 Karanvir Gill
Karanvir Gill Hyung-Suk Yoo
Hyung-Suk Yoo Harshini Chakravarthy1
Harshini Chakravarthy1- 1Department of Ophthalmology and Visual Sciences, University of British Columbia (UBC), Vancouver, BC, Canada
- 2International Collaboration on Repair Discoveries (ICORD), Vancouver Coastal Health Research Institute, University of British Columbia, Vancouver, BC, Canada
- 3Department of Pathology and Laboratory Medicine, University of British Columbia, Vancouver, BC, Canada
Age-related macular degeneration (AMD), a prevalent and progressive degenerative disease of the macula, is the leading cause of blindness in elderly individuals in developed countries. The advanced stages include neovascular AMD (nAMD), characterized by choroidal neovascularization (CNV), leading to subretinal fibrosis and permanent vision loss. Despite the efficacy of anti-vascular endothelial growth factor (VEGF) therapy in stabilizing or improving vision in nAMD, the development of subretinal fibrosis following CNV remains a significant concern. In this review, we explore multifaceted aspects of subretinal fibrosis in nAMD, focusing on its clinical manifestations, risk factors, and underlying pathophysiology. We also outline the potential sources of myofibroblast precursors and inflammatory mechanisms underlying their recruitment and transdifferentiation. Special attention is given to the potential role of mast cells in CNV and subretinal fibrosis, with a focus on putative mast cell mediators, tryptase and granzyme B. We summarize our findings on the role of GzmB in CNV and speculate how GzmB may be involved in the pathological transition from CNV to subretinal fibrosis in nAMD. Finally, we discuss the advantages and drawbacks of animal models of subretinal fibrosis and pinpoint potential therapeutic targets for subretinal fibrosis.
Introduction
Age-related macular degeneration (AMD) is a progressive degenerative disease of the macula and is among the leading causes of blindness in individuals aged 50 years and older globally (1). With aging populations and longer life expectancies, AMD is becoming an increasingly significant public health concern and is expected to impact 288 million individuals globally by 2040 (2). The early/intermediate stages of AMD are characterized by extracellular apolipoproteins and oxidized proteins, collectively known as drusen, that accumulate and cause disturbance between the basal lamina of the retinal pigment epithelium (RPE) and the Bruch’s membrane (3, 4). The disease can further advance and cause more severe vision loss in its later stages, classified into an atrophic or dry form and a neovascular or wet form of AMD (nAMD). In dry AMD, there are slowly expanding circumscribed areas of atrophy, leading to the loss of cells such as photoreceptors, RPE, and the choriocapillaris, known as geographic atrophy (5). On the other hand, wet AMD involves choroidal neovascularization (CNV), which is also known as macular neovascularization. The neovascularization in wet AMD can be categorized into three subtypes. Type 1 or occult CNV involves the development of new blood vessels beneath the RPE. Type 2 or classic CNV involves the disruption of Bruch’s membrane and the proliferation of blood vessels into the subretinal space. Type 3 neovascularization, also known as intraretinal angiomatous proliferation, occurs within the neuroretina (6). In every case, these newly formed immature blood vessels lead to blood leakage and hemorrhage, eventually attracting stromal cells and immune cells and triggering the neovascular endothelium to transform into a fibrovascular membrane (7). Although fibrosis may help restrict exudation from CNV, excessive fibrosis can lead to subretinal scarring and damage choriocapillaris, RPE and photoreceptors (Figure 1) (8). Fibrosis demarcates the end-stage of nAMD, resulting in permanent vision loss.
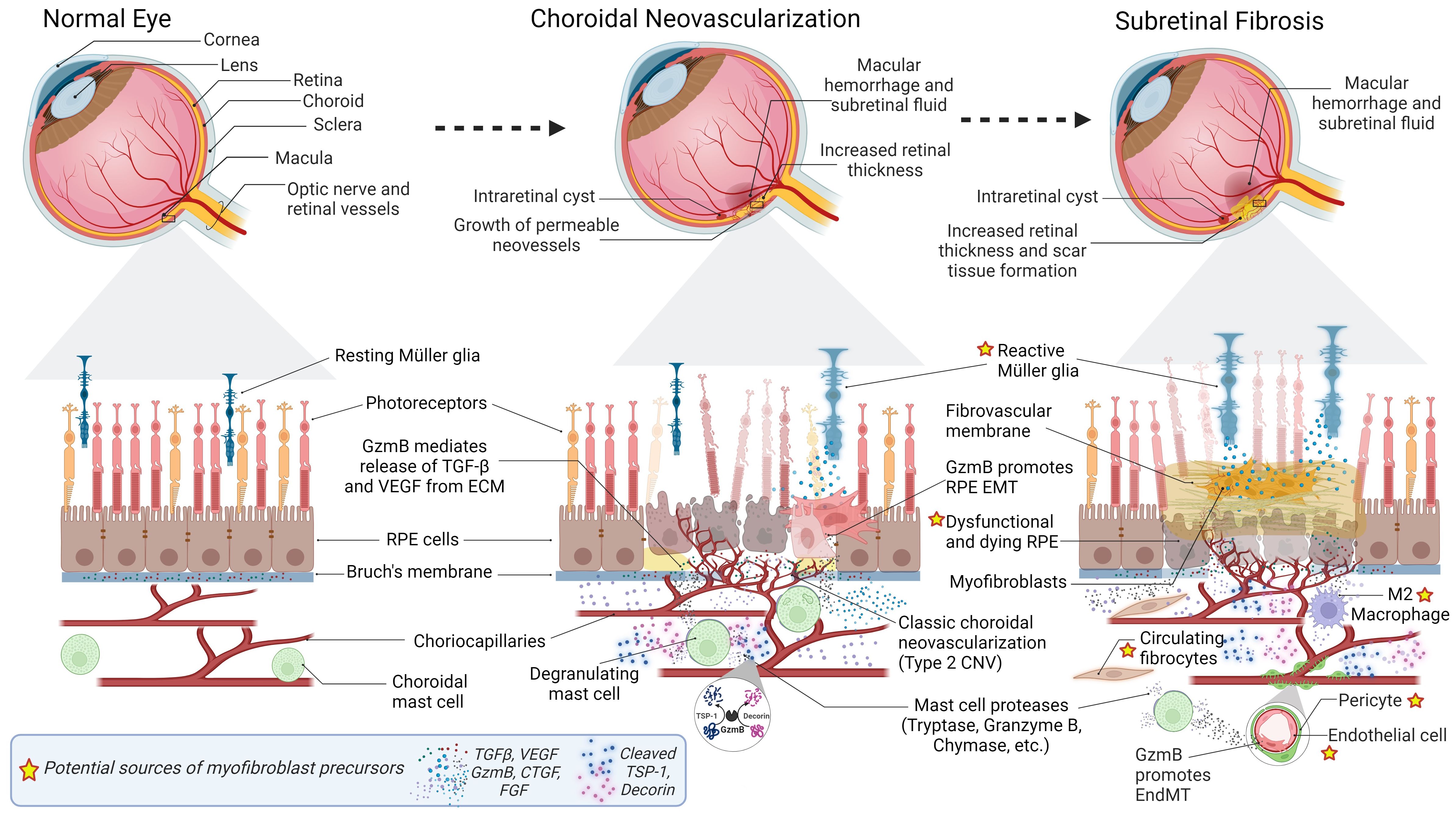
Figure 1 Progression of Subretinal Fibrosis in Age-Related Macular Degeneration (AMD). Normal Eye: Cross-sectional representation of a normal eye. Below, a magnified view depicts the composition of a normal retina, including resting Muller glia, photoreceptors, RPE cells, Bruch’s membrane, choriocapillaris, and choroidal mast cells. Growth factors (TGF- β, VEGF) sequestered in the Bruch’s membrane ECM are depicted. Choroidal Neovascularization (CNV): Pathological changes associated with CNV depicted in the eye cross-section include macular hemorrhage, subretinal fluid, increased retinal thickness, intraretinal cyst, and permeable neovessels. The magnified view of the retina focuses on classic choroidal neovascularization (Type 2 CNV), reactive Muller glia, and dysfunctional and dying RPE. GzmB promotes CNV, inflammation and fibrosis by mediating release of growth factors from the ECM, and fragmentation of anti-angiogenic proteins DCN and TSP-1. Subretinal Fibrosis: The eye cross-section illustrates scar tissue formation, intraretinal cyst, macular hemorrhage, and subretinal fluid. The corresponding details of the magnified retina reveal reactive Muller glia, fibrovascular membrane, dysfunctional and dying RPE, myofibroblasts, circulating fibrocytes, M2 macrophages, pericytes, endothelial cells, Labels with a star indicate potential sources of myofibroblast precursors. Increased secretion of growth factors (VEGF, TGF-β, CTGF, FGF), and mast cell proteases (tryptase, Granzyme B, chymase) are also depicted. GzmB promotes degradation of tight junction proteins, leading to EMT of RPE cells and EndMT of endothelial cells lining the choroidal vasculature. GzmB-mediated fragmentation of DCN and TSP-1 may also contribute to myofibroblast transdifferentiation.
If left untreated, nAMD typically leads to a swift and permanent decline in central vision, which manifests through a growing difficulty to read, drive, and recognize faces (9). The vision loss associated with AMD also has a significant psychosocial impact, which is evident from studies demonstrating that individuals rate this symptom as one of the most severe health outcomes. In fact, it is considered more debilitating than cancer, acquired immune deficiency syndrome, or Alzheimer’s disease (10). Furthermore, AMD is linked to a reduced quality of life, diminished mobility and independence, and an increased risk of falls and depression (10). Considering the severe burden of AMD, it is critical to investigate effective therapeutic targets for AMD and facilitate the preclinical development of new drugs for AMD.
The primary treatment for nAMD is currently the administration of anti-vascular endothelial growth factor (VEGF) drugs via intravitreal injection (11). Although anti-VEGF therapy can stabilize or improve vision, studies have demonstrated that even after treatment, subretinal fibrosis developed in approximately 45-70% of eyes (12, 13), indicating that the conventional treatment for nAMD does not address subretinal fibrosis. During the development of subretinal fibrosis, different types of cells, including RPE, glial cells, fibroblasts, and macrophages, undergo proliferation and/or infiltration. These cells become stimulated by inflammatory cytokines and growth factors and participate in the remodeling of the extracellular matrix (ECM) (14). Due to the complexity of the cellular and molecular processes of fibrosis, it is challenging to develop effective therapeutic interventions, and no such therapies currently exist. Recently, granzyme B (GzmB) has been shown to have a critical role in the pathogenesis of nAMD, and it is emerging as a promising therapeutic target to improve vision in nAMD patients. In this review, we will discuss 1) the clinical features and underlying mechanisms of subretinal fibrosis in nAMD, 2) the emerging role of GzmB in CNV and subretinal fibrosis of nAMD and 3) GzmB-related therapeutic targets for inhibiting subretinal fibrosis.
Clinical manifestations and risk factors of subretinal fibrosis in nAMD
Subretinal fibrosis can be classified into fibrotic and non-fibrotic scars (12). Fibrotic scars are typically characterized by a raised, white or yellowish area within or beneath the neuroretina (15), often containing blood vessels (16). Whereas non-fibrotic scars are typically flat, unpigmented lesions with varying degrees of peripheral dark pigmentation (12).Fibrotic and non-fibrotic scars can both develop with similar occurrence rates (24.7% and 20.6%, respectively) in nAMD patients after 2 years of anti-VEGF treatment (12).
Histopathological research has revealed that the degree of harm to photoreceptors corresponds with the spatial extent of subretinal fibrosis in nAMD patients (17). These findings strengthen the clinical observation that subretinal fibrosis is the most critical determinant of long-term visual acuity (18). Furthermore, spectral domain optical coherence tomography (SD-OCT) investigations have revealed that subretinal hyper-reflective material (SHRM) in the RPE is a diagnostic biomarker for subretinal fibrosis in nAMD, correlating with the formation of a retinal scar associated with subretinal fibrosis and a poor visual prognosis (19, 20). The location, border definition, and thickness of SHRM can predict the likelihood of subretinal fibrosis (21). Recently, Souied et al. (2020) demonstrated by using SD-OCT that fibrosis is not only found within subretinal space but also underneath the RPE and within the neuroretina in nAMD patients (22). This suggests that there may be a spectrum of fibrosis in nAMD.
Several additional phenotypic risk factors can increase the risk of developing subretinal fibrosis. A prospective cohort study by Daniel et al. (2014) found that eyes with type 2 or classic CNV lesions that penetrate the RPE monolayer and grow in the subretinal space have a higher tendency to develop scar formation than those with type 1 or occult CNV lesions, which are typically restricted to the space beneath the RPE (12). These findings suggest that a subretinal lesion with extensively damaged and scattered RPE is more likely to progress to fibrosis, and this increased risk may be linked to the presence of transdifferentiated RPE cells in surgically extracted CNV fibrous membranes. Furthermore, Kim et al. (2020) discovered that approximately 32% of type 3 CNV patients also exhibited fovea-related fibrosis, emphasizing the importance of recognizing different CNV subtypes and their potential contribution to subretinal fibrosis (23, 24).
