 Yunjuan Jiao1,2†
Yunjuan Jiao1,2† Yuanhao Lv1,2†Mingjie Liu1Yun Liu2Miaomiao Han2Xiwen Xiong3Hongyan Zhou4
Yuanhao Lv1,2†Mingjie Liu1Yun Liu2Miaomiao Han2Xiwen Xiong3Hongyan Zhou4 Jiateng Zhong1,2,5*Xiaohong Kang6*
Jiateng Zhong1,2,5*Xiaohong Kang6* Wei Su1,5*
Wei Su1,5*- 1Department of Pathology, The First Affiliated Hospital of Xinxiang Medical University, Xinxiang, China
- 2Department of Pathology, Xinxiang Medical University, Xinxiang, China
- 3Henan Health Commission Key Laboratory of Gastrointestinal Cancer Prevention and Treatment, The First Affiliated Hospital of Xinxiang Medical University, Xinxiang, China
- 4Xinxiang Key Laboratory of Precision Diagnosis and Treatment for Colorectal Cancer, Xinxiang First People’s Hospital, Xinxiang, China
- 5Xinxiang Engineering Technology Research Center of Digestive Tumor Molecular Diagnosis, The First Affiliated Hospital of Xinxiang Medical University, Xinxiang, China
- 6Department of Oncology, The First Affiliated Hospital of Xinxiang Medical University, Xinxiang, China
Histone methylation can affect chromosome structure and binding to other proteins, depending on the type of amino acid being modified and the number of methyl groups added, this modification may promote transcription of genes (H3K4me2, H3K4me3, and H3K79me3) or reduce transcription of genes (H3K9me2, H3K9me3, H3K27me2, H3K27me3, and H4K20me3). In addition, advances in tumor immunotherapy have shown that histone methylation as a type of protein post-translational modification is also involved in the proliferation, activation and metabolic reprogramming of immune cells in the tumor microenvironment. These post-translational modifications of proteins play a crucial role in regulating immune escape from tumors and immunotherapy. Lysine methyltransferases are important components of the post-translational histone methylation modification pathway. Lysine methyltransferase 2C (KMT2C), also known as MLL3, is a member of the lysine methyltransferase family, which mediates the methylation modification of histone 3 lysine 4 (H3K4), participates in the methylation of many histone proteins, and regulates a number of signaling pathways such as EMT, p53, Myc, DNA damage repair and other pathways. Studies of KMT2C have found that it is aberrantly expressed in many diseases, mainly tumors and hematological disorders. It can also inhibit the onset and progression of these diseases. Therefore, KMT2C may serve as a promising target for tumor immunotherapy for certain diseases. Here, we provide an overview of the structure of KMT2C, disease mechanisms, and diseases associated with KMT2C, and discuss related challenges.
Introduction
Post-translational modification (PTM) of proteins is the covalent modification of amino acid side chains in translated proteins. It can expand the functional diversity of proteins by regulating protein folding, activity, stability, localization, signal transduction and binding under physiological and pathological conditions (1). Its main forms include ubiquitination, phosphorylation, methylation, acetylation, glycosylation and succinylation (2). It is closely related to immune cell activation, signal regulation, immune response and tumor metabolic reprogramming (3–5). It can directly or indirectly affect the efficacy of immunotherapy by modulating immune checkpoints or remodeling the tumor immune microenvironment (6–8). Numerous studies have demonstrated that aberrant post-translational modifications of proteins can affect cancer development by regulating tumor metabolic reprogramming (9). Histone modification is the process by which histones are methylated, acetylated, phosphorylated, adenylated, ubiquitinated, ADP-ribosylated, etc. by the action of relevant enzymes. Histone methylation occurs predominantly on lysine or arginine residues in H3 and H4 and regulates cellular metabolic processes by activating or repressing gene expression (10, 11).
Mutations and translocations of histone lysine methyltransferases (KMTs) and lysine demethylases (KDMs) are common mechanisms driving tumorigenesis (12–17). Thus, both KMTs and KDMs are potential therapeutic targets for human cancer (18–21). KMTs were classified into six subfamilies based on major amino acid sequences and substrate specificity (22). SET1A, SET1B and MLL1-4 belong to the KMT2 family and catalyze mono-, di- and trimethylation of histone H3 lysine position 4, which is proposed to be involved in the positive regulation of gene transcription (23, 24). This family of enzymes was originally isolated biochemically from yeast as a macromolecular complex named COMPASS (Complex Proteins Associated with Set1) (24, 25).
Histone-lysine N-methyltransferase 2 (KMT2) family genes, also known as MLL genes, are frequently mutated in various types of cancer (26). The KMT2 complex methylates histone 3 lysine 4 (H3K4) to regulate DNA accessibility and transcription. Unlike other COMPASS family members that trimethylated H3K4, MLL3 (KMT2C) and MLL4 catalyze histone H3K4 monomethylation at the enhancer (27, 28) and are the most common mutant histone modifiers in human cancer (29). KMT2C mutations have been detected in a variety of tumors, including hepatocellular carcinoma (30), breast cancer (31), colon cancer (32), bladder cancer (33), myelodysplastic syndromes and acute myeloid leukemia (AML) (34). In a PI3K-driven breast tumor model, KMT2C interacts with the FOXA1 transcription factor and mutational inactivation leads to increased mammary stem cell activity and accelerated tumor progression (35, 36). In addition, chromosome fragmentation leading to rearrangement of KMT2C has been reported in colon cancer (37). In this review, we will focus on KMT2C, the major mammalian histone methyltransferase. Its established importance in gene regulation and mutation frequency in developmental diseases and cancers warrants an exploration of the literature to stimulate further research and development of new therapeutic approaches. Thus, we focus on the structure of KMT2C as well as the cancers associated with KMT2C and its mechanism of action in cancer, and discuss the related challenges in the hope of providing possible application value.
The structure of KMT2C
KMT2C, also known as MLL3, localized to chromosome 7q36.1, is a member of the TRX/MLL gene family and is a histone methyltransferase that specifically catalyzes the monomethylation of histone H3 lysine K4 in enhancer regions (38, 39), thought to be involved in tissue growth regulation, tumorigenesis and transcriptional co-activation (40). As shown in Figure 1, the gene encodes 4911 amino acids and contains seven plant homologous structural domains (PHD), a high mobility group structural domain, two FY (phenylalanine tyrosine) enriched regions, and a SET (Su(var)3-9, Enhancer-of-zeste,Trithorax) structural domain. PHD and SET structural domain proteins are chromatin regulators, some of which are altered in cancer (41). Researchers find KMT2C PHD mutations can expose vulnerability to EZH2 inhibitor therapy by deregulating tumor suppressors (42).

Figure 1 The structure of KMT2C (UniProt ID: Q8NEZ4). PHD, the plant homeodomain (PHD) fingers; HMG, high mobility group domains; FYRN, “FY-rich” domain, N-terminal region; FYRC, “FY-rich” domain, C-terminal region; SET, SET(Su(var)3-9, Enhancer of zeste, Trithorax) domain. KMT2C PHD fingers 1–3 act as “readers” of the histone methylation status, recognizing monomethylated H3K4 (H3K4me1), while the SET domain, located in the Cterminus, is the “writer” that adds methyl- groups to complete the methylation process.
The KMT2C protein binds and methylates the enhancer region of histone 3 lysine 4 (H3K4), promoting the recruitment of transcriptional activators involved in DNA replication restarting (43, 44). Studies have shown that KMT2C has an oncostatic effect and is frequently found in solid tumors inactivated by deletion or mutation of this gene. Deletion of the catalytic core of the SET structural domain in KMT2C induces cell hyperproliferation and uroepithelial tumor formation (45), but knockout of the entire gene is lethal in mice (46). Recently, several studies on polymorphisms in the KMT2C gene have found positive associations with colorectal and pancreatic cancers (47, 48). Furthermore, the KMT2C gene is associated with TKI resistance and may be a potential biomarker for predicting the prognosis of cancer development (49, 50).
KMT2C regulates cellular biobehavioral functions in cancer development
Figure 2 demonstrates the biological function of KMT2C in promoting cancer development.
