 Kerem Abacar
Kerem Abacar Tom Macleod
Tom Macleod Haner Direskeneli
Haner Direskeneli Dennis McGonagle
Dennis McGonagle- 1Department of Internal Medicine, Division of Rheumatology, Marmara University School of Medicine, Istanbul, Türkiye
- 2Leeds Institute of Rheumatic and Musculoskeletal Medicine, University of Leeds, Leeds, United Kingdom
- 3National Institute for Health Research, Leeds Biomedical Research Centre, Leeds Teaching Hospitals, Leeds, United Kingdom
Historically inflammation against self was considered autoimmune which stems back to the seminal observations by Ehrlich who described serum factors, now known to be autoantibodies produced by B lineage cells that mediate “horror autotoxicus”. The 20th century elucidation of B- and T-cell adaptive immune responses cemented the understanding of the key role of adaptive immune responses in mediating pathology against self. However, Mechnikov shared the Nobel Prize for the discovery of phagocytosis, the most rudimentary aspect of innate immunity. Fast forward some 100 years and an immunogenetic understanding of innate immunity led to the categorising of innate immunopathology under the umbrella term ‘auto inflammation’ and terminology such as “horror autoinflammaticus” to highlight the schism from the classical adaptive immune understanding of autoimmunity. These concepts lead to calls for a two-tiered classification of inflammation against self, but just as innate and adaptive immunity are functionally integrated, so is immunopathology in many settings and the concept of an autoimmune to autoinflammation continuum emerged with overlaps between both. Herein we describe several historically designated disorders of adaptive immunity where innate immunity is key, including rheumatoid arthritis (RA), systemic lupus erythematosus (SLE), systemic juvenile idiopathic arthritis (sJIA) and adult-onset Still's disease (AOSD) where the immunopathology phenotype is strongly linked to major histocompatibility complex (MHC) class II associations and responds to drugs that target T-cells. We also consider MHC-I-opathies including psoriasis and Behcet's disease(BD) that are increasingly viewed as archetype CD8 T-cell related disorders. We also briefly review the key role of barrier dysfunction in eczema and ulcerative colitis (UC) where innate tissue permeability barrier dysfunction and microbial dysbiosis contributes to prominent adaptive immune pathological mechanisms. We also highlight the emerging roles of intermediate populations of lymphocytes including gamma delta (γδ) and mucosal-associated invariant T (MAIT) cells that represent a blend of adaptive immune plasticity and innate immune rapid responders that may also determine site specific patterns of inflammation.
Introduction
Inflammation represents both a repair and a defence mechanism against perceived threats to self (1). More than a century has passed since Paul Ehrlich, the 1908 Nobel laureate, posited that inflammation mistakenly directed against self would trigger what he termed “Horror Autotoxicus.” With the discovery of autoantibodies, the concept of autoimmunity emerged including a multitude of mechanisms driving immunopathology (2).
Commencing in the second half of the 20th century, the genetic architecture of the murine major histocompatibility complex (MHC) that underpinned transplant rejection and immunity began to emerge (3). A near identical model was discovered in humans that were termed as human leukocyte antigens (HLA) (4). Subsequently, the classification of HLA molecules into class I and class II and the relationships between T- and B-cells and MHC peptide presentation, a compelling basis for adaptive immune system, cellular and humoral immunity emerged along with abnormal lymphocytic reactions to self-antigens that firmly underpinned autoimmunity (5, 6). Such is the veracity of the autoimmunity concept that many diseases can be well controlled by specific targeting of B- or T-cells. The veracity of the concept also resulted in immunological overreach with the attribution of virtually all non-infectious inflammatory pathologies as autoimmune.
Mechnikov shared the Nobel Prize with Ehrlich for the discovery of phagocytosis, one of the basic functions of innate immunity (7). About a century later, the autoinflammation concept emerged with the identification of ectodomain mutations in the p55 tumour necrosis factor (TNF) receptor in patients with fever and diffuse inflammation (TNF-receptor-associated periodic syndrome, TRAPS) where discernible autoimmunity was lacking (8). This discovery followed fast on the heels of the demonstration that familial Mediterranean fever (FMF), a monogenic disease characterised by excessive neutrophilic inflammatory responses, was due to gain of function mutations in the MEFV (Mediterranean fever) gene which encodes pyrin, a critical activator of the inflammasome expressed in myeloid lineage cells (9, 10). This emergent understanding of innate immunity led to the categorising of innate immunopathology under the umbrella term ‘autoinflammation’ and terminology such as “horror autoinflammaticus” to highlight distinctions from the classical adaptive immune understanding of autoimmunity (11). Self-directed inflammatory pathologies in which local factors in disease-specific sites lead to the activation of innate immune cells and cause target tissue damage, such as impaired homeostasis of canonical cytokine cascades (as in periodic fevers) or abnormal bacterial perception (as in Crohn’s disease), were defined within the scope of autoinflammatory disease (2).
Although classified separately, the innate and adaptive immune systems are functionally integrated and in constant interaction with each other (Table 1). When discussing innate immunity as a driver for disease there are two key considerations - the innate immune cells themselves that reside in or circulate to the target tissue and the target tissue non-immune compartment. For example, barrier disruption in the skin compartment best characterised by filaggrin single nucleotide polymorphisms (SNP) leading to disruption of barrier integrity allows antigenic access that ultimately culminates in T-helper (Th) 2 immune responses and eczema or atopic dermatitis (AD) (12). Although we conceptualise the therapy of AD through immunomodulation, for example with interleukin (IL) 4/13 or Janus Kinase (JAK) inhibition, it is fascinating that children with AD can be treated with “wet wraps” that at the simplest level restore epidermal barrier homeostasis (13). Herein, we further describe these complex interactions in rheumatic disease settings, where the integration of both the innate and adaptive immune system work in unison to determine phenotypes that are too often pigeonholed into either autoimmunity or autoinflammation where in reality there is functionally integrated immunopathology.
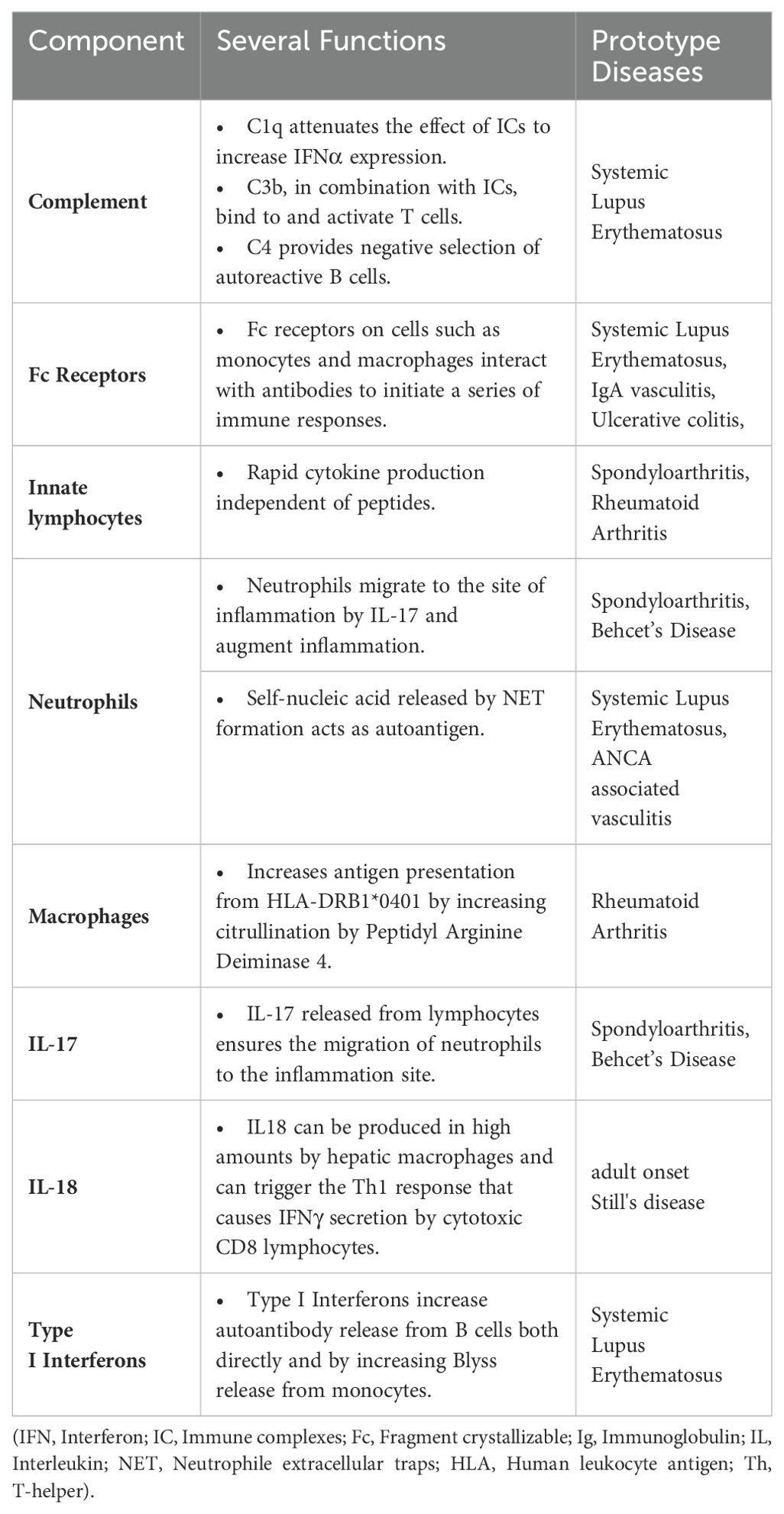
Table 1. Links between innate and adaptive immunity.
Rheumatoid arthritis
It is now well established that both human and murine experimental immunopathology is heterogeneous and clinical disease phenotypes may be predominantly autoimmune or predominantly innate immune (14). Rheumatoid arthritis (RA), for example, has shared epitope positive, rheumatoid factor (RF) and anti-citrullinated protein antibody (ACPA) positive cases that respond to B- or T-cell targeting with agents such as Rituximab or Abatacept which are very much in the autoimmune spectrum (15, 16). However, it is also clear from monogenic disorders including FMF and Blau syndrome which develop arthritis and synovitis that closely resemble that of RA, that an RA phenotype can be exclusively innate immune mediated with neutrophilic or macrophage driven inflammation and responds to therapies including colchicine or anti-cytokine blockers (17). Nonetheless, in RA the autoantibodies are associated with immune complex formation that is thought to engage macrophages, inducing the production of cytokines including tumour necrosis factor (TNF) in particular. Indeed, this may underscore the seminal translational immunology studies that transformed disease when TNFα blockade was found to be effective (18) and further demonstrates the effectiveness of both innate and adaptive therapies in RA (19). Another fascinating feature of RA is the innate immune barrier dysregulation in the gingiva and in the lung with non-specific citrullination that paves the way for the later autoimmunity in the joint (20). Thus, going forward, it may be possible to envisage targeting innate immunity to prevent ACPA + RA and then adaptive immunity for clinically established disease. An obvious iteration of this is to block non-specific macrophage mediated innate immune citrullination of peptides to target disease.
