 Qin Ao
Qin Ao Huan Hu3
Huan Hu3
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 15 July 2024
Sec. Autoimmune and Autoinflammatory Disorders: Autoinflammatory Disorders
Volume 15 - 2024 | https://doi.org/10.3389/fimmu.2024.1438803
Ferroptosis is an iron-dependent mode of cell death distinct from apoptosis and necrosis. Its mechanisms mainly involve disordered iron metabolism, lipid peroxide deposition, and an imbalance of the antioxidant system. The endoplasmic reticulum is an organelle responsible for protein folding, lipid metabolism, and Ca2+ regulation in cells. It can be induced to undergo endoplasmic reticulum stress in response to inflammation, oxidative stress, and hypoxia, thereby regulating intracellular environmental homeostasis through unfolded protein responses. It has been reported that ferroptosis and endoplasmic reticulum stress (ERS) have an interaction pathway and jointly regulate cell survival and death. Both have also been reported separately in rheumatoid arthritis (RA) mechanism studies. However, studies on the correlation between ferroptosis and ERS in RA have not been reported so far. Therefore, this paper reviews the current status of studies and the potential correlation between ferroptosis and ERS in RA, aiming to provide a research reference for developing treatments for RA.
Rheumatoid arthritis (RA), a chronic autoimmune disease, is characterized by synovial proliferation and progressive bone destruction. Its pathogenesis involves tumor-like proliferation of synovial fibroblasts (FLS), local infiltration of immune cells, and the release of inflammatory cytokines that exacerbate joint inflammation and bone destruction (1, 2). Therefore, inhibiting synovial cell inflammatory proliferation and regulating the immune-inflammatory response are the main strategies for current RA treatment (3). Disease-modifying anti-rheumatic drugs (DMARDs) are routinely used for RA; however, adverse effects, drug resistance, and poor efficacy are often observed in initial treatment. Thus, further understanding of the pathogenesis of RA is urgently needed, and finding safer and more effective therapeutic targets is crucial.
Ferroptosis is a mode of iron-dependent cell death that is morphologically and biochemically distinct from apoptosis, necrosis, and pyroptosis (4, 5). It was discovered by Dion’s team in 2012 when they studied the mechanism of Erastin against tumor RAS mutations and named ferroptosis. The main features of ferroptosis are abnormal iron accumulation, lipid peroxidation, and dysregulation of the antioxidant system (6). It has been reported for many years that iron metabolism is abnormal in RA, with iron content in the synovial membrane and synovial fluid significantly higher than in peripheral blood. Additionally, the serum soluble transferrin receptor (sTFR) level is positively correlated with RA disease activity (7, 8). The localized accumulation of iron ions in the joints can promote the production of large amounts of reactive oxygen species (ROS). Coupled with the dysregulation of the antioxidant system, this can lead to bone destruction, abnormal proliferation of RA-FLS, and the massive production of inflammatory mediators (9).
The endoplasmic reticulum (ER), an important organelle in eukaryotic cells, is involved in protein synthesis, folding, structural maturation, lipid metabolism, and Ca2+ regulation. Inflammation, oxidative stress, Ca2+ dysregulation, and hypoxia can induce cellular endoplasmic reticulum stress (ERS) by overloading the ER’s protein folding mechanism (10). This subsequently stimulates the cell to initiate the unfolded protein response (UPR) to regulate the ER’s protein folding capacity (11). However, sustained and high-intensity ERS can lead to cell death (12). RA, an autoimmune disease, primarily involves the activation of immune cells, which produce large amounts of autoantibodies that attack normal tissues and trigger an inflammatory cascade through the release of inflammatory mediators, exacerbating disease progression. This process involves the endoplasmic reticulum. The inflammatory milieu of RA activates ERS, while protein synthesis is accelerated through the UPR, enabling RA-FLSs to adapt to and tolerate the milieu, exhibiting strong resistance to ERS-induced apoptosis. This may also be responsible for the tumor-like transformation or dedifferentiation of RA-FLS (13).
Some studies have found that ferroptosis and ERS share a common pathway, especially in tumor diseases, and disease trends can be altered by controlling the ERS or ferroptosis pathway (14–17). However, there is no description of the mutual crosstalk between ERS and ferroptosis in the current studies of RA pathogenesis. Therefore, this paper summarizes the roles of ferroptosis and ERS in RA and their potential relevance in the current research status, which provides a direction for exploring the mechanism of RA and new therapeutic targets.
With the study of RA disease mechanisms, it has been found that the aberrant activation of RA-FLSs, destruction of articular cartilage, overactivation of immune cells, and sustained expression of inflammatory factors are associated with ferroptosis. Targeting RA ferroptosis as a potential therapeutic strategy (18).
Iron plays an important role in human growth and development, immune system function, and energy metabolism (19). Abnormalities in iron metabolism can affect gene regulation, enzymatic reactions, redox reactions, and other important processes (20, 21). There are two forms of iron in the human body: Fe2+ and Fe3+. Circulating iron (Fe3+) binds to transferrin (Tf), which then binds to specific transferrin receptor 1 (TFR1) and transfers to the cell. Inside the cell, it is deoxygenated and reduced to Fe2+ by iron oxide reductase. In the cytoplasm, Fe2+ is mainly bound to ferritin to form storage iron, with a small portion stored in the cytoplasm’s labile iron pool (LIP) (21). Due to the instability and high oxidizability of Fe2+, it can generate hydroxyl radicals through the Fenton reaction. These radicals can directly catalyze the oxidation of polyunsaturated fatty acid (PUFA) in the cytosol and plasma membrane under the action of lipoxygenase (LOX), generating a large amount of lipid ROS. This process causes oxidative damage to lipid membranes, proteins, and DNA, ultimately leading to cell death (22). Among the three major mechanistic pathways of ferroptosis, the abnormal accumulation of iron is the initial part of its cascade reaction. The large accumulation of Fe2+ in LIP increases the sensitivity of cells to ferroptosis.
Abnormal iron metabolism in RA is closely related to disease progression. A study showed that serum sTFR levels were significantly higher and serum iron levels were significantly lower in patients with RA compared to healthy controls. sTFR was also found to be significantly and positively correlated with parameters of inflammatory activity and autoimmune disease (23). Later, Wu J et al. (24)found that iron levels in synovial fluid and lipid peroxides in the synovium were significantly higher in RA patients with high disease activity than in those with moderate disease activity, while iron levels in peripheral blood were decreased. This suggests that iron distribution and metabolism are disordered in RA. Iron overload can inhibit osteoblast activity and promote osteoclast differentiation to a certain extent (25).It inhibits precursor cells (MC3T3-E1) from differentiating into osteoblasts by promoting ROS production, blocking the phosphatidylinositol 3-kinase (PI3K/AKT) and Jak/Stat3 signaling pathways, and activating the p38 MAPK, which induces G1-phase blockade and autophagy in osteogenic MC3T3-E1 cells (26). Meanwhile, iron overload also induces osteoclast differentiation and accelerates articular bone destruction by activating JNK, ERK, and NF-kB signaling pathways (27, 28). As a major effector cell of RA arthropathy, elevated ROS and lipid oxidation triggered by iron overload can also lead to the abnormal proliferation of RA-FLSs (24).
Iron, as a regulator of the immune response, affects the phagocytosis of monocytes and macrophages. Iron overload alters the number and viability of T-lymphocyte subpopulations and the distribution of lymphocytes in different compartments of the immune system (29). Under the same ferrous stimulus, anti-inflammatory M2-like macrophages undergo significant ferroptosis, while pro-inflammatory M1-like macrophages experience less ferroptosis. Additionally, iron overload leads to the release of high mobility group protein 1 (HMGB1) in the damage-associated molecular patterns (DAMPs) released from cells. HMGB1 can act as an adjuvant to activate the immune system and promote the secretion of tumor necrosis factor-α (TNF-α) and interleukin-6 (IL-6) by macrophages. The release of inflammatory mediators increases the uptake of iron by monocytes, increasing their sensitivity to ferroptosis (30). Iron overload can also promote synovial and vascular opacification growth and aggravate the progression of synovitis by regulating C-myc and Mdn2 gene expression (31).
In relation to the treatment of RA, it has been found that the iron ion chelator desferrioxamine (DFO) has been used to prevent synovial damage and improve RA anemia (32). Additionally, herbal components such as Icariin, Quercetin, and Galangin have also been shown to chelate iron ions and regulate iron metabolism, thereby reducing oxidative damage and immune dysfunction caused by iron overload (28, 33).
Lipid peroxidation is central to ferroptosis. The diallyl hydrogen of PUFA has low bond dissociation energy, making it prone to lipid peroxidation (34). However, free PUFA is not directly conducive to ferroptosis. It is first converted into acyl-CoA (CoA) through the catalytic action of CoA synthase long-chain family (ACSL) located in the mitochondria and the ER. CoA is then inserted into membrane phospholipids under the catalysis of lysophosphatidylcholine acyltransferase 3 (LPCAT3) and combined with phosphatidylethanolamine (PE) to form polyunsaturated fatty acid phospholipids (PUFA-PE). PUFA-PE generates lipid peroxides mediated by LOX and produces ROS through interaction with Fe2+, which then extracts hydrogen from adjacent acyl chains in the lipid membrane environment, propagating the lipid peroxidation process (35).
