 Xinyu Zhang
Xinyu Zhang Yujing Zhang
Yujing Zhang Shiying Yuan
Shiying Yuan Jiancheng Zhang
Jiancheng Zhang
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 08 July 2024
Sec. Inflammation
Volume 15 - 2024 | https://doi.org/10.3389/fimmu.2024.1434688
Sepsis is described as a life-threatening organ dysfunction and a heterogeneous syndrome that is a leading cause of morbidity and mortality in intensive care settings. Severe sepsis could incite an uncontrollable surge of inflammatory cytokines, and the host immune system's immunosuppression could respond to counter excessive inflammatory responses, characterized by the accumulated anti-inflammatory cytokines, impaired function of immune cells, over-proliferation of myeloid-derived suppressor cells and regulatory T cells, depletion of immune effector cells by different means of death, etc. In this review, we delve into the underlying pathological mechanisms of sepsis, emphasizing both the hyperinflammatory phase and the associated immunosuppression. We offer an in-depth exploration of the critical mechanisms underlying sepsis, spanning from individual immune cells to a holistic organ perspective, and further down to the epigenetic and metabolic reprogramming. Furthermore, we outline the strengths of artificial intelligence in analyzing extensive datasets pertaining to septic patients, showcasing how classifiers trained on various clinical data sources can identify distinct sepsis phenotypes and thus to guide personalized therapy strategies for the management of sepsis. Additionally, we provide a comprehensive summary of recent, reliable biomarkers for hyperinflammatory and immunosuppressive states, facilitating more precise and expedited diagnosis of sepsis.
Sepsis is a life-threatening organ dysfunction caused by a dysregulated host response to infection (1). Sepsis is a major cause of worldwide morbidity and mortality (2). A national-level observational study assembled epidemiological data on sepsis in mainland China, showing that sepsis affects one-fifth of patients in the intensive care unit (ICU), with a 90-day mortality rate of 35.5% (3).
Due to the heterogeneity of sepsis, the management of septic patients relies on rapid identification and ICU-based life-supporting, including fluid resuscitation, appropriate antibiotics, and emergency resuscitation (4). Following the unsatisfactory clinical results of anti-inflammatory medicines for sepsis, researchers have revisited the investigation strategies for appropriate immune-boosting therapies (5). Several immunomodulatory enhancement drugs have shown benefits in animal experiments and clinical trials, including Thymosin α1, Nivolumab,and so on (6, 7). The continuous immunosuppression of sepsis enhances the longer-term risk of secondary infections and readmission rates (8).
The comprehension of sepsis began from the hyper-inflammation to the realization of the indelible role of immunosuppression, and now the appearance of precise endotypes. In this review, we discuss in detail the role of immune cells, organs, and cell death patterns in the complex organismal homeostasis of sepsis. We also try to explore the future direction of sepsis diagnosis and therapy in conjunction with modern technological developments, i.e., combining with artificial intelligence to excavate multi-omics data.
Immunocytes are dramatically altered during the initiation and progression of sepsis. Leukocytes (neutrophils, monocytes, macrophages, natural killer cells, etc.) burst into “cytokine storm” to clear pathogens, while the over-inflammation impairs the stability of the immunological environment (9). The adaptive immune cells, on the other hand, are ineffectively activated and numerically reduced in persistent septic immunosuppression, and suppressive immune cells dominate in survivors. Hence, different immunocytes have central roles in the hyperinflammatory and immunosuppressive phases of sepsis.
Neutrophils, the hub of the “cytokine storm”, release high levels of pro-inflammatory cytokines and proteases in the initial phase of sepsis (9). During sepsis, neutrophils originating from bone marrow (BM) experienced an expansion and consisted of immature and delayed-apoptosis neutrophils (10). Apart from inflammation burst in the early stage of sepsis, neutrophils continually present impaired capacity of chemotaxis and antibacterial, reduced oxidative burst, decreased intracellular myeloperoxidase, and excessive neutrophil extracellular traps (NETs) (11). Programmed cell death ligand 1 (PD-L1)-expressed neutrophils in sepsis inhibit T cell activation, induce T cell apoptosis, and stimulate regulatory T cell proliferation by direct connection (11). A whole blood single-cell map (272,993 cells, n = 39 individuals) of sepsis has demonstrated that immunosuppressive neutrophils partly form the functional basis of the extreme response endotype of sepsis (12). This map offers opportunities for personalized medicine.
NETs are a double-edged sword in sepsis, contributing to pathogen clearance while over-released NETs induce endothelial damage and multiple organ dysfunction syndrome (12). NETs were composed of antimicrobial proteins including myeloperoxidase, histones, and a meshwork of chromatin fibers. Histones cause thrombosis (13), while cell-free DNA extruded from NETs can stimulate clot formation and impair fibrinolysis (14). NETosis occurs via suicidal NETosis, vital NETosis, and mitochondrial NETosis (15). Research has revealed that NETosis is associated with other forms of cell death. NETs activated caspase-1-dependent macrophage pyroptosis to enhance inflammatory responses in cecal ligation and puncture (CLP)-induced sepsis in mice (16). Pyroptotic macrophages as well triggered NET formation during both inhaled-lipopolysaccharide (LPS)- and CLP-induced mouse model of sepsis (17). Platelet pyroptosis exacerbates NET formation which releases S100A8/A9 to promote Gasdermin D (GSDMD)-dependent pyroptosis in mice with CLP-induced sepsis (18). GSDMD also participates in NET release and consequent multiple organ damage in the CLP mouse model (19). Autophagy is involved in the formation of NETs during CLP-induced sepsis to protect mice against lethality (20). These referred experiments demonstrate that the detrimental impact of NETs in sepsis extends beyond mere self-compromise, encompassing the demise of other immune cell populations.
Monocytes/macrophages play important roles in sepsis-induced endotoxin tolerance (ET) and antigen presentation (21). Long-term endotoxin stimulations (e.g., LPS, bacterial compounds) with monocytes/macrophages impaired their capability to upregulate inflammatory cytokines (e.g., tumor necrosis factor-α (TNF-α), IL-6) when facing secondary attack (21). Additionally, anti-inflammatory cytokine IL-10 upregulated by phagocytes represses the proliferation of T cells as well as fosters the proliferation of immunosuppressive cells as MDSCs (22). Metabolic reprogramming participates in the differentiation of BM-derived immune memory monocytes, resulting in either trained immunity (protective response to secondary infections) or immune tolerance (the maladaptive state characterized by consistent immune paralysis) (23). Immunotolerant monocytes signify heightened vulnerability to secondary infections in septic patients (23). The high expression of NOD-like receptor family CARD domain-containing-3 (NLRC3) is associated with ET, and BM-specific NLRC3 deficiency in mice improves the metabolic reprogramming inducibility of ET in macrophages (24). Long-term ET is dependent on epigenetic and metabolic reprogramming, it is also the key to rescue monocyte/macrophage dysfunction.
Reduced expression of human leukocyte antigen on monocytes is associated with immunoparalysis, a state of immunodeficiency observed in sepsis (25). Also, low monocyte human leukocyte antigen-DR (mHLA-DR) is an acknowledged hallmark of monocyte anergy in sepsis. The decreased mHLA-DR reduces the ability to stimulate lymphocyte proliferation and polarization due to the reduced capability to present antigens to the adaptive immune system (26, 27). Clinically, low mHLA-DR is linked to an increased risk of secondary nosocomial infections and mortality after sepsis (28). Therefore, decreased mHLA-DR levels are highly recommended to assess sepsis immunosuppressive severity (29). In 2020, a single-cell RNA-sequencing decodes the subpopulations of peripheral blood mononuclear cells (PBMCs) from septic patients, identifying a new cluster of monocytes (MS1) (30). MS1 fraction was validated as a classifier for sepsis in 11 datasets, with a summary AUC of 0.9. MS1-B module is related to the anti-inflammatory and subsequently immunosuppressive state. Last year, another unique monocyte subset characterized by HLA-DRlowS100Ahigh was identified in sepsis, and S100A9+ monocytes exhibited immunosuppressive function by impairing the immunity of CD4+ T cells (31).
Macrophage polarization is associated with immune homeostasis. The M1 phenotype synthesizes pro-inflammatory cytokines to eliminate pathogens while the M2 macrophage meditates the anti-inflammatory response and tissue repair (32). M2 polarization predominates in late-stage sepsis and increases the proportion of anti-inflammatory cytokines to elicit immunoparalysis (33). Interestingly, macrophages could undergo “re-polarization” due to changes in their microenvironmental stimuli (33). Increasing the proportion of M1 macrophages or decreasing the proportion of M2 macrophages to reach ratio equilibrium has the potential to reverse sepsis-induced immune imbalance (34).
The apoptosis of dendritic cells (DCs) is commonly observed in sepsis and their reduction in the secondary lymphoid organs correlates with an increased risk of secondary infection (35). DCs play a crucial role in triggering adaptive immunity. DCs fail to activate T cells is an indispensable component of septic homeostatic imbalance (36). Roquilly et al. demonstrated that DCs turned into immunoparalysis with disabled antigen presentation function and reduced the secretion of IL-12 after the resolution of Escherichia coli inhalation-induced primary pneumonia (37). Moreover, the ‘‘paralyzed’’ DCs highly express a transcription factor Blimp, which is linked to tolerogenic functions and outcomes in ICU patients (37). The decreased secretion of IL-12 by DCs contributes to activation incompetence, but rising IL-10 level dampens the differentiation of interferon-γ (IFN-γ)-producing T helper type 1 (Th1) cells and the activation of natural killer (NK) cells in CLP-induced sepsis (38). Conversely, CD8+ T cells originating from septic BM elevate the differentiation of DCs with increased production of IL-12 (38). Epigenetically, histone modification enzymes constantly suppress DCs from producing IL-12, persisting for at least 6 weeks after CLP (39), leading to a chronic risk of deadly consequences for the surviving mice. As vital antigen-presenting cells, the population and functional loss of DCs in sepsis compromises the responsiveness of T cells to infection. As reported, Flt3L remarkably improved the number and function of DCs in CLP mice and restored primary CD4+ and CD8+ T cells (36, 40). Moreover, clinical reports have examined the efficacy of recombinant Flt3L (CDX-301), demonstrating that rescuing DCs could be a promising therapeutic target for sepsis (41).
NK cells originate from a similar lineage with innate lymphoid cells but lack the expression of specific T‐cell receptors. This feature benefits rapid, non‐specific immune response for NK cells to intracellular pathogens intrusion (42). In the hyper-inflammatory phase of sepsis, excessive derivation of IFN-γ leads to anomalies in NK cell activation and can initiate cytokine storms through a positive feedback loop, causing severe organ damage (42). One of NK cells’ defects is the main cytotoxic dysfunction, another is the shrinkage of IFN‐γ rendering post-septic patients more susceptible to a second attack (42). Tregs mediate NK cells’ tolerance in sepsis and increased PD-L1 in NK cells contributes to the poor outcome of septic patients (27, 43). The deficiency of NK cells during the immunosuppressive phase of sepsis could be rescued by the adoptive transfer of a homogeneous population or treatment with multiple cytokines such as IL‐2, IL‐12, or IL‐18 (42).
Lymphopenia is a predictor of both early and late mortality and could serve as a reliable biomarker for sepsis (44). T cells are particularly susceptible to rapid and profound lymphopenia, and their essential role in septic immunosuppression has been firmly established (45). Even if the counts of T cell may recover, the compartment is transformed with significant phenotypic and functional changes (45). The persistent immunosuppressive microenvironment in sepsis causes T-cell exhaustion, as evidenced by defective cytokine production, the loss of memory T cell self-renewal capacity via homeostatic proliferative signals, and the continuous expression of multiple inhibitory receptors (46). Immunosuppressive cytokines, such as IL-10 and transforming growth factor beta (TGF-β), are markedly elevated and induce polarization towards Th2 and Th17 in septic patients with poor outcomes. Persistently increased Th2/Th1 ratio has been demonstrated to correlate with the highest 28-day mortality (47.1%) and incidence of ICU-acquired infections (64.7%) (47). T-lymphocyte dysfunction is implicated in persistent inflammation-immunosuppression and catabolism syndrome (PICS)-related immunosuppression (48).
Immune checkpoint (IC) receptors are specific immune homeostasis membrane molecules expressed on T lymphocytes. In sepsis, ICs induce T cell impotency and exhaustion by providing co-inhibitory signals and attenuating co-stimulatory signals (49). Programmed death-1 (PD-1) is a representative negative regulatory molecular in sepsis, and anti-PD-1 antibody administration has been shown to attenuate T-cell dysfunction and improve survival in CLP-induced sepsis in mice (49). Phase I clinical trials to evaluate the safety and tolerability of PD-L1 inhibitor Nabolutumab and BMS-1 in septic patients have been completed, and further evaluation of PD-1 signal pathway inhibitors is expected (6, 50). In addition to PD-1, other ICs with ligands, including CD28, CTLA-4, OX40, and 4–1BB, have shown great translational potential in animal and preclinical studies (49). Given the transformational value, the intervention of ICs may be effective for the therapeutic exploration of sepsis.
Memory T cells in sepsis survivors exhibit immunoparalysis in reaction to secondary infection and memory T cells’ compartments alter. Jensen et al. identified that memory T cell compartments are biased towards central memory CD8+ T cells (TCM) in mice with CLP-induced sepsis. The ability of circulatory TCM to address re-infection was impaired relating to the transcriptional landscape and chromatin accessibility change (51). TCM overrepresentation impacts the re-immune ability for secondary striking and interferes with replenishment from effector memory T cells (TEM) to the tissue (52).
