 Huige Li
Huige Li Ning Xia
Ning Xia- Department of Pharmacology, University Medical Center, Johannes Gutenberg University, Mainz, Germany
Recent evidence suggests that adaptive immune cells are important contributors to metabolic dysfunction–associated steatotic liver disease (MASLD, formerly non-alcoholic fatty liver disease, NAFLD). In liver biopsies from MASLD patients, the accumulation of intrahepatic B cells is positively correlated with the MASLD activity score. Hepatic B-cell infiltration is observed in experimental models of metabolic dysfunction-associated steatohepatitis (MASH, formerly non-alcoholic steatohepatitis, NASH). Intrahepatic B2 cells have been shown to contribute to MASLD/MASH by activating T cells, macrophages and hepatic stellate cells, and by producing pathogenic IgG antibodies. In mice fed a MASH diet, selective depletion of B2 cells reduces steatohepatitis and fibrosis. Intestinal B cells are metabolically activated in MASH and promote T-cell activation independently of TCR signaling. In addition, B cells have been shown to contribute to liver fibrosis by activating monocyte-derived macrophages through the secretion of IgA immunoglobulins. Furthermore, our recent study indicates that certain B cell subsets, very likely regulatory B cells, may play a protective role in MASLD. This review summarizes the molecular mechanisms of B cell functions and discusses future research directions on the different roles of B cells in MASLD and MASH.
1 Introduction
With worldwide increasing prevalence of obesity, metabolic dysfunction–associated steatotic liver disease (MASLD, formerly non-alcoholic fatty liver disease, NAFLD) has become a global health problem affecting up to one-third of the world’s adult populations (1–3). Metabolic dysfunction-associated steatohepatitis (MASH, formerly non-alcoholic steatohepatitis, NASH) is the aggressive form of MASLD and characterized by steatosis, inflammation, hepatocellular injury, and fibrosis. MASH can progress to cirrhosis and/or hepatocellular carcinoma and has become the leading cause of liver-related morbidity and mortality worldwide, and a primary indication for liver transplantation (4, 5).
As the population ages and the duration of exposure to the disease increases, the majority of models predict a significant rise in the prevalence of MASLD-related cirrhosis and end-stage liver disease (6). MASLD is the most rapidly growing contributor to liver-related mortality and morbidity (1). Thus, there is an urgent need to develop new, effective pharmacological therapies for MASH/MASLD and its complications.
Inflammation is key in the progression from hepatic lipid accumulation to the more severe MASH (7). In fact, the onset of the inflammatory response is an early event that precedes the progression toward MASH (8). Importantly, MASLD is a progressive disease. Although immune mechanisms contribute to all stages of MASLD, the immune response may differ in the initial phase of lipid accumulation than in the stage of MASH. For instance, CD8+ T cells promote metabolic dysregulation in the early stage of steatosis whereas they cause hepatocyte death at later stages (9, 10).
Immune responses are a major part of the inflammatory process in MASH (9). Both innate and adaptive immune cells are implicated in the pathogenesis of MASLD (11). The innate immune response in MASH has been the focus of research and it is well established that innate immune cells make a major contribution to inflammation in MASH (12–14). In addition to Kupffer cells, further innate immune cells involved in MASLD include neutrophils, monocytes/macrophages, dendritic cells, natural killer T cells, mucosal-associated invariant T cells, and innate lymphoid cells (ILCs) (11, 13). Especially monocyte-derived macrophages (MoMFs) can directly activate hepatic stellate cells (HSC) and are of particular importance in the context of liver injury and fibrosis in MASH (15, 16). ILCs represent the most recently discovered innate immune cell family and consists of 5 subsets: natural killer (NK) cells, type 1, 2 and 3 ILCs and lymphoid tissue inducer cells (17, 18). Liver type ILC1s are expanded in cirrhotic livers of patients (19) and their roles in liver disease are subject of current research (11, 17). Nevertheless, growing evidence indicates that adaptive immune cells also play an essential role in MASLD (11, 13, 14, 20). The present review article focuses on the role of B lymphocytes.
2 General mechanisms of B cell development, activation and function
2.1 B cell development
During early fetal development, human B-lineage cells are found in various fetal tissues. However, starting from midgestation, bone marrow becomes the sole site for B lymphopoiesis (21). In the bone marrow, common lymphoid progenitor (CLP) cells can differentiate into early-pro-B cells, and then into pro-B cells. A process of gene recombination occurs through the rearrangement of genes coding for segments V (Variable), D (Diversity), and J (Joining) of the heavy chain (H chain) together with that of the genes for segments V and J of the light chain (L chain) of the membrane-bound immunoglobulin (mIg). The early-pro-B cells are typically characterized by DJH rearrangement initiation, and pro-B cells by VDJH rearrangement. A proper VDJH rearrangement is necessary for pro-B cells to differentiate into pre-B cells. Pro-B cells without functional VDJH rearrangement will cease to develop, either undergoing apoptosis or being phagocytized by bone marrow macrophages (21). Pro-B cells express no surface immunoglobulin molecules, whereas pre-B cells express the so-called surrogate light chains paired with the µH chain forming the pre-BCR on the membrane (22). The pre-BCR is essential for pre-B cell proliferation and further differentiation into immature B cells expressing IgM as the complete BCR (22).
Bone marrow-derived immature B cells can mature in bone marrow, or migrate from the bone marrow to the spleen, and differentiate, through transitional stages of maturation (T1 and T2 B cells), into either follicular B cells or marginal zone B cells expressing IgM and IgD. Now they are considered mature B cells or naïve B cells (23).
2.2 B cell activation
Naïve B cells get activated in secondary lymphoid organs (spleen, lymph nodes, Peyer’s patches, tonsils, and mucosal tissue) by antigen sensing via the B cell receptor (BCR) (24). In addition to BCR, complex antigens also engage co-receptors on B cells, such as toll-like receptors (TLRs) or complement receptors (CR2), leading to potentiation of the BCR signal. If the signaling via co-receptors is strong, B cells start to proliferate without the need for T cell help, resulting in a T cell-independent type 1 response. Alternatively, B cells can be activated via a T cell-independent type 2 response, if the BCR stimulation by highly multivalent antigens alone is strong enough (24). These T-independent processes can be enhanced by cytokines like BAFF and APRIL (24).
T cell-dependent antigens such as foreign proteins usually activate B cells with the help of T cells (24). After binding to BCR, the antigen is taken up by B cells, degraded, and presented on the cell membrane as peptide pieces in complex with MHC-II (25). The complex is recognized by T helper cells, and bound by T cell receptor (TCR), leading to expression CD40 ligand (CD40L) on the T cell surface. CD40L binds CD40 on B cell surface providing a co-stimulation for B cell activation. In addition, T helper cells secrete cytokines, such as IL-4 and IL-21, promoting B cell proliferation, class switch recombination, and differentiation into antibody-producing cells or germinal center B cells (24).
2.3 B cell function
Class switch recombination allows activated B cells to change the isotype of their BCR from IgM and IgD to IgG, IgA, or IgE, leading to the generation of antibody-secreting B cells (plasmablasts and plasma cells) (22, 24, 25). Antibodies can mediate their effector functions by binding to Fc receptors on target cells or by activating the complement system (24). In addition to antibody production, B cells also function via antigen presentation, cell-cell interaction, and cytokine secretion (22).
3 Genetic mouse models of B cell-deficiency
To study the role of B cells, both therapeutic and genetic approaches are available. Using an anti-IgM depleting antibody, 90-95% mature B cells can be deleted (26). However, plasma cells are not depleted by this antibody. The approach applying anti-CD20 antibodies is more efficient, resulting in 95% B cell depletion (27). Anti-CD20 antibodies don’t deplete plasma cells either, as CD20 is not expressed on plasma cells (28). In contrast, complete B cell-deficiency can be reached in genetically modified mice. Currently, there are several B cell-deficient mouse lines used in research (Table 1).