Moreover, the risk of developing subretinal fibrosis during anti-VEGF treatment is higher in eyes with refractory intraretinal cysts (25), macular hemorrhage (26), as indicated by blocked fluorescence on fluorescein angiography (FA), large basal lesions (15), increased retinal thickness (27), and foveal sub-retinal fluid (28). Although anti-VEGF therapy does not prevent fibrosis in nAMD (26, 29), early intervention with anti-VEGF still offers benefits by limiting poor visual outcomes associated with CNV expansion (15).
While phenotypic risk factors are significant predictors of disease progression later on, genetic and environmental risk factors are more valuable in the earlier stages of the disease (30). Among the environmental risk factors, the most consistently associated ones are age and smoking (30). Diet also seems to be an important contributing factor, as high adherence to the Mediterranean diet has been shown to reduce the risk of developing late AMD, implicated by the presence of fibrous subretinal scar tissues (31). Likewise, physical activity has been shown to suppress CNV in nAMD patients and mice, possibly by inhibiting AIM2 inflammasome in myeloid cells (32). Genetic studies have revealed complement factor H on chromosome 1 and age-related maculopathy susceptibility 2 and high-temperature requirement A serine peptidase 1 (ARMS2/HTRA1) genes on chromosome 10 as candidate genes associated with the risk for nAMD (33–35). Studies have also found that subretinal fibrosis in patients with nAMD is linked to elevated levels of C3a, C4a, and C5a in the plasma (36). Additionally, lower serum concentrations of 25-hydroxyvitamin D are also associated with subretinal fibrosis (37–39). Although studies generally suggest that a combination of genetic and environmental factors can contribute to chronic inflammation and subsequent CNV and subretinal fibrosis, the exact pathophysiological mechanisms of subretinal fibrosis and CNV-to-subretinal fibrosis transition have yet to be determined.
Pathophysiology and cellular constituents of subretinal fibrosis
The development of fibrotic tissue, characterized by the excessive buildup of ECM components such as collagen and fibronectin, is a natural and crucial part of tissue repair in all organs (40). Following tissue injury, activated local fibroblasts increase their contractility, release inflammatory mediators, and produce ECM components (40). When the injury is repetitive, chronic, or severe, ECM components continue to accumulate and disrupt tissue architecture, resulting in excessive fibrotic scarring (40, 41).
Subretinal fibrosis observed in nAMD exhibits similar pathological characteristics. The initial stimulus for the development of CNV in nAMD is the disruption of Bruch’s membrane, which can result from inflammatory and degenerative processes (42). CNV eventually breaks through the Bruch’s membrane and enters the subretinal space. These newly formed and permeable vessels contribute to chronic tissue damage (43). The subsequent recruitment, activation, and proliferation of various cell types, including immune cells and myofibroblasts, lead to excessive deposition and remodeling of the ECM, a prominent characteristic of fibrotic healing (Figure 1) (44). Over time, the neovascular lesion may advance towards a fibrovascular complex and ultimately macular fibrosis, a pathological transition referred to as the angiofibrotic switch (20, 45).
Recently, Souied et al. (2020) revealed in their longitudinal analysis of SD-OCT data that there could be three progression pathways from CNV to fibrosis in patients with AMD (22). All types of neovascularization in nAMD may transition into fibrovascular pigment epithelial detachment (PED), which can transform into subretinal fibrosis and then a fibroglial lesion partially occupying the neuroretina. Alternatively, fibrovascular pigment epithelial detachment may fail to develop into a fibrotic lesion and turn into a fibroatrophic lesion. Finally, Type 2 or classic CNV can transition into subretinal fibrosis and then a fibroglial lesion. Although the pathophysiological mechanisms of these angiofibrotic progression pathways have yet to be determined, transdifferentiation of RPE cells, endothelial cells, macrophages and macroglial cells into collagen-producing myofibroblasts likely occurs, leading to a predominance of the fibrotic component within the lesion.
Before the availability of anti-VEGF therapy, surgical removal of choroidal neovascular membranes (CNVMs) was a common treatment for patients with nAMD (46). Histological studies of excised tissues from patients have revealed that CNVMs comprise various connective tissues, including ECM, as well as cellular components such as endothelial cells, pericytes, RPE, and macrophages (47–50, 51).
Generally, fibrosis in the central nervous system consists of two distinct types of scars: the glial scar and the fibrotic scar (52). The glial scar is mainly composed of reactive astrocytes, which surround the injured area and separate it from healthy tissue (53). In contrast, the fibrotic scar is located in the core of the lesion and consists of fibroblasts and fibroblast-like cells that deposit ECM proteins (54). Both glial and fibrotic scars are likely involved in nAMD, as evidenced by patient and rodent studies (22, 55–57, 58). The fibrotic scar is primarily characterized by the presence of myofibroblasts, which are not normally found in healthy adult tissues (59). Although the exact cellular origins of these myofibroblasts continue to be a subject of ongoing debate, the following section summarizes the likely sources of myofibroblast precursors that may contribute to subretinal fibrosis (60, 61). These potential sources of myofibroblast precursors are depicted in Figure 1.
Potential sources of myofibroblast precursors and inflammatory mechanisms involved in myofibroblast transdifferentiation
RPE cells
The retinal pigmented epithelium (RPE) is a highly polarized and terminally differentiated monolayer positioned between photoreceptors and the choroid, which exhibits distinct morphological features such as apically arranged microvilli and tight junctions (62). The RPE is separated from the choroidal vasculature by the Bruch’s membrane, which is basally located and composed of various ECM proteins, including collagen-IV, laminin, and fibronectin (63). The RPE plays a critical role in maintaining normal retinal physiology and participates in signaling cascades vital for vision (64). Normally, cell-cell contact inhibition, mediated by homotypic adhesion of cadherins to adjacent cells prohibits RPE cells from proliferating (64). The RPE also acts as an outer blood-retinal barrier (oBRB) (62) and releases growth factors essential for the survival of retinal neurons (65). Furthermore, RPE cells produce thrombospondin-1 (TSP-1) and pigment epithelium-derived factor (PEDF), both play a significant role in suppressing CNV (66, 67).
As part of the CNV process in nAMD, detachment and dissociation of the RPE occurs (68). Proteases, such as extracellular GzmB, facilitate the degradation of RPE tight junctions, leading to loss of cell-cell adhesion and increased mobility of RPE cells (69). Upon loss of cell–cell adhesions and apical–basal polarity, RPE cells can transform into mesenchymal cells through epithelial-mesenchymal transition (EMT), as indicated by the decrease in epithelial markers, such as E-cadherin, and increase in mesenchymal markers, such as N-cadherin, vimentin, α-smooth muscle actin (α-SMA) (70, 71). RPE cells undergoing EMT are considered among the major contributors of myofibroblasts within subretinal fibrotic lesions (59).
Endothelial cells and pericytes
The development of CNV occurs through the formation of neovessels from the choroid. The process of endothelial-mesenchymal transition (EndMT) in choroidal endothelial cells may contribute to the population of mesenchymal cells in subretinal fibrotic lesions (72). These leaky neovessels also play a role in retinal edema, and hemorrhage, thereby further exacerbating the pathological wound healing response (59). During the EndMT process, endothelial cells undergo a loss of their endothelial markers, such as vascular endothelial-cadherin (VE-cadherin), CD31, tyrosine kinase with immunoglobulin-like and EGF-like domains (TIE-1, TIE-2), and von Willebrand Factor (vWF), while acquiring mesenchymal markers including fibroblast-specific protein-1 (FSP-1), α-SMA, N-cadherin, and vimentin (73). Along with endothelial cells, pericytes surrounding the choroidal capillaries can differentiate into myofibroblasts through a process known as pericyte-myofibroblast transition (PMT) and contribute to ECM deposition and subretinal fibrosis (74–77).
Müller glial cells
Müller glial cells play a crucial role in maintaining the homeostasis of the neuroretina, recycling of neurotransmitters, forming the inner blood-retinal barrier, and regulating immune and inflammatory responses within the neuroretina (78). Under pathological conditions, Müller cells undergo reactive gliosis, exhibiting cellular hypertrophy, proliferation, cytoplasmic extension, and increased glial fibrillary acidic protein (GFAP) expression (79–81). These activated Müller cells can undergo transdifferentiation into myofibroblasts via glial-mesenchymal transition (GMT), contributing to tissue traction (79, 82). The transdifferentiation of Müller cells into myofibroblasts is a key process in pathological tissue repair, closely associated with the development of fibrosis due to excessive deposition of ECM in the presence of persistent inflammation (83). During the glial-mesenchymal transition, there is an upregulation of myofibroblast markers, including α-SMA (84), and a downregulation of Müller glial cell markers such as glutamine synthetase (GS) and cellular retinaldehyde binding protein (CRALBP) (85).
Macrophages
In an earlier study, it was observed that 61% of human CNV lesions contained macrophages (86). In the experimental mouse model of CNV, macrophages account for approximately 20% of all cells, with approximately 70% of infiltrating macrophages originating from the bone marrow (87). Macrophages are considered to have a significant impact on the development of macular fibrosis not only due to their involvement in immune response and tissue repair but also their ability to transition to myofibroblasts (88). Studies indicate that macrophages can undergo transdifferentiation into myofibroblast-like cells, known as macrophage to myofibroblast transition (MMT) (88–90). It has been reported that M2, especially CD206+ macrophages, rather than M1 macrophages, undergo the transition (89). This process has been shown to contribute to fibrosis in various organs, including the kidneys, lungs, and heart (89–91). Xu and colleagues have shown that MMT is also involved in subretinal fibrosis (88, 92).
Circulating fibrocytes
Originating from bone marrow, circulating fibrocytes produce collagen-1 and can be found in the blood, spleen, and peripheral tissues (93). Following an injury, circulating fibrocytes are recruited to the injury site and differentiate into myofibroblasts, contributing to the wound-healing process (93). While there is no consensus on fibrocyte-specific markers, they are typically identified by their hematopoietic origin (expressing CD34 and CD45) and their ability to produce collagen-1/3 (93). Circulating fibrocytes have been identified in CNVs and may be an important source of myofibroblasts (50, 51). Indeed, Yi et al. (2023) recently reported that an age-dependent increase in circulating fibrocytes can augment the pro-fibrotic properties of macrophages (94).
Inflammatory mechanisms involved in myofibroblast transdifferentiation
While the sources of myofibroblast precursors that can contribute to subretinal fibrosis are diverse, the common underlying process that promotes the recruitment and subsequent transdifferentiation of these precursors is inflammation. The presence of CNV in nAMD is linked to the regulation of myeloid cells, inflammatory cytokines, and activation of the complement system. Moreover, inflammation is believed to play an important role in subretinal fibrosis. In the aging retina and RPE/choroid, a state of low-grade chronic inflammation known as para-inflammation exists, which may promote a profibrotic response during the healing of subretinal wounds (95, 96). The inflammatory response involves various innate and adaptive immune cells that establish a microenvironment that attracts and activates fibroblasts within the subretinal space (97). Infiltrating macrophages are thought to have a pivotal role in mouse models of laser-induced CNV and the associated subretinal fibrosis (88, 98, 99). Following damage to the RPE/Bruch’s membrane complex in laser-induced CNV, retinal microglia and choroidal macrophages constitute the initial wave of infiltrating immune cells (89, 100). While infiltrating macrophages primarily serve to clear debris and initiate retinal repair, they can also contribute to subretinal fibrosis during chronic inflammation through various mechanisms (97). In addition to transdifferentiating into myofibroblasts through MMT, macrophages can secrete pro-angiogenic and pro-fibrotic factors, which can facilitate the recruitment and activation of fibroblasts or trigger mesenchymal transition in endothelial or RPE cells (101). Finally, in the presence of persistent tissue damage, macrophages can contribute to the progression of subretinal inflammation by releasing cytokines and activating the complement system.
In the context of fibrosis, pro-inflammatory cytokines interleukin (IL)-2, IL-6 and anti-inflammatory cytokine IL-10 are major regulators (102). IL-2 plays a role in facilitating RPE cell migration and synthesis of the ECM, suggesting significant contributions towards subretinal fibrosis (103). Similarly, IL-6 serves as a major mediator in the promotion of subretinal fibrosis (104). Furthermore, the inflammasome has been found to serve as a link between the detection of pathogen and danger signals and the activation of pro-IL-1β. Specifically, the Nod-like receptor family pyrin domain-containing 3 (NLRP3) inflammasome is closely connected to the maturation of IL-1β, which in turn plays a crucial role in macrophage recruitment and activation of IL-6 (105–107). IL-10, along with the downstream activation of STAT3 signaling, plays a pivotal role in regulating macrophages during the aging process, primarily promoting an M2 phenotype and facilitating ocular angiogenesis (108). However, IL-10 secreted by RPE cells upon HSP70-mediated cellular stress has been shown to effectively mitigate the development of subretinal fibrosis (109). This dual role of IL-10 underscores its complexity, serving as both pro-angiogenic and anti-fibrotic factors. IL-13, primarily generated by Th2 cells and monocytes/macrophages, inhibits the proliferation of ARPE-19 cells in vitro and promotes EMT (110).