ROS interfere with various biological processes in vivo, including signaling, gene expression, and cellular homeostasis (36). Earlier studies by Blake DR, Roberts D, et al. found that intravenous use of iron dextrose exacerbated synovitis in RA, possibly because iron dextrose worsens synovial inflammation by promoting lipid peroxidation (37, 38). The accumulation of ROS in RA not only promotes cellular ferroptosis but also exhibits different effects on various cells. Excessive ROS are detrimental to non-synovial fibroblasts (N-FLSs); they can promote chondrocyte death by activating the NOX4/p38 MAPK pathway, inducing the expression of matrix metalloproteinases (MMPs), and degrading the cartilage matrix (39). However, ROS accumulation promotes aberrant proliferation, migration, and the release of inflammatory factors in RA-FLSs, exacerbating arthropathy (40).Additionally, ROS can promote the receptor activator of nuclear factor-κB ligand (RANKL) expression and induce osteoclast differentiation by activating the Janus kinase (JAK)2/STAT3 pathway, directly or indirectly, through upregulation of hypoxia-inducible factor-1 (HIF-1a) expression (41). They also participate in key pathological processes, such as angiogenesis and proliferation, by activating the NF-kB pathway to transcribe the vascular endothelial growth factor (VEGF) gene (42). In immune cells, ROS mediate neutrophil formation of neutrophil extracellular traps (NETs) to drive inflammatory responses (43). An overview of the mechanisms of ferroptosis and RA is given in (Figure 1).
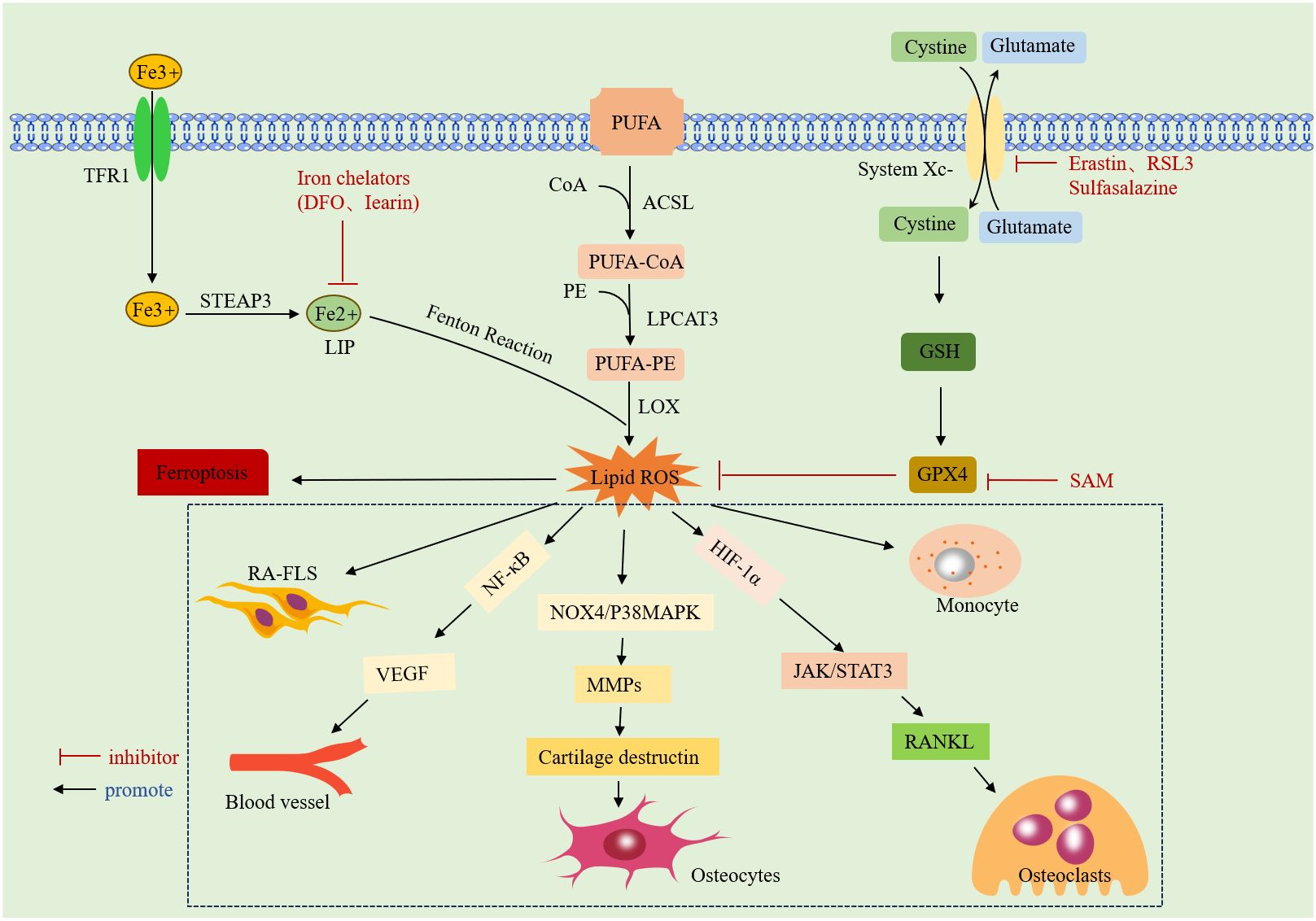
Figure 1 Mechanism of ferroptosis: it due to iron overload-mediated lipid peroxidation and antioxidant dysregulation. Specific introduction to the mechanism of action of ROS on RA.TFR1: transferrin receptor protein 1;STEAP3:lysophosphatidylcholine acyltransferase 3;DFO: deferoxamine;LIP:labile iron pool;CoA:acyl coenzyme A;ACSL: CoA synthase long-chain family;LPCAT3: lysophosphatidylcholine acyltransferase 3;PUFA:polyunsaturated fatty acid;PE:phosphatidylethanolamine; LOX:lipoxygenase;ROS:reactive oxygen species;System Xc-:cystine/glutamate counter transporter protein; RSL3: An inhibitor of GPX4;GSH:glutathione;GPX4: glutathione peroxidase 4; SAM:S-adenosylmethionine;HIF-1a:hypoxia inducible factor 1a;VEGF:vascular endothelial growth factor.NF-κB: nuclear factor kappa B; MMPs: matrix metalloproteinase; RANKL: receptor activator of nuclear factor-κ B ligand.
When lipid peroxides accumulate, cells can resist oxidation and prevent ferroptosis through various regulatory mechanisms, the most important of which is the antioxidant regulatory system composed of the cystine/glutamate antitransporter (System Xc-) and phospholipid hydroperoxidase glutathione peroxidase 4 (GPX4) (6, 44). System Xc- is an amino acid transporter located on the cell membrane, composed of member 11 of the light chain solute carrier family (SLC7A11, xCT) and member 2 of the heavy chain solute carrier family (SLC3A2), which regulates the exchange of glutamate (Glu) and extracellular cystine (Cys) in cells. Cys is reduced to cysteine as a substrate for the synthesis of glutathione (GSH) (45). By blocking System Xc- or blocking glutamate metabolic pathways, more ROS-induced lipid peroxidation and ferroptosis can be triggered (46). Therefore, GPX4/SLC7A11 is considered a key regulator of ferroptosis and a classic pathway of ferroptosis.
With the progress of research, in addition to the GPX4 antioxidant pathway, three other new pathways unrelated to GPX4 in ferroptosis have been identified: ferroptosis inhibition protein 1 (FSP1)/CoQ10, GTP cyclic hydrolase 1 (GCH1)/tetrahydrobiopterin (BH4), and dihydroorotate dehydrogenase (DHODH). These pathways reduce lipid peroxidation, remodel lipid membranes, and prevent cell ferroptosis through direct or indirect means (47).
Compared to healthy individuals, RA patients exhibit lower expression of ACSL4 and increased levels of ferritin heavy chain 1 (FTH1), GPX4, and SLC7A11 in the synovium and FLS. Although both oxidized and antioxidant levels of lipids are higher in RA-FLS than in healthy controls, the increase in antioxidant levels is slightly higher than that of oxidation, indicating that RA-FLS is resistant to ferroptosis (48). Additionally, GPX4 is involved in the regulation of immune cell function; it is associated with B-cell development and function, and GPX4 deficiency inhibits IgM antibody production (49). Furthermore, T-cell activation is dependent on Cys, which is supplied to T-cells by the Xc- system, and overexpression of FSP1 or GPX4 protects CD8+ T-cells from ferroptosis (50, 51).
