 Bing Yu†
Bing Yu† Hao Yang
Hao Yang- Department of the Colorectal Anal Surgery, The Affiliated Taian City Centeral Hospital of Qingdao University, Tai’an, Shandong, China
Colorectal cancer is the third most common cancer and the second most lethal cancer in the world. The main cause of the disease is due to dietary and behavioral factors. The treatment of this complex disease is mainly based on traditional treatments, including surgery, radiotherapy, and chemotherapy. Due to its high prevalence and high morbidity, more effective treatments with fewer side effects are urgently needed. In recent years, immunotherapy has become a potential therapeutic alternative and one of the fastest-developing treatments. Immunotherapy inhibits tumor growth by activating or enhancing the immune system to recognize and attack cancer cells. This review presents the latest immunotherapies for immune checkpoint inhibitors, cell therapy, tumor-infiltrating lymphocytes, and oncolytic viruses. Some of these have shown promising results in clinical trials and are used in clinical treatment.
1 Introduction
Colorectal cancer (CRC) is the third most common malignancy in the world and the second leading cause of cancer deaths (approximately 1.9 million cases) and, thus, is one of the most serious public health problems in the world (1). Early diagnosis and treatment can achieve a satisfactory therapeutic effect. In contrast, survival is only 13.1% if diagnosed in the late metastatic stages. The incidence of CRC is increasing due to changes in the human lifestyle, such as reduced physical activity and increased consumption of high-fat foods (2). New cases of CRC will be more than 2.2 million worldwide, and 1.1 million deaths are estimated to occur by 2030, posing a serious threat to human health.
CRC is a genetically heterogeneous disease with different molecular pathways involved in tumor formation and metastasis (3). The development of CRC usually transforms from normal epithelial cells to uncontrolled proliferative epithelial cells that form polyps and carcinoma, respectively (4). Histologically, adenocarcinoma is the most common variant of CRC. Conventional treatment modalities include radiotherapy, chemotherapy, and surgical interventions (5). To prolong patient survival, the clinician usually adopts a variety of combined treatments depending on the location of the tumor and the mode of infiltration. For localized tumors, surgical treatment may be the best option. However, cancer cells are not completely removed, and residual cancer cells in local tissue, blood, and lymphatics usually lead to tumor recurrence. In recent years, immunotherapy has grown rapidly and relies on innate immunity and adaptive immunity, postoperative adjuvant chemotherapy, and immunotherapy to identify and remove residual cancer cells, with satisfactory therapeutic effects (6).
There are three main types of genetic instabilities in CRC, including chromosomal instability, CPG island methylation, and microsatellite instability (MSI) (7). The microsatellite refers to a class of short tandem repeat DNA sequences composed of 1~6 nucleotides in the genome, which are evenly distributed within the genome, rich in polymorphism information, and easy to detect (8). Normally, microsatellites are relatively conservative, also known as microsatellite stability (microsatellite stable, MSS). However, in disease states, such as tumors, the factors of double-stranded DNA replication can lead to the insertion or deletion of repeats, and replication errors form new microsatellite alleles (9). MSI is one of the most well-studied molecular markers in CRC. MSI indicates inactivation of the mismatch repair (MMR) gene and is usually associated with a CpG island methylation phenotype, while microsatellite stabilization (MSS) is associated with chromosomal instability (CIN). Approximately 15% of CRC patients presented had MSI, while the rest had MSS. MSI CRC also contains more point mutations than MSS CRC, but they have not been fully studied. Most mutations are for short nucleotide repeats, small insertions, and deletions (insertion deletions). Genes that confer cell growth advantage through loss-of-function mutations in microsatellites or MSI target genes have been extensively studied, and many have been published as candidate targets, thus being considered tumor suppressors. The heterogeneity of microsatellite status is widely used as a biomarker for the classification, treatment, and prognosis of genetic diseases and multiple tumors.
Immunotherapy is a therapeutic approach that activates and enhances the immune system to recognize and eliminate cancer cells to inhibit tumor growth (10). The immune system is composed of a variety of cells, tissues, and organs throughout the body to protect the body and remove pathogens, foreign bodies, and abnormal cells (11). Immunotherapy includes immune checkpoint inhibitors (ICIs), chimeric antigen receptor (CAR)-T cells, tumor-infiltrating lymphocytes (TILs), and oncolytic viral therapy (OVT) (12, 13). Studies have shown that melanoma, lung cancer, bladder cancer, and some types of blood cancer respond well to immunotherapy. This review introduces four immunotherapy methods for the treatment of CRC, explains their mechanisms, and discusses opportunities and challenges in the process of immunotherapy research. The aim is to let CRC patients understand the immunotherapy strategies, access the hope of cure, and expand the research ideas for the researchers.
2 Immunotherapy
2.1 Immune checkpoint inhibitors
Immune checkpoints are molecules expressed in T cells that inhibit T cells during the immune response and prevent the autoimmune response (14). Immune checkpoint inhibitors (ICIs) bind to receptors to disrupt immunosuppressive signals between antigen-presenting cells (APCs), tumor cells, and T cells, thus activating T cells, releasing killer factors, and killing tumor cells (15). CRC is highly heterogeneous at both the genetic and molecular levels, so treatment must be targeted to individual patients based on their unique molecular characteristics (16). Microsatellites are highly polymorphic repetitive DNA sequences in the human genome. The Cancer Genome Atlas (TCGA) project used comprehensive molecular analysis (chip-based sequencing technology) to classify CRC into two molecular pathological categories. These include microsatellite instability (MSI) and microsatellite stability (MSS) CRC (17). Approximately 13% of tumors are characterized by genomic instability of cancer cells due to lack of MMR. MSI CRC accounts for 15% of all sporadic CRC and can be divided into MSI-high (MSI-H) and MSI-low (MSI-L) based on the frequency of microsatellite marker instability (18). MSI-H typically has a sustained response to ICIs, including selective monoclonal antibodies against programmed cell death-1 (PD-1), programmed cell death ligand-1 (PD-L1), and cytotoxic T lymphocyte-associated antigen-4 (CTLA-4) (Table 1) (19). In contrast, approximately 85% of patients with CRC harboring microsatellite stable (MSS) tumors typically lack response to ICI. Currently, the most famous immune checkpoints of CRC immunotherapy include CTLA-4 and PD-1 (20, 21).
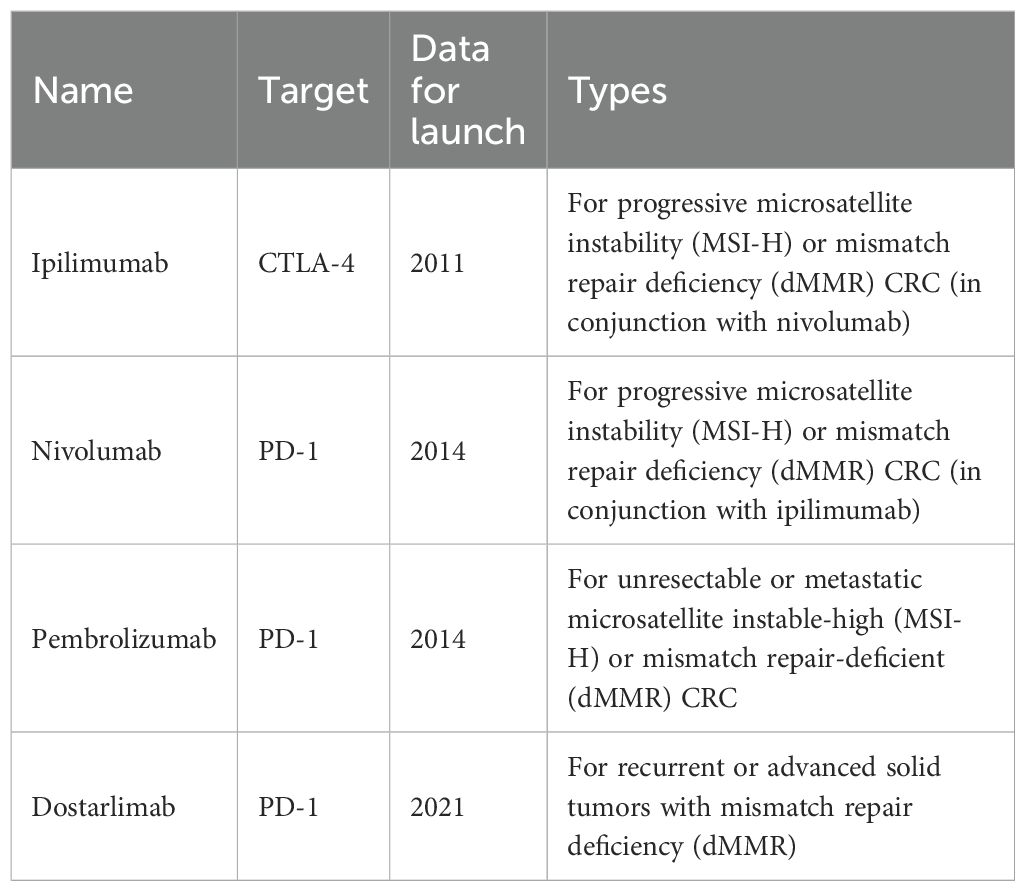
Table 1. FDA-approved monoclonal antibodies to treat colorectal cancer.
