 Yiwen Wang
Yiwen Wang Rui Yang
Rui Yang Rui Liu
Rui Liu Ruoyu Yang1
Ruoyu Yang1 Aili He
Aili He- 1Department of Hematology, The Second Affiliated Hospital of Xi’an JiaoTong University, Xi’an, Shaanxi, China
- 2Xi’an Key Laboratory of Hematological Diseases, Xi’an, Shaanxi, China
The treatment of multiple myeloma (MM) has significantly advanced; however, the underlying genetic mechanisms remain elusive. Clonal events and genetic alterations are recognized as pivotal in the pathogenesis of MM. It is now understood that a multitude of gene mutations, including those affecting RAS, TP53, RB1, and 1q21 amplification, are prevalent in this disease. The incorporation of several high-risk genetic factors into the Second Revision of the International Staging System (R2-ISS) underscores the prognostic significance of genetic aberrations in MM. The retinoblastoma gene (RB1), located in 13q14, encodes the retinoblastoma protein (pRB), a tumor suppressor that regulates cell cycle progression. Deletion of RB1, which is a frequent event in MM, contributes to tumorigenesis by disrupting cell cycle control. In this respect, RB1 loss has been implicated in the progression of MM through its influence on interleukin-6 (IL-6) secretion and cell proliferation. This review comprehensively summarizes the role of RB1 in MM and expounds on the potential of targeting RB1 as a therapeutic strategy for this malignancy.
1 Introduction
Multiple myeloma (MM) is a malignant proliferative neoplasm originating from plasma cells within the bone marrow. Despite significant advancements in therapy that have improved patient outcomes, a comprehensive understanding of the gene mutations and their underlying mechanisms in MM remains crucial for the development of more effective treatments. To date, numerous genes have been implicated in the prognosis of MM, including deletions of chromosomes 13 and 17p (1). Among them, the retinoblastoma susceptibility gene (RB1) was the first tumor suppressor gene identified. There is growing consensus suggesting that RB1 mutations play a critical role in the development of various cancers, including breast and prostate cancer (2, 3). Located on chromosome 13, RB1 deletions in the 13q14.2 region are frequently associated with tumorigenesis (4). In addition, loss of the retinoblastoma (RB) protein (pRB) has been strongly linked to deletions in the 13q chromosomal region, encompassing RB1 (5). Notably, RB1 deletions have been detected in approximately 50% of MM cases, predominantly due to complete monosomy 13 (85%), and are correlated with poorer prognosis (6). Consequently, RB1 is considered a tumor suppressor gene in MM (7). This review aimed to summarize the function of RB1 in MM and to explore the potential of targeting RB1 for MM treatment.
2 Roles of cytogenetic abnormalities in the pathogenesis of multiple myeloma
MM is a malignancy derived from plasma cells characterized by the secretion of monoclonal immunoglobulins. Clonal events, which occur in virtually all myeloma cells, including monoclonal gammopathy of undetermined significance (MGUS) and smoldering multiple myeloma (SMM), are pivotal in disease initiation. Acquired genetic alterations such as copy number gains, secondary translocations, and somatic mutations contribute to the progression of myeloma by disrupting cell cycle regulation (8).
Current evidence suggests that abnormal copy numbers can result in the gain or loss of chromosomal regions, contributing to the initiation and progression of malignant processes. Deletions of tumor suppressor genes, including but not limited to CDKN2C, FAF1, and FAM46C [due to del(1p)], BIRC2 and NIRC3 [due to del(11q)], RB1 and DIS3 [due to del(13q)], and TP53 [due to del(17p)], can occur through the deletion of the chromosomal arms (q or p). Among these, the RB1 gene is a critical tumor suppressor with a significant role in the pathogenesis of MM.
Common secondary translocations are simple, reciprocal exchanges that frequently involve breakpoints within or adjacent to the immunoglobulin heavy chain (IgH) switch region. These translocations are believed to arise from aberrant IgH class switch recombination. However, given the infrequent occurrence of IgH class switch recombination in plasma cell tumors and the association of complex MYC gene translocations and insertions with advanced disease stages, MYC rearrangement is considered a potential mechanism underlying genomic instability. MYC rearrangement often positions the MYC gene near potent immunoglobulin enhancers, contributing to genomic instability and potentially leading to secondary ectopic insertions (9, 10).
Patients with myeloma frequently harbor somatic mutations within genes of the RAS/MAPK pathway (9). In addition, the pathogenesis of myeloma has been implicated in the dysregulation of the NF-κB and PI3K pathways (11). Moreover, aberrant apoptotic pathways, such as those resulting from the t(11;14) translocation leading to BCL2 dependence in malignant plasma cells or MCL1 dependence in other patients, have been proposed to contribute to the development of myeloma (12).
Subsequent acquired genetic events have a significant relationship with the development of MM. Major genetic alterations occur earlier in the disease progression, and the differences in genetic changes between SMM and MM were less pronounced than those between SMM and MGUS. This suggests that additional factors, such as the bone marrow microenvironment, may contribute to disease pathogenesis. MM evolves through a complex interplay between clonal expansion and the bone marrow microenvironment. As the disease progresses, there is an increase of tumor-promoting immune cells and a concomitant loss of anti-tumor immune cells (13).
Currently, the traditional prognostic score for MM is determined using clinical and biochemical parameters, known as the International Staging System (ISS). However, the more recent R2-ISS incorporates cytogenetic features, including del(17p) and 1q21 amplification (14, 15). Genomic analysis of patients with MM revealed significant heterogeneity both inter- and intra-patient (16). Moreover, numerous gene mutations have been documented in newly diagnosed patients, with uneven distribution among subclonal units (16). Despite significant advancements in MM treatment, 25% of newly diagnosed patients exhibit a less than 3-year overall survival (17). High-risk cytogenetic abnormalities (HRCAs) warrant specific treatment regimens, such as three or four-drug induction therapy (PI/IMiD/Dex or PI/IMiD/Dex/anti-CD38 antibody), autologous transplantation, and lenalidomide ± PI consolidation/maintenance (18). The increased detection of common gene mutations has improved the clinical risk scores and offered more reliable prognostication (19). These findings underscore the prognostic significance of certain gene mutations and the need for tailored treatment approaches for patients with poor prognoses. While treatment advances have yielded excellent outcomes for some high-risk myeloma patients, a lot of newly diagnosed individuals still experience treatment failure. Therefore, it is imperative to refine the high-risk myeloma definitions, delineate the MM subtypes, and develop more precise, biologically driven treatment strategies. Elucidating the mechanisms underlying the different gene mutations and the pathogenesis of MM is crucial for optimizing patient care.
