 Hong Xi Liao1,2†
Hong Xi Liao1,2† Naijian Wang
Naijian Wang Dickson Kofi Wiredu Ocansey
Dickson Kofi Wiredu Ocansey Fei Mao
Fei Mao- 1Key Laboratory of Medical Science and Laboratory Medicine of Jiangsu Province, School of Medicine, Jiangsu University, Zhenjiang, Jiangsu, China
- 2Department of Laboratory Medicine, Lianyungang Clinical College, Jiangsu University, Lianyungang, Jiangsu, China
- 3The People’s Hospital of Danyang, Affiliated Danyang Hospital of Nantong University, Zhenjiang, Jiangsu, China
- 4Department of Laboratory Medicine, Danyang Blood Station, Zhenjiang, Jiangsu, China
Inflammatory bowel disease (IBD), a condition of the digestive tract and one of the autoimmune diseases, is becoming a disease of significant global public health concern and substantial clinical burden. Various signaling pathways have been documented to modulate IBD, but the exact activation and regulatory mechanisms have not been fully clarified; thus, a need for constant exploration of the molecules and pathways that play key roles in the development of IBD. In recent years, several protein post-translational modification pathways, such as ubiquitination, phosphorylation, methylation, acetylation, and glycolysis, have been implicated in IBD. An aberrant ubiquitination in IBD is often associated with dysregulated immune responses and inflammation. Mesenchymal stem cells (MSCs) play a crucial role in regulating ubiquitination modifications through the ubiquitin-proteasome system, a cellular machinery responsible for protein degradation. Specifically, MSCs have been shown to influence the ubiquitination of key signaling molecules involved in inflammatory pathways. This paper reviews the recent research progress in MSC-regulated ubiquitination in IBD, highlighting their therapeutic potential in treating IBD and offering a promising avenue for developing targeted interventions to modulate the immune system and alleviate inflammatory conditions.
1 Introduction
Over the past 20 years, the incidence and prevalence of Inflammatory bowel disease (IBD) have increased in the newly industrialized countries of Asia, South America, the Middle East, and Africa, and the rate is particularly significant in South America and East Asia. Thus, IBD is gradually expanding to become a global disease of public health concern1. According to China’s epidemiological data, the incidence of IBD, ulcerative colitis (UC) and Crohn’s disease (CD), in the north of China is lower than in the south. For instance, the age-standardized incidence of IBD in Daqing City (Heilongjiang Province), Wuhan, and Guangzhou are 1.77/100,000, 1.96/100,000, and 3.14/100,000 respectively (1, 2). According to a study (3), the number of IBD patients in China will reach 1.5 million by 2025. Similar observations have been reported, with an estimated 6·8 million cases of IBD globally, where the USA had the highest age-standardized prevalence rate (464·5 [438·6–490·9] per 100 000 population), followed by the UK (449·6 [420·6–481·6] per 100 000) (4).
IBD has evolved from a common and fatal condition to a manageable chronic condition (5), which is divided into two broad categories, mainly UC and CD (6, 7). The main clinical manifestations are persistent abdominal pain, diarrhea, and bloody stool, accompanied by malnutrition, weight loss, mental distress, and other symptoms. If the course of the disease is prolonged, there is a possibility of colorectal cancer, a severe threat to human health. It has been observed that people with a history of digestive disease, a family history of IBD, and a functional imbalance in the body’s immune system are more likely to develop IBD than the general population (8). As a chronic non-specific intestinal inflammatory disease, IBD is associated with environmental, gut microbial, genetic, and immune factors. If IBD is not correctly diagnosed and treated in time, the disease becomes severe and lesions accumulate in organs of the body, with complications for disease such as colon cancer, coronary heart disease, primary sclerosing cholangitis, and phlebitis.
Ubiquitination is an important post-translational modification of proteins, and the enzymes involved mainly include the E1 ubiquitin-activating enzyme (E1), E2 ubiquitin binding enzyme (E2), and E3 ubiquitin-ligase (E3). Ubiquitin modification can regulate the localization and function of proteins in cells, degrade proteins, and regulate life activities such as signal transmission, gene expression regulation, cell proliferation, differentiation, apoptosis, inflammation and immunity (9). Abnormal ubiquitination can cause cancer, metabolic syndrome, neurodegenerative diseases, autoimmune diseases, inflammatory diseases, infections, and muscular dystrophy. Recent studies have reported associations between ubiquitination and/or deubiquitination and the onset and development of IBD (10). A genome-wide association analysis (GWAS) study showed that rare variants of E3 ligase RNF186 were associated with IBD (11). At the same time, the expression of ubiquitin mRNA in the colonic tissue of experimental colitis rats was significantly higher than that in the normal group. Therefore, abnormal ubiquitination may be one of the important mechanisms of colonic inflammation and immune damage in IBD.
In recent years, extracellular vesicles (Evs), as a strategy of “cell-free therapy,” have become a “new favorite” in research. With the development of various related technologies, more and more researchers have turned their attention to the use of Evs for disease diagnosis, prognosis, and therapeutic clinical applications. Mesenchymal stem cells (MSCs) and their derived exosomes (MSC-Exs) have been shown to play a significant role in the repair of various diseases, including IBD (12). MSCs and MSC-Exs play an important role in the information transmission process between damaged intestinal cells and can be involved in the regulation of intestinal inflammation and damage repair, showing great potential in the treatment of IBD (13). The main mechanism by which mesenchymal stem cells-derived extracellular vesicles (MSC-Evs) inhibit the activation of colon macrophages depends on the inhibition of NF-κB and iNOS transduction signals. After injection of MSC-Evs, the expression of NF-κB p65 in colon macrophages can be downregulated, and the production of NO, IL-1β, and IL-18 can be reduced, thus alleviating the symptoms of colitis (14, 15). At the same time, ubiquitination plays an essential role in the regulation of multiple biological functions, including inflammation. Studies in mice with colitis found that ubiquitin protein from inflammatory tissue is upregulated, and ubiquitin (Ub) is involved in the signaling pathway that regulates the expression of inflammatory factors (mTOR signaling pathway) (16). Wu et al. found that human umbilical cord mesenchymal stem cells-derived exosome (hucMSC-Ex) can down-regulate the expression level of ubiquitin protein. This, in turn, reduces NF-KB and mTOR activation (17). In addition, hucMSC-Ex can modulate the expression of polyubiquitination, including K48. It is concluded that MSC-Ex may play an anti-inflammatory role by regulating the level of ubiquitin modification. Currently, the product development and clinical translational application of MSCs and MSC-Exs are a hot topic in drug development, and cell-free therapy plays a unique role as a breakthrough clinical therapy technology. However, no literature currently summarizes the research progress of MSCs regulating ubiquitin modification to repair IBD. This review can provide a theoretical basis for Cell-free therapy to treat IBD through ubiquitin modification, which has important research value.
2 Mesenchymal stem cells
MSCs are a class of pluripotent stem cells with the function of self-renewal, self-proliferation, and multi-differentiation (18, 19). They positively express CD73, CD90, and CD105 and negatively express CD19, and CD45 (20). MSCs are derived from a wide range of sources, including bone marrow, adipose tissue, endometrial polyps, umbilical cord, amniotic fluid, and placenta (21). They generally possess the functions of inducing regeneration, maintaining general tissue homeostasis, and homing at target sites, which are their inherent characteristics (22). However, the differentiation and proliferation potential of MSCs from different sources may be very different (23).
MSCs can interact with cells of both the innate and adaptive immune systems. Evidence has shown that MSCs exert immunomodulatory functions by regulating the activation, proliferation, and differentiation of immune effector cells, including natural killer cells (NK), macrophages (Mø), dendritic cells (DC), B lymphocytes, and T lymphocytes (24). MSCs can down-regulate NKp30 and natural-killer group 2, member D (NKG2D), to inhibit the cytotoxic activity of resting NK cells, and the latter is an activated receptor involved in NK cell activation and target cell killing (25). MSCs can regulate Th1/Th2 balance (T helper cells) by influencing the levels of interleukin-4 (IL4) and interferon (IFN-γ) in effector T cells (26). MSCs can also reduce inflammation, improve tissue damage, and prevent infection by secreting a variety of immunomodulatory factors, including IFN-γ, prostaglandin E2 (PGE2), and growth factors (TGF-β, VEGF) (27). In conclusion, MSCs have strong immunomodulatory properties but are less immunogenic.
At present, MSCs are widely used in the research and treatment of various human diseases such as cardiovascular diseases, osteoarthritis, metabolic disease, etc. (28)). LAI et al. believe that MSCs play a role through their secreted products (29). It has been reported that MSCs can secrete a variety of Evs, including exosomes (30). In recent years, exosomes, as a promising substitute for MSCs, have attracted more attention, and have great research significance for experimental and clinical applications.
3 Biogenesis, composition, and characteristics of MSC-derived exosomes
Extracellular vesicles (Evs) secreted by cells are divided into apoptotic bodies, microvesicles, and exosomes based on their size, content, and formation mechanism (31). Table 1 presents the classification and function of Evs. Evs are released by almost all living cells and are found in blood, urine, and bronchoalveolar lavage fluid (32). At present, exosomal research is the most attractive and constantly expanding. In 1983, the research team of Rose M. Johnstone (33), a professor in the Department of Biochemistry of McGill University in Canada, first found exosomes in sheep reticulocytes, which were considered to be cell follicles that could transport nonessential proteins between cells and were considered the “garbage” of cell metabolism. With further study, Johnstone named these small vesicles as exosomes in 1987.
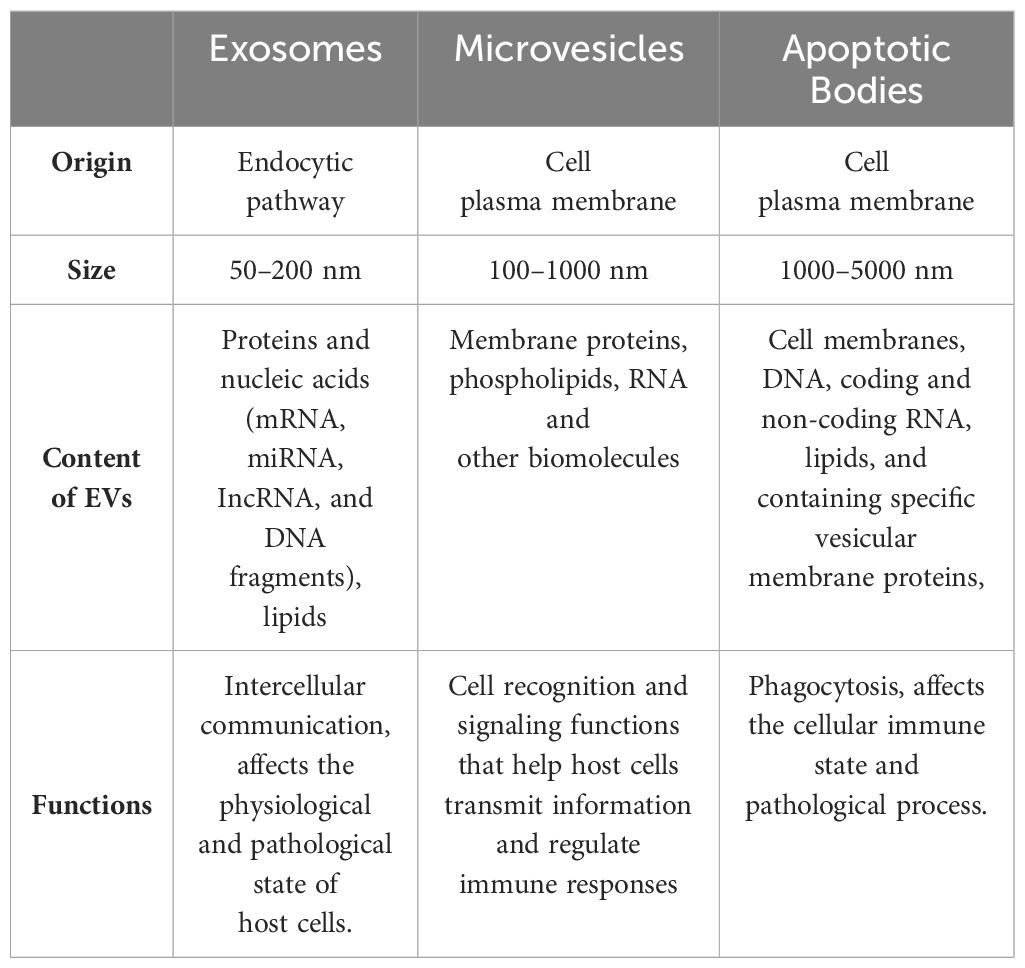
Table 1. Classification and function of Evs.
Exosomes, uniform in size and 50 to 200nm in diameter, have no cellular structure and are highly stable (34–36). Exosomes facilitate intercellular communication by carrying bioactive substances including mRNA, miRNA, IncRNA, DNA fragments, proteins, and lipids from parent cells, thereby regulating the activities of target cells. They positively express markers such as the tetraspanins protein family (CD63, CD81, and CD9), MVB biogenic proteins (Alix, TSG101, and ESCRT Complex), membrane transporters and heat shock proteins (HSP70, HSP90), lipid-associated proteins, etc. (37).
Exosome biogenesis includes three main stages of endosomes, multivesicular body (MVBs) formation and exosome release (Figure 1), which involve double invagination of the plasma membrane (38, 39). Exosomes originate in the endosomal system of cells. Extracellular substances first enter the cell through membrane invagination and endocytosis, fuse with early endosomes (ESEs), and gradually develop and mature into late endosomes (LSEs). Late endosomal invagination leads to the emergence of intracavitary vesicles (ILVs), and multiple ILVs aggregate to form MVBs. MVBs are fused with the cell membrane and released outwards as exosomes in lipid bilayers.
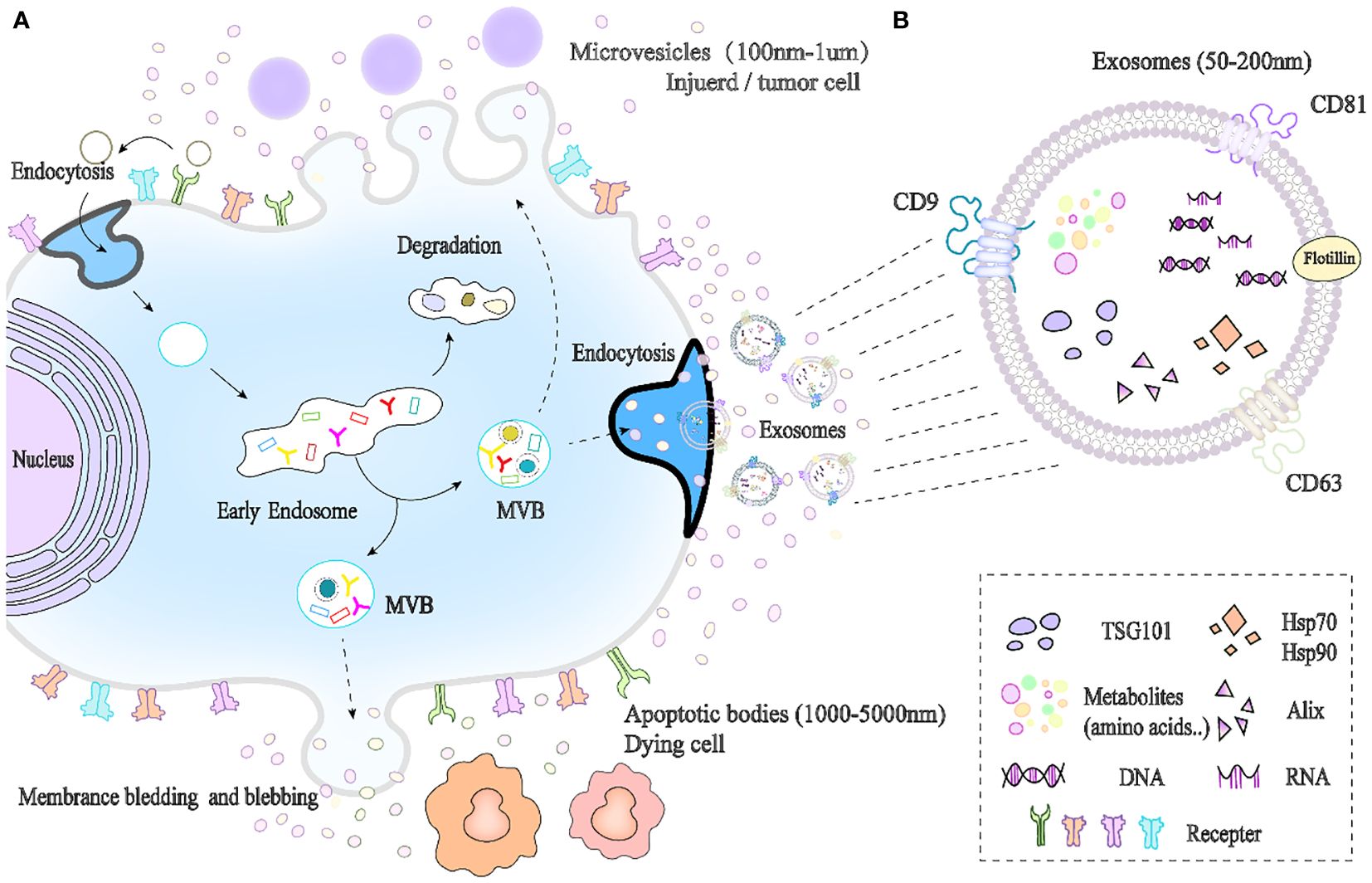
Figure 1. Biogenesis and composition of exosomes. (A) Exosomes originate from endosomal pathways, and extracellular substances enter cells through membrane invagination and endocytosis and then develop into early endosomes (ESEs), late endosomes (LSEs), and intracavitary vesicles (ILVs). Multiple intracavitary vesicles (ILVs) aggregate to form MVBs. MVBs can fuse with lysosomes to degrade and release content into the cytoplasm or be released outside the cell by budding through the cell membrane, and the latter is called exosomes. (B) Exosome composition.
