 Kexin Duan1†
Kexin Duan1† Shujing Wang
Shujing Wang Xinsheng Chen
Xinsheng Chen- 1The Second Clinical Medical College, Guangzhou University of Chinese Medicine, Guangzhou, China
- 2The First Clinical Medical College, Guangzhou University of Chinese Medicine, Guangzhou, China
- 3Department of Dermatology, Guangdong Provincial Hospital of Traditional Chinese Medicine, Guangzhou, China
Background: Numerous observational studies have identified associations between both psoriasis (PsO) and psoriatic arthritis (PsA), and autoimmune diseases (AIDs); however, the causality of these associations remains undetermined.
Methods: We conducted a bidirectional two-sample Mendelian Randomization study to identify causal associations and directions between both PsO and PsA and AIDs, such as systemic lupus erythematosus (SLE), Crohn’s disease (CD), ulcerative colitis (UC), multiple sclerosis (MS), uveitis, bullous pemphigoid (BP), Hashimoto’s thyroiditis (HT), rheumatoid arthritis (RA), vitiligo, and ankylosing spondylitis (AS). The causal inferences were drawn by integrating results from four regression models: Inverse Variance Weighting (IVW), MR-Egger, Weighted Median, and Maximum Likelihood. Furthermore, we performed sensitivity analyses to confirm the reliability of our findings.
Results: The results showed that CD [IVW odds ratio (ORIVW), 1.11; 95% confidence interval (CI), 1.06-1.17; P = 8.40E-06], vitiligo (ORIVW, 1.16; 95% CI, 1.05-1.28; P = 2.45E-03) were risk factors for PsO, while BP may reduce the incidence of PsO (ORIVW, 0.91; 95% CI, 0.87-0.96; P = 1.26E-04). CD (ORIVW, 1.07; 95% CI, 1.02-1.12; P = 0.01), HT (ORIVW, 1.23; 95% CI, 1.08-1.40; P = 1.43E-03), RA (ORIVW, 1.11; 95% CI, 1.02-1.21, P = 2.05E-02), AS (ORIVW, 2.18; 95% CI, 1.46-3.27; P = 1.55E-04), SLE (ORIVW, 1.04; 95% CI, 1.01-1.08; P = 1.07E-02) and vitiligo (ORIVW, 1.27; 95% CI, 1.14-1.42; P = 2.67E-05) were risk factors for PsA. Sensitivity analyses had validated the reliability of the results.
Conclusions: Our study provides evidence for potential causal relationships between certain AIDs and both PsO and PsA. Specifically, CD and vitiligo may increase the risk of developing PsO, while CD, HT, SLE, RA, AS, and vitiligo may elevate the risk for PsA. Additionally, it is crucial to closely monitor the condition of PsO patients with specific AIDs, as they have a higher likelihood of developing PsA than those without AIDs. Moving forward, greater attention should be paid to PsA and further exploration of other PsO subtypes is warranted.
1 Introduction
Psoriasis (PsO), a chronically relapsing dermatological condition with immune involvement, is predominantly featured by erythematous plaques with a scaly appearance (1), affecting approximately 0.11%-1.88% of the global population (2). Since 2014, the World Health Organization has defined PsO by five characteristics: chronic, non-communicable, painful, disfiguring, and disabling (3), exerting a significant psychosocial and economic burden on patients. Approximately 30% of patients with PsO, who only present with skin lesions, will progress to psoriatic arthritis (PsA) (4). PsA is a seronegative inflammatory arthritis, clinically characterized by synovitis with peripheral joint osteolysis, axial involvement, sacroiliitis, nail disorders, and tendinitis (5). In 85% of PsA patients, skin manifestations precede joint symptoms, with joint damage typically appearing about 10 years after the disease onset (6). Furthermore, PsA also shares immunological features with PsO (1), with the IL-23/Th17 pathway playing a central role in the immune regulation and inflammation. The activation of Th1 and Th17 cells and the resultant cytokines they produce, such as IL-17, TNF, and IL-22, drive an inflammatory response that stimulates abnormal proliferation and keratinization of keratinocytes, and exacerbate inflammation in the joints and surrounding soft tissues, leading to cutaneous and skeletal damage (1, 6).
Numerous observational studies have found that PsO and PsA could be comorbid with autoimmune diseases (AIDs) (7, 8). Specifically, research indicated that the risk of comorbidities with AIDs in PsO patients was five times greater than that of the general population (9). Additionally, a South Korean study found a significantly higher risk of rheumatoid arthritis (RA), ankylosing spondylitis (AS), Crohn’s disease (CD), and systemic lupus erythematosus (SLE) in 321,354 PsO patients compared to 321,354 healthy individuals (10). Sardu C et al. selected 25,885 individuals from Sardinia, Italy, to assess the prevalence of 12 AIDs, finding that PsO/PsA ranked second in prevalence within the population (11). Additionally, a retrospective study demonstrated a direct correlation between comorbidity incidence and disease severity in PsO patients, with PsA patients experiencing a significantly higher incidence of comorbidities than those with PsO (12). Although observational studies are widely used for the preliminary etiological investigations, their susceptibility to confounding factors and challenges in determining the temporal sequence of causality make these relationships unclear (13).
Therefore, we performed a Mendelian randomization (MR) analysis to delve deeper into the causal relationships between PsO, PsA, and AIDs such as SLE, multiple sclerosis (MS), CD, ulcerative colitis (UC), uveitis, bullous pemphigoid (BP), Hashimoto’s thyroiditis (HT), RA, AS, and vitiligo. Its principle is founded on Mendel’s Second Law, which states that during meiosis for gamete production, the parental alleles are allocated to the progeny randomly, uninfluenced by environmental, socioeconomic, or other confounding factors. MR analysis utilizes genetic variations as instrumental variables (IVs) to infer causality between exposures and outcomes, thus mitigating the interference from reverse causation and weak confounders (14). It has been extensively applied in exploring complex etiologies of diseases.
2 Methods
2.1 Study design
We conducted a bidirectional two-sample MR analysis based on the Genome-Wide Association Studies (GWAS) database to explore the causal links between PsO, PsA, and AIDs. This MR study followed three fundamental assumptions: (1) the correlation assumption: the IVs should be strongly correlated with the exposure; (2) the exclusivity assumption: the IVs have no direct relation to the outcome; (3) the independence assumption: the IVs shouldn’t be related to any confounder that affects the exposure-outcome relationship (15, 16) (Figure 1).
