 Brandon Johnson
Brandon Johnson Lihong Cheng
Lihong Cheng Jennifer Koenitzer
Jennifer Koenitzer Ian M. Catlett
Ian M. Catlett Peter Schafer
Peter Schafer- 1Translational Medicine, Immunology, Cardiovascular & Neuroscience Disease Team, Bristol Myers Squibb, Princeton, NJ, United States
- 2Immunology Thematic Research Centers (TRC), Bristol Myers Squibb, Princeton, NJ, United States
Translational medicine provides insight into novel drugs and predicts unwanted effects. In well-characterized pathways (e.g., cytokine-Janus kinase [JAK]-signal transducers and activators of transcription [STAT]), a variety of in vitro assessments were used to estimate selectivity of effects on different potential targets (i.e., JAK1, JAK2, JAK3, and tyrosine kinase 2 [TYK2]). Several approved drugs were characterized as selective for the JAK family. These assessments are challenged by a lack of compounds that only inhibit one JAK family member. Deucravacitinib is a first-in-class, oral, selective, allosteric inhibitor of TYK2, a kinase required for IL-12, IL-23, and Type I interferon signaling. Unlike deucravacitinib, which selectively binds to the TYK2 regulatory domain, JAK1,2,3 inhibitors target the catalytic domain, contributing to nonselective targeting of JAK1,2,3. Cytokines associated with JAK1,2,3 signaling are required for both immune and nonimmune functions. A similar laboratory abnormality profile was observed in clinical trials using JAK1,2,3 inhibitors that has not been observed with deucravacitinib. In vitro testing of JAK1,2,3 inhibitors has relied upon assays of signal transduction, such as those measuring STAT phosphorylation, for estimates of potency and selectivity. These assay systems can be effective in estimating in vivo efficacy; however, they may not provide insight into downstream outcomes of receptor signaling, which may be more relevant for evaluating safety aspects. Assay systems assessing functional outcomes from cells may yield a more useful translational evaluation. Here, deucravacitinib was assessed for potency and selectivity versus three representatives of the JAK inhibitor class (tofacitinib, baricitinib, and upadacitinib) based on functional assays. JAK inhibitors had suppressive activity against JAK2-dependent hematopoietic colony-forming assays modeling thrombopoiesis, erythropoiesis, and myelopoiesis; however, deucravacitinib did not. Deucravacitinib had limited potency against NK cells, cytotoxic T cells, T-helper cells, and regulatory T cells activated by JAK1/JAK3-dependent common gamma chain cytokines. These data are consistent with the biologic role of JAK1,2,3 and pharmacodynamic changes in clinical laboratory abnormalities. Against TYK2-dependent cytokines, deucravacitinib selectively inhibited Type I interferon stimulation of monocytes and dendritic cells and was a more potent inhibitor than JAK inhibitors. IL-12 and IL-23 functional outputs were similarly potently inhibited by deucravacitinib. Results are consistent with deucravacitinib selectively inhibiting TYK2.
1 Introduction
Tyrosine kinase 2 (TYK2) is an intracellular enzyme required for signal transduction by Type I interferons (IFNs), interleukin (IL)-12/IL-23, IL-10, and Type III IFN cytokine families. TYK2 is a member of the Janus kinase (JAK) family, which includes JAK1, JAK2, and JAK3. JAKs work in obligate pairs to affect the biologic functions of the various cytokines and growth factors that use each pair. Specific biologic functions absolutely require specific cytokines and both members of a JAK pair. Kinase pairs are dictated by the cytokine receptor and the constitutively associated kinase; pairs include JAK1/JAK2, JAK1/JAK3, JAK2/JAK2, JAK1/TYK2, and JAK2/TYK2. JAK2 is used by hematopoietic cytokines and a wide range of immune and non-immune pathways (i.e., erythropoietin (EPO), IFNγ, and IL-6, respectively). Alternatively, JAK1 and JAK3 mediate signal transduction by the common gamma chain cytokines: IL-2, IL-4, IL-7, IL-9, IL-15, and IL-21. These are mediators of immune cell development, homeostasis, or activation. T-cell, natural killer (NK) cell, and regulatory T-cell development and homeostasis are controlled by IL-7 (1), IL-15 (2), and IL-2 (3), respectively. Many cytokines and growth factors utilize the JAK1/JAK2 pairing and are involved in a variety of physiologic functions (e.g., IL-6 uses this pair and is involved in inflammatory processes and lipid homeostasis).
Several drugs that target members of the JAK family are approved (4, 5). The established pharmaceutical class, JAK inhibitor, is ATP-competitive, active site, catalytic (kinase) domain binding inhibitor. Molecules of this class inhibit multiple members of the JAK family, primarily JAK1 or JAK3; however, varying degrees of JAK2 inhibition have also been observed in clinical trials. This class has a common set of laboratory changes resulting from both on-target and off-target effects. Class effects include on-target effects (i.e., immunosuppression) and off-target effects (i.e., decreased hemoglobin, decreased neutrophils, platelet modulation, liver enzyme increases, creatine phosphokinase increases, decreased lymphocytes, and cholesterol increases) (6–9). This profile can be linked to the range of JAK family members inhibited and the degree of inhibition for each family member. JAK2 is critically involved in hematopoiesis, and effects of inhibition can be observed through measures of red blood cell levels. Dose-dependent decreases in hemoglobin observed with the JAK inhibitors tofacitinib (10, 11), baricitinib (12), and upadacitinib (13) are evidence of the impact on JAK2 kinase. Thus, pharmacodynamic changes in laboratory tests reveal the in vivo impact of inhibition of specific JAK family members. This nonspecificity for JAK family members has limited the dose levels tolerated for optimal therapeutic effect (11). Selective inhibitors of only one family member may convey a better ratio of desired pathway targeting while sparing unwanted effects.
TYK2-dependent cytokines are critical mediators of inflammatory diseases, including IL-23 in psoriasis (14) and inflammatory bowel disease (15), or Type I IFNs in systemic lupus erythematosus (SLE) (16) and Sjögren’s syndrome (17). Partial loss of function mutations in TYK2 have been associated with protection in these disease states and others (18–20). Furthermore, complete loss of function in TYK2 results in recurrent infections linked to these cytokines but not to developmental or homeostatic dysfunction (21). Thus, TYK2 is an attractive target for therapeutic intervention. Deucravacitinib, an oral, selective, allosteric TYK2 inhibitor, is approved for the treatment of adults with moderate to severe plaque psoriasis who are candidates for systemic therapy (22, 23). Deucravacitinib uniquely binds to the regulatory (pseudokinase) domain of TYK2 rather than to the catalytic (kinase) domain where JAK1,2,3 inhibitors bind (24, 25), driving its selectivity and representing the first in a new class of oral drugs. Deucravacitinib shows no inhibition of JAK1, JAK2, or JAK3 at clinically relevant concentrations (25). In clinical trials, deucravacitinib does not have the same pharmacodynamic profile of laboratory changes and abnormalities as JAK1,2,3 inhibitors (26, 27).
JAK1,2,3 inhibitor activity is typically characterized using in vitro assays to measure proximal phosphorylation of signal transduction and activation of transcription (pSTAT) clinically (28–30). However, the translational value of assays used to approximate pharmacokinetic profiles and clinical effects, such as dose-limiting off-target effects of hemoglobin decrease, is unclear. Cellular assays of downstream functional outcomes, as opposed to proximal measures of kinase activity, may have greater translational value to predict a clinical profile.
In this report, deucravacitinib and approved JAK inhibitors were compared in cytokine-mediated assays representative of cellular functions. The aim of this study was to assess whether assays integrating inputs of cellular signaling with functional response might provide better estimates of the kinase inhibition profile. Assays with translational value should provide better estimates of targeted and nontargeted pathway inhibition in clinical trials and more accurately identify relevant risks and the risk-associated exposures.
2 Materials and methods
2.1 Drug compounds and dosing
Deucravacitinib and tofacitinib were synthesized internally by Bristol Myers Squibb. Baricitinib and upadacitinib were acquired from MedChemExpress (Monmouth Junction, NJ). Each compound was resuspended in 100% dimethyl sulfoxide (DMSO) (MilliporeSigma, Burlington, MA) for a 10 mM stock solution. Compounds were then prepared in 2x solutions and mixed 1:1 with media containing cells. For whole blood (WB) assays, 20x solutions in cell media were created before addition of a small volume to blood. Subsequently, a small volume (2 µL) of cytokines was added to the total volume after compound pre-incubation. Dose concentration ranged from 0.5 or 1 nM minimal concentration to 5 or 10 µM maximal concentration in an 8- to 10-point semi-log dilution.
2.2 PBMC functional assays
Peripheral blood mononuclear cells (PBMCs) of normal healthy volunteers (NHVs) from commercially procured leukopaks (Biological Specialty Company, Colmar, PA, and StemExpress, Folsom, CA) were processed, isolated using standard Ficoll gradient-based separation methods, and cryopreserved. On the day of the assay, cells were thawed, suspended in complete media (RPMI 1640 + 10% heat-inactivated fetal bovine serum (FBS) + 5 µg/mL gentamicin), and seeded in technical duplicate at 1-2 × 105 cells per well, then rested for 1 hour. Cells were pretreated with deucravacitinib, tofacitinib, baricitinib, or upadacitinib for ~30 minutes before the addition of recombinant IL-2 (BioLegend, San Diego, CA), recombinant IL-7 (BioLegend), recombinant IL-15 (BioLegend), recombinant IFN-α2 (R&D Systems, Minneapolis, MN), recombinant IL-18 (BioLegend), recombinant IL-12 (R&D Systems), or recombinant IFNγ (R&D Systems) for 18 hours to 6 days. For measurement of proliferation, cells were labeled with CellTrace Far Red (ThermoFisher, Philadelphia, PA) and cultured for 6 days in the presence or absence of cytokines. At endpoint, cells were collected and stained for lineage and activation markers for fluorescence-activated cell-sorting (FACS) analysis. Proliferation was used as an assessment of cellular function in response to IL-15 and IL-7. BCL-2 expression was used as a surrogate marker of functional survival for IL-2− and IL-7−treated cells. The upregulation of GARP was used as representation of T-regulatory cell (Treg) functional response to IL-2. Expression of Treg lineage markers was used to confirm the maintenance/expansion of these cells in culture and was considered a functional output. Cell supernatant was collected for enzyme-linked immunosorbent assays (ELISAs). Supernatants were quantified for concentrations of CXCL9, CXCL10, CXCL11, and/or IFNγ per manufacturer’s protocol as a representation of cellular function to inflammatory JAK1/TYK2 cytokines. All ELISA kits were sourced from R&D Systems.
2.3 Phosphorylation of STAT signaling assays
PBMCs were isolated via a Ficoll gradient from human WB drawn into ethylenediaminetetraacetic acid (EDTA) tubes. PBMCs, which were obtained from NHV at Bristol Myers Squibb with approval of Bristol Myers Squibb Environmental Health & Safety and with written informed consent from the donors, were used for pSTAT assays. Additional pSTAT assays were performed on PBMCs from NHV of commercial leukopaks. Here, cryopreserved PBMCs were thawed in RPMI 1640 containing 10% FBS at 37°C before assay stimulation. After a 1-hour rest period, PBMCs or WB samples were preincubated with inhibitors at least 20 minutes prior to the addition of recombinant IFNα (PBL Assay Science, Piscataway, NJ), recombinant thrombopoietin (TPO; PeproTech, Cranbury, NJ), recombinant IL-7, or recombinant IL-15. After 15 minutes, cell stimulation was stopped with warm Lyse/Fix Buffer before proceeding to FACS staining.
2.4 Monocyte-derived dendritic cell functional assays
Leukopaks from NHV were commercially purchased and negatively selected for CD14+ monocytes (StemCell Technologies, Vancouver, BC) and cryopreserved. Monocytes were thawed in RPMI 1640 + 10% FBS at 1 × 106 cells/mL and rested for 1 hour at 37°C. Six million monocytes were plated in six-well dishes with 100 ng/mL IL-4 (Miltenyi Biotec, Gaithersburg, MD) and 100 ng/mL granulocyte-macrophage colony-stimulating factor (GM-CSF; Miltenyi Biotec) and incubated for 6 to 7 days. Cells were fed fresh media with additional IL-4 and GM-CSF every 3 days prior to assay. Non-adherent immature monocyte-derived dendritic cells were collected, washed, and resuspended at 1 × 106 cells/mL in complete media. The 1 × 105 cells were plated in technical duplicate wells and a nine-point, semi-log dilution of deucravacitinib, tofacitinib, baricitinib, or upadacitinib was added ~30 minutes prior to incubation with recombinant IFN-α2 for 48 hours. At Day 2, cell supernatants were removed for quantification of CXCL9 and CXCL10 by ELISA.
2.5 NK-cell functional assays
Cryopreserved PBMCs were thawed and suspended in RPMI 1640 + 10% FBS and rested for 1 hour at 37°C. NK cells were isolated by negative bead selection (StemCell Technologies) in accordance with the manufacturer’s protocol. Purified NK cells were counted, centrifuged, and seeded in technical duplicate at 1 × 105 cells per well. A nine-point, semi-log dilution of deucravacitinib, tofacitinib, baricitinib, or upadacitinib was added to the cells for preincubation before the addition of recombinant IL-23 (BioLegend) and recombinant human IL-1β (R&D Systems). Cells were collected at 18 hours and stained for lineage and activation marker for FACS analysis. Supernatants were collected for quantification of IFNγ by ELISA.