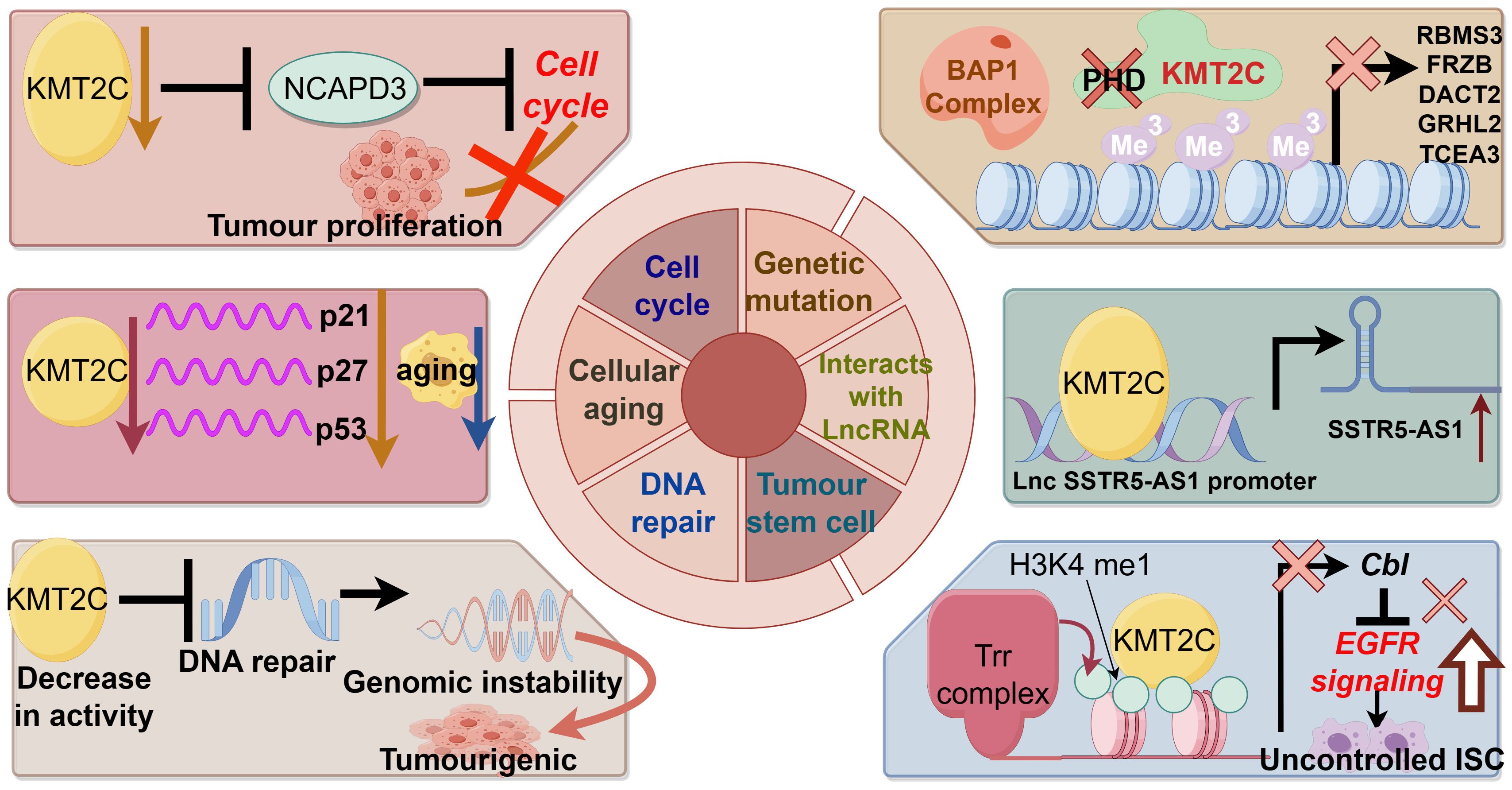
Figure 2 The biological function of KMT2C in promoting cancer development.
Genetic mutation
The MLL family is known as myeloid/lymphoid or mixed-spectrum leukemia proteins, and mutations, deletions and low expression of the gene KMT2C (MLL3) appear to play an important role in the development of leukemia. Chromosomal translocations are the most common form of MLL mutation. Chromosomal translocation fuses the MLL gene with a chaperone gene to form a new fusion protein that promotes leukemia (51). Other studies suggest that loss of KMT2C may contribute to the development of myelodysplastic syndromes and acute myelocytic leukemia (AML) by promoting myelopoiesis (52). In addition, the reduction of KMT2C gene expression can synergize with other factors at chromosome 7q to promote AML (34). Abnormalities in DNA methylation caused by germline mutations in the KMT2C gene were found in Chinese AML and colorectal cancer families and may be responsible for the pathogenesis of patients in the family lines (32, 53). Recent second-generation sequencing has also identified a heterozygous deletion due to a nonsense mutation in KMT2C that promotes human T-cell virus-induced acute T-cell leukemia (54).
Interaction with long non-coding RNA and RNA interacting with PIWI protein
Non-coding RNAs can interact with KMT2C to regulate gene expression. Wang et al. found that LncRNA SSTR5-AS1 increased the enrichment of KMT2C and H3K4me3 in the promoter region of the growth inhibitory receptor 5 by interacting with KMT2C and induced the transcription of the oncogene growth inhibitory receptor 5 in laryngeal squamous carcinoma (55). In addition, the LncRNA HOTAIR, which is highly expressed in tumors such as breast cancer, can mediate oncogene silencing by interacting with KMT2C, and its mechanism of action is also related to promoter activity (56). He et al. also found that piRNA effectively up-regulated the transcription of apoptosis-inducing ligands related to the pro-apoptotic protein tumor necrosis factor (TNF) by inducing the methylation of H3K4/demethylation of H3K27, thereby inhibiting tumor growth (57).
Cellular aging
The absence of cellular senescence mechanisms has been suggested as a possible reason for the unlimited proliferation of tumor cells. Xia et al. found that KMT2C promoted senescence of esophageal squamous carcinoma cells, and its mRNA level was down-regulated in esophageal squamous carcinoma tissues. Knockdown of KMT2C down-regulated senescence factors p21, p27 and p53 mRNA levels, while KMT2C overexpression up-regulated senescence factor levels (58). The mutation rate of KMT2C was found to be higher in breast cancer patients >50 years of age than in those <50 years of age, which may reflect the correlation between KMT2C mutations and cellular senescence (59).
Cell cycle
Disturbance of the cell cycle is an important mechanism of tumorigenesis, and abnormalities of various molecules that regulate the cell cycle can cause tumorigenesis. Dawkins et al. experimented with eight human pancreatic cell lines and showed that cell proliferation was reduced in the presence of methyltransferase depletion, possibly due to cell cycle arrest. Enrichment analyses of the gene sets following KMT2C and KMT2D knockdown showed significant down-regulation of genes related to cell cycle and growth (60). Yuan et al. found that the proliferation of human hepatocellular carcinoma cells HepG2 was significantly inhibited after the expression of KMT2C was down-regulated by the interference of small interfering RNAs, and flow cytometry showed that the cells were mainly blocked in the S phase. KMT2C is an H3K4 monomethyltransferase, and silencing of this gene blocks the normal modification of histone proteins, which may result in cell cycle arrest in S-phase, thus inhibiting cell proliferation.
DNA repair
DNA repair is a cellular reaction in which damaged DNA molecules are restored to their normal DNA sequence structure and genetic information is maintained relatively stable by the action of various enzymes. Abnormalities in the DNA repair system can lead to tumor development. Bladder cancer cells with low KMT2C activity lack homologous recombination-mediated DNA repair of double-strand breaks, resulting in greater endogenous DNA damage, leading to genomic instability and promoting tumorigenesis (61). Another experiment showed that, in order to resist Adriamycin-induced DNA damage, MLL3/4 collaborated with the ASCOM complex to increase the methylation level of H3K4, activate p53, increase the expression of endogenous p53 target genes, and participate in the tumor-suppressing pathway of p53 (45).
Tumor stem cells
Tumor stem cells are important for tumor survival, proliferation, metastasis and recurrence. Tumor stem cells maintain the viability of the tumor cell population through self-renewal and unlimited proliferation. KMT2C-related H3K4 methylation is thought to be associated with stem cell self-renewal (62). KMT2C also regulates the proliferation and transformation of tumor stem cells, and Gervais found that KMT2C regulates the proliferation of intestinal stem cells, and that its absence leads to an increase in the level of EGFR proteins, which promotes the self-renewal of intestinal stem cells and the overgrowth of tumor-like stem cells (63). Another study found that knockdown of KMT2C in gastric epithelial cells promoted epithelial-to-mesenchymal transition and enhanced the expression of mesenchymal transition-associated proteins, and that migration and invasion of gastric epithelial cells were increased 47-88-fold by knockdown of KMT2C (64).
In conclusion, mutations in the KMT2C gene alter the epigenetic state of chromatin and affect gene transcription. In the case of KMT2C wild type, epigenetic regulation of chromatin is in equilibrium. The KMT2C complex can be recruited to the enhancer by the BAP1 complex to catalyze H3K4me1 and enhance gene transcription. However, in tumors with KMT2C mutations, the KMT2C complex is not recruited to BAP1-dependent enhancers, and the increased level of repressive H3K27me3 leads to an imbalance in the epigenetic state of enhancer chromatin, increased chromosome stability, and reduced transcriptional activity, which may result in silencing of tumor suppressor genes (42).
The role of KMT2C in cancer
In recent years, more and more studies have shown the correlation between KMT2C and tumors. According to cBioportal, KMT2C is frequently mutated in a variety of cancer types such as breast cancer (~ 12%), melanoma (~ 45%), colon cancer (~ 14%) and hepatocellular carcinoma (50%) (30). KMT2C was found to be aberrantly expressed in cell lines and tumor tissues of many tumor patients, suggesting that KMT2C is involved in many cancer-related signaling pathways (Figure 3).