Because exosomes can be detected in body fluids, they are considered noninvasive or minimally invasive biomarkers for disease diagnosis. Studies have implicated exosomes in the pathophysiology of several diseases (40). For example, Gui et al. found that compared with healthy controls, the expressions of miR-1 and miR19b-3p in CSF exosomes of Parkinson’s disease (PD) patients were down-regulated, while miR-153, miR-409–3p, and miR-10a5p were up-regulated (41). The exosomal miRNAs significantly correlated with the severity of PD and may be an effective biomarker for evaluating disease development in clinical PD patients. Exosomes can also be used as diagnostic markers of cancer. Roccaro et al. showed that the expression of miRNA-15a in bone marrow MSC-exosomes (BMMSC-Ex) of multiple myeloma patients is significantly down-regulated, which is closely related to the characteristics of multiple myeloma (42). In addition, the presence of high levels of Evs expressing TGF-β2 in the breast milk of normally lactating women can induce breast cancer. What is interesting is that exosomes can play a therapeutic role by delivering drugs themselves or as functional cargo for drug delivery (43, 44). For example, MSC-Exs are used in the treatment of graft-versus-host disease (GVHD), with significant therapeutic effects observed after repeated injection without serious side effects (45, 46).
MSC-Exs have similar biological functions to MSCs, playing an important role in improving tissue repair, immune regulation, inhibiting inflammatory response, and reducing apoptosis (47). They exhibit no tumorigenicity, and are easier to extract, modify, and store (48). MSC-Ex is a natural, non-toxic vesicle that can deliver mRNA, miRNA, and protein. Therefore, it has received extensive attention as a cell-free therapeutic carrier in treating autoimmune diseases, including IBD (49). MSC-Ex research has made substantial progress in the treatment of multiple sclerosis, type-1 diabetes, and other diseases (50–52) (Table 2 shows the application of MSCs and MSC-Ex in various clinical diseases). In the DSS-induced IBD model, hucMSC-Exs treatment reduces the infiltration of macrophages in colon tissue and inhibits the expression of IL-7 (74). Moreover, hucMSC-Exs alleviate insulin secretion function in T2DM by reversing peripheral insulin resistance and alleviating β cell destruction, providing a new approach for T2DM treatment (60). In clinical treatment, MSC-Exs have obvious advantages over MSCs and may completely replace MSC therapy in the future.
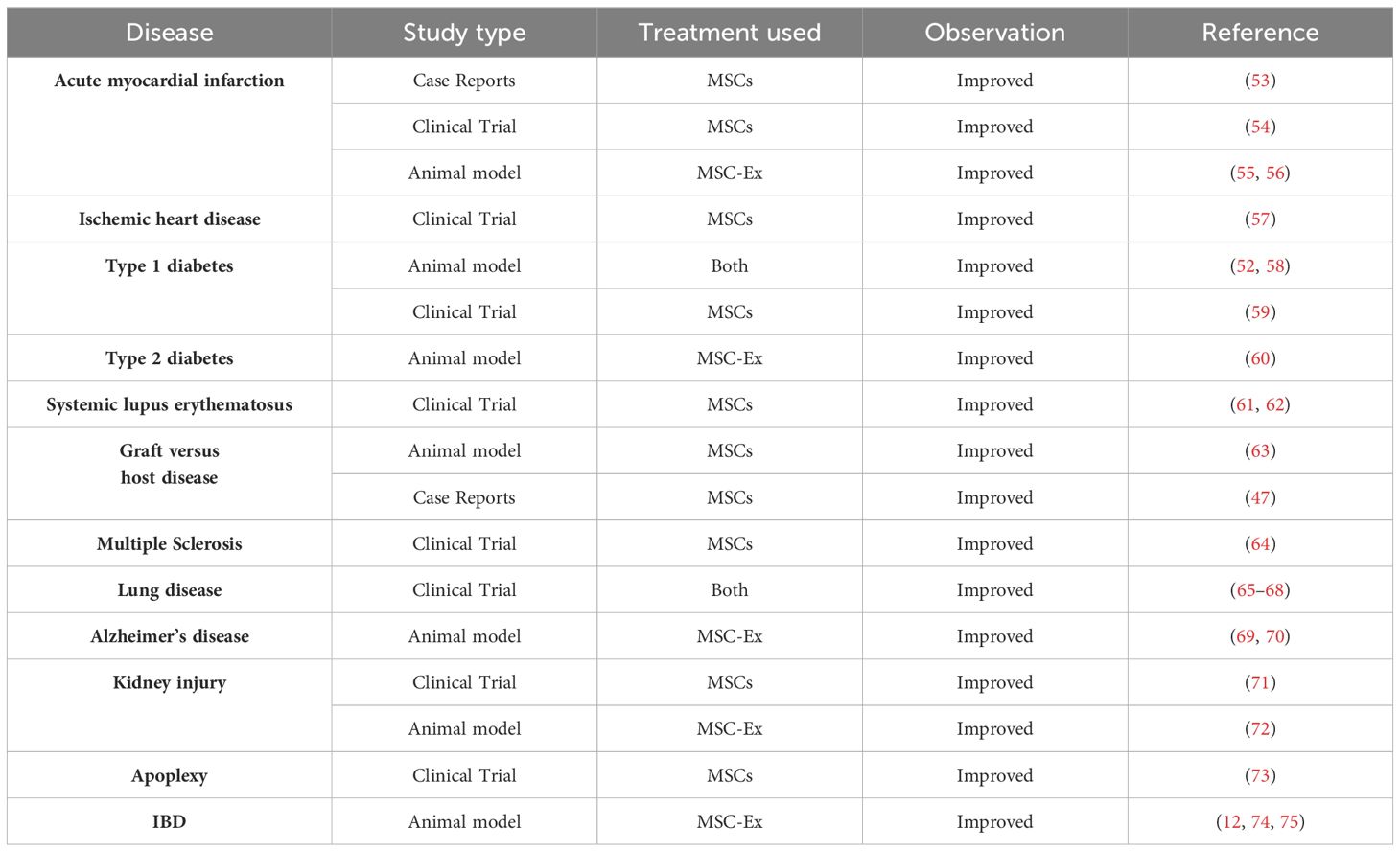
Table 2. Applications of MSCs and MSC-Ex in various clinical diseases.
4 Ubiquitination
Ubiquitination is an important process that regulates the normal expression of genes, along with phosphorylation, glycosylation, acetylation, amidation, etc. (76, 77) Ubiquitination modifies post-translated proteins (PTMs), including protein degradation, signal transduction, and DNA damage repair (78, 79). Ubiquitination modulates various cellular activities involved in inflammatory responses, innate or adaptive immune responses, and ribosomal functions, which are essential for many cell life processes (80).
Cellular processes depend on ubiquitin (Ub) and ubiquitin-like proteins (UBLs) in ubiquitination systems. Ub, a highly conserved small protein in eukaryotes, contains 76 amino acid residues and is a significant part of regulating the everyday life activities of biological proteins (81). The UBLs found so far include NEDD8 (neural-precursor-cell expressed developmentally down-regulated 8) and SUMO (1–5, small ubiquitin-like modifier). Although they have similar functions to Ub, the receptors and signaling molecules they contact are not the same and play different roles. Related studies have shown that NEDD8 which has the same homologous sequence (> 50%) as Ub, binds to Cullin, the subunit cullin of Cullin-ring ligase (the largest multi-unit E3s ubiquitase family, CRLs). After overactivation, it promotes the degradation of tumor suppressor factors (p21, p27) and the occurrence and evolution of cancer (82). Yang W et al. experimentally demonstrated that increased SUMOylating has a neuroprotective effect and seems necessary for survival, at least under certain conditions (ischemia) (83). This theory was further validated in animal models, which found that cerebral ischemia in mice leads to a significant increase in SUMO2/3 conjugates in the hippocampus and cerebral cortex, and a neuroblastoma cell model undergoing hypoxia/glucose deprivation followed by a short period of reoxygenation under the same conditions also exhibits significant increases in SUMO2/3 conjugation (84, 85).
From a more microscopic point of view, the Ub molecule itself contains seven lysine (Lys) residues, and the amino terminus of the Lys residues (K6, K11, K27, K29, K33, K48, and K63) on the substrate protein monopeptide can be labeled for monoubiquitination. Table 3 summary of the different types of ubiquitination and their functions. When the Ub molecule forms a specific isopeptide bond with the carboxyl terminus of another Ub molecule through Lys residues, it is further coupled to form a multiubiquitin chain. Ubiquitin can also bind to non-lysine residues, such as cysteine (Cys). Ubiquitin chains with different links have different cellular functions, constituting the “ubiquitin code” of diversity and complexity (86). Monoubiquitination is generally associated with receptor internalization, while polyubiquitination is usually associated with proteasome degradation signaling (87). Related studies have found that K48 and K11 connected ubiquitin chains mediate substrate proteins to be digested into amino acids by soluble peptidase (26S proteasome complex) in cytoplasm or nucleus through a series of enzymatic reactions (88, 89). Many experimental studies have shown that the polyubiquitin chain of K63 is associated with non-proteasome functions, such as DNA repair, protein sorting, immune modulation, and regulation of the activation of the NF-κB signaling pathway, and also protects target proteins from multiple signaling pathway functions, including T cell receptors, Toll-like receptors (TLRs) and RIG-I-like receptor-mediated signal transduction (90–93). The mechanisms determining whether a protein is monoubiquitinated or polyubiquitinated are not fully understood and require further studies.
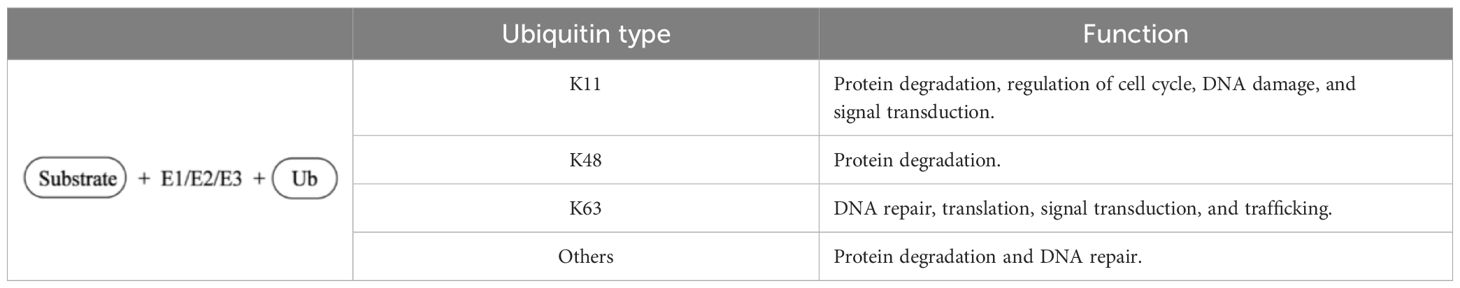
Table 3. Summary of the different types of ubiquitination and their functions.
Ubiquitination of proteins is accomplished through a series of continuous enzymatic reactions (94–96) (Table 4). For the enzymes required for ubiquitination, it is currently estimated that eukaryotic organisms have two E1 enzymes (UBA1 and UBA6), with approximately 30–50 E2 enzymes and more than 600 E3 enzymes. In a tertiary E1-E2-E3 enzyme-linked reaction in mammals, any two members of the E1 family can label all E2 with Ub, and 40 known E2 enzymes can further transmit Ub to the E3s family. Auxiliary E4 enzymes have been explored (97); When the substrate protein signals, the E1 enzyme activates ubiquitin and starts the ubiquitination process, which requires ATP to provide energy. Then, the E3 ubiquitin ligase specifically recognizes the substrate protein and guides the E2 conjugation-carried Ub to covalently bind to the substrate protein (Figure 2) (98). In the ubiquitination system, these three different types of enzymes cooperate to complete the task of modifying proteins. The ubiquitin activator E1 binds to the tail of the ubiquitin molecule, and regulates the downstream of the ubiquitination reaction. Ubiquitin coupling E2 enzyme controls the length and connection type during the assembly of ubiquitin chains, and K48 and K63-mediated polyubiquitination regulates the inflammatory development of the NF-κB signaling pathway. The specificity of the ubiquitin chain is widely believed to be determined by E2-E3 (RING-E3s) matching or substrate-E3 (HECT and with E6-APC) complexes (99).

Table 4. Ubiquitination involves several enzymes: ubiquitin-activating enzyme (E1), ubiquitin-coupling enzyme (E2), ubiquitin-ligase (E3), and deubiquitination enzyme (DUBs).
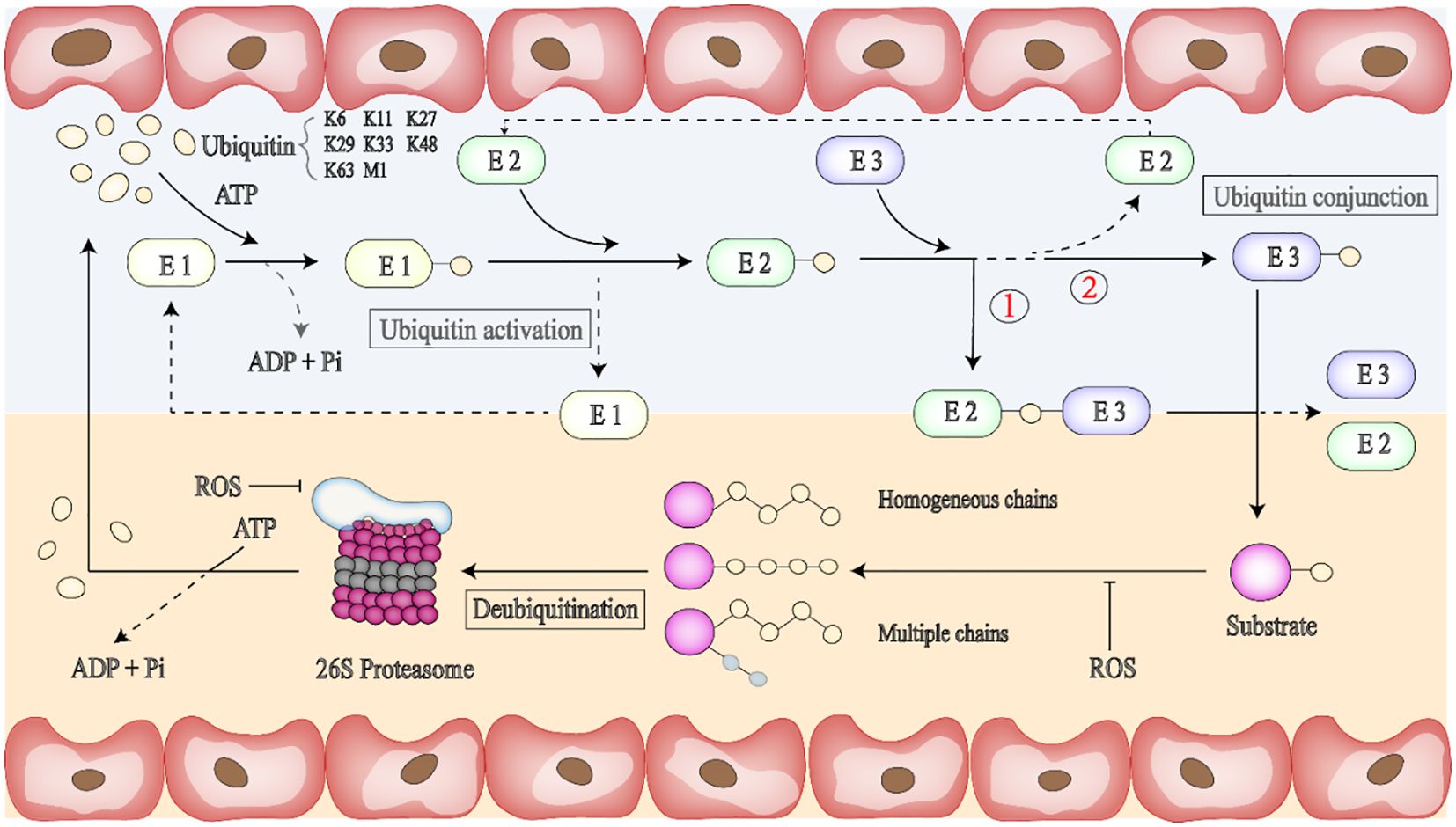
Figure 2. Binding of ubiquitin to protein substrates is a multi-step process. Ubiquitin molecules can be attached to ubiquitin or proteases containing lysine residues such as K6, K11, K27, K29, K33, K48, and K63. First, ATP provides energy, and the E1 enzyme activates ubiquitin and initiates the ubiquitination process. E2 enzyme can provide ubiquitin directly to the protein substrate through lysine (K) residues in the target protein. The third step is to bind the E2 ubiquitase-linked ubiquitin to the E3 ligase-linked target protein, and there are two binding forms, respectively,① E2-Ub-substrate protein-E3 or ② Ub substrate protein-E3. Ubiquitin protein ligase (E3) can promote the interaction with substrate proteins. Ubiquitination is a reversible post-translational protein modification; thus, the substrate carrying the ubiquitin chain can be deactivated by the UPS (Ubiquitin-proteasome system) or DUBs (Deubiquitinating enzymes), and free ubiquitin monomers can be re-recruited for the next round of ubiquitination. The ubiquitin chain usually determines the fate of ubiquitinated proteins.