RA presents a wide range of classification patterns including clinical and pathogenetically different mechanisms and manifestations. Although these all fall under the umbrella of RA, it is obvious that they are different entities (17). Based on the interaction of B-and T-cells, MHC-II associated ACPA-driven disease is a picture that meets the classical definition of autoimmune RA with its unique response to B-cell targeted therapies (21). Antigen presentation of citrullinated proteins produced by endogenous or exogenous factors that may be ACPA targets occurs through MHC-II related mechanisms, and this constitutes one of the most important steps in going from autoimmunity to a frank autoimmune arthritis (22). Citrullination is a post-translational modification process in which arginine in peptides are transformed to citrulline via “peptidyl arginine deiminase” (PAD) (23). The antigenic potency of arginine residues enzymatically modified to citrulline by PAD, a myeloid cell derived enzyme at sites of inflammation, underscores the key early autoinflammatory component. In this context, macrophages, monocytes and neutrophils make an important contribution to this classic autoimmunity of RA with their key role in PAD4 production (24). Indeed, calcium flux leading to increased cytosolic Ca2+, an event that occurs during macrophage activation, NETosis in neutrophils and during apoptosis, activates PADs by inducing essential conformational changes after Ca2+ binding (25, 26). The reason for such increased antigenic effects is the difference in binding potential of citrullinated peptides to MHC peptides in antigen presenting cells (APCs). In particular, the interaction of citrulline vimentin with the P4 pocket of HLA-DRB1*04:01/04, which is specific for RA and called the shared epitope, increased detection of CD4+ T-cells against citrullinated vimentins in the peripheral blood of RA patients and presence of plasma cells producing ACPA are the route of the classical RA autoimmunity (27, 28). In addition, exposure to some viral or bacterial agents, especially Porphyromonas gingivalis, triggers ACPA formation with cross-reactivity due to molecular mimicry (Figure 1) (29).
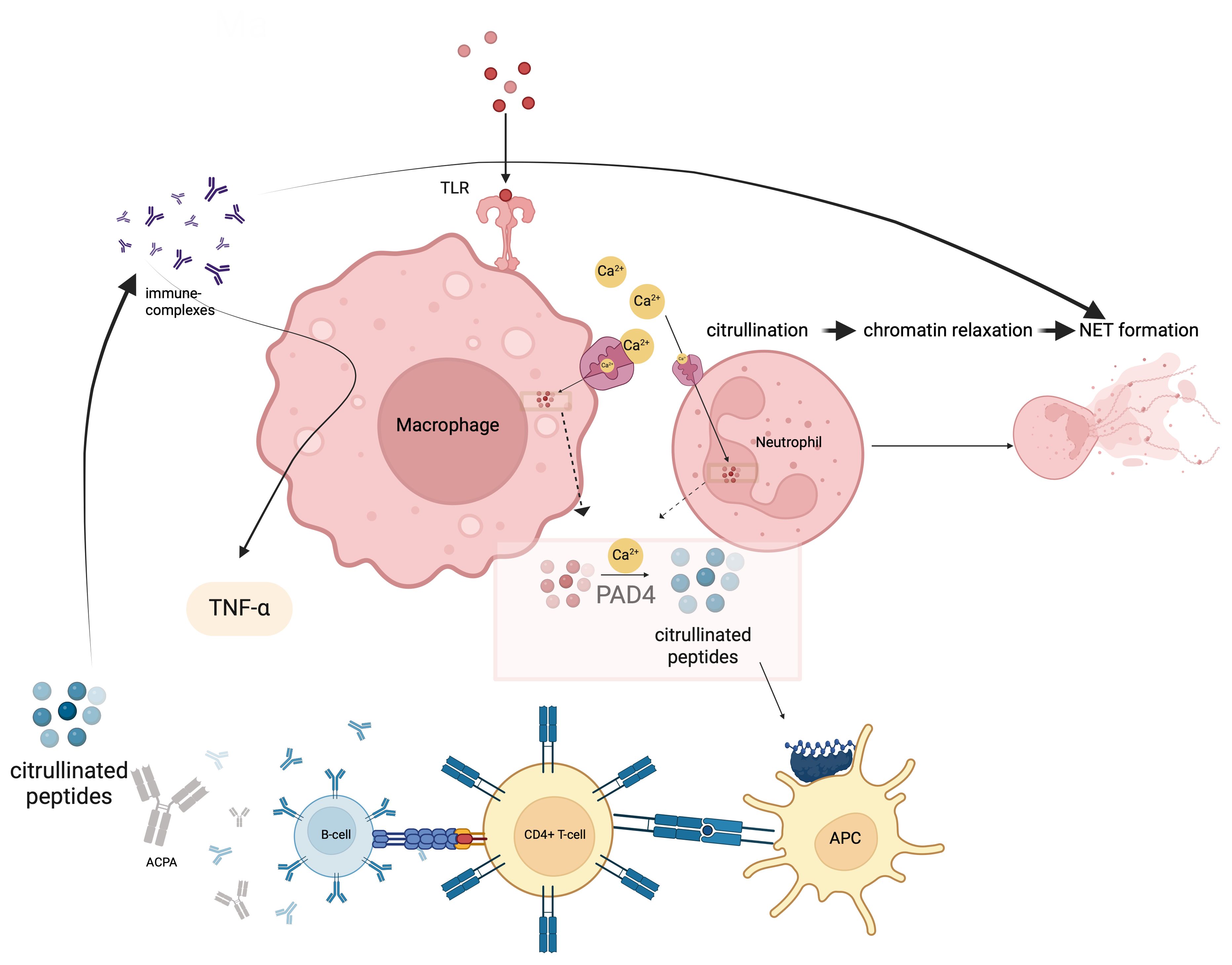
Figure 1. Citrullination occurs via the enzyme peptidyl arginine deiminase-4 (PADI4) and likely predates autoimmunity and occurs at sites of non-specific inflammation such as the gums and lungs where macrophages and neutrophils contribute to such citrullination processes. Afterwards, this innate immune phase leads citrullinated peptides presentation to CD4 Tmphocytes via MHC-II peptide presentation. ACPA production is induced after T and B lymphocyte interactis With citrullinated proteins immune complex formation and FcR binding on macrophages, TNF and other cytokine release occurs. In effect autoimmunity is sandwiched between Innate immune Initiation events and innate immunity terminal effector events. This is likely why RA does not cross the placenta and mediated neonatal autoimmunity.
There are also some components of the innate immune system that upregulate PAD activity and thus increase the antigenic burden (30). Recognition of pathogen-associated molecular patterns (PAMPs) by pattern recognition receptors (PRRs), especially in macrophages, can activate PADs by inducing extracellular Ca2+ influx (31, 32). The production of neutrophil extracellular traps (NETs), extracellular web-like fibre network consisting of nucleic acids that can bind to microorganisms including pathogens that is an important element of the innate immune system, is also evident in RA (33). Increased neutrophil counts in the synovial fluid confirms the contribution of NETosis to the pathogenesis (34, 35). NETosis was associated with increased local PAD4 activity, whereby proteins citrullinated by PAD4 expand an autoantigen pool that fosters autoimmunity (Figure 1) (36–39).
In the continuation of the pathogenesis after the citrullination and formation of ACPA, there are some returns to the innate immune system components. ACPA generates immune complexes (IC) with citrullinated peptides and these ICs can stimulate TNFα secretion in macrophages via the toll-like receptor (TLR) 4/myeloid differentiation primary response (MyD) 88 pathway (40, 41). ACPA also provides M1 polarization in macrophages via interferon regulatory factor (IRF) 4 and 5 and increases their proinflammatory effects (42). In addition, ACPA, both via IC formation and alone, induces NETosis in neutrophils (35). Some the classical and critical autoimmunity of RA is sandwiched between innate immune initiation and effector mechanisms.
Systemic lupus erythematosus
It is broadly agreed that B-cells are central to the pathogenesis of systemic lupus erythematosus (SLE) as the source of the autoantibodies responsible for the disease and SLE is the paradigmatic multi-organ autoimmune disease (43). An SLE hyperfunctioning B-cell compartment is suggested by genome-wide association studies (GWAS) in humans and functional studies in mice (44, 45). In just under two decades, it has been firmly established that innate immune pattern recognition of nucleic acids via TLR7 and TLR9 in particular is foundational in the genesis of experimental and human SLE. Such DNA and RNA bound to nuclear autoantigens appears to underpin the disease proclivity for autoantibodies against nuclear antigens (46). The pivotal role of nucleic acids in SLE phenotypes emerged from monogenic disorders due to mutations in genes linked to nucleic acid metabolism and these phenotypes are classified under the umbrella of interferonopathies (47). In the classical autoantibody-based pathogenesis cascade, the innate immune TLR pathway dysregulation serves as a bridge to autoimmunity via multifaceted type I interferons (IFN), B cell activation and disease.
The IFNs were discovered over 50 years ago by Isaacs and Lindemann and their pervasive roles in immune functions unravelled thereafter (48). Three types of interferon systems have been defined in mammals, depending on their chromosomal location, signal transduction mechanisms, and receptors: type 1, type 2 and type 3 (49). IFNα belongs to the type I IFN family and plays one of the most dominant roles in the pathogenesis of SLE (50, 51). Although IFNα is produced by many cells, the main producer in the pathogenesis of SLE was previously believed to be the plasmacytoid dendritic cell (pDC) (52). Detection of PAMPs by PRRs at the cell surface, endosomal and cytoplasmic regions initiate type I IFN production in the cell via the phosphorylation of IFN regulatory factors (IRFs) which positively regulate genes encoding type I IFNs (53).
Rather than pDCs being the key orchestrators of type 1 IFN production, it has recently emerged that the target tissues including the skin are the major producers of type 1 IFN thus pointing towards an initial innate immune barrier tissue dysregulation (54). However, clues showing that systemic inflammation is triggered in addition to skin involvement as a result of ultraviolet exposure led to the discovery of new circulating sources of type I IFN in addition to local type IFN production. Neutrophils may induce stimulator of interferon genes complex (STING)-dependent interferon signature as a result of NET formation, and also may cause neutrophil-based damage to the kidney by reverse migration, which cannot be demonstrated in pDCs (55).