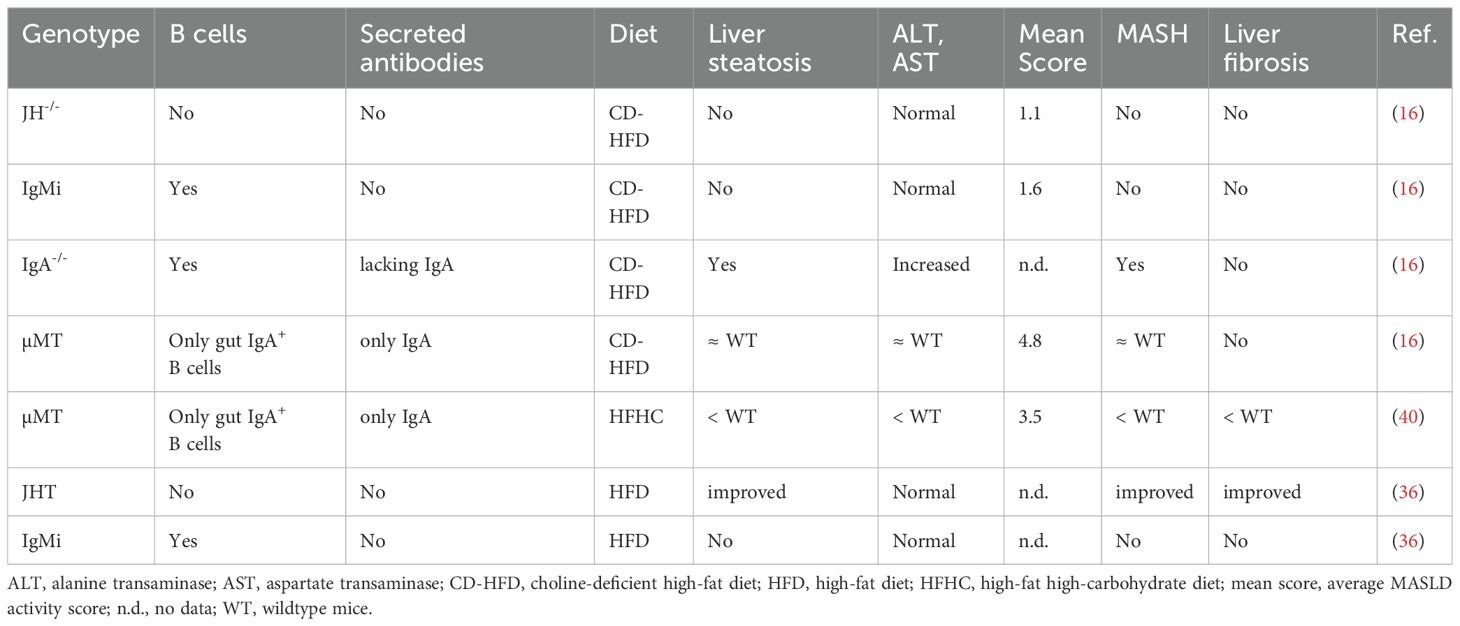
Table 1. Selected findings in genetically modified mice.
3.1 The µMT mice
The µMT mice were generated by disrupting one of the membrane exons of the gene encoding the µ-chain constant region (29). µ-chain is the heavy chain of IgM antibodies and the heavy chain of the pre-BCR expressed on the membrane of developing B cells. This pre-BCR is essential for pre-B cell differentiation. Homozygous mice with µ-chain gene disruption lack immature and mature B cells. The mutants have large pre-B cells, but not small pre-B cells, indicating that B cell development is arrested at the stage of pre-B cell maturation (29). Although BALB/c mice with the μMT mutation are “leaky” and generate small numbers of mature B cells, this phenomenon is not observed in mutant mice on the C57BL/6 background (28, 30). Interestingly, µMT mice develop IgA+ B cells in the absence of IgM, independent of genetic background (31). IgA+ cells are found in the spleen and ileum, with no other B cell populations in µMT spleen or bone marrow. It is known that IgD can substitute for IgM in early phase of B cell development. However, the generation of IgA+ B cells in µMT mice is independent of IgM or IgD (31). Rather, it requires the specialized nature of the gut, where extrafollicular B cells are the major producers of IgA (32). It is proposed that some of the early B cell precursors leave the bone marrow and receive their switching signals (TGF-β and gut commensal bacterial antigens) in gut lymphoid tissues such as Peyer’s patches, and thus bypassing the requirement for IgM or IgD expression (31, 32).
3.2 The JHT mice
The JHT mice were generated by deleting the loci for the heavy chain joining region (JH) and the Eµ intron enhancer (JH–Eµ) of the IgH gene using a Cre-loxP recombination system (33). The rearrangement of the VH-DH-JH rearrangement requires the presence of both JH and Eµ (34). As a result, the formation of heavy chain V region genes through somatic VH-DH-JH rearrangement is impossible in the JHT mice. The mutant mice cannot make functional immunoglobulin molecules and the animals are completely avoid of mature B cells in all tested genetic background (28, 33). Without membrane-bound immunoglobulin molecules, the development of B cells is blocked at the pre-B cell stage in the homozygous mice, because pre-BCR is essential for B cell development (29, 33).
3.3 The JH-/- mice
The JH-/- mice were generated at a different laboratory (34). In these mutant mice, the JH gene segments (but not the Eµ intron enhancer) in the IgH locus are deleted. As a result, B cell development is blocked at the precursor stage, with increased number of CD43+ pro-B and pre-B cells in the bone marrow, but lacking the more mature CD43- pro-B cells. The homozygous mutant mice show a complete absence of mature B cells in the bone marrow, spleen or peripheral blood (34). The IgH gene assembly is initiated by joining of a DH to a JH gene segment, followed by rearrangement of a VH to the DJH complex (34). In the homozygous mutant mice with JH gene segment deletion, however, the VH to D-JH joining doesn’t occur, indicating that D-JH rearrangement is required for VH to D-JH joining, and JH gene segment essential for IgH gene assembly (34). As mentioned above, pro-B cells cannot develop further without functional VDJH rearrangement (21).
3.4 The IgMi mice
The IgMi mice were generated by deleting the polyA responsible for the secreted form of antibodies, but allowing the expression of immunoglobulin molecules on B cell surface (i.e. BCR). Two polyA sites are present in the constant region coding for the heavy chain of IgM. The first polyA site is essential for the maturation of the RNA for the secreted form of the IgM heavy chain, whereas the second polyA site is responsible for the maturation of the RNA for membrane-form of the IgM heavy chain. In the IgMi mice, only the first polyA site is deleted while the second polyA site is kept intact. As a result, these mice express IgM as BCR on the B cell surface enabling B cell development. However, class switch recombination is impossible in these B cells and the cells cannot differentiate into antibody-producing cells. The IgMi mice represent a unique tool to distinguish the B cell roles as antibody producer from other B cell functions (28, 35). The IgMi mice don’t secrete antibodies, but they have normal B cell number of B cells (36, 37), produce cytokines and their adaptive immune responses remain largely intact (16).
4 Intrahepatic B2 cells in MASLD and MASH
Lymphocyte infiltration is one of the histological features of MASLD. In murine models of MASLD, B cells are among the first to accumulate in response to high-fat diet (HFD) feeding (38, 39). In liver biopsies from MASLD patients, intrahepatic B cell accumulation positively correlates with the MASLD activity score (40). In patients with MASH, focal aggregates of B cells and T cells are formed, which resemble ectopic lymphoid structures (13, 41). These aggregates’ size and frequency show a direct association with lobular inflammation and fibrosis scores (41). Hepatic infiltration by B cells is also evident in various experimental models of MASH (41, 42).
B cells are among the most frequent immune cell types in the liver and comprise as much as 20% - 30% of all CD45+ immune cells in the mouse liver under normal chow conditions (40), although much fewer B cells are found in human liver (12, 43). In response to high-fat high-carbohydrate (HFHC) diet feeding, the percentage of B cells among the immune cells remains unchanged, but the total number of immune cells increases significantly. As a result, the absolute number of B cells in the mouse liver almost doubles after 15 weeks of HFHC (40). The majority of the intrahepatic B cells are IgM+ IgD+ B2 cells (CD3− NK1.1− CD19+ CD23+ CD5− B220hi) producing TNF-α and IL-6, and the number of B2 cells is increased in MASH mice (40). Consistently, intrahepatic B cell accumulation is also evident in liver biopsies from MASLD patients (40).
4.1 Activation
T cell-independent mechanism seems to play an important role in the activation of intrahepatic B cells during MASLD/MASH. It is likely that gut-derived microbial antigens or gut microbiota metabolites, such as LPS, activate B cell by binding to BCR and TRL4 simultaneously (40). TLR4 activation in MASH is not only limited to Kupffer cells or hepatocytes (44), but B cells can respond to pathogen-derived ligands through TLR4 signaling as well (45). TRL4 serves as a co-receptor for B cell activation through the MYD88 pathway (40). Consistently, B cell–specific depletion of MyD88 ameliorates MASH progression (40). T-independent B cell-activation can be enhanced by cytokines like BAFF and APRIL (24). Indeed, BAFF levels are increased in MASH patients (46). B cells from HFHC-fed mice show MyD88-dependent upregulation of MHC I, MHC II and CD86, indicating activation and increased capability of antigen presentation (40).
