 Mohamed N. Abdelnabi
Mohamed N. Abdelnabi Ghada S. Hassan
Ghada S. Hassan Naglaa H. Shoukry
Naglaa H. Shoukry- 1Centre de Recherche du Centre hospitalier de l’Université de Montréal (CRCHUM), Montréal, QC, Canada
- 2Département de microbiologie, infectiologie et immunologie, Faculté de médecine, Université de Montréal, Montréal, QC, Canada
- 3Département de médecine, Faculté de médecine, Université de Montréal, Montréal, QC, Canada
Metabolic dysfunction-associated steatotic liver disease (MASLD) comprises a spectrum of liver diseases that span simple steatosis, metabolic dysfunction-associated steatohepatitis (MASH) and fibrosis and may progress to cirrhosis and cancer. The pathogenesis of MASLD is multifactorial and is driven by environmental, genetic, metabolic and immune factors. This review will focus on the role of the type 3 cytokines IL-17 and IL-22 in MASLD pathogenesis and progression. IL-17 and IL-22 are produced by similar adaptive and innate immune cells such as Th17 and innate lymphoid cells, respectively. IL-17-related signaling is upregulated during MASLD resulting in increased chemokines and proinflammatory cytokines in the liver microenvironment, enhanced recruitment of myeloid cells and T cells leading to exacerbation of inflammation and liver disease progression. IL-17 may also act directly by activating hepatic stellate cells resulting in increased fibrosis. In contrast, IL-22 is a pleiotropic cytokine with a dominantly protective signature in MASLD and is currently being tested as a therapeutic strategy. IL-22 also exhibits beneficial metabolic effects and abrogates MASH-related inflammation and fibrosis development via inducing the production of anti-oxidants and anti-apoptotic factors. A sex-dependent effect has been attributed to both cytokines, most importantly to IL-22 in MASLD or related conditions. Altogether, IL-17 and IL-22 are key effectors in MASLD pathogenesis and progression. We will review the role of these two cytokines and cells that produce them in the development of MASLD, their interaction with host factors driving MASLD including sexual dimorphism, and their potential therapeutic benefits.
Introduction
Non-alcoholic fatty liver disease (NAFLD) is a growing epidemic characterized by fat accumulation in the liver and associated with metabolic disorders and obesity. It affects ~30% of the adult population worldwide with the highest prevalence observed in South America (44.37%) while the lowest rates are seen in Europe (25.10%) (1–3). Consensus from major liver societies have recently coined the umbrella term “steatotic liver disease (SLD)” to include the various etiologies of steatosis (4). NAFLD was renamed metabolic dysfunction-associated steatotic liver disease (MASLD) to account for metabolic dysfunction associated with the development of steatosis. MASLD encompasses a spectrum of liver diseases, ranging from simple fat accumulation or metabolic dysfunction-associated steatotic liver (MASL) to specific pattern of fat accumulation in the liver (hepatic steatosis) and necroinflammatory histologic changes, previously termed non-alcoholic steatohepatitis (NASH) and now metabolic dysfunction-associated steatohepatitis (MASH), and progressing into fibrosis that may eventually lead to complications such as cirrhosis and hepatocellular carcinoma (HCC) (5). Hereinafter, we will use the new nomenclature of MASLD and MASH to refer to NAFLD and NASH, respectively. The progression of MASLD into fibrosis varies among patients, with different degrees of severity and clinical presentations. It is not until recently that the FDA approved a therapeutic agent for the treatment of patients with MASH (6). Resmetirom, a liver-directed thyroid hormone receptor beta–selective agonist demonstrated promising results in a phase 3 trial of patients with MASH progressing to fibrosis (6).
The multifactorial nature of MASLD involves metabolic abnormalities, tissue necrosis, immuno-inflammatory mechanisms, and fibrosis (5, 7, 8). In the context of immune and inflammatory processes, type 3 cytokines are important players in the pathogenesis and/or resolution of liver disease. Namely, IL-17A and IL-22 have significant effector functions during acute liver injury, however their role during the different stages of MASLD remains understudied. In this article, we will provide an overview of MASLD and MASH, and their underlying immune-related mechanisms from host factors to cells. We will focus on the type 3 cytokines IL-17A and IL-22, their roles during the different stages of liver disease in general and MASLD in particular and their therapeutic potential.
MASLD predisposing conditions
MASLD is promoted by several metabolic and non-metabolic risk factors. Metabolic syndrome (MetS) is considered the strongest metabolic predisposing condition to MASLD, featuring dyslipidemia, obesity, type 2 diabetes mellitus (T2DM), and systemic hypertension (9). Non-metabolic risk factors, including genetic polymorphisms, sex, and age, are also strong influencers of MASLD and its progression. Several genetic variants have been identified in MASLD patients and are reviewed in detail elsewhere (10, 11). Importantly, a specific single nucleotide polymorphism in the patatin-like phospholipase domain-containing protein 3 (PNPLA3) gene (rs738409) is strongly correlated with MASLD, disrupting the normal regulation of hepatic triglyceride lipolysis and causing fat accumulation in the liver (12, 13). Sex influences MASLD in the general population and in individuals with MetS, though this link is complex and can be affected by age, MetS, and hepatic inflammation (2, 14). For example, MASLD is more common in men than women, especially before menopausal age (typically ≤ 50-60 years), whereas, after the fifth decade, the prevalence of MASLD becomes similar among men and women or possibly even higher in women (14). Sexual dimorphism holds significant importance in MASLD and warrants particular attention as will be discussed in later sections.
Animal models of MASLD
The increasing prevalence of MASLD, its association with life-threatening complications and the scarcity of treatment options calls for deeper investigation into its underlying mechanisms using various animal models. Given the multifactorial nature of MASLD, there is no ideal animal model that fully recapitulates the clinical aspects of the disease in humans. Nevertheless, animal models enabled and will further provide mechanistic insights for understanding the disease, and for testing potenital therapeutics (15). Each model has its pros and cons, and the model(s) used in different studies should be considered carefully in data interpretation. Development of MASLD in animal models, mainly mice, could be induced by one of the following triggers: diet, genetic predisposition, or toxins. Different forms of diets (Table 1), gene deletions (Table 2) or chemical insults (Table 3) promote the initiation and progression of MASLD with different degrees of severity or prevalence of a certain feature or phenotype. In some models, a combination of stimuli is needed, for example injecting mice with the chemicals carbon tetrachloride (CCl4) or thioacetamide (TAA) together with feeding a high-fat diet accelerates disease progression. Tables 1–3 summarize the most commonly used models in each category and provide a concise description of their main symptoms, advantages and limitations.
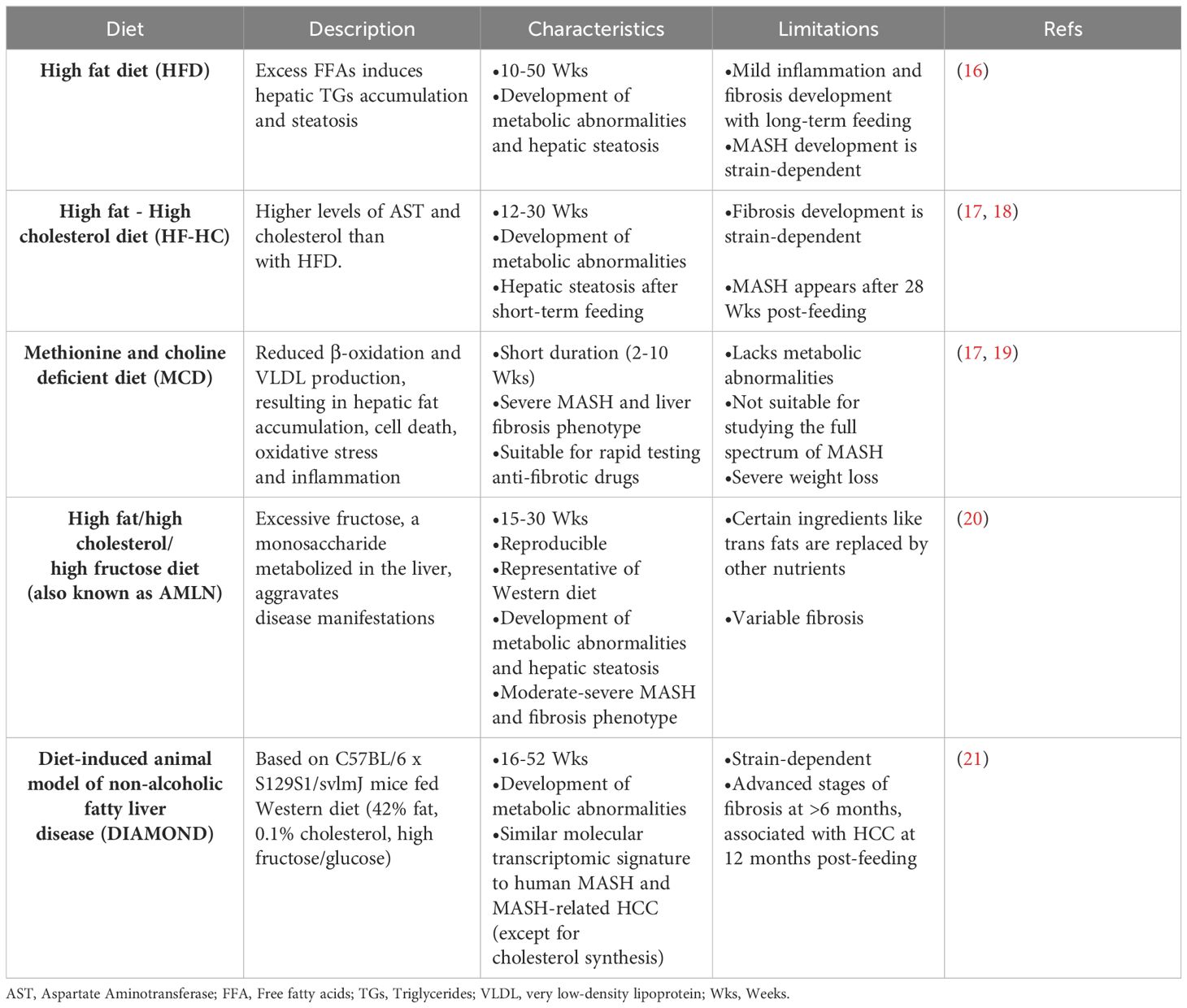
Table 1 Diet-induced MASLD mouse models.
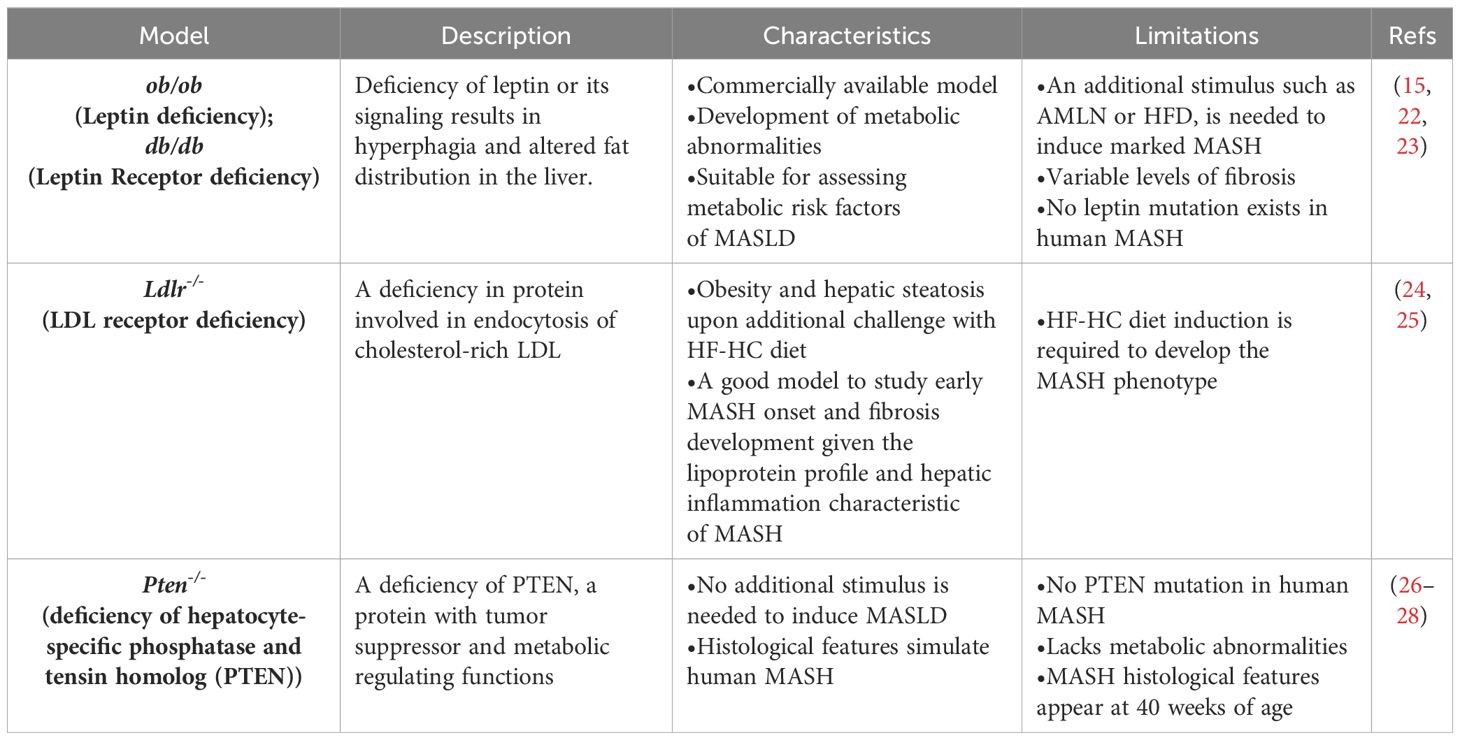
Table 2 Genetic mouse models of MASLD.
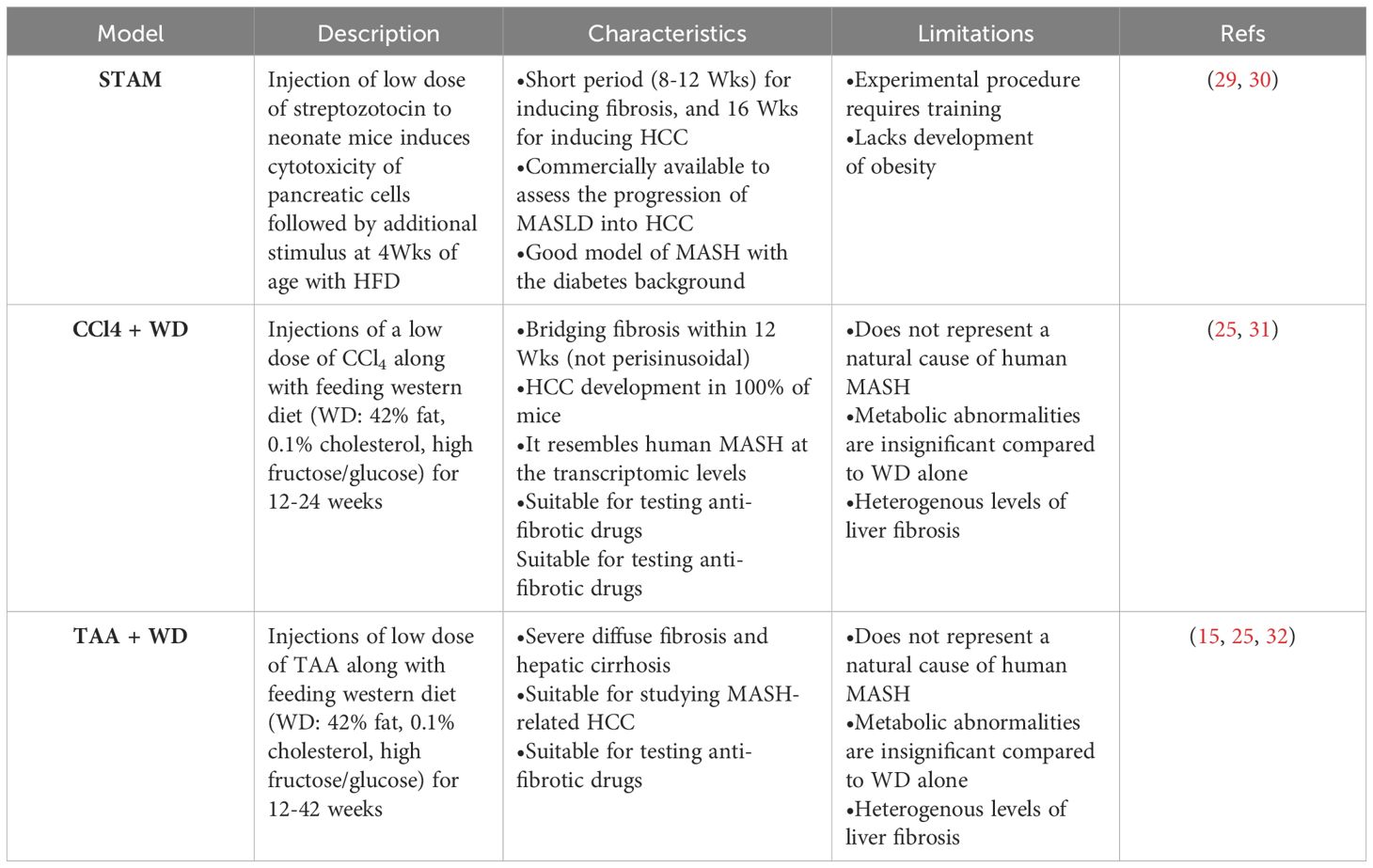
Table 3 Chemically-induced MASLD mouse models.
Pathophysiology of MASLD
In the past decade, there has been significant progress in understanding the pathogenesis of MASLD. Pathogenic drivers that promote MASLD development are heterogeneous, including various hepatic and extrahepatic factors. For example, insulin resistance, impaired adipose tissue function, lipotoxicity, and the interplay of diverse immune cells, are all pivotal pathogenic factors, that may interact together or in combination with other factors such as gut dysbiosis and genetic predisposition to influence the onset and advancement of MASH (5, 33).
Insulin resistance impairs lipid regulation and enhances accumulation of proinflammatory macrophages in adipose tissues, thus inducing lipolysis and promoting free fatty acids (FFAs) trafficking towards the liver (33, 34). MASL is initiated by disrupted regulation of the FFA metabolism that usually takes place in the normal liver, leading to fat accumulation in this organ (33, 35). An excessive accumulation of hepatic FFA, originating from disrupted adipose tissue, de novo lipogenesis, and/or dietary fat intake, exceeds the liver metabolic capacity. This results in lipotoxic metabolite formation and reactive oxygen species (ROS) overproduction inducing hepatocyte death via predominantly apoptosis, and to a lesser extent necroptosis, or pyrotosis (5, 36–38). Consequently, the persistence of hepatocyte death (a hallmark of MASLD progression) induces continuous liver injury, chronic inflammation (MASH), fibrosis progression, and increases the risk of development of HCC (39). Accordingly, understanding this metabolic inflammation and its critical role in the initiation and progression of MASH is crucial for unraveling the underlying mechanisms in MASLD pathogenesis and identifying potential therapeutic targets. Indeed, this inflammatory response is a complex interplay involving numerous innate and adaptive immune cells, each with its unique role. In this section, we briefly discuss the roles of these immune cells and their contributions to the inflammatory aspects of MASH progression.
Immune cells implicated in MASLD
Cells of the innate immune system in MASLD
The infiltration of innate immune cells, mainly neutrophils and macrophages into fatty livers is one of the hallmarks of MASH and highly contributes to disease progression (5, 40). These cells detect damage-associated molecular patterns (DAMPs) and pathogen-associated molecular patterns (PAMPs) from stressed hepatocytes and gut dysbiosis-related bacterial products, respectively (41), and engage in phagocytosis and antigen removal leading to hepatocyte apoptosis and pro-inflammatory cytokine production. Of note, pattern recognition receptors (PRRs) on innate cells are highly increased in MASH, and positively linked to hepatocyte death and disease progression (42, 43).
Liver-resident macrophages, known as Kupffer cells (KCs), play a pivotal role in MASH development through various mechanisms. For example, KCs accelerate hepatic fat accumulation by inhibiting fatty acid oxidation in IL-1α- and TNF-α-dependent mechanisms (44, 45). Recent studies, employing single-cell RNA sequencing technology, have unveiled the heterogeneity and functional diversity of macrophages in MASLD, challenging their conventional dichotomous classification as proinflammatory M1 versus anti-inflammatory/pro-repair M2 subtypes (40, 45, 46). Two distinct subsets of KCs in MASH have been identified based on the expression of the triggering receptor expressed on myeloid cells (TREM)−2. TREM2low KCs were more common in healthy livers, while TREM2high KCs were primarily enriched in MASH livers, and played a protective role against fat accumulation and fibrosis development (47, 48).
