
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 07 November 2024
Sec. Viral Immunology
Volume 15 - 2024 | https://doi.org/10.3389/fimmu.2024.1439328
 Luca Baroncini
Luca Baroncini Christina K. S. Muller
Christina K. S. Muller Nicole P. Kadzioch
Nicole P. Kadzioch Rebekka WolfensbergerDoris RussenbergerSimon BredlTafadzwa Mlambo
Rebekka WolfensbergerDoris RussenbergerSimon BredlTafadzwa Mlambo Roberto F. Speck*
Roberto F. Speck*Introduction: Very little is known about the role of macrophages as immune mediators during natural HIV infection. Humanized mice are an extremely valuable in vivo model for studying HIV pathogenesis. However, the presence of murine mononuclear phagocytes in these models represents a significant limitation for studying their human counterpart. Therefore, we have developed a novel humanized mouse model that allows selective depletion of human myeloid cells at a time point of our choosing.
Methods: We genetically engineered human hematopoietic stem and progenitor cells (HSPCs) to express an inducible caspase-9 (iCas9) suicide system under a synthetic myeloid promoter. Using these HSPCs, we generated humanized mice. iCasp9 induction in vivo resulted in selective human myeloid cell death in this inducible human myeloid depletion (iHMD) mouse model. In addition, we co-cultured monocyte-derived macrophages with ex vivo HIV-infected PBMCs to further mechanistically investigate the effect of macrophages on HIV replication using flow cytometry, cytokine analysis, and RNA sequencing of both macrophages and CD4+ T cells.
Results: HIV infection induced a pro-inflammatory phenotype in HIV-infected humanized NSG mice during the early and late stages of HIV infection. Myeloid cell depletion in HIV-infected iHMD-NSG mice resulted in a rapid increase in HIV RNA replication, which was accompanied by a loss of pro-inflammatory cytokines. Co-culture of macrophages with ex vivo HIV-infected PBMCs reproduced their anti-HIV effects observed in vivo. Transcriptomic data showed macrophages upregulate antiviral cytokines and chemokines in co-culture, while inducing CD4+ T cells to upregulate HIV restriction factors and downregulate pathways involved in protein expression and cell replication.
Discussion: This study describes a novel role of macrophages as effector cells, both ex vivo and in vivo, acting against HIV replication and limiting disease progression.
Macrophages play a crucial role in the innate immune response as well as in maintaining tissue homeostasis and facilitating repair (1). These vastly different tasks require a tremendous plasticity orchestrated by various receptors responding to their cognate ligands, which eventually directs the macrophage differentiation (2). Macrophages are distributed virtually in every organ, where they act as the first line of defense against pathogens, among which viruses (3, 4). Macrophage numbers increase during the early stages of viral infections, including hepatitis C virus (HCV), hepatitis B (HBV) and SARS-CoV2 infection (5–7). These events are usually followed by an upregulation of pro-inflammatory cytokines, resulting in the recruitment of effector cells to the infected tissue and antigen presentation to T cells (8). Macrophages themselves play a central role by killing infected cells and restricting viral infection (9). However, different viruses alter macrophages’ phenotype and functionality, leading to aberrant responses, including immunopathology. For example, macrophages contribute to the insurgency of liver fibrosis during HCV and HBV infection via TGF-1β, IL-1β and TNFα production (5, 6). The alteration in number and functionality of alveolar macrophages are associated with cytokine storm and severe disease prognosis during SARS-CoV-2 infection (10).
In human immunodeficiency virus (HIV) type-1 infection, macrophages have long been viewed as just another HIV-permissive cell that functions as an HIV producer but rarely experiences viral cytopathicity (11). Notably, macrophages can be infected via direct membrane fusion or upon engulfment of HIV-infected CD4+ T cells (12). Furthermore, HIV can hijack macrophages by accumulating in virus-containing compartments (VCCs). These VCCs release HIV-free virions when interacting with activated CD4+ T cells (13). As the HIV infection progresses, HIV quasi-species within the host become increasingly more macrophage-tropic (14). The ability of HIV strains to replicate in macrophages in the absence of T cells was corroborated by in vivo studies in non-human primates and the “macrophage-only humanized mouse model” (15, 16).
Macrophages, such as those found in mucosal tissue, are now considered key cells in virus transmission during sexual contact and then in virus dissemination (17). Depending on the tissue origin, macrophages have a variable half-life ranging from just a few weeks to over four years in the brain (18, 19). Together with their self-renewal capacity (20), this suggests an involvement of these cells in the formation of the HIV reservoir. In fact, latently HIV-infected macrophages are now considered a significant part of the HIV reservoir in people living with HIV and must be targeted to achieve control without combined antiretroviral therapy (cART).
But what about macrophages’ anti-HIV activity? The cells with well-documented anti-HIV activity are cytotoxic CD8+ T cells and natural killer (NK) cells (21, 22). Macrophages’ sensing of HIV during the early phases of infection triggers an inflammatory cascade, polarizing macrophages toward a pro-inflammatory state (23, 24). As HIV infection progresses toward the chronic stage, macrophages shift toward an anti-inflammatory state (25). This switch suggests a complex interplay within the immune system. However, there is currently no in vivo study exploring the potential inhibitory role of macrophages in HIV pathogenesis.
Humanized (hu) mice are based on the transplantation of human hematopoietic progenitor and stem cells (HSPCs) into immunocompromised mice, which results in the reconstitution of human immune cells (26). When infected with HIV, these mice can maintain disseminated infection and represent an invaluable tool for HIV research (27). In this study, we investigated whether macrophages have an anti- or pro-viral effect on HIV replication in HIV-infected hu NOD scid Gamma (NSG) mice by creating a new hu mouse model, in which we were able to selectively deplete human myeloid cells without perturbing any other immune subset. We found that myeloid cell depletion resulted in a significant increase in HV replication in these HIV-infected mice. Using co-culture assays, cytokine analysis and transcriptomic analysis, we showed that macrophages upregulate IFN-stimulated genes (ISGs) and production of HIV co-receptor antagonists which may be the reason for the restriction of HIV replication. These data suggest a novel role of macrophages in HIV infection, which has long been overlooked.
Hu mice were generated as previously described by Audige et al. (28). In brief, immunodeficient NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice were obtained from Charles River Laboratories, and bred and maintained at the Laboratory Animal Service Center of the University of Zurich. Newborn mice were irradiated with 1Gy 1-3 days after birth and subsequently transplanted with 1.5 ± 0.5 x105 CD34+ cells via intrahepatic injection. We checked the degree of human cell engraftment at approximately 16 weeks of age by staining peripheral blood for the pan-human marker CD45.
HEK 293T cells (ATCC, Cat#: CRL-1573) and TZM-bl cells (NIH AIDS Reagents Program, GenBank: ARP-5011) were cultured in Dulbecco’s Modified Eagle Medium (DMEM) (Sigma-Aldrich, Cat#: D5796) supplemented with 10% fetal bovine serum (FBS) (Gibco, Cat#: 26140), 1% 2mM L-glutamine and 1% (Thermo Fischer Scientific, Cat#: 25030081), Penicillin/Streptomycin (Thermo Fischer Scientific 15140148)). PBMCs were isolated from buffy coats via gradient centrifugation, using Greiner Bio-One LeucoSEP™ Polypropylene Tubes (Thermo Fisher scientific, Cat#: Z642843) filled with 15 ml Lymphoprep™ (Stemcell technologies, Cat#: 07861). For both CD8+ T cell and NK cell depletion (Supplementary Figure S14), we performed a negative selection with magnetic bead sorting, either CD8 MicroBeads, human (Miltenyi biotec, Cat#: 130-045-201) or NK cell isolation kit, human (Miltenyi biotec, Cat#: 130-092-657) according to manufacturer protocol (29). Non targeted fraction was used for experimentation. PBMCs were cultured in RPMI-1640 medium supplemented with 10% FBS, interleukin-2 (rhIL-2) (Immunotools, Cat#: 11340027) at 10 U/ml, 2mM L-glutamine, and 1% Penicillin–Streptomycin. All cells were incubated at 37°C and 5% CO2.
Both the CCR5-tropic HIV YU-2 and the CXCR4-tropic HIV NL4-3 molecular clones from NIH AIDS Reagent Program were transfected into HEK 293T cells using polyethylenimine (PEI, Thermo Fisher Scientific, Cat# BMS1003-A). Culture supernatants were harvested 48 hrs later, filtrated (0.22µM, Steriflip, EMD Millipore, Cat#: SE1M179M6) and then stored at -80°C until further use. Tissue culture infectious dose 50 (TCID50) of the viral stocks was determined by infecting TZM-bl cells. 48 hrs after infection, cells were lysed with Passive Lysis Buffer (Cat#: E1941, Promega) and used for measuring the emitted luminescence (Cat#: E1501, Promega) with GloMax Navigator Microplate Luminometer (Promega, Cat#: GM2000). The cut-off for counting a well as HIV+ was 2.5x the background relative light units (RLU). Virus controls had to reach ≥10x the background RLU. TCID50 was calculated according to the Reed Muench method (30).
HuNSG mice with human engraftment level of CD45+ cells > 5% were infected with HIV-1 YU-2 at a TCID50 of 2x105 via intraperitoneal (i.p.) injection approximately one week after checking human engraftment level. Viral dissemination and replication were investigated in plasma of infected mice starting from 4 weeks after infection.
In 6-well plates, PBMCs from 3 different donors, were plated at a density of 1x106 per well, in triplicate. PBMCs were activated for 3 days in RPMI-1640 supplemented with 2 ng/ml phytohemagglutinin (PHA-P) (ThermoFisher, Cat#: 543613) and 50 ng/ml rhIL-2, 10% FBS, 1% penicillin-streptomycin. After 3 days, the medium was changed to RPMI-1640 supplemented with 10 ng/ml rhIL-2, 10% FBS, 1% penicillin-streptomycin. Activated PBMCs were infected with HIV-1 NL4-3 at a multiplicity of infection (MOI) of 0.01 for 12 hrs and cultured for an additional 2 days before co-cultivation with autologous MDMs. For obtaining the autologous MDMs, monocytes were isolated from PBMCs via magnetic-beads sorting (CD14 MicroBeads, human) (Miltenyi Biotec, Cat#: 130-050-201), according to manufacturer protocol (31). CD14 monocytes were differentiated for 6 days in RPMI-1640 medium supplemented with 10% human AB serum (Sigma Aldrich, Cat#: H4522) at a density of 1x106 per well in a F-bottom 6-well plate (Starstedt, Cat#: 83.3920). Pro-inflammatory M1 polarization was achieved by adding 20 ng/ml rhIFNγ (Immunotools, Cat#: 11343534) and 100 ng/ml LPS (Sigma Aldrich, Cat#: L2387) for an additional 2 days, and anti-inflammatory M2 polarization by adding 20 ng/ml rhIL-4 (Immunotools, Cat#: 11340043). In parallel, we kept unstimulated MDMs (M0) in order to have resting macrophages. Ex vivo HIV-1 infected PBMCs were eventually co-cultured with either pro- or anti-inflammatory, or resting macrophages, derived from the same donor. Co-cultures were performed in two separate ways, either in direct contact (cell-to-cell) or separated by a trans well with 1μm pores (Starstedt TC inserts, Cat#: 83.3920).
TZM-bl cells were seeded at a density of 1x104 cells per well in a 96-well F-bottom microplate (Greiner Bio-One, Cat#: 655090). 50 μl of co-culture supernatant was added in triplicate, per well, per sample. After 48 hrs incubation, Luciferase Assay System (Promega, Cat#: E1501) was used according to manufacturer protocol (32), for mammalian cells. Results were acquired using the GloMax Navigator Microplate Luminometer (Promega, Cat#: GM2000), setting exposure to 1 sec.
Depletion of murine and human phagocytes was performed via intraperitoneal (i.p.) injections of clodronate liposomes. The loading dose was 0.03 mg/g body weight, followed by weekly injections with a maintenance dose of 0.015 mg/g body weight.
Third generation lentiviral particles (33) were produced in HEK 293T cells. Briefly, transfection mix was prepared in Opti-MEM medium (ThermoFisher Scientific, Cat#: 11058021) by adding 14.5 µg of pDEST_SP107-iCasp9GFP_pGK-NGFR (Supplementary Figure S7A) lentiviral plasmid, 8.8 µg pSYNGP plasmid, 4 µg RSV-rev plasmid and 4.8 µg pVSV plasmid per ml of transfection mix. The polycation, PEI was added, to the mix in a 1:6 dilution. The transfection mix was incubated 20 min at room temperature (RT) and added to the cells. Medium was changed after 6 hrs with Opti-MEM supplemented with 5mM sodium butyrate, harvested 48 hrs after transfection and spun down at 300g for 5 min. to remove cell debris, and sterile filtered. Lentiviral particles were concentrated via Amicon Centrifugal Filter Unit (Sigma Aldrich, Cat#: UFC901096), according to manufacturer protocols (34). Lentiviral production was validated via Lenti-X GoStix Plus (Takara, Cat#: 631280), according to manufacturer protocol (35), and stored at -80°C. Lentiviral titration was performed by transduction of 6 serial dilutions in HEK 293T cells.
PBMCs were transduced with lentiviral particles by seeding at a MOI of 5 for 12 hrs at a density of 2x105 cells per well in a 24-well plate. To promote transduction, we also added polybrene (Sigma Aldrich, Cat#: TR-1003) at a 1:1000 dilution. Transduced PBMCs were cultured 36 hrs prior to transduction evaluation via flow cytometry and treatment with Rimiducid (AP1903) (MedChemExpress, Cat#: 195514-63-7).