2.6 Functional assay staining and FACS analysis
For functional assays, cells were washed twice with Dulbecco’s phosphate buffered saline (DPBS) and resuspended in 50 µL PBS containing 1:1000 diluted Fixable Near-IR Dead Cell Stain Kit (ThermoFisher) for 30 minutes. Cells were washed twice in Cell Staining Buffer (BioLegend) and stained with FITC CD3 (clone OKT3; eBiosciences, San Diego, CA), Brilliant™ Blue (BB)700 CD3 (clone HIT3α; BD Biosciences, Franklin Lakes, NJ), Brilliant™ Violet (BV)510 CD56 (clone HP-3G10; BD Biosciences), BV786 NKG2D (clone 1D11; BioLegend), PE NKp30 (clone p30-15; BD Biosciences), TruStain FcX (BioLegend), BV421 CD107a (clone R35-38; BD Biosciences), Brilliant™ UltraViolet (BUV)395 CD8 (clone RPA-T8; BD Biosciences), BV510 CD56 (clone 5.1H11; BioLegend), BUV496 CD4 (clone SK3; BD Biosciences), BB515 CD45RO (clone UCHL1; BD Biosciences), BV786 CD25 (clone M-A251; BD Biosciences), BV421 CCR7 (clone G043H7; BioLegend), APC CD127 (clone 40131; R&D Systems), PE-Cy7 CD3 (clone UCHT1; BD Biosciences), BB700 CD45RO (clone UCHL1; BD Biosciences), BV421 CD4 (clone RPA-T4; BD Biosciences), PE CD8 (clone RPA-T8; BD Biosciences), V450 CD14 (clone M5E2; BD Biosciences), APC CD11c (clone BU15; eBioscience), PE CD80 (clone 2D10.4; eBioscience), V500 HLA-DR (clone G46-6; BD Biosciences), BUV395 CD40 (clone 5C3; BD Biosciences), BB515 CD54 (clone HA58; BD Biosciences), BV785 CD19 (clone HIB19; BioLegend), CD69 (clone FN50; BD Biosciences), PE-CF594 CD56 (clone NCAM16.2; BD Biosciences), BUV395 GARP (clone 7B11; BD Biosciences), BV421 NKG2D (clone 1D11; BioLegend), BV480 NKp30 (clone p30-15; BD Biosciences), BUV395 CD69 (clone FN50; BD Biosciences), BV510 CD19 (clone HIB19; BioLegend), BV605 CD8 (clone RPA-T8; BioLegend), BV605 NKp30 (clone p30-15; BD Biosciences), PE CD218a (clone H44; BioLegend), APC CD56 (clone NCAM16.2; BD Biosciences), FITC CD69 (clone FN50; BD Biosciences), BV421 CD14 (clone M5E2; BD Biosciences), BV711 CD11c (clone B-ly6; BD Biosciences), or BV605 CD86 (clone 2331 [FUN-1]; BD Biosciences) for 30 minutes in Cell Staining Buffer containing Brilliant Stain Buffer (BD Biosciences). In instances of intracellular staining, cells were washed and resuspended in FOXP3 Transcription Factor Fixation Buffer (ThermoFisher) for 30 minutes. Cells were washed twice with Permeabilization Buffer (ThermoFisher) before resuspension in Permeabilization Buffer containing PE BCL-2 (clone Bcl-2/100; BD Biosciences), AF647 FOXP3 (clone 236A/E7; BD Biosciences), PE-CF594 FOXP3 (clone 236A/E7; BD Biosciences), AF647 T-bet (clone 04-46; BD Biosciences), and/or PE-CF594 T-bet (clone 04-46; BD Biosciences) for 30 minutes. Cells were washed twice and resuspended in Cytofix Fixation Buffer (BD Biosciences) for 30 minutes. Cells were washed and resuspended in Cell Staining Buffer before flow cytometry analysis on an LSRFortessa.
2.7 Signaling assay staining and FACS analysis
For signaling assays, after treatment with inhibitor and/or cytokine, stimulation was stopped by thoroughly mixing cells or WB with 3x volume of Lyse/Fix Buffer (BD Biosciences) for 10 minutes at 37°C. Fixed cells were then washed with Cell Staining Buffer twice and stained with surface antibodies of TruStain FcX, FITC CD61 (clone VI-PL2; BD Biosciences), PE-Cy7 CD3, BB700 CD45RO, BV786 CD25, BV421 CD4, and/or PE CD8 for 30 minutes in Cell Staining buffer at room temperature. Cells were washed twice with Cell Staining Buffer before the addition of 3x volume of ice-cold Permbuffer III (BD Biosciences). Cells were incubated on ice for 30 minutes, then washed twice with 3x volume of Cell Staining Buffer. PE pSTAT3 (clone 4/P-STAT3; BD Biosciences), AF647 pSTAT5 (clone 47/Stat5(pY694); BD Biosciences), AF647 pSTAT2 (clone D3P2P; Cell Signaling), AF488 FOXP3 (clone 259D/C7; BD Biosciences), and/or PE-CF594 CD56 (clone NCAM16.2; BD Biosciences) in Cell Staining Buffer were incubated with cells for 30 minutes at room temperature. Cells were washed twice and resuspended in Cytofix Fixation Buffer before flow cytometry analysis on an LSRFortessa. The amount of pSTAT expression was quantified by median fluorescence intensity (MFI) after gating on parent population and comparing with fluorescent minus one and unstimulated samples. Gating strategy of cell populations and example plots of functional (Supplementary Figures 1A, B, D, E) and signaling (Supplementary Figure 1C) expression are provided.
2.8 Hematopoietic differentiation assays
Cryopreserved normal human bone marrow light density cells (Lonza Bioscience, Walkersville, MD, and StemCell Technologies) were thawed into Iscove’s Modified Dulbecco’s Medium + 2% FBS containing DNase I (StemCell Technologies). Cells were washed and resuspended in Iscove’s Modified Dulbecco’s Medium + 2% FBS and counted. For erythroid and myeloid progenitor evaluation, 1-1.5 × 104 cells were suspended in MethoCult™ media (StemCell Technologies) containing eight-point dilutions of deucravacitinib, tofacitinib, baricitinib, or upadacitinib. Cell mixtures were then plated into SmartDish™ for 13 to 15 days at 37°C. Following this incubation period, erythroid and myeloid progenitors were evaluated for morphology by the STEMvision™ automated colony-forming unit assay reader and scored. For megakaryocyte progenitor evaluation, 1-1.5 × 105 cells were suspended in MegaCult-C™ media (StemCell Technologies) containing eight-point dilutions of deucravacitinib, tofacitinib, baricitinib, or upadacitinib. Collagen was added to cell mixtures, then plated onto double chamber slides and incubated at 37°C. After 10 to 12 days, slides were dehydrated, fixed, and stained with anti-CD41 (StemCell Technologies). Cells were then enumerated and scored based on size and morphology.
2.9 IC50 calculation and modeling WB potency
Percent inhibition was determined by the following equation:
Kcon is the readout of cytokine-stimulated cells with kinase inhibitor at any given concentration, Un is the readout for the DMSO vehicle control, and Ct is the readout for the cytokine stimulation with DMSO vehicle. Half-maximal inhibitory concentration (IC50) values were determined by curve fitting using nonlinear regression analysis on GraphPad Prism for each donor’s response. Zero inhibition was set at 10-11 M. Model was constrained to maximal inhibition (100% of treatment control) only when appropriate. IC50 was determined by the geometric mean of the donors. The WB IC50 values for the inhibitors from measured in vitro culture media IC50 was determined using the following equation (30):
The unbound free fraction (fu) for deucravacitinib was 0.184 and the blood:plasma ratio was 1.26. The fu for tofacitinib, baricitinib, and upadacitinib were 0.61, 0.59, and 0.56, respectively (30). The blood:plasma ratio for tofacitinib, baricitinib, and upadacitinib were 1.20, 1.32, and 1.16, respectively. Possible serum-bound fraction in in vitro culture medium containing FBS was not used in the determination of culture media IC50.
2.10 Statistical analysis
A statistical analysis was performed using GraphPad PRISM 9. One-way ANOVA was performed to determine differences between groups. Each JAK1,2,3 inhibitor matched donor-to-donor response was compared to deucravacitinib using a paired two-tailed Student’s t test. Data are represented by geometric mean of at least two independent experiments containing multiple donors, where the ‘n’ is representative of donor numbers. Error bars represent ± standard error of the mean and P values are indicated by *P < 0.05, **P < 0.01, ***P < 0.001.
3 Results
Clinically relevant dose must be considered when framing the effect of a kinase inhibitor on a given pathway. Here, we classified inhibitors as being highly potent if the IC50 was determined to be less than or equal to the maximal WB drug concentration (Cmax) of the highest approved maintenance dose (28). Modest potency was considered IC50 <3x Cmax, mild potency was considered <5x Cmax, and minimal to no potency was considered >5x Cmax.
Deucravacitinib does not bind to or inhibit JAK2 in biochemical assays (25) and STAT phosphorylation assays (24). Similarly in the present study, there was no effect of deucravacitinib against pSTAT3 or pSTAT5 signaling in TPO-stimulated platelets (Figure 1A). The JAK1/JAK3 inhibitor tofacitinib had modest to high potency against pSTAT5 (IC50 = 628 nM) and pSTAT3 (IC50 = 201 nM). Baricitinib, known to be potent against JAK1 and JAK2 (31), was highly potent against pSTAT5 (IC50 = 124 nM) and pSTAT3 (IC50 = 31 nM) signaling in this assay. Upadacitinib, a reported JAK1 inhibitor, was also highly potent (pSTAT5 IC50 = 169 nM; pSTAT3 IC50 = 40 nM) in this signaling assay (Figure 1A). Functional models of megakaryocyte differentiation may improve the predictive power of in vitro inhibition assays. Hematopoietic stem cells differentiate into megakaryocyte progenitors with the addition of a mixture of JAK2-dependent cytokines that includes TPO. Unlike TPO pSTAT signaling, differentiation assays were less sensitive to the JAK inhibitors tofacitinib, baricitinib, and upadacitinib by only minimally inhibiting cell differentiation. Deucravacitinib consistently did not inhibit either signaling or functional assays of TPO (Figure 1B; Table 1). Hematopoietic stem cells stimulated with a cocktail of JAK2 cytokines induced differentiation into erythroid, myeloid, or granulocyte progenitors. It was previously reported that JAK1,2,3 inhibitors, but not deucravacitinib, were highly potent against EPO signaling in an erythroleukemic cell line (24). Here, erythroid progenitors were not observed to be functionally inhibited by deucravacitinib at concentrations exceeding 10 µM. Tofacitinib (IC50 = 704 nM), baricitinib (IC50 = 156 nM), and upadacitinib (IC50 = 355 nM) had modest potency (Figure 1C; Table 1). No loss of erythroid, myeloid, or granulocyte colony forming clusters (CFCs) at concentrations <500 nM, or loss in megakaryocyte colony forming units (CFUs) at concentrations <1000 nM were seen in deucravacitinib cultures. In contrast, fewer CFCs and CFUs in tofacitinib, and sparse CFCs and CFUs observed in baricitinib and upadacitinib at these concentrations (Figures 1D, E).