3 Biological characteristics of the RB1 gene
The RB1 gene was the first tumor suppressor gene identified, which was originally linked to the development of retinoblastoma (20, 21).
RB1 is located on chromosome 13q14, spans approximately 180 kb, and comprises 27 exons expressed in most tissues. Studies of RB1-related pathways have revealed striking similarities in the DNA sequence characteristics across tumor types with diverse genetic and epigenetic profiles (20). These findings suggest that modulating the RB1 pathways through targeted interventions based on the biological nature of regulatory RB1 mutations could offer a promising avenue for precise disease treatment.
RB1 exerts its tumor suppressor function through three primary mechanisms. Firstly, it regulates early 2 factor (E2F) transcription factors, inhibiting the transcription of cell cycle genes. The pRB–E2F complex further suppresses pluripotent genes, such as SOX2 in lung cancer (22) and EZH2 in prostate cancer (23). Secondly, RB1 contributes to epigenome stability by inhibiting the cell division cycle, allowing for proper epigenome establishment post-replication. Finally, RB1 is integral to heterochromatin organization and maintenance (24), which are essential for both cell differentiation and chromosomal integrity, including telomere stability. Consequently, RB1 loss disrupts cell differentiation and compromises chromosome segregation and telomere maintenance (Figure 1).
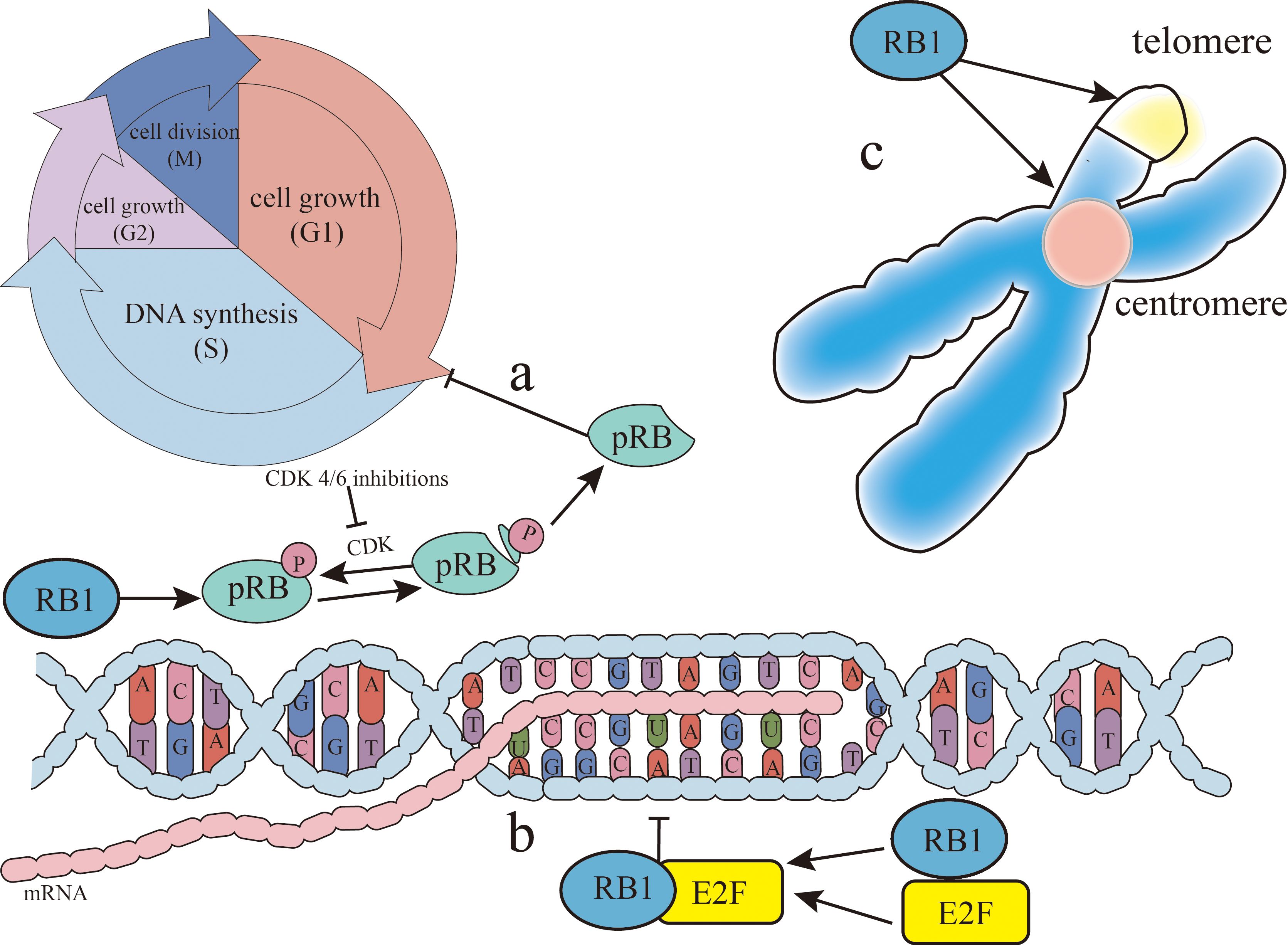
Figure 1. RB1 (retinoblastoma gene) function. There are three main ways RB1 can inhibit cancer. (A) The retinoblastoma protein (pRB), the gene product of RB1, inhibits cancer by dephosphorylation as an active state, blocking the gap 1 (G1)/synthesis (S) phases of the cell cycle. Cyclin-dependent kinase (CDK) can phosphorylate pRB, thereby limiting this process and allowing cancer to progress. CDK4/CDK6 inhibitors can relieve the phosphorylation of pRB, so that pRB can re-inhibit the G1/S phases of the cell cycle to achieve cancer treatment. (B) RB1 inhibits the transcription of the cell cycle genes by binding to early 2 factor (E2F) and regulating the E2F transcription factors. (C) RB1 is associated with the organization and maintenance of heterochromatin, contributing to both stable gene expression for cell differentiation and chromosomal structure at centromeres and telomeres.
The RB1 gene encodes pRB, a key regulator of the cell cycle whose function is modulated by phosphorylation. As a multifunctional chromatin-associated protein, pRB negatively regulates cell proliferation by interacting with chromatin regulators to inhibit transcription (25). As a multifunctional chromatin-related protein, pRB can inhibit transcription by interacting with chromatin regulators, stabilizing the complexes that suppress transcription. The hypophosphorylated form of pRB predominantly exists in quiescent cells, while the phosphorylated form appears at the onset of DNA synthesis (26). Dephosphorylation or the inhibition of pRB phosphorylation can trap cells in the gap 1 (G1)/synthesis (S) phases (27), indicating that the hypophosphorylated form is the active state of pRB. During the cell cycle, cyclin-dependent kinase (CDK) phosphorylates pRB, thereby attenuating its activity. The hyperphosphorylation of pRB during the G1/S transition releases its inhibition of E2F, lifting the cell cycle restrictions and initiating processes that can lead to tumorigenesis. In addition, pRB is implicated in chromosome domain organization and gene activation (28). In summary, pRB, the RB1 gene product, primarily functions as a tumor suppressor through its hypophosphorylated active form, which can arrest the cell cycle. However, mutations in RB1 or aberrant pRB phosphorylation can disrupt cell cycle control, promoting cancer development or progression. Consequently, the detection of RB1 and its associated products is critical for prognostic assessment in cancer.