We isolated white blood cells from the umbilical cord blood using Greiner Bio-One LeucoSEP™ Polypropylene Tubes filled with 15 ml Lymphoprep™ and then CD34+ cells via magnetic beads sorting (CD34 MicroBeads, human) (Miltenyi biotec, Cat#: 130-046-702) according to manufacturer protocol (29). CD34+ cells were either stored in liquid nitrogen in 90% FBS and 10% DMSO or were immediately transduced. For the transduction, CD34+ cells were cultured in Isove’s modified eagle’s medium (Thermo Fisher Scientific, Cat#: 12440053) supplemented with 20% BIT 9500 serum substitute (Stemcell technologies, Cat#: 09500), 10 μg/ml human low-density lipoprotein (Stemcell technologies, Cat#: 02698), 0.1mM 2-mercaptoethanol (Sigma Aldrich, Cat#: 63689-25ML-F), 0.1mM Glutamax 500 (Glibco, Cat#: 35050087), 100 ng/ml FLT3-L, 100 ng/ml SCF, 20 ng/ml IL-3, 20 ng/ml IL-6, 20 ng/ml G-CSF (all Immunotools, Cat#: 11343303, 11343325, 11340033, 11340064, 11343133) and UM171 35nM. CD34+ cells were transduced 12 hrs after isolation at a MOI of 5, in the presence of LentiBlast Premium (1:500) (OZ Biosciences, Cat#: LBPX500) and medium was changed 16 hrs later. Transduced CD34+ cells were cultured for 5 days. NGFR+, CD34+ cells were sorted via magnetic beads sorting (CD271 (NGFR) MicroBeads, human) (Miltenyi biotec, Cat#: 130-099-023). Sorted cells were kept at -80°C in 90% FBS and 10% dimethyl sulfoxide (DMSO (Sigma Aldrich, Cat#: 67-68-5) until further use.
AP1903 was reconstituted in DMSO with a final concentration of 10mM for cell culture experiments, as per manufacturer protocol (36). Cells were treated with 10nM. AP1903 for in vivo treatment, reconstituted in 10% DMSO and 90% corn oil to achieve a concentration of 2.5 mg/ml, as per manufacturer protocol (36). iHMD-NSG mice were treated intravenously (i.v.) via tail vain injection at a concentration of 5 mg/kg. All dilutions of the stock solution were made in phosphate buffered saline (PBS) (Gibco, Cat#: 10010023).
60µl of plasma was collected from the blood by low-speed centrifugation and used for RNA isolation (QIAmp viral isolation kit, Qiagen, Cat#: 52906). Isolated RNA was eluted in 30µl AVE buffer of which 12µl was used for reverse transcription (RT) (iScript Select cDNA synthesis kit, BioRad, Cat#: 1708897). The reverse primer used for cDNA synthesis was skcc (TACTAGTAGTTCCTGCTATGTCACTTCC), which binds to the gag region. qPCR was carried out by using DreamTaq™ Hot Start PCR Master Mix (Thermo Fisher Scientific, Cat#: K9012), skcc (reverse) and ts5’gag (CAAGCAGCCATGCAAATGTTAAAAGA) (forward) primers, and FAM-BHQ1 probe mf319 (TGCAGCTTCCTCATTGATGGT). 10µl of viral cDNA was used in the qPCR reaction. Standards (7.13x107 – 200 copies per ml) were generated by using an HIV-1 culture supernatant with a known copy number (determined using the COBAS TaqMan HIV-1 Test (Roche)).
Serum concentration of cytokines and soluble cytokine receptors were evaluated in serum samples from iHMD-NSG and cell culture supernatant. The following cytokines were evaluated: IL-1α, IL-1RA, IL-1β, IL-2, IL-4, IL-5, IL-6, IL-7, IL-9, IL-10, IL-12p70, IL-13, IL-15, IL-17A, IL-18, IL-21, IL -22, IL-23, IL-27, IL-31, IFN-α, IFN-γ and TNF, chemokines, i.e., CCL2, CCL3, CCL4, CCL5, CCL11, CXCL1, CXCL8, CXCL9, CXCL10, CXCL12, CXCL13 and TNF-β and growth factors, i.e., NGF-β, BDNF, EGF, FGF-2, HGF, LIF, PDGF-BB, PlGF-1, SCF, VEGF-A, VEGF-D, BAFF, GM-CSF and G-CSF. Cytokine concentrations were determined by multiplex bead assay as previously described by Amelio at al. (37). The upper normal values for each marker were defined based on the results obtained in the 450 sera collected from healthy individuals (mean + 2 standard deviations).
All Cat# for all antibodies listed in Supplementary Table S1.
HSPCs were stained for human cell surface markers CD34, CD3, NGFR. PBMCs were stained for human cell surface markers CD3, CD4, CD8, CD14, CD19, NGFR (all BioLegend). Immune phenotyping of MDMs was performed on polarized macrophages by staining for human surface markers HLA-DR, CD38, CD86, CD80, CD11c, CD209, D206, CD163, CD33, CD3, CD19, CD56, CD66b (all BioLegend). Macrophages were detached from the culture plate by pipetting with ice-cold PBS supplemented with 2mM EDTA (Gibco, Cat#: 17892) for approximately 10 min per well. Compensation controls were performed using Ultra Comp eBeads (Thermo Fisher Scientific, Cat#: 01-2222-42). Washing and reagent dilutions were done in PBS or FACS buffer (PBS containing 2% FBS and 0.05% sodium azide). Before acquisition, cells were fixed with 1% paraformaldehyde (PFA) (Sigma, Cat#: P6148)
Immunophenotypic analysis of peripheral blood, splenocytes, hepatocytes, bone marrow leukocytes, lung leucocytes and peritoneal cells was performed to monitor and determine human engraftment, immune activation and the phenotype of multiple cell lineages. Peripheral blood was collected into EDTA tubes. 20μl whole blood were used to determine white blood cell counts. For immunophenotypic analysis, 80-100μl of whole blood were used. Erythrocyte removal from stained whole blood was performed with BD FACS Lysing Solution (BD Biosciences, Cat#: 349202). At necropsy, 500μl of blood were withdrawn. In this instance, leukocytes were extracted by gradient centrifugation in Lymphoprep™. Bone marrow cells were obtained from mouse femurs by flushing the bone marrow out with a 300μl syringe. Single cell suspensions of splenocytes, hepatocytes, lung leukocytes and lymph node leukocytes were obtained by pressing the organs through 70µm nylon mesh. Gradient centrifugation of splenocytes, hepatocytes and lung lymphocytes, was performed with Lymphoprep™, for separation from erythrocytes. In order to extract tissue resident macrophages, lung, liver and spleen tissue samples were reduced and incubated in Collagenase solution (HBSS) (Thermo Fisher, Cat#: 88284), 10% FBS, Collagenase type 4 (0.4 mg/ml) (Sigma Aldrich, Cat#: C4-22-1G)), for 45 min at 37°CC, prior to homogenization through a 70μl cell strainer. 20μl of cell suspension was used to determine the total tissue cell count. 1x106 cells per tissue were stained for flow cytometry. Different staining protocols were used in different experiments. Depending on the staining, the following surface markers were used: murine CD45, F4/80, CD11b, Ly6C, Ly6G; human CD45, CD3, CD4, CD8, CD19, CD33, NKp46, CD56, CD14, CD16, CD19, CD86, CD11b, NGFR, CD66b, HLA-DR, CD38, CD86, CD80, CD11c, CD209, CD206, CD163, CD33 (all BioLegend, Supplementary Table S1). Furthermore, splenocytes and bone marrow cells were stained for human cell surface markers CD45, CD3, CD4, CD8, CD11b (all BioLegend) and for HIV-1 core antigen intracellular marker p24 (Beckman Coulter). Intracellular staining was performed with fixation/permeabilization kit (BD Biosciences), according to manufacturer protocol (38). Compensations for intracellular p24 signal were performed in ACH-2 cells (NIH AIDS program) activated via TNFα stimulation (0.5 ng/ml). The Zombie NIR fixable Viability Kit (BioLegend, Cat#: 423107) was included in all multicolor staining panels, and compensation controls were performed using Ultra Comp eBeads. Washing and reagent dilutions were done in PBS or FACS buffer. Before acquisition, cells were fixed with 1% paraformaldehyde. All samples were acquired at BD LSR II Fortessa (BD Bioscience). Data were analyzed using FlowJo software 10.8.1 (TreeStar).
RNA sequencing was performed on either CD4+ T cells (isolated from PBMCs) or M1-polarized MDMs, from either ex vivo co-culture or monocultures. In fact, we followed exactly the same experimental design as described in the paragraph “Co-culture of MDMs with ex vivo HIV-infected PBMCs”. As the most potent HIV inhibition was observed at 72 hours after co-culturing, we harvested the two cell types at this time point. We performed this experiment with three donors, with three replicates from each donor. For CD4+ T cells, both HIV infected and uninfected cells were isolated after 72 hours of either monoculture or co-culture with MDMs via magnetic bead sorting (CD4 MicroBeads, human, Miltenyi biotec, Cat #: 130-097-048), according to manufacturer protocol (29). We performed two cycles of magnetic beads sorting in parallel, aiming to maximize population purity. Plates with adherent MDMs were washed 3 times with PBS to remove PBMCs residues. RNA from both CD4+ T cells and MDMs was isolated with RNeasy mini kit (Qiagen, Cat#: 74106), according to manufacturer protocol (39). All samples were sequenced paired-end 150bp on the NovaSeqX. The library prep method used is “Illumina Stranded mRNA Prep, Ligation” as per manufacturer protocol.
The Novaseq X Plus (Illumina, Inc, California, USA) was used for cluster generation and sequencing according to standard protocol. Sequencing were paired end at 2 X150 bp or single end 100 bp.
The quality of the isolated RNA was determined with a Fragment Analyzer (Agilent, Santa Clara, California, USA). Only those samples with a 260 nm/280 nm ratio between 1.8–2.1 and a 28S/18S ratio within 1.5–2 were further processed. Illumina Stranded mRNA Prep, Ligation (Illumina, Inc, California, USA) was used in the succeeding steps. Briefly, total RNA samples (100-1000 ng) were poly A enriched and then reverse-transcribed into double-stranded cDNA. The cDNA samples were fragmented, end-repaired and adenylated before ligation of TruSeq adapters containing unique dual indices (UDI) for multiplexing. Fragments containing TruSeq adapters on both ends were selectively enriched with PCR. The quality and quantity of the enriched libraries were validated using the Fragment Analyzer (Agilent, Santa Clara, California, USA). The product is a smear with an average fragment size of approximately 260 bp. The libraries were normalized to 10nM in Tris-Cl 10 mM, pH8.5 with 0.1% Tween 20.
The raw reads were first cleaned by removing adapter sequences and poly-x sequences (> 9 nt used for detection) using fastp (Version 0.23.4) (40). Reads with length <18nt after trimming were additionally filtered out. Sequence pseudo alignment of the resulting high-quality reads to the Human reference genome (build GRCh38.p13) and quantification of gene level expression (gene model definition from GENCODE release 42) was carried out using Kallisto (Version 0.46.1) (41). To detect differentially expressed genes we used the glm approach implemented in the software package DESeq2 (R version: 4.3.2, DESeq2 version: 1.42.0) (42). For comparisons with additional baselines, the glm approach implemented in EdgeR was used (R version: 4.3.2, EdgeR version: 4.0.1) (43). Terms with a false-discovery rate (FDR) < 0.05 were considered significant. Genes showing altered expression with FDR-adjusted p-value <= 0.01 and log2 ratio >= 0.5 were considered significant. Over-representation analysis (ORA) was conducted based on differentially expressed genes with FDR-adjusted p-value < 0.01 using clusterProfiler (Version 4.10.0) (44). Raw data can be access in GEO public repository: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?&acc=GSE269285.
All statistical analyses beside bulk RNA sequencing were performed with GraphPad Prism 8.0 software (GraphPad Software, Inc., La Jolla, CA). The individual statistical test is indicated in each respective figure legend.
All animal experiments were approved by the Cantonal Veterinary Office (license ZH181/17; Amendment 28395 of ZH243/16; ZH081/21). Human cord blood collection was approved by the Ethical Committee of the Kanton Zurich (#EK1103) and collected with the informed consent from the donors. Buffy coats were anonymously provided by the Blood Donation Service Zurich, Swiss Red Cross, Schlieren.
Natural HIV infection in humans results in an activated and exhausted phenotype in immune cells, which has been extensively shown for T-cells (45). However, in vivo data on myeloid cell activation during HIV infection are scarce. Therefore, we performed a phenotypical characterization of human mononuclear phagocytes in multiple organs of HIV-infected and uninfected huNSG mice at four (early HIV-1 infection) and twelve weeks (late HIV-1 infection) post-infection. We found an upregulation of pro-inflammatory markers, i.e., HLA-DR, CD38, CD86, and CD80, during early HIV-1 infection stage in several organs (Figure 1A, Supplementary Figure S1), which persisted into the late infection stages (Figure 1B, Supplementary Figure S2). In contrast, we found no differences in anti-inflammatory markers, CD206, CD163 and CD209, in early HIV infection between the two groups, while they increased in the later infection stage (Figures 1A, B, Supplementary Figures S2, S3). Furthermore, we observed a higher number of specific subsets of human mononuclear phagocytes defined by their expression of pro-inflammatory markers, HLA-DR, CD38, CD80 and CD86 (Supplementary Figure S3), in HIV-infected mice during acute and chronic infection in bone marrow, peritoneum, lungs and spleen (Figures 1C, D). An increase in human mononuclear phagocytes expressing anti-inflammatory markers, CD163, CD206 and CD209, was observed in all organs, but only during late HIV infection (Figures 1E, F). In addition, we observed a higher number of monocytes, only in specific tissues, during both early and late HIV infection (Supplementary Figure S4A), while no difference was observed for dendritic cells (Supplementary Figure S4B). Taken together, these results showed that HIV infection induces strong activation of human mononuclear phagocytes in humanized NSG mice and underscored their plasticity and functionality in huNSG mice. We also noted differences in immune activation at early vs late time points, regardless of HIV infection. This difference was actually expected since the human lymphatic system of humanized mice is known to undergo dynamic changes overtime (46).