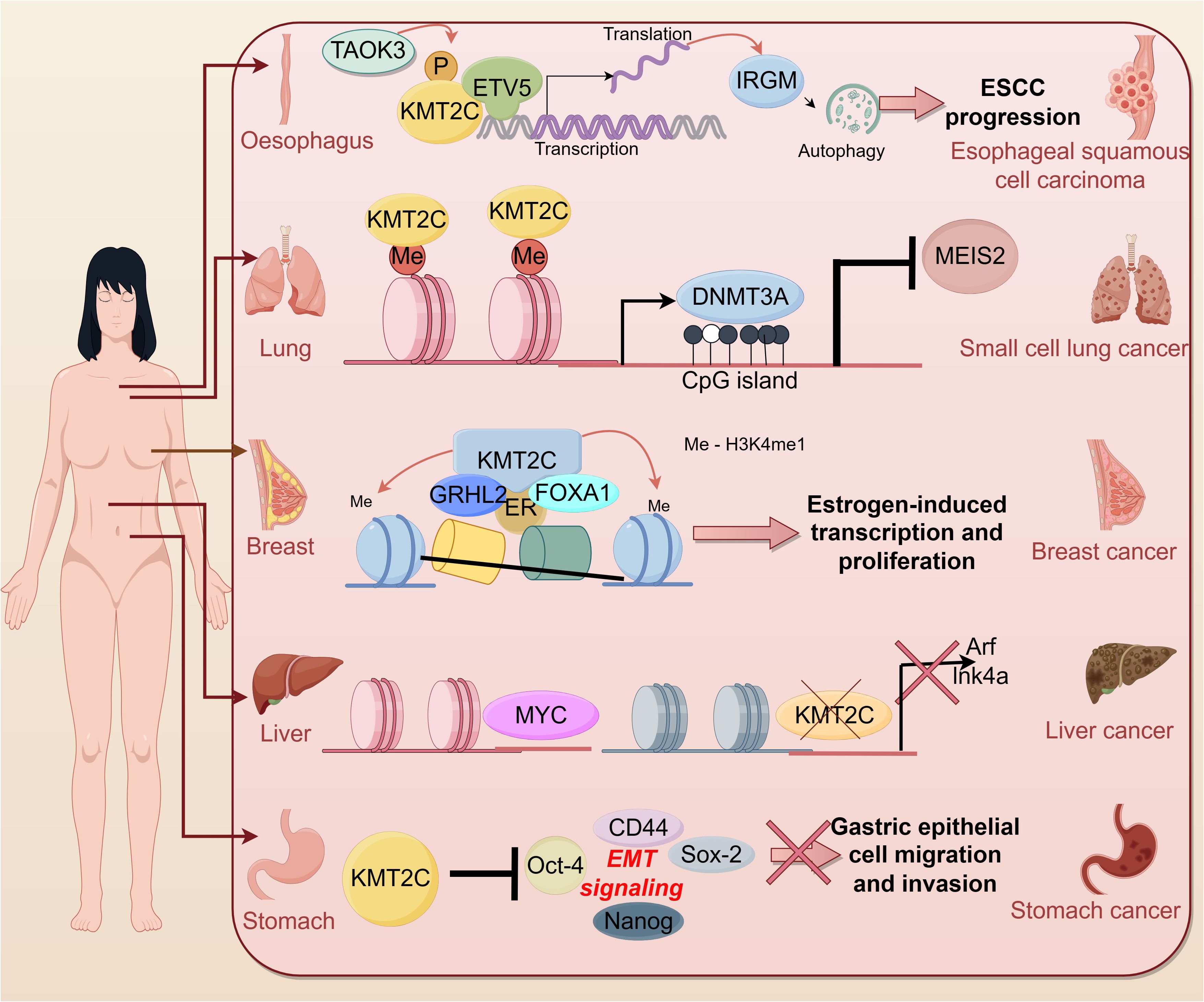
Figure 3 Role of KMT2C in cancer.
Digestive system cancer
Liver cancer
One study identified HMT genes with significant rates of genetic alteration in 360 hepatocellular carcinoma (HCC) samples. Among them, KMT2C had the highest mutation rate in HCC samples at 5.6 (65), and is associated with a poor prognosis (66). In particular, higher KMT2C mutation rates were detected in adolescent and young adult patients with advanced cholangiocarcinoma, which may suggest that cholangiocarcinoma is more aggressive in adolescent and young adult patients (67).
KMT2C, a histone methyltransferase involved in transcriptional co-activation and co-repression, is seen as a silent/intronic mutation or structural alteration in hepatic metastases from breast, melanoma, and colon cancer. Mutations in KMT2C may enhance homing and habituation of hepatocellular carcinoma cells in the liver (68). Thus, common KMT2C mutations/rearrangements may be a driver not only for primary tumors but also for liver metastases. Researchers find KMT2C, which is mutated in >50% of hepatocellular carcinomas, is also mutated in liver metastases (68).
In a recent study, Zhu et al. concluded that KMT2C likely acts as a tumor suppressor to limit Myc-driven hepatocellular carcinomas, identifying multiple tumor suppressor programs regulated by KMT2C including cell-autonomous mechanisms (cellular metabolism) and non-autonomous mechanisms (interactions with the extracellular matrix and the immune system) (69). Mechanistically, the CDKN2A locus is a genomic and transcriptional target of KMT2C in hepatocellular carcinoma cells, and KMT2C mediates oncogene-induced apoptosis in a CDKN2A-dependent manner.
In addition to its role in hepatocellular cancers, KMT2C regulates bile acid (BA) homeostasis by modulating the expression of Farnesoid X Receptor (FXRs) and p53-dependent small heterodimeric chaperone receptors (SHPs) (70).
Gastric cancer
Mutations and low expression of the KMT2C gene are associated with a high risk of progression and poor prognosis in gastric cancer patients and are independent predictors of disease recurrence (71–74). In addition, it has been shown that KMT2C has a high mutation rate in different metastatic sites occurring in gastric cancer patients: peritoneal metastasis (42%), haematogenous metastasis (67%), and distant lymph node metastasis (35%) (75). Zhou et al. found that KMT2C mutations were predominantly detected in HER2 + gastric cancer samples (7/10) and were also associated with the lysine degradation pathway (76).
Cho et al. showed that in vitro knockdown of KMT2C promotes migration and invasion of gastric epithelial cells by promoting the EMT signaling pathway (64). Specifically, there is an increase in KMT2C mutations, which show higher levels of “clock-like” mutational features, increased genome-wide doubling, chromosomal instability (especially copy number loss), reprogrammed microenvironments, enriched cell-cycle pathways, Myc activation, and impaired immune responses (77).
Evidence that KMT2C mutations are potential predictors of immunotherapy response in solid malignancies (78). However, in contrast to the high mutation rate of KMT2C, which promotes migration and invasion of gastric epithelial cells, the high mutation rate of KMT2C tends to show a better prognosis in patients treated with immune checkpoint inhibitors (ICIs), and the KMT2C mutation is significantly and positively correlated with higher intratumoural infiltration of CD8+ lymphocytes, CD163+ tumor-associated macrophages, and PD-L1 in gastric cancer tumors (79).
Pancreas cancer
Cancers with high TMB have been shown to respond better to ICI, such as pancreatic adenocarcinoma (PAAD) (80). PAAD patients with KMT2C mutations have a higher severity of tumor mutational burden (TMB) and a worse prognosis, mainly related to the fact that mutations in the KMT2C gene affect the composition of immune cells in PAAD patients and positively regulate metabolic and protein-related pathways in PAAD (81). These all suggest that KMT2C mutations may serve as biomarkers for predicting the prognosis of PAAD patients and guiding anti-PAAD immunotherapy.
Notably, in a 2016 article, Dawkins et al. suggest that targeting KMT2C in PDAC may also have therapeutic benefit, especially in those patients who exhibit higher KTM2C expression (60). They found by biopsy that low expression of KMT2C also demonstrated a better prognosis, and experiments on eight human pancreatic cell lines showed that when KMT2C was depleted, cell cycle arrest and reduced progression from G0-G1 led to attenuated cell proliferation.
Furthermore, KRAS mutation is a marker for pancreatic ductal adenocarcinoma (PDAC) and is associated with key aspects of its biology, such as inflammation, immune escape and metabolic alterations (82–85). Mutant KRAS is a major oncogenic driver of PDAC and an attractive therapeutic target (86, 87). However, KRAS wild-type (WT) is present in a small proportion of PDAC, and Philip et al. (88) and Yoon et al. (89) found that KMT2C is frequently mutated in KRAS wild-type tumor samples of pancreatic ductal adenocarcinomas, providing a new promising therapeutic target for the targeted treatment of KRAS wild-type PDAC patients. Tumor mutation burden (TMB), a new biomarker, has shown its potential as a predictive biomarker for a variety of applications, including the correlation between different levels of TMB and the response of patients with various types of cancers to immune checkpoint inhibitors (ICIs) (90).
Colorectal cancer
KMT2C is considered an oncogene in colorectal cancer. It has been found to be frequently mutated in colorectal cancer (91) and is commonly mutated in both primary CRC and peritoneal metastases (92) and has been associated with CRC prognosis, and can be used to predict OS and PFS in patients with CRC (93, 94) It is a potential candidate for potentially identifying patients with high-risk colorectal cancer (95).
Mutations in KMT2C may be involved in the transition of non-dysplastic cells to a dysplastic phenotype in patients with long-standing ulcerative colitis (UC) and have a high risk of progression to colorectal tumors (96). Watanabe et al. found that code-shifting mutations in KMT2C in CRC cells and primary tumors were more common in cases of microsatellite instability. In addition, the CpG island-associated promoter of the KMT2C gene is not DNA methylated in CRC cells, nor is it DNA methylated in primary tumors or normal colon, and this region is highly homologous to a pseudogene for age-associated DNA methylation (psi TPTE22) (97). Meanwhile, restoration of KMT2C inhibited CRC cell growth and enhanced genome-wide histone H3 position 4-methylation deposition on enhancers; however, this effect varied depending on the histone H3 position 4-methylation status of the KMT2C-deficient cells. The results suggest that KMT2C inactivation may promote colorectal cancer development through transcriptional dysregulation of several pathways known to be associated with cancer (98). Importantly, it has been found that KMT2C loss-of-function variants (LOF) are associated with higher TMB, and specifically, KMT2C LOF variants are associated with decreased regulatory T cells and increased levels of CD8+T cells, activated NK cells, M1-type macrophages and M2-type macrophages in colorectal cancer (99).
Respiratory system cancers
KMT2C mutations are very common in early stage lung adenocarcinoma and their low expression is associated with shorter overall survival time (100, 101), and the mutation frequency of KMT2C in lung adenocarcinoma patients was shown to be significantly higher in smokers than in non-smokers (102), and more importantly, variants in the gene were associated with younger lung adenocarcinoma patients (103).
