 Hui Pi1,2†
Hui Pi1,2† Guangliang Wang1†
Guangliang Wang1† Yu Wang1
Yu Wang1 Ming Zhang1Qin He2
Ming Zhang1Qin He2 Xilong Zheng3Kai Yin4*
Xilong Zheng3Kai Yin4* Guojun Zhao1*Ting Jiang1*
Guojun Zhao1*Ting Jiang1*- 1Affiliated Qingyuan Hospital, Guangzhou Medical University (Qingyuan People’s Hospital), Qingyuan, Guangdong, China
- 2Department of Microbiology and Immunology, Dali University, Dali, Yunnan, China
- 3Departments of Biochemistry and Molecular Biology and Physiology and Pharmacology, Cumming School of Medicine, University of Calgary, Calgary, AB, Canada
- 4Department of General Practice, The Fifth Affiliated Hospital of Southern Medical University, Guangzhou, Guangdong, China
Atherosclerosis serves as the primary catalyst for numerous cardiovascular diseases. Growing evidence suggests that the immune response is involved in every stage of atherosclerotic plaque evolution. Rapid, but not specific, innate immune arms, including neutrophils, monocytes/macrophages, dendritic cells (DCs) and other innate immune cells, as well as pattern-recognition receptors and various inflammatory mediators, contribute to atherogenesis. The specific adaptive immune response, governed by T cells and B cells, antibodies, and immunomodulatory cytokines potently regulates disease activity and progression. In the inflammatory microenvironment, the heterogeneity of leukocyte subpopulations plays a very important regulatory role in plaque evolution. With advances in experimental techniques, the fine mechanisms of immune system involvement in atherosclerotic plaque evolution are becoming known. In this review, we examine the critical immune responses involved in atherosclerotic plaque evolution, in particular, looking at atherosclerosis from the perspective of evolutionary immunobiology. A comprehensive understanding of the interplay between plaque evolution and plaque immunity provides clues for strategically combating atherosclerosis.
1 Introduction
Atherosclerotic cardiovascular disease (ACD) is a prominent global cause of mortality (1). Atherosclerosis is a slow process, characterized by multifocal structural changes in the vascular wall of large and medium- sized arteries, which result in the development of atherosclerotic plaques (2). Atherosclerotic plaques are the pathophysiological basis of almost all arterial vascular diseases. Advanced plaques may rupture, triggering thrombosis that blocks arteries and disrupts blood flow, leading to an array of life-threatening clinical outcomes called major adverse cardiovascular events (MACEs) (3). Past epidemiologic studies have revealed many risk factors for atherosclerosis, among which traditional risk factors include dyslipidemia, hypertension, hyperhomocysteinemia, hyperfibrinogenemia, diabetes mellitus, smoking, obesity, and genetic predisposition. In recent years, non-traditional drivers such as sleep disorders, lack of exercise, air pollution, environmental stress, as well as inflammation and clonal hematopoiesis associated with the immune system have also received attention (3, 4).
The evolution of atherosclerotic plaques can be broadly categorized into three stages: initiation, progression and complications. It has been shown that fatty streaks are the initial marker of atherosclerosis, which develops in four steps: low-density lipoprotein (LDL) cholesterol uptake, endothelial cell (EC) activation, leukocyte activation and foam cell formation (5). During the development of fibrous plaques, atherosclerotic plaques experience a shift from fatty streaks to intimal growth, and this step is marked by the formation of a lipid-rich necrotic core covered by a fibrous cap. The fibrous cap is composed of vascular smooth muscle cells (VSMCs) which migrate to the side of the arterial lumen and VSMC-derived extracellular matrix (ECM). The atherosclerotic plaques can rupture at the point where the fibrous cap is thinnest exposing the material inside to blood tissue triggering thrombosis. If the lumen is blocked, the narrowing of the diseased artery can also lead to other complications such as heart and brain infarction (6, 7).
Among the many factors that influence the evolution of atherosclerotic plaques, the immune system plays a significant role. During the formation and development of atherosclerotic plaques, the local microenvironment undergoes a series of complex changes, accompanied by the infiltration of multiple immune cells. Evidence suggests that circulating monocytes and resident vascular macrophages are the earliest immune cells recruited into early atherosclerotic plaques (8). Following this, various immune cells such as neutrophils, natural killer (NK) cells, DCs, T cells and B cells gradually infiltrate the plaque and perform their regulatory functions. Subpopulations of leukocytes in the arterial wall are heterogeneous they play a pro-inflammatory or regulatory role in atherosclerotic plaque formation. Most patients with atherosclerosis are immunocompetent individuals (9). The key takeaway about the impact of immune function is that it is complex, and immunomodulation can positively or negatively affect the evolution of atherosclerotic plaques.
The molecular signals that regulate leukocyte recruitment to atherosclerotic lesion sites are complex, and chemokines and their receptors play a key role and have received much attention in the study of atherosclerosis. Chemokines are a class of small, secreted cytokines with chemotactic properties. Depending on the location of their cysteine residues, they can be divided into four subclasses: C, CC, CXC, and CX3C. They act mainly by binding to specific G-protein-coupled chemokine receptors (10). In atherosclerotic lesions, chemokines and many of their receptors are expressed in endothelial cells, leukocytes, and smooth muscle cells, with highest expression especially in regions near the necrotic core. They are widely involved in all stages of atherosclerosis by promoting immune cell adhesion, migration, infiltration, differentiation, and homing to the lesion site (11).
With the advent and advancement of various experimental techniques, we have gained an understanding of the mechanisms involving the immune system in plaque evolution. Here, we view atherosclerotic plaque lesions from the perspective of evolutionary immunobiology, specifically reviewing the immunologic aspects of plaque evolution, including the immune responses involved, the effects of immune cell heterogeneity, and plaque dynamics in the inflammatory microenvironment. The goal is to provide insight into the search for immunotherapy for atherosclerosis.
2 Understanding atherosclerotic plaque evolution
In 1859, Charles Darwin coined the phrase “natural selection” to describe the process of evolution as observed by the different survival outcomes of individuals under environmental stress owing to phenotypic differences. Natural selection has provided a crucial force in evolution (12). It also happens in the evolution of atherosclerotic plaques, as the same mechanism applies to immune cells infiltrating the plaque. The host immune system is a major source of selection pressure during the evolution of atherosclerotic plaques. The immune system has different cell types, states, and positions. The complex networks, interactions and reactions of immune cells give rise to a cellular ecosystem consisting of numerous cell types accompanied by the genetic diversity of antigen receptors (13). Under selective pressure from the immune system, immune cells exhibit differential phenotypes that distinctively influence the evolution of plaques.
Many believe that atherosclerosis is an inevitable progressive process that develops over time, but current evidence supports a more dynamic and discontinuous evolution of the atherosclerotic plaques (14, 15). Fatty streaks are the earliest form of atherosclerotic plaque lesions. Regardless of the prevalence of coronary artery disease, fatty streaks are observed in the aorta of children in all countries. These early pathologies may progress to more advanced lesions or recede. The fatty streaks progress to fibrous plaques, occurring late in the second decade of life and early in the third decade. Atherosclerotic plaque regression is unlikely to occur during this phase. The progression of fibrous plaques is associated with various complications: calcification, internal bleeding, ulceration, and the release of embolic fragments, as well as the formation of blot clots. Thrombosis causes acute events like myocardial infarction and ischemic stroke (4, 6, 7). Generally, plaques take decades to form and do not develop consistently or steadily. Moreover, once fibrous plaques develop, they are more prone to a variety of complications, which can be life-threatening in severe cases.
3 Immune responses in atherosclerotic plaques
The immune system is categorized into innate and adaptive responses. As the first line of defense against invading pathogens, innate immunity is characterized by its ability to produce rapid and nonspecific responses. The main types of cells in the innate immune system involve neutrophils, monocytes/macrophages and DCs (16). Innate immune cells are capable of recognizing pathogen-associated and damage-associated molecular patterns by means of pattern recognition receptors, encompassing scavenger receptors (SRs) and toll-like receptors (TLRs) (17). Adaptive immunity is more specific, but slower. Adaptive immunity is mainly composed of T cells and B cells that identify the specific epitopes on pathogens as well as antigens for which they produce a variety of antigen-specific T cell receptors and immunoglobulins (18). The evolution of immune cells plays a key role in the development of atherosclerotic plaques, actively promoting plaque formation and development (Figure 1).
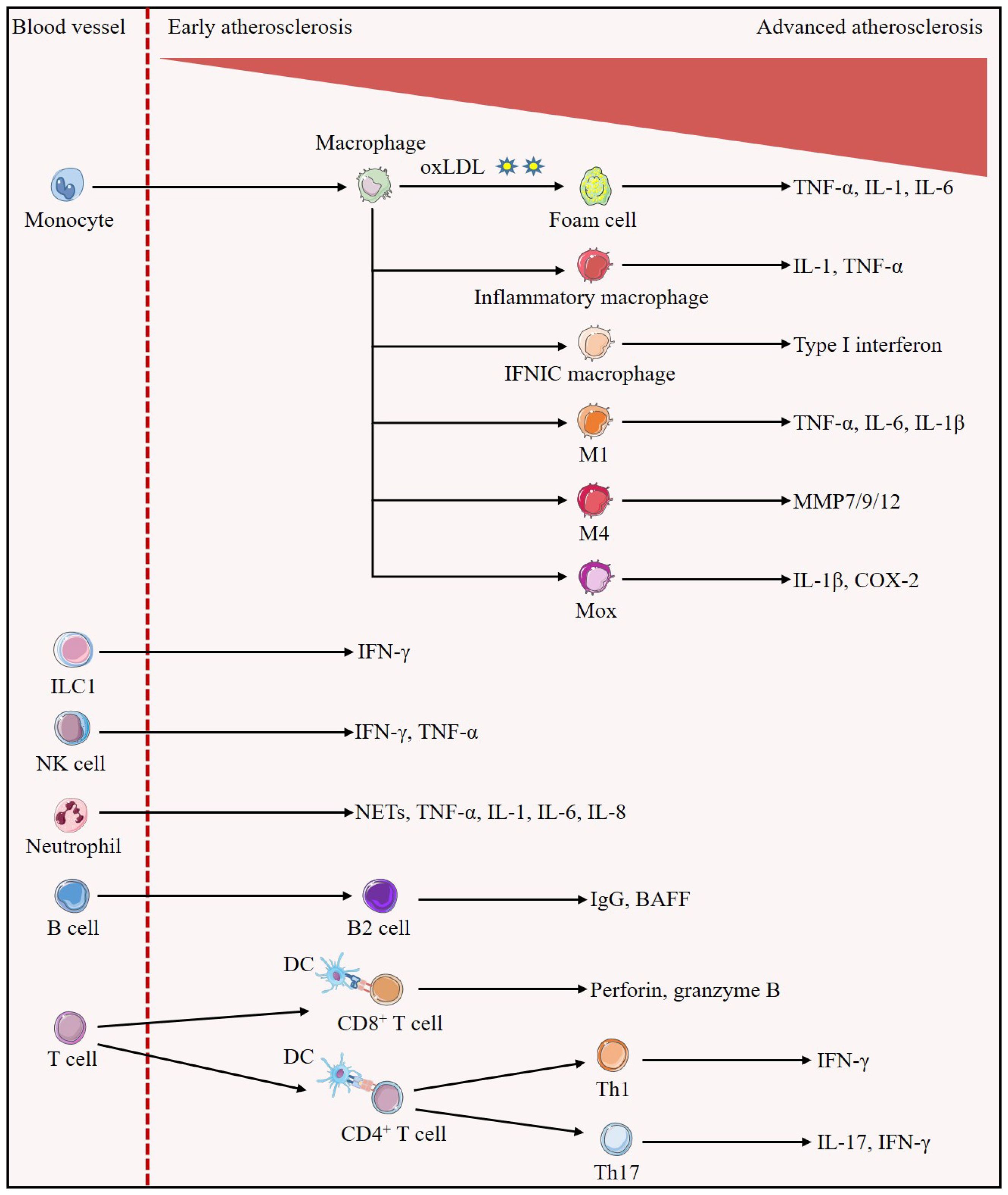
Figure 1. Evolution of immune cells in the development of atherosclerotic plaques. Early in the lesion, monocytes/macrophages predominate. Monocytes differentiate into macrophages, which can uptake oxLDL to become foam cells, and also differentiate into inflammatory macrophages and IFNICs macrophages, which secrete IL-1, TNF-α, and type I interferon to exert pro-inflammatory effects, respectively; in addition, they can also be polarized into the M1, M4, and Mox phenotypes, which secrete pro-inflammatory cytokines such as IL-1β, TNF-α, IL-6, or MMP, promoting atherosclerosis. In all stages of atherosclerosis, ILC1 is identified as the most abundant subset of ILCs and is involved in reinforcing plaque formation through the production of IFN-γ. Atherosclerotic lesions trigger migration and activation of NK cells as well as release of pro-atherogenic factors IFN-γ and TNF-α. Neutrophils secrete pro-inflammatory mediators such as NETs, TNF-α, IL-1, IL-6 and IL-8, increasing plaque inflammation and susceptibility to rupture. A large number of B cells with a structure similar to tertiary lymphoid organs are found in the adventitial layer of blood vessels, in which B2 cells secrete mediators that aggravate atherosclerosis such as IgG and BAFF. In advanced atherosclerosis, numerous T cells homing to the plaque. Activated DCs present cognate peptides on MHC class I molecules to CD8+T cells and on MHC class II molecules to CD4+T cells. Activated CD8+T cells exert atherogenic effects by secreting perforin and granzyme B and inducing cell death. CD4+T cells are polarized into different subsets, and Th1 cells are able to further stimulate foam cell formation and exacerbate atherosclerosis through the secretion of their major cytokine, IFN-γ. IL-17 secreted by Th17 cells may accompany the increase of IFN-γ to promote plaque inflammatory response and play pro-atherogenic roles.
3.1 The arterial wall
The arterial wall is composed of three main layers: the intima, the media, and the adventitia. The intima is the innermost layer in contact with blood flow and is covered by a single layer of ECs on one side of the lumen (vascular endothelium) and an inner elastic layer on the peripheral side. The media consists mainly of VSMCs surrounded by ECM. The adventitia is composed of fibroblasts and connective tissue (19). The vascular endothelium is the initial obstacle for circulating molecules, cells, or pathogens in the bloodstream (20). Disruption of intravascular homeostatic regulatory mechanisms leads to endothelial dysfunction (21), which facilitates the development and progression of atherosclerotic plaques and can be considered as an independent vascular danger factor.
In addition to endothelial dysfunction, perivascular adipose tissue (PVAT) dysfunction is also a key factor in plaque formation. PVAT, a mixture of brown and white adipose tissue that surrounds most of the vascular system, is thought to be an active component of the vascular wall that regulates vascular homeostasis and influences the pathogenesis of atherosclerosis (22). Under physiological conditions, PVAT exhibits potent anti-atherosclerotic properties through its thermogenic capacity and secretion of several bioactive molecules such as adiponectin and NO. Under the influence of pathological states, such as obesity, diabetes and hypertension, which are metabolic diseases, PVAT becomes dysfunctional and secretes high levels of pro-inflammatory factors such as leptin, resistin, inflammatory cytokines tumor necrosis factor alpha (TNF-α) and interleukin (IL) -6, and chemokines such as monocyte chemotactic protein-1 (MCP-1), which induce endothelial dysfunction, immune cell infiltration and inflammation, and ultimately promote the development of atherosclerosis (23, 24).
Atherosclerotic plaque formation begins with endothelial dysfunction followed by LDL accumulation and its modification in the intima (25). As a result of impaired endothelial barrier function and elevated blood levels of plasma lipids, LDL infiltrates the endothelium and deposits on the arterial wall via endocytosis and is retained in the intima by ECM macromolecules (26). As soon as they enter the subendothelial space, the captured LDL particles are oxidatively modified to activate ECs and VSMCs. Activated ECs induce selective monocytes to be recruited to the intima, rapidly differentiating into tissue macrophages and transforming into foam cells through accumulation of trapped and modified lipoproteins. Notably, a small proportion of foam cells are derived from VSMCs and ECs (27, 28). The accumulation of foam cells is a driving factor in plaque growth. Overall, the combination of endothelial cell injury, oxidative modification of LDL, and macrophage activation lead to increased LDL uptake, which promotes the formation and progression of atherosclerosis.
3.2 Innate immunity
Lipid accumulation stimulates the production of inflammatory mediators and cytokines by ECs. EC activation triggers the release of leukocyte adhesion molecules (AMs) and chemokines that stimulate inflammatory monocyte migration and infiltration. Infiltrated monocytes in the intima are stimulated to mature into macrophages by macrophage colony-stimulating factor (M-CSF) and granulocyte-macrophage colony-stimulating factor (GM-CSF) produced by activated ECs (29). Macrophages express a range of SRs, including CD36, CD68, lectin-type oxidized LDL receptor 1 (LOX-1), SR-A, and SR-B1, which mediate ligand internalization, degradation, and antigen presentation. In contrast, TLRs can directly induce an inflammatory response (17). Many TLR family members such as TLR1, TLR2, TLR4, and TLR5 that are detected in human plaque and atherosclerosis mouse models are mainly expressed by macrophages and ECs (30). Of these, TLR4 is noteworthy as it is highly expressed in human atherosclerotic plaques and its expression is upregulated by oxidized LDLs (oxLDLs) (31). Myeloid differentiation factor 88 (MyD88) is a critical adaptor protein involved in TLR4 signaling. Apolipoprotein E -deficient (ApoE-/-) mice lacking both TLR4 or MyD88 show reduced atherosclerosis (32). Overall, SRs and TLRs play a significant role in the evolution of atherosclerotic plaques.
Other innate immune cells such as neutrophils, Innate lymphoid cells (ILCs) and DCs can also influence the evolution of atherosclerotic plaques. Neutrophils are the major responders to tissue damage and infection, releasing pro-inflammatory mediators to neutralize dangers such as toxins (33). Neutrophils aggravate endothelial dysfunction, induce monocytes to move into atherosclerotic lesions, activate macrophages and promote foam cell formation by releasing granule proteins. Analysis of human plaque specimens suggests that neutrophil-derived proteases and reactive oxygen species (ROS) contribute to plaque destabilization (34). In addition, cholesterol crystals trigger neutrophils to release neutrophil extracellular traps (NETs), complex structures composed of nuclear chromatin and proteins sourced from the nucleus, granules, and cytoplasm. NETs cause macrophages to release cytokines that activate Th17 cells, expanding the recruitment of immune cells in atherosclerotic plaques (35, 36). Therefore, neutrophils play a facilitating role in the evolution of plaques.
ILCs are a unique type of lymphocytes that have been newly discovered in recent years. Five distinct subsets have been identified: ILC1s, ILC2s, ILC3s, lymphoid tissue-inducing cells and NK cells. ILCs are mainly localized in barrier tissues such as skin, intestinal mucosa, and lung, and play an important role in innate immune response to infections, lymphangiogenesis, and remodeling of damaged tissues by responding to microenvironmental signals released from surrounding tissue (37, 38). ILC1s are the predominant subset of ILCs in mouse and human atherosclerotic plaques and are involved in enhancing plaque formation by producing IFN-γ in response to stimulation by the transcription factor T-bet; conversely, removal of ILC1 attenuated plaque formation in ApoE-/- mice (39). ILC2s require the transcription factor GATA3 and are protective against atherosclerosis through the production of type 2 cytokines that promote B1 cell proliferation and IgM production as well as modulate diseased macrophages (40). ILC3s are the least abundant lymphocyte subset in atherosclerosis, and its dependence on the transcription factor RORγt for the production of IL-17 and IL-22 on the effects of atherosclerosis has been inconsistently investigated, leading to our lack of clarity on the role of ILC3s in atherosclerosis (41). Because of the overlap in gene expression between ILCs and T cells and the limitations of high-dimensional single-cell techniques, our information on the role of ILCs in atherosclerosis is limited, and the exact mechanisms require more research.