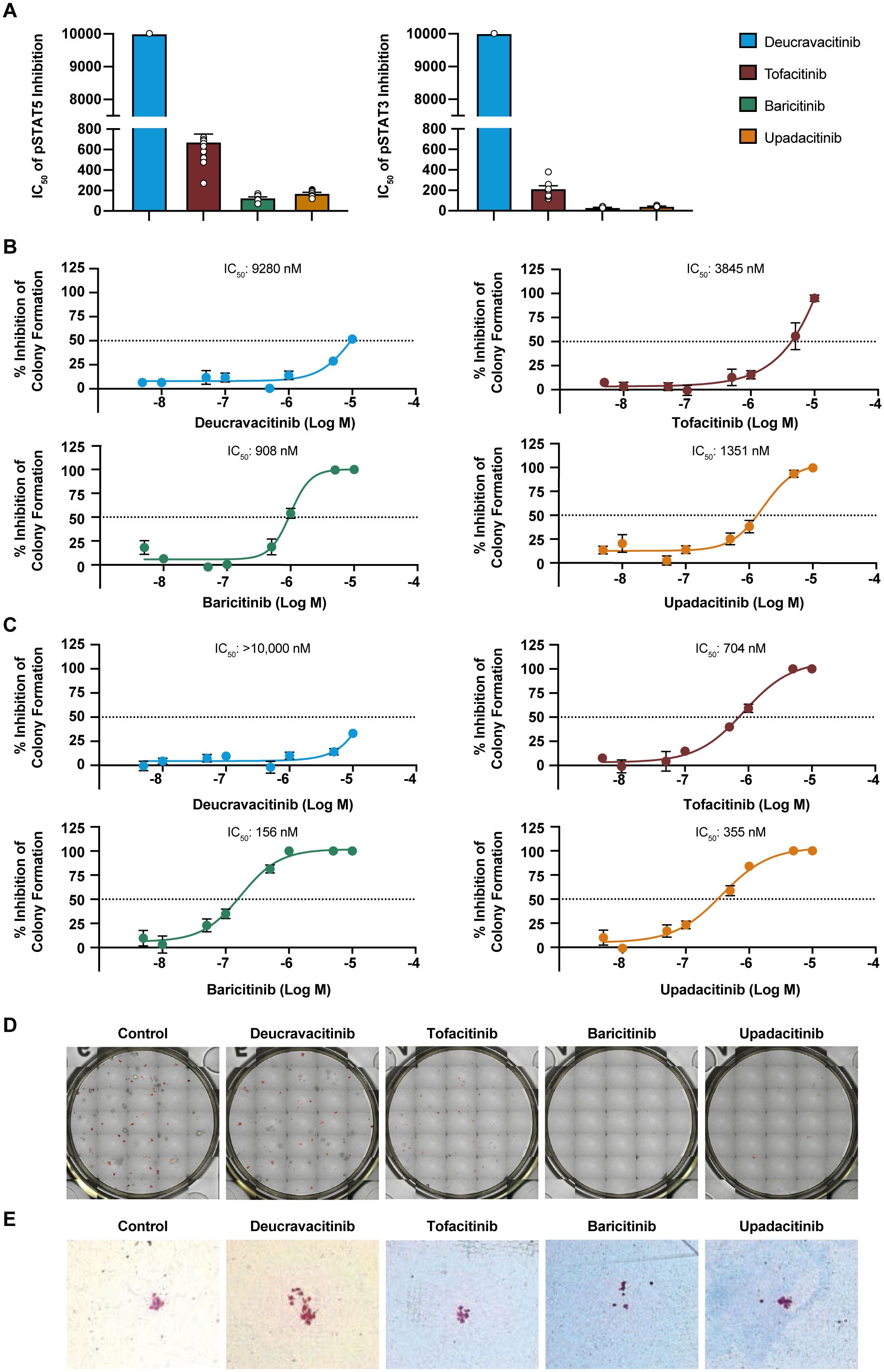
Figure 1. JAK inhibitors, but not deucravacitinib, have potency in JAK2-reliant hematopoietic assays. (A) Measured IC50 values of kinase inhibitors against thrombopoietin (50 ng/mL)-induced pSTAT5 and pSTAT3 of platelets from normal healthy volunteer human whole blood (n = 6-12 donors). Note that pSTAT3 data are adapted from Chimalakonda et al. (28), and graphed here for comparison. (B) Dose titration of kinase inhibitors on total megakaryocyte progenitor colony formation (Total CFU-Mk) from human pluripotent stem cell-cultured MegaCult™ containing thrombopoietin (50 ng/mL), IL-3 (10 ng/mL), and IL-6 (10 ng/mL) for 10 to 12 days (n = 4 donors). (C) Dose titration of kinase inhibitors on total erythroid progenitor colony formation from human pluripotent stem cell-cultured in MethoCult™ containing stem cell factor, GM-CSF, IL-3, G-CSF, and thrombopoietin for 13 to 15 days. (D) Representative images of total colony forming cluster (Total CFC) in MethoCult™ colony formation at 500 nM of kinase inhibitors. (E) Representative images of small CFU in MegaCult™ cultures at 1000 nM of kinase inhibitors.
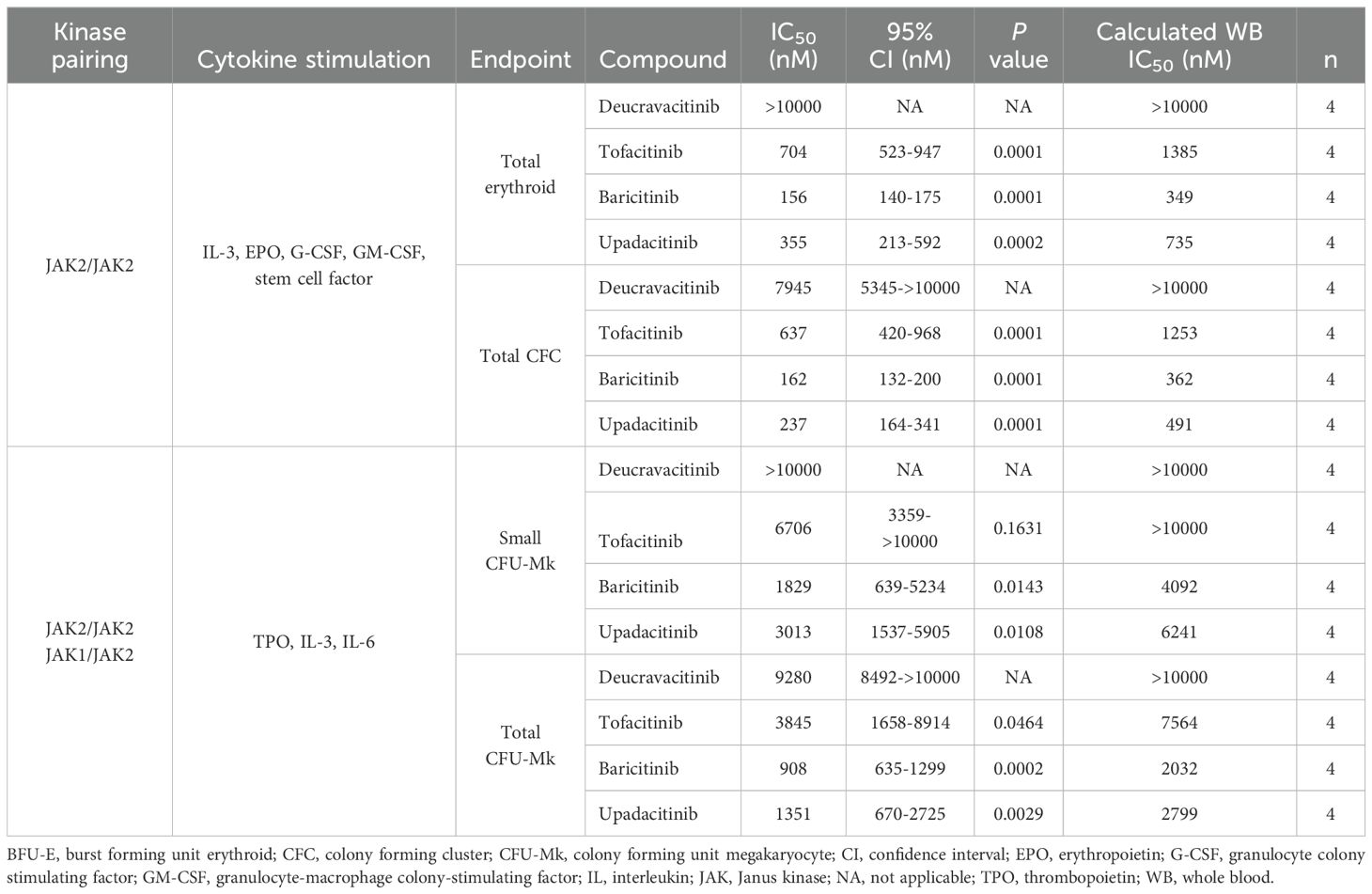
Table 1. Potencies of inhibitors in JAK2 hematopoietic assays.
JAK1 and JAK3 are required for the function of common gamma chain cytokines, IL-2, IL-4, IL-7, IL-9, IL-15, and IL-21. IL-7, IL-15 and IL-2 have vital homeostatic functions in T cells and NK cells, and IL-15 plays a role in NK-cell development (32) and CD8 function (33–35). IL-15–induced proliferation of NK and CD8 T cells that was strongly inhibited by JAK1,2,3 inhibitors and was only mildly to modestly inhibited by deucravacitinib (Figures 2A, B). IL-7 is required for thymic T-cell development (36), and T-cell survival in the periphery through expression of anti-apoptotic factor BCL-2 (37). Thus, BCL-2 expression was used as a marker of functional response to IL-7. Deucravacitinib had modest to no potency against IL-7, although differences were observed between the functional and signaling readouts in total CD4 T cells (Table 2). JAK1,2,3 inhibitors were highly potent against IL-7 and were consistently more potent in pSTAT5 signaling assays (Figure 2C) than against assays measuring BCL-2 upregulation (Figure 2D; Table 2). This observation was similarly reflected in IL-15 assays of NK cell activation by NKG2D upregulation compared with pSTAT5 expression (Table 2). We also studied the differences in kinase inhibitor potency against different immune cells and subsets. Interestingly, we find examples where differences between immune cell subsets are observed in common functional outputs but not in signaling outputs. In Figure 2C, inhibition of IL-7–mediated STAT phosphorylation was consistent in different subpopulations of CD4 T cells for each compound. Alternatively, BCL-2 expression was more potently inhibited in memory T cells than in STAT phosphorylation. Functional expression assays of BCL-2 within these donors show memory cells to be more prone to kinase inhibition than naive cells (Figure 2D; Table 2).
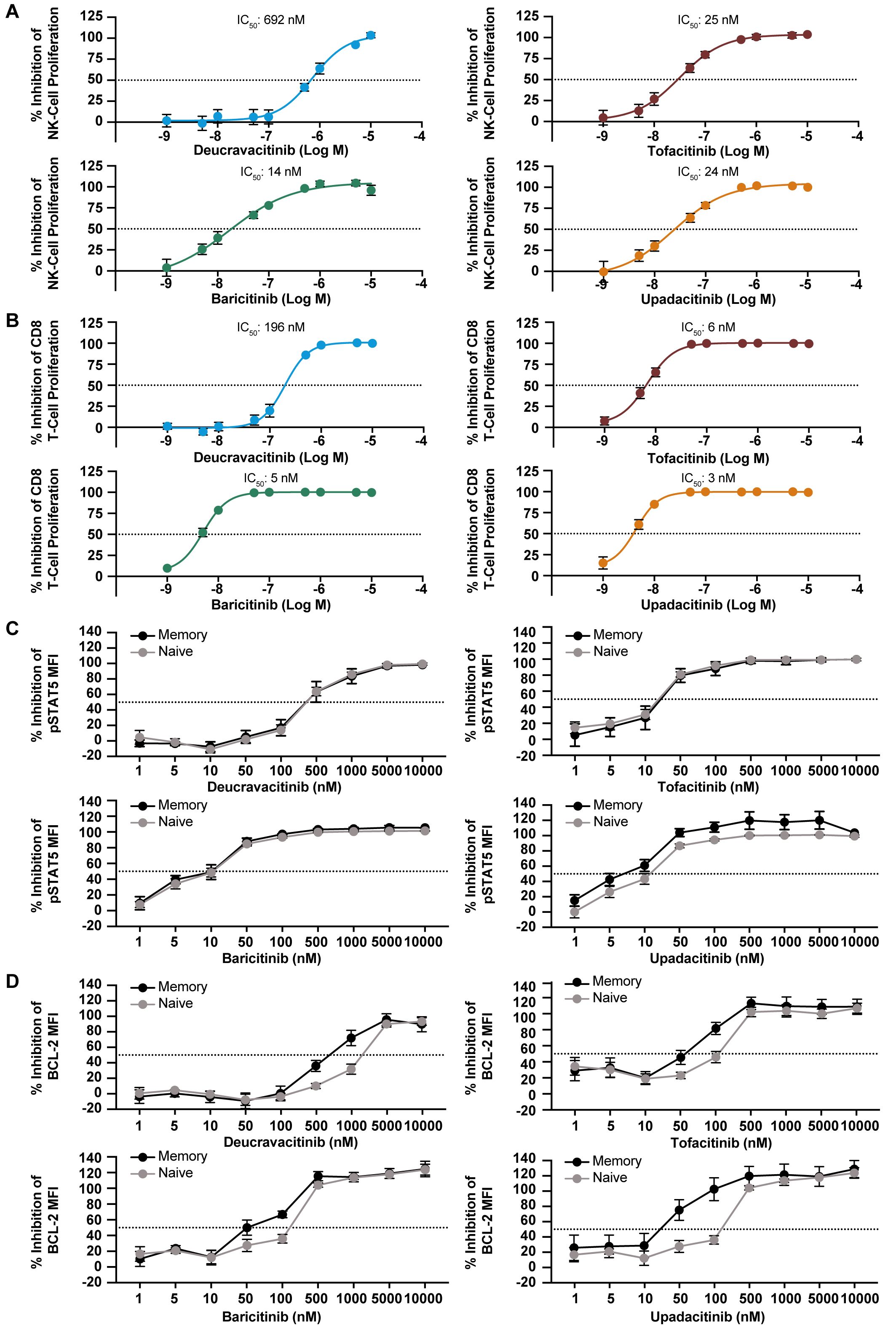
Figure 2. Homeostatic gamma chain cytokines are inhibited by JAK inhibitors but not deucravacitinib. PBMCs from normal healthy volunteers were treated with IL-15 (10/ng/mL) for 6 days in the presence or absence of dose-ranging kinase inhibitor. Inhibitory effect on proliferation was evaluated in: (A) NK cells and (B) CD8+ T cells (n = 6 donors). PBMCs from normal healthy volunteers were treated with IL-7 (50 ng/mL) for 15 min (C) or overnight (D) in the presence or absence of dose-ranging kinase inhibitor. Changes in BCL-2 and pSTAT5 median fluorescent intensity (MFI) were used to evaluate functional inhibitory effect in memory and naive CD4+ T cells (n = 5-6 donors).