Another key target is CTLA-4, which is the first immune checkpoint to be discovered by James Allison in the 1990s. Studies showed that CTLA-4 can competitively bind to costimulator B7 with CD28, and the immunosuppressive effect after binding was significantly stronger than that of CD28 alone (Figure 1) (22). In 2011, ipilimumab, called Yervoy, which was a whole human monoclonal antibody against CTLA-4, was approved for the treatment of melanoma as the first CTLA-4 mab (23). Subsequently, researchers found that ipilimumab showed good therapeutic effect in metastatic colorectal cancer (mCRC) (24). However, in the course of clinical application, it was found that after CRC patients applied ipilimumab, there were severe immune-mediated adverse reactions, including fatigue, diarrhea, musculoskeletal pain, and rash. The therapeutic effect of ipilimumab attracts many researchers who are trying to reduce side effects by combining multiple drugs. A phase 2 clinical trial of 141 individuals showed that nivolumab combined with low-dose ipilimumab as first-line treatment for patients with high/mismatch repair deficiency (MSI-H/dMMR) (mCRC) demonstrated robust and durable clinical benefit with good tolerance (25). The results of the 4-year follow-up of the study also further confirmed the effectiveness of this combination treatment strategy (26).
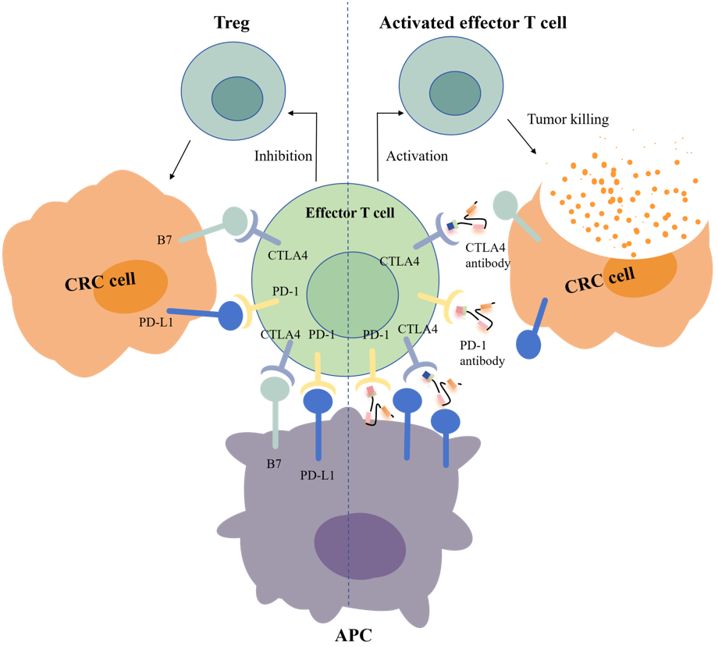
Figure 1. Mechanisms of action of PD-1 and CTLA-4 antibodies.
Programmed cell death protein 1 (PD-1) is the most important receptor for activating T-cell expression and mediating immunosuppression, while programmed cell death ligand 1 (CD274, PD-L1) is involved in programmed death, leading to apoptosis or inactivation of T cells (27). A phase III trial showed that PD-1 blockade was associated with significantly longer progression-free survival and fewer treatment-related adverse events in MSI-H or dMMR CRC than chemotherapy (28). In 2002, evidence of PD-1 pathway-mediated tumor immunity was first reported: after PD-1 binds to PD-L1, tumor cells used recognition of T-cell receptors to further suppress immunity and evade immune surveillance, and then significantly promoted tumorigenesis and invasion (Figure 1) (29). PD-1/PD-L1 inhibitors can prevent T-cell apoptosis and dysfunction, thus further enhancing T-cell activation. However, only a small proportion of CRC patients with defective mismatch repair/high level of microsatellite instability (dMMR/MSI-H) showed a response to anti-PD-1/PD-L1 treatment. In 2014, the FDA approved two monoclonal antibodies PD-1 (nivolumab and pembrolizumab) for the treatment of dMMR/MSI-H CRC, showing good and stable therapeutic effects (12). The KEYNOTE-177 study comparing the efficacy of pembrolizumab with standard chemotherapy in the first-line treatment of dMMR/MSI-H mCRC showed that median progression-free survival (PFS) was 16.5 months (95% CI 5.4–38.1) with pembrolizumab and 8.2 months (6.1–10.2) (HR 0.59, 95% CI 0.45–0.79) with chemotherapy (30). Thirty-three of 153 patients (22%) with pembrolizumab treatment and 95 of 143 patients (66%) with chemotherapy had grade 3 or worse treatment-related adverse events. Compared with chemotherapy, pembrolizumab monotherapy was associated with longer PFS, higher objective and complete responses, and fewer treatment-related adverse events in patients with MSI-H/dMMR mCRC. Pembrolizumab or nivolumab alone or in combination with ipilimumab is recommended as a first-line treatment option in patients with dMMR/MSI-H mCRC in the 2021 National Comprehensive Cancer Network (NCCN) guidelines (31).
ICI therapy is an effective therapeutic strategy after surgical resection, chemotherapy, radiotherapy, and targeted therapy, with great potential in the treatment of CRC. ICIs have fundamentally altered the prognosis of MSI CRC patients. In an open-label phase III study (KEYNOTE-177), 83% of pembrolizumab-treated patients with metastatic MSI-H-dMMR CRC had a sustained response at 24 months compared to 35% in the chemotherapy group. Despite these advances, MSI-H-dMMR CRC represents only a small subset of CRC, and ICI is largely ineffective in metastatic MSS-pMMR CRC, which represents the majority of patients (32). A phase II CheckMate 142 study reported an objective response rate of 69% with nivolumab combined with low-dose ipilimumab, and its effectiveness warrants first-line dual ICI therapy in a randomized study (25). At the 2024 American Society of Clinical Oncology (ASCO) Digestive Oncology Symposium, the research institute announced the study of CheckMate-8HW (NCT04008030). CheckMate-8HW is a randomized, open-label phase III clinical trial that evaluates the efficacy of nivolumab + ipilimumab versus nivolumab monotherapy or chemotherapy (mFOLFOX-6 or FOLFIRI) with or without bevacizumab/cetuximab in patients with mCRC with high microsatellite instability (MSI-H) or mismatch repair-deficient (dMMR) phenotypes. The results showed that nivolumab + ipilimumab reduced the risk of disease progression or death by 79% in patients with MSI-high or mismatch repair-deficient metastatic colorectal cancer. This study helps define the additional benefit of nivolumab plus ipilimumab versus nivolumab alone and helps clinicians determine the best treatment for their patients.
The FDA has approved multiple monoclonal antibodies for the treatment of CRC, including cetuximab, bevacizumab, panitumumab, ramocumab, ipilimumab, and pembrolizumab, which respond well to cancer. Supplementary Table 1 shows the clinical trials completed with ICI for CRC. These include several combination treatment strategies that have shown great promise in improving the overall clinical outcome of patients. Despite some progress, challenges remain in the widespread use of monoclonal antibody (mAb) in CRC therapy. First, the status of MMR/MSI, RAS, and BRAF before CRC treatment and the detection of the mutation status are important to guide clinical medication, develop personalized medication regimens, and benefit more patients. Second, as an emerging treatment method with great potential, its safety profile cannot be ignored. The question of how to reduce the side effects of treatment and reduce patients’ pain is one of the research emphases. Therefore, clinicians try to combine several different immunotherapy drugs to reduce the concentration and side effects of a single drug. Third, radiotherapy, chemotherapy, and ICI, combined with immunotherapy in CRC, are utilized to achieve the best effect and the least side effects. Issues such as patient selection, biomarker identification, and resistance mechanisms must be addressed to optimize the use of mAbs in clinical practice. In conclusion, ICI can change the therapeutic prospects of CRC, allowing more patients to benefit from treatment.
2.2 Adoptive cell therapy
Cellular immunotherapy for cancer is also known as adoptive cell therapy (ACT). It is a type of immunotherapy in which the cells of the body’s own immune system are genetically modified to express a CAR or a T-cell receptor (TCR) to eliminate cancer (33). ACT has played an important role in the treatment of many types of tumors. ACT offers several advantages over other cancer immunotherapies. Large numbers of antitumor T cells can be grown in vitro, and their antigen affinity can enhance autoimmunity (34). Following the gradual deepening of the TIL research, cells with antitumor activity were isolated from the tumors of patients with melanoma and showed good therapeutic effects. Despite the use of similar techniques, TILs grown from most CRC tissues do not appear to recognize tumor antigens. Further application of ACT led to the development of techniques to introduce antitumor TCRs into autologous lymphocytes for therapeutic use (35). CARs with antitumor specificity can be introduced into normal lymphocytes to enhance antitumor activity (36). CAR T-cell therapy uses gene transfer technology to reprogram a patient’s own T cells to stably express CAR, thereby combining antibody specificity with the potent cytotoxic and memory capabilities of T cells (37). In early-phase clinical trials, CD19-targeted CAR T cells produced complete sustained remissions in populations of patients with refractory B-cell malignancies and, more specifically, showed complete response rates of approximately 90% in patients with relapsed or refractory acute lymphoblastic leukemia.