The stability, functional activity, and interaction of the modified proteins may change, but the modification is a reversible PTM process that can be removed by the UPS (Ubiquitin-proteasome system) or DUBs (Deubiquitinating enzymes) (100). The UPS can remove proteins that have been damaged or are no longer needed in cells (tumor suppressor proteins, cell cycle regulatory proteins, etc.), which is an energy-consuming but highly efficient way to degrade proteins. When the substrate has four or more UB or UBLs, the 26s protease hydrolyzes it into peptide chains, releasing ubiquitin monomers that can be recycled (101). DUBs catalytic deubiquitination modification makes the ubiquitination process maintain the dynamic balance of the cellular process. After DUBs are bound to the ubiquitin substrate protein complex, the ubiquitin chain is broken by severing the isopeptide bond between Lys and the C-terminal of ubiquitin, and the ubiquitin monomer is free and can be collected, thus starting the next round of ubiquitination.
There are approximately over 100 types of DUBs, including ubiquitin-specific proteases (USP), ubiquitin-c-terminal hydrolases (UCH), and so on (102, 103).Different DUBs play different roles in inflammation and cancer development by affecting their substrates’ protein stability, enzyme activity, or subcellular localization (104). The USPs family is the most commonly studied DUB family. USPs regulate protein activation by dissociating single or multiple ubiquitin chains from ubiquitinated substrates (105). In malignant tumors, USP18 is elevated and activates AKT/mTOR signaling, promotes phosphorylated AKT (p-AKT) and p-mTOR protein expression, leading to cancer cell proliferation and migration (106). USP11 controls its stability by promoting deubiquitination of a residual protein (VGLL, a tumor suppressor) and exerts its tumor suppressor effect through the VGLL4/YAP-TEAD regulatory ring (107). Overexpression of USP14 inhibits I-κB and increases NF-κB phosphorylation while increasing cancer cell migration, invasion, and EMT (108). Under selective conditions, USP7, USP10, USP29, USP42, and other DUBs regulate p53 ubiquitination levels (109).
5 The relationship between ubiquitination, MSC/MSC-Ex, and IBD
The occurrence of IBD is mainly due to the abnormal amplification of the immune response of the intestinal mucosal immune system to microbial antigens from the gut in some specific populations carrying susceptible genes under certain circumstances, resulting in inflammatory damage of the intestinal mucosa. In 2001, NOD2 was identified as a susceptibility gene for CD due to its polymorphism (110). In addition, approximately 240 gene loci have been found to be associated with IBD susceptibility and occurrence, of which about 30 are CD and UC (111, 112). The loss of balance between proinflammatory and anti-inflammatory factors leads to the activation of NF-κB, TNFα, NLR, and TLR pathways to expand the range of inflammation and promote the development of IBD (Figure 3). According to mechanistic studies, a variety of signaling pathways have regulatory effects on IBD, but the specific activation, effects, and regulatory mechanisms largely remain unclear. Therefore, further exploratory studies on the pathogenesis of IBD, new targets for its treatment, and diagnostic biomarkers are needed.
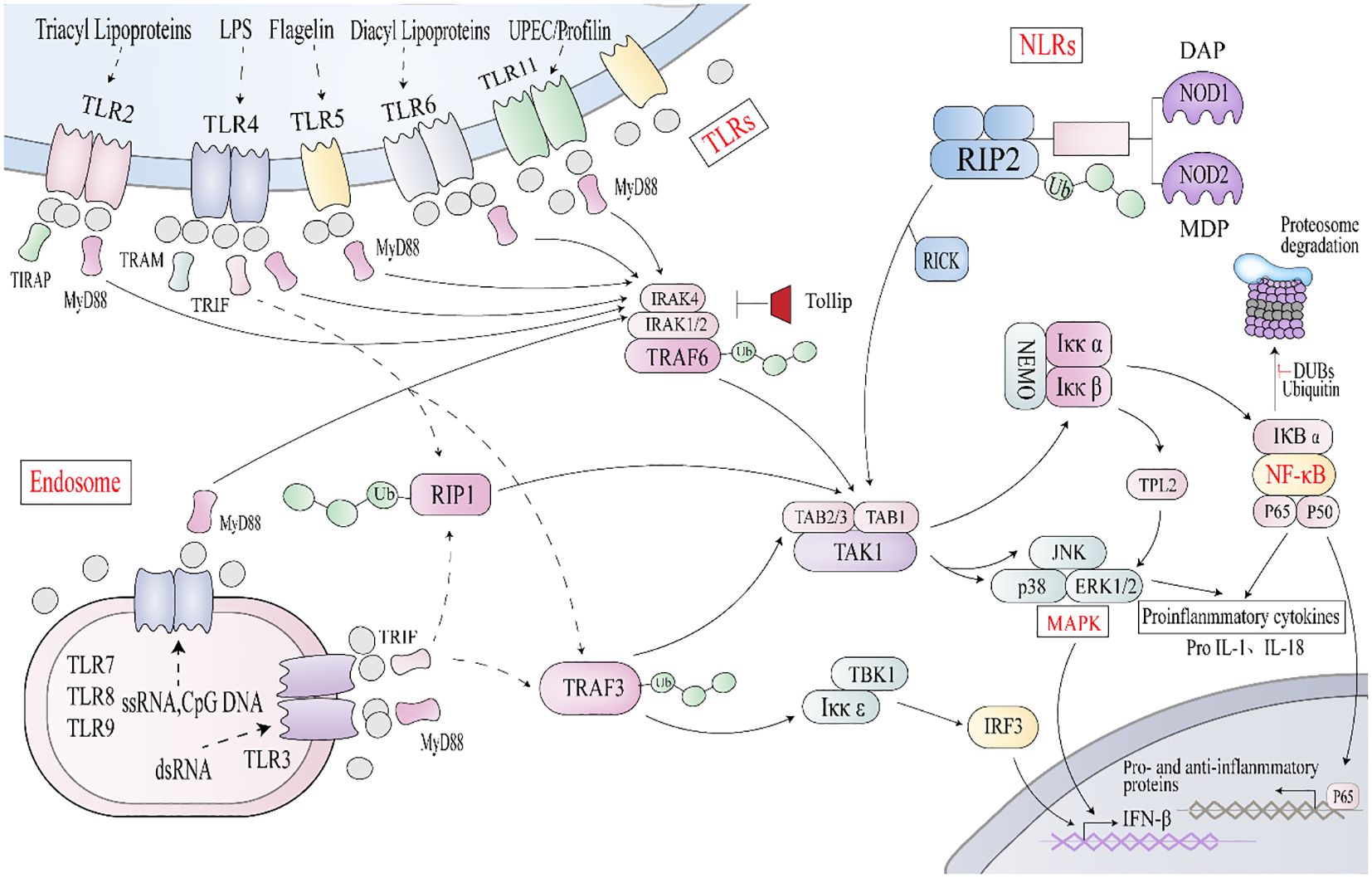
Figure 3. Key signaling pathways associated with IBD ① TLR stimulation triggers MyD88 to interact with IRAK4 (interleukin-1 receptor-associated kinase 4) and ② NLRs interact with RIP2, causing TRAF3, TRAF6, and Lys63 polyubiquitinizes RIP1, to recruit TAK1/TAB2/3 complex or IkK complex. This triggers the activation of NF-κB and mitogen-activated protein kinase (MAPK) signaling pathways, promoting the transcription of proinflammatory and anti-inflammatory genes. Endosomal TLRs transmit signals through a TRIF-dependent pathway, and TRIF, along with RIP1 and TRAF3, activates TAK1 or IKKϵ, leading to phosphorylation of IRF3 and expression of interferon β.
The immune system is mainly composed of the innate and adaptive systems (113, 114). Ubiquitination plays a vital role in the regulation of innate immune signal transduction, but so far, only Lys residues have been identified as ubiquitination sites in innate immune signal molecules. Whether ubiquitination of non-lysine residues plays a role in innate immune signal transduction needs further study. Adaptive immunity relies on specific immune cells (T and B lymphocytes), mediating humoral and cellular immunity, and is characterized by the presence of highly specific antigen recognition receptors, namely T cell receptors (TCR) and B cell receptors (BCR). The regulatory effects of ubiquitination on immune cells are varied and not fully understood. MSCs and MSC-Exs have been shown to target the treatment of inflammatory diseases, including IBD, asthma, and rheumatoid arthritis (115, 116). Ubiquitination is involved in the inflammatory response in IBD, serving as a potential therapeutic target through the modulation of MSC and MSC-Ex.
5.1 NF-κB signaling, ubiquitination, and MSC/MSC-Ex
Protein ubiquitination can regulate various signal-mediated inflammatory responses and plays an important role in the occurrence, development, and outcome of inflammatory diseases such as IBD. As a key component of the innate immune response, the NF-κB signaling pathway is one of the ultimate targets for regulating multiple upstream signaling pathways. Research has shown that the NF-κB signaling pathway is crucial in inducing pro-inflammatory gene expression, regulating inflammasome, activating inflammatory T lymphocytes, and innate immune cell differentiation in IBD patients. The activation of the NF-κB signaling pathway requires RIP2 and TAK1-mediated polyubiquitination of NEMO (NF-κB essential regulator kinase, also known as IKγ) and phosphorylation of the kappaB kinase inhibitor (IKK) complex consisting of NEMO, IKKα, and IKKβ. IKKs can be activated by bacterial lipopolysaccharide (LPS), tumor necrosis factor-α (TNF-α), IL-1β, and various physical and chemical stresses (117). Phosphorylated IκB family proteins become targets of K48 polyubiquitination-dependent proteome degradation, releasing active NF-κB molecules into the nucleus. The classical NF-κB pathway is dominated by the action of IKKβ, which phosphorylates IKKβ family members such as IKKα and P105. When stimulated by LPS, IKK promotes nuclear translocation of p65 and p50 and induces expression of inflammatory factors such as TNF-α, IL-1β, IL-6, and IL-12, further leading to tissue damage (118).
Ub plays a crucial role in regulating the activation of NF-κB signaling. Relevant studies have shown that ubiquitin modification can affect intestinal mucosal inflammatory injury and intestinal epithelial cells’ permeability and apoptosis by regulating the NF-κB signaling pathway. TRAF6, one of the E3 ubiquitin ligases, contains a highly conserved RING domain important for activating the NF-κB pathway (119). Chen et al. report that TRAF6 could only activate IKK under the condition of K63 polyubiquitination and that RING domain mutation results in the loss of ubiquitin ligase activity of TRAF6 and the failure to activate IKK (120). NIK is a central kinase in the non-classical NF-κB pathway and is also involved in the classical NF-κB pathway (121). Inhibition of the E3 ligase TRIM16 effectively increases the formation of K48-linked polyubiquitin chains on NIK (122). NEDD4 is associated with chronic inflammatory diseases, and a single rs8032158 transcription variant (TV3) in the NEDD4 genome has been found in keloid patients to activate the NF-κB signaling pathway by binding to the connexin RIP and highly selectively expressed (123, 124).
MSC and MSC-Ex regulate the NF-κB signaling pathway to influence the treatment of IBD. According to the current literature, the degradation of IκB is mainly dependent on neddylation, and when cullin1 activation is blocked, the accumulation of IκB leads to inhibition of NF-κB activity. Wang et al. verified that miR-326 in hucMSC-Ex inhibits the binding of free NEDD8 and substrate protein cullin1, preventing the expression of E1, E2, and E3 enzymes during neddylation formation (125). The hucMSC-Ex also inhibits the activation of the NF-κB signaling pathway, alleviating IBD. In addition, Qi et al. showed that serum-preconditioned adipose-derived MSCs (CM-AcMSC) significantly prevent the phosphorylation of p65 and IκB in colon cells of DSS-induced colitis model in rats, up-regulate the expression of MUC2 and tight-junction proteins such as ZO-1, claudin-1, and occludin, and protect the integrity of colon mucus (126). These results suggest that CM-AcMSC can significantly mitigate inflammation in colitis rats. A similar study showed that bone marrow MSCs (BM-MSCs) down-regulate the expression of NF-κB p65 mRNA in the colonic mucosa, suggesting that BM-MSCs may influence TNBS-induced colitis by regulating NF-κB mediated proinflammatory response (127). DC-derived exosomes activate the NF-κB signaling pathway via exosomal miR-146b to improve intestinal barrier function in DSS-induced colitis (128). Thus, a number of studies indicate that MSC and MSC-Ex play a role in the treatment of IBD by down-regulating NF-κB signaling, but the specific mechanism related to their regulation of NF-κB signaling via ubiquitination needs further study.
5.2 TLR signaling, ubiquitination, and MSC/MSC-Ex
TLRs are pattern recognition receptors. As an important part of the innate immune system, TLRs activate a series of downstream signals after recognizing pathogen-associated molecular patterns (PAMPs), inducing the secretion of inflammatory cytokines, chemokines, and type I interferons (129, 130). After binding to different stimuli, most TLRs initiate signal transduction by recruiting the adaptor protein MyD88, which contains the TIR domain and is a common important adaptor protein of most TLRs and a variety of envelope receptor-mediated signaling pathways. It plays a role in recruiting downstream kinases and regulating signal transmission; ① In the MyD88-dependent pathway, MyD88 recruits IL-1 receptor-associated kinase-4 (IRAK4) to attract TLR, and the MyD88-IRAK4 complex recruits IRAK4 substrate IRAK2 or related IRAK1 to realize Myddosome (131, 132). This protein interacts with TRAF6, self-ubiquitination modification of TRAF6, activates TAK1, and ultimately stimulates NF-κB and JNK/P38/ERK signaling pathways, which participate in the colon inflammation in IBD; ② MyD88 signaling pathway can also be used as another pathway for TLR to induce inflammation. After TLR activation, it can activate the toll-like receptor-associated activator of interferon (TRIF) and TRAF3, resulting in NF-κB inhibiting the recruitment of protein kinase ϵ/tank-binding kinase 1 (IKϵ/TBK1), inducing the phosphorylation of IRF3 and the expression of interferon-β, and playing an antiviral role. TRIF- and MyD88-mediated signaling involve a series of ubiquitination events. TRAF3 and TRAF6, as members of the E3 ubiquitin ligase family, play an essential regulatory role in MyD88-dependent and TRIF- (non-MyD88) dependent signal transduction, which can be either a “positive signal” or a “negative signal”. Moreover, TRAF3 and TRAF6 can regulate the activation of inflammation-related signaling pathways through ubiquitin modification effects and initiate the gene transcription of many proinflammatory cytokines, such as IL-1, IL-6, TNF-α, and other transcription factors to activate a variety of immune responses, thereby playing a role in immune defense or relieving inflammation. Nrdp1 directly binds to and polyubiquitinates MyD88 and TBK1 while promoting TLR-triggered macrophages to inhibit the production of proinflammatory cytokines. In addition, Nrdp1 and Smurfp1/2 can catalyze the ubiquitination modification of MyD88 or remove the ubiquitin chain for negative regulation (133, 134). A20 and SIGIRR can also adjust the duration and/or strength of the TLR signal (135–137).