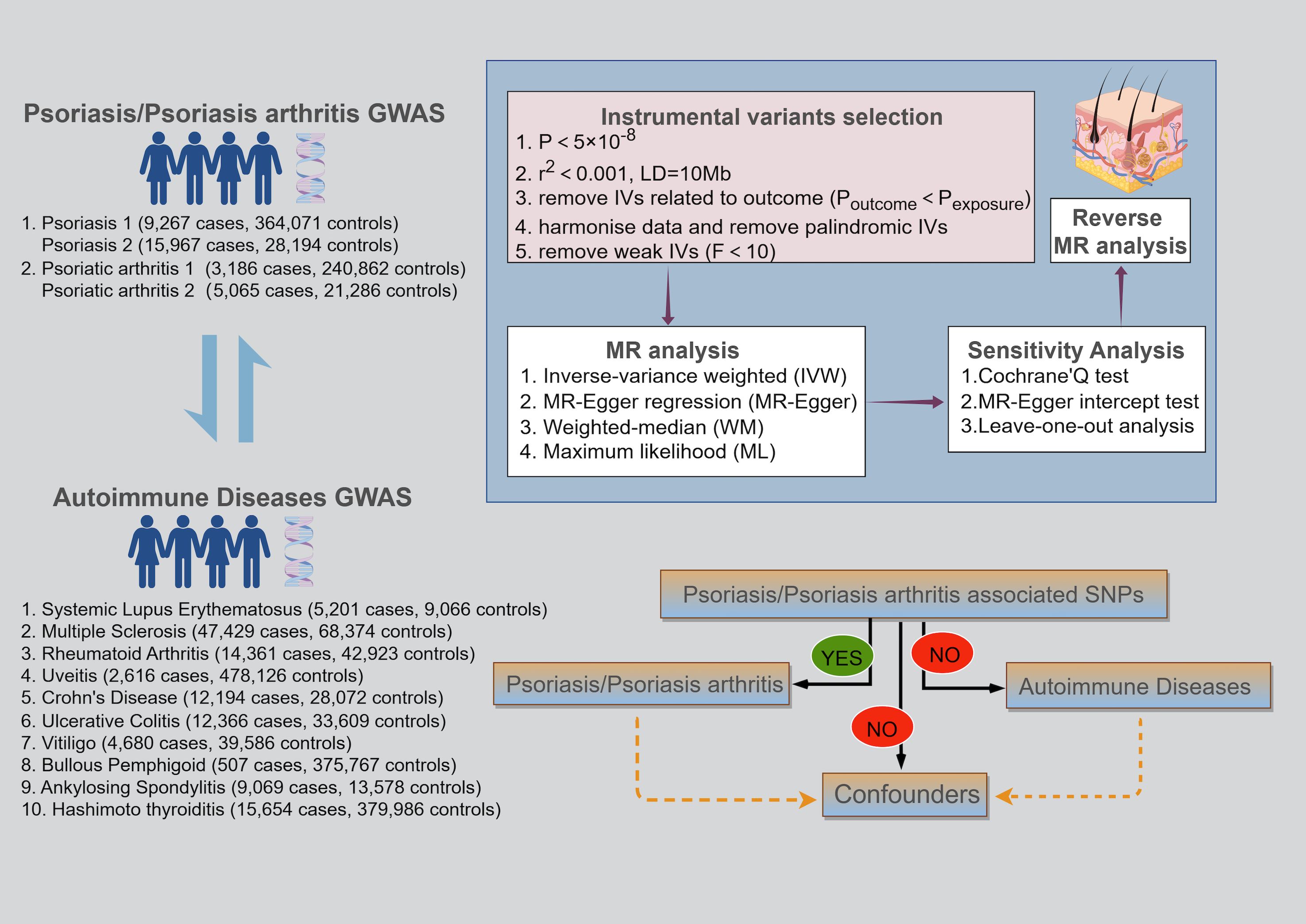
Figure 1 Schematic of the three key assumptions of Mendelian randomization studies and the research flowchart.
2.2 Data sources
The GWAS databases for PsO (9,267 cases and 364,071 controls), PsA (3,186 cases and 240,862 controls), and BP (507 cases, 375,767 controls) all originated from the FinnGen Consortium, which is a large-scale biomedical research project based in Finland that aims to uncover new biomarkers and therapeutic targets by analyzing the genetic information and health data of Finnish participants. To address the sample overlap among PsO, PsA, and BP, additional GWAS databases were selected for PsO and PsA. Specifically, the GWAS data for PsO (15,967 cases and 28,194 controls) were derived from a cross-ethnic investigation conducted by Stuart PE et al., which compared PsO susceptibility between South Asians and Europeans (17). The GWAS data for PsA (5,065 cases and 21,286 controls) were sourced from a study by Soomro M et al. (18), which developed a database to examine genetic markers differentiating PsA from PsO without arthritis. GWAS data for SLE (5,201 cases and 9,066 controls), MS (47,429 cases and 68,374 controls), CD (12,194 cases and 28,072 controls), UC (12,366 cases and 33,609 controls), uveitis (2,616 cases and 478,126 controls), AS (9,069 cases and 13,578 controls), RA (14,361 cases and 33,609 controls), and HT (15,654 cases and 379,986 controls) were sourced from the IEU database (https://gwas.mrcieu.ac.uk). Moreover, GWAS data for vitiligo (4,680 cases and 39,586 controls) were sourced from the most extensive meta-analyses available (19). The comprehensive GWAS data information is summarized in Supplementary Table S1.
2.3 Instrumental variables selection
Selecting appropriate IVs requires adherence to six steps. Firstly, a single nucleotide polymorphism (SNP) must demonstrate a strong correlation with the exposure (p < 5×10-8). Secondly, we set a linkage disequilibrium (LD) threshold (r2) of 0.001 and a 10 Mb clumping window to guarantee the independence of each SNP (20). Should the number of selected SNPs be insufficient, the thresholds for p and r2 can be adjusted to a minimum of p < 5×10-6 and r2 < 0.01, respectively. Thirdly, we exclude the SNPs that exhibit a strong correlation with the outcome variable (poutcome < pexposure). Fourthly, harmonize the data between SNPexposure and SNPoutcome to ensure alignment of allelic directions and compatibility for analysis. Fifthly, confounders are eliminated through the Phenoscanner website to mitigate potential pleiotropic effects (21). Finally, the association strength between IVs and the exposure is measured using F-statistics, computed as F=R2/(1-R2)×(N-K-1)/K, with R2 = 2×MAF×(1-MAF)×β2. In the absence of MAF values, R2 is determined by R2=β2/(β2+SE2×N) (22–24), where R2 indicates the variance in exposure explained by the IVs, N represents the total sample size of the exposure GWAS, MAF denotes the frequency of minor allele, K refers to the number of IVs, and β signifies the SNP’s effect size on exposure. If the F-statistic exceeds 10, it implies that MR analyses are unlikely to be biased by weak IVs. Conversely, SNPs with F-statistics below this threshold should be excluded.
2.4 Mendelian randomization analysis
MR analysis primarily employs Inverse Variance Weighted (IVW) to determine the existence of causality, while MR Egger (ME), Weighted Median (WM), and Maximum Likelihood (ML) serve as supplementary analytical approaches. IVW is characterized by its disregard for the intercept term, fitting the data using the inverse of the outcome variance as weights (25). Unlike IVW, ME incorporates the intercept term within its regression model, also applying the inverse of outcome variance as weights for fitting (26). WM is the median of the distribution function derived from sorting all SNP effect values by their weights. It delivers a reliable causal effect estimate, despite having up to 50% invalid IVs (27). ML, grounded in principles of probability theory, estimates unknown parameters by identifying model parameters that maximize the likelihood of the observed data (28). If P < 0.05 for IVW, a causal link between exposure and outcome is inferred when all five conditions are met simultaneously: (1) at least one other statistical method yields a p-value < 0.05; (2) the odds ratios (OR) from IVW, WM, and ML consistently indicate the same direction of effect; (3) there is no significant evidence of horizontal pleiotropy (P > 0.05); (4) all error lines of the leave-one-out analysis plot are all on the same side of zero; (5) these conditions still hold after adjusting for heterogeneity. This study conducted MR analysis using R software (version 4.2.3), utilizing the R packages TwoSampleMR and RadialMR.
2.5 Sensitivity analyses
We employed Cochran’s Q test, leave-one-out analysis, and the MR-Egger intercept for sensitivity analysis. The objectives of the sensitivity analyses are threefold: first, to evaluate the dependability of the MR analysis outcomes; second, to explore potential biases, such as genetic pleiotropy and data heterogeneity; and third, to determine whether a specific SNP significantly affects the outcome. We employed Cochran’s Q test to assess the extent of heterogeneity. When significant heterogeneity occurs (P < 0.05), we employ MR radial analysis to remove outliers and correct the estimates to verify the reliability of the findings. Leave-one-out analysis determines the combined effect of the remaining SNPs by sequentially excluding each SNP, with all error lines located consistently on one side of zero, suggesting dependable outcomes. Furthermore, MR studies should primarily focus on the horizontal pleiotropy to avoid genetic variants influencing the outcome through exposure. If the intercept of the MR-Egger regression is significantly different from zero (P < 0.05), it indicates the presence of horizontal pleiotropy, and we use MR radial analysis to correct the estimates by excluding outliers.
