 Cordelia Dunai1,2*
Cordelia Dunai1,2* Claire Hetherington2Sarah A. Boardman2Jordan J. Clark3†Parul Sharma3
Claire Hetherington2Sarah A. Boardman2Jordan J. Clark3†Parul Sharma3 Krishanthi Subramaniam3Kukatharmini Tharmaratnam4
Krishanthi Subramaniam3Kukatharmini Tharmaratnam4 Edward J. Needham5
Edward J. Needham5 Robyn Williams6†Yun Huang2Greta K. Wood2
Robyn Williams6†Yun Huang2Greta K. Wood2 Ceryce Collie2Andrew Fower6
Ceryce Collie2Andrew Fower6 Hannah Fox6Mark A. Ellul2Marie Held7Franklyn N. Egbe2
Hannah Fox6Mark A. Ellul2Marie Held7Franklyn N. Egbe2 Michael Griffiths2
Michael Griffiths2 Tom Solomon1,2,8Gerome Breen9,10
Tom Solomon1,2,8Gerome Breen9,10 Anja Kipar3,11
Anja Kipar3,11 Jonathan Cavanagh12Sarosh R. Irani8†
Jonathan Cavanagh12Sarosh R. Irani8† Angela Vincent6
Angela Vincent6 James P. Stewart3
James P. Stewart3 Leonie S. Taams13‡David K. Menon14‡Benedict D. Michael1,2,8*‡
Leonie S. Taams13‡David K. Menon14‡Benedict D. Michael1,2,8*‡- 1NIHR Health Protection Research Unit in Emerging and Zoonotic Infections, Liverpool, United Kingdom
- 2Clinical Infection Microbiology and Immunology, Institute of Infection Ecology and Veterinary Sciences, University of Liverpool, Liverpool, United Kingdom
- 3Department of Infection Biology and Microbiomes, Institute of Infection, Veterinary and Ecological Sciences, University of Liverpool, Liverpool, United Kingdom
- 4Department of Health Data Science, Institute of Population Health, Faculty of Health and Life Sciences, University of Liverpool, Liverpool, United Kingdom
- 5Department of Clinical Neurosciences, University of Cambridge, Cambridge, United Kingdom
- 6Nuffield Department of Clinical Neurosciences, Medical Sciences Division, University of Oxford, Oxford, United Kingdom
- 7Centre for Cell Imaging, Faculty of Health and Life Sciences, University of Liverpool, Liverpool, United Kingdom
- 8Department of Neurology, The Walton Centre NHS Foundation Trust, Liverpool, United Kingdom
- 9Department of Social, Genetic & Developmental Psychiatry Centre, School of Mental Health & Psychological Sciences, King’s College London, London, United Kingdom
- 10NIHR Maudsley Biomedical Research Centre, King’s College London, London, United Kingdom
- 11Laboratory for Animal Model Pathology, Institute of Veterinary Pathology, Vetsuisse Faculty, University of Zurich, Zurich, Switzerland
- 12College of Medical, Veterinary and Life Sciences, University of Glasgow, Glasgow, United Kingdom
- 13Centre for Inflammation Biology and Cancer Immunology, Department of Inflammation Biology, School of Immunology & Microbial Sciences, Faculty of Life Sciences & Medicine, King’s College London, London, United Kingdom
- 14Division of Anaesthesia, Addenbrooke’s Hospital, Cambridge University Hospitals, Cambridge, United Kingdom
Neurological complications, including encephalopathy and stroke, occur in a significant proportion of COVID-19 cases but viral protein is seldom detected in the brain parenchyma. To model this situation, we developed a novel low-inoculum K18-hACE2 mouse model of SARS-CoV-2 infection during which active viral replication was consistently seen in mouse lungs but not in the brain. We found that several mediators previously associated with encephalopathy in clinical samples were upregulated in the lung, including CCL2, and IL-6. In addition, several inflammatory mediations, including CCL4, IFNγ, IL-17A, were upregulated in the brain, associated with microglial reactivity. Parallel in vitro experiments demonstrated that the filtered supernatant from SARS-CoV-2 virion exposed brain endothelial cells induced activation of uninfected microglia. This model successfully recreates SARS-CoV-2 virus-associated para-infectious brain inflammation which can be used to study the pathophysiology of the neurological complications and the identification of potential immune targets for treatment.
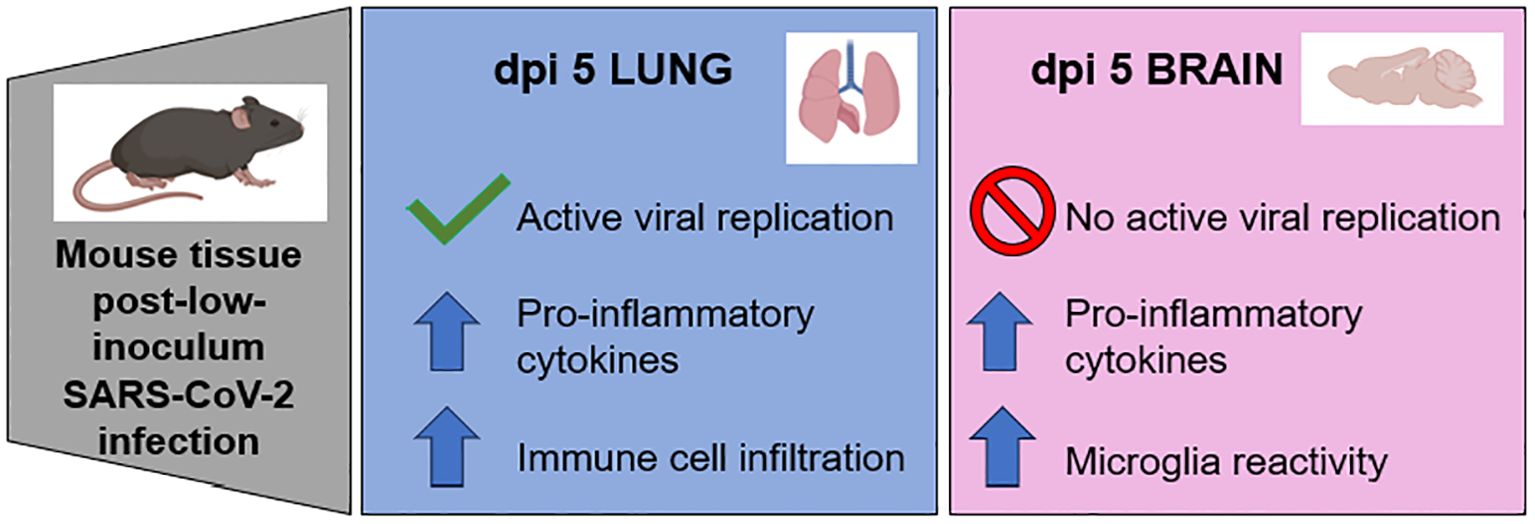
Graphical Abstract. The low inoculum SARS-CoV-2 mouse model of para-infectious brain effects in the absence of active viral replication had increased cytokines (CCL4, IFNγ and IL-17A) and microglia reactivity (increased Iba1 expression and ramification index).
Introduction
SARS-CoV-2 infection has been associated with a range of neurological complications. Although their incidence has decreased with widespread vaccination and effective anti-viral and anti-inflammatory treatments, they remain a significant clinical issue (1). A large retrospective study with contemporary controls found neurological complications were more common in people who had experienced COVID-19 but were also found in non-hospitalised/mild cases of COVID-19, revealing a large healthcare burden left in the wake of the pandemic (2). The different neurological complications, ranging from loss of smell to encephalitis, caused by SARS-CoV-2 infection most likely have very different aetiologies, but viral protein is seldom found in the brain parenchyma strongly suggesting that indirect effects of the virus, such as immune-mediated pathologies, are a likely potential cause (3–6). Indeed, we and others previously found that COVID-19 patients with neurological complications had elevated serum immune mediators and cytokines (IL-6, IL-12p40, IL1-RA, M-CSF, CCL2, and HGF) which correlated with serum brain injury markers (7–10).
Mouse models are important to systematically study early changes in disease and disease progression, but most models of SARS-CoV-2 have involved systemic dissemination with brain pathology that may not reflect the clinical scenarios (11–14). We established a mouse model using an inoculum of virus ten times lower than that which earlier studies have used, and looked for evidence of brain immune activation in the absence of viral replication in the brain. We also developed an in vitro assay to investigate how exposure of endothelial cells to viral protein can lead to cytokine-mediated indirect effects on microglia, which we hypothesize is the most common clinical scenario for how SARS-CoV-2 affects the brain (15).
Methods
Mouse studies of infection with SARS-CoV-2
An AWERB-approved protocol was followed for the mouse studies (University of Liverpool Animal Welfare and Ethical Review Body, UK Home Office Project Licence PP4715265). Mice were maintained under SPF barrier conditions in individually ventilated cages. Male and female 2-4 month old heterozygote hACE2-transgenic C57BL/6 mice (Charles River Laboratory) were infected intranasally with 1x103 or 1x104 plaque-forming units (PFU) of a human isolate of SARS-CoV-2 (Pango B lineage hCoV-2/human/Liverpool/REMRQ0001/2020) under isoflurane anaesthesia. Mice were euthanized on day 5 post infection. Brains were perfused with 30mL PBS w/1mM EDTA, then one hemisphere fixed in formaldehyde-containing PLP buffer overnight at 4°C. Brains were then subjected to a sucrose gradient– 10 and 20% sucrose for 1hr each and then 30% sucrose O/N at 4°C. Brains were then frozen in OCT by submerging moulds in a beaker of 2-methylbutane on dry ice. The other hemisphere was divided in two sagittal sections and half preserved in 4%PFA for histology and half in trizol for RNA and protein extraction. Sera was collected and frozen and then heat-inactivated at 56°C for 30 mins prior to be moved from CL3 to CL2 lab. Lung tissue was preserved in 4%PFA for histology, in trizol for RNA and protein extraction, and in PLP for cryosectioning. Cytokines were measured from tissue protein extract with Bio-rad reagents on a Bio-plex 200 following the manufacturer's protocol.
qPCR of SARS-CoV-2 genes and mouse cytokines
Gene expression was measured from trizol isolated RNA (Invitrogen cat# 15596018, manufacturer’s protocol) using Promega’s GoTaq Probe 1-Step RT-qPCR system (cat#A6120, manufacturer’s protocol) on an Agilent AriaMx. Primers and FAM probes for SARS-CoV-2, cytokines, and housekeeping genes were purchased from IDT (Tables 1, 2) with standard IDT qPCR primer/probe sets for the mouse cytokines.