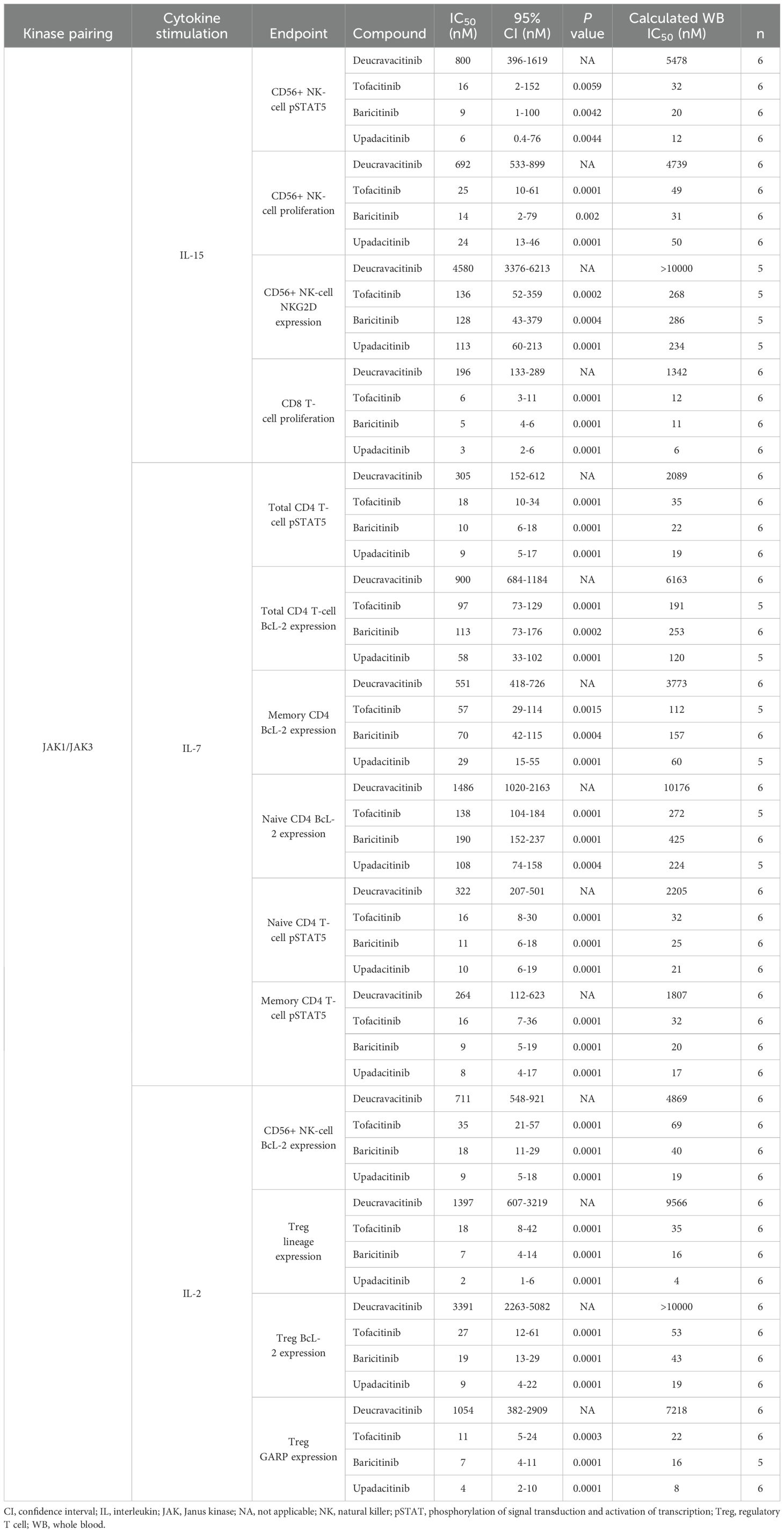
Table 2. Potencies of inhibitors common to gamma chain cytokine assays.
IL-2 is an effector cytokine for T cells (38) and an important NK-cell survival factor (39) but also necessary for Treg survival and the restraint of lethal autoimmunity (40, 41). Deucravacitinib and JAK1,2,3 inhibitors were evaluated against functional effects of IL-2 on Treg. GARP is upregulated upon Treg activation and enhances suppressive function (42). Other representative indicators of IL-2 function on Treg are BCL-2 expression and maintenance of lineage markers expression. Deucravacitinib did not inhibit survival, activation, or lineage marker expression of Tregs, whereas JAK1,2,3 inhibitors potently inhibited IL-2 function in Tregs (Table 2; Supplementary Table 1). A large magnitude of difference in potency was observed between deucravacitinib and JAK1,2,3 inhibitors, particularly upadacitinib, which was >158-fold more potent than deucravacitinib against all measured Treg outputs (Figure 3A). Similarly, IL-2 activity was potently inhibited in NK cells by JAK1,2,3 inhibitors (Figure 3B; Table 2).
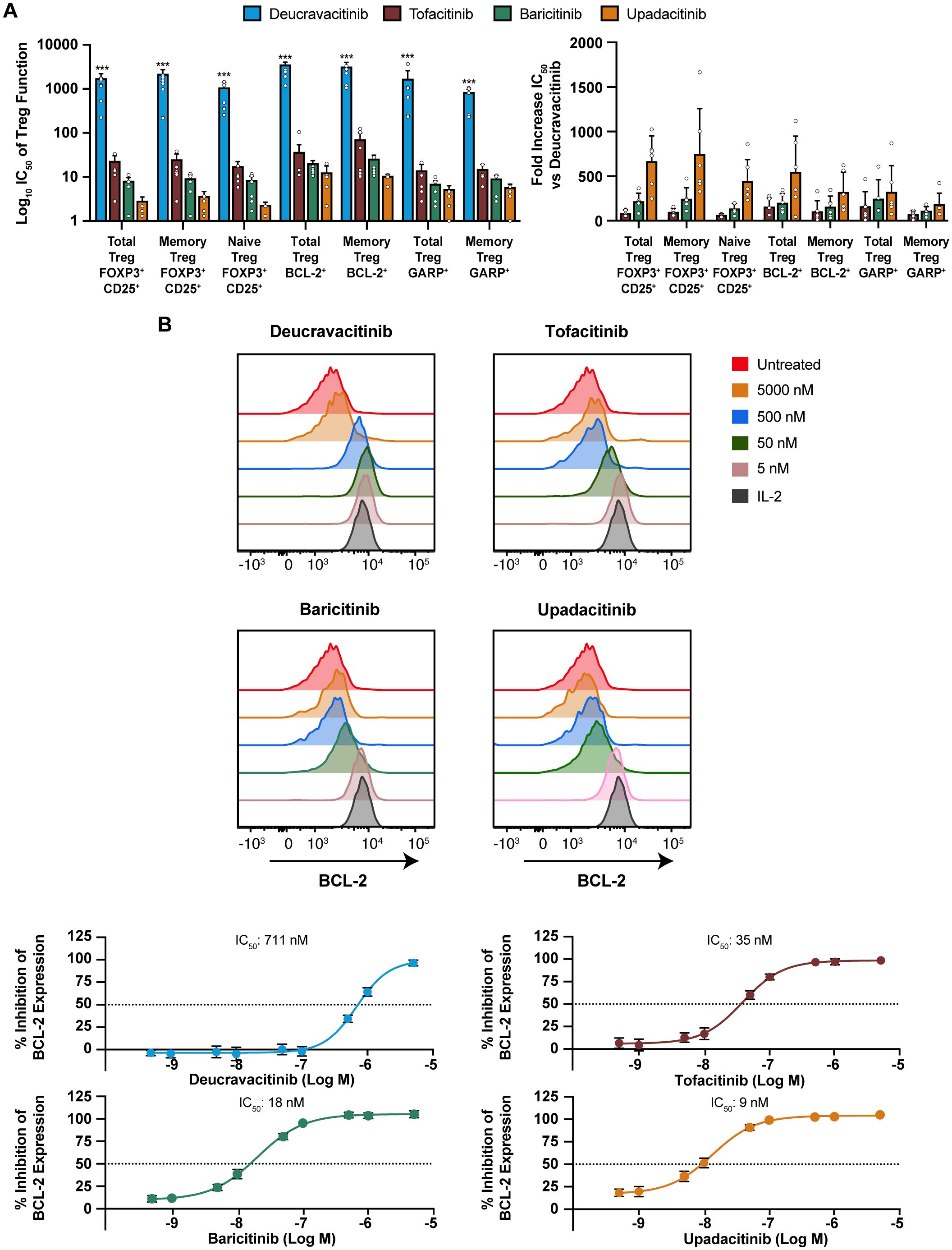
Figure 3. IL-2 effects in Treg and NK cells are potently inhibited by JAK inhibitors but not deucravacitinib. PBMCs from normal healthy volunteers were treated with low-dose IL-2 (2 ng/mL) for 6 days in the presence or absence of dose-ranging kinase inhibitor. (A) IC50 values were determined against functional expression and activation markers and fold difference in potency from deucravacitinib in Treg (n = 5-6 donors). Tregs were delineated as CD3+CD4+FOXP3+CD25+. Memory Tregs or naive Tregs were determined by the presence or absence of CD45RO. (B) Representative histograms of CD3–CD56+ NK BCL-2 response to PBMCs treated for 6 days with low-dose IL-2 in the presence of decreasing kinase inhibitor (n = 6 donors). Statistical significance of deucravacitinib from each JAK1,2,3 inhibitor was determined by paired two-tailed t-test. ***P < 0.001.
In vitro assays are performed in culture media with limited plasma proteins and do not account for the free fraction of drug in WB. Conversions of culture media IC50 to WB equivalents are required for adequate estimates of in vivo pharmacodynamic effect. IL-7 signaling assays were used as a benchmark to test the accuracy of WB conversions of kinase inhibitors (Supplementary Table 2). Inhibitory potency on JAK2 and common gamma chain assays with culture media and WB conversions are summarized in Tables 1, 2, respectively. Collectively, these data show that deucravacitinib has minimal to no potency against signaling or functional assays of JAK2 and common gamma chain in WB. JAK inhibitors show lesser potency in functional JAK2 assays in contrast to high potency in signaling assays and modest to high potency against common gamma chain pathways. Further evaluations of deucravacitinib and JAK1,2,3 inhibitor potencies are shown in Supplementary Table 1. IFNα (JAK1/TYK2) is a proinflammatory cytokine important for antiviral response and is dysregulated in several immune-mediated diseases, including SLE (43). We have previously observed some differences in potency against IFNα pSTAT assays in PBMCs favoring deucravacitinib, which targets TYK2, over tofacitinib, baricitinib, and upadacitinib, which target JAK1 (24). Testing of WB IFNα pSTAT2 supports this observation, with deucravacitinib showing up to a 5.3-fold increase in potency in total T cells, a 5.6-fold increase in B cells, and a 4.3-fold increase in monocytes compared with the JAK1,2,3 inhibitors measured (Figure 4A). Upadacitinib was found to have similar potency to deucravacitinib in the pSTAT2 assay compared with less potent tofacitinib and baricitinib. A striking difference in potency was observed in a functional assay of IFNα-activated monocytes (Figure 4B). Deucravacitinib was observed to be 7-fold, 13-fold, and 60-fold more potent than upadacitinib, baricitinib, and tofacitinib, respectively, showing a difference between targeting TYK2 over JAK1 in the Type I IFN pathway (Table 3). IFNγ is a Type II IFN with both overlapping and distinct yet critical functions from Type I IFN (44). Additionally, the IFNγ receptor pairs with JAK1/JAK2 to initiate downstream signaling and therefore is not an immediate target of deucravacitinib. Activation markers of monocytes and B cells induced by IFNγ, rather than IFNα, were not inhibited by deucravacitinib at relevant concentrations, indicating that it is a functional inhibitor of Type I IFN but not of Type II IFN (Figure 4B; Supplementary Figure 2). CXCL10 production in IFNα-treated monocyte-derived dendritic cells similarly shows a high preferential bias toward inhibition of TYK2 over JAK1. Interestingly, CXCL9, a related IFN-regulated chemokine, shows less of a TYK2-to-JAK1 bias in these same conditions (Figure 4C). Additional measures of chemokine production in total PBMC (Table 3) support observations of functional differences in TYK2 and JAK targeting within Type I IFN signaling. IL-12 and IL-23 are proinflammatory cytokines important for driving the maintenance of dysfunctional T-helper type 1 (Th1) and Th17 immune responses in certain inflammatory diseases (45–48). IL-12 and IL-23 signal through TYK2 via their shared p40 subunit, and JAK2 signals via p35 or p19, respectively. These cytokines are targets of deucravacitinib and baricitinib but not of tofacitinib or upadacitinib. IL-12 is a key regulator of IFNγ production. Previous works report deucravacitinib to show greater potency than JAK1,2,3 inhibitors against IL-12 + IL-18 (24). In this study, we find deucravacitinib to be highly potent and differentiated from JAK1,2,3 inhibitors against IL-12–, IL-12 + IL-18–, and IL-23–induced IFNγ production. In contrast, JAK1,2,3 inhibitors showed a range of potencies, from high to no potency against IL-12 and IL-23, possibly due to both on-target JAK1 and off-target JAK2 effects (Figures 4D, E; Table 3). IL-12–induced effects are largely thought to be synonymous with its downstream effector cytokine IFNγ whose effect is potentiated by JAK1/JAK2. Indeed, certain effects of IL-12 appear to be entirely driven by IFNγ, including chemokines that directly correlate with quantities of IFNγ produced (Supplementary Figure 3). However, we observed other inflammatory effects specifically driven by IL-12 signaling and independent of IFNγ (Figure 4F). Figure 4G shows IL-12–specific potency of deucravacitinib relative to JAK1,2,3 inhibitors, including a more than eight-fold increase in potency advantage of targeting TYK2 (deucravacitinib) than JAK2 (baricitinib) (Table 3).