The complement system is also a significant component of the innate immune system, and compelling evidence supports the pivotal involvement of complement abnormalities in the development of AMD (111–113). Excessive activation of the complement system can potentially contribute to retinal damage either through direct harm to retinal cells via the membrane attack complex (MAC, C5b-9), or indirectly by modulating inflammation through complement fragments such as C3a, C3b, and C5a (114–116). Apart from their pro-inflammatory characteristics, both C3a and C5a are also involved in tissue regeneration and fibrosis (117, 118). Lechner et al. (2016) previously reported a significant increase in plasma levels of C3a and C5a in patients with nAMD, particularly those with macular fibrosis (36). Complement activation has also been found to contribute to subretinal fibrosis through C5a/C5aR-mediated EMT in RPE cells, which likely operate in conjunction with C3a-induced MMT (119).
In addition to interleukins and complement proteins, growth factors are known to play a critical role in inflammation. Transforming growth factor-β (TGF-β) is a pleiotropic growth factor with pro- and anti-inflammatory activity. In particular relevance to fibrosis, TGF-β is the most extensively studied inducer of mesenchymal transition of epithelial cells, endothelial cells, glial cells, macrophages and pericytes. TGF-β is expressed in surgically excised AMD-related CNVMs, and of three TGF-β isoforms, TGF-β2 is expressed at much higher levels in the vitreous humor of patients with retinal fibrosis. TGF-β signaling is conventionally mediated by Smad2/3/4 multimeric complex, and it is known to induce expression of TGF-β-dependent genes involved in myofibroblasts transdifferentiation, such as α-SMA and snai1. Furthermore, TGF-β-mediated Smad-dependent pathways are known to induce the expression of most extracellular matrix components and enzymes involved in matrix reorganization and maturation (120–122).
TGF-β signaling can induce expression of other growth factors that are involved in inflammation and fibrosis, such as VEGF and connective tissue growth factor (CTGF). CTGF is a pro-inflammatory growth factor that is involved in cardiac, renal, hepatic and pulmonary fibrosis. CTGF also may play a critical role in subretinal fibrosis, as it is expressed in surgically excised AMD-related CNVMs with moderate or extensive fibrosis, and RPE cells and choroidal endothelial cells upregulate CTGF in response to exogenous TGF-β and VEGF. Additionally, in a laser-induced CNV rat model, administration of intravitreal anti-CTGF was found to significantly reduce subretinal fibrosis compared to the control and anti-VEGF groups (120–122).
TGF-β signaling can also transactivate epidermal growth factor receptor (EGFR) signaling, which is necessary for the migration and proliferation of pericytes and fibroblasts before they transdifferentiate into myofibroblasts (120, 121). EGF, the major ligand for EGFR, is elevated in the aqueous humor of nAMD patients, which suggests a pathological role of EGFR signaling in nAMD. In support of this idea, EGFR activation has been shown to facilitate RPE EMT in response to cigarette smoke, suggesting that it may have a role in subretinal fibrosis.
Many different types of immune cells are capable of producing and releasing these growth factors, and they all may have important roles in the development of subretinal fibrosis (122–125). However, accumulating evidence shows that mast cells play a key role in AMD, and one of their proteases, GzmB, is a significant contributor to CNV and also likely subretinal fibrosis (69, 126, 127).
Potential role of mast cells in CNV and subretinal fibrosis
Although numerous immune cells are known to be involved in nAMD, the role of mast cells is relatively underexplored despite their presence during the early and late stages of nAMD (97, 102, 128). Mast cells, integral components of the immune system influenced by their microenvironment, accumulate at injury sites associated with various conditions, including wound healing, particularly near blood vessels (129). Mast cells are generally found throughout normal connective tissue, and they are usually next to blood vessels and peripheral nerves or beneath the epithelial layer (130, 131). In the eye, they are predominantly found in the choroid of the eye but are absent in the neuroretina (132).. Studies have demonstrated an increased number and degranulation of mast cells in all forms of age-related macular degeneration, including nAMD (126, 127). Mast cells produce a wide array of pro-inflammatory mediators, proteases, and growth factors that exhibit pro-fibrotic effects, either by directly influencing fibroblasts and fibrocytes or indirectly by recruiting and activating diverse inflammatory cell types. Stored mediators such as histamine, heparin, tryptase, and chymase have various biological effects that modulate fibrosis, including fibroblast proliferation, collagen synthesis, differentiation, chemotaxis, contractility, and ECM degradation (133).
Of all mast cell proteases, tryptase, the major mast cell-specific protease, is well known for its contribution to inflammation and angiogenesis in the RPE (134). Arai et al. (2017) reported that tryptase not only enhances the production of IL-8 and VEGF by RPE but also promotes the migration of RPE (134). Furthermore, Chen et al. (2017) reported that mast cell proteases, tryptase and chymase, stimulate endothelial cell proliferation, facilitate vascular tube formation, and degrade the connective tissue matrix, creating space for neovascular growth (135). In addition to inflammatory angiogenesis, tryptase also plays a pivotal role in various aspects of fibrotic processes. Gailit et al. (2001) conducted studies using the HMC-1 human mast cell line, demonstrating that purified human tryptase induces α-smooth muscle actin expression and contributes to fibroblast contraction (136). Tryptase, when inhibited, reduces this response, highlighting its significance as an active mast cell mediator (136). Furthermore, in idiopathic pulmonary fibrosis (IPF), the severity of the disease correlates with an increase in mast cells expressing both tryptase and chymase, as seen in lung biopsies, with plasma tryptase levels directly correlating with IPF severity (137). Studies have also emphasized the pro-fibrotic role of mast cells, noting that tryptase and chymase contribute to the activation of TGF-β1, a key pro-fibrotic factor (138, 139).
The intricate relationship between inflammation and fibrosis is highlighted by Chen et al. (2014) demonstrating that blocking mast cell activation can effectively reduce both scar formation and inflammation in the skin (140). A study on mast cells’ influences on collagen maturation in the context of periodontal disease also found that chronic inflammation can stimulate increased collagen synthesis by fibroblasts, thereby leading to tissue fibrosis (141). Similarly, the increased presence of activated mast cells that express pro-fibrotic growth factors in the context of idiopathic pulmonary fibrosis highlights the potential role of mast cells in fibroblast dysfunction (133). Additionally, mast cells’ ability to activate fibroblasts through cell-to-cell communication via gap junctions further underscores the complex biological effects modulating fibrosis (142). However, it is important to note that mast cells also produce molecules with antifibrotic properties, such as IL-10, making the exact contribution of individual mediators in regulating fibrosis challenging to predict (143). Considering the general role of mast cells and tryptase in angiogenesis and fibrosis, they are likely involved in CNV and subretinal fibrosis of nAMD (Figure 1). In support of this, we have found that another mast cell protease, GzmB, has a significant role in CNV development. Our mechanistic studies on the role of GzmB in CNV and previous studies on the role of GzmB in fibrosis suggest that mast cells and GzmB are also likely involved in subretinal fibrosis. In the next sections, we discuss our findings on the role of GzmB in CNV and explain how GzmB may be involved in the angiofibrotic switch in nAMD.
The role of granzyme B in CNV
Of all the proteases produced and released by mast cells, GzmB has been at the forefront in recent years due to its extracellular activity to disrupt cellular structure and subsequently induce biochemical changes in cells. GzmB, traditionally known for its intracellular role in immune-mediated apoptosis, has recently gained attention for its extracellular functions. In 2020, we first reported that GzmB in the outer retinal layers is elevated in older normal donor eyes (> 65 years old) compared to younger normal donor eyes (< 55 years old). Interestingly, GzmB in the choroid was significantly elevated in wet AMD eyes with CNV compared to dry AMD eyes with geographic atrophy. GzmB was also significantly elevated in the RPE cells of early AMD eyes with soft drusen. Notably, extracellular GzmB was found in the basement membrane and intercellular spaces of RPE cells in human and non-human primate retina. Similarly, GzmB was also present in the same locations in mouse eyes, and its levels increased in the RPE and choroid layers in an age-dependent manner. Within the choroid of human donor eyes, the GzmB+ cells were confirmed to be mast cells by using toluidine blue staining. Based on these findings, we hypothesized that GzmB may promote CNV by cleaving its substrates in the outer retinal layers. Indeed, we found that exogenous GzmB is capable of cleaving tight junctional proteins in cultured RPE cells, namely ZO-1, occludin and JAM-A. Additionally, exogenous GzmB can cleave ECM proteins that are known to be produced and deposited into the Bruch’s membrane by RPE, such as fibronectin, laminin-5 and collagen IV (69). It should be noted that tryptase is not capable of degrading ZO-1 and JAM-A in cultured RPE cells; however, it can stimulate RPE cells to produce VEGF (134). This suggests that while tryptase may upregulate genes in RPE cells that are involved in CNV, GzmB may cause structural changes in RPE cells and the Bruch’s membrane that facilitate the formation of CNV.
To further test the hypothesis that GzmB may promote CNV, we performed choroid sprouting assay (CSA) experiments where we treated mouse RPE/choroid explants with exogenous GzmB. We observed that exogenous GzmB significantly increased vascular sprouting from the explant compared to the control group, suggesting its crucial role in CNV (126, 144). To further decipher the mechanism of GzmB-mediated CNV, we investigated how GzmB substrates are altered in the CSA supernatant. We identified thrombospondin-1 (TSP-1) as a novel substrate for GzmB and found that it is cleaved by exogenous GzmB in the CSA supernatant. TSP-1 is known as an anti-angiogenic factor, and it is significantly reduced in wet AMD eyes with CNV compared to age-matched healthy eyes. Additionally, exogenous TSP-1 can suppress vascular sprouting in the CSA explants (144). Based on these findings, we speculate that extracellular GzmB degrades TSP-1, suppresses the anti-angiogenic activity of TSP-1 and leads to CNV in nAMD. Another anti-angiogenic factor decorin (DCN) in the CSA supernatant is cleaved by exogenous GzmB, and this suggests that GzmB may promote CNV by degrading additional anti-angiogenic factors in the outer retina (126). Exogenous GzmB can also increase the release of ECM-sequestered growth factors, namely VEGF and TGF-β, into the CSA supernatant (126). This suggests that GzmB may also induce the release of these ECM-sequestered growth factors in the outer retina and promote CNV via the activation of signaling pathways involving VEGF and TGF-β. Considering that mast cells are one of the major sources of GzmB in the choroid of the outer retina, we treated the CSA explants with the mast cell degranulation compound, 48/80. We observed that 48/80 can induce vascular sprouting in the CSA explants, but 48/80-mediated vascular sprouting can be inhibited by the mast cell stabilizer, ketotifen fumarate. Furthermore, a GzmB-specific inhibitor, VTI-1002, can also suppress vascular sprouting in the CSA explants. These findings indicate that GzmB from mast cells is involved in CNV formation (126).
After deciphering the mechanisms of GzmB-mediated vascular sprouting ex vivo, we used a well-established laser-induced mouse model of CNV to confirm the role of GzmB in CNV in vivo. Based on our OCT-based angiography (OCTA) and immunohistochemical data, wild-type mice exhibit significantly larger CNV lesions compared to GzmB knockout (KO) mice, suggesting that GzmB does indeed play a critical role in CNV. Although we have yet to comprehensively decipher the mechanisms of how GzmB deficiency suppresses CNV in vivo, we noticed that there are fewer Iba1+ immune cells within CNV lesions in GzmB KO mice, suggesting that GzmB may play a role in pro-inflammation and the recruitment of macrophages and/or microglia towards CNV lesions. Additionally, CNV lesions in GzmB KO have much less immunolabeling of IL-6, implying that GzmB deficiency likely reduces pro-inflammation by preventing the recruitment of Iba1+ immune cells towards CNV lesions (126). Overall, GzmB plays a critical role in the development of CNV, and pharmacological inhibition of its extracellular activity and/or mast cell degranulation may be a promising therapeutic strategy for suppressing CNV.
