 Jianjun Wang
Jianjun Wang Hui Cao2†
Hui Cao2† Nan Wang
Nan Wang Yiwei Weng
Yiwei Weng Hao Luo
Hao Luo- 1Department of Clinical Laboratory, Kunshan Hospital Affiliated to Jiangsu University, Suzhou, China
- 2Department of Food and Nutrition Safety, Jiangsu Provincial Center for Disease Control and Prevention, Nanjing, Jiangsu, China
- 3Department of Clinical Laboratory, Suzhou BOE Hospital, Suzhou, Jiangsu, China
- 4Department of Clinical Laboratory, The Fourth People’s Hospital of Kunshan, Suzhou, Jiangsu, China
- 5Department of Clinical Laboratory, The Second People's Hospital of Kunshan, Suzhou, China
CD36 is a scavenger receptor that has been reported to function as a signaling receptor that responds to pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) and could integrate metabolic pathways and cell signaling through its dual functions. Thereby influencing activation to regulate the immune response and immune cell differentiation. Recent studies have revealed that CD36 plays critical roles in the process of lipid metabolism, inflammatory response and immune process caused by Mycobacterium tuberculosis infection. This review will comprehensively investigate CD36’s functions in lipid uptake and processing, inflammatory response, immune response and therapeutic targets and biomarkers in the infection process of M. tuberculosis. The study also raised outstanding issues in this field to designate future directions.
1 Introduction
Mycobacterium tuberculosis (M. tuberculosis) as an intracellular pathogen is a threat to global public health and infects approximately a third of the world’s population and it causes approximately 2 million deaths every year around the world (1, 2). The pathogenic mycobacteria has the potential to adapt to the hostile intracellular environment of macrophages and make it latent to infect cells in the surrounding environment (3–5). M. tuberculosis involves host trafficking pathways by regulating maturation pathways such as phagosomal/endosomal to generate a protected niche for itself, the mycobacterial phagosome (6–8). Furthermore, M. Tuberculosis regulates the specific metabolic pathways to subvert the host signaling response, thus regulating the host immune response and enabling access to nutrients, which ultimately favor infection establishment (9, 10). The macrophages are critical immune cells in M. tuberculosis infection and the foam-like macrophages have been found in the granulomatous structures in both M. tuberculosis-infected human disease and animal models (11–13). The foam of macrophages in an M. tuberculosis-infected environment reflected the disruption of intracellular lipid regulatory mechanisms through the observation of infected pathological conditions. Numerous studies have shown that during M. tuberculosis infection, newly generated lipid bodies are often architecturally unique cytoplasmic organelles involved in the manufacture of lipid mediators with immunomodulatory effects (14, 15). In addition, the induction and targeting of liposomes by M. tuberculosis may provide an escape mechanism during infection due to the downregulation of the immune response and/or nutrient uptake in macrophages, promoting the survival and replication in host cells (16, 17). Interestingly, the scavenger receptor CD36 is closely related to the formation of foam-like macrophages and their lipid metabolism during the infection of M. tuberculosis.
Cell surface glycoprotein CD36 is a scavenger receptor present in various cells and functions in various roles in lipid metabolism, mediating lipid acquisition, immune recognition, inflammation, molecular adhesion and apoptosis (18–20). It has associations with angiogenesis, atherothrombotic disease, metabolic disorders, diabetes, obesity and other conditions. By sensing a range of microbial components and endogenous ligands, it may operate as a pattern recognition receptor mediating innate immune or inflammatory responses to a variety of pathogens (21, 22). Researches have shown that CD36 may also function as a co-receptor with the Toll-like receptor (TLR) 2/6 complex, which is involved in innate sensing, lipoteichoic acid binding, and Staphylococcus aureus phagocytic clearance (23, 24). Additionally, a growing body of research has revealed that patients with active tuberculosis (ATB) exhibit reversible changes in CD36 on their peripheral macrophages/monocytes (25). All in all, the significant role of CD36 is to recognize the ligands from the pathogen or host itself to induce the appropriate immune responses to protect the host itself and eliminate the pathogens.
However, the tangible molecular mechanisms and signaling pathways which regulate lipid body biogenesis during M. tuberculosis infection and the contribution it has to tuberculosis’ pathophysiology are not clear. This study will delve into and review the functions and specific signaling mechanisms of CD36 in M. tuberculosis, providing comprehensive materials for the study of CD36 in M. tuberculosis infection
2 The biological functions of CD36 molecular
As a 55 kDa heavily glycosylated protein, the membrane glycoprotein CD36 has two distinct structural parts including extracellular ectodomain and cytosolic domain respectively (26). For a specific collection of adaptor proteins, the extracellular ectodomain serves as a docking location where the signal is relayed for functional outcomes during the signaling pathway initiated at the cell surface via CD36-ligand contact (27). Transmembrane sections link the protein’s ectodomain and cytosolic (C- or N-terminal) portions (Figure 1). Following ligand-receptor contact, CD36 is internalized with or without ligand and the reaction is amplified by the internalized CD36-ligand complex (28).
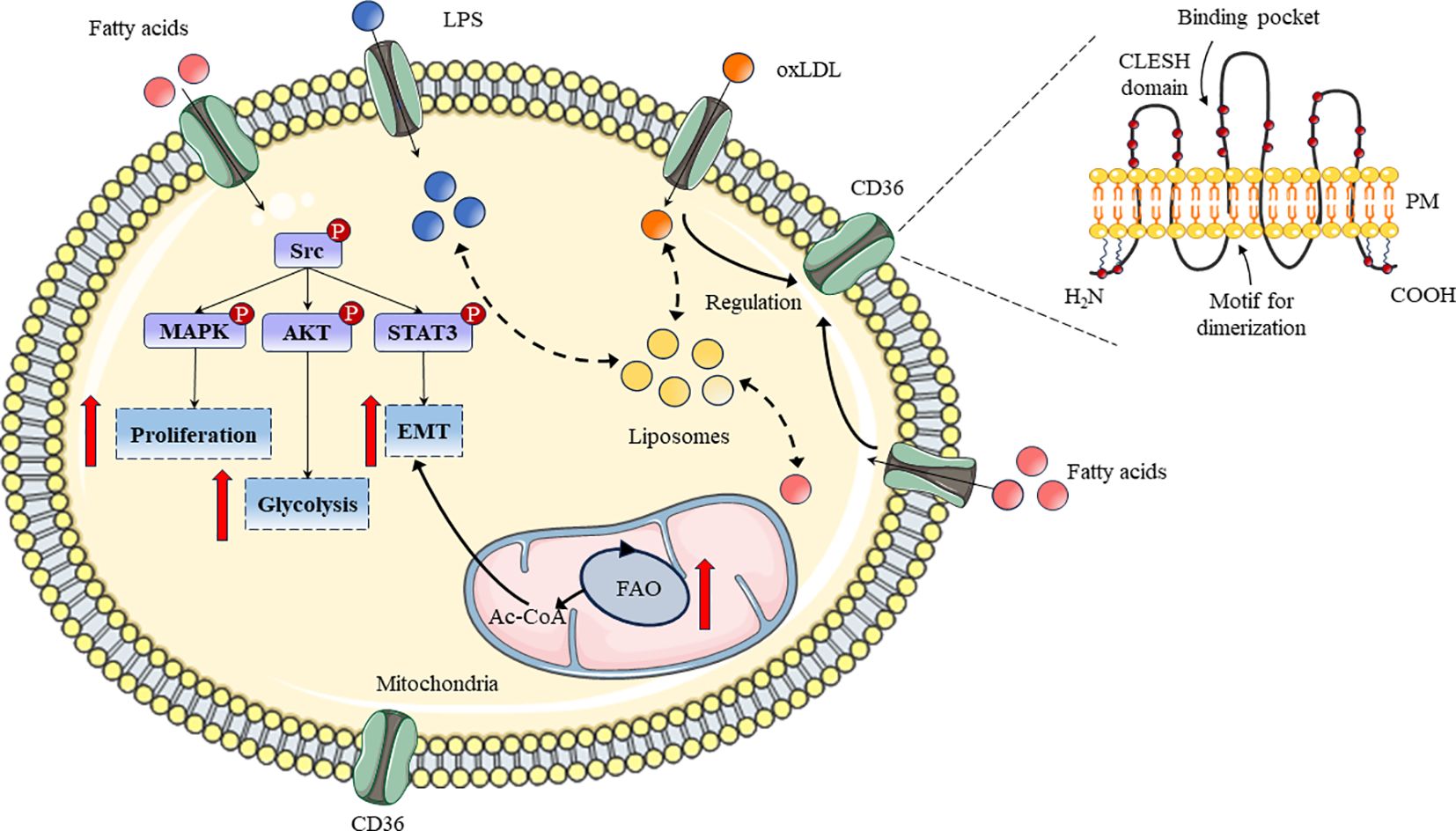
Figure 1 Schematic diagram of CD36 structure and its roles in lipid metabolism process. The structure and related functional regions of CD36 in bilayer lipids were displayed. Meanwhile, the pathways, related signaling pathways, and functions of CD36 in lipid metabolism have been demonstrated.