2.2.1 CAR T cell
Chimeric antigen receptor T (CAR T)-cell therapy is an important breakthrough therapeutic tool for cancer research and is a personalized therapy that has achieved great success in the treatment of hematological malignancies. CAR T therapy isolates patient lymphocytes from peripheral blood and genetically modifies autologous T cells. Lentiviral vectors or retroviruses are used to modify T cells in vitro by genetic engineering technology to produce specific receptors for tumor antigens. Modified T cells can identify cancerous antigens independently of MHC and produce a specific antitumor immune response (Figure 2) (38). CAR T cells express synthetic receptors and redirect to the tumor surface antigen, release perforin, and granulin B to kill tumor cells directly. Through the release of cytokines, endogenous immune cells kill tumor cells and achieve the purpose of treating a tumor. CAR T cells can form immune memory T cells to obtain a specific long-term antitumor activity (39). In recent years, CAR T technology has developed rapidly, and innovative manufacturing processes and transformation strategies have gradually increased the stability and effectiveness of CAR T and have reduced costs and side effects. The CAR comprises a target-binding extracellular region with antigen specificity usually based on antibody fragments from a single-chain variable region (scFv), hinge regions, transmembrane regions, and an intracellular domain that mediates T-cell activation, mainly via the CD3 ζ signaling chain (40). Since the first generation, CAR T-cell production has been optimized to the fifth generation, six CAR T-cell products have been approved worldwide, and countless products are in preclinical and clinical trials. The fifth generation of CAR T tries to break through the limitations of individuals and is used for large-scale production and treatment (Figure 3) (41, 42).
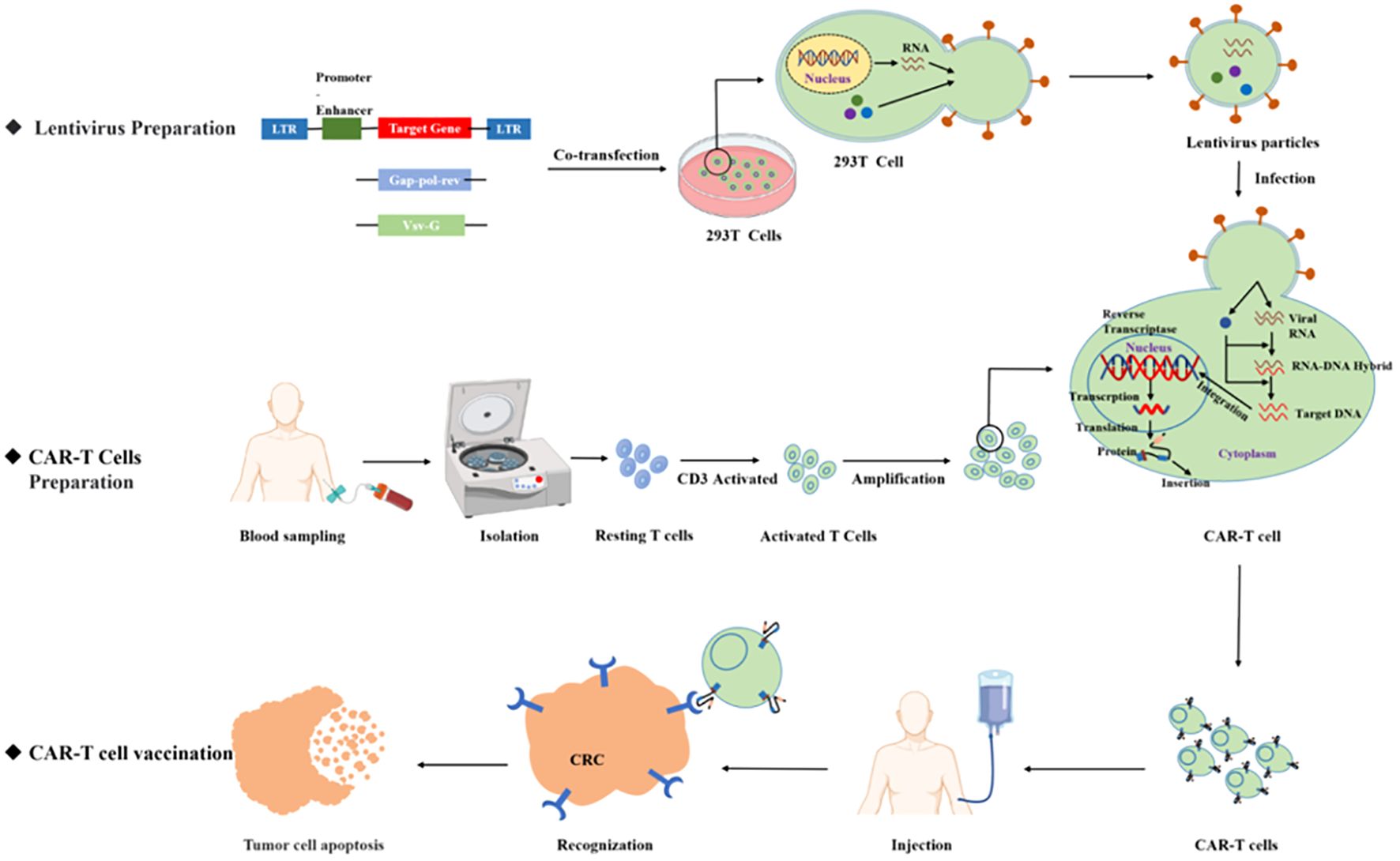
Figure 2. CAR T product preparation process.
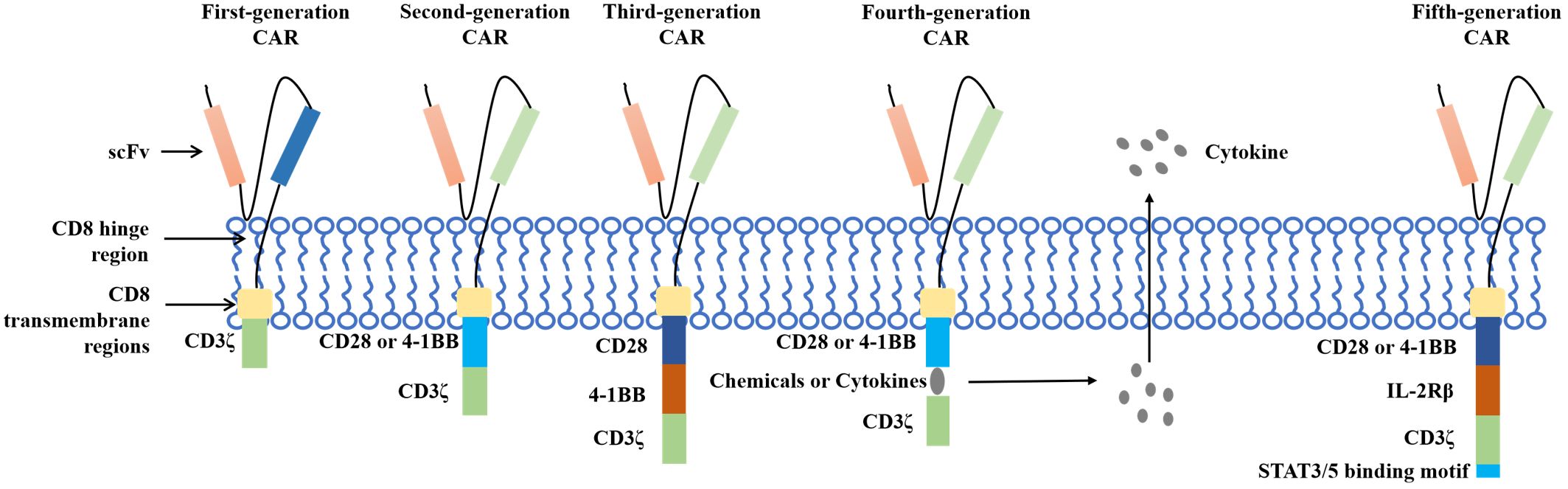
Figure 3. The development of CAR T.
CAR T-cell immunotherapy is a very promising anticancer strategy that has attracted many researchers to explore modification strategies for CRC. Several preclinical and clinical studies are still in the primary stage (phase I/II clinical trials) to evaluate the efficacy, safety, dose levels, and maximum tolerated dose of CAR T cells against various overexpressed molecular targets in CRC (Supplementary Table 2). ClinicalTrials.gov showed that the targets used most frequently in CAR T-cell therapy in CRC studies were CEA and NKG2DL, followed by EGFR and HER-2. In 2017, a phase I increase in the doses of CAR T therapy targeting CEA mCRC was conducted. The results indicated that two patients had stable disease for more than 30 weeks and two patients had significant tumor reduction (43). Whether these findings reveal a good treatment effect and can be applicable to all patients remains to be verified by further clinical trials.
