 Danbao Ma
Danbao Ma Yun Feng
Yun Feng Xiang Lin
Xiang Lin- 1School of Chinese Medicine, The University of Hong Kong, Hong Kong, Hong Kong SAR, China
- 2Department of Ophthalmology, Peking University Third Hospital, Beijing, China
- 3Department of Chinese Medicine, the University of Hong Kong-Shenzhen Hospital (HKU-SZH), Shenzhen, China
Sjögren’s syndrome (SS) or Sjögren’s disease (SjD) is a systemic autoimmune disease clinically manifested as sicca symptoms. This disease primarily impacts the functionality of exocrine glands, specifically the lacrimal and salivary glands (SG). SG fibrosis, an irreversible morphological change, is a severe consequence that occurs in the later stages of the disease due to sustained inflammation. However, the mechanism underlying SG fibrosis in SS remains under-investigated. Glandular fibrosis may arise from chronic sialadenitis, in which the interactions between infiltrating lymphocytes and epithelial cells potentially contributes to fibrotic pathogenesis. Thus, both immune and non-immune cells are closely involved in this process, while their interplays are not fully understood. The molecular mechanism of tissue fibrosis is partly associated with an imbalance of immune responses, in which the transforming growth factor-beta (TGF-β)-dependent epithelial-mesenchymal transition (EMT) and extracellular matrix remodeling are recently investigated. In addition, viral infection has been implicated in the pathogenesis of SS. Viral-specific innate immune response could exacerbate the autoimmune progression, resulting in overt inflammation in SG. Notably, post-COVID patients exhibit typical SS symptoms and severe inflammatory sialadenitis, which are positively correlated with SG damage. In this review, we discuss the immune and non-immune risk factors in SG fibrosis and summarize the evidence to understand the mechanisms upon autoimmune progression in SS.
1 Introduction
Sjögren’s syndrome (SS) or Sjögren’s disease (SjD) is an autoimmune disease that primarily causes dryness of the exocrine glands, in which the salivary and lacrimal glands are predominantly affected. Consistent with other autoimmune diseases, SS displays an unbalanced gender ratio, with a male-to-female ratio of approximating 1:20 according to the latest research, and predominantly occurs in middle-aged women (1). Primary SS (pSS) is defined as the occurrence of SS independently, without the concurrent presence of other rheumatic diseases (2). In contrast, secondary SS is consistently associated with other autoimmune complications, such as rheumatoid arthritis (RA), and systemic lupus erythematosus (SLE), and multiple sclerosis. The disease typically manifests with sicca symptoms, including reduced salivary volume, and systemic manifestations including fatigue, joint, and muscle pain. In later stages, SS can progress to B-cell lymphoma, which is one of the leading causes of disease-associated mortality. Other mortality risks include older age, male gender, vasculitis, cryoglobulinemia and interstitial lung disease (ILD) (3). SS was initially recognized as autoimmune sialadenitis, due to the characteristic infiltration of massive lymphocytes in the salivary gland (SG) and the subsequent development of localized chronic inflammation. Immune activation in the epithelium and the accumulation of lymphocytic infiltrates result in the formation of focal lymphocytic sialadenitis (FLS) within the SG. The identification of Sjögren’s lymphocytic sialadenitis serves as the gold standard for the assessment of SS (4). In patients with SS, aberrant T cell and B cell responses to autoantigens not only lead to the overproduction of inflammatory cytokines and pathogenic autoantibodies but also drive chronic inflammation in the epithelium of the exocrine glands. Furthermore, prolonged inflammation may result in irreversible damage or even more severe organ damage to the SG, such as atrophy and fibrosis.
Fibrosis is a common feature of connective tissue diseases associated with autoimmune disorders. Tissue fibrosis represents a remodeling process of the extracellular matrix in response to injury from acute or chronic stimulation. This involves the replacement of healthy parenchymal tissue with fibrous connective tissue. In the absence of disease, the accumulation of collagen fibers leads to tissue hardening and scarring. However, excessive deposition can initiate the loss of tissue architecture, which may ultimately result in organ dysfunction and failure (5). Non-specific chronic sialadenitis (NSCS) may result in pathological fibrosis in the SG of patients with SS and is likely to be associated with focal lymphocyte infiltration (6). The prevailing hypothesis is that chronic inflammation is the primary driver of local damage and the progressive fibrotic transformation of tissue. Fibrosis has been investigated in several organs in patients with SS, including the lungs, livers, and kidneys, but less is understood about fibrosis in the SG. In fact, this organic lesion results in permanent destruction of the SG, causing significant distress to the patient and necessitating appropriate attention. This pathological feature tends to worsen with age and is difficult to reverse, thus severely impacting the quality of life for the elderly with dental damage and reduced salivary flow (7–9). Early evidence has documented the age-related fibrosis of the SG in patients with SS (10).
Indeed, age is associated with reduced acini, ductal dilation, fatty infiltration and fibrosis (11), but it is not the only factor that contributes to SG fibrosis. As a common consequence of tissue injury and inflammation, fibrosis has been found to be positively correlated with the focus score in minor SG of patients with SS (12). Consequently, the presence of focal lymphocytes plays an important role in the context of fibrosis. Pro-inflammatory cytokines and chemokines, including type I interferon (IFN), tumor necrosis factor alpha (TNFα), transforming growth factor-beta (TGF-β), C-X-C motif chemokine ligand 10 (CXCL10), CXCL13, chemokine (C-C motif) ligand 19 (CCL19) and CCL21, are highly expressed in SGs and are responsible for recruiting immune cells. Focal accumulations of lymphocytes, macrophages and plasma cells typically form fibrosis-related ectopic lymphoid structures (ELS) in SG, supporting the activity of autoreactive plasma cells and the persistent inflammatory stimulation in SGs. Collectively, these immune infiltrates contribute to the fibrotic pathology.
The pathogenicity of immune cells, either directly or indirectly involved in SG fibrosis, remains incompletely understood, while recent evidence has begun to shed light on the contributions of various immune cell populations to SG fibrosis. While chronic inflammation in SS does not inevitably result in fibrosis, once this morphological change has occurred, it is irreversible. Currently, there is no effective therapeutic approach available, largely due to a lack of acknowledgement of the underlying fibrotic mechanisms. This review will discuss the pro-fibrotic role of both immune and non-immune cells, as well as the underlying mechanisms of SG fibrosis in the context of autoimmune progression and infection.
2 The pro-fibrotic role of immune cells in salivary gland of pSS
2.1 T cells
A variety of immune cells are observed to infiltrate the SG, including CD4+ T cells, CD8+ T cells, B cells, macrophages, dendritic cells (DCs), and mast cells. Additionally, non-immune cells such as fibroblasts are also present. Self-reactive immune cells respond to glandular-specific antigens, resulting in the onset of inflammation and subsequent damage to the SG, which ultimately leads to the development of fibrosis (Figure 1). In the initial stages of fibrosis, there is an observable expansion of T cells, particularly CD4+ T cells, in dysfunctional glands. Upon examination of histopathological specimens of SG, it was found that clonally expanded CD4+ T cells from memory T cells were found to be positively correlated with the grade of fibrotic damage (13). A single-cell analysis approach was employed by researchers, who analyzed data and discovered that the proliferation of CD4+ T cells in inflammatory lesions was specifically driven by SG autoantigens from unrelated SS individuals. Moreover, two distinct functional T cell receptor α (TCRα) chains were identified in these hyperactive SG CD4+ T cells, in contrast to those observed in peripheral blood of patients with SS. Given that dual TCR-expressing cells are typically present in autoimmune diseases (14), the enrichment of overactive CD4+ T cells implicates a heightened autoimmune response in the SG and increased severity of SS. Further investigation is warranted into the role of CD4+ T cell subsets in SG fibrosis. It is noteworthy that the majority of local resident T cells within inflammatory site express the CD45RO memory marker, as well as effector hallmarks such as the IL-2 receptor (15). The activation of memory T cells and the proliferation of autoreactive CD4+ T cells together indicate that the process is antigen-driven in situ. Indeed, the abundance of antigen-experienced CD4+CD45RA− T cells have been demonstrated to have a positive correlation with the levels of serum autoantibodies Ro, biopsy focus score and the SG fibrosis (16). The study analyzed the gene expression profile of these CD4+CD45RA- memory T cells and characterized their distinct T follicular helper (Tfh) signature in the SG of pSS cases, using two independent gene set enrichment analysis (GSEA) approaches. Notably, not only do CD4+ memory T cells play a role, but the absolute number of CD8+ memory T cells also correlates with fibrotic morphology, suggesting the complexity of infiltrates in inflamed salivary tissues.
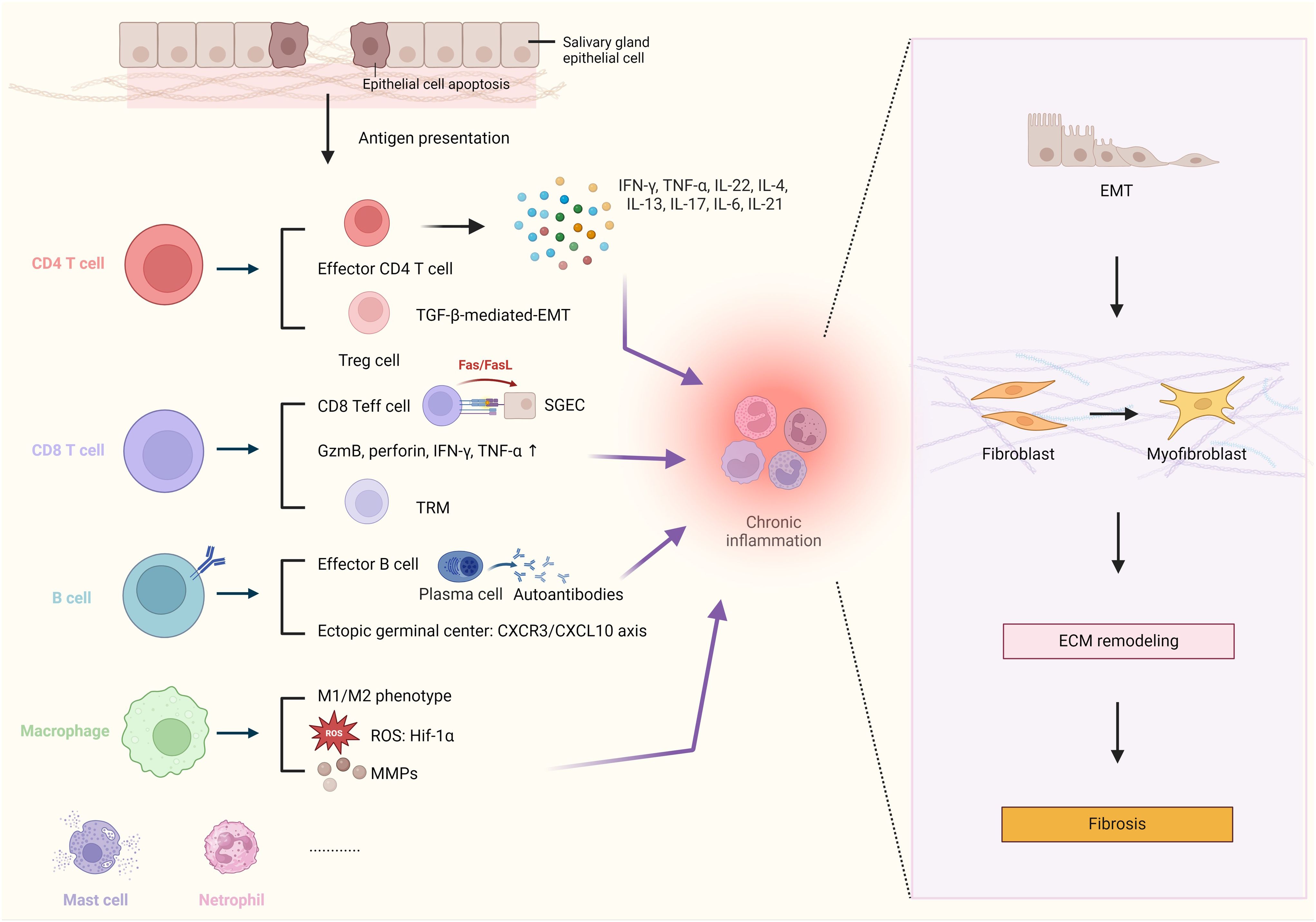
Figure 1. Schematic Illustration of Immune Mediators Involved in Modulation of Fibrosis Pathogenesis in Salivary Glands. Chronic inflammation, caused by various types of immune cells, plays a crucial role in the process of salivary gland (SG) fibrosis, driving the epithelial-mesenchymal transition (EMT) program, myofibroblast differentiation, and extracellular matrix (ECM) remodeling. In patients with Sjögren’s Syndrome, the salivary epithelium is often damaged, and numerous epithelial cells undergo apoptosis. The autoimmune reaction in situ is initiated by autoantigens released by apoptotic salivary gland epithelial cells (SGECs). The chronic inflammation in the SG is primarily caused by pro-inflammatory cytokines from effector T cells, such as interferon-gamma (IFN-γ), tumor necrosis factor-alpha (TNF-α), interleukin-21 (IL-21), and interleukin-6 (IL-6). Transforming growth factor-beta (TGF-β), secreted by regulatory T cells (Treg cells), is the key regulator of EMT-driven fibrosis. CD8+ T cells can exacerbate tissue damage by inducing SGECs apoptosis in contact manner, via the Fas/Fas ligand (FasL) pathway and other effector molecules e.g granzyme B, perforin, IFN-γ, and TNF-α. B cells could differentiate into plasma cells and participate in the formation of ectopic germinal centers via the C-X-C motif chemokine receptor 3/chemokine ligand 10 (CXCR3/CXCL10) axis. Macrophages contribute to fibrosis by producing matrix metalloproteinases (MMPs), associated with enhanced the expression of the reactive oxygen species (ROS)-related gene hypoxia-inducible factor 1-alpha (Hif-1α). Created by BioRender.com.
2.1.1 Th1 cells
The exact involvement of Th1 cells in the pathogenesis of SG fibrosis is not well understood, as there have been only a limited number of studies addressing this issue. Th1 cells represent the predominant population responsible for secreting IFN-γ and IL-2. IFN-γ functions as an anti-fibrotic cytokine in idiopathic pulmonary fibrosis (IPF) (17). The prevalence of Th2-type response over Th1 may be indicative of the severity of IPF. In general, Th1 cells play an anti-fibrotic role in organ fibrosis, including lung, kidney, and liver (18). However, this subset may also exhibit opposite functions in SS, although downregulation of IFN-γ can promote the occurrence of autoimmune diseases (19–21). Elevated Th1 population versus Th2 has been observed in SS (22, 23). In contrast to anti-fibrotic effects of IFN-γ, it has been proposed that a distinct Th1 signature is associated with the severity of SS (24). The imbalance of Th1/Th2 in the pathogenesis of SS has long garnered interest. Th1 cells have been shown to highly express STAT-4 and T-bet, in the local SG of SS patients compared to non-SS individuals (25). Th1-associated IFN-γ act as anti-fibrotic agents due to their inhibitory effect on fibroblast and collagen accumulation. Conversely, they induce proinflammatory macrophages and promote local inflammation in the SG (26, 27). Interactions with these macrophages further give rise to Th1 cell response and the production of additional pro-inflammatory cytokines, which may indirectly contribute to chronic inflammation and SG fibrosis. The overproduction of IFN-γ promotes CXCL10 expression, which in turn facilitates the recruitment of immune cells to the site of inflammation by binding to the receptor CXCR3 (28, 29). CXCL10 and other CXCR3 ligands can attract B cells into the inflamed epithelium (30, 31). The accumulation of inflamed tissue might progress into fibrosis in SG.
IFN-γ is able to activate JAK1/2-induced phosphorylation of STAT1 by binding to IFN-γ receptor-1 and -2 (32). It is interesting that IFN-γ can induce ferroptosis of salivary gland epithelial cells (SGECs) via JAK/STAT1 signaling and contribute to the pathogenicity of SS (33). Inhibition of IFN-γ signaling by using the IFN-γ monoclonal antibody (XMG1.2) in non-obese diabetic (NOD) mice can induce glutathione (GSH) and glutathione peroxidase 4 (GPX4), and relieve the SGEC ferroptosis and progression of SS (33). This provides evidence for the controversial functions of IFN-γ in the pathogenesis of SS. The large amount of the programmed death of SGEC such as ferroptosis may ultimately lead to SG fibrosis.
2.1.2 Th2 cells
Active Th2 transcripts predominantly appear in the later stages of SS (34). A distinctive Th2 profile is associated with the formation of germinal centers and marked B cell infiltration in the SG (24). The activation of GATA-3 and STAT-6 regulates the polarization of naive T cells into the Th2 phenotype, subsequently initiating the production of Th2 cytokines, including IL-4, IL-5, IL-10, and IL-13 (23). Some of these cytokines are capable of inducing fibrosis in tissues (35, 36). The role of type 2 response-dependent fibrosis in tissue repair has been recently elucidated (36). Pathogens, including parasites, helminths, and allergens, can provoke type 2 inflammation to support tissue repair and regeneration (36). However, excessive type 2 immunity may lead to the development of allergic diseases and pathological fibrosis (36, 37). Type 2 cytokines indirectly contribute to fibrotic pathology by interacting with associated immune cells. IL-4 and IL-13 activate tissue-repair macrophages and initiate tissue regeneration (36). These tissue-resident macrophages play a pivotal role in pathological fibrosis following injury, as they secrete cytokines such as TGF-β1, which induce the differentiation of fibroblasts into myofibroblasts (37). The activation of myofibroblast further facilitates the synthesis of extracellular matrix (ECM) (38). Local fibroblasts may be influenced by tissue-specific macrophages in the context of a chronic infection in SG, with the mediation of the fibrotic cascade occurring under the regulation of Th2 cytokines (36, 37, 39, 40). Additionally, fibroblasts can also respond directly to IL-4 and IL-13 signaling, which stimulates their transformation into myofibroblasts (36, 41). This process consequently increases the deposition of collagen and α-smooth muscle actin (α-SMA), thereby supporting wound healing and subsequent fibrosis (36). In recent studies, it has been demonstrated that IL-13 and IL-22 act in concert to prime and stimulate fibroblasts (42). IL-22 originates from a variety of sources, including lymphocytes and non-lymphocytes (43). However, its production by Th2 cells is rarely documented. Both IL-13 and IL-22 facilitate the proliferation of SG-infiltrated fibroblasts, thereby promoting the establishment of tertiary lymphoid structures (TLS) and the expansion of the fibroblastic network (43–47). Notably, the early priming of resident fibroblasts is regulated by IL-13. Together, type 2 cytokines are involved in SG fibrosis through multiple mechanisms.