Unconventional T cells such as gamma-delta (γδ) T cells, mucosal-associated invariant T (MAIT) cells, and natural killer T (NKT) cells are not constrained by antigen recognition of class MHC I and II molecules, they also play fantastic parts in sepsis. The protective functions of γδ T cells in CLP-induced sepsis are shown on bacterial clearance and trigger neutrophil recruitment (53). The reduced adhesion function of γδ T cells in septic patients impairs γδ T cells’ role as professional antigen-presenting cells (54). In clinical, septic patients’ mortality was associated with a significant decrease of γδ T cells (55). With the increasing severity of sepsis, cell death of CD3+ CD56+ γδ T cells progressively increased, especially in non-survivors (55). MAIT cells exhibited a dramatic decrease in peripheral blood during the early stage of sepsis by streptococcal infections, the MAIT cell compartment may gradually return to normal without re-striking (56). Despite current observation, the mechanism of MAIT cells to protect sepsis remains to be fully articulated. A gene set enrichment analysis has revealed that the early upregulation of IFN-γ following administration of high-dose endotoxemia drives septic immunosuppression (57). This immunosuppression is mediated part by NKT cells as they partially prompt post-septic immunosuppression (57). Several studies highlighted the pathogenic role of invariant natural killer T (iNKT) cells in sepsis and IFN-γ is the central mediator of iNKT cell impairment. In contrast, the protective role of variant natural killer T cells in sepsis is indirectly evidenced by sulfatide, although further contributions remain unclear (56).
B cells undergo a reduction in septic shock patients. The absolute decrease in immature B cells (IM), naive B cells, and resting memory B cells (RM) is responsible for the increased percentage of the other two subsets, tissue-like memory B cells, and activated memory B cells (58). Lower counts of IM and RM, which respectively act as immediate precursors of mature B cells and immune memory maintainers, indicate a poorer prognosis in sepsis survivors (58). Memory B cell subsets were preferentially depleted among B cells of septic patients (59). In septic conditions, regulatory B (Breg) cells have an increasing circulation number and negatively regulate the innate immune responses mainly by secreting anti-inflammatory cytokines such as IL-10 and promote Treg cell reaction in sepsis (27). CD39high plasmablasts, an expanded B cell subpopulation in CLP-induced sepsis, elevate extracellular adenosine, impair macrophages’ bactericidal activity, and enhance IL-10 production via the A2A adenosine receptor (60). The differentiation of B cells to plasma cells is blocked due to reduced Th1 cells, resulting in decreased immunoglobulin secretion (61). Although not studied as extensively as T cells, excessive apoptosis of memory B cell compartment and amplification of heterogeneous suppressive subpopulations both contribute to sustainable immune disequilibrium in sepsis (59, 60).
MDSCs are a heterogeneous population that proliferates in sepsis and is comprised of immature neutrophils and monocytes with strong immunosuppressive activity (62). MDSCs can be categorized into granulocytic/polymorphonuclear MDSCs and monocytic MDSCs (M-MDSCs), with the latter subset having more immunosuppressive properties in polymicrobial sepsis (63). The immunosuppression capacity of MDSCs has been demonstrated in several aspects like secretion of immunosuppressive cytokines and activation of Tregs (26). Degradation of L-arginine can damage T cell functions by reducing CD3 zeta-chain expression and the NK cell number during sepsis (62). Although MDSCs are rarely found in healthy individuals, MDSCs are pluripotent when induced by environmental signals, so researchers considered altering the differentiation of MDSCs into mature cells (64). While the early expansion of MDSCs may be beneficial in limiting host inflammation, the prolonged suppression of T cells’ immune responses by MDSCs leads to an unfavorable prognosis (64). Therefore, induction of differentiation maturity or reduced MDSCs from BM in the terminal phase of sepsis is expected to facilitate precision immunotherapy. MDSCs are well-known to induce Tregs amplification, as Lee demonstrated that Tregs could also regulate the immunosuppressive functions and amount of M-MDSCs (65). Crosstalk between these two groups of immunosuppressive cells exists and may lead to immunosuppressive synergy (Figure 1).
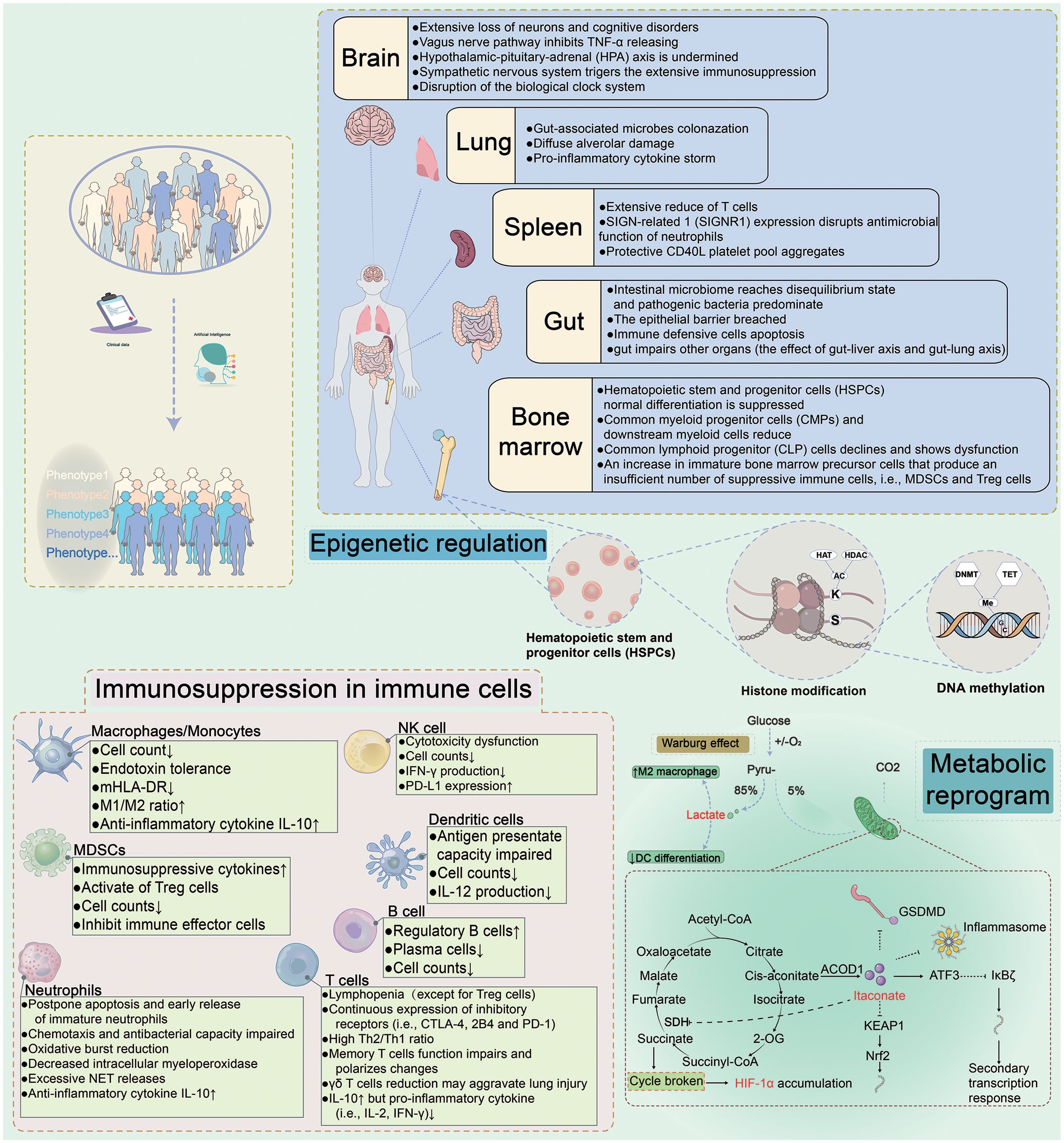
Figure 1 Immune Dysregulation During Sepsis. Sepsis is a sophisticated heterogeneous syndrome. With the development of artificial intelligence, the phenotypes for distinguishing septic patients basing on big data are more accurate. The clinical data of septic patients are gathered and ultimately personalized using artificial intelligence to synthesize the data for precise treatment. Upon prompt identification and classification of septic patients, individualized therapies would be administrated to targeted septic patients. For example, immunostimulatory regimes may be used for immunosuppressive patients, while anti-inflammatory treatments may be effective for septic patients with a hyperinflammatory immunological microenvironment. The underlying mechanisms of sepsis are complex and are interrelated. Different organs (brain, spleen, gut, and bone marrow) contribute to different damage. Metabolic reprogramming and epigenetic regulation in sepsis and immunosuppression present in innate and adaptive immune cells. HAT, histone lysine acetyltransferase; HDAC, histone deacetylase; DNMT, DNA methyltransferase; SDH, succinate dehydrogenase; ACOD1, aconitate decarboxylase 1; ATF3, activating transcription factor 3; HIF-1α, hypoxia-inducible factor 1-α; KEAP1, kelch-like ECH-associated protein 1; Nrf2, nuclear factor erythroid2-related factor 2.
Tregs suppress the proliferation of other effector T cell subsets and divert the phenotypical shift through inhibitory cytokines (27). Tregs also impact innate immunity through the mediation of LPS-stimulated monocytes via the apoptotic Fas/FasL pathway and the direct connection to DCs to downregulate the expression of costimulatory molecules (66). The ratio of Tregs increases in sepsis due to the number of other T cell subsets decreasing. The expression of the anti-apoptotic proteins on Treg makes them more resistant to sepsis-induced apoptosis than other functional T cell subpopulations (66). Th17 and Tregs lie on opposite sides of regulation for the proliferation and polarization of T cell subpopulations, and their ratios serve as reliable biomarkers for predicting sepsis prognosis. The percentage of Th17/Tregs positively correlated with the Sequential Organ Failure Assessment score, as a higher ratio may predict a worse prognosis of sepsis (5). The functions of Tregs vary at different stages of sepsis. The escalation of the Treg cluster may contribute to restoring over-inflammatory eruption during the early stage of sepsis (67). The negative immunomodulatory functions of enhanced Tregs contribute significantly to subsequent sepsis induced-immunosuppression (67).
The process of sepsis is intricately allied to the plummet of immune cell count. These cells are eliminated through various modes of cell death such as apoptosis, pyroptosis, autophagy, NETosis, and ferroptosis to eliminate pathogens and clear damaged cells (Figure 2) (68). However, the excessive "sacrifice" of immune cells may not necessarily confer a beneficial defensive function for septic patients. Cell death pathways interact with others and prompt the development of sepsis.