Macrophages are considered classic cells in TLR studies (138). They are the most critical cells in inducing colon inflammation, releasing large amounts of DAMP in damaged intestinal epithelial cells and activating NF-κB in intestinal macrophages signaling pathways that promote the secretion of inflammatory factors such as TNF-α and IL-1β, lymphocytes and monocytes, recruit chemokines (CCL-17 and CCL-24) and NO, and promote colon damage. Duan et al. demonstrated that LIM domain 7 (LMO7) is an important molecule that regulates macrophage polarization and inhibits intestinal inflammation in a DSS-induced IBD model (139). When proinflammatory activates macrophages, PFKFB3(6-phosphofructose-2-kinase/fructose-2,6bisphosphatase 3) promotes glycolysis by increasing the activity of phosphofructokinase-1 (PFK1). LMO7 promotes the degradation of PFKFB3 through K48-related ubiquitination, thereby effectively preventing excessive inflammatory response of macrophages and protecting tissues from inflammatory damage.
In the regulation of IBD by MSC and MSC-Ex, Liotta et al. demonstrated that the binding of TLR3 or TLR4 to MSCs can modulate their immunosuppressive activity against T lymphocyte proliferation, thereby restoring an effective T cell response during infections (140). Studies have shown that MSCs can inhibit the LPS/TLR4 signaling pathway, thereby reducing the release of inflammatory factors, improving intestinal symptoms of IBD, and reducing parenteral complications (141). Liu’s team reports that metallothionein-2 in MSC-Exs, a key negative regulator of macrophage inflammatory response, plays an anti-inflammatory role in conjunction with other components of MSC-Exs to maintain intestinal barrier integrity and reduce experimental colitis in mice (142). Other studies indicate that MSC-Ev inhibits the activation of proinflammatory M1 macrophages and promotes their polarization to M2 macrophages, alleviating the inflammatory response and DSS-induced IBD (143). Deng et al. developed a technique that can sustainably release MSC-Exs for regenerative purposes using an in situ synthetic biotin-modified MSC-Ex (Bio-Ex) self-assembled biotinylation (144). The Bio-Ex can be taken up by macrophages and play an immunomodulatory role similar to MSC-Ex, promoting the polarization of macrophages to the M2 phenotype. Perhaps ubiquitin is involved in alleviating IBD through the TLR signaling pathway, or the regulation of macrophage inflammatory response by MSCs and MSC-Exs are linked with TLR/ubiquitination. However, the direct link to this hypothesis remains to be proven.
5.3 NLR signaling, ubiquitination, and MSC/MSC-Ex
Nucleotide-binding and oligomeric domain (NOD)-like receptors (NLRs) are a type of PRRs that are primarily distributed in the cytoplasm and have four broad classes of functions: inflammasome assembly, signal transduction, transcriptional activation, and autophagy (145–147). Cumulative data suggest that NLRs play a vital role in a variety of autoimmune diseases, such as IBD, multiple sclerosis (MS), and systemic lupus erythematosus (SLE) (148).
The C-terminal of NLRs (except NLRP10) contains a leucine-rich repeat sequence (LRR) that specifically recognizes the PAMPs’ or DAMPs’ molecular pattern. Based on the unique functional features of the N-terminal effector domain, NLRs can be divided into five subfamilies: NLRA, NLRB, NLRC, NLRP, and NLRX1. The structure of the NLRA subfamily includes CIITA (class II transcription activator), the activation of which is dynamically regulated by a series of post-translational modifications, such as acetylation, phosphorylation and ubiquitination.
Ubiquitination is involved in activating and terminating NOD signaling cascades. After NOD1 and/or NOD2 activation, the oligomerization of the NACHT domain between the NLRs N-terminal and the LRR domain activates and recruits the interacting proteins to form a semallome including RIP2(also known as RICK). E3 ubiquitin ligase cIAP1 forms a ubiquitin chain with RIP2, catalyzes the ubiquitination of RIP2, induces the activation of the TAK1 complex, triggers the activation of NF-κB and MAPK signaling pathway, and promotes the transcription of proinflammatory genes (IL1β, IL-18). Studies have shown that when the NOD1-RIP2 signaling pathway is activated, hybrid ubiquitin chains containing M1-Ub and K63-Ub bonds are rapidly produced, and hybrid ubiquitin chains may affect the deubiquitination rate of K63-Ub and M1-Ub chains. This affects the duration of the innate immune response (149). After NOD2-RIP2 activation, it promotes K)63-linked polyubiquitination of NEMO, thereby promoting the recruitment of TAK1 and activating the NF-κB signaling pathway (150, 151). TRAF4 is an E3 ligase that has been shown to negatively regulate NOD2 signaling (152). Moreover, autophagy-associated protein 16-like-1 (ATG16L1) negatively regulates NOD-driven inflammatory responses by interfering with RIP2 junction polyubiquitination (153).
In addition to NOD-mediated activation of NF-κB and MAPK, inflammatory bodies (NLRs) can also regulate inflammatory responses through ubiquitination modification (154). The NLRP3 inflammasome (NOD-, LRR- and pyrin domain protein 3) is the most studied, an intracellular polymeric protein signaling complex that participates in the innate immune system and plays an important role in maintaining intestinal homeostasis and preventing colitis (155, 156). NLR proteins such as NLRP3 detect pathogens or danger signals and trigger the assembly of caspase-1 inflammasome, leading to the processing and secretion of IL-1β and IL-18, promoting the proliferation and differentiation of pro-inflammatory macrophages and tissue damage. It also contributes to pyrosis, either directly or through ASC junction proteins. A study found that in the GVHD model, activated NLRP3 inflammasome stimulates choline metabolized TMAO (trimethylamine N-oxide) to induce M1 macrophage polarization, leading to the differentiation of T1 and T17, which aggravated the disease (157). Ubiquitination also plays a vital role in the NLR signaling pathway. According to Xu et al. found that E3 ubiquitin ligase gp78 mediates the mixed ubiquitination of NLRP3 and inhibits the activity of NLRP3 by preventing the oligomerization and subcellular translocation of the NACHT domain of NLRP3, thereby reducing the activation of inflammasome and harmful effects (158). Mai et al. confirmed that promoting mitochondrial autophagy driven by E3 (Parkin) and inhibiting the activation of colonic NLRP3 inflammasome has an inhibitory effect on mouse DSS-induced colitis (159). The E3 ligase TRIM31 binds NLRP3 directly and promotes K48-linked NLRP3 ubiquitination and proteasome degradation, maintaining low NLRP3 expression and preventing unwanted inflammasome activation (160). Moreover, interference with A20 inhibits macrophage proliferation and M2-like polarization by activating the NLRP3 inflammasome pathway (161).
Studies report that MSC-Ex miR-378a-5p targets and blocks NLRP3 inflammasome activation in macrophages, leading to Caspase-1 cleavage and IL-1β and IL-18 reduction, delaying pyroptosis cell death and improving IBD (162). MSC-Ex alleviates colitis by increasing FXR in the colon, which binds to the NLRP3 inflammasome and inhibits the activation of inflammasome components (163, 164). The regulation of IBD inflammation by ubiquitination and the treatment of IBD with MSCs and MSC-Exs also involve NOD and NLR signaling pathways, and there may be some unknown relationships that require further studies.
5.4 T cell activation, ubiquitination, and MSC/MSC-Ex
When microbes and metabolites interact with pattern recognition receptors (PRR), such as pregnane X receptor (PXR) and TLR, there is activation of signaling pathways and key proteins that control mucosal barrier and intestinal immune functions. When pathogens invade the human body, TLRs guide mucosin-2 (MUC2) in the intestinal mucus layer to prevent intestinal pathogens and their secretions from penetrating the mucosa, thus playing an essential role in preventing inflammation (165–167). During the progression of IBD, activated T lymphocytes infiltrate the inflamed site and produce a variety of cytokines, further aggravating intestinal inflammation. T cells in IBD patients are composed of pro-inflammatory effector subsets (Th1, Th2, and Th17) and/or regulatory T (Treg) cells that have immunosuppressive effects and maintain intestinal homeostasis (168, 169). The proportion of CD8+ T suppressor cells and CD4+ T helper cells in the lamina propria and epithelium of the intestinal tract of IBD patients is usually normal, but the activated cells showed an increasing trend. The cytokine IL-13 secreted by Th2 cells plays a role in UC, and Th17 is involved in the pathogenesis of IBD through the production of IL-17A. On the other hand, Treg cells suppress the immune response and prevent self-hyperimmunity, and IL-10 produced by Treg cells inhibits the proinflammatory cell Th17 in the gut. In contrast, the elimination of IL-10 receptors in Treg cells leads to Th17 dysregulation and colitis (170).
Both programmed death protein 1 (PD-1) and cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) appear to be key markers for controlling T cell tolerance and have negative regulatory effects on T cell immune function (171, 172). CTLA-4 is mainly involved in the early stages of T cell immune responses in lymph nodes (173, 174), while PD-1 is mainly involved in the late stages of T cell immune responses in peripheral tissues. PD-1 is expressed by activated T cells and downregulates T cell effector function after binding to its ligands PD-L1 and PD-L2 on antigen-presenting cells (175). Intestinal epithelial cells of IBD patients overexpress PD-L1 and PD-L2 (176), and PD-1 blocking can reverse the in vitro inhibition of effector T cells mediated by Treg. Most studies have shown that Tregs constitutively express CTLA-4, which is considered important for its inhibitory function (177).In a FoxP3+ conditional knockout mouse model with CTLA-4 deletion, Wing et al. clearly demonstrated that CTLA-4 deficiency in FoxP3+ Treg cells impairs its inhibitory function (178). Experimental data of Takahashi et al. showed that the signals sent by TCR and CTLA-4 may activate CD25+CD4+ regulatory T cells, thereby transmitting negative signals of activation and proliferation to other T cells. These possibilities are currently being investigated (179).Therefore, maintaining a balanced ratio of Th17 and Treg cell populations is essential for maintaining the intestinal immune system (180).
There is growing evidence that ubiquitination-degrading proteins play a role in suppressing immune responses by targeting the destruction of signaling proteins and pro-transcription factors. Dysfunction of E3s ubiquitin ligase, the final step in catalyzing ubiquitin attachment to substrate proteins, can lead to abnormal T cell activation and loss of tolerance to autoantigens, resulting in immune dysfunction (181), and mice lacking these proteins show significant inflammation and/or autoimmune-like symptoms. Several E3 ubiquitin ligases, most notably Itch, Roquin, and CBI-b, have been shown to regulate T cell activation. Ramon et al. found that NEDD4 family interacting protein 1(Ndfip1) is a regulatory protein of Itch, and the combination of the two can make JunB ubiquitination (182). JunB is a transcription factor that promotes the expression of Th2 cytokines IL-4 and IL-5, Therefore, blocking the production of IL-4 and IL-5 may be one of the targeted mechanisms of Th-cell associated gastroenteritis and IBD. In addition, deubiquitinating enzymes USP22 and UCHL1 have been shown to deubiquitinate and stabilize PD-L1 protein (183, 184). Proteolytic targeting chimeras (PROTACs) can recruit the E3 ligase RNF43 to the lysosomes that induce PD-L1, promoting its ubiquitination and subsequent degradation (185). Fujiwara et al. first demonstrated that Cbl-b deficiency in mice leads to functional resistance of T cells and NK cells to PD-L1/PD-1-mediated immune regulation and mild autoimmune reactions (186). To date, there has been no report of any E3 directly ubiquitinating CTLA-4 or CTLA-4 ubiquitination sites. However, multiple studies have shown a strong correlation between CTLA-4 ligation and the function and expression of Cbl-b (187). It is currently known that the main CTLA-4 inhibitory function is mediated by key T cell inhibitory E3 ligases, namely Cbl-b, Itch, and GRAIL (188). Overall, Cbl-b not only mediates CTLA-4 signaling but also mediates PD-1-induced immunosuppression.
Exosome-mediated immune response has been shown in the pathophysiology of IBD (189). The effect of MSCs on T cells seems to depend on the MSC/T cell ratio: a high MSC/T cell ratio has a strong inhibitory effect, while a low ratio may enhance T cell proliferation. It is known that T-box (T-BET) and retinol-associated orphan receptor γ(t) in T cells (RORγT), as central regulators of Th1 and Th17 cells, respectively, are pathogenic factors for IBD progression, while MSCs may down-regulate Th1-Th17driven autoimmune and inflammatory responses by influencing the expression of T-bet and RORγt (190). HucMSC prevents experimental colitis by increasing the number of CD5+ B cells and CD5+Bregs that produce IL-10, and restores Treg/Th17/Th1 imbalances (191). Olfactory Ecto-Mesenchymal Stem Cell-Derived Exosomes (OE-MSC-Exs) regulate T cell responses and have a significant inhibitory function on CD4+ T cells, presenting a novel cell-free therapy for IBD and other inflammatory diseases (192).
5.5 TGF-β signaling, ubiquitination, and MSC/MSC-Ex
Impairment of the TGF-β signaling pathway is associated with the development of intestinal inflammation in experimental models and IBD patients. TGF-β is an immunosuppressive cytokine produced by various cell types (immune cells, nonhematopoietic cells) and activated by integrins. There are three types of TGF-β in mammals: TGF-β1, TGF-β2, and TGF-β3. TGF-β receptors are widely expressed in body tissues and cells; therefore, members of the TGF-β family regulate the proliferation, differentiation, apoptosis, and inflammation of many cells. TGF-β signaling requires two transmembrane receptors, RI and RII types, that have serine/threonine kinase activity. When the active TGF-β ligand binds to the RII receptor, it activates the kinase activity of the RI receptor and the recruitment of Smad protein, inducing Smad complex formation, nuclear transport, and Smad DNA binding (193). Smad works with universal transcription factors (GTF), -determining transcription factors (LDTF), and other driving factors or helper proteins to regulate target gene transcription.
The TGF-β signaling pathway has been studied for its effective regulatory and inflammatory activity (194). In intestinal immunity, TGF-β inhibits intestinal bacterial antigens’ inflammatory response and helps induce immune tolerance. IBD is characterized by abnormal TGF- β signaling (195). High expression of Smad7 in CD4+ T cells is associated with severe colitis (196). Studies have shown that sCYLD (a short splicing form of CYLD) mediates ubiquitination of K63 junctions and nuclear translocation of Smad7 and that the sCYLD-Smad7 complex inhibits TGF-βsignaling in CD4+ T cells (197). It is worth noting that Smurf1 and Smurf2, members of the E3 ubiquitase family, are critical negative regulators of the TGF-β signaling pathway (198). Smad7 recruits E3 ubiquitin ligases such as Smurf1, Smurf2, and NEDD4L to TGF-β receptors, promoting their ubiquitin-mediated degradation. RNF11 may mediate AMSH ubiquitination through the formation of the Smurf2/RNF11 complex, leading to its degradation by the 26S proteasome, negatively regulating TGF-β signaling (199).
TGF-β1 in exosomes is thought to have therapeutic potential in IBD. Exosomes produced by TGFβ1 gene-modified DCs can inhibit the development of IBD by inhibiting Th17 (200). Another study showed that TGF-β1-modified exosomes (TGF-β1-Exs) induce CD4+ Foxp3+ Tregs, reducing the proportion of Th17 in lymphocytes at the site of inflammation, mitigating the inflammatory response in a mouse model of colitis (201). In experimental models and IBD patients, impaired TGF-β signaling pathways have been associated with the development of intestinal inflammation. OE-MSC-Exs can regulate cell proliferation, decrease the levels of inflammatory cytokines IL-17 and IFN-γ, and increase the inhibitory cytokines TGF-β and IL-10, suggesting that OE-MSC-Exs may effectively alleviate the severity of experimental colitis by inhibiting effector T cells and enhancing regulatory T cells (192). Ma ZJ et al. found that MSC-Ex decreased the concentrations of IFN-γ, TNF-α, and IL-1β and increased the secretion of TGF-β1 and IL-10 by up-regulating anti-inflammatory response and down-regulating inflammatory response (202). It is suggested that MSC-Ex shows therapeutic power in a mouse model of DSS-induced colitis by inhibiting inflammatory mechanisms. Pd-MSC-Ev inhibits TGF-β1-induced inflammatory cytokine secretion and fibrotic marker expression, suggesting that MSC-EVs is expected to be a promising anti-fibrotic drug (203).
5.6 Deubiquitase and E3 ubiquitin ligase in IBD and MSC/MSC-Ex modulation
A variety of deubiquitases and E3 ubiquitin ligases are involved in regulating the process of IBD. USP15 binds to Lys48-linked ubiquitin chains to achieve deubiquitination, inhibiting the degradation of TAB2 and TAB3, thus hindering the selective autophagy degradation mediated by autophagy cargo receptor 1 (204). The proteasomal-associated deubiquitinase USP14 is involved in the negative regulation of the type 1 IFN signaling pathway and can also inhibit the activation of the NF-κB signaling pathway by deubiquitinating K63-linked retinoid-inducing gene I (RIG-I) (205, 206). USP19 negatively regulates the activation of TAK1-TAB1 dependent NF-κB signaling pathway by specifically removing TAK1 coupled Lys63 and Lys27 linked polyubiquitin chains, resulting in impaired TAK1 activity and destruction of the TAK1-TAB2/3 complex (207). The E3 ubiquitin ligase TRIM56 induced by type I IFN interacts with STING and targets STING for K63 junction polyubiquitination, recruitment of TBK1, and induction of IFN-β to induce innate immune response (208).TRIM32 can also interact with STING on mitochondria and ER, facilitating STING interaction with TBK1 (9). Upregulation of surface MHC Class II (MHCII) and co-stimulatory molecules such as CD80 and CD86 leads to DC maturation, E3 ligase membrane-associated RING-CH-1 (MARCH1) promotes endocytosis and lysosomal degradation of MHCII and CD86 by ubiquitinating them. Thus limiting the antigen-presenting capacity of dendritic cells (209–211).