Scavenger receptors have the special ability to collect cellular waste or dead/senescent cells which are then disposed of in the target spleen (29). Moreover, CD36’s potential is dependent on its ability to bind with ligands found in cells or cellular debris, as well as how well this interaction is eliminated (30). The physiochemical aspects of topology, charge and other characteristics of the CD36 ectodomain enable it to recognize a wide variety of ligands, including phosphatidylserine, oxidized low-density lipoprotein (oxiLDL), lipopolysaccharide (LPS), long-chain fatty acids (LCFAs) and so on (31–34). CD36 surface chemistry and ectodomain topology could also facilitate the ligands binding that have diversified unknown origins.
CD36 could be expressed on the surface of various cell types including monocytes, macrophages, cancer cells, endothelial cells and platelets (35). Circulating monocytes in the blood are transmigrated into the arterial intima and differentiated into macrophages, but CD36 expression in macrophages is regulated by various factors (36). In addition, multitudinous chemical compounds such as curcumin, astaxanthin, quercitrin and kaempferol could regulate CD36 expression through erythroid-related factor 2 (Nrf2) and peroxisome proliferator-activated receptor-gamma (PPAR-γ) signaling pathways in macrophages (37–39). In cancer cells, CD36 could also regulate cell proliferation, glycolysis, and Epithelial-Mesenchymal Transition (EMT) through the MAPK, AKT, and STAT3 signaling pathways to determine the physiological and pathological processes of cells (Figure 1) (40–42). In addition, to reduce the parasite burden, a combination of CD36 ligands generates pro-inflammatory cytokines; however, this improper cytokine production leads to pathogenic alterations (43). Undoubtedly, CD36 and its downstream signaling may offer useful resources for developing appropriate adjuvant treatments that aid in the recovery from pathology linked to the disease (44). Furthermore, specific downstream signaling can be triggered by a suitable chemical substance acting as a ligand which will generate an appropriate immune response to eradicate the infection (45).
Macrophages have high expression of CD36 which is intimately associated with their multitude of functions. Because monocyte/macrophage CD36 may bind to and increase oxLDL endocytosis as well as take part in the generation of foam cells, it plays a crucial role in the development of atherosclerotic lesions (46). Macrophages could bind and ingest ox-LDL with the assistance of CD36, and the internalized ox-LDL activates PPAR-γ to upregulate CD36 expression, which facilitates the uptake of ox-LDL further (35, 47, 48). Foam cells and the accumulation of esterified cholesterol in macrophages are caused by elevated ox-LDL (49). Additionally, the uptake of ox-LDL by CD36 sets off cytokine production and immune cell recruitment to the artery intima, which leads to arterial narrowing and the advancement of atherosclerotic vascular disease (50). A significant amount of foam cells are formed in vitro and in vivo due to CD36 and blocking CD36 expression or downstream signaling prevents animals from absorbing ox-LDL which limits the progression of experimental atherosclerosis in mice. A link between carotid atherosclerosis and the plasma levels of soluble CD36 (sCD36) was discovered by Handberg et al. (51).
The function of CD36 is to identify ligands from the pathogen or the host, at which point it triggers the right innate immune response that kills the infection and produces inflammatory cytokines (52). In the immune process initiated by pathogenic infections, the recognition of TLR2 agonists is affected by some accessory receptors, such as CD36, CD11b/CD18 and CD14 (53, 54). Moreover, the identification of co-receptors that promote TLR functions reveals that TLRs combine with other cell surface molecules to link PAMP recognition to the initiation of inflammatory responses (55). Hoebe et al. demonstrated that TLR2/TLR6 heterodimers require CD36 and the heterotypic binding of TLR2/6 to CD36 is not pre-formed, but ligand-induced, revealing the heterotypic association of TLR2 (56, 57). Moreover, deposition of the altered self-components oxLDL and amyloid-beta induce inflammatory response through CD36-triggered the heterodimer activation of TLR4-TLR6 in Alzheimer’s disease and atherosclerosis, indicating a novel model of TLR heterodimerization dominated by co-receptor signaling factors (58). These studies indicated that the function of CD36 cannot be achieved without the coordination of the TLR receptor family.
Recent research on CD36’s function in infectious disorders has shown that it plays a pathological role in infections with viruses, tuberculosis, pneumonia and Staphylococcus aureus. In influenza virus-mediated pneumonia, the expression of CD36 was downregulated and since the host is more susceptible to infection when CD36 is absent, CD36-mediated phagocytosis is required to eliminate germs (59). Moreover, the residues of the cytoplasmic domain (Y463 and C464) were critical and involved in TLR2/6 signaling (60). Meanwhile, CD36-deficient macrophages failed to eliminate the bacteria and decreased the secretion of TNF main components revealed that the cytoplasmic domain of the CD36 receptor is required for the phagocytosis (60). Additional findings revealed that CD36 expression may be associated with HIV infections, influenza and hepatitis virus (61, 62). HBV replication is facilitated by the store-operated Ca2+ channel, which is mediated by the Src-kinase-mediated signaling pathway and is mediated by CD36 (63). The Nef protein of HIV downregulated CD36 which impacted the infected cells’ ability to be phagocytosed and added to the host’s pathology by pointing to the TNF-α and co-interactions of the infected cells (64).
Currently, an increasing number of studies have shown abnormal expression of CD36 in Mycobacterium infection, but the function of CD36 in Mycobacterium infection is still insufficient and ambiguous. Fink et al. discovered via gene sequencing that CD36 is down-regulated in zebrafish infected with M. marinum; conversely, the knockdown of CD36 defective zebrafish larvae resulted in a higher bacterial burden during such infection (65). Bazzi et al. reported downregulating CD36 expression, reducing CCL2 spontaneous release and increasing CCL5, CXCL8/IL-8, IL-6 and TNF-α spontaneous secretion in M. obuense infected macrophages (66). Park et al. found that macrophages expressed CD14, CD36 and TLR2, and function the most active responses against M. avium subsp. paratuberculosis (MAP) infection, which includes the expression of proinflammatory cytokines and chemokines such as CCL4, CCL3, IL-1b, IL-8, and CCL20 (67). CD36 plays an indispensable role in macrophage resistance against Mycobacterium infection, and the abnormality of CD36 is closely related to the inflammatory response of cells and the phagocytosis of tuberculosis.
3 The function of CD36 in M. tuberculosis infection
After infecting host cells, M. tuberculosis generated intracellular phagosome and fused with lysosomes, then the components of M. tuberculosis were presented to immune cells through vesicles. Immune cells regulate host cells to produce immunosuppressive responses by uptake of liposomes of M. tuberculosis in vesicles through CD36 (Figure 2). CD36 has been shown to promote lipid uptake in cells which may serve as a source of carbon to support the cell’s development (68). CD36 acts as a transporter of long chain fatty acid and function its effect on glycolysis and the trichloroacetic acid (TCA) cycle in M. tuberculosis infection (69). During this infection, this pathogen promotes the expression of CD36 and make host cells take up more long-chain fatty acids to produced sufficient ATP through glycolysis or the tricarboxylic acid cycle, allowing it to lurk in host cells for a long time and infect surrounding normal cells (70). M1 macrophages require rapid generation of ATP to activate inflammation through glycolysis via the inhibition of OXPHOS and TCA cycle in mitochondria (71, 72). These findings provided a novel target to treat the infection of M. tuberculosis.
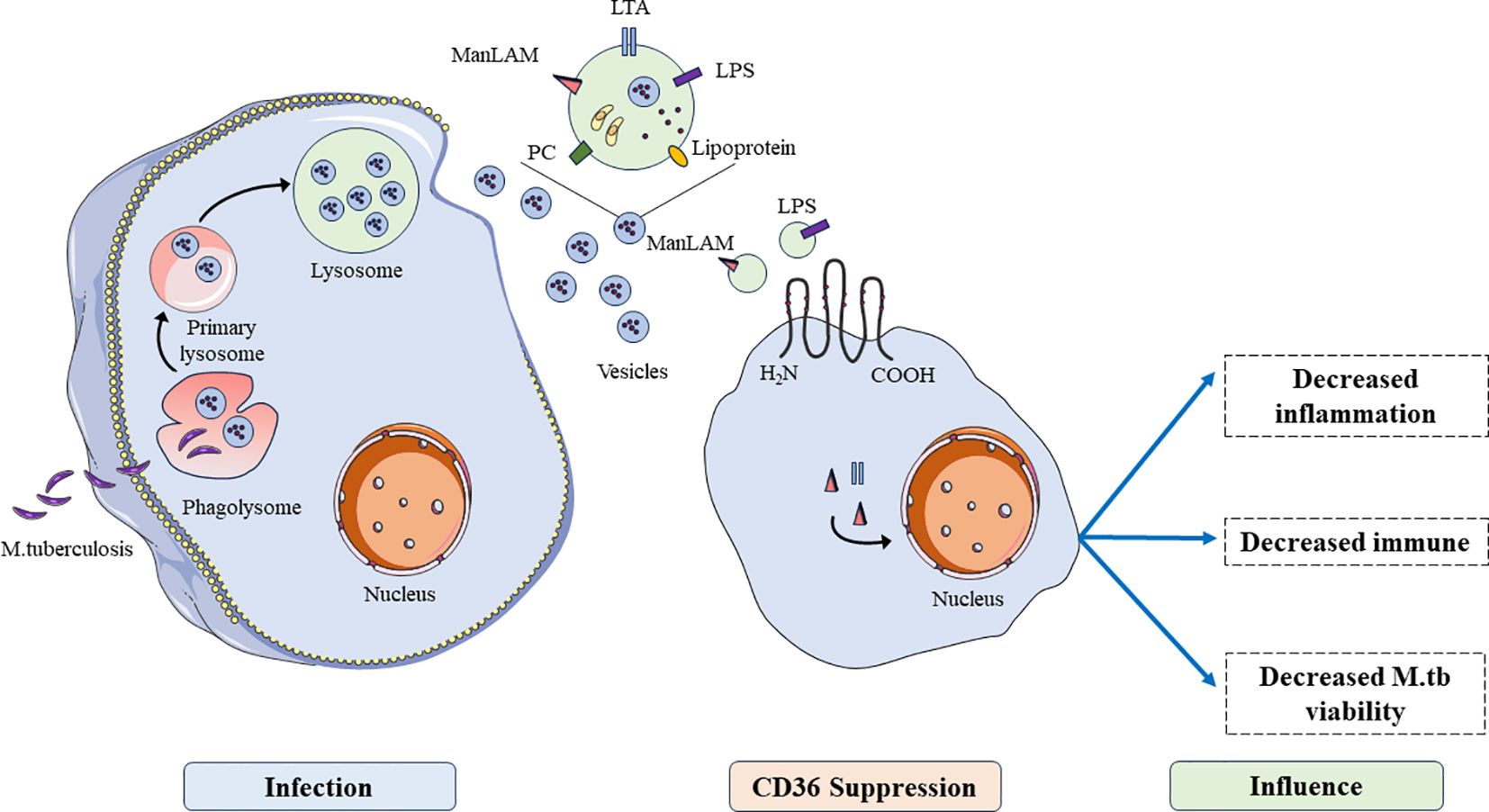
Figure 2 The functions of CD36 in the process of M. tuberculosis infection. After being engulfed by host cells and lysosomal lysis, antigens of M. tuberculosis are presented to immune cells through vesicles, inhibiting immune cell functions such as inhibiting inflammation, immune function, and promoting viability of M. tuberculosis.