3 Results
3.1 Results of selection of instrumental variables
Following the described selection process, we identified IVs (Supplementary Tables S7-S10). Specifically, in studying the effects of BP and uveitis on PsO, we established strong correlation and LD for BP (P < 5×10-6, r2 < 0.01) and uveitis (P < 5×10-6, r2 < 0.001) to extract a sufficient number of SNPs. Furthermore, in investigating the effect of CD on PsA, we set P < 5×10-9, r2 < 0.001 to eliminate horizontal pleiotropy. Ultimately, we calculated that the F value corresponding to each SNP (Supplementary Tables S7-S10) or to all SNPs (Supplementary Table S11) was greater than 10, indicating that the results were not biased by weak IVs.
3.2 Impact of PsO and PsA on AIDs
When considering PsO and PsA as exposures, there were no causal relationships between them and AIDs (Supplementary Figures S1, S2 Supplementary Tables S2, S5).
3.3 Impact of AIDs on PsO and PsA
When PsO and PsA were considered as outcomes, genetically predicted CD [IVW odds ratio (ORIVW), 1.11; 95% confidence interval (CI), 1.06-1.17; P = 8.40E-06], vitiligo (ORIVW, 1.16; 95% CI, 1.05-1.28; P = 2.45E-03) were risk factors for PsO, while BP may reduce the incidence of PsO (ORIVW, 0.91; 95% CI, 0.87-0.96; P = 1.26E-04). Furthermore, no causal link was found between the remaining AIDs and PsO. CD (ORIVW, 1.07; 95% CI, 1.02-1.12; P = 0.01), HT (ORIVW, 1.23; 95% CI, 1.08-1.40; P = 1.43E-03), RA (ORIVW, 1.11; 95% CI, 1.02-1.21; P = 2.05E-02), AS (ORIVW, 2.18; 95% CI, 1.46-3.27; P = 1.55E-04), SLE (ORIVW, 1.04; 95% CI, 1.01-1.08; P = 1.07E-02) and vitiligo (ORIVW, 1.27; 95% CI, 1.14-1.42; P = 2.67E-05) were risk factors for PsA. There were no causal associations found between the remaining AIDs and PsO/PsA (Figures 1–3; Supplementary Tables S3, S4).
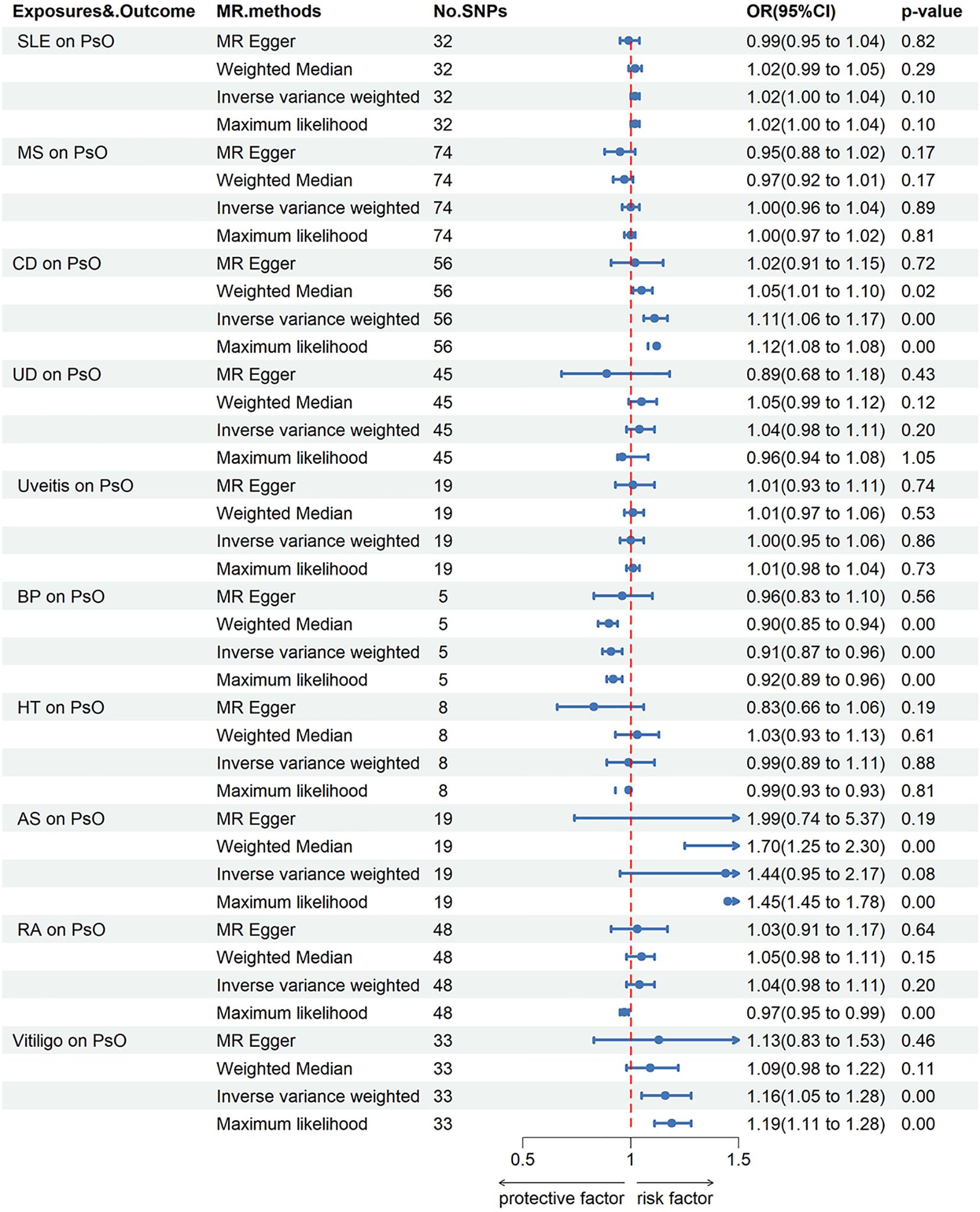
Figure 2 Forest plots utilized four methods to visualize the causal effects of AIDs on PsO risk. PsO, psoriasis; SLE, Systemic lupus erythematosus; MS, multiple sclerosis; RA, Rheumatoid arthritis; UD, Crohn’s disease; UC, Ulcerative colitis; Vitiligo; BP, Bullous pemphigoid; AS, Ankylosing spondylitis; HT, Hashimoto thyroiditis;AIDs, Autoimmune diseases.