2.1.3 Treg cells
Treg cells, a subset of T cells, play an essential role in maintaining immune tolerance. They are of great importance for regulating T cell activity and maintaining immune homeostasis as well as preventing autoimmune responses (48). The differentiation of naïve CD4 T cells into Treg cells requires IL-2 and TGF-β (49). Conversely, TGF-β could be produced by Treg cells. Although TGF-β signaling is indispensable for immunosuppression, it also functions in various other physiological processes. Treg-derived TGF-β, particularly TGF-β1, is a key pro-fibrotic cytokine and serves as the primary driver of epithelial-mesenchymal transition (EMT)-dependent fibrosis in most chronic inflammatory diseases (50) (Figure 1). The phenomenon of inflammatory EMT has been observed during the process of wound healing and pathological fibrosis of the SG (51). In patients with primary SS, TGF-β1 contributes to SG fibrosis via the canonical TGF-β1/SMAD/Snail signaling pathway (52). Snail is a downstream transcriptional factor of phosphorylated SMAD 2/3 that promotes the expression of genes associated with EMT. In the context of an inflamed SG (52), TGF-β1 primarily originates from local SGECs, Treg cells, and macrophages. In TGF-β1-exposed SGECs, intracellular signaling cascades result in SMAD 2/3 phosphorylation and further induce EMT-related gene expression, simultaneously leading to the activation of myofibroblasts and ECM reshaping (53). The expression of epithelial markers, such as E-cadherin, on SGECs is down-regulated, while the expression of mesenchymal markers, such as vimentin and type I collagen, is significantly increased. SG inflammation is positively correlated with the acquisition of mesenchymal properties (51). TGF-β1-treated healthy SGECs showed altered morphology with loosened cell-cell adhesion and an elongated shape (54). The effect of TGF-β1 signaling in SG fibrosis may be manifested by the transition of SGECs into mesenchymal cells. These resident mesenchymal cells specifically exhibit a myofibroblast-like phenotype that is associated with progressive SG fibrosis.
Given that TGF-β1 is a vital regulator in SG fibrosis, local targeting TGF-β1 may offer a promising avenue for the treatment of SG fibrosis. The TGF-β1 inhibitor SB-431542 demonstrated significant inhibitory effects on the expression of fibrosis phenotypic markers vimentin and type I collagen in SGECs (52). It has been revealed that the level of SG inflammation is highly connected to EMT-dependent fibrosis (54–56). With regard to the TGF-β-mediated EMT pathway, several studies have mentioned that the pro-inflammatory cytokines IL-6, IL-17, and IL-22, which share this pathway, are also involved in the fibrotic mechanism of SG (53, 55, 57). In addition, TGF-β/IL-17-induced EMT can trigger the fibrotic program of SG via the canonical SMAD pathway or the non-canonical ERK-mediated pathway (53). During EMT, IL-17 stimulates the phosphorylation of SMAD 2/3 and Erk1/2 in SGECs (53). The subsequent section will discuss the functions of IL-17 in greater depth. Nevertheless, the impact of the imbalance between Treg cells and Th17 cells on the SG microenvironment and its role in fibrotic pathogenesis is necessary to be further explored. In the presence of TGF-β and the absence of IL-6, the Treg phenotype is more encouraged than the Th17 phenotype (58). However, it is possible for Treg cells to transdifferentiate into Th17 cells in the presence of pro-inflammatory factors (59). Intriguingly, upon IL-2 stimulation, there is a lower frequency of Treg cells with STAT5 phosphorylation in the peripheral blood of patients with SS (60). STAT5 is a signal transducer that modulates the stabilization of Treg cells. The malfunction and imbalance of Treg cells in SS may be correlated with the severity of the disease. Collectively, Treg cells have the capacity to suppress the development of autoimmune disease as immune suppressors, yet they also produce TGF-β to support fibrotic pathology. Controversially to decreased Treg numbers in autoimmune mouse models, the elevated levels of TGF-β in the local SG indicate the cellular source of TGF-β other than Treg cells, including SGECs and macrophages. However, a study has reported transcriptomic analysis in SG of patients with pSS, which may enhance the Treg profile with a reduced Th17 signature (16). Therefore, the complex interplay between resident Treg cells and SGECs in the context of autoimmune disorders requires further investigation to elucidate, with a particular focus on SG fibrosis.
2.1.4 Th17 cells
Extensive investigation has revealed a correlation between Th17 cells and autoimmune disorders (61, 62). In the combination of IL-6 or IL-21, TGF-β derived from DCs serves as a trigger for Th17 differentiation (63, 64). Indeed, local expression of IL-6, IL-17, and IL-23 increases with the severity of inflammatory lesions in the minor SG of pSS patients (65). Pro-inflammatory IL-17A is the signature cytokine secreted by Th17 cells. Although IL-17 is important in the immune response to extracellular pathogens in mucosal tissues, it induces the production of other inflammatory cytokines and related chemokines that critically contribute to the pathogenesis of autoimmune diseases including pSS. Increased IL-17 mRNA levels have been detected in the SG of patients with SS, concomitant with a reduction in saliva flow rate (65). An augmentation of salivary IL-17 was also detected in SG from mice with experimental Sjögren’s syndrome (ESS) (66). IL-17 knockout mice immunized with SG proteins did not develop SS (64). The pivotal function of IL-17 in SG inflammation has been extensively studied. However, little is known regarding its effect on SG fibrosis.
Early studies have reported that IL-17 and IL-22 are engaged in the process of EMT (53). Mesenchymal signatures replace the epithelial features during EMT-dependent SG fibrosis in SGECs under the control of IL-17 and IL-22 (53). SGECs treated with IL-17 and IL-22 showed a reduction in cell-cell contacts and adopted a more stretched morphological structure. Furthermore, the levels of IL-17 and IL-22 are associated with focus scores in patients with pSS (67). These findings provide evidence for the importance of IL-17 and IL-22 in the pathogenetic fibrosis of SG. As previously described, IL-17 modulates EMT of epithelial cells depending on TGF-β1 via the SMAD pathway or the noncanonical TGF-β1/Erk1/2/EMT pathway. Among the various subsets of IL-17, IL-17A is known to activate the myofibroblast phenotype that is the initial step in the fibrotic process (68). Proposed treatments for pSS have included those targeting Th17 cells. A proteasome inhibitor bortezomib (BTZ), which targets low-molecular-weight protein 7 (LMP7), demonstrated a notable reduction in IL-17+ Th17 cells in ESS mice (69). The monoclonal antibody rituximab has been confirmed to be effective in decreasing the levels of IL-17 (70). Unfortunately, there is a paucity of research focusing on the direct treatment of SG fibrosis via the targeting of Th17 cells and related cytokines.
Similar to IL-17, IL-22 secreted by Th17 cells is also initiated by TGF-β1 with the presence of IL-6 and other cytokines (71). With respect to the function of IL-22 derived from Th17 cells in fibrosis, it was found that this cytokine induces the accumulation of TGF-β1, α-SMA and collagen that favor liver fibrosis (2). IL-22 primarily functions on stromal cells and epithelial cells, playing an important role in tissue regeneration. More importantly, IL-22 also participates in the formation of TLS through the activation of autoreactive B cells (72). IL-22 upregulates the expression of CXCL13 in stromal fibroblasts and CXCL12 in epithelial cells (43). These chemokines promote the B cell aggregation and autoantibody production, which in turn facilitates the TLS assembly. These findings render IL-22 noticeable in SG fibrosis. In addition to IL-17 and IL-22, Th17 cells simultaneously stimulate the release of IL-6, TNF and matrix metalloproteinase 9 (MMP9) in the inflamed SG, exacerbating the local damage and leading to SG fibrosis (73, 74). Moreover, Th17 cells differentiate into a Th17.1 subpopulation that produces IL-17 and IFN-γ or a Th1-like subset, thereby rendering Th17 cells more responsible for the chronic inflammation and fibrosis of SG (75).
2.1.5 Tfh cells
The Tfh subset helps B cell migration to germinal centers and the formation of TLS, which may indirectly support the development of fibrotic structures within the SG (16, 76). In patients with pSS, accumulated data shows that the frequency of Tfh cells in the peripheral blood and in SG is positively linked to disease progression (61, 77). Tfh cells are characterized by the expression of CD40L, CXCR5, PD-1, and inducible co-stimulator (ICOS) (78). An increase in circulating Tfh cells has been observed in patients with IPF associated with SS (16, 79). The pathogenesis of pSS posits that CD4+T cells largely differentiate into Tfh cells to augment the number of autoreactive B cells and encourage the generation of ectopic germinal centers (76). The emergence of these germinal centers indicates a significant immune cell infiltration and underlies SG fibrosis. Therefore, further investigation is warranted to elucidate the pro-fibrotic role of Tfh cells in SG. IL-21 is a Tfh-associated cytokine that has been proven to enhance the expression of IL-4Rα and IL-13Rα on macrophages (40). This may serve to reinforce the pro-fibrotic activity of macrophages. In patients with systemic sclerosis (SSc), skin fibrosis could be effectively controlled under the treatment with anti-IL-21 and anti-ICOS antibodies (80, 81).
The blockade of ICOS in SG results in a reduction of both pro-inflammatory IL-6 and TNF-α (82). IL-6 provides essential Tfh-trophic signals and induces IL-21 production (83). In a study by Pontarini et al. (2020), it was demonstrated that IFN-γ is produced by PD1+ICOS+Tfh-like cells, apart from Th1 cells in patients with pSS (82). The indirect effect of circulating Tfh cells in SG fibrosis can be reflected by the secretion of IL-17, given that IL-17 is highly correlated with SG fibrosis (84, 85). These Tfh-like cells were characterized by CCR9+CD4+ and CXCR5+CD4+ phenotype (86). CCR9+ Tfh cells specifically express Bcl-6, ICOS and other inflammatory mediators, including IL-7, IL-21, and IFN-γ (86). In ectopic germinal centers, CXCR5highCCR7low Tfh cells are likely to migrate to the CXCL13-enriched B-cell follicles, thereby favoring the expansion of autoreactive B cells (86–88). This specific population also shows the ability to regulate the transition from fibroblast to myofibroblast (86). The principal functions of Tfh cells in SG pathological inflammation are manifested in the maintenance of B cell hyperactivity and the induction of memory B cells via IL-21, CD40L, PD-1, Bcl-6 and BAFF (78, 89). This may also suggest their potential involvement in SG fibrosis. T follicular regulatory (Tfr) cell represents a subtype of Treg cells that inhibit the activity of B cells and Tfh cells (78). Recent investigations have also indicated that the ratio of Tfr/Tfh cells may be closely relevant to the severity of pSS through regulation of the formation of ELS (90).
2.1.6 CD8+T cells
Although the majority of the infiltrated T cells in SG of SS patients are CD4 positive, the frequency of CD4+ T cells is decreased along with the progression of the disease (16). Meanwhile, the number of CD8+ T cells was identified to be either stable or even slightly elevated in severe tissue damage (16). This suggests that CD8+ T cells may play a role in the inflammatory process of pSS. Indeed, the SG of SS patients displays overactivity of CD8+ T cells (91). CD8+ T cells are known to exert cytotoxic functions, which eliminate the infection via the Fas/Fas ligand (FasL) pathway, leading to the large-scale cell apoptosis and tissue damage (Figure 1), including acini injury (91). In the vicinity of apoptotic epithelial acinar cells, CD8+ T cells accumulate in an aberrant manner and release cytotoxic-related molecules, including IFN-γ, TNF-α, perforin, and granzyme B (92). The recruitment of activated CD8+ T cells may owe to CXCR3-mediated chemotaxis, which drives the CD8+T cells’ migration into the inflamed SG in patients with pSS (92).
CD8+ T cells with tissue-resident memory (TRM) signature have been recently highlighted in the contribution to tissue fibrosis in aged mice (93). Single-cell transcriptomics of SG-infiltrated and peripheral blood immune cells have depicted that CD8+CD9+ T cells constitute the tissue-resident memory population infiltrated in SG biopsies of pSS patients (94). In both ESS mouse models and patients with pSS, tissue-specific CD8+CD103+CD69+ TRM was identified as being involved in sialadenitis, with a significant increase in IFN-γ production, in which CD69 and CD103 are the tissue residence markers (95). This TRM population was found to be remarkably expanded and activated in the local SG of ESS mice and patients with pSS (95). When performing anti-CD103 treatment in experimental mice, the collected data showed the restoration of SG functions and a reduction in immune cell aggregations, as evidenced by the focus score (P<0.01) (95). Concomitantly, the release of CD8-related cytokines was also significantly reduced. Interestingly, the results of immunohistochemistry staining presented a negative correlation between TRM proliferation and the expression of epithelial phenotypic markers, such as E-cadherin. This finding may indicate a close association between CD8+-specific TRM and SG fibrosis.
Recent studies by Gao et al. used P40-/-CD25-/-mice as a murine model of SS to investigate the contribution of CD8+ T cells (96, 97). P40 chain is an essential component of IL-12 and IL-23 (98). Both of them are essential for maintaining the equilibrium between Th1/Th17 (98–100). IL-12 is engaged in the Th1 response, while IL-23 is responsible for the Th17 response. P40-/-CD25-/-mice were identified as being able to develop SS and exhibiting distinct lymphocytic foci, atrophy and fibrosis in SG (96). In this mouse model, CD8+ T cells are the predominant infiltrating lymphocytes in the inflammatory site, and are accompanied by TRM features and high IFN-γ production (96). CD8 knockout mice exhibited a significant reduction in the severity of the inflammatory lesion in SG including fibrosis (96). Consequently, it can be postulated that this CD8+ TRM subpopulation may be responsible for fibrotic morphological change in P40-/-CD25-/-mice. However, this finding may be limited to the specific mouse model, given the fundamental functions of P40 and CD25 in immune tolerance. The absence of CD4+ T lymphocytes in mice is an atypical phenomenon and cannot mimic the real situation in humans.
A recent breakthrough in research has revealed that the single-cell gene expression profile of granzyme K+ (GZMK+) CXCR6+CD8+ T cells in peripheral blood is comparable to that of CD69+CD103–CD8+ TRM in SG of pSS (101). The circulating GZMK+ CXCR6+CD8+ T cells, particularly those with high expression of CD122, may be the reservoir of CD8+ TRM subpopulation in tissue (101). IL-15 is a key factor in the differentiation of CD8+ T cells into GZMK+ CXCR6+CD8+ T cells. Importantly, with respect to CD8+CD69+ TRM, the CD103- subset appears to be more pertinent to chronic inflammation of SG due to its elevated level of GZMK, in comparison to the CD103+ subset (101). Moreover, the CD103- subset exhibits heightened sensitivity to T cell receptor signaling and is capable of producing greater quantities of pro-inflammatory cytokines such as IFN-γ and TNF-α, which are associated with the severity of inflammation in SG as well as fibrosis.
2.2 B cells
Numerous studies identified autoreactive B cells as important participants in the pathogenetic process in SS. Based on the current knowledge, they are predominantly involved in the later stage of the disease. Abnormal B cell response to autoantigens Ro/SSA and La/SSB renders B cells differentiation into plasma cells and produces excessive autoantibodies (102). The accumulation of these autoimmune-related antibodies further advances chronic inflammation in the epithelium of the exocrine glands, which may outcome glandular fibrosis (103, 104). The formation of lymphoepithelial lesions (LELs) is initiated by T cell infiltration in the ductal epithelium (105). The majority of B cells in duct epithelium of SG appear after T cells and are derived from the recruitment of T helper cells. Attracted B cells are present in approximately 60% of SG in pSS patients (105). However, in contrast to T cells that exist in almost all ducts of SG in both pSS and non-SS patients, the B cells that infiltrate the ducts with LELs are more specific to this particular type of lesion (105). The diverse functions of B cell subsets have been acknowledged in the context of autoimmune pathogenesis (106). Epithelium-associated Fc Receptor-Like 4 (FcRL4)+ B cells have been identified as a pathogenic cell type that contributes to the development of pSS, with an increased secretion of pro-inflammatory factors that support the formation of LELs, such as IL-6 (107). Age-associated CD11c+ B cells have also been observed to produce SS-related cytokines, including IL-1β, IFN-γ and IL-10 (106). Marginal zone B cells (MZ B) have been found to accumulate in SGs and are linked to the production of autoantibodies that cause epithelial destruction (108, 109). The autoreactive BCR may be associated with the MZ phenotype (110). In addition, multiple regulatory mechanisms have been proposed to explain the migration and infiltration of B cells into inflamed glands. CXCR3 and CXCL10 attract B cells into an ectopic germinal center (28, 29) (Figure 1). Ectopic germinal center is the local center for the activation and maturation of autoreactive B cells. A substantial number of immune cells within ectopic germinal center contributes to SG chronic inflammation and is considered a defining feature of SG fibrosis (81). In patients with SS, serum levels of IL-22 are closely associated with saliva flow and autoantibody production (44). IL-22 can activate the CXCL13 in stromal fibroblasts, and subsequently facilitate the ELS assembly via the CXCR5/CXCL13 axis (43). As Tfh cells mainly express the CXCR5, IL-22 indirectly influences the regulation of T cell-dependent B cell hyperactivity by inducing CXCR5+ Tfh cell migration to B cell follicles (43, 44). Tfh cells critically support B cell survival, maturation, and the aggregation of memory B cells, mediated by CD40L, IL-21, and BAFF (102, 111). Similarly, IL-17 has been shown to orchestrate the establishment of the autoreactive germinal center in autoimmune mouse models (112). IL-17, when present alone or in conjunction with BAFF, can support the expansion of autoreactive B cells (113). Collectively, several factors involved in the interaction of B cells and ectopic germinal centers simultaneously promote the development of chronic inflammation-dependent fibrosis in SG.