Activated neutrophils recruited during liver injury express inflammatory mediators such as myeloperoxidase (MPO) and elastase, and release neutrophil extracellular traps (NETs), all involved in MASH progression (49). MPO, a pro-oxidant enzyme, stimulates the generation of ROS molecules causing hepatocyte death and/or direct activation of hepatic stellate cells (HSCs), and thus enhancing the progression of MASH-related liver fibrosis (50, 51). Neutrophil elastase, a serine protease enzyme, promotes MASH pathology by enhancing the generation of lipotoxic ceramides and upregulation of the inflammatory IL-6 cytokine (52, 53). NET structures which consist of nucleic acids, histones, and antimicrobial peptides, are responsible for containing pathogens and maintaining homeostasis under physiological conditions. NET activation and release, a process known as NETosis is dysregulated in certain pathological contexts, including MASH, mediating detrimental effects such as inducing suppressive regulatory T cell (Treg) differentiation and hence promoting MASH-HCC progression (54). Additional innate immune cells exist in the healthy liver, including dendritic cells (DCs), natural killer (NK), and natural killer T (NKT) cells. They are responsible for various immune functions ranging from maintaining liver tolerance to eliminating antigens of pathogenic origin or malignant nature. However, their role in MASH remains controversial, with opposing functions and divergent effects in liver inflammation and fibrosis, as demonstrated by different studies, highlighting the heterogeneity of these cells and their complex role in MASH (55, 56).
Innate lymphoid cells (ILCs) originate from common lymphoid progenitors (CLPs) but, as innate immune cells, they do not express antigen-specific receptors. Although ILCs are less abundant than other lymphocytes in human and mouse tissues, they are a heterogeneous group. ILCs function similarly to CD4+ T helper cells and are considered their innate counterparts. Based on their cytokine profiles, ILCs are classified into three subsets: ILC1, ILC2, and ILC3, corresponding to Th1-, Th2-, and Th17-like profiles, respectively (57). In the liver, ILC1 is the most prevalent subset and is characterized by the expression of the transcription factor T-bet and the production of IFN-γ (57). The role of ILCs in MASLD is not well understood and remains to be fully elucidated. This is likely due to the lack of robust methods to clearly delineate the subsets of ILCs, particularly distinguishing ILC1 from NK cells. Furthermore, in some models deficient in NK cells, there was also a concurrent deficiency in ILCs. Consequently, no clear role for ILCs has been definitively demonstrated (57).
Other innate immune cells are also implicated in MASH pathogenesis. The γδ T is an unconventional T cell type with limited MHC-dependent antigen recognition capacity. In response to pathogens, γδ T cells secrete IL-17A and IFN-γ (58). In a MASH animal model, IL-17A-producing γδ T cells were shown to expand, contributing to enhanced inflammation and liver injury (59). Mucosal-associated invariant T (MAIT) cells, are usually enriched in the healthy liver and involved in its tolerance, they have been understudied with respect to their role in MASH development. These cells could mediate protective effects, specifically at the early stage of MASH, promoting an anti-inflammatory state in macrophages, and hence delaying MASH progression (60). However, at the late stage, MAIT exacerbates MASH-related fibrosis by producing the proinflammatory cytokine IL-17A (61).
Cells of the adaptive immune system in MASLD
Growing evidence demonstrated the involvement of adaptive immunity in MASH. This is evident through the formation of focal lymphocyte aggregates, T and B cells, in livers of MASH patients and murine models, contributing to increased lobular inflammation and liver injury (38, 62, 63). Indeed, an enrichment of intrahepatic B cells has been reported in MASH, coinciding with elevated levels of the B cell-activating factor (BAFF) implicated in B cell survival and maturation (62, 64). The role of B cells in MASH involves the release of proinflammatory cytokines that activate both macrophages and HSCs (65, 66). However, these findings are primarily based on in vivo animal studies, and their clinical significance remains unclear.
T cells also contribute to MASH pathogenesis. CD8+ T cells are enriched in the liver compared to peripheral blood and are the major conventional (CD3+) T cell population in the liver (67), in particular, tissue-resident memory CD8+ T cells (68, 69). CD8+ T cells are involved in maintaining liver tolerance and homeostasis under static conditions (67). In human and murine MASH, there is an expansion of intrahepatic CD8+ T cells, primarily driven by IFN-α responses (38, 63, 70). Both pathogenic as well as protective roles have been attributed to CD8+ cells in MASH (63, 71), but more research is needed to fully characterize their functions in this context. CD4+ T helper cells (Th), include various subsets, i.e. Th1, Th2, Th17 and Tregs, that are capable of mediating different immune responses, with plasticity in their roles based on the signals they receive (72, 73). Th17 cells, responsible for production of IL-17A and IL-22 cytokines, are mainly involved in the protection against infections and in tissue repair (58, 74) while Tregs, producing IL-10 and TGF-β cytokines, maintain immune tolerance and prevent tissue damage during infections (58). A dysregulation of Th17 and Tregs has been reported in MASH and associated with disease development and progression (38, 58). Other helper T cell subsets, their cytokines and functions in health and disease, including MASH, are reviewed elsewhere (58, 75).
In summary, both innate and adaptive immunity contribute to MASH progression and to liver cirrhosis and HCC. Neutrophils and macrophages are key innate drivers, while adaptive immunity likely sustains inflammation and liver fibrosis evolution. Understanding the role of these immune cells is crucial for developing new treatments. In this review, we focus on type 3 immunity and its effector cytokines, IL-17A and IL-22, and their roles in MASLD and MASH-associated fibrosis.
Type 3 cytokines
Type 3 immunity is a recently introduced term as an additional cell-mediated effector immune response (compared to type 1 and 2) mediated by cells producing the inflammatory cytokines IL-17 and IL-22 (76) and exemplified by IL-17-producing CD4+ T cells, known as T helper 17 cells (Th17). Besides Th17 cells, other immune cells are also type 3 cytokine producers, including Tc17, Th22, Tc22, γδ-T cells, MAIT cells, ILC3s, neutrophils, NK-T cells, macrophages and mast cells, under the control of the retinoic acid receptor-related orphan receptor-gamma-t (RORγt) and the aryl hydrocarbon receptor (AhR) transcription factors (77). Type 3 immunity mainly mediates the protection of the epithelial barriers in different tissues, including the liver, against extracellular bacteria and fungi (78). This immunity is also implicated in pathogenic and protective processes in various diseases ranging from inflammatory and autoimmune diseases to cancer (74, 78, 79). There are six members of the IL-17 family (IL-17A-F). Both IL-17A and F have similar biological activities as they share target tissues, cellular sources, and the receptor (IL-17RA/IL-17RC subunits) (80). IL-17B, interacts with the IL-17RB subunit to mediate neutrophil migration, indicating a pro-inflammatory role, and has been associated with poor prognosis in patients with pancreatic and lung cancers (80–82). IL-17C interacts with the IL-17RE subunit, mediating antimicrobial responses and maintaining epithelial barriers in both the skin and intestine (80, 83). However, in psoriatic mouse models, IL-17C exacerbates psoriatic inflammation (84). IL-17D signaling and biological activity remain poorly understood, with limited reports suggesting a proinflammatory effect through the induction of IL-6 and IL-8 (80, 85). Unlike other IL-17 cytokines, IL-17E uniquely promotes a type 2 response involving IL-4, IL-15, and IL-13, which are associated with dampening Th17-driven inflammation observed in autoimmune disorders (86).
Th17 cells also produce IL-22 (discussed in detail in this review) and IL-26, thus all are termed type 3 cytokines. IL-26 is predominantly produced by activated Th1 and Th17 memory CD4+ T cells. Specifically, IL-26-producing pro-inflammatory Th17 cells are found in chronically inflamed tissues such as chronic hepatitis C and inflammatory bowel diseases (IBD) (87–89). IL-26 signals through its receptor IL-26R (IL-10R2/IL-20R1 dimer) and can directly kill extracellular bacteria through membrane-pore formation. Additionally, IL-26 activates immune cells, especially plasmacytoid dendritic cells (pDCs), by aiding in the internalization of bacterial DNA from dying bacteria, subsequently triggering a type 1 interferon response via toll-like receptor 9 (TLR9) (87, 90). However, the role of IL-26 in chronic inflammation remains unclear. Finally, Th17 cells also produce type 1 cytokines such as IL-21. IL-21 signals through a heterodimer receptor composed of the IL-21 receptor (IL-21R) and the common cytokine receptor γ-chain (γc) and is involved in the differentiation, expansion, and survival of CD4+ and CD8+ T cells, as well as B cells (91).
In summary, IL-17A and IL-22 are particularly significant among type 3 cytokines due to the wide investigation of their roles in the initiation, progression, and resolution of liver disease, notably MASLD, and their therapeutic potential and will thus be the focus of this review. IL-17B-F cytokine members, IL-21, and IL-26, are extensively reviewed elsewhere (80, 87, 91).
The IL-17/IL-17R axis
The most studied members of the IL-17 family of cytokines are IL-17A and IL-17F. These cytokines exhibit 55% homology in their amino acid sequence which is reflected in their comparable functions, sources and targets (92). We will focus on IL-17A, hereinafter referred to as IL-17. Th17 polarization relies on antigenic stimulation, IL-6 and TGF-β, and is stabilized by IL-23 signaling. It is regulated by the activation of several transcription factors including signal transducers and activators of transcription 3 (STAT3), RORγt and AhR, but negatively regulated by Tbet and FOXP3 which promote differentiation of Th1 and Tregs, respectively (78, 93, 94). IL-10-expressing Tregs suppress the differentiation, proliferation, and effector functions of Th17 cells (58, 78). On the other hand, Th17 cells can undergo trans-differentiation to acquire an anti-inflammatory Treg-transcriptional profile, involving the canonical TGF-β signaling and AhR (95).
IL-17 signals through the IL-17 receptor, IL-17R, expressed on epithelial cells, fibroblasts, macrophages, and endothelial cells (78, 96). This interaction activates downstream intracellular signaling, including NF-κB, C/EBP, and MAPKs/p38/JNK phosphorylation, as well as a non-canonical pathway that eventually stabilizes mRNA transcripts of target chemokines or cytokines (97, 98). Consequently, the expression of several downstream genes is enhanced, including that of neutrophil chemoattractants (e.g., CXCL1) (Figure 1), antimicrobial peptides (AMPs), and angiogenic factors (99). Under physiological conditions, these effects of IL-17 protect mucosal surfaces of different organs, including the liver, from external pathogens like bacteria and fungi (76, 78). Numerous clinical and experimental studies have investigated the role of IL-17 in both acute and chronic liver injuries, but several unanswered questions remain.
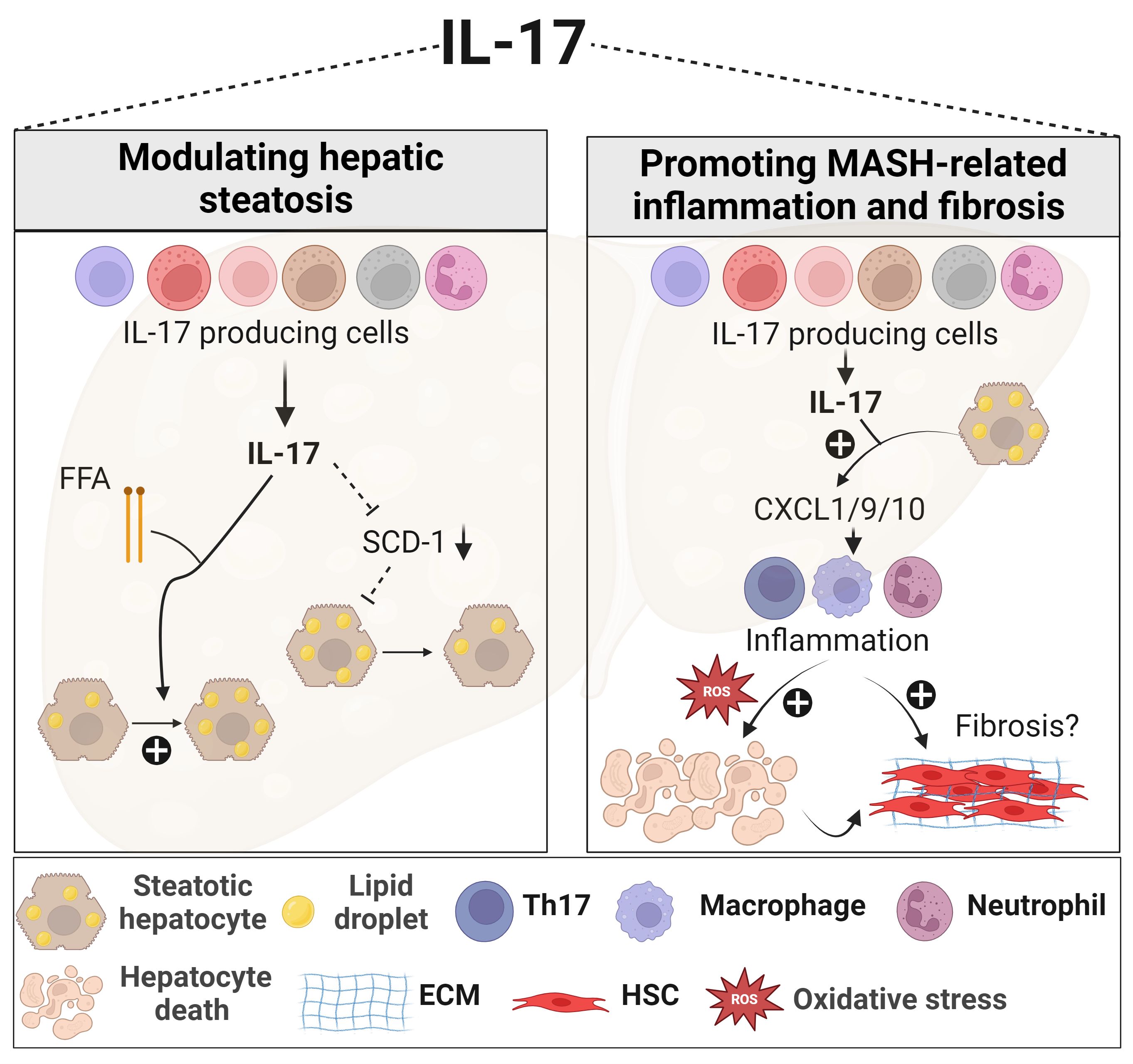
Figure 1 Role of IL-17 in MASLD. In the presence of elevated FFA, IL-17 enhances lipid accumulation within the liver. IL-17 is also associated with reduced hepatic steatosis through downregulating SCD-1, an important enzyme for de novo lipogenesis. IL-17 further exacerbates MASH-related inflammation by actively recruiting inflammatory immune cells, including neutrophils, macrophages, and Th17 cells, via promoting hepatocyte release of chemokines such as CXCL1, CXCL9, and CXCL10. These recruited immune cells contribute to oxidative stress, hepatocyte death, and the progression of fibrosis. However, the effect of IL-17 on liver fibrosis remains contradictory. Generated using Biorender.com. ECM, Extracellular matrix; FFA, free fatty acid; HSC, hepatic stellate cell; SCD-1, stearoyl-CoA desaturase-1.
Role of IL-17 in acute and chronic liver injuries
Acute liver injury (ALI) is induced by various insults, such as drugs, toxins, alcohol, and infections and initiates a self-resolving wound-healing response, encompassing three overlapping phases: inflammation, proliferation/repair, and tissue remodeling (78, 100). IL-17 has been implicated in all phases of ALI, mediating different functions. During the inflammation phase, IL-17 in synergy with other cytokines such as TNF-α and IL-6 induces hepatocytes and fibroblasts to release pro-inflammatory chemokines, facilitating the recruitment of monocytes, macrophages, and neutrophils to the injury site (101–104). Genetic deletion or pharmacological blockade of IL-17 ameliorated ALI underscoring the proinflammatory impact of IL-17 in acute hepatitis (101, 105, 106). A hepatoprotective effect of IL-17 in ALI was also reported. Mice with α-galactosylceramide (αGalCer)-induced hepatitis demonstrated an exacerbated inflammation upon neutralization of IL-17. In the hepatitis B virus surface antigen transgenic mouse (HBs-Tg) treated with ConA, depleting γδ T cells, one of the major cellular source of IL-17 aggravated liver damage and pro-inflammatory events (107, 108).
IL-17 also contributes to the tissue repair phase of the wound healing response to injury. IL-17 indirectly enhances the regeneration of hepatocytes and the differentiation of murine liver progenitor cells (LPCs) into hepatocytes by activating macrophages and DCs to produce IL-6 or IL-27 (109, 110). Given that HSCs express the IL-17R, IL-17 can directly activate HSCs promoting their proliferation and increased expression of collagen and alpha-smooth muscle actin (α-SMA) (103, 111), or indirectly via activating other pro-inflammatory cells in the liver microenvironment (101–103, 109). We demonstrated that IL-17 alone does not activate HSCs in vitro but sensitizes their response to suboptimal doses of TGF-β by upregulating their expression of the TGF-β receptor, TGF-β-RII (112).
IL-17 could be also involved in the tissue remodeling phase usually characterized by increased expression of matrix metalloproteinases (MMPs) and decreased tissue inhibitor of metalloproteinases (TIMPs) (113). Indeed, IL-17 mediates the expression of MMPs such as MMP-1, - 2 and -9 in inflammatory conditions and cancers (114, 115). Taken together, the role of IL-17 is multifaceted in acute hepatitis, acting as a proinflammatory cytokine in some contexts, while mediating repair in others, and warrants further investigation into its implication in the different phases of ALI.
During chronic liver injury, induced by constant damaging insult and characterized by persistent inflammation and hepatic fibrogenesis (78, 116), the effects of IL-17 have been extensively investigated both in humans and in animal models. The hepatic Th17/IL-17 axis is commonly elevated in patients and mice with chronic liver disease (CLD) and correlates with disease severity (102, 103, 117–119). Indeed, a common process seen in various CLDs is the production of chemokines, such as CXCL9, CXCL10, and CCL20 from damaged hepatocytes that attract Th17 cells expressing CXCR3 and CCR6 receptors (120–122). Other specific triggers of Th17 include hepatitis B virus (HBV) antigens and thymic stromal lymphopoietin from hepatitis C virus (HCV)-infected hepatocytes that stimulate antigen-presenting cells (APCs) to release IL-6 and IL-23, promoting Th17 differentiation (119, 123, 124). In this context, antiviral therapies reduce Th17 response and viral load, resulting in improved liver function in chronic viral hepatitis (CVH) (125, 126). Furthermore, a particular characteristic of CLDs, is an imbalanced Th17/Tregs ratio (127), evident in CVH as well as other CLDs such as alcoholic steatohepatitis (ASH) (102, 128), autoimmune hepatitis (129), and MASH, further discussed below (130–133), exacerbating inflammation and liver damage. Mechanistically, IL-17 promotes inflammation by recruiting and activating myeloid cells (macrophages, and neutrophils), inducing them to produce proinflammatory cytokines, e.g., TNF-α, and exacerbating hepatocyte injury. This was further supported by studies using IL-17 neutralizing antibodies or genetic knockout models, where IL-17 deficiency mitigated inflammation and fibrosis progression by reducing neutrophil infiltration and expression of proinflammatory cytokines (111, 134). In addition to its proinflammatory role, IL-17 can also modulate liver fibrosis progression in CLDs. As previously mentioned, IL-17 can exert a pro-fibrogenic function by acting either directly or indirectly on HSCs, leading to their activation and ECM deposition (103, 111). For example, IL-17 induces fibrosis development by stimulating the proliferation and activation of HSCs and triggering them to release IL-8 and CXCL-1, thus enhancing scar tissue deposition and neutrophil recruitment (102, 135). On the other hand, IL-17 also induces monocytes and KCs to produce pro-inflammatory (e.g., IL-6) and profibrogenic (e.g., TGF-β) cytokines that promote collagen deposition by HSCs (103, 136). Conversely, KCs and HSCs themselves can promote Th17 differentiation through an IL-6-dependent mechanism (137, 138), and IL-17 release via a TLR3-dependent pathway (139, 140), respectively. Pharmacological or genetic inhibition of IL-17 production or its signaling pathway (Il17a-/- or Il17ra-/- mice, respectively) in various liver fibrosis models led to reduced HSC activation and limited liver fibrosis progression (103, 111, 141). Altogether, IL-17 seems to be highly implicated in detrimental inflammatory and fibrogenic effects, contributing to CLD development and progression.
Role of IL-17 in MASLD
The metabolic effects of IL-17
The Th17/IL-17 axis exerts diverse effects, encompassing metabolic, proinflammatory, and profibrogenic responses in MASLD (Figure 1). Regarding the metabolic effects, disrupting the IL-17 pathway through genetic deletion (Il17a-/-) or blocking its signaling (Il17ra-/-) resulted in increased weight gain and adiposity in models of diet-induced obesity and MASLD (132, 133, 142). Indeed, IL-17 suppresses adipogenesis by downregulating pro-adipogenic transcription factors such as C/EBP-α and PPARγ (143, 144). A protective effect for IL-17 in the development of hepatic steatosis in MASLD was reported. The lack of IL-17 in various MASLD murine models was associated with increased fat accumulation in the liver (Figure 1), affecting mainly the expression of stearoyl-CoA desaturase-1 (SCD-1), a regulator of lipogenesis (132, 145–147).