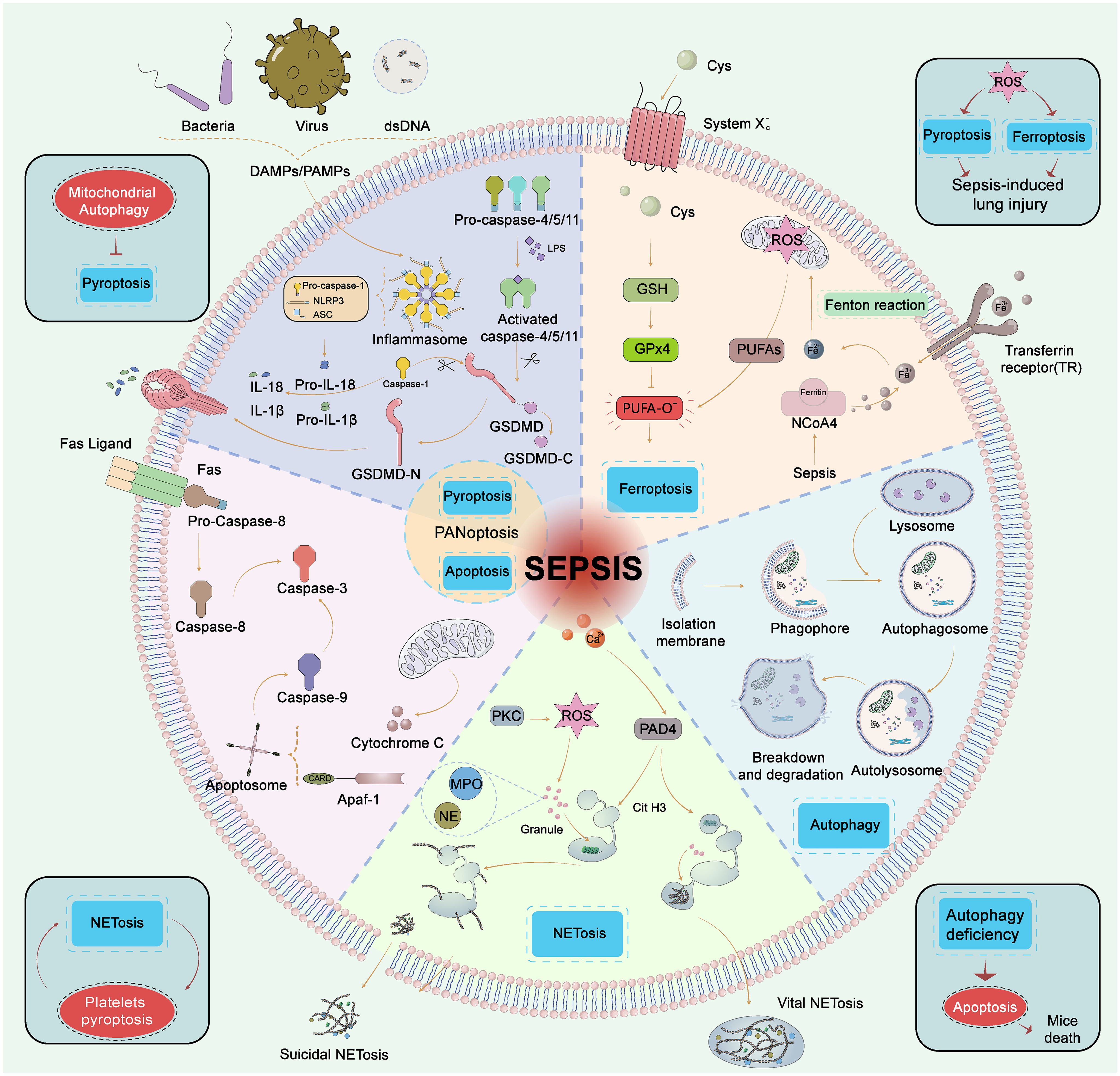
Figure 2 Five types of cell death in sepsis. Pyroptosis, GSDMD is cleaved by caspase-1 (classic pytoptosis pathway) or caspase−4, 5, and 11 (noncanonical pytoptosis pathway) and subsequently integrated into the cell membrane forming a pore. Caspase−1 promotes the release of pro−inflammatory cytokines such as IL−1β and IL−18. Apoptosis, Apoptosis is a programmed process of cell death, which is activated by caspase-8/caspase-3, 9 through extrinsic and intrinsic pathways, respectively. The extrinsic pathway contains the death receptor Fas triggered by the Fas ligand after infection, then Fas activates caspase−8 followed by activation of caspase−3 to trigger the execution pathway of apoptosis. In the intrinsic pathway, cytochrome C is released from mitochondria and then forms an apoptosome with Apaf−1. The apoptosome activates caspase−9 which also activates caspase−3. PANoptosis, PANoptosis is a novel concept in the realm of cell death, that aims to encompass the overlapping and interdependent mechanisms of scorching, apoptotic, and necroptotic apoptosis. Ferroptosis, Ferroptosis is the iron-dependent lipid peroxidation of cell death. The pathways that regulate ferroptosis include both conventional GPX4-dependent and GPX4-independent pathways. GPX4 is reduced in activity and the oxidation of lipids cannot be metabolized by the GPX4-catalyzed glutathione reductase. Subsequent oxidation of lipids by divalent iron ions generates ROS and thus contributes to the onset of ferroptosis. Autophagy, Autophagy is a self-defense mechanism. The autophagosome fuses with the lysosome and further forms a complete autolysosome. Compromised organelles and pathogens are degraded by lysosomal enzymes in autolysosomes and enter the recycling process. NETosis, NETosis is an inflammatory cell death modality of neutrophils and composes of antimicrobial proteins, including MPO, histones, and a network of chromatin fibers. NETosis is activated by alterations in Ca2+ and the production of ROS. PAD4 is activated by high Ca2+ and drives NETosis via guanylate histones. NETosis was divided into suicidal NETosis and vital NETosis, and suicidal NETosis is accompanied by cell rupture and death, while the vital NETosis is accompanied by cell survival. GSDMD, gasdermin−D; Atg, autophagy−related gene; ROS, reactive oxygen species; PUFA, polyunsaturated fatty acid; GPX4, glutathione peroxidase 4; HMGB1, high mobility group box 1; NE, neutrophil elastase; MPO, myeloperoxidase; PAD4, peptide-arginine deaminase 4; PKC, protein kinase C.
Apoptosis is a programmed process of cell death, activated by caspase-8, 9 through extrinsic and intrinsic pathways to cleave executioner caspase-3, 6, 7 (5). During sepsis, the caspase family was activated significantly, while the expression of Bcl-2 (inhibitor of apoptosis) was suppressed (5). The difference between apoptosis and pyroptosis is that apoptosis would not release inflammatory mediators (69). T cells are over-apoptotic and the immune defensive capability is compromised. Inhibition of lymphocyte apoptosis has been demonstrated to lower sepsis mortality (70). However, delayed neutrophil apoptosis negatively contributed to the severity of sepsis due to the expression of anti-apoptotic factors, and the abnormal long persistence of neutrophils may increase the likelihood of organ damage (71). Altogether, these results indicate that inhibition of apoptosis of mature immune cells may be a strategy for restoring immune function to resist infection.
Pyroptosis refers to programmed cell death that is mediated by the gasdermin family, with a prevailing destructive impact on the host during sepsis. GSDMD is cleaved by caspase (classic and noncanonical pyroptosis pathway) and subsequently integrated into the cell membrane (72). Caspases also activate inflammasomes that cleave inflammatory cytokine precursors to release IL-1β, IL-18, and high mobility group box 1 (HMGB1) (72). Wang et al. discovered that Mg2+ can shield against LPS-induced sepsis by impeding GSDMD N-terminal-induced pyroptosis (73). HMGB1 triggers further pyroptosis by re-transporting LPS into the cell via the receptor of advanced glycation end-products, thereby creating an inflammatory loop (49). Researchers have been focusing on the inhibition of canonical pyroptosis in neutrophils and monocytes/macrophages by NOD-, LRR-, and pyrin domain containing 3 (NLRP3) inflammasome, which could rescue lethal sepsis in different septic animal models (74). Nowadays researchers have also focused on lymphocytes and found that inhibition of T cell pyroptosis could increase survival after lethal CLP-induced sepsis (75). Over pyroptosis has been demonstrated to promote the aggravation of sepsis. Therefore, effective and specific blockade of cellular pyroptosis may be a valuable therapeutic intervention to rescue sepsis.
Autophagy is a self-defense mechanism for cell survivorship, forming double-membrane autophagosomes and degrading substrates such as compromised organelles and pathogens (76). In the hyperinflammatory phase of sepsis, autophagy exerts a protective function by eliminating pathogens, neutralizing toxins, preserving mitochondrial integrity, and regulating cytokine secretion (76). However, autophagy efficiency reduction or procedure disturbances may cause damage. For example, the blockade of CD4+ T cell autophagy is associated with increased IL-10 production. Additionally, autophagy-related gene 4B- deficient mice exhibit high mortality and severe ALI in response to LPS stimulation (76). Impaired autophagic flux accumulates high levels of autophagosomes and can't recycle damaged mitochondria (77). Furthermore, the overproduction of outer membrane vesicles causes excessive activation of the inflammasome, resulting in the destruction of autophagosome-lysosome fusion (78). Autophagy in non-immune cells has also been associated with sepsis. The preservation of endothelial cell permeability depends on the stabilization of adherent junctions and endothelial barriers through complete autophagy, thus reducing sepsis-related endothelial damage (79). Therefore, curative activation of complete autophagic flux may be an effective target for sepsis (76).
Ferroptosis is the iron-dependent lipid peroxidation of cell death. Ferroptosis is involved in LPS-injected ALI (80). Ferroptosis is initiated in immune cells due to an imbalance in the antioxidant system of glutathione peroxidase 4 (GPX4), leading to an increase in lipid peroxide levels (81). Thus, the ferroptosis program not only provides nutrients to bacteria but undoubtedly reduces the immune function of hosts. Regulation of antioxidant molecules (e.g., GPX4, glutathione) and targeting the regulatory pathways of ferroptosis could be a new approach for the treatment of sepsis (81).
The effects of the various forms of cell death caused by sepsis are not independent. Autophagy deficiency could lead to immune cell dysfunction and accelerated apoptosis, resulting in reduced immunity and increased mortality in septic mice (77). Mitochondrial autophagy inhibits pyroptosis by reducing the production of reactive oxygen species (ROS), also known as NLRP3 activators (74). The induction of ferroptosis in pulmonary epithelial cells by NETs has been shown to exacerbate CLP-induced ALI in mice, mechanically through the implementation of methyltransferase-like 3-induced m6A modification of GPX4 (82). Both pyroptosis and ferroptosis participate in CLP-induced ALI in mice, which is induced by ROS (83). Apoptotic caspase-7 cleaved GSDMB (a potential proinflammatory mediator in sepsis) to block non-canonical pyroptosis to avoid over-inflammation in sepsis (84). PANoptosis encompasses the overlapping and interdependent mechanisms of pyroptosis, apoptosis, and necroptosis (85). PANoptosome serves as a death complex, a multi-protein molecular framework that recruits downstream inflammatory cell death components such as NLRP3 inflammasome, the RIPK1/RIPK3 complex, and more (86). Moderate PANoptosis plays a crucial role in efficiently scavenging infectious agents and activating immune cells. However, excessive PANoptosis may result in tissue damage. Therefore, achieving a balanced regulation of this process represents a critical direction for future research into PANoptosis (85). The interplay and equilibrium among several cell death patterns remain a challenge for future research.
Recently, researchers utilized spatial transcriptomics to measure gene expression in 9 organs following sepsis, creating a dynamic, organism-wide map. Interestingly, they verified pairwise cytokines' effects on nearly 200 cells and found that non-lymphoid tissues recover to transcriptional homeostasis earlier than lymphoid tissues. This may help explain why post-septic patients are in a state of prolonged immunosuppression (87). We reviewed several important organs and their role in sepsis.
Gut is the main source of infection, leading to secondary strikes on sepsis survivors, hindering the immune system, and elevating readmission rates and mortality (88). Metabolites with anti-inflammatory effects such as short-chain fatty acids and Granisetron decrease, and Granisetron reduces CLP-induced-septic damage of the gut-liver axis (89, 90). Colonization of pathogenic and opportunistic pathogens in the gut takes a dominant position but the growth of commensal organisms was depressed, resulting in the disruption of the immune barrier and the reduction of beneficial metabolites (91). Regeneration of intestinal barrier cells is inhibited and apoptotic cells increase during sepsis, with a reduction in luminal coverage, thickness, and adhesion. Inflammatory cytokines upregulate claudin 2 and junctional adhesion molecules but downregulate claudin 5 and zonula occludens-1, rendering the intestinal hyperpermeability (92). In conclusion, the mechanical, immune, and biological barriers of the septic gut are disturbed after sepsis. The massive proliferation of pathogens and endotoxins could be transferred to the systemic circulation via the mesenteric lymphatics or portal vein (93). Our viewpoint is focusing on the importance of gut among multiple organs, because many other organ axes are associated with the gut. The gut may predispose to more severe secondary infections in septic survivors.
Both biliary and portal circulations continuously defend against invading bacteria. Pathogens can impair liver function, primarily by reducing bacterial clearance, disrupting detoxification, and interfering with the release of inflammatory cytokines during sepsis (94). When the intestinal barrier is compromised, pathogen-associated molecular patterns and damage-associated molecular pattern molecules intrude via two circulations into the liver, where they recognize pattern recognition receptors on Kupffer cells (KCs) or stellate cells, resulting in inappropriate immunogenicity initiation or overwhelming inflammation response (95). One of the features of the immunogenic response is an increase in acute phase proteins (APPs) and the induction of immunosuppression and ET associated with MDSCs by APPs (95). Sepsis modulates the polarization of KCs towards the M1 phenotype, which contributes to the immunoinflammatory response and acute liver injury (96). Peng et al. found that pharmacological inhibition of M1 polarization could alleviate LPS/D-galactosamine-induced liver injury in mice (97). Sepsis-induced cholestasis commonly results in impaired hepatic glycolipid metabolism and apoptotic necrosis (96). Decreased bile acids in the enterohepatic circulation also facilitate intestinal bacterial translocation that increases mortality (98).
The lung is particularly susceptible to damage from sepsis, with approximately half of septic patients developing acute respiratory distress syndrome (ARDS) in the later stage, leading to a further increase in patient mortality (94). Notably, the presence of gut-associated microbes has been demonstrated in the lung of septic patients (99). Carbapenemase-producing Enterobacteriaceae intestinal colonized mice have a notable decrease in pulmonary alveolar macrophages and DCs, and the outcome after secondary Pseudomonas aeruginosa lung strikes is worsened (100). Dickson et al. showed that enterobacteria can metastasize across the intestinal mucosa and even into the lungs in septic-ARDS patients, as verified by analysis of the ecology of lung-associated bacteria in both LPS and CLP models of sepsis (99). The "mesenteric lymphatic hypothesis" has also been supported by multiple lines of evidence. Transfer of gut lymph from critically ill mice to healthy mice causes lung injury (101). Consequently, the attenuation of injury associated with the gut-lung axis in ARDS merits further investigation.
BM serves as the primary lymphoid organ and provides a microenvironment for hematopoietic stem and progenitor cells (HSPCs). A humanized mice model stimulated with CLP/endotoxemia induces HSPCs expansion with a concomitant shrinkage of the downstream progenitor cells in the BM (27). Wang et al. found that the frequency of HSPCs in the peripheral blood of septic patients on day 4 after sepsis is significantly higher than healthy controls (P < 0.05) (102). The expansion of HSPC at early time points in septic patients had a negative correlation with immune cell counts (P < 0.05) (102). Through subpopulation analysis, the most pronounced change in HSPCs is the common myeloid progenitors (CMPs) (102). Deviation of CMPs from normal differentiation to pathological activation may increase the population of peripheral blood CD34+ CD38+ HSPCs. Sepsis-induced BM niche with ablation of osteoblasts associated with reduction of common lymphoid progenitor cells and dysfunction of hematopoietic stem cells (64). Lymphopenia may also be partly caused by the decline in common lymphoid progenitor cells. Macrophage function is impaired partly due to the epigenetic modification of BM progenitor cells with lower expression of mixed lineage leukemia protein-1 (103). Disruption of the homeostatic balance of self-renewal and differentiation of HSPCs ultimately leads to depletion of the progenitor compartment. The infusion of donor HSPCs to a group A Streptococcus-induced sepsis mouse model has increased overall survival (104).
