 and HLA-DRB1*15:02 with gastrointestinal symptoms; HLA-A*29:02, HLA-A*30:01, HLA-B*44:02 and HLA-DRB1*04:01 with cytopenias (Supplementary Tables 2-6).
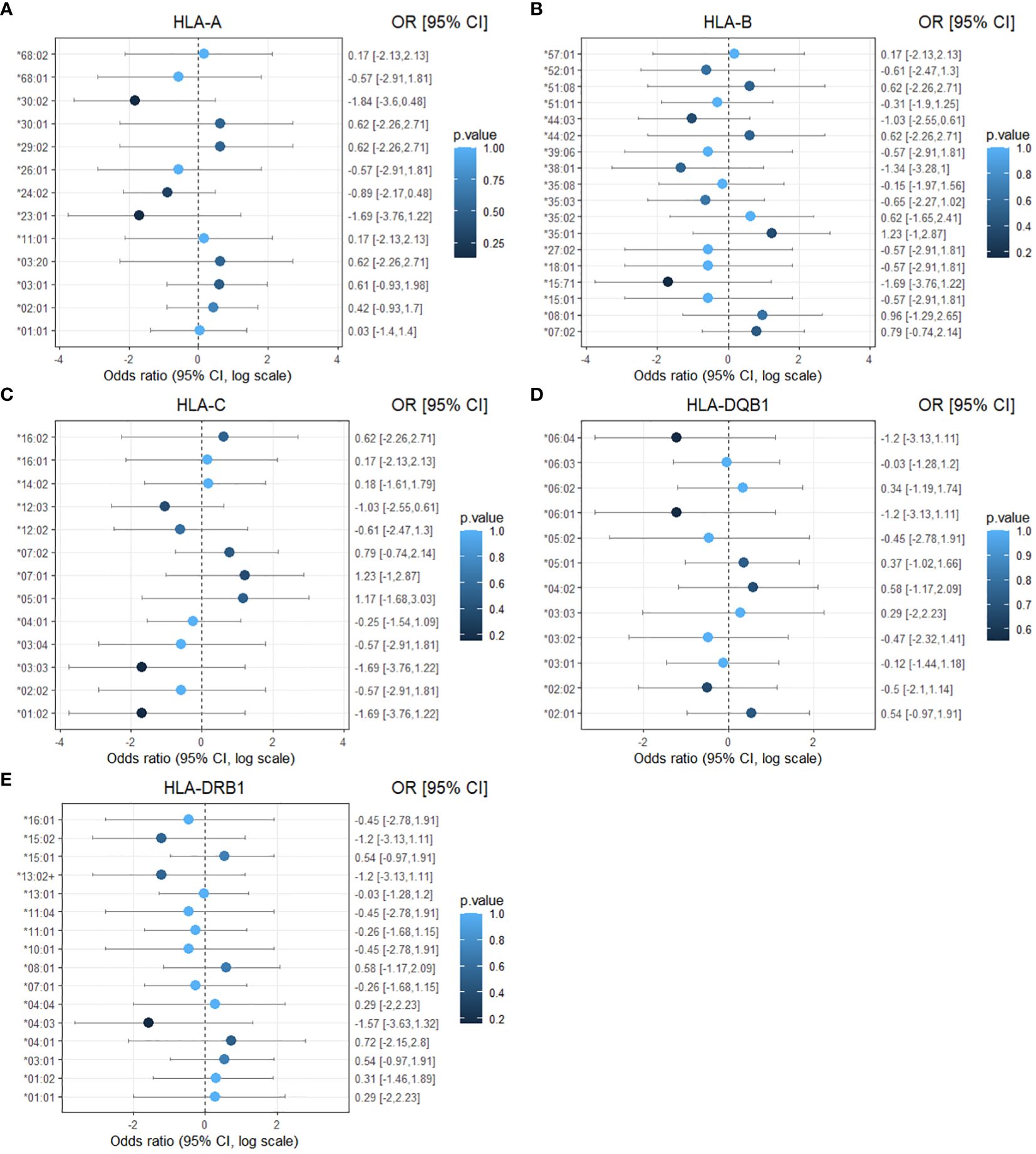
Figure 2. Forest plot of the association of HLA-I and HLA-II alleles with affected status of CTLA4 mutation carriers. The results are displayed separately for five classes of HLA alleles: (A) HLA-A (B) HLA-B (C) HLA-C (D) HLA-DQB1 (E) HLA-DRB1. Dots represent the odds ratio and continuous lines represent the 95% confidence intervals in a logarithmic scale. Calculations were performed using the Haldane and Anscombe correction, and the adjusted Wald method for the confidence intervals. p-values are represented as color intensity.

