 Jingyuan Zhang
Jingyuan Zhang Yuanbo Kang
Yuanbo Kang Zenan Xia2
Zenan Xia2 Yuming Chong
Yuming Chong Xiao Long
Xiao Long Min Shen
Min Shen- 1Department of Rare Diseases, Peking Union Medical College Hospital (PUMCH), Chinese Academy of Medical Sciences & Peking Union Medical College; State Key Laboratory of Complex Severe and Rare Diseases, PUMCH; Department of Rheumatology and Clinical Immunology, PUMCH; National Clinical Research Center for Dermatologic and Immunologic Diseases (NCRC-DID), Ministry of Science & Technology; Key Laboratory of Rheumatology and Clinical Immunology, Ministry of Education, Beijing, China
- 2Department of Plastic Surgery, Peking Union Medical College Hospital, Chinese Academy of Medical Sciences, Beijing, China
Macrophage activation syndrome (MAS), is a severe and fatal complication of various pediatric inflammatory disorders. Kabuki syndrome (KS), mainly caused by lysine methyltransferase 2D (KMT2D; OMIM 602113) variants, is a rare congenital disorder with multi-organ deficiencies. To date, there have been no reported cases of MAS in patients with KS. This report describes a case of a 22-year-old male with Kabuki syndrome (KS) who developed MAS. This unique case not only deepens the understanding of the involvement of KMT2D in immune regulation and disease, but expands the phenotype of the adult patient to better understand the natural history, disease burden, and management of patients with KS complicated with autoimmune disorders.
1 Introduction
Macrophage activation syndrome (MAS) is a lethal complication of various inflammatory disorders and is often associated with primary immunodeficiencies (PIDs) (1). Kabuki syndrome (KS) is a rare congenital disorder hallmarked by dysmorphic facial features (including arched eyebrows, long palpebral fissures with eversion of the lower eyelid, and large protuberant or cupped ears), intellectual disability, growth retardation, and other structural and functional defects. Genetic anomalies in lysine methyltransferase 2D (KMT2D) and lysine demethylase 6A (KDM6A) are implicated in KS. Interestingly, although patients with KS can also exhibit impaired adaptive immunity with significant hypogammaglobulinemia and display autoimmune disorders (2, 3), there are no reported cases of MAS in patients with KS. Here, we present the first report of a KS patient complicated with MAS, indicating a potential association between de novo pathogenic variants of KMT2D and MAS.
2 Case description
A 22-year-old Chinese Han man, clinically diagnosed with common variable immunodeficiency (CVID), was referred to the department of rheumatology and immunology in our hospital due to MAS relapse after medication withdrawal. The patient was susceptible to inspiratory infections during childhood. At the first unprovoked onset of 13, he developed a fever, recurrent respiratory infections, arthritis, and rash. Notably increased ferritin (>2000ng/mL), aspartate aminotransferase (AST) (1930U/L), C-reactive protein (CRP) (66.34mg/L), and erythrocyte sedimentation rate (ESR) (66mm/h) were observed. He was initially diagnosed with systemic juvenile idiopathic arthritis and suspected MAS in the local hospital and treated with methylprednisolone, cyclosporin A, and hydroxychloroquine sulfate. The symptoms improved significantly, with ferritin (288.9ng/mL) and ESR (4mm/h) significantly decreasing. However, pneumocystis pneumonia occurred during steroids, combined with hypogammaglobulinemia in the context, that the patient was suspected of CVID. He began regular intravenous immunoglobulin (IVIG) therapy every three weeks while continuing methylprednisolone, cyclosporine A, and hydroxychloroquine sulfate. He remained stable and was followed up at the local hospital.
At 15, He visited the Department of Pediatrics of our hospital for further evaluation and treatment adjustment. He was considered CVID as consistently low serum IgG ranging from 3.89-4.59g/L. He continued IVIG, methylprednisolone, cyclosporin, and hydroxychloroquine sulfate, and his IgG levels were maintained at 7-8 g/L. During the following 2 years, he remained stable without fever, and regular follow-ups were mainly performed in the local hospital. He gradually discontinued methylprednisolone, cyclosporin, and hydroxychloroquine sulfate in 1.5 years.
Since the age of 17, the patient has been admitted to the local hospital every 2 years for recurrent high fever, erythema of the trunk and limbs (Figure 1A), and arthritis, while maintaining regular IVIG every 3-5 weeks as maintenance treatment. Laboratory data showed several elevated ferritin (>1650ng/mL), increased liver enzyme and decreased platelet (PLT) (<100*109/L). Temporary relief was achieved and ferritin returned to normal through pulse therapy of methylprednisolone (500mg/d*3days) and gradual tapering of methylprednisolone plus cyclosporin or tacrolimus over 1-1.5 years, but symptoms recurred six months after withdrawal.