Notably, regulatory B cells (Breg) are defined as immune suppressors of autoimmune diseases in an IL-10-dependent manner (114). The role of Breg cells in the development of sialoadenitis fibrosis remains largely unclear. Breg cells are capable of inhibiting the trophic signals of Th1 and Th17 responses which are indispensable in the pathogenesis of autoimmunity (115). Bregs also stimulate the expansion of Treg cells and suppress inflammation (114). More importantly, IL-10-producing Breg cells have been identified as markedly repressing the Tfh response by stimulating the STAT5 phosphorylation, either in patients with pSS or ESS in mice (115). Together, Breg cells are able to effectively regulate the development of inflammation and germinal center formation by inhibiting Th1, Th17 and Tfh responses, thus exerting an anti-fibrotic effect. However, they are also capable of promoting Treg proliferation, which may lead to upregulation of the expression of the pro-fibrotic factor TGF-β. More research is needed to evaluate the function of Breg cells in SG fibrosis.
2.3 Macrophages
Macrophages are a critical cell type in the programming of tissue repair and fibrosis. They play a dual role in the initiation of the repair process and the maintenance of ECM within the tissue (116). In response to tissue injury, macrophages differentiate into versatile phenotypes, including tissue resident macrophages, which are responsible for wound healing (39). Systemic repair signals instruct macrophages to rebuild tissue with normal architectural structure (37). Nevertheless, prolonged tissue damage can result in abnormalities of macrophages, which may lead to irreversible morphological changes and pathological fibrosis. Macrophages have the plasticity to transition between M1 and M2 phenotypes, contingent upon the prevailing immune milieu. For instance, IL-4Rα signaling stimulates the M2 phenotype that determines liver inflammation and fibrosis in SSc (81, 117). Tissue injury frequently drives the polarization of macrophages into the pro-inflammatory M2 phenotype, which is characterized by the release of large amounts of cytokines, such as IFN-γ, TNF-α, IL-4, IL-13, IL-10 and TGF-β (36, 116, 118–120). These cytokines have been confirmed to contribute to the SG fibrosis by enhancing the local chronic inflammatory environment and inducing the activation of myofibroblasts, especially the macrophage-derived TGF-β (121).
TGF-β is involved in the EMT-dependent fibrotic signaling pathway in SG, namely the TGF-β1/SMAD/Snail pathway as previously described. This pathway is a key factor in the progression of fibrogenesis. Hypoxia-inducible factor-1α (HIF-1α) has been identified as being engaged in the fibrogenesis triggered by TGF-β1 (122). HIF-1α is a transcription factor that regulates oxygen homeostasis. This finding generates a link between fibrosis and tissue hypoxia (Figure 1). The knockout of the HIF-1α gene resulted in a reduction in TGF-β1 expression in alveolar macrophages, thereby alleviating bleomycin-induced fibrosis (37). The activation of latent TGF-β stored in the ECM by CD14+ monocyte-derived macrophages via integrin αvβ8-mediated pathways has been demonstrated (123). In addition, macrophages are able to influence heterogeneity of myofibroblasts through interactions with the ECM (39). Macrophages are also the primary source of MMPs, a family of enzymes responsible for ECM degradation in tissue (124). MMPs are important contributors in fibrosis and their functions will be discussed in the following section. To this end, elucidating the mechanism by which macrophages induce pathological fibrosis is advantageous for future therapies to regulate the progression of SG inflammation and fibrosis in patients with SS.
2.4 Mast cells
Mast cells are a type of resident immune cell that are mainly found in connective tissues. They primarily play a role in allergic reactions by releasing inflammatory mediators such as histamine and heparin (125, 126). Numerous studies have indicated their functions on tissue repair and chronic inflammation in autoimmune diseases. Their pro-fibrotic function in SG has recently been revealed (12). The interplay between mast cells and fibroblasts stimulates MMPs production, which have been shown to be positively associated with tissue damage (127). The infiltration of mast cells is elevated in the fibrotic zone of SG tissue from pSS patients (125). In this study, the researchers observed a significant correlation between the number of mast cells and the levels of fatty infiltration and fibrosis in minor SG of 19 patients with pSS and 10 healthy individuals (125). Mast cells respond to the acute phase of pSS and release mediators such as IL-1β, TNFα and IL-33 (126), which enhance the focal inflammation in SG. In addition to its capacity to stimulate the Th2 response, IL-33 exhibits pro-inflammatory properties that favor pathological development (118, 128). Moreover, mast cells can interact with fibroblasts through IL-33 (129). This interaction may exacerbate the autoreactive T cell response in the local tissue. The potential role of mast cells in the fibrogenic program could be exploited therapeutically.
2.5 Neutrophils
Multiple recent studies implicated that neutrophils are essential in the pathogenesis of autoimmune disorders, including RA, SLE, and pSS (130). The release of inflammatory cytokines by neutrophils has been identified as a key mechanism by which these cells contribute to the progression of autoimmune diseases (130). An increased basal adherence and increased random migration of neutrophils have been observed in patients with pSS (131), indicating the activation of blood neutrophils and a correlation with an elevated risk for recurrent bacterial infections (132). Upon encountering pathogens, neutrophils undergo degranulation and produce reactive oxygen species (ROS), subsequently entering the NETosis process, which is a form of programmed cell death (133). It is also important that neutrophils have the capacity to migrate from the circulation into resident tissue, where they produce MMPs as one of the antimicrobial peptides that prevent pathogen invasion (134). This may indicate their involvement in ECM degradation. In patients with pSS, neutrophils produce ROS-related molecules during the respiratory burst (135), which may be relevant to the occurrence of inflammation and fibrosis. The results of RNA sequencing and qPCR from a recent study have been corroborated by the observation that neutrophils in patients with pSS exhibited aberrantly elevated expression of genes associated with the type I interferon pathway (136). Moreover, the hyperactivity of type I IFN genes results in mitochondrial damage and is accompanied by ROS activation which favors the formation of neutrophil extracellular traps (NETs) (136, 137). NETs are the components released from NETosis. The generation of NETs may increase the production of autoantibodies (137–139) and cause tissue damage in the context of autoimmune response (137, 138). In conclusion, the impact of neutrophils on SG fibrosis may be attributed to the activation of the type I IFN pathway and MMP-related or ROS-related tissue damage.
3 The pro-fibrotic role of non-immune cells in salivary gland of pSS
3.1 Fibroblasts
Fibroblasts manage the tissue regeneration and homeostasis through the production of connective tissue components, including fibronectin and collagens (140). The differentiation of fibroblasts into myofibroblasts is widely recognized as an essential step in driving tissue fibrogenesis in autoimmunity (141). In particular, fibroblasts in SG have been reported to elicit immunomodulatory functions in pSS (Figure 2). SG-resident fibroblasts participate in the organization of TLS, which is relevant to the level of inflammation and the severity of pSS (42). The TLS structure is well-organized, exhibiting distinct immune cell aggregations and high endothelial venules (72). The fibroblastic network in SG has been proven to be initiated and expanded by the IL-13 and IL-22. Myofibroblasts are typically contractile and α-SMA positive, enabling the release of pro-fibrotic factors such as TGF-β1, IL-1β, IL-6, vascular endothelial growth factor (VEGF), and IL-33 (38). Myofibroblasts regulate not only the deposition of ECM but also the EMT process, thereby driving tissue fibrosis.
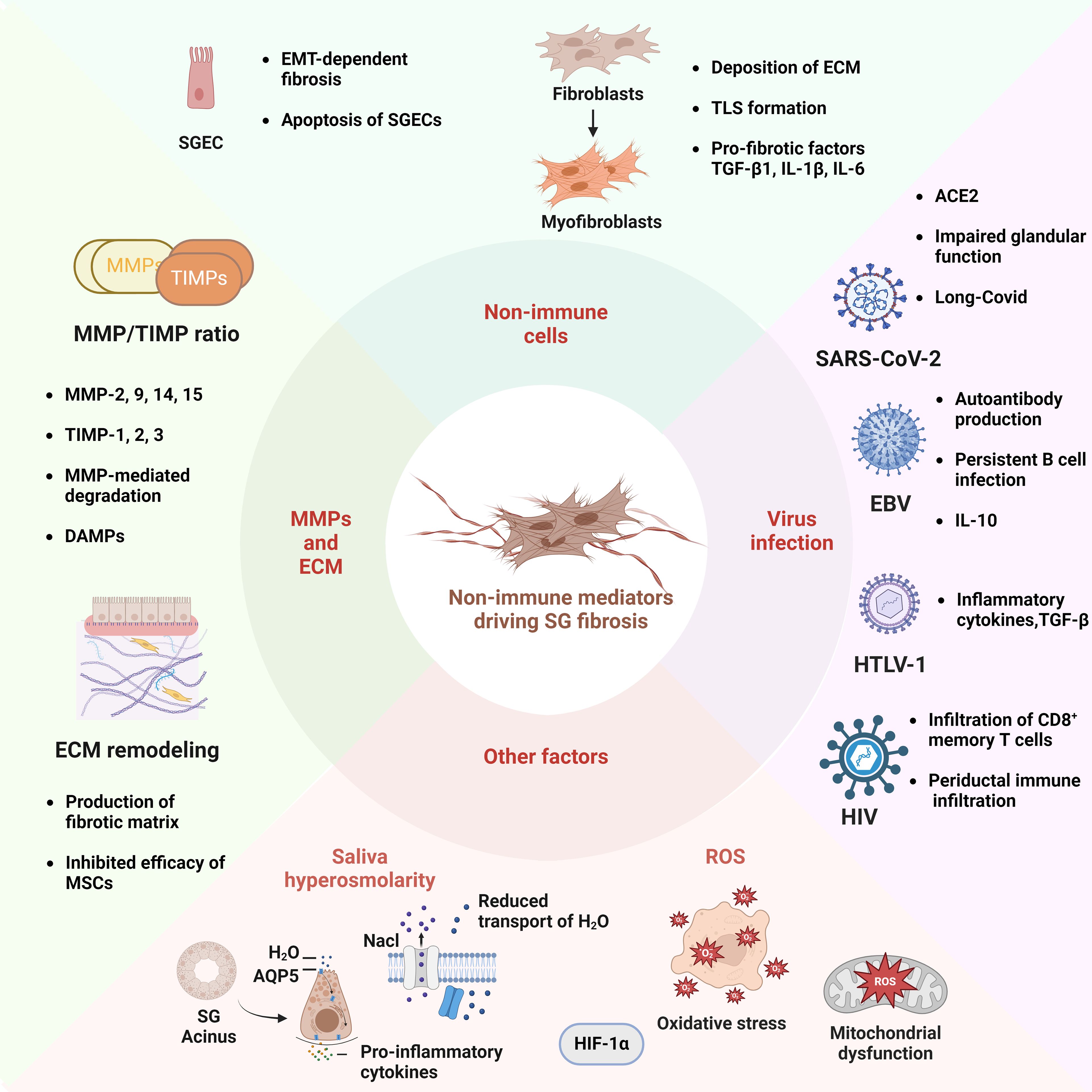
Figure 2. Schematic Illustration of Non-Immune Mediators Associated to the Fibrotic Pathogenesis of Sjögren’s Syndrome. In terms of non-immune factors, the EMT and apoptosis of SGECs are critical processes in the development of salivary gland fibrosis. The transformation of fibroblasts into myofibroblasts is an important step in the development of SG fibrosis. This process involves the deposition of ECM, the formation of TLS, and the accumulation of inflammatory cytokines. The equilibrium between matrix MMPs, such as MMP-2 and -9, and tissue inhibitors of metalloproteinases (TIMPs), including TIMP-1 and -2, influences the degradation of ECM and facilitates the accumulation of fibronectin. The mis-localization of the aquaporin 5 (AQP5) water channel protein in SGECs may result in increased saliva osmolarity, which in turn impairs water transport, while simultaneously enhancing the production of pro-inflammatory cytokines. Viral infection represents a significant etiological factor in SS, with the SARS-CoV-2 virus, Epstein-Barr virus (EBV), human T-lymphotropic virus (HTLV-1), and human immunodeficiency virus (HIV) serving as prominent examples. Additionally, the process of SG fibrosis is influenced by other mechanisms, including oxidative stress and mitochondrial dysfunction. The diagram was created using BioRender.com.
In a single-cell gene expression profile of the SG in pSS patients, CXCL10+ CCL19+ fibroblasts interact specifically with immune cells to establish TLS (142). CXCL10+ CCL19+ immuno-fibroblasts have a similar effect as podoplanin (pdpn)+ CD34− CCL19 expressing fibroblasts in the context of TLS formation (142). The interplay between T cells and dendritic cells is promoted by pdpn-expressing fibroblastic reticular cells in conjunction with pdpn+ stromal fibroblasts, facilitating the transition from innate to adaptive immunity within TLS (38, 142, 143). In patients with SS, CXCL10+ CCL19+ fibroblasts may participate in the development of inflammatory lesions by providing signals such as IFN, IL-1, and TNF, which could exacerbate SG lymphocytic infiltration and favor the resemblance of TLS (31, 38, 142). Single-cell sequencing data has also revealed the existence of another distinct pathology-associated fibroblast subtype in chronic inflammatory diseases, namely secreted protein acidic and cysteine-rich (SPARC)+ COL3A1+ fibroblast (142). SPARC+ COL3A1+ fibroblasts can interact with vascular endothelium and thereby support the ECM restoration. The attenuation of fibrosis via SPARC siRNA has been documented (144). Thus, therapeutic strategies aiming at targeting pathogenic, inflammation-associated fibroblasts are considerable to alleviate the SG fibrosis process.
3.2 SGECs
In patients with pSS, the loss of epithelial character and acquisition of a mesenchymal signature have been well documented during chronic inflammation and fibrogenesis of SG (51). TGF-β is able to induce epithelial cell plasticity in response to inflammatory signals (50). Given the importance of TGF-β in the advancement of EMT-dependent fibrosis in pSS, as a vital fibrogenic mediator, it activates and phosphorylates the canonical TGF-β1/Smad2/3 or non-canonical TGF-β1/Erk1/2 pathway in SGECs, thus mediating their transition to mesenchymal phenotype with a myofibroblast-like morphology. It is noteworthy that IL-6 has been depicted to markedly induce EMT-dependent SG fibrosis in pSS (56). When human SGECs are cultured with IL-6 for 24 to 72 hours, a downregulation of epithelial markers, an upregulation of mesenchymal markers, and a clear morphological change to a mesenchymal phenotype can be observed (56). Additionally, it has been identified that IL-7, secreted by local SGECs, enhances Th1 responses (145). IL-7 also modulates the level of TNF-α, a prototypical Th1 cytokine that contributes to the perpetuation of chronic inflammation in SG (146–148).
As atypical antigen-presenting cells, SGECs play a crucial role in mediating local autoimmune responses (149). SGECs interact with B cells to coordinate differentiation into plasma cells and promote the production of autoantibodies (150). Abnormal interactions between SGECs and immune cells contribute to the exacerbation of local tissue injury in SG. The presence of TNF and IFN-γ in pSS induces apoptosis of SGECs via the Fas/FasL pathway (151). IFN-γ-induced or poly (I:C)-induced anoikis results in the impairment of secretory gland function and high titers of autoimmune antibodies in serum (152). Apoptotic cells also release exosomes that cause sustained damage to the epithelium (153). In conclusion, SGECs are deeply implicated in the pathogenesis of chronic inflammation in SG and may also influence SG fibrosis (Figure 2).
4 Other pro-fibrotic mechanisms in salivary gland of pSS
It is evident that additional molecular mechanisms may contribute to the development of fibrosis in SG of pSS. In the following sections, three distinct factors that contribute to SG fibrosis will be mainly discussed in detail: MMPs and ECM remodeling, salivary hyperosmolarity, and viral infection. It should be noted, however, that there are other molecules and pathways that are relevant to SG fibrosis, although they have been previously only briefly mentioned in the immune cell section.