Figure 3. Clinical symptoms according to HLA class I or II in CTLA4 mutation carriers. Bar graphs depict number of individuals with presence (pink) or absence (blue) of lymphoproliferation, cytopenias, respiratory, gastrointestinal, dermatological or neurological symptoms specific to HLA type.
We did genetic linkage analysis to assess the possibility that there is linkage to the HLA locus in the absence of allele-specific association. Linkage without association can occur if different (HLA) alleles are co-inherited with the diseases in different families. The power to detect different LOD scores was calculated for four different settings of the frequency of the disease-associated allele at the modifier locus, using a single 5-allele marker locus (Table 5). While the power to detect a statistically significant LOD score > 3.0 is low for single markers, the power to detect a positive score > 1.0 is high and this can be boosted by using multi-marker analysis.
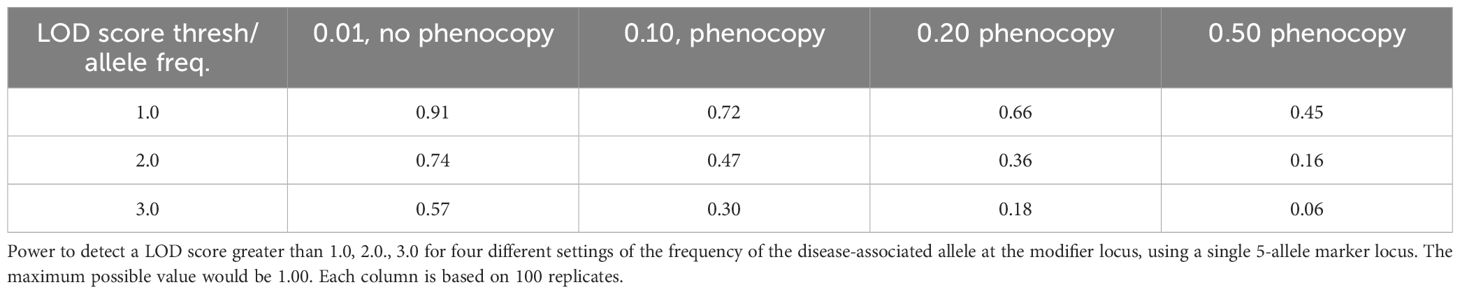
Table 5. Power to detect LOD scores.
All LOD scores were negative, indicating the absence of genetic linkage (Table 6). Therefore, for simplicity, we show the scores at a recombination fraction of 0 between the putative modifier locus and the marker locus. It is not surprising that the observed LOD scores for four out of five marker loci are almost identical because the marker loci are linked and hence have equivalent patterns of inheritance of the marker alleles. The HLA-C locus is less informative than the other four loci and hence the negative scores at HLA-C have a lower magnitude, but also indicate the absence of linkage.