The potential role of GzmB in the angiofibrotic switch in nAMD
Although the exact role of GzmB in subretinal fibrosis has yet to be determined, accumulating evidence shows that GzmB has a dual role in angiogenesis and fibrosis. GzmB has been shown to release VEGF from fibronectin and endothelial cell-derived matrix and results in the induction of vascular permeability, an essential step towards pathological angiogenesis (145). Interestingly, GzmB has been shown to also play a critical role in fibrosis in the heart and skin. In a mouse model of angiotensin II infusion-mediated cardiac fibrosis, GzmB was elevated in fibrotic lesions, and the genetic deletion of GzmB significantly reduced the size of collagen+ fibrotic lesions and mRNA levels of type 1 and 3 collagen, TGF-β and CTGF in the heart. The genetic deletion of perforin cannot attenuate cardiac fibrosis, implying that extracellular and perforin-independent activity of GzmB is responsible for cardiac fibrosis. GzmB deficiency also significantly reduces expression of vimentin and α-SMA within fibrotic lesions, indicating that GzmB does have pro-fibrotic activities (146). In support of these findings in the heart, VTI-1002-mediated inhibition of GzmB in a mouse model of diabetic burn wound healing has been shown not only to significantly reduce scar formation in the skin but also to reduce α-SMA expression within the burn wound. Additionally, VTI-1002 treatment also enhanced the expression of anti-fibrotic protein DCN within the burn wounds (147). In further support of these findings, double knockout of apolipoprotein E (ApoE) and GzmB has been shown to result in improved pressure wound healing in comparison to ApoE KO. The double knockout significantly reduces the area of the pressure wound and the number of GzmB-expressing mast cells within the wound. It also reduces α-SMA expression and simultaneously elevates DCN expression within the pressure wound, indicating that GzmB is a key player in fibrosis (148).
Given the clear role of GzmB in both angiogenesis and fibrosis, GzmB is likely involved in the angiofibrotic switch in nAMD. However, it remains unclear how GzmB can promote the pathological transition from CNV to subretinal fibrosis. Based on our findings thus far, we propose that: 1) GzmB may lead to loss of tight junctional contacts in RPE cells, thereby promoting RPE EMT and myofibroblast transdifferentiation, 2) GzmB may release ECM-sequestered growth factors that can induce the pathological transition from CNV to subretinal fibrosis, and 3) GzmB-mediated fragmentation of TSP-1 and DCN may promote CNV and subretinal fibrosis simultaneously. These proposed mechanisms are depicted in Figure 1.
As described above, GzmB can degrade RPE tight junctional proteins, such as ZO-1, JAM-A and occludin, and it is possible that GzmB-mediated degradation of these proteins leads to RPE EMT. Indeed, in vivo knockdown of ZO-1 in RPE cells has been shown to promote proliferation of RPE cells and upregulate genes that are involved in EMT, such as vimentin, snai1 and α-SMA (149). GzmB may also degrade tight junctions between choroidal endothelial cells, and this could lead to both CNV and EndMT, as the loss of endothelial tight junctions can lead to vascular permeability and EndMT (150). RPE EMT and EndMT will eventually lead to the accumulation of myofibroblasts within the subretinal fibrotic lesion and may contribute to the continual expansion of the lesion and the progressive decline in visual acuity, which are both evident in nAMD patients with subretinal fibrosis (7).
Based on our studies, GzmB is capable of degrading ECM proteins in RPE cells and promote the release of ECM-sequestered growth factors, such as VEGF and TGF-β. While VEGF is known to be strictly involved in CNV, TGF-β has a dual role in both CNV and subretinal fibrosis (88, 151). It may be possible that TGF-β signaling initially promotes CNV along with VEGF signaling and eventually causes a pathological shift towards subretinal fibrosis by promoting myofibroblast transdifferentiation. TGF-β signaling is known to induce CTGF expression and EGFR transactivation, both of which are involved in EMT and the development of fibrosis (152, 153). Hence, GzmB-mediated release of ECM-sequestered growth factors may ultimately lead to diverse signaling cascades that induce the pathological transition from CNV to subretinal fibrosis. It would be interesting to investigate if GzmB can modify the ECM of various sources of myofibroblasts, including macrophages, choroidal fibroblasts and Müller cells, and induce them to transdifferentiate into myofibroblasts.
In addition to releasing ECM-sequestered growth factors, GzmB also degrades anti-angiogenic factors TSP-1 and DCN, ultimately leading to vascular sprouting in CSA explants (126). Although fragments of TSP-1 and DCN may lack the function of the full-length proteins, they have been shown to promote inflammation and fibrosis. Full-length TSP-1 consists of multiple domains, and one of the GzmB cleavage sites exists in the N-terminal domain of TSP-1 (144). The N-terminal domain of TSP-1 has been shown to interact with a variety of receptors that are involved in inflammation (e.g.,integrin receptors α3β1, α4β1 and calreticulin) (154). Integrin receptors facilitate the migration of immune cells, including macrophages and mast cells, and this could explain why GzmB deficiency results in reduced immunolabeling of IL-6 and accumulation of Iba1+ immune cells within CNV lesions (126, 155, 156). In addition to its role in inflammation, the N-terminal domain of TSP-1 is also known to promote angiogenesis by interacting with integrin receptors on endothelial cells, which may further support how GzmB-mediated fragmentation of TSP-1 leads to vascular sprouting in the CSA explants (126, 157). Interestingly, the N-terminal domain is also known to promote collagen expression and matrix deposition by interacting with calreticulin on fibroblasts, suggesting that it may be involved in activation of fibroblasts and therefore myofibroblast transdifferentiation (158). Given that GzmB cleaves the N-terminal domain of TSP-1, GzmB likely promotes the antifibrotic switch by generating the fragments of TSP-1 N-terminals in the outer retina that can simultaneously promote CNV and subretinal fibrosis. DCN fragments are also known to promote inflammation by functioning as damage-associated molecular patterns (DAMPs), and GzmB-mediated fragmentation of DCN may be one mechanism associated with pro-inflammation (159, 160). Fragmentation of DCN may also promote both angiogenesis and fibrosis, as DCN is known to inhibit TGF-β and EGFR signaling, both of which are involved in angiogenesis and fibrosis (161, 162). Overall, GzmB-mediated fragmentation of TSP-1 and DCN in the outer retina is likely another key mechanism involved in the angiofibrotic switch in nAMD.
Animal models of subretinal fibrosis
Various animal models have been established and used to investigate the pathogenesis and treatment of nAMD, providing additional insights into the cellular mechanisms of CNV formation and fibrosis. Rodent models offer significant advantages due to the availability of genetically modified lines, recombinant proteins, and antibodies, along with shorter procedure times and lower costs compared to larger animal models (163). Table 1 summarizes the main experimental models used in nAMD research, highlighting their advantages and drawbacks. In the traditional laser-induced CNV mouse model, injury associated with the laser application leads to RPE injury and the rupture of Bruch’s membrane, triggering an immediate inflammatory response characterized by immune cell recruitment and subsequent development of CNV (166–168). As observed in humans, the transition to a fibrotic phenotype is characterized by the upregulation of certain biomarkers, such as α-SMA. Furthermore, Jo et al. (2011) discovered that the injection of peritoneal macrophages into the subretinal space immediately after the laser-induced CNV resulted in an even larger size of subretinal fibrosis (80). However, this model has not been highly utilized due to the technical challenges. Little et al. (2020) introduced a laser-induced mouse model of subretinal fibrosis wherein second laser burns are applied to CNV lesions one week after the first laser burns, to reflect the clinical situation in nAMD patients with hemorrhage or sustained leakage, which are recognized risk factors for macular fibrosis (164). This results in lesions containing CNV, fibrosis, and hemorrhage, all of which play a role in inflammatory and fibrotic pathways (164). Since it is well established that angiogenesis precedes fibrosis, as in the skin (169), the two-stage laser model can help to study the angiofibrotic switch associated with nAMD and thereby identify therapeutic targets that can halt ongoing CNV and promote early prevention of subretinal fibrosis in nAMD. However, a drawback of this model is that it becomes difficult to separate subretinal fibrosis from CNV, and the effects of anti-fibrotic agents may not solely be attributed to changes in fibrosis when CNV and hemorrhage are still present. To address this issue, Zandi et al. (2023) recently developed a novel subretinal fibrosis model in which CNV is not active from Day 21 post-laser and onwards, yet fibrosis develops from that point forward (165). The late stage in this model is considered ideal for specifically studying the mechanisms of subretinal fibrosis, as it is possible to isolate fibrosis from angiogenesis and investigate molecular changes only related to fibrosis (170). Although the two-stage model by Little et al. (2020) may be more useful for studying the role of GzmB in the angiofibrotic switch and the role of the ‘first responder’ immune cells, especially mast cells, in subretinal fibrosis, the single-laser model by Zandi et al. (2023) would provide valuable insights into the specific role of GzmB in subretinal fibrosis.
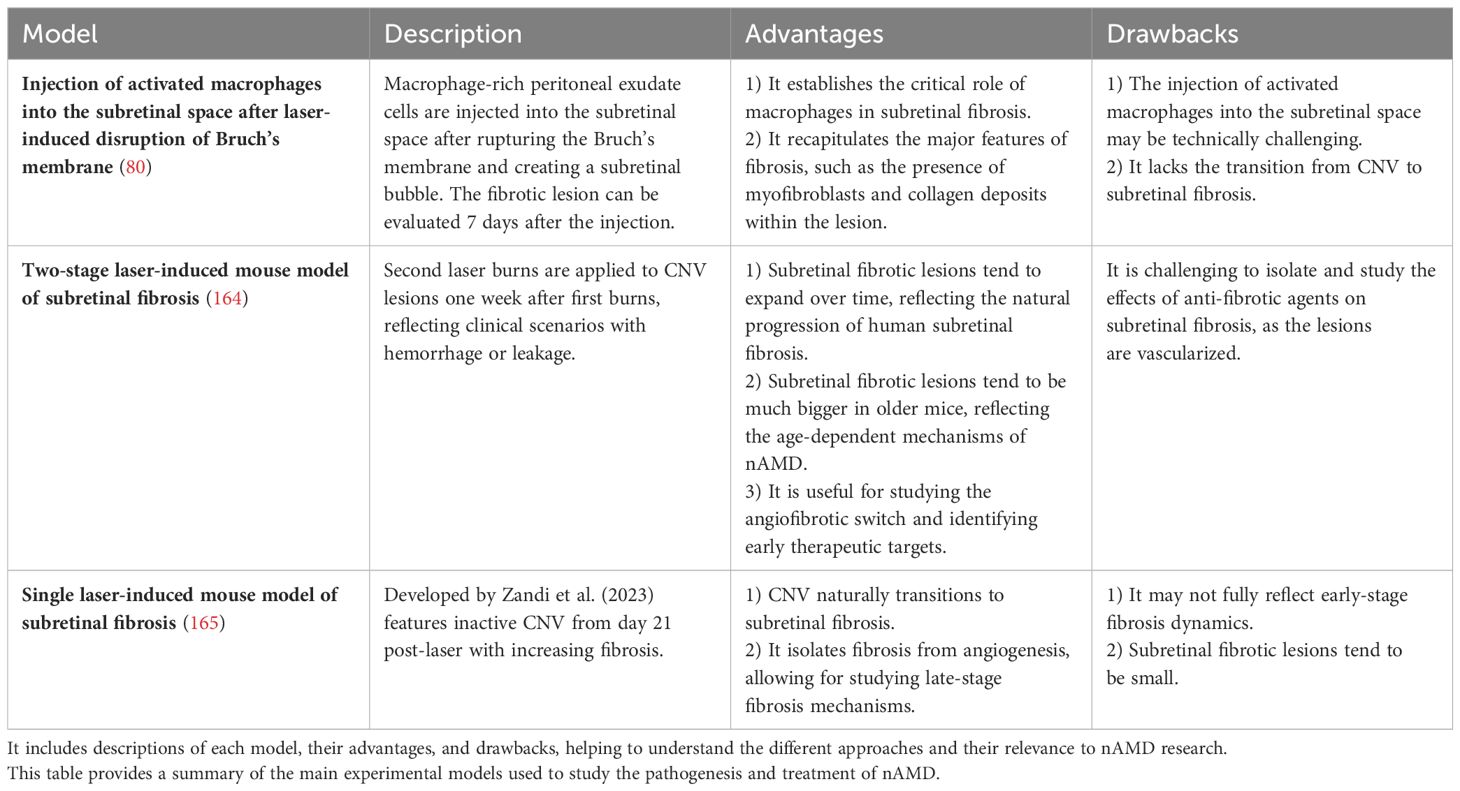
Table 1 Overview of Experimental Models for subretinal fibrosis in nAMD.
Therapeutic targets
Anti-VEGF therapy is the gold standard treatment for nAMD, designed to inhibit angiogenesis and vascular permeability (171). Timely initiation of anti-VEGF therapy has been suggested to aid in preventing fibrosis, as the therapy’s inhibitory effects on angiogenesis and vasopermeability reduce immune cell infiltration and dampen the inflammatory response (15). However, a significant proportion of nAMD patients exhibit an inadequate response to anti-VEGF therapy and develop progressive subretinal fibrosis regardless of anti-VEGF therapy (12, 15). Currently, there is a lack of available treatments specifically designed to target subretinal fibrosis in nAMD patients. There are potential anti-fibrotic drugs that may be effective against subretinal fibrosis; Tenbrock et al. (2022) provided an overview of selected anti-fibrotic drugs for experimental subretinal fibrosis treatment (7).