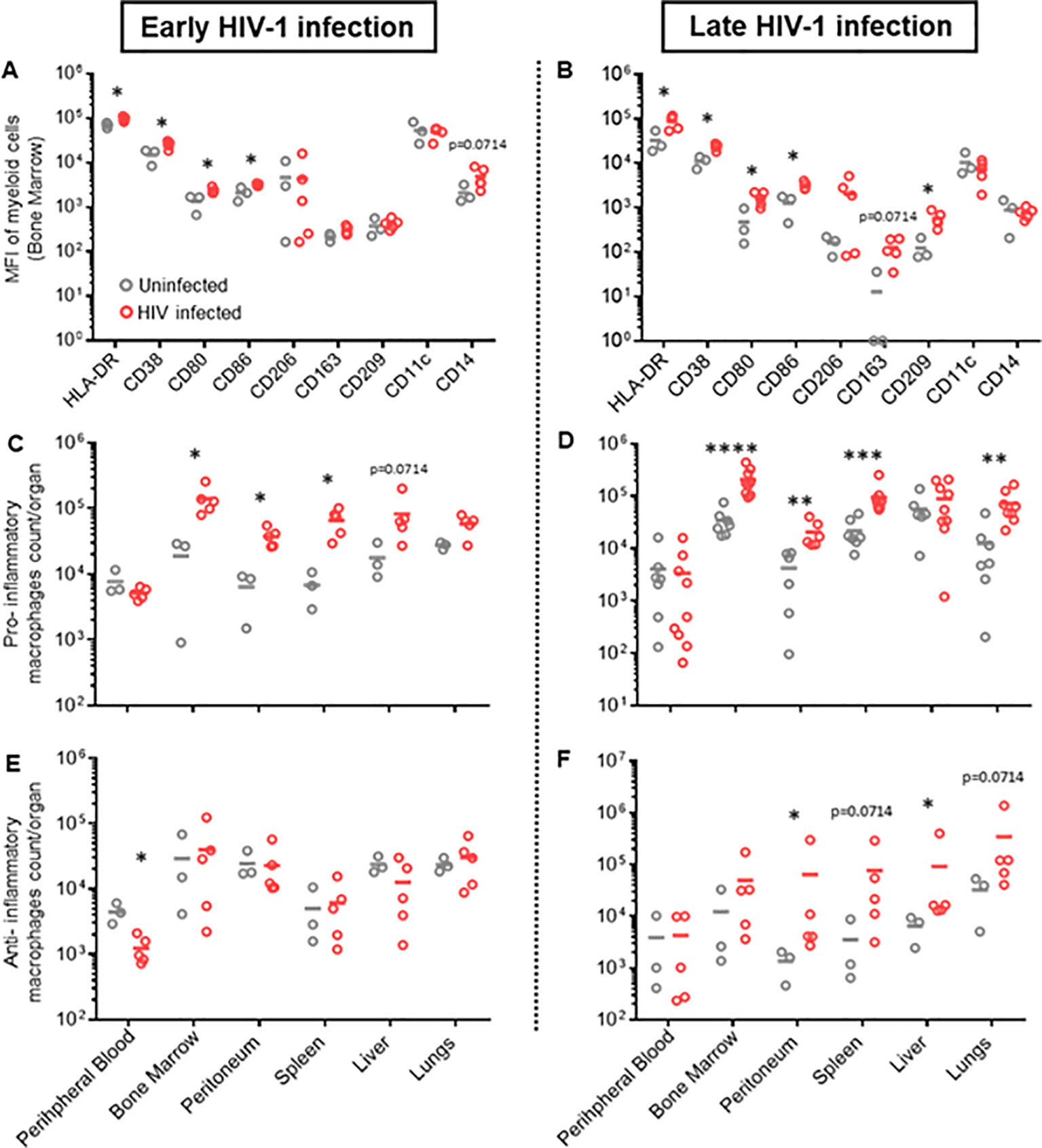
Figure 1. Different phenotypic patterns of human myeloid cells in early and late HIV infection in hu NSG mice. Humanized NSG mice were either infected with HIV-1 YU-2 at TCID50 2*105 (HIV+) or left uninfected (HIV-). (A, C, E). Half of the mice from each group were euthanized either 4 weeks (early HIV-1 infection, left panel) or (B, D, F) 12 weeks after infection (late HIV infection, right panel). Leukocytes were isolated from bone marrow, peritoneum, spleen, liver, lungs and peripheral blood. Cells were characterized via flow cytometry. (A, B) MFI of multiple myeloid surface markers, calculated for myeloid cells of bone marrow for early and late HIV infection (Mean; n HIV-=3; n HIV+=5) (see also Supplementary Figure S3). (C, D) Total count/organ of human CD45+ cells with phenotype CD33+, CD3-, CD19-, CD56-, HLA-DR+, CD38+, CD80+, CD86+, were calculated by using cell population frequencies, obtained by flow cytometry, on total cells processed per sample (Mean) (C) n HIV-=3; n HIV+=5; (D) n HIV-=6; n HIV+=9). (E, F) Total count/organ of human CD45+ cells with phenotype CD33+, CD3-, CD19-, CD56-, CD206+, CD163+, were calculated by using cell population frequencies, obtained by flow cytometry, on total cells processed per sample (Mean; n HIV-=3; n HIV+=5). Statistics by Mann-Whitney (A–F). p-value: *<0.05; **<0.01; ***<0.001; ****<0.0001.
In order to evaluate the role of activated mononuclear phagocytes in HIV infection in huNSG mice, we depleted these cells in HIV-infected mice using clodronate-liposomes (47). One week after treatment, HIV viral load increased up to three logs in the clodronate-treated animals and remained higher than in the control group throughout the experiment (Figure 2A), suggesting that phagocytes are involved in the control of HIV-1 replication over time. Notably, hu mice harbor murine and human macrophages (48) (Supplementary Figure S5A). However, clodronate-mediated depletion is not species-specific (Figures 2C, D) making it impossible to conclude the role of human phagocytes in the system.
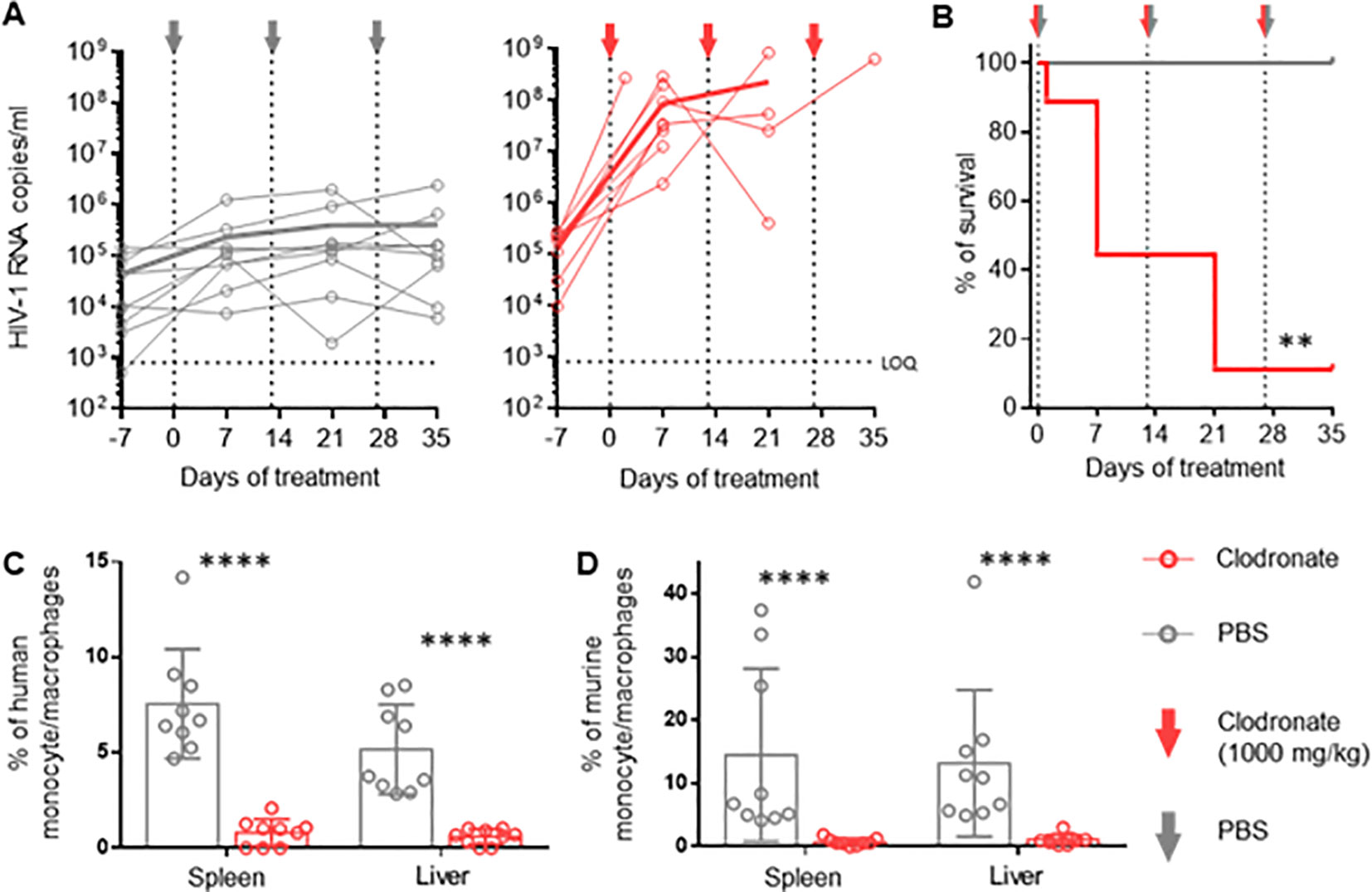
Figure 2. Vigorous increase in HIV replication in hu mice following clodronate mediated depletion of macrophages RNA was extracted from plasma obtained from HIV-1-infected humanized NSG mice treated with clodronate-liposomes (n=9) or PBS (n=9) at day 7, 21 and 35. (A) Each circle represents an individual mouse; HIV-1 RNA copies were measured via in-house RT-qPCR. Thick lines represent the mean of each respective group. (B) Treatment toxicity was established via Kaplan-Meier survival analysis. (C, D) Flow cytometric analysis of leukocytes from spleen and liver of clodronate and mock treated animals. Both human (hCD45+, hCD3-, hCD19-, hCD33+, hCD68+, hCD163) (C) and murine (mCD45+, Ly6G-, Ly6C-, CD11b+, F4/80+) (D) myeloid cells were analyzed (Mean and standard deviation). Mantel-Cox test (B); Mann-Whitney (C, D). p-value: **<0.01; ****<0.0001.
Interestingly, we observed higher CD4+ T cell counts (contrary to CD8+ T cells and NK cells) in the spleen and liver of clodronate-treated animals compared to control animals (Supplementary Figures S6C–E). It is conceivable that the depletion of murine macrophages liberated space for human T-cell expansion, which in turn provided a larger HIV target cell population, at least partially explaining the increase in HIV viral load. Furthermore, clodronate treatment showed high toxicity very early on, resulting in a very low survival rate during the first two weeks (Figure 2B). The extent to which toxicity leads to immune activation and, thus, higher HIV replication remains unanswered as well. The specific role of human macrophages herein remains to be elucidated.
To investigate the role of mononuclear phagocytes in HIV infection in vivo, we created a novel hu mouse model that allows for selective depletion of human myeloid cells. The basic concept was to transplant NSG mice with HSPCs carrying the gene of an apoptosis inducible caspase 9 (iCasp9), which is only active in myeloid cells. Therefore, we generated a lentiviral-based construct that encodes the iCasp9 (49) under the control of a myeloid-specific synthetic promoter, p47-SP107 (50) (Supplementary Figure S7A). iCasp9 apoptosis activity is dependent upon its homodimerization, which is triggered by the otherwise inert molecule AP1903 (49). To identify cells expressing iCasp9, we cloned a GFP downstream of iCasp9, separated by a cleavage peptide. Furthermore, we inserted the truncated neural growth factor receptor (NFGR) under the control of the constitutive promoter pGK to identify and sort the successfully transduced HSCs, which were used for transplanting newborn mice.
We first tested the construct ex vivo in peripheral blood mononuclear cells (PBMCs). Upon transduction, we observed NGFR expression in all cell types, while GFP expression was mainly observed in monocytes (Supplementary Figures S7B, C). This result confirmed the myeloid specificity of the p47-SP107 promoter. Upon treatment with AP1903, >90% of GFP+ cells had undergone apoptosis, demonstrating the high efficiency of iCasp9. We speculate that the higher NGFR expression in monocytes, compared to other cell types, is a result of the high p47-SP107 activity in myeloid cells (50). NGFR expression in B and T cells, as well as their absolute count remained unchanged irrespective of AP1903 (Supplementary Figure S7D). Next, HSPCs were transduced with the lentiviral construct and sorted for the NGFR+ HSPCs (Supplementary Figure S8A). We obtained a population of >95% of genetically modified cells, which were used for the humanization of NSG mice (Figure 3A). Human engraftment level in these mice transplanted was lower when compared to mice transplanted with wild-type HSCPs (Supplementary Figure S8B). However, the human leukocyte distribution was independent of using genetically modified or wild-type HSPCs (Supplementary Figure S8C). As we expected, we observed high GFP expression in human myeloid cells (71.7% ± 13.49%), very low expression in NK cells (2.90% ± 2.53%), and negligible expression in T cells, B cells and murine cells obtained from peripheral blood samples (Figure 3B). These results confirmed the specificity of our lentiviral construct. The GFP expression corresponded to the average 10% of myeloid cells typically present in the total leukocyte population (51).