The role of pRB in regulating the cell cycle primarily involves three key aspects (29). Firstly, pRB significantly influences the cell cycle by inhibiting the E2F transcription factors (30). Beyond cell cycle control, pRB also plays a crucial role in DNA damage response by regulating E2F transcription. Loss of pRB leads to E2F activation and the subsequent transcription of target genes, impacting DNA damage by affecting the expression of repair factors such as MSH2, BRCA1, and PCNA (31, 32). In addition, pRB modulates the DNA repair pathways, including non-homologous end joining (NHEJ) and homologous recombination (HR). By interacting with Ku70 and Ku80, pRB facilitates NHEJ-related chromatin modifications (33). In HR, pRB collaborates with E2F to recruit BRG1, a key enzyme in DNA end processing and repair, to double-strand break sites (34). Collectively, these findings highlighted the substantial role of pRB in DNA damage response and repair. Secondly, pRB can induce apoptosis, notably through mitochondrial pathways, by promoting tumor necrosis factor (TNF) activity (35). Furthermore, pRB loss in a mouse bladder cancer model resulted in a decreased p53 expression and the downregulation of apoptotic genes including BAX, BAK, BID, and APAF1 (36), demonstrating its multifaceted influence on apoptosis. Finally, pRB is essential in metabolic regulation. As mentioned above, pRB inhibits E2F, a key regulator of the cell cycle. Importantly, E2F also participates in numerous metabolic pathways, including nucleotide biosynthesis, glucose oxidation, and mitochondrial function (29). E2F directly promotes nucleotide biosynthesis by upregulating thymidine kinase (TK1) and dihydrofolate reductase (DHFR) (29). In a Drosophila model with pRB deficiency, an increased nucleotide metabolism, particularly involving glutamine and glutathione, was observed, suggesting a critical role for pRB in metabolic homeostasis (37). This is further supported by pRB’s upregulation of the glutamine transporter (ASCT2) and glutaminase (GLS1) (38). Moreover, the association of pRB with c-Myc, a transcriptional regulator of metabolic enzymes (39), underscores its significance in metabolic control (Figure 2).
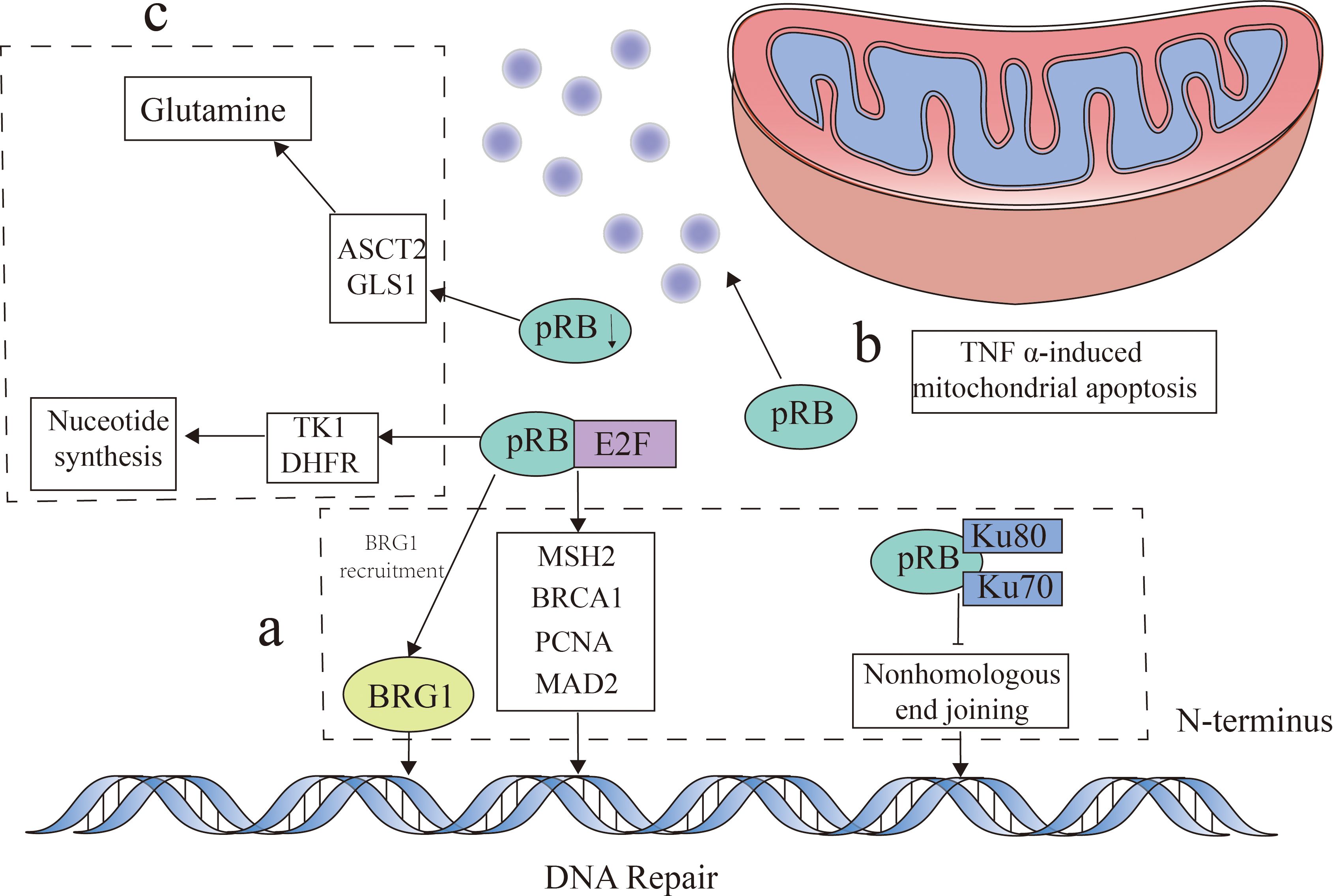
Figure 2. Retinoblastoma protein (pRB) function. pRB exerts its influence through multiple mechanisms. (A) This protein regulates DNA damage response by inhibiting early 2 factor (E2F) transcription factors and by modulating the expression of DNA repair factors such as MSH2, BRCA1, and PCNA, as well as facilitating BRG1 recruitment for DNA end repair. In addition, pRB interacts with Ku70 and Ku80 to influence the DNA repair pathways through non-homologous end joining (NHEJ). (B) Beyond this, pRB plays a role in the regulation of TNFα-induced mitochondrial apoptosis. (C) Moreover, pRB impacts metabolism by inhibiting E2F function, which affects nucleotide biosynthesis through its influence on thymidine kinase (TK1) and dihydrofolate reductase (DHFR). The absence of pRB can lead to the upregulation of the glutamine transporter (ASCT2) and glutaminase (GLS1), consequently affecting glutamine metabolism.