Furthermore, the archetype of local IFN producers outside the hematopoietic system is IFNκ production by keratinocytes (56). This production was also detected in non-lesional skin keratinocytes (57). The key roles of type I IFNs in the process between the UV exposure of the skin and the development of the cutaneous lupus phenotype is the increase of cytokines and other chemoattractant mediators as a result of type I IFN signalling (Figure 2) (58). Specifically, keratinocytes increase the production of proinflammatory cytokines such as IL-6 in response to IFNκ in lupus and conversely neutralization of IFNκ suppresses their IL-6 production (59). It is still controversial whether the local production of type I IFN in keratinocytes is an inducer of the systemic involvement of the disease. However, upregulation of IFN-inducible genes in keratinocytes from non-lesional skin of lupus nephritis patients compared to keratinocytes isolated from healthy controls indicates that systemic inflammation may also be induced by innate immune system-mediated mechanisms in the skin (60). Although aforementioned studies provide important data on the potential role of keratinocytes as a source of type I IFN, the effect of local production on the diversity of the phenotypic presence of the disease is still unpredictable. Keratinocytes, as local activators of photosensitivity and cutaneous lupus involvement, appear as potential predictors and treatment targets of systemic involvement with IFN gene upregulations found in patients with systemic involvement, even if they are not exposed to sunlight. In addition, the high levels of IFNα expression in tubular epithelial cells in lupus nephritis and the evidence that astrocytes are the main source of IFNα in neuro-lupus suggest that local production may be the main driving mechanism in some clusters of the disease (61, 62).
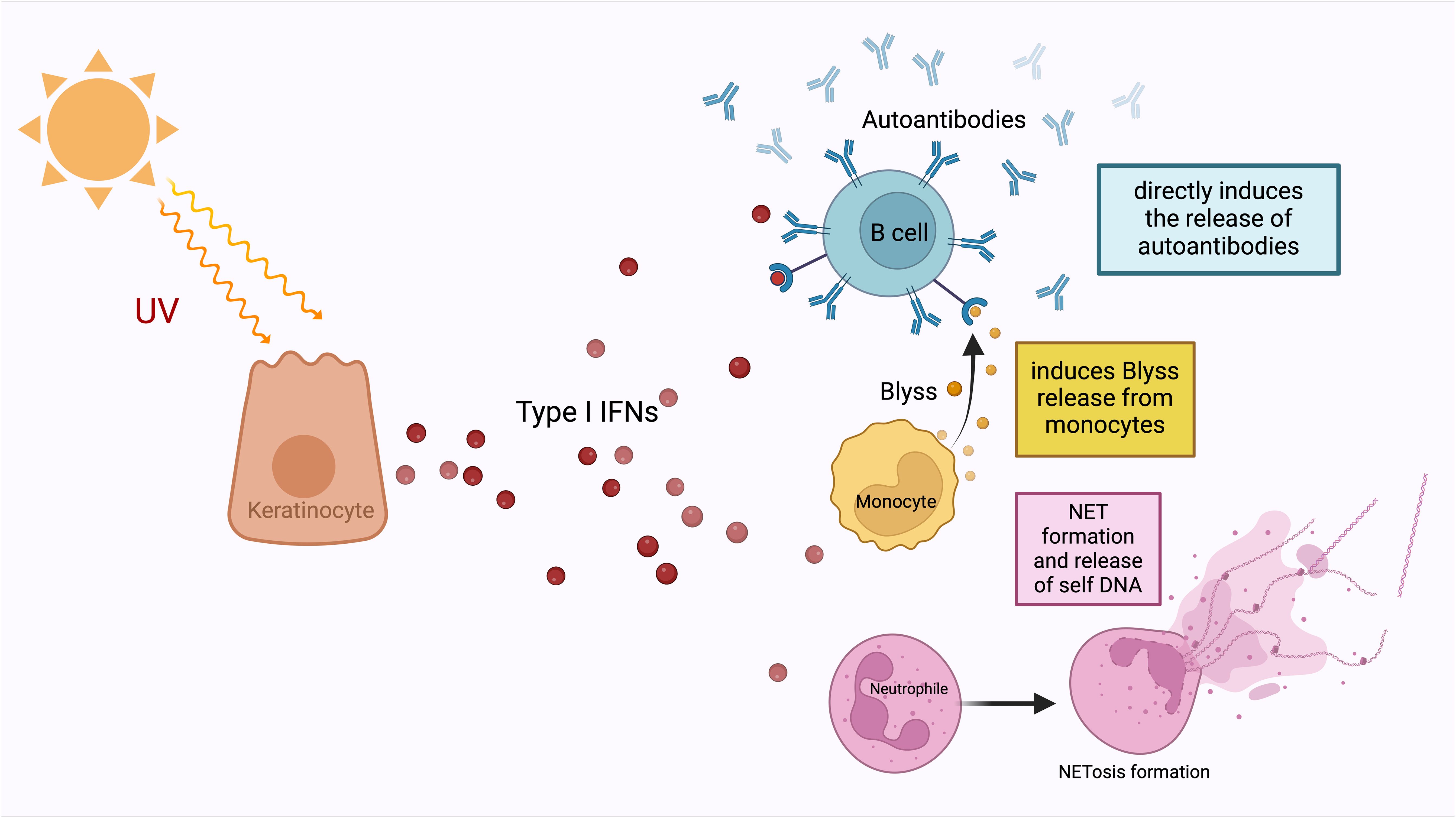
Figure 2. Type I IFNs are released from keratinocytes under the influence of ultraviolet light. They enable the development of autoantibodies from B-lymphocytes, which is the most important step in the pathogenesis of SLE, either directly or by stimulating the release of Blyss from monocytes. In addition to this autoantibody development, type I IFNs also increase NET formation and release nucleic acids from neutrophils. NETosis, which is one of the most important defence mechanisms of neutrophils, is an extracellular web-like fibre network consisting of nucleic acids that can bind to microorganisms including pathogens. The most important contribution of NET formation to the SLE pathogenesis is that free DNA structures that induce autoantibody formation assume the necessary autoantigen role.
The pathway from Type I IFNs to antibody production by B-cells via B lymphocyte stimulator (Blyss) is one of the main determinants of the transition route from the innate immune system to the adaptive immune system within the framework of SLE pathogenesis (63, 64). Type I IFNs can directly ensure the antibody production through Blyss, as well as increase the number of autoantigens, which are the raw material of the MHC system in various ways (Figure 2) (64). Type I IFNs reduce the clearance of apoptotic cells, which is an important source of autoantigen, via decreasing the activation of splenic marginal zone macrophages (65). They also increase NET formation and release (66). The most obvious contribution of NET formation to the pathogenesis of SLE is through providing a source of antigenic free DNA structures that induce autoantibody formation (67). NETosis is found to be associated with autoantibody levels in SLE (67). Hence the old concept of lupus as the paradigm for autoimmunity has been flipped on its head with the understanding that tissue specific IFN dysregulation is capable of driving immunopathology.
Systemic juvenile idiopathic arthritis and adult-onset Still's disease
The connections between the innate and adaptive immune system in systemic juvenile idiopathic arthritis (sJIA) that initiate in childhood and in adult-onset Still's disease (AOSD), which is considered to be the continuation of the same disease pattern, are particularly striking in the context of macrophage activation syndrome (MAS), in which autoinflammatory hyperactivation occurs (68). AOSD exists at the crossroads of autoinflammatory and auto immune diseases because of its pathogenesis encompassing both the innate and the adaptive immune system. Although the innate immune system is the main driver of the disease through cytokines such as IL-1 and IL-6, the fact that the identified genetic susceptibility is strongly associated with the MHC-II immune system highlights the importance of the interconnections of these two systems in AOSD (69–73). HLA-DRB1*11 variants are the alleles most associated with sJIA risk (73). Again, within the MHC class II gene cluster, the incidence of AOSD was found to be more strongly associated with HLA-DR*2, DR*4, DR*7, DRB1*12 and DRB1*15 (74). MHC-II genetic predisposition reinforces the role of antigen presentation and T-cell activation in the pathogenesis (73). In addition, elevation of the biomarkers such as soluble IL-2 receptor-α subunit (also known as soluble CD25) also strongly suggest T-cell hyperactivation (75). Chimeric antigen receptor (CAR) T-cell therapy developed against specific tumour antigens triggers cytokine storm and MAS, demonstrating that gain of function of the adaptive immune system may be sufficient for the MAS phenotype. Also, the monophasic pattern of MAS developing with CAR T-cell therapy for lymphoma suggests that the phenotype might improve with the decrease in T-cell hyperactivation. It also supports the notion that an immune hypersensitivity reaction resulting from an antigen driven adaptive CD4+ T-cell response can trigger MAS (76, 77).
In addition, the cytotoxic function of natural killer (NK) cells was found to be impaired in patients with AOSD. After treatment, both the frequency and cytotoxic functions of NK cells improved. Some proinflammatory cytokines, especially IL-18, are thought to suppress NK cell activity. Indeed, during cytokine storms observed in MAS, NK cytotoxicity decreases (78). IL-18 was increased in the synovial tissue, serum, and lymph nodes of AOSD patient compared to healthy controls. It has also been shown that IL-18 can be produced in high amounts by hepatic macrophages and can trigger the Th1 response that causes IFNγ secretion by cytotoxic CD8+ T lymphocytes (79–81). Type I interferon is a key cytokine in murine models of lymphocytic choriomeningitis virus-induced MAS. In addition, these models suggest that type I interferon may be an important bridging cytokine for MAS developing in the SLE setting.
MHC-1-opathy including Behcet’s disease
Recognising the role of tissue specific factors leading to innate immune dysregulation i.e., pathergy responses and the role of T-cells as suggested by the HLA-B*51 association we originally classified Behcet’s Disease (BD) as an intermediate between innate and adaptive immunity (2). Given the strong overlaps with other MHC-I associated conditions including the seronegative spondyloarthritis (SpA) spectrum including ankylosing spondylitis (AS), psoriasis and acute anterior uveitis (AAU), we termed these related conditions as MHC-I-opathies (82). Barrier dysfunction in the skin and inappropriate innate immune reactions at sites of mechanical stress causes secondary adaptive immune responses with neutrophilic inflammation that provoke the exacerbation and recurrence of these diseases (82).
BD and MHC-I-opathies did not fit the classic female-dominated autoimmune disease concept with autoantibodies playing a leading role but instead exhibit convergent immunology around CD8+ T-cells and a close link to neutrophilic inflammation (83). However, although the absolute importance of MEFV mutation and neutrophils was demonstrated, it is not a typical autoinflammatory disease like FMF either (84, 85). The immune pathway of BD consists of an organizational continuum, rather than a dichotomic system, especially due to the interplays, which provides the bridges between the innate and adaptive arms (86).
Neutrophils and monocytes are hyperactive in BD, suggesting an overactive innate response, most clearly observed in pathergy (skin prick) reaction. This response is dampened by microbial sterilization and decreased traumatic needle use in skin, yet exacerbated when pneumococcal antigens are given together with needle prick, clearly implicating the innate immune system (87). However, T-cell responses to local antigens such as Retinal-S antigen cross-reactive with HLA-B*27 peptides are also shown in BD patients with posterior uveitis (88). Conventional immunosuppressives such as azathioprine or cyclosporin-A are also effective in BD unlike in autoinflammatory diseases, showing an important pathological role for the adaptive immune system as well.
It is not surprising that BD is also included in the concept of MHC-I-opathies, along with SpA and psoriasis, in which significant pathogenetic schemata similarities have been identified. Barrier dysfunction in the skin, oral mucosa and inappropriate innate immune reactions at sites of mechanical stress causes secondary adaptive immune responses with neutrophilic inflammation that provoke the exacerbation and recurrence of these diseases (82). Recently, an HLA-B*51 restricted, endoplasmic reticulum aminopeptidase (ERAP)-1-Hap10 associated CD8+ T-cell response has been described in peptidomes generated from BD patients, similar to B*27 restricted peptidome responses in SpA patients (89).