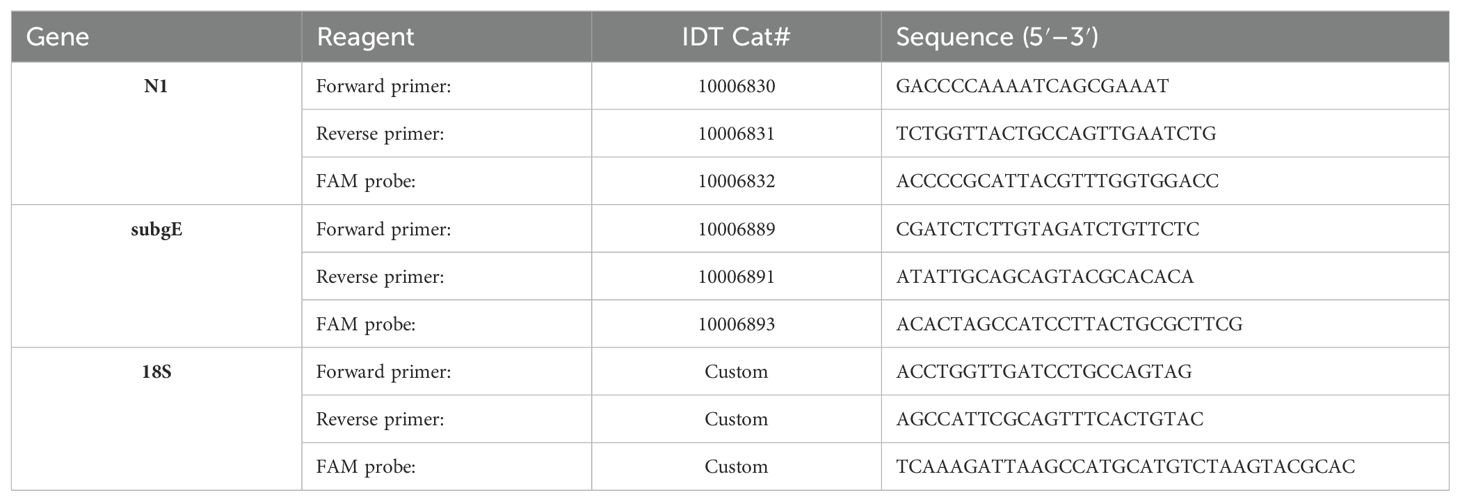
Table 1. Primers and probes for viral genes and normalization.
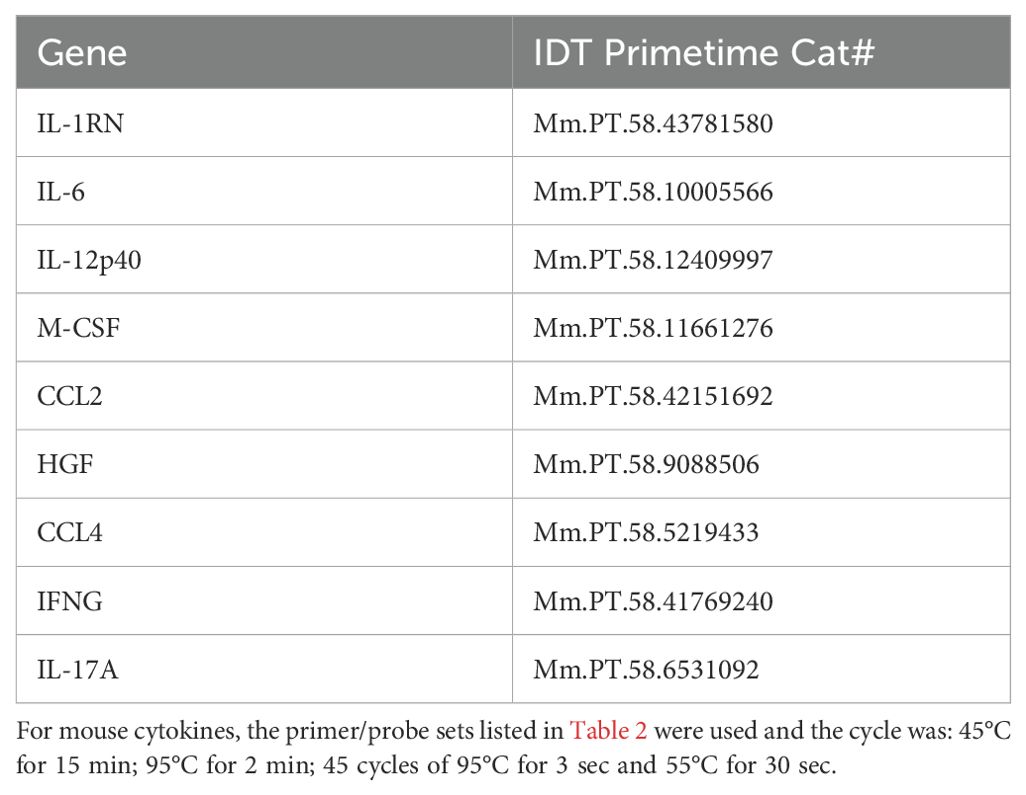
Table 2. Primers and probes for mouse cytokines.
The thermal cycle for N1 and the mouse cytokines was: 45°C for 15min 1x, 95°C for 2 min, then 45 cycles of 95°C for 3 secs followed by 55°C for 30 sec.
For subgenomic E: 45°C for 15 min 1x, 95°C for 2 min, then 45 cycles of 95°C for 15 secs followed by 58°C for 30 sec.
For 18S it was: 45°C for 15 min, 95°C for 2 min and 40 cycles of 95°C for 15s, 60°C for 1 min.
Histology and confocal microscopy
Paraffin-embedded formalin-fixed tissue was sectioned to 4 um sections. Slides were baked at 60°C for 30 minutes and then stained with H&E in an autostainer. H&E slides were imaged on a Leica microscope. For immunofluorescent staining and confocal microscopy, OCT-embedded frozen tissue was sectioned to 12 um or 30 um sections (thicker sections needed for the Z-stack imaging and microglia ramification/reactivation quantification). 100% acetone was used for antigen retrieval (10 mins at room temperature). After air-drying, then PBS washing, tissue sections were permeabilized with 0.1% Triton X-100/PBS (20 mins at room temperature). After rinsing with PBS, tissue sections were blocked with Dako block (5 minutes at room temperature). After another PBS wash, primary antibodies were added at dilutions listed in table for an overnight incubation at 4°C in a humidified chamber. Tissue sections were washed twice with PBS for 5 minutes each wash. Secondary antibody (as described in Table 3) was added for a 2 hr room temperature incubation, followed by two 5-minute PBS washes. DAPI-mounting medium was used for coverslipping. Imaging was performed with Andor Dragonfly spinning disk confocal microscope. Marker fluorescence and microglia counts, intensity, and reactivation indices [method based on previous work (16, 17)] were quantified with Fiji (confocal microscope set up in Table 4 and macros downloadable from the public Github repository). The reactivation index is the area of the cell divided by the projection area (the whole polygon covered by the cell). Reactivation indices of 0-1 of objects with a threshold size of 19 mm2 were quantified from three Z-stack images/mouse and two mice/group.
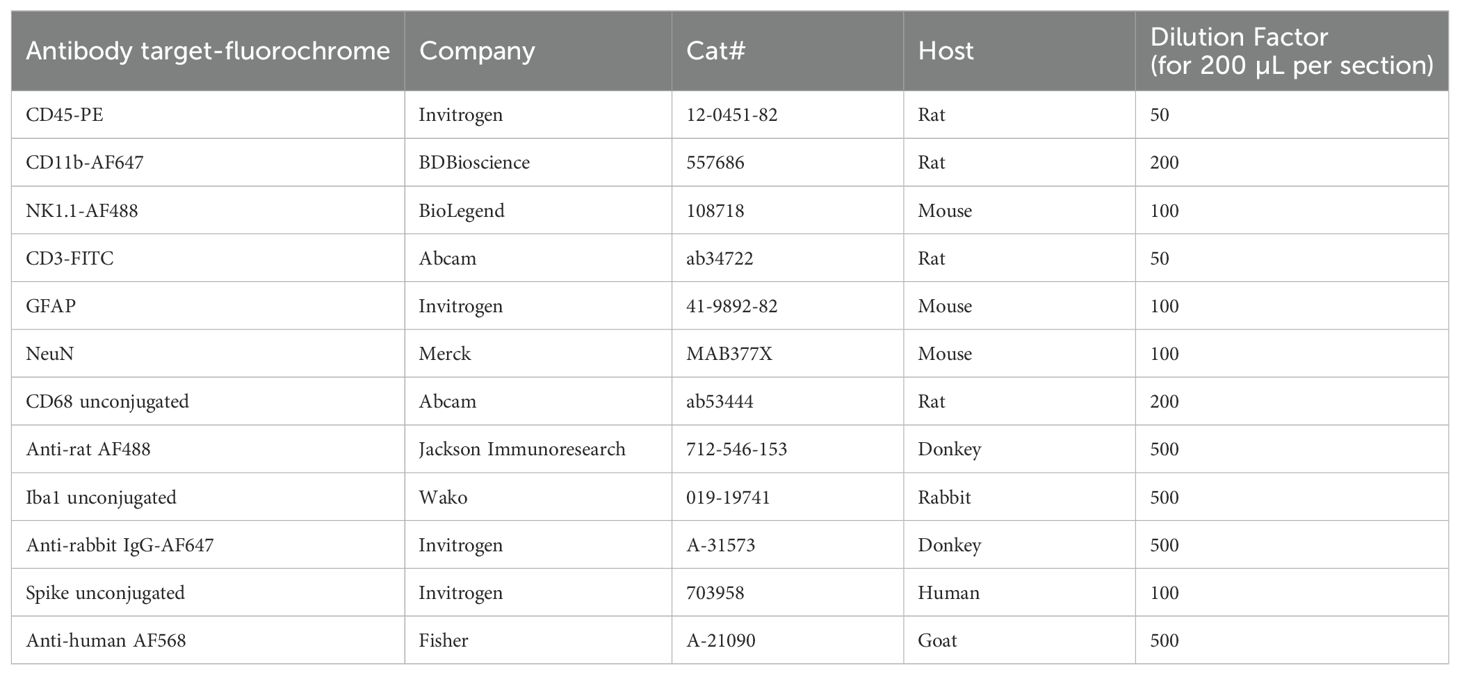
Table 3. Antibodies for immunofluorescent stain and confocal imaging.