4.1 MMPs and ECM remodeling
A glandular response to tissue disruption typically gives rise to an altered extracellular matrix. In the context of pSS progression, SG destruction is common due to the chronic inflammatory environment and ultimately leads to the formation of fibrotic matrix. MMPs are a family of enzymes that are capable of degrading matrix. MMP-2, 9, 14, 15 and tissue inhibitors of metalloproteinases (TIMPs)-1, 2, 3 are all involved in the homeostasis of SG ECM (127, 154, 155). The dysregulation of MMPs drives ECM remodeling and SG degeneration following sequential pathological events, inducing the production of fibrotic matrix (156). The fibrotic basal lamina displays a disorganized pattern of laminin expression (157). A fibrotic matrix impedes the transportation of nutrients and oxygen to the cells and blocks the infiltration of progenitor cells, which are required for tissue renewal (158). Indeed, fibrotic ECM impacts the efficacy of SG regenerative therapies, especially the therapeutic effects of mesenchymal stem cells (MSCs) (158, 159). The activity of MMPs can be regulated by TIMPs (127) (Figure 2). Among these MMPs and TIMPs, the upregulation of MMP-3 and MMP-9 and downregulation of TIMP1 are positively correlated with acinar damage and disease development in pSS patients (127, 160). The ratio of MMPs/TIMPs in human SG is one of the hallmarks of ECM remodeling. A number of pro-inflammatory cytokines are responsible for mediating the balance between MMPs and TIMPs, including IL-1α, IL-6, TNF-α, and IFN-γ (124, 160, 161). The process of anoikis of cells also enables the activation of these enzymes. Additionally, MMP-mediated degradation in SG produces the damage-associated molecular patterns (DAMPs), which subsequently activate the Toll-like receptor 2 (TLR2) and TLR4 pathways (162). Consequently, the nuclear factor kappa B (NF-κB) pathway is activated, further sustaining the chronic B cell response in situ. For example, the small proteoglycans, biglycan (Bgn) and decorin (Dcn) are present in the damaged SG basal lamina in SS mice (162). As components of the ECM and typical DAMPs, these two proteoglycans can bind to TLR4, which subsequently elicits MyD88-dependent signaling cascades, thus exacerbating the downstream autoimmune responses (162). Increased anti-Bgn and -Dcn autoantibodies in serum were found in experimental mice of SS and were correlated with SG dysfunction (163). Besides, aging may also be risk factor in ECM remodeling in SG. Recent studies have revealed that accumulated MMP-2 and MMP-9 are strongly connected with age-related fibrotic remodeling, particularly in aged mice exhibiting a senescence-associated secretory phenotype (SASP) (158, 164).
4.2 Saliva hyperosmolarity
Dysfunctional exocrine glands are unable to produce saliva of the requisite quality and quantity. The displacement of the aquaporin-5 (AQP-5) water channel protein suggests that water is not properly and adequately transported out of the apical membrane (165, 166) (Figure 2). In contrast, the transport of NaCl to the acini lumen results in the formation of saliva with a high osmotic gradient (167). Since acinar cell apoptosis could cause a reduction of salivary volume, the formation of a hyperosmolar environment within the salivary gland could potentially lead to further remodeling of ECM and severe lymphocytic infiltration of SG (167). Notably, gene enrichment analysis of acinar SG cells in response to hyperosmolar stimulation showed robust pro-fibrotic gene expression such as IFN-γ, IL-17, MMP-3, and MMP-13 (167). SGECs from pSS patients showed overexpression of MMP genes that were related to ECM regeneration including COL3A1 and MMP-24, compared to healthy controls (142, 167). Of note, this was associated with upregulation of Snail in SGECs by RNA-seq analysis, which indicates the EMT process in SG. These findings suggest a possible implication of hyperosmolar stress in the pathogenesis of fibrosis.
4.3 Viral infection
It is well known that the tissue fibrosis can be the result of prolonged inflammation in response to a multitude of stimuli, including viral infections, radiation and immune hyperactivity. In studies about the etiology of pSS, several virus infections have been proposed as potential drivers of chronic persistent inflammation and autoimmune response in SG (168, 169). These include the Epstein-Barr virus (EBV), hepatitis C virus (HCV) and human T-lymphotropic virus (HTLV-1) (Figure 2) (169–171). The ability of viral antigens to drive autoimmunity may be, at least in part, due to their molecular mimicry of autoantigens (172–174). These viral antigens possess analogous sequences and structures, favoring cross-reactivity of immune cells with autoantigens and the initiation or promotion of overproduction of autoantibodies (174). In vivo studies have reported the role of viral infection in the mouse salivary gland model. Replication-deficient adenovirus-5 (Adv5) was introduced into the submandibular glands of C57BL/6 mice by retrograde cannulation (175). Adv5 infection may result in reduced saliva, exocrine dysfunction, autoantibody production and TLS formation. The upregulation of CXCL13, CCL19 and CCL21 mRNA accelerated the development of ELS (176). This finding suggests that viral infection indeed impairs SG function and contributes to the organ-specific autoimmune pathogenesis.
The precise role of various viral infections in the pathogenesis of fibrosis in the salivary glands of patients with SS remains incompletely understood. EBV is particularly associated with the onset and progression of SS, as it has been demonstrated to trigger or promote autoimmune responses and autoantibody production (174, 177). EBV is capable of infecting both naive and memory B cells, and it can persistently infect those B cells (178). EBV has been demonstrated to promote B cell differentiation into plasma cells and to increase the production of autoantibodies (179). Furthermore, sustained EBV infection can damage the SG due to IL-10 production, which also results in the production of autoantibodies (180). Taken together, it remains unclear whether EBV plays a pro-fibrotic role, but it is evident that it can accelerate the progression of SS. Additionally, other viruses, including HCV, Hepatitis B Virus (HBV), and HTLV-1, have been identified as potential contributors to the pathogenesis of SS. HCV infection is primarily associated with hepatic impairment in SS (181, 182). While hepatitis and liver fibrosis are well-documented consequences of HCV infection, the pro-fibrotic effect of HCV on the SG is less frequently observed. HBV is more closely associated with liver involvement. HTLV-1 infection has been identified to enhance the production of inflammatory cytokines, including TGF-β, which is associated with fibrosis in SG tissues of SS patients (174, 183). Interestingly, a reduction in ectopic germinal centers has been observed in HTLV-1-seropositive SS patients, which may be attributed to its inhibitory effect on B cell-related cytokines, such as BAFF (184). Human immunodeficiency virus (HIV)-related SS is characterized by the infiltration of CD8+ memory T cells and the presence of viral proteins, including LMP-EBV and p-24, in ductal cells (185). A recent histochemical study demonstrated that acinar atrophy, ductal dilation, and fibrosis, accompanied by collagen deposition in the site of periductal immune infiltration, are hallmarks of HIV-related SS (186, 187). In general, viral infections may contribute to the development of fibrosis in SG through various mechanisms, including the induction of cytokines, chemokines, and interaction with immune cells.
Interestingly, SARS-CoV-2 may also contribute to the pathological process of SS (Figure 2). Angiotensin-converting enzyme 2 (ACE2) transgenic mice were infected with SARS-CoV-2. The researchers found distinct SS-like signatures such as impaired glandular function, elevated anti-SSB/La and severe lymphocytic infiltrates in the SG of infected mice and individuals diagnosed with SARS-CoV-2 infection (188, 189). Clinical data have shown that the 2019 novel coronavirus (2019-nCoV) is detectable in the saliva of 91.7% of infected patients (190). ACE2 is the target receptor of SARS-CoV-2 and is present in the SG. As evidenced by research, ACE2 is mainly expressed in epithelial cells within SG (191). Thus, it can be concluded that SGECs are the target cells during SARS-CoV-2 infection. More importantly, SARS-CoV-2-mediated tissue pathology is frequently accompanied by lymphocytic lesions due to the local auto-reaction (192, 193). These findings imply a potential role for SARS-CoV-2 infection in the development of pathogenic fibrosis within the salivary gland. While there have been reports of lung fibrosis associated with SARS-CoV-2 infection, there is a paucity of research on the topic of salivary gland fibrosis. Therefore, further investigation with a larger cohort of patients with chronic inflammation is necessary to gain a deeper understanding of the relationship between SARS-CoV-2 infection and the development of fibrosis.
4.4 Other potential factors
In addition to the above mechanisms, there are other non-immune factors that contribute to SG fibrosis, such as oxidative stress and mitochondrial dysfunction. Cyclophosphamide (CP), a chemotherapeutic agent which is also used as an immunosuppressant causes oxidative stress (194, 195). However, it can affect SG and reduce saliva production in a long-term manner. CP can increase the expression of oxidative genes and at the same time induce the morphological changes such as parenchymal atrophy and acinar apoptosis of SG (195). In addition, inflammatory markers such as TNF-α and IL-1β were increased in the periductal area induced by CP application. 5-Fluorouracil (5-FU) is also a drug used to treat solid tumors (196). Similar to CP, it can activate the oxidative system and lead to the lesions and morphological alterations of SG such as atrophy and necrosis (195, 196). The treatments of CP and 5-FU may induce inflammation in SG via oxidative stress, and ultimately aggregate the progression of SG fibrosis. ROS are mainly generated by mitochondria via oxidative phosphorylation. Damaged mitochondria produce more ROS and increase the levels of oxidative stress markers such as 8-OHdG and HEL in patients with pSS (197). Furthermore, the impairment of the cell leads to DAMPs molecules released by mitochondria, following the immune reaction cascades (198). Impaired mitochondrial respiratory chain produces less ATP, which exacerbates cell apoptosis and recruits immune cells (198). Thus, mitochondrial dysfunction may play a role in the immune cell infiltration and inflamed microenvironment of pSS, increasing the oxidative stress and resulting in SG fibrosis.
5 Therapeutic strategies
A variety of treatments are available for the alleviation of sicca symptoms, including the use of saliva substitutes. Systemic secretagogues pilocarpine and cevimeline, which act as muscarinic receptor agonists, are capable of relieving symptoms of dry mouth (1). Several monoclonal antibodies specifically targeting B cells have shown favorable outcomes, including rituximab (a CD20specific antibody), epratuzumab (a B cell receptor CD22-specific antibody) and belimumab (a BAFF-blocking antibody) (199–201). With regard to the treatment for SG fibrosis, research is now concentrating on tissue regeneration and anti-inflammation. An siRNA was effective in knocking down the transcription factor of MMP-9, namely ETS1, showing beneficial effects in MMP-mediated degradation (202). A chimeric antigen receptor (CAR)-T cell targeting fibroblast-activating protein (FAP) has been proposed as a potential treatment for oral submucous fibrosis (OSMF) (203). FAP+ fibroblasts possess a greater potential for conversion into myofibroblasts. Therefore, this CAR-T cell may be an effective means of potentiating an anti-fibrotic approach. In addition, microRNAs targeting the various components of the TGF-β1/Smad pathway have been shown to be effective in inhibiting TGF-β1-dependent tissue fibrosis (204).
To date, regenerative therapies remain as the mainstay in the treatment of tissue fibrosis. MSCs treatment has been demonstrated in numerous studies to effectively suppress pathogenic processes in SS and influence the SG microenvironment by replacing the fibrotic matrix (158, 160, 205–208). A number of different studies have demonstrated the efficacy of MSC administration in the treatment of SG (205–208). Injection of MSCs resulted in a significant increase in salivary flow rate and decreased lymphocytic infiltration in the SG in a mouse model of the disease (205, 209–211). According to the study by Xu et al., the levels of anti-SSA/Ro and anti-SSB/La antibodies were downregulated after injection of umbilical cord MSCs in 24 patients with SS (206). In patients with SS, administration of MSCs effectively reduces the level of IL-12, a pro-inflammatory cytokine produced by DCs and implicated in the development of SS (212). Treatment with MSCs may affect the differentiation and maturation of DCs, resulting in the suppression of IL-12 (213). Furthermore, intravenous injection of MSCs in 38 patients showed increased IL-27 levels and decreased ratio of Th17/Treg cells (211). The elevated Treg cells among PBMCs indicated alleviation of SS symptoms (211). These studies provided the clinical evidence for MSC treatment in patients with SS. Moreover, the homing ability to site of tissue regeneration and inflammation makes MSCs remarkable in the treatment of SG impairment. MSCs, as the reservoir of progenitor cells, have the capacity to regulate the SG regeneration. Immunoregulatory cytokines secreted by MSCs reduce B-cell activation and proliferation by inhibiting the overactivation of T cells and dendritic cells (205, 214). MSC can also interact with fibroblasts, thereby exerting an anti-inflammatory effect (215). These cells have the potential to reduce the expression of inflammatory molecules such as intercellular adhesion molecule 1 (ICAM1) and vascular cell adhesion protein 1 (VCAM1) as well as ECM-degrading MMPs (160, 216). Together, these findings suggest anti-fibrotic properties of MSCs by suppressing inflammation and ECM degradation. To investigate the anti-fibrotic activity of MSCs, a previous study conducted by van der Helm et al. (2018) injected bone marrow-derived MSCs (BMMSCs) into a zebrafish embryo model (208). The results demonstrated that liver fibrosis in zebrafish was attenuated in parallel with decreased collagen and TGF-β levels following administration of BMMSCs. Another study reported reduced IL-1β expression and inhibited pro-fibrotic signaling in a cardiac ischemia mouse model treated with MSC exosomes (217). In both mouse models and pSS patients, BMMSCs can promote the immunosuppressive activities of Treg, thereby inhibiting the autoimmune progression and improving the glandular function (208). In an SS-like NOD/Ltj mouse model, treatment with allogeneic bone marrow-derived MSCs showed a therapeutic effect in alleviating SS symptoms and improving the performance of SG (206). The immunoregulatory activities of MSCs are evident in directing the fate of CD4+ T cells towards Treg and Th2, while simultaneously suppressing the Th17 and Tfh pathways (206). In particular, researchers have revealed that the migration of BMMSCs to the site of inflammation is controlled by the stromal cell-derived factor-1 (SDF-1)/CXCR4 axis, which determines the functions of MSCs against SS autoimmunity (119, 206, 208). Collectively, these findings suggest the anti-fibrotic capacity of MSCs in diminishing ECM degradation and SG fibrosis. However, MSC therapy is not without limitations and potential adverse effects. Resident MSCs have the potential to differentiate into myofibroblasts. The accumulation of myofibroblasts derived from MSCs may lead to massive ECM deposition and organ failure (218). TGF-β1 secretion by external MSCs can promote myofibroblast populations and immune system abnormalities (219). Furthermore, Ben-David et al. revealed the possibility of tumor formation after MSC transplantation (220). The researchers conducted an analysis of the MSC-specific chromosomal aberrations that may be responsible for the increase in tumorigenicity. In the study by Liang et al., they evaluated the adverse effects of allogeneic MSCs in 404 patients with autoimmune diseases including SLE and SS. They found that the rate of infections caused by MSCs transplantation was 29.5% and the risk of mortality was 11.1% after 9 years of follow-up (221). It is therefore imperative that the safety and fine-tuning of MSCs therapy in SG fibrosis urgently requires further exploration.
6 Discussion
This review examines the contributions of immune and non-immune cells to the pathophysiological processes of fibrosis in the SG of patients with SS. Although a large number of studies have proposed risk factors for this autoimmune disease, the mechanism of fibrogenesis and the interplays between SGECs and immune cells in SS remain poorly understood. This morphological alteration is typically irreversible and associated with organ dysfunction. In the early stage of SG fibrosis, reduced acini, architectural malformation and ductal dilation could be observed under the microscope. More tissue injury may induce more FLS formation. The large number of FLS is the standard to evaluate the severity of this disease. These may be the morphologic indicators of SG fibrosis. The molecular mechanism causing these pathomorphologic changes is indispensable with abnormal immune response in the context of SS. An excessive autoreaction in the SG of patients with SS or SjD leads to chronic inflammation in the local area. The persistent inflammation observed in the SG is attributed to detrimental immune infiltration and the accumulation of inflammatory cytokines. TGF-β-mediated EMT may be one of the significant mechanisms contributing to the development of fibrosis in SG. Aberrant T-cell and B-cell responses lead to the overproduction of autoantibodies and aggravate tissue damage. It is therefore evident that immune cells underscore their role in exacerbating chronic inflammation of the SG to the point of progressive fibrosis. As for the non-immune cells, they interact with varying extents with fibroblasts and regulate their transformation into myofibroblasts, the culprits of tissue fibrosis. Among the non-immune fibrogenic mediators, MMPs play a critical role in ECM remodeling. MMPs facilitate the replacement of healthy components of the ECM with fibrosis-associated tissue in response to damage signals. Therefore, the development of SG fibrosis represents a deleterious consequence of an excessive reparative response to damage in the collaboration of immune cells and tissue-resident cells. This response is characterized by the production of pro-inflammatory cytokines and enzymes that degrade the ECM and cause local chronic inflammation. Surprisingly, viral infection, including that caused by EBV, HCV, HTLV-1 and SARS-CoV-2 virus, may also trigger the pathogenic processes in patients with SS. Long-term consequences following recovery from SARS-CoV-2 infection include oral complications such as glandular atrophy and fibrosis. The effect of SARS-CoV-2 on SG gland was unexpected and requires further investigation. As of yet, the influence of oral manifestations and the mechanism of dysfunctional SG in the context of COVID-19 remains unclear.
In fact, SG fibrosis is implied in long-COVID or post-COVID conditions, as fibrous connective tissues are formed to repair the glandular damage (189). Excessive formation of hyperplastic fibrous scar tissue results in the obstruction of the gland. A seminal study by Alfaifi et al. elucidated the long-term oral sequelae in post-COVID patients including salivary reduction, glandular destruction and taste dysfunction (222). Both minor and major SGs displayed SS-like features. These included architectural malformation, atrophy, fibrosis and ductal disruption following SARS-CoV-2 infection (188). An antimicrobial peptide called histatin-5 (Hst-5) is involved in the innate immune response during infection. Decreased salivary histatin-5 production has been implicated in SARS-CoV-2-mediated SG damage. The low level of Hst-5 may lead to prolonged infection and oral inflammation (222). Concurrently, the Th17 response is initiated in the post-infected gland, accompanied by elevated levels of cytokine production including IL-17, IL-22 and IL-23 (222, 223). This finding suggests that the pro-fibrotic factors that contributed to the pathological development of pSS also play a critical role in the post-inflammatory process associated with SARS-CoV-2 infection. In light of these observations, it seems reasonable to hypothesize that long-term oral sequelae in post-COVID patients may be capable of causing secondary SS and triggering SG fibrosis.