Pharmacologically inhibiting extracellular GzmB could be an effective approach to address the pathologic ECM remodeling and the subsequent angiogenic activity observed in the outer retina in nAMD (172). We have shown that both pharmacological inhibition of GzmB and genetic deletion of GzmB can be effective against CNV ex vivo and in vivo, respectively. Hence, the GzmB-specific inhibitor VTI-1002 could be a promising option for suppressing CNV. Aubert et al. (2022) recently provided a comprehensive summary of in vitro and in vivo studies highlighting the therapeutic potential of VTI-1002 to treat inflammatory diseases and facilitate wound healing (173). Intravitreal injections of VTI-1002, either on its own or in combination with anti-VEGF biologics, could be an effective therapeutic option for nAMD. However, inhibiting extracellular GzmB derived from unhealthy RPE and mast cells with VTI-1002 by intravitreal injections may be challenging to fully suppress CNV. Mast cell stabilizers, such as ketotifen fumarate, have been studied in the context of reducing degranulation in the choroid and protecting RPE in a rat model of Geographic Atrophy and is an important avenue to pursue (126, 174). Although the role of GzmB in promoting subretinal fibrosis is not yet fully understood, the emerging similarities between choroidal subretinal fibrosis and wound healing in skin and other organs point towards the possibility that GzmB inhibitors may suppress the pro-angiogenic and pro-fibrotic activity associated with the formation of CNV and subretinal fibrosis in nAMD.
The pathophysiology of subretinal fibrosis in nAMD is complex and it involves multiple cell types and inflammatory mediators that promote the recruitment of immune cells and myofibroblast transdifferentiation. Given the complexity of the disease, the addition of potential therapeutics, such as inhibitors of extracellular GzmB or inhibitors of mast cell degranulation discussed here, may not be sufficient to fully suppress the pathological events in nAMD. Future studies on identifying the sequelae of cellular events associated with the angiofibrotic switch will further expand the therapeutic options for choroidal neovascularization and subretinal fibrosis in nAMD (126).
Author contributions
KG: Writing – original draft, Writing – review & editing. HY: Writing – review & editing. HC: Writing – review & editing. DG: Funding acquisition, Writing – review & editing. JM: Funding acquisition, Conceptualization, Supervision, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. We thank Fighting Blindness Canada, the Vancouver Coastal Hospital Research Institute, the Natural Sciences and Engineering Research Council and the Canadian Institutes of Health Research for funding.
Acknowledgments
We thank Faculty of Medicine at UBC, UBC Research Excellence Cluster in Vision, Fighting Blindness Canada, Vancouver Coastal Hospital Research Institute, Natural Sciences and Engineering Research Council and the Canadian Institutes of Health Research.
Conflict of interest
Author DG is a Co-Founder and serves as the Chief Scientific Officer for viDA Therapeutics.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. GBD 2019 Blindness and Vision Impairment Collaborators, Vision Loss Expert Group of the Global Burden of Disease Study. Causes of blindness and vision impairment in 2020 and trends over 30 years, and prevalence of avoidable blindness in relation to VISION 2020: The Right to Sight: An analysis for the Global Burden of Disease Study. Lancet Glob Health. (2021) 9:e144–60. doi: 10.1016/S2214-109X(20)30489-7
2. Wong WL, Su X, Li X, Cheung CM, Klein R, Cheng CY, et al. Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: A systematic review and meta-analysis. Lancet Glob Health. (2014) 2:e106–16. doi: 10.1016/S2214-109X(13)70145-1
3. Ferris FL 3rd, Wilkinson CP, Bird A, Chakravarthy U, Chew E, Csaky K, et al. Clinical classification of age-related macular degeneration. Ophthalmology. (2013) 120:844–51. doi: 10.1016/j.ophtha.2012.10.036
4. Waldstein SM, Vogl WD, Bogunovic H, Sadeghipour A, Riedl S, Schmidt-Erfurth U. Characterization of drusen and hyperreflective foci as biomarkers for disease progression in age-related macular degeneration using artificial intelligence in optical coherence tomography. JAMA Ophthalmol. (2020) 138:740. doi: 10.1001/jamaophthalmol.2020.1376
5. Sarks JP, Sarks SH, Killingsworth MC. Evolution of geographic atrophy of the retinal pigment epithelium. Eye (Lond). (1988) 2:552–77. doi: 10.1038/eye.1988.106
6. Spaide RF, Jaffe GJ, Sarraf D, Freund KB, Sadda SR, Staurenghi G, et al. Consensus nomenclature for reporting neovascular age-related macular degeneration data: consensus on neovascular age-related macular degeneration nomenclature study group. Ophthalmology. (2020) 127:616–36. doi: 10.1016/j.ophtha.2019.11.004
7. Tenbrock L, Wolf J, Boneva S, Schlecht A, Agostini H, Wieghofer P, et al. Subretinal fibrosis in neovascular age-related macular degeneration: current concepts, therapeutic avenues, and future perspectives. Cell Tissue Res. (2022) 387:361–75. doi: 10.1007/s00441-021-03514-8
8. Ferris FL, Fine SL, Hyman L. Age-related macular degeneration and blindness due to neovascular maculopathy. Arch Ophthalmol. (1984) 102:1640–2. doi: 10.1001/archopht.1984.01040031330019
9. Guymer RH, Campbell TG. Age-related macular degeneration. Lancet. (2023) 401:1459–72. doi: 10.1016/S0140-6736(22)02609-5
10. Scott AW, Bressler NM, Ffolkes S, Wittenborn JS, Jorkasky J. Public attitudes about eye and vision health. JAMA Ophthalmol. (2016) 134:1111–8. doi: 10.1001/jamaophthalmol.2016.2627
11. Damkondwar DR, Srinivasan R, Raman R, Kulothungan V, Sharma T. Morphological and functional retinal changes in neovascular age-related macular degeneration treated with intravitreal bevacizumab. Indian J Ophthalmol. (2022) 70:4376–82. doi: 10.4103/ijo.IJO_1184_22
12. Daniel E, Toth CA, Grunwald JE, Jaffe GJ, Martin DF, Fine SL, et al. Risk of scar in the comparison of Age-related Macular Degeneration Treatments Trials. Ophthalmology. (2014) 121:656–66. doi: 10.1016/j.ophtha.2013.10.019
13. Wolff B, Macioce V, Vasseur V, Castelnovo L, Michel G, Nguyen V, et al. Ten-year outcomes of anti-vascular endothelial growth factor treatment for neovascular age-related macular disease: A single-centre French study. Clin Exp Ophthalmol. (2020) 48:636–43. doi: 10.1111/ceo.13742
14. Ishikawa K, Kannan R, Hinton DR. Molecular mechanisms of subretinal fibrosis in age-related macular degeneration. Exp Eye Res. (2016) 142:19–25. doi: 10.1016/j.exer.2015.03.009
15. Bloch SB, Lund-Andersen H, Sander B, Larsen M. Subfoveal fibrosis in eyes with neovascular age-related macular degeneration treated with intravitreal ranibizumab. Am J Ophthalmol. (2013) 156:116–124.e1. doi: 10.1016/j.ajo.2013.02.012
16. Miere A, Semoun O, Cohen SY, El Ameen A, Srour M, Jung C, et al. Optical coherence tomography angiography features of subretinal fibrosis in age-related macular degeneration. Retina. (2015) 35:2275–84. doi: 10.1097/IAE.0000000000000819
17. Green WR, Enger C. Age-related macular degeneration histopathologic studies. The 1992 Lorenz E. Zimmerman Lecture. Ophthalmology. (1993) 100:1519–35. doi: 10.1016/S0161-6420(93)31466-1
18. Cheung CMG, Grewal DS, Teo KYC, Gan A, Mohla A, Chakravarthy U, et al. The evolution of fibrosis and atrophy and their relationship with visual outcomes in asian persons with neovascular age-related macular degeneration. Ophthalmol Retina. (2019) 3:1045–55. doi: 10.1016/j.oret.2019.06.002
19. Willoughby AS, Ying GS, Toth CA, Maguire MG, Burns RE, Grunwald JE, et al. Subretinal hyperreflective material in the comparison of age-related macular degeneration treatments trials. Ophthalmology. (2015) 122:1846–1853.e5. doi: 10.1016/j.ophtha.2015.05.042
20. Roberts PK, Zotter S, Montuoro A, Pircher M, Baumann B, Ritter M, et al. Identification and quantification of the angiofibrotic switch in neovascular AMD. Invest Ophthalmol Vis Sci. (2019) 60:304–11. doi: 10.1167/iovs.18-25189
21. Alex D, Giridhar A, Gopalakrishnan M, Indurkhya S, Madan S. Subretinal hyperreflective material morphology in neovascular age-related macular degeneration: A case control study. Indian J Ophthalmol. (2021) 69:1862–6. doi: 10.4103/ijo.IJO_3156_20
22. Souied EH, Addou-Regnard M, Ohayon A, Semoun O, Querques G, Blanco-Garavito R, et al. Spectral-domain optical coherence tomography analysis of fibrotic lesions in neovascular age-related macular degeneration. Am J Ophthalmol. (2020) 214:151–71. doi: 10.1016/j.ajo.2020.02.016
23. Kim JH, Kim JW, Kim CG, Lee DW. Long-term treatment outcomes in type 3 neovascularization: focus on the difference in outcomes between geographic atrophy and fibrotic scarring. J Clin Med. (2020) 9:1145. doi: 10.3390/jcm9041145
24. Cui J, Maberley D, Samad A, Ma P, Ning A, Matsubara JA, et al. Expression of integrins on human choroidal neovascular membranes. J Ocul Biol Dis Infor. (2009) 2:12–9. doi: 10.1007/s12177-009-9015-9
25. Gianniou C, Dirani A, Jang L, Mantel I. Refractory intraretinal or subretinal fluid in neovascular age-related macular degeneration treated with intravitreal ranizubimab: functional and structural outcome. Retina. (2015) 35:1195–201. doi: 10.1097/IAE.0000000000000465
26. Hwang JC, Del Priore LV, Freund KB, Chang S, Iranmanesh R. Development of subretinal fibrosis after anti-VEGF treatment in neovascular age-related macular degeneration. Ophthalmic Surg Lasers Imaging. (2011) 42:6–11. doi: 10.3928/15428877-20100924-01
27. Evans RN, Reeves BC, Maguire MG, Martin DF, Muldrew A, Peto T, et al. Associations of variation in retinal thickness with visual acuity and anatomic outcomes in eyes with neovascular age-related macular degeneration lesions treated with anti-vascular endothelial growth factor agents. JAMA Ophthalmol. (2020) 138:1043–51. doi: 10.1001/jamaophthalmol.2020.3001
28. Teo KYC, Joe AW, Nguyen V, Invernizzi A, Arnold JJ, Barthelmes D, et al. Prevalence and risk factors for the development of physician-graded subretinal fibrosis in eyes treated for neovascular age-related macular degeneration. Retina. (2020) 40:2285–95. doi: 10.1097/IAE.0000000000002779
29. Barikian A, Mahfoud Z, Abdulaal M, Safar A, Bashshur ZF. Induction with intravitreal bevacizumab every two weeks in the management of neovascular age-related macular degeneration. Am J Ophthalmol. (2015) 159:131–7. doi: 10.1016/j.ajo.2014.10.005
30. Heesterbeek TJ, Lorés-Motta L, Hoyng CB, Lechanteur YTE, den Hollander AI. Risk factors for progression of age-related macular degeneration. Ophthalmic Physiol Optics. (2020) 40:140–70. doi: 10.1111/opo.12675
31. Merle BMJ, Colijn JM, Cougnard-Grégoire A, de Koning-Backus APM, Delyfer MN, Kiefte-de Jong JC, et al. Mediterranean diet and incidence of advanced age-related macular degeneration: the EYE-RISK consortium. Ophthalmology. (2019) 126:381–90. doi: 10.1016/j.ophtha.2018.08.006
32. Cui B, Guo X, Zhou W, Zhang X, He K, Bai T, et al. Exercise alleviates neovascular age-related macular degeneration by inhibiting AIM2 inflammasome in myeloid cells. Metabolism. (2023) 144:155584. doi: 10.1016/j.metabol.2023.155584
33. Seddon JM, Reynolds R, Yu Y, Rosner B. Three new genetic loci (R1210C in CFH, variants in COL8A1 and RAD51B) are independently related to progression to advanced macular degeneration. PloS One. (2014) 9:e87047. doi: 10.1371/journal.pone.0087047
34. Wang JJ, Rochtchina E, Smith W, Klein R, Klein BE, Joshi T, et al. Combined effects of complement factor H genotypes, fish consumption, and inflammatory markers on long-term risk for age-related macular degeneration in a cohort. Am J Epidemiol. (2009) 169:633–41. doi: 10.1093/aje/kwn358
35. Miyake M, Yamashiro K, Tamura H, Kumagai K, Saito M, Sugahara-Kuroda M, et al. The contribution of genetic architecture to the 10-year incidence of age-related macular degeneration in the fellow eye. Invest Ophthalmol Vis Sci. (2015) 56:5353–61. doi: 10.1167/iovs.14-16020
36. Lechner J, Chen M, Hogg R. E, Toth L, Silvestri G, Chakravarthy U, et al. Higher plasma levels of complement C3a, C4a and C5a increase the risk of subretinal fibrosis in neovascular age-related macular degeneration: Complement activation in AMD. Immun Ageing. (2016) 13:4. doi: 10.1186/s12979-016-0060-5
37. Singh A, Falk MK, Subhi Y, Sørensen TL. The association between plasma 25-hydroxyvitamin D and subgroups in age-related macular degeneration: a cross-sectional study. PloS One. (2013) 8:e70948. doi: 10.1371/journal.pone.0070948
38. Merle BMJ, Silver RE, Rosner B, Seddon JM. Associations between vitamin D intake and progression to incident advanced age-related macular degeneration. Invest Ophthalmol Vis Sci. (2017) 58:4569–78. doi: 10.1167/iovs.17-21673
39. Kim KL, Park SP. Association between serum vitamin D deficiency and age-related macular degeneration in Koreans: Clinical case-control pilot study. Medicine. (2018) 97:e11908. doi: 10.1097/MD.0000000000011908
40. Henderson NC, Rieder F, Wynn TA. Fibrosis: from mechanisms to medicines. Nature. (2020) 587:555–66. doi: 10.1038/s41586-020-2938-9
41. Friedlander M. Fibrosis and diseases of the eye. J Clin Invest. (2007) 117:576–86. doi: 10.1172/JCI31030
42. Spraul CW, Grossnihlatis HE. Characteristics of drusen and bruch’s membrane in postmortem eyes with age-related macular degeneration. Arch Ophthalmol. (1997) 115:267–73. doi: 10.1001/archopht.1997.01100150269022
43. Kimura H, Yasukawa T, Tabata Y, Ogura Y. Drug targeting to choroidal neovascularization. Adv Drug Delivery Rev. (2001) 52:79–91. doi: 10.1016/S0169-409X(01)00190-9
44. Wynn TA. Common and unique mechanisms regulate fibrosis in various fibroproliferative diseases. J Clin Invest. (2007) 117:524–9. doi: 10.1172/JCI31487
45. Klaassen I, van Geest RJ, Kuiper EJ, van Noorden CJF, Schlingemann RO. The role of CTGF in diabetic retinopathy. Exp Eye Res. (2015) 133:37–48. doi: 10.1016/j.exer.2014.10.016
46. Penn S. The role of submacular surgery in the treatment of choroidal neovascular membranes. Clin Eye Vis Care. (2000) 12:37–50. doi: 10.1016/S0953-4431(99)00044-2
47. Lopez PF, Lambert HM, Grossniklaus HE, Sternberg P. Well-defined subfoveal choroidal neovascular membranes in age-related macular degeneration. Ophthalmology. (1993) 100:415–22. doi: 10.1016/S0161-6420(93)31657-X
48. Lopez PF, Sippy BD, Lambert HM, Thach AB, Hintonf DR. Transdifferentiated retinal pigment epithelial cells are immunoreactive for vascular endothelial growth factor in surgically excised age-related macular degeneration-related choroidal neovascular membranes. Invest Ophthalmol Vis Sci. (1996) 37:855–68.