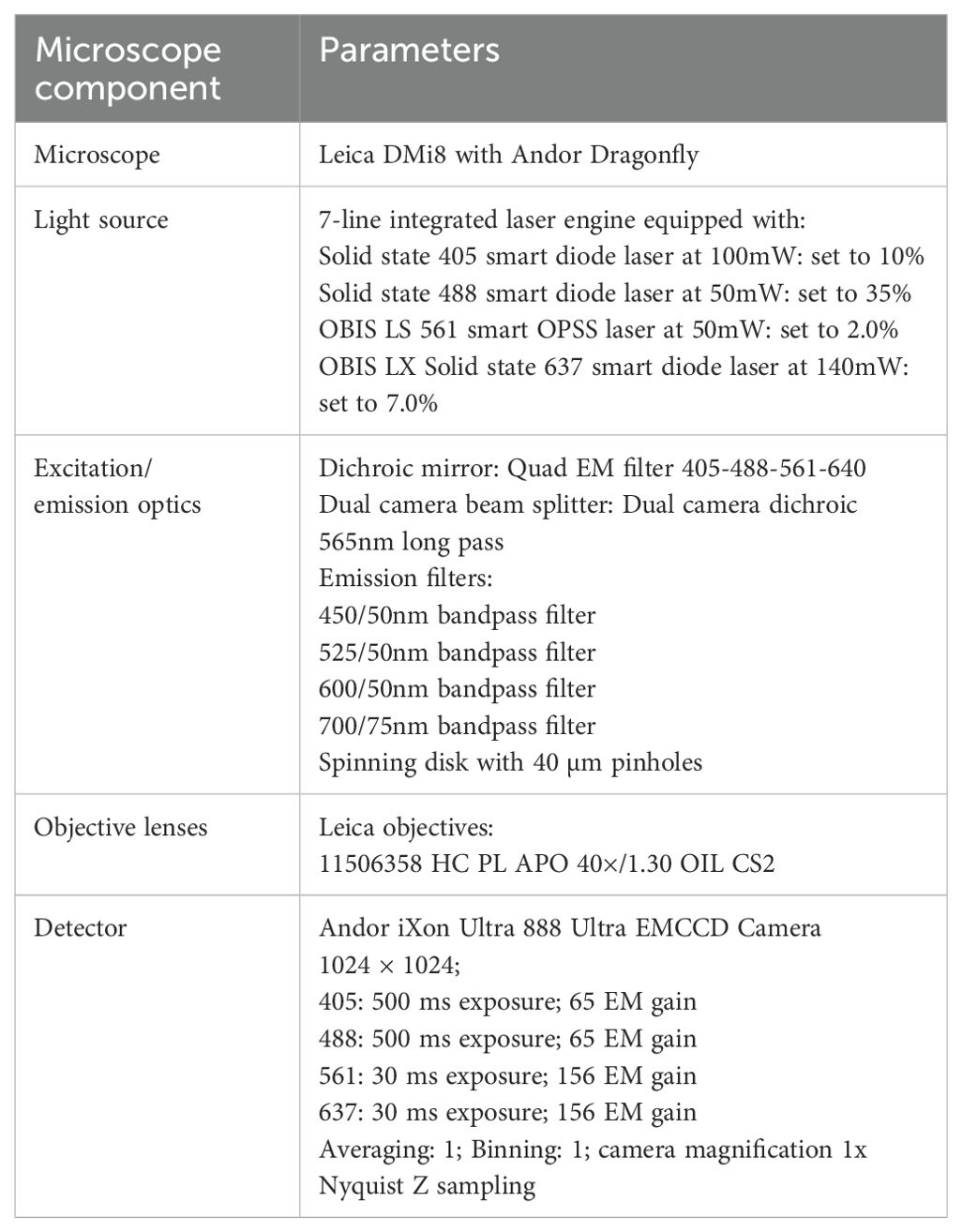
Table 4. Confocal microscopy settings.
In vitro cell culture assays
The investigated cell lines were cultured according to the manufacturer’s recommendations. The mouse brain endothelial cell line, bEnd.3 (ECACC 96091929) was used between passage 22 and 29. The cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 2mM Glutamine, 5µM 2-Mercaptoethanol (2ME), 1mM Sodium Pyruvate (NaP), 1% Non Essential Amino Acids (NEAA), 10% Foetal Bovine Serum (FBS) and 1X penicillin/streptomycin (P/S). Primary mouse microglia (ScienCell, SC-M1900-57) were cultured on poly-L-lysine (ScienCell 0413) coated flasks in Microglia medium (ScienCell 1901) supplemented with 1% FBS, 1x microglia growth supplement (MGS, ScienCell 1952) and 1 x P/S. Primary mouse astrocytes were cultured on poly-L-lysine (ScienCell 0413) coated flasks in Astrocyte Medium (ScienCell 1801) supplemented with 2% FBS, 1x astrocyte growth supplement (AGS, ScienCell 1852) and 1 x P/S.
In vitro incubation with inactivated virus and endothelial supernatants
Acid/heat inactivated SARS-CoV-2 (B.1.1.7) (NIBSC 101027) of known titre was used for all experiments. For incubation with inactivated virus, cells were seeded into either 24 well plates or 6 well plates at 125,000 cells per cm2 and allowed to adhere overnight. Virus dilutions were then prepared to give a ratio of 1 copy (MOI 1), 0.1 copies (MOI 0.1) or 0.01 copies (MOI 0.01) per cells plated in the culture medium. For bEnd.3 experiments, cells were incubated for 24 hours with inactivated virus, 20 µg/mL polyI:C (Merck PL530) or with untreated culture medium. For astrocyte and microglia experiments, cells were incubated for 2 hours with inactivated virus, polyI:C, untreated medium, or supernatant from bEnd.3 cells previously exposed for 24 hours to MOI 1 virus dilution. Supernatant from bEnd.3 cells was filtered with a 20 nm syringe filter directly prior to treating microglia and astrocytes to remove viral particles. After 2 hours, treatments were removed, cells were washed with 1X phosphate-buffered saline (PBS) (Capricorn Scientific CSR154) three times and then the normal culture medium was replaced. For each treatment condition (virus or supernatant) 6-7 individual well replicates were performed and for control conditions (poly:IC or untreated) 3-4 replicates were performed. After 24 hours, the supernatant from these cells was collected and cells were fixed with 4% paraformaldehyde (Sigma 16005).
ELISA of in vitro supernatants
Supernatants were collected into 1 mL cryovials and stored at 4°C for no longer than 1 week prior to cytokine levels being assessed using ELISA. Kits were purchased from Invitrogen for IL-6 (88-7064), CCL2 (88-7391), IFNγ (88-7314) and performed according to manufacturer’s protocol.
Immunostaining of in vitro samples
Microglia were fixed with 4% paraformaldehyde and washed 3x with 1X PBS with calcium and magnesium (PBS +/+) (Capricorn Scientific CSR1576). Cells were then incubated for 1 hour with Dakoblock (Agilent X090930-2). Cells were then washed 1x with PBS and incubated with primary antibody (Iba1 Wako Chemicals 019-19741; CD45-PE Thermo Fisher 12-0451-83) diluted in antibody diluent (1X PBS +/+, 1% BSA, 10% donkey serum (Sigma D9663), 0.1% Triton-X (Sigma X100)) overnight at 4°C. Cells were then washed 3x with 1x PBS +/+ and incubated with secondary antibody (Donkey anti-rabbit AF657, Thermo Fisher A31573) and DAPI (Thermofisher P36962) in antibody diluent and incubated in the dark for 2 hours at room temperature. Cells were then washed 3x with PBS +/+.
In vitro confocal microscopy and reactivation/ramification index quantitation
Microglia were imaged at 25x magnification on a Leica DMi8 on an Andor Dragonfly spinning disk confocal microscope. 16 images per well were taken and stitched into a tilescan image using Fusion imaging software. Tilescan images were then processed in Fiji/ImageJ by thresholding the PE/647 channel and gating for individual cells (excluding cell clusters) and then analysing particles between 500-1000 µm for solidity.
Statistical analyses
Prism software (version 9.4.1, GraphPad Software Inc.) was used for graph generation and statistical analysis. The Shapiro-Wilk normality test used to check the normality of the distribution. Data are expressed as mean ± S.E.M. The difference between two or more non-normally distributed groups was tested using Mann-Whitney U or Kruskal-Wallis tests, respectively. P ≤ 0.05 was considered statistically significant.
Results
Low inoculum intranasal SARS-CoV-2 infection in human ACE2 transgenic C57BL/6 mice does not cause viral replication in the brain parenchyma
We established the mouse model by comparing intranasal infection with a low (1x103 PFU) and a high (1x104 PFU) inoculum of SARS-CoV-2, collecting serum, brain and lung tissues at day 5 post-infection (Figure 1A). At this stage, none of the mice showed weight loss (Supplementary Figure 1A). Both levels of infection caused pathology in the lung, with viral loads evidenced by qPCR of N1 with both low and high inocula (Figure 1B). There was also evidence of active viral replication in the lung, as defined by the surrogate readout of qPCR of subgenomic E which correlates with infectivity (18) (Figure 1C). SARS-CoV-2 N1 transcripts were detected in four out of four brains of mice that had received high inoculum of SARS-CoV-2 and in six of nine that received low inoculum (Figure 1D; first experiment displayed, n=4 group). However, whilst three of four high inoculum mice showed active viral transcription in the brain by the detection of subgenomic E, this was only detectable in two of nine low inoculum mouse brains (tissue from these two animals were not included in subsequent ex vivo experiments, Figure 1E; Supplementary Figures 1E–G).