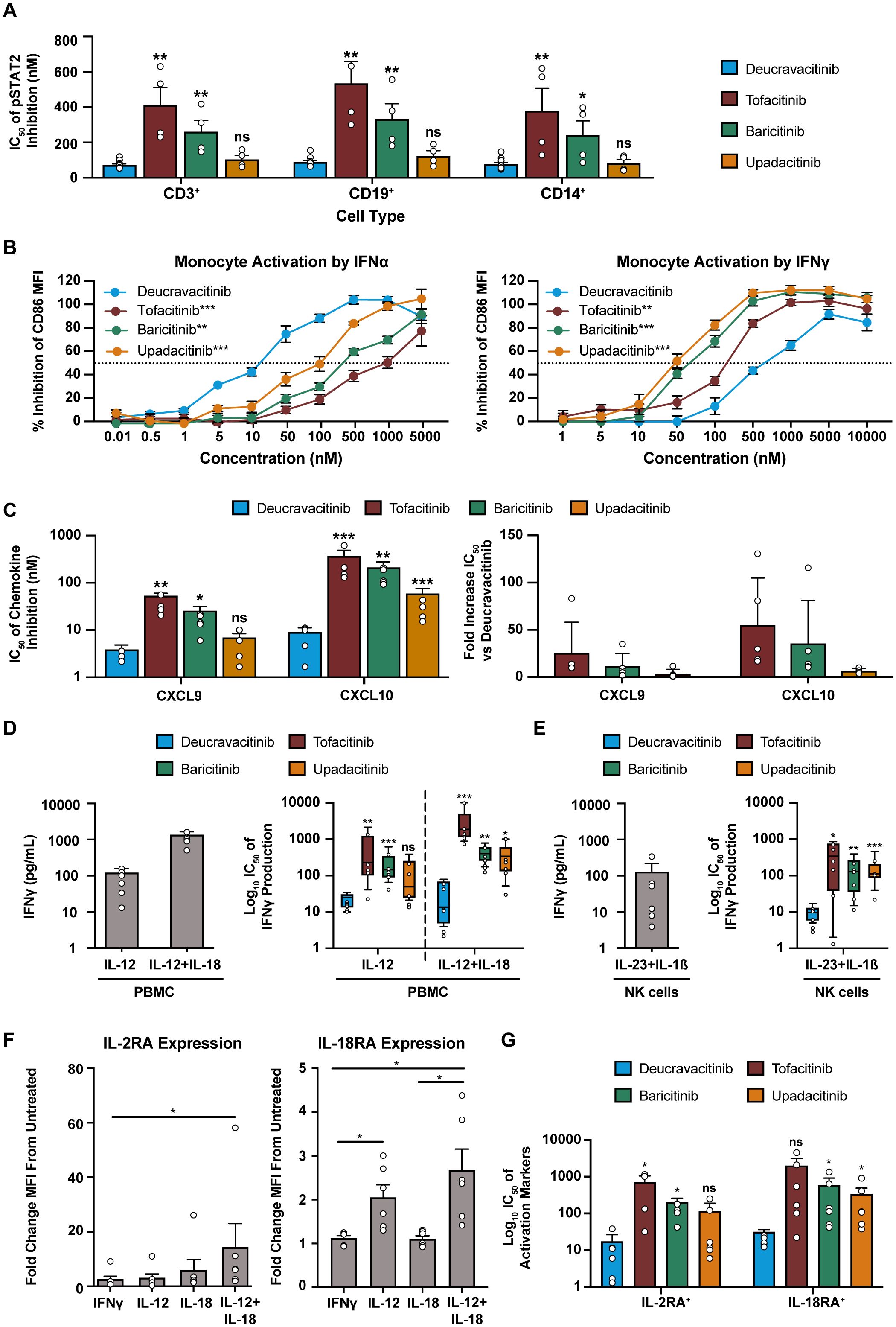
Figure 4. Deucravacitinib potently inhibits functional assays of Type I and Type II IFN. (A) Measured IC50 values of kinase inhibitors against IFNα (1000 U/mL)-induced pSTAT2 of CD3+ T cells, CD19+ B cells, and CD14+ monocytes from normal healthy volunteer human WB (n=4-12 donors). (B) PBMCs from normal healthy volunteers were treated with IFNα (1000 U/mL) or IFNγ (25 ng/mL) overnight in the presence or absence of dose-ranging kinase inhibitor and measured for the inhibition of CD86 expression by median fluorescence intensity on CD14+ monocytes (n = 6 donors). (C) Monocytes differentiated into monocyte-derived dendritic cells (mDC) were treated with IFNα (1000 U/mL) for 48 hours in the presence or absence of dose-ranging kinase inhibitors and measured for chemokine inhibition. IC50 values are represented in fold decreased potency from deucravacitinib (n = 5 donors). (D) PBMCs from normal healthy volunteers were treated with IL-12 (10 ng/mL) or IL-12 (2 ng/mL) + IL-18 (5 ng/mL)overnight in the presence or absence of dose-ranging kinase inhibitor and measured for the inhibition of IFNγ. (E) Negatively selected CD56+ NK cells from normal healthy volunteers were treated with IL-23 (50 ng/mL) + IL-1β (10 ng/mL) overnight in the presence or absence of dose-ranging kinase inhibitor and measured for the inhibition of IFNγ. (F) PBMCs from normal healthy volunteers were treated with indicated cytokines at described doses overnight. Gated CD56+ NK cells were evaluated for activation markers. (G) IL-12 + IL-18 treated PBMC cultures were additionally titrated with kinase inhibitor and evaluated for inhibitory potential against the activation markers of gated CD56+ NK cells. Statistical significance for expression analysis was determined by one-way ANOVA and significance of deucravacitinib from each JAK1,2,3 inhibitor was determined by paired two-tailed t-test. *P<0.05, **P<0.01, ***P<0.001. ns, non-significant.
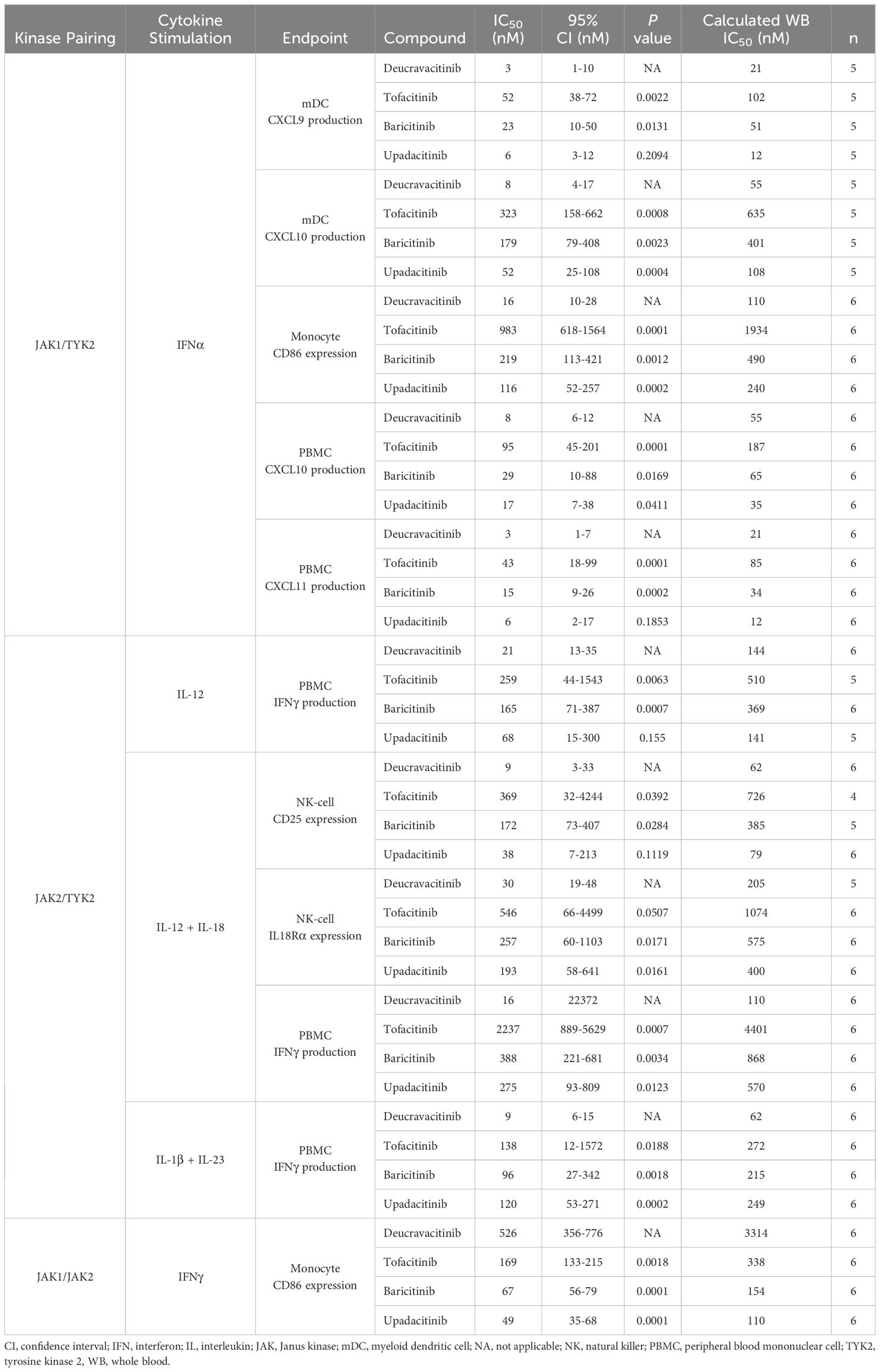
Table 3. Potencies of inhibitors on inflammatory cytokines.
4 Discussion
The introduction of JAK1,2,3 inhibitors has advanced autoimmune disease treatment but raised safety concerns due to lack of selectivity and resulting off-target effects among approved inhibitors. EPO, TPO and GM-CSF, essential for hematopoiesis, thrombopoiesis and myelopoiesis signal through JAK2 (49–51). JAK2 inhibition, an effective strategy for treating malignancy, results in anemia and thrombocytopenia (49, 50). Clinical trials of JAK1,2,3 inhibitors (baricitinib, tofacitinib, upadacitinib) aimed at autoimmune indications show similar effects, including platelet modulation and reductions in erythrocytes and neutrophils (9, 11, 52–54).
Deucravacitinib targets TYK2 through an allosteric mechanism by binding to the regulatory domain, which avoids the catalytic domain’s active site shared with other JAKs (55). Clinical trials of deucravacitinib have not shown clinical laboratory changes or safety signals indicative of JAK2 inhibition (26, 27, 56, 57), nor do in vitro STAT signaling assays reveal any potency against TPO or other JAK2-dependent cytokines (24, 25). JAK2 signaling assays for tofacitinib, baricitinib, and upadacitinib have produced variable potency results (24, 28, 29, 58–60), complicating the pharmacokinetic prediction of daily JAK2 inhibition (28, 30, 58).
Reductions of erythrocytes, platelets and granulocytes observed in clinical trials of JAK1,2,3 are likely due to decreased production. In vitro evaluations of JAK2/STAT signaling inhibition have focused on cell lines or non-proliferative mature myeloid cells and granulocytes and may not be accurate models of blood cell count abnormalities in patients. Instead, as a better model of clinical JAK2 inhibition liability, we have elected to use human pluripotent stem cells (HPSCs) which differentiate into specific lineages in response to EPO, TPO, G-CSF, stem cell factor, and GM-CSF (61–64). Deucravacitinib had no effect on megakaryocyte differentiation, consistent with stable clinical platelet count. JAK1,2,3 inhibitors affect megakaryocyte differentiation and their rank order (baricitinib > upadacitinib > tofacitinib) was consistent with the potency against JAK2 and inhibition in STAT assays.
Clinical studies show that tofacitinib decreases platelets that stabilize over time (52), while baricitinib and upadacitinib show an increase during the first month of treatment followed by a sharp decrease and eventual stabilization (13, 53, 65, 66). TPO levels in blood are balanced by platelet binding (67). Low platelet levels create excess TPO, which causes bone marrow HPSC to differentiate into megakaryocytes and produce more platelets (68). Hence, differences between tofacitinib, baricitinib and upadacitinib are likely due to complex TPO and megakaryocyte biology as well as differing drug distribution between bone marrow and blood.
Deucravacitinib does not affect neutrophils or red blood cell counts (26, 27, 56), while JAK1,2,3 inhibitors do impact these populations. In clinical trials baricitinib and upadacitinib reduce hemoglobin and neutrophils (12, 13), while tofacitinib reduces neutrophils but shows hemoglobin reduction only at higher doses (6, 69, 70). These differences are likely due to varying potency against bone marrow progenitor cells and the contribution of inhibiting JAK1 cytokines IL-6 and OSM to neutropenia and thrombocytopenia (71–73). Overall clinical, and in vitro data support that deucravacitinib lacks JAK2 activity, whereas tofacitinib, baricitinib, and upadacitinib exhibit JAK2 inhibitory activity in vivo.