COVID-mediated SS and SG fibrosis can be explained by several potential mechanisms (223, 224). Utilizing the aforementioned experimental mouse model with Adv5-cannulated SG, Barone et al. demonstrated the beneficial effect of IL-22 in TLO assembly and autoimmunity (43). IL-22 upregulates the expression of CXCL13 in fibroblasts and CXCL12 in epithelial cells, thus exerting a pro-fibrotic effect (43). Similar to Adv5, it can be postulated that the fibrotic process observed in the SG in the context of the COVID-19 may also be dependent on the presence of IL-22, given the evidence that the Th17 pathway is hyperactivated during the course of infection with the SARS-CoV-2 virus (224). Additionally, approximately 30% of COVID-19 survivors develop COVID-associated pulmonary fibrosis (188, 190, 224). The molecular mechanisms underlying COVID-19-induced pulmonary fibrosis have been elucidated (224). EMT, TGF-β, hyperproliferation of myofibroblasts and high levels of pro-inflammatory IL-2, IL-7, and IFN-γ, are extensively involved in pulmonary fibrosis during SARS-CoV-2 infection (223). Intriguingly, these mechanisms were likewise found in the development of SG fibrosis. These findings may provide novel insight of the pathogenesis of SG fibrosis in COVID patients.
In addition to the effects associated with age, elevated serum titers of rheumatoid factor (RF) and anti-cyclic citrullinated peptide (CCP) have been identified as a significant risk factor for fibrosis in the labial SG, as reported by Katona et al. (225). Follicle dendritic cells contribute to the formation of ectopic germinal centers by promoting B cell differentiation and expansion in a complement-dependent manner (226). Plasmacytoid dendritic cells (pDCs) primarily produce type I interferons and have also been found in ELS (227). pDC cells were closely involved in the SS development due to their IFN-α production (19). The trafficking of circulating pDC cells to the ectopic germinal center of the SG has been documented (15), thereby attracting B cells that home in on the ELS by activating the expression of CXCL13 on macrophages (228). NKp44+ NK cells have been linked to focus score and SS severity, although the underlying mechanism was not clear (46). Nonetheless, NKp30+ NK cells can interact with SGECs and have pro-inflammatory functions through the secretion of IFN-γ (229).
To date, there is no established diagnostic marker for fibrosis of the SG, which limits the ability to accurately assess its prognosis. In the later stages of the disease, the current therapeutic options for treating fibrosis are limited in their efficacy. Thus, it is imperative to prioritize the prevention of this pathological process requires more attention than treatment. However, there is a possibility that inflamed tissue in SS lesions may not progress into fibrosis during degeneration, even in cases of heavy immune infiltration (230). Kapsogeorgou et al. conducted a 10-year follow-up of SS patients and observed no significant alteration of lesions in their minor salivary glands. The heterogeneity between patients indicates the complexity of the pathogenesis of SS and often results in delays in disease diagnosis (231). This emphasizes the necessity of studying the underlying mechanisms. The acknowledgement of the processes involved in the formation of fibrosis is not yet fully understood, this review may provide valuable insights that will facilitate further exploration of the pathological processes of SS and may provide insights for the development of new therapeutic strategies.
Author contributions
XL: Conceptualization, Funding acquisition, Writing – original draft, Writing – review & editing. DM: Conceptualization, Writing – original draft, Writing – review & editing, Visualization. YF: Funding acquisition, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was funded by grants through National Key Research and Development Program of China (2023YFE0203100), Mainland-Hong Kong Joint Funding Scheme (MHP/104/22), General Research Fund, Hong Kong Research Grants Council (17116521, 27111820 and 17109123), Health and Medical Research Fund (19201121 and 20212601).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Brito-Zerón P, Baldini C, Bootsma H, Bowman SJ, Jonsson R, Mariette X, et al. Sjögren syndrome. Nat Rev Dis Primers. (2016) 2:16047. doi: 10.1038/nrdp.2016.47
2. Gao Y, Ren H, Meng F, Li J, Cheung E, Li H, et al. Pathological roles of interleukin-22 in the development of recurrent hepatitis C after liver transplantation. PloS One. (2016) 11:e0154419. doi: 10.1371/journal.pone.0154419
3. Huang H, Xie W, Geng Y, Fan Y, Zhang Z. Mortality in patients with primary Sjogren's syndrome: A systematic review and meta-analysis. Rheumatol (Oxford). (2021) 60:4029–38. doi: 10.1093/rheumatology/keab364
4. Fisher BA, Jonsson R, Daniels T, Bombardieri M, Brown RM, Morgan P, et al. Standardisation of labial salivary gland histopathology in clinical trials in primary Sjögren's syndrome. Ann Rheum Dis. (2017) 76:1161–8. doi: 10.1136/annrheumdis-2016-210448
5. Eckes B, Eming SA. Tissue fibrosis: A pathomechanistically unresolved challenge and scary clinical problem. Exp Dermatol. (2017) 26:135–6. doi: 10.1111/exd.13165
6. Liao R, Yang HT, Li H, Liu LX, Li K, Li JJ, et al. Recent advances of salivary gland biopsy in Sjögren's syndrome. Front Med (Lausanne). (2021) 8:792593. doi: 10.3389/fmed.2021.792593
7. Bookman AA, Shen H, Cook RJ, Bailey D, McComb RJ, Rutka JA, et al. Whole stimulated salivary flow: correlation with the pathology of inflammation and damage in minor salivary gland biopsy specimens from patients with primary Sjögren's syndrome but not patients with sicca. Arthritis Rheum. (2011) 63:2014–20. doi: 10.1002/art.30295
8. Lee K-A, Kim S-H, Kim H-R, Kim H-S. Impact of age on the diagnostic performance of unstimulated salivary flow rates and salivary gland ultrasound for primary Sjögren's syndrome. Front Med. (2022) 9:968697. doi: 10.3389/fmed.2022.968697
9. Li N, Ye Y, Wu Y, Li L, Hu J, Luo D, et al. Alterations in histology of the aging salivary gland and correlation with the glandular inflammatory microenvironment. iScience. (2023) 26:106571. doi: 10.1016/j.isci.2023.106571
10. Syrjänen S. Age-related changes in structure of labial minor salivary glands. Age Ageing. (1984) 13:159–65. doi: 10.1093/ageing/13.3.159
11. Llamas-Gutierrez FJ, Reyes E, Martínez B, Hernández-Molina G. Histopathological environment besides the focus score in Sjögren's syndrome. Int J Rheum Dis. (2014) 17:898–903. doi: 10.1111/1756-185x.12502
12. Leehan KM, Pezant NP, Rasmussen A, Grundahl K, Moore JS, Radfar L, et al. Minor salivary gland fibrosis in Sjögren's syndrome is elevated, associated with focus score and not solely a consequence of aging. Clin Exp Rheumatol. (2018) 36 Suppl 112:80–8.
13. Joachims ML, Leehan KM, Lawrence C, Pelikan RC, Moore JS, Pan Z, et al. Single-cell analysis of glandular T cell receptors in Sjögren's syndrome. JCI Insight. (2016) 1(8):e85609. doi: 10.1172/jci.insight.85609
14. Ni PP, Solomon B, Hsieh CS, Allen PM, Morris GP. The ability to rearrange dual TCRs enhances positive selection, leading to increased allo- and autoreactive T cell repertoires. J Immunol. (2014) 193:1778–86. doi: 10.4049/jimmunol.1400532
15. Goules AV, Kapsogeorgou EK, Tzioufas AG. Insight into pathogenesis of Sjögren's syndrome: dissection on autoimmune infiltrates and epithelial cells. Clin Immunol. (2017) 182:30–40. doi: 10.1016/j.clim.2017.03.007
16. Joachims ML, Leehan KM, Dozmorov MG, Georgescu C, Pan Z, Lawrence C, et al. Sjögren's syndrome minor salivary gland CD4(+) memory T cells associate with glandular disease features and have a germinal center T follicular helper transcriptional profile. J Clin Med. (2020) 9(7):2164. doi: 10.3390/jcm9072164
17. Deng L, Huang T, Zhang L. T cells in idiopathic pulmonary fibrosis: crucial but controversial. Cell Death Discovery. (2023) 9:62. doi: 10.1038/s41420-023-01344-x
18. Wynn TA. Cellular and molecular mechanisms of fibrosis. J Pathol. (2008) 214:199–210. doi: 10.1002/path.2277
19. Yao Y, Liu Z, Jallal B, Shen N, Rönnblom L. Type I interferons in Sjögren's syndrome. Autoimmun Rev. (2013) 12:558–66. doi: 10.1016/j.autrev.2012.10.006
20. Hong SM, Lee J, Jang SG, Lee J, Cho ML, Kwok SK, et al. Type I interferon increases inflammasomes associated pyroptosis in the salivary glands of patients with primary Sjögren's syndrome. Immune Netw. (2020) 20:e39. doi: 10.4110/in.2020.20.e39
21. Szczerba BM, Rybakowska PD, Dey P, Payerhin KM, Peck AB, Bagavant H, et al. Type I interferon receptor deficiency prevents murine Sjogren's syndrome. J Dent Res. (2013) 92:444–9. doi: 10.1177/0022034513483315
22. Mitsias DI, Tzioufas AG, Veiopoulou C, Zintzaras E, Tassios IK, Kogopoulou O, et al. The Th1/Th2 cytokine balance changes with the progress of the immunopathological lesion of Sjogren's syndrome. Clin Exp Immunol. (2002) 128:562–8. doi: 10.1046/j.1365-2249.2002.01869.x
23. de Souza TR, de Albuquerque Tavares Carvalho A, Duarte ÂP, Porter SR, Leão JC, Gueiros LA. Th1 and Th2 polymorphisms in Sjögren's syndrome and rheumatoid arthritis. J Oral Pathol Med. (2014) 43:418–26. doi: 10.1111/jop.12149
24. Witas R, Gupta S, Nguyen CQ. Contributions of major cell populations to Sjögren's syndrome. J Clin Med. (2020) 9(9):3057. doi: 10.3390/jcm9093057
25. van Woerkom JM, Kruize AA, Wenting-van Wijk MJ, Knol E, Bihari IC, Jacobs JW, et al. Salivary gland and peripheral blood T helper 1 and 2 cell activity in Sjögren's syndrome compared with non-Sjögren's sicca syndrome. Ann Rheum Dis. (2005) 64:1474–9. doi: 10.1136/ard.2004.031781
26. Mustafa W, Zhu J, Deng G, Diab A, Link H, Frithiof L, et al. Augmented levels of macrophage and Th1 cell-related cytokine mRNA in submandibular glands of MRL/lpr mice with autoimmune sialoadenitis. Clin Exp Immunol. (1998) 112:389–96. doi: 10.1046/j.1365-2249.1998.00609.x
27. Greenwell-Wild T, Moutsopoulos NM, Gliozzi M, Kapsogeorgou E, Rangel Z, Munson PJ, et al. Chitinases in the salivary glands and circulation of patients with Sjögren's syndrome: macrophage harbingers of disease severity. Arthritis Rheum. (2011) 63:3103–15. doi: 10.1002/art.30465
28. Zhang J, Zhang X, Shi X, Liu Y, Cheng D, Tian Q, et al. CXCL9, 10, 11/CXCR3 axis contributes to the progress of primary Sjogren's syndrome by activating GRK2 to promote T lymphocyte migration. Inflammation. (2023) 46:1047–60. doi: 10.1007/s10753-023-01791-9
29. Ruffilli I. Sjögren's syndrome and chemokines. Clin Ter. (2014) 165:e464–9. doi: 10.7417/ct.2014.1793
30. Ogawa N, Ping L, Zhenjun L, Takada Y, Sugai S. Involvement of the interferon-gamma-induced T cell-attracting chemokines, interferon-gamma-inducible 10-kd protein (CXCL10) and monokine induced by interferon-gamma (CXCL9), in the salivary gland lesions of patients with Sjögren's syndrome. Arthritis Rheum. (2002) 46:2730–41. doi: 10.1002/art.10577
31. Hasegawa H, Inoue A, Kohno M, Muraoka M, Miyazaki T, Terada M, et al. Antagonist of interferon-inducible protein 10/CXCL10 ameliorates the progression of autoimmune sialadenitis in MRL/lpr mice. Arthritis Rheum. (2006) 54:1174–83. doi: 10.1002/art.21745
32. Li H, Gade P, Xiao W, Kalvakolanu DV. The interferon signaling network and transcription factor C/EBP-beta. Cell Mol Immunol. (2007) 4:407–18.
33. Cao T, Zhou J, Liu Q, Mao T, Chen B, Wu Q, et al. Interferon-gamma induces salivary gland epithelial cell ferroptosis in Sjogren's syndrome via JAK/STAT1-mediated inhibition of system Xc(). Free Radic Biol Med. (2023) 205:116–28. doi: 10.1016/j.freeradbiomed.2023.05.027
34. De Vita S, Boiocchi M, Sorrentino D, Carbone A, Avellini C, Dolcetti R, et al. Characterization of prelymphomatous stages of B cell lymphoproliferation in Sjögren's syndrome. Arthritis Rheum. (1997) 40:318–31. doi: 10.1002/art.1780400217
35. Moriyama M, Hayashida JN, Toyoshima T, Ohyama Y, Shinozaki S, Tanaka A, et al. Cytokine/chemokine profiles contribute to understanding the pathogenesis and diagnosis of primary sjögren's syndrome. Clin Exp Immunol. (2012) 169:17–26. doi: 10.1111/j.1365-2249.2012.04587.x
36. Gieseck RL, Wilson MS, Wynn TA. Type 2 immunity in tissue repair and fibrosis. Nat Rev Immunol. (2018) 18:62–76. doi: 10.1038/nri.2017.90
37. Wynn TA, Vannella KM. Macrophages in tissue repair, regeneration, and fibrosis. Immunity. (2016) 44:450–62. doi: 10.1016/j.immuni.2016.02.015
38. Kendall RT, Feghali-Bostwick CA. Fibroblasts in fibrosis: novel roles and mediators. Front Pharmacol. (2014) 5:123. doi: 10.3389/fphar.2014.00123
39. Vasse GF, Nizamoglu M, Heijink IH, Schlepütz M, van Rijn P, Thomas MJ, et al. Macrophage–stroma interactions in fibrosis: biochemical, biophysical, and cellular perspectives. J Pathol. (2021) 254:344–57. doi: 10.1002/path.5632
40. Pesce J, Kaviratne M, Ramalingam TR, Thompson RW, Urban JF Jr., Cheever AW, et al. The IL-21 receptor augments Th2 effector function and alternative macrophage activation. J Clin Invest. (2006) 116:2044–55. doi: 10.1172/jci27727
41. Dimitriou ID, Kapsogeorgou EK, Moutsopoulos HM, Manoussakis MN. CD40 on salivary gland epithelial cells: high constitutive expression by cultured cells from Sjögren's syndrome patients indicating their intrinsic activation. Clin Exp Immunol. (2002) 127:386–92. doi: 10.1046/j.1365-2249.2002.01752.x
42. Kerstin K. Fibroblasts in Sjögren’s syndrome. In: Mojca Frank B, Katja L, editors. Fibroblasts. IntechOpen, Rijeka (2021).
43. Barone F, Nayar S, Campos J, Cloake T, Withers DR, Toellner K-M, et al. Il-22 regulates lymphoid chemokine production and assembly of tertiary lymphoid organs. Proc Natl Acad Sci. (2015) 112:11024–9. doi: 10.1073/pnas.1503315112
44. Lavoie TN, Stewart CM, Berg KM, Li Y, Nguyen CQ. Expression of interleukin-22 in Sjögren's syndrome: significant correlation with disease parameters. Scand J Immunol. (2011) 74:377–82. doi: 10.1111/j.1365-3083.2011.02583.x
45. Zhou J, Onodera S, Hu Y, Yu Q. Interleukin-22 exerts detrimental effects on salivary gland integrity and function. Int J Mol Sci. (2022) 23(21):12997. doi: 10.3390/ijms232112997
46. Ciccia F, Guggino G, Rizzo A, Ferrante A, Raimondo S, Giardina A, et al. Potential involvement of IL-22 AND IL-22-producing cells in the inflamed salivary glands of patients with Sjogren's syndrome. Ann Rheum Dis. (2012) 71:295–301. doi: 10.1136/ard.2011.154013
47. Belle I, Mahlios J, McKenzie A, Zhuang Y. Aberrant production of IL-13 by T cells promotes exocrinopathy in Id3 knockout mice. Cytokine. (2014) 69:226–33. doi: 10.1016/j.cyto.2014.06.009
48. Li X, Li X, Qian L, Wang G, Zhang H, Wang X, et al. T regulatory cells are markedly diminished in diseased salivary glands of patients with primary Sjögren's syndrome. J Rheumatol. (2007) 34:2438–45.