With the development of cell technology, researchers no longer focused on T cells and began to test other CAR immune cells and even the combination of multiple immune methods. In 2019, researchers fused the extracellular domain of cell receptor NKG2D of natural killer (NK) cells with DAP12 to enhance NK cell tumor response. The preclinical trial showed good therapeutic effects in CRC tumor-bearing mice, and a preliminary clinical trial (NCT03415100) was conducted (44). Preliminary verification showed that NKG2D CAR-NK cells can identify tumor cells and exhibit antitumor effector functions in patients with mCRC. In 2020, researchers constructed CYAD-101, using a non-gene-edited peptide-based technology (TIM) in combination with NKG2D-based CAR to control graft-versus-host disease (GvHD) (45). Preclinical findings showed that CYAD-101 still maintained the CAR-directed antitumor activity in the absence of induced GvHD. Subsequently, a phase I alloSHRINK clinical study (NCT03692429) enrolled 15 patients with refractory mCRC who had previously failed at least first-line treatment with oxaliplatin, showing 2 patients in partial remission and 9 with stable results. This study demonstrated the attempt of allogeneic CAR T-cell therapies to overcome the limitations of autologous CAR T and was presented at the American Society of Clinical Oncology Gastrointestinal Cancer Symposium 2021 (ASCO-GI). In 2023, researchers secreted bispecific PD-1-TREM2 scFv antibodies into the tumor microenvironment (TME) which could simultaneously target PD-1, TAM, and MDSC. In the CRC mouse model, CAR T cells for BsAb PD-1-TREM2 scFv secretion were shown to exhibit a stronger antitumor potential (46). This study innovatively combined CAR T cells and BsAb into a single immunotherapy platform with greater antitumor efficacy in tumor-bearing mice, prompting new research for the study of CAR T. Despite the impressive success of CAR T-cell therapy in hematological malignancies, particularly CD19-positive B-cell malignancies, the development of CAR T-cell therapy in solid tumors has stalled (47). Ongoing efforts are advancing basic research in this field, with several studies progressing to clinical trials and multiple combination strategies proposed to further improve efficacy and safety (Supplementary Table 2). Each strategy has a mechanism of action and various advantages or limitations. A variety of targets are available for CAR treatment of CRC, and many promising therapeutic strategies have been proposed and shown to be successful in preclinical models. The important role of CAR therapy in solid tumors has been demonstrated in various studies (48). In addition to engineering with T cells, NK cells have been considered an alternative vehicle for CAR constructs and are thought to be less susceptible to GvHD. CAR-NK cells can expand the therapeutic scope of solid tumors and extend to allogeneic CAR therapy (49). CAR-related toxicity often presents acutely, and control mechanisms should ideally allow the rapid control of CAR T-cell activity (50).
CAR T cells are one of the most studied and promising methods among ACTs, and although clinical trials are still in the early stages, the results have shown promising therapeutic effects. Some barriers and limitations must be addressed during the study. First, the TME is an important limitation of CAR T-cell therapy. The TME presents local tissue hypoxia, nutrient metabolism disorders, and more acid products from hypermetabolism. These factors affect T-cell survival, proliferation, and activation and also limit the inhibitory effect of CAR T cells on tumors. Whether personalized modification of CAR T cells, such as integration with antitumor cytokines, inoculation mode, and dose control, can reduce the impact of the TME still needs further exploration. Second, CAR T-cell therapy can cause many toxic effects. One of the most common is the cytokine release syndrome (CRS), which is the CAR T-cell infusion cytokine secretion reaction and causes other systemic toxicity. Patients may have respiratory circulation disorders, liver dysfunction, gastrointestinal reaction, and neurotoxic reaction, even in a short period of time, which can be life-threatening. Researchers are also constantly trying to minimize side effects and benefit more CRC patients. The selection of CAR T cells should also be adapted to the type of target tumor, as tissue-specific vascularization can prevent adequate biodistribution, concentration, and persistence of CAR T cells in affected organs (51). Positive results from clinical trials are now expected to provide hope for this emerging cell-based therapy.
2.2.2 Tumor-infiltrating lymphocytes
TILs are an ACT which extract immune cells from tumor tissue, enhance their antitumor vitality in vitro, and are reinjected into the TME to enhance the immune activity to inhibit tumor growth (52). TILs generally represent a heterogeneous population of αβ T cells present in the TME, consisting of CD4+ and CD8+ subpopulations. These cells differentiate into killer cells that release perforin and express the apoptosis inducer FASL after amplification. Perforin disrupts the cell membrane and helps the granzyme enter the cell. Then, caspase precursors will undergo cleavage and induce apoptosis of the tumor cells (53). Since the distributions of TILs are different in different types of TME, it is particularly important to choose which kind of TILs. Researchers are trying to explore the relationship between TILs and rehabilitation to screen for TILs for cancer treatment. However, the production and responsiveness of TIL products to solid tumors vary due to individual differences (54, 55). Studies have suggested that TILs are an important prognostic factor in infiltrating or peripheral CRC (56, 57). Studies have shown that CD8+ T-cell infiltration was consistently higher than infiltration of CD4+ T cells in the CRC TME and was associated with a better prognosis in CRC. CD8+ TILs mediate the tumor rejection response by recognizing the tumor-associated antigens (TAAs) and directly killing transformed cells (58). Effector CD8 T cells in the TME produce IL-2, IL-12, and IFN-γ to enhance the cytotoxic potential of CD8 TILs to target the tumor cells (59).
Fewer TILs are present in CRC than in other kinds of cancers; therefore, collecting and increasing the amount of treatment of TILs is one of the challenges of TILs for the treatment of CRC (60). Currently, researchers are using the traditional collection and in-vitro expansion methods for the rapid expansion protocol (REP) of the T cell. Under anesthesia, tumors are excised from the patient and are cut into small pieces or digested enzymatically to obtain a single-cell suspension. Tumor fragments are then cultured alone in the presence of high-dose IL-2 (6,000 IU/mL) for 3–5 weeks prior to REP. The selected pure lymphocyte cultures will then be co-cultured with lymphocytes and tumor cells in the presence of irradiated feeder lymphocytes (an antibody targeting ϵ subunits within human CD3) and IL-2 to rapidly expand for 5 to 6 weeks. The patient will then be transfused with cells (Figure 4).
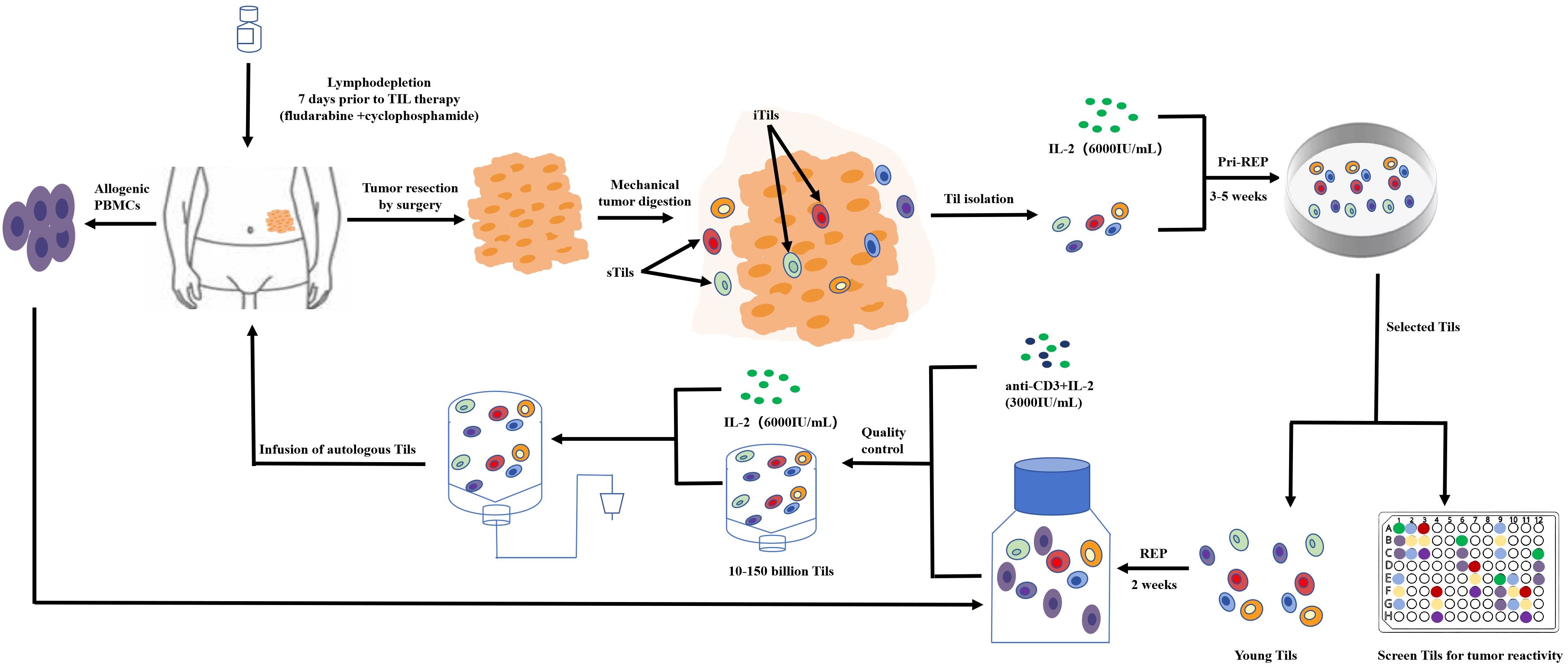
Figure 4. TIL product preparation process.
Researchers have successfully designed an in-vitro amplification model for TILs (61). Tumor tissues from 12 patients undergoing surgery for primary CRC surgery were cut for pathological examination and transferred to a one-time perfusion bioreactor with a starting medium containing IL-2 and IL-12. The expanded TILs consisted mainly of (73%) the ACT-relevant CD3+/CD8+ effector memory phenotype (CD45RO+/CCR7), confirming that the amplified TILs showed high functional potential by measuring non-specific stimulation (interferon-γ, tumor necrosis factor-α cytokine assay), taking an important step in the immunotherapy of TILs.
How to expand effector T cells more efficiently and make them exert their antitumor effect is also one of the difficulties to overcome in TIL therapy. Preclinical studies have shown that CD8-dominated TIL produced a stronger antitumor capacity when using anti-4-1BB and CD3 antibody agonists in early isolated TIL culture (62). This single-center TIL was treated for a phase II trial (NCT03610490) for patients with CRC, PDAC, and OVCA refractory to standard therapy. The results showed that the DCR is 62.5% but the ORR is 0% and the median PFS and OS were 2.53 months and 18.86 months, respectively. The single-arm study was unable to conclude the efficacy of TIL compared to standard second- or third-line treatment options in different cohorts. However, the results of this experiment showed the effect of TIL therapy in inhibiting solid tumors. More studies are needed to identify host factors associated with resistance to TIL therapy.