Sun et al. showed that USP11 plays a catalytic and non-catalytic role in regulating the stability of IκBα, thereby negatively regulating TNF-α induced NF-κB activation (212). Deubiquitination of USP26 has been found to stabilize SMAD7, leading to reduced TGF-β signaling (213). Moreover, RNF182 promotes the degradation of cytoplasmic p65 through K48 ubiquitination, inhibiting inflammatory responses (214). In patients with IBD, up-regulated USP16 expression levels can be found in macrophages. When stimulated by LPS or TNF-α, USP16 specifically removes Lys33-linked IKKs polyubiquitin chains and promotes IKK-β-mediated phosphorylation of P105, leading to autoimmune responses and the development of IBD (215). Li Y et al. confirmed that CVMSC-Exs promote trophoblast migration and proliferation by up-regulating TRIM72 expression, thereby promoting P53 ubiquitination, proteasome degradation, and reducing cell apoptosis (216). Patients with inflammatory bowel disease (IBD) have higher levels of angiotensin-converting enzyme 2 (ACE2) expression in the gut (217).ACE2 deubiquitination mediated by deubiquitination enzyme UCHL1 and ACE2 SUMO mediated by E3 SUMO protein ligase PIAS4 can increase ACE2 protein levels, while AP2-mediated lysosomal degradation can decrease ACE2 protein levels (218, 219). NEDD4 has been shown to be involved in Ev biosynthesis (exosome production), and NEDD4 is a novel GSDMD interacting protein. Bulek et al. ‘s data suggest that GSDMD uses selective autophagy components (including LC3+ vesicles) to mediate NEDD4-dependent sEV biosynthesis for IL-1β output, thereby supporting the interaction between autophagy and exosome biosynthesis (220), Regardless of the proposed mechanism, exosomes can alleviate DSS-induced colitis in mice by controlling ubiquitin modification levels (17). However, at present, there is little literature on the clear association between deubiquitin/E3 ubiquitin ligase and MSC/MSC-EX in IBD disease, and the specific mechanism still needs to be further explored (17, 221–223).
6 Conclusion
Scientists have been exploring more effective and easier ways to treat IBD, mainly focusing on the interaction between genetic, environmental, immunological, and gut microbial factors, to discover further fundamental mechanisms of IBD occurrence and preventive strategies. The aim is to reduce patients’ medical and disease burden and improve the quality of life of affected individuals (224). Despite the best efforts, the application of MSCs and MSC-Exs remains a black box filled with unknown secrets. Studies have reported that treatment with MSCs or similar cells may promote the likelihood of cancer in patients; although this risk is unlikely to exist, there is still a chance of occurrence (225). As research continues to deepen and clinical trials advance, we hope to better understand the risks and potential benefits of exosome treatment for patients. Thus, the search for new, safe, efficient, and low-cost treatments for IBD, including MSC-Exs, is still underway.
Several experimental conclusions have proved that ubiquitin modification is involved in regulating important signaling pathways, such as NF-κB, NOD, TGF-β, and TNF-α. In addition, the dysregulation of components of the ubiquitination system often leads to various diseases such as cancer, IBD, and other autoimmune diseases. Some ubiquitin enzymes are known to directly regulate various inflammation-related transcription factors from the Smad, p53, Jun, and other families, and the ubiquitination-mediated degradation of signaling intermediates is an essential means to terminate inflammatory responses (226). However, how ubiquitination mediates the transmission and function of inflammatory signals to trigger the occurrence of IBD is largely unexplored and requires further studies. Moreover, the link between ubiquitination, IBD, and MSCs/MSC-Exs could provide an experimental basis for a novel therapeutic target and subsequent clinical application. More exploratory studies are needed in this area.
Author contributions
HL: Writing – original draft. XM: Writing – review & editing. LW: Writing – review & editing. NW: Writing – review & editing. DO: Writing – review & editing. BW: Writing – review & editing. FM: Conceptualization, Writing – original draft.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was sponsored by the National Natural Science Fund of China (Grant no. 82250410378), 2022 Jiangsu Excellent postdoctoral program (Grant no. 2022ZB634), Zhenjiang key research and development plan (social development) (Grant no. SH2022062, SH2022091 and SH2023050) and Project of Suzhou Science and Technology (Grant no. SKY2022027).
Acknowledgments
The author is grateful to the members of the research team for the time and energy spent in writing the manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Yang H, Li Y, Wu W, Sun Q, Zhang Y, Zhao W, et al. The incidence of inflammatory bowel disease in Northern China: a prospective population-based study. PloS One. (2014) 9:e101296. doi: 10.1371/journal.pone.0101296
2. Zeng Z, Zhu Z, Yang Y, Ruan W, Peng X, Su Y, et al. Incidence and clinical characteristics of inflammatory bowel disease in a developed region of Guangdong Province, China: a prospective population-based study. J Gastroenterol Hepatol. (2013) 28:1148–53. doi: 10.1111/jgh.12164
3. Kaplan GG. The global burden of IBD: from 2015 to 2025. Nat Rev Gastroenterol Hepatol. (2015) 12:720–7. doi: 10.1038/nrgastro.2015.150
4. GBD 2017 Inflammatory Bowel Disease Collaborators. The global, regional, and national burden of inflammatory bowel disease in 195 countries and territories, 1990-2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet Gastroenterol Hepatol. (2020) 5:17–30. doi: 10.1016/S2468-1253(19)30333-4
5. Zhao M, Gönczi L, Lakatos PL, Burisch J. The burden of inflammatory bowel disease in europe in 2020. J Crohns Colitis. (2021) 15:1573–87. doi: 10.1093/ecco-jcc/jjab029
6. Kostic AD, Xavier RJ, Gevers D. The microbiome in inflammatory bowel disease: current status and the future ahead. Gastroenterology. (2014) 146:1489–99. doi: 10.1053/j.gastro.2014.02.009
7. Mulder DJ, Noble AJ, Justinich CJ, Duffin JM. A tale of two diseases: the history of inflammatory bowel disease. J Crohns Colitis. (2014) 8:341–8. doi: 10.1016/j.crohns.2013.09.009
8. Xu F, Dahlhamer JM, Zammitti EP, Wheaton AG, Croft JB. Health-risk behaviors and chronic conditions among adults with inflammatory bowel disease - United States, 2015 and 2016. MMWR Morb Mortal Wkly Rep. (2018) 67:190–5. doi: 10.15585/mmwr.mm6706a4
9. Hu H, Sun SC. Ubiquitin signaling in immune responses. Cell Res. (2016) 26:457–83. doi: 10.1038/cr.2016.40
10. Zou M, Zeng QS, Nie J, Yang JH, Luo ZY, Gan HT. The role of E3 ubiquitin ligases and deubiquitinases in inflammatory bowel disease: friend or foe? Front Immunol. (2021) 12:769167. doi: 10.3389/fimmu.2021.769167
11. Beaudoin M, Goyette P, Boucher G, Lo KS, Rivas MA, Stevens C, et al. Deep resequencing of GWAS loci identifies rare variants in CARD9, IL23R and RNF186 that are associated with ulcerative colitis. PloS Genet. (2013) 9:e1003723. doi: 10.1371/journal.pgen.1003723
12. Yu H, Yang X, Xiao X, Xu M, Yang Y, Xue C, et al. Human adipose mesenchymal stem cell-derived exosomes protect mice from DSS-induced inflammatory bowel disease by promoting intestinal-stem-cell and epithelial regeneration. Aging Dis. (2021) 12:1423–37. doi: 10.14336/AD.2021.0601
13. da Costa Gonçalves F, Paz AH. Cell membrane and bioactive factors derived from mesenchymal stromal cells: Cell-free based therapy for inflammatory bowel diseases. World J Stem Cells. (2019) 11:618–33. doi: 10.4252/wjsc.v11.i9.618
14. Wu H, Fan H, Shou Z, Xu M, Chen Q, Ai C, et al. Extracellular vesicles containing miR-146a attenuate experimental colitis by targeting TRAF6 and IRAK1. Int Immunopharmacol. (2019) 68:204–12. doi: 10.1016/j.intimp.2018.12.043
15. Yang J, Liu XX, Fan H, Tang Q, Shou ZX, Zuo DM, et al. Extracellular Vesicles Derived from Bone Marrow Mesenchymal Stem Cells Protect against Experimental Colitis via Attenuating Colon Inflammation, Oxidative Stress and Apoptosis. PloS One. (2015) 10:e0140551. doi: 10.1371/journal.pone.0140551
16. Fujimoto K, Kinoshita M, Tanaka H, Okuzaki D, Shimada Y, Kayama H, et al. Regulation of intestinal homeostasis by the ulcerative colitis-associated gene RNF186. Mucosal Immunol. (2017) 10:446–59. doi: 10.1038/mi.2016.58
17. Wu Y, Qiu W, Xu X, Kang J, Wang J, Wen Y, et al. Exosomes derived from human umbilical cord mesenchymal stem cells alleviate inflammatory bowel disease in mice through ubiquitination. Am J Transl Res. (2018) 10:2026–36.
18. He H, Zhao ZH, Han FS, Liu XH, Wang R, Zeng YJ. Overexpression of protein kinase C ε improves retention and survival of transplanted mesenchymal stem cells in rat acute myocardial infarction. Cell Death Dis. (2016) 7:e2056. doi: 10.1038/cddis.2015.417
19. Kim KW, Moon SJ, Park MJ, Kim BM, Kim EK, Lee SH, et al. Optimization of adipose tissue-derived mesenchymal stem cells by rapamycin in a murine model of acute graft-versus-host disease [published correction appears in Stem Cell Res Ther. Stem Cell Res Ther. (2015) 6:202. doi: 10.1186/s13287-015-0197-8
20. Park JY, Jeon HJ, Kim TY, Lee KY, Park K, Lee ES, et al. Comparative analysis of mesenchymal stem cell surface marker expression for human dental mesenchymal stem cells. Regener Med. (2013) 8:453–66. doi: 10.2217/rme.13.23
21. Ding DC, Shyu WC, Lin SZ. Mesenchymal stem cells. Cell Transplant. (2011) 20:5–14. doi: 10.3727/096368910X
22. Li Z, Hu X, Zhong JF. Mesenchymal stem cells: characteristics, function, and application. Stem Cells Int. (2019) 2019:8106818. doi: 10.1155/2019/8106818
23. Maleki M, Ghanbarvand F, Reza Behvarz M, Ejtemaei M, Ghadirkhomi E. Comparison of mesenchymal stem cell markers in multiple human adult stem cells. Int J Stem Cells. (2014) 7:118–26. doi: 10.15283/ijsc.2014.7.2.118
24. Djouad F, Charbonnier LM, Bouffi C, Louis-Plence P, Bony C, Apparailly F, et al. Mesenchymal stem cells inhibit the differentiation of dendritic cells through an interleukin-6-dependent mechanism. Stem Cells. (2007) 25:2025–32. doi: 10.1634/stemcells.2006-0548
25. Uccelli A, Moretta L, Pistoia V. Mesenchymal stem cells in health and disease. Nat Rev Immunol. (2008) 8:726–36. doi: 10.1038/nri2395
26. Musiał-Wysocka A, Kot M, Majka M. The pros and cons of mesenchymal stem cell-based therapies. Cell Transplant. (2019) 28:801–12. doi: 10.1177/0963689719837897
27. Zhang X, He J, Wang W. Progress in the use of mesenchymal stromal cells for osteoarthritis treatment. Cytotherapy. (2021) 23:459–70. doi: 10.1016/j.jcyt.2021.01.008
28. Murphy MB, Moncivais K, Caplan AI. Mesenchymal stem cells: environmentally responsive therapeutics for regenerative medicine. Exp Mol Med. (2013) 45:e54. doi: 10.1038/emm.2013.94
29. Lai RC, Yeo RW, Lim SK. Mesenchymal stem cell exosomes. Semin Cell Dev Biol. (2015) 40:82–8. doi: 10.1016/j.semcdb.2015.03.001
30. Pathan M, Fonseka P, Chitti SV, Kang T, Sanwlani R, Van Deun J, et al. Vesiclepedia 2019: a compendium of RNA, proteins, lipids and metabolites in extracellular vesicles. Nucleic Acids Res. (2019) 47:D516–9. doi: 10.1093/nar/gky1029
31. Srivastava A, Rathore S, Munshi A, Ramesh R. Extracellular vesicles in oncology: from immune suppression to immunotherapy. AAPS J. (2021) 23:30. doi: 10.1208/s12248-021-00554-4
32. Jeyaram A, Jay SM. Preservation and storage stability of extracellular vesicles for therapeutic applications. AAPS J. (2017) 20:1. doi: 10.1208/s12248-017-0160-y
33. Johnstone RM, Adam M, Hammond JR, Orr L, Turbide C. Vesicle formation during reticulocyte maturation. Association of plasma membrane activities with released vesicles (exosomes). J Biol Chem. (1987) 262:9412–20. doi: 10.1016/S0021-9258(18)48095-7
34. Kahlert C, Kalluri R. Exosomes in tumor microenvironment influence cancer progression and metastasis. J Mol Med (Berl). (2013) 91:431–7. doi: 10.1007/s00109-013-1020-6
35. Lin J, Li J, Huang B, Liu J, Chen X, Chen XM, et al. Exosomes: novel biomarkers for clinical diagnosis. ScientificWorldJournal. (2015) 2015:657086. doi: 10.1155/2015/657086
36. Simons M, Raposo G. Exosomes–vesicular carriers for intercellular communication. Curr Opin Cell Biol. (2009) 21:575–81. doi: 10.1016/j.ceb.2009.03.007
37. Joo HS, Suh JH, Lee HJ, Bang ES, Lee JM. Current knowledge and future perspectives on mesenchymal stem cell-derived exosomes as a new therapeutic agent. Int J Mol Sci. (2020) 21:727. doi: 10.3390/ijms21030727
38. Kalluri R, LeBleu VS. The biology, function, and biomedical applications of exosomes. Science. (2020) 367:eaau6977. doi: 10.1126/science.aau6977
39. Maas SLN, Breakefield XO, Weaver AM. Extracellular vesicles: unique intercellular delivery vehicles. Trends Cell Biol. (2017) 27:172–88. doi: 10.1016/j.tcb.2016.11.003