Distant organ metastases are still the leading cause of cancer-related deaths (104), and the KMT2C gene, a high-frequency mutated gene common to both primary and metastatic foci of lung cancer, may provide a potential biomarker for immune checkpoint blockade in LUAD with metastases to different organs (105).
For lung cancer, the most common sites of metastasis are the contralateral lung, brain, bone, and liver (106, 107), and the study of Gao et al. (104) found that in the whole cohort of primary (PR) and metastatic foci (MT - liver, MT - bone, and MT - brain), the frequency of mutations in the KMT2C gene was high and the mutations were common to PR and metastatic foci (MT - liver, MT - bone, and MT - brain), and in the MT - bone, LRP1B mutations co-occurred with KMT2C mutations. Meanwhile, Liu et al. found that KMT2C mutations were associated with lung cancer metastasis to the brain by whole exome sequencing (108).
The development of specific antibodies against the programmed death (PD1) receptor, its ligand PD-L1 (programmed death ligand-1), and the cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) receptor in first- or second-line therapeutic strategies for patients with non-small-cell lung cancer has led to an unprecedented prolongation of patient survival in the last decade (109). Therefore, there is an urgent need for effective biomarkers to predict the response of non-small cell lung cancer (NSCLC), especially NSCLC with low tumor mutation load, to immune checkpoint block (ICB) therapy (110). Gu et al. found that KRAS and KMT2C co-mutations improved the response to immunotherapy in patients with non-small cell lung cancer (111). The study by Bai et al. highlighted the potential predictive value of KMT2C for immunotherapeutic benefit in non-squamous NSCLC, and furthermore, the combination of KMT2C and PD-L1 could serve as the best partner for guiding therapeutic decisions based on anti-PD-(L)1 (112). In a recent study, researchers found that mutations in three chromatin remodeling-associated genes, KMT2C, BCOR and KDM5C, were associated with ICB responses in NSCLC, including NSCLC with low TMB levels. Moreover, the combination of KMT2C mutation with TMB or PD - L1 expression further improved this correlation. These data suggest the potential of KMT2C mutation status as a predictive biomarker for ICB therapy in NSCLC. KMT2C mutations were shown to have potential as predictive biomarkers for ICB therapy in NSCLC alone or in combination with PD - L1 expression or TMB (110).
Urogenital system cancers
Bladder cancer
KMT2C plays a repressive role in bladder cancer and is one of the most commonly mutated genes in BCa patients (113). Specifically, downregulation of KMT2C in bladder cancer cells results in widespread changes in epigenetic status and expression of DNA damage response and DNA repair genes. More specifically, cells with low KMT2C activity lack homologous recombination-mediated DNA repair of double-strand breaks, and as a result, these cells suffer much higher levels of endogenous DNA damage and genomic instability. Finally, these cells appear to be heavily dependent on PARP1/2 for DNA repair and treatment with the PARP1/2 inhibitor Olaparib results in synthetic lethality, suggesting that low KMT2C-expressing cancer cells are an attractive target for PARP1/2 inhibitor therapy (61).
Prostate cancer
KMT2C has been described as the most mutated epigenetic regulator and driver in PCa tumor tissues (114).Coelho et al. suggest that the KMT2C gene may play as important a role in tumor suppression in PCa patients as BRCA2 (115). Moreover, alterations in KMT2C are more likely to coincide with alterations in TP53, suggesting a more aggressive phenotype in PCa, which correlates with sensitivity to treatment with poly ADT-ribose polymerase (PARP) inhibitors (116). KMT2C mutations indicate rapid progression during conventional combined antiandrogen blockade (CAB) therapy and may serve as a potential biomarker for predicting response to prostate cancer therapy (117).
Currently, high-risk human papillomavirus (hr HPV) infection is also considered a risk factor for PCa (118), and KMT2C, KMT2D and ERCC2 mutations are more frequent in HPV-positive tumors (119). The frequency of mutations in PREX2, PTEN, AGO2, and KMT2C was significantly higher in patients with a history of smoking than in nonsmokers. Smokers (p = 0.006) had a significantly higher overall mortality rate (28.5% versus 22.8%) (120). In a recent study, the authors found that KMT2C mutations were associated with PCa metastasis, and in addition, they found that KMT2C mutations were associated with reduced PCa disease-free survival, and that inhibition of the MYC signaling axis may be a viable therapeutic option for patients with KMT2C truncation (121).
Breast cancer
KMT2C is a gene with a high number of mutations found in breast cancer, some of which are associated with chromatin function, affecting transcriptional mechanisms identified in breast tumorigenesis and development (122) and, in addition, KMT2C mutations observed in circulating tumor DNA (ctDNA) from six-month postoperative samples may be indicative of breast cancer recurrence and prognosis (123). Alterations in KMT2C are significantly enriched in metastatic populations compared to primary breast cancers (124). KMT2C is mutated at a higher rate in HR +/HER2 type breast cancers than in other subtypes (59). KMT2C mutations have also been shown to be critical for ER α regulation, which can lead to hormone-driven proliferation of breast cancer cells (125).
Role of KMT2C as a key chromatin regulatory protein in enhancer elements and as a factor that promotes histone H3 position 4-methylation deposition in these enhancers. The precursor factor FOXA1 interacts with the chromatin-modifying factor KMT2C to promote monomethylation of H3K4 on enhancer elements, thereby generating the potential for transcription from these enhancer regions (35). In the presence of E2, KMT2C and KMT2D, together with ER, regulate the expression of the HOXC10 gene and promote breast cancer progression (126).
Ovarian cancer
Huang et al. found that overexpression of XIST enhanced the anticancer sensitivity of paclitaxel on ovarian cancer cells, and its effect may be related to the up-regulation of KMT2C. XIST affects the expression of KMT2C in ovarian cancer by enhancing the stability of KMT2C mRNA through the direct targeting of mi -93-5p. The results of this study suggest that the miR-93-5p/XIST/KMT2C signaling axis may provide new potential therapeutic targets for ovarian cancer treatment and play an important role in future ovarian cancer therapy (127).
Blood system cancers
KMT2C is localized at 7q36 and was first described as a chromosomal region frequently missing in myeloid leukemia (128).
KMT2C levels decrease during progression of chronic granulocytic leukemia and correlate with different clinical stages. After treatment of the imatinib mesylate (IM)-sensitive CML cell line KCL22S with Dasatinib or Nilotinib, a restoration of KMT2C gene expression and a higher rate of apoptosis and enhanced expression of p21 (CDKN1A) compared to the control group was observed, accompanied by a decrease in the expression of CDK2, CDK4, and Cyclin B1 (CCNB1), which suggests that the p53 regulatory pathway is involved in the regulation of cancer by KMT2C (49).
Nervous system cancers
KMT2C was one of the first few recurrently mutated genes identified in a sequencing study of early medulloblastoma (129). In the adult medulloblastoma cohort in the Jones et al. study, KMT2C was one of the most commonly mutated genes, with 30% of mutations detected in cases of different ages, sexes, histological types, and molecular typologies, again demonstrating the central importance of chromatin modifications in the pathophysiology of medulloblastomas (130) and highlighting the fact that a more comprehensive review of the adult medulloblastoma epigenetic landscape is the need for a more comprehensive assessment (131). In another study, researchers identified inactivating mutations in the histone lysine N -methyltransferase genes KMT2C or KMT2D in 16% of patients with childhood medulloblastoma (MB) (129).
Alterations in KMT2C at the gene level or at the protein level may disrupt the epigenetic programme and lead to malignant transformation of gliomas (132).Kleefstra et al. (133) found that an autosomal dominant nonsense mutation (p. Arg1481 *) in KMT2C leads to neurodevelopmental disorders manifested by mental retardation, growth retardation, mild dysmorphic features (including prominent eyebrows, thick ear whorls, and misaligned teeth), and neuropsychiatric traits, including hyperactivity and aggression. Mutations in KMT2C in neurodevelopmental disorders have also been described by Vallianatos et al. (134).
Future prospects and conclusions
Post-translational modifications are typical biochemical reactions that covalently bind (poly)peptide chains, chemical parts, lipids or carbohydrates to amino acids of a target molecule during or after translation. PTMs occur in most known proteins, and virtually all amino acids can be changed by one or more of these reactions. Modified proteins gain uncommon amino acids that can have a significant impact on their structure and function. Post-translational modifications diversify the proteome by altering the structure, location, interactions, and function of proteins and their regulation, thereby affecting various functional aspects of the cell. Methylation is an important post-translational modification that regulates various biological functions of cells by modifying proteins. In recent years, due to increased research on tumor progression and treatment, more and more researchers have begun to focus on methylation in the search for effective anti-tumor therapy.
KMT2C is the writer of the methylation process that is widespread in eukaryotes. it plays an essential role in histone methylation modification. KMT2C is frequently mutated in a variety of human cancers, including hepatocellular carcinoma, non-small-cell lung cancer, and breast cancer. KMT2C stimulates the development, progression and metastasis of human tumors by regulating different signaling pathways associated with human tumors, as well as a variety of proteins that are not involved in the above signaling pathways. The EMT signaling pathway promotes tumor cell migration and invasion, and is an important tumor-promoting factor. KMT2C inhibits the expression of proteins associated with the EMT signaling pathway, which may provide a direction for the development of new anticancer drugs. However, the regulation of GSK3b/p65, DSB repair and other signaling pathways by KMT2C needs to be further investigated, and the specific mechanism of KMT2C’s action in other tumors and its potential as a novel anti-tumor therapy need to be further studied. Although previous findings have shown that KMT2C is a promising target for the treatment of cervical and bladder cancers, the specific molecular mechanisms involved in the regulation of KMT2C in cervical and bladder cancers remain to be further investigated. We suggest that future studies focus on the pathways that promote cancer progression activated by KMT2C mutations, develop inhibitors of the corresponding pathways and demonstrate their efficacy and safety in treating these diseases.