As the largest peripheral immune organ, T-cell exhaustion and suppressor cell expansion in the spleen may present important immunosuppressive mechanisms in sepsis (105). Splenic macrophages in red marrow and marginal zone express CD169 and the receptor SIGN-related 1. The CD169 macrophages disrupt neutrophil antimicrobial function, deplete mature neutrophils and promote T cell apoptosis, ultimately leading to a reduction in both innate and adaptive immunity in systemic candidiasis (106). Spleen enriches the CD40L ligand platelet population during cecal slurry injection-induced sepsis in mice, potentially facilitating improved bacterial clearance (possibly via increased NETs), reducing organ injury, and improving the survival of mice (107). Various cell subpopulations of the spleen exert different influences in sepsis. Thus, the hematopoietic and immune functions of the spleen in sepsis merit further exploration.
The central nervous system (CNS) modulates the immune response and is responsible for the collapse of the peripheral immune system. Neuroimmune reflexes such as the vagus nerve pathway, hypothalamic-pituitary-adrenal (HPA) axis, and central circadian system are disturbed by sepsis, resulting in extensive loss of neurons and cognitive disorders. The formed vicious cycle between sepsis-associated encephalopathy and turbulent immunity leads to poor outcomes in septic patients (108). Constitutive activation of the vagus nerve after CLP in mice blocked the release of TNF-α and was incompetent to the second LPS challenge, and the α7 nicotinic acetylcholine receptor plays an important role in sepsis neural regulation (109). The anti-inflammatory HPA axis inhibits the production of proinflammatory cytokines and reaches ephemeral endogenous glucocorticoids peak in sepsis (108). However, long-stay ICU patients with suppressed HPA axis suffer central (secondary) adrenal insufficiency (110). The target tissues of many septic patients show resistance to glucocorticoids and fail to maintain immune tolerance (111). The suprachiasmatic nucleus modulates the cyclic expression of circadian clock gene programs. It was demonstrated that the core biological clock gene Bmal1 trans-regulates immune checkpoint PD-L1 on macrophages and T-cell apoptosis in CLP-induced sepsis (112). Similarly, mice deficient in Per2 are more resistant to LPS-induced septic shock. The CNS is a major predecessor in the regulation of the release of the inflammatory cytokines. Dysregulated hormonal secretion rhythms and immune system disorders occur in the early stages of sepsis. Most of the immune cells may express catecholamine receptors and rhythmic genes, so maintenance of CNS is necessary to control the immune imbalance.
Epigenetic modifications activate or deactivate transcriptional regulation without altering the DNA sequence, and have been proven to regulate sepsis ER (8). DNA methylation, non-coding RNA, and histone modifications within immune cells contribute to the pathophysiological mechanism of sepsis. Histone H3 lysine 9 dimethylation in late sepsis inactivates the promoter of TNF and IL-1β transcription, downregulating immune sensitivity to endotoxin stimulation (111). The high dynamics of histone deacetylases (HDACs) overwhelm the acetylation of histone acetyltransferases (HATs) to progress immunosuppression and HDAC inhibitors have been shown to improve sepsis progression. Panobinostat as a pan-HDAC inhibitor could inhibit IL-10 production in LPS-stimulated macrophages (113). Although HDAC inhibitors have been clinically approved, it remains to be elusive to locate the precise timing to intervene in gene silencing during immunoparalysis. DNA methylation has a more precise and stable function than histone modifications, connected with the stabilization of the transcriptional state. Binnie et al. aimed to identify significant DNA methylome change in tolerized monocytes in septic patients and highlighted that DNA methylation is determined by the altered levels of inflammatory cytokines (114). Other researches also demonstrate that DNA methylation is involved in monocyte dysregulation during sepsis. The absence of RNA m6A demethylase alkB homolog 5 in neutrophils inhibits mRNA expression of chemokine receptor CXCR2, thus injuring the neutrophil migration, as observed in CLP-induced mouse model of sepsis (115). MiR-221, miR-579, and miR125b tend to directly prevent TNF translation for the existence of binding sites for the 3’ untranslated region of TNF, while miR146a acts indirectly (116). The increased expression of specific microRNAs (e.g., miR-220, miR-221, miR-222) in macrophages correlates with immunoparalysis in septic patients and serves as biomarkers of immunosuppression (117). MiR-210 coordinates M1 macrophages and increases glycolysis in response to LPS stimulation at the expense of oxidative phosphorylation (OXPHOS) (117). Epigenetic modification agents show promise as therapeutic tools in animal models of sepsis (118). With the numerous mechanisms already discussed, epigenetic regulation occupies the center of the cellular immune metabolism and immunologic responses of sepsis.
The altered metabolic pathways of immune cells and the abnormal accumulation of the metabolites in sepsis both play vital regulation roles. Immune cells convert from OXPHOS to glycolysis as the main metabolic source to rapidly produce high quantities of ATP and activate the immunological effect during sepsis (119). However, patients in the late stage presented mitochondrial dysfunction and oxidative imbalance. This has been demonstrated by a reduction in cytokine production and impaired phagocytosis in PBMCs with immune tolerance (119, 120). Leukocytes in the immunosuppression phase of sepsis present extensive immunometabolic defects, with impaired glycolysis and oxidative metabolism (120). It is promising that the immunostimulatory therapy, recombinant IFN-γ, has been demonstrated to partially reverse metabolic paralysis in septic patients, and it can promote polarization of macrophages towards the M1 phenotype (120). Intact mitochondrial complexes are integral to support the long-term survival and functions of macrophages (121). A recent study has indicated that mitochondrial STAT3 drives FAO to worsen sepsis in LPS-injected mice (122). Mitochondrial dysfunction in sepsis is unable to provide adequate biological energy and associated with unfavorable clinical endpoints (121).
LPS stimulation compromises macrophage activity and results in high glycolysis and abnormal accumulation of tricarboxylic acid (TCA) cycle intermediates (120). Pyruvate kinase M2 (PKM2) is upregulated in activating macrophages and the tetrameric form of PKM2 resists tyrosine phosphorylation, which attenuates glycolytic biosynthetic intermediates accumulation. The tetramer form prevents nuclear translocation-induced transcription of hypoxia-inducible factor-1α (HIF-1α) and IL-1β, but promotes M2 phenotype and ET (123). As a glycolytic key metabolite, PKM2 not only promotes the release of HMGB1 in sepsis, and its inhibition also decreases the production of another vital metabolite, lactate.
Lactate has been considered as a metabolic waste, but in recent years lactate has been discovered as signaling molecule, biomarker, and modifier of lactation in sepsis. High lactate induces the release of HMGB1 into circulation by lactylation/acetylation via exosome to disrupt endothelium integrity and vascular permeability in CLP-induced septic mice (124). Lactate overproduction is correlated with the high morbidity and poor prognosis of septic patients with acute kidney injury (AKI). The results have shown that lactylation at lysine 20 of Fis1 may be the mechanism of lactate to cause CLP-induced AKI (123). High circulation lactate levels promote the inactivation of immune cells and the production of immunosuppressive cells. Hyper-lactate also increases the expression of anti-inflammatory genes TGF-β and IL-10, promoting M2 macrophage generation and inhibiting DC differentiation (121).
Itaconate is a macrophage immunometabolite with anti-inflammatory and immunomodulatory properties after sepsis (125). The main effects of itaconate are the anti-oxidative stress pathway of nuclear factor E2-related factor 2 (Nrf2) as well as the influence on succinate dehydrogenase (SDH) and NLRP3 inflammasomes (125, 126). Dimethyl itaconate (itaconate derivative) ameliorates survival within 24h and reduces ALI in septic mice (127). And 4-OI reduces inflammatory cytokine and lactate production in mice exposed to lethal endotoxemia to prolong survival (128). Nrf2 is generally known to control oxidative stress by mediating glutathione synthase. In CLP-induced sepsis in mice, Nrf2 may also function as a transcriptional suppressor to inhibit PD-L1 expression (129). Researchers have discovered an additional facet of itaconate's function. Wu et al. found that the CDK2-aconitate decarboxylase 1 axis triggers the inflammatory cytokines storm, with its expression levels correlating with sepsis severity in patients (130). Similarly, Chen et al. found that MYD88-STING1-mediated itaconate production exacerbates CLP- and LPS-triggered sepsis (131). Interestingly, β-glucan bypasses the inhibition of SDH by itaconate and reconstitutes macrophage metabolism and immunity (132). Therefore, we hypothesize that the reductions in itaconate production could restore TCA circulation activity, thereby improving immune tolerance. Currently, we are uncertain whether itaconate is favorable for the restoration of metabolic paralysis in sepsis and it deserves further investigation (132).
No universally accepted gold standard was conceded for the treatment of sepsis during the past years. With the development of bioinformatics, artificial intelligence is combined with disease diagnosis and prediction. Machine learning (MI) is trained to build more accurate models on medical data and assist clinicians in the early classification and diagnosis of sepsis (133). In contrast to the algorithms built on structured data, the SERA algorithm has extracted and synthesized unstructured data from electronic medical records, and the algorithm predicted sepsis 12 hours in advance with high sensitivity and specificity (AUC = 0.94) (134). MI is not just to accurately predict and diagnose sepsis. MI is also used to make antibiotic recommendations for sepsis. After training and validation with two intensive care testing sets ( the MIMIC-III and AmsterdamUMCdb), the antibiotic treatment strategy recommended by the T4 model is proven to be effective in decreasing the mortality rate (135). The COMPOSER model was trained for the early identification of patients with a high risk of sepsis and abandoned the prediction of false predictions to improve accuracy (AUC = 0.925–0.953) (136). Recently this model was identified to be associated with an increase in the bundle compliance of sepsis (137).
Hyperinflammation and immunosuppression fail to precisely characterize the mechanisms for disease progress in septic patients. MI with multi-omics databases would classify patients into subgroups within similar pathological dynamics (133). Group-based trajectory modeling (GBTM) was utilized to model the evolution of physiology repeated-measured temperature trajectory with septic patients to classify sub-phenotypes. Four temperature trajectory groups were identified. The investigators speculated on the association of immunological phenotypes with temperature subtypes, with hypothermic subjects being possibly more relevant to immunosuppression (138). MI quantified similarities between septic individuals and differentiated patients into phenotypes basing not only on temperature or organ dysfunction, but also fluid responsiveness (139). However, the most prevalent technique is to deeply mine whole blood immune cell transcriptomics with MI (Table 1) (133). How to downsize multiple dimensions and integrate sepsis heterogeneity are future challenges, the key biomarkers might fill the vacuum of the clinical and transcriptomic data (144). The ideal biomarker should meet the following criteria: accurate identification, prognosis prediction, and easy detection. However, individual biomarkers may not be fully satisfied. The combined application of biomarkers may compensate for the limitations of diagnostic specificity or sensitivity and potentially yield higher diagnostic value. Most high inflammatory biomarkers are used to diagnose early sepsis, while immunosuppressive biomarkers can predict early or late prognosis in septic patients. The challenge lies in correctly combining the appropriate biomarkers to achieve optimal diagnostic results. Tables 2, 3
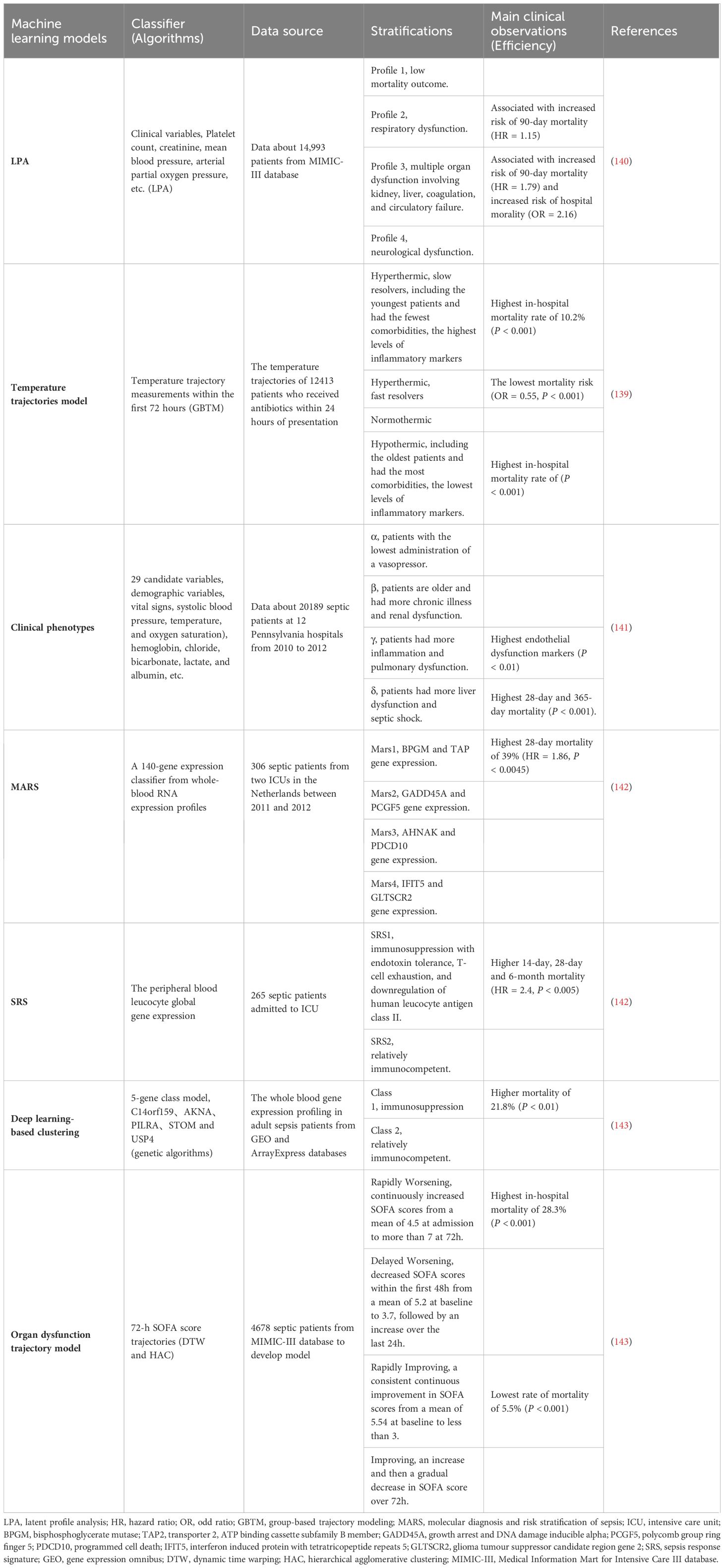
Table 1 Machine learning models for sepsis phenotypes.