49. Hinton DR, He S, Lopez PF. Apoptosis in surgically excised choroidal neovascular membranes in age-related macular degeneration. Arch Ophthalmol. (1998) 116:203–9. doi: 10.1001/archopht.116.2.203
50. Grossniklaus HE, Hutchinson AK, Capone A, Woolfson J, Lambert HM. Clinicopathologic features of surgically excised choroidal neovascular membranes. Ophthalmology. (1994) 101:1099–111. doi: 10.1016/S0161-6420(13)31216-0
51. Grossniklaus HE, Green WR, S. S. T. R. Group. Histopathologic and ultrastructural findings of surgically excised choroidal neovascularization. Arch Ophthalmol. (1998) 116:745–9. doi: 10.1001/archopht.116.6.745
52. Kawano H, Kimura-Kuroda J, Komuta Y, Yoshioka N, Li HP, Kawamura K, et al. Role of the lesion scar in the response to damage and repair of the central nervous system. Cell Tissue Res. (2012) 349:169–80. doi: 10.1007/s00441-012-1336-5
53. Yoshioka N, Hisanaga SI, Kawano H. Suppression of fibrotic scar formation promotes axonal regeneration without disturbing blood-brain barrier repair and withdrawal of leukocytes after traumatic brain injury. J Comp Neurol. (2010) 518:3867–81. doi: 10.1002/cne.22431
54. Soderblom C, Luo X, Blumenthal E, Bray E, Lyapichev K, Ramos J, et al. Perivascular fibroblasts form the fibrotic scar after contusive spinal cord injury. J Neurosci. (2013) 33:13882–7. doi: 10.1523/JNEUROSCI.2524-13.2013
55. Wu KHC, Madigan MC, Billson FA, Penfold PL. Differential expression of GFAP in early v late AMD: a quantitative analysis. Br J Ophthalmol. (2003) 87:1159. doi: 10.1136/bjo.87.9.1159
56. Salas A, Badia A, Fontrodona L, Zapata M, García-Arumí J, Duarri A. Neovascular progression and retinal dysfunction in the laser-induced choroidal neovascularization mouse model. Biomedicines. (2023) 11:2445. doi: 10.3390/biomedicines11092445
57. Ishida T, Yoshida T, Shinohara K, Cao K, Nakahama KI, Morita I, et al. Potential role of sirtuin 1 in Müller glial cells in mice choroidal neovascularization. PloS One. (2017) 12:e0183775. doi: 10.1371/journal.pone.0183775
58. Im S, Han JW, Park EJ, Bang JH, Shin HJ, Chang HS, et al. Suppression of choroidal neovascularization and epithelial-mesenchymal transition in retinal pigmented epithelium by adeno-associated virus-mediated overexpression of CCN5 in mice. PloS One. (2022) 17:e0269937. doi: 10.1371/journal.pone.0269937
59. Shu DY, Butcher E, Saint-Geniez M. EMT and endMT: emerging roles in age-related macular degeneration. Int J Mol Sci. (2020) 21:1–26. doi: 10.3390/ijms21124271
60. Ayazi M, Zivkovic S, Hammel G, Stefanovic B, Ren Y. Fibrotic scar in CNS injuries: from the cellular origins of fibroblasts to the molecular processes of fibrotic scar formation. Cells. (2022) 11:2371. doi: 10.3390/cells11152371
61. Wanner IB, Anderson MA, Song B, Levine J, Fernandez A, Gray-Thompson Z, et al. Glial scar borders are formed by newly proliferated, elongated astrocytes that interact to corral inflammatory and fibrotic cells via STAT3-dependent mechanisms after spinal cord injury. J Neurosci. (2013) 33:12870–86. doi: 10.1523/JNEUROSCI.2121-13.2013
62. Naylor A, Hopkins A, Hudson N, Campbell M. Tight junctions of the outer blood retina barrier. Int J Mol Sci. (2019) 21:211. doi: 10.3390/ijms21010211
63. Murali A, Krishnakumar S, Subramanian A, Parameswaran S. Bruch’s membrane pathology: A mechanistic perspective. Eur J Ophthalmol. (2020) 30:1195–206. doi: 10.1177/1120672120919337
64. Yang X, Chung JY, Rai U, Esumi N. Cadherins in the retinal pigment epithelium (RPE) revisited: P-cadherin is the highly dominant cadherin expressed in human and mouse RPE in vivo. PloS One. (2018) 13:e0191279. doi: 10.1371/journal.pone.0191279
65. Sheedlo HJ, Li L, Turner JE. Effects of RPE-cell factors secreted from permselective fibers on retinal cells in vitro. Brain Res. (1992) 587:327–37. doi: 10.1016/0006-8993(92)91015-7
66. Miyajima-Uchida H, Hayashi H, Beppu R, Kuroki M, Fukami M, Arakawa F, et al. Production and accumulation of thrombospondin-1 in human retinal pigment epithelial cells. Invest Ophthalmol Vis Sci. (2000) 41:561–7.
67. Tombran-Tink J, Shivaram SM, Chader GJ, Johnson LV, Bok D. Expression, secretion, and age-related downregulation of pigment epithelium-derived factor, a serpin with neurotrophic activity. J Neurosci. (1995) 15:4992–5003. doi: 10.1523/JNEUROSCI.15-07-04992.1995
68. Paterson C, Cannon J, Vargis E. The impact of early RPE cell junction loss on VEGF, Ang-2, and TIMP secretion in vitro. Mol Vis. (2023) 29:87.
69. Matsubara JA, Tian Y, Cui JZ, Zeglinski MR, Hiroyasu S, Turner CT, et al. Retinal distribution and extracellular activity of granzyme B: A serine protease that degrades retinal pigment epithelial tight junctions and extracellular matrix proteins. Front Immunol. (2020) 11:574. doi: 10.3389/fimmu.2020.00574
70. Hirasawa M, Noda K, Noda S, Suzuki M, Ozawa Y, Shinoda K, et al. Transcriptional factors associated with epithelial-mesenchymal transition in choroidal neovascularization. Mol Vis. (2011) 17:1222–30.
71. Ghosh S, Shang P, Terasaki H, Stepicheva N, Hose S, Yazdankhah M, et al. A role for βA3/A1-crystallin in type 2 EMT of RPE cells occurring in dry age-related macular degeneration. Invest Ophthalmol Vis Sci. (2018) 59:AMD104–13. doi: 10.1167/iovs.18-24132
72. Sun JX, Chang TF, Li MH, Sun LJ, Yan XC, Yang ZY, et al. SNAI1, an endothelial–mesenchymal transition transcription factor, promotes the early phase of ocular neovascularization. Angiogenesis. (2018) 21:635–52. doi: 10.1007/S10456-018-9614-9/FIGURES/8
73. Medici D, Kalluri R. Endothelial-mesenchymal transition and its contribution to the emergence of stem cell phenotype. Semin Cancer Biol. (2012) 22:379. doi: 10.1016/j.semcancer.2012.04.004
74. Luo X, Yang S, Liang J, Zhai Y, Shen M, Sun J, et al. Choroidal pericytes promote subretinal fibrosis after experimental photocoagulation. DMM Dis Models Mech. (2018) 11:dmm032060. doi: 10.1242/DMM.032060/258736/AM/CHOROIDAL-PERICYTES-PROMOTE-SUBRETINAL-FIBROSIS
75. Zhao Z, Zhang Y, Zhang C, Zhang J, Luo X, Qiu Q, et al. TGF-β promotes pericyte-myofibroblast transition in subretinal fibrosis through the Smad2/3 and Akt/mTOR pathways. Exp Mol Med. (2022) 54:673–84. doi: 10.1038/s12276-022-00778-0
76. Xu YH, Feng YF, Zou R, Yuan F, Yuan YZ. Silencing of YAP attenuates pericyte-myofibroblast transition and subretinal fibrosis in experimental model of choroidal neovascularization. Cell Biol Int. (2022) 46:1249–63. doi: 10.1002/cbin.11809
77. Siedlecki J, Asani B, Wertheimer C, Hillenmayer A, Ohlmann A, Priglinger C, et al. Combined VEGF/PDGF inhibition using axitinib induces αSMA expression and a pro-fibrotic phenotype in human pericytes. Graefe’s Arch Clin Exp Ophthalmol. (2018) 256:1141–9. doi: 10.1007/S00417-018-3987-8/FIGURES/6
78. Reichenbach A, Bringmann A. Glia of the human retina. Glia. (2020) 68:768–96. doi: 10.1002/glia.23727
79. Bringmann A, Iandiev I, Pannicke T, Wurm A, Hollborn M, Wiedemann P, et al. Cellular signaling and factors involved in Müller cell gliosis: Neuroprotective and detrimental effects. Prog Retin Eye Res. (2009) 28:423–51. doi: 10.1016/j.preteyeres.2009.07.001
80. Jo YJ, Sonoda KH, Oshima Y, Takeda A, Kohno R, Yamada J, et al. Establishment of a new animal model of focal subretinal fibrosis that resembles disciform lesion in advanced age-related macular degeneration. Invest Ophthalmol Vis Sci. (2011) 52:6089–95. doi: 10.1167/iovs.10-5189
81. Yoo HS, Shanmugalingam U, Smith PD. Harnessing astrocytes and müller glial cells in the retina for survival and regeneration of retinal ganglion cells. Cells. (2021) 10:1339. doi: 10.3390/cells10061339
82. Wu D, Kanda A, Liu Y, Noda K, Murata M, Ishida S. Involvement of müller glial autoinduction of TGF-β in diabetic fibrovascular proliferation via glial-mesenchymal transition. Invest Ophthalmol Vis Sci. (2020) 61:29. doi: 10.1167/iovs.61.14.29
83. da Silva RA, Roda VMP, Akamine PS, da Silva DS, Siqueira PV, Matsuda M, et al. Blockade of the TGF-β pathway by galunisertib inhibits the glial-mesenchymal transition in Müller glial cells. Exp Eye Res. (2023) 226:109336. doi: 10.1016/j.exer.2022.109336
84. Guidry C. Isolation and characterization of porcine Müller cells. Myofibroblastic dedifferentiation in culture. Invest Ophthalmol Vis Sci. (1996) 37:740–52.
