
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 02 July 2024
Sec. Inflammation
Volume 15 - 2024 | https://doi.org/10.3389/fimmu.2024.1428920
This article is part of the Research Topic Crosstalk in Ferroptosis, Immunity & Inflammation View all 25 articles
 Yu Cao1††
Yu Cao1†† Chang Lu1††
Chang Lu1†† Narasimha M. Beeraka1,2,3†
Narasimha M. Beeraka1,2,3† Sergey Efetov1†Mikhail Enikeev1†Yu Fu1†
Sergey Efetov1†Mikhail Enikeev1†Yu Fu1† Xinyi Yang1†
Xinyi Yang1† Basappa Basappa4†
Basappa Basappa4† Mingze He1*†
Mingze He1*† Zhi Li1*†
Zhi Li1*†Ferroptosis induces significant changes in mitochondrial morphology, including membrane condensation, volume reduction, cristae alteration, and outer membrane rupture, affecting mitochondrial function and cellular fate. Recent reports have described the intrinsic cellular iron metabolism and its intricate connection to ferroptosis, a significant kind of cell death characterized by iron dependence and oxidative stress regulation. Furthermore, updated molecular insights have elucidated the significance of mitochondria in ferroptosis and its implications in various cancers. In the context of cancer therapy, understanding the dual role of anastasis and ferroptosis in chemoresistance is crucial. Targeting the molecular pathways involved in anastasis may enhance the efficacy of ferroptosis inducers, providing a synergistic approach to overcome chemoresistance. Research into how DNA damage response (DDR) proteins, metabolic changes, and redox states interact during anastasis and ferroptosis can offer new insights into designing combinatorial therapeutic regimens against several cancers associated with stemness. These treatments could potentially inhibit anastasis while simultaneously inducing ferroptosis, thereby reducing the likelihood of cancer cells evading death and developing resistance to chemotherapy. The objective of this study is to explore the intricate interplay between anastasis, ferroptosis, EMT and chemoresistance, and immunotherapeutics to better understand their collective impact on cancer therapy outcomes. We searched public research databases including google scholar, PubMed, relemed, and the national library of medicine related to this topic. In this review, we discussed the interplay between the tricarboxylic acid cycle and glycolysis implicated in modulating ferroptosis, adding complexity to its regulatory mechanisms. Additionally, the regulatory role of reactive oxygen species (ROS) and the electron transport chain (ETC) in ferroptosis has garnered significant attention. Lipid metabolism, particularly involving GPX4 and System Xc- plays a significant role in both the progression of ferroptosis and cancer. There is a need to investigate the intricate interplay between anastasis, ferroptosis, and chemoresistance to better understand cancer therapy clinical outcomes. Integrating anastasis, and ferroptosis into strategies targeting chemoresistance and exploring its potential synergy with immunotherapy represent promising avenues for advancing chemoresistant cancer treatment. Understanding the intricate interplay among mitochondria, anastasis, ROS, and ferroptosis is vital in oncology, potentially revolutionizing personalized cancer treatment and drug development.
Chemoresistance promotes cancer stemness by facilitating the release of exosomes containing peptides, nucleic acids, and various small molecules in multiple cancer types. This process converts drug-sensitive cells into cancer stem cells (1, 2). These cells represent a distinct subpopulation within tumor tissues, exhibiting self-renewal capabilities and giving rise to diverse tumor cell phenotypes (3). Chemoresistance could induce tumor heterogeneity (4–7), contributing significantly to tumor progression (8, 9). Chemoresistance can be modulated by the ferroptosis (9).
Cell death is a critical event within the physiological milieu and ferroptosis represents a significant kind of iron-dependent regulated cell death (RCD), distinct from established mechanisms such as apoptosis, pyroptosis, necrosis, and autophagy (10, 11). Ferroptosis is characterized by the generation of ROS and iron ion-dependent lipid peroxidation. Within cellular environments, ferroptosis is mediated by compounds like erastin or rat sarcoma virus oncogene homolog-selective lethal 3 (RSL3), which inhibit the expression of glutathione peroxidase 4 (GPX4) (12, 13). GPX4 is a selenoprotein that specifically catalyzes the conversion of lipid peroxide into lipids using glutathione. Consequently, reduced GPX4 levels disrupt the cellular oxidation balance. The Fenton reaction between ferrous ions and endogenous hydrogen peroxide generates hydroxyl radicals, which, in conjunction with polyunsaturated fatty acids, undergo multi-step free radical chain reactions, resulting in the formation of lipid peroxides (14). In a complete cellular system, ROS can stimulate mitochondria to open the permeability transition pore, amplifying the ROS signal through positive feedback. This positive feedback loop can be potentially regulated by mitochondria (15, 16). Consequently, when mitochondrial membrane integrity is compromised, the regulatory mechanism becomes impaired, leading to increased ROS production. After lipid peroxidation, modifications in the cell membrane is observed subsequently cause the generation of cytotoxic byproducts including malondialdehyde and 4-hydroxynonanaldehyde are generated, ultimately leading to the induction of ferroptotic cell death (17, 18).
When the term “anastasis” was introduced in 2012, it was associated solely with the reversal of apoptosis. This study explore novel cellular recovery phenomenon involving the reversal of ferroptosis in addition to anastasis, chemoresistance in cancer research.
Ferroptosis is a kind of programmed cell death reported in diseases including cancers (19, 20). Although the regulatory mechanisms and implications of ferroptosis continue to be elucidated, its potential resembles that of apoptosis in either augmenting cell death during cancer therapy or attenuating cell death to safeguard susceptible cells such as neurons, cardiomyocytes, and hepatocytes at the time of tissue damage (21).
For a significant duration, cell death was predominantly categorized into two main types: programmed cell death (PCD), exemplified by apoptosis mediated by cysteine aspartate-specific proteinase (caspase), and non-programmed cell death modalities such as necrosis (22–24) (Table 1). However, with advancements in molecular research within cell biology, an expanding repertoire of PCD modalities, including autophagy-dependent cell death, pyroptosis, and more recently, ferroptosis and cuproptosis, have been elucidated (47–49). Ferroptosis, a novel variant of programmed non-apoptotic cell death exerts a crucial regulatory influence on cancer progression. Growing evidence indicates ferroptosis induction as a promising avenue for cancer therapy. In recent years, several experimental reports described the role of mitochondria in various regulated cell death (RCD) processes, encompassing apoptosis, necrosis, pyroptosis, and ferroptosis (Figure 1). Thus, this study of ferroptosis reversibility offers novel insights for potential therapeutic interventions aimed at modulating cell death and survival through its reversible nature.
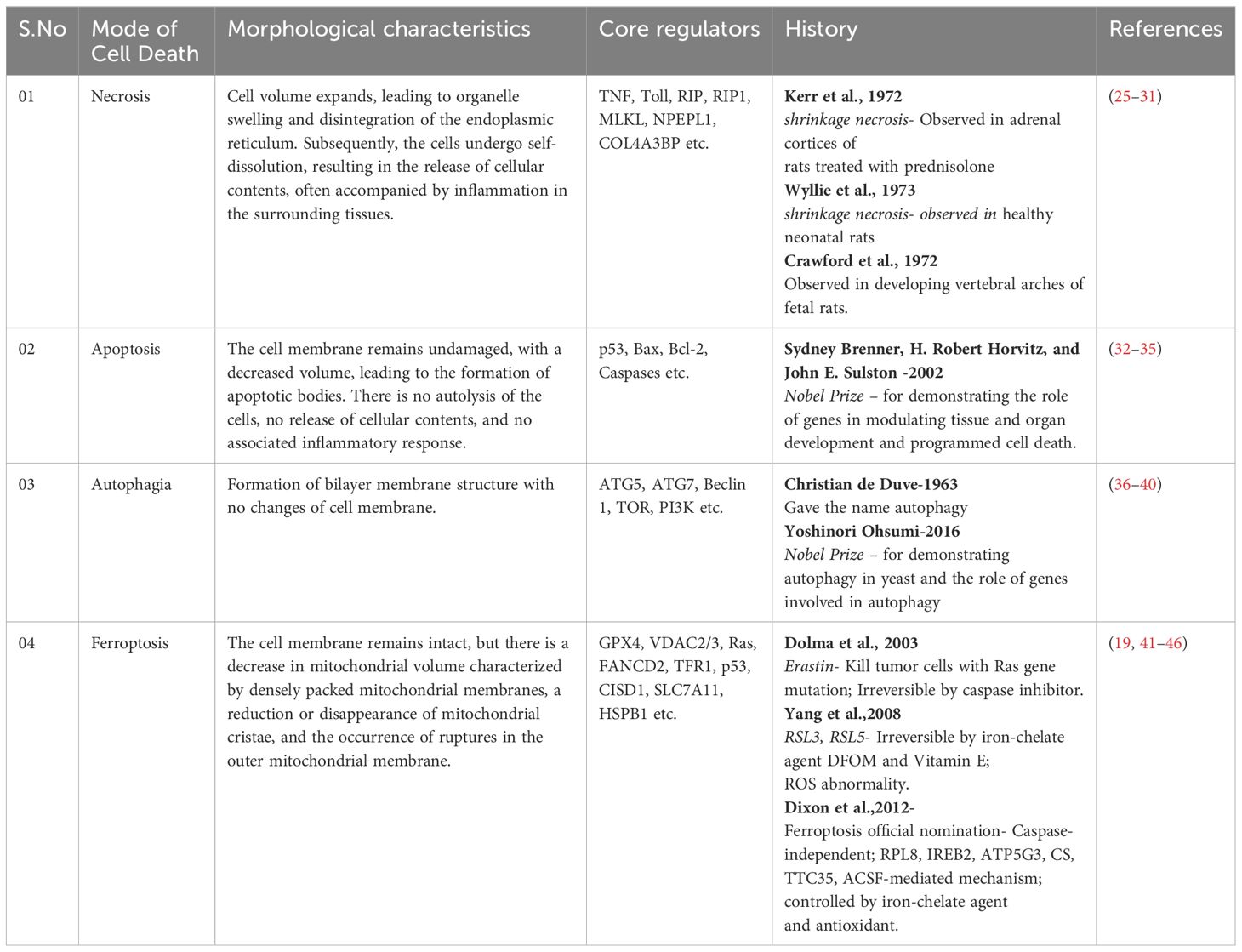
Table 1 History of classification and differences of cell death mode implicated in cell physiology.
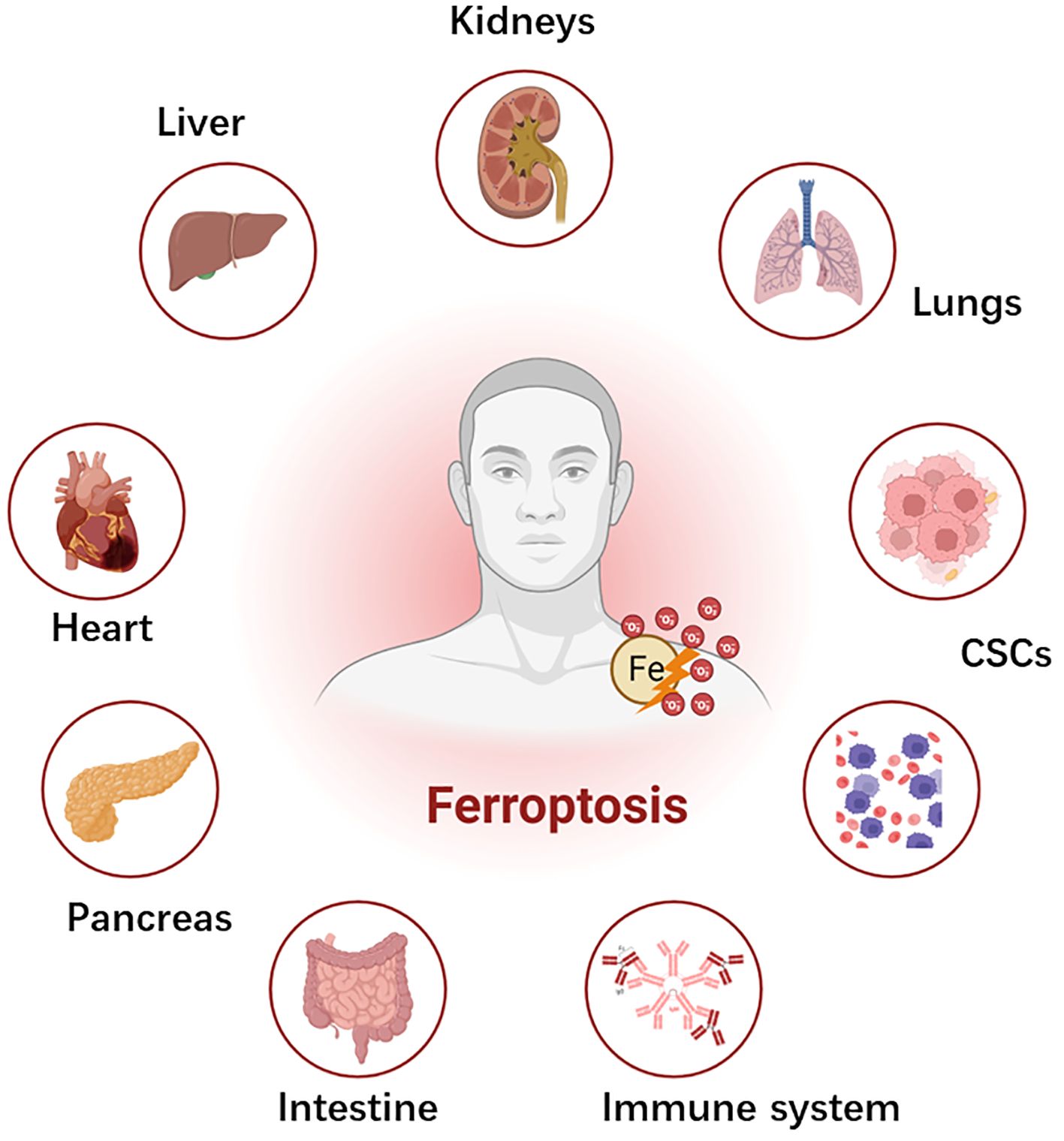
Figure 1 Schematic depiction of ferroptosis and systemic toxicities. Ferroptosis plays a pivotal role in several diseases across different organs. Ferroptosis has significant implications in the pathophysiology of chemoresistance and the development of cancer stem cells (CSCs), which yet require future studies in chemoresistant cancers (?). In the liver, it contributes to the development and progression of hepatocellular carcinoma, liver fibrosis, and ischemia-reperfusion injury. Within the cardiovascular system, it is implicated in ischemia reperfusion injury, transplantation, and atherosclerosis. Gastrointestinal system disorders, including gastric cancer and colorectal cancer, are influenced by ferroptosis. In the pancreas, it is associated with pancreatic cancer and type I diabetes mellitus. In the kidney, ferroptosis is involved in acute kidney injury, ischemia-reperfusion injury, and clear cell renal cell carcinoma. Lung diseases, such as lung cancer and acute lung injury, typically have ferroptosis-mediated pathophysiology. Hematological malignancies in the blood system and the suppression of T- cell immunological function in the immune system are also associated with ferroptosis.
Mitochondria play dual roles as key contributors to oxidative phosphorylation and primary generators of intracellular ROS (50, 51). Furthermore, they function as central hubs for various metabolic and signaling pathways, particularly fatty acid metabolism (52). Several studies have demonstrated that mitochondrial iron content, assessed through selective fluorescent iron indicators or electron paramagnetic resonance, constitutes a substantial portion, ranging from 20% to 50%, of the total cellular iron, depending on cell type (53–55). Mitochondrial iron plays a critical role in the formation of iron-sulfur clusters and heme synthesis. Notably, redox-active iron pools within mitochondria contribute to the accumulation of mitochondrial (MitoROS) (56–58). A distinctive feature of ferroptosis is its ability to evoke specific immune responses. During ferroptotic cell death, intracellular components are released into the extracellular milieu, triggering an immune reaction (59, 60). This response subsequently activates neighboring immune cells, such as macrophages, and facilitates the recruitment of additional immune cells to the site. It’s noteworthy that other forms of RCD may also induce immune responses, although the nature of the immune response varies depending on the type of RCD involved (61). Induction of ferroptosis in cells using erastin result in the reduced mitochondrial size compared to normal cells (62, 63).
In this review, we described a comprehensive exploration of the complex relationships and interactions between mitochondria, ROS, and ferroptosis-mediated chemoresistance and subsequently described the need for investigating the interplay of anastasis, EMT and mitochondrial ROS-mediated ferroptosis, and comparative efficacy of anticancer immunotherapeutics in metastatic chemoresistant cancers. This study is beneficial to several oncologists, molecular biologists, clinicians to develop personalized cancer treatment strategies to promote novel drug development.
We undertook a significant literature analysis, sourcing data from different databases including Pubmed, Medline, eMedicine, Scopus, Google Scholar, the National Library of Medicine (NLM), and ReleMed. Our focus encompassed published reports and articles investigating the role of mitochondria, ROS, anastasis, ferroptosis-mediated chemoresistance, and immunotherapeutics in metastatic cancers.
Iron in dietary sources primarily exists as Fe (III) form, which undergoes reduction to Fe (II) within the intestinal lumen facilitated by brush-border membrane ferrireductase, duodenal cytochrome b (DCYTb), and other iron-reducing enzymes (64, 65). The resulting Fe (II) is then transported across the apical membrane of small intestinal epithelial cells (IECs) via divalent metal transporter 1 (DMT1/SLC11A2) and is subsequently sequestered within the labile iron pool (LIP) (66, 67). Intracellular Fe (II) is exported from IECs into the bloodstream via basolateral membrane ferroportin (FPN/SLC11A3), promoted through poly(rC)-binding proteins, and re-oxidized to Fe (III) by hephaestin (HEPH) (68, 69). Fe (III) is then bound to transferrin (TF) to form transferrin-bound iron (TBI) complexes, enabling circulation for transport to various tissues and organs. Within cells, TBI binds to transferrin receptor 1 (TfR1) and undergoes endocytosis, where Fe (III) is released into the cytoplasm, reduced back to Fe (II) by six-transmembrane epithelial antigen of prostate (STEAP3), and transported via DMT1/SLC11A2. A portion of cytoplasmic Fe (II) is stored in ferritin (FT) as the Fe2+-FT complex, while the remainder contributes to the LIP. Heme also plays a significant role in iron metabolism, entering cells through heme carrier protein 1 (HCP1) and feline leukemia virus subgroup C receptor 2 (FLVCR2), with subsequent release of Fe (II) catalyzed by heme oxygenase 1 (HO-1) for transport to the LIP (69–72). In addition, non-transferrin-bound irons (NTBI) in the cytoplasm of different tissues are transported to the cells through different NTBI transporters such as Zrt/Irt-like protein 8/14 (ZIP8/14, SLC39A8/SLC39A14), L-type and T-type calcium channels after which they are stored in the LIP (73–75). The process of detachment and release of Fe (II) from FT in Fe2+-FT is referred to as ferritinophagy, and it is regulated by nuclear receptor coactivator 4 (NCOA4). FT has heavy chain (FTH) and light chain (FTL) subunits. NCOA4 binds specifically to FTH1 and induces Fe2+-FT degradation, which separates FT from Fe (II) and thus allows Fe (II) release (76–80). A portion of intracellular Fe (II) enters the mitochondria through the MFRN1 and MFRN2 channels where they are then involved in the synthesis of heme and iron-sulfur cluster (ISC). ISC is an important inorganic cofactor that is widely engaged in a variety of biological functions including electron transfer which is the fundamental basis for cellular ATP production and protein synthesis. Meanwhile, recent studies have shown that ISC is tightly associated with DNA regulation (81–83).
Mitochondrial Ferritin (FtMt) is a protein responsible for iron storage within cell mitochondria, sharing structural and functional similarities with cytoplasmic H-ferritin (84, 85). FtMt expression is confined to the mitochondria of the central nervous system and select oxygen-dependent tissues. Studies have indicated that erastin induces upregulation of Voltage-Dependent Anion Channel (VDAC) proteins on the mitochondrial membrane, thereby triggering ferroptosis (86–88). However, investigations on FtMt-SY5Y cells treated with erastin have shown unchanged levels of VDAC2 and VDAC3, suggesting a specific protective mechanism of FtMt against ferroptosis (89, 90). Upon erastin treatment, there is an upregulation of NADPH oxidase (NOX) proteins, critical for ROS generation in ferroptosis (91, 92). Interestingly, erastin-treated cells overexpressing FtMt exhibit a minimal increase in NOX2 levels, indicating inhibition of ROS generation by FtMt during ferroptosis (92, 93). Additionally, the mitochondrial glutaminolysis-tricarboxylic acid cycle-electron transport chain (TCA-ETC) axis plays a pivotal role in initiating ferroptosis. Within this pathway, ROS generation leads to ferroptosis through lipid peroxidation (94, 95) (Figure 2).