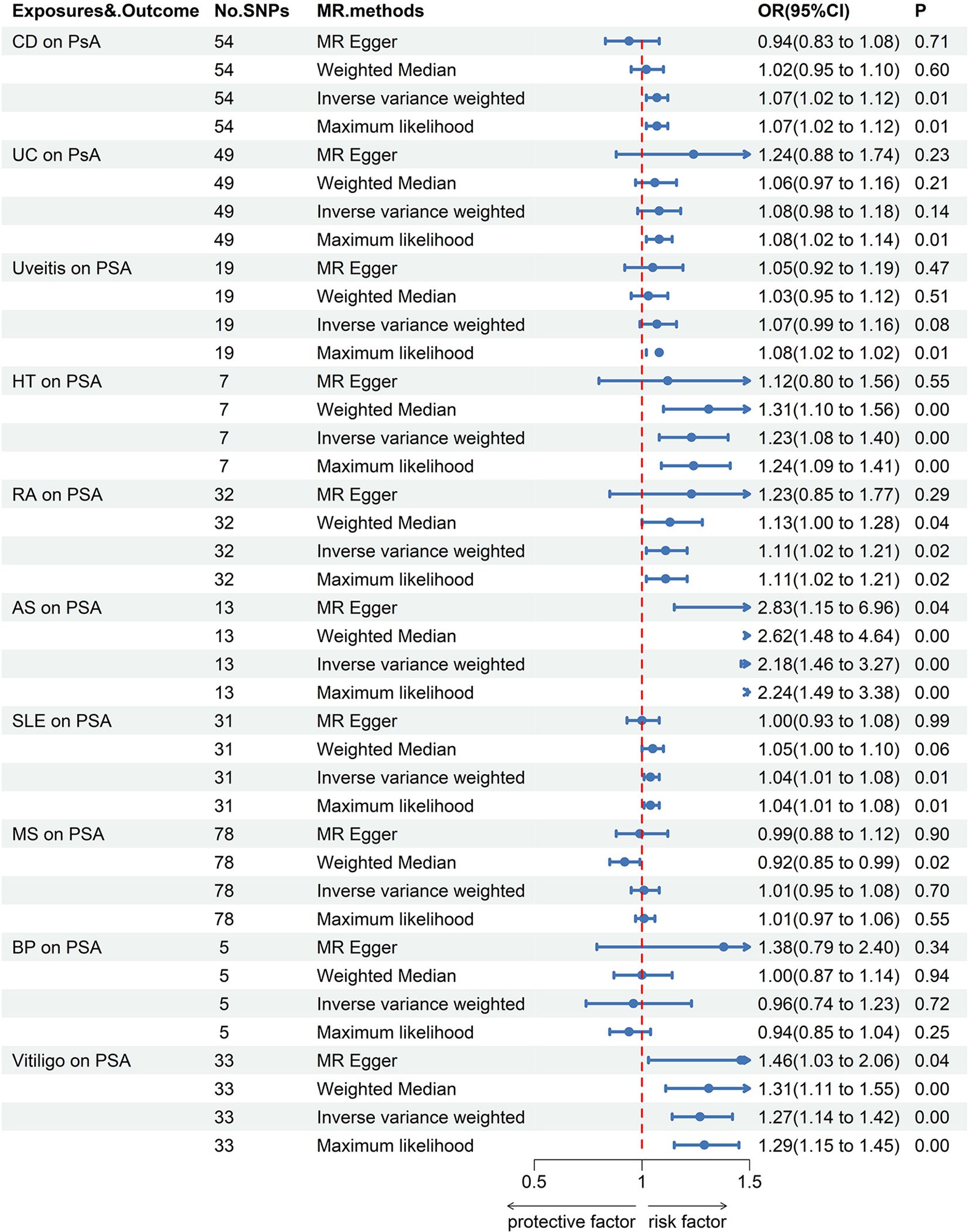
Figure 3 Forest plots utilized four methods to visualize the causal effects of AIDs on PsA risk. PsA, psoriasis arthritis; SLE, Systemic lupus erythematosus; MS, multiple sclerosis; RA, Rheumatoid arthritis; UD, Crohn’s disease; UC, Ulcerative colitis; Vitiligo; BP, Bullous pemphigoid; AS, Ankylosing spondylitis; HT, Hashimoto thyroiditis;AIDs, Autoimmune diseases.
3.4 Results of sensitivity analysis
In addition to the previously mentioned pleiotropy between CD and PsA, we also found pleiotropy between SLE and PsO (P = 0.038). Therefore, we utilized the MR-Radial to remove five outlier SNPs—rs389884, rs4274624, rs4388254, rs58688157, and rs58721818—which effectively mitigated pleiotropy. In studies with causal relationships, significant heterogeneity was observed when CD and vitiligo affected PsO. Therefore, we used MR-Radial to reduce heterogeneity and found that the positive results remained stable (Supplementary Figure S19, Supplementary Table S6). Furthermore, in a leave-one-out analysis, excluding each SNP individually did not result in significant variation in the estimated causal effects (Supplementary Figures S3-S18). In summary, our study findings are deemed reliable and valid.
4 Discussions
To date, this study represents the first large-scale MR analysis to explore the causal associations between both PsO and PsA and ten types of AIDs. Genetic prediction results indicate that BP might lower the risk of developing PsO, while CD and vitiligo may increase it. Additionally, certain AIDs including CD, AS, RA, HT, vitiligo, and SLE are more likely to induce PsA. This study’s findings align with those observed in an earlier cohort study, which revealed that most AIDs were diagnosed prior to PsO (9).
CD is an inflammatory bowel disorder characterized by abdominal pain, diarrhea, and bloody stool (29). A meta-analysis incorporating nine observational studies indicated a significantly elevated chance of developing CD and UC in patients with PsO (30). Nonetheless, a United States cohort study showed a significantly higher CD risk in female PsA patients, with no increased UC risk, matching our genetic predictions (31). Genetic susceptibility is a key determinant of the underlying risk of both PsO and CD, with strong associations confirmed at the IL23R, IL12B, REL, and TYK2 loci for both conditions (32). Moreover, both PsO and CD exhibit dysbiosis of the gut microbiota, which can disrupt the immune balance between effector T cells and regulatory T (Treg) cells. This imbalance could also increase intestinal permeability, allowing gut bacteria and their metabolites to enter the skin through the bloodstream, thereby triggering inflammatory skin responses (33, 34). Additionally, dysbiosis can increase the levels of hydrogen sulfide in the gut while inhibiting the production of protective metabolites such as butyrate and propionate, thus potentially triggering the onset of CD (32). Furthermore, the TNF pathway (35), IL-23/IL-17 pathway (36), JAK-STAT pathway (37), and ROR-γT/Th17 axis (38) represent common pathogenic pathways between PsO and inflammatory bowel diseases.
BP is primarily characterized by tense, non-rupturing large blisters (39). A cohort study conducted in Taiwan revealed that the risk of developing PsO among patients with BP was significantly higher than that in the healthy population (40). Ohata et al. conducted a study on patients diagnosed with both psoriasis and autoimmune blistering diseases (AIBD). The results indicated that BP was the most prevalent form of AIBD, affecting 63.4% of the patients (41). Some scholars believe that certain treatment modalities for PsO, such as corticosteroids, anti-IL-17A monoclonal antibodies, tumor necrosis factor-alpha (TNF-α) antagonists, and ultraviolet light therapy, might trigger the onset of BP (42–44). Furthermore, the release of a large amount of neutrophil chemotactic factors by PsO could initiate a cascade of reactions leading to BP (45); additionally, the disruption or even absence of laminin in the psoriatic skin lesions affects the differentiation and proliferation of keratinocytes, potentially inducing BP through antibodies (such as those targeting the basement membrane zone) (46). Lastly, the involvement of Th17 cells and IL-17 promotes the production of pro-inflammatory cytokines and matrix metalloproteinases, culminating in blister formation (47). However, our study results do not align with the findings from epidemiological research. This discrepancy may be attributed to the small sample size of the GWAS for BP and the insufficient strength of the IVs used (P < 5×10-6, r2 < 0.01). Therefore, caution should be exercised when interpreting the Mendelian Randomization results between BP and PsO.
Vitiligo is an immune-mediated depigmenting disease (48). Previous literature has shown that vitiligo often precedes the onset of PsO (49, 50), aligning with our findings on the causal sequence. A retrospective study on the prevalence of comorbid conditions in vitiligo patients revealed that out of 2,441 vitiligo patients, 565 (23%) had comorbid AIDs, with thyroid diseases and psoriasis being the most common (51). A meta-analysis encompassing ten observational studies revealed a notably higher incidence of PsO among vitiligo patients compared to the general population (52). In terms of genetic susceptibility, Zhu et al. identified that rs9468925 within the HLA-C/HLA-B locus is associated with both PsO and vitiligo (53). In the pathogenesis of vitiligo, CD8+ T cells play a predominant role (54), while the Th17 pathway and IL-17 also exert multiple effects (55): firstly, IL-17 attracts CD8+ T cells into the surrounding tissue, directly causing the destruction of melanocytes. Secondly, IL-17 enhances the inflammatory response of endothelial cells and keratinocytes, promoting the migration and infiltration of inflammatory cells such as neutrophils and T cells. Neutrophils can increase the production of ROS, inducing oxidative stress and further damaging melanocytes. Moreover, IL-17 promotes melanocyte apoptosis by inhibiting the expression of MITF and downregulating BCL2 (56). NB-UVB treatment alleviates oxidative stress responses and improves the condition by reducing IL-17 levels (57).