Pepino et al. found macrophages pretreated with surfactant lipids increased the CD36 expression and induced the translocation of CD36 from the cytosol to the membrane (27). According to Dodd et al., surfactant lipids and/or surfactant protein A (SP-A) can cause preformed CD36 to be redistributed in the cell membrane, which can increase the production of CD36 (68). Advanced glycation end products (AGEs), which are also CD36 ligands, and oxiLDL are produced as a result of oxidative stress (73). The expression of CD36 in cells involved in the inflammation and immune response is dependent on the recognition and the uptake of lipid and metabolic components of M. tuberculosis (74, 75). Furthermore, CD36 plays a role in the uptake of surfactant lipids by human macrophages the CD36 knockdown decreases the uptake of dipalmitoylphosphatidylcholine (DPPC) and pre-infection exposure of human macrophages to surfactant lipids promotes M. tuberculosis growth in a manner that is CD36-dependent. These findings suggest that CD36 mediates the uptake of surfactant lipids by human macrophages, a function that M. tuberculosis exploits for growth. When M. tuberculosis is phagocytosed by macrophages, the immune system produces cytokines that target the phagocytes by secreting antimicrobial compounds, attracting PMNs to the infection site and producing granulomas (76). Oxidative stress is produced by granulomas made of immobilized M. tuberculosis and infiltrating macrophages to destroy the immobilized bacteria (77).
Hematopoietic stem cells (HSCs) also require CD36-mediated free fatty acid (FFA) uptake to compensate for metabolic requirements during acute infections (78). Once the components or metabolites of M. tuberculosis circulate in the peripheral blood, HSCs rapidly produce a large number of monocytes, T lymphocytes, and dendritic cells, promoting the transformation of monocytes into macrophages to cope with M. tuberculosis infection (79). Mistry et al. revealed the significant roles of CD36 in HSC metabolism in response to acute infections, which is impaired if CD36-mediated FFA uptake is absent and leads to increased mortality in HSCs (80). Meanwhile, M. tuberculosis not only could alter hematopoiesis by directly infecting the bone marrow niche harboring HSC, but also induce systemic cytokines such as TPO, SCF, IL-3, IL-6, TGF-β, MIP-1α and so on during chronic infection (81, 82).
However, monocytes and macrophages are crucial in the formation of granuloma.
Castaño et al. reported that monocytes infected by M. tuberculosis produced fewer granules and decreased the number of cytoplasmic projections and the expression of CD36, CD86 and CD68 compared to monocytes differentiated in the absence of mycobacteria (25). Monocytes treated with recombinant IL-1b prevented the increased expression of HLA-II, CD86 and CD36 observed with differentiation into macrophages (25). Moreover, infected monocytes suppressed the secretion of IL-6, TNF-a, IL-10, IL-12p70, but promoted the expression of IL-1b in response to LPS and purified protein M. tuberculosis-derived (25). According to a recent work by Baranova et al., CD36 mediates signaling triggered by gram-negative bacteria and LPS via a JNK-mediated signaling pathway in a manner that is TLR2/4-independent and serves as a phagocytic receptor for a range of bacteria (83). Furthermore, Józefowski et al. demonstrated that an antibody-neutralizing scavenger receptor SR-PSOX/CXCL16 partially reversed the augmentation of LPS-induced TNF-a generation by dextran sulphate, whereas an antibody-neutralizing CD36 reversed the stimulatory effect of deacylated ManLAM (84). According to this study, NO generation is regulated by unidentified scavenger receptors, whereas CD36 regulates the activity of ManLAM and its deacylated form that results in TNF-a release in LPS-stimulated J774 cells and peritoneal murine macrophages (84). Accordingly, M. fortuitum which was unable to synthesize ManLAM was not bound by murine CD36 expressed in HEK293 cells, indicating that CD36 is required for the uptake of mycobacteria (85). Depending on the route of infection, CD36 may have distinct effects on interactions between mycobacteria and distinct populations of macrophages that mycobacteria first encounter in vivo. Alemán et al. found that the phagocytosis of M. tuberculosis-induced neutrophils by immature dendritic cells (iDCs) leads to lymphoproliferation, which is significantly reduced by blocking CD36 and not DC-SIGN on iDCs (86). However, Hedlund et al. revealed that dendritic cells (DC) from M. tuberculosis-induced apoptotic neutrophils contained almost all stimulatory capacity, and the cell contact-dependent activation required binding of CD11b/CD18 to the DC via DC-SIGN but did not involve CD36 indicating that the cell interaction is crucial for DC activation (87). Of course, these studies reflected differences in CD36 expression and other M. tuberculosis sensing receptors, determining macrophage responses to M. tuberculosis.
CD36 is also a lysosomal membrane protein and plays critical roles in lysosomal enzyme trafficking and uptake of pathogens, respectively and generally in host cell defences against intracellular pathogens (88). LmpB is a functional homologue of CD36 and could specifically mediate the uptake of mycobacteria and lysosome biogenesis (89). This evidence indicated the functions of LmpA and LmpB as ancestors of the family of LIMP-2 and CD36, in lysosome biogenesis and host cell defence (89). Almeida et al. revealed that the synergistic interactions between CD36 and TLR2 are responsible for the altered lipid metabolism of host cells caused by infection. This data showed that CD36-TLR2 cooperation and signaling compartmentalization inside rafts, via PPAR-γ dependent and NF-κb independent pathways, reroute host response signaling, thereby promoting lipid accumulation and down-regulating the response of BCG-infected macrophages (90) (80). BCG induced CD36 expression and CD36’s roles were further validated by the inhibition of BCG-induced lipid body formation in vitro and in vivo in macrophages from CD36 deficient animals. Hawkes et al. showed CD36-/- mice showed decreased levels of inflammatory factors, the density of granuloma and the burden of M. tuberculosis in the spleen and liver during M. tuberculosis infection (91). The viable bacteria in macrophages from CD36-/- mice and the growth of M. tuberculosis were decreased significantly (91). These results fully demonstrated that CD36 regulated cellular lipid uptake and metabolism through various signaling pathways containing TLR receptors, PPAR-γ and NF-κb pathways, as well as eliminating intracellular mycobacteria through lysosomes and endocytosis.
In summary, after infection with M. tuberculosis, CD36 protein could induce the transformation of mononuclear cell into macrophages, polarization of M2 macrophages, promotion of cytokine secretion, inhibition of macrophage migration and T cell activation, resulting in inflammatory response and specific cellular immune suppression (Figure 3). Preliminary studies at the cellular and animal model levels have shown the functional role of CD36 in the immune response to tuberculosis infection, but the specific molecular mechanisms still need to be further investigated.