In studying TNF-α, an important inflammatory mediator in the pathogenesis of RA, it was found that small doses of TNF-α could enhance Cys uptake and GSH synthesis by activating the NF-κB pathway. This upregulated the functional subunits of SLC7A11 and System Xc-, protecting RA-FLSs from oxidative stress and excess iron, thereby inhibiting ferroptosis. However, this process could be blocked by a low dose of imidazole ketone erastin (IKE) in combination with the TNF antagonist etanercept (24). In contrast, treatment with IL-6, another important inflammatory mediator, increased intracellular iron levels and decreased SLC40A1 and ferritin expression, but did not alter GSH expression, making RA-FLSs more susceptible to ferroptosis. In other studies targeting ferroptosis, Ling H et al. (48)found that glycine enhanced ferroptosis via S-Adenosylmethionine(SAM) mediated GPX4 promoter methylation and ferritin decrease. The ferroptosis inducer RSL3 can induce ferroptosis by decreasing the expression of SLC2A3 and affecting the glycolytic metabolism of RA-FLS (52). Additionally, the active peptide G1dP3 promotes RA-FLS ferroptosis through a p53/SLC7A11 axis-dependent mechanism (53). Sulfasalazine, an important biological agent, was found to inhibit System Xc- (54). Beyond the classical ferroptosis antioxidant pathways, the CoQ10 pathway, which is unrelated to GPX4, was found to act as a ROS scavenger and anti-inflammatory substance. This pathway could regulate Th17 differentiation and IL-17 secretion via the STAT3 pathway, thereby inhibiting arthritis progression in CIA mice (17). Clinical trials targeting key molecules of ferroptisis is given in (Table 1).
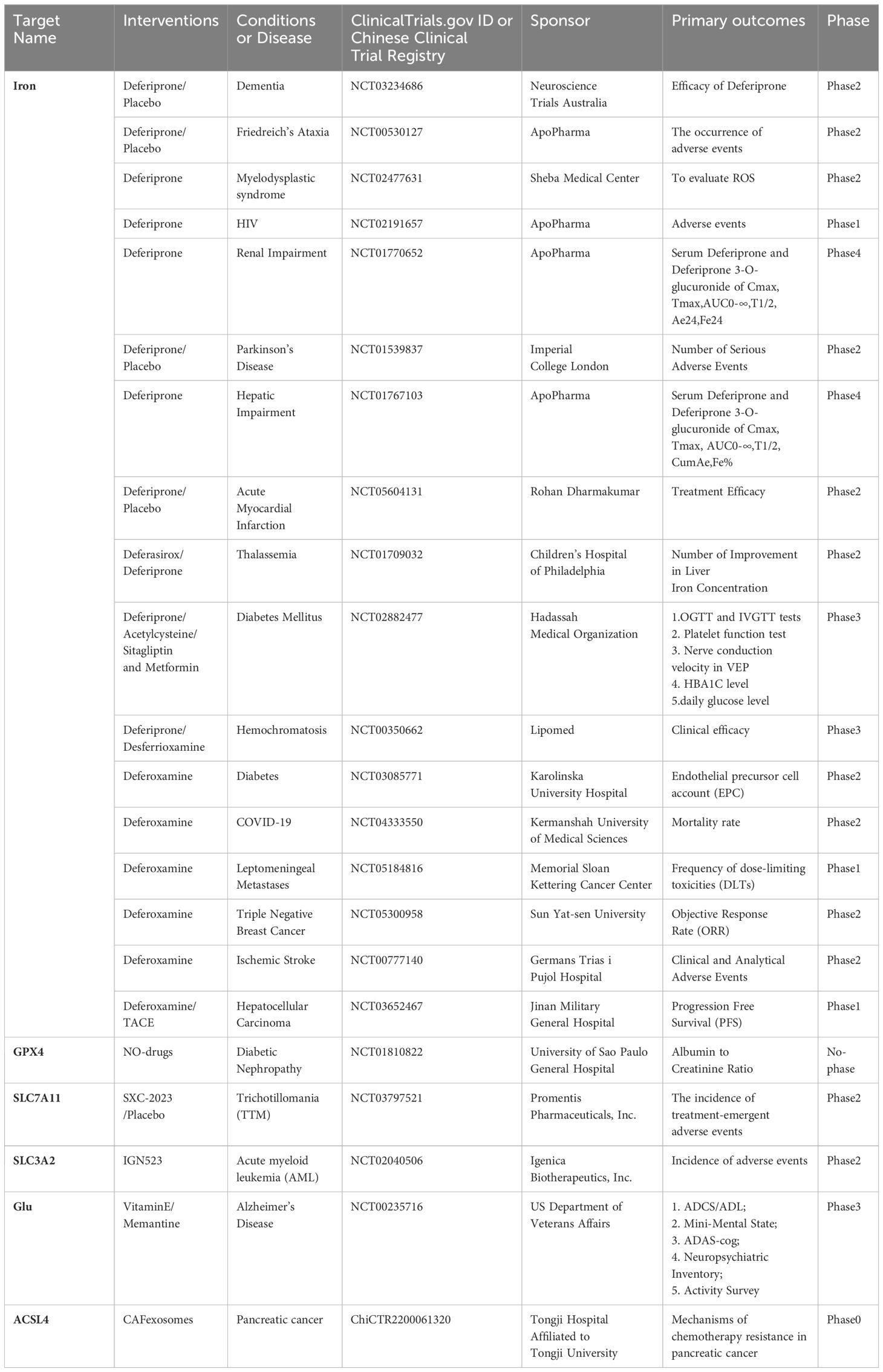
Table 1 Clinical trials targeting key molecules of ferroptisis.
Cells stimulated by internal and external injury factors can induce ERS and initiate the UPR. The UPR signaling pathway is mainly mediated by protein kinase RNA-like ER kinase (PERK), inositol-requiring enzyme 1 (IRE1), and activating transcription factor 6 (ATF6). In a physiological state, PERK, IRE1, and ATF6 are non-covalently bonded to glucose-regulated protein 78 (GRP78). When ERS occurs, GRP78 binds to misfolded proteins and dissociates from PERK, IRE1, and ATF6, activating downstream signaling pathways to inhibit DNA transcription and translation, prevent protein synthesis, and accelerate the degradation of aberrant proteins, thus regulating the adaptation of the intracellular environment. However, UPR can also activate downstream apoptotic signaling pathways and promote apoptosis when the damage is persistent and severe or when the extent of the crosstalk network exceeds the resolution capacity of ER loading aberrations (55). An overview of the mechanisms of ERS is given in (Figure 2).
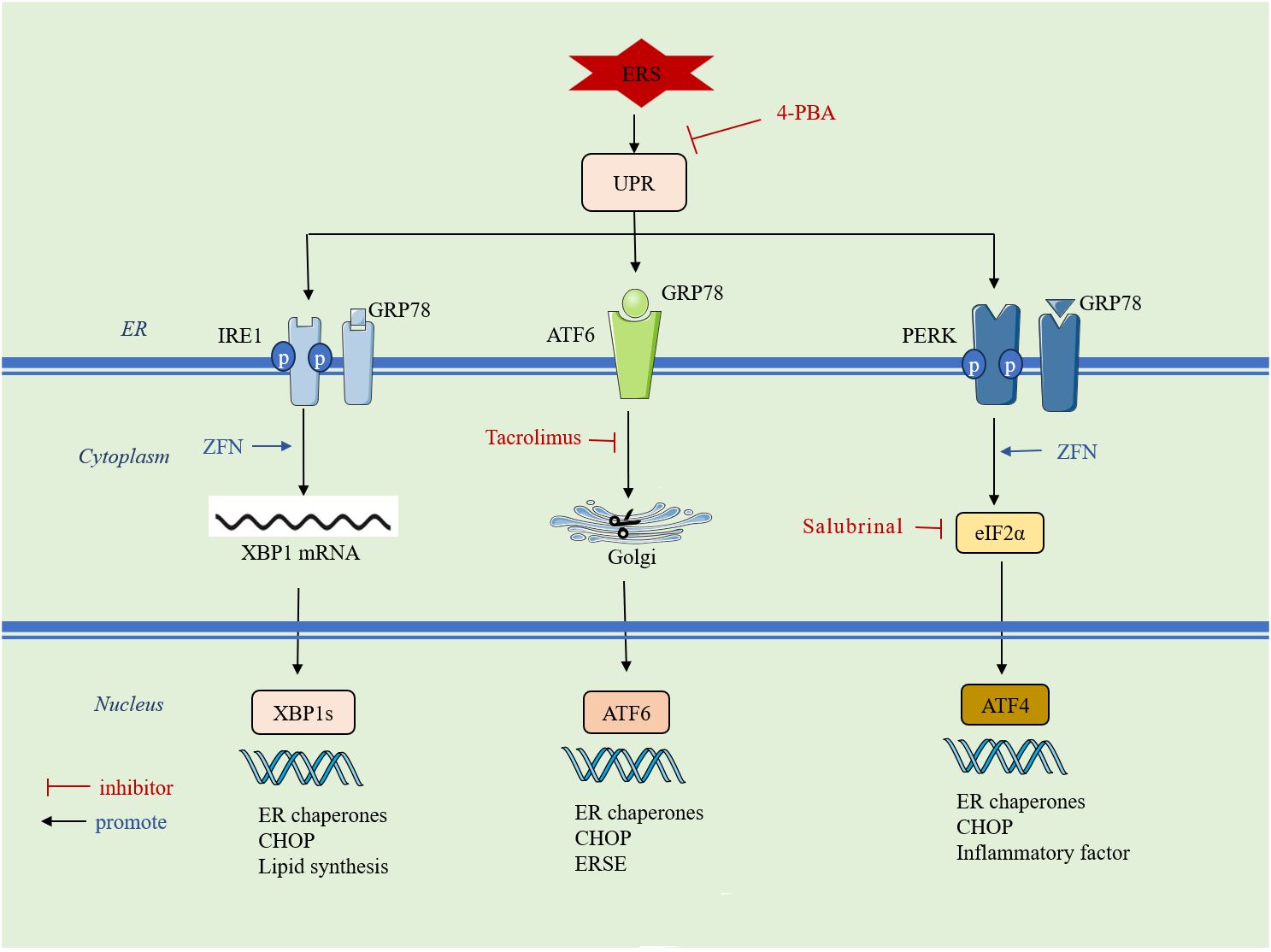
Figure 2 ERS initiates the UPR: it is mainly mediated by three signaling pathways, PERK, IRE1 and ATF6. ERS: endoplasmic reticulum stress; UPR: unfolded protein response; 4-PBA: 4-phenylbutyric acid; GRP78: glucose-regulated protein 78; IRE1: inositol requirement protein 1; PERK: protein kinase RNA-like ER kinase;ATF: activating transcription factor; ZFN: zinc ferrate nanoparticles;eIF2α: eukaryotic translation initiation factor 2α; XBP1: homeostasis transcription factor X-box protein 1; CHOP: C/EBP-homologous protein;ERSE:endoplasmic reticulum stress element.