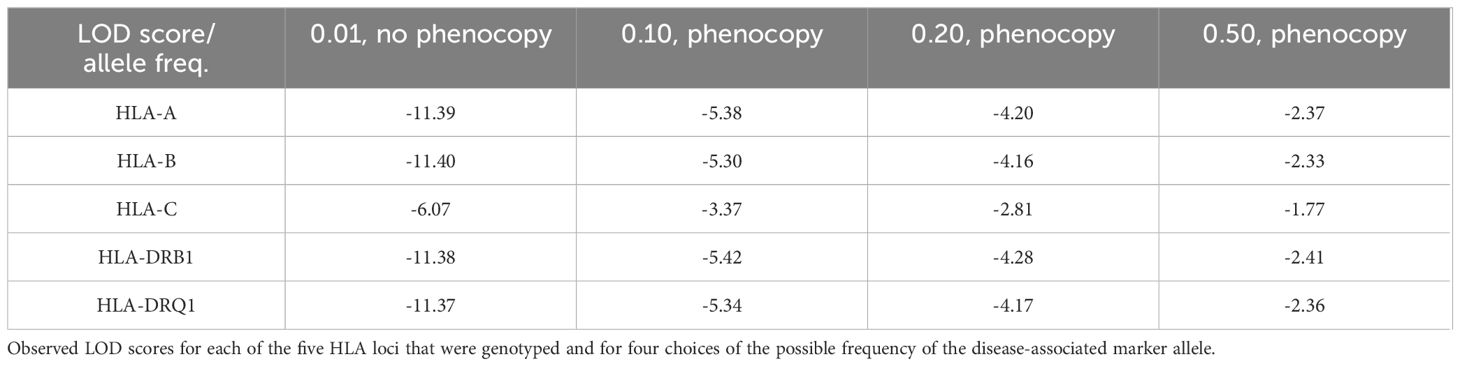
Table 6. Observed scores.
In this exploratory study, we evaluated the frequency of HLA class I and class II alleles as a potential genetic disease modifier factor in CTLA-4 insufficiency. In particular, we analyzed the frequency of HLA alleles A, B, C, DRB1, and DQB1 in 43 CTLA4 mutation carriers coming from seven different families. Our findings indicated no statistically significant associations between the frequency of HLA alleles and the disease onset, or the disease severity observed in individuals carrying genetic variants in CTLA4. However, we found potential positive associations of HLA-DQB1*05:01 and HLA-DRB1*01:02 alleles with respiratory tract involvement. It is noteworthy that all individuals with the HLA-DRB1*01:02 allele were from the same family, suggesting that other genetic or environmental factors may be influencing these findings. Importantly, HLA-DRB1*01:02 has been associated with susceptibility to rheumatoid arthritis (27), and HLA-DQB1*05:01 with a cluster of severe Guillain-Barré syndrome (28). Additionally, a potential positive association of HLA-C*05:01 with neurological involvement was observed, an allele associated with COVID-19 mortality (29). Interestingly, HLA-DRB1*01:01 showed a protective association with gastrointestinal symptoms. This allele has been reported to be protective for multiple sclerosis (30) and for the development of antibodies against administered factor VIII therapy in hemophilia A (31). Further investigation of HLA type I and II alleles in a larger sample size is warranted to confirm any significant correlation.
It is important to acknowledge various critical limitations in our study. First, the small number of CTLA-4 insufficient families analyzed resulted in a low statistical power in both, the single HLA allele frequencies association and the genetic linkage analysis methods, indicating that there is only a small chance of detecting a true effect of the HLA alleles. Furthermore, the odds ratio calculations did not account for the familial clustering of some individuals, which could have influenced the results. Second, the small number of HLA alleles analyzed in our patient cohort. HLA genes have a high number of polymorphisms that can be better detected using a genome wide association study (GWAS) instead of four-digit HLA typing. Finally, the lack of a long-term follow-up of the CTLA4 mutations carriers is a limitation, as they might change their disease status over time. However, it is worth noting that these constraints are common in studies involving rare diseases such as CTLA-4 insufficiency, where, to our knowledge, only 222 patients have been published to date (32) making these challenges exceedingly difficult to overcome.