NK cells are innate immune system effector lymphocytes that sense pathologic alterations or stressed cells by activating receptors and play a cell-mediated cytotoxic role through the release of granzymes and perforin or cytokines, as in the case of interferon-gamma (IFN-γ) (42). NK cells have been detected in both human and mouse atherosclerotic plaque lesions (43, 44). Nevertheless, there is some controversy regarding the role of NK cells in atherosclerotic plaques. An early study of the role of NK cells in atherosclerosis using beige LDL receptor-/- (Ldlr-/-) mice showed that NK cells have atheroprotective effects (45). Another study investigating the impact of NK cell loss and gain of function in atherosclerosis development demonstrated that NK cells possess a pro-atherogenic role. Their production of interferon-γ, perforin, and granzyme B, as well as the expansion of the necrotic core within the lesion, facilitates the evolution of atherosclerotic plaques (46). Therefore, additional research is necessary to elucidate the precise role of NK cells in atherosclerotic plaques.
DCs are present in healthy arteries and can accumulate in atherosclerotic lesions. They are especially enriched in rupture-prone areas within atherosclerotic plaques, which can lead to plaque destabilization, and are involved in a variety of pathogenic and protective mechanisms during plaque formation. In the earliest stages of plaque formation, DCs may uptake lipids and form a foam-cell-like appearance, as well as regulate lipid metabolism through other mechanisms (47). DCs can also produce many pro-inflammatory cytokines, including TNF, IL-6, and IL-12, which are all pro-atherosclerotic (48). In addition, DCs in the arterial wall also express TLR4, which performs an essential role during the formation and development of atherosclerotic plaques. DCs as specialized antigen-presenting cells link innate and adaptive immunity by presenting antigens to T cells. Thereby, DCs significantly influence the occurrence and advancement of atherosclerotic lesions.
3.3 Adaptive immunity
The antigen-specific adaptive immune responses are triggered after the innate immune response and are mediated by T and B cells (49). The hypothesis that adaptive immunity plays a role in human atherosclerosis has been long studied.
T cells are recruited into atherosclerotic plaques through a mechanism that resembles monocyte recruitment via the receptor C-X-C motif chemokine receptor 3 (CXCR3), which is expressed by all CD4+ T lymphocytes in the same lesion (50). By inhibiting CXCR3 and blocking the migration of T cells from the circulation into the atherosclerotic plaque, plaque formation can be reduced (51). In addition to CXCR3, other chemokine receptors have been found to be responsible for T cell recruitment. Galkina et al. (52) found that CXCR6 expressed on multiple T cell subsets can facilitate T cell aggregation in the aorta and thus promote the development of atherosclerosis. Li et al. (53) demonstrated that CD4+ T cells homing to atherosclerotic plaques specifically required CC chemokine receptor 5 (CCR5), and blocking or knocking down CCR5 significantly prevented T cell recruitment. Similarly, CCR2 deficiency limits T cells entering the arterial intima and suppresses atherosclerosis (54). Recent studies have found that deletion of CCR7 not only leads to reduced plaque content in mice, but also results in disturbed access of T cells to and from the site of plaque inflammation (55).The ApoE-/- mice lacking CD4+ T cells show reduced atherosclerosis (56). CD4+ T cells from ApoE-/- mice were transferred to ApoE-/- mice crossed with severe combined immunodeficiency (SCID) strains of mice, leading to greatly increased fatty streak lesions compared to immunocompetent ApoE-/- mice (57). These studies indicate that CD4+ T cells play a pro-atherogenic role in the early stages of atherosclerotic plaque formation.
The CD4+ T cells that play a pro-atherosclerotic role are mainly T helper (Th) cells, especially Th1 cells, while regulatory T cells (Tregs) have been identified to play an anti-atherosclerotic role. CCL1 is a potent leukocyte chemotactic agent that plays an essential role in recruiting Tregs by binding to CCR8. Disruption of the CCL1-CCR8 axis facilitates atherosclerosis by inhibiting IL-10 production and Tregs recruitment and function (58). Likewise, CCL17 acts as a chemoattractant for CCR4+ T cells that inhibits Tregs migration and expansion, further limiting their function. Blocking CCL17 increases Tregs infiltration and reduces atherosclerosis (59). In addition, a study by Shao et al. (60) found that in ApoE-/- mice, CCR5 mediated Tregs homing to the aorta in the presence of IL-35, which ultimately inhibited atherosclerosis by maintaining the suppressive functions of Tregs.
Various studies have come to distinct conclusions about the effects of CD8+ T cells on atherosclerosis. In early human atherosclerotic plaques, CD8+ cytotoxic T cells (CTLs) are not as plentiful as CD4+ T cells, but in late human plaques they represent up to 50% of the leukocytes. CTLs are activated during hypercholesterolemia and can facilitate plaque inflammation and instability in lesions (61). Mice lacking antigenic peptide transporter 1 (TAP1) have lower numbers of CD8+ T cells and ApoE-/-Tap1-/- mice exhibit the same atherosclerotic lesions, immune cell infiltration and lipid accumulation as ApoE-/- mice (62). This indicates that CD8+ T cells have no or minimal effects on atherosclerotic plaques. However, other studies using the transfer of CD8+ T cells lacking perforin, granzyme B or TNF-α have shown that CD8+ T cells contribute to the evolution of atherosclerotic plaques by perforin- and granzyme B-mediated cytotoxicity and TNF-α-mediated inflammation by promoting apoptosis in macrophages, ECs and VSMCs and the development of the necrotic core (63). Thus, CD8+ T cells may play a secondary role in the early stages of atherosclerotic plaque evolution, but they may be triggered by intracellular infection, which then promotes the accumulation of atherosclerotic plaque or regulates the later stages of atherosclerosis.
Atherosclerotic plaque formation is intensely influenced by diverse arms of the immune system, including B lymphocytes (64). B cells are detected in atherosclerotic lesions, which have a structural organization similar to tertiary lymphoid organs and contribute to atherosclerotic plaque formation (65). Two major subsets of B cells have been found in atherosclerotic plaques. B1 cells spontaneously produce natural IgM antibodies to protect against atherosclerosis. Conventional B2 lymphocytes produce pro-atherogenic IgG, IgA, and IgE antibodies (64). In fact, it was discovered 40 years ago that atherosclerotic plaques contain immunoglobulins, particularly IgM and IgG, which are present in all stages of lesion development (66, 67). In addition, specific monoclonal antibodies against different epitopes of oxLDL, now referred to as oxidation-specific epitopes, have been discovered both in human and mouse plasma and in atherosclerotic plaques (68). B cells regulate plaque evolution by releasing antigen-specific antibodies that mediate humoral immune responses.
The earliest evidence that B cells are involved in atherosclerosis in mice came from Caligiuri et al. (69) who indicated that increased atherosclerosis in ApoE-/- mice after splenectomy was reversed through adoptive transfer of splenocytes, supporting an atheroprotective role. In addition, Major et al. (70) demonstrated that transplantation of B-cell-deficient mouse bone marrow to Ldlr-/- mice caused an increase in atherosclerotic plaque formation, clearly demonstrating the protective effect of B cells against atherosclerotic lesions. Further, injection of polyclonal immunoglobulin preparations into ApoE-/- mice resulted in a reduction in fatty streaks and fibrous plaques, indicating that atherosclerosis could be inhibited (71). Consistent with these results, the use of bone marrow transplants, whereby Ldlr-/- mice were reconstituted with wild-type or IL-5-/- bone marrow, resulted in reduced secretion of T15/EO6 clonotype natural IgM antibodies and accelerated atherosclerosis (72). The above studies suggest that humoral immunity and B cells have properties that inhibit the evolution of atherosclerotic plaques. In contrast, the use of CD20-specific monoclonal antibody-mediated depletion of mature B cells reduced the size of atherosclerotic plaque lesions in ApoE-/- and Ldlr-/- mice (73). Furthermore, relay transfer of conventional B2 cells into lymphocyte-deficient ApoE-/- mice and B cell-deficient ApoE-/- mice effectively promoted the development of atherosclerotic plaques (74). These studies, in turn, show that B cells have a pro-atherosclerotic role. Overall B cells have both a protective and pathogenic role in atherosclerosis depending on the subclass, but more research is required to elucidate the role of each subclass.
4 Evolutionary mechanism for untreated atherosclerotic plaque under immunomodulation
Atherosclerosis is now recognized as a chronic inflammatory disease, with inflammation having an essential role in all stages of the pathogenic process, encompassing plaque formation, progression, and rupture. Inflammation is the response to the existence of exogenous and endogenous antigens by the immune system. In general, driven by the immune system, inflammation profoundly influences the evolutionary trajectory of atherosclerotic plaques.
4.1 Inflammation is the driving factor of atherosclerotic plaque initiation
Inflammation underlies atherosclerotic plaque formation. Plaque inflammation is driven by cytokines, chemokines, AMs, inflammatory signaling pathways, bioactive lipids, and immune cells (75, 76). Nearly all conventional risk factors for atherosclerosis are associated with and actively contribute to inflammatory processes.
Infiltration and retention of oxLDL in the arterial wall serve as critical initiating events that trigger the inflammatory response and facilitate atherosclerotic plaque formation (77). It has been demonstrated that atherosclerotic plaques in both humans and experimental animals contain oxidatively modified LDL (78). OxLDLs contain oxidized lipids, and the products of their degradation make them critical inflammatory components that contribute to the development of atherosclerotic plaques. According to the level of LDL oxidation, oxLDLs are divided into minimally modified LDL (mmLDL) or extensively oxLDL (79). The extensively oxLDL and mmLDL can trigger pro-inflammatory responses in ECs and macrophages, leading to endothelial dysfunction and recruitment of leukocytes to the site of the lesion. Furthermore, mmLDL induces upregulation of TLR2 and TLR4 expression in monocytes/macrophages, and increased TNF-α secretion promotes plaque inflammatory responses (80). In addition, oxLDL and corresponding antibodies bind to form immune complexes (ICs) that have pro-atherogenic and pro-inflammatory properties and ICs activate the inflammasome through signaling at multiple receptors (81, 82). NOD-like receptor family pyrin domain-containing protein 3 (NLRP3) is the most broadly studied of the numerous inflammasomes and an influential regulator in the pathogenesis of cardiovascular disease. A characteristic feature of atherosclerotic plaques is increased expression of the NLRP3 inflammasome component (83), and inhibition of the NLRP3 inflammasome using MCC950 inhibitor reduces the development of atherosclerotic plaques in ApoE-/- mice (84). In conclusion, oxLDL promotes plaque inflammatory responses by interacting with immune components.
SRs are distributed on arterial vessel wall cells and vascular ECs, and oxLDL binds to the corresponding receptors to promote the evolution of atherosclerotic plaques through multiple mechanisms. CD36 is a pattern recognition receptor expressed on multiple cell types and is a part of the class B family of SRs. CD36 is not only involved in the uptake of oxLDL and foam cell formation, but also in atherosclerotic plaque formation by interacting with oxLDL and triggering an inflammatory response (85). LOX-1 belongs to the C-type hemagglutinin family and is a specific receptor for oxLDL. LOX-1 is expressed by ECs, macrophages and VSMCs in various stages of atherosclerotic lesions (86). Overexpression of LOX-1 facilitates endothelial dysfunction, vascular inflammation, and plaque formation, and participates in the destabilization of atherosclerotic plaques in vivo (87, 88). In contrast, LOX-1 deficiency maintains endothelial function and reduces atherosclerosis development (89). Altogether, oxLDL recognizes the corresponding receptors and promotes the development of plaque inflammation.
4.2 Inflammation accelerates the evolution of atherosclerotic plaque
The accumulation of oxLDL and its binding to receptors initiate plaque inflammation, while the infiltration of immune cells, the activation of plaque-associated inflammatory signaling pathways, and the response to host tissues and microorganisms maintain the development of inflammation, thereby further accelerating the evolution of plaque.
Infiltration and accumulation of pro-inflammatory and anti-inflammatory leukocytes in the intima of the arterial wall are hallmarks of atherosclerotic plaque formation and progression, as well as drivers of atherosclerotic lesion growth. Immune cell infiltration is triggered by chemokines and AMs (90). Members of the chemokine and AM families mediate the recruitment of immune cells to infiltrate the lesion and accelerate the development of the plaque inflammatory response. In addition, they modulate cellular homeostasis, leading to endothelial dysfunction as well as involvement in thrombosis (91, 92). Many previous studies have focused on P-selectin glycoprotein ligand-1 (PSGL-1), an adhesion ligand which is expressed on leukocytes and ECs. Knockdown of PSGL-1 in an ApoE-/- mouse model reduced monocyte infiltration and leukocyte adhesion, decreased atherosclerotic plaque area and was protective against atherosclerosis (93, 94). Furthermore, systemic low-grade chronic inflammation triggered by the common risk factor obesity is also capable of promoting immune cells infiltration, including lymphocytes and macrophages, which impairs systemic metabolism by exacerbating adipose tissue inflammation, leading to cholesterol deposition in the blood vascular wall, with the ensuing immune process supporting the growth of atherosclerotic plaques and contributing to cardiovascular complications (95).Overall, the development and growth of plaques in atherosclerosis cannot occur without the infiltration of immune cells.
Signaling pathways that are mediated by immune, inflammatory mediators are associated with the evolution of atherosclerotic plaques. Nuclear factor-κB (NF-κB) serves as the primary transcription factor in the inflammatory response. The NF-κB pathway is induced and activated by pro-inflammatory cytokines, adhesion molecules, chemokines and growth factors and exerts a pivotal role in atherosclerotic plaque inflammation (96). Activated NF-κB is observed in several cells including macrophages, VSMCs and ECs in atherosclerotic lesions. Studies have shown that activated NF-κB is involved in many features of atherosclerosis, including mediating foam cell formation, enhancing vascular inflammation, stimulating VSMCs proliferation and migration, exacerbating vascular calcification, promoting plaque formation and destruction, and regulating vascular apoptosis (97). In Ldlr-/- mice fed an atherogenic diet for a prolonged period, the NF-κB signaling pathway in the endothelium was activated, leading to increased atherosclerotic plaque formation (98). Furthermore, TLRs are expressed in all atherosclerotic plaque immune cells and are engaged in the inflammatory response to plaques. The TLRs signaling pathway can be broadly divided into MyD88-dependent and non-MyD88-dependent pathways. MyD88 activates NF-κB and MAPK signaling pathways and attracts the expression of inflammatory cytokines. The non-MyD88-dependent pathway, TIR-domain-containing adapter protein-inducible interferon (IFN)-β (TRIF)-dependent pathway, activates IFN regulatory factor 3 (IRF3) and induces the expression of type I IFNs (99, 100). Among these, TLR4 signaling plays an important role in activating atherosclerotic plaque inflammation and lipid accumulation. Knockdown of Adenosine triphosphate-binding cassette transport increases TLR4 and MyD88/TRIF signaling in macrophages, enhances expression of inflammatory cytokines and accelerates plaque inflammation (101). In general, TLRs can induce the synthesis of pro-inflammatory mediators as well as promote plaque inflammation through activating complex cell signaling pathways. Next, janus kinase (JAK)-signal transducer and activator of transcription (STAT) signaling pathways exert pro-inflammatory effects in atherosclerotic plaques by inducing diverse pro-inflammatory cytokines such as IL-6, TNF-α and IL-1 (102). The JAK/STAT pathway is activated when various cytokine receptors on the cell membrane bind to the corresponding ligands. The JAK family is composed of four members: JAK1, JAK2, JAK3, and TYR2, while the STAT family is composed of seven members: STAT1, STAT2, STAT3, STAT4, STAT5a, STAT5b, and STAT6. Gharavi et al. (103) found that activated STAT3 was predominantly present in ECs and inflammatory cells in inflammatory areas of atherosclerotic plaques, with less in non-inflammatory areas. Mice with STAT3 knockout in the endothelium showed reduced fatty streak formation and decreased macrophage content compared to wild-type mice. In addition, there is growing evidence that the Wnt pathway is involved in multiple processes in the evolution of atherosclerotic plaques, including regulation of endothelial dysfunction, VSMC proliferation and migration, regulation of inflammation and foam cell formation, and pathological angiogenesis and calcification, all of which are key processes in plaque formation and stabilization (104). In conclusion, in the plaque inflammation microenvironment, crosstalk between signaling pathways further accelerates inflammation and plays a role in plaque evolution.
Research into atherosclerotic plaque development has also focused on the host tissues and microorganisms that may influence this process. Endothelial-mesenchymal transition (EndMT) is present in atherosclerotic plaques, where ECs can obtain a mesenchymal phenotype, with loss of expression of EC markers and function, and gain of expression of mesenchymal cell markers and function, directly contributing to plaque evolution (105). In addition to this, the brown adipocyte-specific PPARγ knockout mice were crossed with ApoE-/- mice and the resulting double knockout showed increased vascular and systemic inflammation and significantly increased atherosclerotic lesions (106). Thus, host cells and tissues can contribute to the development of plaque lesions by increasing inflammation.
Furthermore, the presence of microorganisms in atherosclerotic plaques is well recognized. Pathogenic microorganisms are able to infect arterial wall cells, facilitate local inflammation of the plaque, evoke an adaptive immune response and influence plaque progression. For example, Chlamydia pneumoniae infection enhances monocyte adhesion, induces foam cell formation, activates LDL receptors, and triggers inflammation and atherosclerosis (107). Porphyromonas gingivalis induces endothelial cell dysfunction, promotes VSMC proliferation, migration, and calcification and the formation of foam cells, which also leads to an imbalance of Tregs and Th cells, suppresses T cell immunity, facilitates inflammatory responses, and ultimately promotes the evolution of atherosclerotic plaques (108). Moreover, a study showed that gut microbiota (GM) are key environmental factors in regulating inflammation in atherosclerotic plaques (109). GM can influence the evolution of atherosclerotic plaques through direct invasion of plaques and by regulating cholesterol metabolism and the production of harmful metabolites. An animal study showed that transplantation of the pro-inflammatory GM from caspase1-/- mice to Ldlr-/- mice enhanced systemic inflammation and increased atherosclerosis (110). In conclusion, microorganisms can regulate plaque progression by influencing inflammatory responses through multiple mechanisms.
In addition to bacteria, some viruses can also influence the evolution of atherosclerotic plaques. For example, cytomegalovirus (CMV) is capable of infecting almost all plaque immune cells, expressing viral cytokines and chemokines to promote an inflammatory environment, and triggering plaque formation through mechanisms such as induction of endothelial damage, increased lipid deposition, and VSMC proliferation and migration (111). ApoE-/- mice infected with CMV increases lesion size and IFN-γ levels, contributing to the development of atherosclerotic plaques (112). Other viruses including Epstein-Barr virus, herpes simplex virus type I, hepatitis virus, and human immunodeficiency virus have been reported to be associated with atherosclerosis, but the exact mechanisms need to be further studied. An in-depth study of the effects of pathogenic microorganisms on plaque evolution may also bring insights for the prevention and treatment of atherosclerosis.
5 Immune cell heterogeneity contributes to atherosclerotic plaque evolution
Leukocyte subsets accumulate at different stages of plaques and are involved in the immune response in the atherosclerotic lesion process. Single-cell RNA sequencing (scRNA-Seq) reveals heterogeneity of immune cells in atherosclerotic plaques (113). Studying immune cell heterogeneity could provide new insights into the evolutionary mechanisms of atherosclerotic plaques.
5.1 Monocyte/macrophage heterogeneity
Monocytes are heterogeneous populations and human blood monocytes are classified into three subpopulations according to differences in the expression of the lipopolysaccharide (LPS) receptor CD14 and the FcγIII receptor CD16: classical CD14++CD16-, intermediate CD14++CD16+ and non-classical CD14+CD16++ monocytes (114). In mice, monocytes are categorized into two major subpopulations depending on Ly-6C expression: Ly6ChiCCR2+CX3CR1low and Ly6ClowCCR2-CX3CR1hi (115) (Figure 2). Similarly, macrophages are a heterogeneous cell population arising from the heterogeneity of monocytes and the stimulation of the inflammatory microenvironment of plaques. In recent years, the concept of macrophage heterogeneity has been better explored, but in atherosclerosis the traditional classification of M1 and M2 subsets continues to be used (116). A full understanding of the heterogeneous expression of plaque monocytes/macrophages will help to uncover the role of different functional phenotypes on plaque evolution.