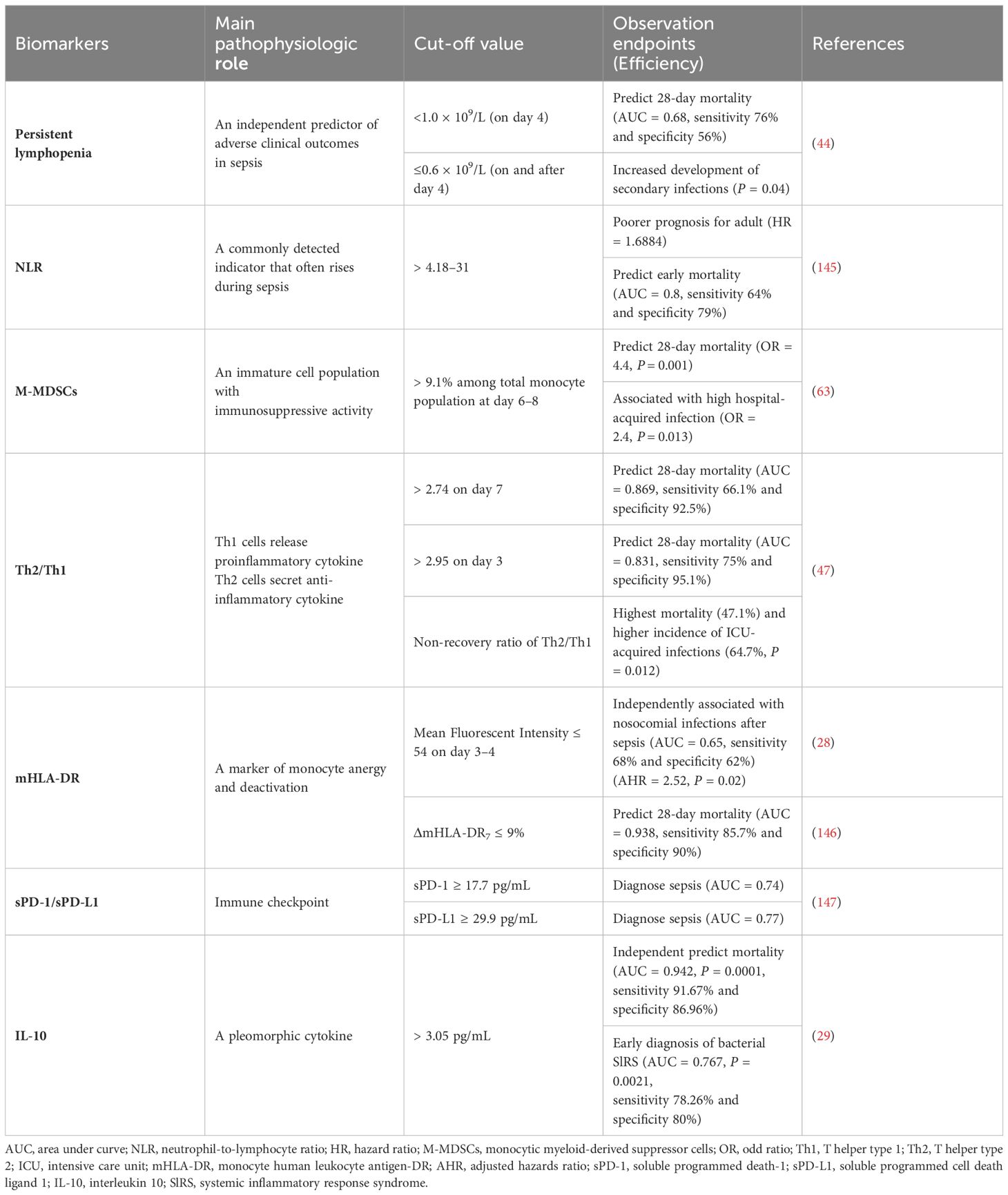
Table 2 Biomarkers for immunosuppression in sepsis.
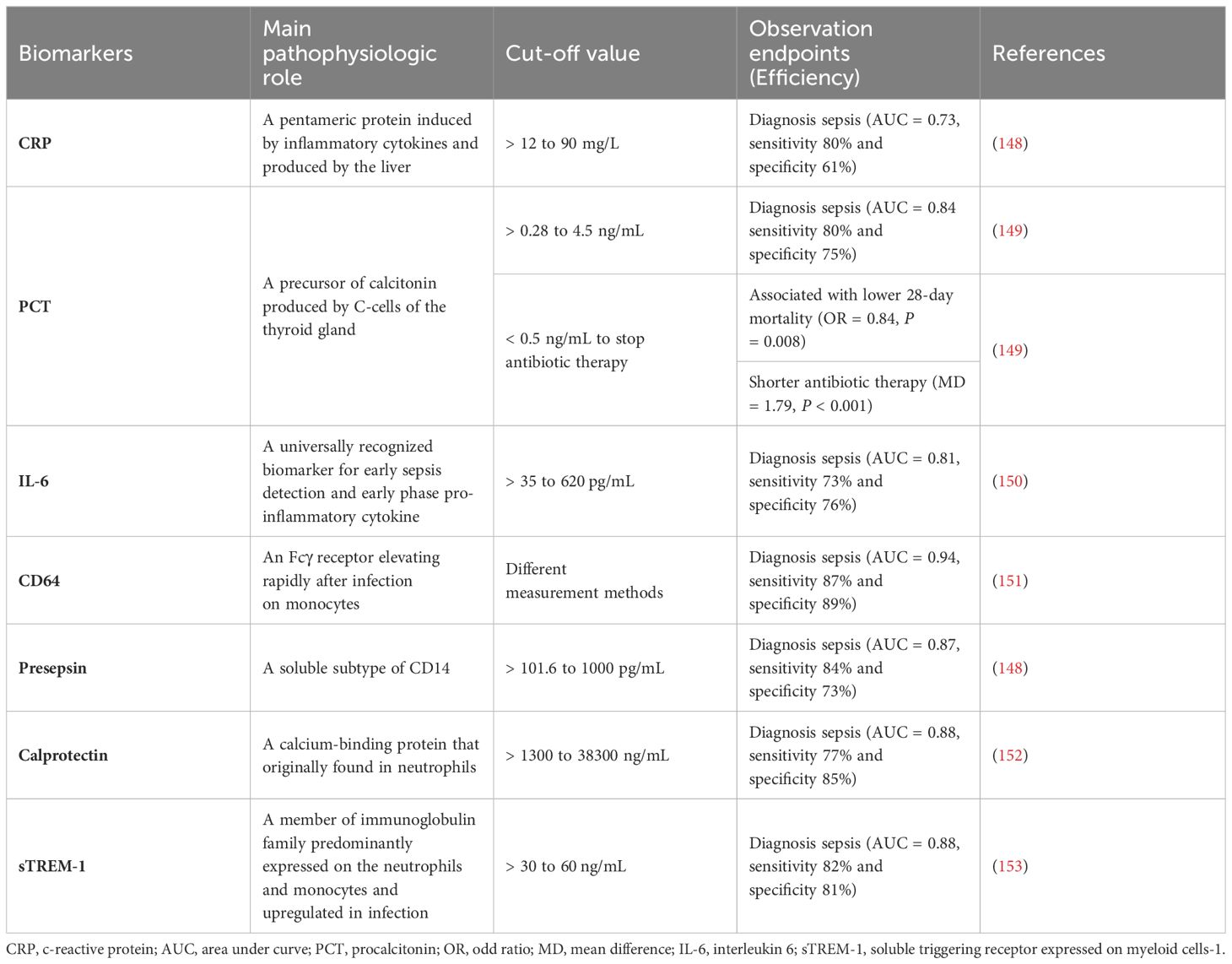
Table 3 Biomarkers for pro-inflammation in sepsis.
We have profiled the mechanism of sepsis from multiple aspects. Hyperinflammation in sepsis is accompanied by prolonged immunosuppression. Excessive immunosuppression is the foundation of PICS, high readmission rates, and increased secondary mortality. Owing to the heterogeneity of sepsis, clinicians can’t treat patients with “one-size-fits-all” therapy. The establishment of a therapeutic regime for sepsis would require an investigation into the mechanisms of the host immune disorder, in particular the intricate network among epigenetic alterations and metabolic reprogramming, and other mechanisms. Primarily, researchers require the identification of a uniformly approved set of biomarkers, which is accessible and easily testable. Secondly, the standardized sepsis subgroups and endotypes could help clinicians identify patients who develop immunosuppression. Finally, personalized precision therapy potentially could be based on artificial algorithms which in anticipation would dynamically mimic the course of septic patients. Overall, continued research in sepsis has the potential to greatly improve our understanding and treatment of this devastating condition.
XZ: Writing – original draft. YZ: Writing – original draft. SYY: Conceptualization, Funding acquisition, Writing – review & editing. JZ: Conceptualization, Funding acquisition, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the National Key Research and Development Program of China (2021YFC2501800 (to SYY)), and the National Natural Science Foundation of China (Grant No. 82071480 (to JZ); Grant No. 82272231 (to SYY); Grant No. 82302471 (to YZ)).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
ICU, intensive care unit; BM, bone marrow; NETs, neutrophil extracellular traps; PD-1, programmed death-1; PD-L1, programmed cell death ligand 1; Tregs, regulatory T cells; MODS, multiple organ dysfunction syndrome; GSDMD, gasdermin D; ROS, reactive oxygen species; CLP, cecal ligation and puncture; ET, endotoxin tolerance; LPS, lipopolysaccharide; NLRC3, NOD-like receptor family CARD domain-containing-3; mHLA-DR, monocyte human leukocyte antigen-DR; PBMCs, peripheral blood mononuclear cells; DCs, dendritic cells; STAT3, signal transducer and activator of transcription 3; IFN-γ, interferon-γ; Th1, T helper type 1; Th2, T helper type 2; NK, natural killer; PICS, persistent inflammation-immunosuppression and catabolism syndrome; TCM, memory CD8+ T cells; TEM, effector memory T cells; MAIT cells, mucosal-associated invariant T cells; NKT cells, natural killer T cells; ALI, acute lung injury; iNKT, invariant natural killer T cells; IM, immature B cells; RM, resting memory B cells; MDSCs, myeloid-derived suppressor cells; M-MDSCs, monocytic MDSCs; Breg cells, regulatory B cells; HMGB1, high mobility group box 1; NLRP3 inflammasome, NOD-, LRR-, and pyrin domain containing 3 inflammasome; GPX4, glutathione peroxidase 4; KCs, kupffer cells; APPs, acute phase proteins; HSPCs, hematopoietic stem and progenitor cells; CMPs, common myeloid progenitors; HPA, hypothalamic-pituitary-adrenal; HDACs, histone deacetylases; HAT, histone acetyltransferases; OXPHOS, oxidative phosphorylation; FAO, fatty acid oxidation; TCA, tricarboxylic acid cycle; PKM2, pyruvate kinase M2; HIF-1α, hypoxia-inducible factor-1α; Nrf2, nuclear factor E2-related factor 2; SDH, succinate dehydrogenase; IL-10, interleukin 10; IL-6, interleukin 6; GBTM, group-based trajectory modeling.
1. Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, et al. The third international consensus definitions for sepsis and septic shock (Sepsis-3). JAMA. (2016) 315:801–10. doi: 10.1001/jama.2016.0287
2. Rudd KE, Johnson SC, Agesa KM, Shackelford KA, Tsoi D, Kievlan DR, et al. Global, regional, and national sepsis incidence and mortality, 1990–2017: analysis for the Global Burden of Disease Study. Lancet. (2020) 395:200–11. doi: 10.1016/S0140-6736(19)32989-7
3. Xie J, Wang H, Kang Y, Zhou L, Liu Z, Qin B, et al. The epidemiology of sepsis in chinese ICUs: A national cross-sectional survey. Crit Care Med. (2020) 48:e209. doi: 10.1097/CCM.0000000000004155
4. Marik PE, Linde-Zwirble WT, Bittner EA, Sahatjian J, Hansell D. Fluid administration in severe sepsis and septic shock, patterns and outcomes: an analysis of a large national database. Intensive Care Med. (2017) 43:625–32. doi: 10.1007/s00134-016-4675-y
5. Liu D, Huang S-Y, Sun J-H, Zhang H-C, Cai Q-L, Gao C, et al. Sepsis-induced immunosuppression: mechanisms, diagnosis and current treatment options. Military Med Res. (2022) 9:56. doi: 10.1186/s40779-022-00422-y
6. Hotchkiss RS, Colston E, Yende S, Crouser ED, Martin GS, Albertson T, et al. Immune checkpoint inhibition in sepsis: a Phase 1b randomized study to evaluate the safety, tolerability, pharmacokinetics, and pharmacodynamics of nivolumab. Intensive Care Med. (2019) 45:1360–71. doi: 10.1007/s00134-019-05704-z
7. Pei F, Guan X, Wu J. Thymosin alpha 1 treatment for patients with sepsis. Expert Opin Biol Ther. (2018) 18:71–6. doi: 10.1080/14712598.2018.1484104
8. Hotchkiss RS, Monneret G, Payen D. Immunosuppression in sepsis: a novel understanding of the disorder and a new therapeutic approach. Lancet Infect Dis. (2013) 13:260–8. doi: 10.1016/S1473-3099(13)70001-X
9. van der Poll T, Shankar-Hari M, Wiersinga WJ. The immunology of sepsis. Immunity. (2021) 54:2450–64. doi: 10.1016/j.immuni.2021.10.012
10. Margraf A, Lowell CA, Zarbock A. Neutrophils in acute inflammation: current concepts and translational implications. Blood. (2022) 139:2130–44. doi: 10.1182/blood.2021012295
11. Qi X, Yu Y, Sun R, Huang J, Liu L, Yang Y, et al. Identification and characterization of neutrophil heterogeneity in sepsis. Crit Care. (2021) 25:50. doi: 10.1186/s13054-021-03481-0
12. Kwok AJ, Allcock A, Ferreira RC, Cano-Gamez E, Smee M, Burnham KL, et al. Neutrophils and emergency granulopoiesis drive immune suppression and an extreme response endotype during sepsis. Nat Immunol. (2023) 24:767–79. doi: 10.1038/s41590-023-01490-5
13. Xu J, Zhang X, Pelayo R, Monestier M, Ammollo CT, Semeraro F, et al. Extracellular histones are major mediators of death in sepsis. Nat Med. (2009) 15:1318–21. doi: 10.1038/nm.2053
14. Gould TJ, Vu TT, Stafford AR, Dwivedi DJ, Kim PY, Fox-Robichaud AE, et al. Cell-free DNA modulates clot structure and impairs fibrinolysis in sepsis. Arteriosclerosis Thrombosis Vasc Biol. (2015) 35:2544–53. doi: 10.1161/ATVBAHA.115.306035
15. Huang Z, Zhang H, Fu X, Han L, Zhang H, Zhang L, et al. Autophagy-driven neutrophil extracellular traps: The dawn of sepsis. Pathol - Res Pract. (2022) 234:153896. doi: 10.1016/j.prp.2022.153896
16. Chen L, Zhao Y, Lai D, Zhang P, Yang Y, Li Y, et al. Neutrophil extracellular traps promote macrophage pyroptosis in sepsis. Cell Death Dis. (2018) 9:597. doi: 10.1038/s41419-018-0538-5
17. Kuang L, Wu Y, Shu J, Yang J, Zhou H, Huang X. Pyroptotic Macrophage-Derived Microvesicles Accelerate Formation of Neutrophil Extracellular Traps via GSDMD-N-expressing Mitochondrial Transfer during Sepsis. Int J Biol Sci. (2024) 20:733–50. doi: 10.7150/ijbs.87646
18. Su M, Chen C, Li S, Li M, Zeng Z, Zhang Y, et al. Gasdermin D-dependent platelet pyroptosis exacerbates NET formation and inflammation in severe sepsis. Nat Cardiovasc Res. (2022) 1:732–47. doi: 10.1038/s44161-022-00108-7
19. Silva CMS, Wanderley CWS, Veras FP, Sonego F, Nascimento DC, Gonçalves AV, et al. Gasdermin D inhibition prevents multiple organ dysfunction during sepsis by blocking NET formation. Blood. (2021) 138:2702–13. doi: 10.1182/blood.2021011525
20. Park SY, Shrestha S, Youn Y-J, Kim J-K, Kim S-Y, Kim HJ, et al. Autophagy primes neutrophils for neutrophil extracellular trap formation during sepsis. Am J Respir Crit Care Med. (2017) 196:577–89. doi: 10.1164/rccm.201603-0596OC
21. Venet F, Demaret J, Gossez M, Monneret G. Myeloid cells in sepsis-acquired immunodeficiency. Ann N Y Acad Sci. (2021) 1499:3–17. doi: 10.1111/nyas.14333
22. Bah I, Kumbhare A, Nguyen L, McCall CE, El Gazzar M. IL-10 induces an immune repressor pathway in sepsis by promoting S100A9 nuclear localization and MDSC development. Cell Immunol. (2018) 332:32–8. doi: 10.1016/j.cellimm.2018.07.003
23. Bomans K, Schenz J, Sztwiertnia I, Schaack D, Weigand MA, Uhle F. Sepsis induces a long-lasting state of trained immunity in bone marrow monocytes. Front Immunol. (2018) 9:2685. doi: 10.3389/fimmu.2018.02685
24. Xu J, Gao C, He Y, Fang X, Sun D, Peng Z, et al. NLRC3 expression in macrophage impairs glycolysis and host immune defense by modulating the NF-κB-NFAT5 complex during septic immunosuppression. Mol Ther. (2022) 31(1): 154–73. doi: 10.1016/j.ymthe.2022.08.023
25. Leentjens J, Kox M, Koch RM, Preijers F, Joosten LAB, van der Hoeven JG, et al. Reversal of immunoparalysis in humans in vivo: a double-blind, placebo-controlled, randomized pilot study. Am J Respir Crit Care Med. (2012) 186:838–45. doi: 10.1164/rccm.201204-0645OC
26. Schrijver IT, Théroude C, Roger T. Myeloid-derived suppressor cells in sepsis. Front Immunol. (2019) 10:327. doi: 10.3389/fimmu.2019.00327
27. Venet F, Monneret G. Advances in the understanding and treatment of sepsis-induced immunosuppression. Nat Rev Nephrol. (2018) 14:121–37. doi: 10.1038/nrneph.2017.165
28. Landelle C, Lepape A, Voirin N, Tognet E, Venet F, Bohé J, et al. Low monocyte human leukocyte antigen-DR is independently associated with nosocomial infections after septic shock. Intensive Care Med. (2010) 36:1859–66. doi: 10.1007/s00134-010-1962-x
29. Matera G, Puccio R, Giancotti A, Quirino A, Pulicari MC, Zicca E, et al. Impact of interleukin-10, soluble CD25 and interferon-γ on the prognosis and early diagnosis of bacteremic systemic inflammatory response syndrome: a prospective observational study. Crit Care. (2013) 17:R64. doi: 10.1186/cc12596
30. Reyes M, Filbin MR, Bhattacharyya RP, Billman K, Eisenhaure T, Hung DT, et al. An immune-cell signature of bacterial sepsis. Nat Med. (2020) 26:333–40. doi: 10.1038/s41591-020-0752-4
31. Yao R-Q, Zhao P-Y, Li Z-X, Liu Y-Y, Zheng L-Y, Duan Y, et al. Single-cell transcriptome profiling of sepsis identifies HLA-DRlowS100Ahigh monocytes with immunosuppressive function. Military Med Res. (2023) 10:27. doi: 10.1186/s40779-023-00462-y
32. Viola A, Munari F, Sánchez-Rodríguez R, Scolaro T, Castegna A. The metabolic signature of macrophage responses. Front Immunol. (2019) 10:1462. doi: 10.3389/fimmu.2019.01462
33. Chen X, Liu Y, Gao Y, Shou S, Chai Y. The roles of macrophage polarization in the host immune response to sepsis. Int Immunopharmacol. (2021) 96:107791. doi: 10.1016/j.intimp.2021.107791
34. Rackov G, Hernández-Jiménez E, Shokri R, Carmona-Rodríguez L, Mañes S, Álvarez-Mon M, et al. p21 mediates macrophage reprogramming through regulation of p50-p50 NF-κB and IFN-β. J Clin Invest. (2016) 126:3089–103. doi: 10.1172/JCI83404
35. Grimaldi D, Louis S, Pène F, Sirgo G, Rousseau C, Claessens YE, et al. Profound and persistent decrease of circulating dendritic cells is associated with ICU-acquired infection in patients with septic shock. Intensive Care Med. (2011) 37:1438–46. doi: 10.1007/s00134-011-2306-1
36. Strother RK, Danahy DB, Kotov DI, Kucaba TA, Zacharias ZR, Griffith TS, et al. Polymicrobial sepsis diminishes dendritic cell numbers and function directly contributing to impaired primary CD8 T cell responses. In Vivo. J Immunol. (2016) 197:4301–11. doi: 10.4049/jimmunol.1601463
37. Roquilly A, McWilliam HEG, Jacqueline C, Tian Z, Cinotti R, Rimbert M, et al. Local modulation of antigen-presenting cell development after resolution of pneumonia induces long-term susceptibility to secondary infections. Immunity. (2017) 47:135–147.e5. doi: 10.1016/j.immuni.2017.06.021
38. Antoni A-C, Pylaeva E, Budeus B, Jablonska J, Klein-Hitpaß L, Dudda M, et al. TLR2-induced CD8+ T-cell deactivation shapes dendritic cell differentiation in the bone marrow during sepsis. Front Immunol. (2022) 13:945409. doi: 10.3389/fimmu.2022.945409
39. Wen H, Dou Y, Hogaboam CM, Kunkel SL. Epigenetic regulation of dendritic cell-derived interleukin-12 facilitates immunosuppression after a severe innate immune response. Blood. (2008) 111:1797–804. doi: 10.1182/blood-2007-08-106443
40. Patil NK, Bohannon JK, Luan L, Guo Y, Fensterheim B, Hernandez A, et al. Flt3 ligand treatment attenuates T cell dysfunction and improves survival in a murine model of burn wound sepsis. Shock. (2017) 47:40–51. doi: 10.1097/SHK.0000000000000688
41. Anandasabapathy N, Breton G, Hurley A, Caskey M, Trumpfheller C, Sarma P, et al. Efficacy and safety of CDX-301, recombinant human Flt3L, at expanding dendritic cells and hematopoietic stem cells in healthy human volunteers. Bone Marrow Transplant. (2015) 50:924–30. doi: 10.1038/bmt.2015.74
42. Wang F, Cui Y, He D, Gong L, Liang H. Natural killer cells in sepsis: Friends or foes? Front Immunol. (2023) 14:1101918. doi: 10.3389/fimmu.2023.1101918
43. Jiang W, Li X, Wen M, Liu X, Wang K, Wang Q, et al. Increased percentage of PD-L1+ natural killer cells predicts poor prognosis in sepsis patients: a prospective observational cohort study. Crit Care. (2020) 24:617. doi: 10.1186/s13054-020-03329-z
44. Drewry AM, Samra N, Skrupky LP, Fuller BM, Compton SM, Hotchkiss RS. Persistent lymphopenia after diagnosis of sepsis predicts mortality. Shock. (2014) 42:383–91. doi: 10.1097/SHK.0000000000000234
45. Jensen IJ, Sjaastad FV, Griffith TS, Badovinac VP. Sepsis-induced T cell immunoparalysis: the ins and outs of impaired T cell immunity. J Immunol. (2018) 200:1543–53. doi: 10.4049/jimmunol.1701618
46. Huang S, Liu D, Sun J, Zhang H, Zhang J, Wang Q, et al. Tim-3 regulates sepsis-induced immunosuppression by inhibiting the NF-κB signaling pathway in CD4 T cells. Mol Ther. (2022) 30:1227–38. doi: 10.1016/j.ymthe.2021.12.013
47. Xue M, Xie J, Liu L, Huang Y, Guo F, Xu J, et al. Early and dynamic alterations of Th2/Th1 in previously immunocompetent patients with community-acquired severe sepsis: a prospective observational study. J Transl Med. (2019) 17:57. doi: 10.1186/s12967-019-1811-9
48. Darden DB, Kelly LS, Fenner BP, Moldawer LL, Mohr AM, Efron PA. Dysregulated immunity and immunotherapy after sepsis. J Clin Med. (2021) 10:1742. doi: 10.3390/jcm10081742
49. McBride MA, Patil TK, Bohannon JK, Hernandez A, Sherwood ER, Patil NK. Immune checkpoints: novel therapeutic targets to attenuate sepsis-induced immunosuppression. Front Immunol. (2021) 11:624272. doi: 10.3389/fimmu.2020.624272
50. Hotchkiss RS, Colston E, Yende S, Angus DC, Moldawer LL, Crouser ED, et al. Immune checkpoint inhibition in sepsis: A phase 1b randomized, placebo-controlled, single ascending dose study of antiprogrammed cell death-ligand 1 antibody (BMS-936559). Crit Care Med. (2019) 47:632–42. doi: 10.1097/CCM.0000000000003685