To date, several additional modifiers in the context of CTLA-4 insufficiency have been suggested, but none has shown a positive association. For instance, infections with specific pathogens reported no difference between affected and unaffected CTLA4 mutation carriers, specifically for the seroprevalence for EBV (EBNA-1) IgG, CMV IgG, parvovirus B19 IgG, HSV-1/2 IgG and Toxoplasma gondii. Similar negative associations were found with EBV-specific IgG antibodies measured by FcgRIII/CD16A activation (8). Interestingly, HLA class II plays an important role in the entry of EBV into B cells. In fact, HLA-DQ isotypes show variable binding affinity for the EBV envelope glycoprotein gp42, supporting the association between susceptibility and resistance to EBV according to the HLA-DQ isotype (33). As previous reports revealed a particular susceptibility of CTLA-4 insufficient patients to EBV infections (34), we cannot exclude the hypothesis that interaction between a specific HLA type and an infectious pathogen trigger the disease onset or disease severity in CTLA-4 insufficiency.
Fifty years since the first report on the association between HLA-B and Hodgkin’s lymphoma, HLA alleles have been associated with a great number of human diseases, proving that HLA molecules are essential for protective immunity and for deleterious, disease-causing autoimmune reactivity (9). The current molecular understanding behind this association lays on two major mechanisms. The first one is the specificity of HLA-peptide-TCR interaction, which is governed by several mechanisms such as alternate TCR docking, low-affinity-mediated thymic escape, TCR stabilization of a weak peptide-HLA complexes, and altered presentation for peptides. The second major mechanism that might also generate and activate autoreactive T cells is epitope variation that includes, molecular mimicry, post-translational modification of antigenic peptides, generation of hybrid peptides, and differential HLA expression and stability (35). Both mechanisms generate autoreactive T cells that escape negative selection in the thymus and in the periphery, infiltrating organs and causing tissue damage. However, it remains to be determined whether these autoreactive T cells are a consequence of altered multiple mechanisms in a single patient, or whether certain molecular mechanisms are shared in a particular diseases, or in patient subgroups carrying the same HLA risk alleles. Moreover, it remains challenging to identify a particular self-peptide that becomes immunogenic during lifetime and causes activation of autoreactive T cells in a specific autoimmune response. Several technical approaches addressing this challenge have been developed, including the identification of clonally expanded and presumably pathogenic CD4+ or CD8+ T cells by single cell TCR analysis in the organ-infiltrated tissue, or characterization of the peptide recognition motif of the TCRs of interest by peptide library, or screening of the immunopeptidome. Analysis of the TCR repertoire of tissue-infiltrating T cells in CTLA-4 insufficient patients will be beneficial.
In addition to genetic disease triggers, several researchers have proposed the potential effect of non-genetic modifiers such as epigenetic changes or environmental factors like observed in other IEI. For instance, hypermethylation of various B-cell genes were found as a disease factor in a pair of monozygotic twins discordant for CVID (36). Moreover, Ramirez et al., identified different chromatin landscapes involving binding sites for transcription factors (TF) known for their importance in the germinal center reaction, in patients with diagnosis of CVID and mutations in TNFRSF13B from healthy donors (37). Gut dysbiosis is another possible disease modifier in CTLA-4 insufficiency. Several studies have shown a positive association between gut dysbiosis and systemic inflammation in several IEI, including chronic granulomatous disease (CGD), selective IgA deficiency, X-linked lymphoproliferative disease type-2 (XLP) and in CVID (38–40). It is thought that dysfunction of gut barriers due to damaged cell tight junctions, result in increased exposure of HLA to specific pathogens in the gut (41).
In conclusion, our results suggest that HLA-A, -B, -C, DRB1, DQB1 are unlikely to trigger the disease onset in CTLA-4 insufficient individuals and are not correlated with disease severity. However, our investigation raises the possibility that HLA-DQB1*05:01, HLA-DRB1*01:02, HLA-C*05:01 and HLA-DRB1*01:01 may influence the development of certain symptoms. It is important to note that this study is exploratory, and future studies with larger cohorts may either refute these findings or uncover other associations with specific clinical presentations. Additionally, further research, including epigenetic and microbiome analyses in a larger cohort of CTLA4 mutation carriers, will contribute to understand the pathogenicity of CTLA-4 insufficiency.