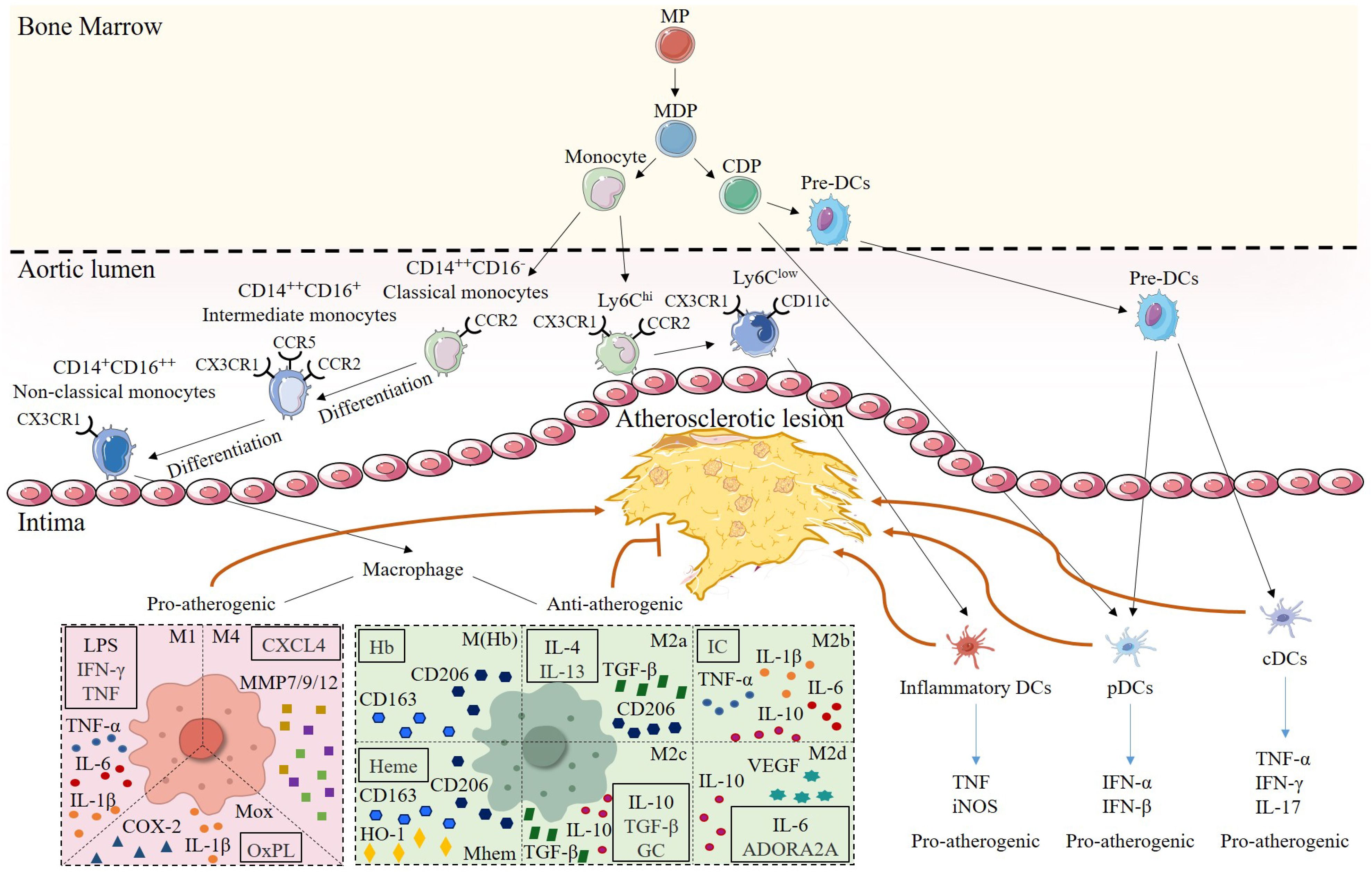
Figure 2. Heterogeneity of monocytes/macrophages and DCs in atherosclerotic lesions. Monocytes and DCs share a common origin in myeloid progenitor (MP). Monocyte-DC precursor (MDP) gives rise to monocytes and common DC precursor (CDP). Among them, mouse monocytes are classified into two subsets according to their surface expression of Ly-6C. Ly6Chi subsets have pro-inflammatory functions and highly express CCR2. Ly6Clow subsets primarily patrol along the vascular endothelium, are engaged in tissue repair, and can express CD11c, and differentiate into inflammatory DCs to promote plaque progression in the plaque inflammatory microenvironment. Human monocytes are subdivided into three subsets based on the expression of CD14 and CD16.CD14++CD16- classical monocytes, which express high levels of CCR2, can differentiate into CD14++CD16+ intermediate monocytes that express CCR5, and then further differentiate into CD14+CD16++ nonclassical monocytes, which are highly expressive of CX3CR1. Once monocytes enter the intima, they can mature into macrophages. Differentiation of different macrophage subsets depends on the stimulation of the inflammatory microenvironment within the lesion. Pro-atherogenic macrophages include M1, M4, and Mox, which are activated by Th1 cytokines, CXCL4, and OxPL, respectively, and secrete pro-inflammatory mediators, such as TNF-α, MMP, and IL-1β, to exert pro-inflammatory effects. Anti-atherogenic macrophages include M(Hb), Mhem, and M2. M(Hb) and Mhem are induced by Hb and Heme and secrete CD163 and CD206 to exert anti-inflammatory effects. M2 macrophages are subdivided into M2a, activated by IL-4 and IL-13; M2b, activated by IC; M2c, activated by IL-10, TGF-β and GC, and M2d, activated by IL-6 and ADORA2A. M2 macrophages mainly secrete Th2 cytokines such as IL-10 as well as TGF-β, which inhibit the development in atherosclerosis. CDP can further differentiate into pDCs and pre-DCs, and pre-DCs enter the arterial intima and become pDCs and cDCs, respectively, and play pro-atherogenic roles by secreting TNF or IFN.
Different monocyte/macrophage subsets play different roles in the evolution of atherosclerotic plaques. Classical CD14++CD16-monocytes account for about 85% of total monocytes, intermediate CD14++CD16+ monocytes for about 5% and non-classical CD14+CD16++ monocytes for about 10% (117). Classical monocytes, as major phagocytes, are capable of producing M1 macrophages and foam cells with strong peroxidase activity, producing ROS and secreting cytokines such as IL-10 in response to LPS and taking up LDL during infection and inflammation. They also exhibit high levels of CCR2 and CD62L, as well as low levels of CX3CR1. Human CD14++CD16+ intermediate monocytes are pro-inflammatory. This subset exhibits the highest ROS production and lowest peroxidase activity and secretes IL-1 and TNF-α in response to LPS associated with the evolution of atherosclerotic plaques. It also expresses CXCR1, CCR2, and CCR5 (118). It has been demonstrated that enzymatic degradation of LDL preferentially binds intermediate monocytes and is able to induce foam cell formation, suggesting that this subset has a pro-atherogenic effect (119). Non-classical monocytes are weak phagocytes that preferentially take up oxLDL and have an inflammatory effect, producing numerous inflammatory cytokines for example TNF-α and IL-1β. Additionally, they also express an elevated level of CX3CR1 and have a strong affinity for ECs, acting more like patrolling immune cells (120). Overall, human monocyte subsets differ not only in their proportions and phenotypes, but also significantly in their functions, but all promote plaque development.
Murine monocytes are the most studied. The Ly6Chi subset resembles human classical monocytes and is referred to as ‘‘inflammatory’’ monocytes, while the patrolling Ly6Clow subset resembles non-classical monocytes in part and is referred to as ‘‘resident’’ monocytes (121). Ly6ChiCCR2+CX3CR1low monocytes rely on CX3CR1, CCR2, and CCR5 for transport to plaques, while Ly6ClowCCR2-CX3CR1hi are recruited to plaques through CCR5. In the arterial intima, Ly6Chi monocytes can differentiate into M1 macrophages, which go on to produce foam cells and produce inflammatory cytokines such as TNF-α, IL-6, ROS, and matrix metalloproteinases (MMPs) that actively contribute to plaque formation and progression (122). Research has demonstrated that the amount of Ly6Chi cells in the blood increases dramatically as atherosclerosis progresses in hypercholesterolemic ApoE-/- mice, suggesting that Ly6Chi monocytes have a crucial role in plaque progression (123). Ly6Chi monocytes can be converted into Ly6Clow cells. Unlike Ly6Chi, Ly6Clow monocytes are recruited less to plaques but preferentially express the DC-associated marker CD11c (124). Ly6Clow monocytes rely on CX3CR1 to patrol healthy tissue in a long-range crawl across the endothelium, which allows them to rapidly invade infected tissue, initiate an early immune response and differentiate into macrophages when early inflammation occurs (125). Monocyte heterogeneity is important in the evolution of atherosclerotic plaques, but the mechanism of action of some subsets is unclear and much future research is needed.
Indeed, macrophage phenotype is influenced by microenvironmental factors within the atherosclerotic plaques. Both M1 and M2 macrophage populations are present and increasing in number throughout the atherosclerotic process (126). Monocytes are induced by GM-CSF and stimulated by Th1 cytokines, for example TNF-α and IFN-γ, to differentiate into M1 macrophages (127), which express various pro-inflammatory cytokines, including IL-13, IL-6, and TNF-α, leading to the recruitment of inflammatory cells and accelerating the development of plaques. In addition, M1 macrophages can also secrete MMPs, which degrade the ECM and cause plaque rupture. M1 macrophages accumulate in the rupture-prone shoulder area of the plaque, and both M1 and M2 macrophages are expressed at the fibrous cap near the necrotic core (126). Unlike M1 macrophages, M2 macrophages are induced to polarize by different cytokines like M-CSF, IL-4, IL-13, and IL-10 (127). Among these, large amounts of IL-13 are produced mainly from ILC2s.Activation of ILC2s has been shown to be associated with reduced atherosclerotic burden. Engelbertsen et al. (128) found that treating Ldlr-/-rag1-/- mice with IL-2/anti-IL-2 complexes resulted in the expansion of CD25+ ILC2s, which lowered very low-density lipoprotein cholesterol and atherosclerosis, and conversely ILC2s depletion led to an acceleration of the atherosclerotic process (129). Mantani et al. (130) also observed that IL-25 treatment of ApoE-/- mice inhibited the development of atherosclerosis by a massive expansion of ILC2s, an increase in IL-5 concentration, and greater numbers of B1 cells and IgM antibodies. Meanwhile, the expansion of ILC2s secreted large amounts of IL-13, which enhanced plaque stability and prevented the development of atherosclerotic lesions by increasing collagen deposition and promoting macrophage polarization towards M2 (131–133). According to the stimulation signal, M2 macrophages are further divided into four different subsets. M2a macrophages are induced by IL-4 and IL-13 to secrete anti-inflammatory cytokines including IL-10 and IL-1, which have strong effects on anti-inflammation, as well as high expression of mannose receptor, pro-fibrotic factors, and transforming growth factor involved in the repair of damaged tissues (134, 135). M2b macrophages can be stimulated by immune complexes, TLR agonists in combination with IL-1 receptor agonists, or LPS to produce and express large numbers of the anti-inflammatory cytokine IL-10, which exerts an immunomodulatory role in atherosclerotic plaques, but also secretes pro-inflammatory cytokines such as TNF-α, IL-1β, and IL-6 (121, 134). M2c macrophages are induced to activate by IL-10, TGF-β or glucocorticoids and they are capable of releasing IL-10 and pentraxin 3 for anti-inflammatory effects, and high expression of Mer receptor kinase for its efficient efferocytosis (136, 137). The process of M2d macrophage polarization is mediated by IL-6 and TLR stimulation of the adenosine A2A receptor, producing large amounts of IL-10 and vascular endothelial growth factor (VEGF) to promote angiogenesis and plaque growth in atherosclerotic plaques (138) (Figure 2). Overall, in addition to M2b macrophages, M2 macrophages produce many anti-inflammatory cytokines that maintain effective efferocytosis in atherosclerotic plaques as well as reducing plaque inflammation.
Recently, many research studies have revealed the presence of other macrophage subsets in atherosclerotic plaques, in addition to M1 and M2 types. M4 macrophages are induced by CXCL4, which is widely expressed in plaques, and exert atherosclerotic effects by producing MMP12, MMP7, and MMP9 leading to increased plaque vulnerability (139–141). The presence of M (Hb) and Mhem-type macrophages in areas of advanced atherosclerotic plaque bleeding is induced by hemoglobin and heme, respectively (142, 143). They characteristically express proteins and receptors such as the SR cysteine-rich type 1 protein M130 and macrophage mannose receptor 1 and activate related signaling pathways to increase liver X-receptor (LXR)-α activity to enhance cholesterol efflux, thereby preventing foam cell formation and exerting anti-atherogenic effects (143–145). Mox macrophages, which lack phagocytosis, account for approximately one-third of all macrophages in mouse advanced atherosclerotic plaques. Their formation is dependent on oxidized phospholipids and the transcription factor Nrf2, and they are capable of secreting pro-inflammatory cytokines such as IL-1β and cyclooxygenase 2, which contribute to plaque evolution and inflammation (146). It follows that macrophages can polarize in the complex microenvironment of the atherosclerotic plaque, and that the great heterogeneity of macrophages has a different impact on plaque evolution.
Single-cell techniques have greatly improved our understanding of macrophage subsets in plaques. Zernecke et al. (147) comprehensively analyzed leukocyte subpopulations identified by scRNA-Seq, and cell counting (CyTOF) and confirmed five macrophage subsets emerging in the aorta of atherosclerotic mice: resident -like macrophages, inflammatory macrophages, Trem2hi foamy macrophages, interferon-inducible cell (IFNIC) macrophages and cavity macrophages. The newly discovered macrophage subsets exhibit heterogeneity in gene expression, with each subset having genes that are more significantly enriched in expression relative to the other subsets. Resident-like macrophages express Lyve1, Timd4, Mrc1, Pf4 and Ccr2 (148, 149). Inflammatory macrophages highly express the inflammatory chemokines Ccl2-4 and Cxcl2 and are also enriched in a large number of classical pro-inflammatory transcripts, such as Il1α, Il1β, and Nlrp3, which play an important role in regulating the progression of atherosclerosis (150). Trem2hi foamy macrophages possess a gene expression profile that partly overlaps with those of inflammatory macrophages and in addition express Lgals3, an atherosclerosis biomarker that promotes the differentiation of monocytes to macrophages and is linked to alternative macrophage activation and plaque evolution (151, 152), as well as expressing the Cd9 and Ctsd, the gene encoding cathepsin D. Cathepsin D is involved in the modification of LDL and facilitates phagocytosis of oxLDL by macrophages, which leads to the formation of foam cells (153). IFNIC macrophages are characterized by type I interferon and express a variety of interferon-inducible genes such as Ifit3, Irf7 and Isg15, it is known that type I interferons usually have pro-atherogenic effects (154). Cavity macrophages express MHCII encoding genes as well as Cd226, Itgax and Ccr2, and the function of the cells is unknown (147). In conclusion, resident-like macrophages have anti-inflammatory properties, IFNIC macrophages and inflammatory macrophages are pro-inflammatory, Trem2hi foam macrophages are not pro-inflammatory but function in lipid metabolism and cholesterol efflux (155, 156). Similarly, integrated analysis of scRNA-Seq data from immune cells in human atherosclerosis revealed that macrophages in human lesions exhibit similar transcriptional status to subsets of mouse aortic macrophages (157). The newly discovered subsets add an important dimension to our study of plaque macrophages, but the impact on plaque evolution and the specific mechanisms of action still need to be clarified by in-depth scientific studies and explorations.
5.2 DC heterogeneity
DCs are heterogeneous blood-borne professional antigen-presenting cells with the ability to capture, process, and present antigens to T cells that recognize antigens and induce antigen-specific immune responses (158). The functional heterogeneity of DCs identified in plaques suggests their complicated and multifaceted roles in atherosclerotic disease pathogenesis. The plaque inflammatory microenvironment and exogenous stimuli affect the DCs phenotype and are able to control the switch to an inflammatory or tolerant phenotype. DCs can be categorized into several subsets based on their origin, location, and function (159). In atherosclerotic plaques, the focus is on myeloid DCs (mDCs), plasmacytoid DCs (pDCs), pre-DCs, conventional DCs (cDCs), and inflammatory DCs (Figure 2).
mDCs are characterized by the expression of CD1c, CD11c, and CD33 and they secrete IL-12. In contrast the pDCs express CD123 and produce type I IFN (160). The differences in the surface markers of mDCs and pDCs results in inconsistent quantitative changes exhibited in atherosclerotic disease. For example, in coronary artery disease patients mDCs precursors, but not pDCs precursors, are significantly reduced, which is contrary to other research showing a marked reduction in the amount of pDCs and a remarkable increase in the amount of mDCs in patients with coronary artery disease (161, 162). In atherosclerosis, mDCs and pDCs were confined to the shoulder of lesions to produce IFN-α, demonstrating their association with plaque instability (163). mDC and pDC coexistence synergized TLR4 and TLR9 ligand inflammatory effects as demonstrated by increased production of TNF-α, IL-12, and MMP9 (164). The development and stereotyping of mDCs are modulated by a variety of transcriptional and hematopoietic growth factors, with CCr7, Zbtb46, and Flt3 representing the core genes responsible for the development as well as functional and phenotypic maintenance of mDCs. mDCs have been shown to be associated with the pathogenesis of atherosclerosis. Various subsets of mDCs may have both atherogenic and atheroprotective activities during atherogenesis. The pro-inflammatory effects of mDCs include pro-inflammatory subsets that produce inflammatory molecules and initiate effector T cells. On the contrary, tolerogenic mDCs combat inflammation by inhibiting the activity of pro-inflammatory T cells and macrophages and by inducing immunosuppressive Tregs (165). Deficiency of FMS-like tyrosine kinase 3 ligand in mice leads to lack of CD103+CD11c+ mDCs and other DC subpopulations as well as advanced atherosclerosis. In Ldlr-/- mice, CD103+CD11c+ mDCs exert immunosuppressive effects through mechanisms that support FoxP3+ Treg homeostasis (166). These findings suggest that CD103+CD11c+ mDCs are atheroprotective. Transplantation of MyD88-deficient CD11c+ DCs into Ldlr-/- mice decreases stimulation of peripheral effector T cells, reduces the aggregation of effector T cells and Tregs in plaques, and results in an increase in plaque size owing to accumulation of myeloid-derived pro-inflammatory cells (167). Overall, mDC subsets play diverse roles in atherogenesis.
Several studies have discussed the role of pDCs in atherosclerotic plaque evolution. In a mouse model of atherosclerosis, depletion of pDCs using multiple antibodies against bone marrow stromal cell antigen 2 (BST2) promotes the accumulation of T cells in plaques, leading to enhanced atherosclerosis in Ldlr-/- mice (168); whereas in ApoE-/- mice, atherosclerosis is reduced by decreasing macrophage infiltration in plaques and stabilizing plaques by increasing collagen content (169, 170). The transcription factor E2-2/Tcf4 is an important regulator of pDCs development. Additional studies have found atheroprotective effects with specific deletion of the transcription factor E2-2/Tcf4 or impaired MHCII antigen presentation in pDCs, suggesting that in atherosclerosis, pDCs also promote MHCII-dependent antigen presentation through T-cell responses (171). Therefore, pDCs can drive immune responses in atherosclerosis and their function is influenced by environmental factors.