40. Yao YF, Qu MW, Li GC, Zhang FB, Rui HC. Circulating exosomal miRNAs as diagnostic biomarkers in Parkinson's disease. Eur Rev Med Pharmacol Sci. (2018) 22:5278–83. doi: 10.26355/eurrev_201808_15727
41. Gui Y, Liu H, Zhang L, Lv W, Hu X. Altered microRNA profiles in cerebrospinal fluid exosome in Parkinson disease and Alzheimer disease. Oncotarget. (2015) 6:37043–53. doi: 10.18632/oncotarget.v6i35
42. Roccaro AM, Sacco A, Maiso P, Azab AK, Tai YT, Reagan M, et al. BM mesenchymal stromal cell-derived exosomes facilitate multiple myeloma progression [published correction appears in J Clin Invest. J Clin Invest. (2013) 123:1542–55. doi: 10.1172/JCI66517
43. Andaloussi S EL, Mäger I, Breakefield XO, Wood MJ. Extracellular vesicles: biology and emerging therapeutic opportunities. Nat Rev Drug Discovery. (2013) 12:347–57. doi: 10.1038/nrd3978
44. Namee NM, O'Driscoll L. Extracellular vesicles and anti-cancer drug resistance. Biochim Biophys Acta Rev Cancer. (2018) 1870:123–36. doi: 10.1016/j.bbcan.2018.07.003
45. Galipeau J, Sensébé L. Mesenchymal stromal cells: clinical challenges and therapeutic opportunities. Cell Stem Cell. (2018) 22:824–33. doi: 10.1016/j.stem.2018.05.004
46. Le Blanc K, Rasmusson I, Sundberg B, Götherström C, Hassan M, Uzunel M, et al. Treatment of severe acute graft-versus-host disease with third party haploidentical mesenchymal stem cells. Lancet. (2004) 363:1439–41. doi: 10.1016/S0140-6736(04)16104-7
47. Shen Z, Huang W, Liu J, Tian J, Wang S, Rui K. Effects of mesenchymal stem cell-derived exosomes on autoimmune diseases. Front Immunol. (2021) 12:749192. doi: 10.3389/fimmu.2021.749192
48. Phinney DG, Pittenger MF. Concise Review: MSC-Derived Exosomes for Cell-Free Therapy [published correction appears in Stem Cells. Stem Cells. (2017) 35:851–8. doi: 10.1002/stem.2575
49. Fang Y, Ni J, Wang YS, Zhao Y, Jiang LQ, Chen C, et al. Exosomes as biomarkers and therapeutic delivery for autoimmune diseases: Opportunities and challenges. Autoimmun Rev. (2023) 22:103260. doi: 10.1016/j.autrev.2022.103260
50. Pusic AD, Pusic KM, Kraig RP. What are exosomes and how can they be used in multiple sclerosis therapy? Expert Rev Neurother. (2014) 14:353–5. doi: 10.1586/14737175.2014.890893
51. Riazifar M, Mohammadi MR, Pone EJ, Yeri A, Lässer C, Segaliny AI, et al. Stem cell-derived exosomes as nanotherapeutics for autoimmune and neurodegenerative disorders. ACS Nano. (2019) 13:6670–88. doi: 10.1021/acsnano.9b01004
52. Mahdipour E, Salmasi Z, Sabeti N. Potential of stem cell-derived exosomes to regenerate β islets through Pdx-1 dependent mechanism in a rat model of type 1 diabetes. J Cell Physiol. (2019) 234:20310–21. doi: 10.1002/jcp.28631
53. Zeinaloo A, Zanjani KS, Bagheri MM, Mohyeddin-Bonab M, Monajemzadeh M, Arjmandnia MH. Intracoronary administration of autologous mesenchymal stem cells in a critically ill patient with dilated cardiomyopathy. Pediatr Transplant. (2011) 15:E183–6. doi: 10.1111/j.1399-3046.2010.01366.x
54. Yang Z, Zhang F, Ma W, Chen B, Zhou F, Xu Z, et al. A novel approach to transplanting bone marrow stem cells to repair human myocardial infarction: delivery via a noninfarct-relative artery. Cardiovasc Ther. (2010) 28:380–5. doi: 10.1111/j.1755-5922.2009.00116.x
55. Arslan F, Lai RC, Smeets MB, Akeroyd L, Choo A, Aguor EN, et al. Mesenchymal stem cell-derived exosomes increase ATP levels, decrease oxidative stress and activate PI3K/Akt pathway to enhance myocardial viability and prevent adverse remodeling after myocardial ischemia/reperfusion injury. Stem Cell Res. (2013) 10:301–12. doi: 10.1016/j.scr.2013.01.002
56. Li X, Hu X, Chen Q, Jiang T. Bone marrow mesenchymal stem cell-derived exosomes carrying E3 ubiquitin ligase ITCH attenuated cardiomyocyte apoptosis by mediating apoptosis signal-regulated kinase-1. Pharmacogenet Genomics. (2023) 33:117–25. doi: 10.1097/FPC.0000000000000499
57. Hare JM, Fishman JE, Gerstenblith G, DiFede Velazquez DL, Zambrano JP, Suncion VY, et al. Comparison of allogeneic vs autologous bone marrow–derived mesenchymal stem cells delivered by transendocardial injection in patients with ischemic cardiomyopathy: the POSEIDON randomized trial [published correction appears in JAMA. JAMA. (2012) 308:2369–79. doi: 10.1001/jama.2012.25321
58. Lachaud CC, Cobo-Vuilleumier N, Fuente-Martin E, Diaz I, Andreu E, Cahuana GM, et al. Umbilical cord mesenchymal stromal cells transplantation delays the onset of hyperglycemia in the RIP-B7.1 mouse model of experimental autoimmune diabetes through multiple immunosuppressive and anti-inflammatory responses. Front Cell Dev Biol. (2023) 11:1089817. doi: 10.3389/fcell.2023.1089817
59. Brandhorst H, Brandhorst D, Abraham A, Acreman S, Schive SW, Scholz H, et al. Proteomic profiling reveals the ambivalent character of the mesenchymal stem cell secretome: assessing the effect of preconditioned media on isolated human islets. Cell Transplant. (2020) 29:963689720952332. doi: 10.1177/0963689720952332
60. Sun Y, Shi H, Yin S, Ji C, Zhang X, Zhang B, et al. Human mesenchymal stem cell derived exosomes alleviate type 2 diabetes mellitus by reversing peripheral insulin resistance and relieving β-cell destruction. ACS Nano. (2018) 12:7613–28. doi: 10.1021/acsnano.7b07643
61. Sun L, Akiyama K, Zhang H, Yamaza T, Hou Y, Zhao S, et al. Mesenchymal stem cell transplantation reverses multiorgan dysfunction in systemic lupus erythematosus mice and humans. Stem Cells. (2009) 27:1421–32. doi: 10.1002/stem.68
62. Liang J, Zhang H, Hua B, Wang H, Lu L, Shi S, et al. Allogenic mesenchymal stem cells transplantation in refractory systemic lupus erythematosus: a pilot clinical study [published correction appears in Ann Rheum Dis. Ann Rheum Dis. (2010) 69:1423–9. doi: 10.1136/ard.2009.123463
63. Guo J, Yang J, Cao G, Fan H, Guo C, Ma YE, et al. Xenogeneic immunosuppression of human umbilical cord mesenchymal stem cells in a major histocompatibility complex-mismatched allogeneic acute graft-versus-host disease murine model. Eur J Haematol. (2011) 87:235–43. doi: 10.1111/ejh.2011.87.issue-3
64. Connick P, Kolappan M, Patani R, Scott MA, Crawley C, He XL, et al. The mesenchymal stem cells in multiple sclerosis (MSCIMS) trial protocol and baseline cohort characteristics: an open-label pre-test: post-test study with blinded outcome assessments. Trials. (2011) 12:62. doi: 10.1186/1745-6215-12-62
65. Wilson JG, Liu KD, Zhuo H, Caballero L, McMillan M, Fang X, et al. Mesenchymal stem (stromal) cells for treatment of ARDS: a phase 1 clinical trial. Lancet Respir Med. (2015) 3:24–32. doi: 10.1016/S2213-2600(14)70291-7
66. Weiss DJ, Casaburi R, Flannery R, LeRoux-Williams M, Tashkin DP. A placebo-controlled, randomized trial of mesenchymal stem cells in COPD. Chest. (2013) 143:1590–8. doi: 10.1378/chest.12-2094
67. Lightner AL, Sengupta V, Qian S, Ransom JT, Suzuki S, Park DJ, et al. Bone marrow mesenchymal stem cell-derived extracellular vesicle infusion for the treatment of respiratory failure from COVID-19: A randomized, placebo-controlled dosing clinical trial. Chest. (2023) 164:1444–53. doi: 10.1016/j.chest.2023.06.024
68. Harrell CR, Miloradovic D, Sadikot R, Fellabaum C, Markovic BS, Miloradovic D, et al. Molecular and cellular mechanisms responsible for beneficial effects of mesenchymal stem cell-derived product "Exo-d-MAPPS" in attenuation of chronic airway inflammation. Anal Cell Pathol (Amst). (2020) 2020:3153891. doi: 10.1155/2020/3153891
69. Yang L, Zhai Y, Hao Y, Zhu Z, Cheng G. The regulatory functionality of exosomes derived from hUMSCs in 3D culture for alzheimer's disease therapy. Small. (2020) 16:e1906273. doi: 10.1002/smll.201906273
70. Lee M, Ban JJ, Yang S, Im W, Kim M. The exosome of adipose-derived stem cells reduces β-amyloid pathology and apoptosis of neuronal cells derived from the transgenic mouse model of Alzheimer's disease. Brain Res. (2018) 1691:87–93. doi: 10.1016/j.brainres.2018.03.034
71. Swaminathan M, Kopyt N, Atta MG, Radhakrishnan J, Umanath K, Nguyen S, et al. Pharmacological effects of ex vivo mesenchymal stem cell immunotherapy in patients with acute kidney injury and underlying systemic inflammation. Stem Cells Transl Med. (2021) 10:1588–601. doi: 10.1002/sctm.21-0043
72. Zhang G, Zou X, Huang Y, Wang F, Miao S, Liu G, et al. Mesenchymal stromal cell-derived extracellular vesicles protect against acute kidney injury through anti-oxidation by enhancing nrf2/ARE activation in rats. Kidney Blood Press Res. (2016) 41:119–28. doi: 10.1159/000443413
73. Honmou O, Houkin K, Matsunaga T, Niitsu Y, Ishiai S, Onodera R, et al. Intravenous administration of auto serum-expanded autologous mesenchymal stem cells in stroke. Brain. (2011) 134:1790–807. doi: 10.1093/brain/awr063
74. Mao F, Wu Y, Tang X, Kang J, Zhang B, Yan Y, et al. Exosomes derived from human umbilical cord mesenchymal stem cells relieve inflammatory bowel disease in mice. BioMed Res Int. (2017) 2017:5356760. doi: 10.1155/2017/5356760
75. Chang X, Song YH, Xia T, He ZX, Zhao SB, Wang ZJ, et al. Macrophage-derived exosomes promote intestinal mucosal barrier dysfunction in inflammatory bowel disease by regulating TMIGD1 via mircroRNA-223. Int Immunopharmacol. (2023) 121:110447. doi: 10.1016/j.intimp.2023.110447
76. Singh V, Ram M, Kumar R, Prasad R, Roy BK, Singh KK. Phosphorylation: implications in cancer. Protein J. (2017) 36:1–6. doi: 10.1007/s10930-017-9696-z
77. Diallo I, Seve M, Cunin V, Minassian F, Poisson JF, Michelland S, et al. Current trends in protein acetylation analysis. Expert Rev Proteomics. (2019) 16:139–59. doi: 10.1080/14789450.2019.1559061
78. Duan G, Walther D. The roles of post-translational modifications in the context of protein interaction networks. PloS Comput Biol. (2015) 11:e1004049. doi: 10.1371/journal.pcbi.1004049
79. Pickart CM. Ubiquitin enters the new millennium. Mol Cell. (2001) 8:499–504. doi: 10.1016/S1097-2765(01)00347-1
80. Higgins R, Gendron JM, Rising L, Mak R, Webb K, Kaiser SE, et al. The unfolded protein response triggers site-specific regulatory ubiquitylation of 40S ribosomal proteins. Mol Cell. (2015) 59:35–49. doi: 10.1016/j.molcel.2015.04.026
81. Hershko A, Ciechanover A. The ubiquitin system. Annu Rev Biochem. (1998) 67:425–79. doi: 10.1146/annurev.biochem.67.1.425
82. Zhou L, Jiang Y, Luo Q, Li L, Jia L. Neddylation: a novel modulator of the tumor microenvironment. Mol Cancer. (2019) 18:77. doi: 10.1186/s12943-019-0979-1
83. Flotho A, Melchior F. Sumoylation: a regulatory protein modification in health and disease. Annu Rev Biochem. (2013) 82:357–85. doi: 10.1146/annurev-biochem-061909-093311
84. Yang W, Sheng H, Warner DS, Paschen W. Transient global cerebral ischemia induces a massive increase in protein sumoylation. J Cereb Blood Flow Metab. (2008) 28:269–79. doi: 10.1038/sj.jcbfm.9600523
85. Yang W, Thompson JW, Wang Z, Wang L, Sheng H, Foster MW, et al. Analysis of oxygen/glucose-deprivation-induced changes in SUMO3 conjugation using SILAC-based quantitative proteomics. J Proteome Res. (2012) 11:1108–17. doi: 10.1021/pr200834f
86. Akutsu M, Dikic I, Bremm A. Ubiquitin chain diversity at a glance. J Cell Sci. (2016) 129:875–80. doi: 10.1242/jcs.183954
87. Chen Y, Zhou D, Yao Y, Sun Y, Yao F, Ma L. Monoubiquitination in homeostasis and cancer. Int J Mol Sci. (2022) 23:5925. doi: 10.3390/ijms23115925
88. Wang Y, Le WD. Autophagy and ubiquitin-proteasome system. Adv Exp Med Biol. (2019) 1206:527–50. doi: 10.1007/978-981-15-0602-4_25
89. Lu Y, Lee BH, King RW, Finley D, Kirschner MW. Substrate degradation by the proteasome: a single-molecule kinetic analysis. Science. (2015) 348:1250834. doi: 10.1126/science.1250834
90. Hoege C, Pfander B, Moldovan GL, Pyrowolakis G, Jentsch S. RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature. (2002) 419:135–41. doi: 10.1038/nature00991
91. Liu P, Gan W, Su S, Hauenstein AV, Fu TM, Brasher B, et al. K63-linked polyubiquitin chains bind to DNA to facilitate DNA damage repair. Sci Signal. (2018) 11:eaar8133. doi: 10.1126/scisignal.aar8133
92. Ohtake F, Saeki Y, Ishido S, Kanno J, Tanaka K. The K48-K63 branched ubiquitin chain regulates NF-κB signaling. Mol Cell. (2016) 64:251–66. doi: 10.1016/j.molcel.2016.09.014
93. Skaug B, Jiang X, Chen ZJ. The role of ubiquitin in NF-kappaB regulatory pathways. Annu Rev Biochem. (2009) 78:769–96. doi: 10.1146/annurev.biochem.78.070907.102750
94. Komander D, Rape M. The ubiquitin code. Annu Rev Biochem. (2012) 81:203–29. doi: 10.1146/annurev-biochem-060310-170328
95. Deshaies RJ, Joazeiro CA. RING domain E3 ubiquitin ligases. Annu Rev Biochem. (2009) 78:399–434. doi: 10.1146/annurev.biochem.78.101807.093809
96. Ye Y, Rape M. Building ubiquitin chains: E2 enzymes at work. Nat Rev Mol Cell Biol. (2009) 10:755–64. doi: 10.1038/nrm2780
97. Hoppe T. Multiubiquitylation by E4 enzymes: 'one size' doesn't fit all. Trends Biochem Sci. (2005) 30:183–7. doi: 10.1016/j.tibs.2005.02.004
98. Luo QF, Chen W, Zhang ST. Identification of Nedd4 as a novel regulator in Hedgehog signaling. Chin Med J (Engl). (2012) 125:3851–5.