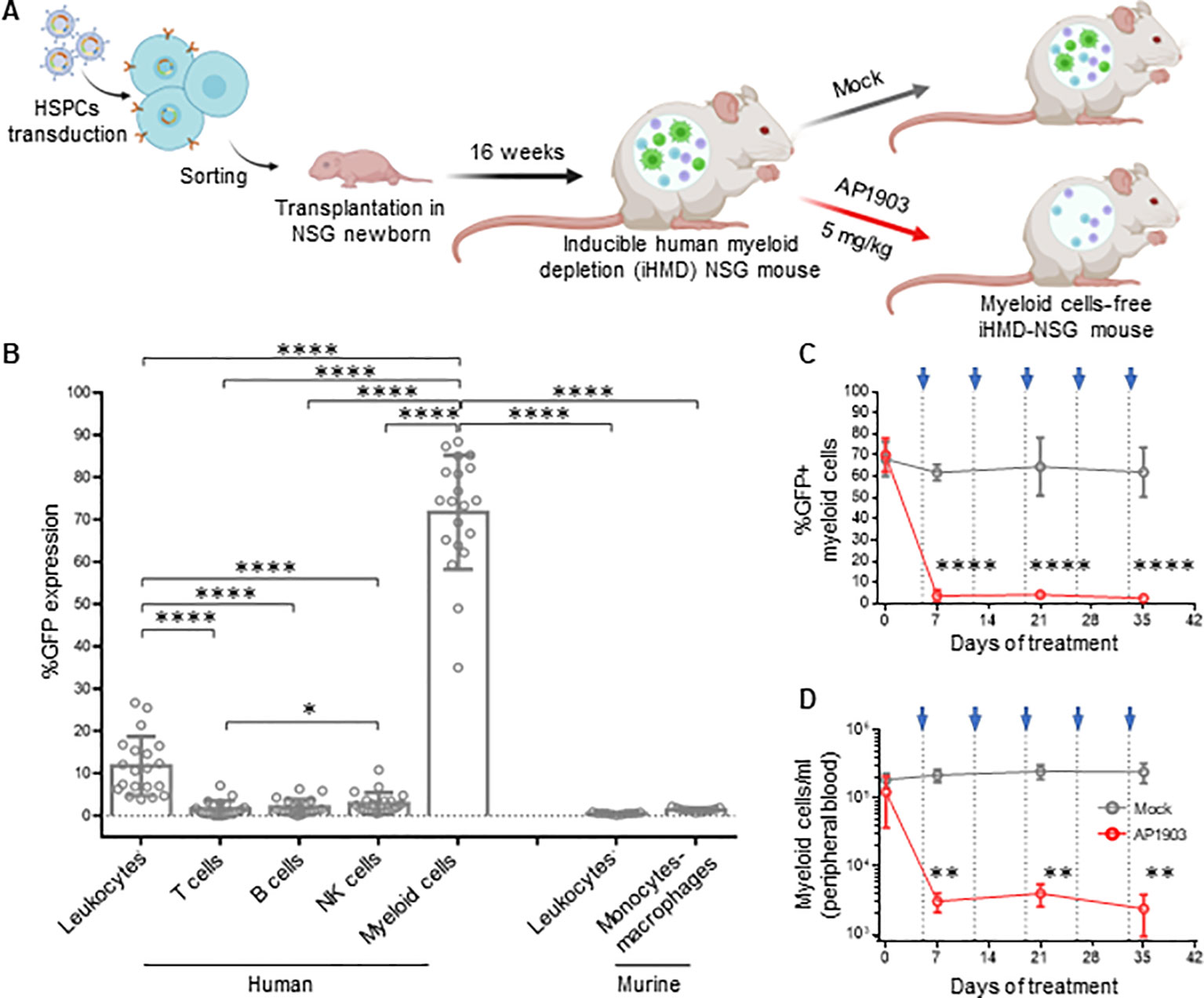
Figure 3. Efficient depletion of myeloid cells in iHMD-NSG mice. (A) Schematics for the generation and functionality testing of iHMD-NSG mice. BioRender.com/g70c130 Engraftment in iHMD NSG mice was evaluated 16 weeks post transplantation. Engrafted mice were treated once a week for 5 weeks. (B) GFP expression in human leukocytes from 3 independent cohorts (n=20) and murine leukocytes from 2 (n=11) (Mean and standard deviation). (C) Flow cytometry analysis of GFP expression in human myeloid cells from peripheral blood of iHMD-NSG mice. AP1903 5mg/kg (red, n=4) and mock (grey, n=3) (Mean and standard deviation). (D) Myeloid cell count overtime for both treatments, was calculated by using cell population frequencies, obtained by flow cytometry, on total cells processed per peripheral blood sample (Mean and standard deviation). Statistics by Mann-Whitney (B). Two-way ANOVA (C, D). p-value: *<0.05; **<0.01; ***<0.001; ****<0.0001.
A single injection of AP1903 triggered apoptosis in >90% of the GFP+ cells within 24h in bone marrow, spleen, liver and peripheral blood (Supplementary Figure S9A). In addition, weekly injections of AP1903 were well tolerated (Supplementary Figure S9B) and resulted in consistently low GFP expression and absolute count of myeloid cells (Figures 3C, D). GFP expression and cell counts in the different organs were comparable to peripheral white blood cells (Supplementary Figures S9C, D). In addition, the longitudinal analysis of human leukocytes showed that AP1903 had no impact on the human graft (Supplementary Figure S9E). Therefore, with this inducible human myeloid depletion (iHMD) model, we were able to selectively deplete human myeloid cells without targeting other human cell types.
We generated iHMD-NSG or huNSG mice with HSPCs from three different donors to consider donor-specific differences. Both iHMD-NSG and huNSG mice were infected with HIV for eight weeks to achieve stable viremia over time. iHMD-NSG mice were then treated with weekly injections of either AP1903 (5mg/kg) or PBS, and huNSG mice with AP1903 (Figure 4A). In fact, we observed a significant increase in HIV RNA copy numbers in iHMD-NSG mice after the first dose of AP1903, which persisted throughout the treatment period (Figure 4C, Supplementary Figure S10A). We confirmed that AP1903 in iHMD-NSG mice resulted in vigorous depletion of myeloid cells in peripheral blood, bone marrow, liver and spleen (Figures 4B, D, Supplementary Figure S10D). No effect was observed in either PBS-treated iHMD-NSG mice or AP1903-treated huNSG mice. Furthermore, we observed a correlation between the initial %GFP expression in myeloid cells and the increase in HIV viral load following depletion of these cells (Figure 4E). This observation indicated that anti-HIV activity was determined by the level of myeloid cells present. Based on these data, we concluded that myeloid cells are a significant determinant in controlling HIV replication in HIV-infected humanized mice.
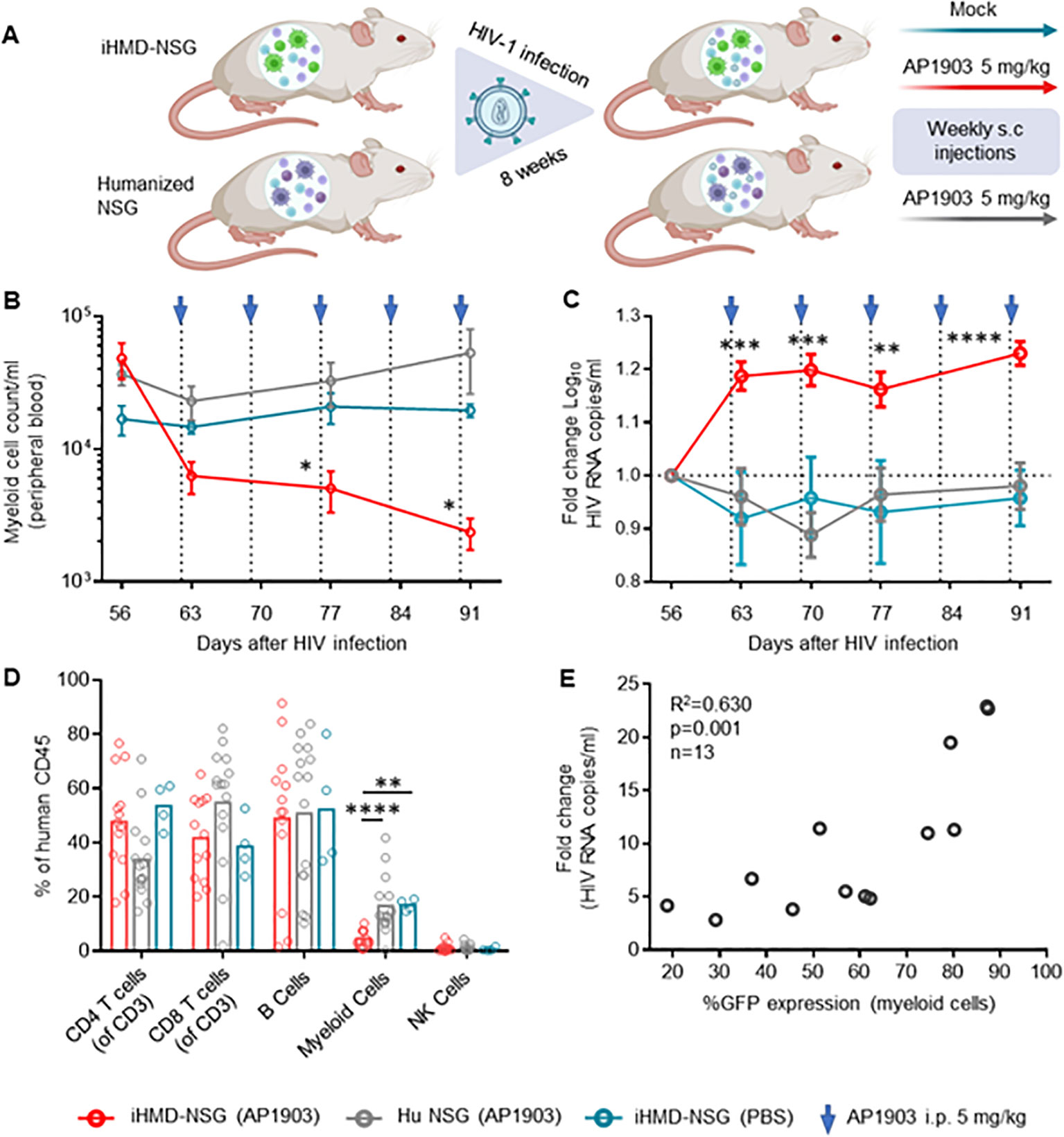
Figure 4. Depletion of myeloid cells results in a vigorous increase of HIV viral load. (A) Schematics of experimental procedure, BioRender.com/m34c552. Engrafted mice were infected with HIV YU-2 for 8 weeks. HIV infected mice were treated once a week for 5 weeks (B) Myeloid cell count overtime for the three treatment regimens overtime, were calculated by using cell population frequencies, obtained by flow cytometry, on total cells processed per sample (Mean and standard error of the mean). (C) Fold change in viral load compared to pre-treatment baseline VL at day 56, via RT-qPCR. Results from 3 independent studies are shown: Hu NSG mice treated with AP1903 (5mg/kg) (n=15), iHMD-NSG treated with either AP1903 (5mg/kg) (n=13) or PBS (n=4) (Mean and standard error of the mean). (D) Leukocytes frequencies distribution in the three treatment regimens, measured vial flow cytometry at euthanasia (Mean). (E) Linear regression analysis between average fold change in HIV RNA copies/ml of peripheral blood and GFP expression in myeloid cells prior AP1903 treatment in iHMD-NSG mice. Statistic by two-way ANOVA (B, C), Mann-Whitney (D). p-value: *<0.05; **<0.01; ***<0.001; ****<0.0001. Linear regression analysis (E).
Longitudinal analysis showed no expansion of non-myeloid populations upon myeloid cell depletion (Figure 5A, Supplementary Figure S10B). However, we did not quantify the number of effector and central memory CD4+ T cells, which are the main target cells of productive HIV infection, and therefore we may have missed their expansion by only counting the total amount of CD4+ T cells. In any case, we detected higher intracellular HIV p24 antigen expression in CD4+ T cells from bone marrow of the AP1903-treated iHMD-NSG compared to the control groups (Figures 5B, C). Furthermore, we performed a cytokine analysis using plasma of iHMD-NSG mice obtained before and after myeloid cell depletion (Figure 5D). We observed downregulation of 19 cytokines after treatment with AP1903. Among these, chemokine receptor ligands and pro-inflammatory cytokines including CXCL12, CCL5, CCL4, IL-16, IL1RA, IL-27, IL-2, IL-10 have known anti-HIV effects (52–54). Taken together, these results suggest macrophages restrict HIV replication via secretion of soluble factors limiting viral production in CD4+ T cells.
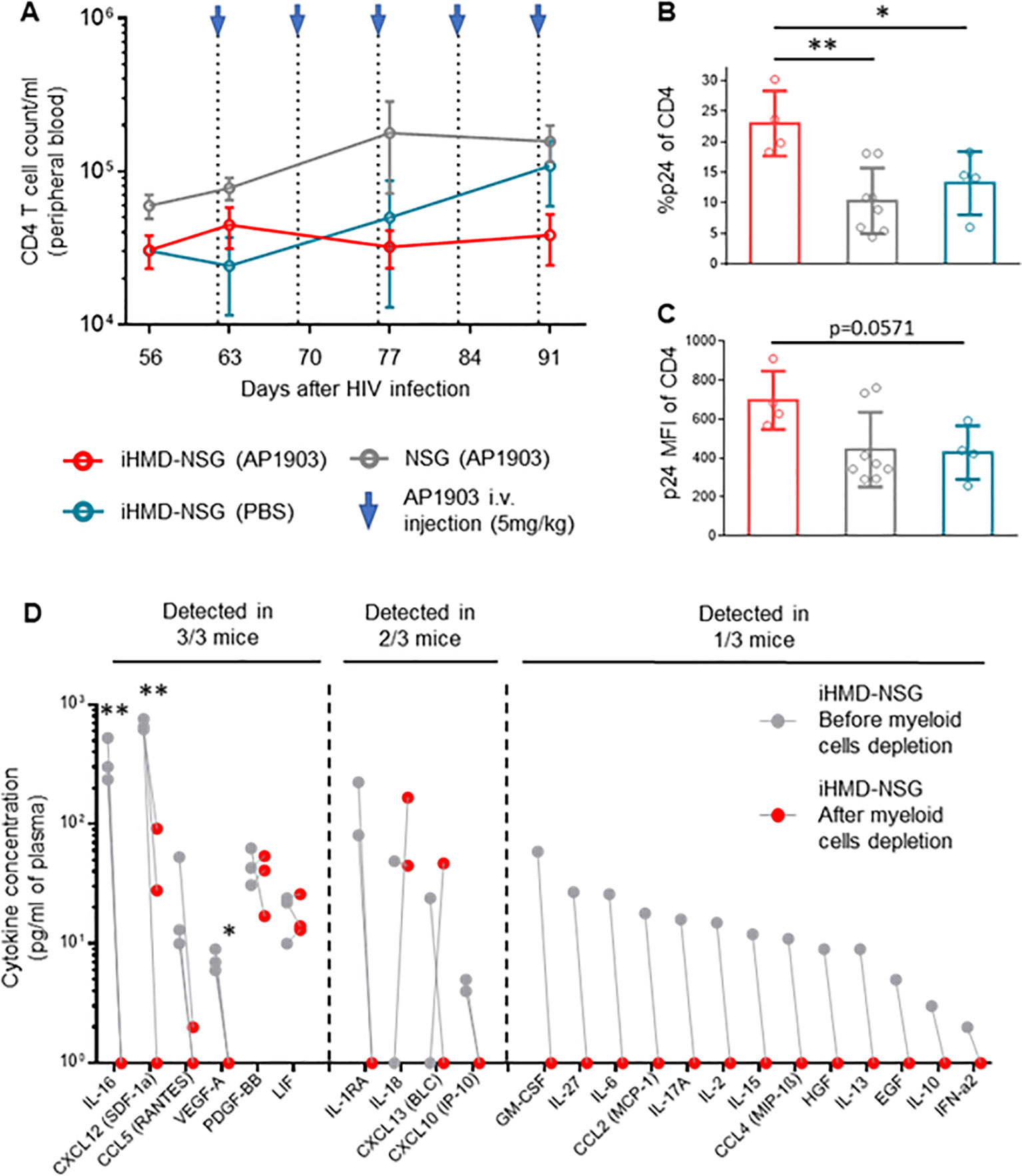
Figure 5. Depletion of myeloid cells results in increase of p24 expression in CD4+ T cells and downregulation of cytokines/chemokines. Hu NSG mice were treated with AP1903 (5mg/kg) (n=15), iHMD-NSG were treated with either AP1903 (5mg/kg) (n=13) or PBS (n=4) weekly, for 5 weeks. All mice were infected with YU-2. (A) CD4+ T cells count over time in the three different groups, calculated by using cell population frequencies, obtained by flow cytometry, on total peripheral blood cells processed per sample (Mean and standard error of the mean). (B, C) p24 intracellular expression in CD4+ T cells, evaluated via flow cytometry in bone marrow of iHMD NSG AP1903 treated mice (n=4), iHMD NSG mock treated mice (n=4), NSG AP1903 treated mice (n=8) (Mean and standard deviation). (D) Luminex multiplex assay for cytokine analysis, performed in plasma samples of 3 AP1903-treated iHMD NSG mice, before and after treatment. Statistic by Two-way ANOVA (A), Mann-Whitney test (B, C), two-tailed Student’s t-test (D) p-value: *<0.05; **<0.01; ***<0.001; ****<0.0001.