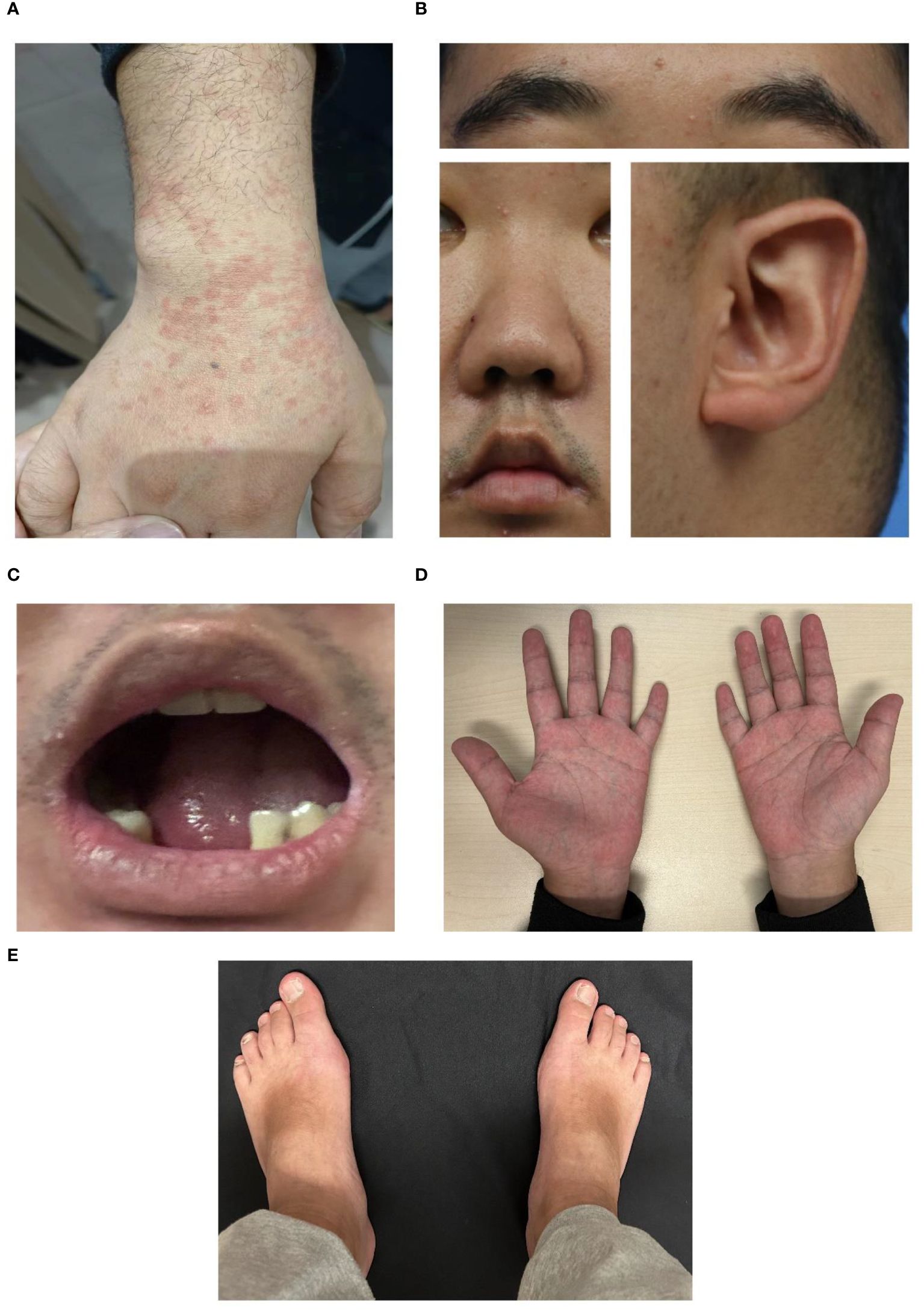
Figure 1 Representative images of the patient with Kabuki syndrome. (A) Erythema during a disease flare. (B) Representative facial images of the patient. An arched eyebrow, epicanthus, short columella with a slightly narrow nose, thick and narrow lips, and prominent cupped ears were observed in the lateral view. (C) Dental anomalies. (D, E) brachydactyly. (D) Short hands with stubby fingers and dustpan-line finger creases, and short fifth digits were displayed.
As recurrent MAS relapse after medication withdrawal, he was transferred to our hospital for further diagnosis at age 22. Laboratory data during this MAS relapse revealed thrombocytopenia (34*109/L, < diagnostic criteria 181*109/L), markedly increased ferritin (37907ng/mL, > diagnostic criteria 684ng/mL), and (AST) (1117U/L, > diagnostic criteria 48U/L). Notably increased Inflammatory indicators ESR (18mm/h, reference value 0-15mm/h) and CRP (31.83mg/L, reference value < 8mg/L) were also observed. Immunologic testing showed decreased serum IgA (0.41g/L, reference value 0.7-4.0g/L), B lymphocytes (21/μL, reference value 180-324/μL), NK cells (77/μL, reference value 175-567/μL), while IgG was normal (15.91g/L, reference value 7-17g/L). The laboratory data of this MAS episode are shown in Figure 2. Surprisingly, we found elevated CD8+T (1823/μL, reference value 404-754/μL), CD8+CD28+ (784/μL, reference value 190-392/μL), CD8+DR+ (1416/μL, reference value 20-178/μL), and CD8+CD38+ cells (1794/μL, reference value 157-385/μL). An abdomen routine scan showed splenomegaly. Tests for autoimmune indicators and viral infections were normal.