Pre-DCs are the final precursor stage in the formation of DCs, and they can develop into DCs without dendrites but require further development to obtain dendritic forms and full DC function. Little cell division is involved in their development into DCs, leading to different types of DCs being produced by different pre-DCs, such as some monocytes, pDCs, and cDCs (172). Some studies have reported increased accumulation of pre-DCs in plaques, which may be attributed to increased levels of circulating chemokines of CCL2, CCL5, and CXCL12 enhancing the entry of pre-DCs into plaques (173, 174). cDCs are also classified as migratory DCs, e.g., Langerhans cells and dermal DCs and lymphoid-tissue-resident DCs, which are not able to migrate to the lymph nodes. Recently, based on different developmental pathways, cDCs have been further classified into cDC type 1 (cDC1), including CD8α+ DCs in lymphoid tissues and CD103+ DCs in nonlymphoid tissues, and CD11b+ cDC2s (47). cDCs can be engaged in interactions with T cells and NKT cells, resulting in increased production of IFN-γ, IL-17, and TNF-α by T cells promoting plaque development. Among them, CD11b+ cDC2s were able to participate in lipid accumulation and foam cell formation (48). cDCs can regulate the evolution of atherosclerotic plaques in an antigen-dependent manner in part by modulating T cell activation and adaptive immune responses. Inflammatory DCs do not exist in a stable state, instead they appear as a result of inflammation (160, 172). CX3CR1 deficiency affects the accumulation of DCs in the aortic wall and significantly attenuates the atherosclerotic burden, which could demonstrate that inflammatory DCs may differentiate primarily from Ly6Clow monocytes and promote plaque evolution (175). The pro-inflammatory microenvironment at the atherosclerotic plaque provides an excellent option for the predominant differentiation of circulating DC precursors and monocytes toward inflammatory DCs, which in turn can promote the differentiation of naïve T cells into inflammatory and pro-atherosclerotic Th subsets, such as Th1 and Th17 (158). Inflammatory DCs act as an important player in the pro-inflammatory response at plaque lesion sites. In conclusion, pre-DCs develop into different subsets of mature DCs under the influence of different factors and function in the evolution of plaques.
5.3 T cell heterogeneity
In atherosclerotic plaques, T cells are less numerous than monocytes/macrophages, but like monocytes/macrophages that exhibit heterogeneity in markers and function, T cells also exhibit heterogeneity and regulate the adaptive immune response to plaque. CD4+ T cells are commonly found in atherosclerotic plaques, and CD4+ T cell subsets are substantially heterogeneous. In response to different stimulus activations, CD4+ T cells differentiate into different Th subsets, including Th1, Th2, Th17 and Tregs (Table 1).
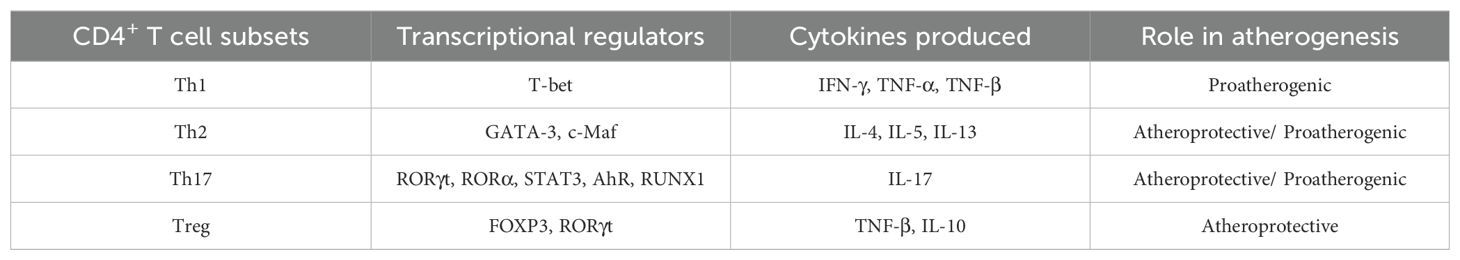
Table 1. Heterogeneity of CD4+ T cell subpopulations.
Th1 is the prominent T cell subtype in atherosclerotic lesions and Th1 cytokines predominate in atherosclerotic mouse models and human plaques. They secrete mainly IFN-γ, which exerts a pro-atherogenic effect (176). IFN-γ is a major inducer of atherosclerotic lesions and promotes plaque development and instability in a number of ways, including endothelial dysfunction, recruitment of inflammatory cells, downregulation of intraplaque cellular cholesterol efflux, promotion of lipid deposition and foam cell formation, and induction of pro-inflammatory cytokine secretion (177, 178). IFN-γ-/- mice crossed with Ldlr-/- mice showed significantly fewer atherosclerotic lesions compared to Ldlr-/- mice after 8 weeks of feeding a cholesterol diet (179). Similarly, ApoE-/- and IFN-γ receptor double knockout mice showed a substantial reduction in atherosclerotic lesions, as evidenced by reduced lipid accumulation and reduced numbers of inflammatory cells, compared to ApoE-/- mice after 3 months of feeding a Western type diet (180), whereas exogenous injection of IFN-γ to ApoE-/- mice resulted in increased lesions (181). All the above studies suggest that IFN-γ plays a facilitating role in the evolution of plaques. T-bet is a transcription factor essential for Th1 cell differentiation, and Ldlr-/- mice lacking T-bet show diminished atherosclerosis (182). This further suggests that Th1 cells promote atherogenesis and also confirms that atherosclerosis is driven by the Th1 response.
Th2 cells are poorly represented in atherosclerotic plaques, and their function in atherosclerosis is still controversial. Th2 cells typically produce cytokines such as IL-4, IL-5, and IL-13. IL-4 can effectively inhibit the Th1 response, Th1 cytokine formation and IFN-γ secretion, and has atheroprotective effects (176, 183). However, other studies have shown that IL-4 deficiency leads to decreased atherosclerotic lesion formation in Ldlr-/- mice, suggesting a pro-atherogenic effect of Th2 cells (184). Furthermore, in angiotensin II-induced atherosclerotic ApoE-/- mice, IL-4 administration showed no effect on plaque development (185). In summary, IL-4 exerts different effects on the development of atherosclerotic plaques. Unlike IL-4, IL-5 and IL-13 have a defined atheroprotective effect. IL-5 induces B1 cells to produce atheroprotective natural IgM antibodies specific for oxLDL. Second, IL-5 deficiency enhances plaque formation (72). Similarly, plaque development is accelerated by IL-13 deficiency, and IL-13 exerts atheroprotective effects by increasing collagen content, reducing monocyte recruitment, and inducing M2-type macrophage polarization (133). Furthermore, ILC2 cells, which are thought to correspond to Th2 cells, have been found to perform class II MHC restricted antigen presentation and to orchestrate Th2-like immune responses (186). IL-25 as well as IL-33 induced by Th2 cells can promote the proliferation of ILC2 cells, and the activation of ILC2 cells in turn promotes the Th2 response, thereby reducing the burden of atherosclerosis (187). In conclusion, the exact role of Th2 cells in atherosclerosis is still unclear. Possible reasons for this include different research teams have various experimental designs, study methods, and focuses; cytokines produced by Th2 cells, especially IL-4, exhibit complex biological properties; most studies have been conducted in animal models, but animal models of atherosclerosis have certain limitations; and there are also complex interactions between immune cells, and Th2 may synergize with or antagonize other immune cells, thereby affecting its role in atherosclerosis. More in-depth and comprehensive studies are needed to elucidate the role of Th2 in atherosclerosis.
The Th17 subset has been identified in atherosclerosis-prone mouse and human atherosclerotic plaques and its role remains controversial (188). Differentiation of Th17 cells is mediated by retinoid-related orphan nuclear receptor (ROR)γt (189) and other transcription factors such as RORα, STAT3 (190), Aryl hydrocarbon receptor, and runt-related transcription factor 1 (191, 192). Unlike classical Th1 and Th2 cells, Th17 cells exhibit significant heterogeneity due to the unstable expression of RORγt and its property of being influenced by environmental cues. Th17 cells mainly secrete the cytokine IL-17. The effect of IL-17 in atherosclerotic plaques would seem to be complex. IL-17 further exerts pro-atherosclerotic effects by inducing IL-6, CXCL8 and CXCL10, and GM-CSF in VSMCs to promote inflammatory activation (193). On the other hand, the atheroprotective effect of IL-17 is associated with inhibition of the pro-atherogenic factor IFN-γ and production of the anti-atherogenic factor IL-10, as well as suppression of the expression of vascular cell adhesion molecule 1 (VCAM-1) leading to a reduction in leukocyte recruitment within the lesion (188). Due to the complexity of IL-17’s action, several related experimental studies have yielded different results. Experiments with functional blockade of IL-17 have demonstrated that IL-17 has pro-atherosclerotic effects, and that inhibition of IL-17 reduces plaque development, decreases plaque vulnerability, and reduces leukocyte infiltration (194). Another study showed that IL-17 exerts atheroprotective effects by promoting collagen cap formation and stabilizing plaques (195). In conclusion, Th17 plays a complex role in atherosclerosis, as demonstrated by its ability to both promote atherosclerosis and inhibit its development.
Tregs are a highly heterogeneous population of CD4+ T cells with immunosuppressive functions. In the pro-inflammatory microenvironment formed by pro-inflammatory factors such as IL-1β, IL-2, and IL-23, CD4+FOXP3+ T cells co-express FOXP3 and RORγt transcription factors and are able to produce IL-17 upon stimulation (196). IL-1β and IL-2 induce the differentiation of the initial FOXP3+ Tregs into Th17 cells, and similarly IL-23 is able to differentiate Tregs into Th17 cells through inducing a high level of RORγt expression and enhancing the loss of FOXP3 (197, 198). Tregs have been clearly demonstrated to be atheroprotective, with reduced numbers and impaired suppressive function linked to the evolution of atherosclerotic plaques (199). The amount of Tregs was reduced in mice that developed atherosclerosis compared to ApoE-/- mice in which no atherosclerotic plaques were detected (200). In a functional study of CD4+FOXP3+ Tregs, it was found that when Tregs were depleted by anti-CD25 antibodies, plaque vulnerability increased, and lesion progression was accelerated (201). Despite the small number of CD4+FOXP3+ Tregs found in mouse and human plaques, their secretion of cytokines, such as TGF-β and IL-10, has a profound impact on plaque evolution. In ApoE-/- mice, specific blockade of TGF-β signaling promotes atherosclerosis, as evidenced by increased macrophage activation, reduced collagen, increased plaque size, and increasing instability. In contrast, TGF-β overexpression reduced plaque vulnerability and atherosclerosis (202). Similarly, IL-10-deficient mice were found to have a substantially increased susceptibility to atherosclerosis compared to normal mice, as evidenced by increased T cell infiltration, high IFN-γ expression, and reduced collagen content (203). Overall, Tregs exert atheroprotective effects on atherosclerosis by secreting cytokines to inhibit plaque-associated inflammatory responses.
Using scRNA-seq screening methods, Winkels et al. (204) found five T cell populations in ApoE-/- mice; Gu et al. (205) identified three T cell populations; and Cochain et al. (150) detected four T cell populations in Ldlr-/- mice. Recently, Winkels et al. (206) synthesised most of the available scRNA-seq studies and outlined four T cell phenotypes that have been more consistently studied: Cxcr6-expressing T cells, where CXCR6 is expressed by CD4+ T cells and NKT cells and is able to direct T cell homing. The overall lack of CXCR6 reduces atherosclerosis and T cell accumulation (52). Naive T cells, expressing Cd28+Ccr7+, increase IL-7 signaling and support T cell survival, differentiation as well as proliferation in more advanced atherosclerosis (207). Cd8+ cytotoxic T cells, in scRNA-seq, about 31% of the T cells were identified as cytotoxic T cells based on the genotype and diet of the tested mice. The cells mainly expressed Cd8a/b,Nkg7, Ms4a4b, Ccl5 and Gzmk. Depletion of CD8+ T cells promotes atherosclerosis and reduces plaque stability (204). Another population of T cells that express both CD4 and CD8 or neither may represent thymocyte-like T cells (204), but this is still controversial.
5.4 B cell heterogeneity
B cells are heterogeneous populations of lymphocytes derived from bone marrow and composed of multiple subsets of cells with distinct localization properties, activation requirements, survival characteristics, and immunoglobulin secretion profiles (64). In mouse atherosclerosis, B cells are subdivided into two main subsets, B1 and B2, where B1 cells are divided into CD5+ B1a cells and CD5- B1b cells according to the expression of the leukocyte differentiation antigen CD5, and B2 cells contain follicular (FO) and marginal zone (MZ) B cells. CyTOF, and scRNA-Seq also confirmed the presence of B1- and B2-like cells in mouse atherosclerotic vessels (204).B1-like cell cluster is enriched for B1 cell genes (Tppp3, S100a6, and Cd9), and the B2-like cell cluster shares gene expression with germinal center and MZ B cells, for example, Fcer2a and Cd23 (147).Various subsets of B cells can contribute to diverse, and sometimes contradictory roles in the development of atherosclerotic plaques.
B1 cells are intrinsic immune cells that are usually found in plasma membrane cavities such as the pleural and peritoneal cavities. Without the need for Th cells, both B1a and B1b cells can secrete IgM under antigenic stimulation, which specifically binds to LDL oxidizing epitopes in atherosclerotic plaques and reduces lipid uptake by macrophages, thus decreases the formation of foam cells. At the same time, IgM specifically binds to apoptotic cells so that inflammatory cells can be eliminated, thus slowing down atherosclerosis progression (64). B1a cells undergo migration to the spleen and differentiate into GM-CSF-producing innate response activator (IRA) B cells stimulated by LPS. In atherosclerotic mice, IRA-B cells congregate in large numbers in secondary lymphoid tissues, activate the Th1 immune response, stimulate extraparenchymal hematopoiesis, and activate DCs to promote atherosclerotic plaque formation (208). Kyaw et al. (209) found that splenectomy in ApoE-/- mice resulted in a decrease in peritoneal B1a cells as well as a substantial reduction in plasma IgM levels; however, transfer of B1a cells to splenectomized mice was effective in attenuating atherosclerotic lesions and reducing plaque necrotic cores and apoptotic cells. However, the role of B1b cells in atherosclerosis remains uncertain and necessitates additional investigation. Overall, B1 cells exert a protective effect against atherosclerosis primarily through the secretion of natural IgM antibodies that bind oxLDL and apoptotic cells.
B2 cells are what we commonly refer to as mature B lymphocytes, produced in the bone marrow and differentiated in secondary lymphoid tissues. Preliminary studies on the role of B2 cells in atherosclerosis have demonstrated their pro-atherogenic effect. The use of a CD20-specific monoclonal antibody to selectively remove B2 cells, but not B1a cells, from ApoE-/- and Ldlr-/- mice reduced atherogenesis and progression of atherosclerosis (73, 74). In addition, the fact that B2 cell depletion was correlated with the reduction of activated splenic CD4+ T cells, T cells proliferation, and diseased T cells suggests that B2 cells through a T cell-dependent mechanism exacerbate atherosclerosis (210). Consistent with the effects of B2 cell depletion, Kyaw et al. (74) found that the introduction of splenic B2 cells, but not B1 cells into lymphocyte-deficient recombinase activating gene 2 (Rag2)-/-γ-chain-/-ApoE-/- or B-cell-deficient/ApoE-/- mice through adoptive transfer exacerbated atherosclerosis. The pro-atherogenic effect of B2 cells was further demonstrated by studies of B cell-activating factor receptor (BAFFR)-deficient effects in atherosclerosis-prone mice. Being a member of the tumor necrosis factor receptor family, BAFFR plays a crucial role in the maintenance of mature B2 cells (211). In a separate study, Kyaw et al. (212) showed that selective inhibition of BAFFR with an anti-BAFFR antibody resulted in depletion of B2 lymphocytes and reduction of atherosclerosis in ApoE-/- mice. Collectively, these studies provide clear evidence that substantial reduction in B2 cell count mitigates the progression of atherosclerosis. In contrast to Kyaw et al., however, Doran et al. (213) discovered that adoptive transfer of ApoE-/- mouse splenic B2 cells into cholesterol-fed μMT/ApoE-/- mice significantly reduced the area of atherosclerotic plaques in mice. This discrepancy may be due to differences in the ratio of FO to MZ B cells or in the genetic background of the mice. Further research has confirmed the specific role of FO and MZ B2 cell in atherosclerosis. Namely, FO B cells promote atherosclerosis primarily through IgG production and activation of Th1 cells (214). Similarly, MZ B cells promote atherosclerotic plaque progression by secreting IgG, activating follicular helper T (Tfh) cells, and inducing the expression of inflammatory factors such as IFN-γ (215).
The variety of functions performed by B cells, such as antibody production, cytokine release, and antigen presentation, as well as the unique ways in which B cells adapt to the inflammatory microenvironment of plaques, have led to different B cell subsets exhibiting complex heterogeneity in the evolution of atherosclerotic plaques.
6 Immunotherapy for atherosclerosis
The evolution of atherosclerotic plaques is regulated by both the immune system and inflammatory responses, and the study of immune as well as anti-inflammatory based therapies for atherosclerosis is of great clinical importance. Canakinumab is a human monoclonal antibody (mAb) that binds to IL-1β and blocks the interplay of IL-1β with IL-1R, preventing the inflammatory response from occurring. IL-1β plays multiple roles in the atherosclerotic process, including promoting adhesion of immune cells to vascular endothelial cells, promoting proliferation of VSMCs, and inducing procoagulant activity (216). A large-scale, double blind, randomized clinical trial, the Canakinumab Anti-Inflammatory Thrombosis Outcome Study (CANTOS), has confirmed for the first time the inflammatory hypothesis of atherosclerosis (217). By dividing 10,061 atherosclerotic patients with prior myocardial infarction and a high-sensitivity C-reactive protein (hs-CRP) ≥2 mg/L in 39 countries into a placebo group and three different doses of canakinumab, administered subcutaneously every three months. The risk of canakinumab on primary endpoint events including nonfatal myocardial infarction, nonfatal stroke, and cardiovascular death was assessed. Ultimately, the 150 mg and 300 mg doses of canakinumab were found to reduce patients’ levels of hs-CRP and other inflammatory markers, the risk of cardiovascular events, and the incidence and mortality of selected cancers (218). It is worth noting that the use of canakinumab is accompanied by certain risks, and further studies are needed to investigate its precise mechanism of action as well as its safety and efficacy in different patients. Overall, the CANTOS study opens up new avenues for immunotherapy in cardiovascular disease.
Advances have also been made in the study of antibody immunotherapies targeting other cytokines that play pro-inflammatory roles in plaque evolution, as well as chemokines and their receptors that also have important roles. In patients with psoriatic arthritis, the use of anti-TNF-α antibodies significantly reduced the size of carotid plaques and inhibited plaque development (219). Additionally, adalimumab treatment steadily reduced the levels of E-selectin, hs-CRP, and IL-22, suppressed systemic inflammation, and decreased the risk of atherosclerosis (220). At chronic kidney disease patients, the anti-IL-6 antibody remarkably decreased biomarkers of atherosclerotic inflammation and thrombosis (221). Blocking the binding of CCR2 to its ligand CCL2 with the highly specific MLN1202 in patients with atherosclerotic cardiovascular risk reduced the levels of hs-CRP (222).