On the other hand, IL-17 promotes insulin resistance and hinders glucose uptake in adipose tissue and the liver (132, 133, 148). Furthermore, stimulating hepatocytes with IL-17 in vitro, in the presence of FFAs, upregulated their lipid content (Figure 1), via inhibition of insulin-signaling pathways, and induced IL-6 production (131, 133). This latter IL-17-mediated effect together with the known role of IL-6, in synergy with TGF-β in enhancing the differentiation and proliferation of Th17 (149), constitute a positive feedback loop between hepatocytes and Th17 cells in the context of MASLD. Similarly, Th17 cells sustain adipose tissue inflammation by promoting IL-6 and IL-1β secretion from adipocytes, monocytes, and macrophages (133, 144, 150).
Despite these paradoxical metabolic effects, the inhibitory function of IL-17 against adipogenesis suggests a regulatory potential of this cytokine to control obesity and limit excess adiposity in MASLD. Nevertheless, chronic low-grade inflammation associated with MASLD, driven by IL-6 or IL-1β cytokines released from adipose tissue macrophages, could counteract these protective effects of IL-17 (151). Further research is essential to elucidate the precise role of IL-17 in MASLD-related hepatic steatosis.
The inflammatory effects of IL-17
The role of IL-17 in inducing MASH-related inflammation and promoting liver injury is well established. Hepatic Th17 or IL-17+ cells and Th17-related genes, including RORγt and IL-23, are enriched in MASH patients (130, 131, 133, 141, 152). Notably, a distinctive Th17 subset, termed inflammatory hepatic CXCR3+ IL-17+ IFN-γ+ TNF-α+ Th17 (ihTh17) was detected in the livers of MASLD patients. This subset exhibited a higher inflammatory profile compared to conventional hepatic CXCR3-Th17 (chTh17) cells and correlated with MASLD severity (152). Moreover, genome-wide association studies (GWAS) revealed single nucleotide polymorphisms (SNPs) in key IL-17 axis pathways, including RORγt, STAT4, and IL-17R that were associated with the severity of human MASLD and hepatobiliary disease (153, 154). Moreover, in obese MASLD patients, an elevated Th17/Treg ratio in the peripheral blood and liver was positively linked to the transition from MASL to MASH, highlighting Th17 involvement at the initiation and the progression of MASH-related inflammation (130). Importantly, the ratio of Th17 to resting Tregs, but not activated Tregs, distinguished MASL from MASH patients and correlated with the hepatocyte death marker cytokeratin18 (130). In the same study, one-year post-bariatric surgery, the hepatic Th17/Treg ratio significantly reversed to normal levels and associated with improved MASH. In line with this, the peripheral and hepatic venous IL-10/IL-17 ratio was markedly reduced in morbidly obese MASH patients, signifying an inflammatory state compared to those without MASH (155).
As described in humans, upregulation of the IL-17 axis and an imbalance of the Th17/Tregs ratio were observed in several murine MASH models and linked to adverse outcomes of the disease (131–133, 146, 147, 152, 156). Interventions targeting IL-17 or its signaling through pharmacological inhibitors, neutralizing antibodies or genetic deletion (Il17a-/- or Il17ra-/-), effectively decreased inflammation in several MASH models by reducing hepatocyte injury, the infiltration of pro-inflammatory immune cells, and the release of chemokines and cytokines (131–133, 145–147, 156). Despite the uncertainty as to the mechanisms underlying IL-17 detrimental effects in MASH, some potential pathways have been suggested. In methyl choline-deficient diet (MCD)- or high fat diet (HFD)-induced MASH models, the Th17/IL-17 axis increased hepatic CXCL-10 via an NF-κB/p65-dependent mechanism (Figure 1), promoting the recruitment of inflammatory macrophages and T cells (145, 146). Furthermore, the upregulated CXCR3-CXCL9/10 axis in the fatty liver environment, attracts ihTh17 cells that then induce macrophage infiltration and hepatocyte ballooning and thus exacerbate MASH progression (Figure 1) (152). Another mechanism that underscores the implication of Th17/IL-17 in MASLD-related inflammation, is its capacity to induce ROS production in a NADPH oxidase-dependent manner (132). Other functions of IL-17 including induction of IL-6 release from hepatocytes in the presence of FFA, further contribute to its inflammatory signature and role in MASLD (131). As to Tregs, some studies reported that their numbers were decreased in MASLD models (157) while others reported that their levels were unchanged (147, 158) or even increased (159, 160). Mechanisms underlying these contradicting observations were not well explored. A deeper investigation into the role of Tregs and their proportion as compared to Th17 cells, is needed in the context of human and mouse models of MASH.
The fibrogenic effects of IL-17
Like other CLDs, the Th17/IL-17 axis contributes to MASH-related fibrosis by enhancing inflammation, hepatocellular injury, and the activation of HSCs (Figure 1). However, evidence supporting the profibrogenic role of IL-17 in MASH is primarily derived from a small number of pre-clinical studies. IL-17 deficiency (Il17a-/-) in the MCD-induced models was shown to reduce liver fibrosis (147), while treating the HFD-induced MASH model with recombinant IL-17 worsened fibrosis development (133). However, divergent findings were observed in another MCD-fed IL-17-deficient model, where similar level of liver fibrosis was detected when compared to wild-type animals (145). Taken together, further research is needed to clarify the fibrogenic role of IL-17 and its modulation of HSCs in MASH.
IL-17 as a therapeutic target in MASLD
Targeting the IL-17 axis is a promising therapeutic intervention to manage the onset and progression of MASLD (161). Direct inhibition of IL-17 through monoclonal antibodies (e.g., secukinumab and ixekizumab) or of IL-17R, (brodalumab) has been approved for treating autoimmune diseases like psoriasis, psoriatic arthritis, and ankylosing spondylitis (161, 162). Indirect inhibition of IL-17 that involves blocking Th17 cell generation and related cytokines was also evaluated for therapeutic potential. Blocking the IL-6 (e.g., tocilizumab), IL-1β (e.g., canakinumab), or IL-23 (e.g., guselkumab) pathways partially reduces Th17 cell differentiation or survival (163–166). Additionally, targeting RORγt/RORc with small molecule antagonists (e.g., S18-000003) or inverse agonists (e.g., SHR168442) restores the Th17/Tregs balance and suppresses Th17-related cytokine production (167). Inhibiting the IL-17 pathway itself has shown promising outcomes in preclinical ASH studies. For example, IL-17 exacerbated alcohol-induced liver/brain axis injury, highlighting its involvement in systemic effects and potential contribution to brain disorders (168). Interestingly, using an anti-IL-17 antibody or an RORγt inhibitor not only reversed alcohol-induced injury of the liver/brain axis in mice with severe ASH but also reduced their voluntary alcohol drinking (168). In an experimental ASH model, genetic ablation of the IL-17RA in steatotic hepatocytes downregulated TNFRI, sterol regulatory element-binding proteins 1/2 (SREBP1/2), and 7-dehydrocholesterol reductase (DHCR7), leading to protection against hepatic steatosis and HCC development (169). In the CCl4-induced chronic liver injury, we observed reduced levels of intrahepatic IL-17+ cells including neutrophils, upon inhibiting RORγt with GSK805 resulting in decreased liver fibrosis (141). Nevertheless, no clinical studies have investigated the efficacy of IL-17 inhibitors in the context of MASLD.
It is important to note, that blocking the IL-17 pathway is linked with adverse outcomes such as injection site reactions and upper respiratory infections given the role of the IL-17 axis in mucosal immunity against bacterial and fungal infections (161). Moreover, patients with Crohn’s disease showed no amelioration or even reactivation of their disease upon treatment with anti-IL-17 drugs (170, 171). Furthermore, inhibiting RORγt has been associated with altered T cell receptor (TCR) α gene rearrangement, reducing T cell diversity and increasing the risk of thymic aberrations (172, 173). Thus, the use of inhibitors of the IL-17 pathway in treating CLDs, including MASLD, still needs further assessment to establish the balance between effectiveness and safety.
The IL-22/IL-22RA1 axis
IL-22, initially known as IL-10-related T cell-derived inducible factor (IL-TIF), belongs to the IL-10 cytokine family (174). Various adaptive and innate immune cells produce IL-22 such as Th17, Th22, CD8+ T (Tc22), γδT, NKT cells, ILC3s, neutrophils, and macrophages (175). IL-22 interacts with its receptor (IL-22R), a heterodimeric receptor composed of IL-22RA1 and IL-10RB2 subunits (176, 177). Notably, IL-22RA1 is predominantly expressed on epithelial cells, but also on fibroblasts and LPCs (178). This unique characteristic highlights the novelty of IL-22 as a cytokine affecting mainly epithelial cells. IL-22 binds to the IL-22R and triggers downstream signaling primarily through STAT3 activation but also via STAT1 and STAT5 pathways (179). IL-22/IL-22R induces phosphorylation of Janus kinase 1 (JAK1) and tyrosine kinase 2 (TYK2) that allow the translocation of STAT3 into the nucleus (175, 179). In addition to the JAK-STAT3 axis, IL-22 downstream signaling also involves activation of MAPK, PI3K, and AKT-induced mTOR pathways (179, 180). Consequently, all these pathways play a crucial role in regulating genes essential for innate immune defense, including the production of AMPs, acute phase proteins, proinflammatory mediators, and tissue regeneration (78, 175).
IL-22 induces the expression of several AMPs like mucus-associated proteins (MUC1), S100A7/8/9, regenerating islet-derived protein family (REG3β), and lipocalin 2 (LCN2) across various epithelial tissues including the liver (181–184). The antimicrobial function of IL-22 is underscored in vivo as it restricts the replication and dissemination of Klebsiella pneumoniae in the liver and lungs of infected mice (181, 185). Moreover, IL-22 signaling promotes the survival of hepatocytes, by inducing the expression of anti-apoptotic molecules like B-cell lymphoma 2 (BCL-2) and B-cell lymphoma-extra-large (BCL-XL) and thus protecting against hepatitis (186–188). In addition, IL-22 elevates acute-phase proteins such as CXCL-1, serum amyloid A (SAA), and LPS-binding proteins in hepatocytes (189). Interestingly, despite the restriction of IL-22RA1 to epithelial cells, IL-22 can indirectly influence the recruitment of immune cells. In the skin and lungs, IL-22 suppresses the recruitment of Th17 or Th2 cells through CCL17 and CCL22 chemokines, respectively (181, 190). In addition, it was shown that the proliferation of LPCs and intestinal stem cells (ISCs) is enhanced through the IL-22-induced STAT3 mechanism, facilitating tissue regeneration in both acute and chronic injuries (191–193). The influence of IL-22 on HSCs is controversial, with reports suggesting either inhibitory or activating effects, as discussed further below (194–196). Altogether, these conflicting roles for IL-22 warrant further investigation into its implication in the physiology and pathology of different anatomical sites.
Regulation of IL-22: Focus on IL-22BP
Several cytokines or transcription factors can either positively or negatively regulate IL-22 expression. IL-23 and IL-1β, primarily produced by DCs and macrophages, are key stimulators of IL-22 production from ILC3s, Th17, γδ T, and NKT cells (175, 197–199). Conversely, TGF-β can directly inhibit IL-22 expression through the c-maf transcription factor or indirectly by inhibiting Th17 differentiation and promoting Treg polarization (197, 200, 201). Additional cytokines like IL-25, IL-27, and IL-38 can inhibit IL-22 production, although their precise mechanisms remain unclear (175). The transcription factors AhR, RORγt, STAT3, and Notch are critical regulators of IL-22 expression. Specifically, AhR, activated by ligands from the diet or microbiome, can directly upregulate IL-22 and influence Th17 and ILC3s development (202, 203).
IL-22 binding protein (IL-22BP) or IL-22RA2 acts as a decoy receptor that modulates IL-22 activity, by specifically blocking IL-22 interaction with IL-22RA1. Its high affinity for IL-22 binding (1000-fold greater than IL-22RA1) suggests tight regulation of the activity of this cytokine (204, 205). IL-22BP is expressed in various epithelia, including the liver, with immune cells like DCs, eosinophils, and CD4+ T cells as its main producers (175, 206, 207). Of note, the production of IL-22BP itself is influenced by various factors. For example, in an inflamed mouse colon, activation of the inflammasome likely inhibits IL-22BP expression by inducing IL-18 (208), while retinoic acid in DCs, enhances IL-22BP expression (209). In the healthy gastrointestinal (GI) tract, IL-22BP regulates IL-22 activity in the follicle-associated epithelium, impacting mucin levels, AMPs production, and proliferation of epithelial cells (208, 210). Although the absence of IL-22BP alters these mucosal effects, its influence on the microbiome is minimal (210). In contrast, mice lacking IL-22 developed dysbiosis in their GI microbiome (211). This suggests that high levels of IL-22, in the absence of IL-22BP, do not influence microbiome composition, while deficiency of IL-22 has a significant impact on mucosal immunity. Interestingly, despite the potent neutralizing activity of IL-22BP, its expression levels, particularly in the GI tract, do not often parallel IL-22 levels. IL-22BP levels are high during homeostasis, though they are downregulated during inflammation. For example, in colitis models or following LPS administration, IL-22 levels significantly increase, while IL-22BP drops (212). However, a prolonged IL-22 rise can subsequently induce IL-22BP elevation, neutralizing IL-22 activity. This suggests that IL-22BP could mediate IL-22 signaling after the initial effects of this latter have been established, ensuring adequate mucosal immunity and tissue regeneration, and preventing detrimental IL-22 effects (208, 212). Nevertheless, in the context of colorectal cancer, the lack of IL-22BP in vivo is linked to disrupted IL-22 signaling, impaired wound healing, and an elevated risk of tumor development (208). The role of IL-22BP/IL-22 axis in liver injury and MASLD is discussed later in this review.
Role of IL-22 in acute and chronic liver injuries
IL-22 is implicated in the three phases of the wound-healing response following an acute insult. Consistent findings from diverse experimental models, including both infectious and non-infectious causes of ALI, converge on IL-22 exerting a hepatoprotective effect during the inflammatory phase (186, 187, 213–216). Specifically, IL-22 induces the expression of glutathione and anti-apoptotic molecules (BCL2, BCL-XL) in hepatocytes, thereby inhibiting hepatocyte death and mitigating oxidative stress (186–188, 214, 215). Other evidence suggests that the ability of IL-22 to reduce hepatocyte apoptosis is likely mediated by activating autophagy, particularly through the involvement of autophagy-related gene (ATG)4 and ATG7 proteins (217). In the proliferation/regeneration phase, IL-22 facilitates the proliferation of both hepatocytes and liver LPCs by inducing the expression of cyclin D and c-myc (186, 187, 193, 214). The implication of IL-22 in the tissue remodeling phase of ALI is not yet fully understood. Indeed, the effect of IL-22 on HSCs and hence the ECM is controversial, with reports suggesting inhibition of HSCs proliferation (194), enhancement of their activation (141, 195), or promotion of their senescence by IL-22 (218). Despite these hepatoprotective effects of IL-22 in ALI, several studies have shown contrasting findings, highlighting a rather pathogenic role for IL-22. In a rat allogeneic liver transplantation model, IL-22 exhibited hepatoprotective effects during the ischemia-reperfusion injury stage (day one), though it appeared to be pathogenic during the acute rejection stage (day 7), exacerbating liver injury (216). This detrimental effect of IL-22 was attributed to the upregulation of pro-inflammatory cytokines and chemokines, leading to an imbalance in Th17/Tregs (216). Consistently, in ischemia-reperfusion and APAP-induced liver injury models using IL-22BP-deficient mice (Il22ra2-/-), the dysregulated IL-22 signaling, marked by upregulated CXCL-10 expression, resulted in increased hepatic inflammatory monocytes and hepatocyte death, exacerbating liver injury (219). In line with the pathogenic signature of IL-22, blocking this cytokine using a neutralizing anti-IL-22 antibody markedly decreased the recruitment of pro-inflammatory immune cells in HBV-transgenic T-cell adoptive transfer model, mitigating ALI (220). Alternatively, the presence of other inflammatory cytokines, such as IL-17, could contribute to the detrimental effects of IL-22 (103, 221). This generally suggests opposite effects of IL-22 rather than a paradox, which is context-dependent and may rely on the specific inflammatory response, the time of exposure, IL-22BP regulation, and the activated signaling pathways in target cells (pro-survival/regeneration vs. pro-inflammatory) (103, 204, 216, 218).
The dual nature of IL-22 functions has also been reported in CLDs. Intrahepatic enrichment of IL-22 in chronic hepatitis B (CHB) or C (CHC) patients, while not directly impacting the replication of HBV and HCV in hepatocytes (120, 214, 220, 222, 223), suggests a crucial role of the cytokine in the progression of liver disease. These elevated IL-22 levels during CHB and CHC infections could contribute to chronic inflammation and increased severity of liver fibrosis (120, 141, 195, 214), by enhancing the recruitment of proinflammatory Th17 cells into the liver and a persistent activation and proliferation of HSCs (120, 195). On the other hand, other studies reported a negative association between intrahepatic IL-22 levels and liver inflammation as well as fibrosis in CHB patients (193, 224), probably due to the capacity of IL-22 to promote the survival of hepatocytes and induce the proliferation of LPCs (224). Similarly in ASH patients, increased levels of circulating IL-22-producing CD4+ T cells, including Th17 cells is associated with favorable short-term prognosis (225). Experimental evidence also supports a protective function for IL-22 against ethanol-induced chronic liver injury in mice (226, 227). In this model, reduced IL-22 production by gut ILC3s contributed to an impaired regenerating islet-derived protein 3 gamma (REG3G) and enhanced gut dysbiosis (228) while the administration of IL-22-producing engineered bacteria or recombinant IL-22 alleviated gut and hepatic symptoms (226, 228).
The role of IL-22 in liver fibrosis appears even more complex and context-dependent, with conflicting findings reported in various studies. An anti-fibrotic role has been attributed for IL-22 via its effect on HSCs, inhibiting their activation or inducing their senescence in a STAT3-dependent mechanism. These results were mainly observed in models with supraphysiological expression of IL-22 (IL-22 transgenic (Tg) mice or upon treatment with recombinant IL-22) (103, 194, 218, 229). In line with the anti-fibrotic function of IL-22, patients with Schistosoma japonicum infection had elevated hepatic IL-22 and decreased IL-22BP transcripts, correlating with reduced fibrosis and portal hypertension (230). On the other hand, other evidence demonstrated rather a pro-fibrogenic function for IL-22 in CLDs. Specifically, upon an in vitro stimulation with IL-22, HSCs exhibited reduced senescence and increased proliferation (195), and showed enhanced sensitivity to TGF-β signaling (141). This fibrogenic effect was confirmed in the chronic CCl4 model, where mice lacking IL-22 signaling (Il22ra1-/-) exhibited reduced hepatic fibrosis compared to their wild-type counterparts (141). These different results as to the role of IL-22 in CLDs might be due to the variation in the animal model employed (IL-22 Tg vs Il22ra1-/-) and to exogenous versus endogenous IL-22. Nevertheless, uncertainty remains regarding whether circulating IL-22 levels accurately mirror hepatic levels of the cytokine in these individuals (231). In summary, despite variations between data from HSC cell lines and in vivo models, the dual role of IL-22 points toward a context-dependent effect regulated at multiple levels.
Role of IL-22 in MASLD
The metabolic effects of IL-22
The protective effects of IL-22 in MASLD fall mainly under its beneficial functions against MetS and liver injury events (Figure 2). In various models of diet-induced MASLD, IL-22 improved glucose tolerance, enhanced insulin sensitivity, and reduced body weight along with decreased adiposity (147, 232–234), via mechanisms dependent on STAT3 and Akt activation (232–234). In addition, IL-22 was shown to inhibit several hepatic lipogenic genes (Figure 2) (e.g., fatty acid synthase (FAS)), thereby reducing liver steatosis as well (234–236). These protective metabolic effects of IL-22 were demonstrated upon administration of exogenous IL-22 or in IL-22 Tg mice, rather than being associated with endogenous levels of the cytokine which are usually low in circulation and livers of HFD-induced MASLD (232, 233, 237).