99. Bedford L, Lowe J, Dick LR, Mayer RJ, Brownell JE. Ubiquitin-like protein conjugation and the ubiquitin-proteasome system as drug targets. Nat Rev Drug Discovery. (2011) 10:29–46. doi: 10.1038/nrd3321
100. Mevissen TE, Hospenthal MK, Geurink PP, Elliott PR, Akutsu M, Arnaudo N, et al. OTU deubiquitinases reveal mechanisms of linkage specificity and enable ubiquitin chain restriction analysis. Cell. (2013) 154:169–84. doi: 10.1016/j.cell.2013.05.046
101. Scheffner M, Nuber U, Huibregtse JM. Protein ubiquitination involving an E1-E2-E3 enzyme ubiquitin thioester cascade. Nature. (1995) 373:81–3. doi: 10.1038/373081a0
102. Clague MJ, Urbé S, Komander D. Breaking the chains: deubiquitylating enzyme specificity begets function [published correction appears in Nat Rev Mol Cell Biol. Nat Rev Mol Cell Biol. (2019) 20:338–52. doi: 10.1038/s41580-019-0099-1
103. Nijman SM, Luna-Vargas MP, Velds A, Brummelkamp TR, Dirac AM, Sixma TK, et al. A genomic and functional inventory of deubiquitinating enzymes. Cell. (2005) 123:773–86. doi: 10.1016/j.cell.2005.11.007
104. Cheng J, Guo J, North BJ, Wang B, Cui CP, Li H, et al. Functional analysis of deubiquitylating enzymes in tumorigenesis and development. Biochim Biophys Acta Rev Cancer. (2019) 1872:188312. doi: 10.1016/j.bbcan.2019.188312
105. Chen R, Pang X, Li L, Zeng Z, Chen M, Zhang S. Ubiquitin-specific proteases in inflammatory bowel disease-related signalling pathway regulation. Cell Death Dis. (2022) 13:139. doi: 10.1038/s41419-022-04566-6
106. Liu M, Gao S, Xu X, Zhang L, Xu B, Lu D. USP18 contributes to the proliferation and migration of ovarian cancer cells by regulating the AKT/mTOR signaling pathway. Acta Biochim Pol. (2022) 69:417–22. doi: 10.18388/abp.2020_5871
107. Zhang E, Shen B, Mu X, Qin Y, Zhang F, Liu Y, et al. Ubiquitin-specific protease 11 (USP11) functions as a tumor suppressor through deubiquitinating and stabilizing VGLL4 protein. Am J Cancer Res. (2016) 6:2901–9. doi: 10.1158/0008-5472.CAN-15-2120
108. Gong X, Jia L, Zhou L, Hu T. USP14 predicts poorer survival outcomes and promotes tumor progression in endometrial carcinoma by activating NF-κB signaling. Aging (Albany NY). (2023) 15:12120–35. doi: 10.18632/aging.v15i21
109. Ke JY, Dai CJ, Wu WL, Gao JH, Xia AJ, Liu GP, et al. USP11 regulates p53 stability by deubiquitinating p53. J Zhejiang Univ Sci B. (2014) 15:1032–8. doi: 10.1631/jzus.B1400180
110. Hugot JP, Chamaillard M, Zouali H, Lesage S, Cézard JP, Belaiche J, et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn's disease. Nature. (2001) 411:599–603. doi: 10.1038/35079107
111. Guan Q. A comprehensive review and update on the pathogenesis of inflammatory bowel disease. J Immunol Res. (2019) 2019:7247238. doi: 10.1155/2019/7247238
112. Jarmakiewicz-Czaja S, Zielińska M, Sokal A, Filip R. Genetic and epigenetic etiology of inflammatory bowel disease: an update. Genes. (2022) 13:2388. doi: 10.3390/genes13122388
113. Xing Y, Hogquist KA. T-cell tolerance: central and peripheral. Cold Spring Harb Perspect Biol. (2012) 4:a006957. doi: 10.1101/cshperspect.a006957
114. Kucuksezer UC, Ozdemir C, Akdis M, Akdis CA. Influence of innate immunity on immune tolerance. Acta Med Acad. (2020) 49:164–80. doi: 10.5644/ama2006-124.295
115. Wang S, Lei B, Zhang E, Gong P, Gu J, He L, et al. Targeted therapy for inflammatory diseases with mesenchymal stem cells and their derived exosomes: from basic to clinics. Int J Nanomedicine. (2022) 17:1757–81. doi: 10.2147/IJN.S355366
116. Grégoire C, Lechanteur C, Briquet A, Baudoux É, Baron F, Louis E, et al. Review article: mesenchymal stromal cell therapy for inflammatory bowel diseases. Aliment Pharmacol Ther. (2017) 45:205–21. doi: 10.1111/apt.13864
117. Yu T, Gan S, Zhu Q, Dai D, Li N, Wang H, et al. Modulation of M2 macrophage polarization by the crosstalk between Stat6 and Trim24. Nat Commun. (2019) 10:4353. doi: 10.1038/s41467-019-12384-2
118. Liu J, Huang X, Hao S, Wang Y, Liu M, Xu J, et al. Peli1 negatively regulates noncanonical NF-κB signaling to restrain systemic lupus erythematosus. Nat Commun. (2018) 9:1136. doi: 10.1038/s41467-018-03530-3
119. Ishida T, Mizushima Si, Azuma S, Kobayashi N, Tojo T, Suzuki K, et al. Identification of TRAF6, a novel tumor necrosis factor receptor-associated factor protein that mediates signaling from an amino-terminal domain of the CD40 cytoplasmic region. J Biol Chem. (1996) 271:28745–8. doi: 10.1074/jbc.271.46.28745
120. Chen ZJ. Ubiquitination in signaling to and activation of IKK. Immunol Rev. (2012) 246:95–106. doi: 10.1111/j.1600-065X.2012.01108.x
121. Pflug KM, Sitcheran R. Targeting NF-κB-inducing kinase (NIK) in immunity, inflammation, and cancer. Int J Mol Sci. (2020) 21:8470. doi: 10.3390/ijms21228470
122. Zhou B, Huang Y, Feng Q, Zhu H, Xu Z, Chen L, et al. TRIM16 promotes aerobic glycolysis and pancreatic cancer metastasis by modulating the NIK-SIX1 axis in a ligase-independent manner. Am J Cancer Res. (2022) 12:5205–25.
123. Fujita M, Yamamoto Y, Jiang JJ, Atsumi T, Tanaka Y, Ohki T, et al. NEDD4 is involved in inflammation development during keloid formation. J Invest Dermatol. (2019) 139:333–41. doi: 10.1016/j.jid.2018.07.044
124. Zhu F, Wu B, Li P, Wang J, Tang H, Liu Y, et al. Association study confirmed susceptibility loci with keloid in the Chinese Han population. PloS One. (2013) 8:e62377. doi: 10.1371/journal.pone.0062377
125. Wang G, Yuan J, Cai X, Xu Z, Wang J, Ocansey DKW, et al. HucMSC-exosomes carrying miR-326 inhibit neddylation to relieve inflammatory bowel disease in mice. Clin Transl Med. (2020) 10:e113. doi: 10.1002/ctm2.113
126. Qi LL, Fan ZY, Mao HG, Wang JB. The Therapeutic Efficacy of Adipose Tissue-Derived Mesenchymal Stem Cell Conditioned Medium on Experimental Colitis Was Improved by the Serum From Colitis Rats [published correction appears in Front Bioeng Biotechnol. Front Bioeng Biotechnol. (2021) 9:694908. doi: 10.3389/fbioe.2021.694908
127. Zuo D, Tang Q, Fan H, Shou Z, Liu X, Cao D, et al. Modulation of nuclear factor-κB-mediated pro-inflammatory response is associated with exogenous administration of bone marrow-derived mesenchymal stem cells for treatment of experimental colitis. Mol Med Rep. (2015) 11:2741–8. doi: 10.3892/mmr.2014.3038
128. Nata T, Fujiya M, Ueno N, Moriichi K, Konishi H, Tanabe H, et al. MicroRNA-146b improves intestinal injury in mouse colitis by activating nuclear factor-κB and improving epithelial barrier function. J Gene Med. (2013) 15:249–60. doi: 10.1002/jgm.2717
129. Liu J, Chen Y, Liu D, Liu W, Hu S, Zhou N, et al. Ectopic expression of SIGIRR in the colon ameliorates colitis in mice by downregulating TLR4/NF-κB overactivation. Immunol Lett. (2017) 183:52–61. doi: 10.1016/j.imlet.2017.01.015
130. Wu G, McBride DW, Zhang JH. Axl activation attenuates neuroinflammation by inhibiting the TLR/TRAF/NF-κB pathway after MCAO in rats. Neurobiol Dis. (2018) 110:59–67. doi: 10.1016/j.nbd.2017.11.009
131. Lin SC, Lo YC, Wu H. Helical assembly in the MyD88-IRAK4-IRAK2 complex in TLR/IL-1R signalling. Nature. (2010) 465:885–90. doi: 10.1038/nature09121
132. Cohen P. The TLR and IL-1 signalling network at a glance. J Cell Sci. (2014) 127:2383–90. doi: 10.1242/jcs.149831
133. Lee YS, Park JS, Kim JH, Jung SM, Lee JY, Kim SJ, et al. Smad6-specific recruitment of Smurf E3 ligases mediates TGF-β1-induced degradation of MyD88 in TLR4 signalling. Nat Commun. (2011) 2:460. doi: 10.1038/ncomms1469
134. Wang C, Chen T, Zhang J, Yang M, Li N, Xu X, et al. The E3 ubiquitin ligase Nrdp1 'preferentially' promotes TLR-mediated production of type I interferon. Nat Immunol. (2009) 10:744–52. doi: 10.1038/ni.1742
135. Yu W, Venkatraman A, Menden HL, Martinez M, Umar S, Sampath V. Short-chain fatty acids ameliorate necrotizing enterocolitis-like intestinal injury through enhancing Notch1-mediated single immunoglobulin interleukin-1-related receptor, toll-interacting protein, and A20 induction. Am J Physiol Gastrointest Liver Physiol. (2023) 324:G24–37. doi: 10.1152/ajpgi.00057.2022
136. Parvatiyar K, Barber GN, Harhaj EW. TAX1BP1 and A20 inhibit antiviral signaling by targeting TBK1-IKKi kinases. J Biol Chem. (2010) 285:14999–5009. doi: 10.1074/jbc.M110.109819
137. Wald D, Qin J, Zhao Z, Qian Y, Naramura M, Tian L, et al. SIGIRR, a negative regulator of Toll-like receptor-interleukin 1 receptor signaling. Nat Immunol. (2003) 4:920–7. doi: 10.1038/ni968
138. Jiang M, Gao T, Liu Y, Cao X, Li Y, Li J, et al. CD14 dictates differential activation of mesenchymal stromal cells through AKT, NF-κB and P38 signals. Biosci Rep. (2019) 39:BSR20190807. doi: 10.1042/BSR20190807
139. Duan S, Lou X, Chen S, Jiang H, Chen D, Yin R, et al. Macrophage LMO7 deficiency facilitates inflammatory injury via metabolic-epigenetic reprogramming. Acta Pharm Sin B. (2023) 13:4785–800. doi: 10.1016/j.apsb.2023.09.012
140. Liotta F, Angeli R, Cosmi L, Filì L, Manuelli C, Frosali F, et al. Toll-like receptors 3 and 4 are expressed by human bone marrow-derived mesenchymal stem cells and can inhibit their T-cell modulatory activity by impairing Notch signaling. Stem Cells. (2008) 26:279–89. doi: 10.1634/stemcells.2007-0454
141. Niu GC, Liu L, Zheng L, Zhang H, Shih DQ, Zhang X. Mesenchymal stem cell transplantation improves chronic colitis-associated complications through inhibiting the activity of toll-like receptor-4 in mice. BMC Gastroenterol. (2018) 18:127. doi: 10.1186/s12876-018-0850-7
142. Liu H, Liang Z, Wang F, Zhou C, Zheng X, Hu T, et al. Exosomes from mesenchymal stromal cells reduce murine colonic inflammation via a macrophage-dependent mechanism. JCI Insight. (2019) 4:e131273. doi: 10.1172/jci.insight.131273
143. Cao L, Xu H, Wang G, Liu M, Tian D, Yuan Z. Extracellular vesicles derived from bone marrow mesenchymal stem cells attenuate dextran sodium sulfate-induced ulcerative colitis by promoting M2 macrophage polarization. Int Immunopharmacol. (2019) 72:264–74. doi: 10.1016/j.intimp.2019.04.020
144. Deng D, Li X, Zhang JJ, Yin Y, Tian Y, Gan D, et al. Biotin-avidin system-based delivery enhances the therapeutic performance of MSC-derived exosomes. ACS Nano. (2023) 17:8530–50. doi: 10.1021/acsnano.3c00839
145. Chen L, Cao SQ, Lin ZM, He SJ, Zuo JP. NOD-like receptors in autoimmune diseases. Acta Pharmacol Sin. (2021) 42:1742–56. doi: 10.1038/s41401-020-00603-2
146. Mukherjee T, Hovingh ES, Foerster EG, Abdel-Nour M, Philpott DJ, Girardin SE. NOD1 and NOD2 in inflammation, immunity and disease. Arch Biochem Biophys. (2019) 670:69–81. doi: 10.1016/j.abb.2018.12.022
147. Kim YK, Shin JS, Nahm MH. NOD-like receptors in infection, immunity, and diseases. Yonsei Med J. (2016) 57:5–14. doi: 10.3349/ymj.2016.57.1.5
148. Liu P, Lu Z, Liu L, Li R, Liang Z, Shen M, et al. NOD-like receptor signaling in inflammation-associated cancers: From functions to targeted therapies. Phytomedicine. (2019) 64:152925. doi: 10.1016/j.phymed.2019.152925
149. Emmerich CH, Bakshi S, Kelsall IR, Ortiz-Guerrero J, Shpiro N, Cohen P. Lys63/Met1-hybrid ubiquitin chains are commonly formed during the activation of innate immune signalling. Biochem Biophys Res Commun. (2016) 474:452–61. doi: 10.1016/j.bbrc.2016.04.141
150. Hasegawa M, Fujimoto Y, Lucas PC, Nakano H, Fukase K, Núñez G, et al. A critical role of RICK/RIP2 polyubiquitination in Nod-induced NF-kappaB activation. EMBO J. (2008) 27:373–83. doi: 10.1038/sj.emboj.7601962
151. Kanayama A, Seth RB, Sun L, Ea CK, Hong M, Shaito A, et al. TAB2 and TAB3 activate the NF-kappaB pathway through binding to polyubiquitin chains. Mol Cell. (2004) 15:535–48. doi: 10.1016/j.molcel.2004.08.008
152. Tigno-Aranjuez JT, Abbott DW. Ubiquitination and phosphorylation in the regulation of NOD2 signaling and NOD2-mediated disease. Biochim Biophys Acta. (2012) 1823:2022–8. doi: 10.1016/j.bbamcr.2012.03.017
153. Sorbara MT, Ellison LK, Ramjeet M, Travassos LH, Jones NL, Girardin SE, et al. The protein ATG16L1 suppresses inflammatory cytokines induced by the intracellular sensors Nod1 and Nod2 in an autophagy-independent manner [published correction appears in Immunity. Immunity. (2013) 39:858–73. doi: 10.1016/j.immuni.2013.10.013
154. Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell. (2002) 10:417–26. doi: 10.1016/S1097-2765(02)00599-3
155. Nazari S, Pourmand SM, Motevaseli E, Hassanzadeh G. Mesenchymal stem cells (MSCs) and MSC-derived exosomes in animal models of central nervous system diseases: Targeting the NLRP3 inflammasome. IUBMB Life. (2023) 75:794–810. doi: 10.1002/iub.2759
156. Varghese GP, Uporova L, Halfvarson J, Sirsjö A, Fransén K. Polymorphism in the NLRP3 inflammasome-associated EIF2AK2 gene and inflammatory bowel disease. Mol Med Rep. (2015) 11:4579–84. doi: 10.3892/mmr.2015.3236
157. Wu K, Yuan Y, Yu H, Dai X, Wang S, Sun Z, et al. The gut microbial metabolite trimethylamine N-oxide aggravates GVHD by inducing M1 macrophage polarization in mice. Blood. (2020) 136:501–15. doi: 10.1182/blood.2019003990
158. Xu T, Yu W, Fang H, Wang Z, Chi Z, Guo X, et al. Ubiquitination of NLRP3 by gp78/Insig-1 restrains NLRP3 inflammasome activation. Cell Death Differ. (2022) 29:1582–95. doi: 10.1038/s41418-022-00947-8
159. Mai CT, Wu MM, Wang CL, Su ZR, Cheng YY, Zhang XJ. Palmatine attenuated dextran sulfate sodium (DSS)-induced colitis via promoting mitophagy-mediated NLRP3 inflammasome inactivation. Mol Immunol. (2019) 105:76–85. doi: 10.1016/j.molimm.2018.10.015
160. Song H, Liu B, Huai W, Yu Z, Wang W, Zhao J, et al. The E3 ubiquitin ligase TRIM31 attenuates NLRP3 inflammasome activation by promoting proteasomal degradation of NLRP3. Nat Commun. (2016) 7:13727. doi: 10.1038/ncomms13727
161. Zheng Y, Wang S, Zhong Y, Huang C, Wu X. A20 affects macrophage polarization through the NLRP3 inflammasome signaling pathway and promotes breast cancer progression. Exp Ther Med. (2023) 25:147. doi: 10.3892/etm.2023.11846
162. Cai X, Zhang ZY, Yuan JT, Ocansey DKW, Tu Q, Zhang X, et al. hucMSC-derived exosomes attenuate colitis by regulating macrophage pyroptosis via the miR-378a-5p/NLRP3 axis. Stem Cell Res Ther. (2021) 12:416. doi: 10.1186/s13287-021-02492-6
163. Ocansey DKW, Zhang Z, Xu X, Liu L, Amoah S, Chen X, et al. Mesenchymal stem cell-derived exosome mitigates colitis via the modulation of the gut metagenomics-metabolomics-farnesoid X receptor axis. Biomater Sci. (2022) 10:4822–36. doi: 10.1039/D2BM00559J