TILs play an important role in identifying and killing target tumor cells and have achieved good results in recent years. However, there are still many challenges in the development process, such as treatment safety, a long production cycle, high production costs, optimization of manufacturing processes, and the use of innovative genetic modification techniques to create more effective TIL cell therapies. Several TIL/CAR T-cell trials have been associated with safety concerns, particularly the development of adverse effects on and off the target, including CAR T-cell-associated encephalopathy syndrome (CRES), extratumoral effects, and acute respiratory distress syndrome due to targeted humoral recognition and killing (63). Furthermore, CRS is the most common side effect of CAR T therapy (64). Although timely pharmacologic intervention is effective in managing most adverse events, ACT can persist for a long time, accompanied by any adverse effects (65). Conversely, tumor-restricted expression of neoantigens driven by somatic mutations ensures the therapeutic generation of cellular therapeutic reactivity against these antigens, which is independent of normal tissue toxicity and is considered an ideal and safe solution for ACT. However, with continuous improvement and advancement of technology, it is likely that effective TILs will be designed in the future to bring hope to CRC patients.
2.3 Oncolytic virotherapy
OVT chooses a small virus as the viral vector and chimeric antitumor genes and immune factors to increase the targeting and immune activity. Oncolytic viruses (OVs) can infect the tumor cells by intratumoral administration, intravenous administration, and cell carrier delivery to target proliferation in the tumor cells. In the process of value-added process, the immune factors or tumor-related antibodies are released to directly induce oncolysis, activate the immune response, and drive immune cells toward the TME to inhibit tumor growth (Figure 5) (66, 67). The release of antiviral cytokines (especially interferons) is used to initiate antiviral responses, which promote the maturation of APCs such as dendritic cells (DC) and stimulate CD8 T cells and NK cells. The lysis of the infected tumor cells will release the viral progeny, DAMP (including host cell proteins), PAMP (viral particles), and TAAs into the TME (68, 69). The virus offspring will infect additional nearby or distal tumor cells. DAMPs and PAMPs stimulate the immune system by activating receptors. TAAs and neoantigens are absorbed by APCs to activate antigen and virus-specific CD8 T-cell responses and, finally, to create an immunostimulatory environment (70). If the OVs are chimeric with VEGF antibodies, VEGF antibodies will be released into the TME, and the VEGF function will be inhibited. Therefore, tumor-nourishing vessels will be inhibited and local blood perfusion will be reduced, resulting in tumor cells lacking oxygen and nutrients needed for growth.
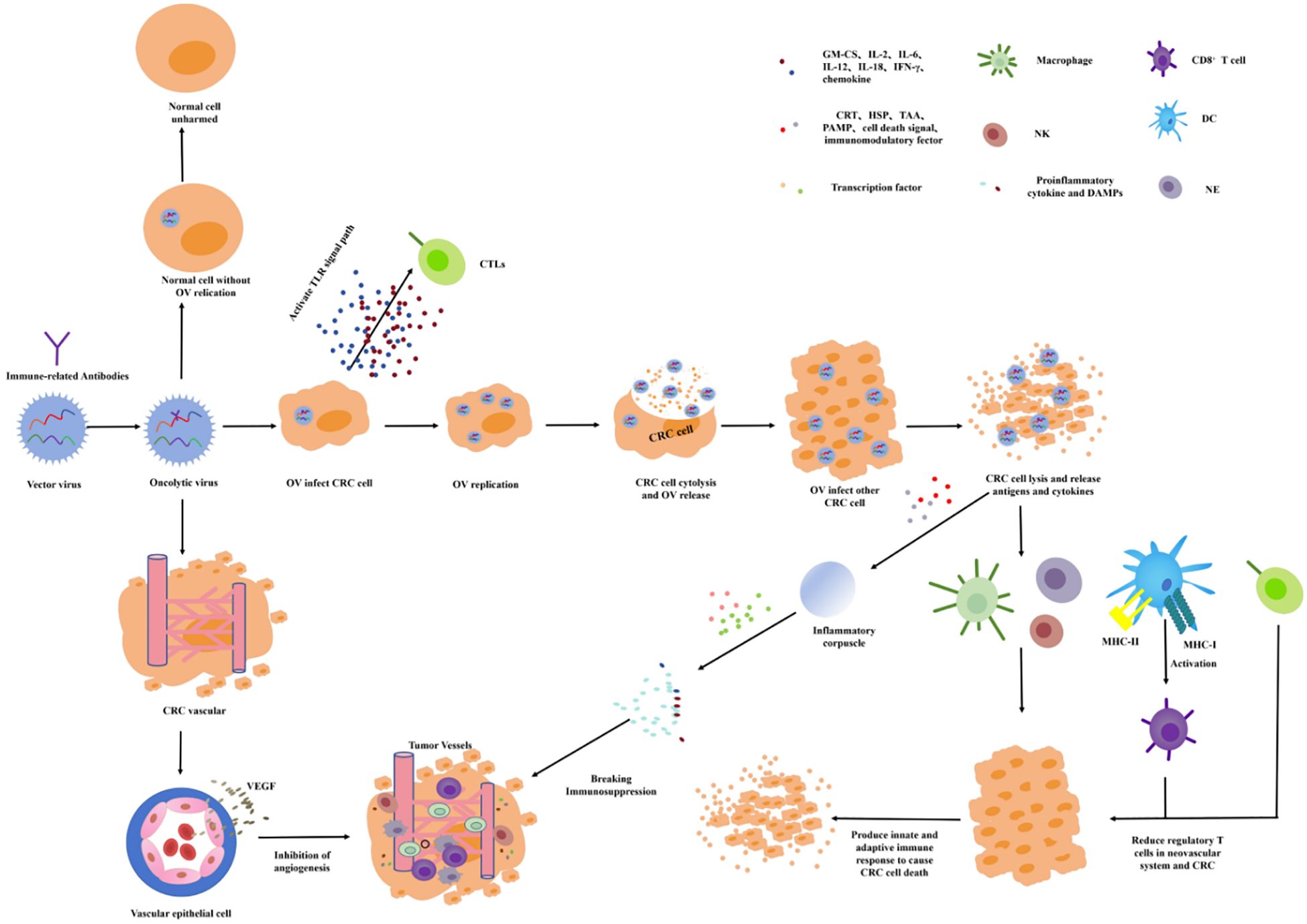
Figure 5. Mechanisms of oncolytic virus action.
OVT is one of the most promising immunotherapies for CRC. In recent years, OVT has achieved satisfactory results at both the cellular and organismal levels, and the research direction is increasingly turning to clinical trials. Currently, the most commonly used viruses for OVT research are the poxvirus, reovirus, herpes simplex virus (HSV), and adenovirus. Some tumor suppressor genes or antibodies that enhance immune response are transformed into viral DNA to play an oncolytic role in the amplification or metabolism of OVs (71). In the study of OVs for the treatment of CRC, researchers have tried to use OV technology to target the human 5T4 gene, CEA, PD-1, and CTLA-4 for the treatment of CRC. Modification of OVs has achieved a good therapeutic effect in preclinical research and is expected to provide a new treatment strategy for the clinical treatment of CRC (72).
The human 5T4 gene is known as the trophoblast glycoprotein. It is located at 6q14.1 and expresses a heavy N-glycosylated protein of 72 kDa. 5T4 is highly expressed in human trophoblast cells and most tumors but is rarely expressed in normal tissues (73). Researchers modified vaccinia Ankara-5T4 to treat inoperable mCRC with chemotherapy. This randomized phase I and phase II clinical trial in patients evaluated the effectiveness of cyclophosphamide in increasing the therapeutic potential of modified vaccinia Ankara-5T4 immunotherapy. The enrolled patients showed better tumor control effects when given cyclophosphamide or MVA-5T4 (74). Although cyclophosphamide failed to enhance the immunogenicity of MVA-5T4, its survival benefit and minimal adverse effects have been demonstrated, thus requiring further investigation.
The CEA subgroup is expressed mainly on the cell membrane. For more than 50 years, CEA has been identified as an important marker of CRC and other malignancies. The CEA gene promoter constructs can strongly inhibit CEA-producing adenocarcinoma cells (75). The insertion of the ST13 tumor suppressor gene and the CEA promoter E1A (Δ24) into an oncolytic adenovirus vector inhibits the growth of the SW620 CRC xenograft in nude mice and prolongs the survival time of mice (76). A team of researchers has already filed a patent protecting the invention (201110319434.4). Researchers constructed a recombinant oncolytic herpes simplex virus type 1 (HSV-1), called VG2025, which uses the dual regulation of transcription and translation (TTDR) of key viral genes to improve viral safety and promote tumor-specific viral replication without reducing virulence (77). VG2025 can efficiently replicate virally in CEA-positive cancer cells, promoting oncolysis and the release of tumor antigens while limiting viral replication in healthy tissues. The CEA promoter in VG2025 can be replaced by other tumor-specific promoters as part of a broader platform to facilitate biomarker-based precision OVT.
OVT has produced promising results for CRC in clinical and preclinical studies. With advances in molecular techniques, the safety and specificity of OVs have been achieved. However, many challenges still hampered the optimal antitumor activity of OVs. How are oncolytic viruses engineered more effectively? The host’s antiviral immunity needs to be taken into consideration, and how to successfully deliver the OVs remains the biggest challenge (78). Currently, the combination of OVs and ICIs has shown promise in multiple clinical trials. This strategy is expected to be a promising therapeutic option for CRC.