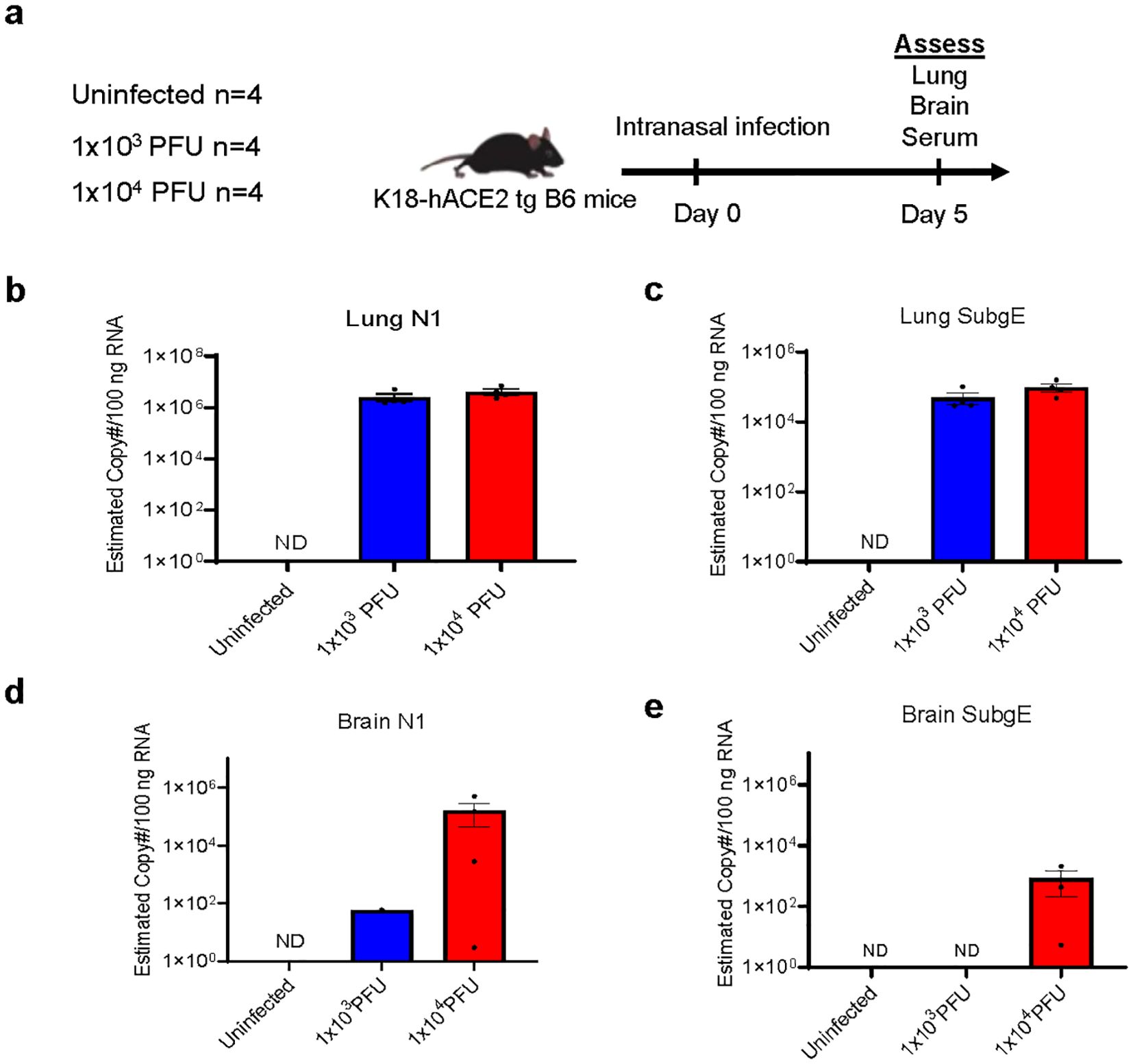
Figure 1. Low inoculum intranasal SARS-CoV-2 infection in human ACE2 transgenic C57BL/6 mice does not cause viral replication in the brain parenchyma. (A) Schema of K18 human-ACE2 transgenic C57B/6 mouse study randomised to no infection, 'low inoculum' infection at 1x103 plaque-forming units (PFU) and 'high inoculum' infection at 1x104 PFU, with endpoint at day 5 post-infection, showing one of two independent experiments, (n=4/group). (B) Real-time polymerase chain reaction (RT-QPCR) identifies SARS-CoV-2 N1 and (C) subgenomic E transcripts in lung homogenate confirming pulmonary infection and viral lytic replication at both inoculum of infection. (D) RT-QPCR of brain homogenate demonstrates that minimal SARS-CoV-2 N1 is present in the perfused brain parenchyma and there are no detectable SARS-CoV-2 subgenomic E transcripts (E) confirming the absence of lytic viral replication in the brain at low-dose infection. ND, not detected.
H&E staining of lung and brain sections from infected mice were assessed for pathology and mononuclear cell clusters (Supplementary Figures 1B–D). The lungs of SARS-CoV-2 infected mice showed signs of pathology including oedema, haemorrhage, and fibrosis (Supplementary Figure 1B). Mononuclear cells were present with both inocula compared with uninfected mice (Supplementary Figure 1B. The brain tissue also had a few clusters of mononuclear cells in the frontal cortex with both inocula of SARS-CoV-2 (Supplementary Figures 1C, D).
Spike protein is present in the lungs, but not brains, of 1x103 PFU SARS-CoV-2 infected mice and there are differences in immune activation
Immunofluorescent staining and confocal microscopy of the lungs showed large amounts of viral spike protein with increased Iba1 expression (Figure 2A). The lungs also showed increases in CD45 and CD11b staining compared with uninfected animals (Figure 3A). Confocal microscopy of ex vivo brain sections from mice, following the low inoculum, demonstrated Iba-1 staining but, in contrast to lung tissue, showed no evidence of staining for SARS-CoV-2 spike protein (Figures 2B, 3B). These brains also showed no increases in numbers of CD3+ or NK1.1+ cells (Supplementary Figures 2A, B), or apoptotic cells (Supplementary Figure 2C).
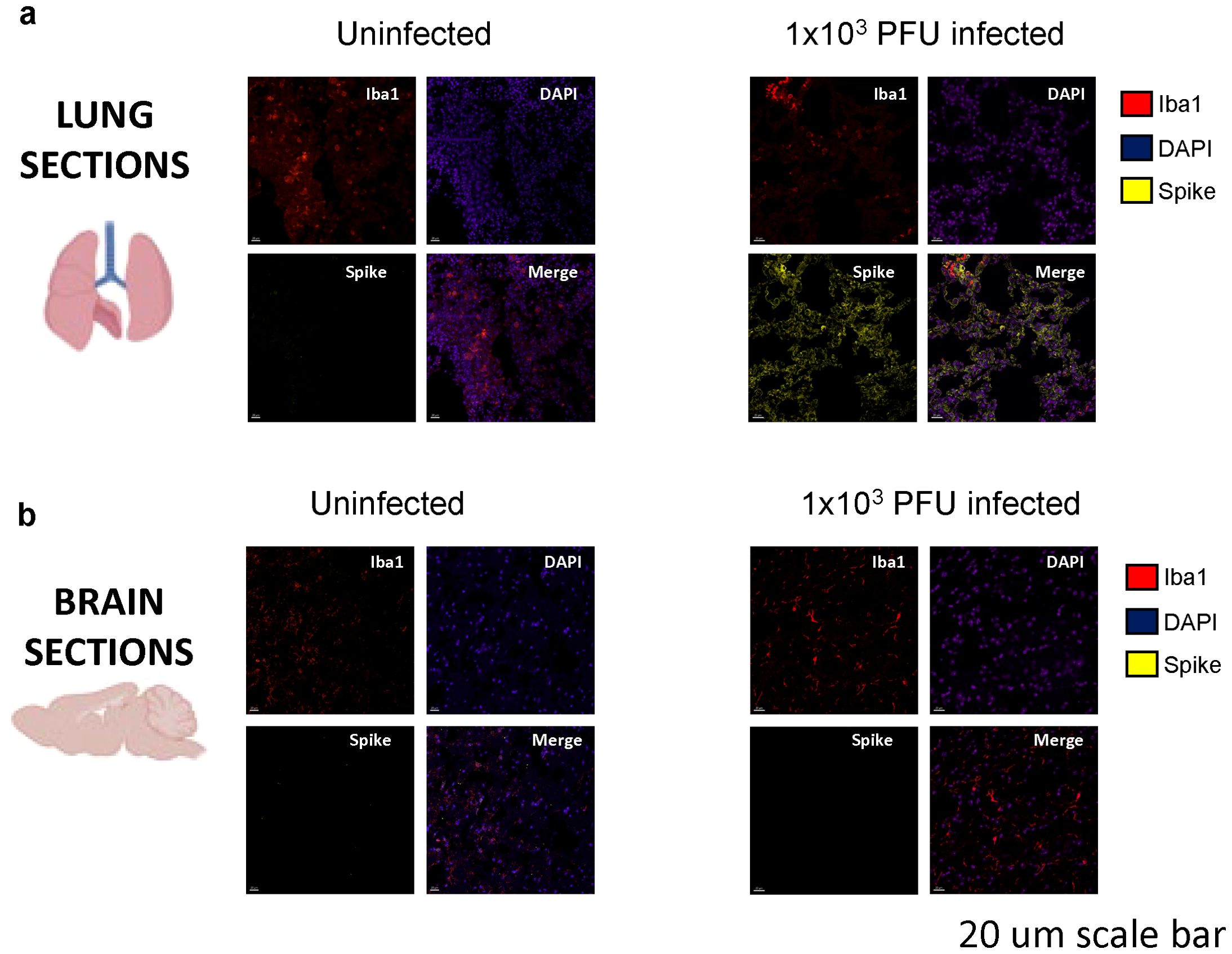
Figure 2. Spike protein is present in the lungs, but not in the brains of low-inoculum SARS-CoV-2 infected mice at day 5 post 1x103 PFU infection. (A) Confocal microscopy of low-inoculum infection in this model confirms the presence of both SARS-CoV-2 spike protein (yellow) and monocyte lineage cells (red) in the lungs. (B) Despite the absence of spike protein (yellow) in the brain in the low-inoculum infection model, there are consistent large areas of accumulation of Iba1+ microglia (red) of perfused brain parenchyma.