The datasets presented in this article are not readily available due to ethical and privacy restrictions. Requests to access the datasets should be directed to Ym9kby5ncmltYmFjaGVyQHVuaWtsaW5pay1mcmVpYnVyZy5kZQ==.
The studies involving humans were approved by Ethics Committee, Medical Centre - University of Freiburg, (registration number 466/18). The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants or the participants’ legal guardians/next of kin.
SP-C: Data curation, Formal analysis, Investigation, Methodology, Software, Visualization, Writing – original draft, Writing – review & editing. NM: Conceptualization, Data curation, Investigation, Methodology, Project administration, Writing – original draft, Writing – review & editing. FE: Investigation, Methodology, Writing – original draft, Writing – review & editing. VP: Data curation, Resources, Writing – original draft, Writing – review & editing. H-ML: Data curation, Resources, Writing – original draft, Writing – review & editing. ON: Data curation, Resources, Writing – original draft, Writing – review & editing. ID: Data curation, Resources, Writing – original draft, Writing – review & editing. KT: Data curation, Resources, Writing – original draft, Writing – review & editing. AAS: Formal analysis, Investigation, Methodology, Software, Writing – original draft, Writing – review & editing. BG: Conceptualization, Funding acquisition, Project administration, Resources, Supervision, Writing – original draft, Writing – review & editing. LG-D: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Software, Supervision, Visualization, Writing – original draft, Writing – review & editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by the funding of the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany´s Excellence Strategy (CIBSS – EXC-2189 Project ID 390939984), and by the collaborative research center SFB 403222702 (SFB 1381/2) to LG-D and SFB1160/3_B5 to BG. In addition, BG received funding from the E-rare program of the EU managed by the DFG, grant code GR1617/14-1/iPAD; and by the German Federal Ministry of Education and Research (BMBF) through a grant to the German Auto-Immunity Network (GAIN), grant code 01GM1910A. LG-D received also funding from the Hans A. Krebs Medical Scientist Program from the Faculty of Medicine, University of Freiburg. This work was supported in part by the Center for Chronic Immunodeficiency (CCI), Freiburg Center for Rare Diseases (FZSE) and by the Intramural Research Program of the National Institutes of Health, NCI. Some samples have been taken from the CCI-biobank, a partner of the Freeze Biobank Freiburg. The article processing charge was funded by the Baden-Wuerttemberg Ministry of Science, Research and Art and the University of Freiburg in the funding program Open Access Publishing.
We would like to thank all patients, family members, and physicians for their participation in this study. We also thank Pavla Mrovecova from our laboratory for her support in sample management and DNA extraction, and Derek Hazard for his valuable advising in biostatistics from the Institute of Medical Biometry and Statistics from the University Medical Center Freiburg.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2024.1447995/full#supplementary-material
CHAI, CTLA-4 haploinsufficiency; CVID, common variable immunodeficiency; CTLA-4, cytotoxic T-lymphocyte antigen 4; EBV, Epstein Barr virus; HLA, human leukocyte antigen; IEI, inborn errors of immunity; LOD, logarithm of odds; MHC, major histocompatibility complex; OR, odds ratio; TCR, T-cell receptor.