Anti-programmed cell death protein 1 (PD-1) mAb has recently been found to exhibit potential therapeutic efficacy in the treatment of atherosclerosis. There are activated subsets of PD-1+ T cells in the plaques that act as pro-inflammatory (223). PD-1 belongs to the immunoglobulin superfamily and is mainly expressed on the surface of T cells. It usually binds to its ligands PD-L1 and PD-L2, mediating immune suppression signals. It plays an important role in regulating T cell functions such as proliferation, survival, cytokine production and other effector functions and maintaining immune system homeostasis (224). A latest study by Fan et al. (225) found that the use of an anti-PD-1 mAb that binds to Fc was effective in reducing the size of human atherosclerotic plaques. The potential mechanism of action is that the Fc-binding ability of the anti-PD-1 mAb enables it to be trapped by the Fcγ receptors, which then interact with PD-1 expressed on the surface of T cells as an alternative PD-1 ligand to inhibit PD-1+ T-cell function in atherosclerotic plaques. This finding suggests that T cell- targeting immunotherapy could be a novel strategy to address atherosclerosis. Here we have only reviewed relevant antibody immunotherapies that have been used in human clinical trial studies (Table 2). It is known that quite a number of immune molecules show considerable therapeutic prospects in animal models. We believe that future studies will reveal more targets and translate them into clinical applications, providing more options and possibilities for the treatment of atherosclerosis.
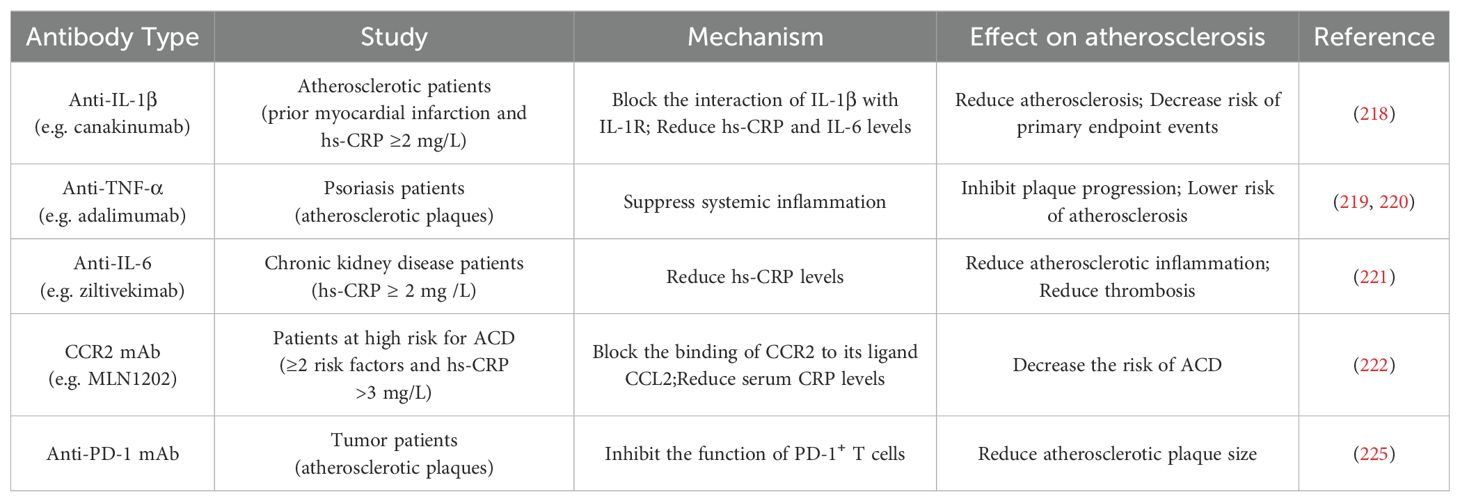
Table 2. Antibody immunotherapy for human atherosclerosis.
Antigen-antibody reactions play a pivotal role in the evolution of plaques. Therefore, the use of immunomodulatory strategies to activate immune responses against relevant antigens has the potential to alter the natural course of atherosclerosis. The purpose of vaccination is to prevent the progression of atherosclerosis by stimulating the body to produce antibodies that block the target antigen. What has received earlier attention from researchers is LDL-related vaccines. OxLDL is thought to be critical in causing intimal inflammation and foam cell formation in atherosclerosis. One of the most studied is the specific malondialdehyde-modified apolipoprotein B100 (MDA-ApoB100), which is a relatively important oligopeptide fragment of oxLDL molecules with strong antigenicity (226). Fredrikson et al. (227) reported that ApoE-/- atherosclerotic mice vaccinated with a peptide vaccine of the MDA-ApoB100 fragment showed a reduction of atherosclerotic plaques by up to 60%, and a significant increase in collagen content in the residual plaques, and a decrease in macrophages, which increased the stability of plaques to some extent and inhibited plaque evolution. In addition to more studies on oxLDL-related vaccines, the results of cholesterol transporter protein (CETP) vaccine studies have been promising. The primary function of CETP in atherosclerosis is to regulate the transport of high-density lipoprotein (HDL) cholesterol to LDL cholesterol (228). CETP-associated vaccines play a role in inhibiting plaque development through mechanisms such as elevating HDL levels, decreasing LDL levels to promote reverse cholesterol transport, and reducing plaque burden (229). Other vaccines such as CD40L and PCSK9 have been found to play an inhibitory role in atherosclerotic plaque evolution (230–233). Plaque evolution is a complex, multifactorial process. From the perspective of developing an anti-atherosclerotic vaccine, not only can we explore the impact of immunotherapy on the mechanism of plaque evolution, but also contribute substantially to the prevention and treatment of atherosclerosis.
Inflammation is a well-studied therapeutic target, and strategies to control inflammation have been successfully applied against many diseases. After many studies it appears that inflammation has also emerged as an important target for the treatment of atherosclerosis. Anti-inflammatory therapeutic agents for atherosclerosis have been extensively studied in recent years. Among the most widely researched and used drugs are statins, which are inhibitors of 3-hydroxy-3-methylglutaryl coenzyme A reductase and are usually used to treat atherosclerosis. They are the drugs of choice for clinical lipid-lowering, with commonly used drugs such as rosuvar, atorval, and simvastatin calcium tablets. Statins exert various anti-inflammatory effects in addition to their lipid-lowering properties. Statins can inhibit macrophage growth, reduce the level of pro-inflammatory cytokines, downregulate the expression of adhesion molecules and chemokines, and prevent monocyte recruitment (234), as well as inhibit T cell activation and attenuate inflammatory responses through direct binding to lymphocyte function-associated antigen-1 (235). Furthermore, statins promote the stabilization of plaques by decreasing MMP expression and inhibiting tissue factor to reduce the risk of thrombotic events (236). In summary, statins can prevent, reduce, and even reverse atherosclerotic plaque burden. Reducing inflammation may be a key mechanism by which statins alter plaque biology and slow disease progression.
In addition to statins, there are several common anti-inflammatory drugs that are also used to treat atherosclerosis. Colchicine is utilized for treating inflammatory diseases such as atherosclerosis due to its anti-inflammatory and antifibrotic activity. Its anti-inflammatory mechanism is mainly through downregulation of multiple inflammatory pathways including inhibition of phospholipase A2, reduction of leukotriene B4 and prostaglandin E2 release from monocytes, inhibition of neutrophil function through microtubule proteins, and reduction of endothelial adhesion, which leads to inhibition of inflammation and increase of plaque stabilization (237–239). Methotrexate is a better non-specific anti-inflammatory agent for treating atherosclerosis due to its lesser effect on the levels of atherosclerosis-related cytokines. Methotrexate performs its anti-inflammatory effect by inhibiting folate metabolism and reducing T-cell proliferation (240). In addition, methotrexate can regulate the expression of intercellular cell adhesion molecule-1, E-selectin, and VCAM-1, suppress cyclooxygenase and lipoxygenase, decrease C-reactive protein levels, and regulate the secretion of IL-6, TNF-α, and metalloproteinases, thereby regulating plaque evolution (241). In summary, targeting inflammatory pathways to prevent and treat atherosclerosis is a promising new avenue.
7 Conclusion
Atherosclerosis is defined as an inflammatory disease, with inflammatory responses throughout the disease progression, and inflammatory mediators, cytokines, through inflammatory signaling pathways exerting different roles in the regulation of the plaque inflammatory microenvironment. Many studies have clearly demonstrated that immune responses mediate the entire process of atherosclerotic plaque evolution, including initiation, progression, and thrombotic complications. Crosstalk between innate and adaptive immune pathways strongly regulates plaque activity and progression, while the heterogeneity of immune cells plays a pivotal role in plaque evolution.
The evolution of atherosclerosis involves complex interactions of immune cells as well as phenotypic plasticity. Omics studies, especially scRNA-seq, have highlighted the specific transcriptional profiles of various cell lineages at the site of atherosclerotic lesions. The detailed map of the cells revealed not only enhances our understanding of the heterogeneity of different cells in the plaque environment, but also deepens our comprehension of the mechanisms of disease occurrence and progression and provides a solid scientific basis for the development of novel precision therapeutic strategies. By combining high-throughput omics data with experimental studies to analyze the gene expression profiles and functional properties of specific cell subsets, it is possible to develop targeted drugs against specific cell subsets or the immune molecules they produce, such as immune checkpoint inhibitors against PD1+ T cells (225), antibodies against pro-inflammatory cytokines or chemokines (218–222). Gene expression products and functional markers may also become important biomarkers for assessing atherosclerosis disease, predicting progression and monitoring treatment effects, and the development of these biomarkers could help to achieve early diagnosis and precise treatment of the disease. Furthermore, adjusting the dose and type of therapeutic drugs according to the number of immune cells and the level of cytokines produced, and developing a more personalized treatment plan can improve the effectiveness of treatment and reduce adverse reactions. In addition, understanding the interactions between different cell subsets and exploring the possibility of combining drugs, such as an integrated treatment strategy combining lipid-lowering and anti-inflammatory drugs, has shown certain efficacy in clinical trials (242, 243), with a view to achieving breakthroughs in reducing plaque formation, stabilizing lesions and preventing MACEs.
However, the vast majority of investigations into the pathophysiologic mechanisms of atherosclerotic plaque pathology have originated from experimental animal models, and there are significant barriers to translating interventions from animal models to the clinic, primarily because mouse models reproduce human atherosclerosis to a very limited extent. It is encouraging that the integration of bioinformatics-, transcriptomics-, and proteomics-based datasets will help us to define the immune profile of human atherosclerosis in a more comprehensive manner. As multidimensional approaches continue to progress, it is expected that the cellular and molecular mediators associated with human atherosclerotic plaques will be characterized in depth, and new potential therapeutic targets will undoubtedly emerge. It is our firm belief that our understanding of immune-mediated processes in atherosclerosis will continue to evolve, and the specific mechanisms of atherosclerotic plaque evolution will be further resolved from an immunologic perspective.
Author contributions
HP: Writing – original draft. GW: Writing – original draft. YW: Writing – original draft. MZ: Writing – original draft. QH: Writing – original draft. XZ: Writing – review & editing. KY: Writing – review & editing. GZ: Writing – review & editing. TJ: Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the National Natural Science Foundation of China (81870337, 82370463, 82060065) and the Natural Science Foundation of Guangdong Province (2021A1515010717).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Barquera S, Pedroza-Tobias A, Medina C, Hernández-Barrera L, Bibbins-Domingo K, Lozano R, et al. Global overview of the epidemiology of atherosclerotic cardiovascular disease. Arch Med Res. (2015) 46(5):328–38. doi: 10.1016/j.arcmed.2015.06.006
2. Camaré C, Pucelle M, Nègre-Salvayre A, Salvayre R. Angiogenesis in the atherosclerotic plaque. Redox Biol. (2017) 12:18–34. doi: 10.1016/j.redox.2017.01.007
3. Libby P, Buring JE, Badimon L, Hansson GK, Deanfield J, Bittencourt MS, et al. Atherosclerosis. Nat Rev Dis Primers. (2019) 5(1):56. doi: 10.1038/s41572-019-0106-z
4. Libby P. The changing landscape of atherosclerosis. Nature. (2021) 592(7855):524–33. doi: 10.1038/s41586-021-03392-8
5. Rafieian-Kopaei M, Setorki M, Doudi M, Baradaran A, Nasri H. Atherosclerosis: process, indicators, risk factors and new hopes. Int J Prev Med. (2014) 5(8):927–46.
6. Jebari-Benslaiman S, Galicia-García U, Larrea-Sebal A, Olaetxea JR, Alloza I, Vandenbroeck K, et al. Pathophysiology of atherosclerosis. Int J Mol Sci. (2022) 23(6):3346. doi: 10.3390/ijms23063346
7. Berenson GS, Srinivasan SR, Freedman DS, Radhakrishnamurthy B, Dalferes ER Jr. Atherosclerosis and its evolution in childhood. Am J Med Sci. (1987) 294(6):429–40. doi: 10.1097/00000441-198712000-00008
8. Camici PG, Rimoldi OE, Gaemperli O, Libby P. Non-invasive anatomic and functional imaging of vascular inflammation and unstable plaque. Eur Heart J. (2012) 33(11):1309–17. doi: 10.1093/eurheartj/ehs067
9. Razeghian-Jahromi I, Karimi Akhormeh A, Razmkhah M, Zibaeenezhad MJ. Immune system and atherosclerosis: Hostile or friendly relationship. Int J Immunopathology Pharmacol. (2022) 36:1190312284. doi: 10.1177/03946320221092188
10. Hughes CE, Nibbs RJB. A guide to chemokines and their receptors. FEBS J. (2018) 285(16):2944–71. doi: 10.1111/febs.14466
11. Gencer S, Evans BR, van der Vorst EPC, Döring Y, Weber C. Inflammatory chemokines in atherosclerosis. Cells. (2021) 10(2):226. doi: 10.3390/cells10020226
12. On the origin of species by means of natural selection, or the preservation of favoured races in the struggle for life. Br Foreign Medico-Chirurgical Rev. (1860) 25(50):367–404.
13. Stubbington M, Rozenblatt-Rosen O, Regev A, Teichmann SA. Single-cell transcriptomics to explore the immune system in health and disease. Science. (2017) 358(6359):58–63. doi: 10.1126/science.aan6828
14. Kubo T, Maehara A, Mintz GS, Doi H, Tsujita K, Choi SY, et al. The dynamic nature of coronary artery lesion morphology assessed by serial virtual histology intravascular ultrasound tissue characterization. J Am Coll Cardiol. (2010) 55(15):1590–7. doi: 10.1016/j.jacc.2009.07.078
15. Deliargyris EN. Intravascular ultrasound virtual histology derived thin cap fibroatheroma now you see it, now you don't. J Am Coll Cardiol. (2010) 55(15):1598–9. doi: 10.1016/j.jacc.2009.10.069
16. Herrero-Fernandez B, Gomez-Bris R, Somovilla-Crespo B, Gonzalez-Granado JM. Immunobiology of atherosclerosis: A complex net of interactions. Int J Mol Sci. (2019) 20(21):5293. doi: 10.3390/ijms20215293
17. Murshid A, Borges TJ, Lang BJ, Calderwood SK. The scavenger receptor SREC-i cooperates with toll-like receptors to trigger inflammatory innate immune responses. Front Immunol. (2016) 7:226. doi: 10.3389/fimmu.2016.00226
18. Roy P, Orecchioni M, Ley K. How the immune system shapes atherosclerosis: roles of innate and adaptive immunity. Nat Rev Immunol. (2022) 22(4):251–65. doi: 10.1038/s41577-021-00584-1
20. Reitsma S, Slaaf DW, Vink H, van Zandvoort MA, oude Egbrink MG. The endothelial glycocalyx: composition, functions, and visualization. Pflugers Archiv: Eur J Physiol. (2007) 454(3):345–59. doi: 10.1007/s00424-007-0212-8
21. Esper RJ, Nordaby RA, Vilariño JO, Paragano A, Cacharrón JL, Machado RA. Endothelial dysfunction: a comprehensive appraisal. Cardiovasc Diabetol. (2006) 5:4. doi: 10.1186/1475-2840-5-4
22. Liu Y, Sun Y, Hu C, Liu J, Gao A, Han H, et al. Perivascular adipose tissue as an indication, contributor to, and therapeutic target for atherosclerosis. Front Physiol. (2020) 11:615503. doi: 10.3389/fphys.2020.615503
23. Qi XY, Qu SL, Xiong WH, Rom O, Chang L, Jiang ZS. Perivascular adipose tissue (PVAT) in atherosclerosis: a double-edged sword. Cardiovasc Diabetology. (2018) 17(1):134. doi: 10.1186/s12933-018-0777-x
24. Nosalski R, Guzik TJ. Perivascular adipose tissue inflammation in vascular disease. Br J Pharmacol. (2017) 174(20):3496–513. doi: 10.1111/bph.13705
25. Mundi S, Massaro M, Scoditti E, Carluccio MA, van Hinsbergh VWM, Iruela-Arispe ML, et al. And cardiovascular risk factors-a review. Cardiovasc Res. (2018) 114(1):35–52. doi: 10.1093/cvr/cvx226
26. Borén J, Williams KJ. The central role of arterial retention of cholesterol-rich apolipoprotein-b-containing lipoproteins in the pathogenesis of atherosclerosis: a triumph of simplicity. Curr Opin Lipidology. (2016) 27(5):473–83. doi: 10.1097/MOL.0000000000000330
27. Shashkin P, Dragulev B, Ley K. Macrophage differentiation to foam cells. Curr Pharm Design. (2005) 11(23):3061–72. doi: 10.2174/1381612054865064
28. Chistiakov DA, Melnichenko AA, Myasoedova VA, Grechko AV, Orekhov AN. Mechanisms of foam cell formation in atherosclerosis. J Mol Med (Berl). (2017) 95(11):1153–65. doi: 10.1007/s00109-017-1575-8
29. Clinton SK, Underwood R, Hayes L, Sherman ML, Kufe DW, Libby P. Macrophage colony-stimulating factor gene expression in vascular cells and in experimental and human atherosclerosis. Am J Pathol. (1992) 140(2):301–16.