Author contributions
YJ: Writing – original draft. YHL: Writing – original draft. ML: Writing – review & editing. YL: Writing – review & editing. MH: Writing – review & editing. XX: Writing – review & editing. HZ: Writing – review & editing. JZ: Conceptualization, Writing – review & editing. XK: Conceptualization, Writing – review & editing. WS: Conceptualization, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the National Natural Science Foundation of China (No. U No. 82074268, 82074231), Henan province young and middle-aged health science and technology innovation talent project (YXKC2021044), Science and Technology Research Projects of Henan province (222102310421).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Czuba LC, Hillgren KM, Swaan PW. Post-translational modifications of transporters. Pharmacol Ther. (2018) 192:88–99. doi: 10.1016/j.pharmthera.2018.06.013
2. Wang R, Wang G. Protein modification and autophagy activation. Adv Exp Med Biol. (2019) 1206:237–59. doi: 10.1007/978-981-15-0602-4_12
3. Fang Y, Xu X, Ding J, Yang L, Doan MT, Karmaus PWF, et al. Histone crotonylation promotes mesoendodermal commitment of human embryonic stem cells. Cell Stem Cell. (2021) 28:748–763.e7. doi: 10.1016/j.stem.2020.12.009
4. Kim E-J, Liu P, Zhang S, Donahue K, Wang Y, Schehr JL, et al. BAF155 methylation drives metastasis by hijacking super-enhancers and subverting anti-tumor immunity. Nucleic Acids Res. (2021) 49:12211–33. doi: 10.1093/nar/gkab1122
5. Kukkula A, Ojala VK, Mendez LM, Sistonen L, Elenius K, Sundvall M. Therapeutic potential of targeting the SUMO pathway in cancer. Cancers (Basel). (2021) 13(17):4402. doi: 10.3390/cancers13174402
6. Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, Merad M, et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med. (2018) 24:541–50. doi: 10.1038/s41591-018-0014-x
7. Dai X, Gao Y, Wei W. Post-translational regulations of PD-L1 and PD-1: Mechanisms and opportunities for combined immunotherapy. Semin Cancer Biol. (2022) 85:246–52. doi: 10.1016/j.semcancer.2021.04.002
8. Yamaguchi H, Hsu J-M, Yang W-H, Hung M-C. Mechanisms regulating PD-L1 expression in cancers and associated opportunities for novel small-molecule therapeutics. Nat Rev Clin Oncol. (2022) 19:287–305. doi: 10.1038/s41571-022-00601-9
9. Baylin SB, Jones PA. Epigenetic determinants of cancer. Cold Spring Harb Perspect Biol. (2016) 8(9):a019505. doi: 10.1101/cshperspect.a019505
10. Chen Y, Ren B, Yang J, Wang H, Yang G, Xu R, et al. The role of histone methylation in the development of digestive cancers: a potential direction for cancer management. Signal Transduct Target Ther. (2020) 5:143. doi: 10.1038/s41392-020-00252-1
11. Tran TQ, Lowman XH, Kong M. Molecular pathways: metabolic control of histone methylation and gene expression in cancer. Clin Cancer Res. (2017) 23:4004–9. doi: 10.1158/1078-0432.CCR-16-2506
12. Morgan MA, Shilatifard A. Chromatin signatures of cancer. Genes Dev. (2015) 29:238–49. doi: 10.1101/gad.255182.114
13. Piunti A, Shilatifard A. Epigenetic balance of gene expression by Polycomb and COMPASS families. Science. (2016) 352:aad9780. doi: 10.1126/science.aad9780
14. Plass C, Pfister SM, Lindroth AM, Bogatyrova O, Claus R, Lichter P. Mutations in regulators of the epigenome and their connections to global chromatin patterns in cancer. Nat Rev Genet. (2013) 14:765–80. doi: 10.1038/nrg3554
15. Kotake Y, Cao R, Viatour P, Sage J, Zhang Y, Xiong Y. pRB family proteins are required for H3K27 trimethylation and Polycomb repression complexes binding to and silencing p16INK4alpha tumor suppressor gene. Genes Dev. (2007) 21:49–54. doi: 10.1101/gad.1499407
16. Hamamoto R, Furukawa Y, Morita M, Iimura Y, Silva FP, Li M, et al. SMYD3 encodes a histone methyltransferase involved in the proliferation of cancer cells. Nat Cell Biol. (2004) 6:731–40. doi: 10.1038/ncb1151
17. Mazur PK, Reynoird N, Khatri P, Jansen PWTC, Wilkinson AW, Liu S, et al. SMYD3 links lysine methylation of MAP3K2 to Ras-driven cancer. Nature. (2014) 510:283–7. doi: 10.1038/nature13320
18. Klaus CR, Iwanowicz D, Johnston D, Campbell CA, Smith JJ, Moyer MP, et al. DOT1L inhibitor EPZ-5676 displays synergistic antiproliferative activity in combination with standard of care drugs and hypomethylating agents in MLL-rearranged leukemia cells. J Pharmacol Exp Ther. (2014) 350:646–56. doi: 10.1124/jpet.114.214577
19. Herz H-M, Morgan M, Gao X, Jackson J, Rickels R, Swanson SK, et al. Histone H3 lysine-to-methionine mutants as a paradigm to study chromatin signaling. Science. (2014) 345:1065–70. doi: 10.1126/science.1255104
20. Kubicek S, O’Sullivan RJ, August EM, Hickey ER, Zhang Q, Teodoro ML, et al. Reversal of H3K9me2 by a small-molecule inhibitor for the G9a histone methyltransferase. Mol Cell. (2007) 25:473–81. doi: 10.1016/j.molcel.2007.01.017
21. Ferguson AD, Larsen NA, Howard T, Pollard H, Green I, Grande C, et al. Structural basis of substrate methylation and inhibition of SMYD2. Structure. (2011) 19:1262–73. doi: 10.1016/j.str.2011.06.011
22. Mohan M, Herz H-M, Shilatifard A. SnapShot: Histone lysine methylase complexes. Cell. (2012) 149:498–498.e1. doi: 10.1016/j.cell.2012.03.025
23. Kouzarides T. Chromatin modifications and their function. Cell. (2007) 128:693–705. doi: 10.1016/j.cell.2007.02.005
24. Shilatifard A. The COMPASS family of histone H3K4 methylases: mechanisms of regulation in development and disease pathogenesis. Annu Rev Biochem. (2012) 81:65–95. doi: 10.1146/annurev-biochem-051710-134100
25. Miller T, Krogan NJ, Dover J, Erdjument-Bromage H, Tempst P, Johnston M, et al. COMPASS: a complex of proteins associated with a trithorax-related SET domain protein. Proc Natl Acad Sci U.S.A. (2001) 98:12902–7. doi: 10.1073/pnas.231473398
26. Rao RC, Dou Y. Hijacked in cancer: the KMT2 (MLL) family of methyltransferases. Nat Rev Cancer. (2015) 15:334–46. doi: 10.1038/nrc3929
27. Herz H-M, Mohan M, Garruss AS, Liang K, Takahashi Y-H, Mickey K, et al. Enhancer-associated H3K4 monomethylation by Trithorax-related, the Drosophila homolog of mammalian Mll3/Mll4. Genes Dev. (2012) 26:2604–20. doi: 10.1101/gad.201327.112
28. Hu D, Gao X, Morgan MA, Herz H-M, Smith ER, Shilatifard A. The MLL3/MLL4 branches of the COMPASS family function as major histone H3K4 monomethylases at enhancers. Mol Cell Biol. (2013) 33:4745–54. doi: 10.1128/MCB.01181-13
29. Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, et al. Mutational landscape and significance across 12 major cancer types. Nature. (2013) 502:333–9. doi: 10.1038/nature12634
30. Fujimoto A, Totoki Y, Abe T, Boroevich KA, Hosoda F, Nguyen HH, et al. Whole-genome sequencing of liver cancers identifies etiological influences on mutation patterns and recurrent mutations in chromatin regulators. Nat Genet. (2012) 44:760–4. doi: 10.1038/ng.2291
31. Ellis MJ, Ding L, Shen D, Luo J, Suman VJ, Wallis JW, et al. Whole-genome analysis informs breast cancer response to aromatase inhibition. Nature. (2012) 486:353–60. doi: 10.1038/nature11143
32. Li W-D, Li Q-R, Xu S-N, Wei F-J, Ye Z-J, Cheng J-K, et al. Exome sequencing identifies an MLL3 gene germ line mutation in a pedigree of colorectal cancer and acute myeloid leukemia. Blood. (2013) 121:1478–9. doi: 10.1182/blood-2012-12-470559
33. Gui Y, Guo G, Huang Y, Hu X, Tang A, Gao S, et al. Frequent mutations of chromatin remodeling genes in transitional cell carcinoma of the bladder. Nat Genet. (2011) 43:875–8. doi: 10.1038/ng.907
34. Chen C, Liu Y, Rappaport AR, Kitzing T, Schultz N, Zhao Z, et al. MLL3 is a haploinsufficient 7q tumor suppressor in acute myeloid leukemia. Cancer Cell. (2014) 25:652–65. doi: 10.1016/j.ccr.2014.03.016
35. Jozwik KM, Chernukhin I, Serandour AA, Nagarajan S, Carroll JS. FOXA1 Directs H3K4 Monomethylation at Enhancers via Recruitment of the Methyltransferase MLL3. Cell Rep. (2016) 17:2715–23. doi: 10.1016/j.celrep.2016.11.028
36. Zhang Z, Christin JR, Wang C, Ge K, Oktay MH, Guo W. Mammary-stem-cell-based somatic mouse models reveal breast cancer drivers causing cell fate dysregulation. Cell Rep. (2016) 16:3146–56. doi: 10.1016/j.celrep.2016.08.048