1. Bretscher PA. A two-step, two-signal model for the primary activation of precursor helper T cells. Proc Natl Acad Sci U S A. (1999) 96:185–90. doi: 10.1073/pnas.96.1.185
2. Qureshi OS, Kaur S, Hou TZ, Jeffery LE, Poulter NS, Briggs Z, et al. Constitutive clathrin-mediated endocytosis of CTLA-4 persists during T cell activation. J Biol Chem. (2012) 287:9429–40. doi: 10.1074/jbc.M111.304329
3. Schubert D, Bode C, Kenefeck R, Hou TZ, Wing JB, Kennedy A, et al. Autosomal dominant immune dysregulation syndrome in humans with CTLA4 mutations. first report on human CTLA-4 insufficiency. Nat Med. (2014) 20:1410–6. doi: 10.1038/nm.3746
4. Kuehn HS, Ouyang W, Lo B, Deenick EK, Niemela JE, Avery DT, et al. Immune dysregulation in human subjects with heterozygous germline mutations in CTLA4. first report on human CTLA-4 insufficiency. Science. (2014) 345:1623–7. doi: 10.1126/science.1255904
5. Mitsuiki N, Schwab C, Grimbacher B. What did we learn from CTLA-4 insufficiency on the human immune system? Immunol Rev. (2019) 287:33–49. doi: 10.1111/imr.12721
6. Egg D, Schwab C, Gabrysch A, Arkwright PD, Cheesman E, Giulino-Roth L, et al. Increased risk for Malignancies in 131 affected CTLA4 mutation carriers. Front Immunol. (2018) 9:2012. doi: 10.3389/fimmu.2018.02012
7. Schwab C, Gabrysch A, Olbrich P, Patiño V, Warnatz K, Wolff D, et al. Phenotype, penetrance, and treatment of 133 cytotoxic T-lymphocyte antigen 4-insufficient subjects. first report on long-term follow-up about human CTLA-4 insufficiency. J Allergy Clin Immunol. (2018) 1932–46. doi: 10.1016/j.jaci.2018.02.055
8. Krausz M, Mitsuiki N, Falcone V, Komp J, Posadas-Cantera S, Lorenz HM, et al. Do common infections trigger disease-onset or -severity in CTLA-4 insufficiency? Front Immunol. (2022) 13:1011646. doi: 10.3389/fimmu.2022.1011646
9. Sic H, Speletas M, Cornacchione V, Seidl M, Beibel M, Linghu B, et al. An activating janus kinase-3 mutation is associated with cytotoxic T lymphocyte antigen-4-dependent immune dysregulation syndrome. Front Immunol. (2017) 8:1824. doi: 10.3389/fimmu.2017.01824
10. Turnbull C, Bones J, Stanley M, Medhavy A, Wang H, Lorenzo AMD, et al. DECTIN-1: A modifier protein in CTLA-4 haploinsufficiency. Sci Adv. (2023) 9:eadi9566. doi: 10.1126/sciadv.adi9566
11. Choo SY. The HLA system: genetics, immunology, clinical testing, and clinical implications. Yonsei Med J. (2007) 48:11–23. doi: 10.3349/ymj.2007.48.1.11
12. Hosomichi K, Shiina T, Tajima A, Inoue I. The impact of next-generation sequencing technologies on HLA research. J Hum Genet. (2015) 60:665–73. doi: 10.1038/jhg.2015.102
13. Prinz JC. Immunogenic self-peptides - the great unknowns in autoimmunity: Identifying T-cell epitopes driving the autoimmune response in autoimmune diseases. Front Immunol. (2022) 13:1097871. doi: 10.3389/fimmu.2022.1097871
14. Forcione DG, Sands B, Isselbacher KJ, Rustgi A, Podolsky DK, Pillai S. An increased risk of Crohn's disease in individuals who inherit the HLA class II DRB3*0301 allele. Proc Natl Acad Sci U.S.A. (1996) 93:5094–8. doi: 10.1073/pnas.93.10.5094
15. Bos WH, van de Stadt LA, Sohrabian A, Ronnelid J, van Schaardenburg D. Development of anti-citrullinated protein antibody and rheumatoid factor isotypes prior to the onset of rheumatoid arthritis. Arthritis Res Ther. (2014) 16:405. doi: 10.1186/ar4511
16. Vorechovsky I, Webster AD, Plebani A, Hammarstrom L. Genetic linkage of IgA deficiency to the major histocompatibility complex: evidence for allele segregation distortion, parent-of-origin penetrance differences, and the role of anti-IgA antibodies in disease predisposition. Am J Hum Genet. (1999) 64:1096–109. doi: 10.1086/302326
17. De La Concha EG, Fernandez-Arquero M, Martinez A, Vidal F, Vigil P, Conejero L, et al. , et al. HLA class II homozygosity confers susceptibility to common variable immunodeficiency (CVID). Clin Exp Immunol. (1999) 116:516–20. doi: 10.1046/j.1365-2249.1999.00926.x
18. Lathrop GM, Lalouel JM, Julier C, Ott J. Strategies for multilocus linkage analysis in humans. Proc Natl Acad Sci USA. (1984) 81:3443–6. doi: 10.1073/pnas.81.11.3443
19. Schaffer AA, Gupta SK, Shriram K, Cottingham RW Jr. Avoiding recomputation in linkage analysis. Hum Hered. (1994) 44:225–37. doi: 10.1159/000154222
20. Cottingham RW Jr., Idury RM, Schaffer AA. Faster sequential genetic linkage computations. Am J Hum Genet. (1993) 53:252–63.