85. Kanda A, Noda K, Hirose I, Ishida S. TGF-β-SNAIL axis induces Müller glial-mesenchymal transition in the pathogenesis of idiopathic epiretinal membrane. Sci Rep. (2019) 9:1–13. doi: 10.1038/s41598-018-36917-9
86. Grossniklaus HE, Ling JX, Wallace TM, Dithmar S, Lawson DH, Cohen C, et al. Macrophage and retinal pigment epithelium expression of angiogenic cytokines in choroidal neovascularization. Mol Vis. (2002) 8:119–26.
87. Espinosa-Heidmann DG, Reinoso MA, Pina Y, Csaky KG, Caicedo A, Cousins SW. Quantitative enumeration of vascular smooth muscle cells and endothelial cells derived from bone marrow precursors in experimental choroidal neovascularization. Exp Eye Res. (2005) 80:369–78. doi: 10.1016/j.exer.2004.10.005
88. Little K, Llorián-Salvador M, Tang M, Du X, Marry S, Chen M, et al. Macrophage to myofibroblast transition contributes to subretinal fibrosis secondary to neovascular age-related macular degeneration. J Neuroinflamm. (2020) 17:355. doi: 10.1186/s12974-020-02033-7
89. Wang YY, Jiang H, Pan J, Huang XR, Wang YC, Huang HF, et al. Macrophage-to-myofibroblast transition contributes to interstitial fibrosis in chronic renal allograft injury. J Am Soc Nephrol. (2017) 28:2053–67. doi: 10.1681/ASN.2016050573/-/DCSUPPLEMENTAL
90. Meng XM, Wang S, Huang XR, Yang C, Xiao J, Zhang Y, et al. Inflammatory macrophages can transdifferentiate into myofibroblasts during renal fibrosis. Cell Death Dis. (2016) 7:e2495–5. doi: 10.1038/cddis.2016.402
91. Liu H, Guan Q, Zhao P, Li J. TGF-β-induced CCR8 promoted macrophage transdifferentiation into myofibroblast-like cells. Exp Lung Res. (2022) 48:86–99. doi: 10.1080/01902148.2022.2055227
92. Yi C, Liu J, Deng W, Luo C, Qi J, Chen M, et al. Macrophage elastase (MMP12) critically contributes to the development of subretinal fibrosis. J Neuroinflamm. (2022) 19:1–16. doi: 10.1186/S12974-022-02433-X/FIGURES/8
93. Reinhardt JW, Breuer CK. Fibrocytes: A critical review and practical guide. Front Immunol. (2021) 12:784401/BIBTEX. doi: 10.3389/FIMMU.2021.784401/BIBTEX
94. Yi C, Liu J, Deng W, Luo C, Qi J, Chen M, et al. Old age promotes retinal fibrosis in choroidal neovascularization through circulating fibrocytes and profibrotic macrophages. J Neuroinflamm. (2023) 20:45. doi: 10.1186/s12974-023-02731-y
95. Chen M, Xu H. Parainflammation, chronic inflammation, and age-related macular degeneration. J Leukoc Biol. (2015) 98:713–25. doi: 10.1189/jlb.3RI0615-239R
96. Lin T, Walker GB, Kurji K, Fang E, Law G, Prasad SS, et al. Parainflammation associated with advanced glycation endproduct stimulation of RPE in vitro: implications for age-related degenerative diseases of the eye. Cytokine. (2013) 62:369–81. doi: 10.1016/j.cyto.2013.03.027
97. Szczepan M, Llorián-Salvador M, Chen M, Xu H. Immune cells in subretinal wound healing and fibrosis. Front Cell Neurosci. (2022) 16:916719. doi: 10.3389/fncel.2022.916719
98. Sakurai E, Anand A, Ambati BK, Van Rooijen N, Ambati J. Macrophage depletion inhibits experimental choroidal neovascularization. Invest Ophthalmol Vis Sci. (2003) 44:3578–85. doi: 10.1167/iovs.03-0097
99. Tsutsumi C, Sonoda KH, Egashira K, Qiao H, Hisatomi T, Nakao S, et al. The critical role of ocular-infiltrating macrophages in the development of choroidal neovascularization. J Leukoc Biol. (2003) 74:25–32. doi: 10.1189/jlb.0902436
100. Liu J, Copland DA, Horie S, Wu WK, Chen M, Xu Y, et al. Myeloid cells expressing VEGF and arginase-1 following uptake of damaged retinal pigment epithelium suggests potential mechanism that drives the onset of choroidal angiogenesis in mice. PloS One. (2013) 8:72935. doi: 10.1371/journal.pone.0072935
101. Zhu L, Fu X, Chen X, Han X, Dong P. M2 macrophages induce EMT through the TGF-β/Smad2 signaling pathway. Cell Biol Int. (2017) 41:960–8. doi: 10.1002/cbin.10788
102. Tan W, Zou J, Yoshida S, Jiang B, Zhou Y. The role of inflammation in age-related macular degeneration. Int J Biol Sci. (2020) 16:2989. doi: 10.7150/ijbs.49890
103. Jing R, Qi T, Wen C, Yue J, Wang G, Pei C, et al. Interleukin-2 induces extracellular matrix synthesis and TGF-β2 expression in retinal pigment epithelial cells. Dev Growth Differ. (2019) 61:410. doi: 10.1111/dgd.12630
104. Sato K, Takeda A, Hasegawa E, Jo YJ, Arima M, Oshima Y, et al. Interleukin-6 plays a crucial role in the development of subretinal fibrosis in a mouse model. Immunol Med. (2018) 41:23–9. doi: 10.1080/09114300.2018.1451609
105. McGeough MD, Pena CA, Mueller JL, Pociask DA, Broderick L, Hoffman HM, et al. Interleukin-6 is a marker of inflammation with no direct role in inflammasome-mediated mouse models. J Immunol. (2012) 189:2707. doi: 10.4049/jimmunol.1101737
106. Gao J, Liu RT, Cao S, Cui JZ, Wang A, To E, et al. NLRP3 inflammasome: activation and regulation in age-related macular degeneration. Mediators Inflammation. (2015) 2015:690243. doi: 10.1155/2015/690243
107. Gao J, Cui JZ, To E, Cao S, Matsubara JA. Evidence for the activation of pyroptotic and apoptotic pathways in RPE cells associated with NLRP3 inflammasome in the rodent eye. J Neuroinflamm. (2018) 15:15. doi: 10.1186/s12974-018-1062-3
108. Nakamura R, Sene A, Santeford A, Gdoura A, Kubota S, Zapata N, et al. IL10-driven STAT3 signalling in senescent macrophages promotes pathological eye angiogenesis. Nat Commun. (2015) 6:7847. doi: 10.1038/ncomms8847
109. Yang Y, Takeda A, Yoshimura T, Oshima Y, Sonoda KH, Ishibashi T. IL-10 is significantly involved in HSP70-regulation of experimental subretinal fibrosis. PloS One. (2013) 8:80288. doi: 10.1371/journal.pone.0080288
110. Fu B, Liu ZL, Zhang H, Gu F. Interleukin-13 and age-related macular degeneration. Int J Ophthalmol. (2017) 10:535–40. doi: 10.18240/ijo.2017.04.06
111. Park YG, Park YS, Kim IB. Complement system and potential therapeutics in age-related macular degeneration. Int J Mol Sci. (2021) 22:6851. doi: 10.3390/ijms22136851
112. Cao S, Wang JC, Gao J, Wong M, To E, White VA, et al. CFH Y402H polymorphism and the complement activation product C5a: effects on NF-κB activation and inflammasome gene regulation. Br J Ophthalmol. (2016) 100:713–8. doi: 10.1136/bjophthalmol-2015-307213
113. Seth A, Cui J, To E, Kwee M, Matsubara J. Complement-associated deposits in the human retina. Invest Ophthalmol Vis Sci. (2008) 49:743–50. doi: 10.1167/iovs.07-1072
114. Mullins RF, Schoo DP, Sohn EH, Flamme-Wiese MJ, Workamelahu G, Johnston RM, et al. The membrane attack complex in aging human choriocapillaris: relationship to macular degeneration and choroidal thinning. Am J Pathol. (2014) 184:3142. doi: 10.1016/j.ajpath.2014.07.017
115. Nozaki M, Raisler BJ, Sakurai E, Sarma JV, Barnum SR, Lambris JD, et al. Drusen complement components C3a and C5a promote choroidal neovascularization. Proc Natl Acad Sci U.S.A. (2006) 103:2328. doi: 10.1073/pnas.0408835103
116. Wang JCC, Cao S, Wang A, To E, Law G, Gao J, et al. CFH Y402H polymorphism is associated with elevated vitreal GM-CSF and choroidal macrophages in the postmortem human eye. Mol Vis. (2015) 21:264–72.
117. Hillebrandt S, Wasmuth HE, Weiskirchen R, Hellerbrand C, Keppeler H, Werth A, et al. Complement factor 5 is a quantitative trait gene that modifies liver fibrogenesis in mice and humans. Nat Genet. (2005) 37:835–43. doi: 10.1038/ng1599
118. Strey CW, Markiewski M, Mastellos D, Tudoran R, Spruce LA, Greenbaum LE, et al. The proinflammatory mediators C3a and C5a are essential for liver regeneration. J Exp Med. (2003) 198:913–23. doi: 10.1084/jem.20030374
119. Llorián-Salvador M, Byrne EM, Szczepan M, Little K, Chen M, Xu H. Complement activation contributes to subretinal fibrosis through the induction of epithelial-to-mesenchymal transition (EMT) in retinal pigment epithelial cells. J Neuroinflamm. (2022) 19:182. doi: 10.1186/s12974-022-02546-3
120. Zhao Y, Ma J, Fan Y, Wang Z, Tian R, Ji W, et al. TGF-β transactivates EGFR and facilitates breast cancer migration and invasion through canonical Smad3 and ERK/Sp1 signaling pathways. Mol Oncol. (2018) 12:305–21. doi: 10.1002/1878-0261.12162
121. Cao S, Pan Y, Terker AS, Arroyo Ornelas JP, Wang Y, Tang J, et al. Epidermal growth factor receptor activation is essential for kidney fibrosis development. Nat Commun. (2023) 14:7357. doi: 10.1038/s41467-023-43226-x
122. Travis MA, Sheppard D. TGF-β activation and function in immunity. Annu Rev Immunol. (2014) 32:51–82. doi: 10.1146/annurev-immunol-032713-120257
123. Levy YA, Fainberg KM, Amidror T, Regev K, Auriel E, Karni A. High and dysregulated secretion of epidermal growth factor from immune cells of patients with relapsing-remitting multiple sclerosis. J Neuroimmunol. (2013) 257:82–9. doi: 10.1016/j.jneuroim.2013.01.012
124. Fuchs L, Mausner-Fainberg K, Luban A, Asseyer SE, Golan M, Benhamou M, et al. CTGF/CCN2 has a possible detrimental role in the inflammation and the remyelination failure in the early stages of multiple sclerosis. J Neuroimmunol. (2022) 371:577936. doi: 10.1016/j.jneuroim.2022.577936
125. Hoeres T, Wilhelm M, Smetak M, Holzmann E, Schulze-Tanzil G, Birkmann J. Immune cells regulate VEGF signalling via release of VEGF and antagonistic soluble VEGF receptor-1. Clin Exp Immunol. (2018) 192:54–67. doi: 10.1111/cei.13090
126. Obasanmi G, Uppal M, Cui JZ, Xi J, Ju MJ, Song J, et al. Granzyme B degrades extracellular matrix and promotes inflammation and choroidal neovascularization. Angiogenesis. (2024), 1–23. doi: 10.1007/S10456-024-09909-9/FIGURES/9
127. Bhutto IA, McLeod DS, Jing T, Sunness JS, Seddon JM, Lutty GA. Increased choroidal mast cells and their degranulation in age-related macular degeneration. Br J Ophthalmol. (2016) 100:720. doi: 10.1136/bjophthalmol-2015-308290
128. Penfold P, Killingsworth M, Sarks S. An ultrastructural study of the role of leucocytes and fibroblasts in the breakdown of Bruch’s membrane. Aust J Ophthalmol. (1984) 12:23–31.