Reductions in NK and T cells, increases in B cells, and lymphopenia are on-target effects of JAK1,2,3 inhibitors (74, 75) but not deucravacitinib (56). Hematopoietic dependency is attributed to specific cytokines, while lymphocyte homeostasis is likely influenced by all common gamma chain cytokines. The relative contribution of each of these toward lymphocyte populations have been determined through studying genetic deficiencies. IL-7 is crucial for thymic T cell development and peripheral maintenance (76), while a defective IL-7 receptor presents as T-cell–deficient (but not NK-cell–deficient) severe combined immunodeficiency (77). IL-2 supports various lymphocyte subtypes, but its loss results in lethal autoimmunity (78). IL-15 is vital for NK cell expansion and maturation (32), and its absence results in a dramatic loss of mature NK cells and memory CD8 T cells (79).
We chose functional assays of NK and CD8 proliferation and activation as a model of IL-15 effect. NK cells were considerably less prone to inhibition from kinase inhibitors than CD8 cells. Differences in kinase inhibitor potency for the same cytokine have been previously reported between cell types in pSTAT assays (29, 30) and our results indicate these differences extend to cell lineage subsets. Naive T cells were more resistant to kinase inhibition than effector and memory T cells in IL-7–induced BCL-2 assays. Both naive and memory T cells depend on IL-7 for survival, with naive cells undergoing apoptosis without IL-7 and memory cells experiencing partial reduction (1, 80). A lower threshold of JAK signaling required for naive versus memory cell survival may reflect how critical the pathway is to T-cell function. The differential effects on naive and memory populations in JAK1,2,3 inhibitor clinical trials remain unknown.
Signaling assays of IL-7 and IL-15 indicate that JAK1,2,3 inhibitors are highly potent, as previously reported with IL-2. Modeling based on IL-2 IC50 predicts up to 94% average daily inhibition at clinical dosage (28), which would suggest severe lymphopenia to be more prevalent with JAK1,2,3 inhibitor use. It is instead likely that functional measures of cytokine signaling, such as BCL-2 employed herein, are more predictive of in vivo biological pharmacodynamic effects. In psoriasis clinical trials, T-cell or NK-cell counts remained stable with deucravacitinib treatment (56), which is in agreement with a lack of effect on this pathway. Conversely, JAK1,2,3 inhibitor trials showed short-term fluctuations, followed by fast declines in NK and slower declines in CD4, and CD8 cells (74, 75). NK cells turn over three to four times faster than T cells (81), which may explain this observation and suggests different cytokine dependencies.
Tregs critically rely on IL-2 for survival and function (3, 41). This work shows deucravacitinib to minimally affect IL-2 and thus spare Tregs, unlike JAK1,2,3 inhibitors whose strong IL-2 potency could explain the decline in circulating peripheral Tregs observed in clinical trials of tofacitinib and baricitinib (75, 82). JAK1,2,3 inhibitors have also been associated with increased risk of serious infections, including herpes zoster (66, 70, 83). Infection rate in baricitinib trials was associated with decreased NK and CD4 T-cell counts (75), whereas for tofacitinib correlates were seen with overall lymphocyte count decreases but no particular subset (74). A similar infection profile is observed during Type I and Type II IFN inhibition when a very high pathway blockade is achieved (84, 85).
The overall infection risk of JAK1,2,3 inhibitors can likely be attributed to broad immunosuppression involving many cytokines and lymphocyte decreases. Deucravacitinib is a potent Type I IFN inhibitor and indirectly inhibits IFNγ; however, infection rates are low relative to JAK1,2,3 inhibitors due to optimized dosing that sufficiently controls inflammation without complete pathway inhibition (26, 28). This is consistent with TYK2 partial loss of function mutations known to be protective against autoimmunity without evidence of susceptibility to infection (86, 87).
Functional assays provide additional value over signaling assays alone. Inhibition of STAT signaling from JAK-regulated cytokines varies between cell types, and non-trivial differences within the same population have been reported (24, 30). Discerning the relative signaling contribution of different STATs is complex and not understood. Deucravacitinib and JAK1,2,3 inhibitors inhibit Type I IFN via TYK2 and JAK1, respectively, with deucravacitinib being more potent. How much more potent depends on the cell type and STAT assay (24). The functional data presented in this report reinforces this observation, while possibly being more meaningful in the context of observed pharmacodynamic effects in vivo. Herein, deucravacitinib was at least six-fold more potent against IFNα-induced CXCL10 than JAK inhibitors. Phase 2 SLE data shows deucravacitinib reduces CXCL10 by 42% at week 12 (88) and the IFN-5 (MX1, HERC5, IFIT1, RSAD2, EIF2AK2) gene signature score by up to 70% at Week 4 (89). Baricitinib reduced CXCL10 by 23% at week 12 and IFN-5 genes up to 32% in SLE patients (90, 91). For upadacitinib, CXCL10 levels fluctuated in SLE patients and did not reduce at week 12, whereas the IFN signature reduced by 29% but only at week 24 and not prior (92). Final evaluation of clinical efficacy for upadacitinib in SLE trials is ongoing. While final conclusions cannot be made from cross-trial comparisons, the data suggest that JAK1,2,3 inhibitors have a less robust effect against Type I IFN than deucravacitinib. It appears that IFN signaling has greater reliance on TYK2 than JAK1, which is in line with models showing the average coverage of JAK1 inhibition by baricitinib and upadacitinib to be as high or higher than deucravacitinib for TYK2 (28, 30).
Potency differences in shared pathways of TYK2 and JAK expand beyond Type I IFN. Baricitinib is equipotent against both JAK1 and JAK2 (31) and assays against common gamma chain cytokines conducted herein confirm high JAK1 potency. However, our assessment of IL-12 or IL-23 function show deucravacitinib is more potent than baricitinib by 20-fold in the TYK2/JAK2 pathway. This suggests that targeting TYK2 in IL-12/23–driven diseases such as psoriasis is more effective, while additionally avoiding JAK1,2,3 inhibitor hematopoietic safety concerns.
The mechanistic reasons for favoring TYK2 inhibition and the variability in functional readouts in specific cytokine pathways are unclear. JAK family member mutations can reduce or enhance cytokine signaling (93, 94), but effects vary by cytokine and cell type (86, 95, 96), suggesting fundamental differences in signaling requirements among JAK family members within receptor pairs. Other differences could be due to the targeting modalities. The pseudokinase domain lacks catalytic activity and regulates kinase signaling by steric inhibition of the kinase domain or by limiting catalytic site flexibility (97). Deucravacitinib inhibits TYK2 by stabilizing the structure of the pseudokinase domain in the inactive confirmation, thereby preventing the active site from having the mobility to phosphorylate (25). This is fundamentally different from tofacitinib, baricitinib, and upadacitinib, which are adenosine triphosphate (ATP) competitive active site inhibitors and work by blocking phosphorylation directly.
Potential limitations of this work include the use of healthy donor immune cells versus those sourced from psoriasis or SLE patients; however, as we investigated fundamental cellular processes, we believe these data to be highly valuable. Further work evaluating immune cell functionality, such as assessing Treg suppression ability, could be performed to expand on the chosen readouts of Treg markers, BCL2, and GARP expression. Killing assays could be performed to more deeply understand functional consequences of JAK1,2,3 inhibition on CD8 and NK cells. Additionally, our observation of pathway inhibition favoring TYK2 over JAK1/2 was limited to studying key inflammatory cytokines (Type I IFN, IL-12, and IL-23). This may or may not be observed in other common pathways (i.e., IL-19, IL-20, IL-22).
The data presented herein have shown the importance of functional analysis alongside signaling assays when studying kinase inhibitors. This work reinforces that deucravacitinib is TYK2 selective with superior target fidelity compared to approved JAK1,2,3 inhibitors, sparing JAK2 and JAK3 homeostatic pathways. Deucravacitinib is more potent in Type I IFN and IL-12/IL-23 pathways than JAK2 inhibitors. These findings support clinical observations that deucravacitinib effectively limits cytokine-driven inflammation in autoimmune disease, without compromising normal immune function.
Data availability statement
Bristol Myers Squibb details their data sharing request process via https://www.bms.com/researchers-and-partners/independent-research/data-sharing-request-process.html.
Ethics statement
The studies involving humans were performed in accordance with the Helsinki Declaration of 1964 and its amendments. Study was approved by Bristol Myers Squibb Environmental Health & Safety. All blood donors provided written informed consent to participate in this study. The Bristol Myers Squibb blood collection program was governed by an internal group equivalent to an ethics committee.
Author contributions
BJ: Writing – original draft, Writing – review & editing, Conceptualization, Data curation, Formal analysis, Investigation, Methodology. LC: Writing – original draft, Writing – review & editing, Data curation, Formal analysis, Methodology. JK: Writing – original draft, Writing – review & editing, Formal analysis, Investigation. IMC: Writing – original draft, Writing – review & editing, Conceptualization, Formal analysis, Investigation, Methodology. PS: Writing – original draft, Writing – review & editing, Investigation.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was sponsored by Bristol Myers Squibb.
Acknowledgments
Editorial assistance was provided by Peloton Advantage, LLC, an OPEN Health company, funded by Bristol Myers Squibb.
Conflict of interest
BJ, LC, JK, and PS are employees of and shareholders in Bristol Myers Squibb. Author IMC was an employee of Bristol Myers Squibb at the time of study conduct.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2024.1437512/full#supplementary-material
References
1. Tan JT, Dudl E, LeRoy E, Murray R, Sprent J, Weinberg KI, et al. IL-7 is critical for homeostatic proliferation and survival of naive T cells. Proc Natl Acad Sci U S A. (2001) 98:8732–7. doi: 10.1073/pnas.161126098
2. Cavazzana-Calvo M, Hacein-Bey S, de Saint Basile G, De Coene C, Selz F, Le Deist F, et al. Role of interleukin-2 (IL-2), IL-7, and IL-15 in natural killer cell differentiation from cord blood hematopoietic progenitor cells and from gamma c transduced severe combined immunodeficiency X1 bone marrow cells. Blood. (1996) 88:3901–9. doi: 10.1182/blood.V88.10.3901.bloodjournal88103901
3. Malek TR, Yu A, Vincek V, Scibelli P, Kong L. CD4 regulatory T cells prevent lethal autoimmunity in IL-2Rbeta-deficient mice. Implications for the nonredundant function of IL-2. Immunity. (2002) 17:167–78. doi: 10.1016/S1074-7613(02)00367-9
4. Megna M, Potestio L, Ruggiero A, Cacciapuoti S, Maione F, Tasso M, et al. JAK inhibitors in psoriatic disease. Clinical cosmetic investigational Dermatol. (2023) 16:3129–45. doi: 10.2147/CCID.S433367
5. Shawky AM, Almalki FA, Abdalla AN, Abdelazeem AH, Gouda AM. A comprehensive overview of globally approved JAK inhibitors. Pharmaceutics. (2022) 14:1001. doi: 10.3390/pharmaceutics14051001
6. Schulze-Koops H, Strand V, Nduaka C, DeMasi R, Wallenstein G, Kwok K, et al. Analysis of haematological changes in tofacitinib-treated patients with rheumatoid arthritis across phase 3 and long-term extension studies. Rheumatology. (2017) 56:46–57. doi: 10.1093/rheumatology/kew329
7. Valenzuela F, Papp KA, Pariser D, Tyring SK, Wolk R, Buonanno M, et al. Effects of tofacitinib on lymphocyte sub-populations, CMV and EBV viral load in patients with plaque psoriasis. BMC Dermatol. (2015) 15:8. doi: 10.1186/s12895-015-0025-y
8. Weinhold KJ, Bukowski JF, Brennan TV, Noveck RJ, Staats JS, Lin L, et al. Reversibility of peripheral blood leukocyte phenotypic and functional changes after exposure to and withdrawal from tofacitinib, a Janus kinase inhibitor, in healthy volunteers. Clin Immunol. (2018) 191:10–20. doi: 10.1016/j.clim.2018.03.002
9. Papp KA, Menter MA, Raman M, Disch D, Schlichting DE, Gaich C, et al. A randomized phase 2b trial of baricitinib, an oral Janus kinase (JAK) 1/JAK2 inhibitor, in patients with moderate-to-severe psoriasis. Br J Dermatol. (2016) 174:1266–76. doi: 10.1111/bjd.2016.174.issue-6
10. Papp KA, Menter A, Strober B, Langley RG, Buonanno M, Wolk R, et al. Efficacy and safety of tofacitinib, an oral Janus kinase inhibitor, in the treatment of psoriasis: a Phase 2b randomized placebo-controlled dose-ranging study. Br J Dermatol. (2012) 167:668–77. doi: 10.1111/j.1365-2133.2012.11168.x
11. Papp KA, Menter MA, Abe M, Elewski B, Feldman SR, Gottlieb AB, et al. Tofacitinib, an oral Janus kinase inhibitor, for the treatment of chronic plaque psoriasis: results from two randomized, placebo-controlled, phase III trials. Br J Dermatol. (2015) 173:949–61. doi: 10.1111/bjd.14018
12. Kay J, Harigai M, Rancourt J, Dickson C, Melby T, Issa M, et al. Changes in selected haematological parameters associated with JAK1/JAK2 inhibition observed in patients with rheumatoid arthritis treated with baricitinib. RMD Open. (2020) 6:e001370. doi: 10.1136/rmdopen-2020-001370
13. Brmester GR, Kremer JM, Van den Bosch F, Kivitz A, Bessette L, Li Y, et al. Safety and efficacy of upadacitinib in patients with rheumatoid arthritis and inadequate response to conventional synthetic disease-modifying anti-rheumatic drugs (SELECT-NEXT): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet. (2018) 391:2503–12. doi: 10.1016/S0140-6736(18)31115-2
14. Harden JL, Krueger JG, Bowcock AM. The immunogenetics of psoriasis: a comprehensive review. J Autoimmun. (2015) 64:66–73. doi: 10.1016/j.jaut.2015.07.008
15. Danese S, Peyrin-Biroulet L. Selective tyrosine kinase 2 inhibition for treatment of inflammatory bowel disease: new hope on the rise. Inflammation Bowel Dis. (2021) 27:2023–30. doi: 10.1093/ibd/izab135
16. Chasset F, Arnaud L. Targeting interferons and their pathways in systemic lupus erythematosus. Autoimmun Rev. (2018) 17:44–52. doi: 10.1016/j.autrev.2017.11.009
17. Del Papa N, Minniti A, Lorini M, Carbonelli V, Maglione W, Pignataro F, et al. The role of interferons in the pathogenesis of Sjögren’s syndrome and future therapeutic perspectives. Biomolecules. (2021) 11:251. doi: 10.3390/biom11020251
18. Couturier N, Bucciarelli F, Nurtdinov RN, Debouverie M, Lebrun-Frenay C, Defer G, et al. Tyrosine kinase 2 variant influences T lymphocyte polarization and multiple sclerosis susceptibility. Brain. (2011) 134:693–703. doi: 10.1093/brain/awr010
19. Contreras-Cubas C, García-Ortiz H, Velázquez-Cruz R, Barajas-Olmos F, Baca P, Martínez-Hernández A, et al. Catalytically impaired TYK2 variants are protective against childhood- and adult-onset systemic lupus erythematosus in Mexicans. Sci Rep. (2019) 9:12165. doi: 10.1038/s41598-019-48451-3
20. Khatri B, Tessneer KL, Rasmussen A, Aghakhanian F, Ragna Reksten T, Adler A, et al. Genome-wide association study identifies Sjögren’s risk loci with functional implications in immune and glandular cells. Nat Commun. (2022) 13:4287. doi: 10.1038/s41467-022-30773-y
21. Kreins AY, Ciancanelli MJ, Okada S, Kong X-F, Ramirez-Alejo N, Sebnem Kilic S, et al. Human TYK2 deficiency: mycobacterial and viral infections without hyper-IgE syndrome. J Exp Med. (2015) 212:1641–62. doi: 10.1084/jem.20140280