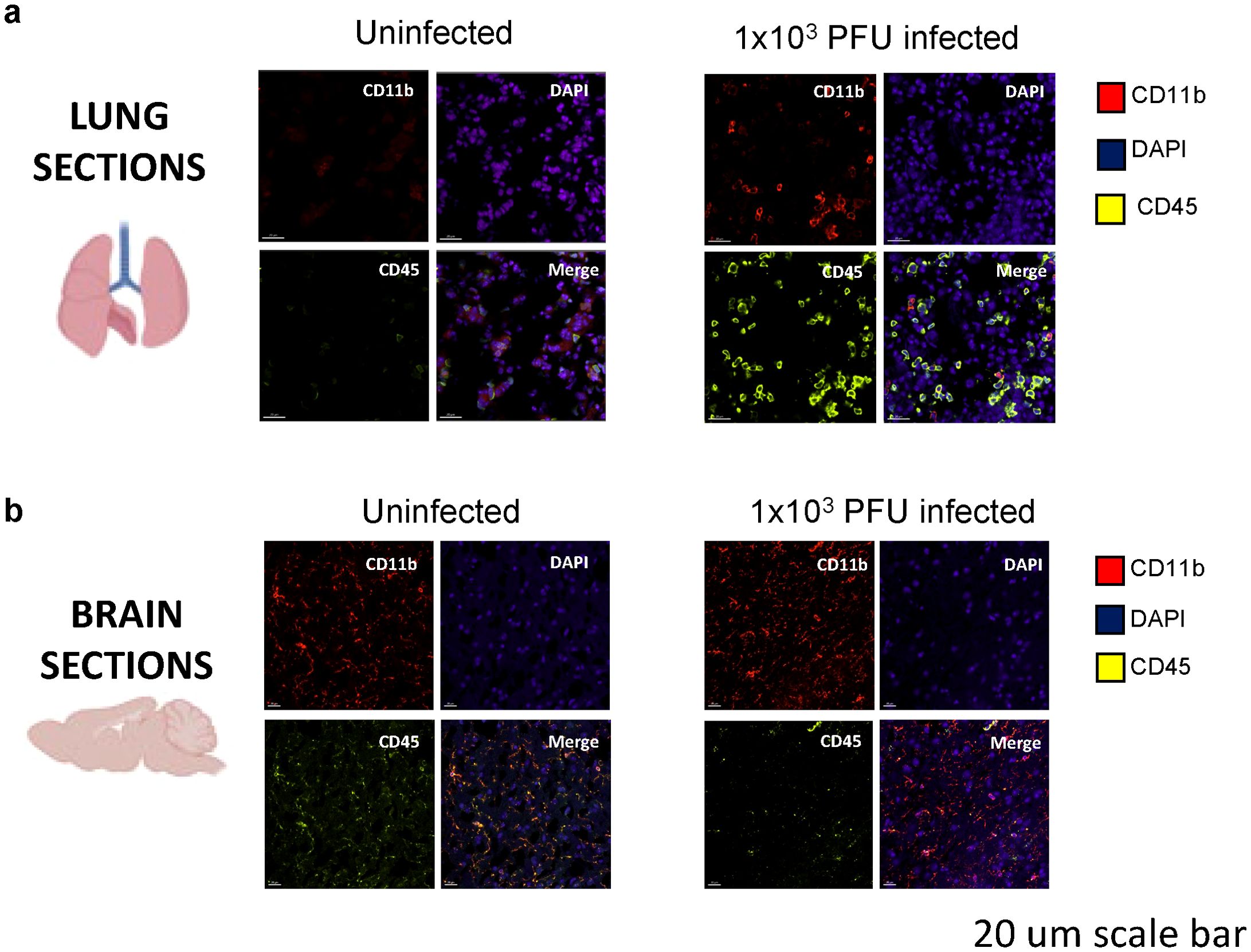
Figure 3. Immune activation is present in the lungs of SARS-CoV-2 infected mice at day 5 post 1x103 PFU infection. (A) Confocal microscopy of perfused organs in the low-inoculum SARS-CoV-2 infection model identifies the presence of CD45 positive leucocytes (yellow) and occasionally CD11b positive cells of macrophage/monocyte lineage (red), shown with DAPI (blue) in the lung. (B) No significant differences in CD45 and CD11b expression were found when comparing uninfected and infected brains.
Several inflammatory mediators are elevated in the lungs and brains of 1x103 PFU SARS-CoV-2 infected mice
In order to understand the mechanisms driving this apparent para-infectious neuropathology, we assessed transcription and protein levels of inflammatory mediators in brains and lung from the seven low-inoculum infected mice that showed no evidence of local viral replication in the brain. Our previous clinical study, comparing serum immune mediators from low vs. normal Glasgow coma scale score COVID-19 patients, demonstrated that six mediators of interest were increased in serum (7). Lung tissue from the mice revealed four of these, IL1-RA, IL-6, CCL2, and IL-12p40, to be upregulated with low-inoculum infection by QPCR (Figures 4A, B).
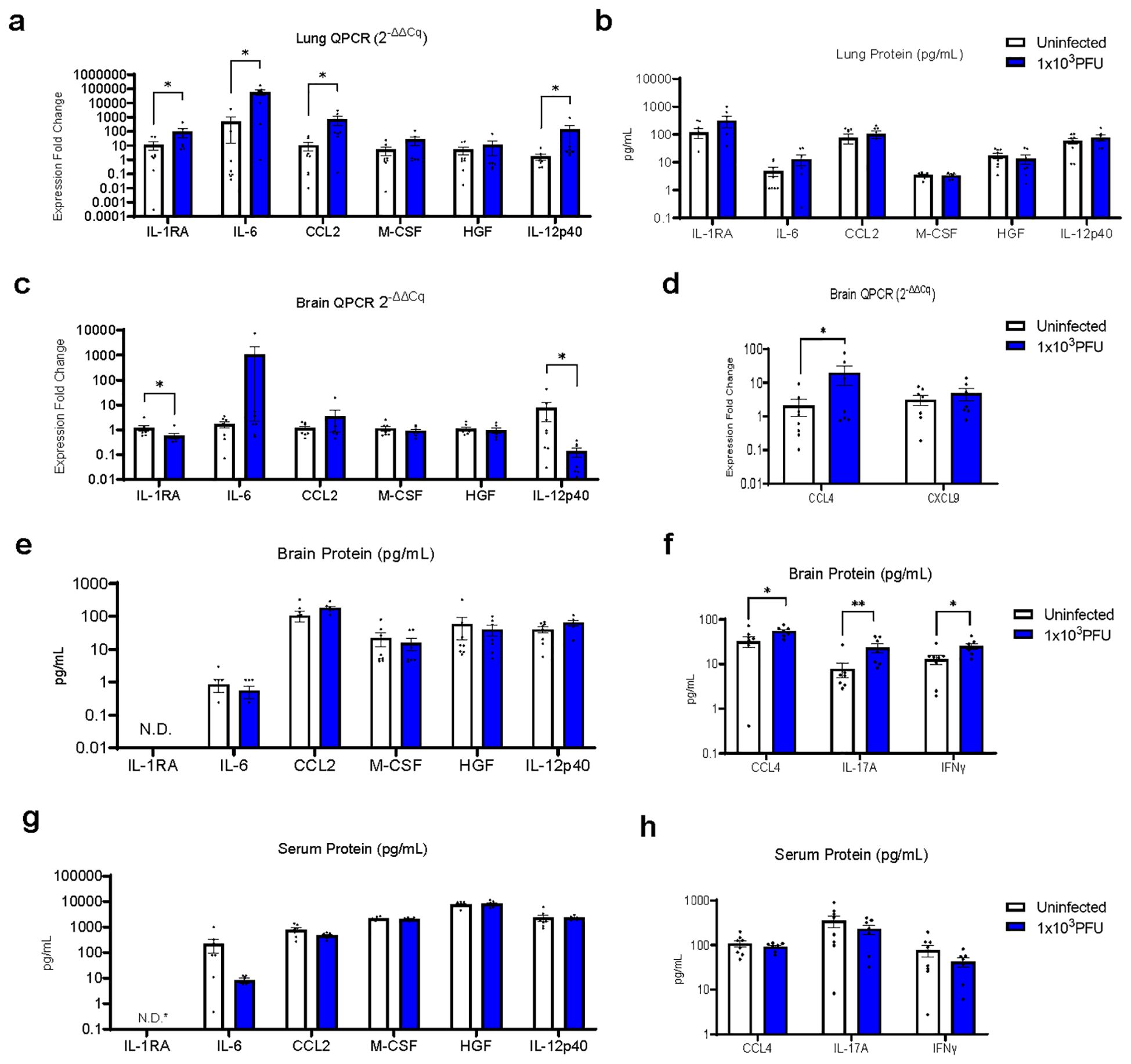
Figure 4. RT-QPCR of perfused lung parenchyma in the low-inoculum infection model (day 5 post 1x103 PFU infection) confirms upregulation of several inflammatory mediators. (A) RT-QPCR of six immune mediators of interest in perfused lung. (B) Luminex was used to check the same six immune mediators of interest in perfused lung. (C–F) In this low-inoculum infection model several pro-inflammatory mediators were found to be increased in the perfused brains, including (D) CCL4 by RT-QPCR and (F) IL-17A and IFNγ by Luminex. (G, H) In contrast, no statistically significant differences were seen in serum cytokine protein levels. *IL-RA undetectable by ELISA in all, but one uninfected serum sample (=3.5 pg/mL). (These data are from two independent experiments combined, n=7-8/group). Pairwise comparisons by Mann-Whitney U test (*p<0.05, **p<0.01).
The brains from low inoculum infected mice showed increased transcripts of CCL4 and decreased levels of IL-1RA and IL-12p40 (Figures 4C, D; Supplementary Table 1). Although many brain cytokine proteins were not different between uninfected and low inoculum mice, CCL4, IFNγ and IL-17A were increased (Figures 4E, F; Supplementary Table 2). Consistent with an intra-cerebral local response, there were no increases in any of these cytokines in the mouse sera (Figures 4G, H).
Brains from low inoculum SARS-CoV-2 infected mice showed immune activation and increases in microglial reactivity despite the absence of active SARS-CoV-2 replication
Brains from low inoculum mice showed no detectable viral proteins (checked by spike staining) and no detectable viral transcription (checked using subgenomic E). However, these brains showed reactive microglia, with increased Iba1 expression (as measured by percentage area, fluorescence intensity and reactivation indices, Figures 5A–F). Clusters of GFAP+ astrocytes were found in the regions of high Iba1 expression in the brain, suggesting concomitant microgliosis and astrogliosis, as has been reported in human post-mortem samples (Supplementary Figures 3A, B). To ask whether parainfectious brain injury with potential blood-brain-barrier damage had taken place, the brain supernatant/serum albumin ratios were measured in uninfected mice and low inoculum infected animals (Supplementary Figure 3C). In humans post-COVID-19, NfL has been found to be raised, but there were no significant differences in the brain injury marker NfL, either by ELISA or Simoa (Supplementary Figure 3D) at this early time point (day 5 post infection) (7).