3 Discussion
Immunotherapy is a therapeutic approach that utilizes various cytokines, antibody drugs, OVs, and immune cells to activate or enhance the immune system to inhibit tumor growth (79). Since the FDA approved the first mAb, more than 100 mAb products have been approved with significant therapeutic efficacy (80). The ultimate goal of different tumor immunotherapies is to improve the precision of targeted therapies and reduce adverse effects while effectively eliminating tumor cells. This review describes the development of ICIs, cell therapy, TILs, and OV therapy in CRC. The mechanisms of several immunotherapies are described and ongoing immunotherapy research is introduced for patients with CRC to provide hope for treatment and to provide new research ideas for researchers.
High infiltration of specific subsets of immune cells in the immune microenvironment of type I CRC has been associated with improved survival and reduced risk of recurrence in patients with stage I/II CRC (81). However, patients with MSS CRC have been reported to have a high mortality risk in multiple cohorts. MSI/MSS subtyping has changed the diagnosis and treatment strategy of CRC. Indeed, patients with MSI-H are not sensitive to fluorouracil treatment; therefore, the combination chemotherapy regimen (FOLFOX) [(fluorouracil, 5-FU), oxaliplatin (oxaliplatin), and folic acid (folinic acid)] is less effective (up to 73.6% insensitive) than in patients with MSS CRC (only 26.6% insensitive) (82). Therefore, it appears that clarifying the microsatellite status of patients before CRC treatment is extremely valuable to guide treatment stratification.
The practical landscape for immunotherapeutic agents in solid tumors of MSI-H is evolving rapidly. Different strategies are under investigation, but the vast majority of them include combinations of anti-PD(L)-1 agents with other immunomodulators. Anti-CTLA-4 plus anti-PD(L)-1 is the only combination to date that has shown better survival in patients with MSI-H cancers (25). The optimal timing of immunotherapy intervention, the duration of immune agents, the appropriate dose of immune drugs, and the combination strategy of preoperative immunotherapy and the cytotoxic profiles of these drugs are unknown (83). A short-course radiotherapy treatment paradigm before CRC surgery followed by 4–5 cycles of anti-PD1 therapy combined with fluorouracil or its derivative chemotherapy appears to be more ideal (84). Screening for patients who may benefit from immunotherapy is still based on MSI/MMR status: while some patients with MSI-H/dMMR CRC do not benefit from ICI treatment, other patients with MSS/pMMR can achieve a good clinical response from immunotherapy (85, 86). With the breakthrough of anti-PD-1 therapies, such as pembrolizumab and nivolumab, targeting the MSI-H:dMMR patient population, we have begun to see the potential of immunotherapy (87). However, in MSI-H tumors, there is also an urgent need to identify new biomarkers to predict the benefit of immunotherapy and possibly the benefit of specific immunotherapy agents. With the advent of cutting-edge technologies such as scRNA-seq, high-parameter flow cytometry, and spatial transcriptomics, our understanding of antitumor immune responses has been revolutionized. As these techniques become more advanced and widely adopted in the future, they have the potential to provide us with important insights into the behavior of immune cells in the TME. This knowledge can be used as a basis for designing more effective immunotherapies.
Tumor immunotherapy has become the main development direction and the trend of development of tumor therapy and is expected to become the final method of tumor treatment. However, the efficacy of these immunotherapies is limited by multiple mechanisms, including the emergence of compensatory inhibitory mechanisms that negatively regulate the antitumor immune response, leading to acquired resistance. The search for new tumor targets, the study of signaling mechanisms, and the development of new technologies are constantly increasing. Due to their novel, complex, and technical nature, these therapies can pose previously untapped risks to public health and individual patients. ICI therapy, TIL cell therapy, CAR T therapy, and OV vaccines were considered to have low to moderate risk. Potential risk factors include bioactive substances used in manufacturing, such as antibodies, cytokines, sera, growth factors, and antibiotics, as well as risks to the stability and viability of the product during storage, freezing, thawing, and cold chain transport. In addition, the product itself can carry inherent risks, such as incomplete removal of tumor cells or other unwanted cells, and potential complications or reduced product activity associated with homing, transplantation, migration, and proliferation. Researchers have tried to combine several drugs to reduce the concentration of each single drug and to reduce adverse drug reactions and have also achieved good experimental results. Combination therapy has shown superiority in some studies. In addition to different combinations of immunotherapy, immunotherapy combined with chemotherapy, radiotherapy, or other drugs has been explored, with a focus on improving efficacy, reversing resistance, and reducing adverse effects. In conclusion, immunotherapy is currently the most promising treatment modality and is expected to be a new therapeutic strategy for patients with CRC.
Author contributions
BY: Conceptualization, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Writing – original draft. JK: Investigation, Methodology, Project administration, Resources, Supervision, Visualization, Writing – original draft, Writing – review & editing. HL: Conceptualization, Data curation, Formal analysis, Methodology, Project administration, Validation, Writing – original draft. ZL: Data curation, Formal analysis, Methodology, Project administration, Supervision, Validation, Writing – original draft. HY: Conceptualization, Data curation, Investigation, Methodology, Software, Writing – original draft. MZ: Conceptualization, Funding acquisition, Investigation, Resources, Supervision, Validation, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2024.1433315/full#supplementary-material
References
1. Dekker E, Tanis PJ, Vleugels JLA, Kasi PM, Wallace MB. Colorectal cancer. Lancet. (2019) 394:1467–80. doi: 10.1016/S0140-6736(19)32319-0
2. Burnett-Hartman AN, Murphy CC, Lee JK. Novel, emerging risk factors for colorectal cancer remain understudied. Gastroenterology. (2022) 163:574–6. doi: 10.1053/j.gastro.2022.06.085
3. Kastrinos F, Samadder NJ, Burt RW. Use of family history and genetic testing to determine risk of colorectal cancer. Gastroenterology. (2020) 158:389–403. doi: 10.1053/j.gastro.2019.11.029
4. Khaliq AM, Erdogan C, Kurt Z, Turgut SS, Grunvald MW, Rand T, et al. Refining colorectal cancer classification and clinical stratification through a single-cell atlas. Genome Biol. (2022) 23:113. doi: 10.1186/s13059-022-02677-z
5. Ghadimi M, Rodel C, Hofheinz R, Flebbe H, Grade M. Multimodal treatment of rectal cancer. Dtsch Arztebl Int. (2022) 119:570–80. doi: 10.3238/arztebl.m2022.0254
6. Fan A, Wang B, Wang X, Nie Y, Fan D, Zhao X, et al. Immunotherapy in colorectal cancer: current achievements and future perspective. Int J Biol Sci. (2021) 17:3837–49. doi: 10.7150/ijbs.64077
7. Lin A, Zhang J, Luo P. Crosstalk between the MSI status and tumor microenvironment in colorectal cancer. Front Immunol. (2020) 11:2039. doi: 10.3389/fimmu.2020.02039
8. Nojadeh JN, Behrouz Sharif S, Sakhinia E. Microsatellite instability in colorectal cancer. EXCLI J. (2018) 17:159–68. doi: 10.17179/excli2017-948.