During the cell cycle, the CDK-mediated phosphorylation of pRB limits its inhibitory function on E2F, leading to an unchecked cell proliferation. The RB1 pathway governs the transition from the G1 to the S phase in both mitotic and carcinogenic cell division (30). Stimulated by various factors, including the D-type cyclins, CDK4 and CDK6 become activated and drive this process (27). Their role in facilitating a smooth G1-to-S-phase transition is well established. Moreover, CDK4/CDK6 are pivotal in the uncontrolled cell cycle proliferation characteristic of MM cells (39). These kinases hyperphosphorylate and inactivate pRB, releasing the brakes on cancer cell proliferation and promoting tumorigenesis. The inactivation of RB1 stimulates downstream gene expression, propelling the cell cycle into the S phase. This process is catalyzed by CDK4 and D1-type cyclins. In contrast, CDKN2A, which encodes the p16INK4a protein, acts as a tumor suppressor, opposing CDK4/CDK6 (40). Therapeutic strategies targeting CDK4/CDK6, such as antibodies and inhibitors, offer potential treatment options. By increasing the inhibitory effects of RB1 on tumor proliferation, CDK4/CDK6 inhibitors have demonstrated efficacy in cancers such as breast cancer (2). Moreover, the inhibition of CDK4/CDK6 has been shown to effectively reduce MM tumor burden (41). Therefore, elucidating the RB1 pathway and the role of pRB is crucial for understanding the mechanisms of cancer development and for identifying more precise treatment approaches.
4 Relationship between RB1 and MM
Numerous studies have established RB1 as a frequent cytogenetic abnormality in MM, with deletions affecting approximately 50% of cases and correlating with poor prognosis (6). Karyotype and fluorescence in situ hybridization (FISH) analyses of 45 newly diagnosed MM patients in Singapore identified fibroblast growth factor receptor 3 (FGFR3) and RB1 deletions as the most common genetic alterations (42). An investigation on the prognostic factors, including gene variants and copy number variations (CNVs), in newly diagnosed MM patients revealed that loss of 13q is associated with adverse survival outcomes. The prognostic impact was exacerbated when combined with 1q gain, 6p gain, and 13q reduction (43). Furthermore, analysis of patients with MM at different disease stages, from initial diagnosis to end-stage secondary plasmacytic leukemia (sPCL), demonstrated that RB1 and ZKSCAN3 mutations emerged exclusively in the final sPCL samples, suggesting a role of RB1 in both myeloma initiation and extramedullary infiltration (44). In addition, loss of RB1 has been linked to an increased risk of secondary primary malignancies (SPMs) in patients with MM (7). Chromosome 13 deletions, including microdeletions affecting RB1-critical exons, have been identified in MM cases with t(4;14) positivity (7, 45). Moreover, single nucleotide polymorphism (SNP) array and whole-genome sequencing studies have confirmed RB1 as a recurrent deletion target in MM (44). As a biallelic deletion tumor suppressor gene, the precise role of RB1 in the pathogenesis of MM and its association with poor prognosis remain to be fully elucidated. A comprehensive understanding of the relationship between RB1 and MM is essential for accurate prognostication and for the development of effective therapeutic strategies.
4.1 RB1 deficiency can increase IL-6 secretion
Interleukin 6 (IL-6) is a cytokine with a pivotal role in inflammation and immunity. It stimulates B-lymphocyte proliferation, differentiation, and antibody production. As early as 1991, studies have explored the potential of blockage of IL-6 signaling for MM treatment (46). While these initial investigations yielded inconclusive results, research into IL-6 antagonists for MM therapy has continued. In myeloma, IL-6-secreting cells can originate from either autocrine or paracrine pathways (47, 48).
IL-6 is a critical factor in myeloma cell proliferation and survival, playing a pivotal role in the pathogenesis of MM. Comparative analysis of IL-6 production between 13q-deficient and non-13q-deficient patients revealed significantly elevated IL-6 levels in the former group. These patients also exhibited resistance to conventional chemotherapy, but sensitivity to thalidomide or bortezomib (49). pRB, the RB1 gene product, downregulates the IL-6 gene expression, inhibiting MM cell proliferation. RB1 mutations or pRB alterations lead to IL-6 gene upregulation, contributing to the development and poor prognosis of MM. Numerous IL-6 autocrine and paracrine MM cell lines have been established (50). Studies examining the RB1 gene transcription and pRB expression in 22 human myeloma cell lines (HMCLs) and in 10 advanced-stage patients did not identify changes in the expression of pRB during the transition from the IL-6 paracrine to autocrine states, suggesting a lack of association between pRB deletion and IL-6 expression in MM. However, the sole HMCL exhibiting an IL-6 autocrine phenotype displayed homozygous RB1 gene deletion without transcription or expression, implying that the influence of RB1 on IL-6 secretion and subsequent MM proliferation occurs via an autocrine IL-6 mechanism. In essence, pRB downregulates IL-6 gene expression, and its absence promotes IL-6 autocrine signaling in MM cells, driving malignant proliferation.
Therefore, targeting pRB to suppress IL-6 could potentially represent an effective therapeutic strategy for MM. Currently, tocilizumab, a humanized anti-human interleukin-6 receptor (IL-6R) antibody, is widely used in autoimmune diseases such as rheumatoid arthritis (51). Studies have demonstrated that tocilizumab can bind to COS-7 cells expressing human IL-6R and inhibit the growth of the IL-6-dependent myeloma cell line, KPMM2 (52). These findings suggest that in vivo IL-6 reduction may mitigate the progression of MM, although clinical evidence supporting this hypothesis remains limited. In addition, siltuximab, a monoclonal antibody targeting IL-6, was used to treat idiopathic multicentric Castleman disease (iMCD) (53) and cytokine release syndrome (CRS) associated with T-cell redirecting bispecific antibody therapy for relapsed/refractory multiple myeloma (RRMM) (54). While one study indicated a potential delay in high-risk SMM progression with siltuximab (55), overall, research and clinical data on IL-6 reduction as a novel MM treatment strategy are scarce. This paucity of evidence may be attributed to the association between RB1 deletion and increased IL-6 autocrine signaling in MM. Consequently, simply decreasing the serum IL-6 levels may not be enough to effectively control disease onset and progression. Our findings underscore the potential of targeting RB1 as a promising therapeutic avenue for MM.