GRP78-specific antibodies were found in up to 63% of RA patients, 7% of patients with other rheumatoid diseases, and only 1% of healthy controls (56). ERS-related genes were highly expressed in RA-FLS and synovial macrophages. In vitro experiments revealed that in synovial cells, TNF and IL-1β increased GRP78 expression. Inhibition of GRP78 expression using siRNA abrogated TNF- or TGF-β-induced proliferation of FLS and up-regulation of cell cycle protein D1, thereby promoting cell death. Elevated GRP78 levels, in turn, lead to synovial proliferation, which promotes further cytokine secretion, creating a vicious cycle. In the same study, it was also found that GRP78 can directly control VEGF 165-induced angiogenesis (57). Recently, azithromycin was found to reduce collagen-induced arthritis (CIA) in mice by inhibiting GRP78 activity (58). Thus, GRP78 is critical for the development of chronic arthritis.
When PERK dissociates from GRP78, it activates dimerization through autophosphorylation, leading to the phosphorylation of eukaryotic translation initiation factor 2α (eIF2α). eIF2α inhibits mRNA translation, suspends or attenuates protein synthesis, and alleviates the folding pressure of new and accumulated proteins in the ER. However, eIF2α only inhibits the majority of the protein translation process while promoting the synthesis of transcription factor 4 (ATF4) (59, 60). ATF4 positively feeds back to promote upstream GRP78 gene expression to accelerate the restoration of endoplasmic reticulum homeostasis and promotes NF-κB activation, which is involved in the synthesis and release of inflammatory factors (61). When ERS intensity increases and persists for a long period, sustained overexpression of ATF4 activates the nucleus-specific transcription factor (CHOP). CHOP is an important signaling molecule for the shift from survival to apoptosis in ERS, participating in Bcl-2/Bcl-Xl apoptosis regulation and promoting programmed cell death (61). In contrast to normal and osteoarthritis patients, GRP78 and p-eIF2α expression are increased in RA synovial tissues (57). Salubrinal, a selective eIF2α dephosphorylation inhibitor, has been shown to reduce the release of pro-inflammatory cytokines (IL-1β, IL-2, IL-13, and TNF) and the expression of the Dusp2 gene in a mouse model of CIA, alleviating arthritis progression (62, 63). The ERS inducer TG increased osteoclastogenesis through RANKL-induced NF-κB activation and ROS production, while IL-1β promoted RANKL-mediated osteoclast formation by upregulating GRP78, PERK, and IRE1. This process could be inhibited by 4-phenylbutyric acid (4-PBA) (64). Zinc ferrate nanoparticles and docosahexaenoic acid (DHA) have been reported to promote RA-FLS cell apoptosis by targeting the PERK-ATF4-CHOP pathway (65, 66).
ATF6 is an ER type II transmembrane protein. When ERS occurs, ATF6 dissociates from GRP78 and translocates to the Golgi apparatus, where its N-terminal portion is cleaved and activated. It then moves to the nucleus, where it binds to the heterodimer of the transcription factor XBP1, initiating the endoplasmic reticulum stress element (ERSE). This induces the expression of genes such as GRP94, GRP78, and ERP57, accelerating the degradation of intracellular unfolded proteins and reducing ERS (67). However, with continuous ERS stimulation, ATF6 induces the expression of apoptotic genes CHOP, promoting apoptosis (68). Studies on the mechanism of ATF6 in RA are relatively limited. High levels of ATF6 expression have been reported in RA synovium, and pro-inflammatory cytokines such as IL-1β and TNF can induce ATF6 expression in RA-FLS (57, 69). Tacrolimus, an important immunologic agent in RA treatment, reduces ATF6 expression and alleviates RA disease progression by inhibiting ERS-mediated osteoclastogenesis and inflammatory response (70).
IRE1 is a type I transmembrane protein on the ER with endonuclease activity. When IRE1 dissociates from GRP78, it undergoes oligomerization and auto-trans-phosphorylation to become IRE1α. This activates ribonucleic acid endonuclease, which shears X-box binding protein 1 (XBP1) mRNA. The sheared XBP1 mRNA then translocates to the nucleus, translating into the active transcription factor XBP1s. In the nucleus, XBP1s can induce the expression of the GRP78 gene, increase the degradation of endoplasmic reticulum-folded proteins, inhibit the synthesis of new proteins, and induce the transcription of the apoptosis gene CHOP (71). The IRE1α-XBP1s axis can splice to induce toll-like receptor-dependent cytokine secretion and maintain the autocrine cycle of FLS activation (72). Flavonoids from Litsea coreana were found to inhibit IRE1/mTORC1/TNF-α-regulated inflammatory responses triggered in peritoneal macrophages of CIA mice and ameliorated arthritis in a dose-dependent manner in CIA mice (73). In a study of peripheral blood mononuclear cells (PBMCs) from RA patients, it was found that the expression of GRP78, IRE1, and XBP1 was significantly increased in RA patients. Additionally, IRE1α activation in synovial macrophages was higher compared to healthy controls, whereas the progression of inflammatory arthritis in mice was prevented by knockdown of IRE1 (74). In exploring the mechanism of action of zinc ferrate nanoparticles on RA-FLS, the IRE1-XBP1 pathway was activated to induce apoptosis in RA-FLS, in addition to the activation of the PERK-ATF4-CHOP pathway (65). Moreover, IRE1 is a central regulator of B-cell differentiation, and both B-lymphocyte maturation and antibody plasma cell differentiation are regulated by the IRE1-XBP1 axis (75, 76).
This suggests that RA-FLS apoptosis resistance and the persistence of chronic inflammation are associated with ERS. Therefore, breaking this resistance relationship may be key to understanding the mechanism of RA.
Studies have found that there is a shared pathway between ferroptosis and ERS, both of which are involved in cell survival processes. However, the mechanisms of ferroptosis and ERS in RA have only been studied separately, with no reports on their combined effects on RA. Therefore, the possible association between the two in RA can be explored through the review of studies on other diseases. An overview of interaction between ferroptosis and ERS is shown in (Figure 3).