21. Schaffer AA, Lemire M, Ott J, Lathrop GM, Weeks DE. Coordinated conditional simulation with SLINK and SUP of many markers linked or associated to a trait in large pedigrees. Hum Hered. (2011) 71:126–34. doi: 10.1159/000324177
22. Krausz M, Uhlmann A, Rump IC, Ihorst G, Goldacker S, Sogkas G, et al. The ABACHAI clinical trial protocol: Safety and efficacy of abatacept (s.c.) in patients with CTLA-4 insufficiency or LRBA deficiency: A non controlled phase 2 clinical trial. Contemp Clin Trials Commun. (2022) 30:101008. doi: 10.1016/j.conctc.2022.101008
23. Hou TZ, Olbrich P, Soto JML, Sanchez B, Sanchez Moreno P, Borte S, et al. Study of an extended family with CTLA-4 deficiency suggests a CD28/CTLA-4 independent mechanism responsible for differences in disease manifestations and severity. case report on CTLA-4 insufficiency (included in 133 cohort). Clin Immunol. (2018) 188:94–102. doi: 10.1016/j.clim.2018.01.001
24. Gonzalez-Galarza FF, McCabe A, Santos E, Jones J, Takeshita L, Ortega-Rivera ND, et al. Allele frequency net database (AFND) 2020 update: gold-standard data classification, open access genotype data and new query tools. Nucleic Acids Res. (2020) 48:D783–8. doi: 10.1093/nar/gkz1029
26. Haldane JB. The estimation and significance of the logarithm of a ratio of frequencies. Ann Hum Genet. (1956) 20:309–11. doi: 10.1111/j.1469-1809.1955.tb01285.x
27. Roark CL, Anderson KM, Aubrey MT, Rosloniec EF, Freed BM. Arthritogenic peptide binding to DRB1*01 alleles correlates with susceptibility to rheumatoid arthritis. J Autoimmun. (2016) 72:25–32. doi: 10.1016/j.jaut.2016.04.006
28. Schirmer L, Worthington V, Solloch U, Loleit V, Grummel V, Lakdawala N, et al. Higher frequencies of HLA DQB1*05:01 and anti-glycosphingolipid antibodies in a cluster of severe Guillain-Barre syndrome. J Neurol. (2016) 263:2105–13. doi: 10.1007/s00415-016-8237-6
29. Sakuraba A, Haider H, Sato T. Population difference in allele frequency of HLA-C*05 and its correlation with COVID-19 mortality. Viruses. (2020) 12:1333. doi: 10.3390/v12111333
30. Mamedov A, Vorobyeva N, Filimonova I, Zakharova M, Kiselev I, Bashinskaya V, et al. Protective allele for multiple sclerosis HLA-DRB1*01:01 provides kinetic discrimination of myelin and exogenous antigenic peptides. Front Immunol. (2019) 10:3088. doi: 10.3389/fimmu.2019.03088
31. Hosseini S, Arabi S, Yari F, Pourfatollah A, Rezaie N, Moazezi S, et al. HLA-DRB1*01:01, but not HLA-DRB1:1503 or HLA-DRB1*11, is associated with decreased inhibitor risk in Iranian hemophilia A patients. Transfus Apher Sci. (2019) 58:669–73. doi: 10.1016/j.transci.2019.08.019
32. Jamee M, Hosseinzadeh S, Sharifinejad N, Zaki-Dizaji M, Matloubi M, Hasani M, et al. Comprehensive comparison between 222 CTLA-4 haploinsufficiency and 212 LRBA deficiency patients: a systematic review. Clin Exp Immunol. (2021) 205:28–43. doi: 10.1111/cei.13600