4.2 The function of pRB
Retinoblastoma protein, the gene product of RB1, is a critical cell cycle regulator. As previously discussed, pRB controls the cell cycle by inhibiting the E2F transcription factors, and its activity is primarily regulated by cyclin-dependent kinases 4 and 6 (CDK4/CDK6). This underscores the pivotal role of pRB as an intermediate within the RB1 pathway. Immunohistochemical staining and Southern blot analysis of a cohort of MM patients revealed the absence of pRB in 34.7% of advanced cases, suggesting that the inactivation or downregulation of pRB contributes to the progression of MM by disrupting cell cycle control (56). Furthermore, investigations into the cyclin D1/pRb/p16INK4A pathway have linked alterations in cyclin D1 and loss of RB1 to poor prognosis in MM (57). However, the predominantly non-proliferative nature of myeloma cells presents challenges for studying the expression of pRB using immunostaining methods. This research gap hinders our understanding of the role of pRB in MM and necessitates the development of myeloma cell lines for future investigations. Indeed, pRB is a key player in the progression of MM. Therefore, a comprehensive assessment of not only RB1 loss but also pRB mutations will provide valuable insights into the pathogenesis of MM.
5 Opportunity to use RB1 targets to treat MM
Current treatments for MM primarily rely on proteasome inhibitors (PIs) and immunomodulatory drugs (IMiDs) (58). However, these therapies are often accompanied by significant side effects, and patient survival remains limited due to the development of drug resistance in many cases (59). Consequently, identifying novel therapeutic approaches for MM represents a critical unmet medical need.
While IL-6 inhibition holds promise as a therapeutic strategy for MM, the efficacy of the current agents targeting IL-6R has been limited in this context. This may be attributed to the observation that the deletion of RB1 drives tumorigenesis and progression through an IL-6 autocrine loop. Consequently, a mere reduction in the serum IL-6 levels is insufficient for effective MM treatment. Therefore, targeting RB1 presents a more promising therapeutic avenue.
The RB1 gene encodes a tumor suppressor protein that inhibits cell proliferation and division. Patients with MM frequently exhibit RB1 mutations or deletions, leading to tumor cell independence from RB1 and promoting uncontrolled proliferation and dissemination. Exploiting the mechanism and pathway of RB1 offers potential therapeutic avenues for MM. During the cell cycle, CDKs hyperphosphorylate pRB, releasing the E2F-mediated cell cycle progression and contributing to tumorigenesis. The RB1 pathway governs the G1-to-S phase transition in both normal and cancerous cell division (27). D-type cyclins activate CDK4 and CDK6, which subsequently phosphorylate and inactivate the RB1 tumor suppressor, thereby derepressing cancer cell proliferation. Consequently, inhibiting CDK4/CDK6 can enhance the tumor-suppressive function of RB1, as demonstrated in breast cancer treatment (2). CDK4/CDK6 have been implicated in MM cell proliferation, and their inhibition has shown promise in reducing the MM tumor burden (60). However, current clinical trials with approved CDK4/CDK6 inhibitors, such as palbociclib and ribociclib, have shown limited success due to severe hematological toxicities (61–63). Recent studies have identified novel CDK4/CDK6 inhibitors with improved efficacy in inhibiting MM cell proliferation and tumor growth (64). In addition, SET domain bifurcated histone lysine methyltransferase 1 (SETDB1) and the E3 ubiquitin ligase tripartite motif-containing protein 28 (TRIM28) regulate the stability of pRB by inhibiting CDK4/CDK6. TRIM28 promotes the polyubiquitination and degradation of pRB in a phosphorylation-dependent manner, while SETDB1, a chromatin regulator, counteracts the CDK4/CDK6-mediated pRB phosphorylation and subsequent TRIM28-dependent degradation. This suggests a potential therapeutic strategy combining CDK4/CDK6 inhibitors with SETDB1 and TRIM28 modulators to stabilize pRB and suppress tumor growth. Although primarily studied in prostate and breast cancer, this approach warrants investigation in MM (65).
6 Conclusion
The deletion of RB1 is associated not only with the development of MM but also with the occurrence of extramedullary disease. Existing research suggests that the deletion of RB1 disrupts the downregulation of IL-6 by its gene product, pRB, leading to elevated IL-6 levels and the subsequent malignant proliferation of myeloma cells. However, the predominant IL-6 secretion pathway is currently considered autocrine. Moreover, the deletion of RB1 contributes to excessive cell proliferation and death. Furthermore, RB1 deletion likely influences the proliferation of myeloma cells through pRB-mediated cell cycle regulation. While the precise mechanisms underlying the impact of RB1 deletion on the prognosis and development of MM remain to be fully elucidated, its critical role in this disease is indisputable. A comprehensive understanding of the relationship between RB1 and MM will facilitate the classification of high-risk MM genetic subtypes and will enable more targeted therapeutic approaches.
7 Outlook
Significant advancements in MM treatment have extended overall survival, with some centers reporting a median survival of over 10 years in newly diagnosed patients (66). However, disparities in patient outcomes persist, which are often attributed to the development of drug resistance. Consequently, there is an urgent need for novel therapeutic strategies. To address this unmet clinical need, comprehensive investigations into the relationship between RB1 loss and MM prognosis are warranted. While extensive research has elucidated the mechanisms underlying the RB family, particularly the role of its gene product, pRB, clinical and mechanistic studies examining the impact of RB1 on the prognosis of MM remain limited. This gap may be attributed to the complex genetic landscape of MM, which is characterized by multiple gene mutations such as TP53 loss and 1q amplification. To clarify the association between RB1 loss and MM prognosis, large-scale studies employing karyotype and FISH analyses are required to identify patient cohorts with RB1 abnormalities. Subsequently, detailed analyses of these patients can elucidate the prognostic implications of RB1 loss. Furthermore, functional studies investigating the role of pRB in MM cell lines are essential to understanding the underlying mechanisms. While the importance of CDK4/CDK6 inhibitors in MM has been established, clinical translation remains challenging. Leveraging CDK4/CDK6 inhibitors to further explore the role of RB1 in MM could provide valuable insights. In addition, considering the potential influence of RB1 on IL-6 expression and signaling, comprehensive analyses of IL-6 alterations in RB1-deficient MM patients are warranted. In conclusion, a more in-depth understanding of the role of RB1 in the pathogenesis and progression of MM is crucial for the development of targeted therapies. By integrating clinical, genetic, and mechanistic studies, we can advance our knowledge of the impact of RB1 on the prognosis of MM and identify novel therapeutic opportunities.