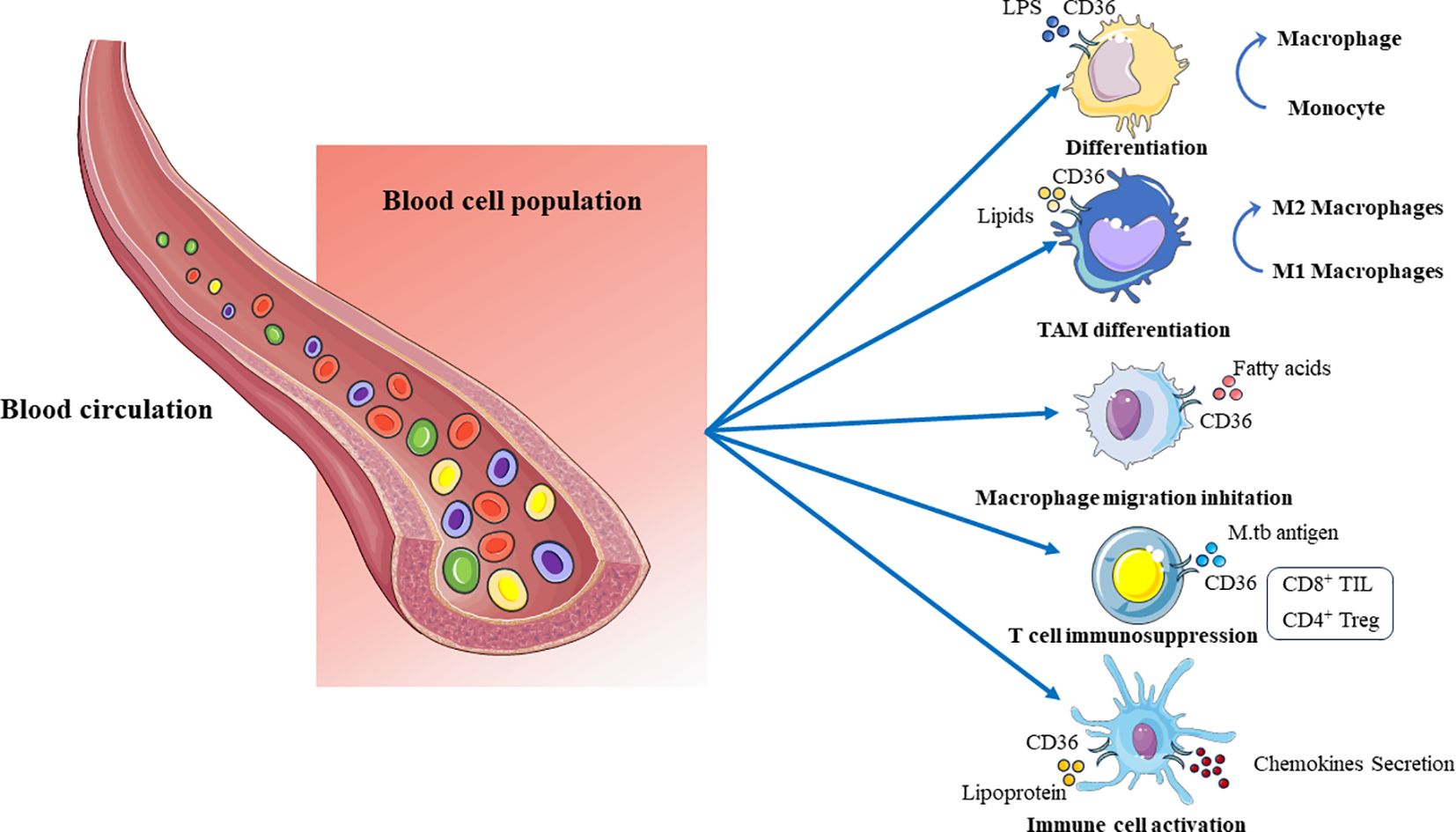
Figure 3 The lipid regulatory function of CD36 on immune cells in peripheral blood cells. CD36 promotes the transformation of mononuclear cells into macrophages during lipid metabolism, M2 polarization of macrophages, inhibition of macrophage migration, promotion of immune cell activation and secretion of cytokines, inhibition of T cell activation, and other functions.
4 The potential function of CD36 in the treatment of tuberculosis
Based on the diverse immune regulatory functions of CD36 in M. tuberculosis infection and its ability to regulate pathogen growth, CD36 has become a potential key target for tuberculosis treatment. Current research on the roles of CD36 in mycobacterial infections has demonstrated the intricate nature of interactions between macrophages and M. tuberculosis. These studies used genetically deficient mice to study the functions of CD36 in anti-mycobacterial host defense, with qualitatively different findings (91, 92). Court et al. reported that the long-term regulation of M. tuberculosis infection is unaffected by the absence of CD36 plus SR-A (92). After being challenged with M. bovis BCG, animals lacking CD36 alone had lower mycobacterial loads and granulomatous reactions, as demonstrated by Hawkes et al. (91). These findings indicated that CD36 is a receptor that mycobacteria use to infiltrate macrophages and demonstrated CD36’s role in cellular scenarios related to granuloma formation, which facilitate early bacterial growth and spread. This evidence indicates studies of CD36 in regulating the survival and growth of intracellular mycobacteria are insufficient and controversial which needs further research and suggests that CD36 may be an important valuable target for controlling or eliminating M. tuberculosis.
The hallmark of M. tuberculosis infection, foam cells, are reduced when stem bromelain-induced macrophages (SBM) cleaves CD36, according to research by Mahajan et al. (93). This creates an environment that facilitates the increased clearance of M. tuberculosis and provides a mechanism for the anti-mycobacterial activity of SBM. ManLAM might be the main CD36 ligand on the surface of M. tuberculosis. Anti-CD36 mAb, but not anti-TLR2 mAb, counteracted the increasing effect of deacylated ManLAM on the LPS-stimulated TNF-a generation, as reported by Baranova et al. (83). This suggests that CD36 can independently initiate intracellular signaling. In macrophages from immunologically naï¨ve guinea pigs infected with M. tuberculosis, Palanisamy et al. demonstrated an increase in CD36 and LOX1, which enhances the uptake of oxidized host macromolecules, including OxLDL (94). The intracellular buildup of OxLDL, the expression of CD36 and LOX1 in macrophages, and the bacterial load were all reduced in guinea pigs immunized with BCG prior to aerosol challenge (94). This study demonstrated that intracellular bacilli survival and persistence were supported by oxidative stress in guinea pigs infected with M. tuberculosis and the possible roles of OxLDL-loaded macrophages. This literature has presented various potential treatments or control strategies for M. tuberculosis infection through CD36 targets including natural compounds, ligands, etc. More importantly, CD36 may play an important role in the immune process of conjugated vaccines, providing a new perspective for subsequent research.
M. tuberculosis infection triggers an immune response that converges on formation of a granuloma, a dynamic and spatially organized tissue structure composed of macrophages, granulocytes, lymphocytes and fibroblasts, and provide the immune microenvironment for M. tuberculosis infection (95). In M. tuberculosis infected microenvironment, additional phagocytes were recruited to the site of infection through the secretion of cytokines and chemokines from above immune cells (96). In addition, the expression of CD36 is regulated at both the transcriptional and posttranslational levels among these different cells. In monocytes, CD36 is upregulated by PPAR-ɣ, as well as by cytokines including CSF, IL-4, and IL-10 that are involved in differentiation toward DCs and reparative “M2”-like phenotypes (97, 98). These findings provide us with new ideas for treating tuberculosis infection. If the expression of CD36 on various immune cells in granulation was inhibited, the energy metabolism of infected cells will be reduced to decrease the latent proliferation of M. tuberculosis. We believe that achieving the goal of treating tuberculosis will not be far away.
5 The functions of CD36 as a biomarker for the diagnosis of tuberculosis
CD36, as an important signaling molecule on the cell surface, is expressed on various cell surfaces, and CD36 has become a diagnostic biomarker for various tumors (99, 100). CD36 also has important potential value in the diagnosis of tuberculosis infection. In vitro, Sánchez et al. also found that M. tuberculosis infection decreased the expression of CD36 correlated with induction of apoptosis indicating that the low expression of CD36 is closely related to tuberculosis infection (101). Shkurupy et al. found CD36, CD11 and CD29 on macrophages increased significantly due to M. tuberculosis infection and participated in macrophages fusion suggesting that the expression of these molecules is a constitutive property of peritoneal (102). CD36 was up-regulated by M. tuberculosis H37Ra infection in macrophages and suppressed in exosomes from H37Ra-infected macrophages indicating that CD36 could be a potential biomarker associated with TB infection (103).
Currently, in vivo, numerous samples of evidence indicate that CD36 also demonstrates its potential as a diagnostic biomarker (Figure 4). Single nucleotide polymorphisms (SNP) in CD36 indicated the risk of pulmonary tuberculosis is decreased in SNPs due to the reduced ability of CD36 to recognize the M. tuberculosis pattern recognition molecules indicated CD36 as important receptors in response to PTB (104). There is evidence indicating that TGF-E down-regulated CD36 expression and the upexpression of TGF-E existing may be responsible for the downexpression and decreased percentage of CD36+ monocytes in TB patients (105, 106). The reduction in CD36+ monocytes suggests that TB patients’ monocytes and macrophages would be less able to identify and eliminate apoptotic cells which would cause the cells to become late necrotic and promote the spread of germs (106). In patients with various clinical manifestations of tuberculosis, Sánchez et al. had previously shown an increased frequency of CD14+ monocytes along with decreased expression of CD36 and HLA-DR, indicating that immature cells were circulating during active TB (107). Flow cytometry was used to evaluate blood mononuclear cells from TB patients with varying clinical phases for the markers CD36, CD14, CD40, CD163 and CD203. However, CD36 and CD14 were found to be reduced in TB patients (107). Zhang et al. found serum exosomal protein CD36 decreased significantly in active TB (ATB) patients, and CD36 was downexpressed in peripheral blood mononuclear cells (PBMCs) of ATB patients through the comprehensive proteomics analysis (108). Similarly, among LTBI individuals with coronary artery disease (CAD), CD36 was down expressed across all monocyte subsets suggesting that the lower expression of CD36 could engender a negative feedback mechanism to counterbalance ongoing inflammation (109).