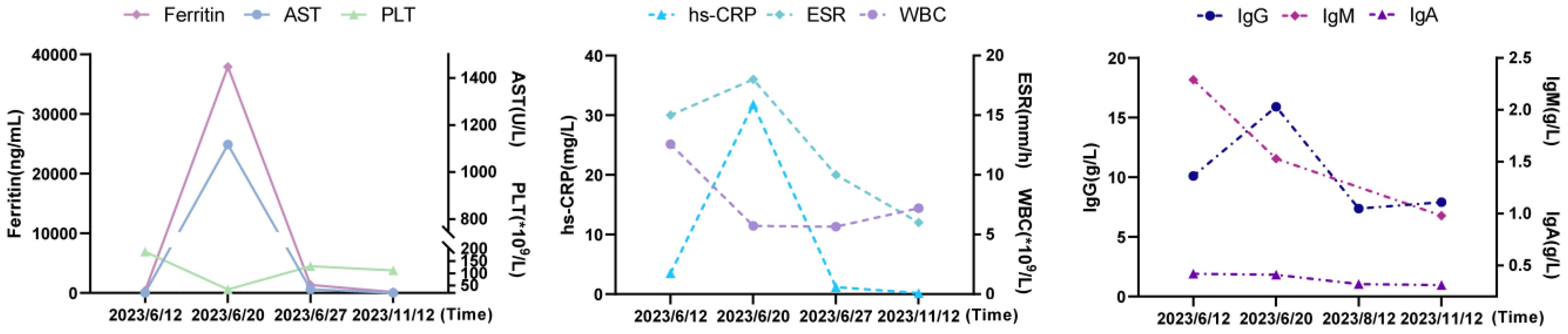
Figure 2 The laboratory data of MAS episode and after-treatment. AST, aspartate aminotransferase; PLT, platelet; hs-CRP, hypersensitive C-reactive protein; ESR, erythrocyte sedimentation rate; WBC, white blood cell; Ig, immunoglobulin.
To ascertain the cause of the recurrent fever, trio-whole exome sequencing (WES) was performed and identified a pathogenic “de novo” (not present in parents and siblings) missense mutation c.15535C>T, p.Arg5179Cys in the KMT2D gene (NM_003482.3, OMIM 602113) (4, 5). Additional questioning revealed that he had hypothyroidism and learning difficulties, with a family history of hypothyroidism but no autoinflammatory diseases, PID, or hemophagocytic lymphohistiocytosis (HLH). Physical examination revealed distinctive features, including arched and broad eyebrows, epicanthus, a shortened and slightly narrow nose, thick and narrow lips, large prominent cupped ears, dental abnormalities, and brachydactyly (Figures 1B-E). Facial quantitative analysis, compared to a 20-29-year-old Northern Chinese population, also revealed a classical Kabuki syndrome (KS) facial appearance characterized by a slightly shorter nose, flat nasal bridge, round and blunt nasal tip, and a small, thick mouth (see Table 1). The patient was ultimately diagnosed as KS with MAS based on diagnostic criteria (2, 6, 7).