Author contributions
YW: Conceptualization, Writing – original draft, Writing – review & editing. RY: Writing – review & editing. RL: Writing – review & editing. RYY: Writing – review & editing. ZL: Writing – review & editing. AH: Resources, Supervision, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Shin SY, Eom HS, Sohn JY, Lee H, Park B, Joo J, et al. Prognostic implications of monosomies in patients with multiple myeloma. Clin Lymphoma Myeloma Leuk. (2017) 17:159–64.e2. doi: 10.1016/j.clml.2016.12.001
2. Palafox M, Monserrat L, Bellet M, Villacampa G, Gonzalez-Perez A, Oliveira M, et al. High p16 expression and heterozygous RB1 loss are biomarkers for CDK4/6 inhibitor resistance in ER(+) breast cancer. Nat Commun. (2022) 13:5258. doi: 10.1038/s41467-022-32828-6
3. Wang ME, Chen J, Lu Y, Bawcom AR, Wu J, Ou J, et al. RB1-deficient prostate tumor growth and metastasis are vulnerable to ferroptosis induction via the E2F/ACSL4 axis. J Clin Invest. (2023) 133(10):e166647. doi: 10.1172/JCI166647
4. Kugalingam N, de Silva D, Abeysekera H, Nanayakkara S, Tirimanne S, Ranaweera D, et al. RB1 screening of retinoblastoma patients in Sri Lanka using targeted next generation sequencing (NGS) and gene ratio analysis copy enumeration PCR (GRACE-PCR). BMC Med Genomics. (2023) 16:279. doi: 10.1186/s12920-023-01721-6
5. Miao C, Tsujino T, Takai T, Gui F, Tsutsumi T, Sztupinszki Z, et al. RB1 loss overrides PARP inhibitor sensitivity driven by RNASEH2B loss in prostate cancer. Sci Adv. (2022) 8:eabl9794. doi: 10.1126/sciadv.abl9794
6. Uryu H, Mishima Y, Ishihara Y, Shirouchi Y, Yamauchi N, Hirano M, et al. Complex karyotype determined using conventional cytogenetic analysis is a poor prognostic factor in patients with multiple myeloma. J Clin Exp Hematop. (2024) 64:10–20. doi: 10.3960/jslrt.23047
7. Wang Y, Wu Y, Xu Z, Zhou H, Chen Y, Zhan R. Cytogenetic abnormalities in patients with newly diagnosed multiple myeloma as a secondary primary Malignancy: a retrospective study. Hematology. (2020) 25:176–80. doi: 10.1080/16078454.2020.1757330
8. Terragna C, Poletti A, Solli V, Martello M, Zamagni E, Pantani L, et al. Multi-dimensional scaling techniques unveiled gain1q&loss13q co-occurrence in Multiple Myeloma patients with specific genomic, transcriptional and adverse clinical features. Nat Commun. (2024) 15:1551. doi: 10.1038/s41467-024-45000-z
9. Walker BA, Mavrommatis K, Wardell CP, Ashby TC, Bauer M, Davies FE, et al. Identification of novel mutational drivers reveals oncogene dependencies in multiple myeloma. Blood. (2018) 132:587–97. doi: 10.1182/blood-2018-03-840132
10. Clarke SE, Fuller KA, Erber WN. Chromosomal defects in multiple myeloma. Blood Rev. (2024) 64:101168. doi: 10.1016/j.blre.2024.101168
11. Ramakrishnan V, Kumar S. PI3K/AKT/mTOR pathway in multiple myeloma: from basic biology to clinical promise. Leuk Lymphoma. (2018) 59:2524–34. doi: 10.1080/10428194.2017.1421760
12. Gomez-Bougie P, Maiga S, Tessoulin B, Bourcier J, Bonnet A, Rodriguez MS, et al. BH3-mimetic toolkit guides the respective use of BCL2 and MCL1 BH3-mimetics in myeloma treatment. Blood. (2018) 132:2656–69. doi: 10.1182/blood-2018-03-836718
13. Bolli N, Maura F, Minvielle S, Gloznik D, Szalat R, Fullam A, et al. Genomic patterns of progression in smoldering multiple myeloma. Nat Commun. (2018) 9:3363. doi: 10.1038/s41467-018-05058-y
14. D'Agostino M, Cairns DA, Lahuerta JJ, Wester R, Bertsch U, Waage A, et al. Second revision of the international staging system (R2-ISS) for overall survival in multiple myeloma: A European myeloma network (EMN) report within the HARMONY project. J Clin Oncol. (2022) 40:3406–18. doi: 10.1200/JCO.21.02614
15. Palumbo A, Avet-Loiseau H, Oliva S, Lokhorst HM, Goldschmidt H, Rosinol L, et al. Revised international staging system for multiple myeloma: A report from international myeloma working group. J Clin Oncol. (2015) 33:2863–9. doi: 10.1200/JCO.2015.61.2267
16. Lohr JG, Stojanov P, Carter SL, Cruz-Gordillo P, Lawrence MS, Auclair D, et al. Widespread genetic heterogeneity in multiple myeloma: implications for targeted therapy. Cancer Cell. (2014) 25:91–101. doi: 10.1016/j.ccr.2013.12.015
17. Kumar SK, Dispenzieri A, Lacy MQ, Gertz MA, Buadi FK, Pandey S, et al. Continued improvement in survival in multiple myeloma: changes in early mortality and outcomes in older patients. Leukemia. (2014) 28:1122–8. doi: 10.1038/leu.2013.313
18. Hanamura I. Multiple myeloma with high-risk cytogenetics and its treatment approach. Int J Hematol. (2022) 115:762–77. doi: 10.1007/s12185-022-03353-5
19. Korde N, Roschewski M, Zingone A, Kwok M, Manasanch EE, Bhutani M, et al. Treatment with carfilzomib-lenalidomide-dexamethasone with lenalidomide extension in patients with smoldering or newly diagnosed multiple myeloma. JAMA Oncol. (2015) 1:746–54. doi: 10.1001/jamaoncol.2015.2010
20. Berry JL, Polski A, Cavenee WK, Dryja TP, Murphree AL, Gallie BL. The RB1 story: characterization and cloning of the first tumor suppressor gene. Genes (Basel). (2019) 10(11):879. doi: 10.3390/genes10110879
21. Viana MC, Tavares WC, Brant AC, Boroni M, Seuánez HN. The human retinoblastoma susceptibility gene (RB1): an evolutionary story in primates. Mamm Genome. (2017) 28:198–212. doi: 10.1007/s00335-017-9689-4