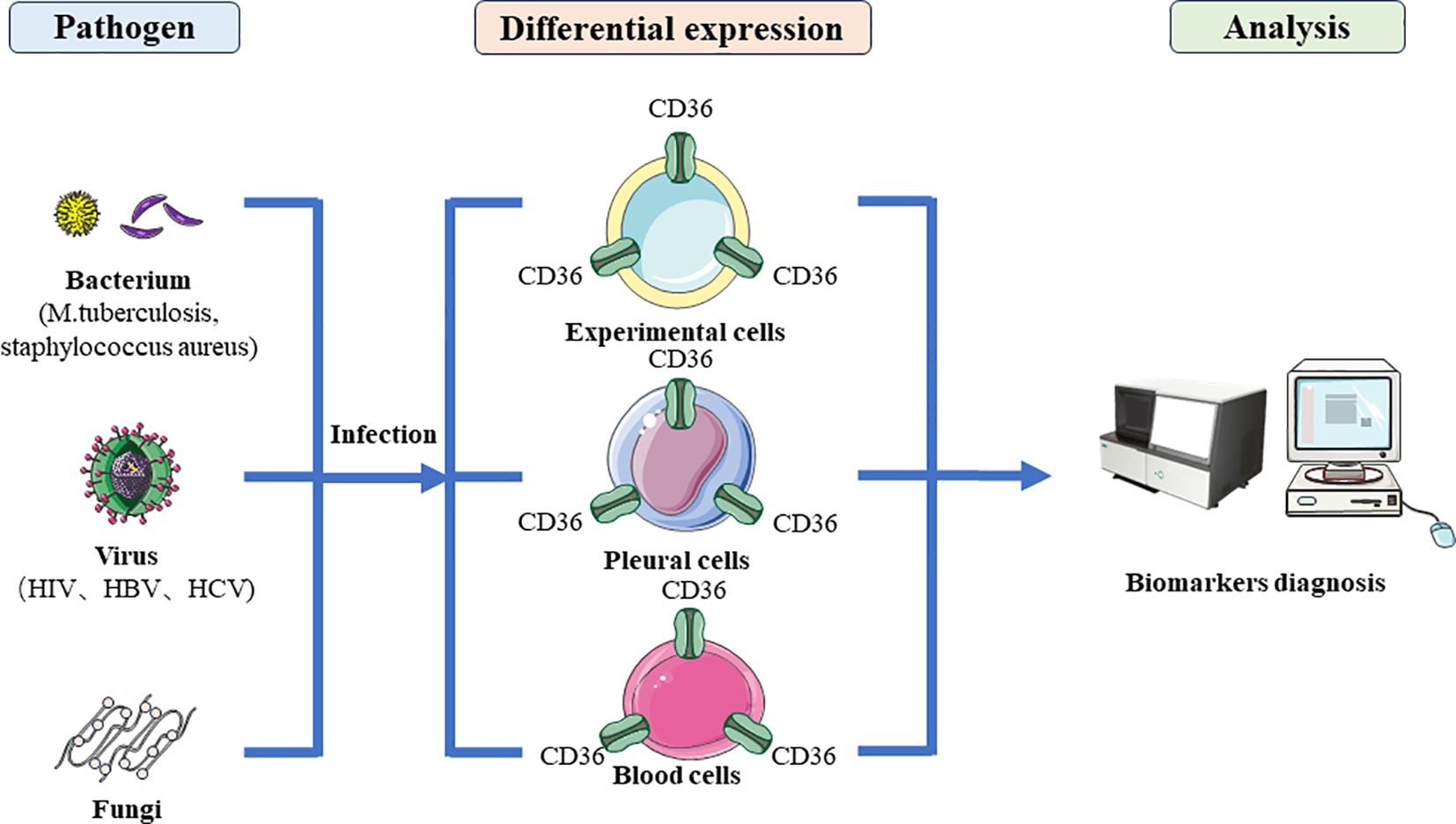
Figure 4 The diagnostic application of CD36 in microbial infections. After bacterial, viral and fungal infection, they regulate the specific expression of CD36 protein in host cells, making CD36 a marker for diagnosing microbial infection.
A large amount of clinical trial data indicated that CD36 is downregulated in the peripheral blood of tuberculosis patients, which is beyond doubt. The abnormal expression of CD36 is only analyzed at the cellular level and peripheral blood in vivo, and it is still far from becoming a diagnostic kit for clinical application. At least the following issues need to be addressed: (1) The abnormal expression of CD36 is specific to M. tuberculosis infection and is not affected by other diseases; (2) The sensitivity and standards of CD36 for detecting M. tuberculosis infection need to be studied; (3) The commercialized CD36 reagent kit needs further optimization. Despite facing numerous challenges, researches on the correlation between CD36 and M. tuberculosis infection have laid an experimental and theoretical foundation for CD36 to become a diagnostic marker of tuberculosis.
6 Future prospects
At present, the scavenger receptor CD36 plays an important role in tumors and lipid metabolism and is a critical hotspot in these fields. However, in recent years, researchers found that CD36 also acted as a significant influence on the recognition of ligands from pathogens especially in M. tuberculosis and host cells. CD36 mainly involves the lipid metabolism, inflammatory response, immune response, vaccine immunity, and diagnostic markers of hosts infected with M. tuberculosis. The functions of CD36 in M. tuberculosis infection has gradually become clear, and numerous studies are conducting animal and drug experiments targeting CD36, hoping to therapy tuberculosis. However, there have been few significant achievements so far, and clinical trials are far from being conducted. At the same time, the clinical diagnosis of tuberculosis infection by CD36 is still in the initial exploration stage both in vivo and in vitro, and requires a large amount of clinical testing to confirm its diagnostic value.
This study comprehensively reviews the relevant roles and functions of CD36 in tuberculosis infection, providing a research foundation for researchers. Although CD36 has a wide range of functions in the process of M. tuberculosis infection in hosts, related studies are still inadequate, and the specific molecular mechanisms in related fields are still not clear enough.
Author contributions
JW: Conceptualization, Data curation, Funding acquisition, Investigation, Writing – original draft, Writing – review & editing. HC: Investigation, Resources, Supervision, Writing – original draft. HY: Data curation, Formal analysis, Writing – original draft. NW: Methodology, Software, Visualization, Writing – review & editing. YW: Formal analysis, Validation, Writing – review & editing. HL: Formal analysis, Project administration, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by grants from the National Natural Science Foundation of China (82002111), the Suzhou Science and Technology Development Plan Project (SKY2022078) and Suzhou Medical Key Discipline Construction Project (SZXK202124).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Koch A, Mizrahi V. Mycobacterium tuberculosis. Trends Microbiol. (2018) 26:555–6. doi: 10.1016/j.tim.2018.02.012
2. Bo H, Moure UAE, Yang Y, Pan J, Li L, Wang M, et al. Mycobacterium tuberculosis-macrophage interaction: Molecular updates. Front Cell Infect Microbiol. (2023) 13:1062963. doi: 10.3389/fcimb.2023.1062963
3. Getahun H, Matteelli A, Chaisson RE, Raviglione M. Latent Mycobacterium tuberculosis infection. N Engl J Med. (2015) 372:2127–35. doi: 10.1056/NEJMra1405427
4. Rahlwes KC, Dias BRS, Campos PC, Alvarez-Arguedas S, Shiloh MU. Pathogenicity and virulence of Mycobacterium tuberculosis. Virulence. (2023) 14:2150449. doi: 10.1080/21505594.2022.2150449
5. Li Y, Qian Y, Wang N, Qiu D, Cao H, Wang Y, et al. The functions and applications of extracellular vesicles derived from Mycobacterium tuberculosis. BioMed Pharmacother. (2023) 168:115767. doi: 10.1016/j.biopha.2023.115767
6. Ehrt S, Schnappinger D, Rhee KY. Metabolic principles of persistence and pathogenicity in Mycobacterium tuberculosis. Nat Rev Microbiol. (2018) 16:496–507. doi: 10.1038/s41579-018-0013-4
7. Smith I. Mycobacterium tuberculosis pathogenesis and molecular determinants of virulence. Clin Microbiol Rev. (2003) 16:463–96. doi: 10.1128/CMR.16.3.463-496.2003
8. Butcher PD. Microarrays for mycobacterium tuberculosis. Tuberculosis (Edinb). (2004) 84:131–7. doi: 10.1016/j.tube.2004.01.002
9. Kim MJ, Wainwright HC, Locketz M, Bekker LG, Walther GB, Dittrich C, et al. Caseation of human tuberculosis granulomas correlates with elevated host lipid metabolism. EMBO Mol Med. (2010) 2:258–74. doi: 10.1002/emmm.201000079
10. Ravesloot-Chávez MM, Van Dis E, Stanley SA. The innate immune response to mycobacterium tuberculosis infection. Annu Rev Immunol. (2021) 39:611–37. doi: 10.1146/annurev-immunol-093019-010426
11. Huang L, Nazarova EV, Russell DG. Mycobacterium tuberculosis: Bacterial Fitness within the Host Macrophage. Microbiol Spectr. (2019) 7:10. doi: 10.1128/microbiolspec.BAI-0001-2019
12. Ahmad F, Rani A, Alam A, Zarin S, Pandey S, Singh H, et al. Macrophage: A cell with many faces and functions in tuberculosis. Front Immunol. (2022) 13:747799. doi: 10.3389/fimmu.2022.747799
13. Weiss G, Schaible UE. Macrophage defense mechanisms against intracellular bacteria. Immunol Rev. (2015) 264:182–203. doi: 10.1111/imr.12266
14. D'Avila H, Almeida PE, Roque NR, Castro-Faria-Neto HC, Bozza PT. Toll-like receptor-2-mediated C-C chemokine receptor 3 and eotaxin-driven eosinophil influx induced by Mycobacterium bovis BCG pleurisy. Infect Immun. (2007) 75:1507–11. doi: 10.1128/IAI.01326-06
15. D'Avila H, Maya-Monteiro CM, Bozza PT. Lipid bodies in innate immune response to bacterial and parasite infections. Int Immunopharmacol. (2008) 8:1308–15. doi: 10.1016/j.intimp.2008.01.035
16. Mattos KA, Lara FA, Oliveira VG, Rodrigues LS, D'Avila H, Melo RC, et al. Modulation of lipid droplets by Mycobacterium leprae in Schwann cells: a putative mechanism for host lipid acquisition and bacterial survival in phagosomes. Cell Microbiol. (2011) 13:259–73. doi: 10.1111/j.1462-5822.2010.01533.x
17. Chandra P, Grigsby SJ, Philips JA. Immune evasion and provocation by Mycobacterium tuberculosis. Nat Rev Microbiol. (2022) 20:750–66. doi: 10.1038/s41579-022-00763-4
18. Yu XH, Fu YC, Zhang DW, Yin K, Tang CK. Foam cells in atherosclerosis. Clin Chim Acta. (2013) 424:245–52. doi: 10.1016/j.cca.2013.06.006
19. Wang J, Li Y. CD36 tango in cancer: signaling pathways and functions. Theranostics. (2019) 9:4893–908. doi: 10.7150/thno.36037
20. Martin C, Chevrot M, Poirier H, Passilly-Degrace P, Niot I, Besnard P. CD36 as a lipid sensor. Physiol Behav. (2011) 105:36–42. doi: 10.1016/j.physbeh.2011.02.029