The effects of B cells can be mediated by interaction with other cell types, by secretion of cytokines, or by antibody production. Indeed, B cells contribute to MASLD and MASH through multiple mechanisms (Figure 1).
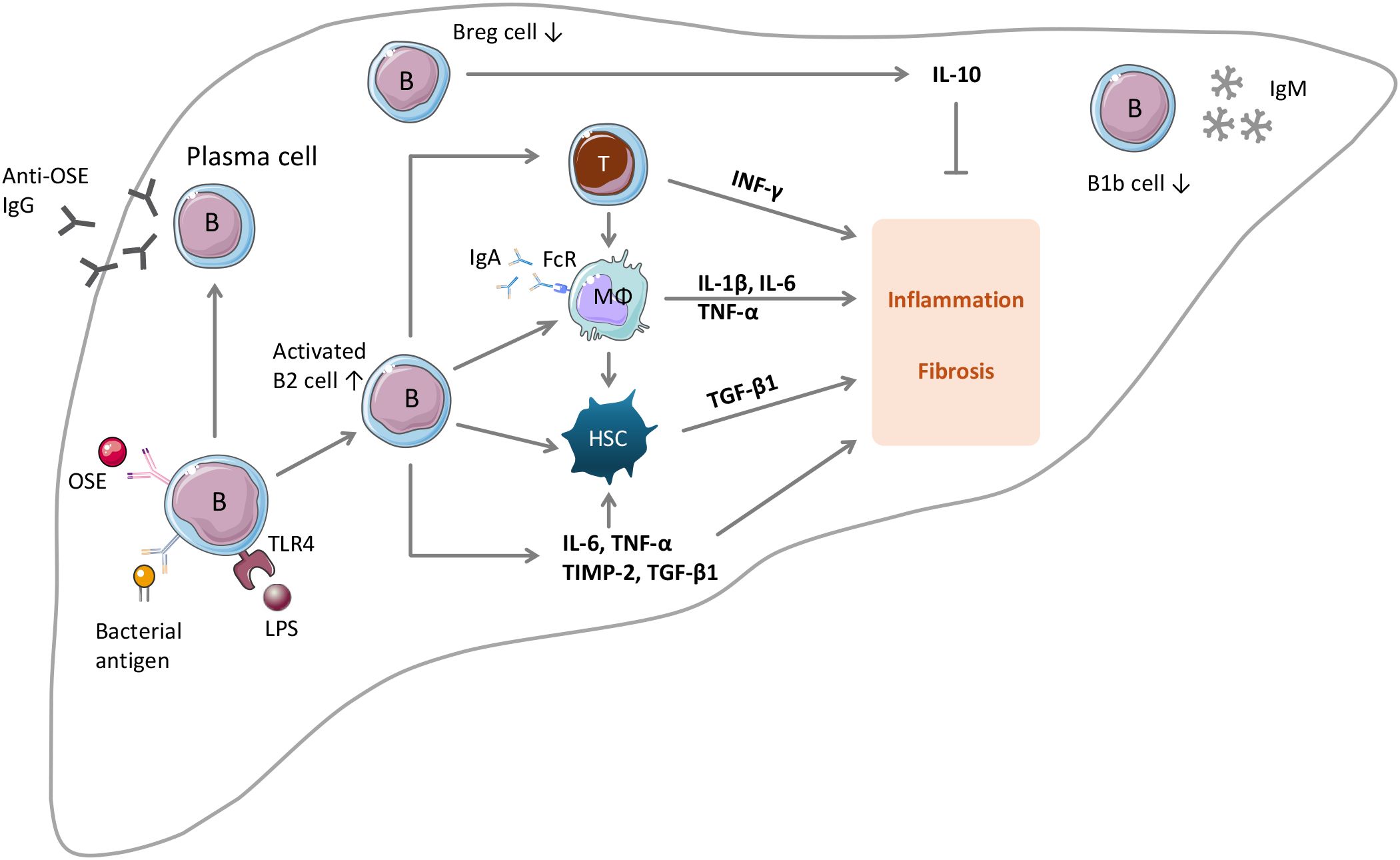
Figure 1. The roles of B lymphocytes in MASLD and MASH. Upon stimulation by OSE, B cells can develop into IgG-producing plasma cells. Intrahepatic B cells can be activated by gut bacterial antigens or by gut microbiota metabolites such as LPS. Under the conditions of MASLD and MASH, the number of B2 cells in the liver increases, whereas the numbers of B1b cells and regulatory B cells (Breg) decrease. Activated B2 cells promote MASLD and MASH through multiple mechanisms: (i) activation of CD4 and CD8 T cells by antigen presentation and by secreting IL-6 and TNFα; (ii) activation of HSC by releasing TNFα; (iii) activation of macrophages in an IgA- and Fc receptor-dependent manner; (iv) by producing pro-inflammatory and profibrotic factors; (v) by releasing anti-OSE IgG antibodies. IL-10 produced by Breg cells has protective effects. The role of B1b cells is unknown.
4.2 Interaction with T cells
The principal T lymphocytes participating in adaptive immunity comprise CD4+ T cells, CD8+ T cells, and γδ T cells. CD4+ T cells can be further classified into subsets such as T helper (Th) 1, Th2, Th17, regulatory T (Treg) cells. Th1 and Th17 subsets promote MASLD progression by producing IFN-γ and IL-17, whereas the roles of Th2 and Treg are incompletely understood (11). Blocking CD4+ T cell recruitment to the liver protects mice from MASLD, supporting the crucial role of these cells (47). In contrast, the role of CD8+ T cells in MASLD pathogenesis varies depending on the context (11). Under the condition of MASH, CD8+ T cells can become auto-aggressive, killing hepatocytes in a MHC-I-independent manner (48). On the other hand, tissue-resident memory CD8+ T cells have been shown to promote the resolution of hepatic inflammation and fibrosis by causing apoptosis of HSCs (49).
The effects of B lymphocytes in MASLD are partly mediated by interaction with T cells.
In patients with MASH, liver biopsies demonstrate the presence of B-cell aggregates within T-cell-rich regions, indicating a potential interaction between these cell types (41). In steatosis (38) and MASH (40), intrahepatic B cells secrete elevated levels of IL-6 and TNFα, which promote the activation of CD4 cells and their differentiation into Th1 cells.
In mouse models of MASH, B-cell activation parallels the onset of steatohepatitis (41). Moreover, the B2 cell response precedes T-cell activation and is accompanied by upregulation of the hepatic expression of BAFF, a cytokine driving B cell survival and maturation. The circulating levels of BAFF are increased in MASH patients, and correlates with the severity of steatohepatitis and fibrosis (46). BAFF-deficient mice are partially protected from MASH (50). Treatment with the BAFF-neutralizing monoclonal antibody Sandy-2 prevents hepatic B2 cell responses and ameliorates established MASH in mice (41). Selective depletion of B2 cells in mice reduces steatohepatitis and fibrosis in mice fed MASH-diets (41).
Theoretically, it requires three signals for B cells to activate T cells: (i) antigen presentation to T cell receptor thereby promoting T cell proliferation and differentiation; (ii) co-stimulatory signals stabilizing the immunological synapse between B cells and T cells, and (iii) cytokine released by B cells to shape the polarization of T cells towards an effector phenotype (51). Indeed, the activation of B cells in MASH is associated with an upregulation of MHC-I and MHC-II indicating enhanced antigen-presenting ability (40, 41). The protein antigens are likely derived from gut bacteria, which are processed by B cells as professional antigen presenting cells. The resulting peptides are presented to T cells by forming complexes with MHC molecules. In addition, the activated B cells show also enhanced expression of CD86 (40, 41). CD86 binding with CD28 on T cells represents the most important costimulatory interaction (51). Finally, activated liver B cells release TNFα and IL-6 (40), further stimulating T cell activation and polarization.