51. Jensen IJ, Li X, McGonagill PW, Shan Q, Fosdick MG, Tremblay MM, et al. Sepsis leads to lasting changes in phenotype and function of memory CD8 T cells. eLife. (2021) 10:e70989. doi: 10.7554/eLife.70989
52. Heidarian M, Griffith TS, Badovinac VP. Sepsis-induced changes in differentiation, maintenance, and function of memory CD8 T cell subsets. Front Immunol. (2023) 14:1130009. doi: 10.3389/fimmu.2023.1130009
53. Chen W, Lai D, Li Y, Wang X, Pan Y, Fang X, et al. Neuronal-activated ILC2s promote IL-17A production in lung γδ T cells during sepsis. Front Immunol. (2021) 12:670676. doi: 10.3389/fimmu.2021.670676
54. Yang X-W, Li H, Feng T, Zhang W, Song X-R, Ma C-Y, et al. Impairment of antigen-presenting function of peripheral γδ T cells in patients with sepsis. Clin Exp Immunol. (2022) 207:104–12. doi: 10.1093/cei/uxab029
55. Andreu-Ballester JC, Arribas MA, Rico M, García-Ballesteros C, Galindo-Regal L, Sorando-Serra R, et al. Changes of CD3+CD56+ γδ T cell number and apoptosis during hospital admission are related to mortality in septic patients. Clin Immunol. (2022) 236:108956. doi: 10.1016/j.clim.2022.108956
56. Szabo PA, Anantha RV, Shaler CR, McCormick JK, Haeryfar SMM. CD1d- and MR1-restricted T cells in sepsis. Front Immunol. (2015) 6:401. doi: 10.3389/fimmu.2015.00401
57. Kim EY, Ner-Gaon H, Varon J, Cullen AM, Guo J, Choi J, et al. Post-sepsis immunosuppression depends on NKT cell regulation of mTOR/IFN-γ in NK cells. J Clin Invest. (2020) 130:3238–52. doi: 10.1172/JCI128075
58. Dong X, Liu Q, Zheng Q, Liu X, Wang Y, Xie Z, et al. Alterations of B cells in immunosuppressive phase of septic shock patients. Crit Care Med. (2020) 48:815–21. doi: 10.1097/CCM.0000000000004309
59. Shankar-Hari M, Fear D, Lavender P, Mare T, Beale R, Swanson C, et al. Activation-associated accelerated apoptosis of memory B cells in critically ill patients with sepsis. Crit Care Med. (2017) 45:875–82. doi: 10.1097/CCM.0000000000002380
60. Nascimento DC, Viacava PR, Ferreira RG, Damaceno MA, Piñeros AR, Melo PH, et al. Sepsis expands a CD39+ plasmablast population that promotes immunosuppression via adenosine-mediated inhibition of macrophage antimicrobial activity. Immunity. (2021) 54:2024–2041.e8. doi: 10.1016/j.immuni.2021.08.005
61. Nedeva C. Inflammation and cell death of the innate and adaptive immune system during sepsis. Biomolecules. (2021) 11:1011. doi: 10.3390/biom11071011
62. Veglia F, Sanseviero E, Gabrilovich DI. Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat Rev Immunol. (2021) 21:485–98. doi: 10.1038/s41577-020-00490-y
63. Waeckel L, Venet F, Gossez M, Monard C, Rimmelé T, Monneret G. Delayed persistence of elevated monocytic MDSC associates with deleterious outcomes in septic shock: a retrospective cohort study. Crit Care. (2020) 24:132. doi: 10.1186/s13054-020-02857-y
64. Rincon JC, Efron PA, Moldawer LL. Immunopathology of chronic critical illness in sepsis survivors: Role of abnormal myelopoiesis. J Leukoc Biol. (2022) 112:1525–34. doi: 10.1002/JLB.4MR0922-690RR
65. Lee C-R, Kwak Y, Yang T, Han JH, Park S-H, Ye MB, et al. Myeloid-Derived Suppressor Cells Are Controlled by Regulatory T Cells via TGF-β during Murine Colitis. Cell Rep. (2016) 17:3219–32. doi: 10.1016/j.celrep.2016.11.062
66. Hotchkiss RS, Monneret G, Payen D. Sepsis-induced immunosuppression: from cellular dysfunctions to immunotherapy. Nat Rev Immunol. (2013) 13:862–74. doi: 10.1038/nri3552
67. Gao Y, Yao Y, Zhang X, Chen F, Meng X, Chen X, et al. Regulatory T cells: angels or demons in the pathophysiology of sepsis? Front Immunol. (2022) 13:829210. doi: 10.3389/fimmu.2022.829210
68. Wen X, Xie B, Yuan S, Zhang J. The “Self-sacrifice” of immuneCells in sepsis. Front Immunol. (2022) 13:833479. doi: 10.3389/fimmu.2022.833479
69. Jiang Y, Gao S, Chen Z, Zhao X, Gu J, Wu H, et al. Pyroptosis in septic lung injury: Interactions with other types of cell death. Biomedicine Pharmacotherapy. (2023) 169:115914. doi: 10.1016/j.biopha.2023.115914
70. Lannes-Costa PS, Pimentel BA da S, Nagao PE. Role of caveolin-1 in sepsis – A mini-review. Front Immunol. (2022) 13:902907. doi: 10.3389/fimmu.2022.902907
71. Meghraoui-Kheddar A, Chousterman BG, Guillou N, Barone SM, Granjeaud S, Vallet H, et al. Two new neutrophil subsets define a discriminating sepsis signature. Am J Respir Crit Care Med. (2022) 205:46–59. doi: 10.1164/rccm.202104-1027OC
72. Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. (2015) 526:660–5. doi: 10.1038/nature15514
73. Wang D, Zheng J, Hu Q, Zhao C, Chen Q, Shi P, et al. Magnesium protects against sepsis by blocking gasdermin D N-terminal-induced pyroptosis. Cell Death Differ. (2020) 27:466–81. doi: 10.1038/s41418-019-0366-x
74. Guo R, Wang H, Cui N. Autophagy regulation on pyroptosis: mechanism and medical implication in sepsis. Mediators Inflammation. (2021) 2021:9925059. doi: 10.1155/2021/9925059
75. Liu Y, Zhang Y, Liu Q, Li T, Wang W, Li H, et al. Inhibition of DDX3X ameliorated CD4+ T cells pyroptosis and improves survival in septic mice. Mol Immunol. (2023) 154:54–60. doi: 10.1016/j.molimm.2022.12.014
76. Chao R, Hui Z, Tian Tian W, Yonming Y. Autophagy: A potential therapeutic target for reversing sepsis-induced immunosuppression. Front Immunol. (2017) 8:1832. doi: 10.3389/fimmu.2017.01832
77. Ho J, Yu J, Wong SH, Zhang L, Liu X, Wong WT. Autophagy in sepsis: Degradation into exhaustion. Autophagy. (2016) 12::1073–1082. doi: 10.1080/15548627.2016.1179410
78. David L, Taieb F, Pénary M, Bordignon P-J, Planès R, Bagayoko S, et al. Outer membrane vesicles produced by pathogenic strains of Escherichia coli block autophagic flux and exacerbate inflammasome activation. Autophagy. (2022) 18:2913–25. doi: 10.1080/15548627.2022.2054040
79. Li Y, Suo L, Fu Z, Li G, Zhang J. Pivotal role of endothelial cell autophagy in sepsis. Life Sci. (2021) 276:119413. doi: 10.1016/j.lfs.2021.119413
80. He R, Liu B, Xiong R, Geng B, Meng H, Lin W, et al. Itaconate inhibits ferroptosis of macrophage via Nrf2 pathways against sepsis-induced acute lung injury. Cell Death Discovery. (2022) 8:43. doi: 10.1038/s41420-021-00807-3
81. Wu J, Liu Q, Zhang X, Tan M, Li X, Liu P, et al. The interaction between STING and NCOA4 exacerbates lethal sepsis by orchestrating ferroptosis and inflammatory responses in macrophages. Cell Death Dis. (2022) 13:653. doi: 10.1038/s41419-022-05115-x
82. Zhang H, Liu J, Zhou Y, Qu M, Wang Y, Guo K, et al. Neutrophil extracellular traps mediate m6A modification and regulates sepsis-associated acute lung injury by activating ferroptosis in alveolar epithelial cells. Int J Biol Sci. (2022) 18:3337–57. doi: 10.7150/ijbs.69141
83. Cao Z, Qin H, Huang Y, Zhao Y, Chen Z, Hu J, et al. Crosstalk of pyroptosis, ferroptosis, and mitochondrial aldehyde dehydrogenase 2-related mechanisms in sepsis-induced lung injury in a mouse model. Bioengineered. (2022) 13:4810–20. doi: 10.1080/21655979.2022.2033381
84. Li X, Zhang T, Kang L, Xin R, Sun M, Chen Q, et al. Apoptotic caspase-7 activation inhibits non-canonical pyroptosis by GSDMB cleavage. Cell Death Differ. (2023) 30:2120–34. doi: 10.1038/s41418-023-01211-3
85. Christgen S, Zheng M, Kesavardhana S, Karki R, Malireddi RKS, Banoth B, et al. Identification of the PANoptosome: A molecular platform triggering pyroptosis, apoptosis, and necroptosis (PANoptosis). Front Cell Infect Microbiol. (2020) 10:237. doi: 10.3389/fcimb.2020.00237
86. Liu X, Tang A-L, Chen J, Gao N, Zhang G, Xiao C. RIPK1 in the inflammatory response and sepsis: Recent advances, drug discovery and beyond. Front Immunol. (2023) 14:1114103. doi: 10.3389/fimmu.2023.1114103
87. Takahama M, Patil A, Richey G, Cipurko D, Johnson K, Carbonetto P, et al. A pairwise cytokine code explains the organism-wide response to sepsis. Nat Immunol. (2024) 25:226–39. doi: 10.1038/s41590-023-01722-8
88. Adelman MW, Woodworth MH, Langelier C, Busch LM, Kempker JA, Kraft CS, et al. The gut microbiome’s role in the development, maintenance, and outcomes of sepsis. Crit Care. (2020) 24:278. doi: 10.1186/s13054-020-02989-1
89. Martin-Gallausiaux C, Marinelli L, Blottiere HM, Larraufie P, Lapaque N. SCFA: mechanisms and functional importance in the gut. Proc Nutr Soc. (2021) 80:37–49. doi: 10.1017/S0029665120006916
90. Gong S, Yan Z, Liu Z, Niu M, Fang H, Li N, et al. Intestinal microbiota mediates the susceptibility to polymicrobial sepsis-induced liver injury by granisetron generation in mice. HEPATOLOGY. (2019) 69:1751–67. doi: 10.1002/hep.30361
91. Wiersinga WJ, van der Poll T. Immunopathophysiology of human sepsis. EBioMedicine. (2022) 86:104363. doi: 10.1016/j.ebiom.2022.104363
92. Klingensmith NJ, Fay KT, Swift DA, Bazzano JM, Lyons JD, Chen C-W, et al. Junctional adhesion molecule-A deletion increases phagocytosis and improves survival in a murine model of sepsis. JCI Insight. (2022) 7:e156255. doi: 10.1172/jci.insight.156255
93. Jin C, Chen J, Gu J, Zhang W. Gut-lymph-lung pathway mediates sepsis-induced acute lung injury. Chin Med J. (2020) 133:2212–8. doi: 10.1097/CM9.0000000000000928
94. Lelubre C, Vincent J-L. Mechanisms and treatment of organ failure in sepsis. Nat Rev Nephrol. (2018) 14:417–27. doi: 10.1038/s41581-018-0005-7
95. Strnad P, Tacke F, Koch A, Trautwein C. Liver — guardian, modifier and target of sepsis. Nat Rev Gastroenterol Hepatol. (2017) 14:55–66. doi: 10.1038/nrgastro.2016.168
96. Zhang X, Liu H, Hashimoto K, Yuan S, Zhang J. The gut-liver axis in sepsis: interaction mechanisms and therapeutic potential. Crit Care. (2022) 26:213. doi: 10.1186/s13054-022-04090-1
97. Peng J, Li J, Huang J, Xu P, Huang H, Liu Y, et al. p300/CBP inhibitor A-485 alleviates acute liver injury by regulating macrophage activation and polarization. Theranostics. (2019) 9:8344–61. doi: 10.7150/thno.30707
98. Shi L, Jin L, Huang W. Bile acids, intestinal barrier dysfunction, and related diseases. Cells. (2023) 12:1888. doi: 10.3390/cells12141888
99. Dickson RP, Singer BH, Newstead MW, Falkowski NR, Erb-Downward JR, Standiford TJ, et al. Enrichment of the lung microbiome with gut bacteria in sepsis and the acute respiratory distress syndrome. Nat Microbiol. (2016) 1:1–9. doi: 10.1038/nmicrobiol.2016.113
100. Le Guern R, Grandjean T, Stabler S, Bauduin M, Gosset P, Kipnis É, et al. Gut colonisation with multidrug-resistant Klebsiella pneumoniae worsens Pseudomonas aeruginosa lung infection. Nat Commun. (2023) 14:78. doi: 10.1038/s41467-022-35767-4
101. Zhang Y-M, Zhang S-K, Cui N-Q. Intravenous infusion of mesenteric lymph from severe intraperitoneal infection rats causes lung injury in healthy rats. World J Gastroenterol. (2014) 20:4771–7. doi: 10.3748/wjg.v20.i16.4771
102. Wang P, Wang J, Li Y-H, Wang L, Shang H-C, Wang J-X. Phenotypical changes of hematopoietic stem and progenitor cells in sepsis patients: correlation with immune status? Front Pharmacol. (2020) 11:640203. doi: 10.3389/fphar.2020.640203
103. Davis FM, Schaller MA, Dendekker A, Joshi AD, Kimball AS, Evanoff H, et al. Sepsis induces prolonged epigenetic modifications in bone marrow and peripheral macrophages impairing inflammation and wound healing. Arterioscler Thromb Vasc Biol. (2019) 39:2353–66. doi: 10.1161/ATVBAHA.119.312754
104. Morales-Mantilla DE, Kain B, Le D, Flores AR, Paust S, King KY. Hematopoietic stem and progenitor cells improve survival from sepsis by boosting immunomodulatory cells. eLife. (2022) 11:e74561. doi: 10.7554/eLife.74561