21. Febbraio M, Hajjar DP, Silverstein RL. CD36: a class B scavenger receptor involved in angiogenesis, atherosclerosis, inflammation, and lipid metabolism. J Clin Invest. (2001) 108:785–91. doi: 10.1172/JCI14006
22. Voll RE, Herrmann M, Roth EA, Stach C, Kalden JR, Girkontaite I. Immunosuppressive effects of apoptotic cells. Nature. (1997) 390:350–1. doi: 10.1038/37022
23. Nilsen NJ, Deininger S, Nonstad U, Skjeldal F, Husebye H, Rodionov D, et al. Cellular trafficking of lipoteichoic acid and Toll-like receptor 2 in relation to signaling: role of CD14 and CD36. J Leukoc Biol. (2008) 84:280–91. doi: 10.1189/jlb.0907656
24. Alva-Murillo N, López-Meza JE, Ochoa-Zarzosa A. Nonprofessional phagocytic cell receptors involved in Staphylococcus aureus internalization. BioMed Res Int. (2014) 2014:538546. doi: 10.1155/2014/538546
25. Castaño D, Barrera LF, Rojas M. Mycobacterium tuberculosis alters the differentiation of monocytes into macrophages in vitro. Cell Immunol. (2011) 268:60–7. doi: 10.1016/j.cellimm.2011.02.006
26. Li Y, Huang X, Yang G, Xu K, Yin Y, Brecchia G, et al. CD36 favours fat sensing and transport to govern lipid metabolism. Prog Lipid Res. (2022) 88:101193. doi: 10.1016/j.plipres.2022.101193
27. Pepino MY, Kuda O, Samovski D, Abumrad NA. Structure-function of CD36 and importance of fatty acid signal transduction in fat metabolism. Annu Rev Nutr. (2014) 34:281–303. doi: 10.1146/annurev-nutr-071812-161220
28. Heit B, Kim H, Cosío G, Castaño D, Collins R, Lowell CA, et al. Multimolecular signaling complexes enable Syk-mediated signaling of CD36 internalization. Dev Cell. (2013) 24:372–83. doi: 10.1016/j.devcel.2013.01.007
29. Fadok VA, Bratton DL, Henson PM. Phagocyte receptors for apoptotic cells: recognition, uptake, and consequences. J Clin Invest. (2001) 108:957–62. doi: 10.1172/JCI200114122
30. Penberthy KK, Ravichandran KS. Apoptotic cell recognition receptors and scavenger receptors. Immunol Rev. (2016) 269:44–59. doi: 10.1111/imr.12376
31. Baranova IN, Bocharov AV, Vishnyakova TG, Kurlander R, Chen Z, Fu D, et al. CD36 is a novel serum amyloid A (SAA) receptor mediating SAA binding and SAA-induced signaling in human and rodent cells. J Biol Chem. (2010) 285:8492–506. doi: 10.1074/jbc.M109.007526
32. Zaidi NE, Shazali NAH, Leow TC, Osman MA, Ibrahim K, Cheng WH, et al. CD36-fatty acid-mediated metastasis via the bidirectional interactions of cancer cells and macrophages. Cells. (2022) 11:3556. doi: 10.3390/cells11223556
33. Xia L, Zhou Z, Chen X, Luo W, Ding L, Xie H, et al. Ligand-dependent CD36 functions in cancer progression, metastasis, immune response, and drug resistance. BioMed Pharmacother. (2023) 168:115834. doi: 10.1016/j.biopha.2023.115834
34. Banesh S, Ramakrishnan V, Trivedi V. Mapping of phosphatidylserine recognition region on CD36 ectodomain. Arch Biochem Biophys. (2018) 660:1–10. doi: 10.1016/j.abb.2018.10.005
35. Park YM. CD36, a scavenger receptor implicated in atherosclerosis. Exp Mol Med. (2014) 46:e99. doi: 10.1038/emm.2014.38
37. Mimche PN, Thompson E, Taramelli D, Vivas L. Curcumin enhances non-opsonic phagocytosis of Plasmodium falciparum through up-regulation of CD36 surface expression on monocytes/macrophages. J Antimicrob Chemother. (2012) 67:1895–904. doi: 10.1093/jac/dks132
38. Inoue M, Tanabe H, Matsumoto A, Takagi M, Umegaki K, Amagaya S, et al. Astaxanthin functions differently as a selective peroxisome proliferator-activated receptor-γ modulator in adipocytes and macrophages. Biochem Pharmacol. (2012) 84:692–700. doi: 10.1016/j.bcp.2012.05.021
39. Choi JS, Bae JY, Kim DS, Li J, Kim JL, Lee YJ, et al. Dietary compound quercitrin dampens VEGF induction and PPARgamma activation in oxidized LDL-exposed murine macrophages: association with scavenger receptor CD36. J Agric Food Chem. (2010) 58:1333–41. doi: 10.1021/jf9032443
40. Sini S, Deepa D, Harikrishnan S, Jayakumari N. High-density lipoprotein from subjects with coronary artery disease promotes macrophage foam cell formation: role of scavenger receptor CD36 and ERK/MAPK signaling. Mol Cell Biochem. (2017) 427:23–34. doi: 10.1007/s11010-016-2895-7
41. Pan J, Fan Z, Wang Z, Dai Q, Xiang Z, Yuan F, et al. CD36 mediates palmitate acid-induced metastasis of gastric cancer via AKT/GSK-3β/β-catenin pathway. J Exp Clin Cancer Res. (2019) 38:52. doi: 10.1186/s13046-019-1049-7
42. Su T, Huang C, Yang C, Jiang T, Su J, Chen M, et al. Apigenin inhibits STAT3/CD36 signaling axis and reduces visceral obesity. Pharmacol Res. (2020) 152:104586. doi: 10.1016/j.phrs.2019.104586
43. Peruń A, Gębicka M, Biedrob R, Skalska P, Józefowski S. The CD36 and SR-A/CD204 scavenger receptors fine-tune Staphylococcus aureus-stimulated cytokine production in mouse macrophages. Cell Immunol. (2022) 372:104483. doi: 10.1016/j.cellimm.2022.104483
44. Banesh S, Layek S, Trivedi V. Hemin acts as CD36 ligand to activate down-stream signalling to disturb immune responses and cytokine secretion from macrophages. Immunol Lett. (2022) 243:1–18. doi: 10.1016/j.imlet.2022.01.004
45. Banesh S, Trivedi V. Therapeutic potentials of scavenger receptor CD36 mediated innate immune responses against infectious and non-infectious diseases. Curr Drug Discovery Technol. (2020) 17:299–317. doi: 10.2174/1570163816666190802153319
46. Liu W, Yin Y, Zhou Z, He M, Dai Y. OxLDL-induced IL-1 beta secretion promoting foam cells formation was mainly via CD36 mediated ROS production leading to NLRP3 inflammasome activation. Inflammation Res. (2014) 63:33–43. doi: 10.1007/s00011-013-0667-3
47. Gautam S, Banerjee M. The macrophage Ox-LDL receptor, CD36 and its association with type II diabetes mellitus. Mol Genet Metab. (2011) 102:389–98. doi: 10.1016/j.ymgme.2010.12.012
48. Yang X, Yao H, Chen Y, Sun L, Li Y, Ma X, et al. Inhibition of glutathione production induces macrophage CD36 expression and enhances cellular-oxidized low density lipoprotein (oxLDL) uptake. J Biol Chem. (2015) 290:21788–99. doi: 10.1074/jbc.M115.654582
49. Chistiakov DA, Melnichenko AA, Myasoedova VA, Grechko AV, Orekhov AN. Mechanisms of foam cell formation in atherosclerosis. J Mol Med (Berl). (2017) 95:1153–65. doi: 10.1007/s00109-017-1575-8
50. Jiang Y, Wang M, Huang K, Zhang Z, Shao N, Zhang Y, et al. Oxidized low-density lipoprotein induces secretion of interleukin-1w by macrophages via reactive oxygen species-dependent NLRP3 inflammasome activation. Biochem Biophys Res Commun. (2012) 425:121–6. doi: 10.1016/j.bbrc.2012.07.011
51. Handberg A, Højlund K, Gastaldelli A, Flyvbjerg A, Dekker JM, Petrie J, et al. Plasma sCD36 is associated with markers of atherosclerosis, insulin resistance and fatty liver in a nondiabetic healthy population. J Intern Med. (2012) 271:294–304. doi: 10.1111/j.1365-2796.2011.02442.x
52. Yimin, Kohanawa M, Zhao S, Ozaki M, Haga S, Nan G, et al. Contribution of toll-like receptor 2 to the innate response against Staphylococcus aureus infection in mice. PloS One. (2013) 8:e74287. doi: 10.1371/journal.pone.0074287