37. Kloosterman WP, Hoogstraat M, Paling O, Tavakoli-Yaraki M, Renkens I, Vermaat JS, et al. Chromothripsis is a common mechanism driving genomic rearrangements in primary and metastatic colorectal cancer. Genome Biol. (2011) 12:R103. doi: 10.1186/gb-2011-12-10-r103
38. Li Y, Han J, Zhang Y, Cao F, Liu Z, Li S, et al. Structural basis for activity regulation of MLL family methyltransferases. Nature. (2016) 530:447–52. doi: 10.1038/nature16952
39. Xue H, Yao T, Cao M, Zhu G, Li Y, Yuan G, et al. Structural basis of nucleosome recognition and modification by MLL methyltransferases. Nature. (2019) 573:445–9. doi: 10.1038/s41586-019-1528-1
40. Dorighi KM, Swigut T, Henriques T, Bhanu NV, Scruggs BS, Nady N, et al. Mll3 and mll4 facilitate enhancer RNA synthesis and transcription from promoters independently of H3K4 monomethylation. Mol Cell. (2017) 66:568–576.e4. doi: 10.1016/j.molcel.2017.04.018
41. Saha V, Chaplin T, Gregorini A, Ayton P, Young BD. The leukemia-associated-protein (LAP) domain, a cysteine-rich motif, is present in a wide range of proteins, including MLL, AF10, and MLLT6 proteins. Proc Natl Acad Sci U.S.A. (1995) 92:9737–41. doi: 10.1073/pnas.92.21.9737
42. Wang L, Zhao Z, Ozark PA, Fantini D, Marshall SA, Rendleman EJ, et al. Resetting the epigenetic balance of Polycomb and COMPASS function at enhancers for cancer therapy. Nat Med. (2018) 24:758–69. doi: 10.1038/s41591-018-0034-6
43. Ruthenburg AJ, Allis CD, Wysocka J. Methylation of lysine 4 on histone H3: intricacy of writing and reading a single epigenetic mark. Mol Cell. (2007) 25:15–30. doi: 10.1016/j.molcel.2006.12.014
44. Ray Chaudhuri A, Callen E, Ding X, Gogola E, Duarte AA, Lee J-E, et al. Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature. (2016) 535:382–7. doi: 10.1038/nature18325
45. Lee J, Kim D-H, Lee S, Yang Q-H, Lee DK, Lee S-K, et al. A tumor suppressive coactivator complex of p53 containing ASC-2 and histone H3-lysine-4 methyltransferase MLL3 or its paralogue MLL4. Proc Natl Acad Sci U.S.A. (2009) 106:8513–8. doi: 10.1073/pnas.0902873106
46. Lee J-E, Wang C, Xu S, Cho Y-W, Wang L, Feng X, et al. H3K4 mono- and di-methyltransferase MLL4 is required for enhancer activation during cell differentiation. Elife. (2013) 2:e01503. doi: 10.7554/eLife.01503
47. Heitzer E, Tomlinson I. Replicative DNA polymerase mutations in cancer. Curr Opin Genet Dev. (2014) 24:107–13. doi: 10.1016/j.gde.2013.12.005
48. Duell EJ, Sala N, Travier N, Muñoz X, Boutron-Ruault MC, Clavel-Chapelon F, et al. Genetic variation in alcohol dehydrogenase (ADH1A, ADH1B, ADH1C, ADH7) and aldehyde dehydrogenase (ALDH2), alcohol consumption and gastric cancer risk in the European Prospective Investigation into Cancer and Nutrition (EPIC) cohort. Carcinogenesis. (2012) 33:361–7. doi: 10.1093/carcin/bgr285
49. Rabello D do A, Ferreira VD da S, Berzoti-Coelho MG, Burin SM, Magro CL, Cacemiro M da C, et al. MLL2/KMT2D and MLL3/KMT2C expression correlates with disease progression and response to imatinib mesylate in chronic myeloid leukemia. Cancer Cell Int. (2018) 18:26. doi: 10.1186/s12935-018-0523-1
50. Sato K, Akimoto K. Expression levels of KMT2C and SLC20A1 identified by information-theoretical analysis are powerful prognostic biomarkers in estrogen receptor-positive breast cancer. Clin Breast Cancer. (2017) 17:e135–42. doi: 10.1016/j.clbc.2016.11.005
51. Ballabio E, Milne TA. Molecular and epigenetic mechanisms of MLL in human leukemogenesis. Cancers (Basel). (2012) 4:904–44. doi: 10.3390/cancers4030904
52. Arcipowski KM, Bulic M, Gurbuxani S, Licht JD. Loss of mll3 catalytic function promotes aberrant myelopoiesis. PloS One. (2016) 11:e0162515. doi: 10.1371/journal.pone.0162515
53. Yang F, Gong Q, Shi W, Zou Y, Shi J, Wei F, et al. Aberrant DNA methylation of acute myeloid leukemia and colorectal cancer in a Chinese pedigree with a MLL3 germline mutation. Tumour Biol. (2016) 37:12609–18. doi: 10.1007/s13277-016-5130-y
54. Yeh C-H, Bai XT, Moles R, Ratner L, Waldmann TA, Watanabe T, et al. Mutation of epigenetic regulators TET2 and MLL3 in patients with HTLV-I-induced acute adult T-cell leukemia. Mol Cancer. (2016) 15:15. doi: 10.1186/s12943-016-0500-z
55. Wang B, Zhao L, Chi W, Cao H, Cui W, Meng W. Aberrant methylation-mediated downregulation of lncRNA SSTR5-AS1 promotes progression and metastasis of laryngeal squamous cell carcinoma. Epigenet Chromatin. (2019) 12:35. doi: 10.1186/s13072-019-0283-8
56. Bhan A, Hussain I, Ansari KI, Kasiri S, Bashyal A, Mandal SS. Antisense transcript long noncoding RNA (lncRNA) HOTAIR is transcriptionally induced by estradiol. J Mol Biol. (2013) 425:3707–22. doi: 10.1016/j.jmb.2013.01.022
57. He X, Chen X, Zhang X, Duan X, Pan T, Hu Q, et al. (GAS5)/SnoRNA-derived piRNA induces activation of TRAIL gene by site-specifically recruiting MLL/COMPASS-like complexes. Nucleic Acids Res. (2015) 43:3712–25. doi: 10.1093/nar/gkv214
58. Xia M, Ling F, Gao F, Tao C. MLL3 promotes the senescence of esophageal squamous cell carcinoma. Onco Targets Ther. (2019) 12:1575–82. doi: 10.2147/OTT.S187540
59. Chen X, Zhang G, Chen B, Wang Y, Guo L, Cao L, et al. Association between histone lysine methyltransferase KMT2C mutation and clinicopathological factors in breast cancer. BioMed Pharmacother. (2019) 116:108997. doi: 10.1016/j.biopha.2019.108997
60. Dawkins JBN, Wang J, Maniati E, Heward JA, Koniali L, Kocher HM, et al. Reduced expression of histone methyltransferases KMT2C and KMT2D correlates with improved outcome in pancreatic ductal adenocarcinoma. Cancer Res. (2016) 76:4861–71. doi: 10.1158/0008-5472.CAN-16-0481
61. Rampias T, Karagiannis D, Avgeris M, Polyzos A, Kokkalis A, Kanaki Z, et al. The lysine-specific methyltransferase KMT2C/MLL3 regulates DNA repair components in cancer. EMBO Rep. (2019) 20(17):1732–7. doi: 10.15252/embr.201846821
62. Sze CC, Cao K, Collings CK, Marshall SA, Rendleman EJ, Ozark PA, et al. Histone H3K4 methylation-dependent and -independent functions of Set1A/COMPASS in embryonic stem cell self-renewal and differentiation. Genes Dev. (2017) 31:1732–7. doi: 10.1101/gad.303768.117
63. Gervais L, van den Beek M, Josserand M, Sallé J, Stefanutti M, Perdigoto CN, et al. Stem cell proliferation is kept in check by the chromatin regulators kismet/CHD7/CHD8 and trr/MLL3/4. Dev Cell. (2019) 49:556–573.e6. doi: 10.1016/j.devcel.2019.04.033
64. Cho S-J, Yoon C, Lee JH, Chang KK, Lin J-X, Kim Y-H, et al. KMT2C mutations in diffuse-type gastric adenocarcinoma promote epithelial-to-mesenchymal transition. Clin Cancer Res. (2018) 24:6556–69. doi: 10.1158/1078-0432.CCR-17-1679
65. Aravena TI, Valdés E, Ayala N, D’Afonseca V. A computational approach to predict the role of genetic alterations in methyltransferase histones genes with implications in liver cancer. Cancer Inform. (2023) 22:11769351231161480. doi: 10.1177/11769351231161480
66. Fujikura K, Akita M, Ajiki T, Fukumoto T, Itoh T, Zen Y. Recurrent mutations in APC and CTNNB1 and activated wnt/β-catenin signaling in intraductal papillary neoplasms of the bile duct: A whole exome sequencing study. Am J Surg Pathol. (2018) 42:1674–85. doi: 10.1097/PAS.0000000000001155
67. Feng H, Tong H, Yan J, He M, Chen W, Wang J. Genomic features and clinical characteristics of adolescents and young adults with cholangiocarcinoma. Front Oncol. (2019) 9:1439. doi: 10.3389/fonc.2019.01439
68. Anjanappa M, Hao Y, Simpson ER, Bhat-Nakshatri P, Nelson JB, Tersey SA, et al. A system for detecting high impact-low frequency mutations in primary tumors and metastases. Oncogene. (2018) 37:185–96. doi: 10.1038/onc.2017.322
69. Zhu C, Soto-Feliciano YM, Morris JP, Huang C-H, Koche RP, Ho Y-J, et al. MLL3 regulates the CDKN2A tumor suppressor locus in liver cancer. Elife. (2023) 12:e80854. doi: 10.7554/eLife.80854
70. Kim D-H, Kim J, Lee JW. Requirement for MLL3 in p53 regulation of hepatic expression of small heterodimer partner and bile acid homeostasis. Mol Endocrinol. (2011) 25:2076–83. doi: 10.1210/me.2011-1198
71. Li B, Liu HY, Guo SH, Sun P, Gong FM, Jia BQ. Association of MLL3 expression with prognosis in gastric cancer. Genet Mol Res. (2014) 13:7513–8. doi: 10.4238/2014.September.12.18
72. Li B, Liu H-Y, Guo S-H, Sun P, Gong F-M, Jia B-Q. A missense mutation (S3660L) in MLL3 gene influences risk of gastric cancer. J BUON. (2014) 19:394–7.