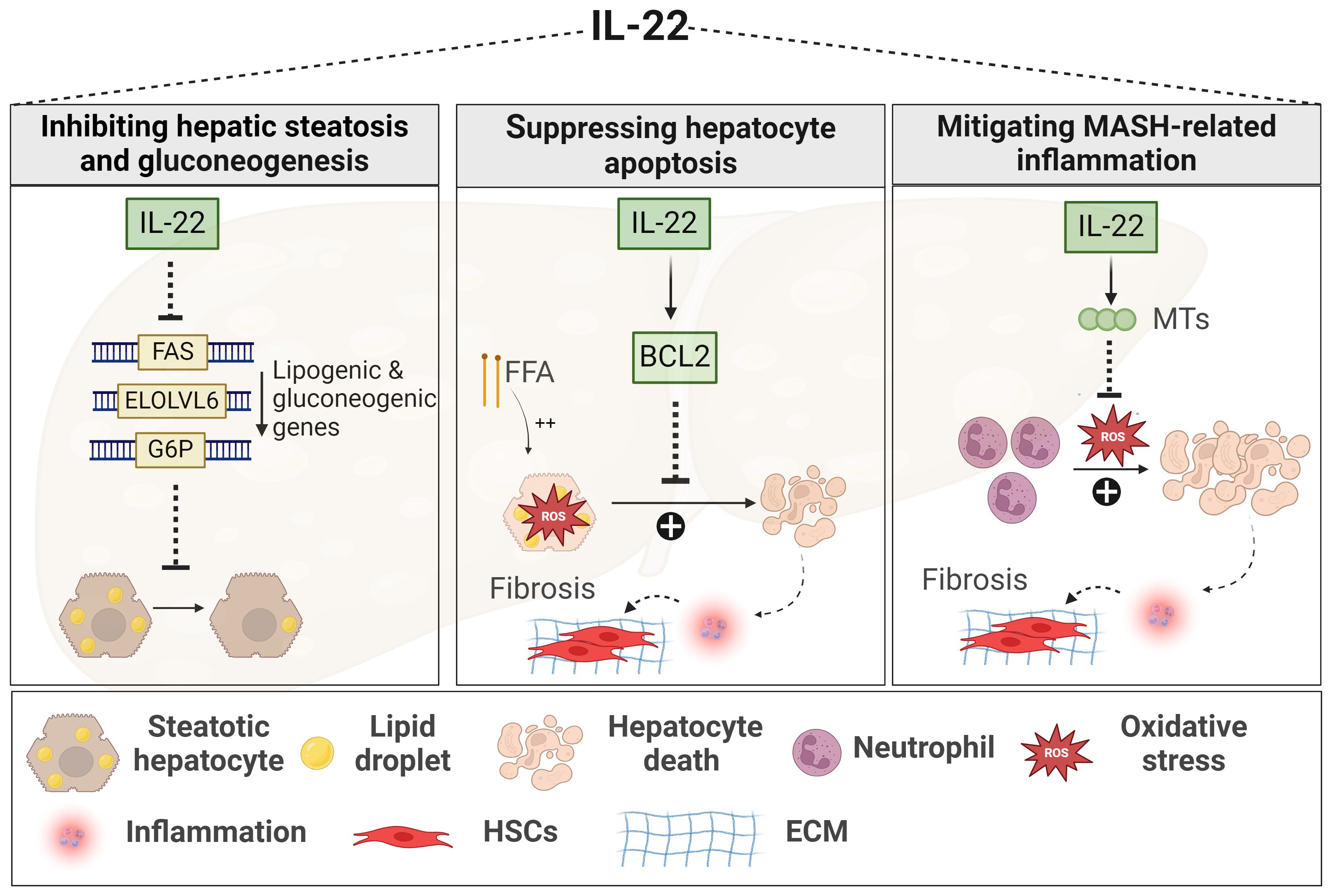
Figure 2 Role of IL-22 in MASLD. IL-22 downregulates the expression of lipogenic genes, such as FAS, ELOLV6, and G6P, thereby limiting lipid accumulation within the liver and reducing hepatic steatosis. IL-22 promotes hepatocyte survival against oxidative stress induced by either FFA or inflammatory neutrophils. This hepatoprotective mechanism involves the induction of anti-apoptotic signals, such as BCL2, and anti-oxidant signals, such as MTs, leading to decreased MASH-related inflammation and fibrosis. Generated using Biorender.com. BCL2, B-cell lymphoma 2; ECM, Extracellular matrix; ELOLV6, elongation of long-chain fatty acid family member 6; FAS, fatty acid synthase; FFA, free fatty acid; G6P, gluconeogenic genes like glucose 6 phosphate; HSC, hepatic stellate cell; MTs, metallothioneins.
The inflammatory and fibrogenic effects of IL-22
IL-22 also exerts protective functions in the context of MASH (147, 238, 239). A human recombinant IL-22 fusion protein, IL-22Fc remarkably ameliorated liver inflammation and fibrosis in neutrophil-driven MASH by suppressing oxidative stress via STAT-3-mediated induction of the antioxidant metallothionein proteins (Figure 2) (MT1 and MT2) (238). In line with this, liver-targeted delivery of the IL-22 gene in an HFD-induced MASH model promoted hepatocyte survival and proliferation (Figure 2) (234). Again here, these effects of IL-22 were observed upon its exogenous uptake (234, 238). Nevertheless, some studies still demonstrated a protective role for endogenous IL-22 against liver inflammation and fibrosis, particularly in MCD-induced MASH, but such effect was contingent on the absence of the IL-17 cytokine (Il17a-/-) (147). Another study reported that elevated levels of IL-22-producing ILC3s in the liver of HFD-induced MASH model were associated with reduced hepatic steatosis, while genetic deficiency of ILC3s worsened liver inflammation and fibrosis progression (Figure 2) (239). Of note, IL-22BP and its regulation of IL-22 activity were not examined in these experimental MASLD studies. In summary, cumulative evidence from in vivo MASLD models suggests multiple beneficial effects of IL-22, providing protection not only against MetS but also against liver damage and fibrosis progression. Yet, limitations in the experimental studies, as mentioned above, and the absence of clinical investigations hinder a conclusive determination of the beneficial role of IL-22 in the context of MASH.
The Therapeutic Potential of IL-22 in MASLD
Phase 1 clinical trials employing IL-22Fc reported good tolerability and safety in healthy human subjects (240, 241). This was associated, even at significantly increased serum IL-22 levels (up to 2000 ng/ml), with induced acute-phase proteins and minimal side effects (240, 241). Importantly, IL-22Fc reduced inflammatory markers and improved clinical scores in a phase 2a open-label trial for moderate to severe ASH (242). Also, in another phase 2 study, IL-22Fc, when combined with systemic corticosteroids, demonstrated safety and efficacy in treating lower gastrointestinal acute graft-versus-host disease (GVHD) (243). The treatment was associated with a distinct fecal microbiota composition, indicating an improvement in GVHD-associated dysbiosis (243). These studies highlight a potential pharmacological advantage of IL-22Fc in both healthy and diseased subjects with minimal adverse events.
Additional evidence supported a mitochondria-protecting function of IL-22Fc against liver injuries induced by HFD or acetaminophen (244, 245). Mechanistically, this was dependent on STAT-3-mediated activation of mTOR and AKT and/or STAT-3-activating AMPK pathways, leading to decreased ROS production, and preserving mitochondrial integrity (244, 245). Moreover, IL-22Fc demonstrated protective effects in acute-on-chronic liver failure (ACLF), a high-mortality syndrome marked by acute decompensated cirrhosis and intense systemic inflammation (246). In an ACLF model, involving chronic CCl4 treatment, an acute high CCl4 dose and an infection with Klebsiella pneumonia, IL-22Fc was capable of reversing the anti-regenerative IFN-γ/STAT1 cascade to the pro-regenerative IL-6/STAT3 pathway. This resulted in reduced bacterial load, enhanced anti-apoptotic signals in hepatocytes, and improved survival rates in mice (246). In contrast, another study demonstrated that progression of ACLF and high mortality rates in diseased patients correlated with elevated serum levels of endogenous IL-22 and a low IL-22BP/IL-22 ratio (247). Nevertheless, this discrepancy in observations may be attributed to the assessment of endogenous versus exogenous IL-22 or of systemic versus hepatic functions of IL-22. Overall, despite promising outcomes indicating the potential efficacy of IL-22 therapy in mitigating MASLD progression, concerns exist regarding its application in CLDs due to its potential to promote inflammation in certain contexts and the risk of adverse off-target effects. Therefore, further clinical studies are needed to thoroughly assess the efficacy and safety of IL-22 therapy in MASLD.
Sexual dimorphism in MASLD
Sexual dimorphism influences immune responses to infections and inflammatory diseases. Females generally exhibit stronger innate (e.g., phagocytic activity of neutrophils) and adaptive (e.g., CD4+/CD8+ T cell ratio) immunity than males (248–250). These sex-specific differences comprise a network of interconnected factors like reproductive hormones, genetics, epigenetics, and environmental elements. Yet, these factors have not been fully characterized to elucidate the molecular mechanisms underlying sexual dimorphism in the liver during health and disease (251). In this section, we will focus on sex hormones, specifically testosterone as an androgen for males and estradiol as an estrogen for females. Other factors are reviewed in depth elsewhere (248, 250).
Sex hormones-related inflammation and fibrosis in MASLDs
The liver itself as well as various innate and adaptive immune cells are major targets for sex hormones as they express androgen (AR) and estrogen receptors (ER). Thus, sex hormones play a fundamental role in MASLDs and CLDs in general, since they modulate liver functions and immune responses (250, 252). Of note, advanced MASH-related fibrosis and/or HCC are generally more prevalent in males than females (253, 254), while autoimmune hepatitis and ASH progress more aggressively in females compared to males (255). Although this could imply that female sex hormones may not often provide resistance against liver injury, it suggests a protective effect in the context of MASH, especially at the premenopausal age.
Experimental studies with estrogen-deficient mice or administration of estradiol into diet-induced obesity (DIO) models revealed that estradiol protects against hepatic steatosis by inhibiting de novo lipogenesis, enhancing free fatty acid (FFA) oxidation, and influencing triglyceride and cholesterol metabolism (256–258). Although these protective effects are more pronounced in livers of females, the conversion of testosterone to estradiol in male livers has also been observed, but to a lesser extent, and correlated with improving hepatic insulin sensitivity and bile acid synthesis (259). In line with this, low testosterone levels in males with MASLD were linked to MetS and hepatic steatosis (260, 261). Other studies reported that male mice lacking androgens (specifically testosterone) or AR to have favorable effects against their hepatic steatosis upon testosterone replacement (251, 262). In contrast, females with polycystic ovary syndrome (PCOS), characterized by excessive androgen production, had increased hepatic steatosis, overweight, and MetS, rendering them more susceptible to developing MASLD (263, 264).
Estradiol exerts an anti-lipogenic influence and multiple in vivo studies also reported an anti-inflammatory implication of this hormone in MASLD models. With respect to the anti-inflammatory role of estradiol, it has been demonstrated that ovariectomized female mice on HFD exhibit increased levels of hepatic macrophage infiltration, along with pro-inflammatory cytokines (IL-1β, IL-6) and chemokine (CCR2), compared to controls (sham-operated mice) (265, 266). Moreover, IL-6 production is suppressed in KCs upon estradiol administration in male mice harboring DEN-induced HCC (267). Estradiol enhanced apoptosis in activated HSCs, suppressing MAPK pathways (ERK and p38) and protecting against liver fibrosis induced by chemicals like CCl4 and thioacetamide in female mice (268–270). Similarly, ovariectomized female mice on a high-glucose and HFD developed significant collagen fibril accumulation as compared to control sham-operated females on the same diet (266). Mechanisms underlying this anti-fibrotic role of estradiol in MASH remain however undefined.
In summary, sex hormones, specifically estradiol, may offer therapeutic advantages in mitigating hepatic steatosis and MASLD-related inflammation/fibrosis in both sexes. Nevertheless, the use of hormonal therapy as a hepatoprotective drug hasn’t been conclusively established, and more studies are needed to explore the impact of sex hormones on immune cells in the liver, further defining their role in MASLD and ultimately identifying potential therapeutic targets for personalized medicine.
Sex-based differences of IL-22 and IL-17 in MASLD
Earlier studies of MASLD have focused primarily on using male mice given the higher prevalence of the disease in males. However, given the important roles that IL-17 and IL-22 play in MASLD and with the increased awareness of the key role of sex-dependent differences observed at the level of these cytokines and progression of MASLD, recent studies integrated males and female to better investigate the contribution of sexual dimorphism to MASLD, and more specifically with respect to the IL-17/IL-22 levels and functions. These studies are summarized in this section and Figure 3.
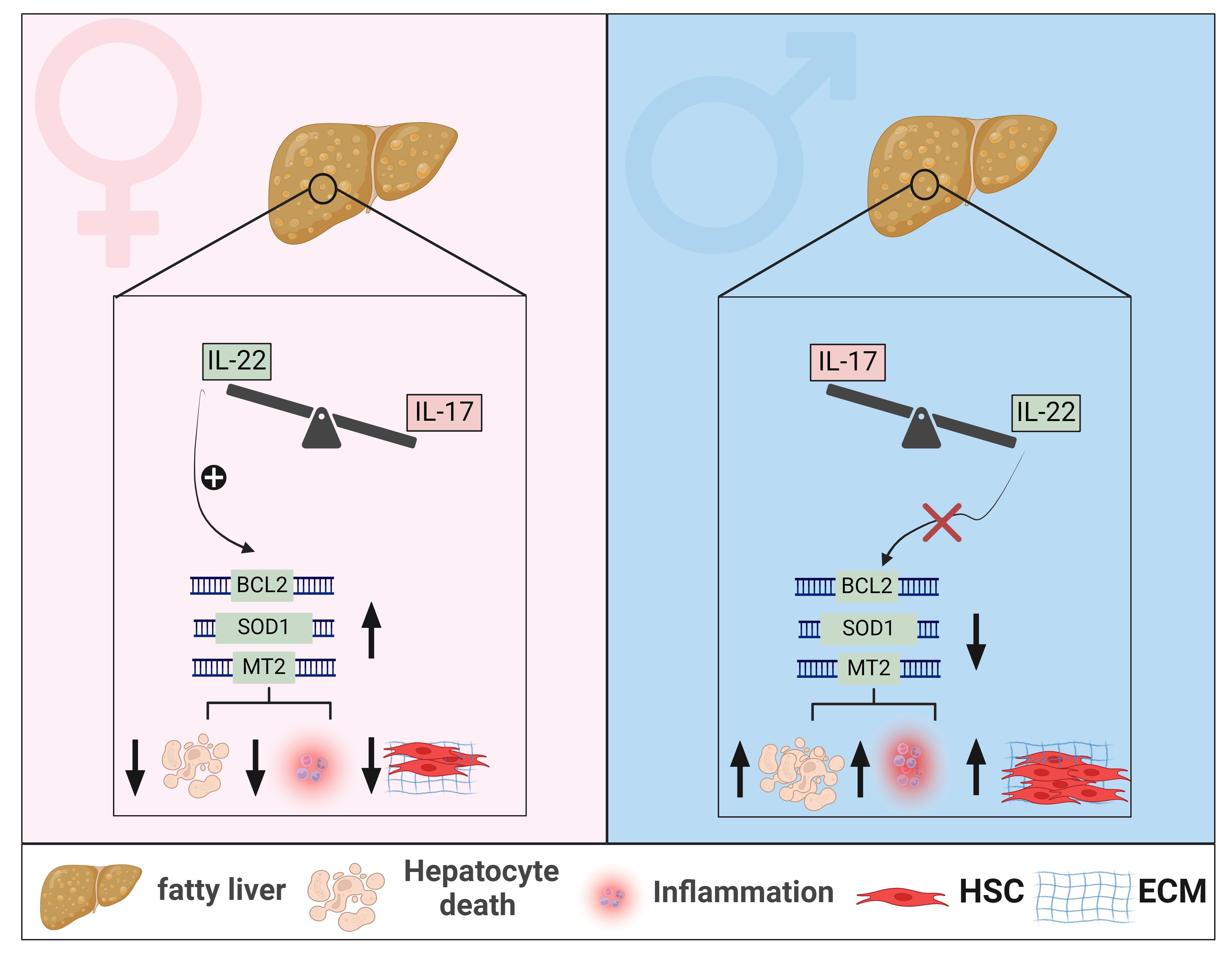
Figure 3 Sexual dimorphism of IL-22 and IL-17 effects in MASLD. In livers of female mice with MASLD, IL-22-producing cells are elevated, whereas in male mice with MASLD, cells producing IL-17 are predominantly enriched. Sexual dimorphism of hepatic IL-22 is associated with increased hepatic expression of anti-apoptotic genes, such as BCL2, and anti-oxidant genes such as SOD1 and MT2, in female mice, leading to mitigated hepatocyte death, reduced MASH-related inflammation, and decreased fibrosis progression. These hepatoprotective effects of IL-22 are absent in livers of male mice with MASLD. Generated using Biorender.com. BCL2, B-cell lymphoma 2; ECM, Extracellular matrix; HSC, hepatic stellate cell; MT2, metallothionein 2; SOD1, Superoxide dismutase 1.
Sex-related differences in the expression and function of IL-22 and IL-17
The sexual dimorphism-related signature of IL-17 in liver diseases has been demonstrated in the context of autoimmune conditions. For example, in an acute cholangitis model, females developed pronounced cholangitis, driven by increased recruitment of endogenous Th17, along with elevated expression of the chemokines CXCL-9 and CXCL-10 (271). Testosterone treatment of these female mice suppressed their inflammatory response while castration of male mice induced their susceptibility to acute cholangitis development (271). These data underscore the crucial role of testosterone in modulating immune responses in autoimmune liver disease. Diabetes induced by HFD/Streptozotocin (STZ, an antibiotic that destruct pancreatic islet β cell), was associated with increased Th17/IL-17 levels in the circulation and brain of female rats compared to males (272). However, the significance of such sexual dimorphic Th17 signature was not investigated. It is important to note here that a sex-specific difference in the Th17/IL-17 axis is also common among other diseases like ankylosing spondylitis (273), Sjögren’s syndrome (274), rheumatoid arthritis (275), and urinary tract infection (276). Taken together, studies across diverse diseases underscore the critical implication of sex hormones, particularly testosterone, in modulating Th17/IL-17 responses (Figure 3). This highlights the necessity for further investigation into the specific role of IL-17 in sex-specific immune dynamics in MASLD.
As to IL-22, most preclinical studies examining its role in MASLD were conducted exclusively in male mice, leaving knowledge gaps in the context of sex-based immunological differences in IL-22 between males and females (147, 232–234, 238, 239). Such an incomplete picture is also partly due to limited research regarding the endogenous hepatic IL-22 in human MASLD and the predominant focus on experimental studies. Our group has demonstrated elevated levels of hepatic IL-22 gene and protein expression in females compared to males in three different MASLD cohorts and in vivo models (277). This was associated with enrichment of hepatic Th17/IL-22, Th22, γδ T cells, and neutrophils as major IL-22 producers in females. Our study aligns with other reports that demonstrated reduced or unchanged levels of hepatic IL-22 expression in males, including humans and mice, with MASLD compared to healthy controls (232, 233, 278). Moreover, a similar signature of sexual dimorphism in IL-22 has been reported in the APAP-induced liver injury model, where females had increased expression of hepatic IL-22 levels compared to males (279). Notably, our results also showed that the endogenous expression of IL-22BP was elevated in livers of female mice and not males, with MASLD (277). This increased IL-22BP expression was not associated with impaired overall bioactivity of IL-22, highlighting a protective function of the IL-22BP itself in the context of MASLD. We further validated the sex-dependent influence of IL-22 in HFD-induced MASLD models and demonstrated that a deficiency of IL-22 receptor signaling (Il22ra1-/- mice) exacerbated liver injury, hepatocyte death, lobular inflammation, and the advancement of MASH-related fibrosis in female but not male mice (277). Therefore, our results support the hepatoprotective effects of the endogenous IL-22/IL-22RA1 axis against MASLD-related inflammation and fibrosis in a sex-dependent manner. This effect was mediated by IL-22-induced antiapoptotic (e.g., BCL2) and antioxidant signals (e.g., MT2), suppressing hepatocyte death (Figures 2, 3).
These novel findings may suggest a potential role of the female sex hormones in mediating hepatoprotective effects through the regulation of IL-22 expression. Although this remains a hypothesis to be tested, it is supported by several observations. In vitro, the synthetic progestin, medroxyprogesterone acetate (MPA), increased IL-22 expression in human Th22 cell clones but did not affect IL-22 production by Th17 cells (280). Additionally, LPS- or anti-CD3/CD28-stimulated splenocytes of female mice produced low levels of IL-22 upon their treatment with testosterone or dihydrotestosterone as compared to cells from males (279). Oral contraceptives increased serum IL-22 levels and the IL-22/IL-22BP ratio in females with polycystic ovary syndrome compared to healthy controls (281). Research, on the other hand, lacks a clear explanation for the reduction of endogenous IL-22 serum or hepatic levels seen in male mice upon diet-induced MASLD or obesity challenge. Importantly, this phenomenon seems not to depend on the type of diet used or the duration of diet induced obesity or MASLD models (232, 233, 237, 277), suggesting that low hepatic IL-22 levels appear stable regardless of the severity of metabolic inflammation. There is extensive evidence of low testosterone levels observed specifically in males with obesity, MetS, and MASLD (282), however little data exist linking testosterone levels with the decreased hepatic IL-22 seen in these patients. Taken together, further studies are essential to unravel the hormonal regulatory mechanisms that contribute to the sex difference in IL-22 expression in MASLD.