To validate our in vivo observations, we explored whether human macrophages exert control of HIV replication on ex vivo infected PBMCs. Therefore, we co-cultured ex vivo HIV-infected PBMCs with autologous M1 (pro-inflammatory), M2 (anti-inflammatory) polarized monocyte-derived macrophages (MDMs) or M0 (resting) MDMs, either directly (cell-to-cell) or separated by a trans-well (Figure 6A). The pore size of 1 µm of the trans-wells blocks cell passage while allowing free exchange of molecules (55).We infected PBMCs with the CXCR4-tropic HIV strain NL4-3, which does not productively infect macrophages. The use of this virus allowed us to examine the effects of macrophages on HIV replication in PBMCs uncoupled from potential productive HIV infection of macrophages – macrophages serving here rather as bystander cells. Polarization resulted in upregulation of CD38, CD86 and CD80 markers for pro-inflammatory MDMs, and CD206, CD209 and CD163 for anti-inflammatory MDMs, when compared to resting MDMs (Figure 6B, Supplementary Figure S11A). We found that MDMs, most notably the pro-inflammatory MDMs, inhibited HIV replication over time (Figure 6C, Supplementary Figure S12). Intriguingly, the anti-HIV effect persisted when the MDMs were physically separated by the trans-wells from the HIV-infected PBMCs (Figure 6D, Supplementary Figures S11B, C), which suggests a molecule-dependent rather than contact-dependent effect. Cytokine analysis resulted in a comparable profile to what we observed in iHMD-NSG mice (Supplementary Figure S13), with increased levels of anti-HIV soluble factors in the presence of pro-inflammatory MDMs. Furthermore, to roll out potential anti-HIV effects of other effector cells present in the co-culture, we depleted either NK cells or CD8+ T cells in both HIV-infected monocultures and co-cultures with pro-inflammatory MDMs (Supplementary Figure S14A). In all instances, no significant difference was observed when effector cells were depleted, strengthening MDMs’ anti-HIV effect. Overall, these ex vivo data are in line with the in vivo results, suggesting that pro-inflammatory macrophages have an HIV-inhibitory effect.
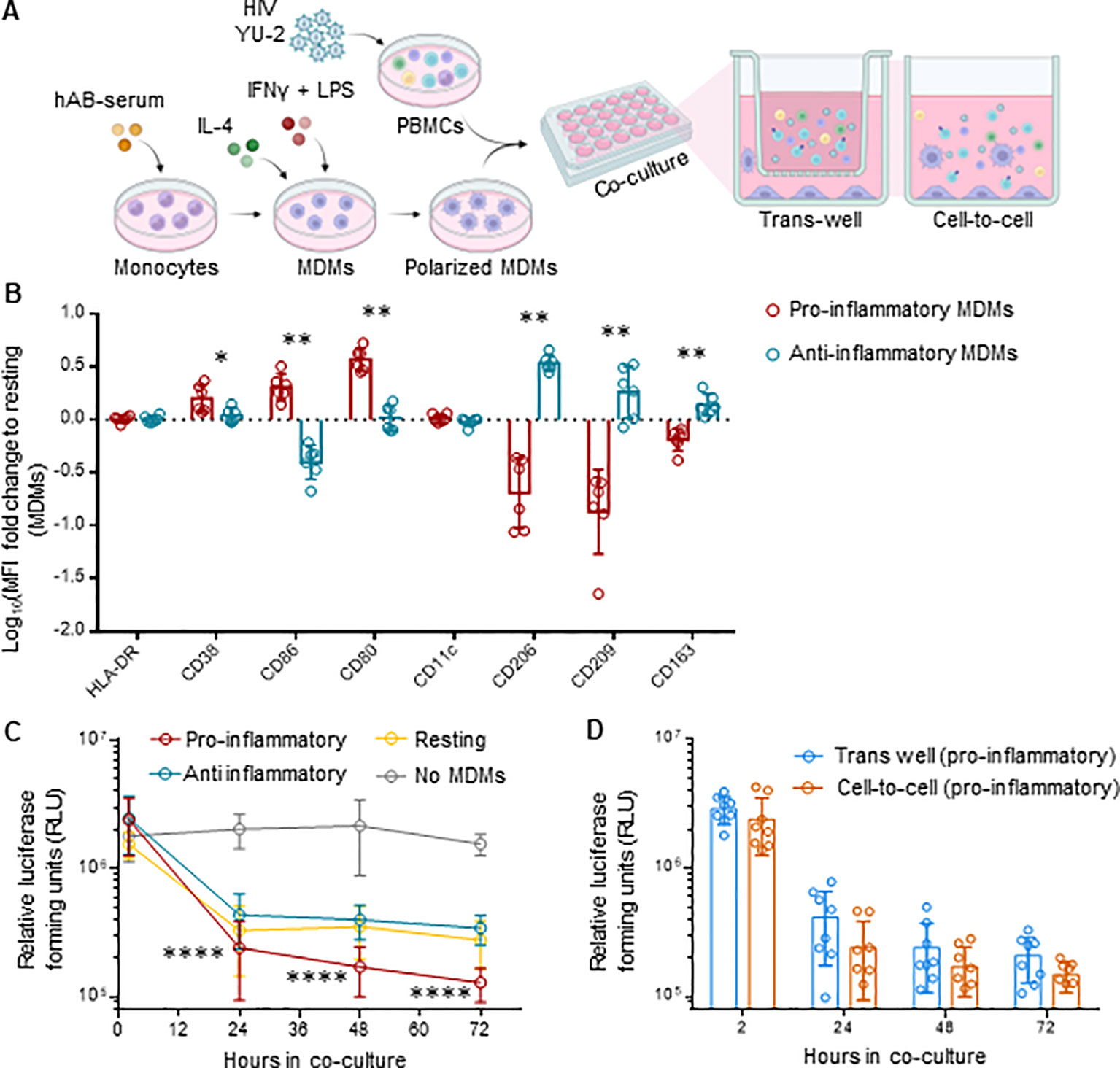
Figure 6. MDMs inhibit HIV replication in ex vivo co-cultures with HIV-infected PBMCs. (A) Schematics of experimental procedure, BioRender.com/u59h395 CD14+ monocytes were differentiated in MDMs for 6 days and polarized in either pro- (M1), anti-inflammatory (M2) or kept as resting (m0) MDMs. Differently polarized MDMs were co-cultured at a 1:1 ratio with autologous activated PBMCs infected with HIV NL4.3 2½ days ago. Co-cultures were either in direct contact (cell-to-cell) or separated by a trans-well (1 μm pore size). HIV infected PMBCs were also kept in monoculture as control. Culture supernatant was harvested at 2, 24, 48, 72 hours after co-culture setup for evaluation of HIV replication for each culture condition. (B) Flow cytometric analysis of polarized MDMs at the end of the experiment, i.e., 72 hours of co-cultivation, including pro-inflammatory (HLA-DR, CD38, CD86, CD80, CD11c) (n=6) and anti-inflammatory (HLA-DR, CD206, CD209, CD163) (n=6) myeloid surface markers. Displayed as logarithmic fold change to MFI of resting MDMs (Mean and standard deviation). (C) Viral replication for every cell-to-cell co-culture condition (n=8 per condition measured via TZM-bl luciferase assay at 2, 24, 48 and 72 hours post co-culture setup (Mean and standard deviation). (D) Viral replication of trans-well (n=8) and cell-to-cell (n=8) co-cultures for pro-inflammatory MDMs-HIV infected PMBCs, measured via TZM-bl luciferase assay at 2, 24, 48 and 72 hours post co-culture setup (Mean and standard deviation). Statistic by and Mann-Whitney (B); two-way ANOVA (C, D). p-value: *<0.05; **<0.01; ****<0.0001.
To better understand the mechanisms behind MDM’s control of HIV replication, we performed bulk RNA sequencing on both MDMs and CD4+ T-cells from mono- and co-cultures maintained for 72 hours, ± HIV infection. Transcriptome analysis of MDMs from HIV-infected co-cultures showed a significant fold increase of 48 genes compared to uninfected control, mainly ISGs (Figure 7A, Supplementary Table S2). Over-representation analysis (ORA) of these genes identified upregulated biological processes involved in defense response to the virus, immune system process and innate immune response (Figures 7A, B, Supplementary Table S4). We observed upregulation of HIV co-receptor ligands independently of HIV infection, i.e., significant increases in CCL3, CCL4, CCL5 and CCL8 (Figure 7C, Supplementary Table S2). Transcriptome analysis of sorted CD4+ T cells from either HIV-infected co-cultures or monocultures showed a consistent clustering of differently expressed features (Supplementary Figures S15A, B). The addition of autologous MDMs to HIV-infected PBMCs induced upregulation of transcripts involved in immune defense mechanisms in CD4+ T cells, including inflammatory response, antigen processing and T cell activation (Figures 8A, B, D). At the same time, we observed marked downregulation of DNA replication, DNA repair, cell division and mitosis (Figures 8A, C, D). Additionally, gene set enrichment analysis (GSEA) showed downregulation of mRNA splicing and rRNA production (Supplementary Figure S15D, Supplementary Table S7). Interestingly, among the upregulated genes in CD4+ T cells, we identified known HIV restriction factors, including SERINC2, SAMHD1, MARCHF1 and MARCHF2 (Figure 8E, Supplementary Table S3) (56). However, upregulation of these restriction factors appears to be dependent on the presence of MDMs but independent of HIV. Taken together, these results indicate that, during HIV infection, the presence of MDMs upregulates antiviral response while downregulating DNA replication in CD4+ T cells, leading to restriction of viral replication.
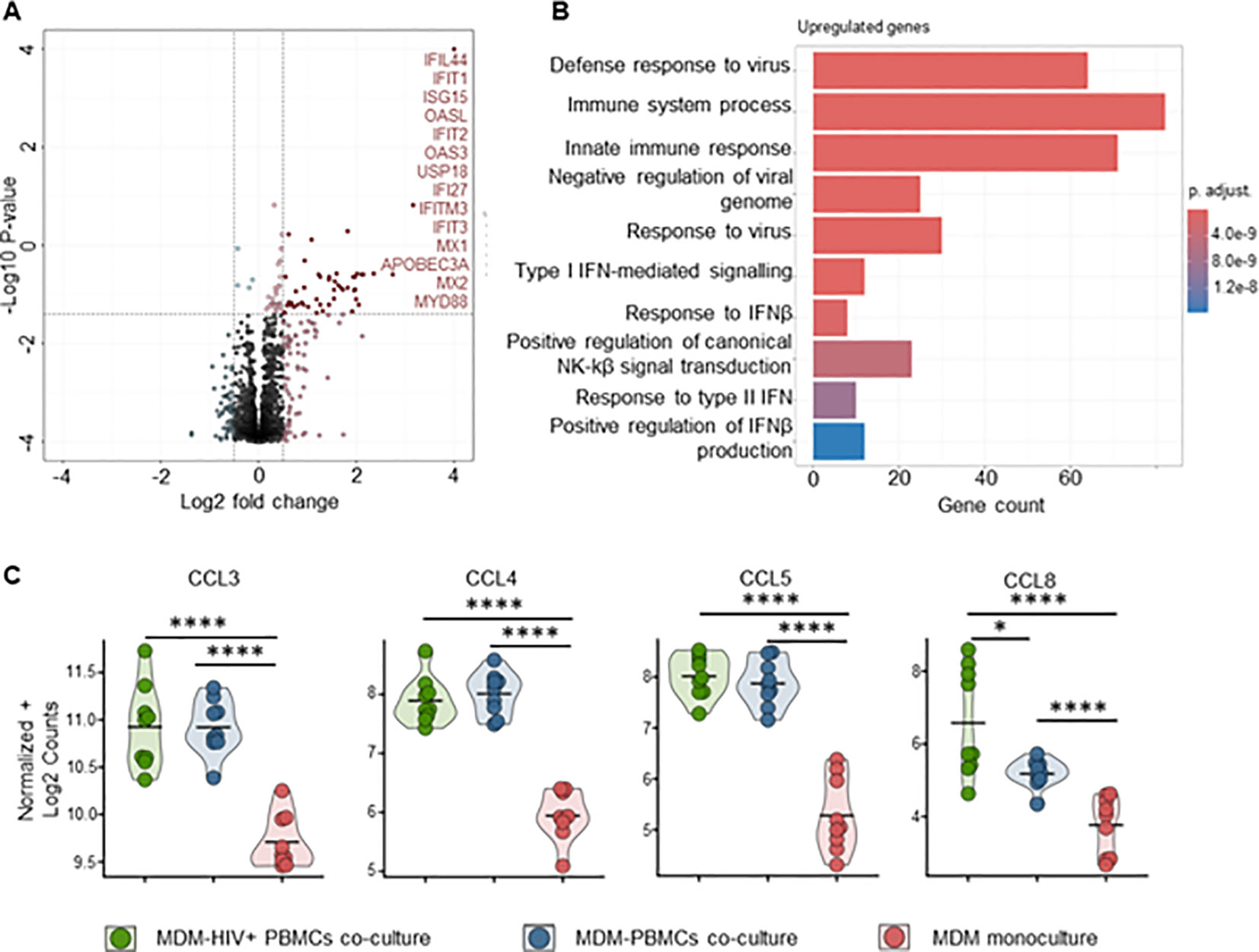
Figure 7. Pro-inflammatory (M1-polarized) MDMs bulk RNA sequencing shows increased antiviral response in the presence of HIV infected PBMCs (A) Volcano plot of differentially expressed genes in MDMs-PBMCs co-culture vs HIV+ MDMs-PBMCs co-culture (72h timepoint). Significant upregulated threshold 0.5 ≤ Log2 fold change ≥ 0.5; P-value ≤ 0.01. Red dots indicate significantly upregulated genes, annotated on the side. (B) Hypergeometric ORA test of upregulated biological processes of MDMs-PBMCs co-culture vs MDMs-HIV+ PBMCs co-culture comparison. (C) Violin plots of expression levels of HIV co-receptors ligands in MDMs in the 3 different culture conditions analyzed (Mean, n=9 per condition). Statistic by DESeq2 (see materials & methods section). FDR-adjusted p-value: *<0.05; ****<0.0001.