As a result, intrahepatic B cells promote T-cell activation during MASH and stimulate the generation of effector memory (EM) CD4 and CD8 T cells, which promotes inflammation during MASLD (40, 52). Compared to HFHC wildtype mice, livers from HFHC µMT mice show reduced percentages of EM CD4 and CD8 T cells and IFN-γ+ CD4 T cells. Similar effect can be achieved by anti-CD20 antibody-mediated B cell-depletion (40). Interestingly, the capability to activate liver T cells is likely to be restricted to intrahepatic B cells but not systemic B cells, as shown in experiments with HFHC-fed µMT recipients reconstituted with B cells from either the liver or spleen of HFHC-fed wildtype donor mice. Compared with recipients of spleen B cells, recipients of liver B cells showed worsened response in glucose tolerance test and insulin tolerance test, and increased frequency of IFN-γ+ CD4 T cells in the liver (40). These results indicate that the pro-inflammatory phenotype of B cells results from a local activation and is not a systemic event (38, 40).
B cell-mediated Th1 activation of CD4 T cells also plays an important role in M1 activation of hepatic macrophages in MASH. CD4 T-cell depletion in MASH mice significantly lowers the hepatic mRNA expression of macrophage M1 markers (42).
4.3 Interaction with macrophages
Macrophages are key players in MASLD development and progression (4). Liver macrophages represent a heterogeneous cell population that can be divided into two main categories: tissue-resident Kupffer cells and monocyte-derived macrophages (MoMFs) (4, 14). Kupffer cells initiate the inflammatory response in the liver and recruit monocytes to the liver. The recruited monocytes differentiate into pro-inflammatory macrophages, contributing to MASLD progression and fibrosis development (4). The pro-fibrotic effects of B2 cells involve the activation of liver macrophages and HSCs (40, 53). In MASLD, macrophages can be activated by a number of factors, including endotoxins and bacterial components due to increased intestinal permeability, factors released by damaged or dying hepatocytes, and nutritional lipids and their metabolites (4). Macrophage activation by B cells involves IgA and Fc receptor signaling (see below).
Activated macrophages promote MASLD progression by releasing proinflammatory cytokines and recruiting further immune cells (4). TNF and IL-1β are key cytokines in MASLD, driving the development of steatosis, inflammation and fibrosis, and the majority TNF and IL-1β molecules in the liver are derived from activated macrophages (54). In addition, macrophages also activate HSCs and myofibroblasts by releasing IL-6 and TGF-β, and are an important source of oxidative stress in MASLD (55). Finally, both Kupffer cells and infiltrating macrophages make a major contribution to MASLD development by recruiting neutrophils, B and T cells (56, 57).
4.4 Interaction with hepatic stellate cells
The activation of quiescent HSCs is a key event in liver fibrogenesis (58). Activated HSCs differentiate into myofibroblasts and contribute to fibrosis by secreting large amounts of collagen and profibrotic factors (53, 59). Interestingly, there exists a positive feedback mechanism in B cell-HSC interaction. Activated B2 cells strongly induce HSC activation by secreting pro-inflammatory cytokines including TNF-α; which is known to have a key role in promoting fibrosis (60). B cells are strong producers of TNF-α (40), and TNFα activates HSC by stimulating TNF-α receptor 1 (61). In turn, activated HSCs support liver B cell survival and maturation to plasma cells as well as B cell cytokine production by secreting retinoid acid (53).
4.5 Cytokine production
It has been known since the 1980s that B cells can be secrete a wide variety of cytokines (62, 63). Cytokine production by B cells is context-dependent; different stimuli lead to the production of different cytokines, with some being pro-inflammatory and others with anti-inflammatory properties (62). In steatosis (38) and MASH (40), intrahepatic B cells secrete elevated levels of IL-6 and TNF-α, which promote the activation of CD4 T cells and their differentiation into Th1 cells. As mentioned above, B cell-derived TNF-α is also involved in HSC activation (40), and TNF-α activates HSC by stimulating TNF-α receptor 1 (61). In addition to stimulating other immune cells, B cells also directly promote disease progression by releasing pro-fibrotic factors such as TGF-β1 and TIMP2 (40). TIMP2 is known to promote liver fibrosis through inhibition of extracellular matrix degradation (64).
4.6 IgG antibodies
Oxidative stress and lipid peroxidation are common characteristics of MASLD and MASH. Lipid peroxidation leads to the production of oxidized phospholipids and reactive aldehydes, including malondialdehyde (MDA) and 4-hydroxynonenal (4-HNE). These molecules activate hepatic inflammation by acting as danger-associated molecular patterns. In addition, they also form antigenic adducts with cellular macromolecules known as oxidation-specific epitopes (also called oxidative stress-derived epitopes, OSEs) (13, 14, 65, 66). MASLD patients have reduced levels of protective IgM natural antibodies against OSEs (see section 7) (67). In addition, elevated titers of pathogenic anti-OSE IgG have been detected in adult MASLD patients and in children with MASH (68). Especially, Anti-MDA IgG antibodies are associated with MASLD/MASH progression (68). High titers of anti-OSE IgG are associated with the severity of lobular inflammation and the prevalence of intrahepatic B/T cell aggregates (68), and are independently associated with increased risk of hepatic decompensation and mortality in MASH patients (69). Anti-OSE IgG is also an independent risk factor for MASH progression to fibrosis (68). In animal experiments, the increase in titers of anti-OSE IgG is associated with the maturation of liver B2 cells to plasma cells (41, 42), whereas reducing lipid peroxidation by supplementation with N-acetylcysteine attenuates steatohepatitis progression in a rat model (70). Conversely, immunization of MASH mice with MDA-adducted proteins stimulates IgG production and enhances transaminase release, lobular inflammation and fibrosis (42). It is likely that the resulting anti-MDA IgG antibodies promote MASH progression by inducing antibody-mediated injury (42).
5 Intestinal IgA+ B cells
In a recent study (16), strikingly different results were obtained in JH-/- and µMT mice. The findings demonstrate that intestine B-cell responses are sufficient to induce MASH in mice (16).
In wildtype mice, choline-deficient high-fat diet (CD-HFD) induced MASH phenotypes. In contrast, JH-/- mice were protected from CD-HFD-induced MASH, with reduced liver steatosis, lower MASLD activity score, normalized ALT levels and decreased hepatic lipid accumulation (16). In addition, fewer CD3+ T cells, CD8+ T cells, activated T cells (CD8+ CD62L-), TNF (CD8+ TNF+)- and IFNγ (CD8+ IFNγ+)-producing CD8+ cells were found in JH-/- CD-HFD livers than in wildtype mice (16). To corroborate the results from these B cell-deficient mice, the authors depleted B cells therapeutically by using an anti-CD20 monoclonal antibody (αCD20 mAb) in wildtype MASH mice. The results from both the genetical and the therapeutical approaches were very similar and indicate a key role of B cell-response in the development of MASH.
Further remarkable results were obtained in µMT mice. These mice selectively develop IgA+ B cells in the gastrointestinal tract, but lacking B cells in the circulation or other organs (16, 31). Strikingly, CD-HFD-induced MASH levels in µMT mice were comparable to that in wildtype mice. CD-HFD µMT mice showed similar MASLD activity score, serum ALT levels, and hepatic CD8 cells accumulation as wildtype CD-HFD mice.
JH-/- mice are a complete B cell-knockout model. In contrast, µMT mice have IgA+ B cells (only) in the intestine (see section 3.1). In wildtype mice, CD-HFD feeding led to an increase in IgA levels in the small intestine. Interestingly, IgA (although at lower levels) were present in the small intestine of µMT CD-HFD but not JH-/- CD-HFD mice. IgM and IgG2b were absent in both µMT and JH-/- mice (16). Importantly, MASH in µMT mice could be reversed by αCD20 antibody treatment, indicating that the susceptibility of µMT mice to MASH development was attributable to IgA+ B cells in the intestine. Indeed, MASH diet-induced accumulation of IgA+ B cells in small intestines was reduced by αCD20 antibody treatment (16).
Mechanistic data reveal that intestine B cells contribute to MASH by inducing CD8+ T cell-hyperactivation. Upon MASH diet intervention, intestinal B cells cluster with CD8+ T cells by a direct, antigen-independent cell-cell interaction stabilized by the co-stimulatory molecules LFA-1 and ICAM1. Moreover, intestinal B and T cells can efficiently migrate from the intestine to the liver. Thus, intestinal B cells are key players in MASH development by promoting T cell activation and transmigration to the liver. Another mechanism of B cells in MASH involves the secretion of IgA (16).