129. Elieh Ali Komi D, Wöhrl S, Bielory L. Mast cell biology at molecular level: a comprehensive review. Clin Rev Allergy Immunol. (2020) 58:342–65. doi: 10.1007/S12016-019-08769-2/FIGURES/5
130. Theoharides TC, Twahir A, Kempuraj D. Mast cells in the autonomic nervous system and potential role in disorders with dysautonomia and neuroinflammation. Ann Allergy Asthma Immunol. (2024) 132:440–54. doi: 10.1016/j.anai.2023.10.032
131. Krystel-Whittemore M, Dileepan KN, Wood JG. Mast cell: A multi-functional master cell. Front Immunol. (2015) 6:620. doi: 10.3389/fimmu.2015.00620
132. McMenamin PG. The distribution of immune cells in the uveal tract of the normal eye. Eye (Lond). (1997) 11:183–93. doi: 10.1038/eye.1997.49
133. Bradding P, Pejler G. The controversial role of mast cells in fibrosis. Immunol Rev. (2018) 282:198–231. doi: 10.1111/imr.12626
134. Arai R, Usui-Ouchi A, Ito Y, Mashimo K, Murakami A, Ebihara N. Effects of secreted mast cell mediators on retinal pigment epithelial cells: focus on mast cell tryptase. Mediators Inflammation. (2017) 2017:3124753. doi: 10.1155/2017/3124753
135. Chen Y, Li C, Xie H, Fan Y, Yang Z, Ma J, et al. Infiltrating mast cells promote renal cell carcinoma angiogenesis by modulating PI3K→AKT→GSK3β→AM signaling. Oncogene. (2017) 36:2879. doi: 10.1038/onc.2016.442
136. Gailit J, Marchese MJ, Kew RR, Gruber BL. The differentiation and function of myofibroblasts is regulated by mast cell mediators. J Invest Dermatol. (2001) 117:1113–9. doi: 10.1046/j.1523-1747.2001.15211.x
137. Shimbori C, Upagupta C, Bellaye PS, Ayaub EA, Sato S, Yanagihara T, et al. Mechanical stress-induced mast cell degranulation activates TGF-β1 signalling pathway in pulmonary fibrosis. Thorax. (2019) 74:455–65. doi: 10.1136/thoraxjnl-2018-211516
138. Tatler AL, Porte J, Knox A, Jenkins G, Pang L. Tryptase activates TGFbeta in human airway smooth muscle cells via direct proteolysis. Biochem Biophys Res Commun. (2008) 370:239–42. doi: 10.1016/j.bbrc.2008.03.064
139. Lindstedt KA, Wang Y, Shiota N, Saarinen J, Hyytiäinen M, Kokkonen JO, et al. Activation of paracrine TGF-beta1 signaling upon stimulation and degranulation of rat serosal mast cells: a novel function for chymase. FASEB J. (2001) 15:1377–88. doi: 10.1096/fj.00-0273com
140. Chen L, Schrementi ME, Ranzer MJ, Wilgus TA, DiPietro LA. Blockade of mast cell activation reduces cutaneous scar formation. PloS One. (2014) 9:e85226. doi: 10.1371/journal.pone.0085226
141. e Ribeiro LSF, dos Santos JN, Rocha CAG, Cury PR. Association between mast cells and collagen maturation in chronic periodontitis in humans. J Histochem Cytochem. (2018) 66:467. doi: 10.1369/0022155418765131
142. Foley TT, Ehrlich HP. Through gap junction communications, co-cultured mast cells and fibroblasts generate fibroblast activities allied with hypertrophic scarring. Plast Reconstr Surg. (2013) 131:1036–44. doi: 10.1097/PRS.0b013e3182865c3f
143. Verma SK, Garikipati VNS, Krishnamurthy P, Schumacher SM, Grisanti LA, Cimini M, et al. Interleukin-10 inhibits bone marrow fibroblast progenitor cell-mediated cardiac fibrosis in pressure-overloaded myocardium. Circulation. (2017) 136:940–53. doi: 10.1161/CIRCULATIONAHA.117.027889
144. Obasanmi G, Zeglinski MR, Hardie E, Wilhelm AC, Turner CT, Hiroyasu S, et al. Granzyme B contributes to choroidal neovascularization and age-related macular degeneration through proteolysis of thrombospondin-1. Lab Invest. (2023) 103:100123. doi: 10.1016/j.labinv.2023.100123
145. Hendel A, Hsu I, Granville DJ. Granzyme B releases vascular endothelial growth factor from extracellular matrix and induces vascular permeability. Lab Invest. (2014) 94:716. doi: 10.1038/labinvest.2014.62
146. Shen Y, Cheng F, Sharma M, Merkulova Y, Raithatha SA, Parkinson LG, et al. Granzyme B deficiency protects against angiotensin II-induced cardiac fibrosis. Am J Pathol. (2016) 186:87–100. doi: 10.1016/j.ajpath.2015.09.010
147. Shen Y, Zeglinski MR, Turner CT, Raithatha SA, Wu Z, Russo V, et al. Topical small molecule granzyme B inhibitor improves remodeling in a murine model of impaired burn wound healing. Exp Mol Med. (2018) 50:1–11. doi: 10.1038/s12276-018-0095-0
148. Turner CT, Bolsoni J, Zeglinski MR, Zhao H, Ponomarev T, Richardson K, et al. Granzyme B mediates impaired healing of pressure injuries in aged skin. NPJ Aging Mech Dis. (2021) 7:6. doi: 10.1038/s41514-021-00059-6
149. Georgiadis A, Tschernutter M, Bainbridge JW, Balaggan KS, Mowat F, West EL, et al. The tight junction associated signalling proteins ZO-1 and ZONAB regulate retinal pigment epithelium homeostasis in mice. PloS One. (2010) 5:e15730. doi: 10.1371/journal.pone.0015730
150. Ciszewski WM, Wawro ME, Sacewicz-Hofman I, Sobierajska K. Cytoskeleton reorganization in endMT-the role in cancer and fibrotic diseases. Int J Mol Sci. (2021) 22:11607. doi: 10.3390/ijms222111607
151. Wang X, Ma W, Han S, Meng Z, Zhao L, Yin Y, et al. TGF-β participates choroid neovascularization through Smad2/3-VEGF/TNF-α signaling in mice with Laser-induced wet age-related macular degeneration. Sci Rep. (2017) 7:9672. doi: 10.1038/s41598-017-10124-4
152. Kasai H, Allen JT, Mason RM, Kamimura T, Zhang Z. TGF-beta1 induces human alveolar epithelial to mesenchymal cell transition (EMT). Respir Res. (2005) 6:56. doi: 10.1186/1465-9921-6-56
153. Murillo MM, del Castillo G, Sánchez A, Fernández M, Fabregat I. Involvement of EGF receptor and c-Src in the survival signals induced by TGF-beta1 in hepatocytes. Oncogene. (2005) 24:4580–7. doi: 10.1038/sj.onc.1208664
154. Tanase C, Enciu AM, Codrici E, Popescu ID, Dudau M, Dobri AM, et al. Fatty acids, CD36, thrombospondin-1, and CD47 in glioblastoma: together and/or separately? Int J Mol Sci. (2022) 23:604. doi: 10.3390/ijms23020604
155. Zhang Y, Wang H. Integrin signalling and function in immune cells. Immunology. (2012) 135:268–75. doi: 10.1111/j.1365-2567.2011.03549.x
156. Pastwińska J, Żelechowska P, Walczak-Drzewiecka A, Brzezińska-Błaszczyk E, Dastych J. The art of mast cell adhesion. Cells. (2020) 9:2664. doi: 10.3390/cells9122664
157. Dias JV, Benslimane-Ahmim Z, Egot M, Lokajczyk A, Grelac F, Galy-Fauroux I, et al. A motif within the N-terminal domain of TSP-1 specifically promotes the proangiogenic activity of endothelial colony-forming cells. Biochem Pharmacol. (2012) 84:1014–23. doi: 10.1016/j.bcp.2012.07.006
158. Sweetwyne MT, Pallero MA, Lu A, Van Duyn Graham L, Murphy-Ullrich JE. The calreticulin-binding sequence of thrombospondin 1 regulates collagen expression and organization during tissue remodeling. Am J Pathol. (2010) 177:1710–24. doi: 10.2353/ajpath.2010.090903
159. Kehlet SN, Bager CL, Willumsen N, Dasgupta B, Brodmerkel C, Curran M, et al. Cathepsin-S degraded decorin are elevated in fibrotic lung disorders - development and biological validation of a new serum biomarker. BMC Pulm Med. (2017) 17:110. doi: 10.1186/s12890-017-0455-x
160. Merline R, Moreth K, Beckmann J, Nastase MV, Zeng-Brouwers J, Tralhão JG, et al. Signaling by the Matrix Proteoglycan Decorin Controls Inflammation and Cancer through PDCD4 and microRNA-21. Sci Signal. (2011) 4:ra75. doi: 10.1126/scisignal.2001868
161. Mohan RR, Tripathi R, Sharma A, Sinha PR, Giuliano EA, Hesemann NP, et al. Decorin antagonizes corneal fibroblast migration via caveolae-mediated endocytosis of epidermal growth factor receptor. Exp Eye Res. (2019) 180:200. doi: 10.1016/j.exer.2019.01.001
162. Boivin WA, Shackleford M, Vanden Hoek A, Zhao H, Hackett TL, Knight DA, et al. Granzyme B cleaves decorin, biglycan and soluble betaglycan, releasing active transforming growth factor-β1. PloS One. (2012) 7:e33163. doi: 10.1371/journal.pone.0033163
163. Grossniklaus HE, Kang SJ, Berglin L. Animal models of choroidal and retinal neovascularization. Prog Retin Eye Res. (2010) 29:500–19. doi: 10.1016/j.preteyeres.2010.05.003
164. Little K, Llorián-Salvador M, Tang M, Du X, O'Shaughnessy Ó, McIlwaine G, et al. A two-stage laser-induced mouse model of subretinal fibrosis secondary to choroidal neovascularization. Transl Vis Sci Technol. (2020) 9. doi: 10.1167/tvst.9.4.3
165. Zandi S, Li Y, Jahnke L, Schweri-Olac A, Ishikawa K, Wada I, et al. Animal model of subretinal fibrosis without active choroidal neovascularization. Exp Eye Res. (2023) 229:109428. doi: 10.1016/j.exer.2023.109428
166. Ishibashi T, Miller H, Orr G, Sorgente N, Ryan SJ. Morphologic observations on experimental subretinal neovascularization in the monkey. Invest Ophthalmol Vis Sci. (1987) 28:1116–30.
167. Tobe T, Ortega S, Luna JD, Ozaki H, Okamoto N, Derevjanik NL, et al. Targeted disruption of the FGF2 gene does not prevent choroidal neovascularization in a murine model. Am J Pathol. (1998) 153:1641–6. doi: 10.1016/S0002-9440(10)65753-7
168. Shah RS, Soetikno BT, Lajko M, Fawzi Amani AA. A mouse model for laser-induced choroidal neovascularization. J Vis Exp. (2015) 106:e53502. doi: 10.3791/53502
169. Shpichka A, Butnaru D, Bezrukov EA, Sukhanov RB, Atala A, Burdukovskii V, et al. Skin tissue regeneration for burn injury. Stem Cell Res Ther. (2019) 10:94. doi: 10.1186/s13287-019-1203-3
170. Ishikawa K, Sreekumar PG, Spee C, Nazari H, Zhu D, Kannan R, et al. αB-crystallin regulates subretinal fibrosis by modulation of epithelial-mesenchymal transition. Am J Pathol. (2016) 186:859–73. doi: 10.1016/j.ajpath.2015.11.014
171. Wolf AT, Harris A, Oddone F, Siesky B, Vercellin AV, Ciulla TA. Disease progression pathways of wet AMD: opportunities for new target discovery. Expert Opin Ther Targets. (2022) 26:5. doi: 10.1080/14728222.2022.2030706
172. Dubchak E, Obasanmi G, Zeglinski MR, Granville DJ, Yeung SN, Matsubara JA. Potential role of extracellular granzyme B in wet age-related macular degeneration and fuchs endothelial corneal dystrophy. Front Pharmacol. (2022) 13:980742. doi: 10.3389/fphar.2022.980742
173. Aubert A, Lane M, Jung K, Granville DJ. Granzyme B as a therapeutic target: an update in 2022. Expert Opin Ther Targets. (2022) 26:979–93. doi: 10.1080/14728222.2022.2161890/SUPPL_FILE/IETT_A_2161890_SM2584.DOCX
Keywords: age-related macular degeneration (AMD), subretinal fibrosis, choroidal neovascularization, inflammation, immunology, granzyme B, mast cell, retinal pigment epithelium (RPE)
Citation: Gill K, Yoo H-S, Chakravarthy H, Granville DJ and Matsubara JA (2024) Exploring the role of granzyme B in subretinal fibrosis of age-related macular degeneration. Front. Immunol. 15:1421175. doi: 10.3389/fimmu.2024.1421175
Received: 22 April 2024; Accepted: 02 July 2024;
Published: 18 July 2024.
Edited by:
Fehim Esen, Istanbul Medeniyet University, TürkiyeReviewed by:
Stuart Mills, University of South Australia, AustraliaPeirong Lu, The First Affiliated Hospital of Soochow University, China
Copyright © 2024 Gill, Yoo, Chakravarthy, Granville and Matsubara. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Joanne A. Matsubara, am9hbm5lLm1hdHN1YmFyYUB1YmMuY2E=