30. Edfeldt K, Swedenborg J, Hansson GK, Yan ZQ. Expression of toll-like receptors in human atherosclerotic lesions: a possible pathway for plaque activation. Circulation. (2002) 105(10):1158–61. doi: 10.1161/circ.105.10.1158
31. Xu XH, Shah PK, Faure E, Equils O, Thomas L, Fishbein MC, et al. Toll-like receptor-4 is expressed by macrophages in murine and human lipid-rich atherosclerotic plaques and upregulated by oxidized LDL. Circulation. (2001) 104(25):3103–8. doi: 10.1161/hc5001.100631
32. Michelsen KS, Wong MH, Shah PK, Zhang W, Yano J, Doherty TM, et al. Lack of toll-like receptor 4 or myeloid differentiation factor 88 reduces atherosclerosis and alters plaque phenotype in mice deficient in apolipoprotein E. Proc Natl Acad Sci United States America. (2004) 101(29):10679–84. doi: 10.1073/pnas.0403249101
33. Silvestre-Roig C, Braster Q, Ortega-Gomez A, Soehnlein O. Neutrophils as regulators of cardiovascular inflammation. Nat Rev Cardiol. (2020) 17(6):327–40. doi: 10.1038/s41569-019-0326-7
34. Soehnlein O. Multiple roles for neutrophils in atherosclerosis. Circ Res. (2012) 110(6):875–88. doi: 10.1161/CIRCRESAHA.111.257535
35. Warnatsch A, Ioannou M, Wang Q, Papayannopoulos V. Inflammation neutrophil extracellular traps license macrophages for cytokine production in atherosclerosis. Sci (New York N.Y.). (2015) 349(6245):316–20. doi: 10.1126/science.aaa8064
36. Döring Y, Soehnlein O, Weber C. Neutrophil extracellular traps in atherosclerosis and atherothrombosis. Circ Res. (2017) 120(4):736–43. doi: 10.1161/CIRCRESAHA.116.309692
37. Vivier E, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, et al. Innate lymphoid cells: 10 years on. Cell. (2018) 174(5):1054–66. doi: 10.1016/j.cell.2018.07.017
38. Klose CSN, Artis D. Innate lymphoid cells as regulators of immunity, inflammation and tissue homeostasis. Nat Immunol. (2016) 17(7):765–74. doi: 10.1038/ni.3489
39. Pertiwi KR, Teunissen MBM, Krebbers G, Willems MCM, Huisman L, Poelen C, et al. Enrichment of type 1 innate lymphoid cells in the course of human atherosclerotic plaque development suggests contribution to atherogenesis. Front Immunol. (2024) 15:1354617. doi: 10.3389/fimmu.2024.1354617
40. Perry HM, Oldham SN, Fahl SP, Que X, Gonen A, Harmon DB, et al. Helix-loop-helix factor inhibitor of differentiation 3 regulates interleukin-5 expression and B-1a B cell proliferation. Arteriosclerosis Thrombosis Vasc Biol. (2013) 33(12):2771–9. doi: 10.1161/ATVBAHA.113.302571
41. Engelbertsen D, Lichtman AH. Innate lymphoid cells in atherosclerosis. Eur J Pharmacol. (2017) 816:32–6. doi: 10.1016/j.ejphar.2017.04.030
42. Vivier E, Tomasello E, Baratin M, Walzer T, Ugolini S. Functions of natural killer cells. Nat Immunol. (2008) 9(5):503–10. doi: 10.1038/ni1582
43. Bobryshev YV, Lord RSA. Identification of natural killer cells in human atherosclerotic plaque. Atherosclerosis. (2005) 180:423–7. doi: 10.1016/j.atherosclerosis.2005.01.046
44. Whitman SC, Rateri DL, Szilvassy SJ, Yokoyama W, Daugherty A. Depletion of natural killer cell function decreases atherosclerosis in low-density lipoprotein receptor null mice. Arteriosclerosis Thrombosis Vasc Biol. (2004) 24(6):1049–54. doi: 10.1161/01.ATV.0000124923.95545.2c
45. Schiller NK, Boisvert WA, Curtiss LK. Inflammation in atherosclerosis: lesion formation in LDL receptor-deficient mice with perforin and lyst(beige) mutations. Arteriosclerosis Thrombosis Vasc Biol. (2002) 22(8):1341–6. doi: 10.1161/01.ATV.0000024082.46387.38
46. Selathurai A, Deswaerte V, Kanellakis P, Tipping P, Toh BH, Bobik A, et al. Natural killer (NK) cells augment atherosclerosis by cytotoxic-dependent mechanisms. Cardiovasc Res. (2014) 102(1):128–37. doi: 10.1093/cvr/cvu016
47. Zernecke A. Dendritic cells in atherosclerosis: evidence in mice and humans. Arteriosclerosis Thrombosis Vasc Biol. (2015) 35(4):763–70. doi: 10.1161/ATVBAHA.114.303566
48. Koltsova EK, Ley K. How dendritic cells shape atherosclerosis. Trends Immunol. (2011) 32(11):540–7. doi: 10.1016/j.it.2011.07.001
49. Miteva K, Madonna R, De Caterina R, Van Linthout S. Innate and adaptive immunity in atherosclerosis. Vasc Pharmacol. (2018) 22:S1537–1891(17)30464-0. doi: 10.1016/j.vph.2018.04.006
50. Karin N, Wildbaum G, Thelen M. Biased signaling pathways via CXCR3 control the development and function of CD4+ T cell subsets. J Leukocyte Biol. (2016) 99(6):857–62. doi: 10.1189/jlb.2MR0915-441R
51. van Wanrooij EJA, de Jager SCA, van Es T, de Vos P, Birch HL, Owen DA, et al. CXCR3 antagonist NBI-74330 attenuates atherosclerotic plaque formation in LDL receptor-deficient mice. Arteriosclerosis Thrombosis Vasc Biol. (2008) 28(2):251–7. doi: 10.1161/ATVBAHA.107.147827
52. Galkina E, Harry BL, Ludwig A, Liehn EA, Sanders JM, Bruce A, et al. CXCR6 promotes atherosclerosis by supporting t-cell homing, interferon-gamma production, and macrophage accumulation in the aortic wall. Circulation. (2007) 116(16):1801–11. doi: 10.1161/CIRCULATIONAHA.106.678474
53. Li J, Mcardle S, Gholami A, Kimura T, Wolf D, Gerhardt T, et al. CCR5+T-bet+FoxP3+ effector CD4 T cells drive atherosclerosis. Circ Res. (2016) 118(10):1540–52. doi: 10.1161/CIRCRESAHA.116.308648
54. Veillard NR, Steffens S, Pelli G, Lu B, Kwak BR, Gerard C, et al. Differential influence of chemokine receptors CCR2 and CXCR3 in development of atherosclerosis in vivo. Circulation. (2005) 112(6):870–8. doi: 10.1161/CIRCULATIONAHA.104.520718
55. Luchtefeld M, Grothusen C, Gagalick A, Jagavelu K, Schuett H, Tietge UJ, et al. Chemokine receptor 7 knockout attenuates atherosclerotic plaque development. Circulation. (2010) 122(16):1621–8. doi: 10.1161/CIRCULATIONAHA.110.956730
56. Zhou X, Robertson AL, Rudling M, Parini P, Hansson GK. Lesion development and response to immunization reveal a complex role for CD4 in atherosclerosis. Circ Res. (2005) 96(4):427–34. doi: 10.1161/01.RES.0000156889.22364.f1
57. Zhou X, Nicoletti A, Elhage R, Hansson GK. Transfer of CD4(+) T cells aggravates atherosclerosis in immunodeficient apolipoprotein E knockout mice. Circulation. (2000) 102(24):2919–22. doi: 10.1161/01.CIR.102.24.2919
58. Vila-Caballer M, González-Granado JM, Zorita V, Abu Nabah YN, Silvestre-Roig C, Del Monte-Monge A, et al. Disruption of the CCL1-CCR8 axis inhibits vascular treg recruitment and function and promotes atherosclerosis in mice. J Mol Cell Cardiol. (2019) 132:154–63. doi: 10.1016/j.yjmcc.2019.05.009
59. Weber C, Meiler S, Döring Y, Koch M, Drechsler M, Megens RT, et al. CCL17-expressing dendritic cells drive atherosclerosis by restraining regulatory T cell homeostasis in mice. J Clin Invest. (2011) 121(7):2898–910. doi: 10.1172/JCI44925
60. Shao Y, Yang WY, Saaoud F, Drummer C 4th, Sun Y, Xu K, et al. IL-35 promotes CD4+Foxp3+ tregs and inhibits atherosclerosis via maintaining CCR5-amplified treg-suppressive mechanisms. JCI Insight. (2021) 6(19):e152511. doi: 10.1172/jci.insight.152511
61. Lichtman AH, Binder CJ, Tsimikas S, Witztum JL. Adaptive immunity in atherogenesis: new insights and therapeutic approaches. J Clin Invest. (2013) 123(1):27–36. doi: 10.1172/JCI63108
62. Kolbus D, Ljungcrantz I, Söderberg I, Alm R, Björkbacka H, Nilsson J, et al. TAP1-deficiency does not alter atherosclerosis development in apoe-/- mice. PloS One. (2012) 7(3):e33932. doi: 10.1371/journal.pone.0033932
63. Kyaw T, Winship A, Tay C, Kanellakis P, Hosseini H, Cao A, et al. Cytotoxic and proinflammatory CD8+ T lymphocytes promote development of vulnerable atherosclerotic plaques in apoE-deficient mice. Circulation. (2013) 127(9):1028–39. doi: 10.1161/CIRCULATIONAHA.112.001347
64. Tsiantoulas D, Sage AP, Mallat Z, Binder CJ. Targeting B cells in atherosclerosis: closing the gap from bench to bedside. Arteriosclerosis Thrombosis Vasc Biol. (2015) 35(2):296–302. doi: 10.1161/ATVBAHA.114.303569
65. Houtkamp MA, de Boer OJ, van der Loos CM, van der Wal AC, Becker AE. Adventitial infiltrates associated with advanced atherosclerotic plaques: structural organization suggests generation of local humoral immune responses. J Pathology. (2001) 193(2):263–9. doi: 10.1002/1096-9896(2000)9999:9999<::AID-PATH774>3.0.CO;2-N
66. Vlaicu R, Rus HG, Niculescu F, Cristea A. Immunoglobulins and complement components in human aortic atherosclerotic intima. Atherosclerosis. (1985) 55(1):35–50. doi: 10.1016/0021-9150(85)90164-9
67. van Leeuwen M, Damoiseaux J, Duijvestijn A, Tervaert JW. The therapeutic potential of targeting B cells and anti-oxLDL antibodies in atherosclerosis. Autoimmun Rev. (2009) 9(1):53–7. doi: 10.1016/j.autrev.2009.03.001
68. Palinski W, Hörkkö S, Miller E, Steinbrecher UP, Powell HC, Curtiss LK, et al. Cloning of monoclonal autoantibodies to epitopes of oxidized lipoproteins from apolipoprotein E-deficient mice. demonstration of epitopes of oxidized low density lipoprotein in human plasma. J Clin Invest. (1996) 98(3):800–14.
69. Caligiuri G, Nicoletti A, Poirier B, Hansson GK. Protective immunity against atherosclerosis carried by B cells of hypercholesterolemic mice. J Clin Invest. (2002) 109(6):745–53. doi: 10.1172/JCI7272
70. Major AS, Fazio S, Linton MF. B-lymphocyte deficiency increases atherosclerosis in LDL receptor-null mice. Arteriosclerosis Thrombosis Vasc Biol. (2002) 22(11):1892–8. doi: 10.1161/01.ATV.0000039169.47943.EE
71. Nicoletti A, Kaveri S, Caligiuri G, Bariéty J, Hansson GK. Immunoglobulin treatment reduces atherosclerosis in apo e knockout mice. J Clin Invest. (1998) 102(5):910–8. doi: 10.1172/JCI119892
72. Binder CJ, Hartvigsen K, Chang M, Miller M, Broide D, Palinski W, et al. IL-5 links adaptive and natural immunity specific for epitopes of oxidized LDL and protects from atherosclerosis. J Clin Invest. (2004) 114(3):427–37. doi: 10.1172/JCI200420479
73. Ait-Oufella H, Herbin O, Bouaziz J, Binder CJ, Uyttenhove C, Laurans L, et al. B cell depletion reduces the development of atherosclerosis in mice. J Exp Med. (2010) 207(8):1579–87. doi: 10.1084/jem.20100155
74. Kyaw T, Tay C, Khan A, Dumouchel V, Cao A, To K, et al. Conventional B2 B cell depletion ameliorates whereas its adoptive transfer aggravates atherosclerosis. J Immunol (Baltimore Md. 1950). (2010) 185(7):4410–9. doi: 10.4049/jimmunol.1000033
75. Yan ZQ, Hansson GK. Innate immunity, macrophage activation, and atherosclerosis. Immunol Rev. (2007) 219:187–203. doi: 10.1111/j.1600-065X.2007.00554.x
76. Zhu Y, Xian X, Wang Z, Bi Y, Chen Q, Han X, et al. Research progress on the relationship between atherosclerosis and inflammation. Biomolecules. (2018) 8(3):80. doi: 10.3390/biom8030080
77. Linton MF, Yancey PG, Davies SS, Jerome WG, Linton EF, Song WL, et al. The role of lipids and lipoproteins in atherosclerosis. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc. (2000–2019).
78. Yla-Herttuala S, Palinski W, Rosenfeld ME, Parthasarathy S, Carew TE, Butler S, et al. Evidence for the presence of oxidatively modified low density lipoprotein in atherosclerotic lesions of rabbit and man. J Clin Invest. (1989) 84(4):1086–95. doi: 10.1172/JCI114271
79. Parthasarathy S, Raghavamenon A, Garelnabi MO, Santanam N. Oxidized low-density lipoprotein. Methods Mol Biol (Clifton N.J.). (2010) 610:403–17. doi: 10.1007/978-1-60327-029-8_24
80. Chávez-Sánchez L, Madrid-Miller A, Chávez-Rueda K, Legorreta-Haquet MV, Tesoro-Cruz E, Blanco-Favela F. Activation of TLR2 and TLR4 by minimally modified low-density lipoprotein in human macrophages and monocytes triggers the inflammatory response. Hum Immunol. (2010) 71(8):737–44. doi: 10.1016/j.humimm.2010.05.005
81. Lopes-Virella MF, Virella G. Pathogenic role of modified LDL antibodies and immune complexes in atherosclerosis. J Atheroscl Thrombosis. (2013) 20(10):743–54. doi: 10.5551/jat.19281
82. Rhoads JP, Lukens JR, Wilhelm AJ, Moore JL, Mendez-Fernandez Y, Kanneganti TD, et al. Oxidized low-density lipoprotein immune complex priming of the Nlrp3 inflammasome involves TLR and FcγR cooperation and is dependent on CARD9. J Immunol (Baltimore Md. 1950). (2017) 198(5):2105–14. doi: 10.4049/jimmunol.1601563
83. Rajamäki K, Mäyränpää MI, Risco A, Tuimala J, Nurmi K, Cuenda A, et al. p38δ MAPK: A novel regulator of NLRP3 inflammasome activation with increased expression in coronary atherogenesis. Arteriosclerosis Thrombosis Vasc Biol. (2016) 36(9):1937–46. doi: 10.1161/ATVBAHA.115.307312
84. van der Heijden T, Kritikou E, Venema W, van Duijn J, van Santbrink PJ, Slütter B, et al. NLRP3 inflammasome inhibition by MCC950 reduces atherosclerotic lesion development in apolipoprotein E-deficient mice-brief report. Arteriosclerosis Thrombosis Vasc Biol. (2017) 37(8):1457–61. doi: 10.1161/ATVBAHA.117.309575
85. Park YM. CD36, a scavenger receptor implicated in atherosclerosis. Exp Mol Med. (2014) 46(6):e99. doi: 10.1038/emm.2014.38
86. Kataoka H, Kume N, Miyamoto S, Minami M, Moriwaki H, Murase T, et al. Expression of lectinlike oxidized low-density lipoprotein receptor-1 in human atherosclerotic lesions. Circulation. (1999) 99(24):3110–7. doi: 10.1161/01.CIR.99.24.3110
87. Akhmedov A, Rozenberg I, Paneni F, Camici GG, Shi Y, Doerries C, et al. Endothelial overexpression of LOX-1 increases plaque formation and promotes atherosclerosis in vivo. Eur Heart J. (2014) 35(40):2839–48. doi: 10.1093/eurheartj/eht532
88. Ishino S, Mukai T, Kume N, Asano D, Ogawa M, Kuge Y, et al. Lectin-like oxidized LDL receptor-1 (LOX-1) expression is associated with atherosclerotic plaque instability–analysis in hypercholesterolemic rabbits. Atherosclerosis. (2007) 195(1):48–56. doi: 10.1016/j.atherosclerosis.2006.11.031
89. Mehta JL, Sanada N, Hu CP, Chen J, Dandapat A, Sugawara F, et al. Deletion of LOX-1 reduces atherogenesis in LDLR knockout mice fed high cholesterol diet. Circ Res. (2007) 100(11):1634–42. doi: 10.1161/CIRCRESAHA.107.149724
90. Kobiyama K, Ley K. Atherosclerosis. Circ Res. (2018) 123(10):1118–20. doi: 10.1161/CIRCRESAHA.118.313816
91. Back M, Weber C, Lutgens E. Regulation of atherosclerotic plaque inflammation. J Intern Med. (2015) 278(5):462–82. doi: 10.1111/joim.12367
92. Yin M, Li C, Jiang J, Le J, Luo B, Yang F, et al. Cell adhesion molecule-mediated therapeutic strategies in atherosclerosis: From a biological basis and molecular mechanism to drug delivery nanosystems. Biochem Pharmacol. (2021) 186:114471. doi: 10.1016/j.bcp.2021.114471
93. Luo W, Wang H, Ohman MK, Guo C, Shi K, Wang J, et al. P-selectin glycoprotein ligand-1 deficiency leads to cytokine resistance and protection against atherosclerosis in apolipoprotein E deficient mice. Atherosclerosis. (2012) 220(1):110–7. doi: 10.1016/j.atherosclerosis.2011.10.012
94. An G, Wang H, Tang R, Yago T, McDaniel JM, McGee S, et al. P-selectin glycoprotein ligand-1 is highly expressed on ly-6Chi monocytes and a major determinant for ly-6Chi monocyte recruitment to sites of atherosclerosis in mice. Circulation. (2008) 117(25):3227–37. doi: 10.1161/CIRCULATIONAHA.108.771048
95. Oishi Y, Manabe I. Integrated regulation of the cellular metabolism and function of immune cells in adipose tissue. Clin Exp Pharmacol Physiol. (2016) 43(3):294–303. doi: 10.1111/1440-1681.12539
96. Gwon WG, Joung EJ, Kwon MS, Lim SJ, Utsuki T, Kim HR. Sargachromenol protects against vascular inflammation by preventing TNF-alpha-induced monocyte adhesion to primary endothelial cells via inhibition of NF-kappaB activation. Int Immunopharmacol. (2017) 42:81–9. doi: 10.1016/j.intimp.2016.11.014
97. Yu XH, Zheng XL, Tang CK. Nuclear factor-kappaB activation as a pathological mechanism of lipid metabolism and atherosclerosis. Adv Clin Chem. (2015) 70:1–30. doi: 10.1016/bs.acc.2015.03.004
98. Hajra L, Evans AI, Chen M, Hyduk SJ, Collins T, Cybulsky MI. The NF-kappa B signal transduction pathway in aortic endothelial cells is primed for activation in regions predisposed to atherosclerotic lesion formation. Proc Natl Acad Sci U S A. (2000) 97(16):9052–7. doi: 10.1073/pnas.97.16.9052
99. Lee CC, Avalos AM, Ploegh HL. Accessory molecules for toll-like receptors and their function. Nat Rev Immunol. (2012) 12(3):168–79. doi: 10.1038/nri3151
100. Leulier F, Lemaitre B. Toll-like receptors–taking an evolutionary approach. Nat Rev Genet. (2008) 9(3):165–78. doi: 10.1038/nrg2303
101. Yvan-Charvet L, Welch C, Pagler TA, Ranalletta M, Lamkanfi M, Han S, et al. Increased inflammatory gene expression in ABC transporter-deficient macrophages: free cholesterol accumulation, increased signaling via toll-like receptors, and neutrophil infiltration of atherosclerotic lesions. Circulation. (2008) 118(18):1837–47. doi: 10.1161/CIRCULATIONAHA.108.793869
102. Zhang X, Chen S, Yin G, Liang P, Feng Y, Yu W, et al. The role of JAK/STAT signaling pathway and its downstream influencing factors in the treatment of atherosclerosis. J Cardiovasc Pharmacol Ther. (2024) 29:2060161234. doi: 10.1177/10742484241248046