164. Garcia-Irigoyen O, Moschetta A. A novel protective role for FXR against inflammasome activation and endotoxemia. Cell Metab. (2017) 25:763–4. doi: 10.1016/j.cmet.2017.03.014
165. Al-Shaibi AA, Abdel-Motal UM, Hubrack SZ, Bullock AN, Al-Marri AA, Agrebi N, et al. Human AGR2 Deficiency Causes Mucus Barrier Dysfunction and Infantile Inflammatory Bowel Disease [published correction appears in Cell Mol Gastroenterol Hepatol. Cell Mol Gastroenterol Hepatol. (2021) 12:1809–30. doi: 10.1016/j.jcmgh.2021.07.001
166. Wlodarska M, Luo C, Kolde R, d'Hennezel E, Annand JW, Heim CE, et al. Indoleacrylic acid produced by commensal peptostreptococcus species suppresses inflammation. Cell Host Microbe. (2017) 22:25–37.e6. doi: 10.1016/j.chom.2017.06.007
167. Goto Y, Kiyono H. Epithelial barrier: an interface for the cross-communication between gut flora and immune system. Immunol Rev. (2012) 245:147–63. doi: 10.1111/j.1600-065X.2011.01078.x
168. Giuffrida P, Corazza GR, Di Sabatino A. Old and new lymphocyte players in inflammatory bowel disease. Dig Dis Sci. (2018) 63:277–88. doi: 10.1007/s10620-017-4892-4
169. Kmieć Z, Cyman M, Ślebioda TJ. Cells of the innate and adaptive immunity and their interactions in inflammatory bowel disease. Adv Med Sci. (2017) 62:1–16. doi: 10.1016/j.advms.2016.09.001
170. Ye M, Joosse ME, Liu L, Sun Y, Dong Y, Cai C, et al. Deletion of IL-6 exacerbates colitis and induces systemic inflammation in IL-10-deficient mice. J Crohn's colitis. (2020) 14:831–40. doi: 10.1093/ecco-jcc/jjz176
171. Buchbinder EI, Desai A. CTLA-4 and PD-1 pathways: similarities, differences, and implications of their inhibition. Am J Clin Oncol. (2016) 39:98–106. doi: 10.1097/COC.0000000000000239
172. Chen RY, Zhu Y, Shen YY, Xu QY, Tang HY, Cui NX, et al. The role of PD-1 signaling in health and immune-related diseases. Front Immunol. (2023) 14:1163633. doi: 10.3389/fimmu.2023.1163633
173. Walunas TL, Lenschow DJ, Bakker CY, Linsley PS, Freeman GJ, Green JM, et al. CTLA-4 can function as a negative regulator of T cell activation. Immunity. (1994) 1:405–13. doi: 10.1016/1074-7613(94)90071-X
174. Krummel MF, Allison JP. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J Exp Med. (1995) 182:459–65. doi: 10.1084/jem.182.2.459
175. Wang C, Thudium KB, Han M, Wang XT, Huang H, Feingersh D, et al. In vitro characterization of the anti-PD-1 antibody nivolumab, BMS-936558, and in vivo toxicology in non-human primates. Cancer Immunol Res. (2014) 2:846–56. doi: 10.1158/2326-6066.CIR-14-0040
176. Rajabian Z, Kalani F, Taghiloo S, Tehrani M, Rafiei A, Hosseini-Khah Z, et al. Over-expression of immunosuppressive molecules, PD-L1 and PD-L2, in ulcerative colitis patients. Iran J Immunol. (2019) 16:62–70. doi: 10.22034/IJI.2019.39407
177. Krummey SM, Ford ML. Braking bad: novel mechanisms of CTLA-4 inhibition of T cell responses. Am J Transplant. (2014) 14:2685–90. doi: 10.1111/ajt.12938
178. Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z, et al. CTLA-4 control over Foxp3+ regulatory T cell function. Science. (2008) 322:271–5. doi: 10.1126/science.1160062
179. Takahashi T, Tagami T, Yamazaki S, Uede T, Shimizu J, Sakaguchi N, et al. Immunologic self-tolerance maintained by CD25(+)CD4(+) regulatory T cells constitutively expressing cytotoxic T lymphocyte-associated antigen 4. J Exp Med. (2000) 192:303–10. doi: 10.1084/jem.192.2.303
180. Ma Y, Wang Z, Zhang A, Xu F, Zhao N, Xue J, et al. Human placenta-derived mesenchymal stem cells ameliorate GVHD by modulating Th17/Tr1 balance via expression of PD-L2. Life Sci. (2018) 214:98–105. doi: 10.1016/j.lfs.2018.10.061
181. Lohr NJ, Molleston JP, Strauss KA, Torres-Martinez W, Sherman EA, Squires RH, et al. Human ITCH E3 ubiquitin ligase deficiency causes syndromic multisystem autoimmune disease. Am J Hum Genet. (2010) 86:447–53. doi: 10.1016/j.ajhg.2010.01.028
182. Ramon HE, Riling CR, Bradfield J, Yang B, Hakonarson H, Oliver PM. The ubiquitin ligase adaptor Ndfip1 regulates T cell-mediated gastrointestinal inflammation and inflammatory bowel disease susceptibility. Mucosal Immunol. (2011) 4:314–24. doi: 10.1038/mi.2010.69
183. Huang X, Zhang Q, Lou Y, Wang J, Zhao X, Wang L, et al. USP22 deubiquitinates CD274 to suppress anticancer immunity. Cancer Immunol Res. (2019) 7:1580–90. doi: 10.1158/2326-6066.CIR-18-0910
184. Mao R, Tan X, Xiao Y, Wang X, Wei Z, Wang J, et al. Ubiquitin C-terminal hydrolase L1 promotes expression of programmed cell death-ligand 1 in non-small-cell lung cancer cells. Cancer Sci. (2020) 111:3174–83. doi: 10.1111/cas.14529
185. Cotton AD, Nguyen DP, Gramespacher JA, Seiple IB, Wells JA. Development of antibody-based PROTACs for the degradation of the cell-surface immune checkpoint protein PD-L1. J Am Chem Soc. (2021) 143:593–8. doi: 10.1021/jacs.0c10008
186. Fujiwara M, Anstadt EJ, Clark RB. Cbl-b deficiency mediates resistance to programmed death-ligand 1/programmed death-1 regulation. Front Immunol. (2017) 8:42. doi: 10.3389/fimmu.2017.00042
187. Augustin RC, Bao R, Luke JJ. Targeting Cbl-b in cancer immunotherapy. J immunotherapy Cancer. (2023) 11:e006007. doi: 10.1136/jitc-2022-006007
188. Gavali S, Liu J, Li X, Paolino M. Ubiquitination in T-cell activation and checkpoint inhibition: new avenues for targeted cancer immunotherapy. Int J Mol Sci. (2021) 22:10800. doi: 10.3390/ijms221910800
189. Xu AT, Lu JT, Ran ZH, Zheng Q. Exosome in intestinal mucosal immunity. J Gastroenterol Hepatol. (2016) 31:1694–9. doi: 10.1111/jgh.13413
190. Chen QQ, Yan L, Wang CZ, Wang WH, Shi H, Su BB, et al. Mesenchymal stem cells alleviate TNBS-induced colitis by modulating inflammatory and autoimmune responses. World J Gastroenterol. (2013) 19:4702–17. doi: 10.3748/wjg.v19.i29.4702
191. Chao K, Zhang S, Qiu Y, Chen X, Zhang X, Cai C, et al. Human umbilical cord-derived mesenchymal stem cells protect against experimental colitis via CD5(+) B regulatory cells [published correction appears in Stem Cell Res Ther. Stem Cell Res Ther. (2016) 7:109. doi: 10.1186/s13287-016-0376-2
192. Tian J, Zhu Q, Zhang Y, Bian Q, Hong Y, Shen Z, et al. Olfactory ecto-mesenchymal stem cell-derived exosomes ameliorate experimental colitis via modulating th1/th17 and treg cell responses. Front Immunol. (2020) 11:598322. doi: 10.3389/fimmu.2020.598322
193. Wakefield LM, Hill CS. Beyond TGFβ: roles of other TGFβ superfamily members in cancer. Nat Rev Cancer. (2013) 13:328–41. doi: 10.1038/nrc3500
194. Perez LG, Kempski J, McGee HM, Pelzcar P, Agalioti T, Giannou A, et al. TGF-β signaling in Th17 cells promotes IL-22 production and colitis-associated colon cancer [published correction appears in Nat Commun. Nat Commun. (2020) 11:2608. doi: 10.1038/s41467-020-16363-w
195. Ihara S, Hirata Y, Koike K. TGF-β in inflammatory bowel disease: a key regulator of immune cells, epithelium, and the intestinal microbiota. J Gastroenterol. (2017) 52:777–87. doi: 10.1007/s00535-017-1350-1
196. de Ceuninck van Capelle C, Spit M, Ten Dijke P. Current perspectives on inhibitory SMAD7 in health and disease. Crit Rev Biochem Mol Biol. (2020) 55:691–715. doi: 10.1080/10409238.2020.1828260
197. Tang Y, Reissig S, Glasmacher E, Regen T, Wanke F, Nikolaev A, et al. Alternative splice forms of CYLD mediate ubiquitination of SMAD7 to prevent TGFB signaling and promote colitis. Gastroenterology. (2019) 156:692–707.e7. doi: 10.1053/j.gastro.2018.10.023
198. Shearwin-Whyatt L, Dalton HE, Foot N, Kumar S. Regulation of functional diversity within the Nedd4 family by accessory and adaptor proteins. Bioessays. (2006) 28:617–28. doi: 10.1002/bies.20422
199. Li H, Seth A. An RNF11: Smurf2 complex mediates ubiquitination of the AMSH protein. Oncogene. (2004) 23:1801–8. doi: 10.1038/sj.onc.1207319
200. Zhang H, Wang L, Li C, Yu Y, Yi Y, Wang J, et al. Exosome-induced regulation in inflammatory bowel disease. Front Immunol. (2019) 10:1464. doi: 10.3389/fimmu.2019.01464
201. Cai Z, Zhang W, Yang F, Yu L, Yu Z, Pan J, et al. Immunosuppressive exosomes from TGF-β1 gene-modified dendritic cells attenuate Th17-mediated inflammatory autoimmune disease by inducing regulatory T cells. Cell Res. (2012) 22:607–10. doi: 10.1038/cr.2011.196
202. Ma ZJ, Wang YH, Li ZG, Wang Y, Li BY, Kang HY, et al. Immunosuppressive effect of exosomes from mesenchymal stromal cells in defined medium on experimental colitis. Int J Stem Cells. (2019) 12:440–8. doi: 10.15283/ijsc18139
203. Zheng W, Bian S, Qiu S, Bishop CE, Wan M, Xu N, et al. Placenta mesenchymal stem cell-derived extracellular vesicles alleviate liver fibrosis by inactivating hepatic stellate cells through a miR-378c/SKP2 axis. Inflammation Regen. (2023) 43:47. doi: 10.1186/s41232-023-00297-z
204. Zhou Q, Cheng C, Wei Y, Yang J, Zhou W, Song Q, et al. USP15 potentiates NF-κB activation by differentially stabilizing TAB2 and TAB3. FEBS J. (2020) 287:3165–83. doi: 10.1111/febs.15202
205. Wang F, Ning S, Yu B, Wang Y. USP14: structure, function, and target inhibition. Front Pharmacol. (2022) 12:801328. doi: 10.3389/fphar.2021.801328
206. Li H, Quan J, Zhao X, Ling J, Chen W. USP14 negatively regulates RIG-I-mediated IL-6 and TNF-α production by inhibiting NF-κB activation. Mol Immunol. (2021) 130:69–76. doi: 10.1016/j.molimm.2020.12.022
207. Lei CQ, Wu X, Zhong X, Jiang L, Zhong B, Shu HB. USP19 inhibits TNF-α- and IL-1β-triggered NF-κB activation by deubiquitinating TAK1. J Immunol. (2019) 203:259–68. doi: 10.4049/jimmunol.1900083
208. Tsuchida T, Zou J, Saitoh T, Kumar H, Abe T, Matsuura Y, et al. The ubiquitin ligase TRIM56 regulates innate immune responses to intracellular double-stranded DNA. Immunity. (2010) 33:765–76. doi: 10.1016/j.immuni.2010.10.013
209. Corcoran K, Jabbour M, Bhagwandin C, Deymier MJ, Theisen DL, Lybarger L. Ubiquitin-mediated regulation of CD86 protein expression by the ubiquitin ligase membrane-associated RING-CH-1 (MARCH1). J Biol Chem. (2011) 286:37168–80. doi: 10.1074/jbc.M110.204040
210. Tze LE, Horikawa K, Domaschenz H, Howard DR, Roots CM, Rigby RJ, et al. CD83 increases MHC II and CD86 on dendritic cells by opposing IL-10-driven MARCH1-mediated ubiquitination and degradation. J Exp Med. (2011) 208:149–65. doi: 10.1084/jem.20092203
211. Baravalle G, Park H, McSweeney M, Ohmura-Hoshino M, Matsuki Y, Ishido S, et al. Ubiquitination of CD86 is a key mechanism in regulating antigen presentation by dendritic cells. J Immunol. (2011) 187:2966–73. doi: 10.4049/jimmunol.1101643
212. Sun W, Tan X, Shi Y, Xu G, Mao R, Gu X, et al. USP11 negatively regulates TNFalpha-induced NF-kappaB activation by targeting on IkappaBalpha. Cell Signal. (2010) 22:386–94. doi: 10.1016/j.cellsig.2009.10.008
213. Kit Leng Lui S, Iyengar PV, Jaynes P, Isa ZFBA, Pang B, Tan TZ, et al. USP26 regulates TGF-β signaling by deubiquitinating and stabilizing SMAD7 [published correction appears in EMBO Rep. EMBO Rep. (2017) 18:797–808. doi: 10.15252/embr.201643270
214. Cao Y, Sun Y, Chang H, Sun X, Yang S. The E3 ubiquitin ligase RNF182 inhibits TLR-triggered cytokine production through promoting p65 ubiquitination and degradation. FEBS Lett. (2019) 593:3210–9. doi: 10.1002/1873-3468.13583
215. Yu JS, Huang T, Zhang Y, Mao XT, Huang LJ, Li YN, et al. Substrate-specific recognition of IKKs mediated by USP16 facilitates autoimmune inflammation. Sci Adv. (2021) 7:eabc4009. doi: 10.1126/sciadv.abc4009
216. Li Y, Wang C, Xi HM, Li WT, Liu YJ, Feng S, et al. Chorionic villus-derived mesenchymal stem cells induce E3 ligase TRIM72 expression and regulate cell behaviors through ubiquitination of p53 in trophoblasts. FASEB J. (2021) 35:e22005. doi: 10.1096/fj.202100801R
217. Shahrokh S, Baradaran Ghavami S, Asadzadeh Aghdaei H, Parigi TL, Farmani M, Danese S, et al. High prevalence of SARS-Coronavirus-2 in patients with inflammatory bowel disease and the role of soluble angiotensin converting Enzyme2. Arch Physiol Biochem. (2024) 130:325–32. doi: 10.1080/13813455.2022.2080228
218. Bednash JS, Johns F, Farkas D, Elhance A, Adair J, Cress K, et al. Inhibiting the deubiquitinase UCHL1 reduces SARS-coV-2 viral uptake by ACE2. Am J Respir Cell Mol Biol. (2023) 68:566–76. doi: 10.1165/rcmb.2022-0331OC
219. Jin S, He X, Ma L, Zhuang Z, Wang Y, Lin M, et al. Suppression of ACE2 SUMOylation protects against SARS-CoV-2 infection through TOLLIP-mediated selective autophagy. Nat Commun. (2022) 13:5204. doi: 10.1038/s41467-022-32957-y
220. Bulek K, Zhao J, Liao Y, Rana N, Corridoni D, Antanaviciute A, et al. Epithelial-derived gasdermin D mediates nonlytic IL-1β release during experimental colitis. J Clin Invest. (2020) 130:4218–34. doi: 10.1172/JCI138103
221. Saadh MJ, Mikhailova MV, Rasoolzadegan S, Falaki M, Akhavanfar R, Gonzáles JLA, et al. Therapeutic potential of mesenchymal stem/stromal cells (MSCs)-based cell therapy for inflammatory bowel diseases (IBD) therapy. Eur J Med Res. (2023) 28:47. doi: 10.1186/s40001-023-01008-7
222. Guo G, Tan Z, Liu Y, Shi F, She J. The therapeutic potential of stem cell-derived exosomes in the ulcerative colitis and colorectal cancer. Stem Cell Res Ther. (2022) 13:138. doi: 10.1186/s13287-022-02811-5
223. Ocansey DKW, Zhang L, Wang Y, Yan Y, Qian H, Zhang X, et al. Exosome-mediated effects and applications in inflammatory bowel disease. Biol Rev Camb Philos Soc. (2020) 95:1287–307. doi: 10.1111/brv.12608
224. Schirmer M, Garner A, Vlamakis H, Xavier RJ. Microbial genes and pathways in inflammatory bowel disease. Nat Rev Microbiol. (2019) 17:497–511. doi: 10.1038/s41579-019-0213-6
225. Prockop DJ, Olson SD. Clinical trials with adult stem/progenitor cells for tissue repair: let's not overlook some essential precautions. Blood. (2007) 109:3147–51. doi: 10.1182/blood-2006-03-013433
Keywords: mesenchymal stem cell, exosome, inflammatory bowel disease, post-translational modification, ubiquitination
Citation: Liao HX, Mao X, Wang L, Wang N, Ocansey DKW, Wang B and Mao F (2024) The role of mesenchymal stem cells in attenuating inflammatory bowel disease through ubiquitination. Front. Immunol. 15:1423069. doi: 10.3389/fimmu.2024.1423069
Received: 25 April 2024; Accepted: 22 July 2024;
Published: 09 August 2024.
Edited by:
Francesca Granucci, University of Milano-Bicocca, ItalyReviewed by:
Byung-Gyu Kim, Case Western Reserve University, United StatesMallika Ghosh, University of Connecticut Health Center, United States
Copyright © 2024 Liao, Mao, Wang, Wang, Ocansey, Wang and Mao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fei Mao, bWFvZmVpMjAwM0B1anMuZWR1LmNu
†These authors have contributed equally to this work