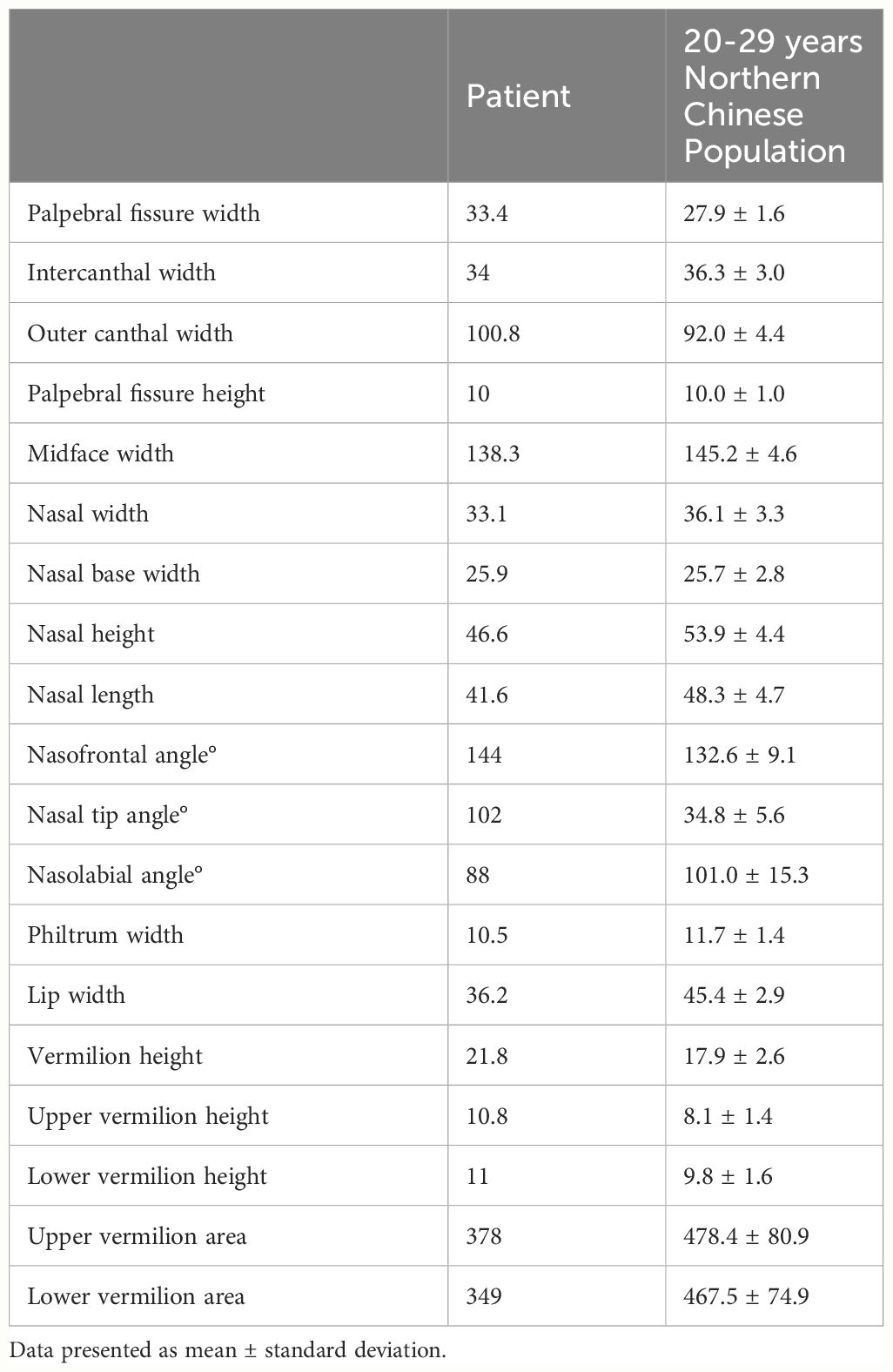
Table 1 Facial quantitative analysis results of the patient.
He responded well to methylprednisolone pulse therapy (500mg/d*3days), tacrolimus 1mg twice daily, and hydroxychloroquine 1mg twice daily with significant symptoms and inflammation indicators improvement (see laboratory data from 27th, June 2023 in Table 2). He remained stable without fever while receiving gradual tapered of methylprednisolone, tacrolimus 1mg twice daily, and hydroxychloroquine sulfate 1mg twice daily, alongside regular IVIG with 25-30mg every 3-5weeks, despite low IgG (7.38g/L, reference