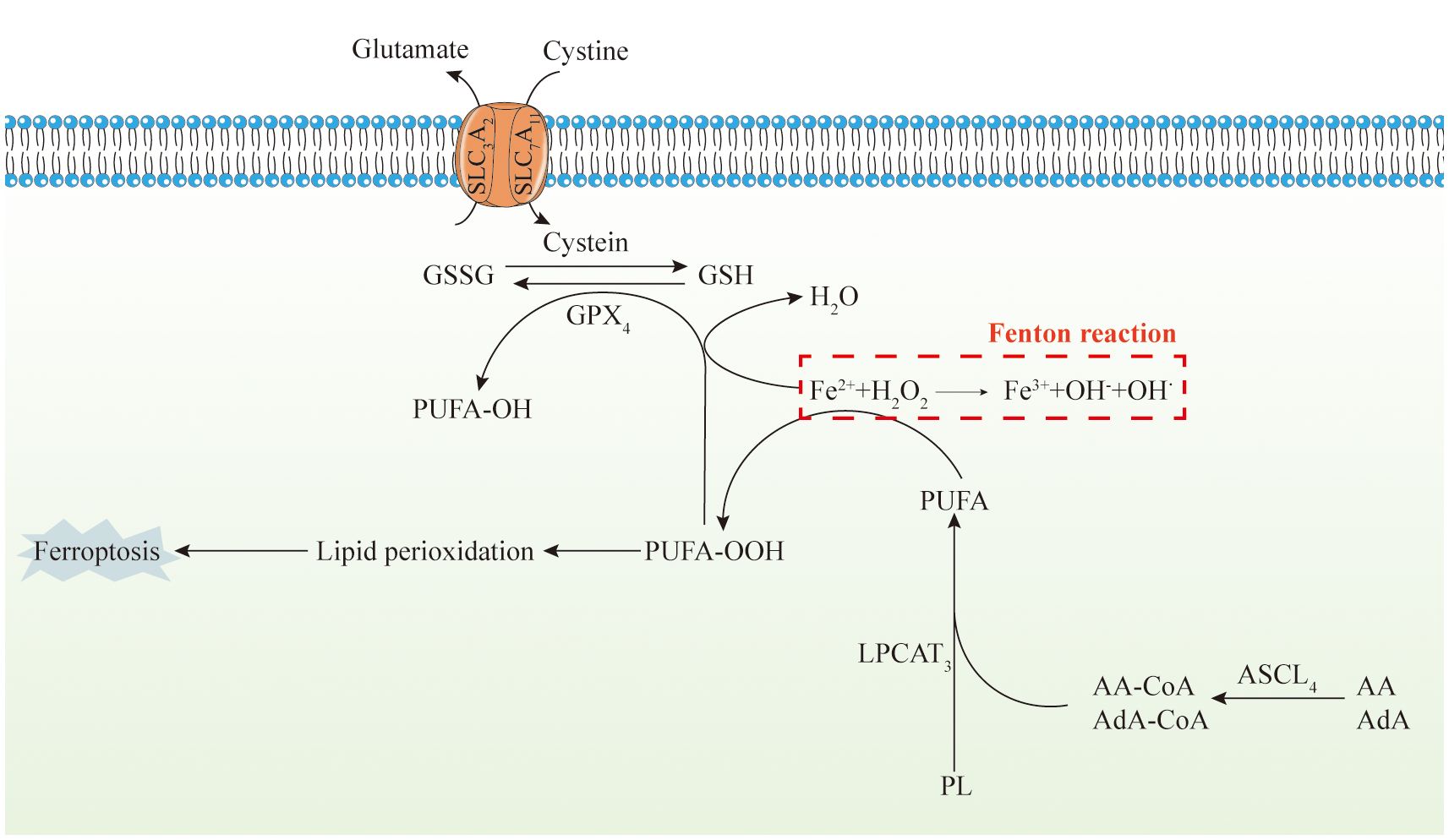
Figure 2 Mitochondria, iron metabolism, and ferroptosis. The diagram depicts the intricate series of events involved in iron metabolism within the human body, alongside ferroptosis, a form of regulated cell death. Initially, dietary iron, predominantly in the Fe(III) state, undergoes a sequence of conversions and transport processes. These processes include its reduction to Fe(II) within the intestinal environment, uptake by small intestinal epithelial cells, sequestration within the labile iron pool, extracellular release facilitated by ferroportin, and subsequent re-oxidation mediated by hephaestin. Subsequently, iron complexes with transferrin (TF), form transferrin-bound iron, which circulates in the bloodstream. TBI is internalized by cells via endocytosis, where it undergoes reduction to Fe(II) and enters the cellular cytoplasm. A fraction of the Fe(II) is stored within ferritin, while the remaining is reincorporated into the LIP. Heme, another significant iron source, follows a comparable pathway. Additionally, non-transferrin-bound iron from various tissues penetrates cells and is sequestered within the LIP. Furthermore, an additional reservoir of LIP is replenished through ferritinophagy, regulated by nuclear receptor coactivator 4. Intracellular free Fe(II) gains access to mitochondria via MFRN1 and undergoes subsequent biochemical transformations, ultimately leading to the onset of ferroptosis. Furthermore, ferroptosis is characterized by its dependence on iron and lipotoxicity. It operates by suppressing the activity of the lipid repair enzyme glutathione peroxidase 4 (GPX4), resulting in the accumulation of lipid hydroperoxides. Genetically, multiple genes modulate ferroptosis. Unlike other types of cell death characterized by extracellular manifestations, ferroptosis primarily unfolds intracellularly. This leads to distinctive cellular alterations, including reduced mitochondrial size, heightened membrane density, damaged cristae, fragmentation of the outer membrane without compromising the cell membrane, and minimal changes in nuclear morphology without chromatin condensation. Biochemically, the deficiency in peroxidation repair capacity primarily originates from the impairment of the phospholipid peroxidase GPX4. This deficiency triggers the acquisition of reactive iron and the oxidation of phospholipids containing polyunsaturated fatty acids (PUFA), ultimately inducing ferroptosis. The decrease in intracellular antioxidant capacity exacerbates lipid ROS accumulation, and cause cellular ferroptosis. Glutathione peroxidase is influenced by various pathways, including the XC−/GSH/GPX4 system, and the ACSL4/LPCAT3/15-LOX and FSP1/CoQ10/NAD(P)H pathways.
Dolma et al. (45) reported a compound within the erastin class that exhibited selective cytotoxicity against tumor cells specific for Ras gene mutations. This compound induced a distinct mode of cell death not previously documented, which was unaffected by caspase inhibitors. Subsequently, Yang et al. (46) identified two additional erastin analogs, RSL3 and RSL5, and demonstrated that changes in ROS levels could induce cell death. This process could be counteracted by iron chelators such as deferoxamine and vitamin E. Dixon et al. (10) formally coined the term “ferroptosis” to elucidate that this novel form of cell death relies on iron but is independent of the caspase activity. This study delineated the ferroptosis regulation by multiple genetic factors and elucidated its mechanistic association with the cystine/glutamate antiporter (System Xc-) (Table 1).
ROS encompasses a heterogeneous group of oxygen-containing reactive molecules, including superoxides, peroxide radicals, hydroxyl radicals, and alkoxyl radicals (96–98). Intracellular ROS are predominantly generated during ATP production within various cell types, representing normal metabolic byproducts of the mitochondrial respiratory chain. Furthermore, enzymes such as NADPH oxidase contribute to ROS production via enzymatic reactions (99, 100).
Polyunsaturated fatty acids (PUFA) constitute integral components of cell membrane phospholipid bilayers, crucial for maintaining membrane fluidity (101). Intracellular iron (II) (Fe (II)) participates in the Fenton reaction with hydrogen peroxide (H2O2), yielding hydroxyl radicals (OH•) that subsequently involved in lipid peroxidation reactions. Fe (II) itself catalyzes lipid peroxidation, promoting the formation of ROS such as PUFA-OOH, thereby causing cellular membrane damage (102–108).
The fundamental mechanism underlying ferroptosis involves the higher ROS generation, which leads to lipid peroxidation and disruption of cellular redox homeostasis, ultimately results in cell death that has significant implications in oncology. Despite the existence of various upstream regulatory pathways, it is widely acknowledged that the activity of glutathione peroxidase 4 (GPX4) directly or indirectly influences the onset of ferroptosis (13, 109–111).
The principal morphological hallmark of ferroptosis is the changes in mitochondrial architecture and function, distinguishing it from other modes of cell death (112). Mitochondria undergoing ferroptosis display distinct alterations in morphology. Upon treatment with erastin, cells lack the characteristic morphological features typically associated with the absence of nuclear condensation, fragmentation, and necrotic lysis characteristic of necrosis; the lack of cellular crumpling, chromatin condensation, and apoptotic vesicle formation typical of apoptosis are changes associated with mitochondria-mediated ferroptosis. In response to erastin treatment, cells exhibit a distinct set of alterations characterized by a higher mitochondrial potential and membrane density. Furthermore, there is a reduction or disappearance of mitochondrial ridges, accompanied by the rupture of the mitochondrial outer membrane. These distinctive alterations serve as discernible criteria for distinguishing ferroptosis from apoptosis, necroptosis, and autophagy (113–118). These morphological alterations should be explored in various chemoresistant cancers.
The tricarboxylic acid (TCA) cycle and glutamine metabolism play integral roles in ferroptosis. While the precise metabolic pathways remain elusive, evidence suggests a close association between TCA cycle metabolites, enzymes, and ferroptotic cell death (119–121). In cellular contexts, blocking the cystine/glutamate antiporter (System Xc (-)) prevents the reverse transport of cystine and glutamic acid, thereby reducing cystine uptake, and causing intracellular ROS accumulation, lipid peroxidation, and initiation of ferroptosis (122). Elevated extracellular glutamate levels impede System Xc (-) function, consequently promoting ferroptosis.
Hence, diminishing glutamine (Gln) availability or inhibiting glutamine metabolism profoundly attenuates ferroptosis. Without adequate glutamine, mitochondrial damage and cell death induced by cystine starvation or erastin treatment are impeded (123, 124). Thus, glutamine is necessary for ferroptosis, and glutamine synthase (GLS) is a key regulatory factor for glutamine decomposition. GLS is divided into two isoforms: GLS1 and GLS2. GLS1 is mainly located in the cytoplasm, while GLS2 is located in the mitochondria (125–127). GLS1 and GLS2 catalyze the conversion of Gln to Glu (128, 129). Tal Hirschhorn et al. (130) found that ferroptosis can be prevented by drugs or gene inhibition of mitochondrial subtype GLS2. GLS2 has previously been shown to have activity consistent with tumor inhibition. GLS2 is a transcriptional target of p53 and upregulates during p53-dependent ferroptosis (131). Suzuki S et al. (132) found that GLS2-mediated glutamate can be converted into glutamic acid in the presence of aspartate aminotransferase (GOT) and glutamate dehydrogenase 1 (GLUD1). α - KG increases the production of ROS to downregulate the antioxidant defense function of cells, increase their sensitivity to iron-dependent cell death, and thus promote iron-dependent cell death. Downstream products of α-KG, such as succinic acid and fumaric acid, can also enhance cysteine depletion and cause cell ferroptosis (133). Several enzymes in the TCA cycle are necessary for iron-mediated ferroptosis caused by cystine starvation or erastin therapy, such as fumarate hydroxylase (FH), aconitase, and citrate synthesis (CS) (134–137). Therefore, reducing glutamine or blocking the glutamine decomposition typically impairs CDI ferroptosis. Without glutamine, cysteine starvation or erastin treatment cannot induce mitochondrial damage and subsequent cell death (123, 124).
Glycolysis serves as a crucial pathway for cells undergoing anaerobic respiration to generate energy. In tumor cells, metabolic rates are typically increased to sustain their rapid and uncontrolled proliferation. Metabolic shift is one of the significant physiological activities occurring in tumor cells to foster uncontrolled proliferation. Mainly, malignant cells rely on glycolysis to fulfill their energy demands while maintaining redox homeostasis to prevent ferroptosis (119, 138, 139). Ibtissam et al. (140) described that glycolytic cells exhibit significant inhibition of mitochondrial OXPHOS activity, mitigating ROS stress. However, this inhibition is reversible, and when glycolysis is suppressed, metabolic reconfiguration toward OXPHOS elevates cellular ROS levels, subsequently disrupting iron homeostasis and promoting lipid peroxidation, thus rendering tumor cells more sensitive to conventional chemotherapeutic agents and ferroptosis inducers. Hence, it is crucial to develop novel ferroptosis inducers to modulate the mitochondrial OXPHOS mainly to inhibit the development of CSCs in TME.
The altered function of VDACs may contribute to glycolytic alterations. Specifically, the opening of VDACs enhance mitochondrial substance exchange, subsequently increasing aerobic respiration efficiency (141, 142). VDACs, comprising VDAC1, VDAC2, and VDAC3 isoforms typically facilitate ATP, adenosine diphosphate, and mitochondrial-cytoplasmic exchanges (141, 142). Studies have indicated (143–145) that VDAC closure by microtubule proteins in a quiescent state restricts respiratory substrate influx into mitochondria, promoting glycolysis in cancer cells (Warburg effect), and this mechanism is yet to be studied in chemoresistant cancer cells. Yagoda et al. (146) revealed that cells activated by the RAS-RAF-MEK pathway can bind to certain VDAC ligands, subsequently enhancing susceptibility to erastin, a pivotal factor in cancer cell development. This heightened sensitivity prevents and reverses VDAC blockade by cytoplasmic free microtubule proteins, leading to VDAC opening and subsequent non-apoptotic cell death. Furthermore, intracellular free microtubulin abundance inhibits VDAC conductivity. N. DeHart et al. (86) described a novel mechanism of cell killing induced by erastin and erastin-like compounds, where the reversal of microtubulin-dependent VDAC inhibition fosters mitochondrial hyperpolarization and oxidative stress, culminating in mitochondrial dysfunction, dissipation of mitochondrial membrane potential (ΔΨm), and cell death. These mitochondrial mechanisms pertinent to the ferroptosis in chemoresistant tumor cells in other different cancer types should be explored typically by several preclinical studies.
Mitochondria is the primary site for oxidative respiration within cells, where ETC activity not only facilitates ATP production but also plays a crucial role in generating ROS necessary for triggering cellular ferroptosis (147–149). For instance, glucose-dependent mitochondrial bioenergetic processes involve the transfer of electrons from complexes I and III in the ETC to molecular oxygen, resulting in ROS generation during catabolism (150–153). ROS, including hydrogen peroxide, superoxide anion, hydroxyl radicals, and peroxynitrite, are predominantly produced via mitochondrial metabolism and act as mediators of intracellular signaling pertinent to various forms of cell death including in tumor cells (16).
Electron leakage from ETC complexes I and III generates O2·-, which is consequently converted to H2O2 by disproportionation in the presence of superoxide dismutase. H2O2 reacts with Fe2+ to form hydroxyl radicals, initiating a chain reaction with PUFAs to induce cell death via the production of PUFA hydroperoxides (PUFA ⁃OOH) (154). Studies by Feng et al. (155) demonstrate that COX7A1, a cytochrome C subunit, inhibits mitochondrial autophagy and promotes susceptibility to ferroptosis in NSCLC cells by modulating the TCA cycle, ETC complex IV activity. This mechanism should be explored in several other cancer types.
Additionally, the inhibition of ETC mitochondrial complexes I, II, III, and IV attenuates ROS accumulation and ferroptosis induced by cysteine starvation or erastin et al. (134). Inhibition of mitochondrial complex III function with S3QEL significantly reduces lipid peroxidation and ferroptosis induced by cysteine starvation, highlighting the crucial role of superoxide produced by complex III in cysteine starvation-induced ferroptosis (156). The implications of cysteine depletion-induced ferroptosis in CSCs should be explored vividly by exploring the different cell signaling mechanisms.
Furthermore, ETC activity could influence the regulation of energy sensor adenosine 5’⁃monophosphate (AMP) and ROS levels. Mitochondrial electron transfer via the ETC complex generates a proton concentration gradient that powers ATP synthesis via ATP synthase. ATP production inhibits AMP-dependent protein kinase (AMPK) activity (157), promoting cellular ferroptosis by decreasing phosphorylation of acetyl-coenzyme A carboxylase (ACC) and increasing lipid synthesis, thereby influencing cellular susceptibility to ferroptosis (157).
Lipids form the structural basis for cell membranes and organelles (mitochondria, endoplasmic reticulum), with lipid metabolism closely linked to cellular ferroptosis sensitivity via multiple pathways (47). System Xc- functions as a reverse cystine/glutamate transporter, comprising the transmembrane transporter proteins SLC3A2 and SLC7A11 (xCT). It transports intracellular glutamate and extracellular cystine, with intracellular cystine being converted to cysteine. Cysteine plays a crucial role in glutathione synthesis, primarily existing as reduced glutathione (GSH) and oxidized glutathione (GSSG), dynamically balanced within cells (158–161). GPX4, a selenoprotein, plays a pivotal role in maintaining this balance by catalyzing the conversion of glutathione to GSSG (122, 162). It effectively reduces phospholipid peroxides, decomposes H2O2 to water with GSSG, and inhibits arachidonic acid activation, thereby suppressing lipid peroxidation and ROS generation (22–24). As we discussed above, PUFAs are major substrates for lipid peroxidation during ferroptosis, damaging membrane structure and function. Enzymes involved in binding PUFA to phospholipids, such as acyl coenzyme A synthetase long-chain family member 4 (ACSL4), are integral to ferroptosis. ACSL4, besides being a sensitive indicator of iron-dependent cell death, regulates lipid metabolism by linking free fatty acids to CoA linkage, ultimately exchanged for phospholipids (163). ACSL4 specifically binds to substrates like arachidonic acid (AA) and adrenergic acid (ADA) (164) leads to the generation of fatty acyl CoA esters, esterified by lysophosphatidylcholine acyltransferase 3 (LPCAT3) into phospholipids like phosphatidyl ethanolamine (AA-ETA), predominantly found in the endoplasmic reticulum. Subsequent oxidation of AA-PE and ADA-PE by 15-lipoxygenase (ALOX15) produces lipid hydroperoxides, and other signaling molecules promoting ferroptosis (162, 165, 166). Recent studies highlight alternative pathways to ferroptosis, indicating that ACSL4/LPCAT3/15-LOX or p53/SLC7A11/12-LOX may cause lipid peroxidation during this process (167, 168). This underscores the complexity of ferroptosis regulation, offering potential therapeutic avenues for modulating cell death pathways to ameliorate chemoresistant-cancers (Figure 3).
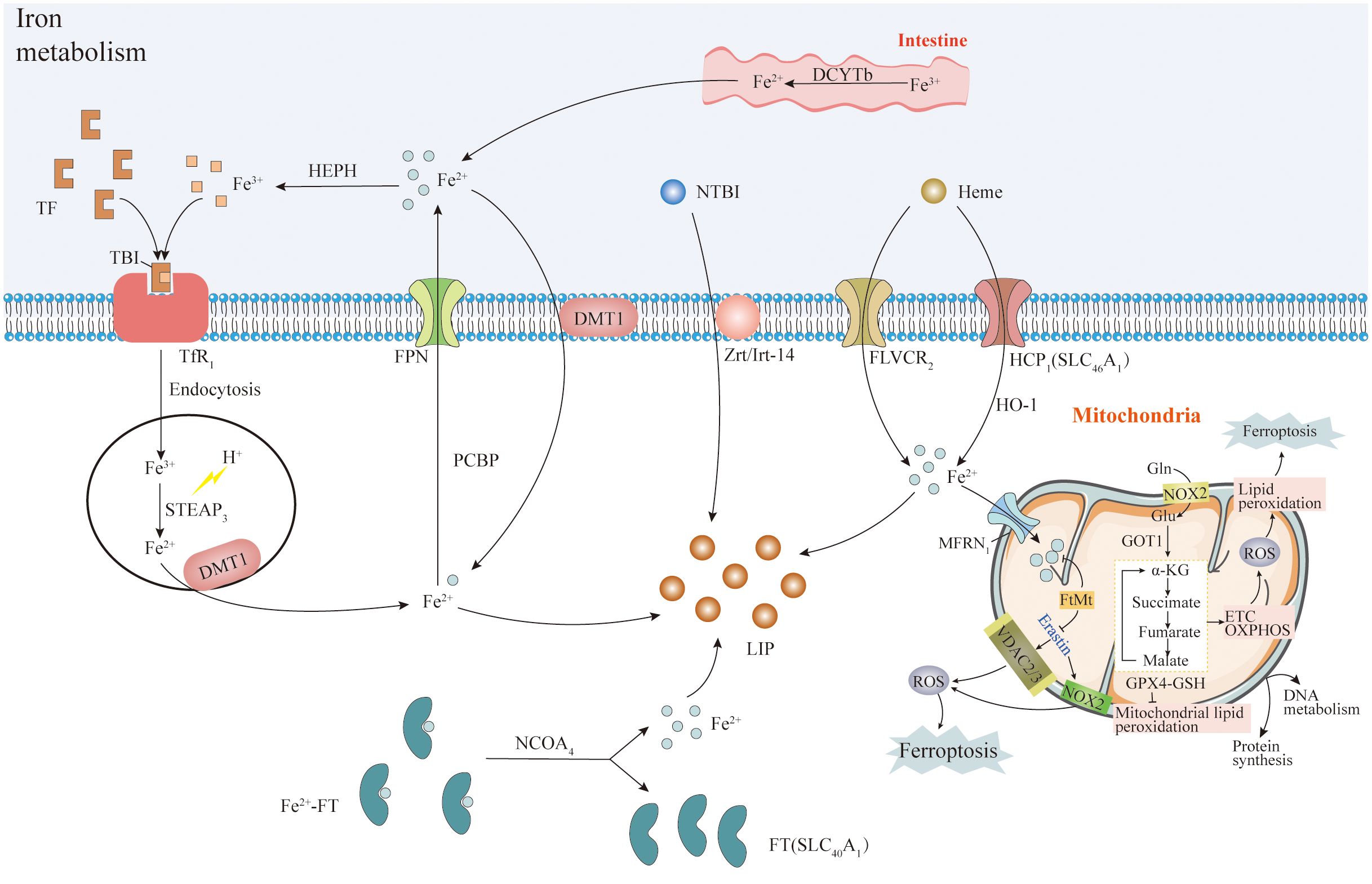
Figure 3 System Xc- and lipid metabolism. System Xc- is a transmembrane transporter composed of SLC3A2 and SLC7A11 (xCT) proteins. It transports intracellular glutamate and extracellular cystine, which is converted into cysteine. Cysteine plays a pivotal role in synthesizing the antioxidant glutathione. Glutathione exists mainly in reduced (GSH) and oxidized (GSSG) forms, maintained in balance by various mechanisms. GPX4, a selenoprotein, converts glutathione to GSSG and efficiently reduces phospholipid peroxides while decomposing H2O2. GPX4 also inhibits arachidonic acid activation and lipid peroxidation (ROS). Long-chain acyl-CoA synthetase 4 (ACSL4) activates arachidonic acid and adrenic acid into their CoA forms, promoting the activation of polyunsaturated fatty acids (PUFA) mediated by LPCAT3. Intracellular Fe(II) undergoes the Fenton reaction with H2O2, generating hydroxyl radicals (OH·), contributing to lipid peroxidation and ROS production, causing cell membrane damage. Note: The complete pathways explanation was given in subheading 3.