53. Chávez-Sánchez L, Garza-Reyes MG, Espinosa-Luna JE, Chávez-Rueda K, Legorreta-Haquet MV, Blanco-Favela F. The role of TLR2, TLR4 and CD36 in macrophage activation and foam cell formation in response to oxLDL in humans. Hum Immunol. (2014) 75:322–9. doi: 10.1016/j.humimm.2014.01.012
54. Triantafilou M, Gamper FG, Haston RM, Mouratis MA, Morath S, Hartung T, et al. Membrane sorting of toll-like receptor (TLR)-2/6 and TLR2/1 heterodimers at the cell surface determines heterotypic associations with CD36 and intracellular targeting. J Biol Chem. (2006) 281:31002–11. doi: 10.1074/jbc.M602794200
55. Strapagiel D, Kasztalska K, Druszczyńska M, Kowalewicz-Kulbat M, Vrba A, Matusiak A, et al. Monocyte response receptors in BCG driven delayed type hypersensitivity to tuberculin. Folia Histochem Cytobiol. (2008) 46:353–9. doi: 10.2478/v10042-008-0044-1
56. Hoebe K, Georgel P, Rutschmann S, Du X, Mudd S, Crozat K, et al. CD36 is a sensor of diacylglycerides. Nature. (2005) 433:523–7. doi: 10.1038/nature03253
57. Triantafilou M, Gamper FG, Lepper PM, Mouratis MA, Schumann C, Harokopakis E, et al. Lipopolysaccharides from atherosclerosis-associated bacteria antagonize TLR4, induce formation of TLR2/1/CD36 complexes in lipid rafts and trigger TLR2-induced inflammatory responses in human vascular endothelial cells. Cell Microbiol. (2007) 9:2030–9. doi: 10.1111/j.1462-5822.2007.00935.x
58. Stewart CR, Stuart LM, Wilkinson K, van Gils JM, Deng J, Halle A, et al. CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat Immunol. (2010) 11:155–61. doi: 10.1038/ni.1836
59. Cooper GE, Pounce ZC, Wallington JC, Bastidas-Legarda LY, Nicholas B, Chidomere C, et al. Viral inhibition of bacterial phagocytosis by human macrophages: redundant role of CD36. PloS One. (2016) 11:e0163889. doi: 10.1371/journal.pone.0163889
60. Stuart LM, Deng J, Silver JM, Takahashi K, Tseng AA, Hennessy EJ, et al. Response to Staphylococcus aureus requires CD36-mediated phagocytosis triggered by the COOH-terminal cytoplasmic domain. J Cell Biol. (2005) 170:477–85. doi: 10.1083/jcb.200501113
61. Feeney ER, McAuley N, O'Halloran JA, Rock C, Low J, Satchell CS, et al. The expression of cholesterol metabolism genes in monocytes from HIV-infected subjects suggests intracellular cholesterol accumulation. J Infect Dis. (2013) 207:628–37. doi: 10.1093/infdis/jis723
62. Berre S, Gaudin R, Cunha de Alencar B, Desdouits M, Chabaud M, Naffakh N, et al. CD36-specific antibodies block release of HIV-1 from infected primary macrophages and its transmission to T cells. J Exp Med. (2013) 210:2523–38. doi: 10.1084/jem.20130566
63. Huang J, Zhao L, Yang P, Chen Z, Ruan XZ, Huang A, et al. Fatty acid translocase promoted hepatitis B virus replication by upregulating the levels of hepatic cytosolic calcium. Exp Cell Res. (2017) 358:360–8. doi: 10.1016/j.yexcr.2017.07.012
64. Olivetta E, Tirelli V, Chiozzini C, Scazzocchio B, Romano I, Arenaccio C, et al. HIV-1 Nef impairs key functional activities in human macro-phages through CD36 downregulation. PloS One. (2014) 9:e93699. doi: 10.1371/journal.pone.0093699
65. Fink IR, Benard EL, Hermsen T, Meijer AH, Forlenza M, Wiegertjes GF. Molecular and functional characterization of the scavenger receptor CD36 in zebrafish and common carp. Mol Immunol. (2015) 63:381–93. doi: 10.1016/j.molimm.2014.09.010
66. Bazzi S, El-Darzi E, McDowell T, Modjtahedi H, Mudan S, Achkar M, et al. Defining genome-wide expression and phenotypic contextual cues in macrophages generated by granulocyte/macrophage colony-stimulating factor, macrophage colony-stimulating factor, and heat-killed mycobacteria. Front Immunol. (2017) 8:1253. doi: 10.3389/fimmu.2017.01253
67. Park HT, Park WB, Kim S, Lim JS, Nah G, Yoo HS. Revealing immune responses in the Mycobacterium avium subsp. paratuberculosis-infected THP-1 cells using single cell RNA-sequencing. PloS One. (2021) 16:e0254194. doi: 10.1371/journal.pone.0254194
68. Dodd CE, Pyle CJ, Glowinski R, Rajaram MV, Schlesinger LS. CD36-mediated uptake of surfactant lipids by human macro-phages promotes intracellular growth of Mycobacterium tu-berculosis. J Immunol. (2016) 197:4727–35. doi: 10.4049/jimmunol.1600856
69. Luo X, Zheng E, Wei L, Zeng H, Qin H, Zhang X, et al. The fatty acid receptor CD36 promotes HCC progression through activating Src/PI3K/AKT axis-dependent aerobic glycolysis. Cell Death Dis. (2021) 12:328. doi: 10.1038/s41419-021-03596-w
70. Kim JS, Kim YR, Yang CS. Host-directed therapy in tuberculosis: targeting host metabolism. Front Immunol. (2020) 11:1790. doi: 10.3389/fimmu.2020.01790
71. Biswas SK, Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat Immunol. (2010) 11:889–96. doi: 10.1038/ni.1937
72. Wang S, Liu G, Li Y, Pan Y. Metabolic reprogramming induces macrophage polarization in the tumor microenvironment. Front Immunol. (2022) 13:840029. doi: 10.3389/fimmu.2022.840029
73. He L, Zhou X, Yin X, Tian L, Yang L, Fan K, et al. Comparative study of the growth and survival of recombinant Mycobacterium smegmatis expressing Mce4A and Mce4E from Mycobacterium bovis. DNA Cell Biol. (2015) 34:125–32. doi: 10.1089/dna.2014.2487
74. Zhu W, Li W, Silverstein RL. Advanced glycation end products induce a prothrombotic phenotype in mice via interaction with platelet CD36. Blood. (2012) 119:6136–44. doi: 10.1182/blood-2011-10-387506
75. Ohgami N, Nagai R, Ikemoto M, Arai H, Miyazaki A, Hakamata H, et al. CD36, serves as a receptor for advanced glycation endprod-ucts (AGE). J Diabetes Complications. (2002) 16:56–9. doi: 10.1016/S1056-8727(01)00208-2
76. Ndlovu H, Marakalala MJ. Granulomas and inflammation: hostdirected therapies for tuberculosis. Front Immunol. (2016) 7:434. doi: 10.3389/fimmu.2016.00434
77. Ehlers S, Schaible UE. The granuloma in tuberculosis: dynamics of a host-pathogen collusion. Front Immunol. (2013) 3:411. doi: 10.3389/fimmu.2012.00411
78. Filippi MD. Hungry hematopoietic stem cells during bacterial infection: fatty acid for food. Immunometabolism. (2022) 4:e220011. doi: 10.20900/immunometab20220011
79. Kaufmann E, Sanz J, Dunn JL, Khan N, Mendonça LE, Pacis A, et al. BCG educates hematopoietic stem cells to generate protective innate immunity against tuberculosis. Cell. (2018) 172:176–90. doi: 10.1016/j.cell.2017.12.031
80. Mistry JJ, Hellmich C, Moore JA, Jibril A, Macaulay I, Moreno-Gonzalez M, et al. Free fatty-acid transport via CD36 drives β-oxidation-mediated hematopoietic stem cell response to infection. Nat Commun. (2021) 12:7130. doi: 10.1038/s41467-021-27460-9
81. Dougan M, Dranoff G, Dougan SK. GM-CSF, IL-3, and IL-5 family of cytokines: regulators of inflammation. Immunity. (2019) 50:796–811. doi: 10.1016/j.immuni.2019.03.022
82. Mirantes C, Passegué E, Pietras EM. Pro-inflammatory cytokines: emerging players regulating HSC function in normal and diseased hematopoiesis. Exp Cell Res. (2014) 329:248–54. doi: 10.1016/j.yexcr.2014.08.017