value 8-18g/L) and IgA (0.31-0.32g/L, reference value 0.7-4.0g/L). Routine blood examination, liver and kidney function, and inflammatory indicators remained normal during the next few months (see laboratory data from 12th, August and 12th, November 2023 in Table 2). Figure 3 provides a timeline detailing the development of the disease and the treatments received.
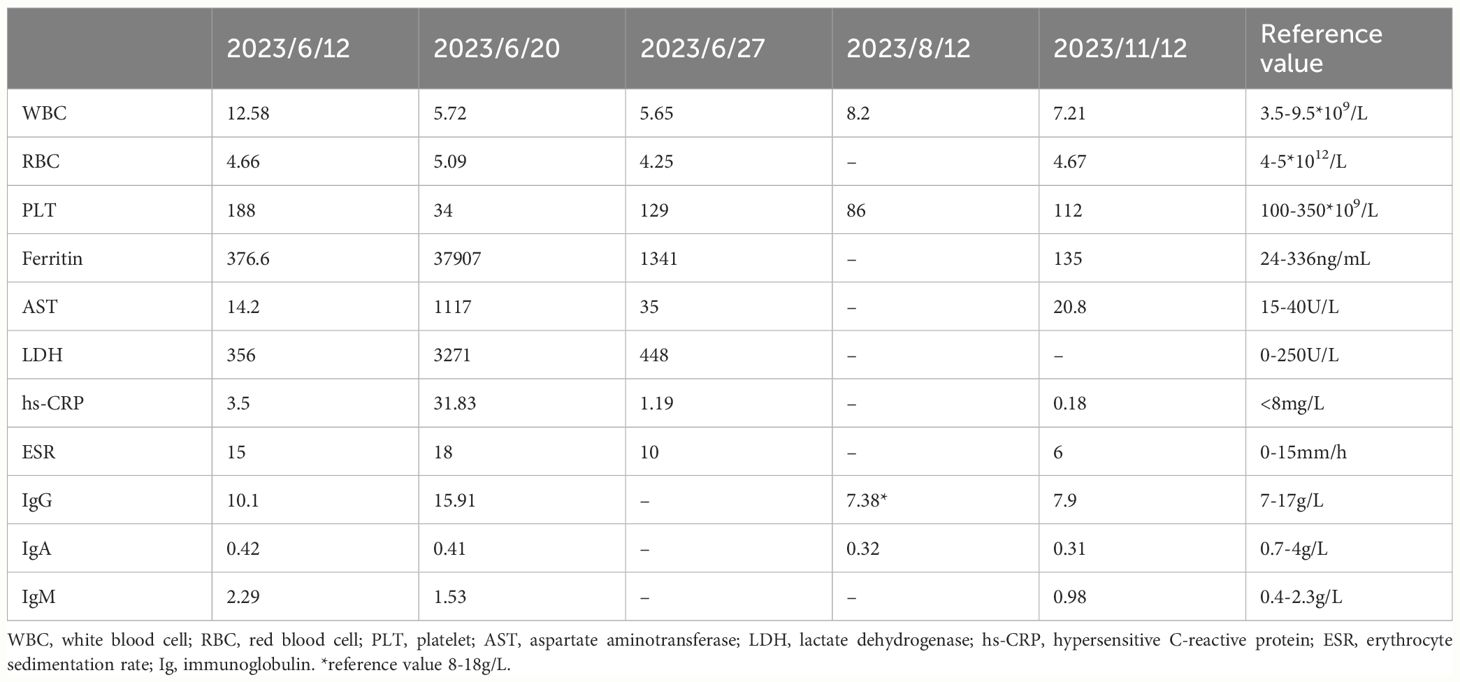
Table 2 Latest follow-up Laboratory data of the patient.
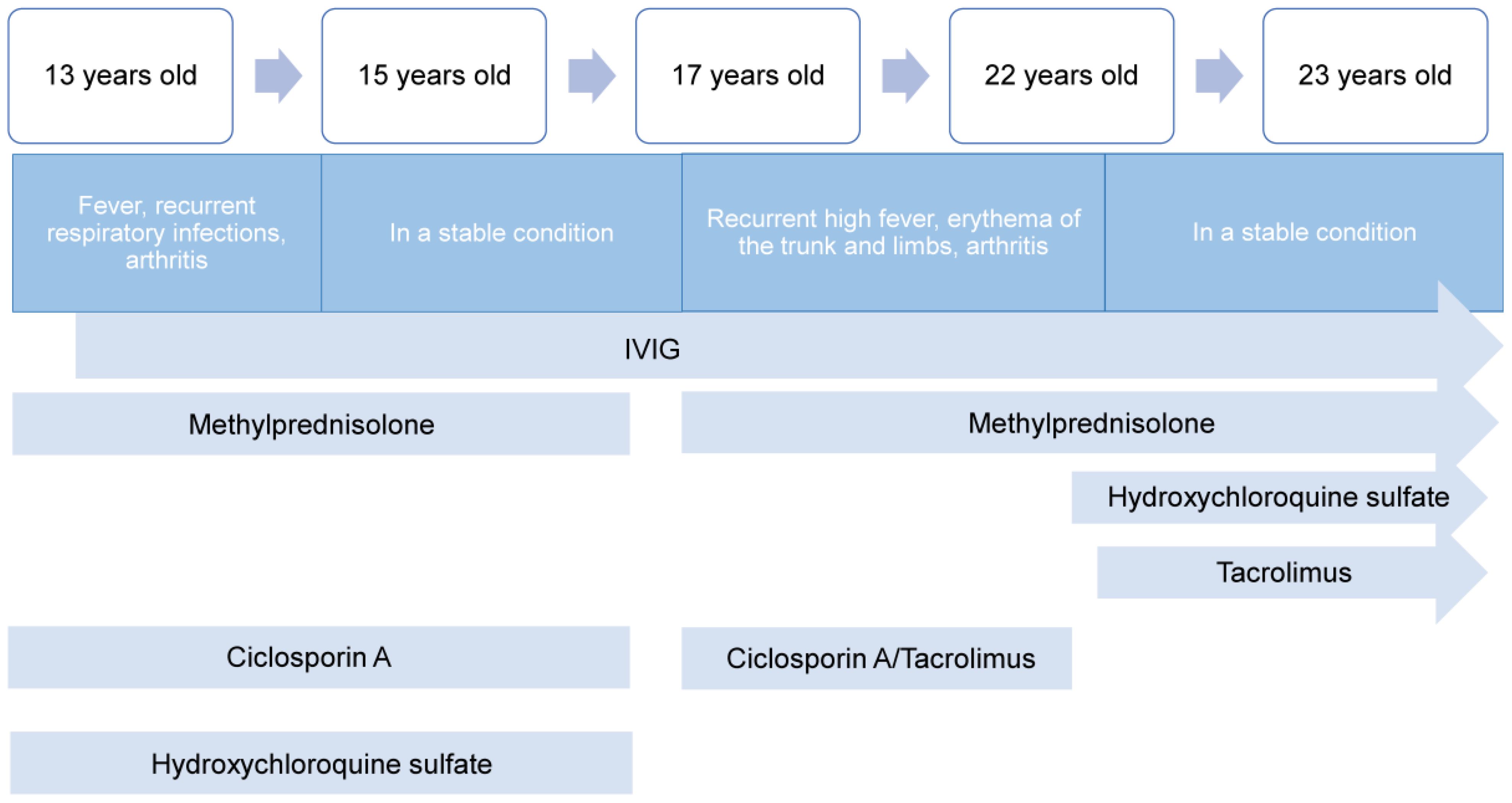
Figure 3 A timeline showing the development of the disease and the treatments received.