It is widely accepted that, as a result of the barrier perturbation that develops due to mechanical stress, the transition from the tissue-resident innate immune system to the adaptive immune system occurs via the IL-23/IL-17 axis (Figure 3) (82). In this cyclical process, the adaptive immune system T-cell responses are closely integrated with innate immune system neutrophilic function. Type 17 T-cells (Th17 cells, CD8+ T-cells, γδ T-cells, NKT-cells) play a lead role in the recruitment of neutrophils to the site both indirectly via IL-17-induced IL-8 and G-CSF (through myeloid and endothelial cells), and directly by GM-CSF production (Figure 3) (90). Besides Th17s, γδ T-cells and NKT-cells are also responsible for increased circulating IL-17 in BD (91, 92). In BD, as in other MHC-I-opathies, IL-17 is thought to be the main disease-driving cytokine produced by lymphocytes to stimulate the innate immune system, particularly neutrophils (93). As far as is known, the main mechanism by which adaptive immune system elements exacerbate BD is by interfering with the innate immune system and inducing peak neutrophilic inflammation. A recent novel insight relevant to BD is that neutrophils, in addition to carrying out symptom-driving tissue damage, can also produce IL-23 which offers a novel mechanism for how innate immunity could maintain the type 17 immunity (83, 94).
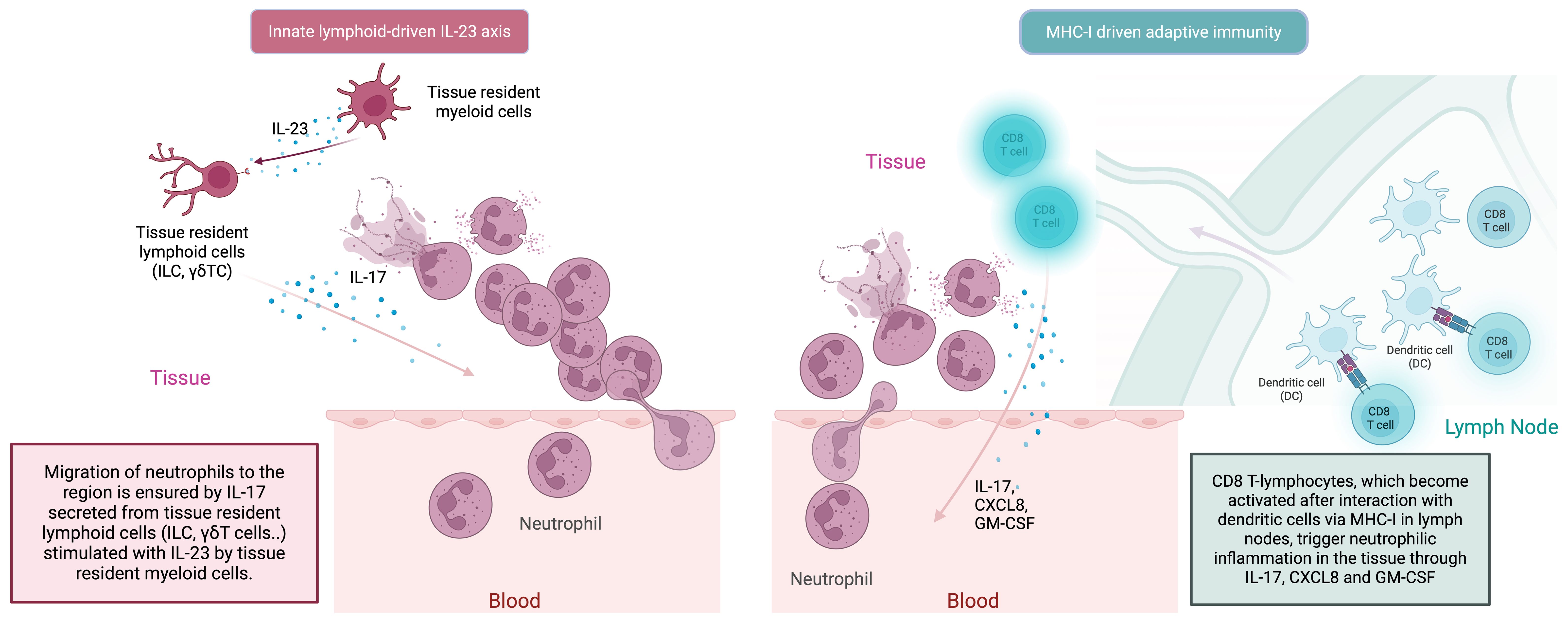
Figure 3. In the MHC-I-opathies, the ultimate aim of CD8 T-cell adaptive immunity is to facilitate neutrophil migration and activation at sites of Inflammation. Initially innate cells including DCs, macrophages and even neutrophils in tissues can produce IL-23. Such IL-23 release stimulates tissue resident lymphoid cells including ILC3 cells and other innate lymphocytes to produce IL-17. In parallel, APC activation of conventional T-cells in the usual MHC-1 driven adaptive pathway dramatically amplifies the release of IL-17, CXCLS and GM-CSF from CD8 T-lymphocytes to which antigen is presented by dendritic cells. This further increases neutrophil recruitment and inflammation. There the IL-23/17 axis is both a key part of both innate and adaptive immune responses at certain tissue specific sites including the skin the gut the enthesis and the uveal tract.
Autoimmunity and barrier defects- inflammatory bowel diseases
Although the exact cause of mucosal damage is not fully known, the overarching hypothesis is that inflammatory bowel disease (IBD) pathogenesis is linked to impaired immune response against bacterial microorganisms in the intestinal mucosa in genetically predisposed individuals (95). This impairment occurs in both the innate and the adaptive immune system (96). Indeed, more than 50 monogenic forms of IBD have been described with both obvious innate and adaptive immune system mutations as drivers (97). Autoinflammatory-manner diseases due to defects in signalling pathways responsible for stabilizing the barrier and stress response, which is the foremost component of intestinal mucosal immunity, immunodeficiencies affecting granulocyte and phagocyte activation, impaired T- and B-cell selection and activation, are examples of the monogenic-based IBD phenotypes (98).
Although the barrier disruption in ulcerative colitis (UC) suggests innate immunity pre-eminence, the fact that therapy with cyclosporine, integrin blockers and S1P (Sphingosine-1 Phosphate) receptor modulators (working by blocking interaction between S1P and S1P1 receptors, which regulate lymphocyte egress from the spleen and lymph nodes into the systemic circulation, decreasing intestinal inflammation in IBD) all of which block T-cell function, attests to the major additional role played by cells of the adaptive immune system (99). However, the susceptibility loci could not explain the majority of the pathogenesis of the patients with IBD. NOD2 (encoding nucleotide oligomerization domain receptor) and CARD9 genes, which are especially important in innate immune system mechanisms, stand out among the defined Crohn’s disease susceptibility genes (100, 101). In addition, there are SNPs defined in IL23R, JAK2, STAT3, and TYK2 genes, which encode the definitive cytokines of the IL-23/Th17 pathway cascade, and in genes that play a role in T-cell response such as TNFSF15 (102–105).
Macrophages and dendritic cells, which both stimulate other cells of the innate immune system and present antigens to lymphocytes via MHC class II, are the main cellular bridges (106). DCs and macrophages contain innate immune receptors (pattern-recognition receptors, PRRs) such as TLRs and NOD proteins that sense pathogen-associated molecular patterns (PAMPs) of extra- and intracellular pathogens, respectively. In the adaptive immune system, triggering of antigen-specific T-cells occurs through antigen presentation by dendritic cells. The interactions of α4β7 integrin expressed on lymphocytes and mucosal address-cell adhesion molecule 1 (MAdCAM-1) in the endothelium of intestinal venules are important for the most critical step, which is the adhesion of effector and memory T-cells in the intestine (107). Hence IBD development often transitions from innate to classical adaptive immune system in disease.
IL-23, which is a vitally important link between innate and adaptive mechanisms in many of these type-17 T-cell related autoimmune diseases, is also one of the key cytokines targeted as a therapeutic agent in IBD (108). IL-23 promotes inflammation by increasing cytokines such as IL-17A, IL-17F, IL-21, IL-22, IL-26 produced from Th17 cells in the blood and intestinal mucosa (109). IL-17 produced by Th17 cells induces the secretion of other proinflammatory cytokines (e.g., IL-1β, IL-6, TNF), growth factors (G-CSF and GM-CSF) and chemokines from stromal, epithelial and myeloid cells and thus supports the migration of neutrophils and macrophages to the region (110). Although IL-6 and TGFβ must be present for naive T-cells to differentiate into Th17 cells, IL-23 also enables these cells to develop and proliferate, as well as secrete IL-17 (111).
Autoimmunity and barrier defects- atopic Th2 diseases
Atopic diseases and most typically eczema or AD is not typically viewed as autoimmune due to the lack of autoantibodies. However, strong evidence incriminates dysregulated adaptive immune responses with Th2 lymphocytic responses via IL-4 and IL-13 mediating the cutaneous manifestations. The use of cyclosporin or anti IL-4/13 or anti-IL-13 attests to the veracity of this adaptive immunopathology (112). Yet, a key factor in the development of AD is a loss of barrier function; frequently as a result of reduced levels of filaggrin. Indeed, mutations within the filaggrin gene are the most significantly associated with AD (113). The breakdown in barrier function facilitates influx of allergens, dysbiosis in microflora which further contributes to allergen production and loss of water retention leading to dry skin and exacerbated lesions (114).
This breakdown in barrier integrity allows activation of the innate immune system and epithelial cells, causing production of Th2-driving cytokines and alarmins such as TSLP, IL-25, and IL-33, ultimately skewing the adaptive immune system to a Th2 response (115–118). Activated Th2 cells subsequently produce IL-4, IL-13 and IL-31, which directly contribute to the pathogenesis of AD by stimulating sensory neurons causing pruritis (119–121). In addition, IL-4 and IL-13 drive IgE class switching and synthesis in B cells, which is a significant driver of atopy facilitating mast cell degranulation following FcϵR binding and recognition of sensitized allergens (122, 123). Again, whilst the adaptive immune system is the crucial driver of pathology in AD following established Th2 skewing, the innate response is critical in initiating and driving Th2 development.