9. Kok M, Chalabi M, Haanen J. How I treat MSI cancers with advanced disease. ESMO Open. (2019) 4:e000511. doi: 10.1136/esmoopen-2019-000511
10. Weng J, Li S, Zhu Z, Liu Q, Zhang R, Yang Y, et al. Exploring immunotherapy in colorectal cancer. J Hematol Oncol. (2022) 15:95. doi: 10.1186/s13045-022-01294-4
11. Franke AJ, Skelton WP, Starr JS, Parekh H, Lee JJ, Overman MJ, et al. Immunotherapy for colorectal cancer: A review of current and novel therapeutic approaches. J Natl Cancer Inst. (2019) 111:1131–41. doi: 10.1093/jnci/djz093
12. Ganesh K, Stadler ZK, Cercek A, Mendelsohn RB, Shia J, Segal NH, et al. Immunotherapy in colorectal cancer: rationale, challenges and potential. Nat Rev Gastroenterol Hepatol. (2019) 16:361–75. doi: 10.1038/s41575-019-0126-x
13. Rastin F, Javid H, Oryani MA, Rezagholinejad N, Afshari AR, Karimi-Shahri M. Immunotherapy for colorectal cancer: Rational strategies and novel therapeutic progress. Int Immunopharmacol. (2024) 126:111055. doi: 10.1016/j.intimp.2023.111055
14. Boukouris AE, Theochari M, Stefanou D, Papalambros A, Felekouras E, Gogas H, et al. Latest evidence on immune checkpoint inhibitors in metastatic colorectal cancer: A 2022 update. Crit Rev Oncol Hematol. (2022) 173:103663. doi: 10.1016/j.critrevonc.2022.103663
15. Xie YH, Chen YX, Fang JY. Comprehensive review of targeted therapy for colorectal cancer. Signal Transduct Target Ther. (2020) 5:22. doi: 10.1038/s41392-020-0116-z
16. Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA Jr., Kinzler KW. Cancer genome landscapes. Science. (2013) 339:1546–58. doi: 10.1126/science.1235122
17. Lizardo DY, Kuang C, Hao S, Yu J, Huang Y, Zhang L. Immunotherapy efficacy on mismatch repair-deficient colorectal cancer: From bench to bedside. Biochim Biophys Acta Rev Cancer. (2020) 1874:188447. doi: 10.1016/j.bbcan.2020.188447
18. Ros J, Balconi F, Baraibar I, Saoudi Gonzalez N, Salva F, Tabernero J, et al. Advances in immune checkpoint inhibitor combination strategies for microsatellite stable colorectal cancer. Front Oncol. (2023) 13:1112276. doi: 10.3389/fonc.2023.1112276
19. Chen JT, Zhou YW, Han TR, Wei JL, Qiu M. Perioperative immune checkpoint inhibition for colorectal cancer: recent advances and future directions. Front Immunol. (2023) 14:1269341. doi: 10.3389/fimmu.2023.1269341
20. Morse MA, Hochster H, Benson A. Perspectives on treatment of metastatic colorectal cancer with immune checkpoint inhibitor therapy. Oncologist. (2020) 25:33–45. doi: 10.1634/theoncologist.2019-0176
21. Liu Y, Wang Z, Hao H, Wang Y, Hua L. Insight into immune checkpoint inhibitor therapy for colorectal cancer from the perspective of circadian clocks. Immunology. (2023) 170:13–27. doi: 10.1111/imm.13647
22. Makaremi S, Asadzadeh Z, Hemmat N, Baghbanzadeh A, Sgambato A, Ghorbaninezhad F, et al. Immune checkpoint inhibitors in colorectal cancer: challenges and future prospects. Biomedicines. (2021) 9:1075. doi: 10.3390/biomedicines9091075
23. Korman AJ, Garrett-Thomson SC, Lonberg N. The foundations of immune checkpoint blockade and the ipilimumab approval decennial. Nat Rev Drug Discovery. (2022) 21:509–28. doi: 10.1038/s41573-021-00345-8
24. Kanani A, Veen T, Soreide K. Neoadjuvant immunotherapy in primary and metastatic colorectal cancer. Br J Surg. (2021) 108:1417–25. doi: 10.1093/bjs/znab342
25. Lenz HJ, Van Cutsem E, Luisa Limon M, Wong KYM, Hendlisz A, Aglietta M, et al. First-Line nivolumab plus low-Dose ipilimumab for microsatellite instability-High/Mismatch repair-Deficient metastatic colorectal cancer: the phase II checkMate 142 study. J Clin Oncol. (2022) 40:161–70. doi: 10.1200/JCO.21.01015
26. Andre T, Lonardi S, Wong KYM, Lenz HJ, Gelsomino F, Aglietta M, et al. Nivolumab plus low-dose ipilimumab in previously treated patients with microsatellite instability-high/mismatch repair-deficient metastatic colorectal cancer: 4-year follow-up from CheckMate 142. Ann Oncol. (2022) 33:1052–60. doi: 10.1016/j.annonc.2022.06.008
27. Lin KX, Istl AC, Quan D, Skaro A, Tang E, Zheng X. PD-1 and PD-L1 inhibitors in cold colorectal cancer: challenges and strategies. Cancer Immunol Immunother. (2023) 72:3875–93. doi: 10.1007/s00262-023-03520-5
28. Andre T, Shiu KK, Kim TW, Jensen BV, Jensen LH, Punt C, et al. Pembrolizumab in microsatellite-Instability-High advanced colorectal cancer. N Engl J Med. (2020) 383:2207–18. doi: 10.1056/NEJMoa2017699
29. Li H, van der Merwe PA, Sivakumar S. Biomarkers of response to PD-1 pathway blockade. Br J Cancer. (2022) 126:1663–75. doi: 10.1038/s41416-022-01743-4
30. Diaz LA Jr., Shiu KK, Kim TW, Jensen BV, Jensen LH, Punt C, et al. Pembrolizumab versus chemotherapy for microsatellite instability-high or mismatch repair-deficient metastatic colorectal cancer (KEYNOTE-177): final analysis of a randomised, open-label, phase 3 study. Lancet Oncol. (2022) 23:659–70. doi: 10.1016/S1470-2045(22)00197-8
31. Benson AB, Venook AP, Al-Hawary MM, Arain MA, Chen YJ, Ciombor KK, et al. Colon cancer, version 2.2021, NCCN clinical practice guidelines in oncology. J Natl Compr Canc Netw. (2021) 19:329–59. doi: 10.6004/jnccn.2021.0012
32. Ros J, Baraibar I, Saoudi N, Rodriguez M, Salva F, Tabernero J, et al. Immunotherapy for colorectal cancer with high microsatellite instability: the ongoing search for biomarkers. Cancers (Basel). (2023) 15:4245. doi: 10.3390/cancers15174245
33. Wang Z, Cao YJ. Adoptive cell therapy targeting neoantigens: A frontier for cancer research. Front Immunol. (2020) 11:176. doi: 10.3389/fimmu.2020.00176
34. Granhoj JS, Witness Praest Jensen A, Presti M, Met O, Svane IM, Donia M. Tumor-infiltrating lymphocytes for adoptive cell therapy: recent advances, challenges, and future directions. Expert Opin Biol Ther. (2022) 22:627–41. doi: 10.1080/14712598.2022.2064711
35. Chan JD, Lai J, Slaney CY, Kallies A, Beavis PA, Darcy PK. Cellular networks controlling T cell persistence in adoptive cell therapy. Nat Rev Immunol. (2021) 21:769–84. doi: 10.1038/s41577-021-00539-6
36. Chen YJ, Abila B, Mostafa Kamel Y. CAR-T: what is next? Cancers (Basel). (2023) 15:663. doi: 10.3390/cancers15030663
37. Ma S, Li X, Wang X, Cheng L, Li Z, Zhang C, et al. Current progress in CAR-T cell therapy for solid tumors. Int J Biol Sci. (2019) 15:2548–60. doi: 10.7150/ijbs.34213
38. Sterner RC, Sterner RM. CAR-T cell therapy: current limitations and potential strategies. Blood Cancer J. (2021) 11:69. doi: 10.1038/s41408-021-00459-7
39. Chen R, Chen L, Wang C, Zhu H, Gu L, Li Y, et al. CAR-T treatment for cancer: prospects and challenges. Front Oncol. (2023) 13:1288383. doi: 10.3389/fonc.2023.1288383
40. Zhang C, Liu J, Zhong JF, Zhang X. Engineering CAR-T cells. biomark Res. (2017) 5:22. doi: 10.1186/s40364-017-0102-y
41. Zhu Y, Feng J, Wan R, Huang W. CAR T cell therapy: remedies of current challenges in design, injection, infiltration and working. Drug Des Devel Ther. (2023) 17:1783–92. doi: 10.2147/DDDT.S413348
42. Qin X, Wu F, Chen C, Li Q. Recent advances in CAR-T cells therapy for colorectal cancer. Front Immunol. (2022) 13:904137. doi: 10.3389/fimmu.2022.904137
43. Zhang C, Wang Z, Yang Z, Wang M, Li S, Li Y, et al. Phase I escalating-Dose trial of CAR-T therapy targeting CEA(+) metastatic colorectal cancers. Mol Ther. (2017) 25:1248–58. doi: 10.1016/j.ymthe.2017.03.010
44. Xiao L, Cen D, Gan H, Sun Y, Huang N, Xiong H, et al. Adoptive transfer of NKG2D CAR mRNA-engineered natural killer cells in colorectal cancer patients. Mol Ther. (2019) 27:1114–25. doi: 10.1016/j.ymthe.2019.03.011
45. Michaux A, Mauen S, Breman E, Dheur MS, Twyffels L, Saerens L, et al. Clinical grade manufacture of CYAD-101, a NKG2D-based, first in class, non-Gene-edited allogeneic CAR T-Cell therapy. J Immunother. (2022) 45:150–61. doi: 10.1097/CJI.0000000000000413
46. Chen J, Zhu T, Jiang G, Zeng Q, Li Z, Huang X. Target delivery of a PD-1-TREM2 scFv by CAR-T cells enhances anti-tumor efficacy in colorectal cancer. Mol Cancer. (2023) 22:131. doi: 10.1186/s12943-023-01830-x
47. Gumber D, Wang LD. Improving CAR-T immunotherapy: Overcoming the challenges of T cell exhaustion. EBioMedicine. (2022) 77:103941. doi: 10.1016/j.ebiom.2022.103941
48. Liu Y, An L, Huang R, Xiong J, Yang H, Wang X, et al. Strategies to enhance CAR-T persistence. biomark Res. (2022) 10:86. doi: 10.1186/s40364-022-00434-9