Although both ferroptotic and apoptotic modes of cell death processes can be reversed, the underlying mechanisms facilitating these cell recovery phenomena remain to be fully elucidated in distinct cancer types. Primarily, both modes of cell death are typically different suggesting that different mechanisms may be required to halt the respective cell death mediators and repair the distinct types of damage for cell recovery. For instance, apoptosis involves the activation of proteases that can cause cleavage of distinct functional proteins, along with DNases like endonuclease G and DNA fragmentation factors, resulting in genomic destruction (169–171). Upon cessation of apoptotic stimulus, the cells that are undergoing recovery could induce upregulation of heat shock proteins and XIAP to impair stimulation of caspases, whereas the ICAD/DFF45 could cause inhibition of DNase activity, and PARP (a DNA repair enzymatic protein) to foster genome repair (172–174). In contrast, ferroptosis is characterized by the accumulation of ROS beyond the cell’s redox balance, which can cause lipid peroxidation followed by cell death (175). Administration of compounds like glutathione (GSH) or Fer-1 can facilitate ferroptosis reversal, potentially by enhancing GPX4 activity to mitigate ROS accumulation (176), while Fer-1 acts as a ROS scavenger to eliminate excessive cytosolic and lipid ROS, which could confer to the redox homeostasis and enabling cell recovery (177, 178).
Though glutathione (GSH) and Fer-1 are considered as inhibitors of ferroptosis, not all ferroptosis inhibitors possess the capacity to facilitate its reversal. For instance, compounds like aminooxyacetic acid (AOA), the iron chelator deferoxamine (DFO), dopamine, and vitamin C are known to inhibit ferroptosis initiation but do not promote cell recovery once ferroptosis has initiated. This limitation may stem from their targeting of upstream pathways involved in ferroptosis initiation, instead of conferring to the modulation of other downstream signaling pathways responsible for cell death. DFO functions as an iron chelator, depleting iron levels and hindering the iron-dependent accumulation of lipid ROS (19). Similarly, AOA inhibits transaminase activity, thereby blocking glutamine metabolism to alpha-ketoglutarate implicated predominantly involved in fatty acid synthesis (179). Dopamine prevents GPX4 degradation, while vitamin C scavenges free radicals in the aqueous phase (180–182). Previous studies (178) on the ferroptosis reversal through GSH and Fer-1 implicate regulators linked to GPX4 activity and lipid peroxidation as potential compounds that can induce ferroptosis reversibility. Unlike GSH and Fer-1, which act on mechanisms associated with removing lipid ROS, these inhibitors predominantly target the initiation of lipid peroxidation. This suggests potential differences in the regulation of preventing reversal of ferroptosis that warrant further investigation.
Several previous studies described the reversal of apoptosis when the removal of apoptotic stimuli is induced in vitro and in vivo studies (172–174, 178, 183–188). In contrast, another study (178) reveal that simply removing ferroptosis-inducing stimuli, including ‘erastin’ from HT-1080 cells or ‘erastin/glutamate’ from HT-22 cells, is insufficient to cause recovery of these ferroptosis-initiated cells. In this study (178), authors demonstrated the need to explore the possibility that certain types of dying cells with robust redox balance restoration capabilities may recover without the supplementation of GSH or Fer-1, a possibility that requires further investigation.
Ferroptosis can be reversed but raises fundamental inquiries that remain unanswered. For instance, can ferroptosis reversal occur within live animals? Tracking ferroptosis in vivo presents technical challenges, as cells recovered from ferroptosis appear morphologically identical to healthy non-ferroptotic cells. During the reversal of ferroptosis or apoptosis, cells encounter distinct forms of cellular damage. The existence of a common master regulator or signaling pathway triggering anastasis remains to be determined. Additionally, the broader physiological & therapeutic implications of this cellular recovery process require further exploration. Ongoing investigations aim to address these questions and identify the molecular regulators involved in these pathways. Understanding the signaling mechanisms and compounds that modulate ferroptosis and the reversal of ferroptosis could generate new approaches to investigate and comprehend the cell recovery process of anastasis (178).
Chemotherapy is a widely used treatment strategy aimed to ameliorate cancer cell proliferation (189, 190); however, some cells can survive chemotherapy, leading to cancer recurrence and metastasis (191–193). Even though various mechanisms, such as alterations in chemotherapeutic metabolism, changes in gene expression, metabolic reprogramming, stemness development, and changes in cell death pathways are significant influential factors which have been implicated in chemotherapy resistance (194–196), however, the precise underlying mechanisms remain unclear.
Apoptosis, a key mechanism targeted by chemotherapy, involves the activation of caspases, a group of cysteine proteases, ultimately leading to cell death. Previously, activation of executioner caspases was thought to be irreversible, marking a “point of no return” in apoptosis (197–199). Previous reports revealed anastasis process, wherein cells could refrain from apoptotic stress although executioner caspase activity is evident (174, 184, 200). Anastasis, or cellular survival following stress-induced activation of executioner caspases, has been documented in a subset of mammalian cell lines following exposure to various chemical stressors and chemotherapeutic agents (172, 173, 184, 201–204).
Studies on different cancer cell types, including breast (108, 205–208), melanoma (117), cervical, and ovarian cancers (209), have shown that anastasis confers new traits upon cancer cells, such as increased drug resistance and migration (172, 173, 201–204, 210). For instance, anastatic breast and cervical cancer cells are associated with enhanced chemoresistance and migration capabilities (203). Similarly, melanoma cells surviving executioner caspase activation caused through the upregulation in the expression of transient tBid or exposure to the dacarbazine (a chemotherapeutic drug) demonstrate elevated cell migration in vitro and increased metastasis in vivo.
According to previous reports (202), a lineage tracing system was employed to identify and isolate cells undergoing executioner caspase activation and their progeny. These studies suggest that anastatic colorectal cancer cells exhibit a higher metastasis, and chemoresistance due to increased upregulation in the expression of cIAP2 and NF-κB activity. According to this study, NF-κB activation and cIAP2 expression conferred by chemotherapeutics are crucial for anastasis (202). Exposure to chemotherapeutic drugs triggers NF-κB activation and upregulation of cIAP2 expression in colorectal cancer cells, promoting anastasis. Therefore, activated NF-κB and cIAP2 establish a positive feedback loop within anastatic cells, further enhancing migration and metastasis (202).
Thus, anastasis cause colorectal cancer cells with heightened migratory potential. Similar observations of higher metastasis following survival from stress-induced executioner caspase activation were evident in various tumor types, including cervical (173), breast (203), melanoma (202), and ovarian cancers (204), suggesting that a higher migration may represent a typical phenotypic alteration associated with anastasis. Furthermore, apoptosis and executioner caspases are typically involved in mediating the metastasis of lymph nodes (211–213). However, anastatic cells maintained enhanced migration independently of apoptotic cells, suggesting that executioner caspase activity is dispensable for sustaining increased migratory capacity in anastasis (214, 215). This observation aligns with previous reports on melanoma cells (202).
Various cancer cells exhibit increased migration following anastasis, with diverse underlying molecular mechanisms. Inhibition of TGF-β signaling moderately mitigated anastasis-induced migration in HeLa cells following brief ethanol exposure (173). Another report showed that the blocking of nuclear export reversed heightened migration in anastatic breast cancer cells (203). Similarly, a previous study (202) concluded that melanoma cells surviving executioner caspase activation resulted in augmented motility through the JNK hyperactivation.
Ru Wang et al., 2013 (216) revealed that cIAP2 enhances migration in anastatic colorectal cancer cells in an NF-κB-dependent manner, emphasizing role of cIAP2 as a positive regulator of migration. According to previous reports, NF-κB has a significant role in proliferating cancer cells and promotes migration or metastasis by modulating the downstream signaling cascades including STAT3 and MMPs (217–219). cIAP2 has previously been associated with drug resistance in pancreatic cancer, colorectal cancer (220, 221), and oral squamous cell carcinoma (222, 223), further supporting its role in mediating chemoresistance. A positive feedback loop between cIAP2 and NF-κB fostered levels of cIAP2 expression and NF-κB activity in anastatic cells, enhancing their migratory potential. Thus, cIAP2/NF-κB signaling and anastasis are critical regulators of post-chemotherapy metastasis (216). Additionally, Ru Wang et al., 2013 revealed (216) demonstrated a cIAP2-dependent increase in chemoresistance in anastatic colorectal cancer cells.
Studies on Drosophila notum epithelium demonstrated that (224) the ability of stressed cells to undergo anastasis may be relied upon executioner caspase activation. By inhibiting caspase-3 and 7, cIAP2 can foster a higher executioner caspase activity (225, 226), thereby promoting anastasis. Molecular mechanisms governing the anastasis regulation through cIAP2, NF-κB, and regulators which were already investigated in previous studies warrant further elucidation in future research for several other chemoresistant cancers.
Drug-resistant cancer cells, along with neighboring stromal cells, may create a protective environment, allowing vulnerable cancer cells to evade ferroptosis and chemotherapy. For example, cancer-associated fibroblasts (CAFs) could foster a reduction in cisplatin accumulation, subsequently conferring resistance to both chemotherapy and ferroptosis (21). Ferroptosis is intricately controlled by a multifaceted network involving epigenetic, pre-transcriptional, post-transcriptional, and post-translational modifications (227). Tumor cells, characterized by heightened sensitivity to signals regulating cell survival and death, consequently cause increased susceptibility to ferroptosis. Various factors that promote tumor growth, the buildup of iron, enhanced lipid synthesis, or undergoing epithelial-to-mesenchymal transition (EMT), can augment susceptibility of tumor cells to ferroptosis. Additionally, numerous chemotherapeutic agents elicit anti-cancer efficacy through the induction of oxidative stress, disrupting the anabolism of genetic components thereby synergizing with ferroptosis to ameliorate cancers. Nonetheless, the precise targeting of tumor cells by promoting ferroptosis induction remains a challenging task in current oncological strategies (9). Tumor drug resistance encompasses several intricate mechanisms, with disruption of redox homeostasis emerging as a pivotal contributor. Tumor cells exhibit greater resistance to oxidative stress by modulating ROS generation, thereby acquiring drug resistance (228). Ferroptosis typically linked with mitochondrial oxidative stress and ROS production, plays a significant role. Alterations in oxidative stress regulation can modulate ferroptosis, consequently influencing tumor cell sensitivity to chemotherapeutics. This association depends upon a nuanced balance between lipid peroxidation and the accumulation of ROS promote ferroptosis, thereby suppressing tumor proliferation; as we discussed the reduction of lipid peroxidation and ROS levels facilitates tumor cell survival, promoting resistance to anti-tumor agents. The dynamic interplay between ROS generation and scavenging mechanisms lies at the core of this relationship, which require future studies pertinent to several chemoresistant cancers (9).
As we discussed previously, chemotherapy remains a significant therapeutic modality treating numerous solid malignancies, but the challenge of chemoresistance significantly limits its effectiveness (229). Interestingly, a significant link has been observed between resistance to cisplatin and resistance to ferroptosis within tumor environments. Malignant cells resistant to cisplatin often exhibit increased expression of genes associated with ferroptosis resistance (230–232). For instance, cisplatin-resistant cancer cells overexpress the Wnt pathway membrane receptor, frizzled homolog 7 (FZD7), leading to activation of TP63 and subsequently enhanced GPX4 expression (233, 234). Various cancer types develop cisplatin resistance by upregulating system Xc− expression and maintaining high levels of GSH (235–237). Recent studies indicate that cisplatin treatment could induce ferroptosis in tumor cells, suggesting the significant efficacy of chemotherapy in triggering ferroptosis (238, 239).
CAFs have been implicated in reducing cisplatin accumulation, thereby promoting resistance to both chemotherapy and ferroptosis by supplying glutathione (GSH) and cysteine to ovarian cancer cells (240). Another study described that upregulation of miR-4443 in NSCLC cells, and undergoes exosomal transfer to sensitive cells, and enables upregulation of FSP1 expression. This elevated FSP1 expression is crucial to evade ferroptosis. Conversely, CD8+ T cells induce the generation of IFN-γ subsequently reduces system Xc− activity in cancer cells, thus fostering reduction in cisplatin resistance through modulation of the JAK/STAT pathway (240).
The significant interplay between chemoresistance and the underlying ferroptosis mechanisms is yet to be explored vividly to develop novel therapeutic modalities against ferroptosis-resistant tumor cells (239). Combinatorial administration of GPX4 inhibitors and system Xc− inhibitors & cisplatin has been shown to overcome chemotherapy resistance, enhancing the anti-cancer response in various tumors (238, 241, 242). However, these mechanisms require future studies in several other chemoresistant cancer types.
Chemoresistance specific to the anti-glioma agent, temozolomide (TMZ) is typically linked to resistance to ferroptosis induced by the overexpression of system Xc− and GSH (243, 244). Moreover, cancer cells can alter the iron homeostasis by lipocalin-2, subsequently causing ferroptosis resistance to 5-fluorouracil (245). The phenotypes of chemoresistant tumors closely mimic resistance to ferroptosis, particularly focusing on crucial targets such as GPX4, system Xc−, FSP1, and NRF2 (6, 246–249). Hence, it is crucial to profile these ferroptosis targets in chemoresistant tumors (Figure 4).
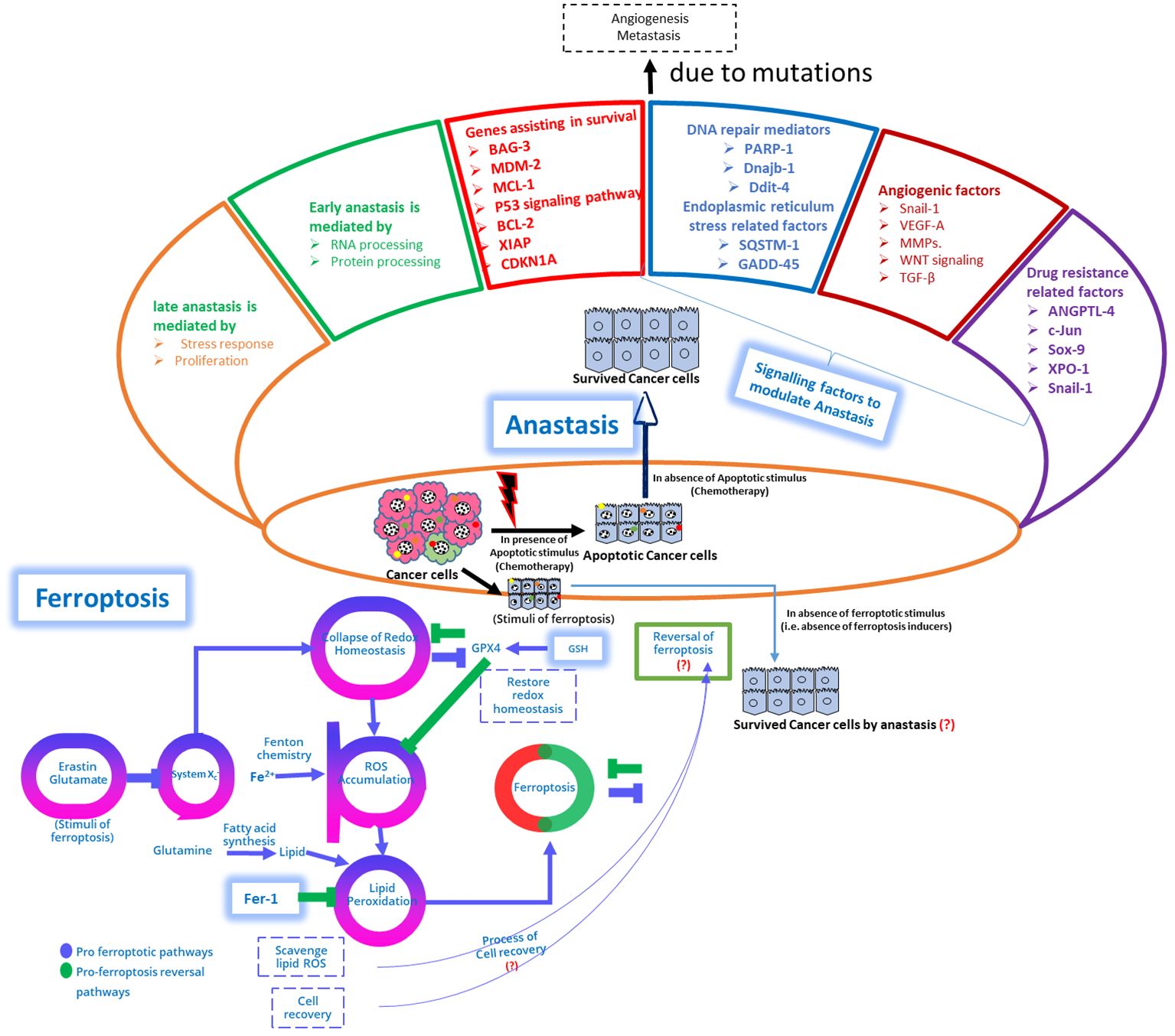
Figure 4 Schematic depiction of genomic variations within genes regulating apoptosis are evident in cancer and contribute to the activation of pro-proliferation and pro-survival pathways, leading to apoptosis resistance. Upper Panel: While apoptosis has traditionally been regarded as irreversible, emerging evidence suggests its potential reversibility, termed anastasis, wherein cells can escape apoptotic signals even after reaching advanced stages. Anastasis poses challenges for chemotherapy development and utilization. This figure demonstrates the multifaceted role of survival mechanisms, metastasis, epithelial-mesenchymal transition (EMT), and DNA damage repair in facilitating anastasis, and the mechanistic interplay between ferroptosis reversal and anastasis to modulate chemoresistance, (?) emphasizing its implications for drug resistance and therapeutic strategies. Lower Panel: A proposed model illustrates the reversal of ferroptosis, and the interplay between pro-ferroptosis reversal pathways and pro-ferroptosis pathways is discussed. The schematic representation depicts structural and molecular changes during anastasis, showing cancer cells recovering from apoptosis by activating anastasis and acquiring mesenchymal features and molecular alterations promoting survival, metastasis, and angiogenesis. Genes associated with anastasis-mediated drug resistance, including ATF3, c-FOS, c-JUN, INBHA, SNAIL-1, ANGPTL4, and SOX-9, are highlighted. Cells recovering from apoptosis through anastasis exhibit pronounced structural changes, alter focal adhesion kinases, activate genes related to the actin cytoskeleton, and elevate proteins associated with chemoresistance. In addition, the interplay between the anastasis, and mitochondrial-ROS modulated ferroptosis proposed mechanisms should be explored to demonstrate the underlying mechanisms pertinent to chemoresistance with anastasis, and ferroptosis reversal (178, 250).
Yet, the adaptive reaction to chemotherapeutics includes the activation of MTDH gene, promoting the mesenchymal phenotype of tumors and augmenting chemoresistance. However, diminishing the expression of GPX4 and system Xc− can increase susceptibility to ferroptosis. Thus it has been demonstrated that the ferroptosis resistance emerges as a new rationale underlying tumor recurrence following chemotherapy (21).
The surgical intervention combined with adjuvant multi-drug chemotherapy is another strategy of therapeutic modality for patients diagnosed with pancreatic ductal adenocarcinoma to enhance survival rates. However, the persistence of gemcitabine resistance in PDAC cells, continues to challenge clinicians (251). Recent insights suggest a correlation between gemcitabine resistance (252, 253) and ferroptosis resistance, primarily driven by the hyperexpression of system Xc−. Notably, inhibiting system Xc− in PDAC cells leads to glutathione (GSH) depletion in vitro, triggering ferroptosis and restoring sensitivity to both gemcitabine and cisplatin (254).