73. Li B, Liu H-Y, Guo S-H, Sun P, Gong F-M, Jia B-Q. Mll3 genetic variants affect risk of gastric cancer in the chinese han population. Asian Pac J Cancer Prev. (2013) 14:4239–42. doi: 10.7314/APJCP.2013.14.7.4239
74. Nemtsova MV, Kalinkin AI, Kuznetsova EB, Bure IV, Alekseeva EA, Bykov II, et al. Mutations in epigenetic regulation genes in gastric cancer. Cancers (Basel). (2021) 13(18):4586. doi: 10.3390/cancers13184586
75. Kung C-Y, Fang W-L, Hung Y-P, Huang K-H, Chen M-H, Chao Y, et al. Comparison of the mutation patterns between tumor tissue and cell-free DNA in stage IV gastric cancer. Aging (Albany NY). (2023) 15:777–90. doi: 10.18632/aging.v15i3
76. Zhou C, Feng X, Yuan F, Ji J, Shi M, Yu Y, et al. Difference of molecular alterations in HER2-positive and HER2-negative gastric cancers by whole-genome sequencing analysis. Cancer Manag Res. (2018) 10:3945–54. doi: 10.2147/CMAR.S172710
77. Wang R, Song S, Harada K, Ghazanfari Amlashi F, Badgwell B, Pizzi MP, et al. Multiplex profiling of peritoneal metastases from gastric adenocarcinoma identified novel targets and molecular subtypes that predict treatment response. Gut. (2020) 69:18–31. doi: 10.1136/gutjnl-2018-318070
78. Zhang R, Wu H-X, Xu M, Xie X. KMT2A/C mutations function as a potential predictive biomarker for immunotherapy in solid tumors. biomark Res. (2020) 8:71. doi: 10.1186/s40364-020-00241-0
79. Sun B, Chen H, Lao J, Tan C, Zhang Y, Shao Z, et al. The epigenetic modifier lysine methyltransferase 2C is frequently mutated in gastric remnant carcinoma. J Pathol Clin Res. (2023) 9:409–22. doi: 10.1002/cjp2.335
80. Varricchi G, Loffredo S, Marone G, Modestino L, Fallahi P, Ferrari SM, et al. The immune landscape of thyroid cancer in the context of immune checkpoint inhibition. Int J Mol Sci. (2019) 20(16):3934. doi: 10.3390/ijms20163934
81. Huang Y, Liu J, Zhu X. Mutations in lysine methyltransferase 2C and PEG3 are associated with tumor mutation burden, prognosis, and antitumor immunity in pancreatic adenocarcinoma patients. Digit Health. (2022) 8:20552076221133699. doi: 10.1177/20552076221133699
82. Mueller S, Engleitner T, Maresch R, Zukowska M, Lange S, Kaltenbacher T, et al. Evolutionary routes and KRAS dosage define pancreatic cancer phenotypes. Nature. (2018) 554:62–8. doi: 10.1038/nature25459
83. Golan T, Hammel P, Reni M, Van Cutsem E, Macarulla T, Hall MJ, et al. Maintenance olaparib for germline BRCA-mutated metastatic pancreatic cancer. N Engl J Med. (2019) 381:317–27. doi: 10.1056/NEJMoa1903387
84. Bryant KL, Mancias JD, Kimmelman AC, Der CJ. KRAS: feeding pancreatic cancer proliferation. Trends Biochem Sci. (2014) 39:91–100. doi: 10.1016/j.tibs.2013.12.004
85. Storz P, Crawford HC. Carcinogenesis of pancreatic ductal adenocarcinoma. Gastroenterology. (2020) 158:2072–81. doi: 10.1053/j.gastro.2020.02.059
86. Bailey P, Chang DK, Nones K, Johns AL, Patch A-M, Gingras M-C, et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature. (2016) 531:47–52. doi: 10.1038/nature16965
87. Buscail L, Bournet B, Cordelier P. Role of oncogenic KRAS in the diagnosis, prognosis and treatment of pancreatic cancer. Nat Rev Gastroenterol Hepatol. (2020) 17:153–68. doi: 10.1038/s41575-019-0245-4
88. Philip PA, Azar I, Xiu J, Hall MJ, Hendifar AE, Lou E, et al. Molecular characterization of KRAS wild-type tumors in patients with pancreatic adenocarcinoma. Clin Cancer Res. (2022) 28:2704–14. doi: 10.1158/1078-0432.CCR-21-3581
89. Yoon K-A, Woo SM, Kim Y-H, Kong S-Y, Lee MK, Han S-S, et al. Comprehensive cancer panel sequencing defines genetic diversity and changes in the mutational characteristics of pancreatic cancer patients receiving neoadjuvant treatment. Gut Liver. (2019) 13:683–9. doi: 10.5009/gnl18355
90. Researchers strive to refine TMB. Cancer Discovery. (2021) 11:1314. doi: 10.1158/2159-8290.CD-ND2021-0107
91. Gimeno-Valiente F, Martín-Arana J, Tébar-Martínez R, Gambardella V, Martínez-Ciarpaglini C, García-Micó B, et al. Sequencing paired tumor DNA and white blood cells improves circulating tumor DNA tracking and detects pathogenic germline variants in localized colon cancer. ESMO Open. (2023) 8:102051. doi: 10.1016/j.esmoop.2023.102051
92. Shin W, Yun J, Han K, Park D-G. Comparison of genetic variation between primary colorectal cancer and metastatic peritoneal cancer. Genes Genomics. (2023) 45:989–1001. doi: 10.1007/s13258-023-01408-3
93. Yang J, Zhao S, Su J, Liu S, Wu Z, Ma W, et al. Comprehensive genomic profiling reveals prognostic signatures and insights into the molecular landscape of colorectal cancer. Front Oncol. (2023) 13:1285508. doi: 10.3389/fonc.2023.1285508
94. Liu K, Wang J-F, Zhan Y, Kong D-L, Wang C. Prognosis model of colorectal cancer patients based on NOTCH3, KMT2C, and CREBBP mutations. J Gastrointest Oncol. (2021) 12:79–88. doi: 10.21037/jgo-21-28
95. Desette A, Guichet P-O, Emambux S, Masliantsev K, Cortes U, Ndiaye B, et al. Deciphering brain metastasis stem cell properties from colorectal cancer highlights specific stemness signature and shared molecular features. Cell Mol Gastroenterol Hepatol. (2023) 16:757–82. doi: 10.1016/j.jcmgh.2023.07.008
96. Chakrabarty S, Varghese VK, Sahu P, Jayaram P, Shivakumar BM, Pai CG, et al. Targeted sequencing-based analyses of candidate gene variants in ulcerative colitis-associated colorectal neoplasia. Br J Cancer. (2017) 117:136–43. doi: 10.1038/bjc.2017.148
97. Watanabe Y, Castoro RJ, Kim HS, North B, Oikawa R, Hiraishi T, et al. Frequent alteration of MLL3 frameshift mutations in microsatellite deficient colorectal cancer. PloS One. (2011) 6:e23320. doi: 10.1371/journal.pone.0023320
98. Larsson C, Cordeddu L, Siggens L, Pandzic T, Kundu S, He L, et al. Restoration of KMT2C/MLL3 in human colorectal cancer cells reinforces genome-wide H3K4me1 profiles and influences cell growth and gene expression. Clin Epigenet. (2020) 12:74. doi: 10.1186/s13148-020-00863-z
99. Liu R, Niu Y, Liu C, Zhang X, Zhang J, Shi M, et al. Association of KMT2C/D loss-of-function variants with response to immune checkpoint blockades in colorectal cancer. Cancer Sci. (2023) 114:1229–39. doi: 10.1111/cas.15716
100. Fagan RJ, Dingwall AK. COMPASS Ascending: Emerging clues regarding the roles of MLL3/KMT2C and MLL2/KMT2D proteins in cancer. Cancer Lett. (2019) 458:56–65. doi: 10.1016/j.canlet.2019.05.024
101. Qian J, Zhao S, Zou Y, Rahman SMJ, Senosain M-F, Stricker T, et al. Genomic underpinnings of tumor behavior in in situ and early lung adenocarcinoma. Am J Respir Crit Care Med. (2020) 201:697–706. doi: 10.1164/rccm.201902-0294OC
102. Shang Y, Li X, Liu W, Shi X, Yuan S, Huo R, et al. Comprehensive genomic profile of Chinese lung cancer patients and mutation characteristics of individuals resistant to icotinib/gefitinib. Sci Rep. (2020) 10:20243. doi: 10.1038/s41598-020-76791-y
103. Yang B, Li J, Li F, Zhou H, Shi W, Shi H, et al. Comprehensive analysis of age-related somatic mutation profiles in Chinese young lung adenocarcinoma patients. Cancer Med. (2019) 8:1350–8. doi: 10.1002/cam4.1839
104. Gao Y, Bado I, Wang H, Zhang W, Rosen JM, Zhang XH-F. Metastasis organotropism: redefining the congenial soil. Dev Cell. (2019) 49:375–91. doi: 10.1016/j.devcel.2019.04.012