Conclusion and future perspective
The pivotal roles of type 3 cytokines, IL-17 and IL-22, emerge prominently in MASLD. Accumulating evidence points toward IL-17 contributing to persistent inflammation and the progression of MASLD-related fibrosis, while IL-22 exerts a hepatoprotective role, mitigating inflammation and fibrosis associated with the disease. Sex-based differences in hepatic endogenous IL-22 levels and its binding protein IL-22BP have been reported in MASLD and ALIs, while sexual dimorphism of IL-17 in these conditions is largely unexplored. The sexual dimorphism of IL-22 is associated with protective effects in females, ameliorating MASLD-related inflammation, and delaying fibrosis progression. Nevertheless, it remains unclear whether this sex-specific IL-22 signature is dependent on estrogen. Additionally, the influence of sex differences on gut microbiota and how it modulates immune responses like type 3 cytokines during MASLD progression is still lacking. This underscores the necessity for further experimental and clinical investigations to explore sexual dimorphism of IL-22 and IL-17, in the context of hormonal regulatory mechanisms, in MASLD. Exploring IL-22 and IL-17 as potential therapeutics for CLDs has shown promising outcomes in clinical trials. However, the efficacy and safety of targeting these pathways in human MASLD remain unknown and necessitate further in-depth investigation. Therapeutic approaches for MASLD treatment should be tested in both sexes and sexual dimorphism in inflammatory responses and disease progression should be considered.
Author contributions
MA: Visualization, Writing – original draft, Writing – review & editing. GH: Writing – original draft, Writing – review & editing. NS: Funding acquisition, Supervision, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. Our research is funded by the Canadian Institutes of Health Research (CIHR) (PJ4-192193 and PJT-175134). M.N.A received the Bourse d’Exemption des Droits de Scolarité Supplémentaires from the Université de Montréal and a doctoral fellowship from the Canadian Network on Hepatitis C (CanHepC). CanHepC is funded by a joint initiative of CIHR and the Public Health Agency of Canada (HPC-178912). The funders had no role in the preparation or the decision to publish this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Eslam M, Sanyal AJ, George J. MAFLD: A consensus-driven proposed nomenclature for metabolic associated fatty liver disease. Gastroenterology. (2020) 158:1999–2014.e1. doi: 10.1053/j.gastro.2019.11.312
2. Ge X, Zheng L, Wang M, Du Y, Jiang J. Prevalence trends in non-alcoholic fatty liver disease at the global, regional and national levels, 1990-2017: a population-based observational study. BMJ Open. (2020) 10:e036663. doi: 10.1136/bmjopen-2019-036663
3. Younossi ZM, Golabi P, Paik JM, Henry A, Van Dongen C, Henry L. The global epidemiology of nonalcoholic fatty liver disease (NAFLD) and nonalcoholic steatohepatitis (NASH): a systematic review. Hepatol (Baltimore Md). (2023) 77:1335–47. doi: 10.1097/HEP.0000000000000004
4. Rinella ME, Lazarus JV, Ratziu V, Francque SM, Sanyal AJ, Kanwal F, et al. A multisociety Delphi consensus statement on new fatty liver disease nomenclature. J Hepatol. (2023) 79:1542–56. doi: 10.1016/j.jhep.2023.06.003
5. Friedman SL, Neuschwander-Tetri BA, Rinella M, Sanyal AJ. Mechanisms of NAFLD development and therapeutic strategies. Nat Med. (2018) 24:908–22. doi: 10.1038/s41591-018-0104-9
6. Harrison SA, Bedossa P, Guy CD, Schattenberg JM, Loomba R, Taub R, et al. A phase 3, randomized, controlled trial of resmetirom in NASH with liver fibrosis. N Engl J Med. (2024) 390:497–509. doi: 10.1056/NEJMoa2309000
7. An J, Sohn JH. Pharmacological advances in the treatment of nonalcoholic fatty liver diseases : focused on global results of randomized controlled trials. Clin Mol Hepatol. (2023) 29:S268–s75. doi: 10.3350/cmh.2022.0437
8. Neuschwander-Tetri BA. Therapeutic landscape for NAFLD in 2020. Gastroenterology. (2020) 158:1984–98.e3. doi: 10.1053/j.gastro.2020.01.051
9. Fahed G, Aoun L, Bou Zerdan M, Allam S, Bou Zerdan M, Bouferraa Y, et al. Metabolic syndrome: updates on pathophysiology and management in 2021. Int J Mol Sci. (2022) 23(2):786. doi: 10.3390/ijms23020786
10. Sookoian S, Pirola CJ. Genetic predisposition in nonalcoholic fatty liver disease. Clin Mol hepatology. (2017) 23:1–12. doi: 10.3350/cmh.2016.0109
11. Eslam M, Valenti L, Romeo S. Genetics and epigenetics of NAFLD and NASH: Clinical impact. J hepatology. (2018) 68:268–79. doi: 10.1016/j.jhep.2017.09.003
12. Singal AG, Manjunath H, Yopp AC, Beg MS, Marrero JA, Gopal P, et al. The effect of PNPLA3 on fibrosis progression and development of hepatocellular carcinoma: a meta-analysis. Am J gastroenterology. (2014) 109:325–34. doi: 10.1038/ajg.2013.476
13. Speliotes EK, Butler JL, Palmer CD, Voight BF, Hirschhorn JN. PNPLA3 variants specifically confer increased risk for histologic nonalcoholic fatty liver disease but not metabolic disease. Hepatol (Baltimore Md). (2010) 52:904–12. doi: 10.1002/hep.23768
14. Lonardo A, Nascimbeni F, Ballestri S, Fairweather D, Win S, Than TA, et al. Sex differences in nonalcoholic fatty liver disease: state of the art and identification of research gaps. Hepatol (Baltimore Md). (2019) 70:1457–69. doi: 10.1002/hep.30626
15. Hansen HH, Feigh M, Veidal SS, Rigbolt KT, Vrang N, Fosgerau K. Mouse models of nonalcoholic steatohepatitis in preclinical drug development. Drug Discovery Today. (2017) 22:1707–18. doi: 10.1016/j.drudis.2017.06.007
16. Ito M, Suzuki J, Tsujioka S, Sasaki M, Gomori A, Shirakura T, et al. Longitudinal analysis of murine steatohepatitis model induced by chronic exposure to high-fat diet. Hepatol Res. (2007) 37:50–7. doi: 10.1111/j.1872-034X.2007.00008.x
17. Montandon SA, Somm E, Loizides-Mangold U, de Vito C, Dibner C, Jornayvaz FR. Multi-technique comparison of atherogenic and MCD NASH models highlights changes in sphingolipid metabolism. Sci Rep. (2019) 9:16810. doi: 10.1038/s41598-019-53346-4
18. Savard C, Tartaglione EV, Kuver R, Haigh WG, Farrell GC, Subramanian S, et al. Synergistic interaction of dietary cholesterol and dietary fat in inducing experimental steatohepatitis. Hepatology. (2013) 57:81–92. doi: 10.1002/hep.25789
19. Rinella ME, Green RM. The methionine-choline deficient dietary model of steatohepatitis does not exhibit insulin resistance. J hepatology. (2004) 40:47–51. doi: 10.1016/j.jhep.2003.09.020
20. Clapper JR, Hendricks MD, Gu G, Wittmer C, Dolman CS, Herich J, et al. Diet-induced mouse model of fatty liver disease and nonalcoholic steatohepatitis reflecting clinical disease progression and methods of assessment. Am J Physiol Gastrointestinal liver Physiol. (2013) 305:G483–95. doi: 10.1152/ajpgi.00079.2013
21. Asgharpour A, Cazanave SC, Pacana T, Seneshaw M, Vincent R, Banini BA, et al. A diet-induced animal model of non-alcoholic fatty liver disease and hepatocellular cancer. J hepatology. (2016) 65:579–88. doi: 10.1016/j.jhep.2016.05.005
22. Kennedy AJ, Ellacott KL, King VL, Hasty AH. Mouse models of the metabolic syndrome. Dis Models Mech. (2010) 3:156–66. doi: 10.1242/dmm.003467
23. Trak-Smayra V, Paradis V, Massart J, Nasser S, Jebara V, Fromenty B. Pathology of the liver in obese and diabetic ob/ob and db/db mice fed a standard or high-calorie diet. Int J Exp pathology. (2011) 92:413–21. doi: 10.1111/j.1365-2613.2011.00793.x
24. Schreyer SA, Vick C, Lystig TC, Mystkowski P, LeBoeuf RC. LDL receptor but not apolipoprotein E deficiency increases diet-induced obesity and diabetes in mice. Am J Physiol Endocrinol Metab. (2002) 282:E207–14. doi: 10.1152/ajpendo.2002.282.1.E207
25. Oligschlaeger Y, Shiri-Sverdlov R. NAFLD preclinical models: more than a handful, less of a concern? Biomedicines. (2020) 8(2):28. doi: 10.3390/biomedicines8020028
26. Varela-Rey M, Embade N, Ariz U, Lu SC, Mato JM, Martinez-Chantar ML. Non-alcoholic steatohepatitis and animal models: understanding the human disease. Int J Biochem Cell Biol. (2009) 41:969–76. doi: 10.1016/j.biocel.2008.10.027
27. Flessa CM, Nasiri-Ansari N, Kyrou I, Leca BM, Lianou M, Chatzigeorgiou A, et al. Genetic and diet-induced animal models for non-alcoholic fatty liver disease (NAFLD) research. Int J Mol Sci. (2022) 23(24):15791. doi: 10.3390/ijms232415791
28. Watanabe S, Horie Y, Kataoka E, Sato W, Dohmen T, Ohshima S, et al. Non-alcoholic steatohepatitis and hepatocellular carcinoma: lessons from hepatocyte-specific phosphatase and tensin homolog (PTEN)-deficient mice. J Gastroenterol Hepatol. (2007) 22 Suppl 1:S96–S100. doi: 10.1111/j.1440-1746.2006.04665.x
29. Fujii M, Shibazaki Y, Wakamatsu K, Honda Y, Kawauchi Y, Suzuki K, et al. A murine model for non-alcoholic steatohepatitis showing evidence of association between diabetes and hepatocellular carcinoma. Med Mol morphology. (2013) 46:141–52. doi: 10.1007/s00795-013-0016-1
30. Kountouras J, Kazakos E, Kyrailidi F, Polyzos SA, Zavos C, Arapoglou S, et al. Innate immunity and nonalcoholic fatty liver disease. Ann Gastroenterol. (2023) 36:244–56. doi: 10.20524/aog.2023.0793
31. Tsuchida T, Lee YA, Fujiwara N, Ybanez M, Allen B, Martins S, et al. A simple diet- and chemical-induced murine NASH model with rapid progression of steatohepatitis, fibrosis and liver cancer. J hepatology. (2018) 69:385–95. doi: 10.1016/j.jhep.2018.03.011
32. Sharma L, Gupta D, Abdullah ST. Thioacetamide potentiates high cholesterol and high fat diet induced steato-hepatitic changes in livers of C57BL/6J mice: A novel eight weeks model of fibrosing NASH. Toxicol Lett. (2019) 304:21–9. doi: 10.1016/j.toxlet.2019.01.001
33. Hardy T, Oakley F, Anstee QM, Day CP. Nonalcoholic fatty liver disease: pathogenesis and disease spectrum. Annu Rev pathology. (2016) 11:451–96. doi: 10.1146/annurev-pathol-012615-044224
34. Sabio G, Das M, Mora A, Zhang Z, Jun JY, Ko HJ, et al. A stress signaling pathway in adipose tissue regulates hepatic insulin resistance. Sci (New York NY). (2008) 322(5907):1539–43. doi: 10.1126/science.1160794
35. Hirsova P, Ibrabim SH, Gores GJ, Malhi H. Lipotoxic lethal and sublethal stress signaling in hepatocytes: relevance to NASH pathogenesis. J Lipid Res. (2016) 57:1758–70. doi: 10.1194/jlr.R066357
36. Feldstein AE, Canbay A, Angulo P, Taniai M, Burgart LJ, Lindor KD, et al. Hepatocyte apoptosis and fas expression are prominent features of human nonalcoholic steatohepatitis. Gastroenterology. (2003) 125:437–43. doi: 10.1016/s0016-5085(03)00907-7
37. Feldstein AE, Canbay A, Guicciardi ME, Higuchi H, Bronk SF, Gores GJ. Diet associated hepatic steatosis sensitizes to Fas mediated liver injury in mice. J hepatology. (2003) 39:978–83. doi: 10.1016/S0168-8278(03)00460-4
38. Sutti S, Jindal A, Locatelli I, Vacchiano M, Gigliotti L, Bozzola C, et al. Adaptive immune responses triggered by oxidative stress contribute to hepatic inflammation in NASH. Hepatol (Baltimore Md). (2014) 59:886–97. doi: 10.1002/hep.v59.3
39. García-Ruiz C, Colell A, Marí M, Morales A, Fernández-Checa JC. Direct effect of ceramide on the mitochondrial electron transport chain leads to generation of reactive oxygen species. Role of mitochondrial glutathione. J Biol Chem. (1997) 272:11369–77. doi: 10.1074/jbc.272.17.11369
40. Cai B, Dongiovanni P, Corey KE, Wang X, Shmarakov IO, Zheng Z, et al. Macrophage merTK promotes liver fibrosis in nonalcoholic steatohepatitis. Cell Metab. (2020) 31:406–21.e7. doi: 10.1016/j.cmet.2019.11.013
41. Schuster S, Cabrera D, Arrese M, Feldstein AE. Triggering and resolution of inflammation in NASH. Nat Rev Gastroenterol hepatology. (2018) 15:349–64. doi: 10.1038/s41575-018-0009-6
42. Mridha AR, Wree A, Robertson AAB, Yeh MM, Johnson CD, Van Rooyen DM, et al. NLRP3 inflammasome blockade reduces liver inflammation and fibrosis in experimental NASH in mice. J hepatology. (2017) 66:1037–46. doi: 10.1016/j.jhep.2017.01.022
43. Csak T, Ganz M, Pespisa J, Kodys K, Dolganiuc A, Szabo G. Fatty acid and endotoxin activate inflammasomes in mouse hepatocytes that release danger signals to stimulate immune cells. Hepatol (Baltimore Md). (2011) 54:133–44. doi: 10.1002/hep.v54.1
44. Tosello-Trampont AC, Landes SG, Nguyen V, Novobrantseva TI, Hahn YS. Kuppfer cells trigger nonalcoholic steatohepatitis development in diet-induced mouse model through tumor necrosis factor-α production. J Biol Chem. (2012) 287:40161–72. doi: 10.1074/jbc.M112.417014
45. Tran S, Baba I, Poupel L, Dussaud S, Moreau M, Gélineau A, et al. Impaired kupffer cell self-renewal alters the liver response to lipid overload during non-alcoholic steatohepatitis. Immunity. (2020) 53:627–40.e5. doi: 10.1016/j.immuni.2020.06.003
46. Seidman JS, Troutman TD, Sakai M, Gola A, Spann NJ, Bennett H, et al. Niche-specific reprogramming of epigenetic landscapes drives myeloid cell diversity in nonalcoholic steatohepatitis. Immunity. (2020) 52:1057–74.e7. doi: 10.1016/j.immuni.2020.04.001
47. Hendrikx T, Porsch F, Kiss MG, Rajcic D, Papac-Miličević N, Hoebinger C, et al. Soluble TREM2 levels reflect the recruitment and expansion of TREM2(+) macrophages that localize to fibrotic areas and limit NASH. J Hepatol. (2022) 77(5):1373–85. doi: 10.1016/j.jhep.2022.06.004
48. Hou J, Zhang J, Cui P, Zhou Y, Liu C, Wu X, et al. TREM2 sustains macrophage-hepatocyte metabolic coordination in nonalcoholic fatty liver disease and sepsis. J Clin Invest. (2021) 131(4):e135197. doi: 10.1172/JCI135197
49. Xu R, Huang H, Zhang Z, Wang FS. The role of neutrophils in the development of liver diseases. Cell Mol Immunol. (2014) 11:224–31. doi: 10.1038/cmi.2014.2
50. Pulli B, Ali M, Iwamoto Y, Zeller MW, Schob S, Linnoila JJ, et al. Myeloperoxidase-hepatocyte-stellate cell cross talk promotes hepatocyte injury and fibrosis in experimental nonalcoholic steatohepatitis. Antioxidants Redox Signaling. (2015) 23:1255–69. doi: 10.1089/ars.2014.6108
51. Koop AC, Thiele ND, Steins D, Michaëlsson E, Wehmeyer M, Scheja L, et al. Therapeutic targeting of myeloperoxidase attenuates NASH in mice. Hepatol Commun. (2020) 4:1441–58. doi: 10.1002/hep4.1566
52. Mirea AM, Toonen EJM, van den Munckhof I, Munsterman ID, Tjwa E, Jaeger M, et al. Increased proteinase 3 and neutrophil elastase plasma concentrations are associated with non-alcoholic fatty liver disease (NAFLD) and type 2 diabetes. Mol Med (Cambridge Mass). (2019) 25:16. doi: 10.1186/s10020-019-0084-3
53. Chen J, Liang B, Bian D, Luo Y, Yang J, Li Z, et al. Knockout of neutrophil elastase protects against western diet induced nonalcoholic steatohepatitis in mice by regulating hepatic ceramides metabolism. Biochem Biophys Res Commun. (2019) 518:691–7. doi: 10.1016/j.bbrc.2019.08.111
54. Papayannopoulos V. Neutrophil extracellular traps in immunity and disease. Nat Rev Immunol. (2018) 18:134–47. doi: 10.1038/nri.2017.105
55. Diedrich T, Kummer S, Galante A, Drolz A, Schlicker V, Lohse AW, et al. Characterization of the immune cell landscape of patients with NAFLD. PloS One. (2020) 15:e0230307. doi: 10.1371/journal.pone.0230307
56. Arrese M, Cabrera D, Kalergis AM, Feldstein AE. Innate immunity and inflammation in NAFLD/NASH. Digestive Dis Sci. (2016) 61:1294–303. doi: 10.1007/s10620-016-4049-x
57. Bourinet M, Anty R, Gual P, Luci C. Roles of innate lymphoid cells in metabolic and alcohol-associated liver diseases. JHEP Rep. (2024) 6:100962. doi: 10.1016/j.jhepr.2023.100962
58. Van Herck MA, Weyler J, Kwanten WJ, Dirinck EL, De Winter BY, Francque SM, et al. The differential roles of T cells in non-alcoholic fatty liver disease and obesity. Front Immunol. (2019) 10:82. doi: 10.3389/fimmu.2019.00082
59. Li F, Hao X, Chen Y, Bai L, Gao X, Lian Z, et al. The microbiota maintain homeostasis of liver-resident γδT-17 cells in a lipid antigen/CD1d-dependent manner. Nat Commun. (2017) 7:13839. doi: 10.1038/ncomms13839
60. Li Y, Huang B, Jiang X, Chen W, Zhang J, Wei Y, et al. Mucosal-associated invariant T cells improve nonalcoholic fatty liver disease through regulating macrophage polarization. Front Immunol. (2018) 9:1994. doi: 10.3389/fimmu.2018.01994
61. Hegde P, Weiss E, Paradis V, Wan J, Mabire M, Sukriti S, et al. Mucosal-associated invariant T cells are a profibrogenic immune cell population in the liver. Nat Commun. (2018) 9:2146. doi: 10.1038/s41467-018-04450-y
62. Bruzzì S, Sutti S, Giudici G, Burlone ME, Ramavath NN, Toscani A, et al. B2-Lymphocyte responses to oxidative stress-derived antigens contribute to the evolution of nonalcoholic fatty liver disease (NAFLD). Free Radical Biol Med. (2018) 124:249–59. doi: 10.1016/j.freeradbiomed.2018.06.015
63. Wolf MJ, Adili A, Piotrowitz K, Abdullah Z, Boege Y, Stemmer K, et al. Metabolic activation of intrahepatic CD8+ T cells and NKT cells causes nonalcoholic steatohepatitis and liver cancer via cross-talk with hepatocytes. Cancer Cell. (2014) 26:549–64. doi: 10.1016/j.ccell.2014.09.003
64. Miyake T, Abe M, Tokumoto Y, Hirooka M, Furukawa S, Kumagi T, et al. B cell-activating factor is associated with the histological severity of nonalcoholic fatty liver disease. Hepatol Int. (2013) 7:539–47. doi: 10.1007/s12072-012-9345-8
65. Xiong X, Kuang H, Ansari S, Liu T, Gong J, Wang S, et al. Landscape of intercellular crosstalk in healthy and NASH liver revealed by single-cell secretome gene analysis. Mol Cell. (2019) 75:644–60.e5. doi: 10.1016/j.molcel.2019.07.028