105. Boomer JS, To K, Chang KC, Takasu O, Osborne DF, Walton AH, et al. Immunosuppression in patients who die of sepsis and multiple organ failure. JAMA. (2011) 306:2594–605. doi: 10.1001/jama.2011.1829
106. Ioannou M, Hoving D, Aramburu IV, Temkin MI, De Vasconcelos NM, Tsourouktsoglou T-D, et al. Microbe capture by splenic macrophages triggers sepsis via T cell-death-dependent neutrophil lifespan shortening. Nat Commun. (2022) 13:4658. doi: 10.1038/s41467-022-32320-1
107. Valet C, Magnen M, Qiu L, Cleary SJ, Wang KM, Ranucci S, et al. Sepsis promotes splenic production of a protective platelet pool with high CD40 ligand expression. J Clin Invest. (2022) 132:e153920. doi: 10.1172/JCI153920
108. Ren C, Yao R-Q, Zhang H, Feng Y-W, Yao Y-M. Sepsis-associated encephalopathy: a vicious cycle of immunosuppression. J Neuroinflamm. (2020) 17:14. doi: 10.1186/s12974-020-1701-3
109. Rana M, Fei-Bloom Y, Son M, La Bella A, Ochani M, Levine YA, et al. Constitutive vagus nerve activation modulates immune suppression in sepsis survivors. Front Immunol. (2018) 9:2032. doi: 10.3389/fimmu.2018.02032
110. Van den Berghe G, Téblick A, Langouche L, Gunst J. The hypothalamus-pituitary-adrenal axis in sepsis- and hyperinflammation-induced critical illness: Gaps in current knowledge and future translational research directions. eBioMedicine. (2022) 84:104284. doi: 10.1016/j.ebiom.2022.104284
111. Torres LK, Pickkers P, van der Poll T. Sepsis-induced immunosuppression. Annu Rev Physiol. (2022) 84:157–81. doi: 10.1146/annurev-physiol-061121-040214
112. Deng W, Zhu S, Zeng L, Liu J, Kang R, Yang M, et al. The circadian clock controls immune checkpoint pathway in sepsis. Cell Rep. (2018) 24:366–78. doi: 10.1016/j.celrep.2018.06.026
113. von Knethen A, Brüne B. Histone deacetylation inhibitors as therapy concept in sepsis. Int J Mol Sci. (2019) 20:346. doi: 10.3390/ijms20020346
114. Lorente-Sorolla C, Garcia-Gomez A, Català-Moll F, Toledano V, Ciudad L, Avendaño-Ortiz J, et al. Inflammatory cytokines and organ dysfunction associate with the aberrant DNA methylome of monocytes in sepsis. Genome Med. (2019) 11:66. doi: 10.1186/s13073-019-0674-2
115. Liu Y, Song R, Zhao L, Lu Z, Li Y, Zhan X, et al. m6A demethylase ALKBH5 is required for antibacterial innate defense by intrinsic motivation of neutrophil migration. Sig Transduct Target Ther. (2022) 7:1–15. doi: 10.1038/s41392-022-01020-z
116. Binnie A, Tsang JLY, Hu P, Carrasqueiro G, Castelo-Branco P, Dos Santos CC. Epigenetics of sepsis. Crit Care Med. (2020) 48:745–56. doi: 10.1097/CCM.0000000000004247
117. Virga F, Cappellesso F, Stijlemans B, Henze A-T, Trotta R, Van Audenaerde J, et al. Macrophage miR-210 induction and metabolic reprogramming in response to pathogen interaction boost life-threatening inflammation. Sci Adv. (2021) 7:eabf0466. doi: 10.1126/sciadv.abf0466
118. Wu D, Shi Y, Zhang H, Miao C. Epigenetic mechanisms of Immune remodeling in sepsis: targeting histone modification. Cell Death Dis. (2023) 14:112. doi: 10.1038/s41419-023-05656-9
119. Liu J, Zhou G, Wang X, Liu D. Metabolic reprogramming consequences of sepsis: adaptations and contradictions. Cell Mol Life Sci. (2022) 79:456. doi: 10.1007/s00018-022-04490-0
120. Cheng S-C, Scicluna BP, Arts RJW, Gresnigt MS, Lachmandas E, Giamarellos-Bourboulis EJ, et al. Broad defects in the energy metabolism of leukocytes underlie immunoparalysis in sepsis. Nat Immunol. (2016) 17:406–13. doi: 10.1038/ni.3398
121. Van den Bossche J, van der Windt GJW. Fatty acid oxidation in macrophages and T cells: time for reassessment? Cell Metab. (2018) 28:538–40. doi: 10.1016/j.cmet.2018.09.018
122. Li R, Li X, Zhao J, Meng F, Yao C, Bao E, et al. Mitochondrial STAT3 exacerbates LPS-induced sepsis by driving CPT1a-mediated fatty acid oxidation. Theranostics. (2022) 12:976–98. doi: 10.7150/thno.63751
123. An S, Yao Y, Hu H, Wu J, Li J, Li L, et al. PDHA1 hyperacetylation-mediated lactate overproduction promotes sepsis-induced acute kidney injury via Fis1 lactylation. Cell Death Dis. (2023) 14:457. doi: 10.1038/s41419-023-05952-4
124. Yang K, Fan M, Wang X, Xu J, Wang Y, Tu F, et al. Lactate promotes macrophage HMGB1 lactylation, acetylation, and exosomal release in polymicrobial sepsis. Cell Death Differ. (2022) 29:133–46. doi: 10.1038/s41418-021-00841-9
125. Hooftman A, Angiari S, Hester S, Corcoran SE, Runtsch MC, Ling C, et al. The immunomodulatory metabolite itaconate modifies NLRP3 and inhibits inflammasome activation. Cell Metab. (2020) 32:468–478.e7. doi: 10.1016/j.cmet.2020.07.016
126. Mills EL, Ryan DG, Prag HA, Dikovskaya D, Menon D, Zaslona Z, et al. Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nature. (2018) 556:113–7. doi: 10.1038/nature25986
127. Zhang S, Jiao Y, Li C, Liang X, Jia H, Nie Z, et al. Dimethyl itaconate alleviates the inflammatory responses of macrophages in sepsis. Inflammation. (2021) 44:549–57. doi: 10.1007/s10753-020-01352-4
128. Liao S-T, Han C, Xu D-Q, Fu X-W, Wang J-S, Kong L-Y. 4-Octyl itaconate inhibits aerobic glycolysis by targeting GAPDH to exert anti-inflammatory effects. Nat Commun. (2019) 10:5091. doi: 10.1038/s41467-019-13078-5
129. Zhang P, Wang Y, Yang W, Yin Y, Li C, Ma X, et al. 4-Octyl itaconate regulates immune balance by activating Nrf2 and negatively regulating PD-L1 in a mouse model of sepsis. Int J Biol Sci. (2022) 18:6189–209. doi: 10.7150/ijbs.74456
130. Wu R, Liu J, Wang N, Zeng L, Yu C, Chen F, et al. Aconitate decarboxylase 1 is a mediator of polymicrobial sepsis. Sci Transl Med. (2022) 14:eabo2028. doi: 10.1126/scitranslmed.abo2028
131. Chen F, Wu R, Liu J, Kang R, Li J, Tang D. The STING1-MYD88 complex drives ACOD1/IRG1 expression and function in lethal innate immunity. iScience. (2022) 25:104561. doi: 10.1016/j.isci.2022.104561
132. Shi X, Zhou H, Wei J, Mo W, Li Q, Lv X. The signaling pathways and therapeutic potential of itaconate to alleviate inflammation and oxidative stress in inflammatory diseases. Redox Biol. (2022) 58:102553. doi: 10.1016/j.redox.2022.102553
133. Giamarellos-Bourboulis EJ, Aschenbrenner AC, Bauer M, Bock C, Calandra T, Gat-Viks I, et al. The pathophysiology of sepsis and precision-medicine-based immunotherapy. Nat Immunol. (2024) 25:19–28. doi: 10.1038/s41590-023-01660-5
134. Goh KH, Wang L, Yeow AYK, Poh H, Li K, Yeow JJL, et al. Artificial intelligence in sepsis early prediction and diagnosis using unstructured data in healthcare. Nat Commun. (2021) 12:711. doi: 10.1038/s41467-021-20910-4
135. Liu R, Buck KH, Caterino JM, Zhang P. Estimating treatment effects for time-to-treatment antibiotic stewardship in sepsis. Nat Mach Intell. (2023) 5:421–31. doi: 10.1038/s42256-023-00638-0
136. Shashikumar SP, Wardi G, Malhotra A, Nemati S. Artificial intelligence sepsis prediction algorithm learns to say “I don’t know. NPJ Digit Med. (2021) 4:134. doi: 10.1038/s41746-021-00504-6
137. Boussina A, Shashikumar SP, Malhotra A, Owens RL, El-Kareh R, Longhurst CA, et al. Impact of a deep learning sepsis prediction model on quality of care and survival. NPJ Digit Med. (2024) 7:14. doi: 10.1038/s41746-023-00986-6
138. Bhavani SV, Carey KA, Gilbert ER, Afshar M, Verhoef PA, Churpek MM. Identifying novel sepsis subphenotypes using temperature trajectories. Am J Respir Crit Care Med. (2019) 200:327–35. doi: 10.1164/rccm.201806-1197OC
139. Papathanakos G, Andrianopoulos I, Xenikakis M, Papathanasiou A, Koulenti D, Blot S, et al. Clinical sepsis phenotypes in critically ill patients. Microorganisms. (2023) 11:2165. doi: 10.3390/microorganisms11092165
140. Zhang Z, Zhang G, Goyal H, Mo L, Hong Y. Identification of subclasses of sepsis that showed different clinical outcomes and responses to amount of fluid resuscitation: a latent profile analysis. Crit Care. (2018) 22:347. doi: 10.1186/s13054-018-2279-3
141. Seymour CW, Kennedy JN, Wang S, Chang C-CH, Elliott CF, Xu Z, et al. Derivation, validation, and potential treatment implications of novel clinical phenotypes for sepsis. JAMA. (2019) 321:2003–17. doi: 10.1001/jama.2019.5791
142. Davenport EE, Burnham KL, Radhakrishnan J, Humburg P, Hutton P, Mills TC, et al. Genomic landscape of the individual host response and outcomes in sepsis: a prospective cohort study. Lancet Respir Med. (2016) 4:259–71. doi: 10.1016/S2213-2600(16)00046-1
143. Xu Z, Mao C, Su C, Zhang H, Siempos I, Torres LK, et al. Sepsis subphenotyping based on organ dysfunction trajectory. Crit Care. (2022) 26:197. doi: 10.1186/s13054-022-04071-4
144. van Amstel RBE, Kennedy JN, Scicluna BP, Bos LDJ, Peters-Sengers H, Butler JM, et al. Uncovering heterogeneity in sepsis: a comparative analysis of subphenotypes. Intensive Care Med. (2023) 49:1360–9. doi: 10.1007/s00134-023-07239-w
145. Wu H, Cao T, Ji T, Luo Y, Huang J, Ma K. Predictive value of the neutrophil-to-lymphocyte ratio in the prognosis and risk of death for adult sepsis patients: a meta-analysis. Front Immunol. (2024) 15:1336456. doi: 10.3389/fimmu.2024.1336456
146. Wu J-F, Ma J, Chen J, Ou-Yang B, Chen M-Y, Li L-F, et al. Changes of monocyte human leukocyte antigen-DR expression as a reliable predictor of mortality in severe sepsis. Crit Care. (2011) 15:R220. doi: 10.1186/cc10457
147. Banerjee D, Monaghan S, Zhao R, Walsh T, Palmisciano A, Phillips GS, et al. Soluble programmed cell death protein-1 and programmed cell death ligand-1 in sepsis. Crit Care. (2018) 22:146. doi: 10.1186/s13054-018-2064-3
148. Kondo Y, Umemura Y, Hayashida K, Hara Y, Aihara M, Yamakawa K. Diagnostic value of procalcitonin and presepsin for sepsis in critically ill adult patients: a systematic review and meta-analysis. J Intensive Care. (2019) 7:22. doi: 10.1186/s40560-019-0374-4
149. Papp M, Kiss N, Baka M, Trásy D, Zubek L, Fehérvári P, et al. Procalcitonin-guided antibiotic therapy may shorten length of treatment and may improve survival-a systematic review and meta-analysis. Crit Care. (2023) 27:394. doi: 10.1186/s13054-023-04677-2
150. Iwase S, Nakada T-A, Hattori N, Takahashi W, Takahashi N, Aizimu T, et al. Interleukin-6 as a diagnostic marker for infection in critically ill patients: A systematic review and meta-analysis. Am J Emerg Med. (2019) 37:260–5. doi: 10.1016/j.ajem.2018.05.040
151. Yeh C-F, Wu C-C, Liu S-H, Chen K-F. Comparison of the accuracy of neutrophil CD64, procalcitonin, and C-reactive protein for sepsis identification: a systematic review and meta-analysis. Ann Intensive Care. (2019) 9:5. doi: 10.1186/s13613-018-0479-2
152. Gao R-Y, Jia H-M, Han Y-Z, Qian B-S, You P, Zhang X-K, et al. Calprotectin as a diagnostic marker for sepsis: A meta-analysis. Front Cell Infect Microbiol. (2022) 12:1045636. doi: 10.3389/fcimb.2022.1045636
Keywords: sepsis, immune cells, cell death mode, machine learning, biomarkers
Citation: Zhang X, Zhang Y, Yuan S and Zhang J (2024) The potential immunological mechanisms of sepsis. Front. Immunol. 15:1434688. doi: 10.3389/fimmu.2024.1434688
Received: 18 May 2024; Accepted: 25 June 2024;
Published: 08 July 2024.
Edited by:
Lynn Xiaoling Qiang, Northwell Health, United StatesReviewed by:
Wei Chong, The First Affiliated Hospital of China Medical University, ChinaCopyright © 2024 Zhang, Zhang, Yuan and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jiancheng Zhang, emhqY2hlbmcxQDEyNi5jb20=; Shiying Yuan, eXVhbl9zaGl5aW5nQDE2My5jb20=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.