Changes in apoptosis-regulating genes are common in cancer, promoting activation of pro-proliferation and pro-survival pathways, leading to resistance against apoptosis. Contrary to apoptosis irreversibility, emerging evidence suggests its potential reversibility, termed anastasis, enabling cells to evade apoptotic signals even at advanced stages, challenging chemotherapy development (178, 250). It is crucial to explore the multifaceted role of survival mechanisms, metastasis, EMT, and DNA damage repair in facilitating anastasis. It also explores the interplay between ferroptosis reversal and anastasis in modulating chemoresistance, highlighting implications for drug resistance and therapeutic strategies. Future studies are warranted to examine ferroptosis reversal, and the interaction between pro-ferroptosis reversal and pro-ferroptosis pathways. The schematic representation Figure 4 delineates structural and molecular changes during anastasis, with cancer cells activating anastasis to recover from apoptosis, acquiring mesenchymal features, and molecular alterations promoting survival, metastasis, and angiogenesis. Genes linked to anastasis-mediated drug resistance, such as ATF3, c-FOS, c-JUN, INBHA, SNAIL-1, ANGPTL4, and SOX-9, are emphasized (178, 250). Cells recovering from apoptosis via anastasis undergo notable structural changes, altering focal adhesion kinases, activating genes associated with the actin cytoskeleton, and increasing chemoresistance-associated proteins. Additionally, exploring the interplay between anastasis and mitochondrial-ROS modulated ferroptosis mechanisms can elucidate underlying mechanisms of chemoresistance with anastasis and ferroptosis reversal (178, 250)
Intricate interplay pertinent to the anastasis, chemoresistance, and ferroptosis resistance in tumors require studies related to therapeutic efficacy of several therapeutic strategies. The recurrence of tumors following chemotherapy may, in part, be attributed to ferroptosis resistance. This underscores the significant chemotherapeutic efficacy of combinatorial regimens. In this context, targeting the tumor microenvironment (TME) and metabolic pathways to enhance ferroptosis sensitivity in malignant cells could emerge as a significant therapeutic strategy for overcoming the challenges posed by chemoresistance (21).
Emerging evidence indicates that ferroptosis can mitigate acquired resistance to several targeted therapies, including lapatinib, erlotinib, trametinib, dabrafenib, and vemurafenib (255–257). Notably, tumor cell lines resistant to these targeted drugs often exhibit markers of EMT and display heightened susceptibility to ferroptosis (255–257). EMT, characterized by the loss of epithelial characteristics and acquisition of mesenchymal properties, is known to confer chemoresistance in tumor cells, driven by transcription factors such as TWIST1 and ZEB1. For instance, β-elemene has been shown to sensitize KRAS mutant colorectal cancer cells to cetuximab through a ferroptotic mode of cell death while blocking EMT (257). Thus, ferroptosis could modulate chemoresistance to the chemotherapeutics and appear intricately linked to EMT processes.
Additionally, agents targeting PD-1 and PD-L1 garnered approval from the FDA for treating various malignancies. Anti-PD-L1 antibodies have been observed to foster lipid peroxide-mediated ferroptosis in proliferating cancer cells, with the efficacy of anti-PD-L1 antibody therapy being compromised by ferroptosis inhibitors (258). Combining anti-PD-L1 antibodies with ferroptosis inducers has shown significant efficacy in inhibiting tumor growth, attributed to the release of interferon-γ by cytotoxic T cells, which activates STAT1 and inhibits xCT expression, thereby promoting ferroptosis (258). Implicating ferroptosis inducers that stimulate STAT1 activation may overcome tumor resistance to immunotherapy, providing a wide range of potential for the clinical implementation of ferroptosis inducers in combination with immune checkpoint inhibitors (ICIs) (9).
Cancer stem cells (CSCs) pose significant challenges for developing new therapies due to their strong association with tumor relapse and chemoresistance (259, 260). Cells that are undergoing anastasis exhibit resistance to various apoptotic phases. Previous reports in cancer metastasis demonstrated that cells recovering from apoptotic phase show a higher CD44 expression, a key marker that signifies chemoresistance, and these cells exhibit properties similar to CSCs. Additionally, the apoptosis reversal induces epigenetic modifications in the gene promoters related to CD44 and CD24 genes (201), further contributing to their stem-like characteristics. It is crucial to examine functional role of anastatic mechanisms for the formation and maintenance of CSCs by the prospective studies. Anastasis harnesses conventional prosurvival and pro-metastatic factors to impair the apoptosis process. Actuation of EMT and the related EMT modulators is considered as crucial aspect to modulate cell survival and for maintaining stemness (261, 262). For instance, among these modulators, SOX-9, a gene involved in tumor growth, angiogenesis, differentiation, and survival, plays a critical role. SOX-9 is also linked to chemoresistance against gemcitabine, underscoring its significance in chemoresistance and the need for exploring its role in anastasis subsequently to identify targeted therapeutic strategies to overcome these challenges (263, 264).
Understanding the molecular signatures of anastasis through preclinical or clinical studies is a significant aspect in cancer research (172). Anastasis was observed in mouse models where primary murine liver cells and NIH3T3 mouse embryonic fibroblast cells were exposed to ethanol to induce apoptosis. Following the treatment, cells were subjected to washing and allowed to recovery phase in apoptosis by exposing them into a fresh media (172, 184). Transcriptomic analysis from the cells obtained from recovery phase of this apoptosis revealed a significant upregulation of genes linked to survival pathways; these genes are BCL-2 family members (BAG-3, BCL-2, and MCL-1), XIAP, MDM2. In addition, the genes related to heat shock proteins including Dnajb1, Hsp90aa1, Hspa1b, and Hspb1 were found to be upregulated in this recovery phase (172, 184) (Figure 4). Notably, residual apoptotic markers dissipated during early phase of anastasis initiation. Inhibiting key survival factors such as Hsp90, MDM2, XIAP, and BCL-2 significantly impaired anastasis, highlighting the significant dependency on these cellular survival mechanisms (172, 184).
Cancer cell survival is not solely dependent on anti-apoptotic gene expression. It also relies on genes involved in regulating cell cycle, and DNA repair; In addition, the survival of these proliferating cancer cells relies on gene expression pertinent to angiogenesis, migration, and TGF-β modulation, and these genes are reported to be upregulated at the time of early anastasis phase (172). Previous reports on HeLa cells recovering from ethanol-induced apoptosis revealed significant increases in c-Fos, c-Jun, Klf4, and Snail-1 expression (Figure 4). These findings imply that the upregulated genes at the recovery phase and post-apoptosis induction are primarily linked to proliferation and pro-survival signaling (173). On the contrary, genes activated during late anastasis predominantly affect actin cytoskeleton rearrangement, and ribosome biogenesis (Figure 4) (173, 200). At the time of anastasis, the involvement of several survival factors includes MDM2 revealing that the p53 signaling implications include CDKs, expression of E2F and cell cycle factors (265). Other investigations into the AP-1 transcription factor actuation, which includes c-Jun and c-Fos; these genes are significantly involved in mediating pro-apoptotic and pro-survival factor expression. Hence, their actuation during anastasis offers insights into the balance between pro-survival and pro-apoptotic signaling which is yet to be explored vividly (266–268).
Many proteins that play roles in promoting cell proliferation and preventing apoptosis are also critical for recovering cells from death, which indicates that cancer cells can trigger survival mechanisms under stress conditions, resulting in significant molecular transformations that restore the cell to its normal state (173, 183). During anastasis, structural changes such as actuation of focal adhesion kinases and reorganization of the actin cytoskeleton (173, 183). Due to these changes, recovered cancer cells undergo migration. Furthermore, Snail-1 expression is significantly increased during the initial anastasis phase. When Snail-1 is silenced using shRNA, the recovery process, including migration, is impaired, leading to increased PARP-1 cleavage and sustained activation of cell death pathways (173, 183). Early phase of cell recovery involves the coordinated actuation of TGF-β signaling, which is crucial for the survival of the cells followed by migration. Additionally, MMP-9, MMP-10, MMP-13, VEGF-A, and Angpt-l4 are other EMT and angiogenic factors that could play a significant functional role in promoting the cell recovery from cell death (173, 183).
Furthermore, EMT plays a crucial role in modulating the process of metastasis, anastasis, and stemness, making it a promising target for combinatorial drug design (81) (250, 269). Salinomycin, mocetinostat, and metformin could effectively target EMT and they were studied significantly for their potential to overcome chemoresistance when used alongside drugs like doxorubicin, 5-FU, and gemcitabine (270–276). Trichostatin A and vorinostat are HDAC inhibitors that can inhibit EMT by downregulating E-cadherin expression and modulating the expression of HIF-1α and NF-κB (277–280). In cancers with EGFR mutations, diindolylmethane (DIM) and its analogs have demonstrated potential in preventing EMT and metastasis, suggesting their suitability for use in combinatorial regimens along with DNA-damaging agents (281, 282). Combinatorial regimen of DIM with camptothecin can prevent drug-induced EMT in the murine models associated with Apc-floxed colorectal cancers (283). Recurrent administration of therapies such as chemo/radio-therapies can cause side effects by co-activating DNA repair followed by cell survival and EMT pathways, which are crucial in anastasis. For instance, the nuclear translocation of nucleases pertinent to apoptosis such as AIF and Endo-G are evident at the time of anastasis when observed during reversal of ethanol-mediated DNA damage (283).
Critical involvement of DNA damage response (DDR) proteins during anastasis yet require future studies for several other cancers. DDR proteins, such as ATM, ATR, and DNA-PK, are known to play pivotal roles in detecting and repairing DNA damage, thereby maintaining genomic stability. Their activation during anastasis suggests a potential mechanism by which cells recover from near-death states and restore proliferation (184). Given the established link between robust DNA repair mechanisms and poor cancer prognosis (284), it is imperative to further investigate how these pathways contribute to anastatic processes and chemoresistance. Elucidating these mechanisms may reveal new therapeutic targets to improve cancer treatment outcomes, particularly in tumors that exhibit high levels of DNA repair activity. Studies should focus on understanding the specific DDR pathways (284) involved in anastasis and their impact on the efficacy of chemotherapeutic agents, potentially leading to the development of novel strategies to overcome resistance.
Cancer cells exert chemoresistance when administered recurrent cytotoxic chemotherapeutics to several cancer types, and EMT plays a pivotal role in mitigating drug susceptibility (270, 285–287). At the time of treating cancer cells with DNA-damaging agents, specifically epithelial-origin cancer cells start expressing mesenchymal markers. As previously discussed, EMT is closely associated with survival factors, in addition, the drug-induced EMT leads to a significant activation of survival factors such as NF-κB, c-FLIP, survivin, as well as AKT. Notably, markers pertinent to early apoptosis were found to occur alongside EMT and cell survival proteins. Moreover, EMT modulator vimentin was shown to hinder process of apoptosis (283). EMT is significantly higher in drug-induced metastasis than in spontaneous metastasis. For instance, autochthonous breast cancer murine model was studied where transgenic mice with a Cre recombinase construct driven by the Fsp-1 promoter (288) which would express Cre recombinase in mesenchymal lineage cells undergoing EMT. This system was designed so that when the Fsp-1 promoter was actuated, the Cre recombinase could induce knock out of RFP in another transgenic construct that included GFP (288). Consequently, cells expressing GFP indicated EMT activation. According to this study, spontaneous lung metastasis had few GFP-positive cells, whereas cyclophosphamide-treated mice had significant GFP expression in metastatic lung nodules (288). GFP-positive cells confined to the regions of metastasis exhibited a higher chemoresistance than the GFP-negative cells (288). These findings highlight the complexity of drug-induced EMT and in vivo actuation of anastasis.
As previously discussed, NF-κB plays a critical role in promoting cell survival, making it a key target in emerging cancer therapies. Bortezomib, for instance, inhibits NF-κB by preventing the degradation of IκB, thereby suppressing NF-κB signaling pathways. This therapeutic approach in combinatorial regimen with temozolomide and paclitaxel resulted in promising results (289). Furthermore, several NF-κB inhibitors such as curcumin, BMS-345541, and bindarit exhibit significant efficacy (290). These compounds could be particularly effective when used as combinatorial regimen with DNA-damaging drug molecules to counteract anastasis, a process through which cells survive and recover from apoptosis phases. This combination strategy aims to enhance the efficacy of cancer treatments by targeting the molecular mechanisms involved in evading apoptosis and chemoresistance. Further research and clinical trials are essential to validate these findings and optimize combination therapies for better clinical outcomes.
Previous studies reported the combinatorial regimen of immunotherapeutics along with novel ferroptosis modulators against cancers. Anti-cancer immunity is higher with GPX4 inhibitors against triple-negative breast cancers (TNBCs) specifically LAR subtype. The synergistic effect of GPX4 inhibition and PD-1 blockade demonstrates a higher efficacy compared to individual therapeutic interventions in vivo settings (291). In glioma, wherein ferroptosis serves as the predominant mechanism of programmed cell death (PCD), it fosters an immunosuppressive tumor microenvironment facilitated by tumor-associated macrophages, thereby promoting malignant progression and unfavorable clinical outcomes. Targeting ferroptosis inhibition is suggested as a promising strategy to augment the immune checkpoint blockade (ICB) efficacy (292).
In melanoma, a predictive model termed the Ferroptosis Score (FPS) was developed utilizing 32 ferroptosis-related genes to prognosticate cancer outcomes. Elevated FPS levels are indicative of a more dynamic tumor immune microenvironment and enhanced responsiveness to immune checkpoint blockade (ICB), such as PD-1 blockade (293). BEBT-908, an inhibitor targeting PI3K and HDAC, induces immunogenic ferroptotic cell death in cancer cells, augmenting ICB therapy efficacy by upregulating MHC class I expression and activating IFN-γ signaling (294). Statins facilitate immunotherapy by inducing ferroptosis in non-small cell lung cancer cells while inhibiting PD-L1 expression (295). Inhibition of Ferroptosis Suppressor Protein 1 (FSP1) significantly induces ferroptosis in cancer cells and promotes immune cell infiltration, including dendritic cells, macrophages, and T cells. Combinatorial therapy with immunotherapy and FSP1 inhibition typically mitigates hepatocellular carcinoma burden in vivo (296). Inflammation-associated ferroptosis biomarkers positively correlate with PD-L1 expression, high microsatellite instability, tumor mutational burden, and ICB response rates (297). Short-term methionine starvation synergistically promotes ferroptosis with CD8+ T cells by stimulating CHAC1 transcription, enhancing ICB therapy. Combined treatment involving intermittent methionine starvation, system xc- inhibition and PD-1 blockade exhibits potent anti-tumor effects (298). Ferritin Light Chain (FTL) promotes cancer cell ferroptosis in glioblastoma, polarizing tumor-associated macrophages towards the M2-type, thus fostering a pro-tumor TME. Inhibiting FTL represents a promising strategy to sensitize glioblastoma to PD-1 blockade (299). However, further investigations are warranted to evaluate the efficacy of these ICBs in combination with ferroptosis modulators against chemoresistant cancers (21).
Recent advancements in cancer research and development have significantly shifted the focus from merely identifying anti-cancer compounds to unraveling the intricate molecular mechanisms underpinning cell survival and drug resistance. Current studies described the molecular signaling pathways between tumors and their microenvironment, examining how these interactions affect cellular survival and resistance to therapy-induced apoptosis. This review highlights various cellular mechanisms that enable cancer cells to evade apoptosis and acquire oncogenic phenotypes. Two pivotal concepts emerging from this research are mitochondria-modulated ferroptosis in cancer cells, anastasis, which introduces a novel perspective on cancer-related therapies by fostering novel investigations on chemotherapeutics or immunotherapeutics to mitigate chemoresistant cancers.
As previously highlighted, ferroptosis is characterized by iron-dependent toxicity, excessive ROS accumulation, and consequent lipid peroxidation, disrupting cellular redox balance and culminating in membrane damage, ultimately triggering cell death (10, 102, 300). Excessive oxidative stress induces irreversible damage to mitochondrial function and integrity, leading to energy depletion and ferroptosis (88). Ferroptosis manifests distinct morphological, genetic, and biochemical features when compared to apoptosis, necrosis, and autophagy (12, 108, 301). Numerous studies have implicated ferroptosis in diverse biological processes and pathogeneses, prominently in cardiovascular diseases, brain lesions, renal impairment, and various malignancies, where it plays a pivotal regulatory role within the tumor microenvironment (63, 302–307).
Additionally, cancer cells exhibit a higher demand for iron metabolism compared to normal cells, rendering them more susceptible to ferroptosis induction (305, 308). Research further suggests a close nexus between ferroptosis and the inhibition of tumor cell growth. Activation of ferroptosis can impede cancer cell proliferation, thereby influencing the efficacy of tumor immunotherapy and patient prognosis (309–313). Consequently, targeting or inducing ferroptosis in cancer cells is emerging as a novel therapeutic strategy (314). This comprehensive review elucidated the intricate interplay between ferroptosis, mitochondria, and iron metabolism, underscoring the potential translational impact of these findings in oncology.
In prostate cancer (PCa), it has been proposed that the ferroptosis initiators erastin and RSL3 could profoundly inhibit PCa cell progression without adverse events (315–317). In pancreatic cancer, gemcitabine is the primary treatment, albeit with unsatisfactory efficacy (167). Heat shock protein family A5 (HSPA5) has been closely linked to the prognosis of gemcitabine-treated patients (318). Zhu et al. (318) suggested that HSPA5-GPX4 pathway modulation to contribute to chemoresistance in pancreatic cancer cells against gemcitabine, and blocking of HSPA5 or GPX4 gene expression potentially reversing this resistance. Ferroptosis has also been implicated in this process.
In hepatocellular carcinoma (HCC), sorafenib remains the principal chemotherapeutic drug. Sun et al. (319) found that sorafenib could induce HCC cell death by increasing oxidative stress. However, the ferroptosis inhibitor ferrostatin-1 could counteract this process, indicating the significance of ferroptosis in the mechanism of action of sorafenib. Furthermore, a combination of erastin, sorafenib, and haloperidol could induce a higher intracellular iron concentration, inducing the Fenton reaction and excessive ROS production, ultimately leading to HCC cell ferroptosis (320).
In TNBC, Ding et al. (321) demonstrated that the inhibitor DMOCPTL ubiquitinated GPX4 through EGR1 regulation, inducing TNBC cell ferroptosis. The fundamental mechanism of action and clinical value of ferroptosis remain incompletely understood. Indeed, potential valid ferroptosis-related biomarkers warrant further investigation. Moreover, it remains unclear in clinical treatment whether inducing ferroptosis in tumor cells would impair liver and kidney functions in patients. Developing ferroptosis-inducing agents capable of selectively eliminating chemoresistant tumor cells while sparing normal cells remains an area requiring further exploration. Basic research and clinical translation of ferroptosis encounters myriad uncertainties and obstacles. As foundational research on ferroptosis progresses, the clinical application of ferroptosis inducers and inhibitors may become viable. Thus, research endeavors focused on ferroptosis hold considerable potential to confer substantial benefits to cancer patients with chemoresistance in the future by exploring the interplay between ferroptosis, anastasis, and chemoresistance. Specifically, can the reversal of ferroptosis manifest in vivo within live animals? Tracking ferroptosis in vivo poses technical challenges, given the morphological similarity between cells recovered from ferroptosis and healthy non-ferroptotic cells. Additionally, while cells undergoing reversal of ferroptosis or apoptosis must address distinct forms of cellular damage, it is plausible that they may share a common master regulator or signaling molecule initiating the process of anastasis. Moreover, elucidating the long-term physiological, pathological, and therapeutic aspects of anastasis modulators remains imperative (178).
The fundamental mechanism underlying ferroptosis involves the interplay of iron metabolism and lipid peroxides. The identification of FSP1 as a negative regulator expands our understanding of ferroptosis. Extensive investigations into ferroptosis have unveiled numerous potential therapeutic targets for combating chemoresistant tumors. These discoveries hold promise for the development of novel drugs and treatment modalities targeting these specific pathways. Presently, resistance to conventional chemotherapy, targeted therapy, or immunotherapy agents represents a major challenge in the management of refractory tumors or recurrent malignancies. Given the ubiquitous nature of chemoresistance mechanisms, tumors that exhibit resistance to one drug often display varying degrees of resistance to others within the same class, with some even demonstrating multidrug resistance, significantly compromising treatment efficacy and patient survival. Multiple studies have demonstrated that ferroptosis can enhance tumor cell death synergistically with anti-tumor drugs, thereby augmenting drug efficacy and potentially reversing drug resistance, offering a promising therapeutic avenue for patients with drug-resistant tumors. Notably, tumor cells exhibit heightened sensitivity to iron overload and ROS accumulation compared to normal cells, rendering targeting of iron ions a feasible approach to enhance treatment efficacy and mitigate side effects.