49. Alunno A, Petrillo MG, Nocentini G, Bistoni O, Bartoloni E, Caterbi S, et al. Characterization of a new regulatory CD4+ T cell subset in primary Sjögren's syndrome. Rheumatol (Oxford). (2013) 52:1387–96. doi: 10.1093/rheumatology/ket179
50. Sisto M, Ribatti D, Lisi S. Organ fibrosis and autoimmunity: the role of inflammation in TGFβ-dependent EMT. Biomolecules. (2021) 11:310. doi: 10.3390/biom11020310
51. Sisto M, Lisi S, Ribatti D. The role of the epithelial-to-mesenchymal transition (EMT) in diseases of the salivary glands. Histochem Cell Biol. (2018) 150:133–47. doi: 10.1007/s00418-018-1680-y
52. Tan Z, Wang L, Li X. Composition and regulation of the immune microenvironment of salivary gland in Sjögren’s syndrome. Front Immunol. (2022) 13:967304. doi: 10.3389/fimmu.2022.967304
53. Sisto M, Lorusso L, Tamma R, Ingravallo G, Ribatti D, Lisi S. Interleukin-17 and -22 synergy linking inflammation and EMT-dependent fibrosis in Sjögren's syndrome. Clin Exp Immunol. (2019) 198:261–72. doi: 10.1111/cei.13337
54. Sisto M, Lorusso L, Ingravallo G, Tamma R, Ribatti D, Lisi S. The TGF-Β1 signaling pathway as an attractive target in the fibrosis pathogenesis of Sjögren's syndrome. Mediators Inflammation. (2018) 2018:1965935. doi: 10.1155/2018/1965935
55. Sisto M, Ribatti D, Ingravallo G, Lisi S. The expression of follistatin-like 1 protein is associated with the activation of the EMT program in Sjögren's syndrome. J Clin Med. (2022) 11(18):5368. doi: 10.3390/jcm11185368
56. Sisto M, Tamma R, Ribatti D, Lisi S. Il-6 contributes to the TGF-Β1-mediated epithelial to mesenchymal transition in human salivary gland epithelial cells. Arch Immunol Ther Exp (Warsz). (2020) 68:27. doi: 10.1007/s00005-020-00591-5
57. Sisto M, Ribatti D, Lisi S. Understanding the complexity of Sjögren's syndrome: remarkable progress in elucidating NF-Κb mechanisms. J Clin Med. (2020) 9(9):2821. doi: 10.3390/jcm9092821
58. Barbi J, Pardoll D, Pan F. Metabolic control of the Treg/Th17 axis. Immunol Rev. (2013) 252:52–77. doi: 10.1111/imr.12029
59. Li B, Xing Y, Gan Y, He J, Hua H. Labial gland-derived mesenchymal stem cells and their exosomes ameliorate murine Sjögren's syndrome by modulating the balance of Treg and Th17 cells. Stem Cell Res Ther. (2021) 12:478. doi: 10.1186/s13287-021-02541-0
60. Keindl M, Davies R, Bergum B, Brun JG, Hammenfors D, Jonsson R, et al. Impaired activation of stat5 upon IL-2 stimulation in Tregs and elevated sIL-2R in Sjögren's syndrome. Arthritis Res Ther. (2022) 24:101. doi: 10.1186/s13075-022-02769-y
61. Psianou K, Panagoulias I, Papanastasiou AD, de Lastic AL, Rodi M, Spantidea PI, et al. Clinical and immunological parameters of Sjögren's syndrome. Autoimmun Rev. (2018) 17:1053–64. doi: 10.1016/j.autrev.2018.05.005
62. Fei Y, Zhang W, Lin D, Wu C, Li M, Zhao Y, et al. Clinical parameter and Th17 related to lymphocytes infiltrating degree of labial salivary gland in primary Sjögren's syndrome. Clin Rheumatol. (2014) 33:523–9. doi: 10.1007/s10067-013-2476-z
63. Kawanami T, Sawaki T, Sakai T, Miki M, Iwao H, Nakajima A, et al. Skewed production of IL-6 and TGFβ by cultured salivary gland epithelial cells from patients with Sjögren's syndrome. PloS One. (2012) 7:e45689. doi: 10.1371/journal.pone.0045689
64. Lin X, Rui K, Deng J, Tian J, Wang X, Wang S, et al. Th17 cells play a critical role in the development of experimental Sjögren's syndrome. Ann Rheum Dis. (2015) 74:1302–10. doi: 10.1136/annrheumdis-2013-204584
65. Katsifis GE, Rekka S, Moutsopoulos NM, Pillemer S, Wahl SM. Systemic and local interleukin-17 and linked cytokines associated with Sjögren's syndrome immunopathogenesis. Am J Pathol. (2009) 175:1167–77. doi: 10.2353/ajpath.2009.090319
66. Xiao F, Du W, Zhu X, Tang Y, Liu L, Huang E, et al. IL-17 drives salivary gland dysfunction via inhibiting TRPC1-mediated calcium movement in Sjögren's syndrome. Clin Transl Immunol. (2021) 10:e1277. doi: 10.1002/cti2.1277
67. Zhang LW, Zhou PR, Wei P, Cong X, Wu LL, Hua H. Expression of interleukin-17 in primary Sjögren's syndrome and the correlation with disease severity: A systematic review and meta-analysis. Scand J Immunol. (2018) 87:e12649. doi: 10.1111/sji.12649
68. Kartasheva-Ebertz D, Gaston J, Lair-Mehiri L, Mottez E, Buivan TP, Massault PP, et al. IL-17a in human liver: significant source of inflammation and trigger of liver fibrosis initiation. Int J Mol Sci. (2022) 23(17):9773. doi: 10.3390/ijms23179773
69. Xiao F, Lin X, Tian J, Wang X, Chen Q, Rui K, et al. Proteasome inhibition suppresses Th17 cell generation and ameliorates autoimmune development in experimental Sjögren's syndrome. Cell Mol Immunol. (2017) 14:924–34. doi: 10.1038/cmi.2017.8
70. Ciccia F, Guggino G, Rizzo A, Alessandro R, Carubbi F, Giardina A, et al. Rituximab modulates IL-17 expression in the salivary glands of patients with primary Sjögren’s syndrome. Rheumatology. (2014) 53:1313–20. doi: 10.1093/rheumatology/keu004
71. Liang SC, Tan XY, Luxenberg DP, Karim R, Dunussi-Joannopoulos K, Collins M, et al. Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J Exp Med. (2006) 203:2271–9. doi: 10.1084/jem.20061308
72. Pipi E, Nayar S, Gardner DH, ColaFrancesco S, Smith C, Barone F. Tertiary lymphoid structures: autoimmunity goes local. Front Immunol. (2018) 9:1952. doi: 10.3389/fimmu.2018.01952
73. Fogli LK, Sundrud MS, Goel S, Bajwa S, Jensen K, Derudder E, et al. T cell-derived Il-17 mediates epithelial changes in the airway and drives pulmonary neutrophilia. J Immunol. (2013) 191(6):3100–11. doi: 10.4049/jimmunol.1301360
74. Iwakura Y, Ishigame H, Saijo S, Nakae S. Functional specialization of interleukin-17 family members. Immunity. (2011) 34:149–62. doi: 10.1016/j.immuni.2011.02.012
75. Verstappen GM, Corneth OBJ, Bootsma H, Kroese FGM. Th17 cells in primary Sjögren's syndrome: pathogenicity and plasticity. J Autoimmun. (2018) 87:16–25. doi: 10.1016/j.jaut.2017.11.003
76. Pedroso R, Ribeiro F, Pires AR, Graca L, Fonseca VR. Identification of human T follicular cells in ectopic lymphoid structures. Methods Mol Biol. (2022) 2380:225–33. doi: 10.1007/978-1-0716-1736-6_19
77. Blokland SLM, van Vliet-Moret FM, Hillen MR, Pandit A, Goldschmeding R, Kruize AA, et al. Epigenetically quantified immune cells in salivary glands of Sjögren's syndrome patients: A novel tool that detects robust correlations of T follicular helper cells with immunopathology. Rheumatol (Oxford). (2020) 59:335–43. doi: 10.1093/rheumatology/kez268
78. Qi J, Liu C, Bai Z, Li X, Yao G. T follicular helper cells and T follicular regulatory cells in autoimmune diseases. Front Immunol. (2023) 14:1178792. doi: 10.3389/fimmu.2023.1178792
79. Fonseca VR, Romão VC, Agua-Doce A, Santos M, López-Presa D, Ferreira AC, et al. The ratio of blood T follicular regulatory cells to T follicular helper cells marks ectopic lymphoid structure formation while activated follicular helper T cells indicate disease activity in primary Sjögren's syndrome. Arthritis Rheumatol. (2018) 70:774–84. doi: 10.1002/art.40424
80. Papadimitriou T-I, van Caam A, van der Kraan PM, Thurlings RM. Therapeutic options for systemic sclerosis: current and future perspectives in tackling immune-mediated fibrosis. Biomedicines. (2022) 10:316. doi: 10.3390/biomedicines10020316
81. Sisto M, Lisi S. Immune and non-immune inflammatory cells involved in autoimmune fibrosis: new discoveries. J Clin Med. (2023) 12:3801. doi: 10.3390/jcm12113801
82. Pontarini E, Murray-Brown WJ, Croia C, Lucchesi D, Conway J, Rivellese F, et al. Unique expansion of IL-21+ Tfh and Tph cells under control of ICOS identifies Sjögren’s syndrome with ectopic germinal centres and malt lymphoma. Ann Rheumatic Dis. (2020) 79:1588–99. doi: 10.1136/annrheumdis-2020-217646
83. Chen FY, Gaylord E, McNamara N, Knox S. Deciphering molecular and phenotypic changes associated with early autoimmune disease in the aire-deficient mouse model of Sjögren's syndrome. Int J Mol Sci. (2018) 19(11):3628. doi: 10.3390/ijms19113628
84. Benchabane S, Boudjelida A, Toumi R, Belguendouz H, Youinou P, Touil-Boukoffa C. A case for IL-6, IL-17a, and nitric oxide in the pathophysiology of Sjögren's syndrome. Int J Immunopathol Pharmacol. (2016) 29:386–97. doi: 10.1177/0394632016651273
85. Wei L, Xiong H, Li W, Li B, Cheng Y. Upregulation of IL-6 expression in human salivary gland cell line by IL-17 via activation of P38 MAPK, ERK, PI3K/Akt, and NF-ΚB pathways. J Oral Pathol Med. (2018) 47:847–55. doi: 10.1111/jop.12765
86. Blokland SLM, Hillen MR, Kruize AA, Meller S, Homey B, Smithson GM, et al. Increased CCL25 and T helper cells expressing CCR9 in the salivary glands of patients with primary Sjögren's syndrome: potential new axis in lymphoid neogenesis. Arthritis Rheumatol. (2017) 69:2038–51. doi: 10.1002/art.40182
87. Kramer JM, Klimatcheva E, Rothstein TL. CXCL13 is elevated in Sjögren's syndrome in mice and humans and is implicated in disease pathogenesis. J Leukoc Biol. (2013) 94:1079–89. doi: 10.1189/jlb.0113036
88. Kurosawa M, Shikama Y, Furukawa M, Arakaki R, Ishimaru N, Matsushita K. Chemokines up-regulated in epithelial cells control senescence-associated T cell accumulation in salivary glands of aged and Sjögren's syndrome model mice. Int J Mol Sci. (2021) 22(5):2302. doi: 10.3390/ijms22052302
89. Hernandez-Molina G, Soto-Abraham V, Zamora-Legoff V, Furuzawa-Carballeda J. Differential Th follicular cell subsets in minor salivary glands of patients with primary Sjögren's syndrome and systemic lupus erythematosus associated with Sjögren's syndrome. Clin Exp Rheumatol. (2021) 39 Suppl 133:49–56. doi: 10.55563/clinexprheumatol/yuj21y
90. Xiao F, Han M, Rui K, Ai X, Tian J, Zhang W, et al. New insights into follicular helper T cell response and regulation in autoimmune pathogenesis. Cell Mol Immunol. (2021) 18:1610–2. doi: 10.1038/s41423-021-00688-7
91. Zhou H, Yang J, Tian J, Wang S. CD8+ T lymphocytes: crucial players in Sjögren’s syndrome. Front Immunol. (2021) 11:602823. doi: 10.3389/fimmu.2020.602823
92. Caldeira-Dantas S, Furmanak T, Smith C, Quinn M, Teos LY, Ertel A, et al. The Chemokine Receptor CXCR3 Promotes Cd8+ T Cell accumulation in uninfected salivary glands but is not necessary after murine cytomegalovirus infection. J Immunol. (2018) 200:1133–45. doi: 10.4049/jimmunol.1701272
93. Goplen NP, Wu Y, Son YM, Li C, Wang Z, Cheon IS, et al. Tissue-resident CD8(+) T cells drive age-associated chronic lung sequelae after viral pneumonia. Sci Immunol. (2020) 5(53):eabc4557. doi: 10.1126/sciimmunol.abc4557
94. Chang L, Zheng Z, Xiao F, Zhou Y, Zhong B, Ni Q, et al. Single-cell clonal tracing of glandular and circulating T cells identifies a population of CD9+CD8+T cells in primary Sjogren's syndrome. J Leukoc Biol. (2023) 115(5):804–18. doi: 10.1093/jleuko/qiad071
95. Mauro D, Lin X, Guggino G, Chong D, Raimondo S, Ma D, et al. Op0042 blocking of CD103+ Tissue resident memory T cells (TRM) as a therapeutic strategy in Sjogren’s syndrome. Ann Rheumatic Dis. (2021) 80:22–. doi: 10.1136/annrheumdis-2021-eular.2649
96. Gao CY, Yao Y, Li L, Yang SH, Chu H, Tsuneyama K, et al. Tissue-resident memory CD8+ T cells acting as mediators of salivary gland damage in a murine model of Sjögren's syndrome. Arthritis Rheumatol. (2019) 71:121–32. doi: 10.1002/art.40676
97. Yao Y, Yang W, Yang YQ, Ma HD, Lu FT, Li L, et al. Distinct from its canonical effects, deletion of IL-12p40 induces cholangitis and fibrosis in interleukin-2rα(-/-) mice. J Autoimmun. (2014) 51:99–108. doi: 10.1016/j.jaut.2014.02.009
98. Lyakh L, Trinchieri G, Provezza L, Carra G, Gerosa F. Regulation of interleukin-12/interleukin-23 production and the T-helper 17 response in humans. Immunol Rev. (2008) 226:112–31. doi: 10.1111/j.1600-065X.2008.00700.x
99. Nanke Y, Kobashigawa T, Yago T, Kawamoto M, Yamanaka H, Kotake S. Detection of IFN-Γ+IL-17+ Cells in salivary glands of patients with Sjögren's syndrome and Mikulicz's disease: potential role of Th17•Th1 in the pathogenesis of autoimmune diseases. Nihon Rinsho Meneki Gakkai Kaishi. (2016) 39:473–7. doi: 10.2177/jsci.39.473
100. Nguyen CQ, Hu MH, Li Y, Stewart C, Peck AB. Salivary gland tissue expression of interleukin-23 and interleukin-17 in Sjögren's syndrome: findings in humans and mice. Arthritis Rheum. (2008) 58:734–43. doi: 10.1002/art.23214
101. Xu T, Zhu H-X, You X, Ma J-F, Li X, Luo P-Y, et al. Single-cell profiling reveals pathogenic role and differentiation trajectory of granzyme K+CD8+ T cells in primary Sjögren’s syndrome. JCI Insight. (2023) 8(8):e167490. doi: 10.1172/jci.insight.167490
102. Youinou P, Saraux A, Pers JO. B-lymphocytes govern the pathogenesis of Sjögren's syndrome. Curr Pharm Biotechnol. (2012) 13:2071–7. doi: 10.2174/138920112802273100
103. Gordon TP, Bolstad AI, Rischmueller M, Jonsson R, Waterman SA. Autoantibodies in primary Sjögren's syndrome: new insights into mechanisms of autoantibody diversification and disease pathogenesis. Autoimmunity. (2001) 34:123–32. doi: 10.3109/08916930109001960
104. Klein A, Klein J, Chacham M, Kleinman S, Shuster A, Peleg O, et al. Acinar atrophy, fibrosis and fatty changes are significantly more common than Sjogren's syndrome in minor salivary gland biopsies. Medicina (Kaunas). (2022) 58(2):175. doi: 10.3390/medicina58020175
105. van Ginkel MS, van der Sluis T, Bulthuis MLC, Buikema HJ, Haacke EA, Arends S, et al. Digital image analysis of intraepithelial B-lymphocytes to assess lymphoepithelial lesions in salivary glands of Sjogren's syndrome patients. Rheumatol (Oxford). (2022) 62:428–38. doi: 10.1093/rheumatology/keac212
106. Du W, Han M, Zhu X, Xiao F, Huang E, Che N, et al. The multiple roles of B cells in the pathogenesis of Sjogren's syndrome. Front Immunol. (2021) 12:684999. doi: 10.3389/fimmu.2021.684999
107. Verstappen GM, Ice JA, Bootsma H, Pringle S, Haacke EA, de Lange K, et al. Gene expression profiling of epithelium-associated FcRL4(+) B cells in primary Sjögren's syndrome reveals a pathogenic signature. J Autoimmun. (2020) 109:102439. doi: 10.1016/j.jaut.2020.102439
108. Daridon C, Pers JO, Devauchelle V, Martins-Carvalho C, Hutin P, Pennec YL, et al. Identification of transitional type II B cells in the salivary glands of patients with Sjogren's syndrome. Arthritis Rheum. (2006) 54:2280–8. doi: 10.1002/art.21936
109. Fletcher CA, Sutherland AP, Groom JR, Batten ML, Ng LG, Gommerman J, et al. Development of nephritis but not sialadenitis in autoimmune-prone baff transgenic mice lacking marginal zone B cells. Eur J Immunol. (2006) 36:2504–14. doi: 10.1002/eji.200636270
110. Kanayama N, Cascalho M, Ohmori H. Analysis of marginal zone B cell development in the mouse with limited B cell diversity: role of the antigen receptor signals in the recruitment of B cells to the marginal zone. J Immunol. (2005) 174:1438–45. doi: 10.4049/jimmunol.174.3.1438
111. Pers JO, Devauchelle V, Daridon C, Bendaoud B, Le Berre R, Bordron A, et al. Baff-modulated repopulation of B lymphocytes in the blood and salivary glands of rituximab-treated patients with Sjögren's syndrome. Arthritis Rheum. (2007) 56:1464–77. doi: 10.1002/art.22603
112. Hsu HC, Yang P, Wang J, Wu Q, Myers R, Chen J, et al. Interleukin 17-producing T helper cells and interleukin 17 orchestrate autoreactive germinal center development in autoimmune BXD2 mice. Nat Immunol. (2008) 9:166–75. doi: 10.1038/ni1552
113. Doreau A, Belot A, Bastid J, Riche B, Trescol-Biemont M-C, Ranchin B, et al. Interleukin 17 acts in synergy with B cell–activating factor to influence B cell biology and the pathophysiology of systemic lupus erythematosus. Nat Immunol. (2009) 10:778–85. doi: 10.1038/ni.1741
114. Chekol Abebe E, Asmamaw Dejenie T, Mengie Ayele T, Dagnew Baye N, Agegnehu Teshome A, Tilahun Muche Z. The role of regulatory B cells in health and diseases: A systemic review. J Inflammation Res. (2021) 14:75–84. doi: 10.2147/jir.S286426
115. Lin X, Wang X, Xiao F, Ma K, Liu L, Wang X, et al. IL-10-producing regulatory B cells restrain the T follicular helper cell response in primary Sjögren’s syndrome. Cell Mol Immunol. (2019) 16:921–31. doi: 10.1038/s41423-019-0227-z
116. Gao R, Tang J, Dong Y, Ming B, Yu Z, Zhong J, et al. The aberrant levels of decorin induce damages of human salivary gland epithelial cells and polarization of macrophages. Mod Rheumatol. (2023) 33:557–65. doi: 10.1093/mr/roac034
117. Cheng D, Chai J, Wang H, Fu L, Peng S, Ni X. Hepatic macrophages: key players in the development and progression of liver fibrosis. Liver Int. (2021) 41:2279–94. doi: 10.1111/liv.v41.10
118. Ishiguro N, Moriyama M, Furusho K, Furukawa S, Shibata T, Murakami Y, et al. Activated M2 macrophages contribute to the pathogenesis of IgG4-related disease via toll-like receptor 7/interleukin-33 signaling. Arthritis Rheumatol. (2020) 72:166–78. doi: 10.1002/art.41052
119. Li N, Gao Z, Zhao L, Du B, Ma B, Nian H, et al. MSC-derived small extracellular vesicles attenuate autoimmune dacryoadenitis by promoting M2 macrophage polarization and inducing tregs via mIR-100-5p. Front Immunol. (2022) 13:888949. doi: 10.3389/fimmu.2022.888949
120. Ogata K, Moriyama M, Kawado T, Yoshioka H, Yano A, Matsumura-Kawashima M, et al. Extracellular vesicles of iPS cells highly capable of producing HGF and TGF-Β1 can attenuate Sjögren's syndrome via innate immunity regulation. Cell Signal. (2023) 113:110980. doi: 10.1016/j.cellsig.2023.110980
121. van Caam A, Vonk M, van den Hoogen F, van Lent P, van der Kraan P. Unraveling SSc pathophysiology; the myofibroblast. Front Immunol. (2018) 9:2452. doi: 10.3389/fimmu.2018.02452
122. Ueno M, Maeno T, Nomura M, Aoyagi-Ikeda K, Matsui H, Hara K, et al. Hypoxia-inducible factor-1α Mediates TGF-Β-induced PAI-1 production in alveolar macrophages in pulmonary fibrosis. Am J Physiology-Lung Cell Mol Physiol. (2011) 300:L740–L52. doi: 10.1152/ajplung.00146.2010
123. Kelly A, Gunaltay S, McEntee CP, Shuttleworth EE, Smedley C, Houston SA, et al. Human monocytes and macrophages regulate immune tolerance via integrin Αvβ8–mediated TGFβ Activation. J Exp Med. (2018) 215:2725–36. doi: 10.1084/jem.20171491
124. Cabral-Pacheco GA, Garza-Veloz I, Castruita-De la Rosa C, Ramirez-Acuña JM, Perez-Romero BA, Guerrero-Rodriguez JF, et al. The roles of matrix metalloproteinases and their inhibitors in human diseases. Int J Mol Sci. (2020) 21(24):9739. doi: 10.3390/ijms21249739
125. Skopouli FN, Li L, Boumba D, Stefanaki S, Hanel K, Moutsopoulos HM, et al. Association of mast cells with fibrosis and fatty infiltration in the minor salivary glands of patients with Sjögren's syndrome. Clin Exp Rheumatol. (1998) 16:63–5.