33. Sathiyamoorthy K, Jiang J, Hu YX, Rowe CL, Möhl BS, Chen J, et al. Assembly and architecture of the EBV B cell entry triggering complex. PloS Pathog. (2014) 10:e1004309. doi: 10.1371/journal.ppat.1004309
34. Rush-Kittle J, Gamez-Diaz L, Grimbacher B. Inborn errors of immunity associated with defects of self-tolerance checkpoints: The CD28 family. Pediatr Allergy Immunol. (2022) 33:e13886. doi: 10.1111/pai.13886
35. Dendrou CA, Petersen J, Rossjohn J, Fugger L. HLA variation and disease. Nat Rev Immunol. (2018) 18:325–39. doi: 10.1038/nri.2017.143
36. Rodriguez-Cortez VC, Del Pino-Molina L, Rodriguez-Ubreva J, Ciudad L, Gómez-Cabrero D, Company C, et al. Monozygotic twins discordant for common variable immunodeficiency reveal impaired DNA demethylation during naive-to-memory B-cell transition. Nat Commun. (2015) 6:7335. doi: 10.1038/ncomms8335
37. Ramirez N, Posadas-Cantera S, Langer N, Caballero Garcia de Oteyza A, Proietti M, Keller B, et al. Multi-omics analysis of naive B cells of patients harboring the C104R mutation in TACI. Front Immunol. (2022) 13:938240. doi: 10.3389/fimmu.2022.938240
38. Chandrasekaran P, Han Y, Zerbe CS, Heller T, DeRavin SS, Kreuzberg SA, et al. Intestinal microbiome and metabolome signatures in patients with chronic granulomatous disease. J Allergy Clin Immunol. (2023) 152:1619–1633 e11. doi: 10.1016/j.jaci.2023.07.022
39. Catanzaro JR, Strauss JD, Bielecka A, Porto AF, Lobo FM, Urban A, et al. IgA-deficient humans exhibit gut microbiota dysbiosis despite secretion of compensatory IgM. Sci Rep. (2019) 9:13574. doi: 10.1038/s41598-019-49923-2
40. Sharma M, Dhaliwal M, Tyagi R, Goyal T, Sharma S, Rawat A. Microbiome and its dysbiosis in inborn errors of immunity. Pathogens. (2023) 12:518. doi: 10.3390/pathogens12040518
Keywords: cytotoxic T-lymphocyte antigen 4 (CTLA-4), immune dysregulation, inborn errors of immunity (IEI), disease modifiers, human leukocyte antigen (HLA), genetic linkage analysis
Citation: Posadas-Cantera S, Mitsuiki N, Emmerich F, Patiño V, Lorenz H-M, Neth O, Dybedal I, Taskén K, Schäffer AA, Grimbacher B and Gámez-Díaz L (2025) The effect of HLA genotype on disease onset and severity in CTLA-4 insufficiency. Front. Immunol. 15:1447995. doi: 10.3389/fimmu.2024.1447995
Received: 12 June 2024; Accepted: 12 July 2024;
Published: 06 January 2025.
Edited by:
Leif Gunnar Hanitsch, Charité University Medicine Berlin, GermanyReviewed by:
Julian Joseph Bosco, The Alfred Hospital, AustraliaCopyright © 2025 Posadas-Cantera, Mitsuiki, Emmerich, Patiño, Lorenz, Neth, Dybedal, Taskén, Schäffer, Grimbacher and Gámez-Díaz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Laura Gámez-Díaz, bGF1cmEuZ2FtZXpAdW5pa2xpbmlrLWZyZWlidXJnLmRl; Bodo Grimbacher, Ym9kby5ncmltYmFjaGVyQHVuaWtsaW5pay1mcmVpYnVyZy5kZQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.