83. Baranova IN, Kurlander R, Bocharov AV, Vishnyakova TG, Chen Z, Remaley AT, et al. Role of human CD36 in bacterial recognition, phagocytosis, and pathogen-induced JNK-mediated signaling. J Immunol. (2008) 181:7147e56. doi: 10.4049/jimmunol.181.10.7147
84. Józefowski S, Sobota A, Pawłowski A, Kwiatkowska K. Mycobacterium tuberculosis lipoarabinomannan enhances LPS-induced TNF-α production and inhibits NO secretion by engaging scavenger receptors. Microb Pathog. (2011) 50:350–9. doi: 10.1016/j.micpath.2011.03.001
85. Philips JA, Rubin EJ, Perrimon N. Drosophila RNAi screen reveals CD36 family member required for mycobacterial infection. Science. (2005) 309:1251e3. doi: 10.1126/science.1116006
86. Alemán M, de la Barrera S, Schierloh P, Yokobori N, Baldini M, Musella R, et al. Spontaneous or Mycobacterium tuberculosis-induced apoptotic neutrophils exert opposite effects on the dendritic cell-mediated immune response. Eur J Immunol. (2007) 37:1524–37. doi: 10.1002/eji.200636771
87. Hedlund S, Persson A, Vujic A, Che KF, Stendahl O, Larsson M. Dendritic cell activation by sensing Mycobacterium tuberculosis-induced apoptotic neutrophils via DC-SIGN. Hum Immunol. (2010) 71:535–40. doi: 10.1016/j.humimm.2010.02.022
88. Huang SC, Everts B, Ivanova Y, O'Sullivan D, Nascimento M, Smith AM, et al. Cell-intrinsic lysosomal lipolysis is essential for alternative activation of macrophages. Nat Immunol. (2014) 15:846–55. doi: 10.1038/ni.2956
89. Sattler N, Bosmani C, Barisch C, Guého A, Gopaldass N, Dias M, et al. Functions of the Dictyostelium LIMP-2 and CD36 homologues in bacteria uptake, phagolysosome biogenesis and host cell defence. J Cell Sci. (2018) 131:jcs218040. doi: 10.1242/jcs.218040
90. Almeida PE, Roque NR, Magalhães KG, Mattos KA, Teixeira L, Maya-Monteiro C, et al. Differential TLR2 downstream signaling regulates lipid metabolism and cytokine production triggered by Mycobacterium bovis BCG infection. Biochim Biophys Acta. (2014) 1841:97–107. doi: 10.1016/j.bbalip.2013.10.008
91. Hawkes M, Li X, Crockett M, Diassiti A, Finney C, Min-Oo G, et al. CD36 deficiency attenuates experimental mycobacterial infection. BMC Infect Dis. (2010) 10:299. doi: 10.1186/1471-2334-10-299
92. Court N, Vasseur V, Vacher R, Frémond C, Shebzukhov Y, Yeremeev VV, et al. Partial redundancy of the pattern recognition receptors, scavenger receptors, and C-type lectins for the long-term control of Mycobacterium tuberculosis infection. J Immunol. (2010) 184:7057e70. doi: 10.4049/jimmunol.1000164
93. Mahajan S, Chandra V, Dave S, Nanduri R, Gupta P. Stem bromelain-induced macrophage apoptosis and activation curtail Mycobacterium tuberculosis persistence. J Infect Dis. (2012) 206:366–76. doi: 10.1093/infdis/jis354
94. Palanisamy GS, Kirk NM, Ackart DF, Obregón-Henao A, Shanley CA, Orme IM, et al. Uptake and accumulation of oxidized low-density lipoprotein during Mycobacterium tuberculosis infection in Guinea pigs. PloS One. (2012) 7:e34148. doi: 10.1371/journal.pone.0034148
95. McCaffrey EF, Donato M, Keren L, Chen Z, Delmastro A, Fitzpatrick MB, et al. The immunoregulatory landscape of human tuberculosis granulomas. Nat Immunol. (2022) 23:318–29. doi: 10.1038/s41590-021-01121-x
96. Sia JK, Rengarajan J. Immunology of mycobacterium tuberculosis infections. Microbiol Spectr. (2019) 7:10. doi: 10.1128/microbiolspec.GPP3-0022-2018
97. Lim HJ, Lee S, Lee KS, Park JH, Jang Y, Lee EJ, et al. PPARgamma activation induces CD36 expression and stimulates foam cell like changes in rVSMCs. Prostaglandins Other Lipid Mediat. (2006) 80:165–74. doi: 10.1016/j.prostaglandins.2006.06.006
98. Szatmari I, Gogolak P, Im JS, Dezso B, Rajnavolgyi E, Nagy L. Activation of PPARgamma specifies a dendritic cell subtype capable of enhanced induction of iNKT cell expansion. Immunity. (2004) 21:95–106. doi: 10.1016/j.immuni.2004.06.003
99. Jiang M, Karsenberg R, Bianchi F, van den Bogaart G. CD36 as a double-edged sword in cancer. Immunol Lett. (2023) 265:7–15. doi: 10.1016/j.imlet.2023.12.002
100. Wang H, Pang J, Zhang S, Yu Q, Chen Y, Wang L, et al. Single-cell and bulk RNA-sequencing analysis to predict the role and clinical value of CD36 in lung squamous cell carcinoma. Heliyon. (2023) 9:e22201. doi: 10.1016/j.heliyon.2023.e22201
101. Borbora SM, Rajmani RS, Balaji KN. PRMT5 epigenetically regulates the E3 ubiquitin ligase ITCH to influence lipid accumulation during mycobacterial infection. PloS Pathog. (2022) 18:e1010095. doi: 10.1371/journal.ppat.1010095
102. Il'in DA, Shkurupy VA. In vitro analysis of the expression of CD11, CD29, CD36, and DC-STAMP molecules during the formation of multinuclear macrophages in BCG-infected mice. Bull Exp Biol Med. (2019) 167:653–5. doi: 10.1007/s10517-019-04591-0
103. Wang N, Yao Y, Qian Y, Qiu D, Cao H, Xiang H, et al. Cargoes of exosomes function as potential biomarkers for Mycobacterium tuberculosis infection. Front Immunol. (2023) 14:1254347. doi: 10.3389/fimmu.2023.1254347
104. Lao W, Kang H, Jin G, Chen L, Chu Y, Sun J, et al. Evaluation of the relationship between MARCO and CD36 single-nucleotide polymorphisms and susceptibility to pulmonary tuberculosis in a Chinese Han population. BMC Infect Dis. (2017) 17:488. doi: 10.1186/s12879-017-2595-2
105. Draude G, Lorenz RL. TGF-beta1 downregulates CD36 and scavenger receptor A but upregulates LOX-1 in human macrophages. Am J Physiol Heart Circ Physiol. (2000) 278:H1042–8. doi: 10.1152/ajpheart.2000.278.4.H1042
106. Toossi Z, Gogate P, Shiratsuchi H, Young T, Ellner JJ. Enhanced production of TGF-beta by blood monocytes from patients with active tuberculosis and presence of TGF-beta in tuberculous granulomatous lung lesions. J Immunol. (1995) 154:465–73. doi: 10.4049/jimmunol.154.1.465
107. Sánchez MD, García Y, Montes C, París SC, Rojas M, Barrera LF, et al. Functional and phenotypic changes in monocytes from patients with tuberculosis are reversed with treatment. Microbes Infect. (2006) 8:2492–500. doi: 10.1016/j.micinf.2006.06.005
108. Zhang M, Xie Y, Li S, Ye X, Jiang Y, Tang L, et al. Proteomics analysis of exosomes from patients with active tuberculosis reveals infection profiles and potential biomarkers. Front Microbiol. (2022) 12:800807. doi: 10.3389/fmicb.2021.800807
Keywords: CD36, Mycobacterium tuberculosis, inflammatory response, immune response, lipid metabolism, biomarker
Citation: Wang J, Cao H, Yang H, Wang N, Weng Y and Luo H (2024) The function of CD36 in Mycobacterium tuberculosis infection. Front. Immunol. 15:1413947. doi: 10.3389/fimmu.2024.1413947
Received: 08 April 2024; Accepted: 20 May 2024;
Published: 31 May 2024.
Edited by:
Veronica Schmitz, Oswaldo Cruz Foundation (Fiocruz), BrazilReviewed by:
Debora Decote-Ricardo, Federal Rural University of Rio de Janeiro, BrazilSahil Mahajan, The Ohio State University, United States
Copyright © 2024 Wang, Cao, Yang, Wang, Weng and Luo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hao Luo, MTAxODkzNzYyOEBxcS5jb20=
†These authors have contributed equally to this work