103. Gharavi NM, Alva JA, Mouillesseaux KP, Lai C, Yeh M, Yeung W, et al. Role of the Jak/STAT pathway in the regulation of interleukin-8 transcription by oxidized phospholipids in vitro and in atherosclerosis in vivo. J Biol Chem. (2007) 282(43):31460–8. doi: 10.1074/jbc.M704267200
104. Tsaousi A, Mill C, George SJ. The wnt pathways in vascular disease: lessons from vascular development. Curr Opin Lipidol. (2011) 22(5):350–7. doi: 10.1097/MOL.0b013e32834aa701
105. Souilhol C, Harmsen MC, Evans PC, Krenning G. Endothelial-mesenchymal transition in atherosclerosis. Cardiovasc Res. (2018) 114(4):565–77. doi: 10.1093/cvr/cvx253
106. Xiong W, Zhao X, Villacorta L, Rom O, Garcia-Barrio MT, Guo Y, et al. Brown adipocyte-specific PPARgamma (Peroxisome proliferator-activated receptor gamma) deletion impairs perivascular adipose tissue development and enhances atherosclerosis in mice. Arterioscler Thromb Vasc Biol. (2018) 38(8):1738–47. doi: 10.1161/ATVBAHA.118.311367
107. Khoshbayan A, Taheri F, Moghadam MT, Chegini Z, Shariati A. The association of chlamydia pneumoniae infection with atherosclerosis: Review and update of in vitro and animal studies. Microb Pathog. (2021) 154:104803. doi: 10.1016/j.micpath.2021.104803
108. Zhang J, Xie M, Huang X, Chen G, Yin Y, Lu X, et al. The effects of porphyromonas gingivalis on atherosclerosis-related cells. Front Immunol. (2021) 12:766560. doi: 10.3389/fimmu.2021.766560
109. Vourakis M, Mayer G, Rousseau G. The role of gut microbiota on cholesterol metabolism in atherosclerosis. Int J Mol Sci. (2021) 22(15):8074. doi: 10.3390/ijms22158074
110. Brandsma E, Kloosterhuis NJ, Koster M, Dekker DC, Gijbels MJJ, van der Velden S, et al. A proinflammatory gut microbiota increases systemic inflammation and accelerates atherosclerosis. Circ Res. (2019) 124(1):94–100. doi: 10.1161/CIRCRESAHA.118.313234
111. Cristescu CV, Alain S, Ruță SM. The role of CMV infection in primary lesions, development and clinical expression of atherosclerosis. J Clin Med. (2022) 11(13):3832. doi: 10.3390/jcm11133832
112. Hsich E, Zhou YF, Paigen B, Johnson TM, Burnett MS, Epstein SE. Cytomegalovirus infection increases development of atherosclerosis in apolipoprotein-e knockout mice. Atherosclerosis. (2001) 156(1):23–8. doi: 10.1016/S0021-9150(00)00608-0
113. Vallejo J, Cochain C, Zernecke A, Ley K. Heterogeneity of immune cells in human atherosclerosis revealed by scRNA-seq. Cardiovasc Res. (2021) 117(13):2537–43. doi: 10.1093/cvr/cvab260
114. Idzkowska E, Eljaszewicz A, Miklasz P, Musial WJ, Tycinska AM, Moniuszko M. The role of different monocyte subsets in the pathogenesis of atherosclerosis and acute coronary syndromes. Scandinavian J Immunol. (2015) 82(3):163–73. doi: 10.1111/sji.12314
115. Geissmann F, Jung S, Littman DR. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity. (2003) 19(1):71–82. doi: 10.1016/S1074-7613(03)00174-2
116. Stremmel C, Stark K, Schulz C. Heterogeneity of macrophages in atherosclerosis. Thromb Haemostasis. (2019) 119(8):1237–46. doi: 10.1055/s-0039-1692665
117. Hristov M, Heine GH. Monocyte subsets in atherosclerosis. Hamostaseologie. (2015) 35(2):105–12. doi: 10.5482/HAMO-14-08-0030
118. Yang J, Zhang L, Yu C, Yang XF, Wang H. Monocyte and macrophage differentiation: circulation inflammatory monocyte as biomarker for inflammatory diseases. biomark Res. (2014) 2(1):1. doi: 10.1186/2050-7771-2-1
119. Kapinsky M, Torzewski M, Büchler C, Duong CQ, Rothe G, Schmitz G. Enzymatically degraded LDL preferentially binds to CD14(high) CD16(+) monocytes and induces foam cell formation mediated only in part by the class B scavenger-receptor CD36. Z]. Arterioscler Thromb Vasc Biol. (2001) 21:1004–10. doi: 10.1161/01.ATV.21.6.1004
120. Cros J, Cagnard N, Woollard K, Patey N, Zhang SY, Senechal B, et al. Human CD14dim monocytes patrol and sense nucleic acids and viruses via TLR7 and TLR8 receptors. Immunity. (2010) 33(3):375–86. doi: 10.1016/j.immuni.2010.08.012
121. Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. (2008) 8(12):958–69. doi: 10.1038/nri2448
122. Chistiakov DA, Grechko AV, Myasoedova VA, Melnichenko AA, Orekhov AN. The role of monocytosis and neutrophilia in atherosclerosis. J Cell Mol Med. (2018) 22(3):1366–82. doi: 10.1111/jcmm.13462
123. Swirski FK, Libby P, Aikawa E, Alcaide P, Luscinskas FW, Weissleder R, et al. Ly-6Chi monocytes dominate hypercholesterolemia-associated monocytosis and give rise to macrophages in atheromata. J Clin Invest. (2007) 117(1):195–205. doi: 10.1172/JCI29950
124. Tacke F, Alvarez D, Kaplan TJ, Jakubzick C, Spanbroek R, Llodra J, et al. Monocyte subsets differentially employ CCR2, CCR5, and CX3CR1 to accumulate within atherosclerotic plaques. J Clin Invest. (2007) 117(1):185–94. doi: 10.1172/JCI28549
125. Quintar A, Mcardle S, Wolf D, Marki A, Ehinger E, Vassallo M, et al. Endothelial protective monocyte patrolling in large arteries intensified by western diet and atherosclerosis. Circ Res. (2017) 120(11):1789–99. doi: 10.1161/CIRCRESAHA.117.310739
126. Stöger JL, Gijbels MJJ, van der Velden S, Manca M, van der Loos CM, Biessen EA, et al. Distribution of macrophage polarization markers in human atherosclerosis. Atherosclerosis. (2012) 225(2):461–8. doi: 10.1016/j.atherosclerosis.2012.09.013
127. Wolfs IMJ, Donners MMPC, de Winther MPJ. Differentiation factors and cytokines in the atherosclerotic plaque micro-environment as a trigger for macrophage polarisation. Thromb Haemostasis. (2011) 106(5):763–71. doi: 10.1160/TH11-05-0320
128. Engelbertsen D, Foks AC, Alberts-Grill N, Kuperwaser F, Chen T, Lederer JA, et al. Expansion of CD25+ innate lymphoid cells reduces atherosclerosis. Arteriosclerosis Thrombosis Vasc Biol. (2015) 35(12):2526–35. doi: 10.1161/ATVBAHA.115.306048
129. Newland SA, Mohanta S, Clément M, Taleb S, Walker JA, Nus M, et al. Type-2 innate lymphoid cells control the development of atherosclerosis in mice. Nat Commun. (2017) 8:15781. doi: 10.1038/ncomms15781
130. Mantani PT, Dunér P, Ljungcrantz I, Nilsson J, Björkbacka H, Fredrikson GN. ILC2 transfers to apolipoprotein E deficient mice reduce the lipid content of atherosclerotic lesions. BMC Immunol. (2019) 20(1):47. doi: 10.1186/s12865-019-0330-z
131. You Y, Zhang X, Wang X, Yue D, Meng F, Zhu J, et al. ILC2 proliferated by IL-33 stimulation alleviates acute colitis in Rag1(-/-) mouse through promoting M2 macrophage polarization. J Immunol Res. (2020) 2020:5018975. doi: 10.1155/2020/5018975
132. Kral M, van der Vorst EPC, Surnov A, Weber C, Döring Y. ILC2-mediated immune crosstalk in chronic (vascular) inflammation. Front Immunol. (2023) 14:1326440. doi: 10.3389/fimmu.2023.1326440
133. Cardilo-Reis L, Gruber S, Schreier SM, Drechsler M, Papac-Milicevic N, Weber C, et al. Interleukin-13 protects from atherosclerosis and modulates plaque composition by skewing the macrophage phenotype. EMBO Mol Med. (2012) 4(10):1072–86. doi: 10.1002/emmm.201201374
134. Martinez FO, Sica A, Mantovani A, Locati M. Macrophage activation and polarization. Front Bioscience A J Virtual Library. (2008) 13:453–61. doi: 10.2741/2692
135. Mahdavian Delavary B, van der Veer WM, van Egmond M, Niessen FB, Beelen RH. Macrophages in skin injury and repair. Immunobiology. (2011) 216(7):753–62. doi: 10.1016/j.imbio.2011.01.001
136. Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. (2004) 25(12):677–86. doi: 10.1016/j.it.2004.09.015
137. Zizzo G, Hilliard BA, Monestier M, Cohen PL. Efficient clearance of early apoptotic cells by human macrophages requires M2c polarization and MerTK induction. J Immunol. (2012) 189(7):3508–20. doi: 10.4049/jimmunol.1200662
138. Ferrante CJ, Pinhal-Enfield G, Elson G, Cronstein BN, Hasko G, Outram S, et al. The adenosine-dependent angiogenic switch of macrophages to an M2-like phenotype is independent of interleukin-4 receptor alpha (IL-4Rα) signaling. Inflammation. (2013) 36(4):921–31. doi: 10.1007/s10753-013-9621-3
139. Pitsilos S, Hunt J, Mohler ER, Prabhakar AM, Poncz M, Dawicki J, et al. Platelet factor 4 localization in carotid atherosclerotic plaques: correlation with clinical parameters. Thromb Haemostasis. (2003) 90(6):1112–20. doi: 10.1160/TH03-02-0069
140. Gleissner CA, Ley K. CXCL4 in atherosclerosis: possible roles in monocyte arrest and macrophage foam cell formation. Thromb Haemost. (2007) 98:917–8. doi: 10.1160/th07-09-0567
141. Chinetti-Gbaguidi G, Colin S, Staels B. Macrophage subsets in atherosclerosis. Nat Rev Cardiol. (2015) 12(1):10–7. doi: 10.1038/nrcardio.2014.173
142. Boyle JJ, Johns M, Kampfer T, Nguyen AT, Game L, Schaer DJ, et al. Activating transcription factor 1 directs mhem atheroprotective macrophages through coordinated iron handling and foam cell protection. Circ Res. (2012) 110(1):20–33. doi: 10.1161/CIRCRESAHA.111.247577
143. Finn AV, Nakano M, Polavarapu R, Karmali V, Saeed O, Zhao X, et al. Hemoglobin directs macrophage differentiation and prevents foam cell formation in human atherosclerotic plaques. J Am Coll Cardiol. (2012) 59(2):166–77. doi: 10.1016/j.jacc.2011.10.852
144. Nielsen MJ, Møller HJ, Moestrup SK. Hemoglobin and heme scavenger receptors. Antioxidants Redox Signaling. (2010) 12(2):261–73. doi: 10.1089/ars.2009.2792
145. De Paoli F, Staels B, Chinetti-Gbaguidi G. Macrophage phenotypes and their modulation in atherosclerosis. Circ J Off J Japanese Circ Society. (2014) 78(8):1775–81. doi: 10.1253/circj.CJ-14-0621
146. Kadl A, Meher AK, Sharma PR, Lee MY, Doran AC, Johnstone SR, et al. Identification of a novel macrophage phenotype that develops in response to atherogenic phospholipids via Nrf2. Circ Res. (2010) 107(6):737–46. doi: 10.1161/CIRCRESAHA.109.215715
147. Zernecke A, Winkels H, Cochain C, Williams JW, Wolf D, Soehnlein O, et al. Meta-analysis of leukocyte diversity in atherosclerotic mouse aortas. Circ Res. (2020) 127(3):402–26. doi: 10.1161/CIRCRESAHA.120.316903
148. Dick SA, Wong A, Hamidzada H, Nejat S, Nechanitzky R, Vohra S, et al. Three tissue resident macrophage subsets coexist across organs with conserved origins and life cycles. Sci Immunol. (2022) 7(67):eabf7777. doi: 10.1126/sciimmunol.abf7777
149. Tiedt R, Schomber T, Hao-Shen H, Skoda RC. Pf4-cre transgenic mice allow the generation of lineage-restricted gene knockouts for studying megakaryocyte and platelet function in vivo. Blood. (2007) 109(4):1503–6. doi: 10.1182/blood-2006-04-020362
150. Cochain C, Vafadarnejad E, Arampatzi P, Pelisek J, Winkels H, Ley K, et al. Single-cell RNA-seq reveals the transcriptional landscape and heterogeneity of aortic macrophages in murine atherosclerosis. Circ Res. (2018) 122(12):1661–74. doi: 10.1161/CIRCRESAHA.117.312509
151. Gleissner CA, Erbel C, Linden F, Domschke G, Akhavanpoor M, Helmes CM, et al. Galectin-3 binding protein, coronary artery disease and cardiovascular mortality: Insights from the LURIC study. Atherosclerosis. (2017) 260:121–9. doi: 10.1016/j.atherosclerosis.2017.03.031
152. Papaspyridonos M, Mcneill E, de Bono JP, Smith A, Burnand KG, Channon KM, et al. Galectin-3 is an amplifier of inflammation in atherosclerotic plaque progression through macrophage activation and monocyte chemoattraction. Arteriosclerosis Thrombosis Vasc Biol. (2008) 28(3):433–40. doi: 10.1161/ATVBAHA.107.159160
153. Zhao CF, Herrington DM. The function of cathepsins b, d, and x in atherosclerosis. Am J Cardiovasc Disease. (2016) 6(4):163–70.
154. Lin J, Nishi H, Poles J, Niu X, Mccauley C, Rahman K, et al. Single-cell analysis of fate-mapped macrophages reveals heterogeneity, including stem-like properties, during atherosclerosis progression and regression. JCI Insight. (2019) 4(4):e124574. doi: 10.1172/jci.insight.124574
155. Kim K, Shim D, Lee JS, Zaitsev K, Williams JW, Kim KW, et al. Transcriptome analysis reveals nonfoamy rather than foamy plaque macrophages are proinflammatory in atherosclerotic murine models. Circ Res. (2018) 123(10):1127–42. doi: 10.1161/CIRCRESAHA.118.312804
156. Spann NJ, Garmire LX, Mcdonald JG, Myers DS, Milne SB, Shibata N, et al. Regulated accumulation of desmosterol integrates macrophage lipid metabolism and inflammatory responses. Cell. (2012) 151(1):138–52. doi: 10.1016/j.cell.2012.06.054
157. Zernecke A, Erhard F, Weinberger T, Schulz C, Ley K, Saliba AE, et al. Integrated single-cell analysis-based classification of vascular mononuclear phagocytes in mouse and human atherosclerosis. Cardiovasc Res. (2023) 119(8):1676–89. doi: 10.1093/cvr/cvac161
158. Yin X, Chen S, Eisenbarth SC. Dendritic cell regulation of T helper cells. Annu Rev Immunol. (2021) 39:759–90. doi: 10.1146/annurev-immunol-101819-025146
159. Chistiakov DA, Sobenin IA, Orekhov AN, Bobryshev YV. Dendritic cells: a double-edge sword in atherosclerotic inflammation. Curr Pharm Design. (2015) 21(9):1118–23. doi: 10.2174/1381612820666141013162528
160. Randolph GJ, Ochando J, Partida-Sánchez S. Migration of dendritic cell subsets and their precursors. Annu Rev Immunol. (2008) 26:293–316. doi: 10.1146/annurev.immunol.26.021607.090254
161. Van Vré EA, Hoymans VY, Bult H, Lenjou M, Van Bockstaele DR, Vrints CJ, et al. Decreased number of circulating plasmacytoid dendritic cells in patients with atherosclerotic coronary artery disease. Coronary Artery Dis. (2006) 17(3):243–8. doi: 10.1097/00019501-200605000-00007
162. Shi H, Ge J, Fang W, Yao K, Sun A, Huang R, et al. Peripheral-blood dendritic cells in men with coronary heart disease. Am J Cardiol. (2007) 100(4):593–7. doi: 10.1016/j.amjcard.2007.03.067
163. Niessner A, Sato K, Chaikof EL, Colmegna I, Goronzy JJ, Weyand CM. Pathogen-sensing plasmacytoid dendritic cells stimulate cytotoxic t-cell function in the atherosclerotic plaque through interferon-alpha. Circulation. (2006) 114(23):2482–9. doi: 10.1161/CIRCULATIONAHA.106.642801
164. Niessner A, Shin MS, Pryshchep O, Goronzy JJ, Chaikof EL, Weyand CM. Synergistic proinflammatory effects of the antiviral cytokine interferon-alpha and toll-like receptor 4 ligands in the atherosclerotic plaque. Circulation. (2007) 116:2043–52. doi: 10.1161/CIRCULATIONAHA.107.697789
165. Chistiakov DA, Sobenin IA, Orekhov AN, Bobryshev YV. Myeloid dendritic cells: Development, functions, and role in atherosclerotic inflammation. Immunobiology. (2015) 220(6):833–44. doi: 10.1016/j.imbio.2014.12.010
166. Choi J, Cheong C, Dandamudi DB, Park CG, Rodriguez A, Mehandru S, et al. Flt3 signaling-dependent dendritic cells protect against atherosclerosis. Immunity. (2011) 35(5):819–31. doi: 10.1016/j.immuni.2011.09.014
167. Subramanian M, Thorp E, Hansson GK, Tabas I. Treg-mediated suppression of atherosclerosis requires MYD88 signaling in DCs. J Clin Invest. (2013) 123(1):179–88. doi: 10.1172/JCI64617
168. Daissormont ITMN, Christ A, Temmerman L, Sampedro Millares S, Seijkens T, Manca M, et al. Plasmacytoid dendritic cells protect against atherosclerosis by tuning t-cell proliferation and activity. Circ Res. (2011) 109(12):1387–95. doi: 10.1161/CIRCRESAHA.111.256529
169. Döring Y, Manthey HD, Drechsler M, Lievens D, Megens RT, Soehnlein O, et al. Auto-antigenic protein-DNA complexes stimulate plasmacytoid dendritic cells to promote atherosclerosis. Circulation. (2012) 125:1673–83. doi: 10.1161/CIRCULATIONAHA.111.046755
170. Macritchie N, Grassia G, Sabir SR, Maddaluno M, Welsh P, Sattar N, et al. Plasmacytoid dendritic cells play a key role in promoting atherosclerosis in apolipoprotein E-deficient mice. Arteriosclerosis Thrombosis Vasc Biol. (2012) 32(11):2569–79. doi: 10.1161/ATVBAHA.112.251314
171. Sage AP, Murphy D, Maffia P, Masters LM, Sabir SR, Baker LL, et al. MHC class II-restricted antigen presentation by plasmacytoid dendritic cells drives proatherogenic T cell immunity. Circulation. (2014) 130(16):1363–73. doi: 10.1161/CIRCULATIONAHA.114.011090
172. Shortman K, Naik SH. Steady-state and inflammatory dendritic-cell development. Nat Rev Immunol. (2007) 7(1):19–30. doi: 10.1038/nri1996
173. Van Vré EA, Van Brussel I, Bosmans JM, Vrints CJ, Bult H. Dendritic cells in human atherosclerosis: from circulation to atherosclerotic plaques. Mediators Inflammation. (2011) 2011:941396. doi: 10.1155/2011/941396
174. Weber C, Schober A, Zernecke A. Chemokines: key regulators of mononuclear cell recruitment in atherosclerotic vascular disease. Arteriosclerosis Thrombosis Vasc Biol. (2004) 24(11):1997–2008. doi: 10.1161/01.ATV.0000142812.03840.6f
175. Liu P, Yu YA, Spencer JA, Johnson AE, Vallanat CT, Fong AM, et al. CX3CR1 deficiency impairs dendritic cell accumulation in arterial intima and reduces atherosclerotic burden. Arteriosclerosis Thrombosis Vasc Biol. (2008) 28(2):243–50. doi: 10.1161/ATVBAHA.107.158675