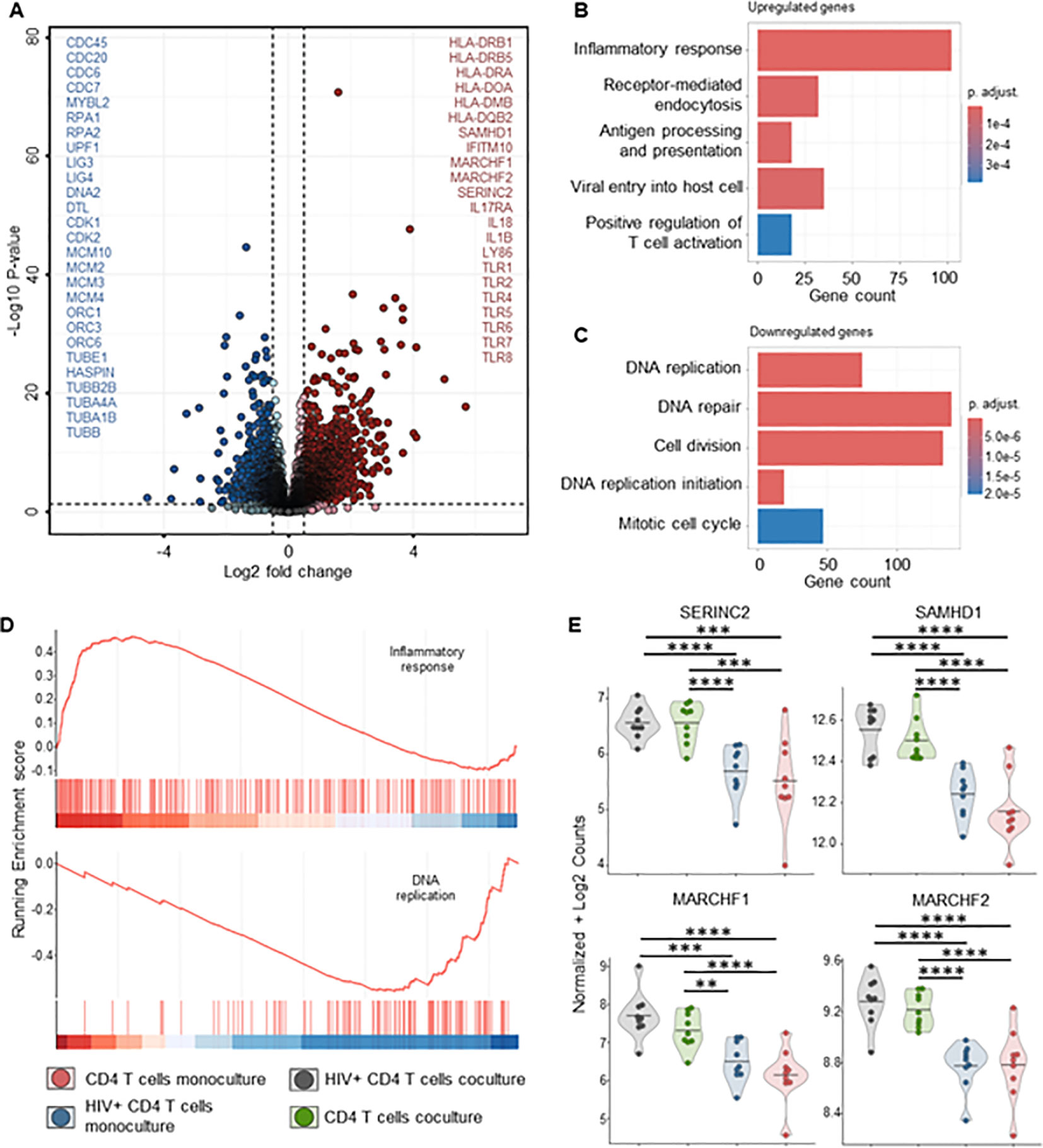
Figure 8. CD4+ T cells bulk RNA sequencing of HIV infected co-cultures of ex vivo HIV-infected PBMCs with macrophages shows increased T cell activation and decreased DNA replication in the presence of MDMs. (A) Volcano plot of differentially expressed genes in sorted CD4+ T cells of HIV infected PBMCs-MDMs co-cultures vs HIV infected PBMCs monoculture (72h time point). Significant upregulated threshold 0.5 ≤ Log2 fold change ≥ 0.5; P-value ≤ 0.01. Red dots indicate significantly upregulated genes in HIV infected PBMCs-MDMs co-cultures. Blue dots indicate significantly downregulated genes in HIV infected PBMCs-MDMs co-cultures. (B) Hypergeometric ORA test of upregulated biological processes of HIV infected PBMCs-MDMs co-cultures vs HIV infected PBMCs monoculture co-culture comparison. (C) Hypergeometric ORA test of downregulated biological processes of HIV infected PBMCs-MDMs co-cultures vs HIV infected PBMCs monoculture co-culture comparison. (D) GSEA showing significant enrichment of inflammatory response and downregulation of DNA replication in CD4+ T cells of HIV infected PBMCs-MDMs co-cultures vs HIV infected PBMCs monoculture. (E) Violin plots of expression levels of HIV restriction factors in MDMs in the 4 different culture conditions analyzed (Mean, n= 9 per condition). Statistic by DESeq2 (see materials & methods section). FDR-adjusted p-value: *<0.05; **<0.01; ***<0.001; ****<0.0001.
Studies of macrophages in HIV infection so far have primarily focused on the contribution of these cells to the viral reservoir. In this study, we showed that macrophages have an anti-HIV effect both in vivo and ex vivo.
We first observed clear phenotypical differences between myeloid cells in HIV-infected and uninfected humanized mice. Upregulation of pro- and anti-inflammatory markers at different stages of infection clearly demonstrated macrophages’ ability to respond to HIV infection and is congruent with phenotypes observed in HIV-infected patients (23). In fact, we found an increased number of macrophages expressing pro-inflammatory markers during early HIV infection in multiple organs. This effect was further increased at the late infection time-point, indicating a constant activation of pro-inflammatory pathways, most likely due to the system’s inability to clear the infection. Different studies on cytokine profiles during HIV infection showed high levels of pro-inflammatory cytokines during acute HIV infection (57, 58). In contrast, an increase in anti-inflammatory cytokine production was observed during chronic infection stages (59, 60), which aligns with our findings of a significant increase of macrophages expressing anti-inflammatory markers at a late infection time point. Our results on myeloid cell phenotyping confirmed plasticity and functionality of macrophages in humanized NSG mice.
By depleting these mononuclear phagocytes, we aimed to understand the specific role of these cells during HIV infection. While knowing that clodronate kills mononuclear cells across species (48), we were intrigued to see a high increase in HIV RNA copy numbers in huNSG treated with clodronate. To note, murine mononuclear phagocytes in humanized mice, despite their defective nature (61), still maintain some functionality (62). In fact, murine macrophages may still phagocytose HIV-infected cells or engulf free virions (63). Furthermore, murine macrophage depletion in immunocompetent mice results in splenomegaly with extensive hematopoiesis (64), which we observed in clodronate-treated mice. Specifically, clodronate-treated mice displayed a significantly higher count of CD4+ T cells in both spleen and liver. Finally, clodronate-associated toxicity may result in T-cell activation, which in turn promotes HIV- replication. Thus, it is rather impossible to draw any firm conclusions on the role of human macrophages from these data.
To explore the role of human mononuclear phagocytes in HIV infection, we needed a hu mouse model that would allow for their specific depletion without any off-target effect. The iHMD model, based on the transplantation of genetically modified HSPCs harboring a myeloid-specific iCasp9, achieved such results. The lower human engraftment level in iHMD-NSG mice compared to plain huNSG mice is most likely a result of the ex vivo manipulation of HSPCs (65, 66). It is important to note that the iHMD-NSG and huNSG mice were comparable in leukocyte distribution. GFP expression, driven by the myeloid-specific promoter, was highest in human myeloid cells, low in NK cells and negligible in T and B cells. Notably, a small fraction of NK cells originate from myeloid progenitor cells, explaining their low GFP expression (67). The less than 100% GFP expression in myeloid cells is likely a result of stochastic epigenetic silencing of the transgene during hematopoiesis (68). In fact, genes introduced by lentiviral transduction randomly integrate into active transcription units of the host cell genome (69) and depending on the integration site, epigenetic silencing may occur. Following AP1903 treatment, we observed a depletion of >95% of GFP+ myeloid cells, similar to what was reported by Stasi et al. (49). The total number of myeloid cells decreased by >90%, which suggests either an extremely high sensitivity of the iCasp9 system or an underestimation of the iCasp9 transgene detected. The latter can be explained by the polycistronic structure of the vector, where a 2A self-cleaving peptide separates the iCasp9 gene and GFP. Liu et al. showed that protein expression decreases the further away from the promoter (70). We also achieved the elimination of GFP+ myeloid cells over time by weekly treatment with AP1903. Thus, the iHMD mouse model permits selective human myeloid cell depletion without off-target effects, while maintaining human engraftment.
AP1903-treatment in HIV-infected iHMD-NSG mice resulted in a rapid and significant increase in HIV load, which persisted over time. Notably, human macrophages in the spleen and bone marrow were HIV infected in huNSG mice (Supplementary Figure S16). However, the half-life of free HIV virions in the peripheral blood is estimated to be about 48h (71, 72). Therefore, the constant and stable increase in viral load cannot be explained by the release of free virions from productively HIV-infected apoptotic macrophages. Although we did not observe CD4+ T cell expansion after macrophage depletion as a cause of the increase in HIV replication, we cannot completely rule it out due to a possible insufficient sample size. Likewise, we cannot rule out that we missed a subtle expansion of effector and/or central memory CD4+ T cells, the main HIV producer cells, since we only counted the number of total CD4+ T cells. In any case, we found a higher frequency of HIV-infected CD4+ T-cells as well as higher intracellular HIV p24 expression, which could explain the increased HIV replication. Interestingly, we observed decreases in several cytokines and chemokines, which were mostly undetectable in iHMD-NSG mice after the depletion of human myeloid cells. Chemokines such as CCL5, CCL4 and CXCL12 act as HIV entry inhibitors by binding to HIV co-receptors (52, 53). Pro-inflammatory cytokines, IL-16, IL1RA, IL-27, IL-2, IL-10 and IFNα, inhibit HIV replication via different mechanisms (54). On the other hand, cytokines, such as IL-6, IL-15 and GM-CSF, were shown to favor HIV replication by inducing expansion of HIV-infected cells (54). Ultimately, the net effect of all the chemokine/cytokine changes will likely shape the anti-HIV activity of macrophages. Our data in iHMD-NSG mice suggest that chemokines/cytokines mediate, at least in part, the HIV inhibitory effects of macrophages. To note, this is the first study ever to demonstrate an anti-HIV-specific effect of human macrophages in vivo through their selective degradation.
To better characterize these mechanisms, we further investigated human macrophages’ anti-HIV activity by co-culturing MDMs with ex vivo HIV-infected PBMCs. Here, we used the CXCR4-tropic strain NL4-3 which does not result in productive infection of macrophages (73). This allowed us to investigate whether macrophages exert an HIV inhibiting effect in PBMCs without being infected themselves. In fact, the addition of MDMs, particularly pro-inflammatory MDMs, led to a significant reduction in HIV replication even in NL4-3 infected co-cultures. Furthermore, finding no differences in HIV replication, regardless of whether the co-cultures were in direct contact or separated by trans-wells argues that soluble factors are at the basis of macrophage’s anti-HIV activity. Cytokine analysis revealed expression patterns comparable to our in vivo analysis, with higher anti-HIV soluble factor concentrations in the presence of pro-inflammatory MDMs. Furthermore, this HIV inhibiting effect requires no productive HIV infection of macrophages. Similarly, other research groups have observed a macrophage-directed anti-viral response via the upregulation of cytokines and chemokines in other virus infections, such as HBV and HCV, Zika virus and Dengue (74–77).
Transcriptomic analysis on MDMs strengthen our observations at a protein level that macrophages are producers of anti-HIV cytokines and chemokines. In fact, MDMs co-cultured with PBMCs, undergo a strong immune activation, together with upregulation of chemokines and cytokines (Supplementary Figure S17A). In particular, we identified the chemokine receptor ligands CCL3, CCL4, CCL5 and CCL8. All of which bind to HIV co-receptor CCR5 and suppress HIV replication in vitro (53, 78–81). In the presence of HIV-infected cells, MDMs showed a transcription profile dominated by anti-viral ISGs (82–85), many of which are known as HIV restriction factors. This signature hints to IFN-α as main trigger of macrophage activation. Macrophages changed the transcriptomic profile of CD4+ T from HIV-infected co-cultures as well. In addition to immune activation, we observed a marked downregulation of genes involved in biological processes ranging from DNA replication to protein production and cell division. Consistent with our data, macrophages were recently described to reduce HIV expression in memory CD4+ T cells in vitro by modulating cell activation (86). The downregulation of DNA transcription and RNA translation in HIV-infected CD4+ T cells can explain the decrease in HIV production from HIV-infected cells. Notably, proliferating CD4+ T cells are the most susceptible one to HIV replication (87). Downregulation of cell division and replication is likely to result in a decrease of new HIV infection of T cells, further promoting the decrease in HIV production.