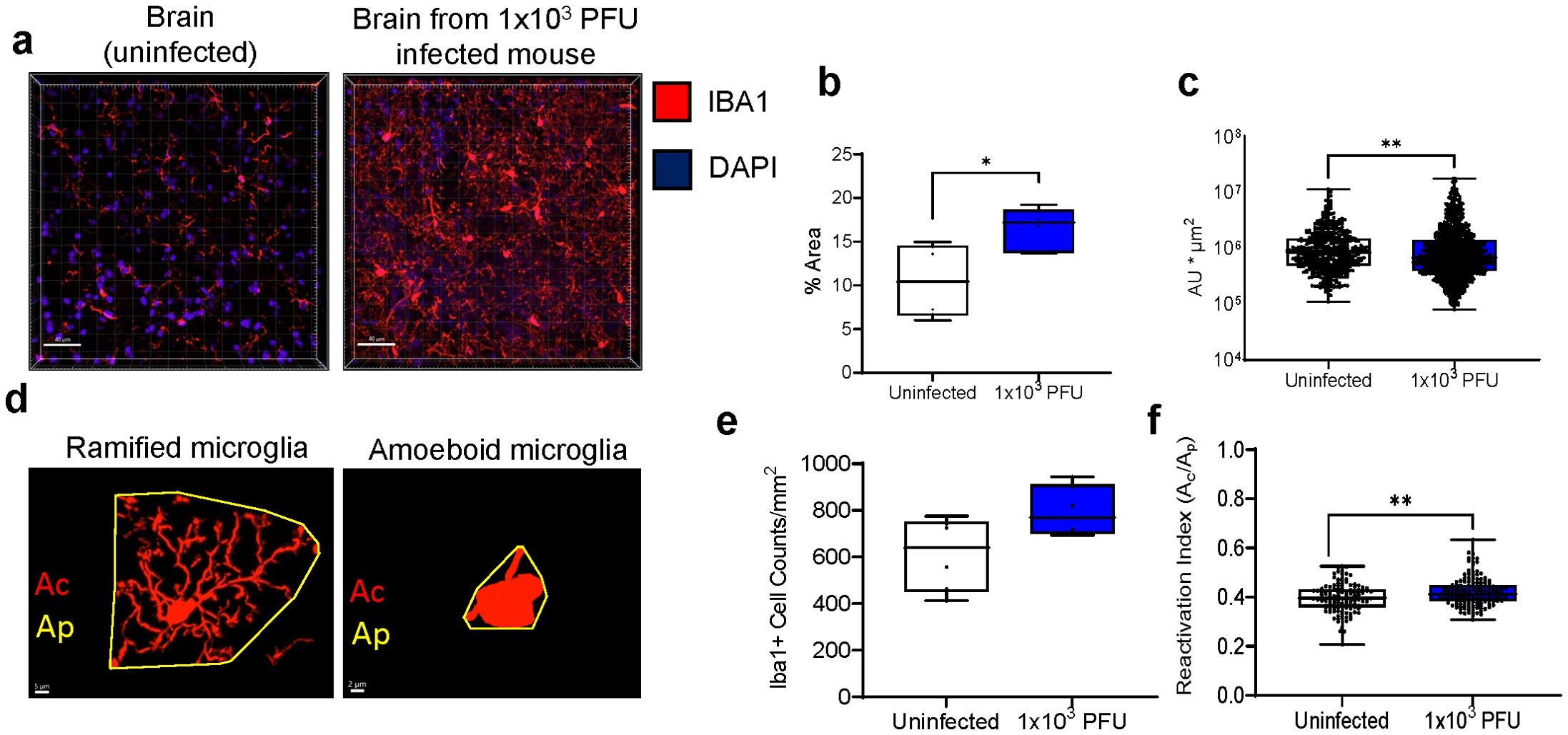
Figure 5. Low inoculum intranasal SARS-CoV-2 infection in mice results in increased pro-inflammatory cytokines in the lung and brain and causes increased microglia reactivity in the brain. (A) Confocal microscopy of 30 μm sections of frontal lobe brain parenchyma following low-inoculum infection reveals reactive microglia with increased expression of Iba1 (red) with nuclei stained by DAPI (purple, 3 images/mouse brain at 40Х, n=2/group). (B) % area that was Iba1-positive, (C) Iba1+ cell counts, (D) Examples of microglia morphology Reactive Iba1+ microglia were quantified by (E) Iba1 fluorescence intensity, and (F) reactivation index between 0 and 1. Ac, cell area; Ap, projection area. Pairwise comparisons by Mann-Whitney U test (*p< 0.05, **p<0.01).
Viral effects on endothelial cells in vitro lead to pro-inflammatory cytokine production and subsequent microglia reactivation
We hypothesized that the cerebral vasculature is the most common route for SARS-CoV-2 infection interacting with neuroglial cells and causing local, not necessarily systemic, inflammation. To study the cascade of events in a controlled environment, we exposed mouse endothelial cells (bEnd.3 cell line) to inactivated SARS-CoV-2 viral particles at three multiplicities of infection (MOI), collected and filtered the supernatant, and measured the cytokines present. The TLR3 agonist polyI:C served as a positive control (Figures 6A, B). The MOIs of 0.1 and 1 used to treat endothelial cells produced significant amounts of pro-inflammatory cytokines IL-6 and CCL2, so the MOI of 1 was chosen for the subsequent experiments as a way to mirror direct infection of brain endothelial cells, but not microglia and astrocytes (Figures 6C–E; Supplementary Figures 4A, B). Primary mouse microglia and astrocytes were exposed to the filtered supernatant from endothelial cells and their cytokine production and for microglia, their reactivation index was measured by Iba1+ morphology (Figures 6D, E; Supplementary Figures 4A, B). The supernatant from MOI=1 exposed endothelial cells resulted in the highest reactivation index indicating a pathway by which SARS-CoV-2 can indirectly affect brain cells and which is consistent with our in vivo data.
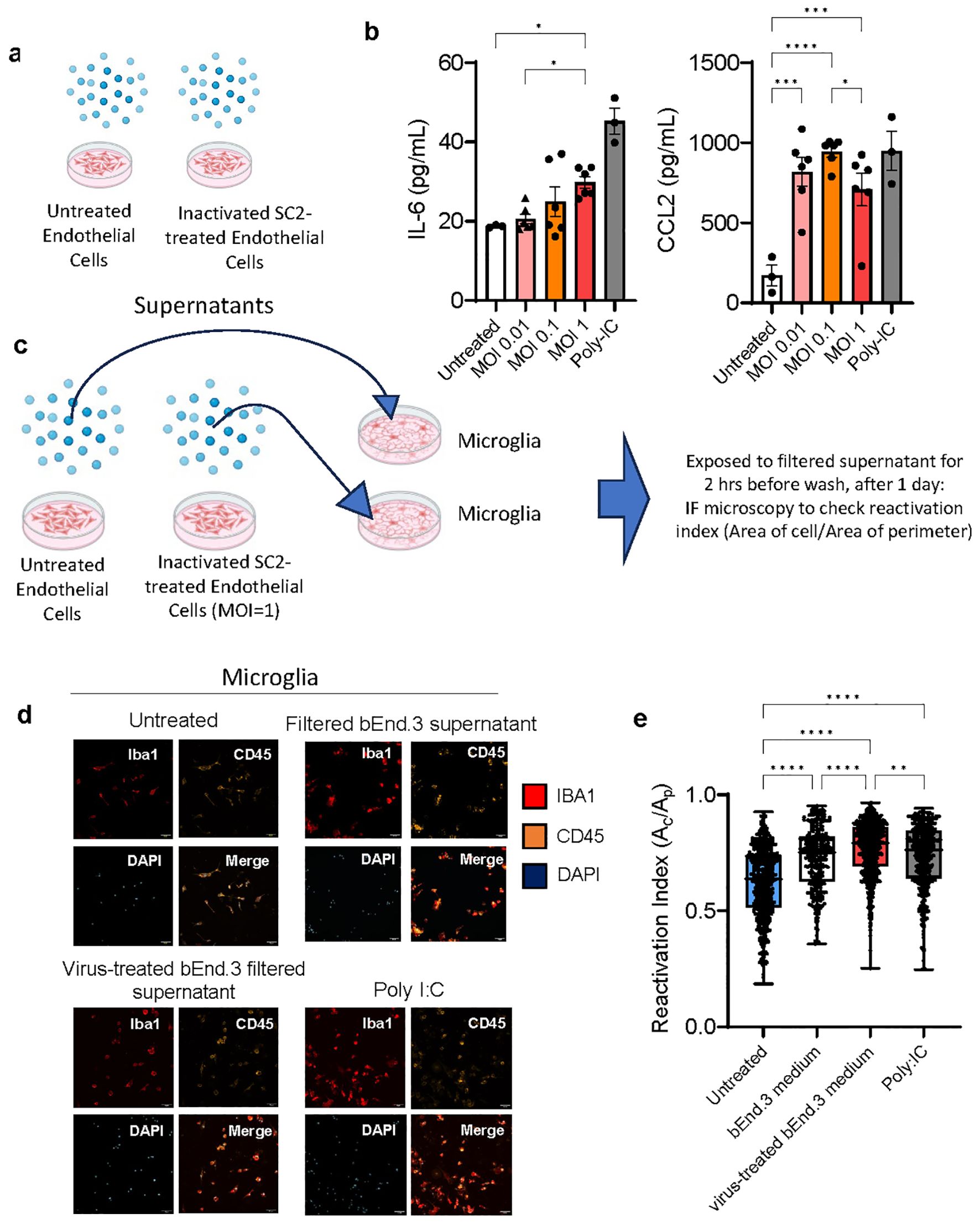
Figure 6. Viral effects on endothelial cells in vitro lead to pro-inflammatory cytokine production and subsequent microglia reactivation. (A, B) bEnd.3 cells were treated with heat and acid inactivated SARS-CoV-2 at a ratio of 0.01,0.1, or 1 virus copies per cell for 24 hours. At 24 hours concentrations of cytokines IL-6 and CCL2 were determined by ELISA and groups compared by ANOVA *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. (C–E) Primary mouse microglia were treated with 20 nm filtered supernatant taken from bEnd. 3 cells incubated with virus at an MOI of 1, for 2 hours, before being washed and culture medium replaced. At 24 hours cells were fixed and immunostained for Ibal and CD45 and imaged by confocal microscopy. 6-7 separate wells were cultured for each treatment condition and 3-4 for each control condition, with 16 images taken per well at 25x magnification. Dots represent individual cells. Groups compared by Kruskal-Wallis *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.