HT, an autoimmune disorder, is characterized by hypothyroidism resulting from thyroid gland dysfunction (58). Numerous studies have identified a link between PsA and autoimmune thyroiditis (59, 60). Bianchi et al. observed that patients with PsA have significantly more thyroid involvement compared to the general population, evidenced by an increased average thyroid volume and higher prevalence rates of anti-microsomal and anti-thyroglobulin antibodies (61). Both HT and PsA are autoimmune diseases mediated by Th1 cell immunity, with Th1 cells, interferon-gamma (IFN-γ), and the chemokine CXCL10 playing pivotal roles in their pathogenesis (6, 62). In HT, Th1 cells produce IFN-γ and TNF-α, stimulating thyroid cells to emit CXCL10. This chemokine binds to receptor CXCR3, attracting Th1 cells to the target tissue, thereby triggering inflammatory responses and thyroid damage (58). Conversely, many PsA patients show increased CXCL10 levels in their serum and synovial fluid (63, 64), capable of attracting plasmacytoid dendritic cells from the blood into the synovial tissue, thereby initiating an inflammatory response (65).
SLE is an autoimmune connective tissue disease that affects multiple organs (66). A case-control study from Israel found that PsA patients exhibited a 2.3-fold higher prevalence of SLE compared to the control group (67). It has been found that PsO and SLE have shared genetic predisposition sites, including PTPN22, TRAF3IP2, and STAT4. Regarding immune mechanisms, the high expression of IL-17/IL-23 axis is crucial for the comorbidity of SLE and PsO. Patients with SLE, due to defects in cell apoptosis, release large amounts of dsDNA and ribonucleoproteins. These substances form nucleic acid immune complexes upon binding with autoantibodies, subsequently activating plasmacytoid dendritic cells (pDCs). Activated pDCs secrete IL-23, stimulating the differentiation and proliferation of Th17 cells and the production of IL-17. IL-17 promotes the proliferation and differentiation of B cells into plasma cells, producing autoantibodies, stimulating the generation of a large number of inflammatory cells, and thereby damaging target organs (68–70). Additionally, a case report detailed the treatment of SLE and PsA with the IL-17 inhibitor secukinumab (71), and a double-blind Phase II trial validated the effectiveness of an IL-23 antagonist in treating SLE. These findings underscore the critical role of the IL-17/IL-23 axis in the development of both SLE and PsA.
RA and AS are autoimmune diseases primarily characterized by bone damage and pain (72). AS predominantly targets the sacroiliac joints, spine, and peripheral joints, whereas RA mainly leads to synovitis, bone erosion, and cartilage damage (72, 73). Prior research has indicated a higher prevalence of AS and RA in patients with PsA compared to those with PsO alone (74, 75). A cohort study from the UK discovered that AS patients experienced a higher risk of comorbidities with PsO, with 4.4% of individuals initially diagnosed with AS also presenting with concurrent PsO (76). There has been ongoing debate regarding whether AS with PsO manifestations inherently constitutes a form of axial PsA (77). This study suggests a causal link between AS and PsA, inferring that AS with PsO manifestations might indeed be an expression of co-occurring PsA and AS. And HLA-B*27 is a shared genetic susceptibility factor for both conditions (78, 79). Moreover, the IL-23/Th17 is also involved in the pathogenesis of AS, with heightened expression of inflammatory cytokines like IL-17 and IL-22 influencing pertinent signaling pathways (80). This leads to enhanced activity of osteoclasts and inhibited function of osteoblasts, resulting in bone damage. Consequently, the IL-17A inhibitor—secukinumab—has shown effectiveness in treating PsA and AS in a phase III randomized clinical trial, further evidencing their overlapping mechanisms of action (81). Previous studies have demonstrated comorbidity of PsA with RA, primarily affecting peripheral joints and manifesting as symmetrical polyarthritis with positive rheumatoid serology. Th17 cells have emerged as research targets in clinical trials for RA (82), with the IL-17A produced by Th17 cells acting synergistically with TNF-α to promote the activation of fibroblasts and chondrocytes (73). Moreover, AS and RA can cause bone and joint damage, which is independently linked to a higher risk of developing PsA (83). This may relate to physical trauma activating innate immunity, leading to the influx of pro-inflammatory molecules into the synovium (84), or activating nerve endings to release neuropeptides, triggering an inflammatory response (85).
In addition, multiple epidemiological studies suggest that PsO and PsA are comorbid with uveitis and MS. 10% of PsO patients and 30% of PsA patients may experience ocular diseases, with uveitis being the most common (86–88). A cohort study from Korea indicated that individuals with severe PsO or PsA exhibit an elevated incidence and recurrence rate of uveitis compared to controls (89). Furthermore, a comprehensive cohort study in Denmark showed that the higher the severity of PsO, the greater the risk of developing MS (90). However, this study did not find a causal link, suggesting that potential confounders or a shared genetic architecture might be the reason behind the epidemiological associations.
In summary, our study has three important implications: (1) It provides evidence of potential causal relationships between certain AIDs and both PsO and PsA, suggesting that increased surveillance of these conditions should be considered in clinical practice; (2) Given that AIDs are more likely to precipitate the occurrence of PsA and most PsA patients initially present only with skin lesions, it can be inferred that patients with PsO and comorbid AIDs are more likely to develop PsA. It indicates that AIDs could be potential risk factors for progressing from PsO to PsA, underscoring the importance of early monitoring for PsA; (3) This study also underscores the critical importance of establishing GWAS database for PsO subtypes, which is vital for future etiological research into PsO. However, our study also presents several limitations. Firstly, the lack of pertinent GWAS data prevents us from exploring the causal links between different subtypes. Secondly, the GWAS database we used primarily targeted European populations, potentially limiting its applicability to other ethnicities. Thirdly, the small GWAS dataset for BP in our study necessitates larger future datasets for validating our findings. Fourthly, although using multiple methods to control confounders, potential horizontal pleiotropy may still exist. Lastly, this study may have overlooked other AIDs are causally linked to PsO and PsA.
5 Conclusions
Certain AIDs are causally associated with PsO and PsA. Furthermore, PsO patients who have certain AIDs are more prone to developing PsA than those without AIDs. Additionally, focusing more on PsO subtypes could enhance our understanding of the condition.
Data availability statement
The detailed data and GWAS data sources referenced in this study are available in the article/Supplementary Materials. Should you require further data, please contact the author directly.
Ethics statement
All data utilized herein were derived from the public genome-wide association studies, thus obviating the need for further ethical consent.
Author contributions
KD: Writing – review & editing, Writing – original draft, Software, Methodology, Formal analysis, Data curation. JRW: Writing – original draft, Data curation, Conceptualization. SC: Writing – original draft, Methodology, Data curation. TC: Writing – original draft, Methodology. JJW: Writing – review & editing, Data curation. SW: Writing – review & editing, Data curation. XC: Writing – review & editing, Supervision, Funding acquisition, Conceptualization.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was supported by the Research Project “Study on the Effects of Subcutaneous Injection and Intravenous Infusion of Adipose-Derived Mesenchymal Stem Cells on Psoriasis Mouse Animal Models(2023KTSCX026)” and “Guangdong Province’s Third Batch of Famous Traditional Chinese Medicine Practitioner Inheritance Projects - Ruiqiang Fan Guangdong Famous Traditional Chinese Medicine Inheritance Studio”.