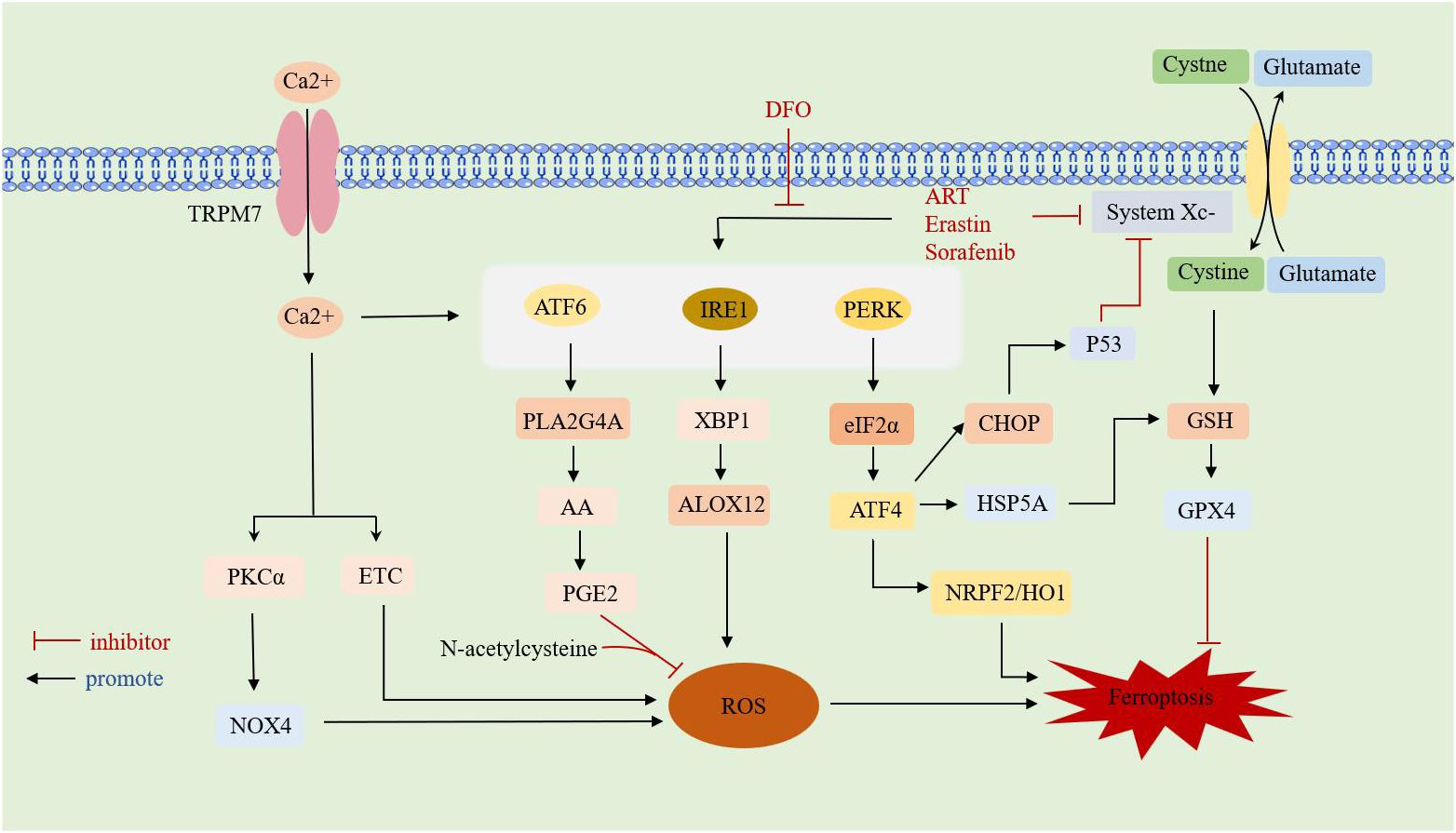
Figure 3 Overview of interaction between ferroptosis and ERS.TRPM7: transient receptor potential cation channel subfamily M member 7;DFO: deferoxamine; ART:artesunate;ATF: activating transcription factor; PERK: protein kinase RNA-like ER kinase; IRE1: inositol requirement protein 1; XBP1: homeostasis transcription factor X-box protein 1; eIF2α: eukaryotic translation initiation factor 2α; HSPA5: heat shock protein family A member 5; CHOP: C/EBP-homologous protein; ALOX12: arachidonate 12-lipoxygenase;GSH: glutathione; GPX4: glutathione peroxidase 4; HO-1: heme oxygenase-1; NRF2: nuclear factor-E2-related factor 2; PLA2G4A: phospholipase A2 group IVA;AA: arachidonic acid;PGE2: Prostaglandin E2;PKCa: calcium-activated protein kinase Cα;ETC: electron transport chain; NOX4: NADPH oxidoreductase; ROS: reactive oxygen species.
It has been reported that Sorafenib inhibits the Xc- system transport of cysteine, leading not only to ferroptosis but also inducing ERS, such as eIF2α phosphorylation, and upregulation of ATF4 and cation transport-regulated homologue 1 (77). ATF4 has been suggested as an important regulator of SLC7A11 expression, and cells are more susceptible to drug-induced ferroptosis when ATF4 is knocked down (78, 79). The ferroptosis inducers ART and erastin both caused a time-dependent increase in GRP78 expression and induced ERS. Additionally, DFO inhibited ART-induced ERS (80). Yang J et al. (17) found that proteasome inhibitors could induce ERS and activate ferroptosis signals while studying the mechanism of drug resistance in liver cancer cells, causing tumor cells to adaptively tolerate ERS.
The activation of the PERK-eIF2α-ATF4-CHOP pathway mediated by ERS promotes p53 expression and is involved in the interaction between ferroptosis and apoptosis. p53, a classical tumor suppressor gene, inhibits the uptake of cystine by System Xc- through downregulation of SLC7A11 expression, leading to a decrease in the antioxidant capacity of cells. However, p53 is overexpressed in RA, where it regulates the balance between Th17 cells and Treg cells and accelerates chondrocyte destruction by blocking the cell proliferation cycle (80–82). Chen D et al. (78) found that ATF4 was significantly elevated in malignant gliomas. Activated ATF4 could transcribe System Xc-, promote GSH expression to elevate antioxidant capacity, and inhibit cellular ferroptosis, whereas knockdown of ATF4 caused ROS accumulation, inducing ferroptosis. Chen Y et al. (83) found that bisartemisinin (DHA), the active ingredient of the traditional Chinese medicine artemisinin, induced ferroptosis in glial cells. However, DHA itself induced ERS in glioma cells by activating PERK-ATF4-HSPA5, increasing the expression and activity of GPX4, thereby neutralizing DHA-induced lipid peroxidation and protecting glioma cells from ferroptosis. Thus, inhibiting the negative feedback pathway may be a therapeutic strategy to enhance the antitumor activity of DHA. In the study of Tagitinin C-induced ferroptosis in colorectal cancer cells, it was found that the mechanism of action of Tagitinin C might be related to the activation of the PERK-Nrf2-HO-1 signaling pathway induced by ER stress, leading to ferroptosis (16). Additionally, ATF4 can promote ferroptosis in tumor cells by increasing tumor microvessel generation and shaping vascular architecture in an SLC7A11-dependent manner (84). Subsequent studies have demonstrated that inhibition of PERK or ATF4 promotes ferroptosis in tumor cells by downregulating SLC7A11 expression, reducing GSH production, and thereby promoting ferroptosis (79, 85). For the cardiotoxic effects of sorafenib (SOR), the upregulation of ATF4 expression attenuated cardiomyocyte ferroptosis (15).
Increased ATF6α expression induced by androgen deprivation in prostate cancer renders prostate cancer (Pca) cells resistant to ferroptosis. This resistance is related to the ATF6α-PLA2G4A-mediated release of arachidonic acid (AA) and the resulting production of prostaglandin E2 (PGE2). PLA2G4A, a member of the cytoplasmic phospholipase family, catalyzes the hydrolysis of membrane phospholipids to release AA, which produces PGE2. Together with N-acetylcysteine, this process prevents heme-induced ferroptosis (86, 87). Studies on the progression of renal tubular epithelial mesenchymal transition (EMT) in diabetic nephropathy found that XBP1 overexpression increased HRD1 expression and inhibited NRF2 expression, enhancing cellular susceptibility to ferroptosis. This indicates that ERS triggers ferroptosis-associated EMT progression via the XBP1-HRD1-NRF2 pathway (14). Additionally, IRE1α-XBP1 induces the transcription of G protein alpha subunit 12 (ALOX12), promoting lipid peroxidation and mediating the onset of ferroptosis in hepatocytes (88).Various anti-solid tumor drugs can increase intracellular ROS and Ca2+, both of which can induce ERS. This is achieved by initiating Ca2+ channels on the tumor cell membrane, leading to the inward flow of extracellular Ca2+. When the concentration of Ca2+ in the cytoplasm reaches a certain level, the calcium storage pool in the endoplasmic reticulum also releases Ca2+, inducing ERS (89, 90). The accumulated ROS in cells interacts with Ca2+, increasing the permeability of the endoplasmic reticulum membrane by promoting redox modification of Ca2+ channels, leading to the release of Ca2+ reserves from the endoplasmic reticulum into the cytoplasm and inducing Ca2+ overload (91). Ca2+ overload regulates NADPH oxidase (NOX) and the electron transport chain (ETC) in mitochondria to promote ROS production (92). TRPM7, a transient membrane potential cationic protein channel with high permeability to Ca2+, is significantly expressed in RA-FLS cells. Inhibiting TRPM7 increases CHOP and calpain expression and increases apoptosis in RA-FLS cells in vitro (93). Notably, inhibition of TRPM7 also reduced articular cartilage destruction and suppressed chondrocyte ferroptosis in adjuvant arthritis rats (AAR). This is achieved by reducing intracellular Ca2+ overload through inhibition of TRPM7, which in turn reduces calcium-activated protein kinase Cα (PKCα)/NOX4 pathway-mediated ROS production, thus attenuating cellular ferroptosis and ERS (94, 95).
These results suggest that there are many interactions between ferroptosis and ERS. However, to determine whether these relationships also exist in RA, further more precise studies are needed to confirm their regulation and interactions in RA.
This paper systematically outlines the current research on the mechanisms of ferroptosis in RA. As an iron-dependent form of cell death, ferroptosis involves three major mechanistic pathways, all contributing to RA pathogenesis. Notably, synovial fibroblasts, osteoblasts, osteoclasts, and immune cells exhibit varying sensitivities to ferroptosis. Therefore, exploring the underlying mechanisms in detail is crucial to understanding its role in RA. Additionally, the paper reviews the role of ERS in RA, highlighting that immune system overactivation, excessive autoantibody production, and apoptosis resistance in RA-FLS are linked to ERS-mediated UPR responses.