105. Feng A, Li Y, Li G, Wang Y, Wen Q, Yang Z, et al. Genomic features of organ-specific metastases in lung adenocarcinoma. Front Oncol. (2022) 12:908759. doi: 10.3389/fonc.2022.908759
106. Hess KR, Varadhachary GR, Taylor SH, Wei W, Raber MN, Lenzi R, et al. Metastatic patterns in adenocarcinoma. Cancer. (2006) 106:1624–33. doi: 10.1002/cncr.21778
107. Riihimäki M, Hemminki A, Fallah M, Thomsen H, Sundquist K, Sundquist J, et al. Metastatic sites and survival in lung cancer. Lung Cancer. (2014) 86:78–84. doi: 10.1016/j.lungcan.2014.07.020
108. Liu Z, Zheng M, Lei B, Zhou Z, Huang Y, Li W, et al. Whole-exome sequencing identifies somatic mutations associated with lung cancer metastasis to the brain. Ann Transl Med. (2021) 9:694. doi: 10.21037/atm-21-1555
109. Reck M, Remon J, Hellmann MD. First-line immunotherapy for non-small-cell lung cancer. J Clin Oncol. (2022) 40:586–97. doi: 10.1200/JCO.21.01497
110. Liu D, Benzaquen J, Morris LGT, Ilié M, Hofman P. Mutations in KMT2C, BCOR and KDM5C predict response to immune checkpoint blockade therapy in non-small cell lung cancer. Cancers (Basel). (2022) 14(11):2816. doi: 10.3390/cancers14112816
111. Gu G, Yu B, Wan H, Lu S, Zhu X, Zhao Y, et al. Molecular characteristics and the effect of KRAS mutation on the prognosis of immunotherapy in non-small cell lung cancer in Xinjiang, China. Onco Targets Ther. (2022) 15:1021–32. doi: 10.2147/OTT.S381825
112. Bai X, Wu D-H, Ma S-C, Wang J, Tang X-R, Kang S, et al. Development and validation of a genomic mutation signature to predict response to PD-1 inhibitors in non-squamous NSCLC: a multicohort study. J Immunother Cancer. (2020) 8(1):e000381. doi: 10.1136/jitc-2019-000381
113. Fantini D, Glaser AP, Rimar KJ, Wang Y, Schipma M, Varghese N, et al. A Carcinogen-induced mouse model recapitulates the molecular alterations of human muscle invasive bladder cancer. Oncogene. (2018) 37:1911–25. doi: 10.1038/s41388-017-0099-6
114. Armenia J, Wankowicz SAM, Liu D, Gao J, Kundra R, Reznik E, et al. The long tail of oncogenic drivers in prostate cancer. Nat Genet. (2018) 50:645–51. doi: 10.1038/s41588-018-0078-z
115. Coelho KBCA, Squire JA, Duarte KG, Sares CTG, Moreda NA, Pereira JL, et al. Germline variants in early and late-onset Brazilian prostate cancer patients. Urol Oncol. (2024) S1078-1439:00016–4. doi: 10.1016/j.urolonc.2024.01.015
116. Liu Z, Guo H, Zhu Y, Xia Y, Cui J, Shi K, et al. TP53 alterations of hormone-naïve prostate cancer in the Chinese population. Prostate Cancer Prostatic Dis. (2021) 24:482–91. doi: 10.1038/s41391-020-00302-3
117. Zhu S, Xu N, Liang J, Zhao F, Wang Z, Ni Y, et al. Mutations in epigenetic regulator KMT2C detected by liquid biopsy are associated with worse survival in prostate cancer patients. Oncol Res. (2023) 31:605–14. doi: 10.32604/or.2023.028321
118. Moghoofei M, Keshavarz M, Ghorbani S, Babaei F, Nahand JS, Tavakoli A, et al. Association between human papillomavirus infection and prostate cancer: A global systematic review and meta-analysis. Asia Pac J Clin Oncol. (2019) 15:e59–67. doi: 10.1111/ajco.13124
119. Lang B, Cao C, Zhao X, Wang Y, Cao Y, Zhou X, et al. Genomic alterations related to HPV infection status in a cohort of Chinese prostate cancer patients. Eur J Med Res. (2023) 28:239. doi: 10.1186/s40001-023-01207-2
120. Elshafei A, Al-Toubat M, Feibus AH, Koul K, Jazayeri SB, Lelani N, et al. Genetic mutations in smoking-associated prostate cancer. Prostate. (2023) 83:1229–37. doi: 10.1002/pros.24554
121. Limberger T, Schlederer M, Trachtová K, Garces de Los Fayos Alonso I, Yang J, Högler S, et al. KMT2C methyltransferase domain regulated INK4A expression suppresses prostate cancer metastasis. Mol Cancer. (2022) 21:89. doi: 10.1186/s12943-022-01542-8
122. Mathioudaki A, Ljungström V, Melin M, Arendt ML, Nordin J, Karlsson Å, et al. Targeted sequencing reveals the somatic mutation landscape in a Swedish breast cancer cohort. Sci Rep. (2020) 10:19304. doi: 10.1038/s41598-020-74580-1
123. Hao S, Tian W, Zhao J, Chen Y, Zhang X, Gao B, et al. Analysis of circulating tumor DNA to predict neoadjuvant therapy effectiveness and breast cancer recurrence. J Breast Cancer. (2020) 23:373–84. doi: 10.4048/jbc.2020.23.e41
124. van Geelen CT, Savas P, Teo ZL, Luen SJ, Weng C-F, Ko Y-A, et al. Clinical implications of prospective genomic profiling of metastatic breast cancer patients. Breast Cancer Res. (2020) 22:91. doi: 10.1186/s13058-020-01328-0
125. Gala K, Li Q, Sinha A, Razavi P, Dorso M, Sanchez-Vega F, et al. KMT2C mediates the estrogen dependence of breast cancer through regulation of ERα enhancer function. Oncogene. (2018) 37:4692–710. doi: 10.1038/s41388-018-0273-5
126. Ansari KI, Hussain I, Kasiri S, Mandal SS. HOXC10 is overexpressed in breast cancer and transcriptionally regulated by estrogen via involvement of histone methylases MLL3 and MLL4. J Mol Endocrinol. (2012) 48:61–75. doi: 10.1530/JME-11-0078
127. Huang R, Zhu L, Zhang Y. XIST lost induces ovarian cancer stem cells to acquire taxol resistance via a KMT2C-dependent way. Cancer Cell Int. (2020) 20:436. doi: 10.1186/s12935-020-01500-8
128. Ruault M, Brun ME, Ventura M, Roizès G, De Sario A. MLL3, a new human member of the TRX/MLL gene family, maps to 7q36, a chromosome region frequently deleted in myeloid leukaemia. Gene. (2002) 284:73–81. doi: 10.1016/S0378-1119(02)00392-X
129. Parsons DW, Li M, Zhang X, Jones S, Leary RJ, Lin JC-H, et al. The genetic landscape of the childhood cancer medulloblastoma. Science. (2011) 331:435–9. doi: 10.1126/science.1198056
130. Jones DTW, Northcott PA, Kool M, Pfister SM. The role of chromatin remodeling in medulloblastoma. Brain Pathol. (2013) 23:193–9. doi: 10.1111/bpa.12019
131. Wong GC-H, Li KK-W, Wang W-W, Liu AP-Y, Huang QJ, Chan AK-Y, et al. Clinical and mutational profiles of adult medulloblastoma groups. Acta Neuropathol Commun. (2020) 8:191. doi: 10.1186/s40478-020-01066-6
132. Zhang L, Liu Y, Wang M, Wu Z, Li N, Zhang J, et al. EZH2-, CHD4-, and IDH-linked epigenetic perturbation and its association with survival in glioma patients. J Mol Cell Biol. (2017) 9:477–88. doi: 10.1093/jmcb/mjx056
133. Kleefstra T, Kramer JM, Neveling K, Willemsen MH, Koemans TS, Vissers LELM, et al. Disruption of an EHMT1-associated chromatin-modification module causes intellectual disability. Am J Hum Genet. (2012) 91:73–82. doi: 10.1016/j.ajhg.2012.05.003
Keywords: histone methylation, KMT2C, biological process, cancer, immunotherapy
Citation: Jiao Y, Lv Y, Liu M, Liu Y, Han M, Xiong X, Zhou H, Zhong J, Kang X and Su W (2024) The modification role and tumor association with a methyltransferase: KMT2C. Front. Immunol. 15:1444923. doi: 10.3389/fimmu.2024.1444923
Received: 06 June 2024; Accepted: 22 July 2024;
Published: 06 August 2024.
Edited by:
Zichuan Liu, Tianjin University, ChinaReviewed by:
Liang Wang, China Medical University, ChinaKai Xu, Huazhong University of Science and Technology, China
Copyright © 2024 Jiao, Lv, Liu, Liu, Han, Xiong, Zhou, Zhong, Kang and Su. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jiateng Zhong, anR6aG9uZ0B4eG11LmVkdS5jbg==; Xiaohong Kang, a3hoaGdkQDE2My5jb20=; Wei Su, aG5zd2VpQDE2My5jb20=
†These authors have contributed equally to this work