Acknowledgments
We extend our appreciation to the researchers and the IEU, GWAS Catalog, and FinnGen Consortium databases for providing the GWAS data for this study. We also thank the Figdraw platform for its assistance in creating charts(ID: PARSPb3bf3).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2024.1422626/full#supplementary-material
References
1. Griffiths CEM, Armstrong AW, Gudjonsson JE, Barker J. Psoriasis. Lancet. (2021) 397:1301–15. doi: 10.1016/S0140-6736(20)32549-6
2. Parisi R, Iskandar IYK, Kontopantelis E, Augustin M, Griffiths CEM, Ashcroft DM. National, regional, and worldwide epidemiology of psoriasis: systematic analysis and modelling study. Bmj. (2020) 369:m1590. doi: 10.1136/bmj.m1590
4. Villani AP, Rouzaud M, Sevrain M, Barnetche T, Paul C, Richard MA, et al. Prevalence of undiagnosed psoriatic arthritis among psoriasis patients: Systematic review and meta-analysis. J Am Acad Dermatol. (2015) 73:242–8. doi: 10.1016/j.jaad.2015.05.001
5. Merola JF, Espinoza LR, Fleischmann R. Distinguishing rheumatoid arthritis from psoriatic arthritis. RMD Open. (2018) 4:e000656. doi: 10.1136/rmdopen-2018-000656
6. Ritchlin CT, Colbert RA, Gladman DD. Psoriatic arthritis. N Engl J Med. (2017) 376:957–70. doi: 10.1056/NEJMra1505557
7. Kamiya K, Kishimoto M, Sugai J, Komine M, Ohtsuki M. Risk factors for the development of psoriasis. Int J Mol Sci. (2019) 20. doi: 10.3390/ijms20184347
8. Bu J, Ding R, Zhou L, Chen X, Shen E. Epidemiology of psoriasis and comorbid diseases: A narrative review. Front Immunol. (2022) 13:880201. doi: 10.3389/fimmu.2022.880201
9. Andersen YMF, Wu JJ, Thyssen JP, Egeberg A. Chronologic order of appearance of immune-mediated inflammatory diseases relative to diagnosis of psoriasis. J Am Acad Dermatol. (2019) 81:1283–91. doi: 10.1016/j.jaad.2019.04.033
10. Jung JM, Kim YJ, Lee WJ, Won CH, Lee MW, Chang SE. Risk of incident autoimmune diseases in patients with newly diagnosed psoriatic disease: a nationwide population-based study. Sci Rep. (2023) 13:16738. doi: 10.1038/s41598-023-43778-4
11. Sardu C, Cocco E, Mereu A, Massa R, Cuccu A, Marrosu MG, et al. Population based study of 12 autoimmune diseases in Sardinia, Italy: prevalence and comorbidity. PloS One. (2012) 7:e32487. doi: 10.1371/journal.pone.0032487
12. Edson-Heredia E, Zhu B, Lefevre C, Wang M, Barrett A, Bushe CJ, et al. Prevalence and incidence rates of cardiovascular, autoimmune, and other diseases in patients with psoriatic or psoriatic arthritis: a retrospective study using Clinical Practice Research Datalink. J Eur Acad Dermatol Venereol. (2015) 29:955–63. doi: 10.1111/jdv.12742
13. Harper S. A future for observational epidemiology: clarity, credibility, transparency. Am J Epidemiol. (2019) 188:840–5. doi: 10.1093/aje/kwy280
14. Davey Smith G, Hemani G. Mendelian randomization: genetic anchors for causal inference in epidemiological studies. Hum Mol Genet. (2014) 23:R89–98. doi: 10.1093/hmg/ddu328
15. Davies NM, Holmes MV, Davey Smith G. Reading Mendelian randomisation studies: a guide, glossary, and checklist for clinicians. Bmj. (2018) 362:k601. doi: 10.1136/bmj.k601
16. Emdin CA, Khera AV, Kathiresan S. Mendelian randomization. Jama. (2017) 318:1925–6. doi: 10.1001/jama.2017.17219
17. Stuart PE, Tsoi LC, Nair RP, Ghosh M, Kabra M, Shaiq PA, et al. Transethnic analysis of psoriasis susceptibility in South Asians and Europeans enhances fine-mapping in the MHC and genomewide. HGG Adv. (2022) 3. doi: 10.1016/j.xhgg.2021.100069
18. Soomro M, Stadler M, Dand N, Bluett J, Jadon D, Jalali-Najafabadi F, et al. Comparative genetic analysis of psoriatic arthritis and psoriasis for the discovery of genetic risk factors and risk prediction modeling. Arthritis Rheumatol. (2022) 74:1535–43. doi: 10.1002/art.42154
19. Jin Y, Andersen G, Yorgov D, Ferrara TM, Ben S, Brownson KM, et al. Genome-wide association studies of autoimmune vitiligo identify 23 new risk loci and highlight key pathways and regulatory variants. Nat Genet. (2016) 48:1418–24. doi: 10.1038/ng.3680
20. Arnold M, Raffler J, Pfeufer A, Suhre K, Kastenmüller G. SNiPA: an interactive, genetic variant-centered annotation browser. Bioinformatics. (2015) 31:1334–6. doi: 10.1093/bioinformatics/btu779
21. Kamat MA, Blackshaw JA, Young R, Surendran P, Burgess S, Danesh J, et al. PhenoScanner V2: an expanded tool for searching human genotype-phenotype associations. Bioinformatics. (2019) 35:4851–3. doi: 10.1093/bioinformatics/btz469
22. Palmer TM, Lawlor DA, Harbord RM, Sheehan NA, Tobias JH, Timpson NJ, et al. Using multiple genetic variants as instrumental variables for modifiable risk factors. Stat Methods Med Res. (2012) 21:223–42. doi: 10.1177/0962280210394459
23. Levin MG, Judy R, Gill D, Vujkovic M, Verma SS, Bradford Y, et al. Genetics of height and risk of atrial fibrillation: A Mendelian randomization study. PloS Med. (2020) 17:e1003288. doi: 10.1371/journal.pmed.1003288
24. Gill D, Efstathiadou A, Cawood K, Tzoulaki I, Dehghan A. Education protects against coronary heart disease and stroke independently of cognitive function: evidence from Mendelian randomization. Int J Epidemiol. (2019) 48:1468–77. doi: 10.1093/ije/dyz200
25. Burgess S, Dudbridge F, Thompson SG. Combining information on multiple instrumental variables in Mendelian randomization: comparison of allele score and summarized data methods. Stat Med. (2016) 35:1880–906. doi: 10.1002/sim.6835
26. Bowden J, Davey Smith G, Burgess S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int J Epidemiol. (2015) 44:512–25. doi: 10.1093/ije/dyv080
27. Bowden J, Davey Smith G, Haycock PC, Burgess S. Consistent estimation in mendelian randomization with some invalid instruments using a weighted median estimator. Genet Epidemiol. (2016) 40:304–14. doi: 10.1002/gepi.21965
28. Xue H, Shen X, Pan W. Constrained maximum likelihood-based Mendelian randomization robust to both correlated and uncorrelated pleiotropic effects. Am J Hum Genet. (2021) 108:1251–69. doi: 10.1016/j.ajhg.2021.05.014