Normally, B cells in the gut-associated lymphoid tissues (GALTs) can be activated by protein antigens derived from gut microbiota bacteria with the help of CD4 T cells, leading to the generation of plasmablasts and plasma cells. In addition, T cell-independent activation by carbohydrate/polysaccharide antigens is also possible (71, 72). The intestinal B cells in the µMT mice on CD-HFD, however, are not plasmablasts or plasma cells (16). Increased intestinal B cells were also observed in CD-HFD-treated germ-free mice (16), indicating that intestinal B cells promote MASH independent of gut bacterial antigens. Rather, the intestinal B cells were activated metabolically carrying enhanced expression of gene related to BCR signaling, B cell-activation and lipid metabolism (16).
6 IgA
IgA is unique among all antibodies because it is produced to be secreted on the mucosal surface (73). As much as several grams of IgA are released into the human gut lumen everyday (71, 72). In the intestine, B cells are activated in the lamina propria via T cell-dependent (stimulated by bacterial antigens) or -independent mechanisms leading to the generation of IgA-producing plasma cells (71, 72). In addition, maternal milk IgA and liver-derived IgA also contribute to gut IgA pool (72). Gut IgA plays a key role in regulating microbiota composition and controlling enteric infections (71–73). The serum level of IgA is higher than that of IgM but much lower than that of IgG (74).
Elevated serum IgA levels have been demonstrated in patients with MASLD (72, 75, 76), and have been identified as an independent predictor of advanced fibrosis (75). MASLD is also accompanied by an accumulation of liver-resident IgA-producing plasma cells (9, 76, 77). However, serum IgA levels do not correlate with MASLD severity in pediatric patients (78). It is still unclear whether this discrepancy is due to the difference in the microbiota composition between children and adults or other reasons.
A recent study has provided evidence showing that IgA can promote the initiation of liver fibrosis by driving the activation of MoMFs in the liver (16).
CD-HFD-induced liver fibrosis is absent in IgA-/- mice lacking secreted IgA (79) or in wildtype mice treated with αCD20 antibody, indicating that the presence of B cells and an intact IgA secretion in the periphery are obligatory for fibrosis development. Moreover, the degree of liver fibrosis correlates with serological IgA levels in several diet-induced MASH models (16).
Macrophages play a crucial role in liver injury and fibrosis in MASH, with MoMFs exhibiting a proinflammatory phenotype that can directly activate HSCs (15). IgA drives MoMF activation via Fc receptor signaling. Diet-induced MASH in wildtype mice is associated with an increased level of MoMFs, and MoMFs are associated in number with the degree of fibrosis. Interestingly, liver MoMFs, but not other immune cell populations, have strongly upregulated FcR genes and genes involved in FcR signaling activation in all tested MASH models (16). Conversely, FcRγ-/- mice on CD-HFD diet develop steatosis, liver damage, and inflammation comparable to wildtype mice, yet liver fibrosis is markedly reduced in these animals (16). Ex vivo experiments demonstrate that serum IgA from MASH mice triggers macrophage activation and induction of profibrogenic genes (IL-1β, IL-6 and TNF) in a manner dependent on Fc receptor signaling (16).
Notably, Fc receptors are also expressed on other immune cells including NK cells (71). Therefore, future studies should investigate whether Fc-mediated mechanisms by IgA also applies to other immune cells.
7 B1 cells and IgM
In addition to B2 cells, a small fraction of B1 cells is also found in the mouse liver (40, 80). These cells are predominantly B1b cells (CD3− NK1.1− CD19+ CD23− CD5− B220lo IgM+ IgD−), whereas CD5+ B1a cells are almost undetectable (40). Upon HFHC feeding, the number of B1b cells is reduced in the MASH mice (40).
B1 cells are named as such because they are derived from the fetal origins and their development is earlier than that of the B2 cells in the bone marrow (81). B1 cells represent the majority of B cell populations in the peritoneal and pleural cavities of mice and their number is maintained by self-renewal. If activated by LPS, IL-5 or IL-10, B1 cells migrate from the body cavities to the spleen or lymph nodes, starting to secrete cytokines or natural IgM antibodies without the need of foreign antigen exposure (81).
Although B1 cells differentiate “spontaneously” into natural antibody-producing cells without requiring foreign antigens or CD4 T cell help, immunization with antigens can lead to dynamic expansion of B1 cell repertoire through BCR-mediated mechanisms (81). The immunization of mice with heat-inactivated pneumococci results in the enhanced production of natural IgM antibodies against bacterial antigens that cross-react with oxidized phosphatidylcholine in LDL molecules (82). The engulfment of oxidized LDL by Kupffer cells is known to promote Kupffer cell activation and hepatic inflammation. Thus, the interaction of IgM with oxidized LDL results in a reduction in their uptake by Kupffer cells, thereby lowering the pro-inflammatory stimuli (83). This may explain why the immunization-induced production of natural IgM leads to a reduction of MASH in LDL receptor-deficient mice receiving a high-fat/cholesterol diet (82).
In patients with MASLD, IgM antibody titers towards different OSE (including MDA and malondialdehydeacetaldehyde) are lower than in healthy control individuals (67). Moreover, the data suggest that obesity/hyperlipidemia is a causal factor for the reduced IgM levels (67). Nevertheless, more studies are needed to decipher the roles of B1 cells and IgM antibodies in MASLD and MASH (65).
8 Regulatory B cells
Notably, the same mouse line can exhibit quite different phenotypes when put on different MASLD- or MASH-inducing diets (Table 1). For example, the MASH phenotype of µMT mice fed CD-HFD is comparable with wildtype mice (16). On HFHC diet, in contrast, µMT mice develop markedly less MASH than wildtype mice (40). Similar situation has been observed for the IgMi mice. In the MASH model induced by CD-HFD, IgMi mice are equally protected from MASH as JH-/- mice (16). In the HFD-induced obesity model, however, we have observed significant differences between IgMi and JHT mice (36).
As detailed in section 3, JHT and JH-/- mice are completely devoid of mature B cells, whereas IgMi mice represent a model of antibody-deficiency. The IgMi mice have normal number of B cells (36, 37), producing cytokines but not secreted antibodies (28, 35). Moreover, the adaptive immune responses remain largely intact (16).
In a recent publication, we put wildtype mice, JHT and IgMi mice on HFD for 21 weeks (36). In wildtype mice, HFD feeding resulted in MASLD with massive macrovesicular steatosis, modest hepatic and adipose tissue inflammation, and incipient liver fibrosis. JHT mice showed reduced HFD-induced MASLD, confirming the pathogenic role of B cells in MASLD (36). Strikingly, the IgMi mice were completely protected from the development of hepatic steatosis, inflammation, and fibrosis. Our study indicates that B cells may also have protective roles, in addition to the pathogenic effects in MASLD.
The common feature of JHT and IgMi mice is that they do not secrete antibodies, whereas HFD feeding in wildtype mice led to increased levels of serum IgG2c (36). The lack of pathogenic antibodies (IgA and IgG) should protect JHT and IgMi mice to a similar extent. Whereas JHT mice have no mature B cells at all, the number of B cells in the IgMi mice remains normal. The fact that the IgMi mice are more protected than the JHT mice indicates that the additional protection observed in the IgMi mice may be attributable to the presence of certain B cell subsets. The results from our study indicate that regulatory B cells (Breg) are likely a candidate subset for these protective B cell populations.
Breg cells represent a minor B cell population with immunomodulatory functions by producing IL-10, IL-35 and TGF-β (51, 84). Although the existence of Breg cells is beyond doubt, it remains difficult to define and identify Breg populations (51). In addition to the IL-10-producing CD1dhighCD5+ cells, various other Breg populations have been reported (23, 51). Until now, there is no unified transcription factor or lineage-specific marker defined for Breg cells, and the identification of Breg cells by surface markers has been controversial (23, 51). Production of IL-10 is considered the major characteristics for the discrimination of Breg from conventional B populations (51).
In our study, CD1dhighCD5+CD19+B220+ Breg cells were found in the liver of both wildtype and IgMi mice. HFD feeding reduced Breg cell number and IL-10 production in the liver of wildtype mice, whereas Breg cell number and IL-10 expression were increased in HFD-fed IgMi mice. Among the CD1dhighCD5+ Breg cells, the number of CD5+ IL-10high cells almost doubled in IgMi mice on HFD. Moreover, livers of patients with advanced liver fibrosis showed abundant deposition of IgG and stromal B cells and low numbers of IL-10 expressing cells, supporting our mouse data (36).