Innate and non-conventional lymphocytes as autoimmunity autoinflammatory bridges
Contrary to their phenotypic and functional resemblance to T-cells, innate lymphoid cells (ILCs) do not contain acquired T-cell antigen receptors. They respond to signals from damaged tissues with the production and secretion of a range of cytokines, but do not undergo clonal selection and expansion. They have an incompletely understood relationship between the microenvironment and components of the adaptive immune response. ILCs are classified into three categories (ILC1s, ILC2s, and ILC3s) that reflect the cytokine expression profiles of classical CD4+ T-helper cell subsets (Th1, Th2, and Th17 respectively) (124). ILCs are stimulated by stress signals, microbial compounds, and cytokines rather than by antigens. Therefore, they tend to take an early role in immune responses. ILC1s are stimulated in response to IL-12, IL-15 and IL-18 produced by myeloid cells in response to intracellular pathogens and produce interferon-gamma. ILC2s are stimulated by epithelial-produced cytokines IL-25, IL-33, IL-4 produced from basophils, secondary to parasitic infections or allergens, leading to the production of IL-4, IL-5 and IL-13. ILC3s mainly respond to IL-1β and IL-23, which are produced by myeloid cells in response to bacterial and fungal infection. In addition, ILC3s produce IL-17 as well as GM-CSF and IL-22 during inflammation (125–132).
ILCs are not antigen-driven yet are similar to other classical T-cells in their potential for cytokine responses. They therefore provide a source of lymphocyte-derived cytokines, such as IFNγ, IL-5, and IL-13, or IL-17 and IL-22, in the local inflammation milieu early in the immune response without the need for antigen specificity. These are first responder equivalents of the classical Th1, Th2 and Th17 paradigms of adaptive immunity (133). In addition, each of these cell types reacts to distinct stimuli. ILC3s express MHC class II on their surface and are capable of presenting antigen to CD4 T-cells (134). Therefore, ILC3s regulate the activity of T-cells specific for microbiota-derived antigens through MHC-II presentation on their surface (135). Also, recently high levels of IL-17-producing ILC3s have been found in psoriatic arthritis and IBD (136, 137). In the shared pathogenesis of disease groups in the MHC-I-opathies concept such as BD, psoriasis or spondylarthritis, some tissue-located ILCs are also thought to be factors that activate the IL-23/IL-17 axis (138). Further research will be required for its other functions in systemic autoimmune diseases.
Non-conventional lymphocytes including mucosa-associated invariant T (MAIT) cells that produce IFNγ, IL-17, and IL-22, invariant NKT (iNKT) cells that produce IFNγ or IL-4, and subsets of γδ-T-cells that produce IFNγ and IL-17 have shared production of some cytokines with innate T-cells in some epithelial or mucosal tissues (139).
γδ T-cells are mostly enriched in various epithelial and intestinal tissues, as well as in the skin. Whilst non-conventional T-cells have functional yet restricted T-cell receptors (TCRs) that recognise conserved antigens, TCR signalling is not necessary for differentiation and function of non-conventional T-cells (140). Most tissue-specific γδ T-cells show a Th1, Th2, Th17, and Treg-like phenotype and are essential in inflammation (141, 142). While γδ T-cells represent a small subset of T-cells in secondary lymphoid organs, they play a unique role in the pathogenesis of IBD as they represent a major subset of intraepithelial lymphocytes (IEL) in intestinal mucosal tissues. Protective γδ T-cells perform tissue repair and epithelial cell healing during intestinal inflammation via IL-22 secretion in response to retinoic acid secreted from DCs yet can have a pathogenic effect through production of IL-17 and IFNγ. DC-induced IL-1β and IL-23 contribute to the differentiation of IL-17-producing γδ T17 cells and additionally to IL-17 and IFNγ-secreting γδ T1/17 cells (143). IL-17 induces matrix metalloproteinase (MMP) secretion by tissue resident fibroblasts, whereas IFNγ induces nitric oxide (NO) production. NO and MMPs evoke inflammation thus contribute to the development of IBD (130, 144). Powerful experimental evidence has shown how γδ T-cells are key players in experimental spondyloarthropathy that was previously thought to be more adaptive immune related or an MHC-1-opathy (145).
Conclusions
In this article we provide translationally relevant insights into some of the key interplays between innate and adaptive immunity that manifests as autoimmune diseases. Whilst autoimmune diseases are often effectively treated by targeting B or T-cells, therapy outcomes may be variable or mixed. Numerous bi-directional links within innate and adaptive immunity could potentially create new therapeutic targets where innate immunopathology is silently smouldering in the background e.g. dysregulation of nucleic acid signalling in SLE. Moreover, each autoimmune disease has variable or even dominant innate immune drivers e.g. MEFV mutations in seronegative RA. Furthermore, environmental factors including bacteria or adjuvants could sculpt overall immune responses in different populations.
Because treatment options targeting innate immune system elements are minimal in the therapeutic armamentarium there is a need to consider adjunct strategies targeting innate immunity in autoimmune disease to more comprehensively treat autoimmunity, reduce adaptive immunity immunosuppression and target cases that are predominantly innate immune driven. The greatest success in innate immunotherapy has likely been colchicine, which target neutrophils, which we have mentioned for its active roles in many diseases. Ultimately, with the data provided by clinical and molecular studies, the autoimmune-autoinflammatory disease classifications designed hitherto will be reshaped and refined with new discoveries in the future.
Author contributions
KA: Conceptualization, Data curation, Investigation, Software, Writing – original draft, Writing – review & editing. TM: Conceptualization, Investigation, Software, Writing – original draft, Writing – review & editing. HD: Conceptualization, Investigation, Software, Writing – original draft, Writing – review & editing. DM: Conceptualization, Investigation, Software, Supervision, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Acknowledgments
All figures were created with BioRender.com.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Chen L, Deng H, Cui H, Fang J, Zuo Z, Deng J, et al. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget. (2018) 9:7204–18. doi: 10.18632/oncotarget.23208
2. McGonagle D, McDermott MF. A proposed classification of the immunological diseases. PloS Med. (2006) 3:e297. doi: 10.1371/journal.pmed.0030297
3. Bach FH, Widmer MB, Segall M, Bach ML, Klein J. Genetic and immunological complexity of major histocompatibility regions. Science. (1972) 176:1024–7. doi: 10.1126/science.176.4038.1024
4. Thorsby E. A short history of hla. Tissue Antigens. (2009) 74:101–16. doi: 10.1111/j.1399-0039.2009.01291.x
5. Handunnetthi L, Ramagopalan SV, Ebers GC, Knight JC. Regulation of major histocompatibility complex class ii gene expression, genetic variation and disease. Genes Immun. (2010) 11:99–112. doi: 10.1038/gene.2009.83
6. Danchin E, Vitiello V, Vienne A, Richard O, Gouret P, McDermott MF, et al. The major histocompatibility complex origin. Immunol Rev. (2004) 198:216–32. doi: 10.1111/j.0105-2896.2004.00132.x
7. Conti AA, Lippi D, Gensini GF. The historical evolution of the concept of apoptosis in rheumatic diseases. Reumatismo. (2005) 57:57–61. doi: 10.4081/reumatismo.2005.57
8. McDermott MF, Aksentijevich I, Galon J, McDermott EM, Ogunkolade BW, Centola M, et al. Germline mutations in the extracellular domains of the 55 kda tnf receptor, tnfr1, define a family of dominantly inherited autoinflammatory syndromes. Cell. (1999) 97:133–44. doi: 10.1016/s0092-8674(00)80721-7
9. Sood R, Blake T, Aksentijevich I, Wood G, Chen X, Gardner D, et al. Construction of a 1-mb restriction-mapped cosmid contig containing the candidate region for the familial mediterranean fever locus (Mefv) on chromosome 16p 13.3. Genomics. (1997) 42:83–95. doi: 10.1006/geno.1997.4629
10. French FMFC. A candidate gene for familial mediterranean fever. Nat Genet. (1997) 17:25–31. doi: 10.1038/ng0997-25
11. Masters SL, Simon A, Aksentijevich I, Kastner DL. Horror autoinflammaticus: the molecular pathophysiology of autoinflammatory disease (*). Annu Rev Immunol. (2009) 27:621–68. doi: 10.1146/annurev.immunol.25.022106.141627
12. Dvornyk V, Ponomarenko I, Belyaeva T, Reshetnikov E, Churnosov M. Filaggrin gene polymorphisms are associated with atopic dermatitis in women but not in men in the caucasian population of central Russia. PloS One. (2021) 16:e0261026. doi: 10.1371/journal.pone.0261026
13. Zhu TH, Zhu TR, Tran KA, Sivamani RK, Shi VY. Epithelial barrier dysfunctions in atopic dermatitis: A skin-gut-lung model linking microbiome alteration and immune dysregulation. Br J Dermatol. (2018) 179:570–81. doi: 10.1111/bjd.16734
14. Szekanecz Z, McInnes IB, Schett G, Szamosi S, Benko S, Szucs G. Autoinflammation and autoimmunity across rheumatic and musculoskeletal diseases. Nat Rev Rheumatol. (2021) 17:585–95. doi: 10.1038/s41584-021-00652-9
15. Mok CC. Rituximab for the treatment of rheumatoid arthritis: an update. Drug Des Devel Ther. (2013) 8:87–100. doi: 10.2147/DDDT.S41645