3 Discussion
Kabuki syndrome is a rare human inborn error of immunity (IEI) involving multi-systems with an estimated frequency ranging from 1:32,000 to 1:86,000 (8, 9) across different ethnicities (10–13). International consensus diagnostic criteria mainly depend on its typical dysmorphic features and pathogenic/likely pathogenic KMT2D or KDM6A variants. However, facial features of KS, most distinctive between ages 3 and 12, become challenging to identify in infants, adolescents, and adults (2, 14). To address this diagnostic challenge, we utilized facial quantitative analysis to enhance the recognition of facial features in older patients and anticipate future applications of this technology for newborn screening and adjunct diagnosis in adults presenting with symptoms like growth retardation, immune deficiency, and recurrent MAS. Our anthropometric data is limited to the northern Chinese population and may have limited reference value for other populations. Multicenter studies on different populations are warranted to promote the application of this model in patients with KS.
The pathogenesis of MAS is closely linked to persistent Epstein-Barr virus (EBV), and cytomegalovirus (CMV) infection, which trigger abnormal immune stimulation, T-cell activation, and excessive cytokine secretion (1). KMT2D plays a key role in terminal B cell differentiation, including somatic hypermutation and class-switch recombination (15, 16). It also epigenetically regulates the gene ITGB7, which mediates leukocyte migration to inflamed and non-inflamed regions to provide immune responses (6). KMT2D pathogenic variants lead to persistent decreased IgA and IgG levels, NK cells, reduced memory (CD27+), and class-switched memory B cells (IgM−) with a predisposition to recurrent infections (15, 17, 18). These immunodeficiencies potentially elevate the risk of developing MAS (19). Interestingly, despite normal counts and distributions of T cells and NK cells in most patients with KS (3), our patient study observed an unusual elevation of CD8+T, CD8+CD28+, CD8+DR+, and CD8+CD38+. CD8+T has been previously reported to be involved in the pathogenesis of MAS (20). Moreover, KMT2D is critical for mediating the survival of activation-induced naïve CD8+T cells (21), and KMT2D variants in colorectal cancer have been linked to increased CD8+T cells (22), more immune cell infiltration, and enrichment of immune-related genes and pathways (23). These findings indicated a potential link between KMT2D mutations and T-cell dysfunction, leading to MAS, which may be validated in cohort studies using more functional experiments.
KS is also closely associated with various autoimmune disorders (14, 24–26), which may increase in prevalence with age (18). In line with these findings, our patient exhibited significant elevation in anti-thyroid peroxidase antibodies (TPO) antibody, s-thyroid stimulating hormone (TSH), alongside decreased free tetraiodothyronine recently. KMT2D encodes for proteins acting in the COMPASS complex, involved in the epigenetic regulation of FOXP3 (27). FOXP3 is crucial to the differentiation of naïve CD4+ T cells into T-regulatory cells, which contribute to the maintenance of peripheral tolerance (3). Therefore, pathogenic KMT2D variants impair the generation, development, and differentiation of regulatory T cells (3), leading to a breakdown of T-cell tolerance, possibly explaining consequent MAS. Further investigation should focus on T cell subsets in a larger cohort of patients with KS to better elucidate the role of KMT2D in the regulation of development and pathogenesis of MAS.
Our patients achieved satisfactory disease control with combination therapy of corticosteroid, tacrolimus, and hydroxychloroquine sulfate. Tacrolimus, known for inhibiting T-cell activation in organ transplants (28), supports the role of T-cell activation in KS with concomitant MAS. Early recognition and initiation of potent immunosuppressive therapy can reduce the severity of organ complications. Despite regular intravenous immunoglobulin administration, our patient continued to exhibit low IgA levels, underscoring the need for vigilant monitoring for potential IgA antibody formation (29, 30). Regular testing of immunoglobulin levels is recommended for patients with KS to ensure effective management of the disease.
4 Conclusions
This report describes the first case of a patient with KS concurrent with MAS. This unique case provides a clue for the association between KMT2D and MAS and the roles of KTM2D in immune regulation and disease. It also expands the phenotype of the adult patient to better understand the natural history, disease burden, and management of patients with KS complicated with autoimmune disorders.