126. Conti P, Stellin L, Caraffa A, Gallenga CE, Ross R, Kritas SK, et al. Advances in mast cell activation by IL-1 and IL-33 in Sjögren's syndrome: promising inhibitory effect of IL-37. Int J Mol Sci. (2020) 21(12):4297. doi: 10.3390/ijms21124297
127. Nagel H, Laskawi R, Wahlers A, Hemmerlein B. Expression of matrix metalloproteinases MMP-2, MMP-9 and their tissue inhibitors TIMP-1,-2, and-3 in benign and Malignant tumours of the salivary gland. Histopathology. (2004) 44:222–31. doi: 10.1111/j.0309-0167.2004.01814.x
128. Furukawa S, Moriyama M, Miyake K, Nakashima H, Tanaka A, Maehara T, et al. Interleukin-33 produced by M2 macrophages and other immune cells contributes to Th2 immune reaction of IgG4-related disease. Sci Rep. (2017) 7:42413. doi: 10.1038/srep42413
129. Xu D, Jiang H-R, Kewin P, Li Y, Mu R, Fraser AR, et al. Il-33 exacerbates antigen-induced arthritis by activating mast cells. Proc Natl Acad Sci. (2008) 105:10913–8. doi: 10.1073/pnas.0801898105
130. Fu X, Liu H, Huang G, Dai SS. The emerging role of neutrophils in autoimmune-associated disorders: effector, predictor, and therapeutic targets. MedComm (2020). (2021) 2:402–13. doi: 10.1002/mco2.69
131. Torsteinsdottir I, Gudbjornsson B, Hakansson L. Enhanced neutrophil and eosinophil adhesion in patients with primary Sjogren's syndrome. Clin Exp Rheumatol. (1998) 16:255–62.
132. Gudbjornsson B, Feltelius N, Hallgren R, Venge P. Neutrophil function in patients with primary Sjogren's syndrome: relation to infection propensity. Ann Rheum Dis. (1991) 50:685–90. doi: 10.1136/ard.50.10.685
133. Jayaprakash K, Demirel I, Khalaf H, Bengtsson T. The role of phagocytosis, oxidative burst and neutrophil extracellular traps in the interaction between neutrophils and the periodontal pathogen porphyromonas gingivalis. Mol Oral Microbiol. (2015) 30:361–75. doi: 10.1111/omi.12099
134. Chakrabarti S, Zee JM, Patel KD. Regulation of matrix metalloproteinase-9 (MMP-9) in TNF-stimulated neutrophils: novel pathways for tertiary granule release. J Leukoc Biol. (2006) 79:214–22. doi: 10.1189/jlb.0605353
135. Rischmueller M, Tieu J, Lester S. Primary Sjogren's syndrome. Best Pract Res Clin Rheumatol. (2016) 30:189–220. doi: 10.1016/j.berh.2016.04.003
136. Peng Y, Wu X, Zhang S, Deng C, Zhao L, Wang M, et al. The potential roles of type I interferon activated neutrophils and neutrophil extracellular traps (Nets) in the pathogenesis of primary Sjogren's syndrome. Arthritis Res Ther. (2022) 24:170. doi: 10.1186/s13075-022-02860-4
137. Wang L, Luqmani R, Udalova IA. The role of neutrophils in rheumatic disease-associated vascular inflammation. Nat Rev Rheumatol. (2022) 18:158–70. doi: 10.1038/s41584-021-00738-4
138. Khandpur R, Carmona-Rivera C, Vivekanandan-Giri A, Gizinski A, Yalavarthi S, Knight JS, et al. Nets are a source of citrullinated autoantigens and stimulate inflammatory responses in rheumatoid arthritis. Sci Transl Med. (2013) 5:178ra40. doi: 10.1126/scitranslmed.3005580
139. Su F, Xiao W, Yang P, Chen Q, Sun X, Li T. Anti-neutrophil cytoplasmic antibodies in new-onset systemic lupus erythematosus. Bras Dermatol. (2017) 92:466–9. doi: 10.1590/abd1806-4841.20175476
140. Konttinen YT, Kangaspunta P, Lindy O, Takagi M, Sorsa T, Segerberg M, et al. Collagenase in Sjögren's syndrome. Ann Rheum Dis. (1994) 53:836–9. doi: 10.1136/ard.53.12.836
141. Klein K. In: Fibroblasts in Sjogren's Syndrome, Fibroblasts-Advances in Infammation, Autoimmunity and Cancer. Bertoncelj M. F., Lakota K. editor. (London, UK: IntechOpen). (2021).
142. Korsunsky I, Wei K, Pohin M, Kim EY, Barone F, Major T, et al. Cross-tissue, single-cell stromal atlas identifies shared pathological fibroblast phenotypes in four chronic inflammatory diseases. Med. (2022) 3:481–518.e14. doi: 10.1016/j.medj.2022.05.002
143. Yamate J, Iwaki M, Kumagai D, Tsukamoto Y, Kuwamura M, Nakatsuji S, et al. Characteristics of a rat fibrosarcoma-derived transplantable tumour line (SS) and cultured cell lines (SS-P and SS-A3-1) showing myofibroblastic and histiocytic phenotypes. Virchows Arch. (1997) 431:431–40. doi: 10.1007/s004280050120
144. Wang J-C, Lai S, Guo X, Zhang X, de Crombrugghe B, Sonnylal S, et al. Attenuation of fibrosis in vitro and in vivo with SPARC siRNA. Arthritis Res Ther. (2010) 12:R60. doi: 10.1186/ar2973
145. Jin JO, Kawai T, Cha S, Yu Q. Interleukin-7 enhances the Th1 response to promote the development of Sjögren's syndrome-like autoimmune exocrinopathy in mice. Arthritis Rheum. (2013) 65:2132–42. doi: 10.1002/art.38007
146. Zhou J, Yu Q. Anti-IL-7 receptor-Α Treatment ameliorates newly established Sjögren's-like exocrinopathy in non-obese diabetic mice. Biochim Biophys Acta Mol Basis Dis. (2018) 1864:2438–47. doi: 10.1016/j.bbadis.2018.04.010
147. Liang Y, Zhang Z, Li J, Luo W, Jiang T, Yang Z. Association between IL-7 and primary Sjögren's syndrome: A single-center study and a systematic scoping review. Int Immunopharmacol. (2022) 108:108758. doi: 10.1016/j.intimp.2022.108758
148. Bikker A, van Woerkom JM, Kruize AA, Wenting-van Wijk M, de Jager W, Bijlsma JW, et al. Increased expression of interleukin-7 in labial salivary glands of patients with primary Sjögren's syndrome correlates with increased inflammation. Arthritis Rheum. (2010) 62:969–77. doi: 10.1002/art.27318
149. Manoussakis MN, Kapsogeorgou EK. The role of epithelial cells in the pathogenesis of Sjögren's syndrome. Clin Rev Allergy Immunol. (2007) 32:225–30. doi: 10.1007/s12016-007-8007-4
150. Kapsogeorgou EK, Tzioufas AG. Interaction of human salivary gland epithelial cells with B lymphocytes: implications in the pathogenesis of Sjögren's syndrome. Mediterr J Rheumatol. (2020) 31:424–6. doi: 10.31138/mjr.31.4.424
151. Nakamura H, Horai Y, Shimizu T, Kawakami A. Modulation of apoptosis by cytotoxic mediators and cell-survival molecules in Sjögren's syndrome. Int J Mol Sci. (2018) 19(8):2369. doi: 10.3390/ijms19082369
152. Hu P, Ming B, Wu X, Cai S, Tang J, Dong Y, et al. Intratracheal poly(I:C) exposure accelerates the immunological disorder of salivary glands in Sjogren's-like NOD/ShiLtJ mice. Front Med (Lausanne). (2021) 8:645816. doi: 10.3389/fmed.2021.645816
153. Kapsogeorgou EK, Abu-Helu RF, Moutsopoulos HM, Manoussakis MN. Salivary gland epithelial cell exosomes: A source of autoantigenic ribonucleoproteins. Arthritis Rheum. (2005) 52:1517–21. doi: 10.1002/art.21005
154. Bonnans C, Chou J, Werb Z. Remodelling the extracellular matrix in development and disease. Nat Rev Mol Cell Biol. (2014) 15:786–801. doi: 10.1038/nrm3904
155. Patel VN, Hoffman MP. Salivary gland development: A template for regeneration. Semin Cell Dev Biol. (2014) 25–26:52–60. doi: 10.1016/j.semcdb.2013.12.001
156. Dinescu ŞC, ForŢofoiu MC, Bumbea AM, Ciurea PL, Busuioc CJ, Muşetescu AE. Histopathological and immunohistochemical profile in primary Sjögren's syndrome. Rom J Morphol Embryol. (2017) 58:409–17.
157. Laine M, Virtanen I, Porola P, Rotar Z, Rozman B, Poduval P, et al. Acinar epithelial cell laminin-receptors in labial salivary glands in Sjögren's syndrome. Clin Exp Rheumatol. (2008) 26:807–13.