16. Favalli EG, Raimondo MG, Becciolini A, Crotti C, Biggioggero M, Caporali R. The management of first-line biologic therapy failures in rheumatoid arthritis: current practice and future perspectives. Autoimmun Rev. (2017) 16:1185–95. doi: 10.1016/j.autrev.2017.10.002
17. McGonagle D, Watad A, Savic S. Mechanistic immunological based classification of rheumatoid arthritis. Autoimmun Rev. (2018) 17:1115–23. doi: 10.1016/j.autrev.2018.06.001
18. Kondo N, Kuroda T, Kobayashi D. Cytokine networks in the pathogenesis of rheumatoid arthritis. Int J Mol Sci. (2021) 22. doi: 10.3390/ijms222010922
19. Smolen JS, Landewe RBM, Bijlsma JWJ, Burmester GR, Dougados M, Kerschbaumer A, et al. Eular recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2019 update. Ann Rheum Dis. (2020) 79:685–99. doi: 10.1136/annrheumdis-2019-216655
20. Nesse W, Westra J, van der Wal JE, Abbas F, Nicholas AP, Vissink A, et al. The periodontium of periodontitis patients contains citrullinated proteins which may play a role in acpa (Anti-citrullinated protein antibody) formation. J Clin Periodontol. (2012) 39:599–607. doi: 10.1111/j.1600-051X.2012.01885.x
21. Smolen JS, Aletaha D, McInnes IB. Rheumatoid arthritis. Lancet. (2016) 388:2023–38. doi: 10.1016/S0140-6736(16)30173-8
22. Fox DA. Citrullination: A specific target for the autoimmune response in rheumatoid arthritis. J Immunol. (2015) 195:5–7. doi: 10.4049/jimmunol.1501021
23. Roudier J, Balandraud N, Auger I. How ra associated hla-dr molecules contribute to the development of antibodies to citrullinated proteins: the hapten carrier model. Front Immunol. (2022) 13:930112. doi: 10.3389/fimmu.2022.930112
24. El Shikh MEM, El Sayed R, Nerviani A, Goldmann K, John CR, Hands R, et al. Extracellular traps and pad4 released by macrophages induce citrullination and auto-antibody production in autoimmune arthritis. J Autoimmun. (2019) 105:102297. doi: 10.1016/j.jaut.2019.06.008
25. Rohrbach AS, Slade DJ, Thompson PR, Mowen KA. Activation of pad4 in net formation. Front Immunol. (2012) 3:360. doi: 10.3389/fimmu.2012.00360
26. Vorobjeva NV, Chernyak BV. Netosis: molecular mechanisms, role in physiology and pathology. Biochem (Mosc). (2020) 85:1178–90. doi: 10.1134/S0006297920100065
27. Carrasco-Marin E, Paz-Miguel JE, Lopez-Mato P, Alvarez-Dominguez C, Leyva-Cobian F. Oxidation of defined antigens allows protein unfolding and increases both proteolytic processing and exposes peptide epitopes which are recognized by specific T cells. Immunology. (1998) 95:314–21. doi: 10.1046/j.1365-2567.1998.00618.x
28. Scally SW, Petersen J, Law SC, Dudek NL, Nel HJ, Loh KL, et al. A molecular basis for the association of the hla-drb1 locus, citrullination, and rheumatoid arthritis. J Exp Med. (2013) 210:2569–82. doi: 10.1084/jem.20131241
29. Sherina N, de Vries C, Kharlamova N, Sippl N, Jiang X, Brynedal B, et al. Antibodies to a citrullinated porphyromonas gingivalis epitope are increased in early rheumatoid arthritis, and can be produced by gingival tissue B cells: implications for a bacterial origin in ra etiology. Front Immunol. (2022) 13:804822. doi: 10.3389/fimmu.2022.804822
30. Stensland ME, Pollmann S, Molberg O, Sollid LM, Fleckenstein B. Primary sequence, together with other factors, influence peptide deimination by peptidylarginine deiminase-4. Biol Chem. (2009) 390:99–107. doi: 10.1515/BC.2009.019
31. Venereau E, Ceriotti C, Bianchi ME. Damps from cell death to new life. Front Immunol. (2015) 6:422. doi: 10.3389/fimmu.2015.00422
32. Asaga H, Yamada M, Senshu T. Selective deimination of vimentin in calcium ionophore-induced apoptosis of mouse peritoneal macrophages. Biochem Biophys Res Commun. (1998) 243:641–6. doi: 10.1006/bbrc.1998.8148
33. Sadeghi M, Dehnavi S, Jamialahmadi T, Johnston TP, Sahebkar A. Neutrophil extracellular trap: A key player in the pathogenesis of autoimmune diseases. Int Immunopharmacol. (2023) 116:109843. doi: 10.1016/j.intimp.2023.109843
34. Lee J, Luria A, Rhodes C, Raghu H, Lingampalli N, Sharpe O, et al. Nicotine drives neutrophil extracellular traps formation and accelerates collagen-induced arthritis. Rheumatol (Oxford). (2017) 56:644–53. doi: 10.1093/rheumatology/kew449
35. Khandpur R, Carmona-Rivera C, Vivekanandan-Giri A, Gizinski A, Yalavarthi S, Knight JS, et al. Nets are a source of citrullinated autoantigens and stimulate inflammatory responses in rheumatoid arthritis. Sci Transl Med. (2013) 5(178):178ra40. doi: 10.1126/scitranslmed.3005580
36. Wegner N, Lundberg K, Kinloch A, Fisher B, Malmstrom V, Feldmann M, et al. Autoimmunity to specific citrullinated proteins gives the first clues to the etiology of rheumatoid arthritis. Immunol Rev. (2010) 233:34–54. doi: 10.1111/j.0105-2896.2009.00850.x
37. Darrah E, Rosen A, Giles JT, Andrade F. Peptidylarginine Deiminase 2, 3 and 4 Have Distinct Specificities against Cellular Substrates: Novel Insights into Autoantigen Selection in Rheumatoid Arthritis. Ann Rheum Dis. (2012) 71:92–8. doi: 10.1136/ard.2011.151712
38. Sohn DH, Rhodes C, Onuma K, Zhao X, Sharpe O, Gazitt T, et al. Local joint inflammation and histone citrullination in a murine model of the transition from preclinical autoimmunity to inflammatory arthritis. Arthritis Rheumatol. (2015) 67:2877–87. doi: 10.1002/art.39283
39. Spengler J, Lugonja B, Ytterberg AJ, Zubarev RA, Creese AJ, Pearson MJ, et al. Release of active peptidyl arginine deiminases by neutrophils can explain production of extracellular citrullinated autoantigens in rheumatoid arthritis synovial fluid. Arthritis Rheumatol. (2015) 67:3135–45. doi: 10.1002/art.39313
40. Elshabrawy HA, Essani AE, Szekanecz Z, Fox DA, Shahrara S. Tlrs, future potential therapeutic targets for ra. Autoimmun Rev. (2017) 16:103–13. doi: 10.1016/j.autrev.2016.12.003
41. Sokolove J, Zhao X, Chandra PE, Robinson WH. Immune complexes containing citrullinated fibrinogen costimulate macrophages via toll-like receptor 4 and fcgamma receptor. Arthritis Rheum. (2011) 63:53–62. doi: 10.1002/art.30081
42. Zhu W, Li X, Fang S, Zhang X, Wang Y, Zhang T, et al. Anti-citrullinated protein antibodies induce macrophage subset disequilibrium in ra patients. Inflammation. (2015) 38:2067–75. doi: 10.1007/s10753-015-0188-z
43. Nashi E, Wang Y, Diamond B. The role of B cells in lupus pathogenesis. Int J Biochem Cell Biol. (2010) 42:543–50. doi: 10.1016/j.biocel.2009.10.011
44. Shlomchik MJ, Madaio MP, Ni D, Trounstein M, Huszar D. The role of B cells in lpr/lpr-induced autoimmunity. J Exp Med. (1994) 180:1295–306. doi: 10.1084/jem.180.4.1295
45. Chan O, Shlomchik MJ. A new role for B cells in systemic autoimmunity: B cells promote spontaneous T cell activation in mrl-lpr/lpr mice. J Immunol. (1998) 160:51–9. doi: 10.4049/jimmunol.160.1.51
46. Herrada AA, Escobedo N, Iruretagoyena M, Valenzuela RA, Burgos PI, Cuitino L, et al. Innate immune cells' Contribution to systemic lupus erythematosus. Front Immunol. (2019) 10:772. doi: 10.3389/fimmu.2019.00772
47. Saulescu I, Ionescu R, Opris-Belinski D. Interferon in systemic lupus erythematosus-a halfway between monogenic autoinflammatory and autoimmune disease. Heliyon. (2022) 8:e11741. doi: 10.1016/j.heliyon.2022.e11741
48. Isaacs A, Lindenmann J. Virus interference. I. The interferon. Proc R Soc Lond B Biol Sci. (1957) 147:258–67. doi: 10.1098/rspb.1957.0048
49. Pestka S, Krause CD, Walter MR. Interferons, interferon-like cytokines, and their receptors. Immunol Rev. (2004) 202:8–32. doi: 10.1111/j.0105-2896.2004.00204.x
50. Chasset F, Arnaud L. Targeting interferons and their pathways in systemic lupus erythematosus. Autoimmun Rev. (2018) 17:44–52. doi: 10.1016/j.autrev.2017.11.009
51. Swiecki M, Colonna M. Type I interferons: diversity of sources, production pathways and effects on immune responses. Curr Opin Virol. (2011) 1:463–75. doi: 10.1016/j.coviro.2011.10.026
52. Swiecki M, Colonna M. The multifaceted biology of plasmacytoid dendritic cells. Nat Rev Immunol. (2015) 15:471–85. doi: 10.1038/nri3865
53. Kumar H, Kawai T, Akira S. Pathogen recognition by the innate immune system. Int Rev Immunol. (2011) 30:16–34. doi: 10.3109/08830185.2010.529976
54. Psarras A, Wittmann M, Vital EM. Emerging concepts of type I interferons in sle pathogenesis and therapy. Nat Rev Rheumatol. (2022) 18:575–90. doi: 10.1038/s41584-022-00826-z
55. Skopelja-Gardner S, Tai J, Sun X, Tanaka L, Kuchenbecker JA, Snyder JM, et al. Acute skin exposure to ultraviolet light triggers neutrophil-mediated kidney inflammation. Proc Natl Acad Sci U.S.A. (2021) 118. doi: 10.1073/pnas.2019097118
56. LaFleur DW, Nardelli B, Tsareva T, Mather D, Feng P, Semenuk M, et al. Interferon-kappa, a novel type I interferon expressed in human keratinocytes. J Biol Chem. (2001) 276:39765–71. doi: 10.1074/jbc.M102502200
57. Sarkar MK, Hile GA, Tsoi LC, Xing X, Liu J, Liang Y, et al. Photosensitivity and type I ifn responses in cutaneous lupus are driven by epidermal-derived interferon kappa. Ann Rheum Dis. (2018) 77:1653–64. doi: 10.1136/annrheumdis-2018-213197
58. Meller S, Winterberg F, Gilliet M, Muller A, Lauceviciute I, Rieker J, et al. Ultraviolet radiation-induced injury, chemokines, and leukocyte recruitment: an amplification cycle triggering cutaneous lupus erythematosus. Arthritis Rheum. (2005) 52:1504–16. doi: 10.1002/art.21034
59. Stannard JN, Reed TJ, Myers E, Lowe L, Sarkar MK, Xing X, et al. Lupus skin is primed for il-6 inflammatory responses through a keratinocyte-mediated autocrine type I interferon loop. J Invest Dermatol. (2017) 137:115–22. doi: 10.1016/j.jid.2016.09.008