These results suggest that B lymphocytes have both detrimental and protective effects in HFD-induced MASLD. IL-10-producing regulatory B cells may represent such a protective B cell subset. Nevertheless, evidence for a causal role of Breg cells is still missing (36). The cytokine IL-10 (62, 85, 86) is a key anti-inflammatory mediator produced not only by Breg cells, but also by various cell types, including B1 and B effector (Be) Be2 cells (62, 81). Therefore, more experimental evidence is needed to verify the identity of the protective B cell subset, and the cellular source of IL-10 in the MASLD liver.
9 Conclusion and future directions
Accumulating evidence supports the pathogenic role of B cells in MASLD and MASH. B2 cells can be activated by oxidation-specific epitopes, by gut microbiota metabolites or bacterial protein antigens. Activated B2 cells promote MASLD and MASH by activating T cells, macrophages, hepatic stellate cells, by producing pathogenic IgG and IgA antibodies, and by releasing pro-inflammatory, pro-fibrotic factors. In addition, there is emerging evidence for a protective role of certain B cell subsets in MASLD and MASH. Nevertheless, more studies are warranted to verify the identity of these cells and the detailed mechanisms involved. Future studies should explore the therapeutic potential of approaches targeting B2 cells or the downstream mechanisms. Conversely, approaches that enhance the protective effects of Breg cells, B1 cells and natural IgM antibodies may also be promising.
Author contributions
HL: Conceptualization, Funding acquisition, Writing – original draft. NX: Conceptualization, Funding acquisition, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. Original works from the authors’ laboratory contributing to this review were supported by a research grant (to HL and NX) from the Boehringer Ingelheim Foundation for the CardioConsortium “Novel and neglected cardiovascular risk factors: molecular mechanisms and therapeutic implications” at the University Medical Center of the Johannes Gutenberg University, Mainz, Germany.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Targher G, Byrne CD, Tilg H. Nafld and increased risk of cardiovascular disease: clinical associations, pathophysiological mechanisms and pharmacological implications. Gut. (2020) 69:1691–705. doi: 10.1136/gutjnl-2020-320622
2. Riazi K, Azhari H, Charette JH, Underwood FE, King JA, Afshar EE, et al. The prevalence and incidence of nafld worldwide: A systematic review and meta-analysis. Lancet Gastroenterol Hepatol. (2022) 7:851–61. doi: 10.1016/S2468-1253(22)00165-0
3. Duell PB, Welty FK, Miller M, Chait A, Hammond G, Ahmad Z, et al. Nonalcoholic fatty liver disease and cardiovascular risk: A scientific statement from the american heart association. Arterioscler Thromb Vasc Biol. (2022) 42:e168–e85. doi: 10.1161/ATV.0000000000000153
4. Kazankov K, Jorgensen SMD, Thomsen KL, Moller HJ, Vilstrup H, George J, et al. The role of macrophages in nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Nat Rev Gastroenterol Hepatol. (2019) 16:145–59. doi: 10.1038/s41575-018-0082-x
5. Wei G, An P, Vaid KA, Nasser I, Huang P, Tan L, et al. Comparison of murine steatohepatitis models identifies a dietary intervention with robust fibrosis, ductular reaction, and rapid progression to cirrhosis and cancer. Am J Physiol Gastrointest Liver Physiol. (2020) 318:G174–G88. doi: 10.1152/ajpgi.00041.2019
6. Estes C, Razavi H, Loomba R, Younossi Z, Sanyal AJ. Modeling the epidemic of nonalcoholic fatty liver disease demonstrates an exponential increase in burden of disease. Hepatology. (2018) 67:123–33. doi: 10.1002/hep.29466
7. Barrow F, Revelo XS. The B side of B cells in nafld. Hepatology. (2022) 76:914–6. doi: 10.1002/hep.32481
8. Schuster S, Cabrera D, Arrese M, Feldstein AE. Triggering and resolution of inflammation in nash. Nat Rev Gastroenterol Hepatol. (2018) 15:349–64. doi: 10.1038/s41575-018-0009-6
9. Huby T, Gautier EL. Immune cell-mediated features of non-alcoholic steatohepatitis. Nat Rev Immunol. (2022) 22:429–43. doi: 10.1038/s41577-021-00639-3
10. Peiseler M, Schwabe R, Hampe J, Kubes P, Heikenwalder M, Tacke F. Immune mechanisms linking metabolic injury to inflammation and fibrosis in fatty liver disease - novel insights into cellular communication circuits. J Hepatol. (2022) 77:1136–60. doi: 10.1016/j.jhep.2022.06.012
11. Sawada K, Chung H, Softic S, Moreno-Fernandez ME, Divanovic S. The bidirectional immune crosstalk in metabolic dysfunction-associated steatotic liver disease. Cell Metab. (2023) 35:1852–71. doi: 10.1016/j.cmet.2023.10.009
12. MacParland SA, Liu JC, Ma XZ, Innes BT, Bartczak AM, Gage BK, et al. Single cell rna sequencing of human liver reveals distinct intrahepatic macrophage populations. Nat Commun. (2018) 9:4383. doi: 10.1038/s41467-018-06318-7
13. Sutti S, Albano E. Adaptive immunity: an emerging player in the progression of nafld. Nat Rev Gastroenterol Hepatol. (2020) 17:81–92. doi: 10.1038/s41575-019-0210-2
14. Barrow F, Khan S, Wang H, Revelo XS. The emerging role of B cells in the pathogenesis of nafld. Hepatology. (2021) 74:2277–86. doi: 10.1002/hep.31889
15. Wen Y, Lambrecht J, Ju C, Tacke F. Hepatic macrophages in liver homeostasis and diseases-diversity, plasticity and therapeutic opportunities. Cell Mol Immunol. (2021) 18:45–56. doi: 10.1038/s41423-020-00558-8
16. Kotsiliti E, Leone V, Schuehle S, Govaere O, Li H, Wolf MJ, et al. Intestinal B cells license metabolic T-cell activation in nash microbiota/antigen-independently and contribute to fibrosis by iga-fcr signalling. J Hepatol. (2023) 79:296–313. doi: 10.1016/j.jhep.2023.04.037
17. Bourinet M, Anty R, Gual P, Luci C. Roles of innate lymphoid cells in metabolic and alcohol-associated liver diseases. JHEP Rep. (2024) 6:100962. doi: 10.1016/j.jhepr.2023.100962
18. Kramer B, Nalin AP, Ma F, Eickhoff S, Lutz P, Leonardelli S, et al. Single-cell rna sequencing identifies a population of human liver-type ilc1s. Cell Rep. (2023) 42:111937. doi: 10.1016/j.celrep.2022.111937
19. Jaeger N, Antonova AU, Kreisel D, Roan F, Lantelme E, Ziegler SF, et al. Diversity of group 1 innate lymphoid cells in human tissues. Nat Immunol. (2024) 25:1460–73. doi: 10.1038/s41590-024-01885-y
20. Deng CJ, Lo TH, Chan KY, Li X, Wu MY, Xiang Z, et al. Role of B lymphocytes in the pathogenesis of nafld: A 2022 update. Int J Mol Sci. (2022) 23:12376. doi: 10.3390/ijms232012376
22. LeBien TW, Tedder TF. B lymphocytes: how they develop and function. Blood. (2008) 112:1570–80. doi: 10.1182/blood-2008-02-078071
23. Sanz I, Wei C, Jenks SA, Cashman KS, Tipton C, Woodruff MC, et al. Challenges and opportunities for consistent classification of human B cell and plasma cell populations. Front Immunol. (2019) 10:2458. doi: 10.3389/fimmu.2019.02458
24. Cyster JG, Allen CDC. B cell responses: cell interaction dynamics and decisions. Cell. (2019) 177:524–40. doi: 10.1016/j.cell.2019.03.016