Integration of cross-disciplinary technologies, such as nano-vehicle agents (322) combined with ferroptosis, may represent a more tailored approach to tumor treatment. However, several challenges must be addressed before clinical application. Firstly, while numerous in vitro methods exist for assessing ferroptosis and anastasis, the development of sensitive and accurate in vivo evaluation techniques remains elusive. Secondly, the specificity and safety profile of compounds targeting ferroptosis regulators must be significantly evaluated in preclinical and clinical settings to minimize adverse effects. Lastly, further elucidation is warranted regarding which tumor types or characteristics, such as specific gene mutations, are particularly susceptible to ferroptosis. Nonetheless, comprehensive investigations into the molecular signaling underlying ferroptosis, anastasis, and tumor drug resistance hold considerable significance to revolutionize tumor diagnosis and treatment, offering new avenues for therapeutic intervention (9). Continued research is warranted to develop personalized combinatorial therapeutic regimens against cancers to modulate these molecular signaling that optimize therapeutic efficacy and overall survival.
YC: Methodology, Investigation, Formal analysis, Data curation, Conceptualization, Writing – review & editing, Writing – original draft. CL: Writing – original draft, Formal analysis, Data curation. NB: Writing – original draft, Validation, Project administration, Data curation, Writing – review & editing, Supervision, Methodology, Investigation, Conceptualization. SE: Writing – review & editing, Project administration, Methodology, Investigation. ME: Writing – review & editing, Methodology, Investigation, Formal analysis. YF: Writing – original draft, Project administration, Methodology. XY: Writing – original draft, Software, Resources. BB: Writing – review & editing, Validation, Project administration. MH: Visualization, Resources, Project administration, Funding acquisition, Writing – review & editing, Supervision, Methodology. ZL: Validation, Software, Resources, Project administration, Methodology, Data curation, Conceptualization, Writing – review & editing, Writing – original draft, Visualization, Supervision, Investigation, Funding acquisition, Formal analysis.
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Fsp-1, fibroblast-specific protein 1; RFP, a red fluorescent protein gene; GFP, green fluorescent protein; CDKs, Cyclin dependent Kinases.
1. Qiu J, Yang G, Feng M, Zheng S, Cao Z, You L, et al. Extracellular vesicles as mediators of the progression and chemoresistance of pancreatic cancer and their potential clinical applications. Mol Cancer. (2018) 17:1–11. doi: 10.1186/s12943-017-0755-z
2. Beeraka NM, Doreswamy SH, Sadhu SP, Srinivasan A, Pragada RR, Madhunapantula SV, et al. The role of exosomes in stemness and neurodegenerative diseases—chemoresistant-cancer therapeutics and phytochemicals. Int J Mol Sci. (2020) 21:6818. doi: 10.3390/ijms21186818
3. Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci. (2003) 100:3983–8. doi: 10.1073/pnas.0530291100
4. Kumari P, Beeraka NM, Tengli A, Bannimath G, Kaur R, Patil M. Recent updates on oncogenic signaling of aurora kinases in chemosensitive, chemoresistant cancers: novel medicinal chemistry approaches for targeting aurora kinases. Curr Med Chem. (2024) 31(23). doi: 10.2174/0929867330666230503124408
5. Liu J, Zheng Q, Beeraka NM, Zhang X, Li T, Song R, et al. Long-term risk of subsequent Malignant neoplasms among childhood and adolescent lymphoma survivors (1975-2013): A population-based predictive nomogram. Oncologist. (2023) 28:e765–73. doi: 10.1093/oncolo/oyad112
6. Wang X, Liu J, Yn LD, Beeraka NM, Zhou R, Lu P, et al. Recent updates on the efficacy of mitocans in photo/radio-therapy for targeting metabolism in chemo/radio-resistant cancers: nanotherapeutics. Curr Med Chem. (2024). doi: 10.2174/0109298673259347231019121757
7. Liu Y, Beeraka NM, Liu J, Chen K, Song B, Song Z, et al. Comparative clinical studies of primary chemoradiotherapy versus S-1 and nedaplatin chemotherapy against stage IVb esophageal squamous cell carcinoma: a multicenter open-label randomized controlled trial. BMJ Open. (2022) 12:e055273. doi: 10.1136/bmjopen-2021-05527
8. Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. (1997) 3:730–7. doi: 10.1038/nm0797-730
9. Nie Z, Chen M, Gao Y, Huang D, Cao H, Peng Y, et al. Ferroptosis and tumor drug resistance: current status and major challenges. Front Pharmacol. (2022) 13:879317. doi: 10.3389/fphar.2022.879317
10. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. et al: Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. (2012) 149:1060–72. doi: 10.1016/j.cell.2012.03.042
11. Jiang X, Stockwell BR, Conrad M. Ferroptosis: mechanisms, biology and role in disease. Nat Rev Mol Cell Biol. (2021) 22:266–82. doi: 10.1038/s41580-020-00324-8
12. Chen X, Kang R, Kroemer G, Tang D. Broadening horizons: the role of ferroptosis in cancer. Nat Rev Clin Oncol. (2021) 18:280–96. doi: 10.1038/s41571-020-00462-0
13. Seibt TM, Proneth B, Conrad M. Role of GPX4 in ferroptosis and its pharmacological implication. Free Radic Biol Med. (2019) 133:144–52. doi: 10.1016/j.freeradbiomed.2018.09.014
14. Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, et al. et al: Regulation of ferroptotic cancer cell death by GPX4. Cell. (2014) 156:317–31. doi: 10.1016/j.cell.2013.12.010
15. Willems PH, Rossignol R, Dieteren CE, Murphy MP, Koopman WJ. Redox homeostasis and mitochondrial dynamics. Cell Metab. (2015) 22:207–18. doi: 10.1016/j.cmet.2015.06.006
16. Cheung EC, Vousden KH. The role of ROS in tumor development and progression. Nat Rev Cancer. (2022) 22:280–97. doi: 10.1038/s41568-021-00435-0
17. von Krusenstiern AN, Robson RN, Qian N, Qiu B, Hu F, Reznik E, et al. et al: Identification of essential sites of lipid peroxidation in ferroptosis. Nat Chem Biol. (2023) 19:719–30. doi: 10.1038/s41589-022-01249-3
18. Feng H, Stockwell BR. Unsolved mysteries: How does lipid peroxidation cause ferroptosis? PloS Biol. (2018) 16:e2006203. doi: 10.1371/journal.pbio.2006203
19. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. (2012) 149:1060–72. doi: 10.1016/j.cell.2012.03.042
20. Stockwell BR, Angeli JPF, Bayir H, Bush AI, Conrad M, Dixon SJ, et al. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell. (2017) 171:273–85. doi: 10.1016/j.cell.2017.09.021
21. Hua Y, Yang S, Zhang Y, Li J, Wang M, Yeerkenbieke P, et al. Modulating ferroptosis sensitivity: environmental and cellular targets within the tumor microenvironment. J Exp Clin Cancer Res. (2024) 43:19. doi: 10.1186/s13046-023-02925-5
22. Liu MR, Zhu WT, Pei DS. System Xc(-): a key regulatory target of ferroptosis in cancer. Invest New Drugs. (2021) 39:1123–31. doi: 10.1007/s10637-021-01070-0
23. Chen X, Yu C, Kang R, Kroemer G, Tang D. Cellular degradation systems in ferroptosis. Cell Death Differ. (2021) 28:1135–48. doi: 10.1038/s41418-020-00728-1
24. Lv H, Zhen C, Liu J, Yang P, Hu L, Shang P. Unraveling the potential role of glutathione in multiple forms of cell death in cancer therapy. Oxid Med Cell Longev. (2019) 2019:3150145. doi: 10.1155/2019/3150145
25. Lee SY, Ju MK, Jeon HM, Jeong EK, Lee YJ, Kim CH, et al. Regulation of tumor progression by programmed necrosis. Oxid Med Cell Longevity. (2018) 2018:3537471. doi: 10.1155/2018/3537471
26. Kaiser WJ, Sridharan H, Huang C, Mandal P, Upton JW, Gough PJ, et al. Toll-like receptor 3-mediated necrosis via TRIF, RIP3, and MLKL. J Biol Chem. (2013) 288:31268–79. doi: 10.1074/jbc.M113.462341
27. Zhao J, Jitkaew S, Cai Z, Choksi S, Li Q, Luo J, et al. Mixed lineage kinase domain-like is a key receptor interacting protein 3 downstream component of TNF-induced necrosis. Proc Natl Acad Sci USA. (2012) 109:5322–7. doi: 10.1073/pnas.1200012109
28. Dhuriya YK, Sharma D. Necroptosis: a regulated inflammatory mode of cell death. J Neuroinflamm. (2018) 15:199. doi: 10.1186/s12974-018-1235-0
29. Kerr J, Searle J. A suggested explanation for the paradoxically slow growth rate of basal-cell carcinomas that contain numerous mitotic figures. J Pathol. (1972) 107:41–4. doi: 10.1002/path.1711070107
30. Wyllie AH, Kerr JF, Currie AR. Cell death in the normal neonatal rat adrenal cortex. J Pathol. (1973) 111:255–61. doi: 10.1002/path.1711110406
31. Crawford AM, Kerr J, Currie A. The relationship of acute mesodermal cell death to the teratogenic effects of 7-OHM-12-MBA in the fetal rat. Br J Cancer. (1972) 26:498. doi: 10.1038/bjc.1972.67
32. Pisani C, Ramella M, Boldorini R, Loi G, Billia M, Boccafoschi F, et al. Apoptotic and predictive factors by Bax, Caspases 3/9, Bcl-2, p53 and Ki-67 in prostate cancer after 12 Gy single-dose. Sci Rep. (2020) 10:7050. doi: 10.1038/s41598-020-64062-9
33. Rahman N, Khan H, Zia A, Khan A, Fakhri S, Aschner M, et al. Bcl-2 Modulation in p53 Signaling Pathway by Flavonoids: A Potential Strategy towards the Treatment of Cancer. Int J Mol Sci. (2021) 22(21):11315. doi: 10.3390/ijms222111315
34. Cui Q, Yu JH, Wu JN, Tashiro S, Onodera S, Minami M, et al. P53-mediated cell cycle arrest and apoptosis through a caspase-3- independent, but caspase-9-dependent pathway in oridonin-treated MCF-7 human breast cancer cells. Acta pharmacologica Sin. (2007) 28:1057–66. doi: 10.1111/j.1745-7254.2007.00588.x
35. Dijkstra C. Nobel Prize for Physiology or Medicine 2002 is awarded for research into the genetic regulation of organ development and programmed cell death. Nederlands tijdschrift voor geneeskunde. (2002) 146:2525–7.
36. Cao L, Walker MP, Vaidya NK, Fu M, Kumar S, Kumar A. Cocaine-mediated autophagy in astrocytes involves sigma 1 receptor, PI3K, mTOR, atg5/7, beclin-1 and induces type II programed cell death. Mol Neurobiol. (2016) 53:4417–30. doi: 10.1007/s12035-015-9377-x
37. Li X, He S, Ma B. Autophagy and autophagy-related proteins in cancer. Mol Cancer. (2020) 19:12. doi: 10.1186/s12943-020-1138-4
38. Debnath J, Gammoh N, Ryan KM. Autophagy and autophagy-related pathways in cancer. Nat Rev Mol Cell Biol. (2023) 24:560–75. doi: 10.1038/s41580-023-00585-z
39. Levine B, Klionsky DJ. Autophagy wins the 2016 Nobel Prize in Physiology or Medicine: Breakthroughs in baker's yeast fuel advances in biomedical research. Proc Natl Acad Sci. (2017) 114:201–5. doi: 10.1073/pnas.1619876114
40. Harnett MM, Pineda MA, de Laté PL, Eason RJ, Besteiro S, Harnett W, et al. From Christian de Duve to Yoshinori Ohsumi: More to autophagy than just dining at home. Biomed J. (2017) 40:9–22. doi: 10.1016/j.bj.2016.12.004
41. Feng Q, Yu X, Qiao Y, Pan S, Wang R, Zheng B, et al. Ferroptosis and acute kidney injury (AKI): molecular mechanisms and therapeutic potentials. Front Pharmacol. (2022) 13:858676. doi: 10.3389/fphar.2022.858676
42. Wang H, Cheng Y, Mao C, Liu S, Xiao D, Huang J, et al. Emerging mechanisms and targeted therapy of ferroptosis in cancer. Mol therapy: J Am Soc Gene Ther. (2021) 29:2185–208. doi: 10.1016/j.ymthe.2021.03.022
43. Tang D, Kang R, Berghe TV, Vandenabeele P, Kroemer G. The molecular machinery of regulated cell death. Cell Res. (2019) 29:347–64. doi: 10.1038/s41422-019-0164-5
44. Chen X, Comish PB, Tang D, Kang R. Characteristics and biomarkers of ferroptosis. Front Cell Dev Biol. (2021) 9:637162. doi: 10.3389/fcell.2021.637162
45. Dolma S, Lessnick SL, Hahn WC, Stockwell BR. Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell. (2003) 3:285–96. doi: 10.1016/S1535-6108(03)00050-3
46. Yang WS, Stockwell BR. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem Biol. (2008) 15:234–45. doi: 10.1016/j.chembiol.2008.02.010
47. Ursini F, Maiorino M. Lipid peroxidation and ferroptosis: The role of GSH and GPx4. Free Radic Biol Med. (2020) 152:175–85. doi: 10.1016/j.freeradbiomed.2020.02.027
48. Toppo S, Vanin S, Bosello V, Tosatto SC. Evolutionary and structural insights into the multifaceted glutathione peroxidase (Gpx) superfamily. Antioxid Redox Signal. (2008) 10:1501–14. doi: 10.1089/ars.2008.2057
49. Maiorino M, Conrad M, Ursini F. GPx4, lipid peroxidation, and cell death: discoveries, rediscoveries, and open issues. Antioxid Redox Signal. (2018) 29:61–74. doi: 10.1089/ars.2017.7115
50. Ismail T, Kim Y, Lee H, Lee DS, Lee HS. Interplay between mitochondrial peroxiredoxins and ROS in cancer development and progression. Int J Mol Sci. (2019) 20(18):4407. doi: 10.3390/ijms20184407
51. Chen K, Zhang J, Beeraka NM, Tang C, Babayeva YV, Sinelnikov MY, et al. et al: advances in the prevention and treatment of obesity-driven effects in breast cancers. Front Oncol. (2022) 12:820968. doi: 10.3389/fonc.2022.820968
52. Zheng J, Conrad M. The metabolic underpinnings of ferroptosis. Cell Metab. (2020) 32:920–37. doi: 10.1016/j.cmet.2020.10.011
53. Rauen U, Springer A, Weisheit D, Petrat F, Korth HG, de Groot H, et al. Assessment of chelatable mitochondrial iron by using mitochondrion-selective fluorescent iron indicators with different iron-binding affinities. Chembiochem. (2007) 8:341–52. doi: 10.1002/cbic.200600311
54. Kholmukhamedov A, Li L, Lindsey CC, Hu J, Nieminen AL, Takemoto K, et al. A new fluorescent sensor mitoferrofluor indicates the presence of chelatable iron in polarized and depolarized mitochondria. J Biol Chem. (2022) 298:102336. doi: 10.1016/j.jbc.2022.102336
55. Chen C, Wang S, Liu P. Deferoxamine enhanced mitochondrial iron accumulation and promoted cell migration in triple-negative MDA-MB-231 breast cancer cells via a ROS-dependent mechanism. Int J Mol Sci. (2019) 20(19):4952. doi: 10.3390/ijms20194952
56. Lill R, Freibert SA. Mechanisms of mitochondrial iron-sulfur protein biogenesis. Annu Rev Biochem. (2020) 89:471–99. doi: 10.1146/annurev-biochem-013118-111540
57. Lv H, Shang P. The significance, trafficking and determination of labile iron in cytosol, mitochondria and lysosomes. Metallomics. (2018) 10:899–916. doi: 10.1039/C8MT00048D
58. Zhang B, Pan C, Feng C, Yan C, Yu Y, Chen Z, et al. Role of mitochondrial reactive oxygen species in homeostasis regulation. Redox report: Commun Free Radical Res. (2022) 27:45–52. doi: 10.1080/13510002.2022.2046423
59. Sun Y, Chen P, Zhai B, Zhang M, Xiang Y, Fang J, et al. et al: The emerging role of ferroptosis in inflammation. Biomedicine pharmacotherapy = Biomedecine pharmacotherapie. (2020) 127:110108. doi: 10.1016/j.biopha.2020.110108
60. Chen X, Kang R, Kroemer G, Tang D. Ferroptosis in infection, inflammation, and immunity. J Exp Med. (2021) 218(6):e2021051. doi: 10.1084/jem.20210518
61. Gao W, Wang X, Zhou Y, Wang X, Yu Y. Autophagy, ferroptosis, pyroptosis, and necroptosis in tumor immunotherapy. Signal Transduct Target Ther. (2022) 7:196. doi: 10.1038/s41392-022-01046-3
62. Xie Y, Hou W, Song X, Yu Y, Huang J, Sun X, et al. Ferroptosis: process and function. Cell Death Differ. (2016) 23:369–79. doi: 10.1038/cdd.2015.158
63. Li J, Cao F, Yin HL, Huang ZJ, Lin ZT, Mao N, et al. Ferroptosis: past, present and future. Cell Death Dis. (2020) 11:88. doi: 10.1038/s41419-020-2298-2
64. Torti SV, Torti FM. Iron and cancer: 2020 vision. Cancer Res. (2020) 80:5435–48. doi: 10.1158/0008-5472.CAN-20-2017
65. Ganasen M, Togashi H, Takeda H, Asakura H, Tosha T, Yamashita K, et al. Structural basis for promotion of duodenal iron absorption by enteric ferric reductase with ascorbate. Commun Biol. (2018) 1:120. doi: 10.1038/s42003-018-0121-8
66. Hsu MY, Mina E, Roetto A, Porporato PE. Iron: an essential element of cancer metabolism. Cells. (2020) 9(12):2591. doi: 10.3390/cells9122591
67. Fiorito V, Chiabrando D, Petrillo S, Bertino F, Tolosano E. The multifaceted role of heme in cancer. Front Oncol. (2019) 9:1540. doi: 10.3389/fonc.2019.01540
68. Kawabata T. Iron-induced oxidative stress in human diseases. Cells. (2022) 11(14):2152. doi: 10.3390/cells11142152
69. Yanatori I, Richardson DR, Toyokuni S, Kishi F. The new role of poly (rC)-binding proteins as iron transport chaperones: Proteins that could couple with inter-organelle interactions to safely traffic iron. Biochim Biophys Acta Gen Subj. (2020) 1864:129685. doi: 10.1016/j.bbagen.2020.129685
70. Zhou L, Zhao B, Zhang L, Wang S, Dong D, Lv H, et al. Alterations in cellular iron metabolism provide more therapeutic opportunities for cancer. Int J Mol Sci. (2018) 19(5):1545. doi: 10.3390/ijms19051545
71. Galaris D, Barbouti A, Pantopoulos K. Iron homeostasis and oxidative stress: An intimate relationship. Biochim Biophys Acta Mol Cell Res. (2019) 1866:118535. doi: 10.1016/j.bbamcr.2019.118535
72. Anderson GJ, Frazer DM. Current understanding of iron homeostasis. Am J Clin Nutr. (2017) 106:1559s–66s. doi: 10.3945/ajcn.117.155804
73. Knutson MD. Non-transferrin-bound iron transporters. Free Radic Biol Med. (2019) 133:101–11. doi: 10.1016/j.freeradbiomed.2018.10.413
74. Angoro B, Motshakeri M, Hemmaway C, Svirskis D, Sharma M. Non-transferrin bound iron. Clin Chim Acta. (2022) 531:157–67. doi: 10.1016/j.cca.2022.04.004
75. He M, Zhang D, Cao Y, Chi C, Zeng Z, Yang X, et al. Chimeric antigen receptor-modified T cells therapy in prostate cancer: A comprehensive review on the current state and prospects. Heliyon. (2023) 9:e19147. doi: 10.1016/j.heliyon.2023.e19147
76. Santana-Codina N, Gikandi A, Mancias JD. The role of NCOA4-mediated ferritinophagy in ferroptosis. Adv Exp Med Biol. (2021) 1301:41–57. doi: 10.1007/978-3-030-62026-4_4
77. Quiles Del Rey M, Mancias JD. NCOA4-mediated ferritinophagy: A potential link to neurodegeneration. Front Neurosci. (2019) 13:238. doi: 10.3389/fnins.2019.00238
78. Fang Y, Chen X, Tan Q, Zhou H, Xu J, Gu Q. Inhibiting ferroptosis through disrupting the NCOA4-FTH1 interaction: A new mechanism of action. ACS Cent Sci. (2021) 7:980–9. doi: 10.1021/acscentsci.0c01592
79. Hou W, Xie Y, Song X, Sun X, Lotze MT, Zeh HJ 3rd, et al. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy. (2016) 12:1425–8. doi: 10.1080/15548627.2016.1187366
80. Ali MY, Oliva CR, Flor S, Griguer CE. Mitoferrin, cellular and mitochondrial iron homeostasis. Cells. (2022) 11(21):3464. doi: 10.3390/cells11213464
81. Read AD, Bentley RE, Archer SL, Dunham-Snary KJ. Mitochondrial iron-sulfur clusters: Structure, function, and an emerging role in vascular biology. Redox Biol. (2021) 47:102164. doi: 10.1016/j.redox.2021.102164
82. Shi R, Hou W, Wang ZQ, Xu X. Biogenesis of iron-sulfur clusters and their role in DNA metabolism. Front Cell Dev Biol. (2021) 9:735678. doi: 10.3389/fcell.2021.735678
83. Zhang M, Liu Z, Le Y, Gu Z, Zhao H. Iron-sulfur clusters: A key factor of regulated cell death in cancer. Oxid Med Cell Longev. (2022) 2022:7449941. doi: 10.1155/2022/7449941
84. Levi S, Ripamonti M, Dardi M, Cozzi A, Santambrogio P. Mitochondrial ferritin: its role in physiological and pathological conditions. Cells. (2021) 10(8):1969. doi: 10.3390/cells10081969
85. Li X, Wang P, Wu Q, Xie L, Cui Y, Li H, et al. The construction and characterization of mitochondrial ferritin overexpressing mice. Int J Mol Sci. (2017) 18(7):1518. doi: 10.3390/ijms18071518
86. DeHart DN, Fang D, Heslop K, Li L, Lemasters JJ, Maldonado EN. Opening of voltage dependent anion channels promotes reactive oxygen species generation, mitochondrial dysfunction and cell death in cancer cells. Biochem Pharmacol. (2018) 148:155–62. doi: 10.1016/j.bcp.2017.12.022
87. Zhao Y, Li Y, Zhang R, Wang F, Wang T, Jiao Y. The role of erastin in ferroptosis and its prospects in cancer therapy. OncoTargets Ther. (2020) 13:5429–41. doi: 10.2147/OTT.S254995
88. Ye L, Lu S, Wu L-L, Yang L, Yang L, Wang J. : The diversified role of mitochondria in ferroptosis in cancer. Cell Death Dis. (2023) 14:519. doi: 10.1038/s41419-023-06045-y
89. Cheng X, Zhang J, Xiao Y, Wang Z, He J, Ke M, et al. Mitochondrial regulation of ferroptosis in cancer therapy. Int J Mol Sci. (2023) 24(12):10037. doi: 10.3390/ijms241210037
90. Ou M, Jiang Y, Ji Y, Zhou Q, Du Z, Zhu H, et al. Role and mechanism of ferroptosis in neurological diseases. Mol Metab. (2022) 61:101502. doi: 10.1016/j.molmet.2022.101502
91. Xie B, Guo Y. Molecular mechanism of cell ferroptosis and research progress in regulation of ferroptosis by noncoding RNAs in tumor cells. Cell Death Discov. (2021) 7:101. doi: 10.1038/s41420-021-00483-3
92. Campanella A, Rovelli E, Santambrogio P, Cozzi A, Taroni F, Levi S. Mitochondrial ferritin limits oxidative damage regulating mitochondrial iron availability: hypothesis for a protective role in Friedreich ataxia. Hum Mol Genet. (2009) 18:1–11. doi: 10.1093/hmg/ddn308
93. Pecchillo Cimmino T, Ammendola R, Cattaneo F, Esposito G. NOX dependent ROS generation and cell metabolism. Int J Mol Sci. 24(3):2086.