60. Der E, Ranabothu S, Suryawanshi H, Akat KM, Clancy R, Morozov P, et al. Single cell rna sequencing to dissect the molecular heterogeneity in lupus nephritis. JCI Insight. (2017) 2. doi: 10.1172/jci.insight.93009
61. Castellano G, Cafiero C, Divella C, Sallustio F, Gigante M, Pontrelli P, et al. Local synthesis of interferon-alpha in lupus nephritis is associated with type I interferons signature and lmp7 induction in renal tubular epithelial cells. Arthritis Res Ther. (2015) 17:72. doi: 10.1186/s13075-015-0588-3
62. van Heteren JT, Rozenberg F, Aronica E, Troost D, Lebon P, Kuijpers TW. Astrocytes produce interferon-alpha and cxcl10, but not il-6 or cxcl8, in aicardi-goutieres syndrome. Glia. (2008) 56:568–78. doi: 10.1002/glia.20639
63. Le Bon A, Thompson C, Kamphuis E, Durand V, Rossmann C, Kalinke U, et al. Cutting edge: enhancement of antibody responses through direct stimulation of B and T cells by type I ifn. J Immunol. (2006) 176:2074–8. doi: 10.4049/jimmunol.176.4.2074
64. Lopez P, Scheel-Toellner D, Rodriguez-Carrio J, Caminal-Montero L, Gordon C, Suarez A. Interferon-alpha-induced B-lymphocyte stimulator expression and mobilization in healthy and systemic lupus erthymatosus monocytes. Rheumatol (Oxford). (2014) 53:2249–58. doi: 10.1093/rheumatology/keu249
65. Li H, Fu YX, Wu Q, Zhou Y, Crossman DK, Yang P, et al. Interferon-induced mechanosensing defects impede apoptotic cell clearance in lupus. J Clin Invest. (2015) 125:2877–90. doi: 10.1172/JCI81059
66. Garcia-Romo GS, Caielli S, Vega B, Connolly J, Allantaz F, Xu Z, et al. Netting neutrophils are major inducers of type I ifn production in pediatric systemic lupus erythematosus. Sci Transl Med. (2011) 3:73ra20. doi: 10.1126/scitranslmed.3001201
67. Gestermann N, Di Domizio J, Lande R, Demaria O, Frasca L, Feldmeyer L, et al. Netting neutrophils activate autoreactive B cells in lupus. J Immunol. (2018) 200:3364–71. doi: 10.4049/jimmunol.1700778
68. Giacomelli R, Ruscitti P, Shoenfeld Y. A comprehensive review on adult onset still's disease. J Autoimmun. (2018) 93:24–36. doi: 10.1016/j.jaut.2018.07.018
69. Church LD, Cook GP, McDermott MF. Primer: inflammasomes and interleukin 1beta in inflammatory disorders. Nat Clin Pract Rheumatol. (2008) 4:34–42. doi: 10.1038/ncprheum0681
70. Jamilloux Y, Gerfaud-Valentin M, Martinon F, Belot A, Henry T, Seve P. Pathogenesis of adult-onset still's disease: new insights from the juvenile counterpart. Immunol Res. (2015) 61:53–62. doi: 10.1007/s12026-014-8561-9
71. Mellins ED, Macaubas C, Grom AA. Pathogenesis of systemic juvenile idiopathic arthritis: some answers, more questions. Nat Rev Rheumatol. (2011) 7:416–26. doi: 10.1038/nrrheum.2011.68
72. Mitrovic S, Fautrel B. New markers for adult-onset still's disease. Joint Bone Spine. (2018) 85:285–93. doi: 10.1016/j.jbspin.2017.05.011
73. Ombrello MJ, Remmers EF, Tachmazidou I, Grom A, Foell D, Haas JP, et al. Hla-drb1*11 and variants of the mhc class ii locus are strong risk factors for systemic juvenile idiopathic arthritis. Proc Natl Acad Sci U.S.A. (2015) 112:15970–5. doi: 10.1073/pnas.1520779112
74. Rao S, Tsang LS, Zhao M, Shi W, Lu Q. Adult-onset still's disease: A disease at the crossroad of innate immunity and autoimmunity. Front Med (Lausanne). (2022) 9:881431. doi: 10.3389/fmed.2022.881431
75. Grom AA, Horne A, De Benedetti F. Macrophage activation syndrome in the era of biologic therapy. Nat Rev Rheumatol. (2016) 12:259–68. doi: 10.1038/nrrheum.2015.179
76. Teachey DT, Rheingold SR, Maude SL, Zugmaier G, Barrett DM, Seif AE, et al. Cytokine release syndrome after blinatumomab treatment related to abnormal macrophage activation and ameliorated with cytokine-directed therapy. Blood. (2013) 121:5154–7. doi: 10.1182/blood-2013-02-485623
77. Giavridis T, van der Stegen SJC, Eyquem J, Hamieh M, Piersigilli A, Sadelain M. Car T cell-induced cytokine release syndrome is mediated by macrophages and abated by il-1 blockade. Nat Med. (2018) 24:731–8. doi: 10.1038/s41591-018-0041-7
78. Villanueva J, Lee S, Giannini EH, Graham TB, Passo MH, Filipovich A, et al. Natural killer cell dysfunction is a distinguishing feature of systemic onset juvenile rheumatoid arthritis and macrophage activation syndrome. Arthritis Res Ther. (2005) 7:R30–7. doi: 10.1186/ar1453
79. Priori R, Barone F, Alessandri C, ColaFrancesco S, McInnes IB, Pitzalis C, et al. Markedly increased il-18 liver expression in adult-onset still's disease-related hepatitis. Rheumatol (Oxford). (2011) 50:776–80. doi: 10.1093/rheumatology/keq397
80. Maruyama J, Inokuma S. Cytokine profiles of macrophage activation syndrome associated with rheumatic diseases. J Rheumatol. (2010) 37:967–73. doi: 10.3899/jrheum.090662
81. ColaFrancesco S, Priori R, Alessandri C, Perricone C, Pendolino M, Picarelli G, et al. Il-18 serum level in adult onset still's disease: A marker of disease activity. Int J Inflam. (2012) 2012:156890. doi: 10.1155/2012/156890
82. McGonagle D, Aydin SZ, Gul A, Mahr A, Direskeneli H. 'Mhc-I-opathy'-unified concept for spondyloarthritis and behcet disease. Nat Rev Rheumatol. (2015) 11:731–40. doi: 10.1038/nrrheum.2015.147
83. Macleod T, Bridgewood C, McGonagle D. Role of neutrophil interleukin-23 in spondyloarthropathy spectrum disorders. Lancet Rheumatol. (2023) 5:e47–57. doi: 10.1016/S2665-9913(22)00334-4
84. Gul A. Pathogenesis of behcet's disease: autoinflammatory features and beyond. Semin Immunopathol. (2015) 37:413–8. doi: 10.1007/s00281-015-0502-8
85. Touitou I, Magne X, Molinari N, Navarro A, Quellec AL, Picco P, et al. Mefv mutations in behcet's disease. Hum Mutat. (2000) 16:271–2. doi: 10.1002/1098-1004(200009)16:3<271::AID-HUMU16>3.0.CO;2-A
86. Perazzio SF, Andrade LEC, de Souza AWS. Understanding behcet's disease in the context of innate immunity activation. Front Immunol. (2020) 11:586558. doi: 10.3389/fimmu.2020.586558
87. Deniz R, Emrence Z, Yalcinkaya Y, Esen BA, Inanc M, Ocal ML, et al. Improved sensitivity of skin pathergy test with polysaccharide pneumococcal vaccine antigens in the diagnosis of behcet disease. Rheumatol (Oxford). (2022) 62(5):1903–1909. doi: 10.1093/rheumatology/keac543
88. Kurhan-Yavuz S, Direskeneli H, Bozkurt N, Ozyazgan Y, Bavbek T, Kazokoglu H, et al. Anti-mhc autoimmunity in behcet's disease: T cell responses to an hla-B-derived peptide cross-reactive with retinal-S antigen in patients with uveitis. Clin Exp Immunol. (2000) 120:162–6. doi: 10.1046/j.1365-2249.2000.01176.x
89. Cavers A, Kugler MC, Ozguler Y, Al-Obeidi AF, Hatemi G, Ueberheide BM, et al. Behcet's disease risk-variant hla-B51/erap1-hap10 alters human cd8 T cell immunity. Ann Rheum Dis. (2022) 81:1603–11. doi: 10.1136/ard-2022-222277
90. Mills KH. Induction, function and regulation of il-17-producing T cells. Eur J Immunol. (2008) 38:2636–49. doi: 10.1002/eji.200838535
91. Yamaguchi Y, Takahashi H, Satoh T, Okazaki Y, Mizuki N, Takahashi K, et al. Natural killer cells control a T-helper 1 response in patients with behcet's disease. Arthritis Res Ther. (2010) 12:R80. doi: 10.1186/ar3005
92. Nishida T, Hirayama K, Nakamura S, Ohno S. Proliferative response of cd8+ Gamma delta+ T cells to streptococcus sanguis in patients with behcet's disease. Ocul Immunol Inflammation. (1998) 6:139–44. doi: 10.1076/ocii.6.3.139.4035
93. Gelmez MY, Cinar S, Cetin EA, Ozcit-Gurel G, Babuna-Kobaner G, Erdugan M, et al. Inflammatory status might direct ilc and nk cells to il-17 expressing ilc3 and nk subsets in behcet's disease. Immunol Lett. (2021) 235:1–8. doi: 10.1016/j.imlet.2021.04.008
94. Stavre Z, Bridgewood C, Zhou Q, Maeda Y, Huang TT, Karman J, et al. A role for neutrophils in early enthesitis in spondyloarthritis. Arthritis Res Ther. (2022) 24:24. doi: 10.1186/s13075-021-02693-7
95. Zhang YZ, Li YY. Inflammatory bowel disease: pathogenesis. World J Gastroenterol. (2014) 20:91–9. doi: 10.3748/wjg.v20.i1.91
96. Kaluzna A, Olczyk P, Komosinska-Vassev K. The role of innate and adaptive immune cells in the pathogenesis and development of the inflammatory response in ulcerative colitis. J Clin Med. (2022) 11. doi: 10.3390/jcm11020400
97. Uhlig HH, Powrie F. Translating immunology into therapeutic concepts for inflammatory bowel disease. Annu Rev Immunol. (2018) 36:755–81. doi: 10.1146/annurev-immunol-042617-053055
98. Uhlig HH. Monogenic diseases associated with intestinal inflammation: implications for the understanding of inflammatory bowel disease. Gut. (2013) 62:1795–805. doi: 10.1136/gutjnl-2012-303956
99. Pang X, He X, Qiu Z, Zhang H, Xie R, Liu Z, et al. Targeting integrin pathways: mechanisms and advances in therapy. Signal Transduct Target Ther. (2023) 8:1. doi: 10.1038/s41392-022-01259-6
100. Liu JZ, van Sommeren S, Huang H, Ng SC, Alberts R, Takahashi A, et al. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat Genet. (2015) 47:979–86. doi: 10.1038/ng.3359
101. Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. (2012) 491:119–24. doi: 10.1038/nature11582
102. de Lange KM, Barrett JC. Understanding inflammatory bowel disease via immunogenetics. J Autoimmun. (2015) 64:91–100. doi: 10.1016/j.jaut.2015.07.013
103. Barrett JC, Hansoul S, Nicolae DL, Cho JH, Duerr RH, Rioux JD, et al. Genome-wide association defines more than 30 distinct susceptibility loci for crohn's disease. Nat Genet. (2008) 40:955–62. doi: 10.1038/ng.175
104. Yamazaki K, McGovern D, Ragoussis J, Paolucci M, Butler H, Jewell D, et al. Single nucleotide polymorphisms in tnfsf15 confer susceptibility to crohn's disease. Hum Mol Genet. (2005) 14:3499–506. doi: 10.1093/hmg/ddi379