25. Nutt SL, Hodgkin PD, Tarlinton DM, Corcoran LM. The generation of antibody-secreting plasma cells. Nat Rev Immunol. (2015) 15:160–71. doi: 10.1038/nri3795
26. Cerny A, Sutter S, Bazin H, Hengartner H, Zinkernagel RM. Clearance of lymphocytic choriomeningitis virus in antibody- and B-cell-deprived mice. J Virol. (1988) 62:1803–7. doi: 10.1128/JVI.62.5.1803-1807.1988
27. Ahuja A, Shupe J, Dunn R, Kashgarian M, Kehry MR, Shlomchik MJ. Depletion of B cells in murine lupus: efficacy and resistance. J Immunol. (2007) 179:3351–61. doi: 10.4049/jimmunol.179.5.3351
28. Waisman A, Croxford AL, Demircik F. New tools to study the role of B cells in cytomegalovirus infections. Med Microbiol Immunol. (2008) 197:145–9. doi: 10.1007/s00430-008-0088-z
29. Kitamura D, Roes J, Kuhn R, Rajewsky K. A B cell-deficient mouse by targeted disruption of the membrane exon of the immunoglobulin mu chain gene. Nature. (1991) 350:423–6. doi: 10.1038/350423a0
30. Hasan M, Polic B, Bralic M, Jonjic S, Rajewsky K. Incomplete block of B cell development and immunoglobulin production in mice carrying the mumt mutation on the balb/C background. Eur J Immunol. (2002) 32:3463–71. doi: 10.1002/1521-4141(200212)32:12<3463::AID-IMMU3463>3.0.CO;2-B
31. Macpherson AJ, Lamarre A, McCoy K, Harriman GR, Odermatt B, Dougan G, et al. Iga production without mu or delta chain expression in developing B cells. Nat Immunol. (2001) 2:625–31. doi: 10.1038/89775
32. Clark EA, Otipoby KL. B cells with the guts to switch. Nat Immunol. (2001) 2:581–2. doi: 10.1038/89721
33. Gu H, Zou YR, Rajewsky K. Independent Control of Immunoglobulin Switch Recombination at Individual Switch Regions Evidenced through Cre-Loxp-Mediated Gene Targeting. Cell. (1993) 73:1155–64. doi: 10.1016/0092-8674(93)90644-6
34. Chen J, Trounstine M, Alt FW, Young F, Kurahara C, Loring JF, et al. Immunoglobulin gene rearrangement in B cell deficient mice generated by targeted deletion of the jh locus. Int Immunol. (1993) 5:647–56. doi: 10.1093/intimm/5.6.647
35. Waisman A, Kraus M, Seagal J, Ghosh S, Melamed D, Song J, et al. Igg1 B cell receptor signaling is inhibited by cd22 and promotes the development of B cells whose survival is less dependent on ig alpha/beta. J Exp Med. (2007) 204:747–58. doi: 10.1084/jem.20062024
36. Karl M, Hasselwander S, Zhou Y, Reifenberg G, Kim YO, Park KS, et al. Dual roles of B lymphocytes in mouse models of diet-induced nonalcoholic fatty liver disease. Hepatology. (2022) 76:1135–49. doi: 10.1002/hep.32428
37. Xia N, Hasselwander S, Reifenberg G, Habermeier A, Closs EI, Mimmler M, et al. B lymphocyte-deficiency in mice causes vascular dysfunction by inducing neutrophilia. Biomedicines. (2021) 9:1686. doi: 10.3390/biomedicines9111686
38. Zhang F, Jiang WW, Li X, Qiu XY, Wu Z, Chi YJ, et al. Role of intrahepatic B cells in non-alcoholic fatty liver disease by secreting pro-inflammatory cytokines and regulating intrahepatic T cells. J Dig Dis. (2016) 17:464–74. doi: 10.1111/1751-2980.12362
39. Wu Z, Xu J, Tan J, Song Y, Liu L, Zhang F, et al. Mesenteric adipose tissue B lymphocytes promote local and hepatic inflammation in non-alcoholic fatty liver disease mice. J Cell Mol Med. (2019) 23:3375–85. doi: 10.1111/jcmm.14232
40. Barrow F, Khan S, Fredrickson G, Wang H, Dietsche K, Parthiban P, et al. Microbiota-driven activation of intrahepatic B cells aggravates nash through innate and adaptive signaling. Hepatology. (2021) 74:704–22. doi: 10.1002/hep.31755
41. Bruzzi S, Sutti S, Giudici G, Burlone ME, Ramavath NN, Toscani A, et al. B2-lymphocyte responses to oxidative stress-derived antigens contribute to the evolution of nonalcoholic fatty liver disease (Nafld). Free Radic Biol Med. (2018) 124:249–59. doi: 10.1016/j.freeradbiomed.2018.06.015
42. Sutti S, Jindal A, Locatelli I, Vacchiano M, Gigliotti L, Bozzola C, et al. Adaptive immune responses triggered by oxidative stress contribute to hepatic inflammation in nash. Hepatology. (2014) 59:886–97. doi: 10.1002/hep.26749
43. Freitas-Lopes MA, Mafra K, David BA, Carvalho-Gontijo R, Menezes GB. Differential location and distribution of hepatic immune cells. Cells. (2017) 6:48. doi: 10.3390/cells6040048
44. Rivera CA, Adegboyega P, van Rooijen N, Tagalicud A, Allman M, Wallace M. Toll-like receptor-4 signaling and kupffer cells play pivotal roles in the pathogenesis of non-alcoholic steatohepatitis. J Hepatol. (2007) 47:571–9. doi: 10.1016/j.jhep.2007.04.019
45. Barrio L, Saez de Guinoa J, Carrasco YR. Tlr4 signaling shapes B cell dynamics via myd88-dependent pathways and rac gtpases. J Immunol. (2013) 191:3867–75. doi: 10.4049/jimmunol.1301623
46. Miyake T, Abe M, Tokumoto Y, Hirooka M, Furukawa S, Kumagi T, et al. B cell-activating factor is associated with the histological severity of nonalcoholic fatty liver disease. Hepatol Int. (2013) 7:539–47. doi: 10.1007/s12072-012-9345-8
47. Rai RP, Liu Y, Iyer SS, Liu S, Gupta B, Desai C, et al. Blocking integrin alpha(4)Beta(7)-mediated cd4 T cell recruitment to the intestine and liver protects mice from western diet-induced non-alcoholic steatohepatitis. J Hepatol. (2020) 73:1013–22. doi: 10.1016/j.jhep.2020.05.047
48. Dudek M, Pfister D, Donakonda S, Filpe P, Schneider A, Laschinger M, et al. Auto-aggressive cxcr6(+) cd8 T cells cause liver immune pathology in nash. Nature. (2021) 592:444–9. doi: 10.1038/s41586-021-03233-8
49. Koda Y, Teratani T, Chu PS, Hagihara Y, Mikami Y, Harada Y, et al. Cd8(+) tissue-resident memory T cells promote liver fibrosis resolution by inducing apoptosis of hepatic stellate cells. Nat Commun. (2021) 12:4474. doi: 10.1038/s41467-021-24734-0
50. Kanemitsu-Okada K, Abe M, Nakamura Y, Miyake T, Watanabe T, Yoshida O, et al. Role of B cell-activating factor in fibrosis progression in a murine model of non-alcoholic steatohepatitis. Int J Mol Sci. (2023) 24:2509. doi: 10.3390/ijms24032509
51. Rastogi I, Jeon D, Moseman JE, Muralidhar A, Potluri HK, McNeel DG. Role of B cells as antigen presenting cells. Front Immunol. (2022) 13:954936. doi: 10.3389/fimmu.2022.954936
52. Ghazarian M, Revelo XS, Nohr MK, Luck H, Zeng K, Lei H, et al. Type I interferon responses drive intrahepatic T cells to promote metabolic syndrome. Sci Immunol. (2017) 2:eaai7616. doi: 10.1126/sciimmunol.aai7616
53. Thapa M, Chinnadurai R, Velazquez VM, Tedesco D, Elrod E, Han JH, et al. Liver fibrosis occurs through dysregulation of myd88-dependent innate B-cell activity. Hepatology. (2015) 61:2067–79. doi: 10.1002/hep.27761
54. Tosello-Trampont AC, Landes SG, Nguyen V, Novobrantseva TI, Hahn YS. Kuppfer cells trigger nonalcoholic steatohepatitis development in diet-induced mouse model through tumor necrosis factor-alpha production. J Biol Chem. (2012) 287:40161–72. doi: 10.1074/jbc.M112.417014
55. Malaguarnera L, Di Rosa M, Zambito AM, dell’Ombra N, Nicoletti F, Malaguarnera M. Chitotriosidase gene expression in kupffer cells from patients with non-alcoholic fatty liver disease. Gut. (2006) 55:1313–20. doi: 10.1136/gut.2005.075697