94. Martínez-Reyes I, Chandel NS. Mitochondrial TCA cycle metabolites control physiology and disease. Nat Commun. (2020) 11:102. doi: 10.1038/s41467-019-13668-3
95. Yoo HC, Yu YC, Sung Y, Han JM. Glutamine reliance in cell metabolism. Exp Mol Med. (2020) 52:1496–516. doi: 10.1038/s12276-020-00504-8
96. Li R, Jia Z, Trush MA. Defining ROS in biology and medicine. React Oxyg Species (Apex). (2016) 1:9–21. doi: 10.20455/ros.2380-2367
97. Chen K, Lu P, Beeraka NM, Sukocheva OA, Madhunapantula SV, Liu J, et al. et al: Mitochondrial mutations and mitoepigenetics: Focus on regulation of oxidative stress-induced responses in breast cancers. Semin Cancer Biol. (2022) 83:556–69. doi: 10.1016/j.semcancer.2020.09.012
98. Chen K, Lu P, Beeraka NM, Sukocheva OA, Madhunapantula SV, Liu J, et al. et al: Corrigendum to "Mitochondrial mutations and mitoepigenetics: Focus on regulation of oxidative stress-induced responses in breast cancers" [Semin. Cancer Biol. 83 (2022) 556-569]. Semin Cancer Biol. (2022) 86:1222. doi: 10.1016/j.semcancer.2020.09.012
99. Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. (2007) 87:245–313. doi: 10.1152/physrev.00044.2005
100. Yang S, Lian G. Correction to: ROS and diseases: role in metabolism and energy supply. Mol Cell Biochem. (2020) 467:13. doi: 10.1007/s11010-020-03697-8
101. Harayama T, Shimizu T. Roles of polyunsaturated fatty acids, from mediators to membranes. J Lipid Res. (2020) 61:1150–60. doi: 10.1194/jlr.R120000800
102. Shan X, Li S, Sun B, Chen Q, Sun J, He Z, et al. Ferroptosis-driven nanotherapeutics for cancer treatment. J Control Release. (2020) 319:322–32. doi: 10.1016/j.jconrel.2020.01.008
103. Valko M, Morris H, Cronin MT. Metals, toxicity and oxidative stress. Curr Med Chem. (2005) 12:1161–208. doi: 10.2174/0929867053764635
104. Koppenol WH, Hider RH. Iron and redox cycling. Do's and don'ts. Free Radic Biol Med. (2019) 133:3–10. doi: 10.1016/j.freeradbiomed.2018.09.022
105. Liu Y, Wang Y, Yang Y, Weng L, Wu Q, Zhang J, et al. Emerging phagocytosis checkpoints in cancer immunotherapy. Signal Transduct Target Ther. (2023) 8:104. doi: 10.1038/s41392-023-01365-z
106. Zhu M, Wang X, Zhou Y, Li M, Wang Y, Wang Y, et al. et al: Breast tumor-targeted drug delivery via polymer nanocarriers: Endogenous and exogenous strategies. J Appl Polymer Sci. (2023) 140:e54227. doi: 10.1002/app.54227
107. Chen K, Zhang J, Beeraka NM, Li J, Sinelnikov MY, Zhang X, et al. et al: novel perspectives on nanotechnological and biomedical implications of monotherapy or combination regimen of lactoferrin. Curr Pharm Des. (2023) 29:1579–91. doi: 10.2174/1381612829666230622140926
108. Cao Y, Efetov SK, He M, Fu Y, Beeraka NM, Zhang J, et al. Updated clinical perspectives and challenges of chimeric antigen receptor-T cell therapy in colorectal cancer and invasive breast cancer. Archivum immunologiae therapiae experimentalis. (2023) 71:19. doi: 10.1007/s00005-023-00684-x
109. Imai H, Matsuoka M, Kumagai T, Sakamoto T, Koumura T. Lipid peroxidation-dependent cell death regulated by GPx4 and ferroptosis. Curr Top Microbiol Immunol. (2017) 403:143–70. doi: 10.1007/82_2016_508
110. Zhao Y, Huang Z, Peng H. Molecular mechanisms of ferroptosis and its roles in hematologic Malignancies. Front Oncol. (2021) 11:743006. doi: 10.3389/fonc.2021.743006
111. Chen M, Shi Z, Sun Y, Ning H, Gu X, Zhang L. Prospects for anti-tumor mechanism and potential clinical application based on glutathione peroxidase 4 mediated ferroptosis. Int J Mol Sci. (2023) 24(2):1607. doi: 10.3390/ijms24021607
112. Wang H, Liu C, Zhao Y, Gao G. Mitochondria regulation in ferroptosis. Eur J Cell Biol. (2020) 99:151058. doi: 10.1016/j.ejcb.2019.151058
113. Huang S, Liu Y, Zhang Y, Zhang R, Zhu C, Fan L, et al. Baicalein inhibits SARS-CoV-2/VSV replication with interfering mitochondrial oxidative phosphorylation in a mPTP dependent manner. Signal Transduct Target Ther. (2020) 5:266. doi: 10.1038/s41392-020-00353-x
114. Liu Y, Chen C, Wang X, Sun Y, Zhang J, Chen J, et al. An epigenetic role of mitochondria in cancer. Cells. (2022) 11(16):2518. doi: 10.3390/cells11162518
115. Liu Y, Shi Y. Mitochondria as a target in cancer treatment. MedComm (2020). (2020) 1:129–39. doi: 10.1002/mco2.16
116. Liu Y, Sun Y, Guo Y, Shi X, Chen X, Feng W, et al. An overview: the diversified role of mitochondria in cancer metabolism. Int J Biol Sci. (2023) 19:897–915. doi: 10.7150/ijbs.81609
117. Liu J, Zhang J, Beeraka NM, Chen K, Sinelnikov MY, Manogaran P, et al. Perspectives on the nanocarriers with miRNAs for targeting melanoma stemness through epigenetic regulation. Pigment Cell Melanoma Res. (2023) 36:268–87. doi: 10.1111/pcmr.13081
118. Chen Y, Li C, Wang N, Wu Z, Zhang J, Yan J, et al. Identification of LINC00654-NINL regulatory axis in diffuse large B-cell lymphoma in silico analysis. Front Oncol. (2022) 12:883301. doi: 10.3389/fonc.2022.883301
119. Yao X, Li W, Fang D, Xiao C, Wu X, Li M, et al. Emerging roles of energy metabolism in ferroptosis regulation of tumor cells. Adv Sci (Weinheim Baden-Wurttemberg Germany). (2021) 8:e2100997. doi: 10.1002/advs.202100997
120. Qiu Y, Cao Y, Cao W, Jia Y, Lu N. The application of ferroptosis in diseases. Pharmacol Res. (2020) 159:104919. doi: 10.1016/j.phrs.2020.104919
121. Tian C, Liu Y, Li Z, Zhu P, Zhao M. Mitochondria related cell death modalities and disease. Front Cell Dev Biol. (2022) 10:832356. doi: 10.3389/fcell.2022.832356
122. Wang L, Liu Y, Du T, Yang H, Lei L, Guo M, et al. ATF3 promotes erastin-induced ferroptosis by suppressing system Xc(). Cell Death Differ. (2020) 27:662–75. doi: 10.1038/s41418-019-0380-z
123. Neitemeier S, Jelinek A, Laino V, Hoffmann L, Eisenbach I, Eying R, et al. BID links ferroptosis to mitochondrial cell death pathways. Redox Biol. (2017) 12:558–70. doi: 10.1016/j.redox.2017.03.007
124. Merkel M, Goebel B, Boll M, Adhikari A, Maurer V, Steinhilber D, et al. Mitochondrial reactive oxygen species formation determines ACSL4/LPCAT2-mediated ferroptosis. Antioxidants (Basel). (2023) 12(8):1590. doi: 10.3390/antiox12081590
125. Villar VH, Merhi F, Djavaheri-Mergny M, Durán RV. Glutaminolysis and autophagy in cancer. Autophagy. (2015) 11:1198–208. doi: 10.1080/15548627.2015.1053680
126. Peyton KJ, Liu XM, Yu Y, Yates B, Behnammanesh G, Durante W. Glutaminase-1 stimulates the proliferation, migration, and survival of human endothelial cells. Biochem Pharmacol. (2018) 156:204–14. doi: 10.1016/j.bcp.2018.08.032
127. He M, Cao Y, Chi C, Zhao J, Chong E, Chin KXC, et al. Unleashing novel horizons in advanced prostate cancer treatment: investigating the potential of prostate specific membrane antigen-targeted nanomedicine-based combination therapy. Front Immunol. (2023) 14:1265751. doi: 10.3389/fimmu.2023.1265751
128. Botman D, Tigchelaar W, Van Noorden CJ. Determination of phosphate-activated glutaminase activity and its kinetics in mouse tissues using metabolic mapping (quantitative enzyme histochemistry). J Histochem Cytochem. (2014) 62:813–26. doi: 10.1369/0022155414551177
129. Grinde MT, Hilmarsdottir B, Tunset HM, Henriksen IM, Kim J, Haugen MH, et al. Glutamine to proline conversion is associated with response to glutaminase inhibition in breast cancer. Breast Cancer research: BCR. (2019) 21:61. doi: 10.1186/s13058-019-1141-0
130. Hirschhorn T, Stockwell BR. The development of the concept of ferroptosis. Free Radic Biol Med. (2019) 133:130–43. doi: 10.1016/j.freeradbiomed.2018.09.043
131. Gnanapradeepan K, Basu S, Barnoud T, Budina-Kolomets A, Kung CP, Murphy ME. The p53 tumor suppressor in the control of metabolism and ferroptosis. Front Endocrinol (Lausanne). (2018) 9:124. doi: 10.3389/fendo.2018.00124
132. Suzuki S, Venkatesh D, Kanda H, Nakayama A, Hosokawa H, Lee E, et al. GLS2 is a tumor suppressor and a regulator of ferroptosis in hepatocellular carcinoma. Cancer Res. (2022) 82:3209–22. doi: 10.1158/0008-5472.CAN-21-3914
133. Gan B. Mitochondrial regulation of ferroptosis. J Cell Biol. (2021) 220(9):e202105043. doi: 10.1083/jcb.202105043
134. Gao M, Yi J, Zhu J, Minikes AM, Monian P, Thompson CB, et al. Role of mitochondria in ferroptosis. Mol Cell. (2019) 73:354–363.e353. doi: 10.1016/j.molcel.2018.10.042
135. Lushchak OV, Piroddi M, Galli F, Lushchak VI. Aconitase post-translational modification as a key in linkage between Krebs cycle, iron homeostasis, redox signaling, and metabolism of reactive oxygen species. Redox Rep. (2014) 19:8–15. doi: 10.1179/1351000213Y.0000000073
136. Linehan WM, Rouault TA. Molecular pathways: Fumarate hydratase-deficient kidney cancer–targeting the Warburg effect in cancer. Clin Cancer Res. (2013) 19:3345–52. doi: 10.1158/1078-0432.CCR-13-0304
137. Lu K, Han Q, Ma Z, Yan Q, Pei Y, Shi P, et al. Injectable platelet rich fibrin facilitates hair follicle regeneration by promoting human dermal papilla cell proliferation, migration, and trichogenic inductivity. Exp Cell Res. (2021) 409:112888. doi: 10.1016/j.yexcr.2021.112888
138. Bando H, Atsumi T, Nishio T, Niwa H, Mishima S, Shimizu C, et al. Phosphorylation of the 6-phosphofructo-2-kinase/fructose 2,6-bisphosphatase/PFKFB3 family of glycolytic regulators in human cancer. Clin Cancer Res. (2005) 11:5784–92. doi: 10.1158/1078-0432.CCR-05-0149
139. Liu Y, Gu W. The complexity of p53-mediated metabolic regulation in tumor suppression. Semin Cancer Biol. (2022) 85:4–32. doi: 10.1016/j.semcancer.2021.03.010
140. Marchiq I, Le Floch R, Roux D, Simon MP, Pouyssegur J. Genetic disruption of lactate/H+ symporters (MCTs) and their subunit CD147/BASIGIN sensitizes glycolytic tumor cells to phenformin. Cancer Res. (2015) 75:171–80. doi: 10.1158/0008-5472.CAN-14-2260
141. Rostovtseva TK, Sheldon KL, Hassanzadeh E, Monge C, Saks V, Bezrukov SM, et al. Tubulin binding blocks mitochondrial voltage-dependent anion channel and regulates respiration. Proc Natl Acad Sci U.S.A. (2008) 105:18746–51. doi: 10.1073/pnas.0806303105
142. Najbauer EE, Becker S, Giller K, Zweckstetter M, Lange A, Steinem C, et al. Structure, gating and interactions of the voltage-dependent anion channel. Eur Biophysics J. (2021) 50:159–72. doi: 10.1007/s00249-021-01515-7
143. Harris RA, Fenton AW. A critical review of the role of M(2)PYK in the Warburg effect. Biochim Biophys Acta Rev Cancer. (2019) 1871:225–39. doi: 10.1016/j.bbcan.2019.01.004
144. Maldonado EN. VDAC-tubulin, an anti-warburg pro-oxidant switch. Front Oncol. (2017) 7:4. doi: 10.3389/fonc.2017.00004
145. Heslop KA, Milesi V, Maldonado EN. VDAC modulation of cancer metabolism: advances and therapeutic challenges. Front Physiol. (2021) 12:742839. doi: 10.3389/fphys.2021.742839
146. Yagoda N, von Rechenberg M, Zaganjor E, Bauer AJ, Yang WS, Fridman DJ, et al. et al: RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature. (2007) 447:864–8. doi: 10.1038/nature05859
147. Idelchik M, Begley U, Begley TJ, Melendez JA. Mitochondrial ROS control of cancer. Semin Cancer Biol. (2017) 47:57–66. doi: 10.1016/j.semcancer.2017.04.005
148. Zong WX, Rabinowitz JD, White E. Mitochondria and cancer. Mol Cell. (2016) 61:667–76. doi: 10.1016/j.molcel.2016.02.011
149. Yang Y, Karakhanova S, Hartwig W, D'Haese JG, Philippov PP, Werner J, et al. Mitochondria and mitochondrial ROS in cancer: novel targets for anticancer therapy. J Cell Physiol. (2016) 231:2570–81. doi: 10.1002/jcp.25349
150. Chen W, Zhao H, Li Y. Mitochondrial dynamics in health and disease: mechanisms and potential targets. Signal Transduct Target Ther. (2023) 8:333. doi: 10.1038/s41392-023-01547-9
151. Moloney JN, Cotter TG. ROS signaling in the biology of cancer. Semin Cell Dev Biol. (2018) 80:50–64. doi: 10.1016/j.semcdb.2017.05.023
152. Yang S, Lian G. ROS and diseases: role in metabolism and energy supply. Mol Cell Biochem. (2020) 467:1–12. doi: 10.1007/s11010-019-03667-9
153. He M, Cao Y, Chi C, Yang X, Ramin R, Wang S, et al. et al: Research progress on deep learning in magnetic resonance imaging-based diagnosis and treatment of prostate cancer: a review on the current status and perspectives. Front Oncol. (2023) 13:1189370. doi: 10.3389/fonc.2023.1189370
154. Liang D, Minikes AM, Jiang X. Ferroptosis at the intersection of lipid metabolism and cellular signaling. Mol Cell. (2022) 82:2215–27. doi: 10.1016/j.molcel.2022.03.022
155. Feng Y, Xu J, Shi M, Liu R, Zhao L, Chen X, et al. et al: COX7A1 enhances the sensitivity of human NSCLC cells to cystine deprivation-induced ferroptosis via regulating mitochondrial metabolism. Cell Death Dis. (2022) 13:988. doi: 10.1038/s41419-022-05430-3
156. Homma T, Kobayashi S, Sato H, Fujii J. Superoxide produced by mitochondrial complex III plays a pivotal role in the execution of ferroptosis induced by cysteine starvation. Arch Biochem Biophys. (2021) 700:108775. doi: 10.1016/j.abb.2021.108775
157. Lee H, Zandkarimi F, Zhang Y, Meena JK, Kim J, Zhuang L, et al. Energy-stress-mediated AMPK activation inhibits ferroptosis. Nat Cell Biol. (2020) 22:225–34. doi: 10.1038/s41556-020-0461-8
158. Snaebjornsson MT, Janaki-Raman S, Schulze A. Greasing the wheels of the cancer machine: the role of lipid metabolism in cancer. Cell Metab. (2020) 31:62–76. doi: 10.1016/j.cmet.2019.11.010
159. Lee JY, Nam M, Son HY, Hyun K, Jang SY, Kim JW, et al. Polyunsaturated fatty acid biosynthesis pathway determines ferroptosis sensitivity in gastric cancer. Proc Natl Acad Sci U.S.A. (2020) 117:32433–42. doi: 10.1073/pnas.2006828117
160. Kim JW, Lee JY, Oh M, Lee EW. An integrated view of lipid metabolism in ferroptosis revisited via lipidomic analysis. Exp Mol Med. (2023) 55:1620–31. doi: 10.1038/s12276-023-01077-y
161. Ni L, Tang C, Wang Y, Wan J, Charles MG, Zhang Z, et al. Construction of a miRNA-based nomogram model to predict the prognosis of endometrial cancer. J Pers Med. (2022) 12(7):1154. doi: 10.3390/jpm12071154
162. Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I, et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol. (2017) 13:91–8. doi: 10.1038/nchembio.2239
163. Xu L, Liu Y, Chen X, Zhong H, Wang Y. Ferroptosis in life: To be or not to be. BioMed Pharmacother. (2023) 159:114241. doi: 10.1016/j.biopha.2023.114241
164. Lee JY, Kim WK, Bae KH, Lee SC, Lee EW. Lipid metabolism and ferroptosis. Biol (Basel). (2021) 10(3):184. doi: 10.3390/biology10030184
165. Küch EM, Vellaramkalayil R, Zhang I, Lehnen D, Brügger B, Sreemmel W, et al. Differentially localized acyl-CoA synthetase 4 isoenzymes mediate the metabolic channeling of fatty acids towards phosphatidylinositol. Biochim Biophys Acta. (2014) 1841:227–39. doi: 10.1016/j.bbalip.2013.10.018
166. Kagan VE, Mao G, Qu F, Angeli JP, Doll S, Croix CS, et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol. (2017) 13:81–90. doi: 10.1038/nchembio.2238
167. Shi JF, Liu Y, Wang Y, Gao R, Wang Y, Liu J. Targeting ferroptosis, a novel programmed cell death, for the potential of alcohol-related liver disease therapy. Front Pharmacol. (2023) 14:1194343. doi: 10.3389/fphar.2023.1194343
168. Chu B, Kon N, Chen D, Li T, Liu T, Jiang L, et al. ALOX12 is required for p53-mediated tumor suppression through a distinct ferroptosis pathway. Nat Cell Biol. (2019) 21:579–91. doi: 10.1038/s41556-019-0305-6
169. Jacobson MD, Weil M, Raff MC. Programmed cell death in animal development. Cell. (1997) 88:347–54. doi: 10.1016/S0092-8674(00)81873-5
170. Riedl SJ, Shi Y. Molecular mechanisms of caspase regulation during apoptosis. Nat Rev Mol Cell Biol. (2004) 5:897–907. doi: 10.1038/nrm1496
171. Taylor RC, Cullen SP, Martin SJ. Apoptosis: controlled demolition at the cellular level. Nat Rev Mol Cell Biol. (2008) 9:231–41. doi: 10.1038/nrm2312
172. Tang HM, Talbot CC Jr., Fung MC, Tang HL. Molecular signature of anastasis for reversal of apoptosis. F1000Research. (2017) 6:43. doi: 10.12688/f1000research.10568.2
173. Sun G, Guzman E, Balasanyan V, Conner CM, Wong K, Zhou HR, et al. A molecular signature for anastasis, recovery from the brink of apoptotic cell death. J Cell Biol. (2017) 216:3355–68. doi: 10.1083/jcb.201706134
174. Tang HM, Tang HL. Anastasis: recovery from the brink of cell death. R Soc Open Sci. (2018) 5:180442. doi: 10.1098/rsos.180442
175. Yang WS, Stockwell BR. Ferroptosis: death by lipid peroxidation. Trends Cell Biol. (2016) 26:165–76. doi: 10.1016/j.tcb.2015.10.014
176. Ursini F, Maiorino M, Gregolin C. The selenoenzyme phospholipid hydroperoxide glutathione peroxidase. Biochim Biophys Acta (BBA)-General Subj. (1985) 839:62–70. doi: 10.1016/0304-4165(85)90182-5
177. Skouta R, Dixon SJ, Wang J, Dunn DE, Orman M, Shimada K, et al. Ferrostatins inhibit oxidative lipid damage and cell death in diverse disease models. J Am Chem Soc. (2014) 136:4551–6. doi: 10.1021/ja411006a
178. Tang HM, Tang HL. Cell recovery by reversal of ferroptosis. Biol Open. (2019) 8(6):bio043182. doi: 10.1242/bio.043182
179. Wise DR, DeBerardinis RJ, Mancuso A, Sayed N, Zhang X-Y, Pfeiffer HK, et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc Natl Acad Sci. (2008) 105:18782–7. doi: 10.1073/pnas.0810199105