66. Zhang F, Jiang WW, Li X, Qiu XY, Wu Z, Chi YJ, et al. Role of intrahepatic B cells in non-alcoholic fatty liver disease by secreting pro-inflammatory cytokines and regulating intrahepatic T cells. J digestive diseases. (2016) 17:464–74. doi: 10.1111/1751-2980.12362
67. Racanelli V, Rehermann B. The liver as an immunological organ. Hepatol (Baltimore Md). (2006) 43:S54–62. doi: 10.1002/hep.21060
68. Pallett LJ, Davies J, Colbeck EJ, Robertson F, Hansi N, Easom NJW, et al. IL-2(high) tissue-resident T cells in the human liver: Sentinels for hepatotropic infection. J Exp Med. (2017) 214:1567–80. doi: 10.1084/jem.20162115
69. Li Y, You Z, Tang R, Ma X. Tissue-resident memory T cells in chronic liver diseases: Phenotype, development and function. Front Immunol. (2022) 13:967055. doi: 10.3389/fimmu.2022.967055
70. Ghazarian M, Revelo XS, Nøhr MK, Luck H, Zeng K, Lei H, et al. Type I interferon responses drive intrahepatic T cells to promote metabolic syndrome. Sci Immunol. (2017) 2(10):eaai7616. doi: 10.1126/sciimmunol.aai7616
71. Bhattacharjee J, Kirby M, Softic S, Miles L, Salazar-Gonzalez RM, Shivakumar P, et al. Hepatic natural killer T-cell and CD8+ T-cell signatures in mice with nonalcoholic steatohepatitis. Hepatol Commun. (2017) 1:299–310. doi: 10.1002/hep4.1041
72. Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, et al. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol. (2005) 6:1123–32. doi: 10.1038/ni1254
73. Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol (Baltimore Md 1950). (1995) 155:1151–64. doi: 10.4049/jimmunol.155.3.1151
74. Weaver CT, Elson CO, Fouser LA, Kolls JK. The Th17 pathway and inflammatory diseases of the intestines, lungs, and skin. Annu Rev pathology. (2013) 8:477–512. doi: 10.1146/annurev-pathol-011110-130318
75. Sutti S, Albano E. Adaptive immunity: an emerging player in the progression of NAFLD. Nat Rev Gastroenterol hepatology. (2020) 17:81–92. doi: 10.1038/s41575-019-0210-2
76. Annunziato F, Romagnani C, Romagnani S. The 3 major types of innate and adaptive cell-mediated effector immunity. J Allergy Clin Immunol. (2015) 135:626–35. doi: 10.1016/j.jaci.2014.11.001
77. Hirota K, Ahlfors H, Duarte JH, Stockinger B. Regulation and function of innate and adaptive interleukin-17-producing cells. EMBO Rep. (2012) 13:113–20. doi: 10.1038/embor.2011.248
78. Molina MF, Abdelnabi MN, Fabre T, Shoukry NH. Type 3 cytokines in liver fibrosis and liver cancer. Cytokine. (2019) 124:154497. doi: 10.1016/j.cyto.2018.07.028
79. Zhao J, Chen X, Herjan T, Li X. The role of interleukin-17 in tumor development and progression. J Exp Med. (2019) 217(1):e20190297. doi: 10.1084/jem.20190297
80. McGeachy MJ, Cua DJ, Gaffen SL. The IL-17 family of cytokines in health and disease. Immunity. (2019) 50:892–906. doi: 10.1016/j.immuni.2019.03.021
81. Wu HH, Hwang-Verslues WW, Lee WH, Huang CK, Wei PC, Chen CL, et al. Targeting IL-17B-IL-17RB signaling with an anti-IL-17RB antibody blocks pancreatic cancer metastasis by silencing multiple chemokines. J Exp Med. (2015) 212:333–49. doi: 10.1084/jem.20141702
82. Yang YF, Lee YC, Lo S, Chung YN, Hsieh YC, Chiu WC, et al. A positive feedback loop of IL-17B-IL-17RB activates ERK/β-catenin to promote lung cancer metastasis. Cancer Lett. (2018) 422:44–55. doi: 10.1016/j.canlet.2018.02.037
83. Ramirez-Carrozzi V, Sambandam A, Luis E, Lin Z, Jeet S, Lesch J, et al. IL-17C regulates the innate immune function of epithelial cells in an autocrine manner. Nat Immunol. (2011) 12:1159–66. doi: 10.1038/ni.2156
84. Johnston A, Fritz Y, Dawes SM, Diaconu D, Al-Attar PM, Guzman AM, et al. Keratinocyte overexpression of IL-17C promotes psoriasiform skin inflammation. J Immunol (Baltimore Md 1950). (2013) 190:2252–62. doi: 10.4049/jimmunol.1201505
85. Starnes T, Broxmeyer HE, Robertson MJ, Hromas R. Cutting edge: IL-17D, a novel member of the IL-17 family, stimulates cytokine production and inhibits hemopoiesis. J Immunol (Baltimore Md 1950). (2002) 169:642–6. doi: 10.4049/jimmunol.169.2.642
86. Kleinschek MA, Owyang AM, Joyce-Shaikh B, Langrish CL, Chen Y, Gorman DM, et al. IL-25 regulates Th17 function in autoimmune inflammation. J Exp Med. (2007) 204:161–70. doi: 10.1084/jem.20061738
87. Larochette V, Miot C, Poli C, Beaumont E, Roingeard P, Fickenscher H, et al. IL-26, a cytokine with roles in extracellular DNA-induced inflammation and microbial defense. Front Immunol. (2019) 10:204. doi: 10.3389/fimmu.2019.00204
88. Dambacher J, Beigel F, Zitzmann K, De Toni EN, Göke B, Diepolder HM, et al. The role of the novel Th17 cytokine IL-26 in intestinal inflammation. Gut. (2009) 58:1207–17. doi: 10.1136/gut.2007.130112
89. Miot C, Beaumont E, Duluc D, Le Guillou-Guillemette H, Preisser L, Garo E, et al. IL-26 is overexpressed in chronically HCV-infected patients and enhances TRAIL-mediated cytotoxicity and interferon production by human NK cells. Gut. (2015) 64:1466–75. doi: 10.1136/gutjnl-2013-306604
90. Meller S, Di Domizio J, Voo KS, Friedrich HC, Chamilos G, Ganguly D, et al. T(H)17 cells promote microbial killing and innate immune sensing of DNA via interleukin 26. Nat Immunol. (2015) 16:970–9. doi: 10.1038/ni.3211
91. Spolski R, Leonard WJ. Interleukin-21: a double-edged sword with therapeutic potential. Nat Rev Drug discovery. (2014) 13:379–95. doi: 10.1038/nrd4296
92. Pappu R, Ramirez-Carrozzi V, Sambandam A. The interleukin-17 cytokine family: critical players in host defence and inflammatory diseases. Immunology. (2011) 134:8–16. doi: 10.1136/gut.2007.130112
93. Bradding P, Pejler G. The controversial role of mast cells in fibrosis. Immunol Rev. (2018) 282:198–231. doi: 10.1111/imr.12626
94. Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, et al. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. (2006) 126:1121–33. doi: 10.1016/j.cell.2006.07.035
95. Gagliani N, Amezcua Vesely MC, Iseppon A, Brockmann L, Xu H, Palm NW, et al. Th17 cells transdifferentiate into regulatory T cells during resolution of inflammation. Nature. (2015) 523:221–5. doi: 10.1038/nature14452
96. Fossiez F, Djossou O, Chomarat P, Flores-Romo L, Ait-Yahia S, Maat C, et al. T cell interleukin-17 induces stromal cells to produce proinflammatory and hematopoietic cytokines. J Exp Med. (1996) 183:2593–603. doi: 10.1084/jem.183.6.2593
97. Sun D, Novotny M, Bulek K, Liu C, Li X, Hamilton T. Treatment with IL-17 prolongs the half-life of chemokine CXCL1 mRNA via the adaptor TRAF5 and the splicing-regulatory factor SF2 (ASF). Nat Immunol. (2011) 12:853–60. doi: 10.1038/ni.2081
98. Monin L, Gaffen SL. Interleukin 17 family cytokines: signaling mechanisms, biological activities, and therapeutic implications. Cold Spring Harbor Perspect Biol. (2018) 10(4):a028522. doi: 10.1101/cshperspect.a028522
99. Amatya N, Garg AV, Gaffen SL. IL-17 signaling: the yin and the yang. Trends Immunol. (2017) 38:310–22. doi: 10.1016/j.it.2017.01.006
100. Eming SA, Wynn TA, Martin P. Inflammation and metabolism in tissue repair and regeneration. Sci (New York NY). (2017) 356(6342):1026–30. doi: 10.1126/science.aam7928
101. Yan S, Wang L, Liu N, Wang Y, Chu Y. Critical role of interleukin-17/interleukin-17 receptor axis in mediating Con A-induced hepatitis. Immunol Cell Biol. (2012) 90:421–8. doi: 10.1038/icb.2011.59
102. Lemmers A, Moreno C, Gustot T, Maréchal R, Degré D, Demetter P, et al. The interleukin-17 pathway is involved in human alcoholic liver disease. Hepatol (Baltimore Md). (2009) 49:646–57. doi: 10.1002/hep.22680
103. Meng F, Wang K, Aoyama T, Grivennikov SI, Paik Y, Scholten D, et al. Interleukin-17 signaling in inflammatory, Kupffer cells, and hepatic stellate cells exacerbates liver fibrosis in mice. Gastroenterology. (2012) 143:765–76.e3. doi: 10.1053/j.gastro.2012.05.049
104. Beringer A, Thiam N, Molle J, Bartosch B, Miossec P. Synergistic effect of interleukin-17 and tumour necrosis factor-α on inflammatory response in hepatocytes through interleukin-6-dependent and independent pathways. Clin Exp Immunol. (2018) 193:221–33. doi: 10.1111/cei.13140
105. Furuya S, Kono H, Hara M, Hirayama K, Sun C, Fujii H. Interleukin 17A plays a role in lipopolysaccharide/D-galactosamine-induced fulminant hepatic injury in mice. J Surg Res. (2015) 199:487–93. doi: 10.1016/j.jss.2015.05.060
106. He J, Lang G, Ding S, Li L. Pathological role of interleukin-17 in poly I:C-induced hepatitis. PloS One. (2013) 8:e73909. doi: 10.1371/journal.pone.0073909
107. Meng Z, Wang J, Yuan Y, Cao G, Fan S, Gao C, et al. γδ T cells are indispensable for interleukin-23-mediated protection against Concanavalin A-induced hepatitis in hepatitis B virus transgenic mice. Immunology. (2017) 151:43–55. doi: 10.1111/imm.12712
108. Wondimu Z, Santodomingo-Garzon T, Le T, Swain MG. Protective role of interleukin-17 in murine NKT cell-driven acute experimental hepatitis. Am J pathology. (2010) 177:2334–46. doi: 10.2353/ajpath.2010.100028
109. Zhao L, Tang Y, You Z, Wang Q, Liang S, Han X, et al. Interleukin-17 contributes to the pathogenesis of autoimmune hepatitis through inducing hepatic interleukin-6 expression. PloS One. (2011) 6:e18909. doi: 10.1371/journal.pone.0018909
110. Guillot A, Gasmi I, Brouillet A, Ait-Ahmed Y, Calderaro J, Ruiz I, et al. Interleukins-17 and 27 promote liver regeneration by sequentially inducing progenitor cell expansion and differentiation. Hepatol Commun. (2018) 2:329–43. doi: 10.1002/hep4.1145
111. Tan Z, Qian X, Jiang R, Liu Q, Wang Y, Chen C, et al. IL-17A plays a critical role in the pathogenesis of liver fibrosis through hepatic stellate cell activation. J Immunol (Baltimore Md 1950). (2013) 191:1835–44. doi: 10.4049/jimmunol.1203013
112. Fabre T, Kared H, Friedman SL, Shoukry NH. IL-17A enhances the expression of profibrotic genes through upregulation of the TGF-beta receptor on hepatic stellate cells in a JNK-dependent manner. J Immunol (Baltimore Md 1950). (2014) 193:3925–33. doi: 10.4049/jimmunol.1400861
113. Duarte S, Baber J, Fujii T, Coito AJ. Matrix metalloproteinases in liver injury, repair and fibrosis. Matrix Biol. (2015) 44–6:147–56. doi: 10.1016/j.matbio.2015.01.004
114. Li J, Lau GK, Chen L, Dong SS, Lan HY, Huang XR, et al. Interleukin 17A promotes hepatocellular carcinoma metastasis via NF-kB induced matrix metalloproteinases 2 and 9 expression. PloS One. (2011) 6:e21816. doi: 10.1371/journal.pone.0021816
115. Cortez DM, Feldman MD, Mummidi S, Valente AJ, Steffensen B, Vincenti M, et al. IL-17 stimulates MMP-1 expression in primary human cardiac fibroblasts via p38 MAPK- and ERK1/2-dependent C/EBP-beta , NF-kappaB, and AP-1 activation. Am J Physiol Heart Circulatory Physiol. (2007) 293:H3356–65. doi: 10.1152/ajpheart.00928.2007
116. Hernandez-Gea V, Friedman SL. Pathogenesis of liver fibrosis. Annu Rev pathology. (2011) 6:425–56. doi: 10.1146/annurev-pathol-011110-130246
117. Zhang JY, Zhang Z, Lin F, Zou ZS, Xu RN, Jin L, et al. Interleukin-17-producing CD4(+) T cells increase with severity of liver damage in patients with chronic hepatitis B. Hepatol (Baltimore Md). (2010) 51:81–91. doi: 10.1002/hep.23273
118. Chang Q, Wang YK, Zhao Q, Wang CZ, Hu YZ, Wu BY. Th17 cells are increased with severity of liver inflammation in patients with chronic hepatitis C. J Gastroenterol hepatology. (2012) 27:273–8. doi: 10.1111/j.1440-1746.2011.06782.x
119. Wang Q, Zhou J, Zhang B, Tian Z, Tang J, Zheng Y, et al. Hepatitis B virus induces IL-23 production in antigen presenting cells and causes liver damage via the IL-23/IL-17 axis. PloS pathogens. (2013) 9:e1003410. doi: 10.1371/journal.ppat.1003410
120. Zhao J, Zhang Z, Luan Y, Zou Z, Sun Y, Li Y, et al. Pathological functions of interleukin-22 in chronic liver inflammation and fibrosis with hepatitis B virus infection by promoting T helper 17 cell recruitment. Hepatol (Baltimore Md). (2014) 59:1331–42. doi: 10.1002/hep.26916
121. Oo YH, Banz V, Kavanagh D, Liaskou E, Withers DR, Humphreys E, et al. CXCR3-dependent recruitment and CCR6-mediated positioning of Th-17 cells in the inflamed liver. J hepatology. (2012) 57:1044–51. doi: 10.1016/j.jhep.2012.07.008
122. Xu Z, Zhang X, Lau J, Yu J. C-X-C motif chemokine 10 in non-alcoholic steatohepatitis: role as a pro-inflammatory factor and clinical implication. Expert Rev Mol Med. (2016) 18:e16. doi: 10.1017/erm.2016.16
123. Li J, Wang FP, She WM, Yang CQ, Li L, Tu CT, et al. Enhanced high-mobility group box 1 (HMGB1) modulates regulatory T cells (Treg)/T helper 17 (Th17) balance via toll-like receptor (TLR)-4-interleukin (IL)-6 pathway in patients with chronic hepatitis B. J Viral hepatitis. (2014) 21:129–40. doi: 10.1111/jvh.12152
124. Lee HC, Sung SS, Krueger PD, Jo YA, Rosen HR, Ziegler SF, et al. Hepatitis C virus promotes T-helper (Th)17 responses through thymic stromal lymphopoietin production by infected hepatocytes. Hepatol (Baltimore Md). (2013) 57:1314–24. doi: 10.1002/hep.26128
125. Hao C, Wang J, Kang W, Xie Y, Zhou Y, Ma L, et al. Kinetics of Th17 cytokines during telbivudine therapy in patients with chronic hepatitis B. Viral Immunol. (2013) 26:336–42. doi: 10.1089/vim.2013.0032
126. Jimenez-Sousa MA, Almansa R, de la Fuente C, Caro-Paton A, Ruiz L, Sanchez-Antolín G, et al. Increased Th1, Th17 and pro-fibrotic responses in hepatitis C-infected patients are down-regulated after 12 weeks of treatment with pegylated interferon plus ribavirin. Eur Cytokine network. (2010) 21:84–91. doi: 10.1684/ecn.2010.0191
127. Xue-Song L, Cheng-Zhong L, Ying Z, Mo-Bin W. Changes of Treg and Th17 cells balance in the development of acute and chronic hepatitis B virus infection. BMC gastroenterology. (2012) 12:43. doi: 10.1186/1471-230X-12-43
128. Kasztelan-Szczerbińska B, Surdacka A, Celiński K, Roliński J, Zwolak A, Miącz S, et al. Prognostic significance of the systemic inflammatory and immune balance in alcoholic liver disease with a focus on gender-related differences. PloS One. (2015) 10:e0128347. doi: 10.1371/journal.pone.0128347
129. Mitra S, Anand S, Das A, Thapa B, Chawla YK, Minz RW. A molecular marker of disease activity in autoimmune liver diseases with histopathological correlation; FoXp3/RORγt ratio. APMIS Acta pathologica microbiologica immunologica Scandinavica. (2015) 123:935–44. doi: 10.1111/apm.12457
130. Rau M, Schilling AK, Meertens J, Hering I, Weiss J, Jurowich C, et al. Progression from nonalcoholic fatty liver to nonalcoholic steatohepatitis is marked by a higher frequency of th17 cells in the liver and an increased th17/resting regulatory T cell ratio in peripheral blood and in the liver. J Immunol (Baltimore Md 1950). (2016) 196:97–105. doi: 10.4049/jimmunol.1501175
131. Tang Y, Bian Z, Zhao L, Liu Y, Liang S, Wang Q, et al. Interleukin-17 exacerbates hepatic steatosis and inflammation in non-alcoholic fatty liver disease. Clin Exp Immunol. (2011) 166:281–90. doi: 10.1111/j.1365-2249.2011.04471.x
132. Harley IT, Stankiewicz TE, Giles DA, Softic S, Flick LM, Cappelletti M, et al. IL-17 signaling accelerates the progression of nonalcoholic fatty liver disease in mice. Hepatol (Baltimore Md). (2014) 59:1830–9. doi: 10.1002/hep.26746
133. Gomes AL, Teijeiro A, Burén S, Tummala KS, Yilmaz M, Waisman A, et al. Metabolic inflammation-associated IL-17A causes non-alcoholic steatohepatitis and hepatocellular carcinoma. Cancer Cell. (2016) 30:161–75. doi: 10.1016/j.ccell.2016.05.020
134. Yu H, Huang J, Liu Y, Ai G, Yan W, Wang X, et al. IL-17 contributes to autoimmune hepatitis. J Huazhong Univ Sci Technol Med Sci = Hua zhong ke ji da xue xue bao Yi xue Ying wen ban = Huazhong keji daxue xuebao Yixue Yingdewen ban. (2010) 30:443–6. doi: 10.1007/s11596-010-0446-0
135. Shi T, Zhang T, Zhang L, Yang Y, Zhang H, Zhang F. The distribution and the fibrotic role of elevated inflammatory th17 cells in patients with primary biliary cirrhosis. Medicine. (2015) 94:e1888. doi: 10.1097/MD.0000000000001888
136. Chen J, Liao MY, Gao XL, Zhong Q, Tang TT, Yu X, et al. IL-17A induces pro-inflammatory cytokines production in macrophages via MAPKinases, NF-κB and AP-1. Cell Physiol Biochem Int J Exp Cell physiology biochemistry Pharmacol. (2013) 32:1265–74. doi: 10.1159/000354525
137. Zhang Y, Wang X, Zhong M, Zhang M, Suo Q, Lv K. MicroRNA let-7a ameliorates con A-induced hepatitis by inhibiting IL-6-dependent Th17 cell differentiation. J Clin Immunol. (2013) 33:630–9. doi: 10.1007/s10875-012-9840-7
138. Gao J, Jiang Z, Wang S, Zhou Y, Shi X, Feng M. Endoplasmic reticulum stress of Kupffer cells involved in the conversion of natural regulatory T cells to Th17 cells in liver ischemia-reperfusion injury. J Gastroenterol hepatology. (2016) 31:883–9. doi: 10.1111/jgh.13163
139. Seo W, Eun HS, Kim SY, Yi HS, Lee YS, Park SH, et al. Exosome-mediated activation of toll-like receptor 3 in stellate cells stimulates interleukin-17 production by γδ T cells in liver fibrosis. Hepatol (Baltimore Md). (2016) 64:616–31. doi: 10.1002/hep.28644
140. Ichikawa S, Mucida D, Tyznik AJ, Kronenberg M, Cheroutre H. Hepatic stellate cells function as regulatory bystanders. J Immunol (Baltimore Md 1950). (2011) 186:5549–55. doi: 10.4049/jimmunol.1003917
141. Fabre T, Molina MF, Soucy G, Goulet JP, Willems B, Villeneuve JP, et al. Type 3 cytokines IL-17A and IL-22 drive TGF-beta-dependent liver fibrosis. Sci Immunol. (2018) 3(28):eaar7754. doi: 10.1126/sciimmunol.aar7754