Moreover, with further research on the relationship between ferroptosis and ERS, studies in hepatocellular carcinoma, rectal carcinoma, glioma, and other tumors have identified a shared pathway between these processes. Ferroptosis can induce ERS and promote apoptosis through multiple pathways, while ERS can also increase ferroptosis sensitivity by interfering with lipid oxidation and antioxidants. However, further research is needed to determine whether the biological relationship between ferroptosis and ERS varies among different cell types and to reveal the mechanisms of death regulation for various cells. Additionally, it remains to be confirmed if the crosstalk between ferroptosis and ERS in RA aligns with research findings in other diseases.
In conclusion, ferroptosis is unlikely to function through a single pathway within the complex network of biological relationships. Consequently, based on the existing crosstalk between ferroptosis and ERS, it may become a new direction for future research, and may also become a new therapeutic target for RA.
QA: Writing – original draft. HH: Writing – review & editing. YH: Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article.This study was supported by the Guizhou University of Chinese Medicine National and provincial science and Technology Innovation talent team cultivation project (GUI TCM TD [2022]004); Science and Technology Fund project of Guizhou Provincial Health Commission(No.gzwkj2023-327).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Rivera J, Colbert RA. Healing the Syk through kinase inhibitors. N Engl J Med. (2010) 363:1362–4. doi: 10.1056/NEJMe1006527
2. Komatsu N, Takayanagi H. Mechanisms of joint destruction in rheumatoid arthritis - immune cell-fibroblast-bone interactions. Nat Rev Rheumatol. (2022) 18:415–29. doi: 10.1038/s41584-022-00793-5
3. McInnes IB, Schett G. Pathogenetic insights from the treatment of rheumatoid arthritis. Lancet. (2017) 389:2328–37. doi: 10.1016/S0140-6736(17)31472-1
4. Magtanong L, Ko PJ, To M, Cao JY, Forcina GC, Tarangelo A, et al. Exogenous monounsaturated fatty acids promote a ferroptosis-resistant cell state. Cell Chem Biol. (2019) 26:420–32.e9. doi: 10.1016/j.chembiol.2018.11.016
5. Tang D, Chen X, Kang R, Kroemer G. Ferroptosis: molecular mechanisms and health implications. Cell Res. (2021) 31:107–25. doi: 10.1038/s41422-020-00441-1
6. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. (2012) 149:1060–72. doi: 10.1016/j.cell.2012.03.042
7. Suominen P, Möttönen T, Rajamäki A, Irjala K. Single values of serum transferrin receptor and transferrin receptor ferritin index can be used to detect true and functional iron deficiency in rheumatoid arthritis patients with anemia. Arthritis Rheum. (2000) 43:1016–20. doi: 10.1002/1529-0131(200005)43:5<1016::AID-ANR9>3.0.CO;2-3
8. Stefanova KI, Delcheva GT, Maneva AI, Batalov AZ, Geneva-Popova MG, Karalilova RV, et al. Pathobiochemical mechanisms relating iron homeostasis to parameters of inflammatory activity and autoimmune disorders in rheumatoid arthritis. Folia Med (Plovdiv). (2016) 58:257–63. doi: 10.1515/folmed-2016-0040
9. Xie Z, Hou H, Luo D, An R, Zhao Y, Qiu C. ROS-dependent lipid peroxidation and reliant antioxidant ferroptosis-suppressor-protein 1 in rheumatoid arthritis: a covert clue for potential therapy. Inflammation. (2021) 44:35–47. doi: 10.1007/s10753-020-01338-2
10. Tabas I, Ron D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat Cell Biol. (2011) 13:184–90. doi: 10.1038/ncb0311-184
11. Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. (2011) 334:1081–6. doi: 10.1126/science.1209038
12. Hetz C, Zhang K, Kaufman RJ. Mechanisms, regulation and functions of the unfolded protein response. Nat Rev Mol Cell Biol. (2020) 21:421–38. doi: 10.1038/s41580-020-0250-z
13. Park YJ, Yoo SA, Kim WU. Role of endoplasmic reticulum stress in rheumatoid arthritis pathogenesis. J Korean Med Sci. (2014) 29:2–11. doi: 10.3346/jkms.2014.29.1.2
14. Liu Z, Nan P, Gong Y, Tian L, Zheng Y, Wu Z. Endoplasmic reticulum stress-triggered ferroptosis via the XBP1-Hrd1-Nrf2 pathway induces EMT progression in diabetic nephropathy. BioMed Pharmacother. (2023) 164:114897. doi: 10.1016/j.biopha.2023.114897
15. Jiang H, Wang C, Zhang A, Li Y, Li J, Li Z, et al. ATF4 protects against sorafenib-induced cardiotoxicity by suppressing ferroptosis. BioMed Pharmacother. (2022) 153:113280. doi: 10.1016/j.biopha.2022.113280
16. Wei R, Zhao Y, Wang J, Yang X, Li S, Wang Y, et al. Tagitinin C induces ferroptosis through PERK-Nrf2-HO-1 signaling pathway in colorectal cancer cells. Int J Biol Sci. (2021) 17:2703–17. doi: 10.7150/ijbs.59404
17. Yang J, Xu H, Wu W, Huang H, Zhang C, Tang W, et al. Ferroptosis signaling promotes the release of misfolded proteins via exosomes to rescue ER stress in hepatocellular carcinoma. Free Radic Biol Med. (2023) 202:110–20. doi: 10.1016/j.freeradbiomed.2023.03.027
18. Zhao T, Yang Q, Xi Y, Xie Z, Shen J, Li Z, et al. Ferroptosis in rheumatoid arthritis: A potential therapeutic strategy. Front Immunol. (2022) 13:779585. doi: 10.3389/fimmu.2022.779585
19. Muckenthaler MU, Rivella S, Hentze MW, Galy B. A red carpet for iron metabolism. Cell. (2017) 168:344–61. doi: 10.1016/j.cell.2016.12.034
20. Galy B, Conrad M, Muckenthaler M. Mechanisms controlling cellular and systemic iron homeostasis. Nat Rev Mol Cell Biol. (2024) 25:133–55. doi: 10.1038/s41580-023-00648-1
21. Zhao H, Tang C, Wang M, Zhao H, Zhu Y. Ferroptosis as an emerging target in rheumatoid arthritis. Front Immunol. (2023) 14:1260839. doi: 10.3389/fimmu.2023.1260839
22. Kajarabille N, Latunde-Dada GO. Programmed cell-death by ferroptosis: antioxidants as mitigators. Int J Mol Sci. (2019) 20(19):4968. doi: 10.3390/ijms20194968
23. Stefanova KI, Delcheva GT, Maneva AI, Batalov AZ, Geneva-Popova MG, Karalilova RV, et al. Pathobiochemical mechanisms relating iron homeostasis with parameters of inflammatory activity and autoimmune disorders in rheumatoid arthritis. Folia Med (Plovdiv). (2018) 60:124–32. doi: 10.1515/folmed-2017-0068
24. Wu J, Feng Z, Chen L, Li Y, Bian H, Geng J, et al. TNF antagonist sensitizes synovial fibroblasts to ferroptotic cell death in collagen-induced arthritis mouse models. Nat Commun. (2022) 13:676. doi: 10.1038/s41467-021-27948-4
25. Xiao W, Beibei F, Guangsi S, Yu J, Wen Z, Xi H, et al. Iron overload increases osteoclastogenesis and aggravates the effects of ovariectomy on bone mass. J Endocrinol. (2015) 226:121–34. doi: 10.1530/JOE-14-0657
26. Cen WJ, Feng Y, Li SS, Huang LW, Zhang T, Zhang W, et al. Iron overload induces G1 phase arrest and autophagy in murine preosteoblast cells. J Cell Physiol. (2018) 233:6779–89. doi: 10.1002/jcp.26405
27. Wang X, Chen B, Sun J, Jiang Y, Zhang H, Zhang P, et al. Iron-induced oxidative stress stimulates osteoclast differentiation via NF-κB signaling pathway in mouse model. Metabolism. (2018) 83:167–76. doi: 10.1016/j.metabol.2018.01.005
28. Jing X, Du T, Chen K, Guo J, Xiang W, Yao X, et al. Icariin protects against iron overload-induced bone loss via suppressing oxidative stress. J Cell Physiol. (2019) 234:10123–37. doi: 10.1002/jcp.27678
29. Walker EM Jr., Walker SM. Effects of iron overload on the immune system. Ann Clin Lab Sci. (2000) 30:354–65.
30. Telfer JF, Brock JH. Proinflammatory cytokines increase iron uptake into human monocytes and synovial fibroblasts from patients with rheumatoid arthritis. Med Sci Monit. (2004) 10:Br91–5.