180. Wang D, Peng Y, Xie Y, Zhou B, Sun X, Kang R, et al. Antiferroptotic activity of non-oxidative dopamine. Biochem Biophys Res Commun. (2016) 480:602–7. doi: 10.1016/j.bbrc.2016.10.099
181. Englard S, Seifter S. The biochemical functions of ascorbic acid. Annu Rev Nutr. (1986) 6:365–406. doi: 10.1146/annurev.nu.06.070186.002053
182. Levine M. New concepts in the biology and biochemistry of ascorbic acid. New Engl J Med. (1986) 314:892–902. doi: 10.1056/NEJM198604033141407
183. Tang H, Yuen K, Tang H, Fung M. Reversibility of apoptosis in cancer cells. Br J Cancer. (2009) 100:118–22. doi: 10.1038/sj.bjc.6604802
184. Tang HL, Tang HM, Mak KH, Hu S, Wang SS, Wong KM, et al. Cell survival, DNA damage, and oncogenic transformation after a transient and reversible apoptotic response. Mol Biol Cell. (2012) 23:2240–52. doi: 10.1091/mbc.e11-11-0926
185. Tang HL, Tang HM, Fung MC, Hardwick JM. In vivo CaspaseTracker biosensor system for detecting anastasis and non-apoptotic caspase activity. Sci Rep. (2015) 5:9015. doi: 10.1038/srep09015
186. Tang HL, Tang HM, Hardwick JM, Fung MC. Strategies for tracking anastasis, a cell survival phenomenon that reverses apoptosis. JoVE (Journal Visualized Experiments). (2015) 2015:e51964. doi: 10.3791/51964
187. Wang S-S, Xie X, Wong CST, Choi PY, Fung MC. HepG2 cells recovered from apoptosis show altered drug responses and invasiveness. Hepatobiliary Pancreatic Dis Int. (2014) 13:293–300. doi: 10.1016/S1499-3872(14)60042-4
188. Ding AX, Sun G, Argaw YG, Wong JO, Easwaran S, Montell DJ. CasExpress reveals widespread and diverse patterns of cell survival of caspase-3 activation during development. vivo. Elife. (2016) 5:e10936. doi: 10.7554/eLife.10936
189. Kaufmann SH. Induction of apoptosis by cancer chemotherapy. Exp Cell Res. (2000) 256:42–9. doi: 10.1006/excr.2000.4838
190. Berry S, Cosby R, Asmis T, Chan K, Hammad N, Krzyzanowska M. Continuous versus intermittent chemotherapy strategies in metastatic colorectal cancer: a systematic review and meta-analysis. Ann Oncol. (2015) 26:477–85. doi: 10.1093/annonc/mdu272
191. Daenen LG, Roodhart JM, van Amersfoort M, Dehnad M, Roessingh W, Ulfman LH, et al. Chemotherapy enhances metastasis formation via VEGFR-1–expressing endothelial cells. Cancer Res. (2011) 71:6976–85. doi: 10.1158/0008-5472.CAN-11-0627
192. Karagiannis GS, Condeelis JS, Oktay MH. Chemotherapy-induced metastasis: molecular mechanisms, clinical manifestations, therapeutic interventions. Cancer Res. (2019) 79:4567–76. doi: 10.1158/0008-5472.CAN-19-1147
193. D’Alterio C, Scala S, Sozzi G, Roz L, Bertolini G. Paradoxical effects of chemotherapy on tumor relapse and metastasis promotion. Semin Cancer Biol. (2020) 60:351–61.
194. Slaby O, Svoboda M, Michalek J, Vyzula R. MicroRNAs in colorectal cancer: translation of molecular biology into clinical application. Mol Cancer. (2009) 8:1–13. doi: 10.1186/1476-4598-8-102
195. Piawah S, Venook AP. Targeted therapy for colorectal cancer metastases: A review of current methods of molecularly targeted therapy and the use of tumor biomarkers in the treatment of metastatic colorectal cancer. Cancer. (2019) 125:4139–47. doi: 10.1002/cncr.32163
196. Das PK, Islam F, Lam AK. The roles of cancer stem cells and therapy resistance in colorectal carcinoma. Cells. (2020) 9:1392. doi: 10.3390/cells9061392
197. Elmore S. Apoptosis: a review of programmed cell death. Toxicologic Pathol. (2007) 35:495–516. doi: 10.1080/01926230701320337
198. Ichim G, Tait SW. A fate worse than death: apoptosis as an oncogenic process. Nat Rev Cancer. (2016) 16:539–48. doi: 10.1038/nrc.2016.58
199. Carneiro BA, El-Deiry WS. Targeting apoptosis in cancer therapy. Nat Rev Clin Oncol. (2020) 17:395–417. doi: 10.1038/s41571-020-0341-y
200. Sun G, Montell DJ. Q&A: Cellular near death experiences—what is anastasis? BMC Biol. (2017) 15:1–5. doi: 10.1186/s12915-017-0441-z
201. Xu Y, So C, Lam H-M, Fung M-C, Tsang S-Y. Apoptosis reversal promotes cancer stem cell-like cell formation. Neoplasia. (2018) 20:295–303. doi: 10.1016/j.neo.2018.01.005
202. Berthenet K, Ferrer CC, Fanfone D, Popgeorgiev N, Neves D, Bertolino P, et al. Failed apoptosis enhances melanoma cancer cell aggressiveness. Cell Rep. (2020) 31(10):107731. doi: 10.1016/j.celrep.2020.107731
203. Seervi M, Sumi S, Chandrasekharan A, Sharma AK, SanthoshKumar T. Molecular profiling of anastatic cancer cells: potential role of the nuclear export pathway. Cell Oncol. (2019) 42:645–61. doi: 10.1007/s13402-019-00451-1
204. Sun L, Yao C, Li X, Wang Y, Wang R, Wang M, et al. Anastasis confers ovarian cancer cells increased Malignancy through elevated p38 MAPK activation. Cell Death Differentiation. (2023) 30:809–24. doi: 10.1038/s41418-022-01081-1
205. Beeraka NM, Zhang J, Uthaiah CA, Anastasia CA, Liu J, Ramachandrappa HVP, et al. Expression patterns and relevance of FN3K, nrf2, and NQO1 in breast cancers. EJMO. (2024) 8(1):88–105. doi: 10.14744/ejmo.2024.61033
206. Beeraka NM, Zhang J, Zhao D, Liu J, Au C, Vikram P, et al. Combinatorial implications of nrf2 inhibitors with FN3K inhibitor. In Vitro Breast Cancer Study. Curr Pharm Design. (2023) 29(30). doi: 10.2174/0113816128261466231011114600
207. Chen K, Zhang J, Beeraka NM, Tang C, Babayeva YV, Sinelnikov MY, et al. Advances in the prevention and treatment of obesity-driven effects in breast cancers. Front Oncol. (2022) 12:820968. doi: 10.3389/fonc.2022.820968
208. Chen K, Lu P, Beeraka NM, Sukocheva OA, Madhunapantula SV, Liu J, et al. Mitochondrial mutations and mitoepigenetics: Focus on regulation of oxidative stress-induced responses in breast cancers. Semin Cancer Biol. (2022) 556–69.
209. Mikhaleva LM, Solomatina AA, Milovanov AP, Beeraka NM, Khovanskaya TN, Chabieva LB, et al. Histomorphological and functional features of the eutopic endometrium in patients with ovarian endometriosis after surgery—a clinical study. Reprod Sci. (2021) 28:2350–8. doi: 10.1007/s43032-021-00508-3
210. Sun G, Ding XA, Argaw Y, Guo X, Montell DJ. Akt1 and dCIZ1 promote cell survival from apoptotic caspase activation during regeneration and oncogenic overgrowth. Nat Commun. (2020) 11:5726. doi: 10.1038/s41467-020-19068-2
211. Naresh KN, Lakshminarayanan K, Pai SA, Borges AM. Apoptosis index is a predictor of metastatic phenotype in patients with early stage squamous carcinoma of the tongue: a hypothesis to support this paradoxical association. Cancer. (2001) 91:578–84. doi: 10.1002/(ISSN)1097-0142
212. Isobe N, Onodera H, Mori A, Shimada Y, Yang W, Yasuda S, et al. Caspase-3 expression in human gastric carcinoma and its clinical significance. Oncology. (2004) 66:201–9. doi: 10.1159/000077996
213. Hu Q, Peng J, Liu W, He X, Cui L, Chen X, et al. Elevated cleaved caspase-3 is associated with shortened overall survival in several cancer types. Int J Clin Exp Pathol. (2014) 7:5057.
214. Zhou M, Liu X, Li Z, Huang Q, Li F, Li CY. Caspase-3 regulates the migration, invasion and metastasis of colon cancer cells. Int J Cancer. (2018) 143:921–30. doi: 10.1002/ijc.31374
215. Cheng YJ, Ch L, YP L, JY H, CC Su, WT C. Yang BC: Caspase-3 enhances lung metastasis and cell migration in a protease-independent mechanism through the ERK pathway. Int J Cancer. (2008) 123:1278–85. doi: 10.1002/ijc.23592
216. Wang R, Wang Y, Liu X, Liu M, Sun L, Pan X, et al. Anastasis enhances metastasis and chemoresistance of colorectal cancer cells through upregulating cIAP2/NFκB signaling. Cell Death Dis. (2023) 14:388. doi: 10.1038/s41419-023-05916-8
217. Patel M, Horgan PG, McMillan DC, Edwards J. NF-κB pathways in the development and progression of colorectal cancer. Trans Res. (2018) 197:43–56. doi: 10.1016/j.trsl.2018.02.002
218. Dolcet X, Llobet D, Pallares J, Matias-Guiu X. NF-kB in development and progression of human cancer. Virchows archiv. (2005) 446:475–82. doi: 10.1007/s00428-005-1264-9
219. Jana A, Krett NL, Guzman G, Khalid A, Ozden O, Staudacher JJ, et al. NFkB is essential for activin-induced colorectal cancer migration via upregulation of PI3K-MDM2 pathway. Oncotarget. (2017) 8:37377. doi: 10.18632/oncotarget.v8i23
220. Siegel RL, Miller KD, Goding Sauer A, Fedewa SA, Butterly LF, Anderson JC, et al. Colorectal cancer statistics, 2020. CA: Cancer J Clin. (2020) 70:145–64. doi: 10.3322/caac.21601
221. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: Cancer J Clin. (2021) 71:209–49. doi: 10.3322/caac.21660
222. Nagata M, Nakayama H, Tanaka T, Yoshida R, Yoshitake Y, Fukuma D, et al. Overexpression of cIAP2 contributes to 5-FU resistance and a poor prognosis in oral squamous cell carcinoma. Br J Cancer. (2011) 105:1322–30. doi: 10.1038/bjc.2011.387
223. Lopes RB, Gangeswaran R, McNeish IA, Wang Y, Lemoine NR. Expression of the IAP protein family is dysregulated in pancreatic cancer cells and is important for resistance to chemotherapy. Int J Cancer. (2007) 120:2344–52. doi: 10.1002/ijc.22554
224. Valon L, Davidović A, Levillayer F, Villars A, Chouly M, Cerqueira-Campos F, et al. Robustness of epithelial sealing is an emerging property of local ERK feedback driven by cell elimination. Dev Cell. (2021) 56:1700–11:e1708. doi: 10.1016/j.devcel.2021.05.006
225. Roy N, Deveraux QL, Takahashi R, Salvesen GS, Reed JC. The c-IAP-1 and c-IAP-2 proteins are direct inhibitors of specific caspases. EMBO J. (1997) 16(23):6914–25. doi: 10.1093/emboj/16.23.6914
226. Eckelman BP, Salvesen GS. The human anti-apoptotic proteins cIAP1 and cIAP2 bind but do not inhibit caspases. J Biol Chem. (2006) 281:3254–60. doi: 10.1074/jbc.M510863200
227. Praticò D, Sung S. Lipid peroxidation and oxidative imbalance: early functional events in Alzheimer's disease. J Alzheimer's Dis. (2004) 6:171–5. doi: 10.3233/JAD-2004-6209
228. Yang C, He L, He P, Liu Y, Wang W, He Y, et al. Increased drug resistance in breast cancer by tumor-associated macrophages through IL-10/STAT3/bcl-2 signaling pathway. Med Oncol. (2015) 32:1–8. doi: 10.1007/s12032-014-0352-6
229. Miller KD, Ortiz AP, Pinheiro PS, Bandi P, Minihan A, Fuchs HE, et al. Cancer statistics for the US Hispanic/Latino population, 2021. CA: Cancer J Clin. (2021) 71:466–87. doi: 10.3322/caac.21695
230. Yu W, Chen Y, Putluri N, Coarfa C, Robertson MJ, Putluri V, et al. Acquisition of cisplatin resistance shifts head and neck squamous cell carcinoma metabolism toward neutralization of oxidative stress. Cancers. (2020) 12:1670. doi: 10.3390/cancers12061670
231. Galluzzi L, Senovilla L, Vitale I, Michels J, Martins I, Kepp O, et al. Molecular mechanisms of cisplatin resistance. Oncogene. (2012) 31:1869–83. doi: 10.1038/onc.2011.384
232. Zhu H, Luo H, Zhang W, Shen Z, Hu X, Zhu X. Molecular mechanisms of cisplatin resistance in cervical cancer. Drug design Dev Ther. (2016) 2016:1885–95. doi: 10.2147/DDDT.S106412
233. Wang GX, Tu H-C, Dong Y, Skanderup AJ, Wang Y, Takeda S, et al. ΔNp63 inhibits oxidative stress-induced cell death, including ferroptosis, and cooperates with the BCL-2 family to promote clonogenic survival. Cell Rep. (2017) 21:2926–39. doi: 10.1016/j.celrep.2017.11.030
234. Wang Y, Zhao G, Condello S, Huang H, Cardenas H, Tanner EJ, et al. Frizzled-7 identifies platinum-tolerant ovarian cancer cells susceptible to ferroptosis. Cancer Res. (2021) 81:384–99. doi: 10.1158/0008-5472.CAN-20-1488
235. Okuno S, Sato H, Kuriyama-Matsumura K, Tamba M, Wang H, Sohda S, et al. Role of cystine transport in intracellular glutathione level and cisplatin resistance in human ovarian cancer cell lines. Br J Cancer. (2003) 88:951–6. doi: 10.1038/sj.bjc.6600786
236. Huang Y, Dai Z, Barbacioru C, Sadée W. Cystine-glutamate transporter SLC7A11 in cancer chemosensitivity and chemoresistance. Cancer Res. (2005) 65:7446–54. doi: 10.1158/0008-5472.CAN-04-4267
237. Chen HH, Kuo MT. Role of glutathione in the regulation of Cisplatin resistance in cancer chemotherapy. Metal-based Drugs. (2010) 2010:430939. doi: 10.1155/2010/430939
238. Guo J, Xu B, Han Q, Zhou H, Xia Y, Gong C, et al. Ferroptosis: a novel anti-tumor action for cisplatin. Cancer Res treatment: Off J Korean Cancer Assoc. (2018) 50:445. doi: 10.4143/crt.2016.572
239. Song Z, Jia G, Ma P, Cang S. Exosomal miR-4443 promotes cisplatin resistance in non-small cell lung carcinoma by regulating FSP1 m6A modification-mediated ferroptosis. Life Sci. (2021) 276:119399. doi: 10.1016/j.lfs.2021.119399
240. Wang W, Kryczek I, Dostál L, Lin H, Tan L, Zhao L, et al. Effector T cells abrogate stroma-mediated chemoresistance in ovarian cancer. Cell. (2016) 165:1092–105. doi: 10.1016/j.cell.2016.04.009
241. Sato M, Kusumi R, Hamashima S, Kobayashi S, Sasaki S, Komiyama Y, et al. The ferroptosis inducer erastin irreversibly inhibits system xc– and synergizes with cisplatin to increase cisplatin’s cytotoxicity in cancer cells. Sci Rep. (2018) 8:968. doi: 10.1038/s41598-018-19213-4
242. Roh J-L, Kim EH, Jang HJ, Park JY, Shin D. Induction of ferroptotic cell death for overcoming cisplatin resistance of head and neck cancer. Cancer Lett. (2016) 381:96–103. doi: 10.1016/j.canlet.2016.07.035
243. Hu Z, Mi Y, Qian H, Guo N, Yan A, Zhang Y, et al. A potential mechanism of temozolomide resistance in glioma–ferroptosis. Front Oncol. (2020) 10:897. doi: 10.3389/fonc.2020.00897
244. Chen L, Li X, Liu L, Yu B, Xue Y, Liu Y. Erastin sensitizes glioblastoma cells to temozolomide by restraining xCT and cystathionine-γ-lyase function. Oncol Rep. (2015) 33:1465–74. doi: 10.3892/or.2015.3712
245. Chaudhary N, Choudhary BS, Shah SG, Khapare N, Dwivedi N, Gaikwad A, et al. Lipocalin 2 expression promotes tumor progression and therapy resistance by inhibiting ferroptosis in colorectal cancer. Int J Cancer. (2021) 149:1495–511. doi: 10.1002/ijc.33711
246. Bi J, Yang S, Li L, Dai Q, Borcherding N, Wagner BA, et al. Metadherin enhances vulnerability of cancer cells to ferroptosis. Cell Death Dis. (2019) 10:682. doi: 10.1038/s41419-019-1897-2
247. Beeraka NM, Bovilla VR, Doreswamy SH, Puttalingaiah S, Srinivasan A, Madhunapantula SV. The taming of nuclear factor erythroid-2-related factor-2 (Nrf2) deglycation by fructosamine-3-kinase (FN3K)-inhibitors-a novel strategy to combat cancers. Cancers. (2021) 13:281. doi: 10.3390/cancers13020281
248. Beeraka NM, Zhang J, Mandal S, Vikram PRH, Liu J, Namintha BM, et al. Screening fructosamine-3-kinase (FN3K) inhibitors, a deglycating enzyme of oncogenic Nrf2: Human FN3K homology modelling, docking and molecular dynamics simulations. PloS One. (2023) 18:e0283705. doi: 10.1371/journal.pone.0283705
249. Zheng Y, Liu J, Beeraka NM, Manogaran P, PR HV, Yn LD, et al. Inflammation and stem cell stochasticity of HPV-induced cervical cancer: epigenetics based biomarkers through microbiome and metabolome for personalized medicine: A systematic review. Curr Med Chem. (2024). doi: 10.2174/0109298673257429231108072717
250. Chakraborty S, Mir KB, Seligson ND, Nayak D, Kumar R, Goswami A. Integration of EMT and cellular survival instincts in reprogramming of programmed cell death to anastasis. Cancer Metastasis Rev. (2020) 39:553–66. doi: 10.1007/s10555-020-09866-x
251. Strobel O, Neoptolemos J, Jaeger D, Buechler MW. Optimizing the outcomes of pancreatic cancer surgery. Nat Rev Clin Oncol. (2019) 16:11–26. doi: 10.1038/s41571-018-0112-1
252. Tang R, Hua J, Xu J, Liang C, Meng Q, Liu J, et al. The role of ferroptosis regulators in the prognosis, immune activity and gemcitabine resistance of pancreatic cancer. Ann Trans Med. (2020) 8(21):1347. doi: 10.21037/atm-20-2554a
253. Lo M, Ling V, Wang Y, Gout P. The xc– cystine/glutamate antiporter: a mediator of pancreatic cancer growth with a role in drug resistance. Br J Cancer. (2008) 99:464–72. doi: 10.1038/sj.bjc.6604485
254. Daher B, Parks SK, Durivault J, Cormerais Y, Baidarjad H, Tambutte E, et al. Genetic ablation of the cystine transporter xCT in PDAC cells inhibits mTORC1, growth, survival, and tumor formation via nutrient and oxidative stresses. Cancer Res. (2019) 79:3877–90. doi: 10.1158/0008-5472.CAN-18-3855
255. Hangauer MJ, Viswanathan VS, Ryan MJ, Bole D, Eaton JK, Matov A, et al. Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature. (2017) 551:247–50. doi: 10.1038/nature24297
256. Viswanathan VS, Ryan MJ, Dhruv HD, Gill S, Eichhoff OM, Seashore-Ludlow B, et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature. (2017) 547:453–7. doi: 10.1038/nature23007
257. Tsoi J, Robert L, Paraiso K, Galvan C, Sheu KM, Lay J, et al. Multi-stage differentiation defines melanoma subtypes with differential vulnerability to drug-induced iron-dependent oxidative stress. Cancer Cell. (2018) 33:890–904:e895. doi: 10.1016/j.ccell.2018.03.017
258. Liu J-L, Fan Y-G, Yang Z-S, Wang Z-Y, Guo C. Iron and Alzheimer’s disease: from pathogenesis to therapeutic implications. Front Neurosci. (2018) 12:632. doi: 10.3389/fnins.2018.00632
259. Peitzsch C, Tyutyunnykova A, Pantel K, Dubrovska A. Cancer stem cells: The root of tumor recurrence and metastases. Semin Cancer Biol. (2017) 10–24.