56. Canbay A, Feldstein AE, Higuchi H, Werneburg N, Grambihler A, Bronk SF, et al. Kupffer cell engulfment of apoptotic bodies stimulates death ligand and cytokine expression. Hepatology. (2003) 38:1188–98. doi: 10.1053/jhep.2003.50472
57. Leroux A, Ferrere G, Godie V, Cailleux F, Renoud ML, Gaudin F, et al. Toxic lipids stored by kupffer cells correlates with their pro-inflammatory phenotype at an early stage of steatohepatitis. J Hepatol. (2012) 57:141–9. doi: 10.1016/j.jhep.2012.02.028
58. Carter JK, Friedman SL. Hepatic stellate cell-immune interactions in nash. Front Endocrinol (Lausanne). (2022) 13:867940. doi: 10.3389/fendo.2022.867940
59. Koyama Y, Brenner DA. Liver inflammation and fibrosis. J Clin Invest. (2017) 127:55–64. doi: 10.1172/JCI88881
60. Chiu YS, Wei CC, Lin YJ, Hsu YH, Chang MS. Il-20 and il-20r1 antibodies protect against liver fibrosis. Hepatology. (2014) 60:1003–14. doi: 10.1002/hep.27189
61. Tarrats N, Moles A, Morales A, Garcia-Ruiz C, Fernandez-Checa JC, Mari M. Critical role of tumor necrosis factor receptor 1, but not 2, in hepatic stellate cell proliferation, extracellular matrix remodeling, and liver fibrogenesis. Hepatology. (2011) 54:319–27. doi: 10.1002/hep.24388
62. de Gruijter NM, Jebson B, Rosser EC. Cytokine production by human B cells: role in health and autoimmune disease. Clin Exp Immunol. (2022) 210:253–62. doi: 10.1093/cei/uxac090
63. Shen P, Fillatreau S. Antibody-independent functions of B cells: A focus on cytokines. Nat Rev Immunol. (2015) 15:441–51. doi: 10.1038/nri3857
64. Brew K, Nagase H. The tissue inhibitors of metalloproteinases (Timps): an ancient family with structural and functional diversity. Biochim Biophys Acta. (2010) 1803:55–71. doi: 10.1016/j.bbamcr.2010.01.003
65. Hoebinger C, Rajcic D, Hendrikx T. Oxidized lipids: common immunogenic drivers of non-alcoholic fatty liver disease and atherosclerosis. Front Cardiovasc Med. (2021) 8:824481. doi: 10.3389/fcvm.2021.824481
66. Hendrikx T, Binder CJ. Oxidation-specific epitopes in non-alcoholic fatty liver disease. Front Endocrinol (Lausanne). (2020) 11:607011. doi: 10.3389/fendo.2020.607011
67. Hendrikx T, Watzenbock ML, Walenbergh SM, Amir S, Gruber S, Kozma MO, et al. Low levels of igm antibodies recognizing oxidation-specific epitopes are associated with human non-alcoholic fatty liver disease. BMC Med. (2016) 14:107. doi: 10.1186/s12916-016-0652-0
68. Albano E, Mottaran E, Vidali M, Reale E, Saksena S, Occhino G, et al. Immune response towards lipid peroxidation products as a predictor of progression of non-alcoholic fatty liver disease to advanced fibrosis. Gut. (2005) 54:987–93. doi: 10.1136/gut.2004.057968
69. De Roza MA, Lamba M, Goh GB, Lum JH, Cheah MC, Ngu JHJ. Immunoglobulin G in non-alcoholic steatohepatitis predicts clinical outcome: A prospective multi-centre cohort study. World J Gastroenterol. (2021) 27:7563–71. doi: 10.3748/wjg.v27.i43.7563
70. Baumgardner JN, Shankar K, Hennings L, Albano E, Badger TM, Ronis MJ. N-acetylcysteine attenuates progression of liver pathology in a rat model of nonalcoholic steatohepatitis. J Nutr. (2008) 138:1872–9. doi: 10.1093/jn/138.10.1872
71. Fleming A, Castro-Dopico T, Clatworthy MR. B cell class switching in intestinal immunity in health and disease. Scand J Immunol. (2022) 95:e13139. doi: 10.1111/sji.13139
72. Inamine T, Schnabl B. Immunoglobulin a and liver diseases. J Gastroenterol. (2018) 53:691–700. doi: 10.1007/s00535-017-1400-8
73. Hand TW, Reboldi A. Production and function of immunoglobulin A. Annu Rev Immunol. (2021) 39:695–718. doi: 10.1146/annurev-immunol-102119-074236
74. Schroeder HW Jr., Cavacini L. Structure and function of immunoglobulins. J Allergy Clin Immunol. (2010) 125:S41–52. doi: 10.1016/j.jaci.2009.09.046
75. McPherson S, Henderson E, Burt AD, Day CP, Anstee QM. Serum immunoglobulin levels predict fibrosis in patients with non-alcoholic fatty liver disease. J Hepatol. (2014) 60:1055–62. doi: 10.1016/j.jhep.2014.01.010
76. Shalapour S, Lin XJ, Bastian IN, Brain J, Burt AD, Aksenov AA, et al. Inflammation-induced iga+ Cells dismantle anti-liver cancer immunity. Nature. (2017) 551:340–5. doi: 10.1038/nature24302
77. Moro-Sibilot L, Blanc P, Taillardet M, Bardel E, Couillault C, Boschetti G, et al. Mouse and Human Liver Contain Immunoglobulin a-Secreting Cells Originating from Peyer’s Patches and Directed against Intestinal Antigens. Gastroenterology. (2016) 151:311–23. doi: 10.1053/j.gastro.2016.04.014
78. Mouzaki M, Bramlage K, Arce-Clachar AC, Xanthakos SA. Serum immunoglobulin a levels do not correlate with liver disease severity in pediatric nonalcoholic fatty liver disease. J Pediatr Gastroenterol Nutr. (2018) 67:631–4. doi: 10.1097/MPG.0000000000002104
79. Harriman GR, Bogue M, Rogers P, Finegold M, Pacheco S, Bradley A, et al. Targeted deletion of the iga constant region in mice leads to iga deficiency with alterations in expression of other ig isotypes. J Immunol. (1999) 162:2521–9. doi: 10.4049/jimmunol.162.5.2521
80. Novobrantseva TI, Majeau GR, Amatucci A, Kogan S, Brenner I, Casola S, et al. Attenuated liver fibrosis in the absence of B cells. J Clin Invest. (2005) 115:3072–82. doi: 10.1172/JCI24798
81. Baumgarth N. A hard(Y) look at B-1 cell development and function. J Immunol. (2017) 199:3387–94. doi: 10.4049/jimmunol.1700943
82. Bieghs V, van Gorp PJ, Walenbergh SM, Gijbels MJ, Verheyen F, Buurman WA, et al. Specific immunization strategies against oxidized low-density lipoprotein: A novel way to reduce nonalcoholic steatohepatitis in mice. Hepatology. (2012) 56:894–903. doi: 10.1002/hep.25660
83. Walenbergh SM, Koek GH, Bieghs V, Shiri-Sverdlov R. Non-alcoholic steatohepatitis: the role of oxidized low-density lipoproteins. J Hepatol. (2013) 58:801–10. doi: 10.1016/j.jhep.2012.11.014
84. Rosser EC, Mauri C. Regulatory B cells: origin, phenotype, and function. Immunity. (2015) 42:607–12. doi: 10.1016/j.immuni.2015.04.005
85. Saraiva M, Vieira P, O’Garra A. Biology and therapeutic potential of interleukin-10. J Exp Med. (2020) 217:e20190418. doi: 10.1084/jem.20190418
Keywords: metabolic dysfunction-associated steatohepatitis, metabolic dysfunction-associated steatotic liver disease, B lymphocytes, NAFLD, NASH
Citation: Li H and Xia N (2024) The multifaceted roles of B lymphocytes in metabolic dysfunction–associated steatotic liver disease. Front. Immunol. 15:1447391. doi: 10.3389/fimmu.2024.1447391
Received: 11 June 2024; Accepted: 05 September 2024;
Published: 20 September 2024.
Edited by:
Haiguang Wang, University of Minnesota Twin Cities, United StatesReviewed by:
Diane Bender, Washington University, St. Louis, United StatesGaetano Vitale, University of Udine, Italy
Copyright © 2024 Li and Xia. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Huige Li, aHVpZ2VsaUB1bmktbWFpbnouZGU=; Ning Xia, eGlhbmluZ0B1bmktbWFpbnouZGU=