105. Kakuta Y, Ueki N, Kinouchi Y, Negoro K, Endo K, Nomura E, et al. Tnfsf15 transcripts from risk haplotype for crohn's disease are overexpressed in stimulated T cells. Hum Mol Genet. (2009) 18:1089–98. doi: 10.1093/hmg/ddp005
106. Bar F, Sina C, Hundorfean G, Pagel R, Lehnert H, Fellermann K, et al. Inflammatory bowel diseases influence major histocompatibility complex class I (Mhc I) and ii compartments in intestinal epithelial cells. Clin Exp Immunol. (2013) 172:280–9. doi: 10.1111/cei.12047
107. O'Keeffe M, Mok WH, Radford KJ. Human dendritic cell subsets and function in health and disease. Cell Mol Life Sci. (2015) 72:4309–25. doi: 10.1007/s00018-015-2005-0
108. Zhou H, Wang F, Wan J, Su S, Shi Y, Li X, et al. Systematic review and meta-analysis of observational studies on the effectiveness and safety of ustekinumab among patients with inflammatory bowel disease in eastern and western countries. J Clin Med. (2023) 12. doi: 10.3390/jcm12051894
109. Raphael I, Nalawade S, Eagar TN, Forsthuber TG. T cell subsets and their signature cytokines in autoimmune and inflammatory diseases. Cytokine. (2015) 74:5–17. doi: 10.1016/j.cyto.2014.09.011
110. Zhu S, Qian Y. Il-17/il-17 receptor system in autoimmune disease: mechanisms and therapeutic potential. Clin Sci (Lond). (2012) 122:487–511. doi: 10.1042/CS20110496
111. Ogino T, Nishimura J, Barman S, Kayama H, Uematsu S, Okuzaki D, et al. Increased th17-inducing activity of cd14+ Cd163 low myeloid cells in intestinal lamina propria of patients with crohn's disease. Gastroenterology. (2013) 145:1380–91 e1. doi: 10.1053/j.gastro.2013.08.049
112. Harris VR, Cooper AJ. Atopic dermatitis: the new frontier. Med J Aust. (2017) 207:351–6. doi: 10.5694/mja17.00463
113. Sandilands A, Sutherland C, Irvine AD, McLean WH. Filaggrin in the frontline: role in skin barrier function and disease. J Cell Sci. (2009) 122:1285–94. doi: 10.1242/jcs.033969
114. Tsakok T, Woolf R, Smith CH, Weidinger S, Flohr C. Atopic dermatitis: the skin barrier and beyond. Br J Dermatol. (2019) 180:464–74. doi: 10.1111/bjd.16934
115. Dainichi T, Kitoh A, Otsuka A, Nakajima S, Nomura T, Kaplan DH, et al. The epithelial immune microenvironment (Eime) in atopic dermatitis and psoriasis. Nat Immunol. (2018) 19:1286–98. doi: 10.1038/s41590-018-0256-2
116. Deleuran M, Hvid M, Kemp K, Christensen GB, Deleuran B, Vestergaard C. Il-25 induces both inflammation and skin barrier dysfunction in atopic dermatitis. Chem Immunol Allergy. (2012) 96:45–9. doi: 10.1159/000331871
117. Cevikbas F, Steinhoff M. Il-33: A novel danger signal system in atopic dermatitis. J Invest Dermatol. (2012) 132:1326–9. doi: 10.1038/jid.2012.66
118. Schmitz J, Owyang A, Oldham E, Song Y, Murphy E, McClanahan TK, et al. Il-33, an interleukin-1-like cytokine that signals via the il-1 receptor-related protein st2 and induces T helper type 2-associated cytokines. Immunity. (2005) 23:479–90. doi: 10.1016/j.immuni.2005.09.015
119. Dillon SR, Sprecher C, Hammond A, Bilsborough J, Rosenfeld-Franklin M, Presnell SR, et al. Interleukin 31, a cytokine produced by activated T cells, induces dermatitis in mice. Nat Immunol. (2004) 5:752–60. doi: 10.1038/ni1084
120. Xiao S, Lu Z, Steinhoff M, Li Y, Buhl T, Fischer M, et al. Innate immune regulates cutaneous sensory il-13 receptor alpha 2 to promote atopic dermatitis. Brain Behav Immun. (2021) 98:28–39. doi: 10.1016/j.bbi.2021.08.211
121. Mack MR, Miron Y, Chen F, Miller PE, Zhang A, Korotzer A, et al. Type 2 cytokines sensitize human sensory neurons to itch-associated stimuli. Front Mol Neurosci. (2023) 16:1258823. doi: 10.3389/fnmol.2023.1258823
122. Punnonen J, Yssel H, de Vries JE. The relative contribution of il-4 and il-13 to human ige synthesis induced by activated cd4+ or cd8+ T cells. J Allergy Clin Immunol. (1997) 100:792–801. doi: 10.1016/s0091-6749(97)70276-8
123. Galli SJ, Tsai M. Ige and mast cells in allergic disease. Nat Med. (2012) 18:693–704. doi: 10.1038/nm.2755
124. Spits H, Di Santo JP. The expanding family of innate lymphoid cells: regulators and effectors of immunity and tissue remodeling. Nat Immunol. (2011) 12:21–7. doi: 10.1038/ni.1962
125. Van Dyken SJ, Mohapatra A, Nussbaum JC, Molofsky AB, Thornton EE, Ziegler SF, et al. Chitin activates parallel immune modules that direct distinct inflammatory responses via innate lymphoid type 2 and gammadelta T cells. Immunity. (2014) 40:414–24. doi: 10.1016/j.immuni.2014.02.003
126. Motomura Y, Morita H, Moro K, Nakae S, Artis D, Endo TA, et al. Basophil-derived interleukin-4 controls the function of natural helper cells, a member of ilc2s, in lung inflammation. Immunity. (2014) 40:758–71. doi: 10.1016/j.immuni.2014.04.013
127. Kim BS, Wang K, Siracusa MC, Saenz SA, Brestoff JR, Monticelli LA, et al. Basophils promote innate lymphoid cell responses in inflamed skin. J Immunol. (2014) 193:3717–25. doi: 10.4049/jimmunol.1401307
128. Hughes T, Becknell B, Freud AG, McClory S, Briercheck E, Yu J, et al. Interleukin-1beta selectively expands and sustains interleukin-22+ Immature human natural killer cells in secondary lymphoid tissue. Immunity. (2010) 32:803–14. doi: 10.1016/j.immuni.2010.06.007
129. Coccia M, Harrison OJ, Schiering C, Asquith MJ, Becher B, Powrie F, et al. Il-1beta mediates chronic intestinal inflammation by promoting the accumulation of il-17a secreting innate lymphoid cells and cd4(+) th17 cells. J Exp Med. (2012) 209:1595–609. doi: 10.1084/jem.20111453
130. Buonocore S, Ahern PP, Uhlig HH, Ivanov II, DR L, KJ M, et al. Innate lymphoid cells drive interleukin-23-dependent innate intestinal pathology. Nature. (2010) 464:1371–5. doi: 10.1038/nature08949
131. Sawa S, Lochner M, Satoh-Takayama N, Dulauroy S, Berard M, Kleinschek M, et al. Rorgammat+ Innate lymphoid cells regulate intestinal homeostasis by integrating negative signals from the symbiotic microbiota. Nat Immunol. (2011) 12:320–6. doi: 10.1038/ni.2002
132. Boehm U, Klamp T, Groot M, Howard JC. Cellular responses to interferon-gamma. Annu Rev Immunol. (1997) 15:749–95. doi: 10.1146/annurev.immunol.15.1.749
133. Mosmann TR, Coffman RL. Th1 and th2 cells: different patterns of lymphokine secretion lead to different functional properties. Annu Rev Immunol. (1989) 7:145–73. doi: 10.1146/annurev.iy.07.040189.001045
134. Hepworth MR, Monticelli LA, Fung TC, Ziegler CG, Grunberg S, Sinha R, et al. Innate lymphoid cells regulate cd4+ T-cell responses to intestinal commensal bacteria. Nature. (2013) 498:113–7. doi: 10.1038/nature12240
135. von Burg N, Chappaz S, Baerenwaldt A, Horvath E, Bose Dasgupta S, Ashok D, et al. Activated group 3 innate lymphoid cells promote T-cell-mediated immune responses. Proc Natl Acad Sci U.S.A. (2014) 111:12835–40. doi: 10.1073/pnas.1406908111
136. Zeng B, Shi S, Ashworth G, Dong C, Liu J, Xing F. Ilc3 function as a double-edged sword in inflammatory bowel diseases. Cell Death Dis. (2019) 10:315. doi: 10.1038/s41419-019-1540-2
137. Blauvelt A, Chiricozzi A. The immunologic role of il-17 in psoriasis and psoriatic arthritis pathogenesis. Clin Rev Allergy Immunol. (2018) 55:379–90. doi: 10.1007/s12016-018-8702-3
138. Leijten EF, van Kempen TS, Boes M, Michels-van Amelsfort JM, Hijnen D, Hartgring SA, et al. Brief report: enrichment of activated group 3 innate lymphoid cells in psoriatic arthritis synovial fluid. Arthritis Rheumatol. (2015) 67:2673–8. doi: 10.1002/art.39261
139. Treiner E, Duban L, Bahram S, Radosavljevic M, Wanner V, Tilloy F, et al. Selection of evolutionarily conserved mucosal-associated invariant T cells by mr1. Nature. (2003) 422:164–9. doi: 10.1038/nature01433
140. Provine NM, Binder B, FitzPatrick MEB, Schuch A, Garner LC, Williamson KD, et al. Unique and common features of innate-like human vdelta2(+) gammadeltat cells and mucosal-associated invariant T cells. Front Immunol. (2018) 9:756. doi: 10.3389/fimmu.2018.00756
141. Paul S, Singh AK, Shilpi LG. Phenotypic and functional plasticity of gamma-delta (Gammadelta) T cells in inflammation and tolerance. Int Rev Immunol. (2014) 33:537–58. doi: 10.3109/08830185.2013.863306
142. Ferrick DA, Schrenzel MD, Mulvania T, Hsieh B, Ferlin WG, Lepper H. Differential production of interferon-gamma and interleukin-4 in response to th1- and th2-stimulating pathogens by gamma delta T cells in vivo. Nature. (1995) 373:255–7. doi: 10.1038/373255a0
143. Ness-Schwickerath KJ, Jin C, Morita CT. Cytokine requirements for the differentiation and expansion of il-17a- and il-22-producing human vgamma2vdelta2 T cells. J Immunol. (2010) 184:7268–80. doi: 10.4049/jimmunol.1000600
144. Do JS, Visperas A, Dong C, Baldwin WM 3rd, Min B. Cutting edge: generation of colitogenic th17 cd4 T cells is enhanced by il-17+ Gammadelta T cells. J Immunol. (2011) 186:4546–50. doi: 10.4049/jimmunol.1004021
Keywords: autoimmunity, autoinflammatory diseases, neutrophil, MHC-I antigen presentation, autoantibodies, immune cells
Citation: Abacar K, Macleod T, Direskeneli H and McGonagle D (2024) How underappreciated autoinflammatory (innate immunity) mechanisms dominate disparate autoimmune disorders. Front. Immunol. 15:1439371. doi: 10.3389/fimmu.2024.1439371
Received: 27 May 2024; Accepted: 04 September 2024;
Published: 20 September 2024.
Edited by:
Christine Gibson Parks, National Institute of Environmental Health Sciences (NIH), United StatesReviewed by:
Zoltan Szekanecz, University of Debrecen, HungaryEsther Erdei, University of New Mexico Health Sciences Center, United States
Copyright © 2024 Abacar, Macleod, Direskeneli and McGonagle. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dennis McGonagle, RC5HLk1jR29uYWdsZUBsZWVkcy5hYy51aw==