260. Dean M, Fojo T, Bates S. Tumor stem cells and drug resistance. Nat Rev Cancer. (2005) 5:275–84. doi: 10.1038/nrc1590
261. Weidenfeld K, Barkan D. EMT and stemness in tumor dormancy and outgrowth: are they intertwined processes? Front Oncol. (2018) 8:381. doi: 10.3389/fonc.2018.00381
262. Wang H, Unternaehrer JJ. Epithelial-mesenchymal transition and cancer stem cells: at the crossroads of differentiation and dedifferentiation. Dev Dynamics. (2019) 248:10–20. doi: 10.1002/dvdy.24678
263. Ruan H, Hu S, Zhang H, Du G, Li X, Li X, et al. Upregulated SOX9 expression indicates worse prognosis in solid tumors: a systematic review and meta-analysis. Oncotarget. (2017) 8:113163. doi: 10.18632/oncotarget.v8i68
264. Higashihara T, Yoshitomi H, Nakata Y, Kagawa S, Takano S, Shimizu H, et al. Sex determining region Y box 9 induces chemoresistance in pancreatic cancer cells by induction of putative cancer stem cell characteristics and its high expression predicts poor prognosis. Pancreas. (2017) 46:1296–304. doi: 10.1097/MPA.0000000000000945
266. Dunn C, Wiltshire C, MacLaren A, Gillespie DA. Molecular mechanism and biological functions of c-Jun N-terminal kinase signaling via the c-Jun transcription factor. Cell signalling. (2002) 14:585–93. doi: 10.1016/S0898-6568(01)00275-3
267. Salameh A, Galvagni F, Anselmi F, De Clemente C, Orlandini M, Oliviero S. Growth factor stimulation induces cell survival by c-Jun· ATF2-dependent activation of Bcl-XL. J Biol Chem. (2010) 285:23096–104. doi: 10.1074/jbc.M109.087221
268. Bossy-Wetzel E, Bakiri L, Yaniv M. Induction of apoptosis by the transcription factor c-Jun. EMBO J. (1997) 16:1695–709. doi: 10.1093/emboj/16.7.1695
269. Ashrafizadeh M, Dai J, Torabian P, Nabavi N, Aref AR, Aljabali AA, et al. Circular RNAs in EMT-driven metastasis regulation: modulation of cancer cell plasticity, tumorigenesis and therapy resistance. Cell Mol Life Sci. (2024) 81:1–25. doi: 10.1007/s00018-024-05236-w
270. Du B, Shim JS. Targeting epithelial–mesenchymal transition (EMT) to overcome drug resistance in cancer. Molecules. (2016) 21:965. doi: 10.3390/molecules21070965
271. Zhou Y, Liang C, Xue F, Chen W, Zhi X, Feng X, et al. Salinomycin decreases doxorubicin resistance in hepatocellular carcinoma cells by inhibiting the β-catenin/TCF complex association via FOXO3a activation. Oncotarget. (2015) 6:10350. doi: 10.18632/oncotarget.v6i12
272. Toden S, Okugawa Y, Jascur T, Wodarz D, Komarova NL, Buhrmann C, et al. Curcumin mediates chemosensitization to 5-fluorouracil through miRNA-induced suppression of epithelial-to-mesenchymal transition in chemoresistant colorectal cancer. Carcinogenesis. (2015) 36:355–67. doi: 10.1093/carcin/bgv006
273. Meidhof S, Brabletz S, Lehmann W, Preca BT, Mock K, Ruh M, et al. ZEB 1-associated drug resistance in cancer cells is reversed by the class I HDAC inhibitor mocetinostat. EMBO Mol Med. (2015) 7:831–47. doi: 10.15252/emmm.201404396
274. Namba T, Kodama R, Moritomo S, Hoshino T, Mizushima T. Zidovudine, an anti-viral drug, resensitizes gemcitabine-resistant pancreatic cancer cells to gemcitabine by inhibition of the Akt-GSK3β-Snail pathway. Cell Death Dis. (2015) 6:e1795–5. doi: 10.1038/cddis.2015.172
275. Zhang R, Zhang P, Wang H, Hou D, Li W, Xiao G, et al. Inhibitory effects of metformin at low concentration on epithelial–mesenchymal transition of CD44+ CD117+ ovarian cancer stem cells. Stem Cell Res Ther. (2015) 6:1–12. doi: 10.1186/s13287-015-0249-0
276. Vazquez-Martin A, Oliveras-Ferraros C, Cufí S, Del Barco S, Martin-Castillo B, Menendez JA. Metformin regulates breast cancer stem cello ntogeny by transcriptional regulation of the epithelial-mesenchymal transition (EMT) status. Cell Cycle. (2010) 9:3831–8. doi: 10.4161/cc.9.18.13131
277. Voon DC, Huang RY, Jackson RA, Thiery JP. The EMT spectrum and therapeutic opportunities. Mol Oncol. (2017) 11:878–91. doi: 10.1002/1878-0261.12082
278. Mrkvicova A, Chmelarova M, Peterova E, Havelek R, Baranova I, Kazimirova P, et al. The effect of sodium butyrate and cisplatin on expression of EMT markers. PloS One. (2019) 14:e0210889. doi: 10.1371/journal.pone.0210889
279. Nagarajan D, Wang L, Zhao W, Han X. Trichostatin A inhibits radiation-induced epithelial-to-mesenchymal transition in the alveolar epithelial cells. Oncotarget. (2017) 8:101745. doi: 10.18632/oncotarget.v8i60
280. Chinnaiyan P, Chowdhary S, Potthast L, Prabhu A, Tsai Y-Y, Sarcar B, et al. Phase I trial of vorinostat combined with bevacizumab and CPT-11 in recurrent glioblastoma. Neuro-oncology. (2012) 14:93–100. doi: 10.1093/neuonc/nor187
281. Thomson CA, Ho E, Strom MB. Chemopreventive properties of 3, 3′-diindolylmethane in breast cancer: evidence from experimental and human studies. Nutr Rev. (2016) 74:432–43. doi: 10.1093/nutrit/nuw010
282. Kim SM. Cellular and molecular mechanisms of 3, 3′-diindolylmethane in gastrointestinal cancer. Int J Mol Sci. (2016) 17:1155. doi: 10.3390/ijms17071155
283. Chakraborty S, Kumar A, Faheem MM, Katoch A, Kumar A, Jamwal VL, et al. Vimentin activation in early apoptotic cancer cells errands survival pathways during DNA damage inducer CPT treatment in colon carcinoma model. Cell Death Dis. (2019) 10:467. doi: 10.1038/s41419-019-1690-2
284. Mazzu YZ, Armenia J, Chakraborty G, Yoshikawa Y, Coggins SAA, Nandakumar S, et al. A novel mechanism driving poor-prognosis prostate cancer: overexpression of the DNA repair gene, ribonucleotide reductase small subunit M2 (RRM2). Clin Cancer Res. (2019) 25:4480–92. doi: 10.1158/1078-0432.CCR-18-4046
285. Arumugam T, Ramachandran V, Fournier KF, Wang H, Marquis L, Abbruzzese JL, et al. Epithelial to mesenchymal transition contributes to drug resistance in pancreatic cancer. Cancer Res. (2009) 69:5820–8. doi: 10.1158/0008-5472.CAN-08-2819
286. Hammond WA, Swaika A, Mody K. Pharmacologic resistance in colorectal cancer: a review. Ther Adv Med Oncol. (2016) 8:57–84. doi: 10.1177/1758834015614530
287. Beretta G L, Gatti L, Perego P, Zaffaroni N. Camptothecin resistance in cancer: insights into the molecular mechanisms of a DNA-damaging drug. Curr medicinal Chem. (2013) 20:1541–65. doi: 10.2174/0929867311320120006
288. Fischer KR, Durrans A, Lee S, Sheng J, Li F, Wong ST, et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature. (2015) 527:472–6. doi: 10.1038/nature15748
289. Amiri KI, Horton LW, LaFleur BJ, Sosman JA, Richmond A. Augmenting chemosensitivity of Malignant melanoma tumors via proteasome inhibition: implication for bortezomib (VELCADE, PS-341) as a therapeutic agent for Malignant melanoma. Cancer Res. (2004) 64:4912–8. doi: 10.1158/0008-5472.CAN-04-0673
290. Godwin P, Baird A-M, Heavey S, Barr M, O’byrne K, Gately K. Targeting nuclear factor-kappa B to overcome resistance to chemotherapy. Front Oncol. (2013) 3:120. doi: 10.3389/fonc.2013.00120
291. Yang F, Xiao Y, Ding J-H, Jin X, Ma D, Li D-Q, et al. Ferroptosis heterogeneity in triple-negative breast cancer reveals an innovative immunotherapy combination strategy. Cell Metab. (2023) 35:84–100:e108. doi: 10.1016/j.cmet.2022.09.021
292. Liu T, Zhu C, Chen X, Guan G, Zou C, Shen S, et al. Ferroptosis, as the most enriched programmed cell death process in glioma, induces immunosuppression and immunotherapy resistance. Neuro-oncology. (2022) 24:1113–25. doi: 10.1093/neuonc/noac033
293. He Y, Dong Y, Chen Y, Zhang G, Zhang H, Lei G, et al. Multi-omics characterization and therapeutic liability of ferroptosis in melanoma. Signal Transduction Targeted Ther. (2022) 7:268. doi: 10.1038/s41392-022-01067-y
294. Fan F, Liu P, Bao R, Chen J, Zhou M, Mo Z, et al. A dual PI3K/HDAC inhibitor induces immunogenic ferroptosis to potentiate cancer immune checkpoint therapy. Cancer Res. (2021) 81:6233–45. doi: 10.1158/0008-5472.CAN-21-1547
295. Mao W, Cai Y, Chen D, Jiang G, Xu Y, Chen R, et al. Statin shapes inflamed tumor microenvironment and enhances immune checkpoint blockade in non–small cell lung cancer. JCI Insight. (2022) 7(18):e161940. doi: 10.1172/jci.insight.161940
296. Cheu JW-S, Lee D, Li Q, Goh CC, Bao MH-R, Yuen VW-H, et al. Ferroptosis suppressor protein 1 inhibition promotes tumor ferroptosis and anti-tumor immune responses in liver cancer. Cell Mol Gastroenterol Hepatol. (2023) 16:133–59. doi: 10.1016/j.jcmgh.2023.03.001
297. Ruan WY, Zhang L, Lei S, Zeng ZR, Yang YS, Cao WP, et al. An inflammation-associated ferroptosis signature optimizes the diagnosis, prognosis evaluation and immunotherapy options in hepatocellular carcinoma. J Cell Mol Med. (2023) 27:1820–35. doi: 10.1111/jcmm.17780
298. Xue Y, Lu F, Chang Z, Li J, Gao Y, Zhou J, et al. Intermittent dietary methionine deprivation facilitates tumoral ferroptosis and synergizes with checkpoint blockade. Nat Commun. (2023) 14:4758. doi: 10.1038/s41467-023-40518-0
299. Li H, Yang C, Wei Y, Li X, Jiang W, Xu Y, et al. Ferritin light chain promotes the reprogramming of glioma immune microenvironment and facilitates glioma progression. Theranostics. (2023) 13:3794. doi: 10.7150/thno.82975
300. Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, et al. ferroptosis: A regulated cell death nexus linking metabolism, redox biology, and disease. Cell. (2017) 171:273–85. doi: 10.1016/j.cell.2017.09.021
301. Wu X, Li Y, Zhang S, Zhou X. Ferroptosis as a novel therapeutic target for cardiovascular disease. Theranostics. (2021) 11:3052–9. doi: 10.7150/thno.54113
302. Zaffaroni N, Beretta GL. Ferroptosis inducers for prostate cancer therapy. Curr Med Chem. (2022) 29:4185–201. doi: 10.2174/0929867329666220111120924
303. Kenny EM, Fidan E, Yang Q, Anthonymuthu TS, New LA, Meyer EA, et al. Ferroptosis contributes to neuronal death and functional outcome after traumatic brain injury. Crit Care Med. (2019) 47:410–8. doi: 10.1097/CCM.0000000000003555
304. Niu X, Chen L, Li Y, Hu Z, He F. Ferroptosis, necroptosis, and pyroptosis in the tumor microenvironment: Perspectives for immunotherapy of SCLC. Semin Cancer Biol. (2022) 86:273–85. doi: 10.1016/j.semcancer.2022.03.009
305. Lane DJR, Ayton S, Bush AI. Iron and alzheimer's disease: an update on emerging mechanisms. J Alzheimers Dis. (2018) 64:S379–s395. doi: 10.3233/JAD-179944
306. Shekhar S, Liu Y, Wang S, Zhang H, Fang X, Zhang J, et al. Novel mechanistic insights and potential therapeutic impact of TRPC6 in neurovascular coupling and ischemic stroke. Int J Mol Sci. (2021) 22(4):2074. doi: 10.3390/ijms22042074
307. Fang X, Zhang J, Roman RJ, Fan F. From 1901 to 2022, how far are we from truly understanding the pathogenesis of age-related dementia? Geroscience. (2022) 44:1879–83. doi: 10.1172/jci.insight.161940
308. Li D, Li Y. The interaction between ferroptosis and lipid metabolism in cancer. Signal Transduct Target Ther. (2020) 5:108. doi: 10.1038/s41392-020-00216-5
309. Zhang Y, Shi J, Liu X, Feng L, Gong Z, Koppula P, et al. BAP1 links metabolic regulation of ferroptosis to tumor suppression. Nat Cell Biol. (2018) 20:1181–92. doi: 10.1038/s41556-018-0178-0
310. Zhao L, Zhou X, Xie F, Zhang L, Yan H, Huang J, et al. Ferroptosis in cancer and cancer immunotherapy. Cancer Commun (Lond). (2022) 42:88–116. doi: 10.1002/cac2.12250
311. Wang W, Green M, Choi JE, Gijón M, Kennedy PD, Johnson JK, et al. CD8(+) T cells regulate tumor ferroptosis during cancer immunotherapy. Nature. (2019) 569:270–4. doi: 10.1038/s41586-019-1170-y
312. Xu X, Zhou N, Lan H, Yang F, Dong B, Zhang X. The ferroptosis molecular subtype reveals characteristics of the tumor microenvironment, immunotherapeutic response, and prognosis in gastric cancer. Int J Mol Sci. (2022) 23(17):9767. doi: 10.3390/ijms23179767
313. Wan RJ, Peng W, Xia QX, Zhou HH, Mao XY. Ferroptosis-related gene signature predicts prognosis and immunotherapy in glioma. CNS Neurosci Ther. (2021) 27:973–86. doi: 10.1111/cns.13654
314. Gu X, Liu Y, Dai X, Yang YG, Zhang X. Deciphering the potential roles of ferroptosis in regulating tumor immunity and tumor immunotherapy. Front Immunol. (2023) 14:1137107. doi: 10.3389/fimmu.2023.1137107
315. Ghoochani A, Hsu EC, Aslan M, Rice MA, Nguyen HM, Brooks JD, et al. Ferroptosis inducers are a novel therapeutic approach for advanced prostate cancer. Cancer Res. (2021) 81:1583–94. doi: 10.1158/0008-5472.CAN-20-3477
316. Wang Y, Ma Y, Jiang K. The role of ferroptosis in prostate cancer: a novel therapeutic strategy. Prostate Cancer Prostatic Dis. (2023) 26:25–9. doi: 10.1038/s41391-022-00583-w
317. Wu Z, Zhang X, Morgan GC, Li B, Wang Y, Wan J, et al. Diagnosis and treatment of inguinal hernias after surgical treatment of prostate cancer, current state of the problem. J Clin Med. (2022) 11(18):5423. doi: 10.3390/jcm11185423
318. Zhu S, Zhang Q, Sun X, Zeh HJ 3rd, Lotze MT, Kang R, et al. HSPA5 regulates ferroptotic cell death in cancer cells. Cancer Res. (2017) 77:2064–77. doi: 10.1158/0008-5472.CAN-16-1979
319. Sun X, Ou Z, Chen R, Niu X, Chen D, Kang R, et al. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology. (2016) 63:173–84. doi: 10.1002/hep.28251
320. Bai T, Lei P, Zhou H, Liang R, Zhu R, Wang W, et al. Sigma-1 receptor protects against ferroptosis in hepatocellular carcinoma cells. J Cell Mol Med. (2019) 23:7349–59. doi: 10.1111/jcmm.14594
321. Ding Y, Chen X, Liu C, Ge W, Wang Q, Hao X, et al. Identification of a small molecule as inducer of ferroptosis and apoptosis through ubiquitination of GPX4 in triple negative breast cancer cells. J Hematol Oncol. (2021) 14:19. doi: 10.1186/s13045-020-01016-8
Keywords: anastasis, ferroptosis, chemoresistance, mitochondria, chemoresistance, EMT
Citation: Cao Y, Lu C, Beeraka NM, Efetov S, Enikeev M, Fu Y, Yang X, Basappa B, He M and Li Z (2024) Exploring the relationship between anastasis and mitochondrial ROS-mediated ferroptosis in metastatic chemoresistant cancers: a call for investigation. Front. Immunol. 15:1428920. doi: 10.3389/fimmu.2024.1428920
Received: 07 May 2024; Accepted: 14 June 2024;
Published: 02 July 2024.
Edited by:
Yu’e Liu, Tongji University, ChinaReviewed by:
Yulu Wang, Xiajin Country People’s Hospital, ChinaCopyright © 2024 Cao, Lu, Beeraka, Efetov, Enikeev, Fu, Yang, Basappa, He and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhi Li, bGktemhpQG1haWwucnU=; Mingze He , aGVtaW5nemU5N0BnbWFpbC5jb20=
†ORCID: Yu Cao, orcid.org/0000-0002-9682-0980
Chang Lu, orcid.org/0009-0005-5739-7357
Narasimha M. Beeraka, orcid.org/0000-0001-6879-7835
Sergey Efetov, orcid.org/0000-0003-0283-2217
Mikhail Enikeev, orcid.org/0000-0002-3007-1315
Yu Fu, orcid.org/0000-0001-9601-4498
Xinyi Yang, orcid.org/0009-0009-2320-0834
Basappa Basappa, orcid.org/0000-0002-8844-468X
Mingze He, orcid.org/0000-0003-0601-4713
Zhi Li, orcid.org/0000-0003-2062-8463
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.