158. Marinkovic M, Tran ON, Wang H, Abdul-Azees P, Dean DD, Chen XD, et al. Extracellular matrix turnover in salivary gland disorders and regenerative therapies: obstacles and opportunities. J Oral Biol Craniofac Res. (2023) 13:693–703. doi: 10.1016/j.jobcr.2023.08.009
159. Genç D, Bulut O, Günaydin B, Göksu M, Düzgün M, Dere Y, et al. Dental follicle mesenchymal stem cells ameliorated glandular dysfunction in Sjögren's syndrome murine model. PloS One. (2022) 17:e0266137. doi: 10.1371/journal.pone.0266137
160. Lozito TP, Jackson WM, Nesti LJ, Tuan RS. Human mesenchymal stem cells generate a distinct pericellular zone of MMP activities via binding of MMPs and secretion of high levels of TIMPs. Matrix Biol. (2014) 34:132–43. doi: 10.1016/j.matbio.2013.10.003
161. Maciejczyk M, Pietrzykowska A, Zalewska A, Knaś M, Daniszewska I. The significance of matrix metalloproteinases in oral diseases. Adv Clin Exp Med. (2016) 25:383–90. doi: 10.17219/acem/30428
162. Ming B, Zhu Y, Zhong J, Dong L. Immunopathogenesis of Sjogren's syndrome: current state of DAMPs. Semin Arthritis Rheum. (2022) 56:152062. doi: 10.1016/j.semarthrit.2022.152062
163. Kiripolsky J, Kasperek EM, Zhu C, Li Q-Z, Wang J, Yu G, et al. Immune-intrinsic Myd88 directs the production of antibodies with specificity for extracellular matrix components in primary Sjögren’s syndrome. Front Immunol. (2021) 12:692216. doi: 10.3389/fimmu.2021.692216
164. Yoshida K, Wang X, Bhawal UK. Dec1 deficiency restores the age-related dysfunctions of submandibular glands. J Physiol Pharmacol. (2021) 72(4):571–581. doi: 10.26402/jpp.2021.4.09
165. Lin X, Shaw PC, Sze SC, Tong Y, Zhang Y. Dendrobium officinale polysaccharides ameliorate the abnormality of aquaporin 5, pro-inflammatory cytokines and inhibit apoptosis in the experimental Sjögren's syndrome mice. Int Immunopharmacol. (2011) 11:2025–32. doi: 10.1016/j.intimp.2011.08.014
166. Xiao L, Ng TB, Feng YB, Yao T, Wong JH, Yao RM, et al. Dendrobium candidum extract increases the expression of aquaporin-5 in labial glands from patients with Sjögren's syndrome. Phytomedicine. (2011) 18:194–8. doi: 10.1016/j.phymed.2010.05.002
167. Rivière E, Chivasso C, Pascaud J, Bechara R, Ly B, Delporte C, et al. Hyperosmolar environment and salivary gland epithelial cells increase extra-cellular matrix remodeling and lymphocytic infiltration in Sjögren’s syndrome. Clin Exp Immunol. (2023) 212:39–51. doi: 10.1093/cei/uxad020
168. Sasaki M, Nakamura S, Ohyama Y, Shinohara M, Ezaki I, Hara H, et al. Accumulation of common T cell clonotypes in the salivary glands of patients with human T lymphotropic virus type I-associated and idiopathic Sjögren's syndrome. J Immunol. (2000) 164:2823–31. doi: 10.4049/jimmunol.164.5.2823
169. Yeh C-C, Wang W-C, Wu C-S, Sung F-C, Su C-T, Shieh Y-H, et al. Association of Sjögrens syndrome in patients with chronic hepatitis virus infection: A population-based analysis. PloS One. (2016) 11:e0161958. doi: 10.1371/journal.pone.0161958
170. Barcelos F, Martins C, Monteiro R, Cardigos J, PRussiani T, Sítima M, et al. Association between EBV serological patterns and lymphocytic profile of SjS patients support a virally triggered autoimmune epithelitis. Sci Rep. (2021) 11:4082. doi: 10.1038/s41598-021-83550-0
171. Nakamura H, Shimizu T, Takagi Y, Takahashi Y, Horai Y, Nakashima Y, et al. Reevaluation for clinical manifestations of HTLV-I-seropositive patients with Sjögren's syndrome. BMC Musculoskelet Disord. (2015) 16:335. doi: 10.1186/s12891-015-0773-1
172. Björk A, Mofors J, Wahren-Herlenius M. Environmental factors in the pathogenesis of primary Sjögren's syndrome. J Intern Med. (2020) 287:475–92. doi: 10.1111/joim.13032
173. Kipps TJ, Tomhave E, Chen PP, Fox RI. Molecular characterization of a major autoantibody-associated cross-reactive idiotype in Sjogren's syndrome. J Immunol. (1989) 142:4261–8. doi: 10.4049/jimmunol.142.12.4261
174. Otsuka K, Sato M, Tsunematsu T, Ishimaru N. Virus infections play crucial roles in the pathogenesis of Sjogren's syndrome. Viruses. (2022) 14(7):1474. doi: 10.3390/v14071474
175. Bombardieri M, Barone F, Lucchesi D, Nayar S, van den Berg WB, Proctor G, et al. Inducible tertiary lymphoid structures, autoimmunity, and exocrine dysfunction in a novel model of salivary gland inflammation in C57BL/6 mice. J Immunol. (2012) 189:3767–76. doi: 10.4049/jimmunol.1201216
176. Bombardieri M, Pitzalis C. Ectopic lymphoid neogenesis and lymphoid chemokines in Sjogren's syndrome: at the interplay between chronic inflammation, autoimmunity and lymphomagenesis. Curr Pharm Biotechnol. (2012) 13:1989–96. doi: 10.2174/138920112802273209
177. Flores-Chavez A, Carrion JA, Forns X, Ramos-Casals M. Extrahepatic manifestations associated with chronic hepatitis C virus infection. Rev Esp Sanid Penit. (2017) 19:87–97. doi: 10.4321/S1575-06202017000300004
178. Mizoguchi I, Ohashi M, Hasegawa H, Chiba Y, Orii N, Inoue S, et al. EBV-induced gene 3 augments IL-23ralpha protein expression through a chaperone calnexin. J Clin Invest. (2020) 130:6124–40. doi: 10.1172/JCI122732
179. Jog NR, James JA. Epstein barr virus and autoimmune responses in systemic lupus erythematosus. Front Immunol. (2020) 11:623944. doi: 10.3389/fimmu.2020.623944
180. Maslinska M, Kostyra-Grabczak K. The role of virus infections in Sjogren's syndrome. Front Immunol. (2022) 13:823659. doi: 10.3389/fimmu.2022.823659
181. Zeron PB, Retamozo S, Bove A, Kostov BA, Siso A, Ramos-Casals M. Diagnosis of liver involvement in primary Sjogren syndrome. J Clin Transl Hepatol. (2013) 1:94–102. doi: 10.14218/JCTH.2013.00011
182. Maitland N, Flint S, Scully C, Crean SJ. Detection of cytomegalovirus and epstein-barr virus in labial salivary glands in Sjogren's syndrome and non-specific sialadenitis. J Oral Pathol Med. (1995) 24:293–8. doi: 10.1111/j.1600-0714.1995.tb01187.x
183. Nakamura H, Horai Y, Tokuyama A, Yoshimura S, Nakajima H, Ichinose K, et al. HTLV-I virological and histopathological analysis in two cases of anti-centromere-antibody-seropositive Sjogren's syndrome. Mod Rheumatol. (2013) 23:133–9. doi: 10.1007/s10165-012-0641-x
184. Nakamura H, Tsukamoto M, Nagasawa Y, Kitamura N, Shimizu T, Kawakami A, et al. Does HTLV-1 infection show phenotypes found in Sjogren's syndrome? Viruses. (2022) 14(1):100. doi: 10.3390/v14010100
185. Rivera H, Nikitakis NG, Castillo S, Siavash H, Papadimitriou JC, Sauk JJ. Histopathological analysis and demonstration of EBV and HIV p-24 antigen but not CMV expression in labial minor salivary glands of HIV patients affected by diffuse infiltrative lymphocytosis syndrome. J Oral Pathol Med. (2003) 32:431–7. doi: 10.1034/j.1600-0714.2003.00061.x
186. Jeffers L, Webster-Cyriaque JY. Viruses and salivary gland disease (SGD): lessons from HIV SGD. Adv Dent Res. (2011) 23:79–83. doi: 10.1177/0022034510396882
187. McArthur CP, Africa CW, Castellani WJ, Luangjamekorn NJ, McLaughlin M, Subtil-DeOliveira A, et al. Salivary gland disease in HIV/AIDS and primary Sjogren's syndrome: analysis of collagen I distribution and histopathology in American and African patients. J Oral Pathol Med. (2003) 32:544–51. doi: 10.1034/j.1600-0714.2003.00159.x
188. Konishi K, Kuwahara H, Fujimoto Y, Nagata K, Takeda J. Severe Covid-19 as a possible mediator of autoimmunity and Sjögren's syndrome. Cureus. (2023) 15:e35290. doi: 10.7759/cureus.35290
189. Shen Y, Voigt A, Goranova L, Abed M, Kleiner DE, Maldonado JO, et al. Evidence of a Sjögren's disease-like phenotype following Covid-19. medRxiv. (2022) 8(24):e166540. doi: 10.1101/2022.10.20.22281265
190. To KK, Tsang OT, Yip CC, Chan KH, Wu TC, Chan JM, et al. Consistent detection of 2019 novel coronavirus in saliva. Clin Infect Dis. (2020) 71:841–3. doi: 10.1093/cid/ciaa149
191. Liu L, Wei Q, Alvarez X, Wang H, Du Y, Zhu H, et al. Epithelial cells lining salivary gland ducts are early target cells of severe acute respiratory syndrome coronavirus infection in the upper respiratory tracts of rhesus macaques. J Virol. (2011) 85:4025–30. doi: 10.1128/jvi.02292-10
192. Channappanavar R, Perlman S. Pathogenic human coronavirus infections: causes and consequences of cytokine storm and immunopathology. Semin Immunopathol. (2017) 39:529–39. doi: 10.1007/s00281-017-0629-x
193. Wang C, Wu H, Ding X, Ji H, Jiao P, Song H, et al. Does infection of 2019 novel coronavirus cause acute and/or chronic sialadenitis? Med Hypotheses. (2020) 140:109789. doi: 10.1016/j.mehy.2020.109789
194. Mostafa OAA, Ibrahim F, Borai E. Protective effects of hesperidin in cyclophosphamide-induced parotid toxicity in rats. Sci Rep. (2023) 13:158. doi: 10.1038/s41598-022-26881-w
195. Ungureanu LB, Gradinaru I, Ghiciuc CM, Amalinei C, Geletu GL, Petrovici CG, et al. Atrophy and inflammatory changes in salivary glands induced by oxidative stress after exposure to drugs and other chemical substances: A systematic review. Medicina (Kaunas). (2023) 59(9):1692. doi: 10.3390/medicina59091692
196. Abdelzaher WY, Nassan MA, Ahmed SM, Welson NN, El-Saber Batiha G, Khalaf HM. Xanthine oxidase inhibitor, febuxostat is effective against 5-fluorouracil-induced parotid salivary gland injury in rats via inhibition of oxidative stress, inflammation and targeting TRPC1/CHOP signalling pathway. Pharm (Basel). (2022) 15(2):232. doi: 10.3390/ph15020232
197. Ryo K, Yamada H, Nakagawa Y, Tai Y, Obara K, Inoue H, et al. Possible involvement of oxidative stress in salivary gland of patients with Sjogren's syndrome. Pathobiology. (2006) 73:252–60. doi: 10.1159/000098211
198. Li N, Li Y, Hu J, Wu Y, Yang J, Fan H, et al. A link between mitochondrial dysfunction and the immune microenvironment of salivary glands in primary Sjogren's syndrome. Front Immunol. (2022) 13:845209. doi: 10.3389/fimmu.2022.845209
199. Álvarez-Rivas N, Sang-Park H, Díaz del Campo P, Fernández-Castro M, Corominas H, Andreu JL, et al. Efficacy of belimumab in primary Sjögren's syndrome: A systematic review. Reumatología Clínica. (2021) 17:170–4. doi: 10.1016/j.reuma.2020.03.003
200. Chen S, Liu Y, Shi G. Anti-CD20 antibody in primary Sjögren's syndrome management. Curr Pharm Biotechnol. (2014) 15:535–41. doi: 10.2174/138920101506140910150431
201. Steinfeld SD, Tant L, Burmester GR, Teoh NK, Wegener WA, Goldenberg DM, et al. Epratuzumab (Humanised anti-CD22 antibody) in primary Sjögren's syndrome: an open-label phase I/II study. Arthritis Res Ther. (2006) 8:R129. doi: 10.1186/ar2018
202. Noll B, Mougeot FB, Brennan MT, Mougeot JC. Regulation of MMP9 transcription by ETS1 in immortalized salivary gland epithelial cells of patients with salivary hypofunction and primary Sjögren's syndrome. Sci Rep. (2022) 12:14552. doi: 10.1038/s41598-022-18576-z
203. Sarode SC, Sharma NK, Sarode G, Sharma M, Radhakrishnan R. Targeting the immune-privileged myofibroblast in oral submucous fibrosis by car T-cell therapy. Med Hypotheses. (2022) 165:110897. doi: 10.1016/j.mehy.2022.110897
204. Meng X-M, Nikolic-Paterson DJ, Lan HY. TGF-Β: the master regulator of fibrosis. Nat Rev Nephrol. (2016) 12:325–38. doi: 10.1038/nrneph.2016.48
205. Chihaby N, Orliaguet M, Le Pottier L, Pers JO, Boisramé S. Treatment of Sjögren's syndrome with mesenchymal stem cells: A systematic review. Int J Mol Sci. (2021) 22(19):10474. doi: 10.3390/ijms221910474
206. Xu J, Wang D, Liu D, Fan Z, Zhang H, Liu O, et al. Allogeneic mesenchymal stem cell treatment alleviates experimental and clinical Sjögren syndrome. Blood. (2012) 120:3142–51. doi: 10.1182/blood-2011-11-391144
207. Ruan GF, Zheng L, Huang JS, Huang WX, Gong BD, Fang XX, et al. Effect of mesenchymal stem cells on Sjögren-like mice and the microrna expression profiles of splenic CD4+ T cells. Exp Ther Med. (2017) 13:2828–38. doi: 10.3892/etm.2017.4313
208. van der Helm D, Groenewoud A, de Jonge-Muller ESM, Barnhoorn MC, Schoonderwoerd MJA, Coenraad MJ, et al. Mesenchymal stromal cells prevent progression of liver fibrosis in a novel zebrafish embryo model. Sci Rep. (2018) 8:16005. doi: 10.1038/s41598-018-34351-5
209. Du ZH, Ding C, Zhang Q, Zhang Y, Ge XY, Li SL, et al. Stem cells from exfoliated Deciduous Teeth alleviate hyposalivation caused by Sjögren syndrome. Oral Dis. (2019) 25:1530–44. doi: 10.1111/odi.13113
210. Khalili S, Liu Y, Kornete M, Roescher N, Kodama S, Peterson A, et al. Mesenchymal stromal cells improve salivary function and reduce lymphocytic infiltrates in mice with Sjögren's-like disease. PloS One. (2012) 7:e38615. doi: 10.1371/journal.pone.0038615
211. Yao G, Qi J, Liang J, Shi B, Chen W, Li W, et al. Mesenchymal stem cell transplantation alleviates experimental Sjögren's syndrome through IFN-Β/IL-27 signaling axis. Theranostics. (2019) 9:8253–65. doi: 10.7150/thno.37351
212. Tian J, Hong Y, Zhu Q, Zhou H, Zhang Y, Shen Z, et al. Mesenchymal stem cell enhances the function of MDSCs in experimental Sjögren syndrome. Front Immunol. (2020) 11:604607. doi: 10.3389/fimmu.2020.604607
213. Shi B, Qi J, Yao G, Feng R, Zhang Z, Wang D, et al. Mesenchymal stem cell transplantation ameliorates Sjögren's syndrome via suppressing IL-12 production by dendritic cells. Stem Cell Res Ther. (2018) 9:308. doi: 10.1186/s13287-018-1023-x
214. Kyurkchiev D, Bochev I, Ivanova-Todorova E, Mourdjeva M, Oreshkova T, Belemezova K, et al. Secretion of immunoregulatory cytokines by mesenchymal stem cells. World J Stem Cells. (2014) 6:552–70. doi: 10.4252/wjsc.v6.i5.552
215. Wang X, Ma Y, Gao Z, Yang J. Human adipose-derived stem cells inhibit bioactivity of keloid fibroblasts. Stem Cell Res Ther. (2018) 9:40. doi: 10.1186/s13287-018-0786-4
216. Bui TM, Wiesolek HL, Sumagin R. ICAM-1: A master regulator of cellular responses in inflammation, injury resolution, and tumorigenesis. J Leukoc Biol. (2020) 108:787–99. doi: 10.1002/jlb.2mr0220-549r
217. Kore RA, Wang X, Ding Z, Griffin RJ, Tackett AJ, Mehta JL. MSC exosome-mediated cardioprotection in ischemic mouse heart comparative proteomics of infarct and peri-infarct areas. Mol Cell Biochem. (2021) 476:1691–704. doi: 10.1007/s11010-020-04029-6
218. Qin L, Liu N, Bao C-L-M, Yang D-Z, Ma G-X, Yi W-H, et al. Mesenchymal stem cells in fibrotic diseases—the two sides of the same coin. Acta Pharmacologica Sin. (2023) 44:268–87. doi: 10.1038/s41401-022-00952-0
219. Salazar KD, Lankford SM, Brody AR. Mesenchymal stem cells produce Wnt isoforms and TGF-Β1 that mediate proliferation and procollagen expression by lung fibroblasts. Am J Physiology-Lung Cell Mol Physiol. (2009) 297:L1002–L11. doi: 10.1152/ajplung.90347.2008
220. Ben-David U, Mayshar Y, Benvenisty N. Large-scale analysis reveals acquisition of lineage-specific chromosomal aberrations in human adult stem cells. Cell Stem Cell. (2011) 9:97–102. doi: 10.1016/j.stem.2011.06.013
221. Liang J, Zhang H, Kong W, Deng W, Wang D, Feng X, et al. Safety analysis in patients with autoimmune disease receiving allogeneic mesenchymal stem cells infusion: A long-term retrospective study. Stem Cell Res Ther. (2018) 9:312. doi: 10.1186/s13287-018-1053-4
222. Alfaifi A, Sultan AS, Montelongo-Jauregui D, Meiller TF, Jabra-Rizk MA. Long-term post-covid-19 associated oral inflammatory sequelae. Front Cell Infect Microbiol. (2022) 12:831744. doi: 10.3389/fcimb.2022.831744
223. Martonik D, Parfieniuk-Kowerda A, Rogalska M, Flisiak R. The role of Th17 response in covid-19. Cells. (2021) 10(6):1550. doi: 10.3390/cells10061550
224. Pi P, Zeng Z, Zeng L, Han B, Bai X, Xu S. Molecular mechanisms of covid-19-induced pulmonary fibrosis and epithelial-mesenchymal transition. Front Pharmacol. (2023) 14:1218059. doi: 10.3389/fphar.2023.1218059
225. Katona K, Farkas N, Kneif M, SütŐ G, Berki T, Balatonyi B, et al. Image analysis of fibrosis in labial salivary glands of patients with systemic autoimmune diseases. Close correlation of lobular fibrosis to seropositive rheumatoid arthritis and increased anti-CCP and RF titres in the serum. Pathology. (2018) 50:418–25. doi: 10.1016/j.pathol.2017.12.339
226. Aloisi F, Pujol-Borrell R. Lymphoid neogenesis in chronic inflammatory diseases. Nat Rev Immunol. (2006) 6:205–17. doi: 10.1038/nri1786
227. Ainola M, Porola P, Takakubo Y, Przybyla B, Kouri VP, Tolvanen TA, et al. Activation of plasmacytoid dendritic cells by apoptotic particles - mechanism for the loss of immunological tolerance in Sjögren's syndrome. Clin Exp Immunol. (2018) 191:301–10. doi: 10.1111/cei.13077
228. Zhao J, Kubo S, Nakayamada S, Shimajiri S, Zhang X, Yamaoka K, et al. Association of plasmacytoid dendritic cells with B cell infiltration in minor salivary glands in patients with Sjögren's syndrome. Mod Rheumatol. (2016) 26:716–24. doi: 10.3109/14397595.2015.1129694
229. Rusakiewicz S, Nocturne G, Lazure T, Semeraro M, Flament C, Caillat-Zucman S, et al. NCR3/NKp30 contributes to pathogenesis in primary Sjögren’s syndrome. Sci Trans Med. (2013) 5:195ra96–ra96. doi: 10.1126/scitranslmed.3005727
230. Kapsogeorgou EK, Christodoulou MI, Panagiotakos DB, Paikos S, Tassidou A, Tzioufas AG, et al. Minor salivary gland inflammatory lesions in Sjogren syndrome: do they evolve? J Rheumatol. (2013) 40:1566–71. doi: 10.3899/jrheum.130256
Keywords: Sjögren’s syndrome, Sjögren’s disease (SjD), salivary gland, fibrosis, infection, epithelial-mesenchymal transition, extracellular matrix remodeling
Citation: Ma D, Feng Y and Lin X (2024) Immune and non-immune mediators in the fibrosis pathogenesis of salivary gland in Sjögren’s syndrome. Front. Immunol. 15:1421436. doi: 10.3389/fimmu.2024.1421436
Received: 22 April 2024; Accepted: 30 September 2024;
Published: 14 October 2024.
Edited by:
Efstathia K. Kapsogeorgou, National and Kapodistrian University of Athens, GreeceReviewed by:
Zhongyu Han, Chengdu University of Traditional Chinese Medicine, ChinaKimberly Julia Jasmer, University of Missouri, United States
Maria-Ioanna (Marianna) Christodoulou, European University Cyprus, Cyprus
Copyright © 2024 Ma, Feng and Lin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiang Lin, bGlueGlhbmdAaGt1Lmhr; Yun Feng, ZmVuZ3l1bkBiam11LmVkdS5jbg==