23. Sotyktu [European summary of product characteristics]. Dublin, Ireland: Bristol Myers Squibb EEIG (2023).
24. Burke JR, Cheng L, Gillooly KM, Strnad J, Zupa-Fernandez A, Catlett IM, et al. Autoimmune pathways in mice and humans are blocked by pharmacological stabilization of the TYK2 pseudokinase domain. Sci Transl Med. (2019) 11:eaaw1736. doi: 10.1126/scitranslmed.aaw1736
25. Wrobleski ST, Moslin R, Lin S, Zhang Y, Spergel S, Kempson J, et al. Highly selective inhibition of tyrosine kinase 2 (TYK2) for the Treatment of autoimmune diseases: discovery of the allosteric inhibitor BMS-986165. J Med Chem. (2019) 62:8973–95. doi: 10.1021/acs.jmedchem.9b00444
26. Armstrong AW, Gooderham M, Warren RB, Papp KA, Strober B, Thaçi D, et al. Deucravacitinib versus placebo and apremilast in moderate to severe plaque psoriasis: efficacy and safety results from the 52-week, randomized, double-blinded, placebo-controlled phase 3 POETYK PSO-1 trial. J Am Acad Dermatol. (2023) 88:29–39. doi: 10.1016/j.jaad.2022.07.002
27. Strober B, Thaçi D, Sofen H, Kircik L, Gordon KB, Foley P, et al. Deucravacitinib versus placebo and apremilast in moderate to severe plaque psoriasis: efficacy and safety results from the 52-week, randomized, double-blinded, Program fOr Evaluation of TYK2 inhibitor psoriasis second phase 3 trial. J Am Acad Dermatol. (2023) 88:40–51. doi: 10.1016/j.jaad.2022.08.061
28. Chimalakonda A, Burke J, Cheng L, Catlett I, Tagen M, Zhao Q, et al. Selectivity profile of the tyrosine kinase 2 inhibitor deucravacitinib compared with janus kinase 1/2/3 inhibitors. Dermatol Ther (Heidelb). (2021) 11:1763–76. doi: 10.1007/s13555-021-00596-8
29. McInnes IB, Byers NL, Higgs RE, Lee J, Macias WL, Na S, et al. Comparison of baricitinib, upadacitinib, and tofacitinib mediated regulation of cytokine signaling in human leukocyte subpopulations. Arthritis Res Ther. (2019) 21:183. doi: 10.1186/s13075-019-1964-1
30. Dowty ME, Lin TH, Jesson MI, Hegen M, Martin DA, Katkade V, et al. Janus kinase inhibitors for the treatment of rheumatoid arthritis demonstrate similar profiles of in vitro cytokine receptor inhibition. Pharmacol Res Perspect. (2019) 7:e00537. doi: 10.1002/prp2.v7.6
31. Fridman JS, Scherle PA, Collins R, Burn TC, Li Y, Li J, et al. Selective inhibition of JAK1 and JAK2 is efficacious in rodent models of arthritis: preclinical characterization of INCB028050. J Immunol. (2010) 184:5298–307. doi: 10.4049/jimmunol.0902819
32. Mrózek E, Anderson P, Caligiuri MA. Role of interleukin-15 in the development of human CD56+ natural killer cells from CD34+ hematopoietic progenitor cells. Blood. (1996) 87:2632–40. doi: 10.1182/blood.V87.7.2632.bloodjournal8772632
33. Mueller YM, Petrovas C, Bojczuk PM, Dimitriou ID, Beer B, Silvera P, et al. Interleukin-15 increases effector memory CD8+ t cells and NK Cells in simian immunodeficiency virus-infected macaques. J Virol. (2005) 79:4877–85. doi: 10.1128/JVI.79.8.4877-4885.2005
34. Van Audenaerde JRM, De Waele J, Marcq E, Van Loenhout J, Lion E, Van den Bergh JMJ, et al. Interleukin-15 stimulates natural killer cell-mediated killing of both human pancreatic cancer and stellate cells. Oncotarget. (2017) 8:56968–79. doi: 10.18632/oncotarget.18185
35. Epardaud M, Elpek KG, Rubinstein MP, Yonekura AR, Bellemare-Pelletier A, Bronson R, et al. Interleukin-15/interleukin-15R alpha complexes promote destruction of established tumors by reviving tumor-resident CD8+ T cells. Cancer Res. (2008) 68:2972–83. doi: 10.1158/0008-5472.CAN-08-0045
36. Roifman CM, Zhang J, Chitayat D, Sharfe N. A partial deficiency of interleukin-7R alpha is sufficient to abrogate T-cell development and cause severe combined immunodeficiency. Blood. (2000) 96:2803–7. doi: 10.1182/blood.V96.8.2803
37. Chetoui N, Boisvert M, Gendron S, Aoudjit F. Interleukin-7 promotes the survival of human CD4+ effector/memory T cells by up-regulating Bcl-2 proteins and activating the JAK/STAT signalling pathway. Immunology. (2010) 130:418–26. doi: 10.1111/j.1365-2567.2009.03244.x
38. Morgan DA, Ruscetti FW, Gallo R. Selective in vitro growth of T lymphocytes from normal human bone marrows. Science. (1976) 193:1007–8. doi: 10.1126/science.181845
39. Henney CS, Kuribayashi K, Kern DE, Gillis S. Interleukin-2 augments natural killer cell activity. Nature. (1981) 291:335–8. doi: 10.1038/291335a0
40. Zemmour D, Charbonnier LM, Leon J, Six E, Keles S, Delville M, et al. Single-cell analysis of FOXP3 deficiencies in humans and mice unmasks intrinsic and extrinsic CD4(+) T cell perturbations. Nat Immunol. (2021) 22:607–19. doi: 10.1038/s41590-021-00910-8
41. Fontenot JD, Rasmussen JP, Gavin MA, Rudensky AY. A function for interleukin 2 in Foxp3-expressing regulatory T cells. Nat Immunol. (2005) 6:1142–51. doi: 10.1038/ni1263
42. Wang R, Wan Q, Kozhaya L, Fujii H, Unutmaz D. Identification of a regulatory T cell specific cell surface molecule that mediates suppressive signals and induces Foxp3 expression. PloS One. (2008) 3:e2705. doi: 10.1371/journal.pone.0002705
43. Miyachi K, Iwamoto T, Kojima S, Ida T, Suzuki J, Yamamoto T, et al. Relationship of systemic type I interferon activity with clinical phenotypes, disease activity, and damage accrual in systemic lupus erythematosus in treatment-naive patients: a retrospective longitudinal analysis. Arthritis Res Ther. (2023) 25:26. doi: 10.1186/s13075-023-03010-0
44. Lee AJ, Ashkar AA. The dual nature of Type I and Type II interferons. Front Immunol. (2018) 9:2061. doi: 10.3389/fimmu.2018.02061
45. Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, et al. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. (2005) 201:233–40. doi: 10.1084/jem.20041257
46. Pawlak M, DeTomaso D, Schnell A, Meyer Zu Horste G, Lee Y, Nyman J, et al. Induction of a colitogenic phenotype in Th1-like cells depends on interleukin-23 receptor signaling. Immunity. (2022) 55:1663–79.e6. doi: 10.1016/j.immuni.2022.08.007
47. Lee E, Trepicchio WL, Oestreicher JL, Pittman D, Wang F, Chamian F, et al. Increased expression of interleukin 23 p19 and p40 in lesional skin of patients with psoriasis vulgaris. J Exp Med. (2004) 199:125–30. doi: 10.1084/jem.20030451
48. Uhlig HH, McKenzie BS, Hue S, Thompson C, Joyce-Shaikh B, Stepankova R, et al. Differential activity of IL-12 and IL-23 in mucosal and systemic innate immune pathology. Immunity. (2006) 25:309–18. doi: 10.1016/j.immuni.2006.05.017
49. Constantinescu SN, Ghaffari S, Lodish HF. The erythropoietin receptor: structure, activation and intracellular signal transduction. Trends Endocrinol Metab. (1999) 10:18–23. doi: 10.1016/S1043-2760(98)00101-5
50. Alexander WS, Roberts AW, Nicola NA, Li R, Metcalf D. Deficiencies in progenitor cells of multiple hematopoietic lineages and defective megakaryocytopoiesis in mice lacking the thrombopoietic receptor c-Mpl. Blood. (1996) 87:2162–70. doi: 10.1182/blood.V87.6.2162.bloodjournal8762162
51. Seymour JF, Lieschke GJ, Grail D, Quilici C, Hodgson G, Dunn AR. Mice lacking both granulocyte colony-stimulating factor (CSF) and granulocyte-macrophage CSF have impaired reproductive capacity, perturbed neonatal granulopoiesis, lung disease, amyloidosis, and reduced long-term survival. Blood. (1997) 90:3037–49. doi: 10.1182/blood.V90.8.3037
52. Yamanaka H, Tanaka Y, Takeuchi T, Sugiyama N, Yuasa H, Toyoizumi S, et al. Tofacitinib, an oral Janus kinase inhibitor, as monotherapy or with background methotrexate, in Japanese patients with rheumatoid arthritis: an open-label, long-term extension study. Arthritis Res Ther. (2016) 18:34. doi: 10.1186/s13075-016-0932-2
53. Kremer J, Huizinga T, Chen L, Saifan C, Issa M, Witt S, et al. Analysis of neutrophils, lymphocytes, and platelets in pooled phase 2 and phase 3 studies of baricitinib for rheumatoid arthritis [abstract FRI0090. Ann Rheum Dis. (2017) 76:512.