49. Watanabe N, Mo F, McKenna MK. Impact of manufacturing procedures on CAR T cell functionality. Front Immunol. (2022) 13:876339. doi: 10.3389/fimmu.2022.876339
50. Rial Saborido J, Volkl S, Aigner M, Mackensen A, Mougiakakos D. Role of CAR T cell metabolism for therapeutic efficacy. Cancers (Basel). (2022) 14:5442. doi: 10.3390/cancers14215442
51. Zhang ZZ, Wang T, Wang XF, Zhang YQ, Song SX, Ma CQ. Improving the ability of CAR-T cells to hit solid tumors: Challenges and strategies. Pharmacol Res. (2022) 175:106036. doi: 10.1016/j.phrs.2021.106036
52. Kumar A, Watkins R, Vilgelm AE. Cell therapy with TILs: training and taming T cells to fight cancer. Front Immunol. (2021) 12:690499. doi: 10.3389/fimmu.2021.690499
53. Paijens ST, Vledder A, de Bruyn M, Nijman HW. Tumor-infiltrating lymphocytes in the immunotherapy era. Cell Mol Immunol. (2021) 18:842–59. doi: 10.1038/s41423-020-00565-9
54. Li B. Why do tumor-infiltrating lymphocytes have variable efficacy in the treatment of solid tumors? Front Immunol. (2022) 13:973881. doi: 10.3389/fimmu.2022.973881
55. Qin M, Chen G, Hou J, Wang L, Wang Q, Wang L, et al. Tumor-infiltrating lymphocyte: features and prognosis of lymphocytes infiltration on colorectal cancer. Bioengineered. (2022) 13:14872–88. doi: 10.1080/21655979.2022.2162660
56. Moreno V, Salazar R, Gruber SB. The prognostic value of TILs in stage III colon cancer must consider sidedness. Ann Oncol. (2022) 33:1094–6. doi: 10.1016/j.annonc.2022.09.155
57. Brummel K, Eerkens AL, de Bruyn M, Nijman HW. Prognostic benefit of TILs independent of clinicopathological and molecular factors. Br J Cancer. (2023) 129:737–8. doi: 10.1038/s41416-023-02335-6
58. Monberg TJ, Borch TH, Svane IM, Donia M. TIL therapy: facts and hopes. Clin Cancer Res. (2023) 29:3275–83. doi: 10.1158/1078-0432.CCR-22-2428
59. Farhood B, Najafi M, Mortezaee K. CD8(+) cytotoxic T lymphocytes in cancer immunotherapy: A review. J Cell Physiol. (2019) 234:8509–21. doi: 10.1002/jcp.27782
60. Bai Z, Zhou Y, Ye Z, Xiong J, Lan H, Wang F. Tumor-infiltrating lymphocytes in colorectal cancer: the fundamental indication and application on immunotherapy. Front Immunol. (2021) 12:808964. doi: 10.3389/fimmu.2021.808964
61. Albrecht HC, Gustavus D, Schwanemann J, Dammermann W, Lippek F, Weylandt KH, et al. Generation of colon cancer-derived tumor-infiltrating T cells (TILs) for adoptive cell therapy. Cytotherapy. (2023) 25:537–47. doi: 10.1016/j.jcyt.2023.01.009
62. Amaria R, Knisely A, Vining D, Kopetz S, Overman MJ, Javle M, et al. Efficacy and safety of autologous tumor-infiltrating lymphocytes in recurrent or refractory ovarian cancer, colorectal cancer, and pancreatic ductal adenocarcinoma. J Immunother Cancer. (2024) 12:e006822. doi: 10.1136/jitc-2023-006822
63. Schubert ML, Schmitt M, Wang L, Ramos CA, Jordan K, Muller-Tidow C, et al. Side-effect management of chimeric antigen receptor (CAR) T-cell therapy. Ann Oncol. (2021) 32:34–48. doi: 10.1016/j.annonc.2020.10.478
64. Guo F, Cui J. CAR-T in solid tumors: Blazing a new trail through the brambles. Life Sci. (2020) 260:118300. doi: 10.1016/j.lfs.2020.118300
65. Zhu X, Li Q, Zhu X. Mechanisms of CAR T cell exhaustion and current counteraction strategies. Front Cell Dev Biol. (2022) 10:1034257. doi: 10.3389/fcell.2022.1034257
66. Ma R, Li Z, Chiocca EA, Caligiuri MA, Yu J. The emerging field of oncolytic virus-based cancer immunotherapy. Trends Cancer. (2023) 9:122–39. doi: 10.1016/j.trecan.2022.10.003
67. Zhu Z, McGray AJR, Jiang W, Lu B, Kalinski P, Guo ZS. Improving cancer immunotherapy by rationally combining oncolytic virus with modulators targeting key signaling pathways. Mol Cancer. (2022) 21:196. doi: 10.1186/s12943-022-01664-z
68. Duan S, Wang S, Qiao L, Yu X, Wang N, Chen L, et al. et al: oncolytic virus-driven biotherapies from bench to bedside. Small. (2023) 19:e2206948. doi: 10.1002/smll.202206948
69. DePeaux K, Delgoffe GM. Integrating innate and adaptive immunity in oncolytic virus therapy. Trends Cancer. (2024) 10:135–46. doi: 10.1016/j.trecan.2023.09.012
70. Huang Z, Guo H, Lin L, Li S, Yang Y, Han Y, et al. Application of oncolytic virus in tumor therapy. J Med Virol. (2023) 95:e28729. doi: 10.1002/jmv.28729
71. Hemminki O, Dos Santos JM, Hemminki A. Oncolytic viruses for cancer immunotherapy. J Hematol Oncol. (2020) 13:84. doi: 10.1186/s13045-020-00922-1
72. Hwang JK, Hong J, Yun CO. Oncolytic viruses and immune checkpoint inhibitors: preclinical developments to clinical trials. Int J Mol Sci. (2020) 21:8627. doi: 10.3390/ijms21228627
73. Stern PL, Harrop R. 5T4 oncofoetal antigen: an attractive target for immune intervention in cancer. Cancer Immunol Immunother. (2017) 66:415–26. doi: 10.1007/s00262-016-1917-3
74. Scurr M, Pembroke T, Bloom A, Roberts D, Thomson A, Smart K, et al. Effect of modified vaccinia ankara-5T4 and low-Dose cyclophosphamide on antitumor immunity in metastatic colorectal cancer: A randomized clinical trial. JAMA Oncol. (2017) 3:e172579. doi: 10.1001/jamaoncol.2017.2579
75. Lee TH, Kim JS, Baek SJ, Kwak JM, Kim J. Diagnostic accuracy of carcinoembryonic antigen (CEA) in detecting colorectal cancer recurrence depending on its preoperative level. J Gastrointest Surg. (2023) 27:1694–701. doi: 10.1007/s11605-023-05761-2
76. Zhou X, Xie G, Wang S, Wang Y, Zhang K, Zheng S, et al. Potent and specific antitumor effect for colorectal cancer by CEA and Rb double regulated oncolytic adenovirus harboring ST13 gene. PloS One. (2012) 7:e47566. doi: 10.1371/journal.pone.0047566
77. Chouljenko DV, Murad YM, Lee IF, Delwar Z, Ding J, Liu G, et al. Targeting carcinoembryonic antigen-expressing tumors using a novel transcriptional and translational dual-regulated oncolytic herpes simplex virus type 1. Mol Ther Oncolytics. (2023) 28:334–48. doi: 10.1016/j.omto.2023.02.003
78. Osali A, Zhiani M, Ghaebi M, Meymanat M, Esmaeilzadeh A. Multidirectional strategies for targeted delivery of oncolytic viruses by tumor infiltrating immune cells. Pharmacol Res. (2020) 161:105094. doi: 10.1016/j.phrs.2020.105094
79. Rui R, Zhou L, He S. Cancer immunotherapies: advances and bottlenecks. Front Immunol. (2023) 14:1212476. doi: 10.3389/fimmu.2023.1212476
80. Dagher OK, Schwab RD, Brookens SK, Posey AD Jr. Advances in cancer immunotherapies. Cell. (2023) 186:1814–1814 e1811. doi: 10.1016/j.cell.2023.02.039
81. Dienstmann R, Villacampa G, Sveen A, Mason MJ, Niedzwiecki D, Nesbakken A, et al. Relative contribution of clinicopathological variables, genomic markers, transcriptomic subtyping and microenvironment features for outcome prediction in stage II/III colorectal cancer. Ann Oncol. (2019) 30:1622–9. doi: 10.1093/annonc/mdz287
82. Taieb J, Karoui M. FOxTROT: are we ready to dance? J Clin Oncol. (2023) 41:1514–7. doi: 10.1200/JCO.22.02108
83. Underwood PW, Ruff SM, Pawlik TM. Update on targeted therapy and immunotherapy for metastatic colorectal cancer. Cells. (2024) 13:245. doi: 10.3390/cells13030245
84. He X, Lan H, Jin K, Liu F. Can immunotherapy reinforce chemotherapy efficacy? a new perspective on colorectal cancer treatment. Front Immunol. (2023) 14:1237764. doi: 10.3389/fimmu.2023.1237764
85. IJ ME, Sanz-Pamplona R, Hermitte F, de Miranda N. Colorectal cancer: A paradigmatic model for cancer immunology and immunotherapy. Mol Aspects Med. (2019) 69:123–9. doi: 10.1016/j.mam.2019.05.003
86. Cabezon-Gutierrez L, Custodio-Cabello S, Palka-Kotlowska M, Diaz-Perez D, Mateos-Dominguez M, Galindo-Jara P. Neoadjuvant immunotherapy for dMMR/MSI-H locally advanced rectal cancer: The future new standard approach? Eur J Surg Oncol. (2023) 49:323–8. doi: 10.1016/j.ejso.2022.10.018
Keywords: colorectal cancer, immunotherapy, immune checkpoint inhibitors, adoptive cell therapy, oncolytic virus
Citation: Yu B, Kang J, Lei H, Li Z, Yang H and Zhang M (2024) Immunotherapy for colorectal cancer. Front. Immunol. 15:1433315. doi: 10.3389/fimmu.2024.1433315
Received: 16 May 2024; Accepted: 30 July 2024;
Published: 21 August 2024.
Edited by:
Zohreh Amoozgar, Massachusetts General Hospital and Harvard Medical School, United StatesReviewed by:
Jeremy Gungabeesoon, Sanofi, United StatesJavier Ros, Vall d’Hebron University Hospital, Spain
Copyright © 2024 Yu, Kang, Lei, Li, Yang and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Meng Zhang, emhhbmdtZW5nMTE3MjAwNkAxNjMuY29t
†These authors have contributed equally to this work and share first authorship