5 Patient perspective
The patient and his mother were fully engaged throughout the treatment and told us that his symptoms improved significantly after treatment and remained stable recently. The patient consented to the publication of this case report and written informed consent was obtained.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding authors.
Ethics statement
The studies involving humans were approved by Peking Union Medical College Hospital. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article. Written informed consent was obtained from the participant/patient(s) for the publication of this case report.
Author contributions
JYZ: Investigation, Writing – original draft. YBK: Investigation, Writing – original draft. ZNX: Investigation, Writing – review & editing. YMC: Investigation, Writing – review & editing. XL: Supervision, Writing – review & editing, Conceptualization, Investigation. MS: Funding acquisition, Supervision, Writing – review & editing, Conceptualization, Investigation.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the National High Level Hospital Clinical Research Funding (grant number 2022-PUMCH-D-002, 2022-PUMCH-B-013).
Acknowledgments
We would like to acknowledge the patient for his consent to participate in the study.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Henderson LA, Cron RQ. Macrophage activation syndrome and secondary hemophagocytic lymphohistiocytosis in childhood inflammatory disorders: diagnosis and management. Pediatr Drugs. (2020) 22:29–44. doi: 10.1007/s40272-019-00367-1
2. Adam MP, Banka S, Bjornsson HT, Bodamer O, Chudley AE, Harris J, et al. Kabuki syndrome: international consensus diagnostic criteria. J Med Genet. (2019) 56:89–95. doi: 10.1136/jmedgenet-2018-105625
3. Stagi S, Gulino AV, Lapi E, Rigante D. Epigenetic control of the immune system: a lesson from Kabuki syndrome. Immunol Res. (2016) 64:345–59. doi: 10.1007/s12026-015-8707-4
4. Dentici ML, Di Pede A, Lepri FR, Gnazzo M, Lombardi MH, Auriti C, et al. Kabuki syndrome: clinical and molecular diagnosis in the first year of life. Arch Dis Child. (2015) 100:158–64. doi: 10.1136/archdischild-2013-305858
5. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
6. Boniel S, Szymanska K, Smigiel R, Szczaluba K. Kabuki syndrome-clinical review with molecular aspects. Genes (Basel). (2021) 12:468. doi: 10.3390/genes12040468
7. Ravelli A, Minoia F, Davi S, Horne A, Bovis F, Pistorio A, et al. 2016 Classification criteria for macrophage activation syndrome complicating systemic juvenile idiopathic arthritis: A European league against rheumatism/American college of rheumatology/Pediatric rheumatology international trials organization collaborative initiative. Arthritis Rheumatol. (2016) 68:566–76. doi: 10.1002/art.39332
8. White SM, Thompson EM, Kidd A, Savarirayan R, Turner A, Amor D, et al. Growth, behavior, and clinical findings in 27 patients with Kabuki (Niikawa-Kuroki) syndrome. Am J Med Genet A. (2004) 127A:118–27. doi: 10.1002/ajmg.a.20674
9. Niikawa N, Kuroki Y, Kajii T, Matsuura N, Ishikiriyama S, Tonoki H, et al. Kabuki make-up (Niikawa-Kuroki) syndrome: a study of 62 patients. Am J Med Genet. (1988) 31:565–89. doi: 10.1002/ajmg.1320310312
10. So PL, Luk HM, Yu KPT, Cheng SSW, Hau EWL, Ho SKL, et al. Clinical and molecular characterization study of Chinese Kabuki syndrome in Hong Kong. Am J Med Genet A. (2021) 185:675–86. doi: 10.1002/ajmg.a.62003
11. Bogershausen N, Gatinois V, Riehmer V, Kayserili H, Becker J, Thoenes M, et al. Mutation update for kabuki syndrome genes KMT2D and KDM6A and further delineation of X-linked Kabuki syndrome subtype 2. Hum Mutat. (2016) 37:847–64. doi: 10.1002/humu.2016.37.issue-9
12. Adam MP, Hudgins L. Kabuki syndrome: a review. Clin Genet. (2005) 67:209–19. doi: 10.1111/j.1399-0004.2004.00348.x
13. Liu S, Hong X, Shen C, Shi Q, Wang J, Xiong F, et al. Kabuki syndrome: a Chinese case series and systematic review of the spectrum of mutations. BMC Med Genet. (2015) 16:26. doi: 10.1186/s12881-015-0171-4
14. Barry KK, Tsaparlis M, Hoffman D, Hartman D, Adam MP, Hung C, et al. From genotype to phenotype-A review of Kabuki syndrome. Genes (Basel). (2022) 13:1761. doi: 10.3390/genes13101761