22. Kareta MS, Gorges LL, Hafeez S, Benayoun BA, Marro S, Zmoos AF, et al. Inhibition of pluripotency networks by the Rb tumor suppressor restricts reprogramming and tumorigenesis. Cell Stem Cell. (2015) 16:39–50. doi: 10.1016/j.stem.2014.10.019
23. Ishak CA, Marshall AE, Passos DT, White CR, Kim SJ, Cecchini MJ, et al. An RB-EZH2 complex mediates silencing of repetitive DNA sequences. Mol Cell. (2016) 64:1074–87. doi: 10.1016/j.molcel.2016.10.021
24. Salvadores M, Supek F. Cell cycle gene alterations associate with a redistribution of mutation risk across chromosomal domains in human cancers. Nat Cancer. (2024) 5:330–46. doi: 10.1038/s43018-023-00707-8
25. Dyson NJ. RB1: a prototype tumor suppressor and an enigma. Genes Dev. (2016) 30:1492–502. doi: 10.1101/gad.282145.116
26. Marković L, Bukovac A, Varošanec AM, Šlaus N, Pećina-Šlaus N. Genetics in ophthalmology: molecular blueprints of retinoblastoma. Hum Genomics. (2023) 17:82. doi: 10.1186/s40246-023-00529-w
27. Duronio RJ, Xiong Y. Signaling pathways that control cell proliferation. Cold Spring Harb Perspect Biol. (2013) 5:a008904. doi: 10.1101/cshperspect.a008904
28. Sanidas I, Lawrence MS, Dyson NJ. Patterns in the tapestry of chromatin-bound RB. Trends Cell Biol. (2024) 34:288–98. doi: 10.1016/j.tcb.2023.07.012
29. Mandigo AC, Tomlins SA, Kelly WK, Knudsen KE. Relevance of pRB loss in human Malignancies. Clin Cancer Res. (2022) 28:255–64. doi: 10.1158/1078-0432.CCR-21-1565
30. Huang MF, Wang YX, Chou YT, Lee DF. Therapeutic strategies for RB1-deficient cancers: intersecting gene regulation and targeted therapy. Cancers (Basel). (2024) 16(8):1558. doi: 10.3390/cancers16081558
31. Geissler F, Nesic K, Kondrashova O, Dobrovic A, Swisher EM, Scott CL, et al. The role of aberrant DNA methylation in cancer initiation and clinical impacts. Ther Adv Med Oncol. (2024) 16:17588359231220511. doi: 10.1177/17588359231220511
32. Chang CN, Feng MJ, Chen YL, Yuan RH, Jeng YM. p15(PAF) is an Rb/E2F-regulated S-phase protein essential for DNA synthesis and cell cycle progression. PloS One. (2013) 8:e61196. doi: 10.1371/journal.pone.0061196
33. Cook R, Zoumpoulidou G, Luczynski MT, Rieger S, Moquet J, Spanswick VJ, et al. Direct involvement of retinoblastoma family proteins in DNA repair by non-homologous end-joining. Cell Rep. (2015) 10:2006–18. doi: 10.1016/j.celrep.2015.02.059
34. Vélez-Cruz R, Manickavinayaham S, Biswas AK, Clary RW, Premkumar T, Cole F, et al. RB localizes to DNA double-strand breaks and promotes DNA end resection and homologous recombination through the recruitment of BRG1. Genes Dev. (2016) 30:2500–12. doi: 10.1101/gad.288282.116
35. Hilgendorf KI, Leshchiner ES, Nedelcu S, Maynard MA, Calo E, Ianari A, et al. The retinoblastoma protein induces apoptosis directly at the mitochondria. Genes Dev. (2013) 27:1003–15. doi: 10.1101/gad.211326.112
36. Wang CY, Xu ZB, Wang JP, Jiao Y, Zhang B. Rb deficiency accelerates progression of carcinoma of the urinary bladder in vivo and in vitro through inhibiting autophagy and apoptosis. Int J Oncol. (2017) 50:1221–32. doi: 10.3892/ijo.2017.3889
37. Nicolay BN, Gameiro PA, Tschöp K, Korenjak M, Heilmann AM, Asara JM, et al. Loss of RBF1 changes glutamine catabolism. Genes Dev. (2013) 27:182–96. doi: 10.1101/gad.206227.112
38. Reynolds MR, Lane AN, Robertson B, Kemp S, Liu Y, Hill BG, et al. Control of glutamine metabolism by the tumor suppressor Rb. Oncogene. (2014) 33:556–66. doi: 10.1038/onc.2012.635
39. Kim S, Armand J, Safonov A, Zhang M, Soni RK, Schwartz G, et al. Sequential activation of E2F via Rb degradation and c-Myc drives resistance to CDK4/6 inhibitors in breast cancer. Cell Rep. (2023) 42:113198. doi: 10.1016/j.celrep.2023.113198
40. Panagiotou E, Gomatou G, Trontzas IP, Syrigos N, Kotteas E. Cyclin-dependent kinase (CDK) inhibitors in solid tumors: a review of clinical trials. Clin Transl Oncol. (2022) 24:161–92. doi: 10.1007/s12094-021-02688-5
41. Baughn LB, di Liberto M, Wu K, Toogood PL, Louie T, Gottschalk R, et al. A novel orally active small molecule potently induces G1 arrest in primary myeloma cells and prevents tumor growth by specific inhibition of cyclin-dependent kinase 4/6. Cancer Res. (2006) 66:7661–7. doi: 10.1158/0008-5472.CAN-06-1098
42. Lim AS, Lim TH, See KH, Ng YJ, Tan YM, Choo NS, et al. Cytogenetic and molecular aberrations of multiple myeloma patients: a single-center study in Singapore. Chin Med J (Engl). (2013) 126:1872–7. doi: 10.3760/cma.j.issn.0366-6999.20123344
43. Lee N, Kim SM, Lee Y, Jeong D, Yun J, Ryu S, et al. Prognostic value of integrated cytogenetic, somatic variation, and copy number variation analyses in Korean patients with newly diagnosed multiple myeloma. PloS One. (2021) 16:e0246322. doi: 10.1371/journal.pone.0246322
44. Egan JB, Shi CX, Tembe W, Christoforides A, Kurdoglu A, Sinari S, et al. Whole-genome sequencing of multiple myeloma from diagnosis to plasma cell leukemia reveals genomic initiating events, evolution, and clonal tides. Blood. (2012) 120:1060–6. doi: 10.1182/blood-2012-01-405977