54. Mease PJ, Lertratanakul A, Anderson JK, Papp K, Van den Bosch F, Tsuji S, et al. Upadacitinib for psoriatic arthritis refractory to biologics: SELECT-PsA 2. Ann Rheum Dis. (2021) 80:312–20. doi: 10.1136/annrheumdis-2020-218870
55. Moslin R, Gardner D, Santella J, Zhang Y, Duncia JV, Liu C, et al. Identification of imidazo[1,2-b]pyridazine TYK2 pseudokinase ligands as potent and selective allosteric inhibitors of TYK2 signalling. MedChemComm. (2017) 8:700–12. doi: 10.1039/C6MD00560H
56. Catlett IM, Hu Y, Gao L, Banerjee S, Gordon K, Krueger JG. Molecular and clinical effects of selective tyrosine kinase 2 inhibition with deucravacitinib in psoriasis. J Allergy Clin Immunol. (2022) 149:2010–20.e8. doi: 10.1016/j.jaci.2021.11.001
57. Lebwohl M, Warren RB, Sofen H, Imafuku S, Paul C, Szepietowski JC, et al. Deucravacitinib in plaque psoriasis: 2-year safety and efficacy results from the phase 3 poetyk trials. Br J Dermatol. (2024) 190:668–79. doi: 10.1093/bjd/ljae014
58. Traves PG, Murray B, Campigotto F, Galien R, Meng A, Di Paolo JA. JAK selectivity and the implications for clinical inhibition of pharmacodynamic cytokine signalling by filgotinib, upadacitinib, tofacitinib and baricitinib. Ann Rheum Dis. (2021) 80:865–75. doi: 10.1136/annrheumdis-2020-219012
59. Parmentier JM, Voss J, Graff C, Schwartz A, Argiriadi M, Friedman M, et al. In vitro and in vivo characterization of the JAK1 selectivity of upadacitinib (ABT-494). BMC Rheumatol. (2018) 2:23. doi: 10.1186/s41927-018-0031-x
60. Meyer DM, Jesson MI, Li X, Elrick MM, Funckes-Shippy CL, Warner JD, et al. Anti-inflammatory activity and neutrophil reductions mediated by the JAK1/JAK3 inhibitor, CP-690,550, in rat adjuvant-induced arthritis. J Inflamm. (2010) 7:41. doi: 10.1186/1476-9255-7-41
61. Hogge D, Fanning S, Bockhold K, Petzer A, Lambie K, Lansdorp P, et al. Quantitation and characterization of human megakaryocyte colony-forming cells using a standardized serum-free agarose assay. Br J Haematol. (1997) 96:790–800. doi: 10.1046/j.1365-2141.1997.d01-2092.x
62. Kaushansky K, Broudy VC, Lin N, Jorgensen MJ, McCarty J, Fox N, et al. Thrombopoietin, the Mp1 ligand, is essential for full megakaryocyte development. Proc Natl Acad Sci U S A. (1995) 92:3234–8. doi: 10.1073/pnas.92.8.3234
63. Dubart A, Feger F, Lacout C, Goncalves F, Vainchenker W, Dumenil D. Murine pluripotent hematopoietic progenitors constitutively expressing a normal erythropoietin receptor proliferate in response to erythropoietin without preferential erythroid cell differentiation. Mol Cell Biol. (1994) 14:4834–42. doi: 10.1128/mcb.14.7.4834
64. Metcalf D. The molecular control of cell division, differentiation commitment and maturation in haemopoietic cells. Nature. (1989) 339:27–30. doi: 10.1038/339027a0
65. Rubbert-Roth A, Enejosa J, Pangan AL, Haraoui B, Rischmueller M, Khan N, et al. Trial of upadacitinib or abatacept in rheumatoid arthritis. N Engl J Med. (2020) 383:1511–21. doi: 10.1056/NEJMoa2008250
66. Smolen JS, Genovese MC, Takeuchi T, Hyslop DL, Macias WL, Rooney T, et al. Safety profile of baricitinib in patients with active rheumatoid arthritis with over 2 years median time in treatment. J Rheumatol. (2019) 46:7–18. doi: 10.3899/jrheum.171361
67. Fielder PJ, Gurney AL, Stefanich E, Marian M, Moore MW, Carver-Moore K, et al. Regulation of thrombopoietin levels by c-mpl-mediated binding to platelets. Blood. (1996) 87:2154–61. doi: 10.1182/blood.V87.6.2154.bloodjournal8762154
68. de Graaf CA, Metcalf D. Thrombopoietin and hematopoietic stem cells. Cell Cycle (Georgetown Tex). (2011) 10:1582–9. doi: 10.4161/cc.10.10.15619
69. Strober B, Buonanno M, Clark JD, Kawabata T, Tan H, Wolk R, et al. Effect of tofacitinib, a Janus kinase inhibitor, on haematological parameters during 12 weeks of psoriasis treatment. Br J Dermatol. (2013) 169:992–9. doi: 10.1111/bjd.2013.169.issue-5
70. Nash P, Coates LC, Kivitz AJ, Mease PJ, Gladman DD, Covarrubias-Cobos JA, et al. Safety and efficacy of tofacitinib in patients with active psoriatic arthritis: interim analysis of OPAL balance, an open-label, long-term extension study. Rheumatol Ther. (2020) 7:553–80. doi: 10.1007/s40744-020-00209-4
71. Genovese MC, McKay JD, Nasonov EL, Mysler EF, da Silva NA, Alecock E, et al. Interleukin-6 receptor inhibition with tocilizumab reduces disease activity in rheumatoid arthritis with inadequate response to disease-modifying antirheumatic drugs: the tocilizumab in combination with traditional disease-modifying antirheumatic drug therapy study. Arthritis Rheumatol. (2008) 58:2968–80. doi: 10.1002/art.v58:10
72. Zeidler C, Kanz L, Hurkuck F, Rittmann KL, Wildfang I, Kadoya T, et al. In vivo effects of interleukin-6 on thrombopoiesis in healthy and irradiated primates. Blood. (1992) 80:2740–5. doi: 10.1182/blood.V80.11.2740.2740
73. Gabay C, Emery P, van Vollenhoven R, Dikranian A, Alten R, Pavelka K, et al. Tocilizumab monotherapy versus adalimumab monotherapy for treatment of rheumatoid arthritis (ADACTA): a randomised, double-blind, controlled phase 4 trial. Lancet. (2013) 381:1541–50. doi: 10.1016/S0140-6736(13)60250-0
74. van Vollenhoven R, Lee EB, Strengholt S, Mojcik C, Valdez H, Krishnaswami S, et al. Evaluation of the short-, mid-, and long-term effects of tofacitinib on lymphocytes in patients with rheumatoid arthritis. Arthritis Rheumatol. (2019) 71:685–95. doi: 10.1002/art.2019.71.issue-5
75. Tanaka Y, McInnes IB, Taylor PC, Byers NL, Chen L, de Bono S, et al. Characterization and changes of lymphocyte subsets in baricitinib-treated patients with rheumatoid arthritis: an integrated analysis. Arthritis Rheumatol. (2018) 70:1923–32. doi: 10.1002/art.2018.70.issue-12
76. Kondrack RM, Harbertson J, Tan JT, McBreen ME, Surh CD, Bradley LM. Interleukin 7 regulates the survival and generation of memory CD4 cells. J Exp Med. (2003) 198:1797–806. doi: 10.1084/jem.20030735
77. Puel A, Ziegler SF, Buckley RH, Leonard WJ. Defective IL7R expression in T(-)B(+)NK(+) severe combined immunodeficiency. Nat Genet. (1998) 20:394–7. doi: 10.1038/3877
78. Suzuki H, Kündig TM, Furlonger C, Wakeham A, Timms E, Matsuyama T, et al. Deregulated T cell activation and autoimmunity in mice lacking interleukin-2 receptor beta. Science. (1995) 268:1472–6. doi: 10.1126/science.7770771
79. Kennedy MK, Glaccum M, Brown SN, Butz EA, Viney JL, Embers M, et al. Reversible defects in natural killer and memory CD8 T cell lineages in interleukin 15-deficient mice. J Exp Med. (2000) 191:771–80. doi: 10.1084/jem.191.5.771
80. Schluns KS, Kieper WC, Jameson SC, Lefrançois L. Interleukin-7 mediates the homeostasis of naive and memory CD8 T cells in vivo. Nat Immunol. (2000) 1:426–32. doi: 10.1038/80868
81. Lutz CT, Karapetyan A, Al-Attar A, Shelton BJ, Holt KJ, Tucker JH, et al. Human NK cells proliferate and die in vivo more rapidly than T cells in healthy young and elderly adults. J Immunol. (2011) 186:4590–8. doi: 10.4049/jimmunol.1002732
82. van Gurp EA, Schoordijk-Verschoor W, Klepper M, Korevaar SS, Chan G, Weimar W, et al. The effect of the JAK inhibitor CP-690,550 on peripheral immune parameters in stable kidney allograft patients. Transplantation. (2009) 87:79–86. doi: 10.1097/TP.0b013e31818bbea7
83. Genovese MC, Fleischmann R, Combe B, Hall S, Rubbert-Roth A, Zhang Y, et al. Safety and efficacy of upadacitinib in patients with active rheumatoid arthritis refractory to biologic disease-modifying anti-rheumatic drugs (SELECT-BEYOND): a double-blind, randomised controlled phase 3 trial. Lancet. (2018) 391:2513–24. doi: 10.1016/S0140-6736(18)31116-4
84. Morand EF, Furie R, Tanaka Y, Bruce IN, Askanase AD, Richez C, et al. Trial of anifrolumab in active systemic lupus erythematosus. N Engl J Med. (2020) 382:211–21. doi: 10.1056/NEJMoa1912196
85. Boedigheimer MJ, Martin DA, Amoura Z, Sánchez-Guerrero J, Romero-Diaz J, Kivitz A, et al. Safety, pharmacokinetics and pharmacodynamics of AMG 811, an anti-interferon-γ monoclonal antibody, in SLE subjects without or with lupus nephritis. Lupus Sci Med. (2017) 4:e000226. doi: 10.1136/lupus-2017-000226
86. Dendrou CA, Cortes A, Shipman L, Evans HG, Attfield KE, Jostins L, et al. Resolving TYK2 locus genotype-to-phenotype differences in autoimmunity. Sci Transl Med. (2016) 8:363ra149. doi: 10.1126/scitranslmed.aag1974
87. Gorman JA, Hundhausen C, Kinsman M, Arkatkar T, Allenspach EJ, Clough C, et al. The TYK2-P1104A autoimmune protective variant limits coordinate signals required to generate specialized T cell subsets. Front Immunol. (2019) 10:44. doi: 10.3389/fimmu.2019.00044
88. Kahlenberg M, Sanz I, Wu C, Hu S, Kim JH, Singhal S, et al. Deucravacitinib reduces interferons, B cell pathways, and serological biomarkers of systemic lupus disease activity: pharmacodynamic analysis from the phase 2 PAISLEY study [abstract 1000. Arthritis Rheumatol. (2022) 74. https://acrabstracts.org/abstract/deucravacitinib-reduces-interferons-b-cell-pathways-and-serological-biomarkers-of-systemic-lupus-disease-activity-pharmacodynamic-analysis-from-the-phase-2-paisley-study/.
89. Morand E, Pike M, Merrill JT, van Vollenhoven R, Werth VP, Hobar C, et al. Deucravacitinib, a tyrosine kinase 2 inhibitor, in systemic lupus erythematosus: a phase II, randomized, double-blind, placebo-controlled trial. Arthritis Rheumatol. (2023) 75:242–52. doi: 10.1002/art.42391
90. Dörner T, Tanaka Y, Dow ER, Koch AE, Silk ME, Ross Terres JA, et al. Mechanism of action of baricitinib and identification of biomarkers and key immune pathways in patients with active systemic lupus erythematosus. Ann Rheum Dis. (2022) 81:1267–72. doi: 10.1136/annrheumdis-2022-222335
91. Dörner T, Tanaka Y, Petri MA, Smolen JS, Wallace DJ, Dow ER, et al. Baricitinib-associated changes in global gene expression during a 24-week phase II clinical systemic lupus erythematosus trial implicates a mechanism of action through multiple immune-related pathways. Lupus Sci Med. (2020) 7:e000424. doi: 10.1136/lupus-2020-000424
92. Gaudreau MC, Fann J, Contrepois S, Friedman A, Sornasse T, Merrill JT. Treatment of systemic lupus erythematosus patients with upadacitinib results in the coordinated inhibition of type 1 IFN-related biomarkers: biomarker analysis of the M19-130 (SLEEK) phase 2 study [abstract POS1133. Ann Rheum Dis. (2023) 82:894–5. doi: 10.1136/annrheumdis-2023-eular.4002
93. Boisson-Dupuis S, Ramirez-Alejo N, Li Z, Patin E, Rao G, Kerner G, et al. Tuberculosis and impaired IL-23-dependent IFN-γ immunity in humans homozygous for a common TYK2 missense variant. Sci Immunol. (2018) 3:eaau8714. doi: 10.1126/sciimmunol.aau8714
94. Lee JW, Kim YG, Soung YH, Han KJ, Kim SY, Rhim HS, et al. The JAK2 V617F mutation in de novo acute myelogenous leukemias. Oncogene. (2006) 25:1434–6. doi: 10.1038/sj.onc.1209163
95. Eletto D, Burns SO, Angulo I, Plagnol V, Gilmour KC, Henriquez F, et al. Biallelic JAK1 mutations in immunodeficient patient with mycobacterial infection. Nat Commun. (2016) 7:13992. doi: 10.1038/ncomms13992
96. Gruber CN, Calis JJA, Buta S, Evrony G, Martin JC, Uhl SA, et al. Complex autoinflammatory syndrome unveils fundamental principles of JAK1 kinase transcriptional and biochemical function. Immunity. (2020) 53:672–84.e11. doi: 10.1016/j.immuni.2020.07.006
Keywords: deucravacitinib, TYK2, JAK, JAK inhibitors, tofacitinib, baricitinib, upadacitinib
Citation: Johnson B, Cheng L, Koenitzer J, Catlett IM and Schafer P (2024) Nonclinical evaluations of deucravacitinib and Janus kinase inhibitors in homeostatic and inflammatory pathways. Front. Immunol. 15:1437512. doi: 10.3389/fimmu.2024.1437512
Received: 23 May 2024; Accepted: 02 September 2024;
Published: 30 September 2024.
Edited by:
Masato Kubo, Tokyo University of Science, JapanReviewed by:
Stephanie Schell, The Pennsylvania State University, United StatesEmira Bousoik, University of Derna, Libya
Copyright © 2024 Johnson, Cheng, Koenitzer, Catlett and Schafer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Brandon Johnson, YnJhbmRvbi5qb2huc29uQGJtcy5jb20=