31. Hakobyan N, Kazarian T, Jabbar AA, Jabbar KJ, Valentino LA. Pathobiology of hemophilic synovitis I: overexpression of mdm2 oncogene. Blood. (2004) 104:2060–4. doi: 10.1182/blood-2003-12-4231
32. Salvarani C, Baricchi R, Lasagni D, Boiardi L, Piccinini R, Brunati C, et al. Effects of desferrioxamine therapy on chronic disease anemia associated with rheumatoid arthritis. Rheumatol Int. (1996) 16:45–8. doi: 10.1007/BF01816434
33. Wang P, Zhang F, He Q, Wang J, Shiu HT, Shu Y, et al. Flavonoid compound icariin activates hypoxia inducible factor-1α in chondrocytes and promotes articular cartilage repair. PloS One. (2016) 11:e0148372. doi: 10.1371/journal.pone.0148372
34. Gaschler MM, Stockwell BR. Lipid peroxidation in cell death. Biochem Biophys Res Commun. (2017) 482:419–25. doi: 10.1016/j.bbrc.2016.10.086
35. Forcina GC, Dixon SJ. GPX4 at the crossroads of lipid homeostasis and ferroptosis. Proteomics. (2019) 19:e1800311. doi: 10.1002/pmic.201800311
36. Rochette L, Dogon G, Rigal E, Zeller M, Cottin Y, Vergely C. Lipid peroxidation and iron metabolism: two corner stones in the homeostasis control of ferroptosis. Int J Mol Sci. (2022) 24(1):449. doi: 10.3390/ijms24010449
37. Roberts D, Davies J. Exacerbation of rheumatoid synovitis by iron-dextran infusion. Lancet. (1987) 1:391. doi: 10.1016/S0140-6736(87)91771-5
38. Blake DR, Lunec J, Ahern M, Ring EF, Bradfield J, Gutteridge JM. Effect of intravenous iron dextran on rheumatoid synovitis. Ann Rheum Dis. (1985) 44:183–8. doi: 10.1136/ard.44.3.183
39. Huang Y, Cai GQ, Peng JP, Shen C. Glucocorticoids induce apoptosis and matrix metalloproteinase-13 expression in chondrocytes through the NOX4/ROS/p38 MAPK pathway. J Steroid Biochem Mol Biol. (2018) 181:52–62. doi: 10.1016/j.jsbmb.2018.03.001
40. Lee HR, Yoo SJ, Kim J, Kang SW. LKB1 regulates inflammation of fibroblast-like synoviocytes from patients with rheumatoid arthritis via AMPK-dependent SLC7A11-NOX4-ROS signaling. Cells. (2023) 12(9):1263. doi: 10.3390/cells12091263
41. Zhu J, Tang Y, Wu Q, Ji YC, Feng ZF, Kang FW. HIF-1α facilitates osteocyte-mediated osteoclastogenesis by activating JAK2/STAT3 pathway in vitro. J Cell Physiol. (2019) 234:21182–92. doi: 10.1002/jcp.28721
42. Zheng S, Zhong ZM, Qin S, Chen GX, Wu Q, Zeng JH, et al. Advanced oxidation protein products induce inflammatory response in fibroblast-like synoviocytes through NADPH oxidase -dependent activation of NF-κB. Cell Physiol Biochem. (2013) 32:972–85. doi: 10.1159/000354500
43. Lood C, Blanco LP, Purmalek MM, Carmona-Rivera C, De Ravin SS, Smith CK, et al. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat Med. (2016) 22:146–53. doi: 10.1038/nm.4027
44. Lewerenz J, Hewett SJ, Huang Y, Lambros M, Gout PW, Kalivas PW, et al. The cystine/glutamate antiporter system x(c)(-) in health and disease: from molecular mechanisms to novel therapeutic opportunities. Antioxid Redox Signal. (2013) 18:522–55. doi: 10.1089/ars.2011.4391
45. Bridges RJ, Natale NR, Patel SA. System xc- cystine/glutamate antiporter: an update on molecular pharmacology and roles within the CNS. Br J Pharmacol. (2012) 165:20–34. doi: 10.1111/j.1476-5381.2011.01480.x
46. Wang L, Liu Y, Du T, Yang H, Lei L, Guo M, et al. ATF3 promotes erastin-induced ferroptosis by suppressing system Xc(). Cell Death Differ. (2020) 27:662–75. doi: 10.1038/s41418-019-0380-z
47. Liu M, Kong XY, Yao Y, Wang XA, Yang W, Wu H, et al. The critical role and molecular mechanisms of ferroptosis in antioxidant systems: a narrative review. Ann Transl Med. (2022) 10:368. doi: 10.21037/atm
48. Ling H, Li M, Yang C, Sun S, Zhang W, Zhao L, et al. Glycine increased ferroptosis via SAM-mediated GPX4 promoter methylation in rheumatoid arthritis. Rheumatol (Oxford). (2022) 61:4521–34. doi: 10.1093/rheumatology/keac069
49. Criscitiello MF, Kraev I, Lange S. Post-translational protein deimination signatures in serum and serum-extracellular vesicles of bos taurus reveal immune, anti-pathogenic, anti-viral, metabolic and cancer-related pathways for deimination. Int J Mol Sci. (2020) 21(8):2861. doi: 10.3390/ijms21082861
50. Levring TB, Hansen AK, Nielsen BL, Kongsbak M, von Essen MR, Woetmann A, et al. Activated human CD4+ T cells express transporters for both cysteine and cystine. Sci Rep. (2012) 2:266. doi: 10.1038/srep00266
51. Drijvers JM, Gillis JE, Muijlwijk T, Nguyen TH, Gaudiano EF, Harris IS, et al. Pharmacologic screening identifies metabolic vulnerabilities of CD8(+) T cells. Cancer Immunol Res. (2021) 9:184–99. doi: 10.1158/2326-6066.CIR-20-0384
52. Xiang J, Chen H, Lin Z, Chen J, Luo L. Identification and experimental validation of ferroptosis-related gene SLC2A3 is involved in rheumatoid arthritis. Eur J Pharmacol. (2023) 943:175568. doi: 10.1016/j.ejphar.2023.175568
53. Hu J, Zhang R, Chang Q, Ji M, Zhang H, Geng R, et al. p53: A regulator of ferroptosis induced by galectin-1 derived peptide 3 in MH7A cells. Front Genet. (2022) 13:920273. doi: 10.3389/fgene.2022.920273
54. Zhao C, Yu Y, Yin G, Xu C, Wang J, Wang L, et al. Sulfasalazine promotes ferroptosis through AKT-ERK1/2 and P53-SLC7A11 in rheumatoid arthritis. Inflammopharmacology. (2024) 32(2):1277–94. doi: 10.1007/s10787-024-01439-6
55. Grootjans J, Kaser A, Kaufman RJ, Blumberg RS. The unfolded protein response in immunity and inflammation. Nat Rev Immunol. (2016) 16:469–84. doi: 10.1038/nri.2016.62
56. Bläss S, Union A, Raymackers J, Schumann F, Ungethüm U, Müller-Steinbach S, et al. The stress protein BiP is overexpressed and is a major B and T cell target in rheumatoid arthritis. Arthritis Rheum. (2001) 44:761–71. doi: 10.1002/(ISSN)1529-0131
57. Yoo SA, You S, Yoon HJ, Kim DH, Kim HS, Lee K, et al. A novel pathogenic role of the ER chaperone GRP78/BiP in rheumatoid arthritis. J Exp Med. (2012) 209:871–86. doi: 10.1084/jem.20111783
58. Zhang Y, Ge L, Song G, Zhang R, Li S, Shi H, et al. Azithromycin alleviates the severity of rheumatoid arthritis by targeting the unfolded protein response component of glucose-regulated protein 78 (GRP78). Br J Pharmacol. (2022) 179:1201–19. doi: 10.1111/bph.15714
59. Bezu L, Sauvat A, Humeau J, Gomes-da-Silva LC, Iribarren K, Forveille S, et al. eIF2α phosphorylation is pathognomonic for immunogenic cell death. Cell Death Differ. (2018) 25:1375–93. doi: 10.1038/s41418-017-0044-9
60. Sun J, Chen W, Li S, Yang S, Zhang Y, Hu X, et al. Nox4 promotes RANKL-induced autophagy and osteoclastogenesis via activating ROS/PERK/eIF-2α/ATF4 pathway. Front Pharmacol. (2021) 12:751845. doi: 10.3389/fphar.2021.751845
61. Hu H, Wang C, Jin Y, Meng Q, Liu Q, Liu Z, et al. Catalpol inhibits homocysteine-induced oxidation and inflammation via inhibiting nox4/NF-κB and GRP78/PERK pathways in human aorta endothelial cells. Inflammation. (2019) 42:64–80. doi: 10.1007/s10753-018-0873-9
62. Boyce M, Bryant KF, Jousse C, Long K, Harding HP, Scheuner D, et al. A selective inhibitor of eIF2alpha dephosphorylation protects cells from ER stress. Science. (2005) 307:935–9. doi: 10.1126/science.1101902
63. Hamamura K, Nishimura A, Chen A, Takigawa S, Sudo A, Yokota H. Salubrinal acts as a Dusp2 inhibitor and suppresses inflammation in anti-collagen antibody-induced arthritis. Cell Signal. (2015) 27:828–35. doi: 10.1016/j.cellsig.2015.01.010
64. Lee EG, Sung MS, Yoo HG, Chae HJ, Kim HR, Yoo WH. Increased RANKL-mediated osteoclastogenesis by interleukin-1β and endoplasmic reticulum stress. Joint Bone Spine. (2014) 81:520–6. doi: 10.1016/j.jbspin.2014.04.012