176. Saigusa R, Winkels H, Ley K. T cell subsets and functions in atherosclerosis. Nat Rev Cardiol. (2020) 17(7):387–401. doi: 10.1038/s41569-020-0352-5
177. Biros E, Reznik JE, Moran CS. Role of inflammatory cytokines in genesis and treatment of atherosclerosis. Trends Cardiovasc Med. (2022) 32(3):138–42. doi: 10.1016/j.tcm.2021.02.001
178. Yu X, Zhang J, Zheng X, Yang YH, Tang CK. Interferon-γ in foam cell formation and progression of atherosclerosis. Clinica Chimica Acta; Int J Clin Chem. (2015) 441:33–43. doi: 10.1016/j.cca.2014.12.007
179. Buono C, Come CE, Stavrakis G, Maguire GF, Connelly PW, Lichtman AH. Influence of interferon-gamma on the extent and phenotype of diet-induced atherosclerosis in the LDLR-deficient mouse. Arterioscler Thromb Vasc Biol. (2003) 23(3):454–60. doi: 10.1161/01.ATV.0000059419.11002.6E
180. Gupta S, Pablo AM, Jiang X, Wang N, Tall AR, Schindler C. IFN-gamma potentiates atherosclerosis in ApoE knock-out mice. J Clin Invest. (1997) 99(11):2752–61. doi: 10.1172/JCI119465
181. Whitman SC, Ravisankar P, Elam H, Daugherty A. Exogenous interferon-gamma enhances atherosclerosis in apolipoprotein E-/- mice. Am J Pathol. (2000) 157(6):1819–24. doi: 10.1016/S0002-9440(10)64820-1
182. Buono C, Binder CJ, Stavrakis G, Witztum JL, Glimcher LH, Lichtman AH. T-bet deficiency reduces atherosclerosis and alters plaque antigen-specific immune responses. Proc Natl Acad Sci U S A. (2005) 102(5):1596–601. doi: 10.1073/pnas.0409015102
183. Seder RA, Paul WE. Acquisition of lymphokine-producing phenotype by CD4+ T cells. Annu Rev Immunol. (1994) 12:635–73. doi: 10.1146/annurev.iy.12.040194.003223
184. King VL, Szilvassy SJ, Daugherty A. Interleukin-4 deficiency decreases atherosclerotic lesion formation in a site-specific manner in female LDL receptor-/- mice. Arteriosclerosis Thrombosis Vasc Biol. (2002) 22(3):456–61. doi: 10.1161/hq0302.104905
185. King VL, Cassis LA, Daugherty A. Interleukin-4 does not influence development of hypercholesterolemia or angiotensin II-induced atherosclerotic lesions in mice. Am J Pathology. (2007) 171(6):2040–7. doi: 10.2353/ajpath.2007.060857
186. Oliphant CJ, Hwang YY, Walker JA, Salimi M, Wong SH, Brewer JM, et al. MHCII-mediated dialog between group 2 innate lymphoid cells and CD4(+) T cells potentiates type 2 immunity and promotes parasitic helminth expulsion. Immunity. (2014) 41(2):283–95. doi: 10.1016/j.immuni.2014.06.016
187. Gurram RK, Wei D, Yu Q, Butcher MJ, Chen X, Cui K, et al. Crosstalk between ILC2s and Th2 cells varies among mouse models. Cell Rep. (2023) 42(2):112073. doi: 10.1016/j.celrep.2023.112073
188. Taleb S, Tedgui A, Mallat Z. IL-17 and Th17 cells in atherosclerosis: subtle and contextual roles. Arteriosclerosis Thrombosis Vasc Biol. (2015) 35(2):258–64. doi: 10.1161/ATVBAHA.114.303567
189. Marks BR, Nowyhed HN, Choi J, Poholek AC, Odegard JM, Flavell RA, et al. Thymic self-reactivity selects natural interleukin 17-producing T cells that can regulate peripheral inflammation. Nat Immunol. (2009) 10(10):1125–32. doi: 10.1038/ni.1783
190. Zhou L, Chong MMW, Littman DR. Plasticity of CD4+ T cell lineage differentiation. Immunity. (2009) 30(5):646–55. doi: 10.1016/j.immuni.2009.05.001
191. Quintana FJ, Basso AS, Iglesias AH, Korn T, Farez MF, Bettelli E, et al. Control of t(reg) and T(H)17 cell differentiation by the aryl hydrocarbon receptor. Nature. (2008) 453(7191):65–71. doi: 10.1038/nature06880
192. Zhang F, Meng G, Strober W. Interactions among the transcription factors Runx1, RORgammat and Foxp3 regulate the differentiation of interleukin 17-producing T cells. Nat Immunol. (2008) 9(11):1297–306. doi: 10.1038/ni.1663
193. Mcgeachy MJ, Cua DJ, Gaffen SL. The IL-17 family of cytokines in health and disease. Immunity. (2019) 50(4):892–906. doi: 10.1016/j.immuni.2019.03.021
194. Erbel C, Chen L, Bea F, Wangler S, Celik S, Lasitschka F, et al. Inhibition of IL-17A attenuates atherosclerotic lesion development in apoE-deficient mice. J Immunol (Baltimore Md. 1950). (2009) 183(12):8167–75. doi: 10.4049/jimmunol.0901126
195. Gisterå A, Robertson AL, Andersson J, Ketelhuth DF, Ovchinnikova O, Nilsson SK, et al. Transforming growth factor-β signaling in T cells promotes stabilization of atherosclerotic plaques through an interleukin-17-dependent pathway. Sci Trans Med. (2013) 5(196):196ra100. doi: 10.1126/scitranslmed.3006133
196. Voo KS, Wang Y, Santori FR, Boggiano C, Wang YH, Arima K, et al. Identification of IL-17-producing FOXP3+ regulatory T cells in humans. Proc Natl Acad Sci United States America. (2009) 106(12):4793–8. doi: 10.1073/pnas.0900408106
197. Deknuydt F, Bioley G, Valmori D, Ayyoub M. IL-1beta and IL-2 convert human treg into T(H)17 cells. Clin Immunol (Orlando Fla.). (2009) 131(2):298–307. doi: 10.1016/j.clim.2008.12.008
198. Bovenschen HJ, van de Kerkhof PC, van Erp PE, Woestenenk R, Joosten I, Koenen HJ. Foxp3+ regulatory T cells of psoriasis patients easily differentiate into IL-17A-producing cells and are found in lesional skin. J Invest Dermatol. (2011) 131(9):1853–60. doi: 10.1038/jid.2011.139
199. Baardman J, Lutgens E. Regulatory T cell metabolism in atherosclerosis. Metabolites. (2020) 10(7):279. doi: 10.3390/metabo10070279
200. Mor A, Planer D, Luboshits G, Afek A, Metzger S, Chajek-Shaul T, et al. Role of naturally occurring CD4+ CD25+ regulatory T cells in experimental atherosclerosis. Arteriosclerosis Thrombosis Vasc Biol. (2007) 27(4):893–900. doi: 10.1161/01.ATV.0000259365.31469.89
201. Ait-Oufella H, Salomon BL, Potteaux S, Robertson AK, Gourdy P, Zoll J, et al. Natural regulatory T cells control the development of atherosclerosis in mice. Nat Med. (2006) 12(2):178–80. doi: 10.1038/nm1343
202. Robertson AL, Rudling M, Zhou X, Gorelik L, Flavell RA, Hansson GK. Disruption of TGF-beta signaling in T cells accelerates atherosclerosis. J Clin Invest. (2003) 112(9):1342–50. doi: 10.1172/JCI18607
203. Mallat Z, Besnard S, Duriez M, Deleuze V, Emmanuel F, Bureau MF, et al. Protective role of interleukin-10 in atherosclerosis. Circ Res. (1999) 85(8):e17–24. doi: 10.1161/01.RES.85.8.e17
204. Winkels H, Ehinger E, Vassallo M, Buscher K, Dinh HQ, Kobiyama K, et al. Atlas of the immune cell repertoire in mouse atherosclerosis defined by single-cell RNA-sequencing and mass cytometry. Circ Res. (2018) 122(12):1675–88. doi: 10.1161/CIRCRESAHA.117.312513
205. Gu W, Ni Z, Tan Y, Deng J, Zhang SJ, Lv ZC, et al. Adventitial cell atlas of wt (Wild type) and apoE (Apolipoprotein E)-deficient mice defined by single-cell RNA sequencing. Arteriosclerosis Thrombosis Vasc Biol. (2019) 39(6):1055–71. doi: 10.1161/ATVBAHA.119.312399
206. Winkels H, Wolf D. Heterogeneity of T cells in atherosclerosis defined by single-cell RNA-sequencing and cytometry by time of flight. Arteriosclerosis Thrombosis Vasc Biol. (2021) 41(2):549–63. doi: 10.1161/ATVBAHA.120.312137
207. Winer H, Rodrigues GOL, Hixon JA, Aiello FB, Hsu TC, Wachter BT, et al. IL-7: Comprehensive review. Cytokine. (2022) 160:156049. doi: 10.1016/j.cyto.2022.156049
208. Hilgendorf I, Theurl I, Gerhardt LMS, Robbins CS, Weber GF, Gonen A, et al. Innate response activator B cells aggravate atherosclerosis by stimulating T helper-1 adaptive immunity. Circulation. (2014) 129(16):1677–87. doi: 10.1161/CIRCULATIONAHA.113.006381
209. Kyaw T, Tay C, Krishnamurthi S, Kanellakis P, Agrotis A, Tipping P, et al. B1a B lymphocytes are atheroprotective by secreting natural IgM that increases IgM deposits and reduces necrotic cores in atherosclerotic lesions. Circ Res. (2011) 109:830–40. doi: 10.1161/CIRCRESAHA.111.248542
210. Perry HM, Mcnamara CA. Refining the role of B cells in atherosclerosis. Arterioscler Thromb Vasc Biol. (2012) 32:1548–9. doi: 10.1161/ATVBAHA.112.249235
211. Sasaki Y, Casola S, Kutok JL, Rajewsky K, Schmidt-Supprian M. TNF family member B cell-activating factor (BAFF) receptor-dependent and -independent roles for BAFF in B cell physiology. J Immunol (Baltimore Md. 1950). (2004) 173(4):2245–52. doi: 10.4049/jimmunol.173.4.2245
212. Kyaw T, Cui P, Tay C, Kanellakis P, Hosseini H, Liu E, et al. BAFF receptor mAb treatment ameliorates development and progression of atherosclerosis in hyperlipidemic ApoE(-/-) mice. PloS One. (2013) 8(4):e60430. doi: 10.1371/journal.pone.0060430
213. Doran AC, Lipinski MJ, Oldham SN, Garmey JC, Campbell KA, Skaflen MD, et al. B-cell aortic homing and atheroprotection depend on Id3. Circ Res. (2012) 110(1):e1–e12. doi: 10.1161/CIRCRESAHA.111.256438
214. Tay C, Liu Y, Kanellakis P, Kallies A, Li Y, Cao A, et al. Follicular B cells promote atherosclerosis via T cell-mediated differentiation into plasma cells and secreting pathogenic Immunoglobulin G. Arteriosclerosis Thrombosis Vasc Biol. (2018) 38(5):e71–84. doi: 10.1161/ATVBAHA.117.310678
215. Upadhye A, Sturek JM, Mcnamara CA. Russell ross memorial lecture in vascular biology: B lymphocyte-mediated protective immunity in atherosclerosis. Arterioscler Thromb Vasc Biol. (2019 2020) 40:309–22. doi: 10.1161/ATVBAHA.119.313064
216. Mai W, Liao Y. Targeting IL-1β in the treatment of atherosclerosis. Front Immunol. (2020) 11:589654. doi: 10.3389/fimmu.2020.589654
217. Libby P. Interleukin-1 beta as a target for atherosclerosis therapy: Biological basis of CANTOS and beyond. J Am Coll Cardiol. (2017) 70(18):2278–89. doi: 10.1016/j.jacc.2017.09.028
218. Ridker PM, Macfadyen JG, Everett BM, Thuren T, Chang WH, Ballantyne C, et al. Relationship of c-reactive protein reduction to cardiovascular event reduction following treatment with canakinumab: a secondary analysis from the CANTOS randomised controlled trial. Lancet (London England). (2018) 391(10118):319–28. doi: 10.1016/S0140-6736(17)32814-3
219. Di Minno MND, Iervolino S, Peluso R, Scarpa R, Di Minno G, CaRRDs study group. Carotid intima-media thickness in psoriatic arthritis: differences between tumor necrosis factor-α blockers and traditional disease-modifying antirheumatic drugs. Arteriosclerosis Thrombosis Vasc Biol. (2011) 31(3):705–12. doi: 10.1161/ATVBAHA.110.214585
220. Gkalpakiotis S, Arenbergerova M, Gkalpakioti P, Potockova J, Arenberger P, Kraml P. Long-term impact of adalimumab therapy on biomarkers of systemic inflammation in psoriasis: Results of a 2 year study. Dermatologic Ther. (2020) 33(6):e14110. doi: 10.1111/dth.14110
221. Ridker PM, Devalaraja M, Baeres FMM, Engelmann MDM, Hovingh GK, Ivkovic M, et al. IL-6 inhibition with ziltivekimab in patients at high atherosclerotic risk (RESCUE): a double-blind, randomised, placebo-controlled, phase 2 trial. Lancet (London England). (2021) 397(10289):2060–9. doi: 10.1016/S0140-6736(21)00520-1
222. Gilbert J, Lekstrom-Himes J, Donaldson D, Lee Y, Hu M, Xu J, et al. Effect of CC chemokine receptor 2 CCR2 blockade on serum c-reactive protein in individuals at atherosclerotic risk and with a single nucleotide polymorphism of the monocyte chemoattractant protein-1 promoter region. Am J Cardiol. (2011) 107(6):906–11. doi: 10.1016/j.amjcard.2010.11.005
223. Fernandez DM, Rahman AH, Fernandez NF, Chudnovskiy A, Amir ED, Amadori L, et al. Single-cell immune landscape of human atherosclerotic plaques. Nat Med. (2019) 25(10):1576–88. doi: 10.1038/s41591-019-0590-4
224. Liang Y, Li L, Chen Y, Xiao J, Wei D. PD-1/PD-L1 immune checkpoints: Tumor vs atherosclerotic progression. Clinica Chimica Acta; Int J Clin Chem. (2021) 519:70–5. doi: 10.1016/j.cca.2021.04.010
225. Fan L, Liu J, Hu W, Chen Z, Lan J, Zhang T, et al. Targeting pro-inflammatory T cells as a novel therapeutic approach to potentially resolve atherosclerosis in humans. Cell Res. (2024) 34(6):407–27. doi: 10.1038/s41422-024-00945-0
226. Lankin VZ, Tikhaze AK, Melkumyants AM. Malondialdehyde as an important key factor of molecular mechanisms of vascular wall damage under heart diseases development. Int J Mol Sci. (2022) 24(1):128. doi: 10.3390/ijms24010128
227. Fredrikson GN, Söderberg I, Lindholm M, Dimayuga P, Chyu KY, Shah PK, et al. Inhibition of atherosclerosis in apoE-null mice by immunization with apoB-100 peptide sequences. Arteriosclerosis Thrombosis Vasc Biol. (2003) 23(5):879–84. doi: 10.1161/01.ATV.0000067937.93716.DB
228. de Grooth GJ, Klerkx AHEM, Stroes ESG, Stalenhoef AF, Kastelein JJ, Kuivenhoven JA. A review of CETP and its relation to atherosclerosis. J Lipid Res. (2004) 45(11):1967–74. doi: 10.1194/jlr.R400007-JLR200
229. García-González V, Delgado-Coello B, Pérez-Torres A, Mas-Oliva J. Reality of a vaccine in the prevention and treatment of atherosclerosis. Arch Med Res. (2015) 46(5):427–37. doi: 10.1016/j.arcmed.2015.06.004
230. Lutgens E, Cleutjens KB, Heeneman S, Koteliansky VE, Burkly LC, Daemen MJ. Both early and delayed anti-CD40L antibody treatment induces a stable plaque phenotype. Proc Natl Acad Sci United States America. (2000) 97(13):7464–9. doi: 10.1073/pnas.97.13.7464
231. Landlinger C, Pouwer MG, Juno C, van der Hoorn JWA, Pieterman EJ, Jukema JW, et al. The AT04A vaccine against proprotein convertase subtilisin/kexin type 9 reduces total cholesterol, vascular inflammation, and atherosclerosis in APOE*3Leiden.CETP mice. Eur Heart J. (2017) 38(32):2499–507. doi: 10.1093/eurheartj/ehx260
232. Wu D, Pan Y, Yang S, Li C, Zhou Y, Wang Y, et al. PCSK9Qβ-003 vaccine attenuates atherosclerosis in apolipoprotein E-deficient mice. Cardiovasc Drugs Ther. (2021) 35(1):141–51. doi: 10.1007/s10557-020-07041-6
233. Tian S, Wang Y, Wan J, Yang M, Fu Z. Co-stimulators CD40-CD40L, a potential immune-therapy target for atherosclerosis: A review. Medicine. (2024) 103(14):e37718. doi: 10.1097/MD.0000000000037718
234. Schönbeck U, Libby P. Inflammation, immunity, and HMG-CoA reductase inhibitors: statins as antiinflammatory agents? Circulation. (2004) 109(21 Suppl 1):II18–26. doi: 10.1161/01.CIR.0000129505.34151.23
235. Weitz-Schmidt G, Welzenbach K, Brinkmann V, Kamata T, Kallen J, Bruns C, et al. Statins selectively inhibit leukocyte function antigen-1 by binding to a novel regulatory integrin site. Nat Med. (2001) 7(6):687–92. doi: 10.1038/89058
236. Hopps E, Caimi G. Matrix metalloproteases as a pharmacological target in cardiovascular diseases. Eur Rev For Med Pharmacol Sci. (2015) 19(14):2583–9.
237. Abu-Fanne R, Stepanova V, Litvinov RI, Abdeen S, Bdeir K, Higazi M, et al. Neutrophil α-defensins promote thrombosis in vivo by altering fibrin formation, structure, and stability. Blood. (2019) 133(5):481–93. doi: 10.1182/blood-2018-07-861237
238. Vaidya K, Tucker B, Kurup R, Khandkar C, Pandzic E, Barraclough J, et al. Colchicine inhibits neutrophil extracellular trap formation in patients with acute coronary syndrome after percutaneous coronary intervention. J Am Heart Assoc. (2021) 10(1):e018993. doi: 10.1161/JAHA.120.018993
239. Akodad M, Sicard P, Fauconnier J, Roubille F. Colchicine and myocardial infarction: A review. Arch Cardiovasc Diseases. (2020) 113(10):652–9. doi: 10.1016/j.acvd.2020.04.007
240. Bertrand M, Tardif J. Inflammation and beyond: new directions and emerging drugs for treating atherosclerosis. Expert Opin Emerging Drugs. (2017) 22(1):1–26. doi: 10.1080/14728214.2017.1269743
241. Tousoulis D, Oikonomou E, Economou EK, Crea F, Kaski JC. Inflammatory cytokines in atherosclerosis: current therapeutic approaches. Eur Heart J. (2016) 37(22):1723–32. doi: 10.1093/eurheartj/ehv759
242. Jiang W, Cen Y, Song Y, Li P, Qin R, Liu C, et al. Artesunate attenuated progression of atherosclerosis lesion formation alone or combined with rosuvastatin through inhibition of pro-inflammatory cytokines and pro-inflammatory chemokines. Phytomedicine Int J Phytotherapy Phytopharmacology. (2016) 23(11):1259–66. doi: 10.1016/j.phymed.2016.06.004
Keywords: atherosclerosis, plaque evolution, immune response, immune cell heterogeneity, inflammatory microenvironment, immunotherapy
Citation: Pi H, Wang G, Wang Y, Zhang M, He Q, Zheng X, Yin K, Zhao G and Jiang T (2024) Immunological perspectives on atherosclerotic plaque formation and progression. Front. Immunol. 15:1437821. doi: 10.3389/fimmu.2024.1437821
Received: 24 May 2024; Accepted: 09 September 2024;
Published: 27 September 2024.
Edited by:
Yvonne Döring, Universitätsspital Bern Departement Herz und Gefässe Universitätsklinik für Angiologie, SwitzerlandReviewed by:
Maria Kral, LMU Munich University Hospital, GermanyMorgan Salmon, University of Michigan, United States
Copyright © 2024 Pi, Wang, Wang, Zhang, He, Zheng, Yin, Zhao and Jiang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ting Jiang, MjAxOTY5NTMwNEBnemhtdS5lZHUuY24=; Guojun Zhao, emhhb2d1b2p1bkBnemhtdS5lZHUuY24=; Kai Yin, Mzc0NzUyMzE3QHFxLmNvbQ==
†These authors have contributed equally to this work