142. Zúñiga LA, Shen WJ, Joyce-Shaikh B, Pyatnova EA, Richards AG, Thom C, et al. IL-17 regulates adipogenesis, glucose homeostasis, and obesity. J Immunol (Baltimore Md 1950). (2010) 185:6947–59. doi: 10.4049/jimmunol.1001269
143. Ahmed M, Gaffen SL. IL-17 inhibits adipogenesis in part via C/EBPα, PPARγ and Krüppel-like factors. Cytokine. (2013) 61:898–905. doi: 10.1016/j.cyto.2012.12.007
144. Shin JH, Shin DW, Noh M. Interleukin-17A inhibits adipocyte differentiation in human mesenchymal stem cells and regulates pro-inflammatory responses in adipocytes. Biochem Pharmacol. (2009) 77:1835–44. doi: 10.1016/j.bcp.2009.03.008
145. Giles DA, Moreno-Fernandez ME, Stankiewicz TE, Cappelletti M, Huppert SS, Iwakura Y, et al. Regulation of inflammation by IL-17A and IL-17F modulates non-alcoholic fatty liver disease pathogenesis. PloS One. (2016) 11:e0149783. doi: 10.1371/journal.pone.0149783
146. Xu R, Tao A, Zhang S, Zhang M. Neutralization of interleukin-17 attenuates high fat diet-induced non-alcoholic fatty liver disease in mice. Acta Biochim Biophys Sinica. (2013) 45:726–33. doi: 10.1093/abbs/gmt065
147. Rolla S, Alchera E, Imarisio C, Bardina V, Valente G, Cappello P, et al. The balance between IL-17 and IL-22 produced by liver-infiltrating T-helper cells critically controls NASH development in mice. Clin Sci (London Engl 1979). (2016) 130:193–203. doi: 10.1042/CS20150405
148. Fabbrini E, Cella M, McCartney SA, Fuchs A, Abumrad NA, Pietka TA, et al. Association between specific adipose tissue CD4+ T-cell populations and insulin resistance in obese individuals. Gastroenterology. (2013) 145:366–74.e1-3. doi: 10.1053/j.gastro.2013.04.010
149. Zhou L, Ivanov II, Spolski R, Min R, Shenderov K, Egawa T, et al. IL-6 programs T(H)-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat Immunol. (2007) 8:967–74. doi: 10.1038/ni1488
150. Eljaafari A, Robert M, Chehimi M, Chanon S, Durand C, Vial G, et al. Adipose tissue-derived stem cells from obese subjects contribute to inflammation and reduced insulin response in adipocytes through differential regulation of the th1/th17 balance and monocyte activation. Diabetes. (2015) 64:2477–88. doi: 10.2337/db15-0162
151. Bechara R, McGeachy MJ, Gaffen SL. The metabolism-modulating activity of IL-17 signaling in health and disease. J Exp Med. (2021) 218(5):e20202191. doi: 10.1084/jem.20202191
152. Moreno-Fernandez ME, Giles DA, Oates JR, Chan CC, Damen M, Doll JR, et al. PKM2-dependent metabolic skewing of hepatic Th17 cells regulates pathogenesis of non-alcoholic fatty liver disease. Cell Metab. (2021) 33:1187–204.e9. doi: 10.1016/j.cmet.2021.04.018
153. Chambers JC, Zhang W, Sehmi J, Li X, Wass MN, van der Harst P, et al. Genome-wide association study identifies loci influencing concentrations of liver enzymes in plasma. Nat Genet. (2011) 43:1131–8. doi: 10.1038/ng.970
154. Namjou B, Lingren T, Huang Y, Parameswaran S, Cobb BL, Stanaway IB, et al. GWAS and enrichment analyses of non-alcoholic fatty liver disease identify new trait-associated genes and pathways across eMERGE Network. BMC Med. (2019) 17:135. doi: 10.1186/s12916-019-1364-z
155. Vonghia L, Magrone T, Verrijken A, Michielsen P, Van Gaal L, Jirillo E, et al. Peripheral and hepatic vein cytokine levels in correlation with non-alcoholic fatty liver disease (NAFLD)-related metabolic, histological, and haemodynamic features. PloS One. (2015) 10:e0143380. doi: 10.1371/journal.pone.0143380
156. Liu Y, She W, Wang F, Li J, Wang J, Jiang W. 3, 3'-Diindolylmethane alleviates steatosis and the progression of NASH partly through shifting the imbalance of Treg/Th17 cells to Treg dominance. Int immunopharmacology. (2014) 23:489–98. doi: 10.1016/j.intimp.2014.09.024
157. He B, Wu L, Xie W, Shao Y, Jiang J, Zhao Z, et al. The imbalance of Th17/Treg cells is involved in the progression of nonalcoholic fatty liver disease in mice. BMC Immunol. (2017) 18:33. doi: 10.1186/s12865-017-0215-y
158. Grohmann M, Wiede F, Dodd GT, Gurzov EN, Ooi GJ, Butt T, et al. Obesity drives STAT-1-dependent NASH and STAT-3-dependent HCC. Cell. (2018) 175:1289–306.e20. doi: 10.1016/j.cell.2018.09.053
159. Dywicki J, Buitrago-Molina LE, Noyan F, Davalos-Misslitz AC, Hupa-Breier KL, Lieber M, et al. The detrimental role of regulatory T cells in nonalcoholic steatohepatitis. Hepatol Commun. (2021) 6(2):320–33. doi: 10.1002/hep4.1807
160. Wang H, Zhang H, Wang Y, Brown ZJ, Xia Y, Huang Z, et al. Regulatory T-cell and neutrophil extracellular trap interaction contributes to carcinogenesis in non-alcoholic steatohepatitis. J hepatology. (2021) 75:1271–83. doi: 10.1016/j.jhep.2021.07.032
161. Beringer A, Miossec P. IL-17 and IL-17-producing cells and liver diseases, with focus on autoimmune liver diseases. Autoimmun Rev. (2018) 17:1176–85. doi: 10.1016/j.autrev.2018.06.008
162. Vu TT, Gooderham M, Papp K. Ixekizumab for treatment of adults with moderate-to-severe plaque psoriasis and psoriatic arthritis. Expert Rev Clin Pharmacol. (2016) 9:1423–33. doi: 10.1080/17512433.2016.1242409
163. Samson M, Audia S, Janikashvili N, Ciudad M, Trad M, Fraszczak J, et al. Brief report: inhibition of interleukin-6 function corrects Th17/Treg cell imbalance in patients with rheumatoid arthritis. Arthritis rheumatism. (2012) 64:2499–503. doi: 10.1002/art.34477
164. Ghoreschi K, Jesson MI, Li X, Lee JL, Ghosh S, Alsup JW, et al. Modulation of innate and adaptive immune responses by tofacitinib (CP-690,550). J Immunol (Baltimore Md 1950). (2011) 186:4234–43. doi: 10.4049/jimmunol.1003668
165. Zong M, Dorph C, Dastmalchi M, Alexanderson H, Pieper J, Amoudruz P, et al. Anakinra treatment in patients with refractory inflammatory myopathies and possible predictive response biomarkers: a mechanistic study with 12 months follow-up. Ann rheumatic diseases. (2014) 73:913–20. doi: 10.1136/annrheumdis-2012-202857
166. Huang X, Shentu H, He Y, Lai H, Xu C, Chen M, et al. Efficacy and safety of IL-23 inhibitors in the treatment of psoriatic arthritis: a meta-analysis based on randomized controlled trials. Immunologic Res. (2023) 71:505–15. doi: 10.1007/s12026-023-09366-4
167. Zhang B, Dömling A. Small molecule modulators of IL-17A/IL-17RA: a patent review (2013-2021). Expert Opin Ther patents. (2022) 32:1161–73. doi: 10.1080/13543776.2022.2143264
168. Xu J, Ma HY, Liu X, Rosenthal S, Baglieri J, McCubbin R, et al. Blockade of IL-17 signaling reverses alcohol-induced liver injury and excessive alcohol drinking in mice. JCI Insight. (2020) 5(3):e131277. doi: 10.1172/jci.insight.131277
169. Ma HY, Yamamoto G, Xu J, Liu X, Karin D, Kim JY, et al. IL-17 signaling in steatotic hepatocytes and macrophages promotes hepatocellular carcinoma in alcohol-related liver disease. J hepatology. (2020) 72:946–59. doi: 10.1016/j.jhep.2019.12.016
170. Colombel JF, Sendid B, Jouault T, Poulain D. Secukinumab failure in Crohn's disease: the yeast connection? Gut. (2013) 62:800–1. doi: 10.1136/gutjnl-2012-304154
171. Ruiz de Morales JMG, Puig L, Dauden E, Canete JD, Pablos JL, Martin AO, et al. Critical role of interleukin (IL)-17 in inflammatory and immune disorders: An updated review of the evidence focusing in controversies. Autoimmun Rev. (2020) 19:102429. doi: 10.1016/j.autrev.2019.102429
172. Guo Y, MacIsaac KD, Chen Y, Miller RJ, Jain R, Joyce-Shaikh B, et al. Inhibition of RORγT skews TCRα Gene rearrangement and limits T cell repertoire diversity. Cell Rep. (2016) 17:3206–18. doi: 10.1016/j.celrep.2016.11.073
173. Guntermann C, Piaia A, Hamel ML, Theil D, Rubic-Schneider T, Del Rio-Espinola A, et al. Retinoic-acid-orphan-receptor-C inhibition suppresses Th17 cells and induces thymic aberrations. JCI Insight. (2017) 2:e91127. doi: 10.1172/jci.insight.91127
174. Dumoutier L, Van Roost E, Ameye G, Michaux L, Renauld JC. IL-TIF/IL-22: genomic organization and mapping of the human and mouse genes. Genes immunity. (2000) 1:488–94. doi: 10.1038/sj.gene.6363716
175. Dudakov JA, Hanash AM, van den Brink MR. Interleukin-22: immunobiology and pathology. Annu Rev Immunol. (2015) 33:747–85. doi: 10.1146/annurev-immunol-032414-112123
176. Bleicher L, de Moura PR, Watanabe L, Colau D, Dumoutier L, Renauld JC, et al. Crystal structure of the IL-22/IL-22R1 complex and its implications for the IL-22 signaling mechanism. FEBS letters. (2008) 582:2985–92. doi: 10.1016/j.febslet.2008.07.046
177. Xie MH, Aggarwal S, Ho WH, Foster J, Zhang Z, Stinson J, et al. Interleukin (IL)-22, a novel human cytokine that signals through the interferon receptor-related proteins CRF2-4 and IL-22R. J Biol Chem. (2000) 275:31335–9. doi: 10.1074/jbc.M005304200
178. Wolk K, Kunz S, Witte E, Friedrich M, Asadullah K, Sabat R. IL-22 increases the innate immunity of tissues. Immunity. (2004) 21:241–54. doi: 10.1016/j.immuni.2004.07.007
179. Lejeune D, Dumoutier L, Constantinescu S, Kruijer W, Schuringa JJ, Renauld JC. Interleukin-22 (IL-22) activates the JAK/STAT, ERK, JNK, and p38 MAP kinase pathways in a rat hepatoma cell line. Pathways that are shared with and distinct from IL-10. J Biol Chem. (2002) 277:33676–82. doi: 10.1074/jbc.M204204200
180. Wolk K, Witte E, Wallace E, Döcke WD, Kunz S, Asadullah K, et al. IL-22 regulates the expression of genes responsible for antimicrobial defense, cellular differentiation, and mobility in keratinocytes: a potential role in psoriasis. Eur J Immunol. (2006) 36:1309–23. doi: 10.1002/eji.200535503
181. Aujla SJ, Chan YR, Zheng M, Fei M, Askew DJ, Pociask DA, et al. IL-22 mediates mucosal host defense against Gram-negative bacterial pneumonia. Nat Med. (2008) 14:275–81. doi: 10.1038/nm1710
182. Zheng Y, Valdez PA, Danilenko DM, Hu Y, Sa SM, Gong Q, et al. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat Med. (2008) 14:282–9. doi: 10.1038/nm1720
183. Liang SC, Tan XY, Luxenberg DP, Karim R, Dunussi-Joannopoulos K, Collins M, et al. Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J Exp Med. (2006) 203:2271–9. doi: 10.1084/jem.20061308
184. Coorens M, Rao A, Grafe SK, Unelius D, Lindforss U, Agerberth B, et al. Innate lymphoid cell type 3-derived interleukin-22 boosts lipocalin-2 production in intestinal epithelial cells via synergy between STAT3 and NF-kappaB. J Biol Chem. (2019) 294:6027–41. doi: 10.1074/jbc.RA118.007290
185. Zheng M, Horne W, McAleer JP, Pociask D, Eddens T, Good M, et al. Therapeutic role of interleukin 22 in experimental intra-abdominal klebsiella pneumoniae infection in mice. Infection immunity. (2016) 84:782–9. doi: 10.1128/IAI.01268-15
186. Zenewicz LA, Yancopoulos GD, Valenzuela DM, Murphy AJ, Karow M, Flavell RA. Interleukin-22 but not interleukin-17 provides protection to hepatocytes during acute liver inflammation. Immunity. (2007) 27:647–59. doi: 10.1016/j.immuni.2007.07.023
187. Radaeva S, Sun R, Pan HN, Hong F, Gao B. Interleukin 22 (IL-22) plays a protective role in T cell-mediated murine hepatitis: IL-22 is a survival factor for hepatocytes via STAT3 activation. Hepatol (Baltimore Md). (2004) 39:1332–42. doi: 10.1002/(ISSN)1527-3350
188. Pan H, Hong F, Radaeva S, Gao B. Hydrodynamic gene delivery of interleukin-22 protects the mouse liver from concanavalin A-, carbon tetrachloride-, and Fas ligand-induced injury via activation of STAT3. Cell Mol Immunol. (2004) 1:43–9.
189. Liang SC, Nickerson-Nutter C, Pittman DD, Carrier Y, Goodwin DG, Shields KM, et al. IL-22 induces an acute-phase response. J Immunol (Baltimore Md 1950). (2010) 185:5531–8. doi: 10.4049/jimmunol.0904091
190. Taube C, Tertilt C, Gyulveszi G, Dehzad N, Kreymborg K, Schneeweiss K, et al. IL-22 is produced by innate lymphoid cells and limits inflammation in allergic airway disease. PloS One. (2011) 6:e21799. doi: 10.1371/journal.pone.0021799
191. Aparicio-Domingo P, Romera-Hernandez M, Karrich JJ, Cornelissen F, Papazian N, Lindenbergh-Kortleve DJ, et al. Type 3 innate lymphoid cells maintain intestinal epithelial stem cells after tissue damage. J Exp Med. (2015) 212:1783–91. doi: 10.1084/jem.20150318
192. Zenewicz LA, Yancopoulos GD, Valenzuela DM, Murphy AJ, Stevens S, Flavell RA. Innate and adaptive interleukin-22 protects mice from inflammatory bowel disease. Immunity. (2008) 29:947–57. doi: 10.1016/j.immuni.2008.11.003
193. Feng D, Kong X, Weng H, Park O, Wang H, Dooley S, et al. Interleukin-22 promotes proliferation of liver stem/progenitor cells in mice and patients with chronic hepatitis B virus infection. Gastroenterology. (2012) 143:188–98.e7. doi: 10.1053/j.gastro.2012.03.044
194. Hu BL, Shi C, Lei RE, Lu DH, Luo W, Qin SY, et al. Interleukin-22 ameliorates liver fibrosis through miR-200a/beta-catenin. Sci Rep. (2016) 6:36436. doi: 10.1038/srep36436
195. Wu LY, Liu S, Liu Y, Guo C, Li H, Li W, et al. Up-regulation of interleukin-22 mediates liver fibrosis via activating hepatic stellate cells in patients with hepatitis C. Clin Immunol (Orlando Fla). (2015) 158:77–87. doi: 10.1016/j.clim.2015.03.003
196. Chen E, Cen Y, Lu D, Luo W, Jiang H. IL-22 inactivates hepatic stellate cells via downregulation of the TGF-β1/Notch signaling pathway. Mol Med Rep. (2018) 17:5449–53. doi: 10.3892/mmr
197. Zheng Y, Danilenko DM, Valdez P, Kasman I, Eastham-Anderson J, Wu J, et al. Interleukin-22, a T(H)17 cytokine, mediates IL-23-induced dermal inflammation and acanthosis. Nature. (2007) 445:648–51. doi: 10.1038/nature05505
198. Sutton CE, Lalor SJ, Sweeney CM, Brereton CF, Lavelle EC, Mills KH. Interleukin-1 and IL-23 induce innate IL-17 production from gammadelta T cells, amplifying Th17 responses and autoimmunity. Immunity. (2009) 31:331–41. doi: 10.1016/j.immuni.2009.08.001
199. Siddiqui KR, Laffont S, Powrie F. E-cadherin marks a subset of inflammatory dendritic cells that promote T cell-mediated colitis. Immunity. (2010) 32:557–67. doi: 10.1016/j.immuni.2010.03.017
200. Zhou L, Lopes JE, Chong MM, Ivanov II, Min R, Victora GD, et al. TGF-beta-induced Foxp3 inhibits T(H)17 cell differentiation by antagonizing RORgammat function. Nature. (2008) 453:236–40. doi: 10.1038/nature06878
201. Rutz S, Noubade R, Eidenschenk C, Ota N, Zeng W, Zheng Y, et al. Transcription factor c-Maf mediates the TGF-β-dependent suppression of IL-22 production in T(H)17 cells. Nat Immunol. (2011) 12:1238–45. doi: 10.1038/ni.2134
202. Lee JS, Cella M, McDonald KG, Garlanda C, Kennedy GD, Nukaya M, et al. AHR drives the development of gut ILC22 cells and postnatal lymphoid tissues via pathways dependent on and independent of Notch. Nat Immunol. (2011) 13:144–51. doi: 10.1038/ni.2187
203. Veldhoen M, Hirota K, Westendorf AM, Buer J, Dumoutier L, Renauld JC, et al. The aryl hydrocarbon receptor links TH17-cell-mediated autoimmunity to environmental toxins. Nature. (2008) 453:106–9. doi: 10.1038/nature06881
204. Lücke J, Sabihi M, Zhang T, Bauditz LF, Shiri AM, Giannou AD, et al. The good and the bad about separation anxiety: roles of IL-22 and IL-22BP in liver pathologies. Semin immunopathology. (2021) 43:591–607. doi: 10.1007/s00281-021-00854-z
205. Jones BC, Logsdon NJ, Walter MR. Structure of IL-22 bound to its high-affinity IL-22R1 chain. Structure (London Engl 1993). (2008) 16:1333–44. doi: 10.1016/j.str.2008.06.005
206. Martin JC, Bériou G, Heslan M, Chauvin C, Utriainen L, Aumeunier A, et al. Interleukin-22 binding protein (IL-22BP) is constitutively expressed by a subset of conventional dendritic cells and is strongly induced by retinoic acid. Mucosal Immunol. (2014) 7:101–13. doi: 10.1038/mi.2013.28
207. Martin JC, Bériou G, Heslan M, Bossard C, Jarry A, Abidi A, et al. IL-22BP is produced by eosinophils in human gut and blocks IL-22 protective actions during colitis. Mucosal Immunol. (2016) 9:539–49. doi: 10.1038/mi.2015.83
208. Huber S, Gagliani N, Zenewicz LA, Huber FJ, Bosurgi L, Hu B, et al. IL-22BP is regulated by the inflammasome and modulates tumorigenesis in the intestine. Nature. (2012) 491:259–63. doi: 10.1038/nature11535
209. Coombes JL, Siddiqui KR, Arancibia-Cárcamo CV, Hall J, Sun CM, Belkaid Y, et al. A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-beta and retinoic acid-dependent mechanism. J Exp Med. (2007) 204:1757–64. doi: 10.1084/jem.20070590
210. Jinnohara T, Kanaya T, Hase K, Sakakibara S, Kato T, Tachibana N, et al. IL-22BP dictates characteristics of Peyer's patch follicle-associated epithelium for antigen uptake. J Exp Med. (2017) 214:1607–18. doi: 10.1084/jem.20160770
211. Sabihi M, Böttcher M, Pelczar P, Huber S. Microbiota-dependent effects of IL-22. Cells. (2020) 9(10):2205. doi: 10.3390/cells9102205
212. Wolk K, Witte E, Hoffmann U, Doecke WD, Endesfelder S, Asadullah K, et al. IL-22 induces lipopolysaccharide-binding protein in hepatocytes: a potential systemic role of IL-22 in Crohn's disease. J Immunol (Baltimore Md 1950). (2007) 178:5973–81. doi: 10.4049/jimmunol.178.9.5973
213. Mastelic B, do Rosario AP, Veldhoen M, Renauld JC, Jarra W, Sponaas AM, et al. IL-22 protects against liver pathology and lethality of an experimental blood-stage malaria infection. Front Immunol. (2012) 3:85. doi: 10.3389/fimmu.2012.00085
214. Park O, Wang H, Weng H, Feigenbaum L, Li H, Yin S, et al. In vivo consequences of liver-specific interleukin-22 expression in mice: Implications for human liver disease progression. Hepatol (Baltimore Md). (2011) 54:252–61. doi: 10.1002/hep.v54.1
215. Xing WW, Zou MJ, Liu S, Xu T, Wang JX, Xu DG. Interleukin-22 protects against acute alcohol-induced hepatotoxicity in mice. Bioscience biotechnology Biochem. (2011) 75:1290–4. doi: 10.1271/bbb.110061