65. Qi W, Jin L, Wu C, Liao H, Zhang M, Zhu Z, et al. Treatment with FAP-targeted zinc ferrite nanoparticles for rheumatoid arthritis by inducing endoplasmic reticulum stress and mitochondrial damage. Mater Today Bio. (2023) 21:100702. doi: 10.1016/j.mtbio.2023.100702
66. Jeong M, Shin JI, Cho J, Jeon YJ, Kim JH, Youn J, et al. DHA induces cell death through the production of ROS and the upregulation of CHOP in fibroblast-like synovial cells from human rheumatoid arthritis patients. Int J Mol Sci. (2023) 24(2):1734. doi: 10.3390/ijms24021734
67. Wang M, Kaufman RJ. Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature. (2016) 529:326–35. doi: 10.1038/nature17041
68. Lima NCR, Melo TQ, Sakugawa AYS, Melo KP, Ferrari MFR. Restoration of rab1 levels prevents endoplasmic reticulum stress in hippocampal cells during protein aggregation triggered by rotenone. Neuroscience. (2019) 419:5–13. doi: 10.1016/j.neuroscience.2019.08.050
69. Connor AM, Mahomed N, Gandhi R, Keystone EC, Berger SA. TNFα modulates protein degradation pathways in rheumatoid arthritis synovial fibroblasts. Arthritis Res Ther. (2012) 14:R62. doi: 10.1186/ar3778
70. Lee WS, Jeong JH, Lee EG, Choi Y, Kim JH, Kim HR, et al. Tacrolimus regulates endoplasmic reticulum stress-mediated osteoclastogenesis and inflammation: In vitro and collagen-induced arthritis mouse model. Cell Biol Int. (2018) 42:393–402. doi: 10.1002/cbin.10861
71. Huang R, Hui Z, Wei S, Li D, Li W, Daping W, et al. IRE1 signaling regulates chondrocyte apoptosis and death fate in the osteoarthritis. J Cell Physiol. (2022) 237:118–27. doi: 10.1002/jcp.30537
72. Savic S, Ouboussad L, Dickie LJ, Geiler J, Wong C, Doody GM, et al. TLR dependent XBP-1 activation induces an autocrine loop in rheumatoid arthritis synoviocytes. J Autoimmun. (2014) 50:59–66. doi: 10.1016/j.jaut.2013.11.002
73. Zhong J, Ma T, Huang C, Liu H, Chen Z, Cao L, et al. Flavonoids from Litsea coreana decreases TNF-α secretion from peritoneal macrophages in adjuvant-induced arthritis rats via UPR pathway. Am J Chin Med. (2014) 42:905–19. doi: 10.1142/S0192415X14500578
74. Ahmadiany M, Alavi-Samani M, Hashemi Z, Moosavi MA, Rahmati M. The increased RNase activity of IRE1α in PBMCs from patients with rheumatoid arthritis. Adv Pharm Bull. (2019) 9:505–9. doi: 10.15171/apb.2019.060
75. Tang CH, Chang S, Paton AW, Paton JC, Gabrilovich DI, Ploegh HL, et al. Phosphorylation of IRE1 at S729 regulates RIDD in B cells and antibody production after immunization. J Cell Biol. (2018) 217:1739–55. doi: 10.1083/jcb.201709137
76. Zhang K, Wong HN, Song B, Miller CN, Scheuner D, Kaufman RJ. The unfolded protein response sensor IRE1alpha is required at 2 distinct steps in B cell lymphopoiesis. J Clin Invest. (2005) 115:268–81. doi: 10.1172/JCI200521848
77. Dixon SJ, Patel DN, Welsch M, Skouta R, Lee ED, Hayano M, et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife. (2014) 3:e02523. doi: 10.7554/eLife.02523
78. Chen D, Fan Z, Rauh M, Buchfelder M, Eyupoglu IY, Savaskan N. ATF4 promotes angiogenesis and neuronal cell death and confers ferroptosis in a xCT-dependent manner. Oncogene. (2017) 36:5593–608. doi: 10.1038/onc.2017.146
79. Gao R, Kalathur RKR, Coto-Llerena M, Ercan C, Buechel D, Shuang S, et al. YAP/TAZ and ATF4 drive resistance to Sorafenib in hepatocellular carcinoma by preventing ferroptosis. EMBO Mol Med. (2021) 13:e14351. doi: 10.15252/emmm.202114351
80. Hong SH, Lee DH, Lee YS, Jo MJ, Jeong YA, Kwon WT, et al. Molecular crosstalk between ferroptosis and apoptosis: emerging role of ER stress-induced p53-independent PUMA expression. Oncotarget. (2017) 8:115164–78. doi: 10.18632/oncotarget.v8i70
81. Ghosh AP, Klocke BJ, Ballestas ME, Roth KA. CHOP potentially co-operates with FOXO3a in neuronal cells to regulate PUMA and BIM expression in response to ER stress. PloS One. (2012) 7:e39586. doi: 10.1371/journal.pone.0039586
82. Takatori H, Kawashima H, Suzuki K, Nakajima H. Role of p53 in systemic autoimmune diseases. Crit Rev Immunol. (2014) 34:509–16. doi: 10.1615/CritRevImmunol.v34.i6
83. Chen Y, Mi Y, Zhang X, Ma Q, Song Y, Zhang L, et al. Dihydroartemisinin-induced unfolded protein response feedback attenuates ferroptosis via PERK/ATF4/HSPA5 pathway in glioma cells. J Exp Clin Cancer Res. (2019) 38:402. doi: 10.1186/s13046-019-1413-7
84. He F, Zhang P, Liu J, Wang R, Kaufman RJ, Yaden BC, et al. ATF4 suppresses hepatocarcinogenesis by inducing SLC7A11 (xCT) to block stress-related ferroptosis. J Hepatol. (2023) 79:362–77. doi: 10.1016/j.jhep.2023.03.016
85. Saini KK, Chaturvedi P, Sinha A, Singh MP, Khan MA, Verma A, et al. Loss of PERK function promotes ferroptosis by downregulating SLC7A11 (System Xc-) in colorectal cancer. Redox Biol. (2023) 65:102833. doi: 10.1016/j.redox.2023.102833
86. Zhao R, Lv Y, Feng T, Zhang R, Ge L, Pan J, et al. ATF6α promotes prostate cancer progression by enhancing PLA2G4A-mediated arachidonic acid metabolism and protecting tumor cells against ferroptosis. Prostate. (2022) 82:617–29. doi: 10.1002/pros.24308
87. Zhou H, Zhang T, Chen L, Cui F, Xu C, Peng J, et al. The functional implication of ATF6α in castration-resistant prostate cancer cells. FASEB J. (2023) 37:e22758. doi: 10.1096/fj.202201347R
88. Tak J, Kim YS, Kim TH, Park GC, Hwang S, Kim SG. Gα(12) overexpression in hepatocytes by ER stress exacerbates acute liver injury via ROCK1-mediated miR-15a and ALOX12 dysregulation. Theranostics. (2022) 12:1570–88. doi: 10.7150/thno.67722
89. King AP, Wilson JJ. Endoplasmic reticulum stress: an arising target for metal-based anticancer agents. Chem Soc Rev. (2020) 49:8113–36. doi: 10.1039/D0CS00259C
90. Cao S, Tang J, Huang Y, Li G, Li Z, Cai W, et al. The road of solid tumor survival: from drug-induced endoplasmic reticulum stress to drug resistance. Front Mol Biosci. (2021) 8:620514. doi: 10.3389/fmolb.2021.620514
91. Zhang Z, Zhang L, Zhou L, Lei Y, Zhang Y, Huang C. Redox signaling and unfolded protein response coordinate cell fate decisions under ER stress. Redox Biol. (2019) 25:101047. doi: 10.1016/j.redox.2018.11.005
92. Hempel N, Trebak M. Crosstalk between calcium and reactive oxygen species signaling in cancer. Cell Calcium. (2017) 63:70–96. doi: 10.1016/j.ceca.2017.01.007
93. Li X, Wang X, Wang Y, Li X, Huang C, Li J. Inhibition of transient receptor potential melastatin 7 (TRPM7) channel induces RA FLSs apoptosis through endoplasmic reticulum (ER) stress. Clin Rheumatol. (2014) 33:1565–74. doi: 10.1007/s10067-014-2599-x
94. Zhou R, Chen Y, Li S, Wei X, Hu W, Tang S, et al. TRPM7 channel inhibition attenuates rheumatoid arthritis articular chondrocyte ferroptosis by suppression of the PKCα-NOX4 axis. Redox Biol. (2022) 55:102411. doi: 10.1016/j.redox.2022.102411
Keywords: ferroptosis, endoplasmic reticulum stress, rheumatoid arthritis, lipid peroxidation, reactive oxygen species, unfolded protein response
Citation: Ao Q, Hu H and Huang Y (2024) Ferroptosis and endoplasmic reticulum stress in rheumatoid arthritis. Front. Immunol. 15:1438803. doi: 10.3389/fimmu.2024.1438803
Received: 26 May 2024; Accepted: 01 July 2024;
Published: 15 July 2024.
Edited by:
Jianan Zhao, Shanghai Guanghua Rheumatology Hospital, ChinaReviewed by:
Zheng Liu, Xiangtan Central Hospital, ChinaCopyright © 2024 Ao, Hu and Huang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ying Huang, MzgzNzc3MzY2MUBxcS5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.