15. Lindsley AW, Saal HM, Burrow TA, Hopkin RJ, Shchelochkov O, Khandelwal P, et al. Defects of B-cell terminal differentiation in patients with type-1 Kabuki syndrome. J Allergy Clin Immunol. (2016) 137:179–87.e110. doi: 10.1016/j.jaci.2015.06.002
16. Borchert GM, Holton NW, Edwards KA, Vogel LA, Larson ED. Histone H2A and H2B are monoubiquitinated at AID-targeted loci. PLoS One. (2010) 5:e11641. doi: 10.1371/journal.pone.0011641
17. Lin JL, Lee WI, Huang JL, Chen PK, Chan KC, Lo LJ, et al. Immunologic assessment and KMT2D mutation detection in Kabuki syndrome. Clin Genet. (2015) 88:255–60. doi: 10.1111/cge.12484
18. Margot H, Boursier G, Duflos C, Sanchez E, Amiel J, Andrau JC, et al. Immunopathological manifestations in Kabuki syndrome: a registry study of 177 individuals. Genet Med. (2020) 22:181–8. doi: 10.1038/s41436-019-0623-x
19. Warnatz K, Voll RE. Pathogenesis of autoimmunity in common variable immunodeficiency. Front Immunol. (2012) 3:210. doi: 10.3389/fimmu.2012.00210
20. De Matteis A, Colucci M, Rossi MN, Caiello I, Merli P, Tumino N, et al. Expansion of CD4dimCD8+ T cells characterizes macrophage activation syndrome and other secondary HLH. Blood. (2022) 140:262–73. doi: 10.1182/blood.2021013549
21. Kim J, Nguyen T, Cifello J, Ahmad R, Zhang Y, Yang Q, et al. Lysine methyltransferase Kmt2d regulates naive CD8(+) T cell activation-induced survival. Front Immunol. (2022) 13:1095140. doi: 10.3389/fimmu.2022.1095140
22. Liu R, Niu Y, Liu C, Zhang X, Zhang J, Shi M, et al. Association of KMT2C/D loss-of-function variants with response to immune checkpoint blockades in colorectal cancer. Cancer Sci. (2023) 114:1229–39. doi: 10.1111/cas.15716
23. Liu C, Jin Y, Zhang H, Yan J, Guo Y, Bao X, et al. Effects of KMT2D mutation and its exon 39 mutation on the immune microenvironment and drug sensitivity in colorectal adenocarcinoma. Heliyon. (2023) 9:e13629. doi: 10.1016/j.heliyon.2023.e13629
24. Gurbuz F, Ozalp Yuregir O, Ceylaner S, Topaloglu AK, Yuksel B. Coexistence of Kabuki syndrome and autoimmune thyroiditis. J Clin Res Pediatr Endocrinol. (2016) 8:105–6. doi: 10.4274/jcrpe
25. Ming JE, Russell KL, McDonald-McGinn DM, Zackai EH. Autoimmune disorders in Kabuki syndrome. Am J Med Genet A. (2005) 132A:260–2. doi: 10.1002/ajmg.a.30332
26. Fujishiro M, Ogihara T, Tsukuda K, Shojima N, Fukushima Y, Kimura S, et al. A case showing an association between type 1 diabetes mellitus and Kabuki syndrome. Diabetes Res Clin Pract. (2003) 60:25–31. doi: 10.1016/S0168-8227(02)00276-0
27. Wei G, Wei L, Zhu J, Zang C, Hu-Li J, Yao Z, et al. Global mapping of H3K4me3 and H3K27me3 reveals specificity and plasticity in lineage fate determination of differentiating CD4+ T cells. Immunity. (2009) 30:155–67. doi: 10.1016/j.immuni.2008.12.009
28. Penninga L, Penninga EI, Moller CH, Iversen M, Steinbruchel DA, Gluud C. Tacrolimus versus cyclosporin as primary immunosuppression for lung transplant recipients. Cochrane Database Syst Rev. (2013), CD008817. doi: 10.1002/14651858.CD008817.pub2
29. Hoffman JD, Ciprero KL, Sullivan KE, Kaplan PB, McDonald-McGinn DM, Zackai EH, et al. Immune abnormalities are a frequent manifestation of Kabuki syndrome. Am J Med Genet A. (2005) 135:278–81. doi: 10.1002/ajmg.a.30722
Keywords: KMT2D, Kabuki syndrome, macrophage activation syndrome, immunodeficiency, T cells
Citation: Zhang J, Kang Y, Xia Z, Chong Y, Long X and Shen M (2024) Case report: Macrophage activation syndrome in a patient with Kabuki syndrome. Front. Immunol. 15:1412084. doi: 10.3389/fimmu.2024.1412084
Received: 04 April 2024; Accepted: 16 July 2024;
Published: 30 July 2024.
Edited by:
Austen Worth, Great Ormond Street Hospital for Children NHS Foundation Trust, United KingdomReviewed by:
Ancuta Lupu, Grigore T. Popa University of Medicine and Pharmacy, RomaniaVictoria R. Dimitriades, University of California, Davis, United States
Copyright © 2024 Zhang, Kang, Xia, Chong, Long and Shen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Min Shen, c2hlbm1wdW1jaEAxNjMuY29t; Xiao Long, cHVtY2xvbmd4aWFvQDEyNi5jb20=
†These authors have contributed equally to this work