216. Zhang Y, Wang X, Mao L, Yang D, Gao W, Tian Z, et al. Dual roles of IL-22 at ischemia-reperfusion injury and acute rejection stages of rat allograft liver transplantation. Oncotarget. (2017) 8:115384–97. doi: 10.18632/oncotarget.23266
217. Shao L, Xiong X, Zhang Y, Miao H, Ren Y, Tang X, et al. IL-22 ameliorates LPS-induced acute liver injury by autophagy activation through ATF4-ATG7 signaling. Cell Death disease. (2020) 11:970. doi: 10.1038/s41419-020-03176-4
218. Kong X, Feng D, Wang H, Hong F, Bertola A, Wang FS, et al. Interleukin-22 induces hepatic stellate cell senescence and restricts liver fibrosis in mice. Hepatol (Baltimore Md). (2012) 56:1150–9. doi: 10.1002/hep.25744
219. Kleinschmidt D, Giannou AD, McGee HM, Kempski J, Steglich B, Huber FJ, et al. A protective function of IL-22BP in ischemia reperfusion and acetaminophen-induced liver injury. J Immunol (Baltimore Md 1950). (2017) 199:4078–90. doi: 10.4049/jimmunol.1700587
220. Zhang Y, Cobleigh MA, Lian JQ, Huang CX, Booth CJ, Bai XF, et al. A proinflammatory role for interleukin-22 in the immune response to hepatitis B virus. Gastroenterology. (2011) 141:1897–906. doi: 10.1053/j.gastro.2011.06.051
221. Sonnenberg GF, Nair MG, Kirn TJ, Zaph C, Fouser LA, Artis D. Pathological versus protective functions of IL-22 in airway inflammation are regulated by IL-17A. J Exp Med. (2010) 207:1293–305. doi: 10.1084/jem.20092054
222. Dambacher J, Beigel F, Zitzmann K, Heeg MH, Göke B, Diepolder HM, et al. The role of interleukin-22 in hepatitis C virus infection. Cytokine. (2008) 41:209–16. doi: 10.1016/j.cyto.2007.11.016
223. Kang YH, Seigel B, Bengsch B, Fleming VM, Billerbeck E, Simmons R, et al. CD161(+)CD4(+) T cells are enriched in the liver during chronic hepatitis and associated with co-secretion of IL-22 and IFN-γ. Front Immunol. (2012) 3:346. doi: 10.3389/fimmu.2012.00346
224. Xiang X, Gui H, King NJ, Cole L, Wang H, Xie Q, et al. IL-22 and non-ELR-CXC chemokine expression in chronic hepatitis B virus-infected liver. Immunol Cell Biol. (2012) 90:611–9. doi: 10.1038/icb.2011.79
225. Støy S, Sandahl TD, Dige AK, Agnholt J, Rasmussen TK, Grønbæk H, et al. Highest frequencies of interleukin-22-producing T helper cells in alcoholic hepatitis patients with a favourable short-term course. PloS One. (2013) 8:e55101. doi: 10.1371/journal.pone.0055101
226. Ki SH, Park O, Zheng M, Morales-Ibanez O, Kolls JK, Bataller R, et al. Interleukin-22 treatment ameliorates alcoholic liver injury in a murine model of chronic-binge ethanol feeding: role of signal transducer and activator of transcription 3. Hepatol (Baltimore Md). (2010) 52:1291–300. doi: 10.1002/hep.23837
227. Liu Y, Verma VK, Malhi H, Gores GJ, Kamath PS, Sanyal A, et al. Lipopolysaccharide downregulates macrophage-derived IL-22 to modulate alcohol-induced hepatocyte cell death. Am J Physiol Cell Physiol. (2017) 313:C305–c13. doi: 10.1152/ajpcell.00005.2017
228. Hendrikx T, Duan Y, Wang Y, Oh JH, Alexander LM, Huang W, et al. Bacteria engineered to produce IL-22 in intestine induce expression of REG3G to reduce ethanol-induced liver disease in mice. Gut. (2019) 68:1504–15. doi: 10.1136/gutjnl-2018-317232
229. Lu DH, Guo XY, Qin SY, Luo W, Huang XL, Chen M, et al. Interleukin-22 ameliorates liver fibrogenesis by attenuating hepatic stellate cell activation and downregulating the levels of inflammatory cytokines. World J gastroenterology. (2015) 21:1531–45. doi: 10.3748/wjg.v21.i5.1531
230. Sertorio M, Hou X, Carmo RF, Dessein H, Cabantous S, Abdelwahed M, et al. IL-22 and IL-22 binding protein (IL-22BP) regulate fibrosis and cirrhosis in hepatitis C virus and schistosome infections. Hepatol (Baltimore Md). (2015) 61:1321–31. doi: 10.1002/hep.27629
231. Kronenberger B, Rudloff I, Bachmann M, Brunner F, Kapper L, Filmann N, et al. Interleukin-22 predicts severity and death in advanced liver cirrhosis: a prospective cohort study. BMC Med. (2012) 10:102. doi: 10.1186/1741-7015-10-102
232. Park O, Ki SH, Xu M, Wang H, Feng D, Tam J, et al. Biologically active, high levels of interleukin-22 inhibit hepatic gluconeogenesis but do not affect obesity and its metabolic consequences. Cell bioscience. (2015) 5:25. doi: 10.1186/s13578-015-0015-0
233. Wang X, Ota N, Manzanillo P, Kates L, Zavala-Solorio J, Eidenschenk C, et al. Interleukin-22 alleviates metabolic disorders and restores mucosal immunity in diabetes. Nature. (2014) 514:237–41. doi: 10.1038/nature13564
234. Zai W, Chen W, Wu Z, Jin X, Fan J, Zhang X, et al. Targeted interleukin-22 gene delivery in the liver by polymetformin and penetratin-based hybrid nanoparticles to treat nonalcoholic fatty liver disease. ACS Appl materials interfaces. (2019) 11:4842–57. doi: 10.1021/acsami.8b19717
235. Zhu J, Zhou M, Zhao X, Mu M, Cheng M. Blueberry, combined with probiotics, alleviates non-alcoholic fatty liver disease via IL-22-mediated JAK1/STAT3/BAX signaling. Food Funct. (2018) 9:6298–306. doi: 10.1039/C8FO01227J
236. Yang L, Zhang Y, Wang L, Fan F, Zhu L, Li Z, et al. Amelioration of high fat diet induced liver lipogenesis and hepatic steatosis by interleukin-22. J hepatology. (2010) 53:339–47. doi: 10.1016/j.jhep.2010.03.004
237. Petersen PS, Lei X, Seldin MM, Rodriguez S, Byerly MS, Wolfe A, et al. Dynamic and extensive metabolic state-dependent regulation of cytokine expression and circulating levels. Am J Physiol Regulatory Integr Comp Physiol. (2014) 307:R1458–70. doi: 10.1152/ajpregu.00335.2014
238. Hwang S, He Y, Xiang X, Seo W, Kim SJ, Ma J, et al. Interleukin-22 ameliorates neutrophil-driven nonalcoholic steatohepatitis through multiple targets. Hepatol (Baltimore Md). (2020) 72:412–29. doi: 10.1002/hep.31031
239. Hamaguchi M, Okamura T, Fukuda T, Nishida K, Yoshimura Y, Hashimoto Y, et al. Group 3 innate lymphoid cells protect steatohepatitis from high-fat diet induced toxicity. Front Immunol. (2021) 12:648754. doi: 10.3389/fimmu.2021.648754
240. Tang KY, Lickliter J, Huang ZH, Xian ZS, Chen HY, Huang C, et al. Safety, pharmacokinetics, and biomarkers of F-652, a recombinant human interleukin-22 dimer, in healthy subjects. Cell Mol Immunol. (2019) 16:473–82. doi: 10.1038/s41423-018-0029-8
241. Rothenberg ME, Wang Y, Lekkerkerker A, Danilenko DM, Maciuca R, Erickson R, et al. Randomized phase I healthy volunteer study of UTTR1147A (IL-22Fc): A potential therapy for epithelial injury. Clin Pharmacol Ther. (2019) 105:177–89. doi: 10.1002/cpt.1164
242. Arab JP, Sehrawat TS, Simonetto DA, Verma VK, Feng D, Tang T, et al. An open-label, dose-escalation study to assess the safety and efficacy of IL-22 agonist F-652 in patients with alcohol-associated hepatitis. Hepatol (Baltimore Md). (2020) 72:441–53. doi: 10.1002/hep.31046
243. Ponce DM, Alousi AM, Nakamura R, Slingerland J, Calafiore M, Sandhu KS, et al. A phase 2 study of interleukin-22 and systemic corticosteroids as initial treatment for acute GVHD of the lower GI tract. Blood. (2023) 141:1389–401. doi: 10.1182/blood.2021015111
244. Chen W, Zai W, Fan J, Zhang X, Zeng X, Luan J, et al. Interleukin-22 drives a metabolic adaptive reprogramming to maintain mitochondrial fitness and treat liver injury. Theranostics. (2020) 10:5879–94. doi: 10.7150/thno.43894
245. Mo R, Lai R, Lu J, Zhuang Y, Zhou T, Jiang S, et al. Enhanced autophagy contributes to protective effects of IL-22 against acetaminophen-induced liver injury. Theranostics. (2018) 8:4170–80. doi: 10.7150/thno.25798
246. Xiang X, Feng D, Hwang S, Ren T, Wang X, Trojnar E, et al. Interleukin-22 ameliorates acute-on-chronic liver failure by reprogramming impaired regeneration pathways in mice. J hepatology. (2020) 72:736–45. doi: 10.1016/j.jhep.2019.11.013
247. Schwarzkopf K, Rüschenbaum S, Barat S, Cai C, Mücke MM, Fitting D, et al. IL-22 and IL-22-binding protein are associated with development of and mortality from acute-on-chronic liver failure. Hepatol Commun. (2019) 3:392–405. doi: 10.1002/hep4.1303
248. Gal-Oz ST, Shay T. Immune sexual dimorphism: connecting the dots. Physiol (Bethesda Md). (2022) 37:55–68. doi: 10.1152/physiol.00006.2021
249. Wikby A, Månsson IA, Johansson B, Strindhall J, Nilsson SE. The immune risk profile is associated with age and gender: findings from three Swedish population studies of individuals 20-100 years of age. Biogerontology. (2008) 9:299–308. doi: 10.1007/s10522-008-9138-6
250. Klein SL, Flanagan KL. Sex differences in immune responses. Nat Rev Immunol. (2016) 16:626–38. doi: 10.1038/nri.2016.90
251. Lefebvre P, Staels B. Hepatic sexual dimorphism - implications for non-alcoholic fatty liver disease. Nat Rev Endocrinology. (2021) 17:662–70. doi: 10.1038/s41574-021-00538-6
252. Grossmann M, Wierman ME, Angus P, Handelsman DJ. Reproductive endocrinology of nonalcoholic fatty liver disease. Endocrine Rev. (2019) 40:417–46. doi: 10.1210/er.2018-00158
253. Ascha MS, Hanouneh IA, Lopez R, Tamimi TA, Feldstein AF, Zein NN. The incidence and risk factors of hepatocellular carcinoma in patients with nonalcoholic steatohepatitis. Hepatol (Baltimore Md). (2010) 51:1972–8. doi: 10.1002/hep.23527
254. Vilar-Gomez E, Calzadilla-Bertot L, Wai-Sun Wong V, Castellanos M, Aller-de la Fuente R, Metwally M, et al. Fibrosis severity as a determinant of cause-specific mortality in patients with advanced nonalcoholic fatty liver disease: A multi-national cohort study. Gastroenterology. (2018) 155:443–57.e17. doi: 10.1053/j.gastro.2018.04.034
255. Xu L, Yuan Y, Che Z, Tan X, Wu B, Wang C, et al. The hepatoprotective and hepatotoxic roles of sex and sex-related hormones. Front Immunol. (2022) 13:939631. doi: 10.3389/fimmu.2022.939631
256. Camporez JP, Jornayvaz FR, Lee HY, Kanda S, Guigni BA, Kahn M, et al. Cellular mechanism by which estradiol protects female ovariectomized mice from high-fat diet-induced hepatic and muscle insulin resistance. Endocrinology. (2013) 154:1021–8. doi: 10.1210/en.2012-1989
257. Zhu L, Martinez MN, Emfinger CH, Palmisano BT, Stafford JM. Estrogen signaling prevents diet-induced hepatic insulin resistance in male mice with obesity. Am J Physiol Endocrinol Metab. (2014) 306:E1188–97. doi: 10.1152/ajpendo.00579.2013
258. Gao H, Bryzgalova G, Hedman E, Khan A, Efendic S, Gustafsson JA, et al. Long-term administration of estradiol decreases expression of hepatic lipogenic genes and improves insulin sensitivity in ob/ob mice: a possible mechanism is through direct regulation of signal transducer and activator of transcription 3. Mol Endocrinol (Baltimore Md). (2006) 20:1287–99. doi: 10.1210/me.2006-0012
259. Phelps T, Snyder E, Rodriguez E, Child H, Harvey P. The influence of biological sex and sex hormones on bile acid synthesis and cholesterol homeostasis. Biol sex differences. (2019) 10:52. doi: 10.1186/s13293-019-0265-3
260. Senmaru T, Fukui M, Okada H, Mineoka Y, Yamazaki M, Tsujikawa M, et al. Testosterone deficiency induces markedly decreased serum triglycerides, increased small dense LDL, and hepatic steatosis mediated by dysregulation of lipid assembly and secretion in mice fed a high-fat diet. Metabolism: Clin experimental. (2013) 62:851–60. doi: 10.1016/j.metabol.2012.12.007
261. Haider A, Gooren LJ, Padungtod P, Saad F. Improvement of the metabolic syndrome and of non-alcoholic liver steatosis upon treatment of hypogonadal elderly men with parenteral testosterone undecanoate. Exp Clin Endocrinol Diabetes. (2010) 118:167–71. doi: 10.1055/s-0029-1202774
262. Nikolaenko L, Jia Y, Wang C, Diaz-Arjonilla M, Yee JK, French SW, et al. Testosterone replacement ameliorates nonalcoholic fatty liver disease in castrated male rats. Endocrinology. (2014) 155:417–28. doi: 10.1210/en.2013-1648
263. Sowers MF, Beebe JL, McConnell D, Randolph J, Jannausch M. Testosterone concentrations in women aged 25-50 years: associations with lifestyle, body composition, and ovarian status. Am J Epidemiol. (2001) 153:256–64. doi: 10.1093/aje/153.3.256
264. Naessén S, Carlström K, Byström B, Pierre Y, Hirschberg AL. Effects of an antiandrogenic oral contraceptive on appetite and eating behavior in bulimic women. Psychoneuroendocrinology. (2007) 32:548–54. doi: 10.1016/j.psyneuen.2007.03.008
265. Kamada Y, Kiso S, Yoshida Y, Chatani N, Kizu T, Hamano M, et al. Estrogen deficiency worsens steatohepatitis in mice fed high-fat and high-cholesterol diet. Am J Physiol Gastrointestinal liver Physiol. (2011) 301:G1031–43. doi: 10.1152/ajpgi.00211.2011
266. Ohashi T, Kato M, Yamasaki A, Kuwano A, Suzuki H, Kohjima M, et al. Effects of high fructose intake on liver injury progression in high fat diet induced fatty liver disease in ovariectomized female mice. Food Chem Toxicol an Int J published Br Ind Biol Res Assoc. (2018) 118:190–7. doi: 10.1016/j.fct.2018.05.006
267. Naugler WE, Sakurai T, Kim S, Maeda S, Kim K, Elsharkawy AM, et al. Gender disparity in liver cancer due to sex differences in MyD88-dependent IL-6 production. Sci (New York NY). (2007) 317:121–4. doi: 10.1126/science.1140485
268. Itagaki T, Shimizu I, Cheng X, Yuan Y, Oshio A, Tamaki K, et al. Opposing effects of oestradiol and progesterone on intracellular pathways and activation processes in the oxidative stress induced activation of cultured rat hepatic stellate cells. Gut. (2005) 54:1782–9. doi: 10.1136/gut.2004.053728
269. Xu JW, Gong J, Chang XM, Luo JY, Dong L, Hao ZM, et al. Estrogen reduces CCL4- induced liver fibrosis in rats. World J gastroenterology. (2002) 8:883–7. doi: 10.3748/wjg.v8.i5.883
270. Lee YH, Son JY, Kim KS, Park YJ, Kim HR, Park JH, et al. Estrogen deficiency potentiates thioacetamide-induced hepatic fibrosis in sprague-dawley rats. Int J Mol Sci. (2019) 20(15):3709. doi: 10.3390/ijms20153709
271. Schwinge D, Carambia A, Quaas A, Krech T, Wegscheid C, Tiegs G, et al. Testosterone suppresses hepatic inflammation by the downregulation of IL-17, CXCL-9, and CXCL-10 in a mouse model of experimental acute cholangitis. J Immunol (Baltimore Md 1950). (2015) 194:2522–30. doi: 10.4049/jimmunol.1400076
272. Jackson L, Li W, Abdul Y, Dong G, Baban B, Ergul A. Diabetic stroke promotes a sexually dimorphic expansion of T cells. Neuromolecular Med. (2019) 21:445–53. doi: 10.1007/s12017-019-08554-6
273. Gracey E, Yao Y, Green B, Qaiyum Z, Baglaenko Y, Lin A, et al. Sexual dimorphism in the th17 signature of ankylosing spondylitis. Arthritis Rheumatol (Hoboken NJ). (2016) 68:679–89. doi: 10.1002/art.39464
274. Voigt A, Esfandiary L, Wanchoo A, Glenton P, Donate A, Craft WF, et al. Sexual dimorphic function of IL-17 in salivary gland dysfunction of the C57BL/6.NOD-Aec1Aec2 model of Sjögren's syndrome. Sci Rep. (2016) 6:38717. doi: 10.1038/srep38717
275. Dimitrijević M, Arsenović-Ranin N, Kosec D, Bufan B, Nacka-Aleksić M, Pilipović I, et al. Sexual dimorphism in Th17/Treg axis in lymph nodes draining inflamed joints in rats with collagen-induced arthritis. Brain behavior immunity. (2019) 76:198–214. doi: 10.1016/j.bbi.2018.11.311
276. Zychlinsky Scharff A, Rousseau M, Lacerda Mariano L, Canton T, Consiglio CR, Albert ML, et al. Sex differences in IL-17 contribute to chronicity in male versus female urinary tract infection. JCI Insight. (2019) 5(13):e122998. doi: 10.1172/jci.insight.122998
277. Abdelnabi MN, Flores Molina M, Soucy G, Quoc-Huy Trinh V, Bédard N, Mazouz S, et al. Sex-dependent hepatoprotective role of IL-22 receptor signaling in non-alcoholic fatty liver disease-related fibrosis. Cell Mol Gastroenterol hepatology. (2022) 14:1269–94. doi: 10.1016/j.jcmgh.2022.08.001
278. Støy S, Laursen TL, Glavind E, Eriksen PL, Terczynska-Dyla E, Magnusson NE, et al. Low interleukin-22 binding protein is associated with high mortality in alcoholic hepatitis and modulates interleukin-22 receptor expression. Clin Trans gastroenterology. (2020) 11:e00197. doi: 10.14309/ctg.0000000000000197
279. Stülb H, Bachmann M, Gonther S, Mühl H. Acetaminophen-induced liver injury exposes murine IL-22 as sex-related gene product. Int J Mol Sci. (2021) 22(19):10623. doi: 10.3390/ijms221910623
280. Piccinni MP, Lombardelli L, Logiodice F, Kullolli O, Maggi E, Barkley MS. Medroxyprogesterone acetate decreases th1, th17, and increases th22 responses via AHR signaling which could affect susceptibility to infections and inflammatory disease. Front Immunol. (2019) 10:642. doi: 10.3389/fimmu.2019.00642
281. Aksun S, Ersal E, Portakal O, Yildiz BO. Interleukin-22/Interleukin-22 binding protein axis and oral contraceptive use in polycystic ovary syndrome. Endocrine. (2023) 81:54–7. doi: 10.1007/s12020-023-03360-4
Keywords: metabolic dysfunction-associated steatotic liver disease, metabolic-dysfunction associated steatohepatitis, liver fibrosis, IL-17, IL-22, sexual dimorphism
Citation: Abdelnabi MN, Hassan GS and Shoukry NH (2024) Role of the type 3 cytokines IL-17 and IL-22 in modulating metabolic dysfunction-associated steatotic liver disease. Front. Immunol. 15:1437046. doi: 10.3389/fimmu.2024.1437046
Received: 23 May 2024; Accepted: 12 July 2024;
Published: 02 August 2024.
Edited by:
Jinghua Peng, Shanghai University of Traditional Chinese Medicine, ChinaReviewed by:
Haiguang Wang, University of Minnesota Twin Cities, United StatesLi Wen, Yale University, United States
Copyright © 2024 Abdelnabi, Hassan and Shoukry. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Naglaa H. Shoukry, bmFnbGFhLnNob3VrcnlAdW1vbnRyZWFsLmNh