29. Torres J, Mehandru S, Colombel JF, Peyrin-Biroulet L. Crohn’s disease. Lancet. (2017) 389:1741–55. doi: 10.1016/S0140-6736(16)31711-1
30. Fu Y, Lee CH, Chi CC. Association of psoriasis with inflammatory bowel disease: A systematic review and meta-analysis. JAMA Dermatol. (2018) 154:1417–23. doi: 10.1001/jamadermatol.2018.3631
31. Li WQ, Han JL, Chan AT, Qureshi AA. Psoriasis, psoriatic arthritis and increased risk of incident Crohn’s disease in US women. Ann Rheum Dis. (2013) 72:1200–5. doi: 10.1136/annrheumdis-2012-202143
32. Teigen LM, Geng Z, Sadowsky MJ, Vaughn BP, Hamilton MJ, Khoruts A. Dietary factors in sulfur metabolism and pathogenesis of ulcerative colitis. Nutrients. (2019) 11. doi: 10.3390/nu11040931
33. O’Neill CA, Monteleone G, McLaughlin JT, Paus R. The gut-skin axis in health and disease: A paradigm with therapeutic implications. Bioessays. (2016) 38:1167–76. doi: 10.1002/bies.201600008
34. Maguire M, Maguire G. The role of microbiota, and probiotics and prebiotics in skin health. Arch Dermatol Res. (2017) 309:411–21. doi: 10.1007/s00403-017-1750-3
35. Olesen CM, Coskun M, Peyrin-Biroulet L, Nielsen OH. Mechanisms behind efficacy of tumor necrosis factor inhibitors in inflammatory bowel diseases. Pharmacol Ther. (2016) 159:110–9. doi: 10.1016/j.pharmthera.2016.01.001
36. Tillack C, Ehmann LM, Friedrich M, Laubender RP, Papay P, Vogelsang H, et al. Anti-TNF antibody-induced psoriasiform skin lesions in patients with inflammatory bowel disease are characterised by interferon-γ-expressing Th1 cells and IL-17A/IL-22-expressing Th17 cells and respond to anti-IL-12/IL-23 antibody treatment. Gut. (2014) 63:567–77. doi: 10.1136/gutjnl-2012-302853
37. Calautti E, Avalle L, Poli V. Psoriasis: A STAT3-centric view. Int J Mol Sci. (2018) 19. doi: 10.3390/ijms19010171
38. Kumar R, Theiss AL, Venuprasad K. RORγt protein modifications and IL-17-mediated inflammation. Trends Immunol. (2021) 42:1037–50. doi: 10.1016/j.it.2021.09.005
39. Bağcı IS, Horváth ON, Ruzicka T, Sárdy M. Bullous pemphigoid. Autoimmun Rev. (2017) 16:445–55. doi: 10.1016/j.autrev.2017.03.010
40. Chen YJ, Wu CY, Lin MW, Chen TJ, Liao KK, Chen YC, et al. Comorbidity profiles among patients with bullous pemphigoid: a nationwide population-based study. Br J Dermatol. (2011) 165:593–9. doi: 10.1111/j.1365-2133.2011.10386.x
41. Ohata C, Ishii N, Koga H, Fukuda S, Tateishi C, Tsuruta D, et al. Coexistence of autoimmune bullous diseases (AIBDs) and psoriasis: A series of 145 cases. J Am Acad Dermatol. (2015) 73:50–5. doi: 10.1016/j.jaad.2015.03.016
42. Zhang X, Mao X, Theresia C, Xu R, Dong J, Pan M, et al. Pemphigus associated with psoriasis vulgaris: A retrospective study of seven patients and a review of the literature. Acta Dermatovenerol Croat. (2018) 26:226–32.
43. Onsun N, Sallahoglu K, Dizman D, Su Ö, Tosuner Z. Bullous pemphigoid during ustekinumab therapy in a psoriatic patient. Eur J Dermatol. (2017) 27:81–2. doi: 10.1684/ejd.2016.2888
44. Ho PH, Tsai TF. Development of bullous pemphigoid during secukinumab treatment for psoriasis. J Dermatol. (2017) 44:e220–e1. doi: 10.1111/1346-8138.13909
45. Dainichi T, Kabashima K. Interaction of psoriasis and bullous diseases. Front Med (Lausanne). (2018) 5:222. doi: 10.3389/fmed.2018.00222
46. Natsumi A, Sugawara K, Yasumizu M, Mizukami Y, Sano S, Morita A, et al. Re-investigating the basement membrane zone of psoriatic epidermal lesions: is laminin-511 a new player in psoriasis pathogenesis? J Histochem Cytochem. (2018) 66:847–62. doi: 10.1369/0022155418782693
47. Maglie R, Hertl M. Pharmacological advances in pemphigoid. Curr Opin Pharmacol. (2019) 46:34–43. doi: 10.1016/j.coph.2018.12.007
48. LeWitt TM, Kundu RV. Vitiligo. JAMA Dermatol. (2021) 157:1136. doi: 10.1001/jamadermatol.2021.1688
49. Wang L, Zhang B. Psoriasis on lesions of vitiligo: A case report. Clin Cosmet Investig Dermatol. (2023) 16:2005–7. doi: 10.2147/CCID.S417290
50. Sandhu K, Kaur I, Kumar B. Psoriasis and vitiligo. J Am Acad Dermatol. (2004) 51:149–50. doi: 10.1016/j.jaad.2003.12.014
51. Sheth VM, Guo Y, Qureshi AA. Comorbidities associated with vitiligo: a ten-year retrospective study. Dermatology. (2013) 227:311–5. doi: 10.1159/000354607
52. Yen H, Chi CC. Association between psoriasis and vitiligo: A systematic review and meta-analysis. Am J Clin Dermatol. (2019) 20:31–40. doi: 10.1007/s40257-018-0394-1
53. Zhu KJ, Lv YM, Yin XY, Wang ZX, Sun LD, He SM, et al. Psoriasis regression analysis of MHC loci identifies shared genetic variants with vitiligo. PloS One. (2011) 6:e23089. doi: 10.1371/journal.pone.0023089
54. Wu J, Zhou M, Wan Y, Xu A. CD8+ T cells from vitiligo perilesional margins induce autologous melanocyte apoptosis. Mol Med Rep. (2013) 7:237–41. doi: 10.3892/mmr.2012.1117
55. Singh RK, Lee KM, Vujkovic-Cvijin I, Ucmak D, Farahnik B, Abrouk M, et al. The role of IL-17 in vitiligo: A review. Autoimmun Rev. (2016) 15:397–404. doi: 10.1016/j.autrev.2016.01.004
56. Kotobuki Y, Tanemura A, Yang L, Itoi S, Wataya-Kaneda M, Murota H, et al. Dysregulation of melanocyte function by Th17-related cytokines: significance of Th17 cell infiltration in autoimmune vitiligo vulgaris. Pigment Cell Melanoma Res. (2012) 25:219–30. doi: 10.1111/j.1755-148X.2011.00945.x
57. Hegazy RA, Fawzy MM, Gawdat HI, Samir N, Rashed LA. T helper 17 and Tregs: a novel proposed mechanism for NB-UVB in vitiligo. Exp Dermatol. (2014) 23:283–6. doi: 10.1111/exd.12369
58. Ragusa F, Fallahi P, Elia G, Rashed LA. Hashimotos’ thyroiditis: Epidemiology, pathogenesis, clinic and therapy. Best Pract Res Clin Endocrinol Metab. (2019) 33:101367. doi: 10.1016/j.beem.2019.101367
59. Fallahi P, Ferrari SM, Ruffilli I, Elia G, Miccoli M, Sedie AD, et al. Increased incidence of autoimmune thyroid disorders in patients with psoriatic arthritis: a longitudinal follow-up study. Immunol Res. (2017) 65:681–6. doi: 10.1007/s12026-017-8900-8
60. Antonelli A, Delle Sedie A, Fallahi P, Ferrari SM, Maccheroni M, Ferrannini E, et al. High prevalence of thyroid autoimmunity and hypothyroidism in patients with psoriatic arthritis. J Rheumatol. (2006) 33:2026–8.