In this study, we investigated the role of macrophages as effector cells both in vivo, using a CCR5-tropic strain, and ex vivo, using a CXCR4-tropic strain. By employing HIV strains with different co-receptor selectivity, we introduced an additional variable, yet both the in vivo and ex vivo experiments produced consistent results. We believe that the observed inhibitory effects of macrophages on HIV are not related to HIV co-receptor selectivity, but this hypothesis needs to be formally tested in future studies. Notably, YU-2 is a primary HIV strain that was directly isolated from the brain and is well characterized for its ability to infect macrophages (88). Global mapping of the HIV-1 transcriptome reveals that both productively infected macrophages and bystander cells exhibit similar activation of the type-1 IFN signaling pathway, suggesting that these two cell populations were exposed to the same triggering signals (89). In contrast, the induction of susceptibility genes related to DNA reorganization required for viral integration and transcription appeared to be specific to productively infected macrophages. Data exploring the impact of CXCR4-tropic strains on macrophages are currently lacking. It is possible that CXCR4-tropic strains trigger the same sensing receptors in the endosomal compartment as CCR5-tropic strains, which is decoupled from productive HIV infection. A particularly intriguing study showed that IFN activation in plasmacytoid dendritic cells depends on the HIV Env–CD4 interaction and the targeting of HIV to early endosomes (90). That might be also the case for macrophages. However, with the current data, we cannot make any definitive statements regarding the detailed mechanisms underlying the anti-HIV effects we observed or to what extent CCR5-tropic HIV infections differ from CXCR4-tropic HIV infections in connection with the inhibitory effect of macrophages. Single-cell RNA sequencing of macrophages isolated from humanized mice infected with HIV strains having distinct co-receptor selectivity is necessary to address these questions. It should be noted that, in addition to the HIV envelope, other HIV genes may influence HIV replication in the various cellular compartments.
Other intriguing questions remain such as the role of mononuclear cells in cART treated subjects that can be addressed using this iHMD-NSG model, how long does it take to reconstitute the myeloid compartment when AP1903 is stopped, and what happens to the HIV replication in that situation and what happens if AP1903 is administered during acute infection vs chronic infection? When interpreting the results of this work, we must be aware of the limitations of the human immune system in humanized NSG mice (91, 92). We would like to emphasize that humanized mice are the best in vivo small animal model to study HIV pathogenesis. It is certainly a very accurate model to study the response of the innate immune system to the HIV challenge. The major shortcoming of humanized mice is the absence of an efficient immune system that of course may have a reciprocal impact on the innate immune response (93). We could imagine that the potential of myeloid cells could be even bigger on the HIV replication in the case of an adaptive immune system. Furthermore, great efforts have been made to improve the human immune system by providing human transgenes (94). It would be interesting to see to what extent a superior myeloid compartment as present for example in humanized MISTRG mice (95) would have resulted in a different outcome. We believe this will not be the case, but that remains speculation.
In summary, our study demonstrated a novel role of macrophages in restricting HIV infection during natural HIV infection. We showed that macrophages produce anti-HIV ISGs and chemokine-ligands, while inducing downregulation of DNA replication in CD4+ T cells which may be the origin of their HIV inhibitory activity. A more in-depth characterization of the anti-HIV effects of these cells could open up new possible immunotherapeutic approaches to HIV infection. While the current focus is primarily on dendritic cell immunotherapy, we believe in the overall HIV-inhibitory potential of myeloid cells coupled with their potential to generate an HIV-specific immune response that could ultimately lead to control of HIV without cART (96).
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: GSE269285 (GEO).
Ethical approval was not required for the studies involving humans because buffy coats were anonymously provided by the Blood Donation Service Zurich, Swiss Red Cross, Schlieren. The studies were conducted in accordance with the local legislation and institutional requirements. The parents provided written informed consent for the use of cord blood that was used in a completely anonymized fashion. The animal study was approved by Cantonal veterinary Office License ZH181/17 Amendment 28395 of Licenses ZH243/16; ZH081/21. The study was conducted in accordance with the local legislation and institutional requirements.
LB: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Validation, Visualization, Writing – original draft, Writing – review & editing. CM: Formal analysis, Investigation, Methodology, Writing – review & editing. NK: Investigation, Writing – review & editing. RW: Investigation, Writing – review & editing. DR: Software, Writing – review & editing. SB: Writing – review & editing. TM: Conceptualization, Supervision, Writing – review & editing. RS: Conceptualization, Funding acquisition, Methodology, Project administration, Resources, Supervision, Visualization, Writing – original draft, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. RS is supported by the Swiss National Science Foundation (SNF #310030-176147), the Swiss Vaccine Research Institute and the Hartmann Müller Stiftung.
We thank Prof. Malcom Brenner for kindly providing the original iCasp9 construct. We are grateful to Prof. Christian Münz and Prof. Melanie Greter for their insight on macrophages and humanized mice. The authors gratefully acknowledge the Functional Genomics Center Zurich (FGCZ) of University of Zurich and ETH Zurich, and in particular Dr. Timothy Sykes, for the support on RNA sequencing experimental design and samples preparation, as well as Falko Noè for his invaluable help during bioinformatics analysis
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2024.1439328/full#supplementary-material
1. Gordon S, Pluddemann A. Tissue macrophages: heterogeneity and functions. BMC Biol. (2017) 15:53. doi: 10.1186/s12915-017-0392-4
2. Roszer T. Understanding the biology of self-renewing macrophages. Cells. (2018) 7. doi: 10.3390/cells7080103
3. Mass E, Ballesteros I, Farlik M, Halbritter F, Gunther P, Crozet L, et al. Specification of tissue-resident macrophages during organogenesis. Science. (2016) 353. doi: 10.1126/science.aaf4238
4. Franken L, Schiwon M, Kurts C. Macrophages: sentinels and regulators of the immune system. Cell Microbiol. (2016) 18:475–87. doi: 10.1111/cmi.12580
5. Yang Y, Tu ZK, Liu XK, Zhang P. Mononuclear phagocyte system in hepatitis C virus infection. World J Gastroenterol. (2018) 24:4962–73. doi: 10.3748/wjg.v24.i44.4962
6. Faure-Dupuy S, Durantel D, Lucifora J. Liver macrophages: Friend or foe during hepatitis B infection? Liver Int. (2018) 38:1718–29. doi: 10.1111/liv.13884
7. Knoll R, Schultze JL, Schulte-Schrepping J. Monocytes and macrophages in COVID-19. Front Immunol. (2021) 12:720109. doi: 10.3389/fimmu.2021.720109
8. Zhou L, Cao X, Fang J, Li Y, Fan M. Macrophages polarization is mediated by the combination of PRR ligands and distinct inflammatory cytokines. Int J Clin Exp Pathol. (2015) 8:10964–74.
9. Yu S, Ge H, Li S, Qiu HJ. Modulation of macrophage polarization by viruses: turning off/on host antiviral responses. Front Microbiol. (2022) 13:839585. doi: 10.3389/fmicb.2022.839585
10. Hu B, Huang S, Yin L. The cytokine storm and COVID-19. J Med Virol. (2021) 93:250–6. doi: 10.1002/jmv.26232
11. Ferreira I, Porterfield JZ, Gupta RK, Mlcochova P. Cell cycle regulation in macrophages and susceptibility to HIV-1. Viruses. (2020) 12. doi: 10.3390/v12080839
12. Dupont M, Sattentau QJ. Macrophage cell-cell interactions promoting HIV-1 infection. Viruses. (2020) 12. doi: 10.3390/v12050492
13. Gaudin R, Berre S, Cunha de Alencar B, Decalf J, Schindler M, Gobert FX, et al. Dynamics of HIV-containing compartments in macrophages reveal sequestration of virions and transient surface connections. PloS One. (2013) 8:e69450. doi: 10.1371/journal.pone.0069450
14. Gorry PR, Churchill M, Crowe SM, Cunningham AL, Gabuzda D. Pathogenesis of macrophage tropic HIV-1. Curr HIV Res. (2005) 3:53–60. doi: 10.2174/1570162052772951
15. Igarashi T, Brown CR, Endo Y, Buckler-White A, Plishka R, Bischofberger N, et al. Macrophage are the principal reservoir and sustain high virus loads in rhesus macaques after the depletion of CD4(+) T cells by a highly pathogenic simian immunodeficiency virus/HIV type 1 chimera (SHIV): Implications for HIV-1 infections of humans. Proc Natl Acad Sci United States America. (2001) 98:658–63. doi: 10.1073/pnas.021551798
16. Honeycutt JB, Thayer WO, Baker CE, Ribeiro RM, Lada SM, Cao Y, et al. HIV persistence in tissue macrophages of humanized myeloid-only mice during antiretroviral therapy. Nat Med. (2017) 23:638–43. doi: 10.1038/nm.4319
17. Woottum M, Yan S, Sayettat S, Grinberg S, Cathelin D, Bekaddour N, et al. Macrophages: key cellular players in HIV infection and pathogenesis. Viruses. (2024) 16. doi: 10.3390/v16020288
18. Ginhoux F, Guilliams M. Tissue-resident macrophage ontogeny and homeostasis. Immunity. (2016) 44:439–49. doi: 10.1016/j.immuni.2016.02.024
19. Reu P, Khosravi A, Bernard S, Mold JE, Salehpour M, Alkass K, et al. The lifespan and turnover of microglia in the human brain. Cell Rep. (2017) 20:779–84. doi: 10.1016/j.celrep.2017.07.004
20. Epelman S, Lavine KJ, Randolph GJ. Origin and functions of tissue macrophages. Immunity. (2014) 41:21–35. doi: 10.1016/j.immuni.2014.06.013
21. Freel SA, Saunders KO, Tomaras GD. CD8(+)T-cell-mediated control of HIV-1 and SIV infection. Immunol Res. (2011) 49:135–46. doi: 10.1007/s12026-010-8177-7
22. Scully E, Alter G. NK cells in HIV disease. Curr HIV/AIDS Rep. (2016) 13:85–94. doi: 10.1007/s11904-016-0310-3
23. Li Q, Smith AJ, Schacker TW, Carlis JV, Duan L, Reilly CS, et al. Microarray analysis of lymphatic tissue reveals stage-specific, gene expression signatures in HIV-1 infection. J Immunol. (2009) 183:1975–82. doi: 10.4049/jimmunol.0803222
24. Herbein G, Varin A. The macrophage in HIV-1 infection: from activation to deactivation? Retrovirology. (2010) 7:33. doi: 10.1186/1742-4690-7-33
25. Burdo TH, Walker J, Williams KC. Macrophage polarization in AIDS: dynamic interface between anti-viral and anti-inflammatory macrophages during acute and chronic infection. J Clin Cell Immunol. (2015) 6.
26. Skelton JK, Ortega-Prieto AM, Dorner M. A Hitchhiker’s guide to humanized mice: new pathways to studying viral infections. Immunology. (2018) 154:50–61. doi: 10.1111/imm.12906
27. Gillgrass A, Wessels JM, Yang JX, Kaushic C. Advances in humanized mouse models to improve understanding of HIV-1 pathogenesis and immune responses. Front Immunol. (2020) 11:617516. doi: 10.3389/fimmu.2020.617516
28. Ivic S, Rochat M-A, Li D, Audigé A, Schlaepfer E, Münz C, et al. Differential dynamics of HIV infection in humanized MISTRG versus MITRG mice. ImmunoHorizons. (2017) 1:162–75. doi: 10.4049/immunohorizons.1700042
29. MiltenyiBiotec. CD34+ cells isolation protocol. Available online at: https://www.miltenyibiotec.com/CH-en/products/cd34-microbead-kit-human.html130-046-702 (Accessed June 12, 2023).
30. Ramakrishnan MA. Determination of 50% endpoint titer using a simple formula. World J Virol. (2016) 5:85–6. doi: 10.5501/wjv.v5.i2.85
31. Biotec M. Monocyte isolation. Available online at: https://www.miltenyibiotec.com/CH-en/products/cd14-microbeads-human.html130-050-201 (Accessed June 03, 2023).
32. Promega. Luciferase assay system protocol. Available online at: https://ch.promega.com/products/luciferase-assays/reporter-assays/luciferase-assay-system/?catNum=E1500protocols (Accessed July 09, 2023).
33. Gill KP, Denham M. Optimized transgene delivery using third-generation lentiviruses. Curr Protoc Mol Biol. (2020) 133:e125. doi: 10.1002/cpmb.125
34. SigmaAldrich. Amicon protein purification system. Available online at: https://www.sigmaaldrich.com/CH/en/technical-documents/technical-article/protein-biology/protein-purification/protein-purification (Accessed June 29, 2023).
35. Takara. Takara Lenti-X GoStix Plus protocol. Available online at: https://www.takarabio.com/products/gene-function/viral-transduction/lentivirus/titration-kits/lenti-x-gostix-plus (Accessed June 29, 2023).
37. Amelio P, Portevin D, Reither K, Mhimbira F, Mpina M, Tumbo A, et al. Mixed Th1 and Th2 Mycobacterium tuberculosis-specific CD4 T cell responses in patients with active pulmonary tuberculosis from Tanzania. PloS Negl Trop Dis. (2017) 11:e0005817. doi: 10.1371/journal.pntd.0005817
38. Biosciences B. BD Cytofix/Cytoperm - cell permeabilization protocol. Available online at: https://www.bdbiosciences.com/content/dam/bdb/products/global/reagents/flow-cytometry-reagents/research-reagents/buffers-and-supporting-reagents-ruo/554714_base/pdf/554714_554715_555028_Book_Website.pdf (Accessed July 02, 2023).
39. QIAGEN. RNA isolation protocol (2023). Available online at: https://www.qiagen.com/us/resources/resourcedetail?id=f646813a-efbb-4672-9ae3-e665b3045b2b&lang=en (Accessed July 01, 2023).