Discussion
To better study the acute host response in SARS-CoV-2, and given that SARS-CoV-2 is rarely identified in the brain parenchyma in clinical samples, we developed a low-inoculum mouse model of COVID-19 which induced pulmonary infection in the absence of replication of virus in the brain. Following intranasal administration, by five days the mice had developed evidence of lung infection and immune cell infiltration (18), with production of inflammatory mediators, including CCL2, IL-6, IL-12p40 and IL-1RA; these cytokines were also elevated acutely in the serum of COVID-19 patients with low Glasgow coma scale scores (7). Despite the absence of viral replication in the brain parenchyma, there was some, albeit limited, production of inflammatory mediators in the brain, including CCL4, IFNγ and IL-17A. In addition, there was an increase in microglial Iba1 staining and microglia reactivation morphology. We had expected to also see increased numbers of CD45+ and CD11b+ cells, but believe that this was limited by the thin cryosections examined. In fact, we needed to examine thicker cryosections in order to quantify the microglia morphology (17). In future studies, flow cytometry would be an important way to quantify immune cell infiltration. Immune infiltrates and microgliosis were also observed in our previous mouse studies (19, 20). Parallel in vitro studies demonstrated that the filtered supernatant from brain endothelial cells exposed to SARS-CoV-2 virions, induced activation of microglia and production of CCL2. Our in vitro model using inactivated virus (due to limitations on CL3 work) delivered viral particles to endothelial cells and we hypothesize that this strongly stimulated them via innate PAMP pathways such as TLRs. Building on this, future studies could apply similar approaches to compare direct and indirect effects of vaccines and active virus on neuroglial cells. Active viruses would stimulate by DAMPs and PAMPs.
Our findings suggest that a primarily pulmonary inflammatory process is rapidly associated with parainfectious immune activation in the brain and the signature of an NK cell and/or T cell response which indicates a cascade of inflammation potentially amenable to treatment. Our mouse model is novel in using a low inoculum of SARS-CoV-2 virus for infection which does not induce lethal brain pathology, allowing us to study the immune activation in the brain in the absence of direct viral invasion. This is congruent with the majority of human autopsy results which show limited virus in the brain, but nevertheless demonstrate inflammation and microglia reactivity (21, 22). Human autopsy studies have studied this concept and found that exposure to lung-derived cytokines is association with microglia activation and that this was reduced by corticosteroid treatment (23). This may also reflect longer term effects, as a hamster model which examined neuropathology at 31 days post-SARS-CoV-2 infection found that Iba1 expression remained elevated (24). Studies of brain organoids have reported that Iba1+ microglia engulf post-synaptic material contributing to synapse elimination (25).
There have been reports of viral encephalitis and neuron degeneration and apoptosis observed in non-human primates (26, 27). Interestingly, in these studies the virus was present at low amounts in the brain and was found predominantly in the vasculature as visualized by co-localization with Von Willebrand Factor (27). Mimicking the clinical scenario, there was no correlation found between neurological markers with severity of respiratory disease. Another study reported increased CCL11 (eotaxin) in mouse serum and CSF that correlated with demyelination (28). That mouse model also lacked direct viral neural invasion by infecting mice that were intratracheally transfected with human ACE2. The demyelination was also observed after intraperitoneal administration of CCL11. Interestingly, clinical studies showed higher plasma levels of CCL11 in the patients who had brain fog (28). We did not observe elevated serum cytokines in our studies and this could be due to severity, timepoint, and/or technical differences. Our negative results in the serum are at least in part contributed to by heat-inactivation of experimental samples, which were part of safety protocols in the CL3 lab at the point when these experiments were conducted (29). Hamster studies have showed that COVID-19 leads to IL-1β and IL-6 expression within the hippocampus and medulla oblongata and is associated with decreased neurogenesis in the hippocampal dentate gyrus which leads to learning and memory deficits (30). This has also been shown in direct in vitro assays—with application of serum from COVID-19 patients with delirium with elevated IL-6 leading to decreased proliferation and increased apoptosis of a human hippocampal progenitor cell line (31). Our in vitro studies enabled us to isolate a potential mechanism by which SARS-CoV-2 indirectly affects brains cells—by studying endothelial cells which express ACE2 and can be directly infected by the virus (15), collecting their supernatants containing pro-inflammatory cytokines, and exposing microglia and astrocytes to them. Spike protein alone has previously been found to cause inflammation and associated cognitive deficits in animal models which is a common and important long-lasting symptom of COVID-19 in humans (32–34).
In conclusion, the low inoculum SARS-CoV-2 mouse model and parallel in vitro studies highlight an approach to study parainfectious effects on the brain and enables characterisation of the neuroglial cells themselves. The cytokine signature and microglia reactivity post infection indicate an acute local immune response including initial inflammation in the absence of active viral replication in the brain that could be amenable to targeted immunosuppression which can direct future studies.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
The animal study was approved by an AWERB-approved protocol was followed for the mouse studies (University of Liverpool Animal Welfare and Ethical Review Body, UK Home Office Project Licence PP4715265. The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
CD: Formal analysis, Investigation, Writing – original draft, Conceptualization, Funding acquisition, Methodology, Visualization. CH: Formal analysis, Investigation, Methodology, Visualization, Writing – review & editing. SB: Formal analysis, Writing – review & editing. JJC: Methodology, Writing – review & editing. PS: Methodology, Writing – review & editing. KS: Methodology, Writing – review & editing. KT: Formal analysis, Writing – review & editing. EN: Writing – review & editing. RW: Writing – review & editing. AF: Writing – review & editing. HF: Writing – review & editing. YH: Writing – review & editing. GW: Writing – review & editing. CC: Writing – review & editing. ME: Writing – review & editing. MH: Formal analysis, Methodology, Visualization, Writing – review & editing. FE: Writing – review & editing. MG: Writing – review & editing. TS: Writing – review & editing. GB: Writing – review & editing. AK: Writing – review & editing. JC: Writing – review & editing. SI: Writing – review & editing. AV: Conceptualization, Investigation, Visualization, Writing – original draft, Writing – review & editing. JS: Funding acquisition, Investigation, Methodology, Project administration, Resources, Writing – review & editing, Supervision. LT: Investigation, Methodology, Writing – review & editing, Supervision. DM: Writing – review & editing, Conceptualization, Investigation, Supervision, Visualization. BM: Writing – original draft, Writing – review & editing, Conceptualization, Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Visualization.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This research was funded by the National Institute for Health and Care Research (NIHR) (CO-CIN-01) and jointly by NIHR and UK Research and Innovation (CV220-169, MC_PC_19059). BDM is supported by the UKRI/MRC (MR/V03605X/1), the MRC/UKRI (MR/V007181/1), MRC (MR/T028750/1) and Wellcome (ISSF201902/3). BDM is also supported by the NIHR Health Protection Research Unit (HPRU) in Emerging and Zoonotic Infections at University of Liverpool in partnership with Public Health England (PHE), in collaboration with Liverpool School of Tropical Medicine and the University of Oxford [award 200907], NIHR HPRU in Respiratory Infections at Imperial College London with PHE [award 200927]. CD is supported by MRC (MC_PC_19044). We acknowledge the Liverpool Centre for Cell Imaging (CCI) for provision of imaging equipment (Dragonfly confocal microscope) and excellent technical assistance (BBSRC grant number BB/R01390X/1). TS is supported by The Pandemic Institute and the NIHR Health Protection Research Unit (HPRU) in Emerging and Zoonotic Infections at University of Liverpool. DM and EN are supported by the NIHR Cambridge Biomedical Centre and by NIHR funding to the NIHR BioResource (RG94028 and RG85445), and by funding from Brain Research UK 201819-20.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Author disclaimer
The views expressed are those of the author(s) and not necessarily those of the UKRI, NHS, the NIHR or the Department of Health and Social Care.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2024.1440324/full#supplementary-material
References
1. Grundmann A, Wu C-H, Hardwick M, Baillie JK, Openshaw PJM, Semple MG, et al. Fewer COVID-19 neurological complications with dexamethasone and remdesivir. Ann Neurol. (2023) 93-1:88–102. doi: 10.1002/ana.26536