61. Bianchi G, Marchesini G, Zoli M, Falasconi MC, Iervese T, Vecchi F, et al. Thyroid involvement in chronic inflammatory rheumatological disorders. Clin Rheumatol. (1993) 12:479–84. doi: 10.1007/BF02231775
62. Antonelli A, Ferrari SM, Corrado A, Di Domenicantonio A, Fallahi P. Autoimmune thyroid disorders. Autoimmun Rev. (2015) 14:174–80. doi: 10.1016/j.autrev.2014.10.016
63. Antonelli A, Fallahi P, Delle Sedie A, Ferrari SM, Maccheroni M, Bombardieri S, et al. High values of alpha (CXCL10) and beta (CCL2) circulating chemokines in patients with psoriatic arthritis, in presence or absence of autoimmune thyroiditis. Autoimmunity. (2008) 41:537–42. doi: 10.1080/08916930802170401
64. Proost P, Struyf S, Loos T, Gouwy M, Schutyser E, Conings R, et al. Coexpression and interaction of CXCL10 and CD26 in mesenchymal cells by synergising inflammatory cytokines: CXCL8 and CXCL10 are discriminative markers for autoimmune arthropathies. Arthritis Res Ther. (2006) 8:R107. doi: 10.1186/ar1997
65. Lande R, Giacomini E, Serafini B, Rosicarelli B, Sebastiani GD, Minisola G, et al. Characterization and recruitment of plasmacytoid dendritic cells in synovial fluid and tissue of patients with chronic inflammatory arthritis. J Immunol. (2004) 173:2815–24. doi: 10.4049/jimmunol.173.4.2815
66. Kiriakidou M, Ching CL. Systemic lupus erythematosus. Ann Intern Med. (2020) 172:Itc81–itc96. doi: 10.7326/AITC202006020
67. Korkus D, Gazitt T, Cohen AD, Feldhamer I, Lavi I, Haddad A, et al. Increased prevalence of systemic lupus erythematosus comorbidity in patients with psoriatic arthritis: A population-based case-control study. J Rheumatol. (2021) 48:207–13. doi: 10.3899/jrheum.190940
68. Martin JC, Baeten DL, Josien R. Emerging role of IL-17 and Th17 cells in systemic lupus erythematosus. Clin Immunol. (2014) 154:1–12. doi: 10.1016/j.clim.2014.05.004
69. López P, Rodríguez-Carrio J, Caminal-Montero L, Mozo L, Suárez A. A pathogenic IFNα, BLyS and IL-17 axis in Systemic Lupus Erythematosus patients. Sci Rep. (2016) 6:20651. doi: 10.1038/srep20651
70. Qu Y, Li D, Liu W, Shi D. Molecular consideration relevant to the mechanism of the comorbidity between psoriasis and systemic lupus erythematosus (Review). Exp Ther Med. (2023) 26:482. doi: 110.3892/etm.2023.12181
71. Yoshinari H, Nakano K, Nakayamada S, Iwata S, Kubo S, Miyagawa I, et al. 110 Successful treatment of refractory lupus nephritis with secukinumab in a patient complicated with psoriasis vulgaris. Lupus Sci Med. (2017) 4:A47–A8. doi: 10.1136/lupus-2017-000215.110
72. Taurog JD, Chhabra A, Colbert RA. Ankylosing spondylitis and axial spondyloarthritis. N Engl J Med. (2016) 374:2563–74. doi: 10.1056/NEJMra1406182
73. McInnes IB, Schett G. The pathogenesis of rheumatoid arthritis. N Engl J Med. (2011) 365:2205–19. doi: 10.1056/NEJMra1004965
74. Veale DJ, Fearon U. The pathogenesis of psoriatic arthritis. Lancet. (2018) 391:2273–84. doi: 10.1016/S0140-6736(18)30830-4
75. Bridgewood C, Newton D, Bragazzi N, Wittmann M, McGonagle D. Unexpected connections of the IL-23/IL-17 and IL-4/IL-13 cytokine axes in inflammatory arthritis and enthesitis. Semin Immunol. (2021) 58:101520. doi: 10.1016/j.smim.2021.101520
76. Stolwijk C, Essers I, van Tubergen A, Boonen A, Bazelier MT, De Bruin ML, et al. The epidemiology of extra-articular manifestations in ankylosing spondylitis: a population-based matched cohort study. Ann Rheum Dis. (2015) 74:1373–8. doi: 10.1136/annrheumdis-2014-205253
77. Feld J, Chandran V, Haroon N, Inman R, Gladman D. Axial disease in psoriatic arthritis and ankylosing spondylitis: a critical comparison. Nat Rev Rheumatol. (2018) 14:363–71. doi: 10.1038/s41584-018-0006-8
78. Eder L, Chandran V, Gladman DD. What have we learned about genetic susceptibility in psoriasis and psoriatic arthritis? Curr Opin Rheumatol. (2015) 27:91–8. doi: 10.1097/bor.0000000000000136
79. Wordsworth P. Genes in the spondyloarthropathies. Rheum Dis Clin North Am. (1998) 24:845–63. doi: 10.1016/S0889-857X(05)70045-4
80. Brown MA, Kenna T, Wordsworth BP. Genetics of ankylosing spondylitis–insights into pathogenesis. Nat Rev Rheumatol. (2016) 12:81–91. doi: 10.1038/nrrheum.2015.133
81. Lubrano E, Perrotta FM. Secukinumab for ankylosing spondylitis and psoriatic arthritis. Ther Clin Risk Manag. (2016) 12:1587–92. doi: 10.2147/TCRM
82. Genovese MC, Van den Bosch F, Roberson SA, Bojin S, Biagini IM, Ryan P, et al. LY2439821, a humanized anti-interleukin-17 monoclonal antibody, in the treatment of patients with rheumatoid arthritis: A phase I randomized, double-blind, placebo-controlled, proof-of-concept study. Arthritis Rheum. (2010) 62:929–39. doi: 10.1002/art.27334
83. Thorarensen SM, Lu N, Ogdie A, Gelfand JM, Choi HK, Love TJ, et al. Physical trauma recorded in primary care is associated with the onset of psoriatic arthritis among patients with psoriasis. Ann Rheum Dis. (2017) 76:521–5. doi: 10.1136/annrheumdis-2016-209334
84. McGonagle D, Lories RJ, Tan AL, Benjamin M. The concept of a “synovio-entheseal complex” and its implications for understanding joint inflammation and damage in psoriatic arthritis and beyond. Arthritis Rheum. (2007) 56:2482–91. doi: 10.1002/art.22758
85. Fearon U, Veale DJ. Pathogenesis of psoriatic arthritis. Clin Exp Dermatol. (2001) 26:333–7. doi: 10.1046/j.1365-2230.2001.00792.x
86. Murray PI, Rauz S. The eye and inflammatory rheumatic diseases: The eye and rheumatoid arthritis, ankylosing spondylitis, psoriatic arthritis. Best Pract Res Clin Rheumatol. (2016) 30:802–25. doi: 10.1016/j.berh.2016.10.007
87. Rehal B, Modjtahedi BS, Morse LS, Schwab IR, Maibach HI. Ocular psoriasis. J Am Acad Dermatol. (2011) 65:1202–12. doi: 10.1016/j.jaad.2010.10.032
88. Rosenbaum JT. Uveitis in spondyloarthritis including psoriatic arthritis, ankylosing spondylitis, and inflammatory bowel disease. Clin Rheumatol. (2015) 34:999–1002. doi: 10.1007/s10067-015-2960-8
89. Kim BR, Choi SW, Choi CW, Lee KH, Kim MJ, Woo SJ, et al. Risk of uveitis in patients with psoriasis in Korea: A nationwide population-based cohort study. J Eur Acad Dermatol Venereol. (2023) 37:1336–43. doi: 10.1111/jdv.19060
Keywords: psoriasis, psoriatic arthritis, autoimmune diseases, Mendelian randomization analysis, genome-wide association studies, single nucleotide
Citation: Duan K, Wang J, Chen S, Chen T, Wang J, Wang S and Chen X (2024) Causal associations between both psoriasis and psoriatic arthritis and multiple autoimmune diseases: a bidirectional two-sample Mendelian randomization study. Front. Immunol. 15:1422626. doi: 10.3389/fimmu.2024.1422626
Received: 24 April 2024; Accepted: 09 July 2024;
Published: 25 July 2024.
Edited by:
Monica Neagu, Victor Babes National Institute of Pathology (INCDVB), RomaniaReviewed by:
Richard Borrelli, University of Turin, ItalyTakemichi Fukasawa, The University of Tokyo Graduate School of Medicine, Japan
Copyright © 2024 Duan, Wang, Chen, Chen, Wang, Wang and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xinsheng Chen, eGluc2hlbmdjaGVuQHNvaHUuY29t
†These authors have contributed equally to this work and share first authorship