45. He Z, O'Neal J, Wilson WC, Mahajan N, Luo J, Wang Y, et al. Deletion of Rb1 induces both hyperproliferation and cell death in murine germinal center B cells. Exp Hematol. (2016) 44:161–5.e4. doi: 10.1016/j.exphem.2015.11.006
46. Klein B, Wijdenes J, Zhang XG, Jourdan M, Boiron JM, Brochier J, et al. Murine anti-interleukin-6 monoclonal antibody therapy for a patient with plasma cell leukemia. Blood. (1991) 78:1198–204. doi: 10.1182/blood.V78.5.1198.1198
47. Gonzalez-Meljem JM, Martinez-Barbera JP. Adamantinomatous craniopharyngioma as a model to understand paracrine and senescence-induced tumourigenesis. Cell Mol Life Sci. (2021) 78:4521–44. doi: 10.1007/s00018-021-03798-7
48. Herbstein F, Sapochnik M, Attorresi A, Pollak C, Senin S, Gonilski-Pacin D, et al. The SASP factor IL-6 sustains cell-autonomous senescent cells via a cGAS-STING-NFκB intracrine senescent noncanonical pathway. Aging Cell. (2024) 23(10):e14258. doi: 10.1111/acel.14258
49. Neemat K, Rania K, Tarek M, Hamdy AA. Effect of 13q deletion on IL-6 production in patients with multiple myeloma: a hypothesis may hold true. Clin Lab. (2014) 60:1393–9. doi: 10.7754/Clin.Lab.2013.130743
50. Zhang XG, Gaillard JP, Robillard N, Lu ZY, Gu ZJ, Jourdan M, et al. Reproducible obtaining of human myeloma cell lines as a model for tumor stem cell study in human multiple myeloma. Blood. (1994) 83:3654–63. doi: 10.1182/blood.V83.12.3654.3654
51. Zhang SX, Chen HR, Wang J, Shao HF, Cheng T, Pei RM, et al. The efficacy and safety of short-term and low-dose IL-2 combined with tocilizumab to treat rheumatoid arthritis. Front Immunol. (2024) 15:1359041. doi: 10.3389/fimmu.2024.1359041
52. Mihara M, Kasutani K, Okazaki M, Nakamura A, Kawai S, Sugimoto M, et al. Tocilizumab inhibits signal transduction mediated by both mIL-6R and sIL-6R, but not by the receptors of other members of IL-6 cytokine family. Int Immunopharmacol. (2005) 5:1731–40. doi: 10.1016/j.intimp.2005.05.010
53. Lang E, van Rhee F. Idiopathic multicentric Castleman disease: An update in diagnosis and treatment advances. Blood Rev. (2024) 64:101161. doi: 10.1016/j.blre.2023.101161
54. Lipe BC, Renaud T. Siltuximab as a primary treatment for cytokine release syndrome in a patient receiving a bispecific antibody in a clinical trial setting. J Oncol Pharm Pract. (2023) 29:1006–10. doi: 10.1177/10781552221140320
55. Brighton TA, Khot A, Harrison SJ, Ghez D, Weiss BM, Kirsch A, et al. Randomized, double-blind, placebo-controlled, multicenter study of siltuximab in high-risk smoldering multiple myeloma. Clin Cancer Res. (2019) 25:3772–5. doi: 10.1158/1078-0432.CCR-18-3470
56. Corradini P, Inghirami G, Astolfi M, Ladetto M, Voena C, Ballerini P, et al. Inactivation of tumor suppressor genes, p53 and Rb1, in plasma cell dyscrasias. Leukemia. (1994) 8:758–67.
57. Krämer A, Schultheis B, Bergmann J, Willer A, Hegenbart U, Ho AD, et al. Alterations of the cyclin D1/pRb/p16(INK4A) pathway in multiple myeloma. Leukemia. (2002) 16:1844–51. doi: 10.1038/sj.leu.2402609
58. Yang Y, Li Y, Gu H, Dong M, Cai Z. Emerging agents and regimens for multiple myeloma. J Hematol Oncol. (2020) 13:150. doi: 10.1186/s13045-020-00980-5
59. Gooding S, Ansari-Pour N, Towfic F, Ortiz Estévez M, Chamberlain PP, Tsai KT, et al. Multiple cereblon genetic changes are associated with acquired resistance to lenalidomide or pomalidomide in multiple myeloma. Blood. (2021) 137:232–7. doi: 10.1182/blood.2020007081
60. Huang X, di Liberto M, Jayabalan D, Liang J, Ely S, Bretz J, et al. Prolonged early G(1) arrest by selective CDK4/CDK6 inhibition sensitizes myeloma cells to cytotoxic killing through cell cycle-coupled loss of IRF4. Blood. (2012) 120:1095–106. doi: 10.1182/blood-2012-03-415984
61. Dhillon S. Palbociclib: first global approval. Drugs. (2015) 75:543–51. doi: 10.1007/s40265-015-0379-9
62. Syed YY. Ribociclib: first global approval. Drugs. (2017) 77:799–807. doi: 10.1007/s40265-017-0742-0
63. Yuan K, Wang X, Dong H, Min W, Hao H, Yang P. Selective inhibition of CDK4/6: A safe and effective strategy for developing anticancer drugs. Acta Pharm Sin B. (2021) 11:30–54. doi: 10.1016/j.apsb.2020.05.001
64. Yuan K, Kuang W, Chen W, Ji M, Min W, Zhu Y, et al. Discovery of novel and orally bioavailable CDK 4/6 inhibitors with high kinome selectivity, low toxicity and long-acting stability for the treatment of multiple myeloma. Eur J Med Chem. (2022) 228:114024. doi: 10.1016/j.ejmech.2021.114024
65. Huang Z, Li X, Tang B, Li H, Zhang J, Sun R, et al. SETDB1 modulates degradation of phosphorylated RB and anticancer efficacy of CDK4/6 inhibitors. Cancer Res. (2023) 83:875–89. doi: 10.1158/0008-5472.CAN-22-0264
Keywords: multiple myeloma, gene mutation, RB1, pRB, CDK4/6 inhibitors
Citation: Wang Y, Yang R, Liu R, Yang R, Lin Z and He A (2024) The significance of RB1 in multiple myeloma. Front. Immunol. 15:1415972. doi: 10.3389/fimmu.2024.1415972
Received: 11 April 2024; Accepted: 11 November 2024;
Published: 27 November 2024.
Edited by:
Hauke Thomsen, Medical School Berlin, GermanyReviewed by:
Katie McKenna, Baylor College of Medicine, United StatesVishal Sindhava, University of Pennsylvania, United States
Copyright © 2024 Wang, Yang, Liu, Yang, Lin and He. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Aili He, aGVhaWxpQHhqdHUuZWR1LmNu