2. Xu E, Xie Y, Al-Aly Z. Long-term neurologic outcomes of COVID-19. Nat Med. (2022), 1–10. doi: 10.1038/s41591-022-02001-z
3. Meinhardt J, Radke J, Dittmayer C, Franz J, Thomas C, Mothes R, et al. Olfactory transmucosal SARS-CoV-2 invasion as a port of central nervous system entry in individuals with COVID-19. Nat Neurosci. (2021) 24:168–75. doi: 10.1038/s41593-020-00758-5
4. Matschke J, Lütgehetmann M, Hagel C, Sperhake JP, Schröder AS, Edler C, et al. Neuropathology of patients with COVID-19 in Germany: a post-mortem case series. Lancet Neurol. (2020) 19:919–29. doi: 10.1016/S1474-4422(20)30308-2
5. Khan M, Yoo S-J, Clijsters M, Backaert W, Vanstapel A, Speleman K, et al. Visualizing in deceased COVID-19 patients how SARS-CoV-2 attacks the respiratory and olfactory mucosae but spares the olfactory bulb. Cell. (2021) 184:5932–5949.e15. doi: 10.1016/j.cell.2021.10.027
6. Dunai C, Collie C, Michael BD. Immune-mediated mechanisms of COVID-19 neuropathology. Front Neurol. (2022) 13. doi: 10.3389/fneur.2022.882905
7. Michael BD, Dunai C, Needham EJ, Tharmaratnam K, Williams R, Huang Y, et al. Para-infectious brain injury in COVID-19 persists at follow-up despite attenuated cytokine and autoantibody responses. Nat Commun. (2023) 14:8487. doi: 10.1038/s41467-023-42320-4
8. Needham EJ, Ren AL, Digby RJ, Norton EJ, Ebrahimi S, Outtrim JG, et al. Brain injury in COVID-19 is associated with dysregulated innate and adaptive immune responses. Brain. (2022), awac321. doi: 10.1093/brain/awac321
9. Frontera JA, Sabadia S, Lalchan R, Fang T, Flusty B, Millar-Vernetti P, et al. A prospective study of neurologic disorders in hospitalized patients with COVID-19 in New York City. Neurology. (2021) 96:e575–86. doi: 10.1212/WNL.0000000000010979
10. Thwaites RS, Sanchez Sevilla Uruchurtu A, Siggins MK, Liew F, Russell CD, et al. Inflammatory profiles across the spectrum of disease reveal a distinct role for GM-CSF in severe COVID-19. Sci Immunol. (2021) 6:eabg9873. doi: 10.1126/sciimmunol.abg9873
11. Leist SR, Dinnon KH, Schäfer A, Tse LV, Okuda K, Hou YJ, et al. A mouse-adapted SARS-CoV-2 induces acute lung injury and mortality in standard laboratory mice. Cell. (2020) 183:1070–1085.e12. doi: 10.1016/j.cell.2020.09.050
12. Carossino M, Montanaro P, O’Connell A, Kenney D, Gertje H, Grosz KA, et al. Fatal neuroinvasion and SARS-CoV-2 tropism in K18-hACE2 mice is partially independent on hACE2 expression. Viruses. (2021) 14(3):535. doi: 10.1101/2021.01.13.425144
13. Golden JW, Cline CR, Zeng X, Garrison AR, Carey BD, Mucker EM, et al. Human angiotensin-converting enzyme 2 transgenic mice infected with SARS-CoV-2 develop severe and fatal respiratory disease. JCI Insight. (2020) 5. doi: 10.1172/jci.insight.142032
14. Kumari P, Rothan HA, Natekar JP, Stone S, Pathak H, Strate PG, et al. Neuroinvasion and encephalitis following intranasal inoculation of SARS-CoV-2 in K18-hACE2 mice. Viruses. (2021) 13:132. doi: 10.3390/v13010132
15. Wenzel J, Lampe J, Müller-Fielitz H, Schuster R, Zille M, Müller K, et al. The SARS-CoV-2 main protease Mpro causes microvascular brain pathology by cleaving NEMO in brain endothelial cells. Nat Neurosci. (2021) 24:1522–33. doi: 10.1038/s41593-021-00926-1
16. Wittekindt M, Kaddatz H, Joost S, Staffeld A, Bitar Y, Kipp M, et al. Different methods for evaluating microglial activation using anti-ionized calcium-binding adaptor protein-1 immunohistochemistry in the cuprizone model. Cells. (2022) 11:1723. doi: 10.3390/cells11111723
17. Heindl S, Gesierich B, Benakis C, Llovera G, Duering M, Liesz A. Automated morphological analysis of microglia after stroke. Front Cell Neurosci. (2018) 12. doi: 10.3389/fncel.2018.00106
18. Santos Bravo M, Berengua C, Marín P, Esteban M, Rodriguez C, del Cuerpo M, et al. Viral culture confirmed SARS-CoV-2 subgenomic RNA value as a good surrogate marker of infectivity. J Clin Microbiol. (2022) 60:e01609–21. doi: 10.1128/JCM.01609-21
19. De Neck S, Penrice-Randal R, Clark JJ, Sharma P, Bentley EG, Kirby A, et al. The stereotypic response of the pulmonary vasculature to respiratory viral infections: findings in mouse models of SARS-CoV-2, influenza A and gammaherpesvirus infections. Viruses. (2023) 15:1637. doi: 10.3390/v15081637
20. Seehusen F, Clark JJ, Sharma P, Subramaniam K, Giuliani SW, Hughes GL, et al. Viral Neuroinvasion and Neurotropism without Neuronal Damage in the hACE2 Mouse Model of COVID-19. Viruses. (2021) 14(5):1020. doi: 10.1101/2021.04.16.440173
21. Thakur KT, Miller EH, Glendinning MD, Al-Dalahmah O, Banu MA, Boehme AK, et al. COVID-19 neuropathology at Columbia University Irving Medical Center/New York Presbyterian Hospital. Brain. (2021) 144:2696–708. doi: 10.1093/brain/awab148
22. Lee MH, Perl DP, Steiner J, Pasternack N, Li W, Maric D, et al. Neurovascular injury with complement activation and inflammation in COVID-19. Brain J Neurol. (2022) 145:2555–68. doi: 10.1093/brain/awac151
23. Grant RA, Poor TA, Sichizya L, Diaz E, Bailey JI, Soni S, et al. Prolonged exposure to lung-derived cytokines is associated with activation of microglia in patients with COVID-19. JCI Insight. (2024) 9:e178859. doi: 10.1172/jci.insight.178859
24. Frere JJ, Serafini RA, Pryce KD, Zazhytska M, Oishi K, Golynker I, et al. SARS-CoV-2 infection in hamsters and humans results in lasting and unique systemic perturbations after recovery. Sci Transl Med. (2022) 14:eabq3059. doi: 10.1126/scitranslmed.abq3059
25. Samudyata, Oliveira AO, Malwade S, Rufino de Sousa N, Goparaju SK, Gracias J, et al. SARS-CoV-2 promotes microglial synapse elimination in human brain organoids. Mol Psychiatry. (2022) 27:3939–50. doi: 10.1038/s41380-022-01786-2
26. Choudhary S, Kanevsky I, Yildiz S, Sellers RS, Swanson KA, Franks T, et al. Modeling SARS-CoV-2: comparative pathology in rhesus macaque and golden Syrian hamster models. Toxicol Pathol. (2022) 50(3):280–93. doi: 10.1177/01926233211072767
27. Rutkai I, Mayer MG, Hellmers LM, Ning B, Huang Z, Monjure CJ, et al. Neuropathology and virus in brain of SARS-CoV-2 infected non-human primates. Nat Commun. (2022) 13:1745. doi: 10.1038/s41467-022-29440-z
28. Fernández-Castañeda A, Lu P, Geraghty AC, Song E, Lee M-H, Wood J, et al. Mild respiratory COVID can cause multi-lineage neural cell and myelin dysregulation. Cell. (2022) 185:2452–2468.e16. doi: 10.1016/j.cell.2022.06.008
29. Xu E, Li T, Chen Q, Wang Z, Xu Y. Study on the effect and application value of heat-inactivated serum on the detection of thyroid function, tumor markers, and cytokines during the SARS-CoV-2 pandemic. Front Med. (2021) 8. doi: 10.3389/fmed.2021.742067
30. Soung AL, Vanderheiden A, Nordvig AS, Sissoko CA, Canoll P, Mariani MB, et al. COVID-19 induces CNS cytokine expression and loss of hippocampal neurogenesis. Brain. (2022), awac270. doi: 10.1093/brain/awac270
31. Borsini A, Merrick B, Edgeworth J, Mandal G, Srivastava DP, Vernon AC, et al. Neurogenesis is disrupted in human hippocampal progenitor cells upon exposure to serum samples from hospitalized COVID-19 patients with neurological symptoms. Mol Psychiatry. (2022), 1–13. doi: 10.1038/s41380-022-01741-1
32. Fontes-Dantas FL, Fernandes GG, Gutman EG, De Lima EV, Antonio LS, Hammerle MB, et al. SARS-CoV-2 Spike protein induces TLR4-mediated long-term cognitive dysfunction recapitulating post-COVID-19 syndrome in mice. Cell Rep. (2023) 42:112189. doi: 10.1016/j.celrep.2023.112189
33. Oh J, Cho W-H, Barcelon E, Kim KH, Hong J, Lee SJ. SARS-CoV-2 spike protein induces cognitive deficit and anxiety-like behavior in mouse via non-cell autonomous hippocampal neuronal death. Sci Rep. (2022) 12:5496. doi: 10.1038/s41598-022-09410-7
Keywords: virology, immunology, SARS-CoV-2, neurology, microglia
Citation: Dunai C, Hetherington C, Boardman SA, Clark JJ, Sharma P, Subramaniam K, Tharmaratnam K, Needham EJ, Williams R, Huang Y, Wood GK, Collie C, Fower A, Fox H, Ellul MA, Held M, Egbe FN, Griffiths M, Solomon T, Breen G, Kipar A, Cavanagh J, Irani SR, Vincent A, Stewart JP, Taams LS, Menon DK and Michael BD (2024) Pulmonary SARS-CoV-2 infection leads to para-infectious immune activation in the brain. Front. Immunol. 15:1440324. doi: 10.3389/fimmu.2024.1440324
Received: 29 May 2024; Accepted: 04 September 2024;
Published: 14 October 2024.
Edited by:
Frédéric Frézard, Federal University of Minas Gerais, BrazilReviewed by:
Zhen Zhao, University of Southern California, United StatesMark Endsley, University of Texas Medical Branch at Galveston, United States
Copyright © 2024 Dunai, Hetherington, Boardman, Clark, Sharma, Subramaniam, Tharmaratnam, Needham, Williams, Huang, Wood, Collie, Fower, Fox, Ellul, Held, Egbe, Griffiths, Solomon, Breen, Kipar, Cavanagh, Irani, Vincent, Stewart, Taams, Menon and Michael. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cordelia Dunai, Y2R1bmFpQGxpdmVycG9vbC5hYy51aw==; Benedict D. Michael, YmVubWljQGxpdmVycG9vbC5hYy51aw==
†Present addresses: Jordan J. Clark, Department of Microbiology, Icahn School of Medicine at Mount Sinai, New York, New York, NY, United States
Center for Vaccine Research and Pandemic Preparedness, Icahn School of Medicine at Mount Sinai, New York, NY, United States
Sarosh R. Irani, Department of Neurology, Mayo Clinic Florida, Jacksonville, FL, United States
Department of Neurosciences, Mayo Clinic Florida, Jacksonville, FL, United States
Robyn Williams, Department of Neurology, Mayo Clinic Florida, Jacksonville, FL, United States
Department of Neurosciences, Mayo Clinic Florida, Jacksonville, FL, United States
‡These authors have contributed equally to this work