40. Chen SF, Zhou YQ, Chen YR, Gu J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. (2018) 34:884–90. doi: 10.1093/bioinformatics/bty560
41. Bray NL, Pimentel H, Melsted P, Pachter L. Near-optimal probabilistic RNA-seq quantification. Nat Biotechnol. (2016) 34:888. doi: 10.1038/nbt.3519
42. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. (2014) 15. doi: 10.1186/s13059-014-0550-8
43. Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. (2010) 26:139–40. doi: 10.1093/bioinformatics/btp616
44. Wu TZ, Hu EQ, Xu SB, Chen MJ, Guo PF, Dai ZH, et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation-Amsterdam. (2021) 2. doi: 10.1016/j.xinn.2021.100141
45. Fenwick C, Joo V, Jacquier P, Noto A, Banga R, Perreau M, et al. T-cell exhaustion in HIV infection. Immunol Rev. (2019) 292:149–63. doi: 10.1111/imr.12823
46. Audige A, Rochat MA, Li D, Ivic S, Fahrny A, Muller CKS, et al. Long-term leukocyte reconstitution in NSG mice transplanted with human cord blood hematopoietic stem and progenitor cells. BMC Immunol. (2017) 18:28. doi: 10.1186/s12865-017-0209-9
47. Nguyen T, Du J, Li YC. A protocol for macrophage depletion and reconstitution in a mouse model of sepsis. STAR Protoc. (2021) 2:101004. doi: 10.1016/j.xpro.2021.101004
48. Thompson HL, van Rooijen N, McLelland BT, Manilay JO. F4/80(+) host macrophages are a barrier to murine embryonic stem cell-derived hematopoietic progenitor engraftment in vivo. J Immunol Res. (2016) 2016:2414906. doi: 10.1155/2016/2414906
49. Di Stasi A, Tey SK, Dotti G, Fujita Y, Kennedy-Nasser A, Martinez C, et al. Inducible apoptosis as a safety switch for adoptive cell therapy. New Engl J Med. (2011) 365:1673–83. doi: 10.1056/nejmoa1106152
50. He W, Qiang M, Ma W, Valente AJ, Quinones MP, Wang W, et al. Development of a synthetic promoter for macrophage gene therapy. Hum Gene Ther. (2006) 17:949–59. doi: 10.1089/hum.2006.17.949
51. Kleiveland CR. Peripheral blood mononuclear cells. impact Food bioactives Health. (2015), 161–7.
52. Dong HF, Wigmore K, Carrington MN, Dean M, Turpin JA, Howard OMZ. Variants of CCR5, which are permissive for HIV-1 infection, show distinct functional responses to CCL3, CCL4 and CCL5. Genes Immunity. (2005) 6:609–19. doi: 10.1038/sj.gene.6364247
53. An P, Nelson GW, Wang L, Donfield S, Goedert JJ, Phair J, et al. Modulating influence on HIV/AIDS by interacting RANTES gene variants. Proc Natl Acad Sci U S A. (2002) 99:10002–7. doi: 10.1073/pnas.142313799
54. Kedzierska K, Crowe SM. Cytokines and HIV-1: interactions and clinical implications. Antivir Chem Chemother. (2001) 12:133–50. doi: 10.1177/095632020101200301
55. Sheard J, Azoidis I, Widera D. Osteogenic and adipogenic differentiation of human mesenchymal stem cells.
56. Colomer-Lluch M, Ruiz A, Moris A, Prado JG. Restriction factors: from intrinsic viral restriction to shaping cellular immunity against HIV-1. Front Immunol. (2018) 9:2876. doi: 10.3389/fimmu.2018.02876
57. Kazer SW, Aicher TP, Muema DM, Carroll SL, Ordovas-Montanes J, Miao VN, et al. Integrated single-cell analysis of multicellular immune dynamics during hyperacute HIV-1 infection. Nat Med. (2020) 26:511–8. doi: 10.1038/s41591-020-0799-2
58. Roberts L, Passmore JA, Williamson C, Little F, Bebell LM, Mlisana K, et al. Plasma cytokine levels during acute HIV-1 infection predict HIV disease progression. AIDS. (2010) 24:819–31. doi: 10.1097/qad.0b013e3283367836
59. Nosik M, Ryzhov K, Rymanova I, Sobkin A, Kravtchenko A, Kuimova U, et al. Dynamics of plasmatic levels of pro- and anti-inflammatory cytokines in HIV-infected individuals with M. tuberculosis co-infection. Microorganisms. (2021) 9. doi: 10.3390/microorganisms9112291
60. Muema DM, Akilimali NA, Ndumnego OC, Rasehlo SS, Durgiah R, Ojwach DBA, et al. Association between the cytokine storm, immune cell dynamics, and viral replicative capacity in hyperacute HIV infection. BMC Med. (2020) 18. doi: 10.1186/s12916-020-01529-6
61. Shultz LD, Schweitzer PA, Christianson SW, Gott B, Schweitzer IB, Tennent B, et al. Multiple defects in innate and adaptive immunologic function in NOD/LtSz-scid mice. J Immunol. (1995) 154:180–91. doi: 10.4049/jimmunol.154.1.180
62. Lossos C, Liu Y, Kolb KE, Christie AL, Van Scoyk A, Prakadan SM, et al. Mechanisms of lymphoma clearance induced by high-dose alkylating agents. Cancer Discovery. (2019) 9:944–61. doi: 10.1158/2159-8290.cd-18-1393
63. Baxter AE, Russell RA, Duncan CJ, Moore MD, Willberg CB, Pablos JL, et al. Macrophage infection via selective capture of HIV-1-infected CD4+ T cells. Cell Host Microbe. (2014) 16:711–21. doi: 10.1016/j.chom.2014.10.010
64. Burnett SH, Kershen EJ, Zhang J, Zeng L, Straley SC, Kaplan AM, et al. Conditional macrophage ablation in transgenic mice expressing a Fas-based suicide gene. J Leukoc Biol. (2004) 75:612–23. doi: 10.1189/jlb.0903442
65. Chagraoui J, Girard S, Spinella JF, Simon L, Bonneil E, Mayotte N, et al. UM171 preserves epigenetic marks that are reduced in ex vivo culture of human HSCs via potentiation of the CLR3-KBTBD4 complex. Cell Stem Cell. (2021) 28:48. doi: 10.1016/j.stem.2020.12.002
66. Ngom M, Imren S, Maetzig T, Adair JE, Knapp DJHF, Chagraoui J, et al. UM171 enhances lentiviral gene transfer and recovery of primitive human hematopoietic cells. Mol Therapy-Methods Clin Dev. (2018) 10:156–64. doi: 10.1016/j.omtm.2018.06.009
67. Grzywacz B, Kataria N, Kataria N, Blazar BR, Miller JS, Verneris MR. Natural killer-cell differentiation by myeloid progenitors. Blood. (2011) 117:3548–58. doi: 10.1182/blood-2010-04-281394
68. Rodrigues CP, Shvedunova M, Akhtar A. Epigenetic regulators as the gatekeepers of hematopoiesis. Trends Genet. (2021) 37:125–42. doi: 10.1016/j.tig.2020.09.015
69. Ciuffi A. Mechanisms governing lentivirus integration site selection. Curr Gene Ther. (2008) 8:419–29. doi: 10.2174/156652308786848021
70. Liu Z, Chen O, Wall JBJ, Zheng M, Zhou Y, Wang L, et al. Systematic comparison of 2A peptides for cloning multi-genes in a polycistronic vector. Sci Rep. (2017) 7:2193. doi: 10.1038/s41598-017-02460-2
71. Frost SD, Martinez-Picado J, Ruiz L, Clotet B, Brown AJ. Viral dynamics during structured treatment interruptions of chronic human immunodeficiency virus type 1 infection. J Virol. (2002) 76:968–79. doi: 10.1128/jvi.76.3.968-979.2002
72. Ho DD, Neumann AU, Perelson AS, Chen W, Leonard JM, Markowitz M. Rapid turnover of plasma virions and CD4 lymphocytes in HIV-1 infection. Nature. (1995) 373:123–6. doi: 10.1038/373123a0
73. Chesebro B, Wehrly K, Nishio J, Perryman S. Mapping of independent V3 envelope determinants of human immunodeficiency virus type 1 macrophage tropism and syncytium formation in lymphocytes. JVirol. (1996) 70:9055–9. doi: 10.1128/jvi.70.12.9055-9059.1996
74. Isorce N, Testoni B, Locatelli M, Fresquet J, Rivoire M, Luangsay S, et al. Antiviral activity of various interferons and pro-inflammatory cytokines in non-transformed cultured hepatocytes infected with hepatitis B virus. Antiviral Res. (2016) 130:36–45. doi: 10.1016/j.antiviral.2016.03.008
75. Carlin AF, Vizcarra EA, Branche E, Viramontes KM, Suarez-Amaran L, Ley K, et al. Deconvolution of pro- and antiviral genomic responses in Zika virus-infected and bystander macrophages. Proc Natl Acad Sci U S A. (2018) 115:E9172–E81. doi: 10.1073/pnas.1807690115
76. Yang D, Chu H, Lu G, Shuai H, Wang Y, Hou Y, et al. STAT2-dependent restriction of Zika virus by human macrophages but not dendritic cells. Emerg Microbes Infect. (2021) 10:1024–37. doi: 10.1080/22221751.2021.1929503
77. Ashhurst TM, Vreden C, Munoz-Erazo L, Niewold P, Watabe K, Terry RL, et al. Antiviral macrophage responses in flavivirus encephalitis. Indian J Med Res. (2013) 138:632–47.
78. Zhao W, Pahar B, Borda JT, Alvarez X, Sestak K. A decline in CCL3-5 chemokine gene expression during primary simian-human immunodeficiency virus infection. PloS One. (2007) 2:e726. doi: 10.1371/journal.pone.0000726
79. Cocchi F, Devico AL, Garzinodemo A, Arya SK, Gallo RC, Lusso P. Identification of rantes, mip-1-alpha, and mip-1-beta as the major hiv-suppressive factors produced by cd8(+) T-cells. Science. (1995) 270:1811–5. doi: 10.1126/science.270.5243.1811
80. Alkhatib G. The biology of CCR5 and CXCR4. Curr Opin HIV AIDS. (2009) 4:96–103. doi: 10.1097/coh.0b013e328324bbec
81. Blanpain C, Migeotte I, Lee B, Vakili J, Doranz BJ, Govaerts C, et al. CCR5 binds multiple CC-chemokines: MCP-3 acts as a natural antagonist. Blood. (1999) 94:1899–905. doi: 10.1182/blood.v94.6.1899
82. Zhou X, Michal JJ, Zhang L, Ding B, Lunney JK, Liu B, et al. Interferon induced IFIT family genes in host antiviral defense. Int J Biol Sci. (2013) 9:200–8. doi: 10.7150/ijbs.5613
83. Perng YC, Lenschow DJ. ISG15 in antiviral immunity and beyond. Nat Rev Microbiol. (2018) 16:423–39. doi: 10.1038/s41579-018-0020-5
84. Schwartz SL, Conn GL. RNA regulation of the antiviral protein 2’-5’-oligoadenylate synthetase. Wiley Interdiscip Rev RNA. (2019) 10:e1534. doi: 10.1002/wrna.1534
85. Berger G, Durand S, Fargier G, Nguyen XN, Cordeil S, Bouaziz S, et al. APOBEC3A is a specific inhibitor of the early phases of HIV-1 infection in myeloid cells. PloS Pathog. (2011) 7:e1002221. doi: 10.1371/journal.ppat.1002221
86. Mutascio S, Mota T, Franchitti L, Sharma AA, Willemse A, Bergstresser SN, et al. CD8 T cells promote HIV latency by remodeling CD4T cell metabolism to enhance their survival, quiescence, and stemness. Immunity. (2023) 56:1132. doi: 10.1016/j.immuni.2023.03.010
87. Zhang ZQ, Wietgrefe SW, Li Q, Shore MD, Duan L, Reilly C, et al. Roles of substrate availability and infection of resting and activated CD4+ T cells in transmission and acute simian immunodeficiency virus infection. Proc Natl Acad Sci U S A. (2004) 101:5640–5. doi: 10.1073/pnas.0308425101
88. Li Y, Kappes JC, Conway JA, Price RW, Shaw GM, Hahn BH. Molecular characterization of human immunodeficiency virus type 1 cloned directly from uncultured human brain tissue: identification of replication-competent and -defective viral genomes. J Virol. (1991) 65:3973–85. doi: 10.1128/jvi.65.8.3973-3985.1991
89. Deshiere A, Joly-Beauparlant C, Breton Y, Ouellet M, Raymond F, Lodge R, et al. Global mapping of the macrophage-HIV-1 transcriptome reveals that productive infection induces remodeling of host cell DNA and chromatin. Sci Rep. (2017) 7:5238. doi: 10.1038/s41598-017-05566-9
90. O’Brien M, Manches O, Wilen C, Gopal R, Huq R, Wu V, et al. CD4 receptor is a key determinant of divergent HIV-1 sensing by plasmacytoid dendritic cells. PloS Pathog. (2016) 12:e1005553. doi: 10.1371/journal.ppat.1005553
91. Tanaka S, Saito Y, Kunisawa J, Kurashima Y, Wake T, Suzuki N, et al. Development of mature and functional human myeloid subsets in hematopoietic stem cell-engrafted NOD/SCID/IL2rgammaKO mice. J Immunol. (2012) 188:6145–55. doi: 10.4049/jimmunol.1103660
92. Andre MC, Erbacher A, Gille C, Schmauke V, Goecke B, Hohberger A, et al. Long-term human CD34+ stem cell-engrafted nonobese diabetic/SCID/IL-2R gamma(null) mice show impaired CD8+ T cell maintenance and a functional arrest of immature NK cells. J Immunol. (2010) 185:2710–20. doi: 10.4049/jimmunol.1000583
93. Baroncini L, Bredl S, Nicole KP, Speck RF. The humanized mouse model: what added value does it offer for HIV research? Pathogens. (2023) 12. doi: 10.3390/pathogens12040608
94. Ye W, Chen Q. Potential applications and perspectives of humanized mouse models. Annu Rev Anim Biosci. (2022) 10:395–417. doi: 10.1146/annurev-animal-020420-033029
95. Rongvaux A, Willinger T, Martinek J, Strowig T, Gearty SV, Teichmann LL, et al. Development and function of human innate immune cells in a humanized mouse model. Nat Biotechnol. (2014) 32:364–72. doi: 10.1038/nbt.2858
Keywords: HIV-1, macrophages, iHMD-NSG mice, humanized mice for HIV, myeloid cells
Citation: Baroncini L, Muller CKS, Kadzioch NP, Wolfensberger R, Russenberger D, Bredl S, Mlambo T and Speck RF (2024) Pro-inflammatory macrophages suppress HIV replication in humanized mice and ex vivo co-cultures. Front. Immunol. 15:1439328. doi: 10.3389/fimmu.2024.1439328
Received: 27 May 2024; Accepted: 04 October 2024;
Published: 07 November 2024.
Edited by:
Guido Poli, Vita-Salute San Raffaele University, ItalyReviewed by:
David W. Gludish, Cornell Univeristy, United StatesCopyright © 2024 Baroncini, Muller, Kadzioch, Wolfensberger, Russenberger, Bredl, Mlambo and Speck. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Roberto F. Speck, Um9iZXJ0by5zcGVja0B1c3ouY2g=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.